
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide hydrogen-bonded organic frameworks†
Thangavel
Vijayakanth
 *a,
Sneha
Dasgupta
b,
Pragati
Ganatra
c,
Sigal
Rencus-Lazar
a,
Aamod V.
Desai
d,
Shyamapada
Nandi
e,
Rahul
Jain
b,
Santu
Bera
f,
Andy I.
Nguyen
*c,
Ehud
Gazit
*ag and
Rajkumar
Misra
*b
*a,
Sneha
Dasgupta
b,
Pragati
Ganatra
c,
Sigal
Rencus-Lazar
a,
Aamod V.
Desai
d,
Shyamapada
Nandi
e,
Rahul
Jain
b,
Santu
Bera
f,
Andy I.
Nguyen
*c,
Ehud
Gazit
*ag and
Rajkumar
Misra
*b
aShmunis School of Biomedicine and Cancer Research, George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv–6997801, Israel. E-mail: vijayakantht@mail.tau.ac.il; ehudg@post.tau.ac.il
bDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Mohali, S.A.S. Nagar (Mohali) 160062, India. E-mail: rkmisra@niper.ac.in
cDepartment of Chemistry, University of Illinois Chicago, Chicago, Illinois 60607, USA. E-mail: andyn@uic.edu
dSchool of Chemistry, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK
eChemistry Division, School of Advanced Sciences, Vellore Institute of Technology, 600127, Chennai, India
fDepartment of Chemistry, Ashoka University, Sonipat, Haryana 131029, India
gSagol School of Neuroscience, Tel Aviv University, 6997801 Tel Aviv, Israel
First published on 7th March 2024
Abstract
Hydrogen-bonded porous frameworks (HPFs) are versatile porous crystalline frameworks with diverse applications. However, designing chiral assemblies or biocompatible materials poses significant challenges. Peptide-based hydrogen-bonded porous frameworks (P-HPFs) are an exciting alternative to conventional HPFs due to their intrinsic chirality, tunability, biocompatibility, and structural diversity. Flexible, ultra-short peptide-based P-HPFs (composed of 3 or fewer amino acids) exhibit adaptable porous topologies that can accommodate a variety of guest molecules and capture hazardous greenhouse gases. Longer, folded peptides present challenges and opportunities in designing P-HPFs. This review highlights recent developments in P-HPFs using ultra-short peptides, folded peptides, and foldamers, showcasing their utility for gas storage, chiral recognition, chiral separation, and medical applications. It also addresses design challenges and future directions in the field.
 Thangavel Vijayakanth | Thangavel Vijayakanth received an MSc from Bharathidasan University, India, and a PhD in chemistry from the Indian Institute of Science Education and Research, Pune, India, in 2020 with Prof. Ramamoorthy Boomishankar. His PhD dissertation was titled “Organo and amino phosphonium cation-derived ferro- and piezo-electric materials and their utility in mechanical energy harvesting applications”. He subsequently joined Tel-Aviv University, Israel, in 2021 for his postdoctoral studies and is currently working with Prof. Ehud Gazit. His current research interests encompass bio-molecular self-assembly, metal–peptide assemblies, porous solids, and glassy peptides, with a focus on applications including catalysis, molecular recognition, energy, and biomedicine. |
 Andy I. Nguyen | Andy Nguyen is an assistant professor at the University of Illinois Chicago. He obtained his BS in chemistry from UC Irvine in 2010 and PhD in inorganic chemistry from UC Berkeley in 2016. While at UCI, he explored redox-active complexes of early transition metals with Alan Heyduk. At UC Berkeley, with T. Don Tilley, he worked on the chemistry of artificial photosynthesis, where he developed cobalt metalloclusters and studied their mechanism of catalytic oxygen evolution. In a postdoctoral fellowship at Lawrence Berkeley National Laboratory with Ron Zuckermann, he gained expertise in peptide and peptidomimetic chemistry. Since 2020, his research group has been involved in integrating peptide design with inorganic chemistry to tackle challenges involving metalloenzyme mimicry, energy-conversion catalysis, and materials design. |
 Ehud Gazit | Ehud Gazit is a Professor and Endowed Chair at the Shmunis School of Biomedicine and Cancer Research and the Department of Materials Science and Engineering at Tel Aviv University. He received his BSc (summa cum laude) after studying at the Special Program for Outstanding Students at Tel-Aviv University and his PhD (highest distinction) from the Weizmann Institute of Science. He has been a faculty member at Tel Aviv University since 2000, following the completion of his post-doctoral studies at Massachusetts Institute of Technology, where he also had held a visiting appointment. He also had visiting appointments at Cambridge University, Fudan University, and Umeå University. He was recently appointed as the 2023 International Solvay Chair in Chemistry. His research interests involve exploring biological and bio-inspired molecular self-assembly. |
 Rajkumar Misra | Rajkumar Misra obtained his PhD degree in 2018 from the Indian Institute of Science Education and Research, Pune. Subsequently, he joined Prof. Darrin Pochan's research group at the University of Delaware as a postdoctoral fellow. After finishing the tenure, he joined Dr. Lihi Adler-Abramovich's research group under the PBC scholarship program for outstanding post-doctoral students at Tel-Aviv University. He is currently an Inspire Faculty Fellow at the National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar. His research interests are the exploration of supramolecular assembly of bioinspired building blocks, artificial peptides, foldamers, and metal–peptide frameworks (MPFs) for catalysis, bio-functional, and advanced medical applications. |
Key learning points(1) Peptide structure and design principles.(2) Peptidomimetics and foldamers. (3) Peptide hierarchical assembly to form functional architectures. (4) Design of hydrogen-bonded crystalline porous frameworks. (5) Applications of porous frameworks. |
1. Introduction
Porous framework materials are crystalline lattices possessing large voids or channels capable of hosting guest molecules. They have very high surface areas and can be used for a number of applications including sensing, gas separation, gas storage, molecular sieving, catalysis, ion-exchange, water-treatment, drug delivery, tissue engineering, capacitors, fuel cells, and batteries.1–5 Zeolites, the most well-known and used porous framework, are utilized in many industrial catalytic and separation processes, especially in the petrochemical sector. Metal–organic and covalent-organic frameworks (MOFs and COFs, respectively) increase the complexity of porous materials due to the introduction of tunable organic linkers.6–8 The augmented complexity of MOFs and COFs relative to zeolites enables structural flexibility, fine-tuning of shape-specific interactions, and access to new topologies. While MOFs and COFs utilize strong bonds for assembly, hydrogen-bonded porous frameworks (HPFs), also known as hydrogen-bonded organic frameworks (HOFs), rely on non-covalent hydrogen bonding interactions to produce porous lattices.9–11 HPFs benefit from the absence of metal nodes, reversibility of hydrogen bond formation, good thermal stability, high surface area, and ease of solution processability.12–16 While crystalline hydrogen-bonded networks have been known since the late 1960s, their realization as porous materials emerged only in 2010.17,18 Since then, there has been considerable research to explore new HPFs that can serve as sophisticated and multifunctional materials with superior properties. Despite significant progress, conventional HPFs are generally achiral, lack biocompatibility, and use hard-to-synthesize building blocks.19,20 Moreover, these structures are often composed of rigid aromatic compounds that are highly hydrophobic (Scheme 1).21–24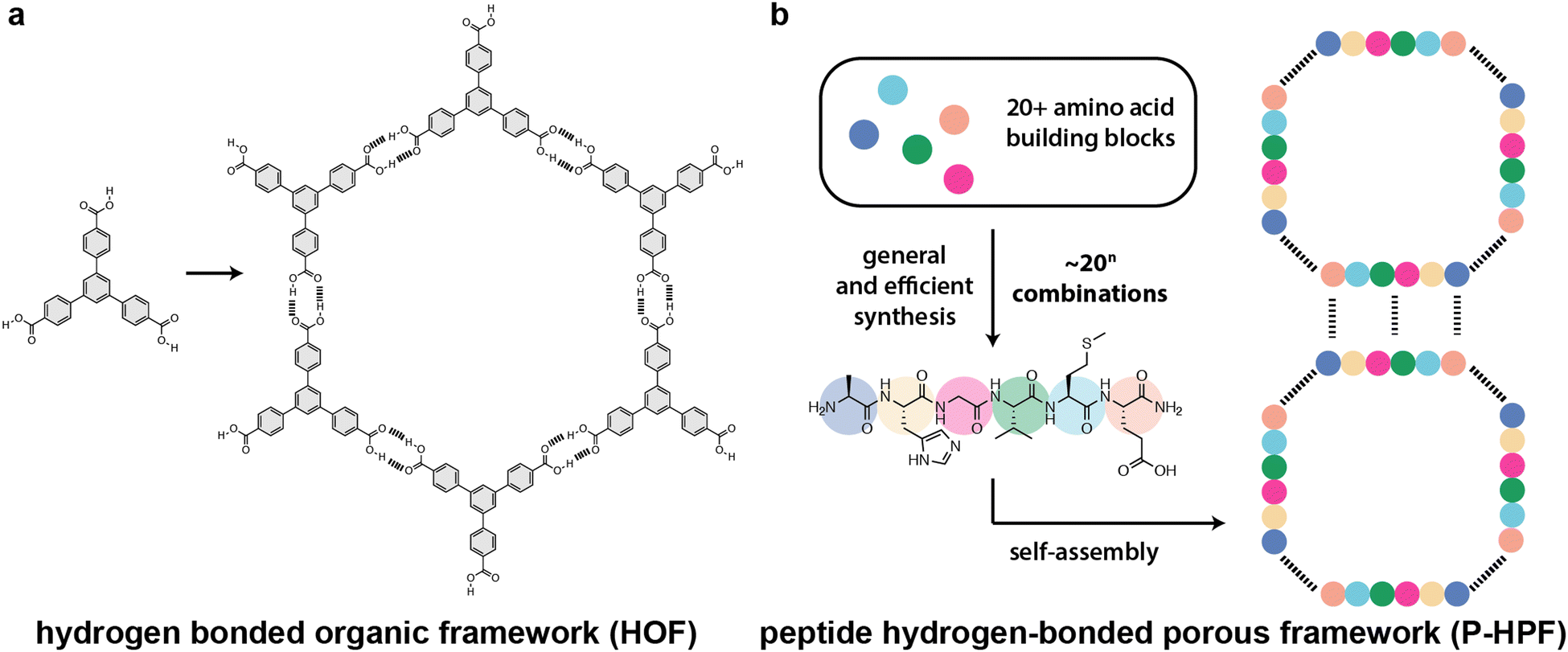 | ||
| Scheme 1 Comparison of (a) hydrogen-bonded frameworks (HOFs or HPFs) and (b) peptide-based hydrogen-bonded porous frameworks (P-HPFs). Hydrogen bonds are shown as dashed lines. | ||
Peptide-based HPFs (P-HPFs) offer significant advantages compared to conventional HPFs, since they are inherently chiral, modular, structurally dynamic, and biocompatible (Scheme 1).25–28 Utilizing a diverse array of amino acids, peptides can be constructed by incorporating both coded and noncoded building blocks. This allows peptides to access a spectrum of chemical properties that can be combined in numerous permutations to achieve diverse functionalities. Here, we will define “peptides” as amino acid sequences fewer than 40–50 residues in length.29–42 Peptides have been mainly explored for application in medicinal chemistry, chemical biology, and soft materials.43–45 The self-assembly of peptides is driven by hydrophobic forces and intermolecular hydrogen bond networks, and is dictated by the amino acid sequence. The diversity and multitude of tunable interactions offer both challenges and opportunities in materials design. Additionally, the ability to incorporate non-natural peptidomimetics (also known as “foldamers”) into a peptide chain offers exciting possibilities for synthetic chemists to develop peptide-based materials. In this tutorial review, we provide a comprehensive overview of crystalline porous frameworks constructed from peptides and foldamers, which only use non-covalent interactions such as hydrogen bonding, π–π stacking, and van der Waals contacts. We place particular emphasis on the importance of design strategies, synthesis, and high-resolution structure characterization. Finally, we highlight the current challenges and promising future directions for peptide- and foldamer-based HPFs in chemistry, materials science, and biotechnology.
