
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineered biological nanoparticles as nanotherapeutics for tumor immunomodulation
Juwita N.
Rahmat†
ab,
Jiayi
Liu†
c,
Taili
Chen
d,
ZhiHong
Li
*ef and
Yong
Zhang
 *g
*g
aDepartment of Biomedical Engineering, College of Design and Engineering, National University of Singapore, Singapore 117585, Singapore
bDepartment of Surgery, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 119074, Singapore
cDepartment of Oncology, The Second Xiangya Hospital, Central South University, Changsha, Hunan Province, China
dDepartment of Oncology, Xiangya Hospital, Central South University, Changsha, Hunan Province, China
eDepartment of Orthopedics, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China. E-mail: lizhihong@csu.edu.cn
fHunan Key Laboratory of Tumor Models and Individualized Medicine, The Second Xiangya Hospital of Central South University, Changsha 410011, China
gDepartment of Biomedical Engineering, College of Engineering, The City University of Hong Kong, Hong Kong SAR. E-mail: yozhang@cityu.edu.hk; Web: https://www.yongzhanglab.com
First published on 8th May 2024
Abstract
Biological nanoparticles, or bionanoparticles, are small molecules manufactured in living systems with complex production and assembly machinery. The products of the assembly systems can be further engineered to generate functionalities for specific purposes. These bionanoparticles have demonstrated advantages such as immune system evasion, minimal toxicity, biocompatibility, and biological clearance. Hence, bionanoparticles are considered the new paradigm in nanoscience research for fabricating safe and effective nanoformulations for therapeutic purposes. Harnessing the power of the immune system to recognize and eradicate malignancies is a viable strategy to achieve better therapeutic outcomes with long-term protection from disease recurrence. However, cancerous tissues have evolved to become invisible to immune recognition and to transform the tumor microenvironment into an immunosuppressive dwelling, thwarting the immune defense systems and creating a hospitable atmosphere for cancer growth and progression. Thus, it is pertinent that efforts in fabricating nanoformulations for immunomodulation are mindful of the tumor-induced immune aberrations that could render cancer nanotherapy inoperable. This review systematically categorizes the immunosuppression mechanisms, the regulatory immunosuppressive cellular players, and critical suppressive molecules currently targeted as breakthrough therapies in the clinic. Finally, this review will summarize the engineering strategies for affording immune moderating functions to bionanoparticles that tip the tumor microenvironment (TME) balance toward cancer elimination, a field still in the nascent stage.
 Juwita N. Rahmat | Juwita Rahmat is currently a Senior Research Fellow in the Department of Surgery, National University of Singapore. After her PhD (2010), she carried out studies on the use of nanoparticles for drug delivery and innovating implantable devices in various animal models. Her current research interests are gut microbiota, nanomedicine, and immunotherapy for the treatment of malignancies. |
 Jiayi Liu | Liu Jiayi is a Research Fellow at the Second Xiangya Hospital of Central South University in China. He obtained his PhD degree from the same institution in 2022. Prior to this, he was a Postdoctoral Researcher in the Department of Biomedical Engineering at the National University of Singapore. His research passions lie in exploring biomaterial applications, developing animal models, and elucidating the mechanisms underlying antitumor activity. |
 Taili Chen | Taili Chen completed her master's degree in Oncology at Xiangya Hospital, Central South University in China. Since 2020, she has been pursuing a PhD degree in the same field. From 2021 to 2023, she studied Biomedical Engineering at the National University of Singapore (NUS) as a visiting student. Her research focuses on the establishment and application of biomaterials using tumour in vivo models. |
 ZhiHong Li | Li Zhihong presently serves as the Vice President of Central South University and holds the position of Director at the National Clinical Evaluation Technology Platform for New Drug Development. His primary research foci encompass fundamental and clinical investigations, as well as the diagnosis and treatment of bone and soft tissue tumors. |
 Yong Zhang | Yong Zhang is currently the Chair Professor of Biomedical Engineering and Head of Department of Biomedical Engineering at City University of Hong Kong. Professor Zhang has authored over 300 peer-reviewed research papers in international journals. His current research interests include nanobiophotonics, nanomedicine, and microfluidic devices. |
1. Introduction
Malignant diseases are on the rise and present a global health problem. The probability of developing cancer increases with advancing age. The conventional management of cancer therapy has centered on the trifecta of surgical resection, chemotherapeutics, and radiation therapy with alternative treatment options, such as cryoablation, immunotherapy, and exploratory clinical trials for refractory, recurrent, or advanced diseases.1 The advent of nanomedicine has achieved progress in providing critical options for patients and physicians facing the dilemma of viable treatment selections after standard care failure. The rise of nanoscience and nanotechnology in the 1980s significantly boosted the synthesis of laboratory-derived nanoformulations for medical purposes. Currently, nanomedicine is considered a viable treatment option, with FDA-approved and exploratory formulations available for clinical trials.2The appeal of nanotechnology for drug delivery and imaging is the enormous potential of targeting the treatment agents to specific targets in the body, including the brain.3 Other advantages of nanoencapsulation include the ability to confer controlled drug release and increased drug stability. Various inorganic and organic materials, such as silver and gold nanoparticles, have been used to fabricate advanced nanoformulations to achieve these end goals.4,5 Organic materials such as liposomes, dendrimers, polymeric micelles, nanoemulsions, and carbon nanomaterials were extensively explored for drug delivery. The appeal of these various nanotechnology systems is that they can encapsulate both hydrophobic and hydrophilic drugs, increasing their stability and bioavailability to target tissues.6 Despite their advantages and increased efficacy, toxicity issues plagued using inorganic nanomaterials. Their potential bioaccumulation and limited knowledge of their long-term effects on the body and the environment limit their clinical utility. Organic nanoparticle systems are limited by low drug-loading capacity, high local absorption, possible drug leakage, rapid elimination via the reticuloendothelial system (RES), and non-specific interactions with biological components, which limits their treatment efficacy.7–11 Thus, the collective disadvantages have hindered the progress of synthetic formulations into clinical translations.
Nanoparticles are defined as small materials ranging from 1–100 nm. Although nanoparticles in biomedical research were used to describe various laboratory-derived synthetics, nanoparticles have existed in nature for millions of years, with sophisticated and precise assembly systems encoded in the genetic material of living organisms.12 These biological nanoparticles (bionanoparticles) are derived and isolated from organic living systems. They utilize their complex assembly machinery to manufacture biological products that could be further engineered in the lab for specific functionalities. Bionanoparticles are pursued due to their diverse function in nature, biocompatibility, non-toxicity, and extended circulatory lifespan. The advent of bionanoparticles in the current landscape is attributable to the impedance of synthetic formulations to clinical fruition, despite the significant efforts, mainly because of their toxicity and biodistribution issues. Hence, bionanoparticles are considered a new paradigm for designing safe and reliable nanomedicine. The immune system is intrinsically able to recognize and eliminate malignant tumor cells. However, a cancerous mass evolved to hide from and escape immune surveillance. Adding immunomodulatory functions to bionanoparticle design, such as promoting M2 to M1 macrophage polarization or inducing inflammatory redox reactions, could potentially enhance nanoparticle-based approaches for cancer therapy by remodeling the tumor microenvironment for immune-mediated tumor eradication. To achieve such an end, the research community has spent effort on two fronts: (1) developing biomimetics that are fabricated with biological molecules on their surface to mimic the immunomodulatory functions of natural biological constituents and (2) engineering bio-immune components to immunologically inert bionanoparticles. In this regard, biomimetic formulation can comprise biological components and synthetic materials. The added biological components to organic or inorganic nanoparticles are vital in the formulation as they confer biocompatibility, specificity, and increased circulation time.
In this review, we will place biomimetic nanoparticles engineered with biologically sourced components under the umbrella of bionanoparticles together with biologically sourced, self-assembling nanoparticle systems. The objectives of this review are threefold. Firstly, it aims to facilitate future bionanoparticle design by presenting the common immune-related aberrations induced in the tumor microenvironment (TME). Secondly, the review will focus on the most applied strategies for achieving nanoimmunomodulatory functions. Finally, the limitations of each approach and potential avenues in the field of bionanoparticle design will be discussed.
2. Tumor-induced aberrations of the immune environment
Genetic instability is an evolving hallmark of cancer, resulting in the expression of abnormal proteins foreign to the immune system.13 Such foreign entities on the cancer cells serve as markers and immunogenic neoantigens that can spontaneously trigger CD8+ T-cell responses, which involve direct killing of the target via granule and cytokine secretion. Hence, the immune system is critical in identifying nascent tumor tissues and responding appropriately to uncontrolled cell division.14 The network of cell, molecular, and metabolic, effectors in the TME involved in tumor control and elimination is complex. Several deviations from the typical immune response can breed dysregulation, leading to molecular processes that drive oncologic pathologies. Recognizing the immune dysregulation within the TME is crucial for developing targeted therapies to disrupt the dynamics of interactions between cancer cells and the microenvironment, restore tissue homeostasis, and enhance antitumor immune responses. This section introduces and summarizes the cellular, molecular, and metabolic effectors involved in tumor-induced immune aberrations to provide a comprehensive view of this topic.2.1. Immunosuppressive cells
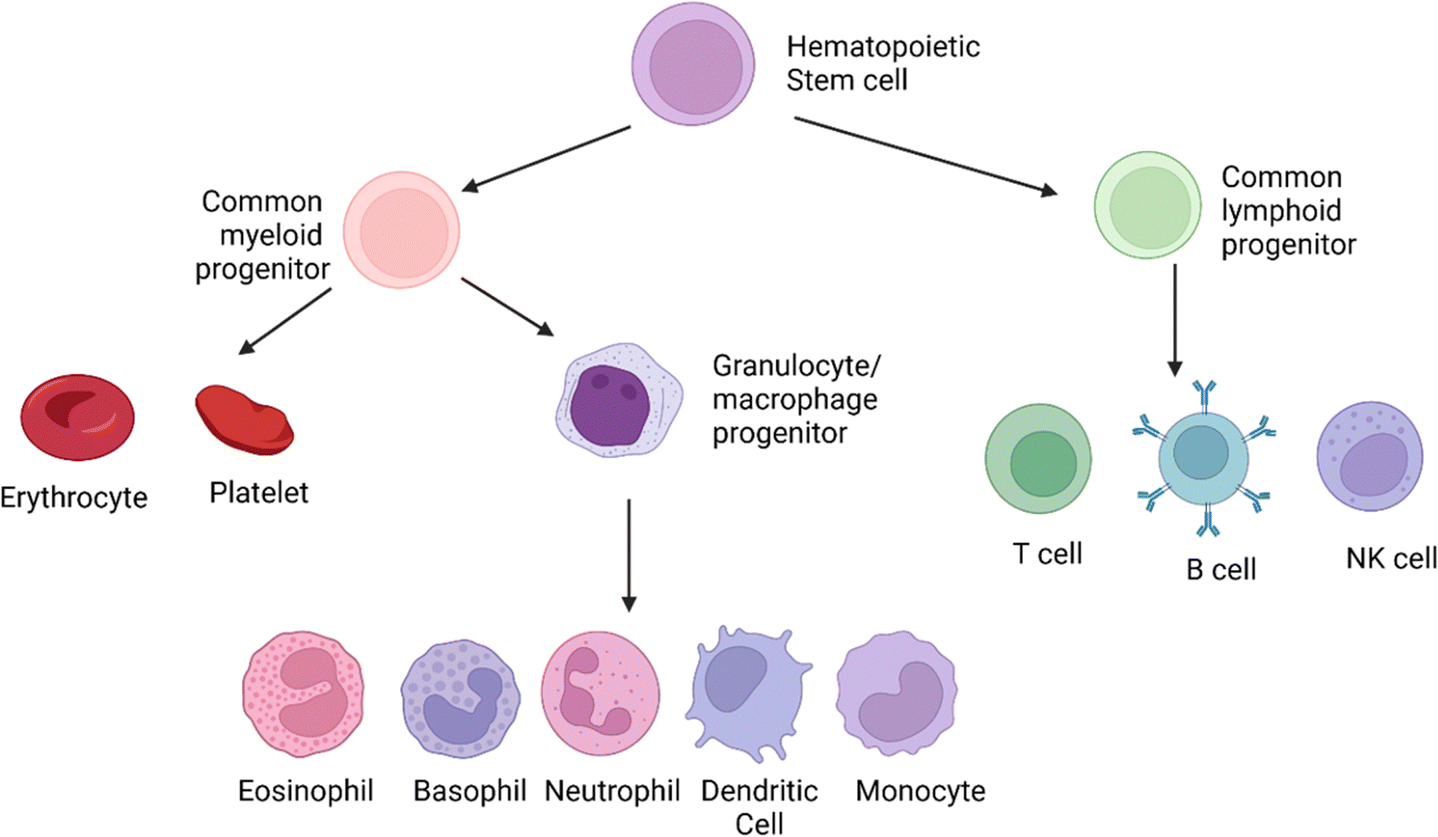 | ||
| Fig. 1 The origins of the cellular elements of the immune system. The cellular components of blood, the adaptive and the innate immune system, arise from the hematopoietic stem cells in the bone marrow. These cells divide to produce two specialized cells, the common myeloid progenitor and the common lymphoid progenitor. The common lymphoid progenitor gives rise to T-cells (CD4+, CD8+, Tregs), B-cells, and the natural killer (NK cells). T-Cells undergo differentiation in the thymus, while B cells differentiate in the bone marrow. Though NK cells are derived from the common lymphoid progenitor, they lack antigen specificity, which is the hallmark of the adaptive immune response. The common myeloid progenitor will give rise to the leukocytes via the intermediary macrophage/granulocyte progenitor. The monocytes (activated to macrophages in the tissues), dendritic cells, and polymorphonuclear granulocytes (eosinophil, basophil, neutrophil) are formed from the intermediary progenitors. The polymorphonuclear granulocytes are distinct in appearance due to their irregularly shaped nuclei.18 This image was created with BioRender.com. | ||
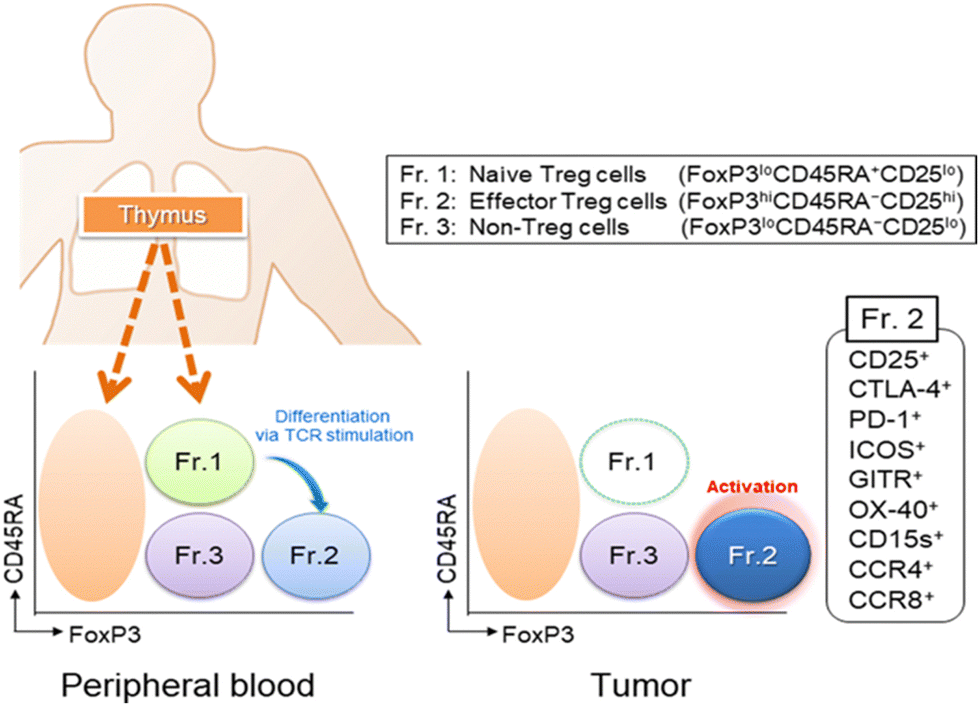 | ||
| Fig. 2 Tregs are classified into three subfractions: Fr.1 naïve/resting Treg cells (nTreg), Fr.2 effector/activated Treg (eTreg), and Fr.3 non-Treg cells. Typical staining patterns of Treg subfractions in peripheral blood and tumor tissues. Treg cells are found in low frequencies in peripheral blood (1–5%) but are higher in the TME (10–50%). Naive/resting Treg cells are hardly detected in the TME. Reproduced from ref. 16 Copyright 2019 with permission from John Wiley & Sons. | ||
Tregs possess multiple chemokine receptors (CCR4, CCR8, CCR10, CXCR3) and are targeted via chemoattractant chemokine gradients (CCL17, CCL22, CCL1, CCL28, CXCL0/10/11) secreted within the TME.19 They can impair the maturation of antigen-presenting cells (APCs) via the expression of checkpoint molecules CTLA-4 and PD1 (discussed in Section 2.2), induced by the transcription factor BATF.20 They also act as an IL-2 cytokine sink through the high expression of CD25 (IL-2 receptor α-chain), resulting in a limited amount of IL-2 in the TME for T-cell proliferation and activation.21 Treg also contributes to the inhibitory immune environment by secreting anti-inflammatory cytokines such as IL-10 and TGF-β, aiding in tumor growth and metastasis.22 The abundance of Tregs in the TME is also attributed to the metabolic pathways that breed a favorable environment for Treg proliferation, survival, and functions (discussed in Section 2.3).
MDSCs are further divided into monocytic (M)-MDSCs and granulocytic/polymorphonuclear (PMN)-MDSCs, defined by specific molecular markers on their surfaces Fig. 3. In mice, the MDSCs are identified functionally by their ability to suppress other immune cells due to the lack of phenotypic cell surface markers that distinguish classical neutrophils/monocytes from MDSCs. In humans, the MDSCs are characterized by the expression of specific markers, such as lectin-type oxidized receptor 1 (LOX1) for PMN-MDSCs and MHCII for M-MDSCs. However, MHCII is an inadequate determinant of M-MDSC in humans. Hence, efforts are ongoing to define the human MDSC subsets further, which will aid in exclusively identifying the suppressor cells in the population and benefit therapeutic strategies aimed at targeting MDSCs within the tumor milieu. MDSCs in the TME contribute to forming the premetastatic niche by facilitating the escape of tumor cells to the circulation and their subsequent engraftment by inducing immune suppression, matrix remodeling, and promoting angiogenesis.27 In the circulatory system, neutrophil PMN-MDSCs escort the circulating tumor cells (CTCs) and protect them from recognition and killing by NK cells.29 PMN-MDSCs also promote the extravasation of CTCs through the vasculature by trapping the CTCs in the microvasculature with an extracellular structure called neutrophil extracellular traps (NETs). NETs can also recruit tumor cells to the premetastatic niche via CDC25.30 Data from both in vitro and in vivo studies suggest the presence of MDSC markers that correlate with cancer conditions in humans.31,32 Translational studies have also demonstrated the predictive value of MDSCs in esophageal squamous cell carcinoma (ESCC) patients.33 However, there is a shortage of published clinical data on MDSC-targeted cancer therapy due to the lack of novel MDSC targets. Moreover, targeting MDSCs is limited by the heterogeneity of the MDSC populations with varying phenotypes and functions, which makes it a challenge to use specific markers and targets that can inhibit all MSDC subsets.
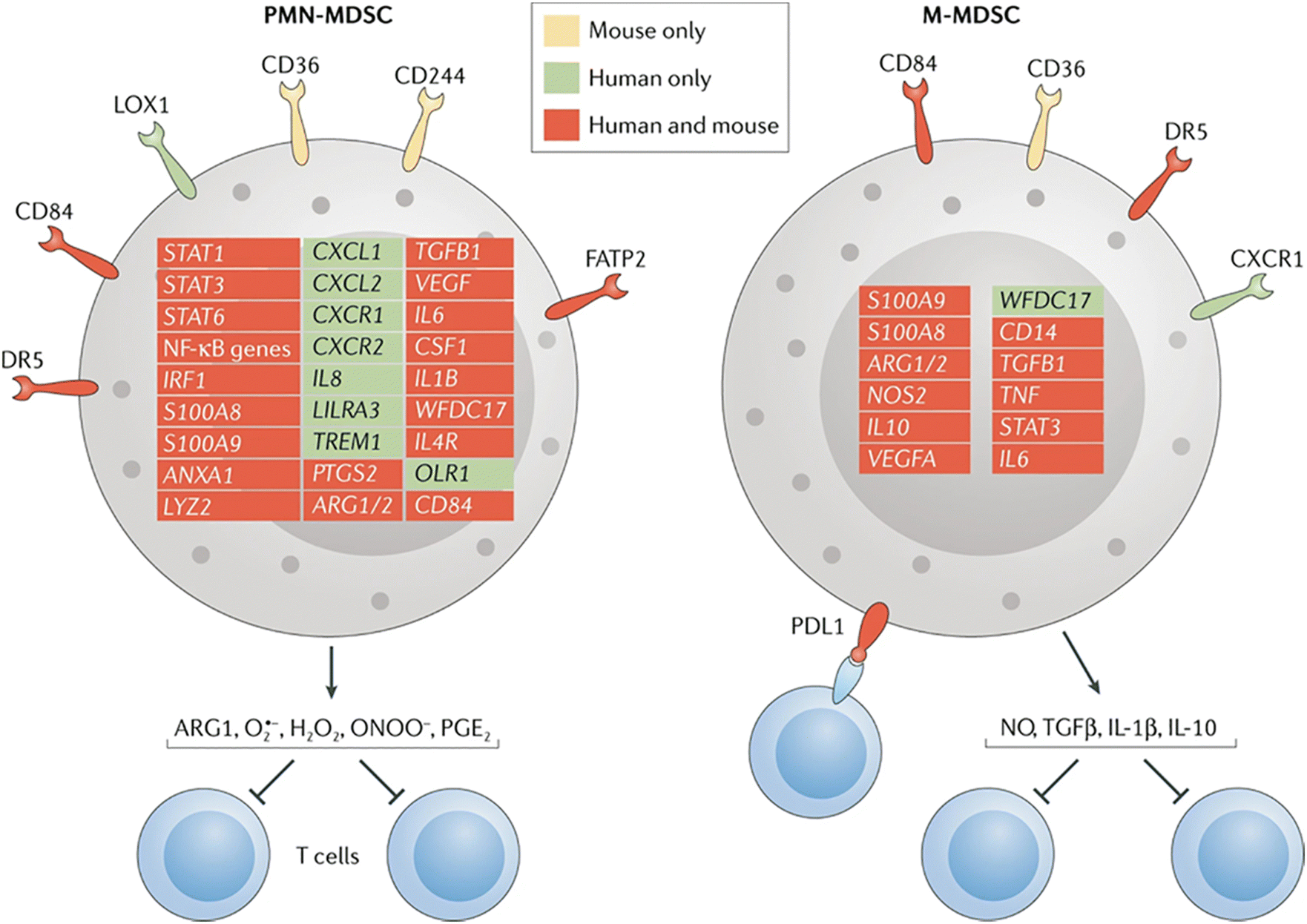 | ||
| Fig. 3 Distinguishing between the MDSCs subtypes. This figure shows the genes and surface molecules that distinguish the PMN_MDSCs and M-MDSCs from classical neutrophils and monocytes. CXCR1, CXC-chemokine receptor 1; FATP2, fatty acid transport protein 2; LOX1, lectin-type oxidized LDL receptor 1; NO, nitric oxide; PGE2, prostaglandin E2. Reproduced from ref. 27 Copyright 2021 with permission from Springer Nature. | ||
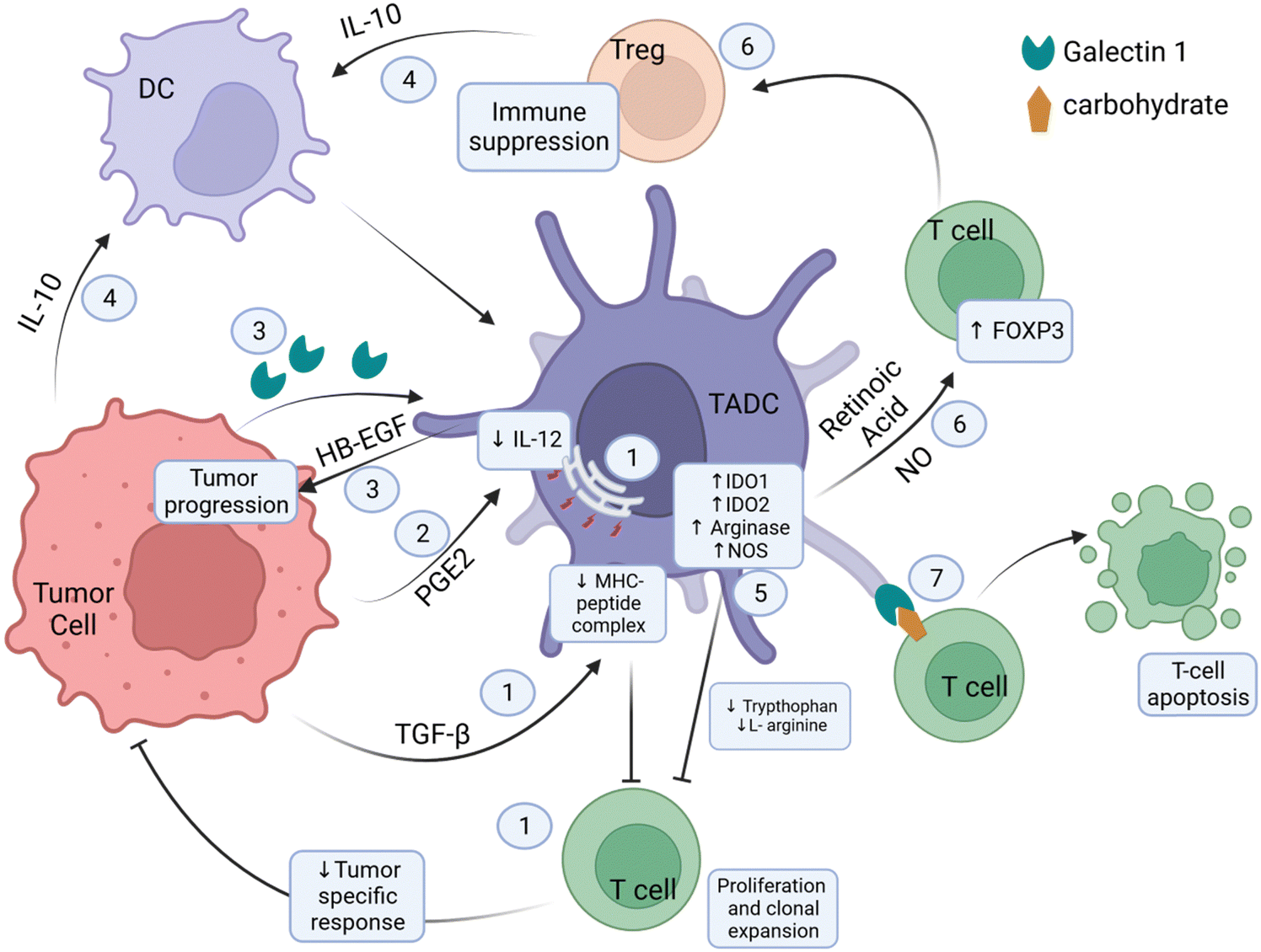 | ||
| Fig. 4 Tumor-DC-T-cell dynamics in the TME. ① The combination of tumor-derived TGF-β and ER stress results in inefficient loading of antigenic peptides on the MHC molecules, reducing the antigen-presenting ability of the DC to the T-cells. Hence, T-cells are unable to activate and mount a tumor-specific immune response. TGF-β limits the ability of the TADC to migrate to the lymph nodes and present antigens to T-cells. ② Tumor-derived PGE2 blocks IL-12 expression, which is essential for T-cell activation. ③ Secretion of galectin 1 by tumor cells induces TADC to secrete HB-EGF, which promotes tumor progression. ④ Tumor cells and Treg cells secrete IL-10, inducing the polarization of DCs to TADCs. ⑤ Overexpression of IDO1, IDO2, and arginase depletes tryptophan and L-arginine, inhibiting T-cell proliferation and clonal expansion. ⑥ Overexpression of iNOS leads to NO build-up. NO and retinoic acid produced by the TADCs induce FOXP3 expression, leading to T-cell differentiation to Treg cells. Treg cells induce and maintain an immunosuppressive environment in the TME. ⑦ Galectin 1 on TADC interacts with the carbohydrates on the surface of T-cells to induce apoptosis. HB-EGF, heparin-binding EGF-like growth factor; FOXP3, forkhead box P3; PGE2, prostaglandin E2; TGF-β, transforming growth factor β; IDO, indoleamine-2,3-dioxygenase; NOS, nitric oxide synthase; NO, nitric oxide; MHC, major histocompatibility complex. This image was created with BioRender.com. | ||
The β-catenin/T-cell factor (TCF) pathway in TADCs facilitates the expression of vitamin A-metabolizing enzymes, which catabolize vitamin A to retinoic acid (RA).37 RA induces Treg responses via Foxp3 activation and stabilizes the Treg phenotype.38 Other metabolic enzymes upregulated in TADCs are those involved in the catabolism of amino acids, namely tryptophan and L-arginine. Upregulated IDO1 and IDO2 degrades tryptophan, reducing the tryptophan levels in the TME. Upregulation of L-arginase catabolizes the breakdown of L-arginine. The catabolic activities deplete these amino acids, essential for T-cell effector function and proliferation.39–41 A separate mechanism also depletes L-arginine in the tumor milieu. TADCs overexpress NOS, and NOS requires L-arginine as a substrate to produce NO, which accumulates in the TME. NO can induce the conversion of CD4+CD25− T-cells to CD4+CD25+ Treg cells, resulting in Treg-induced immune suppressiveness in the TME.42 The expression of iNOS also suppresses DC differentiation into effector APCs.43 Tumor-derived prostaglandin E2 (PGE2) can also induce DC-mediated T-cell tolerance. Additionally, PGE-2 affects DC activity by blocking IL-12 expression and inducing the expression of regulatory molecules in T-cells, specifically CD25 and IDO, that will deter their stimulation.44,45 Functional proteins expressed on the surface of TADCs play various roles in inducing T-cell suppression. One of the surface molecules is Gal-1, which can also be secreted and bind to surface carbohydrates on T-cells to mediate downstream effects of inducing T-cell apoptosis.46 Gal-1 also promotes T-cell differentiation to Foxp3+ Tregs.47,48 TADCs also express inhibitory immune checkpoint ligand PD-L1 on their surface, which will engage surface PD-1 on T-cells, thereby transmitting inhibitory signals.49
Owing to its powerful antigen-presenting ability, the DCs were harnessed as a vaccination tool for cancer treatment in the late 1990s. Ex vivo preparations of tumor antigens-activated DCs were prepared and infused in patients. The first DC-based preparation (sipuleucel-T) for treating prostate cancer was FDA-approved in 2010.50 Combining DC-based therapy with chemotherapy and checkpoint inhibition was also investigated to provide safer and more effective treatment outcomes.51,52 A meta-analysis study of DC-based clinical trials confirms the clinical effectiveness of the regimen in improving mid-term survival for glioblastoma multiforme (GBM) patients and recurrence-free survival in HCC patients.53,54 Despite being promising, DC-based cancer vaccines for cancer immunotherapy are associated with several limitations, such as inconsistent vaccine efficacy due to variations in DC quality and quantity, inefficient loading of tumor antigens into DCs, and the plasticity of the DCs which makes them a challenge for clinical use. These limitations underscore the need for further research and development to optimize the efficacy of DC-based cancer vaccines in clinical settings.
TAMs in the TME are divided into two polarization states, namely M1 and M2 types. A coordinated network of stimuli, signaling pathways, transcriptional, and post-translational factors tightly control the polarization process.56,58 M1 macrophages are defined by the expression of iNOS and the generation of ROS.59 Due to their stimulation by infection and inflammatory events, the M1 phenotype possesses strong antigen presentation via MHCII expression and produces high levels of inflammatory cytokines IL-12 and IL-23.59 M2 phenotypes migrate and are activated in healing tissues that require debris removal, angiogenesis, injury repair, and tissue reconstruction.60 M2 macrophages are characterized by the upregulated expression of scavenger receptors, mannose receptors, dectin-1, DC-SIGN, and chemokine receptors (CCR2, CXCR1, and CXCR2). M2 macrophages express arginase, producing ornithine and polyamines via the arginase pathway.
In the TME, TAMs are generally polarized to the M2 phenotype, which enhances tumor survival by facilitating tissue regeneration and remodeling. However, the M1 and M2 macrophages are present in the TME proportionate to the balance of pro and anti-inflammatory markers in the tumor milieu (Fig. 5). Tumor-derived cytokines such as IL-4, IL-10, TGF-β, macrophage-CSF (MCSF), and CSF-1 have been implicated in M2 phenotype polarization.61 Other factors released by the tumor cells that are M2-polarizing include osteopontin and sonic hedgehog (SHH) protein.62 These factors drive M2 polarization by facilitating monocyte chemotaxis and activating molecular pathways (Hedgehog, Hh) that recruit TAMs and MDSCs for immune modulation.63,64
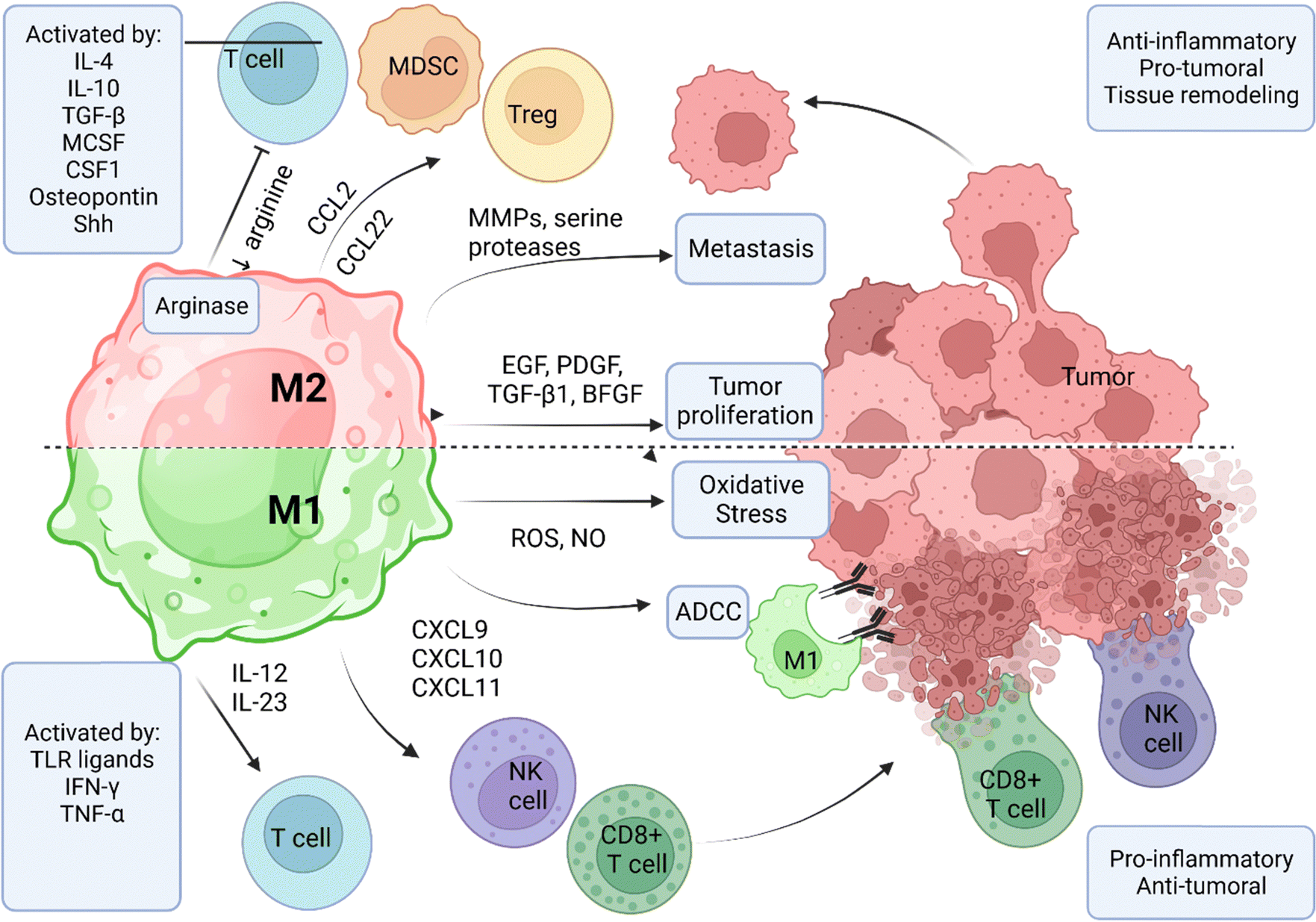 | ||
| Fig. 5 Functional differences between M1 and M2 TAM phenotypes and their role in the tumor microenvironment. M1 TAMs are activated by TLR ligands or pro-inflammatory cytokines such as TNF-α and IFN-γ. M1 TAMS are characterized by high production of inflammatory cytokines, ROS, and NO in the TME, leading to tumor oxidative stress and DNA damage. Inflammatory cytokines CXCL9, CXCL10, and CXCL11 recruits CD8+ T-cells and NK cells. These specialized killer cells can directly eliminate the tumor by secreting pro-apoptotic factors. Production of IL-12 and IL-23 facilitates T-cell activation, proliferation, and tumor-specific immune response. M1 TAMS can directly participate in tumor cell killing via the ADCC process, where their Fcγ receptors will interact with the Fc portion of tumor-specific antibodies bound to the tumor surfaces. Anti-inflammatory cytokines and factors such as IL-4, IL-10, TGF-β, and osteopontin activate M2 TAMS. M2 TAMS facilitates tumor progression by the secretion of proliferative and survival factors such as EGF, PDGF, TGF-β1, and BFGF. The secretion of MMPs and serine proteases enables the breakdown of the extracellular matrix to facilitate tumor metastasis. M2 TAMs secrete CCL22 to create a chemoattractant gradient Treg accrual and activation. Secretion of CCL2 facilitates the recruitment of MDSCs into the tumor milieu. Overexpression of arginase depletes L-arginine from the TME, inhibiting T-cell proliferation and clonal expansion. IL, interleukin; TGF-β, transforming growth factor β; MCSF, macrophage colony-stimulating factor; CSF-1, colony-stimulating factor 1; Shh, sonic hedgehog protein; IFN-γ, interferon-γ; TNF-α, tumor necrosis factor α; MMP, matrix metalloproteinases; EGF, epidermal growth factor; PDGF, platelet-derived growth factor; BFGF, basic fibroblast growth factor; ROS, reactive oxygen species; NO, nitric oxide; ADCC, antibody-dependent cellular cytotoxicity. This image was created with BioRender.com. | ||
M2 phenotype TAMs create a hospitable environment for tumor survival by releasing factors that promote tumor progression and inhibit the effector immune cells. Tumor-proliferating factors expressed by M2 TAMs include the epithelial growth factor (EGF) and other ligands of the epithelial growth factor receptor (EGFR) family, the platelet-derived growth factor (PDGF), TGF-β1, and the basic fibroblast growth factor (BFGF).65 M2 TAMs facilitate metastasis via releasing enzymes and factors that degrade the components of the extracellular matrix, such as MMPs and serine proteases, thereby facilitating the migration of tumor cells into the circulatory system.66 Most importantly, M2 TAMs contribute to the immunoediting of the TME by suppressing T-cell function and inhibiting CD8+ T-cell proliferation facilitated by arginase expression.67 Finally, M2 TAMs can recruit Treg and MDSCs to the TME by secreting CCL22 and CCR2 ligands.68–70 The activation of M1 macrophages is vital for controlling tumor burden via three classical immune-mediated processes: (i) indirect killing by the accrual of other immune cells that can lyse and kill the cancer cells, (ii) direct cytotoxicity by the release of harmful products such as ROS, and (iii) antibody-dependent cellular cytotoxicity (ADCC). M1 phenotype activation is induced by stimuli such as toll-like receptor (TLR) ligands and pro-inflammatory cytokines.58 M1 phenotypes release inflammatory cytokines in the TME, such as CXCL9, CXCL10, and CXCL11, that recruit and activate CD8+ T-cells and natural killer cells.71 M1 TAMs kill tumor cells by releasing cytotoxic factors such as ROS and NO, which cause cell death via oxidative stress and DNA damage.72 M1 macrophages play a crucial role in ADCC by recognizing and eliminating antibody-bound target cells by interacting with the Fc fragment of antibodies via their Fc gamma receptors (FcγR). The receptor interaction triggers phagocytosis of the antibody-bound tumor cells, resulting in their elimination and removal.73
The M1 to M2 phenotype ratio is a proxy indicator for the inflammatory-to-anti-inflammatory factor balance. Various clinical studies have indicated that the M1 to M2 ratio correlates with improved survival, an indication to justify the ratio as a prognostic marker and to target TAM polarization for cancer therapy.74–77 Pre-clinical studies in mice suggest that targeting TAM activation via MHC I molecules,78,79 or reprogramming TAMs into antitumor M1 phenotype might achieve better success in the clinic due to the positive activation effects on antitumor M1-like functions.80,81
Regulatory B-cells. Human B cells derive from the common lymphocyte progenitor lineage in the bone marrow, where they undergo V(D)J recombination to become immature/naïve B-cells expressing B-cell receptors (BCRs) of the IgM/IgD isotype. After encountering their cognate antigen, B-cells undergo affinity maturation and class switch recombination (CSR) to develop into memory B cells or antibody-producing plasma cells with isotypes IgG/A/E. B-Cells can exert anti-tumor functions by activating the complement system and generating neoantigen-specific antibodies that bind to NK cells and macrophages via their Fc receptors.82 The accrual of these effector cells to antibody-targeted tumor cells facilitates recognition and subsequent attack for elimination. DCs and B-cells can recognize these antibodies and internalize the neoantigens for presentation to CD4+ T-cells and CD8+ T-cells. Finally, B-cells release various cytokines and cytotoxic effector molecules that propagate and regulate immune responses.83 Studies using single-cell RNA sequencing (scRNA seq) of tumor biopsies revealed B-cell populations spanning many states and isotype expression in a cancer-dependent fashion.84 Generally, an unswitched naïve-like state and a switched state with a memory-like phenotype are observed with rare populations of germinal center cells and plasma cells.82 However, the exact role of these B-cell states and the antigens they react to remain elusive. These tumor-infiltrating B-cells (TIL-Bs) are usually associated with a positive response and promote anti-tumor immunity via antigen presentation to T-cells.82
Several protein surface markers studies observed regulatory B cells (Bregs) expressing the IgA isotype and secreting IL-10, IL-35, and TGF-β. Bregs are the antithesis of tumor-infiltrating B-cells and are similar to Tregs. Bregs exert strong immunosuppressive functions, but unlike Tregs, which are synonymous with FOXP3 expression, Bregs do not present with a uniform surface marker or transcription factor expression.85 Bregs can exert immunosuppressive effects from a distance by producing antibodies that circulate to the tumor and engage the Fc receptors on macrophages and mast cells, thereby facilitating a pro-tumorigenic state.86 B-Cells can utilize the PD-1/PD-L1 immune checkpoint pathway and express PD-L1 to suppress CD8+ T-cell responses in the TME. Finally, the most commonly reported Breg immunosuppressive mechanism in human studies is the secretion of IL-10.86 IL-10+ Breg cells were detected by immunohistochemistry or flow cytometry in various human cancers.87,88 Breg and Treg expression are also strongly correlated in these studies, suggesting a possible interaction between these two immunosuppressive subtypes.88 Another critical regulatory cytokine secreted by Breg cells is IL-35, which is functionally divergent from IL-10+ Bregs in that IL-35 limits memory differentiation while IL-10 suppresses effector cell functions.85 Nevertheless, these Breg subtypes showed overlapping ability to induce inhibitory receptors such as PD-1 and TIM-3.85
There is congregating evidence that tumor-infiltrating B-cells are prominent in anti-tumor responses. The presence of TIL-Bs is associated with the accrual of effector T-cells, NK cells, and myeloid cells in the TME of ‘hot tumors,’ indicating active antigen recognition and diverse effector functions. However, manipulating TIL-Bs for immunotherapy presents a limitation because their effector functions rely heavily on the nature of their cognate antigens. Hence, ongoing efforts to fully understand their antigenic profiles with methodologies complementing scRNA-seq, enabling the mapping of clonotypes to phenotypes and antigen-specificities. Immunotherapy strategies targeting TIL-B activation (cytokines, antibodies, and vaccines) or antibody-induced Breg depletion were investigated in disease models to delineate the molecular mechanisms involved in treatment efficacy.85,86
Cancer-associated fibroblasts (CAFs). CAFs are activated fibroblastic cells in the TME that present a distinct phenotype from the latent fibroblasts in normal tissues. CAFs can originate from bone marrow-derived precursors/mesenchymal stem cells (MSCs) or tissue-resident fibroblasts, adipocytes, and pericytes.89 Various factors are involved in cancer-induced fibroblast reprogramming, including epigenetic changes, microRNA expression, metabolic determinants, and oxidative stress.90 Generally, CAFs are divided into three functional groups: tissue remodeling (myofibroblastic CAFs), mutual signaling with other cells within the TME, and immune regulation. Currently, there is no single definitive marker that can be used as a determinant hence, both positive and negative markers are used to identify and distinguish latent fibroblasts from CAFs.