2. HPFs derived from ultra-short peptides
2.1 Dipeptides
Ultra-short peptides have drawn significant attention owing to their facile synthesis and reduced conformational flexibility.46–49 Dipeptides are the shortest peptides, and they can self-assemble into well-ordered structures like tubes, spheres, rods, fibers, vesicles, and crystalline materials using non-covalent interactions such as hydrogen bonding, hydrophobic, electrostatic, and van der Waals force contacts.50–57Görbitz and coworkers showed that dipeptides form crystalline porous frameworks with pores of 3–10 Å in diameter (Fig. 1 and Fig. S1, S2, ESI†).25,26 The first family of microporous structures was observed for L-valine–L-alanine (VA) (Fig. 1b and Fig. S2, ESI†).58 VA crystallized in hexagonal space group P61 with six molecules of VA in the unit cell. It is interesting to note that a second new polymorph of VA crystallizes as P63, but the structure has not yet been solved. Crystallographic studies of VA confirm the formation of 3D hydrogen-bonded structures through interactions between the ammonium and the carboxylate groups of the N- and C-termini, respectively. Each N-terminal amino residue forms two head-to-tail hydrogen bonds (NH3+⋯−OOC) with two C-terminal carboxylic acid residues. These two chains are then extended by amino-carbonyl interactions, resulting in hydrophobic porous columnar structures or nano-channels. The porous channels have diameters of 4.7 Å, and can be deployed for a broad range of applications. It is significant to note that the VA-class dipeptide porous structures have a distinctive hydrophobic layer, and even in the absence of guest solvent molecules, the host framework can serve as a robust building block without compromising its crystalline integrity and stability against any deterioration environments. Additionally, given their structural versatility, new guest molecules can be introduced by simply soaking the crystals in a solvent that contains a guest molecule. The framework is robust and can easily capture new solvent guests by replacing the old solvent guests without affecting the framework structure. Acetonitrile solvent was replaced by soaking the crystals in methanol, which could again be replaced by 2-propanol. The majority of dipeptide assemblies have hexagonal symmetry; however, a few examples of monoclinic symmetry were also discovered, demonstrating tunability of the pore structure.59
 | ||
| Fig. 1 (a) Chemical structures of dipeptides containing VA, AV, AI, IA, VI, IV, VV, LS, LL, IL, FL, LF, FG, FF, FW and WG utilized for the generation of microporous frameworks. (b) Molecular structure of the VA dipeptide crystal (left) and a view of the hexagonal crystal packing structure showing the porous hydrophobic channel of 5 Å (right). Reproduced with permission from ref. 58. Copyright 1996, IUCr. (c) Molecular structure of the FF dipeptide crystal (left) and a view of the formation of molecular rings by six FF molecules (right). The hydrophilic channels formed by six FF molecules are shown and the solvent water molecules are removed for structural clarity. Reproduced with permission from ref. 26. Copyright 2001, Wiley-VCH. | ||
Encouraged by these findings, several other peptide derivatives (typically, homo or hetero dipeptides) composed of alanine, valine, serine, methionine, and isoleucine were investigated to modulate the pore size and composition.25,26,59–62 Among various known porous dipeptides, those composed of phenylalanine (F), leucine (L), and tryptophan (W) were demonstrated to exhibit hydrophobic and hydrophilic layers.25,63,64 The hydrophilic columnar channels present in this family of porous peptides distinguish them significantly from the VA class (Fig. 1c).
Sozzani and coworkers reported the utilization of porous dipeptide frameworks for gas absorption applications using L-alanine–L-valine (AV) (Fig. 2a), L-valine–L-alanine (VA), L-isoleucine–L-valine (IV) and L-valine–L-isoleucine (VI), which possess pore channels of diameter 5.0, 4.7, 3.9, and 3.7 Å, respectively.65 The gas sorption analysis of VA showed a high storage capacity of 91 and 50 cm3 g−1 at 1 atm for carbon dioxide (CO2) and methane (CH4) respectively (Fig. 2b). The higher capacity of VA over the other three peptides is due to the complete filling of the guest molecule in the accessible volume. Therefore, crystalline peptide porous nanochannels could be an efficient substitute for porous carbon and zeolites for storing, capturing, and separating gaseous substances.
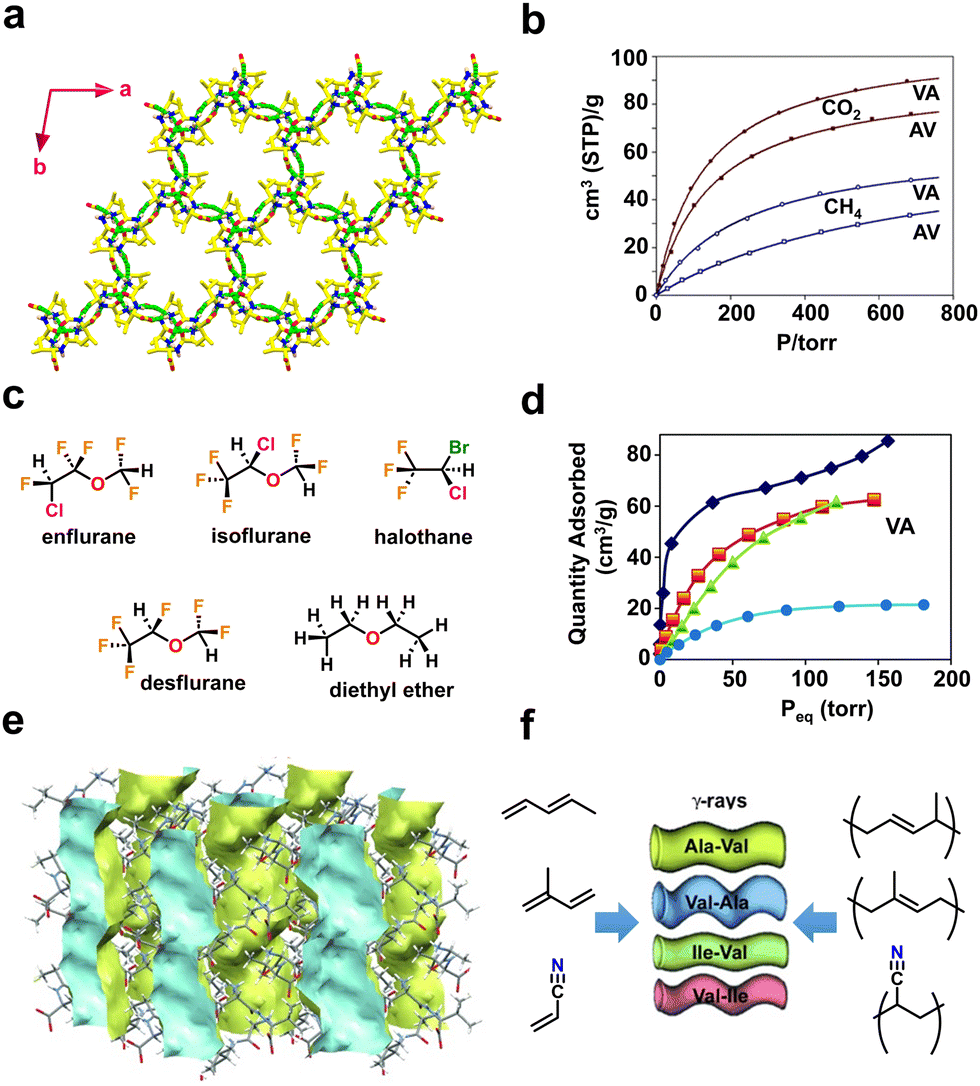 | ||
| Fig. 2 (a) Higher-order crystal structure of AV displaying a porous framework mediated via head-to-tail chain interactions between ammonium (NH3+) and carboxylate (COO−) residues. (b) Comparative gas adsorption isotherms of CO2 (depicted in full circles and squares) and CH4 (open circles and squares) at 195 K for AV and VA. Reproduced with permission from ref. 65. Copyright 2009, Royal Society of Chemistry. (c) Chemical structures of the various halogenated and non-halogenated ethers used as guest molecules. (d) Adsorption isotherms of anesthetics for VA recorded at 298 K. The adsorption curves of guest molecules are depicted in blue diamonds (enflurane), orange squares (isoflurane), green triangles (halothane) and light blue circles (desflurane). Reproduced with permission from ref. 66. Copyright 2018, Royal Society of Chemistry. (e) The crystal structure of AV shows a porous empty 1-dimensional nanochannel along the crystallographic c-axis (depicted in blue and yellow). (f) Schematic representation of the monomers (acrylonitrile, pentadiene, and isoprene) and porous dipeptides (AV, VA, IV and VI) used for the solid-state polymerization process. Reproduced with permission from ref. 67. Copyright 2012, Wiley-VCH. | ||
Volatile fluorinated ethers and alkanes are used as anaesthetics and their storage is extremely challenging. In addition, these compounds are hazardous to the environment; thus it is desirable to adsorb them using porous frameworks. Comotti and coworkers reported a porous framework produced from dipeptides, L-valine–L-alanine (VA), L-alanine–L-isoleucine (AI) and L-valine–L-valine (VV), that effectively hosted fluorinated ethers and alkanes (Fig. 2c).66 The adsorption isotherm demonstrated that VA porous channels can absorb up to 170–200 mmol mol−1 of volatile halogenated ethers and alkanes, revealing the complete loading of the available pore volume (Fig. 2d).
P-HPFs can also mediate polymerization reactions of acrylic or diene monomers (Fig. 2e and f).67 To induce a polymerization reaction, monomers such as acrylonitrile, pentadiene, and isoprene were first adsorbed onto the framework, and then subjected to γ-ray irradiation. The produced products of poly(acrylonitrile), poly(pentadiene), and poly(isoprene) are crystalline and nanostructured, as shown by both powder X-ray diffraction and scanning electron microscopy (SEM) analysis. Furthermore, the isotactic nature and the presence of polymers in the porous system were confirmed by 13C-NMR studies. These findings show the significant influence of a well-defined peptide environment on chemical reactions.