Profiling CAF populations across malignancies using scRNA-seq and flow cytometry established the current knowledge of CAF phenotypes and functions. Biffi et al. summarized the phenotypic and functional yield of these studies in their review article, which profiled CAFs in pancreatic ductal adenocarcinoma (PDAC), breast cancer, lung cancer, melanoma, colon cancer, and head and neck cancer.90 In general, both myofibroblastic (α smooth muscle actin (SMA)-high) and non-myofibroblastic (αSMA-low) CAF populations are present across all cancer types.90–93 Myofibroblastic CAFs are associated with extracellular matrix (ECM) signature, whereas non-myofibroblastic CAFs are typically secretory and inflammatory.94 ECM deposition of CAFs plays distinct roles in the TME, including provision of nutrients, obstructing drug delivery, which leads to hypoperfusion and elevated interstitial fluid pressure, and supporting tumor growth.90 Non-myofibroblastic CAFs create an immunosuppressive TME by preventing T-cell activity and accrual through the secretion of immunosuppressive ligands such as TGF-β and CXCL12.90 CAFs can recruit monocytes, differentiate macrophages, and polarize them into the pro-tumorigenic M2 subtype.95 Additionally, CAFs can recruit immunosuppressive cell populations such as the MDSCs into the tumor stroma. Interestingly, antigen-presenting CAFs (apCAFs) express MHCII proteins but do not express co-stimulatory molecules, which are essential in the induction of T-cell activation and clonal expansion. Hence, it was hypothesized that apCAFs may act as a decoy receptor to detain T-cells and inhibit their response.
Recent studies in PDAC highlighted CAF subtypes that displayed tumor-restraining properties.90 The genetic depletion of αSMA cells and the deletion or pharmacological intervention of sonic hedgehog (SHH) of the hedgehog (Hh) pathway led to reduced survival in preclinical and clinical studies.96,97 The results indicate a tumor-restraining role for these cells. However, genetic approaches will affect other αSMA and SHH-expressing populations, so the assumption that CAF subtypes were involved must be cautiously approached. However, the heterogeneity and diverse roles of CAFs in tumor burden control underscore the need for a unified classification system that can aid in the development of targeted therapies.98
2.2. Immune checkpoint molecules
PD-L1, the ligand binding to PD-1, is a 33 kDa type 1 transmembrane glycoprotein with immunoglobulin domains in its extracellular region. PD-L1 is expressed in various cell types, including lymphocytes, lung tissues, vascular endothelium, mesenchymal stem cells, islet cells, astrocytes, neuronal cells, and keratinocytes.101,102 PD-1 and PD-L1 are crucial immune checkpoint molecules. Their interaction induces inhibitory signaling pathways that fine-tune the activation of effector T-cells during antigen presentation (Fig. 6). However, PD-L1 is also notably overexpressed in tumor cells, contributing to the evasion of immune system-mediated eradication. Its interaction with the PD-1 receptor, expressed on activated T-cells, can suppress T-cell responses, resulting in T-cell anergy, inhibition of cytokine expression, and T-cell apoptosis.103 Thus, overexpression of PD-L1 on tumor cells is one of the essential strategies for evading immune response and eradication. Genomic alterations (amplification or translocation), aberrant signaling pathways (inflammatory and oncogenic signaling), and post-transcriptional/translational modifications (miRNA, methylation, phosphorylation, glycosylation) contributed to the oncogenic overexpression of PD-L1 on tumor and immune cells. The details of such mechanisms have been extensively reviewed by Yadollahi et al.104 Under normal conditions, the interaction of PD-1 with PD-L1 results in a signaling cascade that protects healthy host tissues by promoting Treg development and inhibiting self-reacting T-cells.105 PD-L1 could also be upregulated during multiple inflammatory signaling pathways to restrain T-cell hyperactivity.106 However, the interaction of PD-1 with overexpressed PD-L1 on tumor cells suppresses normal antitumor immunity, protecting the cancerous cells from the host response.
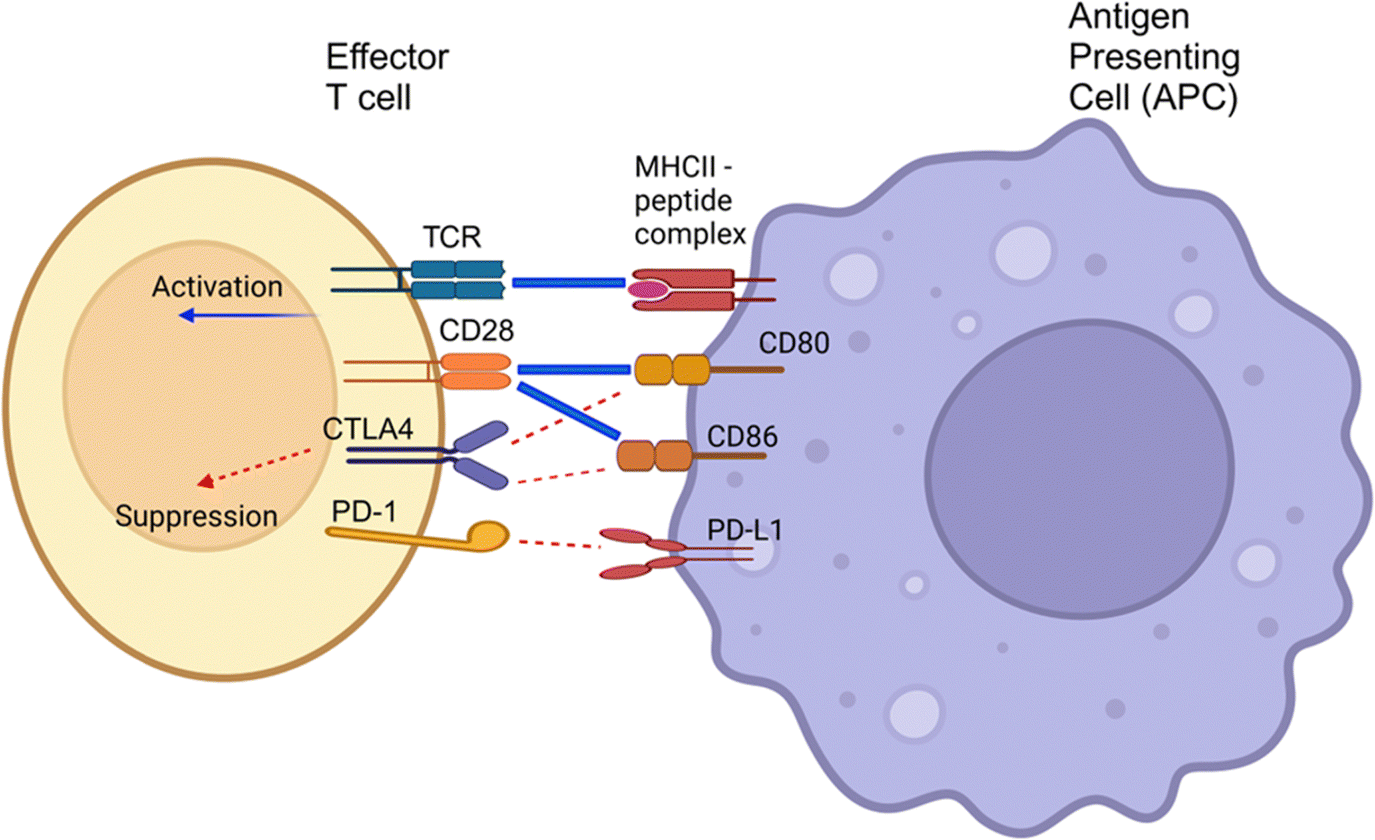 | ||
| Fig. 6 The activation of T-cells by antigen-presenting cells. T-Cell activation requires two activation signals (blue lines). The priming of the T-cell is initiated by the recognition of the MHC I or II molecules, complexed with an antigen peptide. A co-stimulatory signal is generated by the interaction of CD28 molecules with either CD80 (B7-1) or CD86 (B7-2). The activation signals will induce T-cell activation and responses such as proliferation and secretion of cytokines. The degree of activation can be modulated by additional stimulatory (e.g. ICOS-ICOSL, not represented in the schematic) or inhibitory signals. Inhibitory signals are mediated by immune checkpoint molecules, PD-L1/PD-1 and CTLA-4. CTLA-4 binds to CD80 or CD86 with greater affinity than CD28, inhibiting its co-stimulatory function. TCR, T-cell receptor; MHCII, major histocompatibility complex II; CTLA4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1. This image was created with BioRender.com. | ||
The pathways activated by the PD-1/PDL-1 interaction are widely studied and extensively reviewed by Patsoukis et al.107 The cytoplasmic tail of PD-1 contains an immunoreceptor tyrosine-based inhibition motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM).108 The tyrosine residues in these two domains are phosphorylated after PD-1/PD-L1 interaction, leading to the recruitment and activation of Src homology region 2 (SH2) domain-containing tyrosine phosphatase-2 (SHP-2) and the homologous SH2 domain-containing tyrosine phosphatase-1 (SHP-1). These phosphatases will then dephosphorylate CD28, T-cell receptor (TCR), and other costimulatory molecules, inhibiting the signal transduction that leads to T-cell proliferation and cytokine secretion. This sequence of events is known as the canonical “trans” interaction, whereby the interacting PD-L1 and PD-1 are expressed on two different cell types (Fig. 7). However, the molecular interplays vary depending on the co-expression status of PD-L1 and PD-1. When PD-L1 and PD-1 are co-expressed in an APC or cancer cells, it will result in a cis level of interaction where the PD-L1 and PD-1 expressed within the same cell interact and diminish the ability of the PD-L1 to bind to PD-1 expressed on T-cells in trans.109 Hence, co-expressing APCs or cancer cells fails to induce an immunosuppressive effect on PD-1-expressing T-cells. Understanding the mode of PD-1/PD-L1 expression is very important in treatment strategies using PD-1 blockade.
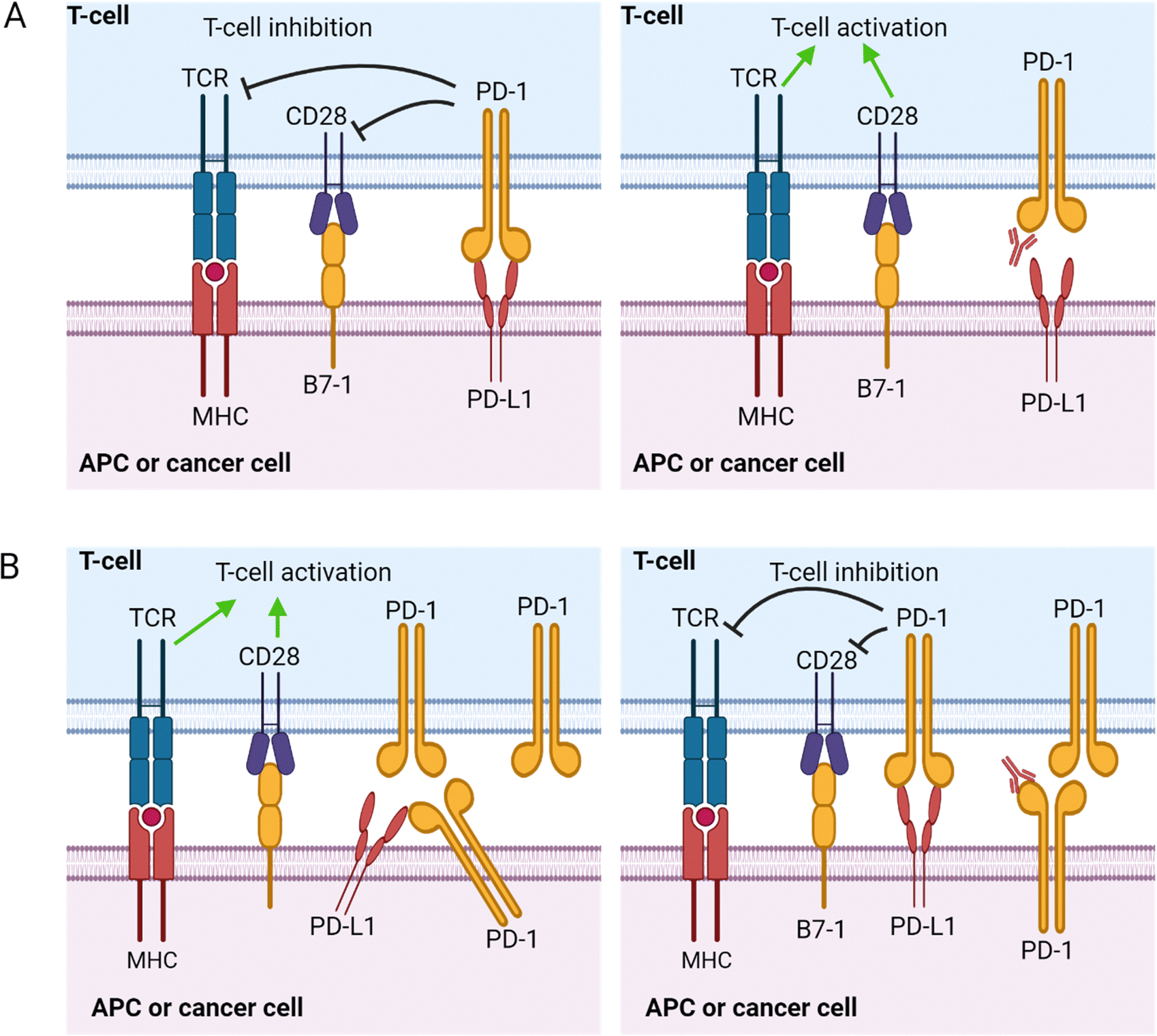 | ||
| Fig. 7 The trans and cis interaction modes of PD-1 and PD-L1. (A) The canonical PD-1/PD-L1 interaction is a trans-mode interaction. PD-L1 expressed on APCs or tumor cells interacts with PD-1 expressed on T-cells in trans. The PD-1/PD-L1 interaction will attenuate the activation of T-cells mediated by the TCR/MHC and B7-1/CD28 stimulation pathways. Checkpoint blockade in trans mode will abrogate the inhibitory signaling of PD-1/PD-L1 interaction and restore T-cell activation. (B) Cis mode interaction occurs when PD-1 and PD-L1 are co-expressed in tumor cells or APCs. Co-expressed PD-1 and PD-L1 will interact, preventing the interaction of PD-L1 on APCs/tumor cells to PD-1 on the T-cells in trans. Checkpoint blockade in cis interaction mode reactivates inhibitory PD-1/PD-L1 interaction in trans. This image was created with BioRender.com. | ||
The prognostic value of PD-L1 and PD-1 in various cancers has been well reported.110 PD-L1 expression has been associated with poor clinical outcomes in colorectal cancer,111 gastric cancers,112 head and neck cancers,113 and breast cancer.114 However, PD-L1 upregulation was associated with better outcomes in an aggressive subtype of breast cancer.115 Such an observation could result from the cis PD-1/PD-L1 interaction that suppresses PD-1 signaling. Hence, PD-1/PD-L1 interaction modes must be factored in for checkpoint blockade therapy eligibility. Clinical trials have demonstrated that PD-L1 or PD-1 blockade therapy successfully abrogates the inhibitory effects, restoring T-cell antitumor activities and functions.116 Tang et al. meticulously studied the data presented from 99 clinical trials to find that preoperative anti-PD-1/PD-L1 combined therapy, particularly with chemotherapy, could achieve better treatment response rates and reduce the number of immune-related adverse events compared to PD-1/PD-L1 monotherapy or dual immunotherapy.117
The role of CTLA-4 in antitumor immunity occurs at the junction between T-cells and APCs, where the innate immune cells present antigenic molecules to adaptive immune effector cells. The interaction between the two components enhances immune response by instructing the recognition of patterns for destroying cells expressing the antigenic molecules, continued surveillance, and memory for long-lasting protection.122 CTLA-4 and CD28 share binding capabilities with two ligands expressed on APCs: CD80 (B7-1) and CD86 (B7-2).118 CTLA-4 interacts with both ligands with higher affinity to CD28, thereby acting as an antagonist of CD28-mediated co-stimulation of T-cell activation, proliferation, and cytokine secretion (Fig. 6).118 The expression of CTLA-4 is primarily localized and stored in intracellular vesicles due to the rapid constitutive endocytosis of CTLA-4 from the plasma membrane. CTLA-4 is rapidly recruited to the cell surface from vesicle storage and new gene expression upon T-cell activation (Fig. 8).118 However, sustained overexpression of CTLA-4 is often induced in tumor-infiltrating T-lymphocytes, contributing to the progression of both solid and hematological cancers via aberrant signaling pathways.123–125
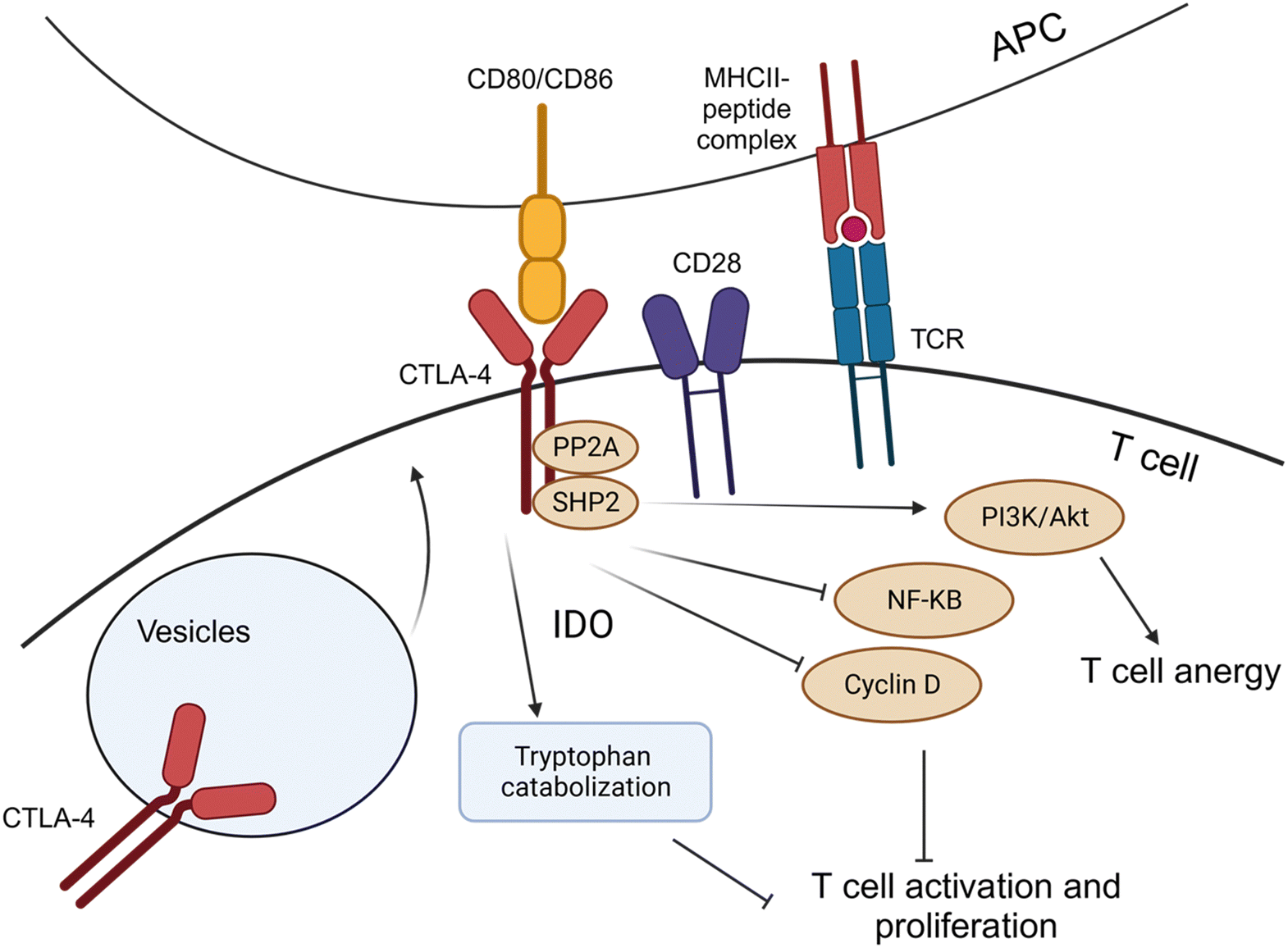 | ||
| Fig. 8 CTLA-4 is usually stored in intracellular vesicles and is rapidly recruited to the cell surface following T-cell activation. CTLA binds to CD80/86 at a higher affinity than CD28, displacing the molecule from the co-stimulatory complex. The interaction with its ligands induces CTLA-4 activation, mediated by the signalosome that includes PP2A and SHP-2. CTLA-4 activation activates the PI3K/Akt pathway to sustain T-cell anergy without immunogenic cell death. NF-KB and cyclin D pathways were inactivated, resulting in the inhibition of T-cell activation and proliferation. Additionally, indoleamine-2,3-dioxygenase (IDO) activity catalyzes the breakdown of tryptophan, increasing the intracellular levels of tryptophan degradation products, which are effector T-cell inhibitors and induce Tregs. This image was created with BioRender.com. | ||
During tumorigenesis, CTLA-4 decreases the T-cell activation by various molecular pathways. After TCR engagement, CTLA-4 expression and recruitment to the membrane were immediately increased, and it competes with the CD28 costimulatory molecule for the CD809/CD86 ligands, leading to a weakened immune response. The interaction of CTLA-4 with the CD80/CD86 molecules in circulating APCs can induce the activity of indoleamine-2,3-dioxygenase (IDO), which leads to catabolization of tryptophan, which is essential for T-cell proliferation, and the generation of inhibitory tryptophan metabolites125 (Fig. 8). Additionally, the signalosome interaction between serine-threonine protein phosphatase 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A (PP2A) and (SHP-2) activates the downstream PI3K/Akt pathway, which helps to sustain T-cell anergy and immune tolerance without inducing antigen-induced cell death.126 The signalosome also inhibits the NF-κB and cyclin D pathways, which are essential regulators of T-cell activation and proliferation.127–129
A (PP2A) and (SHP-2) activates the downstream PI3K/Akt pathway, which helps to sustain T-cell anergy and immune tolerance without inducing antigen-induced cell death.126 The signalosome also inhibits the NF-κB and cyclin D pathways, which are essential regulators of T-cell activation and proliferation.127–129
CTLA-4 is constitutively expressed on FoxP3+ positive cells, unlike conventional T-cells, which express CTLA-4 only after activation, indicating its specific association with Treg cells.130 CTLA-4 blockade treatment with monoclonal antibodies led to the depletion of Treg cell populations in the TME. Studies analyzing the prognostic value of CTLA-4 in cancer treatment yield controversial outcomes.131 Ipilimumab is the first anti-CTLA-4 human monoclonal antibody that was first FDA-approved for treating advanced melanoma patients with a considerable therapeutic effect in renal cell carcinoma.132,133 However, anti-CTLA-4 antibody treatment exhibited a higher incidence of immune-related adverse events (irAEs) than those recorded in PD-1 blockade studies.134 Hence, combinations of CTLA-4 blockade with other therapies, such as chemotherapy, radiotherapy, and double checkpoint blockade (CTLA-4 plus PD-1), were investigated to reduce the severity of irAEs with favorable risk-benefit profiles135,136
Tim-3. TIM-3 was identified 12 years ago as a cell surface molecule selectively expressed on IFN-producing T-cells.137 Now, the expression of Tim-3 has been demonstrated on Treg cells, DCs, NK cells, and monocytes. Tim-3 expression is encoded by the Tim family of genes associated with immune-mediated diseases such as asthma and allergy.138 Binding of Tim3 with Tim-3 ligands (galactin-9: Gal-9, high-mobility group protein: B1HMGB1, carcinoembryonic antigen cell adhesion molecule 1: CEACAM-1, phosphatidylserine: PtdSer) initiate a signaling cascade that results in the activation of nuclear factor of activated T-cells (NFAT) and nuclear factor kB (NF-κB) of the TCR signaling pathway in a normally regulated immune reaction.139 However, in malignant diseases, Tim-3 expression marks dysfunctional or exhausted CD8+ T-cells, which is exacerbated when Tim-3 is doubly expressed with PD-1.140 The first Tim-3 monoclonal antibody, sabatolimab, blocks the binding of Tim-3 to ligands Gal-9 and PtdSer to restore T-cell function by obstructing Tim-3 mediated exhaustion. A phase I/Ib clinical trial of sabatolimab alone and with anti-PD-1 showed that the combination treatment was well-tolerated with preliminary signs of anti-tumor activity.141
Lag-3. Lag-3 was discovered in 1990 as a molecule upregulated on activated T-cells and a subset of NK cells.142 Structurally, Lag-3 resembles CD4 co-receptor and binds to MHCII with higher affinity than CD4.142 Another Lag-3 ligand is LSECtin, a member of the DC-SIGN family of molecules. Lag-3 is a negative regulator of T-cell receptor signaling, but it promotes Treg cell-mediated suppression. The opposing effects of Lag-3 engagement beg the question of the Lag-3 mediated signaling on different effector T-cell subsets to achieve its immunosuppressive effects. At present, Lag-3-related signaling events remain unclear. The known Lag-3 intracellular interaction is crosslinking with CD3 to inhibit calcium flux, T-cell proliferation, and cytokine production.143 Relatlimab is the first commercially developed anti-LAG-3 mAb currently being investigated with the PD-1 inhibitor, nivolumab, to treat unresectable or metastatic melanoma in a phase 2/3 randomized trial. The combination of the two checkpoint inhibitors enhanced progression-free survival than anti-PD-1 therapy alone without any new adverse events.144
TIGIT. TIGIT was identified in 2009 in a genome-wide search for costimulatory or inhibitory molecules on activated T-cells.145 TIGIT is widely expressed in regulatory, memory, and activated T-cells, and its expression levels are upregulated after lymphocyte activation.145 There are five TIGIT ligands: CD155 (also known as PVR), CD112, CD113, Nectin4, and Fab2.139 The binding of TIGIT to its ligands activates immunosuppressive pathways via the interference of T-cell co-stimulation signaling mediated by CD226 or CD96.139 Currently, a phase I anti-TIGIT therapy with IBI939 antibodies is currently ongoing in China for patients with leukemia and solid tumors.146 The investigating team has yet to publish their results.
2.3. Immunosuppressive metabolites
Metabolic pathways release soluble mediators in the tumor microenvironment that influence and shape the immunobiology of many tumor types. This section lists the critical metabolic pathways that lead to profound immunosuppression of effector cell responses.3. Current status of nano-immunotherapy for cancer
Cancer immunotherapy has become a research hotspot during the last two decades. However, current cancer immunotherapy strategies remain inundated with challenges and limitations. A subset of patients fails to respond to the therapy, relapsing after initial treatment. Additionally, the immune-related adverse events that follow are potentially debilitating and life-threatening. Hence, there has been a recent budding pursuit of developing nanotechnology and nanoengineering platforms to narrow the gap in cancer immunotherapy strategies. Examining the repertory of nanoformulations engineered for cancer immunotherapy currently investigated in human clinical trials will provide insights into the strategy, the investigative and evaluation process, and possible avenues for further research endeavors.Due to its FDA approval status, clinical success, and the maturity of the technology, liposomes are the predominant material of choice for developing immunomodulatory formulations for cancer therapeutics. Liposomal platforms are potentially versatile, able to encapsulate various pharmacological agents, and can be functionalized for active targeting of specific tissue sites. An immunotherapy agent for cancer treatment should ideally target tumor-associated antigens (TAAs) to drive the development of a tumor-specific immune response. The most widely investigated liposomal formulation with TAA targeting in the clinic is Tecemotide, formerly known as L-BLP25 or Stimuvax®, which targets adenocarcinomas that express mucin-1 (MUC-1), a member of the membrane-bound 0-glycoprotein mucin.155 FixVac is an RNA–liposome complex formulation comprising RNA encodes for tumor-associated antigens, NY-ESO, and MAGE-3. FixVac is currently investigated in a phase I trial as a potential immunotherapy for melanoma. Interim analysis detected strong CD4+ and CD8+ T-cell responses after vaccination with FixVac.156 The study was completed recently, but the investigative team has not published the results. Lipovaxin-MM is a liposome-based nanotechnology formulated to treat malignant melanoma. It targets DC activation to harness their potent antigen-presenting ability. Very little treatment efficacy was demonstrated when the vaccine was used to treat a small group of patients with malignant myeloma. No humoral or cellular response to the vaccine was observed, indicating that immunosuppressive mechanisms induced by the tumor may have negated the effect of the vaccine.157
Due to careful ethical considerations and calculated risk-to-benefit ratio, exploratory formulations are investigated in advanced, refractory, or recurrent malignancies, which are often a dilemma to manage due to the lack of alternative treatment options. Observing beneficial responses, such as increased overall survival and tolerable toxicity profiles in these clinical cases, are signs of further potential, especially in early malignancies or as preventative measures. Research endeavors should also focus on developing similar biomimetic or bionanoparticle platforms for cancer therapy. For example, exosomes are being investigated in the clinic as prognostic markers (https://www.clinicaltrials.gov). One clinical study was conducted to test the efficacy of a tumor vaccine comprising tumor-antigen-loaded DC-derived exosomes on patients with unresectable NSCLC (ClinicalTrials.gov ID: NCT01159288). However, the results were not published. The outcomes of these studies could provide insights into developing efficient strategies for cancer vaccination and immunotherapy. Nevertheless, the liposome-based cancer immunotherapy studies in the clinic have generated some critical insights that could help direct future growth areas. Table 1 depicts a simple SWOT analysis of the current nano-immunotherapy landscape in the clinic. Based on the study, bionanomaterials have considerable potential to impact this subspecialty of the oncology field.