Görbitz and coworkers also designed hydrophobic dipeptides with noncanonical amino acids like L-2-aminobutanoic acid (Abu, ethyl side chain) and/or L-2-amino-pentanoic acid (L-norvaline, Nva, n-propyl side chain) (Fig. 3 and Fig. S3, ESI†).68 The authors judiciously designed noncanonical amino acids that not only strengthen the structural functionalities of the host framework but also aid in fine-tuning the pore diameter to accommodate various guest molecules (Fig. 3a).
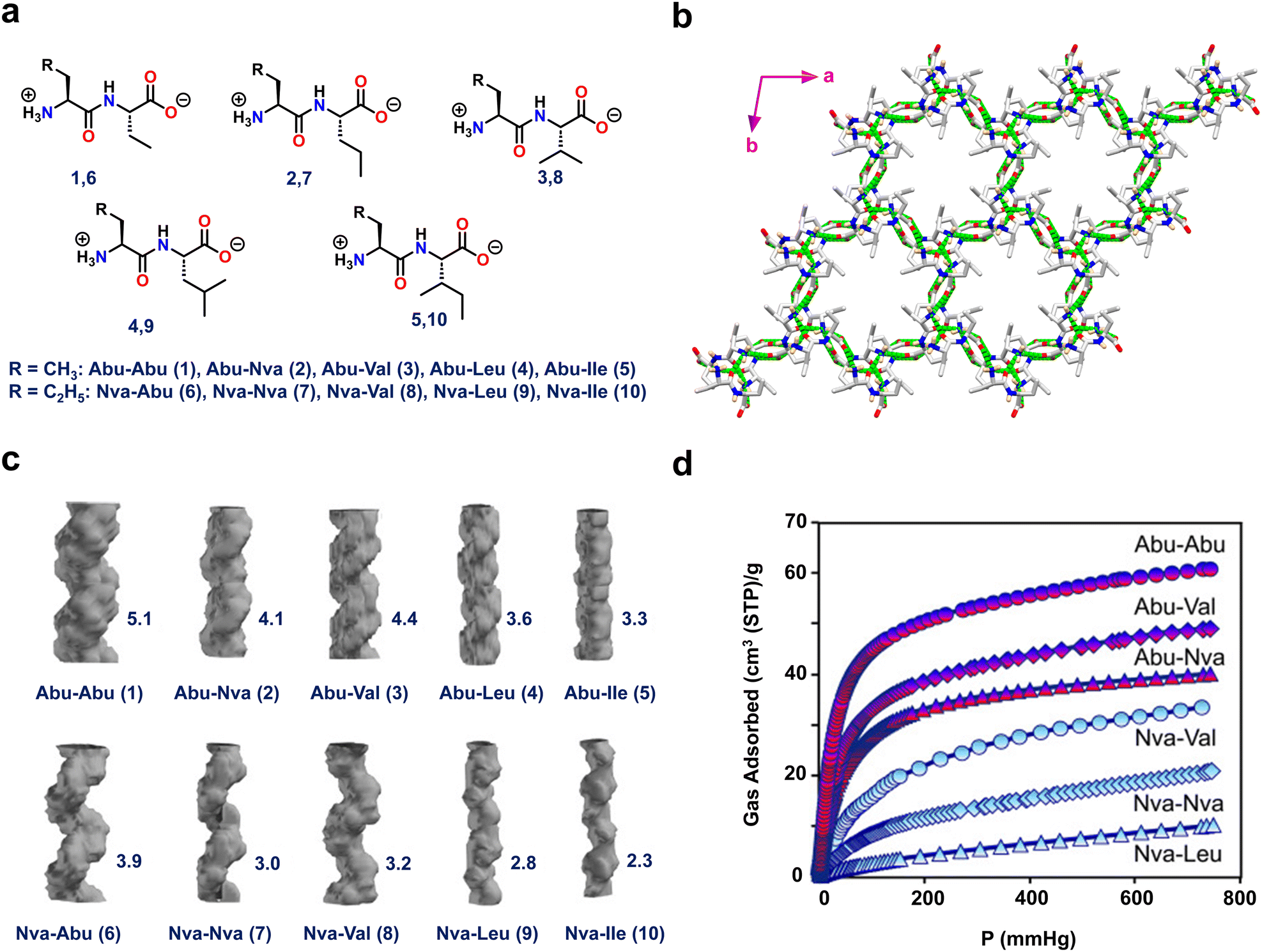 | ||
| Fig. 3 (a) Chemical structures of the ten dipeptides composed of L-2-aminobutanoic acid (Abu) and L-2-amino pentanoic acid (L-norvaline, NVa) residues. (b) Porous crystal structure of Abu–Abu (1) displaying head-to-tail chain interactions between NH3+ and COO− residues. (c) Porous channel-like structures of 1–10 with their corresponding reported cross-sections (Å) on the right-hand side. (d) The CO2 adsorption isotherms for the Abu–Abu, Abu–Val, Abu–Nva, Nva–Val, Nva–Nva, and Nva–Leu dipeptides at 195 K and up to 1 bar pressure. The Abu- and Nva-containing dipeptide curves are indicated by red and blue labels, respectively. Reproduced with permission from ref. 68. Copyright 2015, Wiley-VCH. | ||
Interestingly, single-crystal X-ray diffraction (SC-XRD) analysis of all ten frameworks (Abu–Abu(1), Abu–Nva(2), Abu–Val(3), Abu–Leu(4), Abu–lle(5), Nva–Abu(6), Nva–Nva(7), Nva–Val(8), Nva–Leu(9) and Nva–lle(10)) showed that they all crystallized in the same space group P61, but with varying pore diameters (Fig. 3b). Among the tested peptides, 1 had a distinct directional H-bonded structure composed of supramolecular left-handed double helices linked through head-to-tail chains. Furthermore, the connections between the helical strands were generated via amino and carbonyl linkage of NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C contacts, resulting in a stable and robust porous crystalline framework. The highest pore channel value of 5.1 Å was obtained for 1 (Fig. 3c). These porous peptides were subjected to CO2 and CH4 adsorption studies. Peptide 1 demonstrated the greatest CO2 and CH4 adsorption, of 62 and 38 cm3 g−1, respectively, since it has the largest channel size (Fig. 3d). These findings highlight the utility of non-proteinogenic amino acids to enhance and expand the functional space of peptide materials.
C contacts, resulting in a stable and robust porous crystalline framework. The highest pore channel value of 5.1 Å was obtained for 1 (Fig. 3c). These porous peptides were subjected to CO2 and CH4 adsorption studies. Peptide 1 demonstrated the greatest CO2 and CH4 adsorption, of 62 and 38 cm3 g−1, respectively, since it has the largest channel size (Fig. 3d). These findings highlight the utility of non-proteinogenic amino acids to enhance and expand the functional space of peptide materials.
When optimizing peptide-based HPFs for increased loading of guests, such as gas molecules, several key considerations come into play such as pore size, geometry optimization, surface functionalization, tuning binding sites, structural stability, thermodynamic considerations and kinetics of adsorption.11,16 Generally, larger and well-defined pores facilitate higher gas adsorption capacities. In addition, introducing specific functional groups on the peptide-based framework can enhance its affinity for certain gas molecules. Surface modification can be tailored to promote stronger interactions, leading to increased loading. Furthermore, optimization may involve tuning the hydrogen-bonding sites within the peptide sequences to improve the binding affinity for target gas molecules.69,70 As loading increases, maintaining the structural stability of P-HPFs is crucial. Optimization efforts may focus on enhancing the stability of the framework to prevent collapse or structural degradation under higher gas pressures. In addition, several thermodynamic and kinetic parameters also play an important role in understanding the energetics of the interaction between P-HPFs and gas molecules. Therefore, combining experimental studies with advanced computational techniques allows researchers to tailor P-HPFs with optimal properties for gas adsorption applications.
2.2 Tripeptides
Chen and coworkers developed mechanically robust nanoporous frameworks from tripeptides (Fig. 4a).71 They explored the sequences HYF, YFH, DYF, and YFD. HYF, DYF and YFD rapidly formed single crystals (typically within a few minutes) in phosphate buffer whereas YFH produced amorphous aggregates.71,72 SC-XRD analysis of these tripeptide crystals showed various supramolecular arrangements in the crystal lattice as well as multiple water-containing pore channels of various diameters. Interestingly, HYF crystallized in the trigonal space group R3 and displayed triangular-shaped pores that were filled with ordered water molecules. H-bonding, aromatic stacking, and electrostatic interactions stabilized the overall framework structure (Fig. 4a and Fig. S4, ESI†). The observed parameters for pore volume and edge length for the HYF tripeptide were 2062 Å3 and 15.2 Å, respectively. In contrast, the DYF and YFD tripeptides crystallized in the monoclinic space group P21. Moreover, the crystal packing and the presence of water molecules in these crystals were significantly different compared to HYF, especially the intermolecular interactions between the peptide and water molecules which resulted in a different pore geometry and water occupancy. Compared to DYF and YFD, the ability of HYF to accommodate both ordered and disordered water molecules in pores might be utilized to develop water-responsive materials.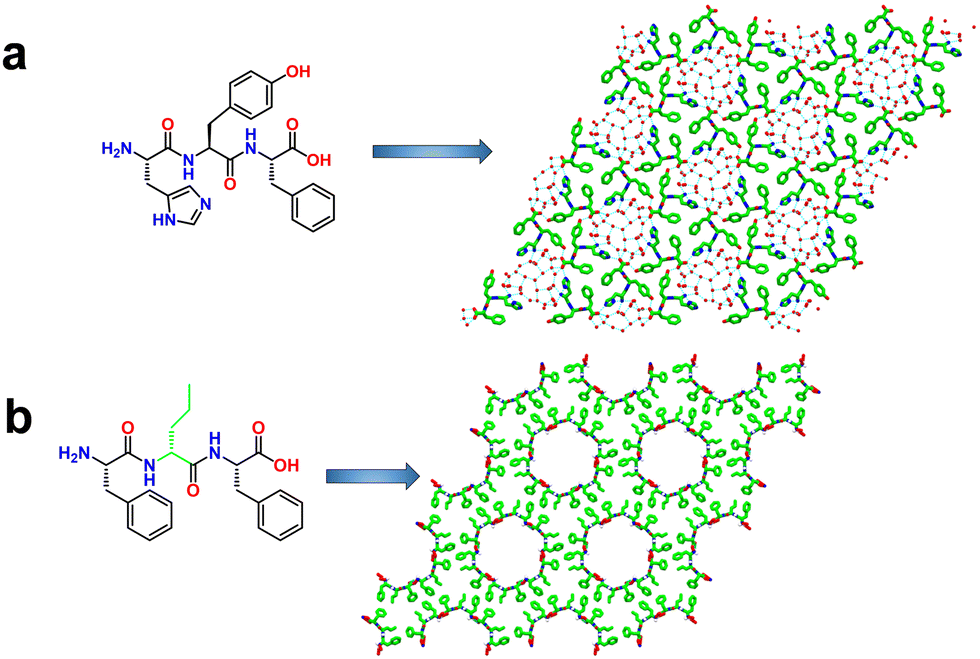 | ||
| Fig. 4 (a) Higher-order crystal packing structure of HYF displaying hydrogen-bonded porous assemblies. The blue lines indicate the water–water, water–HYF and HYF–HYF hydrogen bonds. Reproduced with permission from ref. 72. Copyright 2021, Springer Nature. (b) Single-crystal structures of F-DNva-F showing the porous frameworks with water channels 2.0-nm in diameter. Reproduced with permission from ref. 73. Copyright 2018, Cell Press. | ||
Marchesan and coworkers reported frameworks composed of aliphatic and aromatic amino acids (Fig. 4b and Fig. S5, ESI†).73 A series of LF-DX-LF tripeptides, where DX is an aliphatic D-amino acid, either D-alanine, D-valine, D-norvaline (Nva), D-isoleucine, D-leucine, or D-norleucine (Nle), were synthesized. It was observed that heterochiral sequences self-assembled to form ordered supramolecular hydrogels whereas the homochiral sequences failed to do so. Atomic level structure investigation of these tripeptides revealed the cause of their different self-assembling behavior. For instance, the tripeptide composed of heterochiral LF-DNva-LF displayed amphiphilic conformation with clear separation of hydrophilic and hydrophobic motifs with respect to the peptide backbone. The overall packing produced 2.0-nm wide water channels faced by the hydrophilic components of the peptide, and the interfaces of channels were stabilized through the interactions of hydrophobic units (Fig. 4b). In contrast, the homochiral tripeptide LF-LA-LF was incapable of forming water channels due to a lack of separation between hydrophilic and hydrophobic regions, which resulted in an extended conformation over porous frameworks. These findings illustrate that altering amino acid chirality is a simple yet powerful mechanism to produce new assemblies.