| Strength | Weakness |
|---|---|
| • Inventory of mature technologies for innovation e.g. peptides, humanized monoclonal antibodies, checkpoint inhibitors | • Unclear plans for transitioning laboratory-developed technologies to the clinic |
| • Human-derived bionanoNPs have a better safety profile | • Resource limitations (e.g. lack of suitable TAAs and distinct immune surface markers) |
| • Microbial and viral-like particles have intrinsic immune modulating abilities |
| Opportunities | Threats |
|---|---|
| • Developing computational analysis and AI base technologies that can strategize and predict probability of efficacy | • Debilitating and severe iRAEs |
| • Developing nanovaccines for preventative measure and early-stage malignant disease | • Risk –averse regulatory environment |
| • Unmet needs in hematological malignancies after patients fail CAR-T cell therapy |
4. Strategies for functionalizing bio-nanoparticles for tumor immunomodulation
The tumor microenvironment is complex, shaped intricately by the growing tumor that exhibits heterogeneity, increasing the challenge of standard therapies to provide a complete response. Modulating the immune response towards cancer termination inflammatory phenotypes has significantly impacted clinical practice with the advent of checkpoint inhibitors and CAR-T-cell therapy. Using bionanoparticles to modulate the immune response is a relatively new yet rapidly evolving field with significant potential for clinical translation. Bionanoparticles, such as cell membrane-derived nanoparticles, exosomes, and albumin-based nanoparticles, have garnered attention for their ability to modulate immune responses and deliver therapeutic agents to tumors. The prospect for bionanoparticles in cancer immunotherapy is optimistic, with continued efforts to refine their therapeutic potential and advance their translation into clinical practice. This section focuses on the most utilized bionanoparticles engineered to stimulate an anticancer immune response.4.1. Targeting tumor influx via cell membrane biomimetics
A pivotal area of research focuses on using cell membrane biomimetics to target tumor influx and enhance antigen presentation, thereby overcoming the immune evasion tactics that tumors employ. This section examines the latest advancements in the design and application of biomimetic nanoparticles derived from various cellular membranes, including those of tumor cells, immune cells, and blood cells, each offering unique mechanisms to modulate the tumor microenvironment and potentiate the immune response. Tumor cell membrane-coated nanoparticles are at the forefront of this research, with studies demonstrating their ability to mimic the antigenic profile of tumors and stimulate specific immune responses.158–160 These biomimetic platforms have been engineered to deliver costimulatory signals, activate antigen-presenting cells, and elicit robust CD8+ T-cell responses, showing promise in preclinical and clinical settings. Integrating these nanoparticles with other therapeutic modalities, such as photodynamic, sonodynamic, and photothermal therapies, further amplifies their immunomodulatory effects.161–163Exploring immune cell membrane-coated nanoparticles, particularly those derived from macrophages and T lymphocytes, represents another transformative approach. These nanoparticles are designed to target and disrupt immunosuppressive pathways within the tumor microenvironment, induce immunogenic cell death, and enhance the infiltration and activity of cytotoxic T lymphocytes, thereby restoring the tumoricidal functions of the immune system.164–167 Lastly, the section examines the innovative use of blood-derived membrane biomimetics, including those from red blood cells and platelets, which have been shown to improve drug delivery, evade immune clearance, and modulate the tumor microenvironment. These strategies have demonstrated efficacy in inducing immunogenic cell death, reprogramming immunosuppressive cells, and synergizing with checkpoint blockade therapies to inhibit tumor growth and metastasis.168–171
| Membrane type | Source | Application | Targeting | Immunomodulation mechansim | Animal model | Ref. |
|---|---|---|---|---|---|---|
| Abbreviation: APCs, antigen presentation cells; CTLs, cytotoxic T lymphocytes; ICD, immunogenic cell death; LLC, Lewis lung carcinoma; MSCm, mesenchymal stem cell membranes; NK, natural killer; NSCLC, non-small cell lung cancer; TILs, tumor-infiltrating lymphocytes. | ||||||
| Breast cancer | 4T1 cell line | Drug delivery | Tumor cells | Trigger ICD | Subcutaneous | 181 |
| Breast cancer | 4T1 cell line | Drug delivery | Tumor cells | Increase the penetration of TILs | Metastatic | 175 |
| Breast cancer | 4T1 cell line | Drug delivery | Mitochondria | Induce ICD | Orthotopic | 182 |
| Breast cancer | 4T1 cell line | Antigen-presenting | Tumor cells | Trigger ICD | Orthotopic and residual | 160 |
| Breast cancer | Tumor tissue | Antigen-presenting | Dendritic cells | DC activation and cytokine production | Metastatic | 172 |
| Breast cancer | 4T1 cell line | Drug delivery | Tumor cells | Increse the infiltration of CTLs | Subcutaneous | 195 |
| Breast cancer | Tumor tissue | Antigen-presenting | Tumor cells | Upregulate immunomodulatory cytokines | Metastatic | 174 |
| Colon cancer | CT26 cell line | Antigen-presenting | Macrophages | Trigger CD8+ T lymphocyte responses | Subcutaneous | 178 |
| Colon cancer | MC38-OVA cell line | Drug delivery | Tumor cells | Stimulate human dendritic cells and T lymphocytes | Subcutaneous | 177 |
| Colon cancer | MC38 cell line | Antigen-presenting | Dendritic cells | Stimulate immune checkpoints | Transgenic | 162 |
| Leukemia | C1498-OVA cell line | Antigen-presenting | Tumor cells | Enhance T lymphocyte responses | Metastatic | 159 |
| Lung cancer | LLC cell line | Drug delivery | Dendritic cells | Increase the T lymphocyte infiltrations | Orthotopic | 184 |
| Melanoma | B16-F10 cell line | Drug delivery | Tumor cells | Increase co-uptake of tumor antigen | Immunogenic | 179 |
| Melanoma | B16-F10 cell line | Drug delivery | Tumor cells | Induce dendritic cell maturity | Subcutaneous | 176 |
| Melanoma | B16-OVA cell line | Antigen-presenting | Dendritic cells | Stimulate the maturation of dendritic cells | Subcutaneous | 173 |
| Melanoma | B16 cell line | Drug delivery | Tumor cells | Increases the numbers of M1 macrophages | Subcutaneous | 196 |
| Melanoma | B16F10 cell line | Drug delivery | Tumor cells | Cause PD-L1 DNA sequence breaks | Subcutaneous | 183 |
| Melanoma | B16-F10 cell line | Antigen-presenting | NK cells | Activate NK cells | — | 180 |
| NSCLC | H460 cell line | Drug delivery | Tumor cells | Re-activate lymphocyte cells | Subcutaneous | 197 |
| Prostate cancer | Tumor tissue | Drug delivery | Dendritic cells | Enhance CD8+ T lymphocyte and NK cell infiltration | Subcutaneous | 198 |
| Macrophage | RAW264.7 cell line | Drug delivery | Tumor cells | Induce ICD | Subcutaneous | 199 |
| Macrophage | RAW264.7 cell line | Antigen-presenting | Tumor cells | Induce ICD | Subcutaneous | 164 |
| Macrophage | RAW264.7 cell line | Drug delivery | Tumor cells | Promote lymphocyte infiltration | Immunogenic | 165 |
| Macrophage | Blood | Drug delivery | Tumor cells | Induce CTLs infiltration | Subcutaneous | 185 |
| MSCm | Tissue | Drug delivery | Tumor cells | Promote T lymphocyte infiltration | Subcutaneous | 200 |
| T lymphocyte | CTLL-2 cell line | Antigen-presenting | Tumor cells | Activate cytotoxic T lymphocytes | Subcutaneous | 186 |
| T lymphocyte | CTLL-2 cell line | Antigen-presenting | Tumor cells | Activate cytotoxic T lymphocytes | Subcutaneous | 187 |
| T lymphocyte | Blood | Drug delivery | Tumor cells | T lymphocytes activation | Subcutaneous | 167 |
| Red blood cell | Blood | Drug delivery | Tumor cells | Recruit dendritic cells | Metastatic | 171 |
| Red blood cell | Blood | Drug delivery | Tumor cells | Escape uptake by macrophages | Subcutaneous | 168 |
| Red blood cell | Blood | Drug delivery | Dendritic cells | Induce ICD | Subcutaneous | 188 |
| Platelet | Blood | Drug delivery | Tumor cells | Release aPD-1 antibody | Subcutaneous | 189 |
| Platelet | Blood | Drug delivery | Tumor cells | Repolarize macrophages | Metastatic | 192 |
| Platelet | Blood | Drug delivery | Tumor cells | Enhance cytotoxic T-cell infiltration | Subcutaneous | 190 |
| Platelet | Blood | Antigen-presenting | Tumor cells | Enhance leukemia cells targeting | Orthotopic | 194 |
| Platelet | Blood | Drug delivery | Tumor cells | Enhances local immune activation | Metastatic | 191 |
| Platelet | Blood | Drug delivery | Tumor cells | Sensitize effective ferroptosis | Metastatic | 170 |
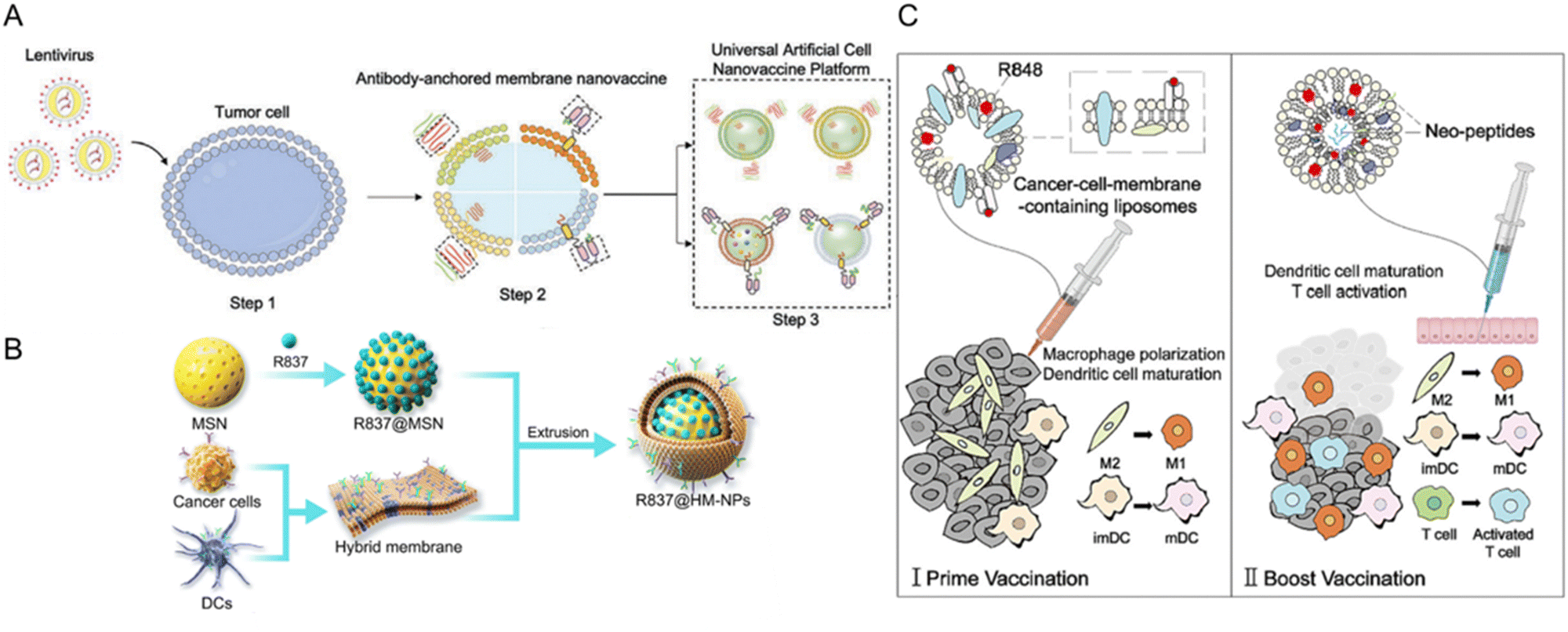 | ||
| Fig. 9 Types of tumor cell membrane-derived nanovaccines fabricated for tumor immunomodulation. (A) Universal nanovaccine platform based on tumor cell-derived components and antibodyanchored membrane (nano-AAM). Reproduced from ref. 162 Copyright 2023 with permission from John Wiley & Sons. (B) A nanovaccines comprising mesoporous silica nanoparticles (MSN) and hybridized membrane components from cancer cells and DCs. MSN is functionalized with imiquimod R837, a TLR7 agonist for immune cell activation (R837@HM-NPs). Reproduced from ref. 158 Copyright 2022 originally published by and used with permission from Dove Medical Press Ltd. (C) Cell membrane-camouflaged liposomes and neopeptide-loaded liposomes for personalized cancer vaccine therapy. The cancer cell membrane-camouflaged liposomes with R848 (Resiquimod,TLR7 and TLR8 Agonist) primes the M2-like macrophages and DCs. Then, liposomes with neopeptides and R848 will boost the immune response by stimulating macrophages, DCs, and T-cells, thereby generating treatment outcomes. Reproduced from ref. 177 Copyright 2023 with permission from Elsevier. | ||
Combining cancer cell membranes and inorganic immune adjuvants is a strategy utilized to elicit a robust and long-lasting immune response. For instance, cancer cell membranes are coated onto layered double hydroxide nanoparticles. These nanoparticles efficiently target antigen-presenting cells and inhibit immune escape, stimulating antigen-presenting cell maturation and tumor-specific CD8+ T-cell responses. This approach has shown significant suppression of tumor growth in vivo.178 Similarly, Gan et al. engineered tumor cell membrane-enveloped aluminum phosphate nanoparticles, stimulating tumor-specific CD8+ T-cell immunity.179 These studies underscore the advantages of using tumor cell membrane-coated nanoparticles to enhance immune cell-mediated cancer immunotherapy.173,174,180
Other immune adjuvants, such as CpG oligonucleotides (ODNs), can be utilized to enhance the immune response of the membrane nanoparticles. Johnson et al. developed an acute myeloid leukemia (AML) cell membrane-coated nanoparticle (AMCNP) vaccine platform with CpG oligonucleotides adjuvant-loaded into the nanoparticles and coated with leukemic cell membrane material (Fig. 10A). The AMNCPs can be recognized by immature antigen-presenting cells (APCs), leading to their maturation and activation. These APCs can then activate circulating T-cells to elicit an adaptive response. AMNCPs vaccination protected mice against AML challenge via intravenous injection of C1498 cells. A rechallenge model tested AMNCPs as an immune adjuvant after a chemotherapy session. The AMNCPs vaccination group received a long-lasting immunity against leukemia re-challenge, with mice surviving up to 12 weeks post-re-challenge. In contrast, mice given C1498 whole cell lysate survived only up to 3 weeks post-re-challenge.159
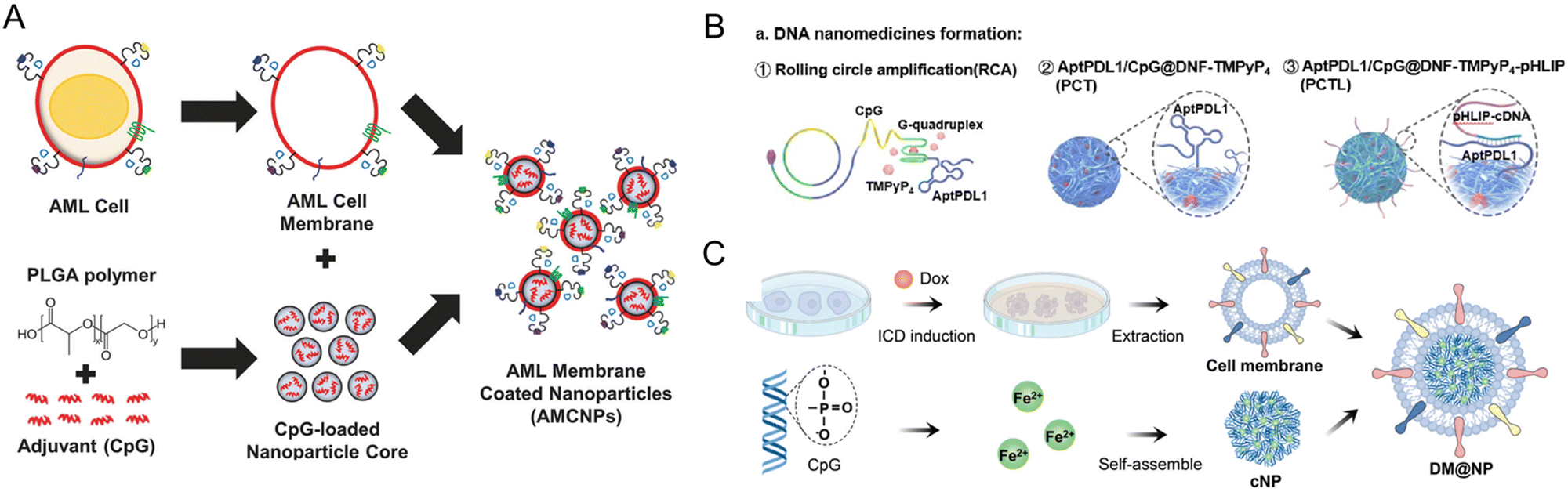 | ||
| Fig. 10 Strategies for generating tumor cell membrane-coated nanoparticles for targeted delivery. (A) CpG oligonucleotides were encapsulated in poly(lactic-co-glycolic acid) (PLGA) polymer nanoparticle cores and loaded into isolated acute myeloid leukemia cell membranes equipped with membrane-associated MHCI restricted antigens (AMCNPs). Immature APCs can recognize the AMCNPs and induce immune activation. Reproduced from ref. 159 Copyright 2021 originally published by and used with permission from Springer Nature. (B) Formation of a DNA nanomedicine composed of PD-L1 aptamers and CpG nanoparticles (PCTL). A photosensitizer (TMPyP4) is inserted into the DNA structure. The PD-L1 aptamers are hidden by the presence of pHLIP-modified cDNA, rendering the formulation non-immunogenic. Reproduced from ref. 183 Copyright 2022 with permission from American Chemical Society. (C) Schematic illustration of the preparation of DM@NP. Tumor cells were pre-induced with doxorubicin to generate membrane particles with damage-associated molecular patterns (DAMPs) from immunogenic cell death. The cell membrane particles coat iron(II)-CpG nanoparticles, an immune adjuvant, and TLR9 agonists. Reproduced from ref. 184 Copyright 2023 with permission from John Wiley & Sons. | ||
Another pivotal aspect in these investigations involves amalgamating membrane coating biomimetics with other techniques for synergistic cancer immunotherapy, such as photodynamic therapy (PDT), sonodynamic treatment (SDT), or photothermal therapy (PTT).160,161,181–184 For example, Wang et al. described the formation of a DNA nanomedicine composed of PD-L1 aptamers and CpG nanoparticles (PCTL, Fig. 10B). A photosensitizer (TMPyP4) was inserted into the DNA structure. The PD-L1 aptamers are hidden by the presence of pHLIP-modified cDNA, rendering the formulation non-immunogenic. Under localized irradiation, the ROS generated will damage the nanostructure and release the internal DNA immunomodulators, providing a stimuli-responsive and spatiotemporal aspect to the nanomedicine. PCTL alone did not exhibit any antitumor activities in vivo but significantly contributed to tumor regression of subcutaneous B16F10 melanoma tumors after irradiation, yielding a prolonged survival time of 71% at 40 days post-treatment. The PCTL plus irradiation group also exhibited marked DC maturation in the tumors with a concomitant increase of CD8+ T-cell activation.183 These methodologies harness nanoparticles' photodynamic or photothermal effects to trigger immunogenic cell death and liberate tumor-associated antigens, subsequently activating immune cells and fostering antitumor immune responses. Tang et al. demonstrated that tumor cell membrane-targeted photosensitive dimers facilitated highly efficient immunogenic cell death (ICD) in tumor cells, propelling cancer immunotherapy.161 Furthermore, Chen et al. designed cancer cell membrane-coated nanoparticles co-loaded with photosensitizer and TLR7 agonist, synergistically enhancing tumor immunotherapy.163 A formulation comprising TLR9 agonist encapsulated with membrane particles extracted from cells pre-induced with doxorubicin (DM@NPs) was formulated and tested in a subcutaneous lung cancer model in mice (Fig. 10C). The complex generated a significant antitumor response that induced the accrual of activated T lymphocytes in the spleen and tumor lesions.184 These examples highlight the importance of targeting immune influx and improving antigen presentation to overcome immune evasion. The immune response can be enhanced by utilizing tumor cell-derived membrane coating biomimetics. These findings provide valuable insights into the development of personalized cancer vaccines and the potential for combination therapies to improve the efficacy of cancer immunotherapy.
| Membrane type | Source | Application | Targeting | Immunomodulation mechanism | Animal model | Ref. |
|---|---|---|---|---|---|---|
| Abbreviation: BMDCs, bone marrow-derived dendritic cells; DCs, dendritic cells; ICD, immunogenic cell death; RBC, red blood cell. | ||||||
| Cancer cell and DC | 4T1 cell line and BMDCs | Drug delivery | Tumor cells | Blockade immune checkpoints | Recurrent | 158 |
| Cancer cell and bacteria | 4T1 cell line and E. coli bacteria | Drug delivery | Tumor cells | Enhance ICD | Metastatic | 203 |
| Cancer cell and RBC | ID8 cell line and blood | Drug delivery | Tumor cells | Activate cytotoxic T lymphocytes | Metastatic | 169 |
| Cancer cell and RBC | 4T1 cell line and blood | Drug delivery | Tumor cells | Deplete tumor-associated macrophages | Subcutaneous | 205 |
| Cancer cell, RBC and macrophage | 4T2 cell line, blood, and marrow | Antigen-presenting | Tumor cells | Polarize macrophages | Metastatic | 204 |
| Cancer cell and DC | 4T1 cell line and BMDCs | Drug delivery | Tumor cells | Express immunological molecules | Bilateral | 202 |
| Cancer cell and DC | 4T1 cell line and BMDCs | Drug delivery | Tumor cells | Activate cytotoxic T lymphocytes | Subcutaneous | 201 |
| Macrophage and thylakoid | RAW264.7 cell line and spinach | Drug delivery | Tumor cells | Induce TAM polarization | Subcutaneous | 166 |
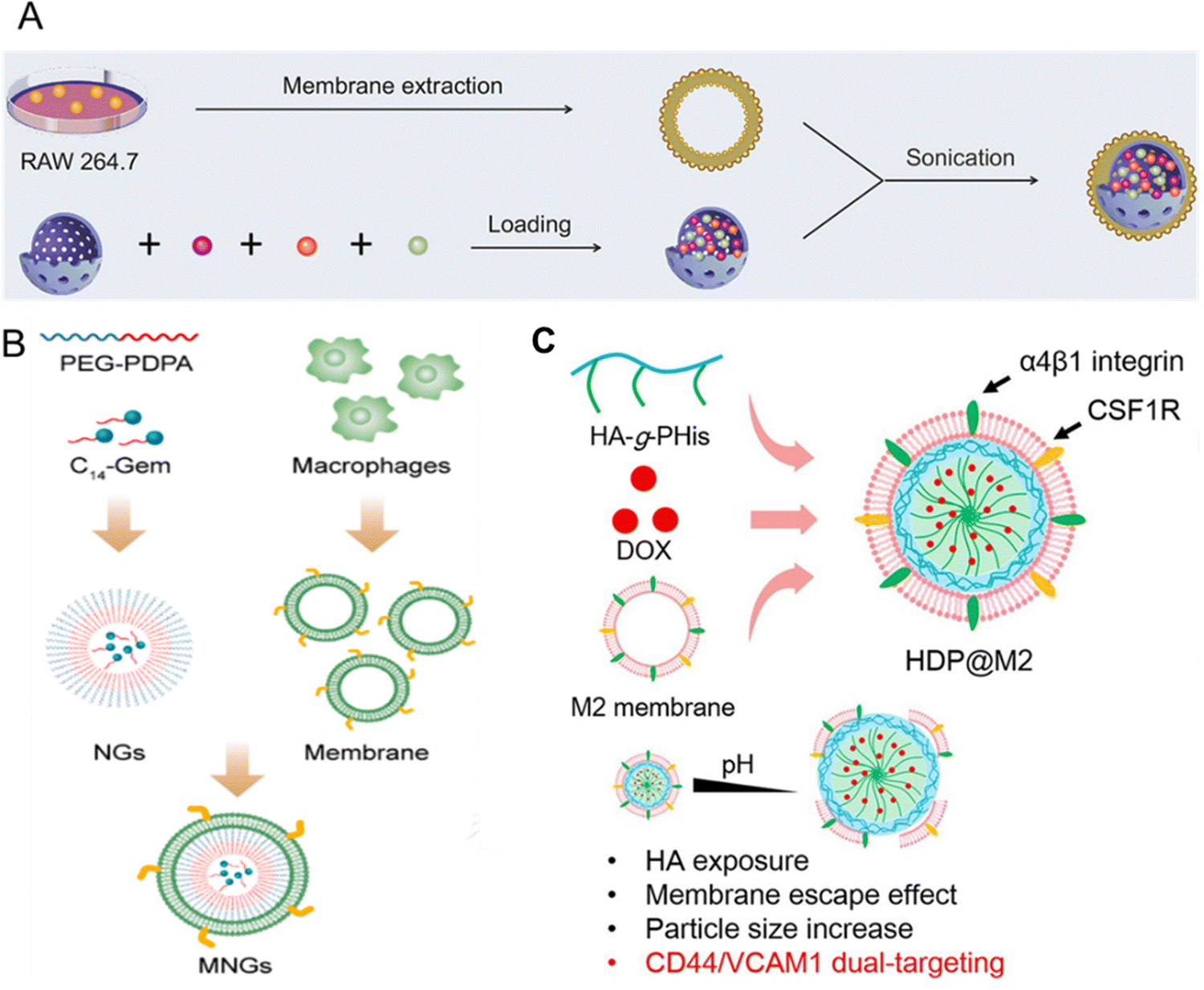 | ||
| Fig. 11 Macrophage-coated nanoplatform. (A) Macrophage membrane-coated mesoporous silica nanoplatform containing catalase (purple dot), doxorubicin (orange dot), and R848 (green dot) encapsulated in the mesoporous silica core. Once endocytosed in tumor cells, the platform releases doxorubicin to induce immunogenic cell death and R848 to induce DC maturation, lymphocyte infiltration, and activation. Reproduced from ref. 164 Copyright 2022 with permission from Elsevier. (B) Macrophage membrane-coated nano gemcitabine (MNGs) for synergistic cancer immunotherapy. The system has an acidic pH drug-releasing capacity to potentiate lymphocyte infiltration and synergize with anti-PD-L1 therapy. Reproduced from ref. 165 Copyright 2023 with permission from American Chemical Society. (C) Tumor-associated macrophage (TAM) membrane-camouflaged nanoparticle for delivery of doxorubicin (DOX). Hyaluronic acid (HA)-g-poly(histidine) was synthesized and loaded with DOX in the inner core of the micelle. TAMs membrane was coated on the surface of the co-polymer to form HDP@M2. Reproduced from ref. 185 Copyright 2022 with permission from Elsevier. | ||
T lymphocyte membrane-decorated nanoinducers also offer a breakthrough solution in the tumor immunomodulation domain. T-lymphocyte membrane-derived exosomes are used to encapsulate ORY-1001, a potent and selective lysine-specific histone demethylase 1A (LSD1) inhibitor (OPEN, Fig. 12A). OPEN can be recognized by PD-L1 expressing cells due to the PD-1 on the surface of the T-lymphocyte membrane and upregulate intratumoral interferons and interferon-stimulated genes, such as MHCI that could enhance neoantigen presentation. Enhancing intratumoral interferons will block the immunosuppression mechanism and increase immune cell recruitment.186 In another study by Li et al., a PBA-modified T-cell membrane was used for cloaking the RCM to generate tumor microenvironment-responsive nanoparticles (Fig. 12B). RCM is a redox-sensitive hyaluronic acid grafted copolymer with antitumor drug curcumin (CUR) incorporated in the core. In the acidic TME, PBA dissociates, releasing the T-cell membrane debris and exposing HA for tumor-targeting. T-cell membrane debris binds with PD-L1 on the tumor cells and blocks immune checkpoint suppression, resulting in CD8+ T-cell activation and infiltration for immunotherapy. The RCM will be endocytosed via HA interaction with CD44 and will release CUR for tumor killing via the breakage of disulfide bonds in the redox environment. These nanoparticles enhance cytokine release and induce immunogenic cell death, elevating CD8+ T-cell levels and providing an effective immune-chemotherapy approach for melanoma.187 Ma et al. integrated CAR-T-cell membranes onto silica nanoparticles for targeted delivery to HCC tumors. CAR-T-cell membrane, specifically recognizing GPC3+ HCC cells, was used to coat mesoporous silica containing IR780 dye for PDT and PTT applications (Fig. 12C). This nanomaterial exhibited photothermal and targeting abilities, providing an alternative strategy for treating HCC, a disease with limited treatment options.167 In summary, immune cell membrane-coated nanoparticles have shown great potential for tumor immunomodulation. These nanoplatforms actively target tumors, inhibit immunosuppressive pathways, induce immunogenic cell death, enhance immune cell infiltration, and restore tumoricidal functions. They offer a promising strategy for improving the efficacy of cancer immunotherapy,
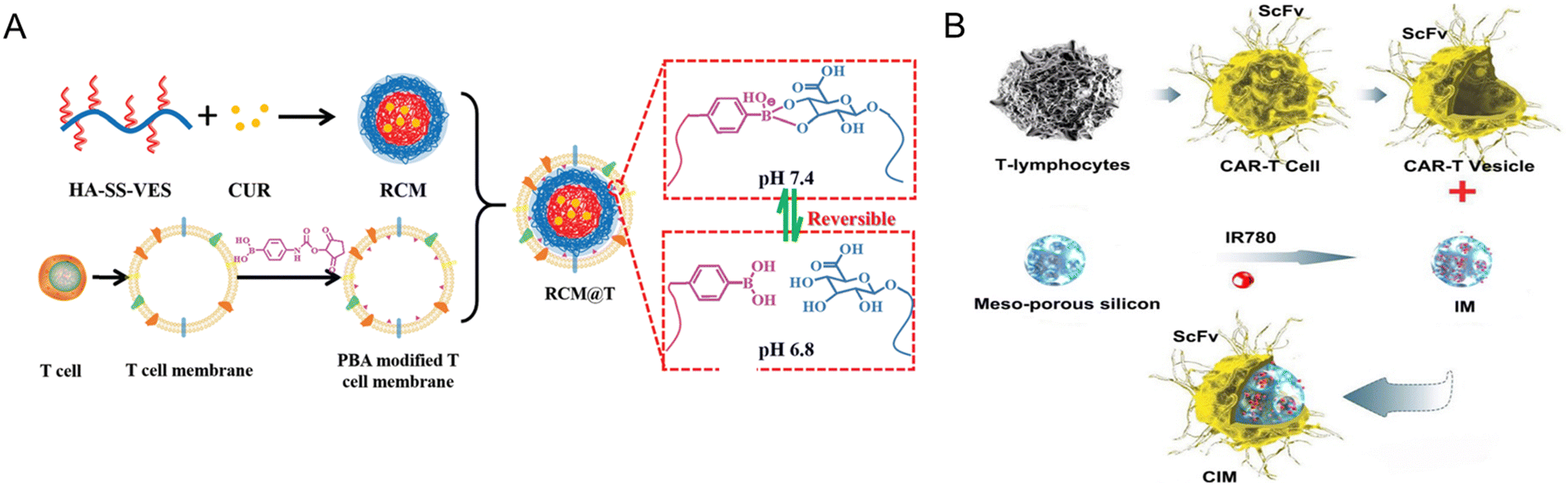 | ||
| Fig. 12 T-cell-coated nano platform. (A) A redox sensitive hyaluronic acid grafted copolymer (HA-grated-disulfide bond-vitamin E succinate, shortened as HA-SS-VES) was incorporated with antitumor drug curcumin (CUR) to prepare RCM. A PBA-modified T-cell membrane was used to cloak the RCM and generate tumor microenvironment-responsive nanoparticles. In the acidic TME, PBA dissociates, releasing the T-cell membrane debris and exposing HA for tumor-targeting. T-Cell membrane debris binds with PD-L1 on the tumor cells and blocks immune checkpoint suppression, resulting in CD8+ T-cell activation and infiltration for immunotherapy. The RCM will be endocytosed via HA interaction with CD44 and will release CUR for tumor killing via the breakage of disulfide bonds in the redox environment. Reproduced from ref. 187 Copyright 2021 with permission Royal Society of Chemistry. (B) CAR-T-cell membrane, specifically recognizing GPC3+ HCC cells, was used to coat mesoporous silica containing IR780 dye for PDT and PTT applications to treat HCC. Reproduced from ref. 167 Copyright 2020 originally published and reused with permission from Ivy Spring International Publisher. | ||
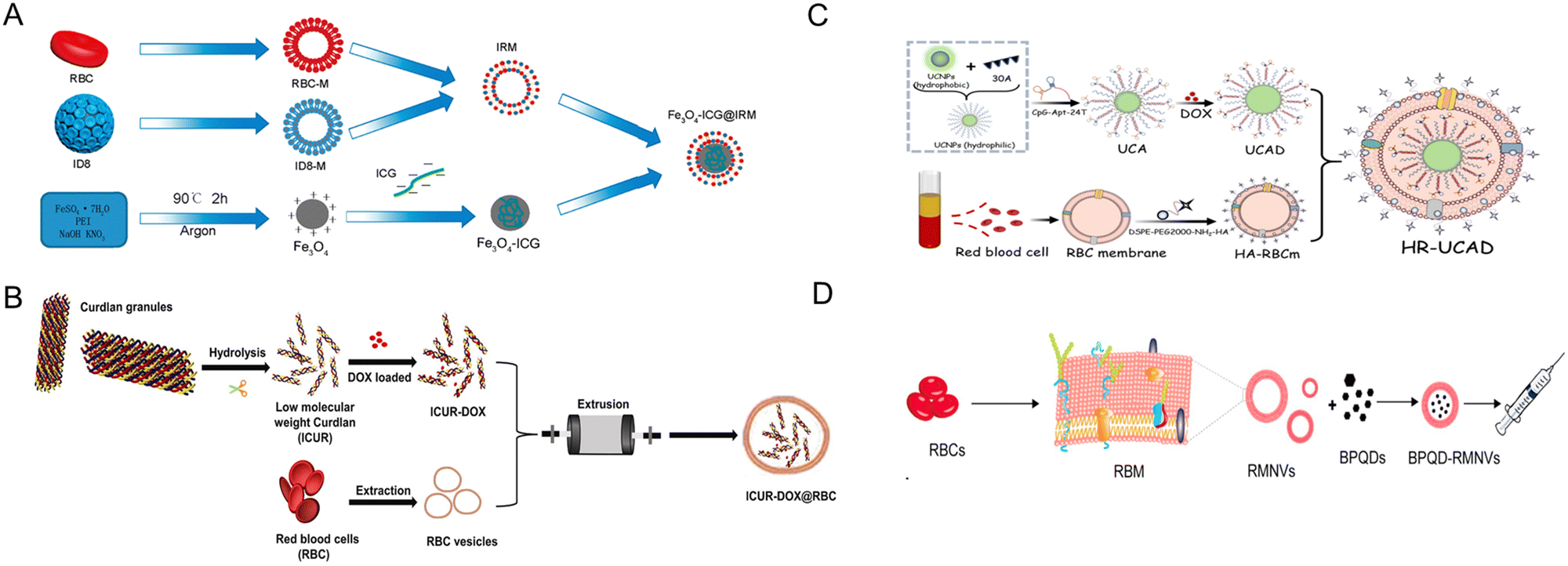 | ||
| Fig. 13 Red blood cell (RBC)-derived bio-nanoparticles. (A) A hybrid membrane comprising ID8 (ovarian cancer) cell membrane (ID8-M) and RBC membrane (RBC-M) was used to coat ICG-loaded magmetic nanoparticles ((Fe3O4-ICG@IRM)). The complex is used for the synergistic PTT-immunotherapy of ovarian cancer. ID8 tumor antigens on the surface can direct to ID8 tumors via homologous homing and the tumor antigens on the IRM can induce antitumor immune response. Reproduced from ref. 169 Copyright 2021 with permission from American Chemical Society. (B) Doxorubicin was encapsulated in the helical structure of the curdlan, a microbial glucan. RBC membrane was then used to wrap the outer layer of the DOX-curdlan using the coextrusion method to generate ICUR-DOX@RBC NPs. Curdlan can interact with dectin-1 receptors on macrophages to induce pro-inflammatory cytokine production and induce M2 → M1 polarization. Reproduced from ref. 168 Copyright 2022 with permission from Elsevier. (C) RBC membrane-camouflaged DNA-functionalized upconversion nanoparticles (UCNPs) for targeted breast cancer chemotherapy and immunotherapy. UCNPs were modified with a 30-mer poly A oligonucleotide hybridized with 24-mer poly T of CpG-aptamer to form a duplex. Dox was introduced into the base pairs of the duplex and the UCNPs@CpG-Apt/DOX was encapsulated within the erythrocyte membrane to form HR-UCAD. Reproduced from ref. 188 Copyright 2023 with permission from Royal Society of Chemistry. (D) A biomimetic black phosphorus quantum dot formulation coated with eryhtrocyte membrane for the PTT of breast cancer (BPQD-RMNV). This formulation was combined with immune checkpoint blockade (anti PD-1) to cure innoculated breast cancer in mice. BPQD-RMNV displayed accumulation in tumor in vivo and increased infiltration of CD8+ T-cells in the tumor. Reproduced from ref. 171 Copyright 2019 with permission from Elsevier. | ||
Another approach using erythrocyte membrane involves camouflaging DNA-functionalized upconversion nanoparticles (UCNPs) with red blood cell membrane for targeted breast cancer chemotherapy and immunotherapy (Fig. 13C). UCNPs were modified for doxorubicin loading followed by membrane coating to yield HR-UCAD. HR-UCAD induced immunogenic cell death of 4T1 cells in vitro, releasing factors that induces the migration and activation of DC2.4 cells. In vivo, the formulation is efficiently inhibited tumor growth with concomitant increase in the percentage of CD8+ and CD4+ T-cell populations.188 Another PTT platform with erythrocyte membrane utility involves use of biomimetic black phosphorus quantum dot formulation coated with eryhtrocyte membrane (BPQD-RMNV, Fig. 13D). Combined with immune checkpoint blockade (anti PD-1), BPQD-RMNV induced regression of innoculated 4T1 breast cancers in mice, inhibited the growth of distant tumors, and increased infiltration of CD8+ T-cells in the tumor lesions.171 These studies exemplify the feasibility of erythrocyte membrane as a cloaking tool to increase the circulation time of various nanoparticle platforms without inducing severe toxicity.
Recent studies highlight the innovative use of platelet-derived membrane-coated nanoparticles (PMCNPs) in cancer therapy (Fig. 14). A novel delivery method employing PMCNPs conjugated with anti-PD-1 antibodies, enhancing tumor-specific thrombosis and improving drug delivery, thus enhancing therapeutic efficacy in breast cancer models.189 Another study focused on modulating the immunosuppressive tumor microenvironment by targeting lactate metabolism using PMCNPs coated with metal–organic frameworks, resulting in enhanced immunogenic cell death and tumor growth inhibition.190 The potential of intratumoral immunotherapy was demonstrated using platelet-cloaked nanoparticles, showcasing tumor regression and metastasis inhibition.191 Similarly, PMCNPs were utilized for pancreatic cancer therapy, encapsulating ferroptosis inducers to disrupt tumor vasculature and inhibit cancer progression.192 PMCNPs camouflaged with magnetic nanoparticles were reported for ferroptosis-enhanced cancer immunotherapy, improving the efficacy of immunotherapy and inducing tumor-specific immune responses.170 Additionally, Yan et al. and Chen et al. utilized PMCNPs for combined chemo-immunotherapy, demonstrating significant inhibition of tumor growth and metastasis in melanoma and acute myeloid leukemia models, respectively.193,194 These studies collectively underscore the potential of PMCNPs in targeting tumors and modulating the tumor microenvironment for enhanced cancer therapy.