3. HPFs derived from folded peptides
While ultra-short peptides discussed above (di- and tripeptides) can form diverse porous assemblies, the lack of a sufficient number of amino acid residues hampers the engineering of their pore environment. Longer peptides have more mutable positions that can be used to fine-tune the pore chemistry, but in general, longer sequences (>3 amino acids) tend to form flexible, random coil structures. However, the judicious design of the sequence can force longer sequences to adopt defined secondary structures through a process known as folding. The folded structures of peptides are mainly held together by diverse non-covalent interactions. Primarily, these supramolecular interactions contribute to the formation of two major protein secondary structures known as α-helix and β-sheet (Fig. 5).74–77 The α-helix has ∼3.6 amino acids per turn and is stabilized by the hydrogen bonding between the N–H hydrogen of i and the carbonyl oxygen of the i + 4 residues (Fig. 5). The N- and C-termini of helices have four exposed N–H donors and CO acceptors, respectively. The intermolecular organizations of α-helices can be further engineered by changing the side chains of the amino acid residues.78,79 Other helical structures are the 310-helix and polyproline-II (PPII) helix. 310-helix has 3 amino acids per turn and is stabilized by the hydrogen bonding between the i and i + 3 residues (Fig. 5). On the other hand, the polyproline-II (PPII) helix is a left-handed helix consisting of repetitive proline residues and is frequently found in the triple-helix structures of collagen. The β-sheet structure is found in a large number of naturally occurring proteinaceous materials, including silk and amyloid fibrils.80 The edges of β-strands have numerous exposed N–H and CO H-bonding groups that promote aggregation into either parallel or antiparallel sheets (Fig. 5). Notably, the transformation of β-sheets into 1-D assemblies called fibrils, known as the amyloidosis process, is related to a variety of non-specific neurodegenerative disorders, including Alzheimer's and Parkinson's disease.81
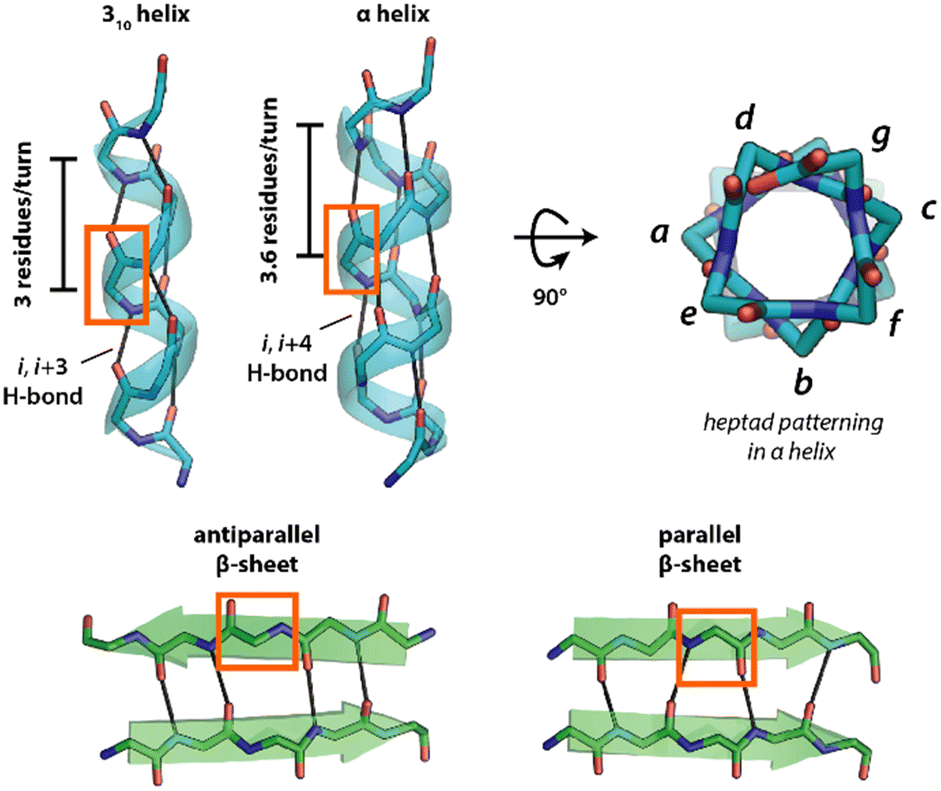 | ||
| Fig. 5 Common secondary structures of peptides. Hydrogen bonds are shown as black lines, and the side chains have been removed for clarity. | ||
Apart from the formation of amyloid-inspired peptide materials, β-sheet structures have also been well-documented for the development of functional materials for various potential applications in the area of biomechanics, tissue engineering and drug design.81–86 In addition to the development of diverse 1D and 2D supramolecular materials via the molecular assembly of these secondary structures, it has been lately established that these secondary structures can also assemble to generate flexible or rigid peptide porous frameworks.
3.1 Helical peptides
Nguyen and co-workers developed porous peptide frameworks from the assembly of short helical peptides (Fig. 6a).87 They designed a 9-residue helical peptide using a heptad patterning, abcdefg, to place hydrophobic and π-stacking groups (SP1). The presence of the Aib residue in the sequence greatly increases the helical propensity of the peptide by limiting the conformation space through the Thorpe–Ingold effect.88 Leu and Aib residues were placed in the a, b and e positions, aiming to promote the association of helices through hydrophobic interactions. A polar Gln residue was incorporated in the g position, mediating higher-order packing through intermolecular hydrogen bonding interactions. The positions c and f were kept mutable for further engineering the pore structures. The two terminal positions of the sequence were capped with rigid, planar bipyridyl moieties to facilitate the growth of the peptide lattice via π–π stacking (Fig. 6a). Circular dichroism (CD), 2D-NMR and SC-XRD demonstrated the helical conformation of the peptide. Further detailed investigation of the crystal structure revealed that the peptide adopted a mixed 310 and α-helical conformation stabilized by i to i + 3 and i to i + 4 intramolecular hydrogen bonding (Fig. 6b). In the higher-order arrangement, the helical peptide further assembled into porous rectangular nanochannels with dimensions of ∼1.5 × 1.1 nm, assisted by strong π–π stacking between terminal bipyridyl residues (Fig. 6c and Fig. S7, ESI†). In particular, the polar Gln residues stabilized the overall porous framework through intermolecular H-bonding with other helices. The authors further functionalized the porous framework by replacing the 4, 5, and 7 residues with other side chains, such as polar hydroxyl (Ser residue), N-donor (His residue), thioether (Met residue), carboxylic acid (Asp residue), and aromatic (4-iodo-Phe) groups. Remarkably, the porous framework was compatible with all these residues, which is normally challenging to achieve in conventional metal–peptide frameworks. The authors also investigated the guest encapsulation capability of the porous channels and found by SC-XRD that three tert-butylbenzene molecules per peptide could be encapsulated into the pore while inducing a noticeable change in the conformation of the peptide framework. The presence of metal-binding bipyridyl residues in the sequence inspired the authors to investigate the coordination chemistry of the helical framework. As a proof-of-concept, they demonstrated that the bipyridyl residues can coordinate Ag+, opening possibilities for designing materials leveraging metal reactivity.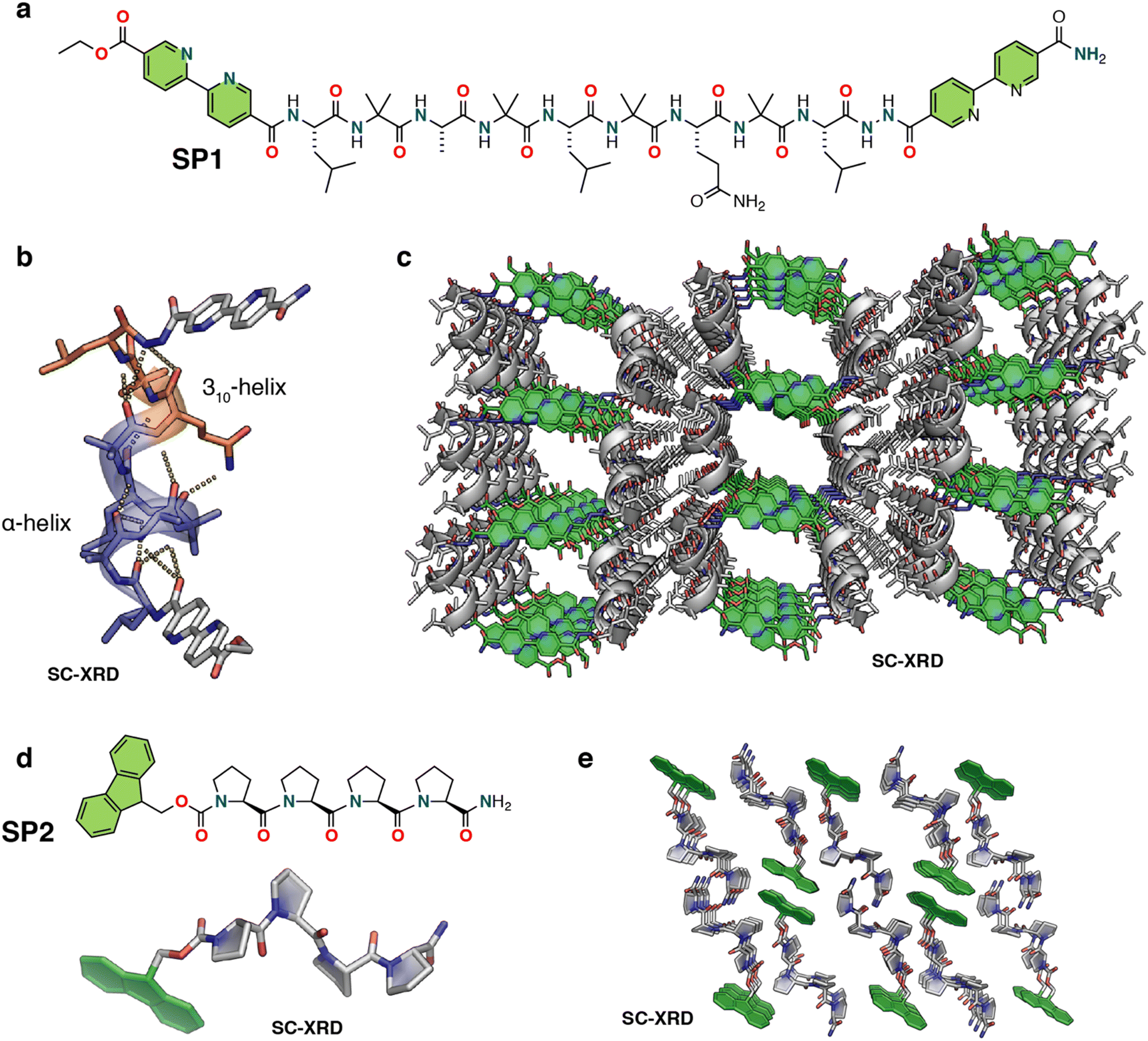 | ||
| Fig. 6 (a) Chemical structure of the nine-residue helical peptide featuring π-stacking bipyridyl residues. (b) X-ray structure of the peptide helix having both 310 and α-helical domains. (c) X-ray crystal structure of the peptide porous framework showing infinite channels. The solvent molecules, both water and acetonitrile, are removed for structural clarity. Reproduced with permission from ref. 87. Copyright 2018, American Chemical Society. (d) Chemical and crystal structure of proline tetramer Fmoc-(Pro)4-NH2 (PP4). (e) Higher-order crystal packing diagram of PP4 displaying solvent-accessible channels (viewed along the b-axis). The solvent ethanol molecules are removed for structural clarity. Reproduced with permission from ref. 90. Copyright 2022, Wiley-VCH. | ||
The authors further extended their design strategy of noncovalent peptide-π-stacking assembly-based frameworks to design thiol-containing porous materials, which are highly challenging to accomplish in MOFs and COFs due to the high reactivity and sensitivity of thiols.89 From the crystal structure of the original peptide, it was assumed that the incorporation of thiol functionality at position 7 would extend the thiol motif towards the channel. Single-crystal X-ray structure analysis of the Cys mutated peptide revealed the formation of pure α-helix conformation rather than the mixed 310-α fold as observed in the original sequence. As the pyridyl rings of BPE shifted towards the cavity, the size of the channel decreased (widths of 4.6 × 4.6 Å).