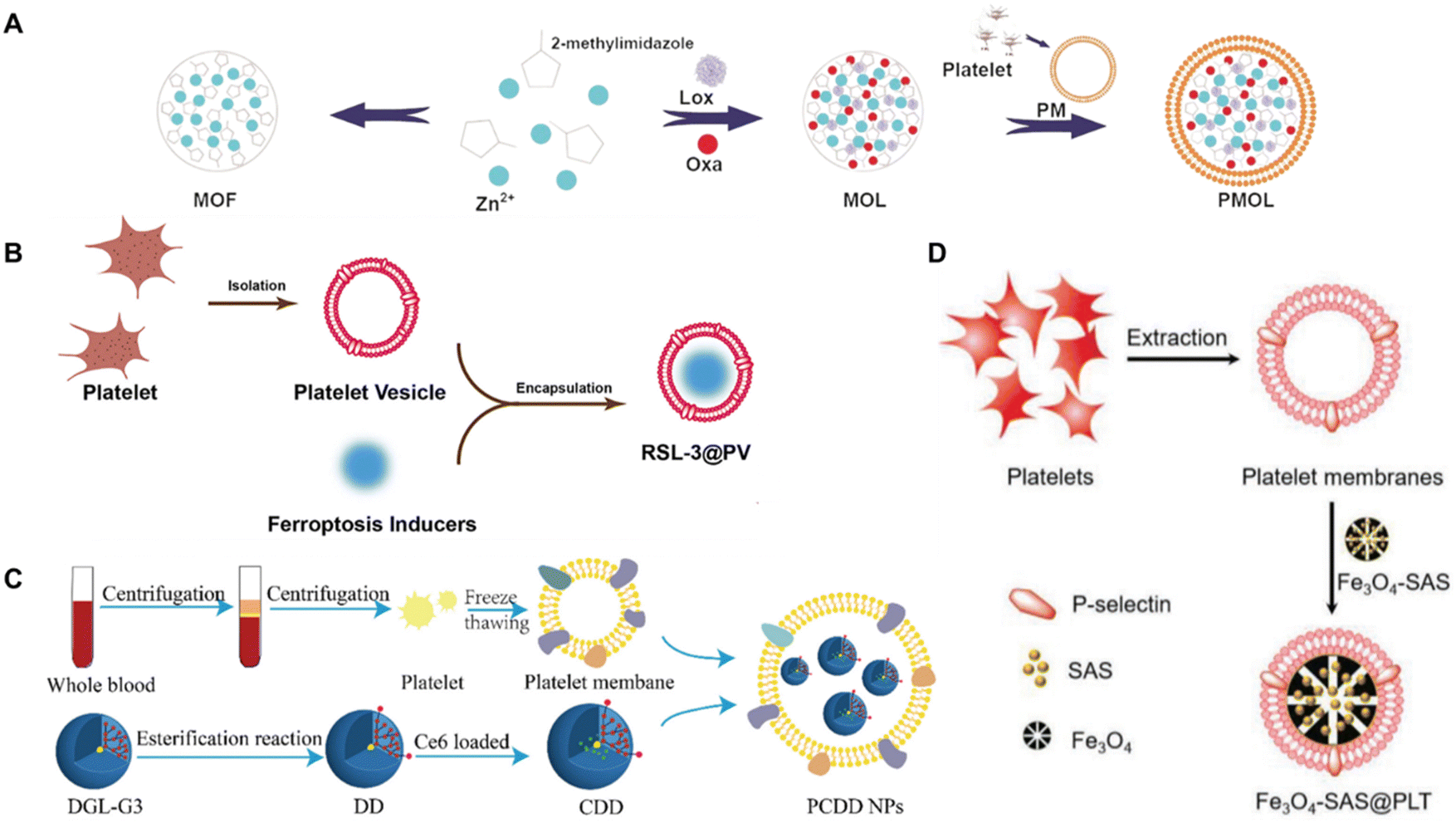 | ||
| Fig. 14 Platelet-derived bio-nanoparticles. (A) Platelet vesicles (PVs) were used to encapsulate a ferroptosis inducer, RSL-3, generating RSL-3@PV nanoparticles. RSL-3@PV nanoparticles were used for treating pancreatic ductal adenocarcinoma (PDAC). Reproduced from ref. 192 Copyright 2022 originally published and reused with permission from frontiers. (B) Novel platelet membrane-coated nanoparticles (PCDD NPs) were constructed for combined chemo-photodynamic- and immunotherapy of melanoma. Reproduced from ref. 193 Copyright 2022 with permission from Royal Society of Chemistry (C) metal–organic frameworks were coated with platelet membranes (PM) for tumor site-specific delivery and rationally designed to carry lactate oxidase which catalytically consumed lactate, while oxaliplatin induced ICD. Reproduced from ref. 190 Copyright 2023 with permission from Elsevier. (D) Sulfasalazine (SAS)-loaded mesoporous magnetic nanoparticles (Fe3O4) were camouflaged with platelet membrane to generate Fe3O4-SAS@PLT nanoparticles. The formulation can trigger ferroptotic cell death, inducing a tumor-specific immune response and repolarize macrophages from M2 → M1 phenotype. Reproduced from ref. 170 Copyright 2020 with permission from John Wiley & Sons. | ||
One approach involves the combination of cancer cell membranes with erythrocyte membranes to create hybrid coatings for nanoparticles, which exhibit immune evasion and homologous tumor-targeting properties. This strategy has been successfully applied to ovarian cancer, where the hybrid membrane-coated nanoparticles activated specific immunity and exhibited synergistic photothermal-immunotherapy effects, leading to the activation of cytotoxic T-cells and reduction of regulatory T-cells.169 A similar concept has been applied using cytomembranes of fused cells derived from dendritic cells (DCs) and cancer cells. This fusion results in nanoparticles that mimic antigen-presenting cells (APCs), displaying tumor antigens and co-stimulatory molecules, which can activate T-cells and induce a powerful antitumor immune response.201 The same group further expanded this approach by engineering nanoplatforms from the cytomembranes of hybrid cells derived from cancer and DCs, which showed significant antitumor effects in mouse models.202
Another innovative strategy is the use of hybrid membranes combining tumor cell membranes with bacterial outer membranes. This approach has been shown to enhance radiosensitivity and trigger antitumor immune responses in breast cancer, thereby amplifying the effects of radiotherapy and immunotherapy.203 Additionally, hybrid cellular membrane nanovesicles have been engineered to amplify macrophage immune responses against cancer recurrence and metastasis by blocking the CD47-SIRPα signaling axis and promoting macrophage repolarization within the tumor microenvironment.204
Moreover, pH-sensitive hybrid membrane-coated nanoparticles have also been developed to reprogram the tumor microenvironment and boost antitumor immunity. These nanoparticles are designed to target the tumor microenvironment and escape from endo/lysosomes after endocytosis, leading to significant tumor inhibition and immune activation.200 Additionally, erythrocyte-cancer cell hybrid membrane camouflaged pH-responsive copolymer micelles have been employed to selectively deliver a CSF-1R inhibitor to tumor-associated macrophages, resulting in TAMs depletion and reversal of the tumor immune microenvironment.205 Lastly, hybrid membrane-coated nanoparticles have been utilized to regulate the immunosuppressive tumor microenvironment and enhance breast cancer immunotherapy by co-delivering immuno-metabolic adjuvant and immune checkpoint inhibitors.206
In conclusion, the fusion of cell membranes to create hybrid biomimetic nanoparticles represents a promising avenue for cancer immunotherapy. These platforms leverage the unique properties of different cell types to target tumors, modulate the immune system, and deliver therapeutic agents, thereby enhancing the overall therapeutic efficacy against various cancers. These studies highlight diverse approaches utilizing cell membrane-camouflaged nanoparticles for tumor immunomodulation. These biomimetic nanosystems aim to optimize drug delivery, prolong circulation, and induce immune responses for effective cancer treatment. Utilizing various cell membranes, such as red blood cells and platelet membranes, to encapsulate therapeutic agents has shown promising outcomes, including inhibiting tumor growth, reprogramming immune cells, inducing immunogenic cell death, and improving cancer immunotherapy outcomes.
4.2. Engineering exosomes for immunomodulation
This section delves into the multifaceted roles of exosomes derived from tumors, immune cells, bacteria, and other sources in modulating the immune landscape of cancer. Tumor-derived exosomes (TDEVs) have been identified as double-edged swords within the tumor microenvironment (TME), capable of both inducing immunosuppression that hampers the efficacy of CAR-T-cells and other immunotherapies and, conversely, being engineered to enhance antitumor immunity.207–210 The narrative then shifts to the promising capabilities of immune cell-derived exosomes. These nanoscale vesicles, sourced from T-cells, macrophages, dendritic cells (DCs), and natural killer (NK) cells, are adept at antigen presentation and can be tailored to deliver specific therapeutic payloads.211–214 Their role in priming the immune system and synergistic effects with existing therapies, such as immune checkpoint inhibitors, are highlighted.Bacterial membrane vesicles, particularly outer membrane vesicles (OMVs) from Gram-negative bacteria, are presented as novel and potent tools for tumor immunomodulation. The section discusses how these OMVs can be harnessed to enhance the infiltration and activation of tumor-specific T-cells, demonstrating synergistic effects with checkpoint blockade therapies.215–217 Lastly, the section examines exosomes derived from alternative sources such as plasma, red blood cells, and stem cells, underscoring their potential as biomarkers for immunotherapy outcomes and as vehicles for targeted drug delivery. These exosomes are poised to reshape the tumor microenvironment and augment the efficacy of immunotherapeutic strategies.218–220
| Exosome type | Source | Application | Targeting | Immunomodulation mechansim | Animal model | Ref. |
|---|---|---|---|---|---|---|
| Abbreviation: CLL, chronic lymphocytic leukemia; HNSCC, head and neck squamous cell carcinoma cells; CTLs, cytotoxic T lymphocytes; ICD, immunogenic cell death; MSCm, mesenchymal stem cell membranes; NSCLC, non-small cell lung cancer; OSCC, oral squamous cell carcinoma; PD-L1, programmed death-ligand 1; PDAC, pancreatic ductal adenocarcinoma; TGF-β, transforming growth factor-beta. | ||||||
| Bladder cancer | MB49 cell line | Antigen-presenting | Macrophages | Inhibit macrophages differentiation | Subcutaneous | 226 |
| Breast cancer | A549 cell line | Drug delivery | Tumor cells | Blockade immune checkpoints | Metastatic | 230 |
| Breast cancer | 4T1 cell line | Drug delivery | Tumor cells | Induce ICD | Orthotopic | 209 |
| Breast cancer | 4T1 cell line | Antigen-presenting | Tumor cells | Increase cytokines secretion | Subcutaneous | 235 |
| Breast cancer | 4T1 cell line | Drug delivery | Tumor cells | Promote macrophage polarization | Subcutaneous | 228 |
| Colon cancer | CT26 cell line | RNA cargo | T lymphocyte | Increase T lymphocyte infiltrations | Subcutaneous | 238 |
| Colon cancer | MC38 cell line | Antigen-presenting | Tumor cells | Blockade immune checkpoints | Transgenic | 233 |
| Colon cancer | MC38 cell line | Antigen-presenting | T lymphocyte | Increase M2-like macrophages | Subcutaneous | 226 |
| CLL | EHEB cell line | Antigen-presenting | T lymphocyte | Impair T-Cell activation | — | 238 |
| Glioblastoma | Body fluids | Antigen-presenting | T lymphocyte | Inhibiti T lymphocyte cycle | Orthotopic | 250 |
| HNSCC | Tumor tissue | Antigen-presenting | Macrophages | Increase cytokines secretion | Subcutaneous | 251 |
| Leukemia | NALM6 cell line | Antigen-presenting | T lymphocyte | Activate CAR-T Cells | Subcutaneous | 221 |
| Lymphoma | Nalm-6 cell line | Antigen-presenting | T lymphocyte | Reprogram T lymphocytes | NA | 222 |
| Leukemia | HL-60 cell line | Antigen-presenting | T lymphocyte | Inhibiti T lymphocyte proliferation | — | 252 |
| Melanoma | B16 cell line | RNA cargo | T lymphocyte | Induce T lymphocyte responses | Transgenic | 236 |
| Melanoma | B16 cell line | Antigen-presenting | T lymphocyte | Blockade immune checkpoints | Metastatic | 234 |
| NSCLC | MRC-5 cell line | RNA cargo | Tumor cells | Induce immunosuppression | Subcutaneous | 237 |
| Neuroblastoma | 9464D cell line | Antigen-presenting | Tumor cells | Sensitize tumors to dinutuximab | Transgenic | 253 |
| OSCC | Tumor tissue | Antigen-presenting | Lymph nodes | Show the distinct immunosuppression | Subcutaneous | 254 |
| Ovarian cancer | HeyA8 cell line | Antigen-presenting | Tumor cells | Induce T lymphocyte exhaustion | Orthotopic | 223 |
| Pancreatic cancer | MIA-PaCa-2 cell line | Antigen-presenting | Dendritic cells | Increase cytokines release | Engrafted | 210 |
| Pancreatic cancer | PDAC cell line | Antigen-presenting | T lymphocyte | Inhibit the efficacy of CAR-T Cells | Transgenic | 207 |
| Dendritic cell | DC2.4 cell line | Antigen-presenting | Tumor cells | Elicit T lymphocyte responses | Metastatic | 213 |
| Dendritic cell | Bone marrow | Drug delivery | Tumor cells | Induced T lymphocyte proliferation | Subcutaneous | 212 |
| Dendritic cell | Bone marrow | Antigen-presenting | Tumor cells | Induce susceptibility to anti-PD-1 | Subcutaneous | 255 |
| Dendritic cell | DC2.4 cell line | Antigen-presenting | Tumor cells | Induce tumor-specific immune responses | Orthotopic | 245 |
| Dendritic cell | DC2.4 cell line | RNA cargo | Tumor cells | Induce anticancer immunity | Subcutaneous | 214 |
| Dendritic cell | Bone marrow | Antigen-presenting | T lymphocytes | Activate tumor-specific CTLs | Metastatic | 256 |
| Dendritic cell | DC2.4 cell line | Antigen-presenting | Tumor cells | Activate T lymphocyte | Subcutaneous | 243 |
| Macrophage | F4/80 cell line | Drug delivery | Tumor cells | Repolarize macrophages | Subcutaneous | 211 |
| Macrophage | RAW264.7 cell line | Antigen-presenting | Tumor cells | Promote macrophage polarization | Subcutaneous | 241 |
| Macrophage | RAW264.7 cell line | Drug delivery | Tumor cells | Induce M1 polarization | Subcutaneous | 247 |
| Macrophage | RAW264.7 cell line | RNA cargo | Tumor cells | Boost intratumoral immune activation | Subcutaneous | 249 |
| T lymphocyte | Spleen and blood | Antigen-presenting | Macrophages | Prime macrophages | Subcutaneous | 257 |
| T lymphocyte | Blood | Antigen-presenting | Tumor cells | Promote T lymphocyte migration | Transgenic | 248 |
| T lymphocyte | Blood | Antigen-presenting | Tumor cells | Target mesothelin | Subcutaneous | 258 |
| T lymphocyte | Blood | Antigen-presenting | Tumor cells | Block PD-L1 and scavenge TGF-β | Subcutaneous | 242 |
| T lymphocyte | Blood | Antigen-presenting | Tumor cells | Exhibit cytotoxic activity | Subcutaneous | 244 |
 | ||
| Fig. 15 Tumor-derived exosomes modulate the tumor microenvironment (A) EVs secreted by HEK293T cells stably expressed CD19 (CD19 EVs). The CD19 EVs were administered to CAR-T cells resulting in enhanced CAR T cell expansion and cytokine secretion, with elevated CAR expression leading to increased cytotoxicity. Reproduced from ref. 229 Copyright 2023 originally published by and used with permission from Dove Medical Press Ltd (B) exosomes derived from immunogenically dying tumor cells can be used to systemically activate the immune system and used for nanovaccine development. The systemically activated immune cells can home to the TME and reprogram the tumor milieu into a hot tumor state, training the immune system in the TME towards tumor eradication. Reproduced from ref. 208 Copyright 2022 with permission from Elsevier. | ||
On the contrary, engineering efforts have explored and opened the potential of TDEVs to enhance cancer immunotherapy.229,230 Zhang et al. engineered HEK293T-derived EVs to present the CD19 antigen as the CAR target (Fig. 15B). In vitro evaluation demonstrated that the CD19-EVs activated the CAR-T cells in an antigen-specific and dose-dependent manner resulting in the expansion and cytokine secretion of the co-cultured CAR-T cells. In vivo, adoptive cell transfer of the CD19-EVs led to the significant regression of subcutaneously implanted Raji tumor cells in mice along with increased CAR-T population in the excised tumors. Excessive cytokine secretion is one of the limitations of CAR-T cell therapy. To ensure that the CD19-EVs did not induce cytokine release syndrome (CRS), serum levels of IFN-γ, IL-2 and TNF-α were measured. No obvious increase in serum cytokine was observed, except for the obvious increase in IFN-γ. This study demonstrates the utility of TDEVs as an antigen presentation vehicle, without the tumor promoting and CRS risks. Furthermore, it also demonstrated that tumor cells themselves can be utilized to reprogram the tumor microenviroment, via sufficient immune activation resulting in immunogenic cell death. Exosomes from the immunologically terminated tumors can hypothetically contain optimum EV cargo for TME reprogramming (Fig. 15C).
The use of standard treatment modality methods that can induce immunogenic cell death is another solution to generating TDEVs with therapeutic value. In these cases, TDEVS are used to encapsulate treatment agents that can be delivered to the tumor site and promote immunogenic cell death. Wang et al. fabricated exosomes loaded with sonosensitizers and immune adjuvants to stimulate anti-tumor immunity, namely ExoCe6+R848 (Fig. 16A).231 ExoCe6+R848 was constructed by simple co-incubation of chlorin e6 and R848 (TLR7 and TLR 8 agonist). After intratumoral injection to the tumor site, ultrasound irradiation was performed two hours later. The treatment significantly inhibited tumor growth and induced significant increase of Il-1β, IL-6, IL12, TNF-α, and IFN-γ in the tumor lysates. Conversely, the secretion of anti-inflammatory cytokines, IL-10 and TGF-β, was significantly reduced.231 Similarly, Hu et al. developed doxorubicin-loaded hybrid nanovesicles comprising liposomes and tumor-derived exosomes (DOX@LINV) fused for combinational immunochemotherapy (Fig. 16B).232 DOX@LINV delivered doxorubicin to the subcutaneous B16F10 subcutaneous tumor sites, eliciting immunogenic cell death. An analysis of the populations in the tumor draining lymph nodes (TDLNs) after immunochemotherapy revealed a decrease in the Foxp3+ Treg, with a concomitant increase in CD8+ and CD4+ T-cell population and elevated granzyme B expression. Furthermore, DOX@LINV exhibited presence of mature DCs and T-cells in the, indicating mature DC migration and priming of T-cells in vivo.232
 | ||
| Fig. 16 Tumor-derived exosomes to enhance cancer immunotherapy. (A) ExoCe6+R848 was constructed by co-incubation of Ce8 and R848 with HEK293T cell-derived exosomes. ExoCe6+R848 can be engulfed by DCs and enhance their maturation after ultrasonic irradiation. These exosomes can also reprogram macrophages from M2 → M1 phenotype. Reproduced from ref. 231 Copyright 2022 originally published by and used with permission from Taylor & Francis Group. (B) DOX-loaded biomimetic hybrid nanoparticles were formed by the cofusion of tumor-derived nanovesicles with artificial liposomes (LIPs). The formed DOX@LINV platform was used for combination immunochemotherapy approach with the ability to target the tumor site via homologous homing and improve the immunogenicity of the tumor. DOX@LINV activated DCs and alleviate the immunosuppresive TME. Reproduced from ref. 232 Copyright 2021 with permission from American Chemical Society. | ||
These studies highlight exosomes' capacity to deliver therapeutic agents to tumor sites, enhancing their anti-tumor effects. The ability of exosomes to modulate the immune response is commonly observed.209,233 However, it is important to reiterate the contrasting nature of these studies according to the context of TDEVs exploration. Some studies explored exosomes' role in overcoming immunotherapy resistance.233 Tumor-derived extracellular vesicles were found to counteract anti-PD-L1 antibodies, leading to therapy resistance.227,234 Conversely, micrometer-sized tumor cell-derived vesicles acted as autologous cancer vaccines, enhancing systemic immune responses and potentially overcoming resistance.208,209,235 Hence, exosomes studies should always be performed in a disease-specific context manner due to the innumerable tumor mechanisms that differ in each disease. Furthermore, naturally occurring TDEVs are useful when tumor cells were induced to undergo immunogenic cell death or are used as a nanoplatform as a vaccine and drug delivery capable of inducing immunogenic cell death, highlighting the importance of the death mechanism in generating therapeutic value. A recurring theme in these studies is the capacity of tumor-derived extracellular vesicles to carry tumor-associated antigens and interact with immune cells.236,237 This interaction alters the phenotype and function of CAR-T cells, promoting the secretion of pro-inflammatory cytokines and upregulating activation-related genes.236–238 In summary, while naturally occurring TDEVs impede immune responses, hindering CAR-T cell efficacy, engineered TDEVs have shown promise in enhancing immunotherapy, modulating immune responses, and potentially overcoming resistance. These findings underscore the dual role of exosomes in cancer immunomodulation and their significance in shaping effective therapeutic strategies.