Mutating BPE with niacin regenerated a porous framework, albeit with a new topology (pore widths 13.3 × 11.0 Å). Extensive investigation was carried out through several single, double and triple Cys mutations at positions 5, 7 and 8, resulting in distinct thiol frameworks with unique pore shapes, different numbers of thiols per peptide, and in various arrangements of thiols. Due to the exceptionally versatile nature of thiol groups, the designed thiol frameworks were explored for several important applications. Upon soaking the single crystals of Cys-containing mutants of these porous frameworks in an aqueous solution of metal ions, many of these peptide frameworks showed differential uptake of Pb2+, Cd2+, Hg2+, and Ag+, indicating their potential ability to selectively remove toxic ions from solution. Moreover, immersion of crystals in 30% H2O2 resulted in the oxidative conversion of thiol frameworks into a chiral strong acid, opening the possibility of their implication in catalysis. Furthermore, it is worth noting that these thiol frameworks exhibited the ability to capture nitric oxide (NO) and subsequently release it at a controlled rate. This represents a compelling proof-of-concept for their potential utility as biocompatible NO slow-release agents, a capability that holds significant relevance in various biomedical applications, including but not limited to cardiovascular disease, wound healing, bacterial infections, and cancer treatment.
Very recently, Palma and coworkers developed a polyproline helix-based hydrogen-bonded porous supramolecular P-HPF, displaying guest molecule encapsulation and enantioselective functions (Fig. 6d, e and Fig. S6, ESI†).90 The porous framework was constructed by linking short helical oligo-proline moieties (SP2) that adopt the polyproline II conformation via hydrogen bonding and fluorene–fluorene interactions (Fig. 6d and e). Single crystal analysis of fluorenylmethoxycarbonyl (Fmoc)-protected tetraproline (PP4) confirmed the polyproline II helical conformation, and further revealed an extended framework containing small porous channels (Fig. 6e). In the higher order packing the peptides arranged in alternating antiparallel rows of PP4, enabling each row of SP2 to be offset from the adjacent row. In parallel, each C-terminal amide Pro4 formed a two-hydrogen-bond structure with adjacent carbonyl Pro2 and Pro3 with bond distances of 2.19 and 2.15 Å, respectively, exhibiting both acceptor and donor characteristics of each SP2, thus forming a 2D sheet that was further stabilized by weaker interactions resulting in the formation of an extended porous framework. Due to the porosity of the framework, the authors performed various guest inclusion studies, crystalline nature analysis and studies on phase changes using NMR, thermogravimetric analysis (TGA), differential thermal analysis (DSC), powder X-ray diffraction and SCXRD analyses. Moreover, the intrinsic chirality of the polyproline II conformation was exploited for chiral separation of racemic (±) 1-phenylethanol (PhEtOH), and it was able to achieve a modest 24% ee for (S)-1-phenylethanol. SP2 also demonstrated iodine encapsulation upon exposure to an iodine solution in hexane solvent, which was supported by SC-XRD analysis.
Lanci et al. employed a computational approach for the design of a “honeycomb-like” framework and hexagonal channels (Fig. 7).91 A de novo peptide sequence consisting of 27 residues per helix was mathematically designed to assemble into a homotrimeric parallel coiled-coil protein (SP3) showing a C3 symmetry and a superhelical pitch of 120 Å. Val and Leu residues were located in the a and d positions of the heptad topology. Except for Cys and Pro, all other amino acids were stepwise considered for the remaining exterior positions to modulate the crystallinity. A single SP3 sequence was then synthesized and crystallized from a standard crystal screen. The crystal contained columnar, hexagonal pores resembling the model, showing, however, deviation in terms of the orientation of adjacent helixes. The protein sequence was then shortened to 26 residues and a series of proteins (SP4, SP5, SP6, SP7) were synthesized. Proteins SP5 and SP6 produced crystals overnight, but SP4 and SP7 failed to do so.
 | ||
| Fig. 7 (a) Sequences of computationally designed proteins that were synthesized and crystallized. (b) Overlap of the computational model (magenta) and crystal structure (cyan) of the SP6 single helix with marked sidechain residues except for Val and Leu for clarity. (c) Hydrogen bonding connections across the interlayer interfaces of SP6 lead to the formation of a framework structure. (d) and (e) Honeycomb-like framework formation in the crystal structure of (d) SP3 and (e) SP6. Reproduced with permission from ref. 91. Copyright 2012, National Academy of Science. | ||
X-ray diffraction data of SP6 showed that the proteins were oriented in an all-parallel polar arrangement. The backbone structures, P6 crystalline lattice and supramolecular assembly into the honeycomb-like framework architecture of SP6 were in excellent agreement with the computational template. The backbone, side-chain interactions and the location of the “glycine zipper” motif GX3G in the sequence played a crucial role in the favourable orientation of neighbouring proteins and the overall crystallinity of the framework. This report showed the great potential of utilizing computational methods for the modular design of de novo protein crystals with on-demand topology.
3.2 β-sheet peptides
The β-sheet secondary structure is prone to self-assembly due to the number of exposed, alternating H-bond donating and accepting sites along the edge of the sheet (Fig. 5). Amyloids are naturally occurring β-sheet fibrils (1D or 2D assemblies) associated with neurological pathologies like Alzheimer's and Parkinson's disease, but their propensity to form extended architectures has also inspired their use in designing new peptide-based materials.92 Over the years, numerous methods, including solid-state NMR and X-ray diffraction, have been employed to investigate the structural aspects of amyloidogenic β-sheets, leading to designed and serendipitous discoveries of various β-sheet mimetic structures. A great effort has been made by Eisenberg and Nowick to crystallize the smaller fragment of an amyloid protein to better understand the atomic level interactions in the higher-order arrangements, revealing the extended peptide conformation which involved intermolecular H-bonds to form an infinite β-sheet.93–99In their ongoing effort to comprehend the supramolecular assembly of β-sheet in more detail,95,96 Nowick and colleagues designed a macrocyclic β-sheet peptide derived from transthyretin (TTR), a tetrameric protein whose dissociation into its monomers forms insoluble fibrils, which are responsible for a variety of amyloid diseases.97 To stabilize folding into a β-sheet, the authors connected both ends with two δ-linked ornithine (δ-Orn) turns to form a macrocycle. Further, an N-methyl group was installed on an alanine in one strand of the macrocycle to control the aggregation of β-sheets. High-resolution crystal structure analysis of the macrocyclic peptide revealed a β-hairpin conformation where two antiparallel strands were connected with intermolecular H-bonding. Further, the β-hairpin self-assembled through intermolecular H-bonding to form extended β-sheets, with higher-order arrangement of such four extended β-sheets leading to the formation of a square channel.
A subsequent study by the same group reported the formation of double-walled nanotubular structures by the higher-order self-assembly of macrocyclic β-sheet with a sequence of β-amyloid peptide, Aβ16-22 (KLVFFAE) (SP8, Fig. 8a).98 This nanotube was comparable, both in size and complexity, to biomolecular assemblies such as viral capsids and microtubules. The designed macrocycle was composed of two heptapeptide strands of a continuous chain of five hydrophobic amino acids, with oppositely charged K16 and E22 residues on the two terminals, namely the E–K strand and the K–E strand. As before, two ornithine residues further connected the ends of the antiparallel β-strands to form a β-hairpin macrocycle, and N-methylation of F19 was used to prevent uncontrolled hydrogen bonding. SC-XRD analysis revealed the six macrocyclic β-hairpin monomers in the asymmetric unit. The relatively flat β-hairpin formed dimers and further engaged with two other dimers through hydrogen bonding and electrostatic interactions, and was responsible for the formation of the inner walls of the nanotubes (Fig. 8b). The more twisted β-hairpins formed tetramers and further organized into β-barrel structures with extensive hydrogen bonding between the strands, forming a central hydrophobic core by the packing of eight side chains of Leu17 residues, which resulted in the formation of the outer wall of the nanotubes (Fig. 8c). The tubular structure was further surrounded by six identical assemblies, with individual inner and outer diameters of 7 nm and 11 nm, respectively, to form honeycomb-like frameworks (Fig. 8d). The junction where three such nanotubes meet is stabilized by aromatic–aromatic interactions between N-methyl-Phe19 and p-iodo-Phe19.
 | ||
| Fig. 8 (a) Chemical structure of macrocyclic β-sheet forming Aβ16–22. (b) X-ray crystallographic structure of the β-hairpin monomers that form the dimer (top) and tetramer (bottom) assembled from the macrocyclic β-sheet peptide. (c) Representation of the β-barrel-like tetramer structure (top view). (d) Honeycomb-like crystal lattice formed by macrocyclic β-sheet showing double-walled peptide nanotube porous structures. Reproduced with permission from ref. 98. Copyright 2017, American Chemical Society. | ||
This result demonstrated the significant potential of Aβ-derived sequences to create complex biomolecular porous assemblies, and future efforts should focus on developing design rules to engineer new structures and functions.
4. HPFs derived from foldamers
Foldamers are sequence-defined oligomers with artificial backbones that can also adopt a secondary structure by a folding process. They offer several advantages compared to natural peptides such as greater folding propensity, increased structural stability, variable backbone geometry, and high proteolytic stability. Moreover, foldamers can assemble into biomimetic supramolecular architectures with unique functions and divergent topologies.99–104 Several complex supramolecular architectures resulting from the higher-order self-assembly of foldamers, including helical bundles, nanofibers, nanosheets and liquid crystals, have been reported. However, the formation of HPFs through the higher-order organization of foldamers has not been validated to a significant extent due to the paucity of high-resolution structural data.103,104 Nevertheless, there are some precedents in the literature that describe the formation of HPFs by foldamers.Guichard and co-workers reported the first example of molecular self-assembly of oligourea foldamers into nanostructures under aqueous conditions.105 The ease of synthesis and the helical propensity of even the short oligourea sequences, coupled with their ability to maintain helicity despite variations of side chains, render them highly desirable candidates for the design of ordered artificial bioinspired materials.
The authors successfully demonstrated that precise manipulation of the foldamer sequence could generate diverse assemblies that can be tailored to encapsulate guest molecules. The authors designed and synthesized two oligourea sequences differing in the sequence of charged and hydrophobic side chains, F1 (Fig. 9a) and F2 (Fig. 9b). SC-XRD analysis revealed the identical helical conformation of the two oligoureas but differences in their quaternary structure. The oligourea sequence F1 assembled into discrete hexameric helical bundles possessing a leucine-rich hydrophobic core with a volume of 495.0 Å3 and a charged, hydrated exterior. The hydrophobic interactions reminiscent of the ‘knobs-into holes’ (KIH)-type packing proved to be the driving force for assembly while salt bridges played no role despite the presence of charged groups.
 | ||
| Fig. 9 (a) and (b) Chemical structure of the oligourea sequences (a) F1 and (b) F2. (c) X-ray crystal structure of the helical conformation of oligourea F2. (d) X-ray crystal structure of the channel-type assembly with a pore diameter of ∼17 Å formed by oligourea F2. (e) Chemical structure of oligourea sequence F3. (f) X-ray crystal structure of the 12-mer oligourea foldamer F3 displaying helical conformation. (g) The densely compacted crystal packing of F3 displays increased internal and external channel dimensions, with pore diameter ∼26 Å, compared with those of F2. Reproduced with permission from ref. 105. Copyright 2015, Nature Springer. | ||
In contrast, the helices of oligourea F2 (Fig. 9c, d) organized into a porous channel-like structure with a highly charged, water-filled interior pore with an internal diameter of ∼17 Å and a completely hydrophobic external surface (Fig. 9d). Unlike oligourea F1, the higher-order arrangement of oligourea F2 into 3D porous structures was primarily held together by polar contacts of extensive salt-bridge networks and hydrophobic interactions between the oligourea helices. The authors were also able to re-engineer the pore size by changing the sequence pattern and length of the oligomers. By applying a similar basic design, alternating the sequence pattern of hydrophobic and hydrophilic side chains with slightly increased length, the designed oligourea F3 assembled into similarly porous structures but with a much larger pore diameter (∼26 Å) (Fig. 9e–g). The result demonstrated an excellent example of controlling the structure–assembly relationship and opening a new route for application in molecular recognition and transportation.
Cai and co-workers reported the higher-order organization of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α/L-sulfono-γ-AA hybrid peptide (F4) into porous hydrogen-bonded organic frameworks (Fig. 10).106 The authors designed and synthesized a hybrid sequence composed of an alternative L-sulfono-γ-AA peptide and α-alanine in a 1:1 repeat pattern. SCXRD analysis of the hybrid peptide revealed a right-handed helical conformation stabilized by intramolecular hydrogen bonding (CO⋯HN) between i + 3 → i residues. Furthermore, higher-order self-assembly of the helical foldamers formed a hydrogen-bonded 1D crystalline framework without permanent porosity. Further, the authors were able to develop a 3D crystalline porous framework by introducing a simple covalent linker to fabricate a dimeric foldamer. The hybrid foldamer F4 was synthesized by dimerization of the hybrid peptide monomer at the third sulfono side chain through covalently connected terephthaloyl dichloride (Fig. 10a). SCXRD analysis of the dimer revealed an identical helical conformation of the two helixes comprising the dimer with similar helical pitch and radius. In the higher-order organization, the hybrid dimer foldamer was further assembled into an extended 2D monolayer sheet through extensive head-to-tail intermolecular hydrogen bonding and the sheets were laterally stacked on top of each other through various non-covalent interactions to form stable 3D supramolecular porous frameworks (Fig. 10b). The authors also investigated the gas adsorption properties of the monomers and dimers. The monomer exhibited very little uptake of N2 and CO2, while the dimeric structure exhibited significant N2 and CO2 uptake, indicating its potential use as a functional porous material (Fig. 10c and d).
:1 α/L-sulfono-γ-AA hybrid peptide (F4) into porous hydrogen-bonded organic frameworks (Fig. 10).106 The authors designed and synthesized a hybrid sequence composed of an alternative L-sulfono-γ-AA peptide and α-alanine in a 1:1 repeat pattern. SCXRD analysis of the hybrid peptide revealed a right-handed helical conformation stabilized by intramolecular hydrogen bonding (CO⋯HN) between i + 3 → i residues. Furthermore, higher-order self-assembly of the helical foldamers formed a hydrogen-bonded 1D crystalline framework without permanent porosity. Further, the authors were able to develop a 3D crystalline porous framework by introducing a simple covalent linker to fabricate a dimeric foldamer. The hybrid foldamer F4 was synthesized by dimerization of the hybrid peptide monomer at the third sulfono side chain through covalently connected terephthaloyl dichloride (Fig. 10a). SCXRD analysis of the dimer revealed an identical helical conformation of the two helixes comprising the dimer with similar helical pitch and radius. In the higher-order organization, the hybrid dimer foldamer was further assembled into an extended 2D monolayer sheet through extensive head-to-tail intermolecular hydrogen bonding and the sheets were laterally stacked on top of each other through various non-covalent interactions to form stable 3D supramolecular porous frameworks (Fig. 10b). The authors also investigated the gas adsorption properties of the monomers and dimers. The monomer exhibited very little uptake of N2 and CO2, while the dimeric structure exhibited significant N2 and CO2 uptake, indicating its potential use as a functional porous material (Fig. 10c and d).
 | ||
| Fig. 10 (a) Dimeric chemical structure of a 1:1 α/L-sulfono-γ-AA hybrid peptide. (b) Representation of a 2D supramolecular network of the dimer formed via 2D self-assembly of intra/intermolecular hydrogen bond contacts. The solvent molecules such as acetonitrile and tetrahydrofuran were removed from the crystal structure for clarity. (c) N2 adsorption isotherms of the monomer and dimer at 77 K. The solid and hollow circles represent the adsorption and desorption isotherms, respectively. (d) CO2 adsorption isotherms of the monomer and dimer at 273 K and 298 K. Reproduced with permission from ref. 106. Copyright 2018, American Chemical Society. | ||
5. Design principles of P-HPFs
Based on the examples in this review, some guidelines for the design of peptide frameworks are emerging. Table S1 (ESI†) analyzes the hydrophobic and hydrophilic content of the peptides discussed in this work. Though H-bonding is a shared interaction in all of these frameworks, the peptides constituting these materials have a significant hydrophobic content of at least 40%. This is not surprising given that the principal force in protein folding is the hydrophobic effect,107 which is an entropic phenomenon driven by expulsion of solvating molecules. Without significant hydrophobicity, solubility of the building blocks may be too high to favor framework formation. The burial of both aliphatic and aromatic residues from solvent is a common structural motif among most examples in this review. H-bonding and other electrostatic forces serve to give directionality and specificity in packing since polar groups will tend to interact with other polar groups. Again, this principle is seen in protein folding as well, where internal H-bonding groups destabilize folding for the benefit of achieving a specific fold.108Going towards frameworks made from longer peptides requires control of backbone flexibility. Folding can be induced locally (at the amino acid level) or globally. SP1 and SP2 exemplify the use of rigid amino acids, Aib and Pro, respectively. SP3 to SP7 showcase global induction of folding, using computational design to pattern hydrophobic residues in such a way that burial of these groups in a coiled-coil requires folding into a helix. The β-sheet peptide, SP8, uses a macrocylization strategy to stabilize the secondary structure, using the noncanonical amino acid ornithine to create an unconventional side-chain to backbone linkage. The shape of the peptide building unit gives some insight into how pores are generated. SP1 for example is built from hydrophobic C-shaped tectons, which in order to maximize burial of the nonpolar groups pack in a way that contains pores. The SP3 series has engineered interacting groups in a trigonal pattern that favors honey-comb lattices. The F-DNva-F building block has a curvature of nearly 120° that propagates into a hexagonal channel. These examples show that there are many ways to achieve pores, and that at a simplified level, shapes that disfavor 1D or 2D tiling may result in porous materials. Outside of peptides, these concepts have been explored in molecular tectonics109 and reticular chemistry,110 but should be generally applicable to the rational design of P-HPFs.
6. Applications of P-HPFs
Peptides possess extensive development prospects due to their nearly limitless sequence possibilities. As peptide-based frameworks are a nascent field, there are only emerging applications of porous peptide materials, many of which parallel those of other framework materials. However, an outstanding feature of peptides is their unique modularity, which is expected to offer a level of fine-tuning over that of traditional porous materials. We have summarized specific peptides involved in these applications in Table 1. The series of ultrashort peptides like VA, AV, VV, AI, IA, VI, IV, and LS exemplify the ability to fine-tune the channel shape through sequence modifications. For example, the sequences AV and VA showcase how a simple permutation could drastically affect the CO2/CH4 selectivity. Frameworks made from longer peptides offer a greater number of mutable positions, enabling more precise fine-tuning of the pore chemistry for the design of diverse applications. SP1 can be mutated at three positions to display a range of functional groups, including acidic and basic groups, while preserving the framework topology.87| Types | Secondary structure | P-HPF name | Crystal system and space group | Pore size [Å] | Functionality | Ref. | |
|---|---|---|---|---|---|---|---|
| Ultrashort peptides | Dipeptides | None | VA | Hexagonal, P61 | 4.7 | CH4, CO2, H2 adsorption, drug delivery system | 65 |
| None | AV | Hexagonal, P61 | 5 | CH4, CO2, H2 adsorption, nanoreactor | 67 | ||
| None | AI | Hexagonal, P61 | 4.7 | Drug delivery system | 66 | ||
| None | IA | Hexagonal, P61 | 3.7 | Drug delivery system | 66 | ||
| None | VI | Hexagonal, P61 | 3.7 | — | 25 | ||
| None | IV | Hexagonal, P61 | 3.9 | — | 25 | ||
| None | VV | Hexagonal, P61 | 4.4 | Drug delivery system | 66 | ||
| None | FF | Hexagonal, P61 | 10 | Bio-materials to medicinal applications | 26 | ||
| None | FL | Orthorhombic, P212121 | 3.5 | — | 26 | ||
| None | LF | Monoclinic, P21 | 3.1 | — | 26 | ||
| None | LL | Orthorhombic, P212121 | 3.3 | — | 26 | ||
| None | IL | Monoclinic, C2 | 3.2 | — | 25 | ||
| None | LS | Hexagonal, P65 | 4.9 | — | 25 | ||
| None | FW | Orthorhombic, P212121 | 3.2 | — | 25 | ||
| None | FA | Orthorhombic, P212121 | 2.5 | — | 25 | ||
| None | WG | Tetragonal, P41 | 4.7 | — | 25 | ||
| None | Abu–Abu | Hexagonal, P61 | 5.1 | CH4 and CO2 storage | 68 | ||
| None | Abu–Nva | Hexagonal, P61 | 4.1 | CH4 and CO2 storage | 68 | ||
| None | Abu–Val | Hexagonal, P61 | 4.4 | CH4 and CO2 storage | 68 | ||
| None | Abu–Leu | Hexagonal, P61 | 3.6 | — | 68 | ||
| None | Abu–Ile | Hexagonal, P61 | 3.3 | — | 68 | ||
| None | Nva–Abu | Hexagonal, P61 | 3.9 | — | 68 | ||
| None | Nva–Nva | Hexagonal, P61 | 3.0 | CH4 and CO2 storage | 68 | ||
| None | Nva–Val | Hexagonal, P61 | 3.2 | CH4 and CO2 storage | 68 | ||
| None | Nva–Leu | Hexagonal, P61 | 2.8 | CH4 and CO2 storage | 68 | ||
| None | Nva–Ile | Hexagonal, P61 | 2.3 | — | 68 | ||
| Tripeptides | β-sheet | HYF | Rhombohedral, R3 | 15.2 | Water responsive nature | 71 | |
| β-sheet | DYF | Monoclinic, P21 | 4.35 | Polymeric pigments | 72 | ||
| β-sheet | YFD | Monoclinic, P21 | 3.47 | Polymeric pigments | 72 | ||
| β-sheet | F-DNva-F | Hexagonal, P63 | 16.1 | Culture and proliferation of fibroblast cell lines | 73 | ||
| HPFs derived from folded peptides | α-310 helix | SP1 | Monoclinic, P21 | 15 | Molecular recognition and host–guest chemistry | 87 | |
| Polyproline II helix | SP2 | Monoclinic, P21 | 4.3 | Host–guest chemistry and chiral separation | 90 | ||
| α helix | SP3 | Hexagonal, P321 | — | — | 91 | ||
| α helix | SP6 | Hexagonal, P6 | — | — | 91 | ||
| β-sheet | SP8 | Hexagonal, P6122 | — | — | 91 | ||
| HPFs derived from foldamers | Helical | F2 | Hexagonal, P6122 | 17 | — | 105 | |
| Helical | F3 | Hexagonal, P622 | 26 | — | 105 | ||
| Helical | F4 | Monoclinic, P2 | 33.4 | N2 and CO2 storage | 106 | ||
This was utilized to engineer higher uptake of a complex dye molecule, Nile Red, as a proof-of-principle for designing hosts for applications like drug delivery or removal of organic pollutants. The structures of these frameworks are also semi-flexible, allowing substrates to induce structural changes that can maximize intermolecular interactions. The binding of t-butyl benzene to SP1 crystals caused the framework channel to morph its shape into a more rugged tunnel that increased dispersion interactions with the substrate. The conformational change was possible due to the presence of rotatable bonds and the noncovalent assembly that allows peptides to slide within the lattice. The intrinsic chirality of P-HPFs can enable separation of chiral molecules, which has significant applications in medicinal chemistry. In 2022, Palma et al. developed polyproline helix containing porous frameworks with stable secondary structures.90SP2 can separate (S)-1-phenylethanol from a racemic mixture of (±) phenylethanol, demonstrating the promising ability of peptide materials in chiral separation.
Folded peptide-based HPFs have recently emerged as a novel class of crystalline porous materials, known for enhanced folding, increased structural stability, variable backbone geometry, and heightened proteolytic stability. In a recent study, Cai and co-workers explored a 1:1 α/L-sulfono-γ-AA hybrid peptide (F4), revealing crystalline porous networks in monomeric and dimeric forms.106 The crystal packing exhibited a distinctive right-handed helix formed through hydrogen bonding. The dimer displayed superior N2 and CO2 adsorption, attributed to its enhanced porous structure. This peptide self-assembly introduces a novel category of materials with promising applications in adsorption, separation, catalysis, and drug delivery.
Overall, the limited examples of P-HPFs already show unique properties over other classes of porous materials. P-HPFs are highly modular, greatly simplifying the process of material exploration and engineering. The semi-flexibility of P-HPFs enables them to subtly adjust their structures to become better hosts for their guests, and this property could allow them achieve better adsorption than rigid materials.
7. Conclusions and future directions in the development of P-HPFs
This perspective comprehensively reviews the current state of P-HPFs, highlighting their immense tunability and potential to address various applications, including molecular recognition, catalysis, gas adsorption, biosynthesis, and medical applications. The interest in P-HPFs can be attributed to their numerous distinguishing characteristics: ease of synthesis and variation, the absence of metal ions, structural adaptability, chirality, biocompatibility, and specific selectivity towards environmentally hazardous guest molecules. Notably, P-HPFs leverage bio-derived building blocks, rendering them more compatible with biological systems. These attributes position them as promising candidates for application in drug delivery. Furthermore, the facile tuning of the cavity size and interior properties via simple amino acid side chain modifications makes P-HPFs attractive for engineering-specific molecular recognition. In comparison to metal-coordinated frameworks, the dynamic and reversible nature of P-HPFs provides a way for designing stimuli-responsive materials. In addition, the intrinsic chirality of P-HPFs makes them ideal candidates for improving chiral separation, which is a major challenge in the pharmaceutical industry.111 More interestingly, chiral P-HPFs could also be applied as asymmetric catalysts for various enantioselective organic transformations.Despite several unique advantages, one perceived weakness of P-HPFs is the higher conformational flexibility of peptide chains compared to rigid aromatic moieties typical of MOFs, COFs, and HOFs. Flexibility can compromise framework stability under extreme conditions, including vacuum, desiccation, high temperature, and high pressure. Nonetheless, P-HPFs are well-suited for applications that predominantly occur in a liquid phase and more ambient temperatures, as is often the case in drug delivery, catalysis, and water purification. To advance the field, the incorporation of abiotic, rigidified foldamer building blocks holds promise for substantially enhancing structural stability. Furthermore, the integration of unnatural backbones offers enhanced resistance to degradation by endogenous proteases, a crucial attribute for in vivo applications like drug delivery.
The design of P-HPFs is still in its nascent stage, with many frameworks being discovered only by serendipity. To develop more intuitive design principles, high-resolution structural data by SC-XRD or other methods (e.g. TEM, cryo-EM) must be actively pursued, from which sequence–structure–function relationships can be extracted. Currently, the rules to design a P-HPF de novo are far less established compared to systems built using rigid aromatic groups, like MOFs, COFs, and HOFs, and the perceived unpredictability of peptide building blocks must be addressed in order to increase their appeal. The examples in this review demonstrate significant and growing progress towards a database of structurally defined P-HPFs that will accelerate the goal of rational design. Structure prediction using computational methods is an emerging area with notable breakthrough success. Drawing upon large databases of crystal structures, the structures of proteins and solid-state materials can be predicted accurately.112,113 The success of machine learning and artificial intelligence for the aforementioned problems is due to the plethora of high-resolution structures from crystallography, NMR spectroscopy, or electron microscopy. Peptides, currently, lack a large structural database, and therefore future efforts to obtain atom-level structural data are invaluable for analysis by computational prediction. If prediction of peptide frameworks is possible, targeted pore geometries and functionalities could be obtained to achieve highly specific separation or catalytic transformations. As peptide synthesis, structural characterization, and computational methods continue to advance in parallel, numerous exciting opportunities await to uncover both fundamental chemistry and practical applications of peptide-based frameworks.
Author contributions
The manuscript was written through the contribution of all the authors. All authors have read the manuscript and given approval for the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the DST Inspire Faculty Fellowship (No. DST/INSPIRE/04/2020/002499) from the Department of Science and Technology, New Delhi. R. M. is also thankful to the National Institute of Pharmaceutical Education and Research, S.A.S. Nagar, for providing the research facilities. T. V. thanks Tel Aviv University for the postdoctoral fellowship. E. G. thanks European Research Council PoC project PepZoPower (101101071). A. I. N. thanks the American Chemical Society Petroleum Research Fund (62285-DNI). S. B. thanks SERB, Govt. of India, for the Ramanujan Fellowship (Ref. no. RJF/2022/000042), and Ashoka University, Sonipat, Haryana, for the infrastructure. S. N. acknowledges Vellore Institute of Technology Chennai for the funding and infrastructure.Notes and references
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dinca, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- M. X. Wu, Y. Wang, G. Zhou and X. Liu, Coord. Chem. Rev., 2021, 430, 213735 CrossRef CAS.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
- J. Andreo, R. Ettlinger, O. Zaremba, Q. Peña, U. Lächelt, R. F. de Luis, R. Freund, S. Canossa, E. Ploetz, W. Zhu, C. S. Diercks, H. Gröger and S. Wuttke, J. Am. Chem. Soc., 2022, 144, 7531–7550 CrossRef CAS PubMed.
- M. D. Allendorf, R. Dong, X. Feng, S. Kaskel, D. Matoga and V. Stavila, Chem. Rev., 2020, 120, 8581–8640 CrossRef CAS PubMed.
- P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef CAS.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- R. B. Lin and B. Chen, Chem, 2022, 8, 2114–2135 CAS.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS PubMed.
- P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737–2738 CrossRef CAS.
- Y. Liu, G. Chang, F. Zheng, L. Chen, Q. Yang, Q. Ren and Z. Bao, Chem. – Eur. J., 2023, 29, e202202655 CrossRef CAS PubMed.
- Z. Zhang, Y. Ye, S. Xiang and B. Chen, Acc. Chem. Res., 2022, 55, 3752–3766 CrossRef CAS PubMed.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- Z. J. Lin, S. A. R. Mahammed, T. F. Liu and R. Cao, ACS Cent. Sci., 2022, 8, 1589–1608 CrossRef CAS PubMed.
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS.
- M. C. Das, S. C. Pal and B. Chen, Joule, 2022, 6, 22–27 CrossRef.
- L. Chen, B. Zhang, L. Chen, H. Liu, Y. Hu and S. Qiao, Mater. Adv., 2022, 3, 3680–3708 RSC.
- Q. Yin, P. Zhao, R. J. Sa, G. C. Chen, J. Lu, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS PubMed.
- I. Hisaki, S. Nakagawa, N. Ikenaka, Y. Imamura, M. Katouda, M. Tashiro, H. Tsuchida, T. Ogoshi, H. Sato, N. Tohnai and M. Miyata, J. Am. Chem. Soc., 2016, 138, 6617–6628 CrossRef CAS PubMed.
- Y. L. Li, E. V. Alexandrov, Q. Yin, L. Li, Z. B. Fang, W. Yuan, D. M. Proserpio and T. F. Liu, J. Am. Chem. Soc., 2020, 142, 7218–7224 CrossRef CAS PubMed.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS PubMed.
- C. H. Görbitz, Chem. – Eur. J., 2007, 13, 1022–1031 CrossRef PubMed.
- C. H. Görbitz, Chem. – Eur. J., 2001, 7, 5153–5159 CrossRef.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- E. Fischer and E. Otto, Ber. Dtsch. Chem. Ges., 1903, 36, 2106–2116 CrossRef CAS.
- M. P. Bas, J. T. Puche and F. Albericio, Chem. Soc. Rev., 2016, 45, 631–654 RSC.
- A. E. Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef PubMed.
- E. Atherton and R. C. Sheppard, Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, Oxford, UK, 1999 Search PubMed.
- G. B. Fields, Solid-Phase Peptide Synthesis (Academic Press, New York, 1997) Search PubMed.
- G. B. Fields, Methods Mol. Biol., 1994, 35, 17–27 CAS.
- K. J. McKnelly, W. Sokol and J. S. Nowick, J. Org. Chem., 2020, 85, 1764–1768 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- T. Bilbrough, E. Piemontese and O. Seitz, Chem. Soc. Rev., 2022, 51, 5691–5730 RSC.
- L. Malik, A. P. Tofteng, S. L. Pedersen, K. K. Sørensen and K. J. Jensen, J. Pept. Sci., 2010, 16, 506–512 CrossRef CAS PubMed.
- H. M. Yu, S. T. Chen and K. T. Wang, J. Org. Chem., 1992, 57, 4781–4784 CrossRef CAS.
- P. G. Vasudev, S. Chatterjee, N. Shamala and P. Balaram, Chem. Rev., 2011, 111, 657–687 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- R. V. Ulijn and R. Jerala, Chem. Soc. Rev., 2018, 47, 3391–3394 RSC.
- T. Vijayakanth, D. J. Liptrot, E. Gazit, R. Boomishankar and C. R. Bowen, Adv. Funct. Mater., 2022, 32, 2109492 CrossRef CAS.
- T. Vijayakanth, S. Shankar, G. Finkelstein-Zuta, S. Rencus-Lazar, S. Gilead and E. Gazit, Chem. Soc. Rev., 2023, 52, 6191–6220 RSC.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- N. Bajpayee, T. Vijayakanth, S. Rencus-Lazar, S. Dasgupta, A. V. Desai, R. Jain, E. Gazit and R. Misra, Angew. Chem., Int. Ed., 2023, 62, e202214583 CrossRef CAS PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- V. Apostolopoulos, J. Bojarska, T. T. Chai, S. Elnagdy, K. Kaczmarek, J. Matsoukas, R. New, K. Parang, O. P. Lopez, H. Parhiz, C. O. Perera, M. Pickholz, M. Remko, M. Saviano, M. Skwarczynski, Y. Tang, W. M. Wolf, T. Yoshiya, J. Zabrocki, P. Zielenkiewicz, M. AlKhazindar, V. Barriga, K. Kelaidonis, E. M. Sarasia and I. Toth, Mol., 2021, 26, 430 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- E. Gazit, Annu. Rev. Biochem., 2018, 87, 533–553 CrossRef CAS PubMed.
- J. Rabone, Y. F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–1057 CrossRef CAS PubMed.
- C. Martí-Gastaldo, D. Antypov, J. E. Warren, M. E. Briggs, P. A. Chater, P. V. Wiper, G. J. Miller, Y. Z. Khimyak, G. R. Darling, N. G. Berry and M. J. Rosseinsky, Nat. Chem., 2014, 6, 343–351 CrossRef PubMed.
- C. Martí-Gastaldo, J. E. Warren, K. C. Stylianou, N. L. O. Flack and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 11044–11048 CrossRef PubMed.
- C. H. Görbitz and E. Gundersen, Acta Crystallogr., 1996, C52, 1764–1767 Search PubMed.
- C. H. Görbitz, CrystEngComm, 2005, 7, 670–673 RSC.
- D. V. Soldatov, I. L. Moudrakovski and J. A. Ripmeester, Angew. Chem., Int. Ed., 2004, 43, 6308–6311 CrossRef CAS PubMed.
- C. H. Görbitz, New J. Chem., 2003, 27, 1789–1793 RSC.
- C. H. Görbitz, M. Nilsen, K. Szeto and L. W. Tangen, Chem. Commun., 2005, 4288–4290 RSC.
- C. H. Görbitz, Chem. Commun., 2006, 2332–2334 RSC.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, G. Distefano and P. Sozzani, Chem. Commun., 2009, 284–286 RSC.
- S. Bracco, D. Asnaghi, M. Negroni, P. Sozzani and A. Comotti, Chem. Commun., 2018, 54, 148–151 RSC.
- G. Distefano, A. Comotti, S. Bracco, M. Beretta and P. Sozzani, Angew. Chem., Int. Ed., 2012, 51, 9258–9262 CrossRef CAS PubMed.
- V. N. Yadav, A. Comotti, P. Sozzani, S. Bracco, T. BongeHansen, M. Hennum and C. H. Görbitz, Angew. Chem., Int. Ed., 2015, 54, 15684–15688 CrossRef PubMed.
- Y. Wang, J. Min, H. Wei, J. Liu, Y. Liang, R. Su, G. Zhang, W. Zhang, Y. Wang and W. Qi, Sci. China Mater., 2023, 66, 470–484 CrossRef CAS.
- Y. F. Han, Y. X. Yuan and H. B. Wang, Molecules, 2017, 22, 266 CrossRef PubMed.
- R. Piotrowska, T. Hesketh, H. Wang, A. R. G. Martin, D. Bowering, C. Zhang, C. T. Hu, S. A. McPhee, T. Wang, Y. Park, P. Singla, T. McGlone, A. Florence, T. Tuttle, R. V. Ulijn and X. Chen, Nat. Mater., 2021, 20, 403–409 CrossRef CAS PubMed.
- A. Lampel, S. A. McPhee, H. A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T. D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- S. Mondal and E. Gazit, ChemNanoMat, 2016, 2, 323–332 CrossRef CAS.
- S. Bera and E. Gazit, Protein Pept. Lett., 2019, 26, 88–97 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- S. Kim, J. H. Kim, J. S. Lee and C. B. Park, Small, 2015, 11, 3623–3640 CrossRef CAS PubMed.
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed.
- Y. B. Lim and M. Lee, J. Mater. Chem., 2008, 18, 723–727 RSC.
- T. H. Larsen, M. C. Branco, K. Rajagopal, J. P. Schneider and E. M. Furst, Macromolecules, 2009, 42, 8443–8450 CrossRef CAS PubMed.
- J. A. Robinson, Acc. Chem. Res., 2008, 41, 1278–1288 CrossRef CAS PubMed.
- S. L. Heinz-Kunert, A. Pandya, V. T. Dang, P. N. Tran, S. Ghosh, D. McElheny, B. D. Santarsiero, Z. Ren and A. I. Nguyen, J. Am. Chem. Soc., 2022, 144, 7001–7009 CrossRef CAS PubMed.
- I. L. Karle and P. Balaram, Biochemistry, 1990, 29, 6747–6756 CrossRef CAS PubMed.
- S. S. Hess, F. Coppola, V. T. Dang, P. N. Tran, P. J. Mickel, J. Oktawiec, Z. Ren, P. Král and A. I. Nguyen, J. Am. Chem. Soc., 2023, 145, 19588–19600 CrossRef CAS PubMed.
- D. F. Brightwell, G. Truccolo, K. Samanta, E. J. Fenn, S. J. Holder, H. J. Shepherd, C. S. Hawes and A. Palma, Chem. – Eur. J., 2022, 28, e202202368 CrossRef CAS PubMed.
- C. J. Lanci, C. M. MacDermaid, S. G. Kang, R. Acharya, B. North, X. Yang, X. J. Qiu, W. F. DeGrado and J. G. Saven, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7304–7309 CrossRef CAS PubMed.
- T. P. J. Knowles and R. Mezzenga, Adv. Mater., 2016, 28, 6546–6561 CrossRef CAS PubMed.
- R. Riek and D. S. Eisenberg, Nature, 2016, 539, 227–235 CrossRef PubMed.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- R. K. Spencer and J. S. Nowick, Isr. J. Chem., 2015, 55, 698–710 CrossRef CAS PubMed.
- P. N. Cheng, J. D. Pham and J. S. Nowick, J. Am. Chem. Soc., 2013, 135, 5477–5492 CrossRef CAS PubMed.
- S. Yoo, A. G. Kreutzer, N. L. Truex and J. S. Nowick, Chem. Sci., 2016, 7, 6946–6951 RSC.
- K. H. Chen, K. A. Corro, S. P. Le and J. S. Nowick, J. Am. Chem. Soc., 2017, 139, 8102–8105 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- S. Hecht and I. Huc, Foldamers: Structure, Properties, and Applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- S. H. Yoo and H. S. Lee, Acc. Chem. Res., 2017, 50, 832–841 CrossRef CAS PubMed.
- G. W. Collie, K. PulkaZiach, C. M. Lombardo, J. Fremaux, F. Rosu, M. Decossas, L. Mauran, O. Lambert, V. Gabelica, C. D. Mackereth and G. Guichard, Nat. Chem., 2015, 7, 871–880 CrossRef CAS PubMed.
- P. Teng, Z. Niu, F. She, M. Zhou, P. Sang, G. M. Gray, G. Verma, L. Wojtas, A. van der Vaart, S. Ma and J. Cai, J. Am. Chem. Soc., 2018, 140, 5661–5665 CrossRef CAS PubMed.
- V. V. Loladze, D. N. Ermolenko and G. I. Makhatadze, J. Mol. Biol., 2002, 320, 343–357 CrossRef CAS PubMed.
- K. J. Lumb and P. S. Kim, Biochemistry, 1995, 34, 8642–8648 CrossRef CAS PubMed.
- M. W. Hossein, Acc. Chem. Res., 2005, 38, 313–323 CrossRef PubMed.
- H. Jiang, D. Alezi and M. Eddaoudi, Nat. Rev. Mater., 2021, 6, 466–487 CrossRef CAS.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. R. Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- A. Merchant, S. Batzner, S. S. Schoenholz, M. Aykol, G. Cheon and E. D. Cubuk, Nature, 2023, 624, 80–85 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cs00648d |
| This journal is © The Royal Society of Chemistry 2024 |