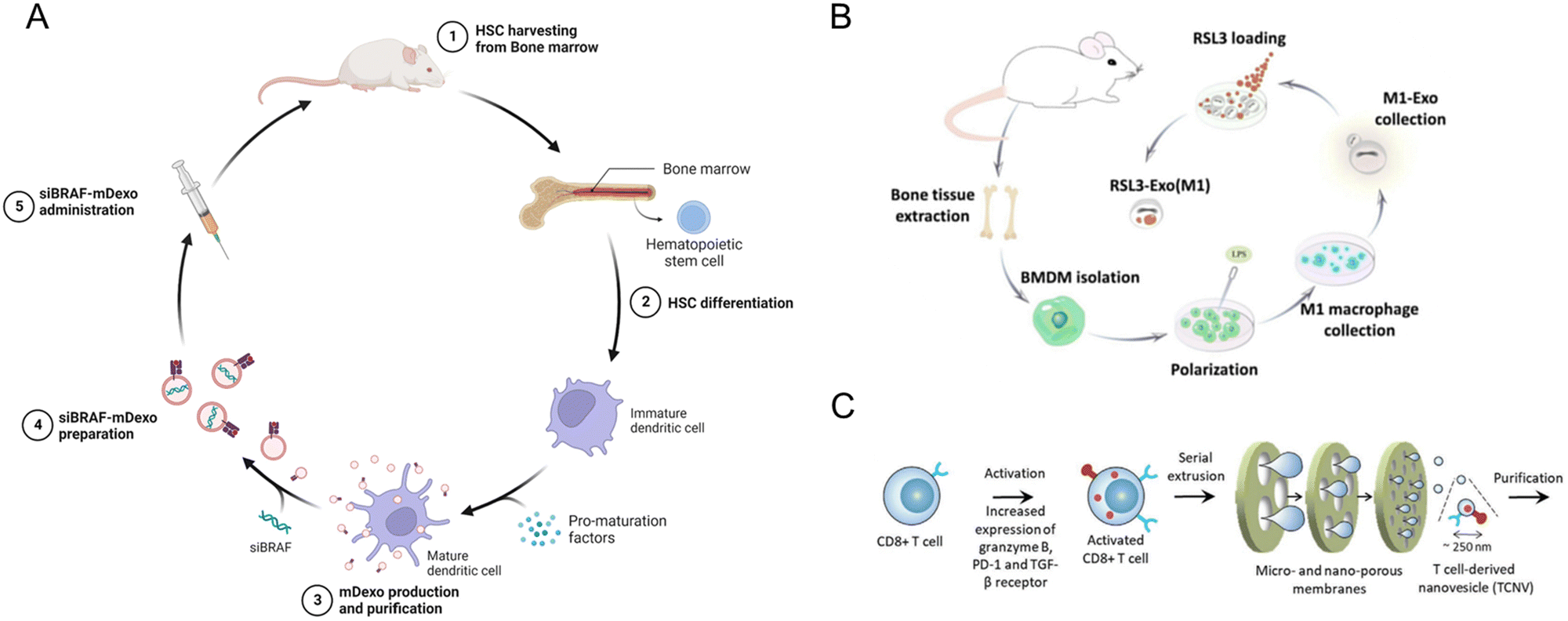 | ||
| Fig. 17 Immune cell-derived exosomes. (A) Construction of DC-derived exosomes for co-delivery of gene therapy and immunotherapy factors, siBRAF-mDexos. Bone marrow cells from the fermus of healthy C57BL/6J mice were prepared for the isolation of hematopoietic stem and progenitor cells. The cells were left to differentiate in media containing 20 ng mL−1 IL-4 and 20 ng mL−1 GM-CSF for 5 days to generate immature DCs. The exosomes (mDexos) were isolated from the supernatant via gradient centrifugation. SiBRAF siRNA was incorporated into the mDexos via electroporation. Reproduced from ref. 212 Copyright 2023 originally published by and used with permission from Dove Medical Press Ltd. (B) Procedure for generating macrophage derived exosomes loaded with ferroptosis factors, RSL3-ExoM1. Bone marrow-derived macrophages were isolated from bone tissue of mice and stimulated by lipopolysaccharide to obtain M1-phenotype macrophages. The exosomes from the M1 macrophages were isolated, followed by M1-Exo production and collection via gradient centrifugation. RSL3, a ferroptosis inducer, was introduced into the exosomes via overnight shaking and excess unincorporated RSL-3 was removed by centrifugation. Reproduced from ref. 241 Copyright 2023 with permission from BMJ Publishing Group Ltd. (C) Preparation of T-cell-derived nanovesicles (TCNVs) for cancer immunotherapy via serial extrusion. TCNVs possess PD-1 protein and TGF-β receptor on their surface. Hence, they can block PD-L1 on cancer cells and scavenge TGF-β in the TME, preventing cytotoxic-T-cell exhaustion. TCNVs can also directly kill tumor cells via granzyme delivery. Reproduced from ref. 242 Copyright 2021 with permission from John Wiley & Sons. | ||
Personalized cancer immunotherapies harness exosomes to deliver patient-specific neoantigens, provoking robust CD8+ T-cell-mediated anticancer immunity.214,243 Dendritic cell-derived exosomes loaded with neoantigens induce potent immune responses.213 Furthermore, exosomes from chimeric antigen receptor T (CAR-T) cells maintain CAR expression, eliciting tailored tumor-specific immune responses and facilitating tumor eradication in preclinical models.244 The exosome vaccines triggered tailored tumor-specific immune responses and tumor eradication in preclinical models.245 Studies also explore innovative approaches.214,246,247 Combining γδ-T-cell-derived exosomes (γδ-T-Exos) with radiotherapy overcomes radioresistance and preserves antitumor activities in immunosuppressive tumor microenvironments.248 A photoactivatable silencing extracellular vesicle (PASEV) merges phototherapy with exosome-mediated small interfering RNA (siRNA) delivery against p21-activated kinase 4 (PAK4), an immune modulator involved in immune exclusion (Fig. 18A). PASEV elicited robust antitumor immunity against B16F10 subcutaneous melanoma tumors. In vivo, PASEV displayed PAK4 silencing and triggered potent antitumor immune response in the lesions and draining lymph nodes.249
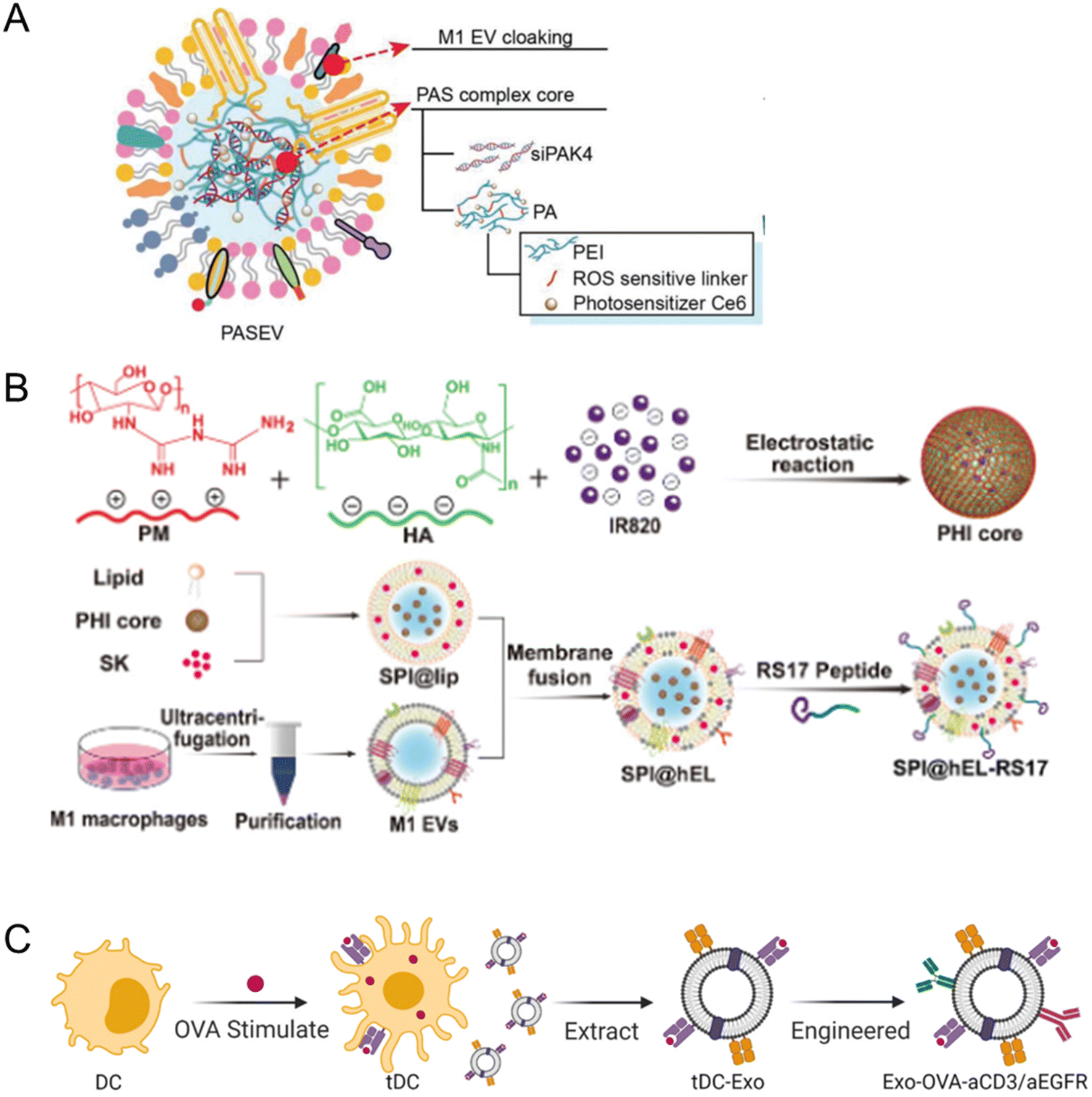 | ||
| Fig. 18 Immune cell-derived personalized exosomes. (A) Photoactivatable silencing exosome sensitizes cancer immunotherapy (PASEV). Small interfering RNAs with, siPAK4 were assembled with a ROS-sensitive linker to form a nanocomplex core, camouflaged with EVs from M1 macrophages. Reproduced from ref. 249 Copyright 2022 with permission from John Wiley & Sons. (B) Immune cell-derived exosomes for amplified CD47 blockade-based cancer immunotherapy. Hybrid nanovesicles derived from M1 macrophages and synthetic liposomes. The formulation was decorated with RS17 peptide (for blocking CD47) metformin, shikonin, and IR820 (a photosensitizer) for a multi-approach treatment of metastatic melanoma. Firstly, the PHI nanoparticle was formed via electrostatic interactions between positively charged polymeric metformin (PM) and negatively charged IR820 and hyaluronic acid (HA). Then the SPI@hEL nanoparticles were formed by membrane fusion with M1 EVs. Reproduced from ref. 211 Copyright 2023 with permission from John Wiley & Sons. (C) Antibody-engineered exosomes from antigen-feeding dendritic cells for precise solid tumor therapy. DC 2.4 cells were activated with ovalbumin and the DC Exo-Ova was purified by gradient centrifugation. The Exo-Ova was functionalized with anti-CD3 and anti-EGFR antibodies via DSPE-PEG-NHS linker to generate Exo-OVA-aCD3/aEGFR. Reproduced from ref. 243 Copyright 2022 with permission from Elsevier. | ||
Immune cell-derived exosomes for amplified CD47 blockade-based cancer immunotherapy is another strategic option. CD47 is found on the surface of many tumors and functions as a ‘don’t eat me’ signal, shielding the tumor growth from immune cell prodding. A hybrid nanovesicles, hEL-RS17, derived from M1 macrophages and synthetic liposomes was formulated to target CD47 blockade (Fig. 18B). The formulation was decorated with RS17 peptide (for blocking CD47) metformin (an immunomodulator), shikonin (SK, a chemotherapeutic agent) and IR820 (a photosensitizer) for multi-approach treatment of metastatic melanoma. This platform represents a “all in one” nanoplatforms designed to tackle the signaling pathway effectors and induce immunogenic cell death via two separate mechanisms that does not exhibit cross-resistance. The platform exhibited potent antitumor efficacy against primary tumors in 4T1 breast tumor and B16F10 melanoma models in mice. An added benefit of lung metastasis inhibition is appealing as the immune response elicited involves protection from tumor recurrence and progression.211
A DC-based platform was engineered for precise solid tumor therapy via DC mediated antigen presentation (Fig. 18C). DC 2.4 cells were activated with ovalbumin as a proxy tumor antigen and was functionalized with anti-CD3 and anti-EGFR antibodies. The formulation functioned as CAR-T cell therapy system with MHC-antigen complexes for antigen presentation and CD86 co-stimulatory molecules for activation and expansion of T-cells. The formulation significantly inhibited the growth subcutaneous B16-Ova lung metastatic melanoma cells and curtailed the number of lung metastases foci in vivo. Significant increase of CD4+ and CD8+ T-cell population was observed in the tumor tissues via immunohistochemical staining.243 In summary, immune cell-derived exosomes represent versatile immunomodulatory tools, offering targeted cargo delivery and profound immune response modulation. Through innovative strategies, they hold promise in reshaping cancer therapy, demonstrating their potential in personalized and combination treatments.
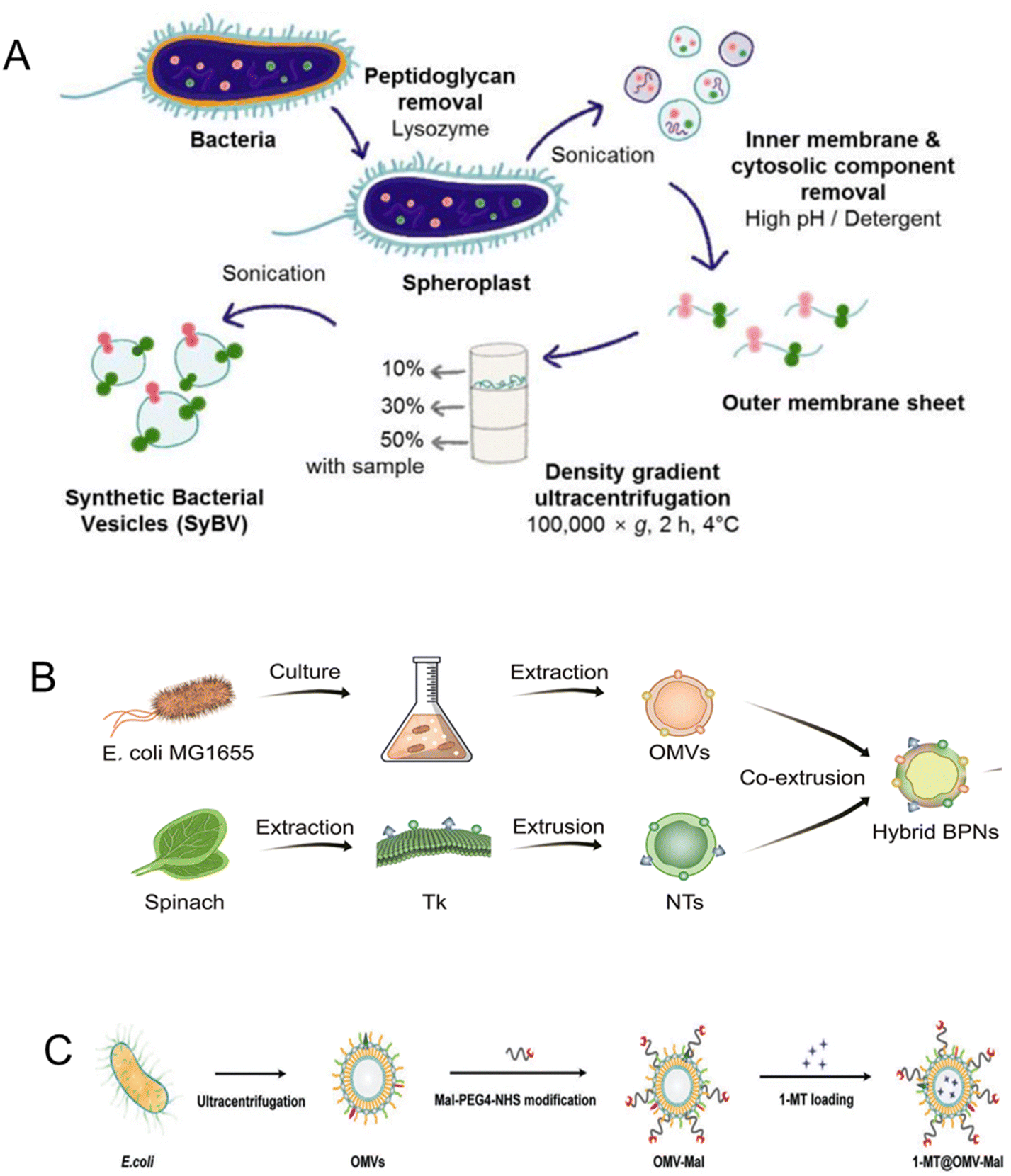 | ||
| Fig. 19 Bacteria-derived exosomes. (A) Schematic diagram of the isolation of bacterial SyBV. E. coli cells were incubated with lysozyme to remove periplasmic components. They were then sonicated to disrupt the cell membranes. The outer membrane vesicles (OMVs) were collected from the interface layer of 10% and 30% iodixanol after buoyant density-gradient ultracentrifugation. Reproduced from ref. 261 Copyright 2021 with permission from John Wiley & Sons. (B) Phytochemically engineered bacterial exosomes for photodynamic effects promoted immunotherapy. The OMVs of E. coli MG1655 was extracted similarly to described in A. Thylakoid nanovesicles (NTs) were prepared from spinach leaves via the extrusion method. The OMVs and NTs were fused using membrane fusion method to obtain a bacteria-plant hybrid vesicles (BPNs). Reproduced from ref. 264 Copyright 2022 with persmission from American Chemical Society. (C) Schematic showing the synthesis of 1-MT@OMV-Mal. E. coli OMVs were isolated by multiple centrifugation and filtration steps. The maleimide (Mal) groups were modified on the surface of the OMV via a reaction between NHS ester in Mal-PEG4-NHS and the amine groups in the membrane proteins to prepare the OMV-Mal. The interior is then loaded with 1-methyl-tryptophan (1-MT, IDO inhibitor) to generate 1-MT@OMV-Mal. Reproduced from ref. 217 Copyright 2022 with permission from John Wiley & Sons. | ||
Additionally, researchers utilized innovative strategies, such as modified E. coli-derived OMVs in versatile nanoplatforms, to enhance cancer immunotherapy. These modified OMVs facilitated targeted delivery, induced immunogenic cell death, and regulated the tumor environment. Bacterial OMVs-based in situ cancer vaccines and biomimetic hybrid nanoplatforms amplified antitumor immune responses and significantly inhibited tumors, offering potential clinical alternatives against tumors with reduced side effects.203,216,264 In an exemplary work by Zhuang et al., hybridized bacteria-plant hybrid vesicles (BPNs) can be generates by membrane fusion method comprising OMVs and thylakoid nanovesicles (NTs) prepared from spinach leaves (Fig. 19B). The efficacy of the BPNs in inducing tumor regression and immune modulation was performed in a xenograft CT26 subcutaneous tumor model in mice. The BPNs increase CD8+ T-cells, reduce Treg populations, and elevated the M1/M2 macrophage ratio in the tumors.264
OMVs can also be utilized as a vaccination or adjuvant platform following a primary treatment. In one example, a multifunctional vaccine based on bacterial OMVs was designed for cancer immunotherapy after photothermal therapy (PTT). The OMV platform, termed 1-MT@OMV-Mal, was loaded with 1-methyl-tryptophan (1-MT), an IDO inhibitor (Fig. 19C). After a one-time ICG-based PTT of subcutaneous CT26 tumors in mice, the platform was used as an immunotherapy vaccine. The vaccination significantly reduced both treated primary tumor and untreated primary tumor volumes compared to PTT alone and induced a potent antitumor immune response involving CD8+ T-cells, B-cells, and macrophages.217 Overall, these studies demonstrate the promising potential of OMVs from Gram-negative bacteria in enhancing cancer immunotherapy through various innovative approaches and combinations with existing immunotherapies.
| Exosome type | Source | Application | Targeting | Immunomodulation mechansim | Animal model | Ref. |
|---|---|---|---|---|---|---|
| BCG, bacillus calmette – guerin; DCs, dendritic cells; ICD, immunogenic cell death; ICI, immune-checkpoint inhibitors; NK, natural killer; PD-L1, programmed death-ligand 1. | ||||||
| Bacteria | Escherichia coli | Antigen-presenting | Tumor cells | Activate T lymphocyte cells | Subcutaneous | 215 |
| Bacteria | Gram-negative bacteria | Antigen-presenting | Tumor cells | Activate NK cells | Subcutaneous | 263 |
| Bacteria | Escherichia coli | Drug delivery | Tumor cells | Induce ICD | Subcutaneous | 216 |
| Bacteria | Mycobacterium bovis BCG | Antigen-presenting | Tumor cells | Induce cytokine responses | Orthotopic | 272 |
| Bacteria | Escherichia coli | Antigen-presenting | Tumor cells | Activate DCs | Metastatic | 264 |
| Bacteria | Escherichia coli | Antigen-presenting | T lymphocytes | Recruit DCs | Metastatic | 273 |
| Bacteria | Gram-negative bacteria | Antigen-presenting | T lymphocytes | Increase T lymphocyte infiltrations | Subcutaneous | 217 |
| Bacteria | Salmonella | Antigen-presenting | T lymphocytes | Increase T lymphocyte infiltrations | Metastatic | 274 |
| Bacteria | Escherichia coli | Antigen-presenting | Tumor exosomes | Induce T lymphocyte responses | Orthotopic | 261 |
| Bacteria | Akkermansia muciniphila | Antigen-presenting | T lymphocytes | Promote macrophage polarization | Subcutaneous | 262 |
| Liquid biopsy | Plasma | Antigen-presenting | Tumor cells | Predict ICI response | — | 267 |
| Liquid biopsy | Blood | DNA cargo | Tumor cells | Determinate circulating PD-L1 | — | 265 and 267 |
| Liquid biopsy | Blood | Antigen-presenting | Tumor cells | Detect exosomal PD-L1 | Subcutaneous | 266 |
| Liquid biopsy | Plasma | Antigen-presenting | Tumor cells | Predict immunotherapeutic outcomes | Subcutaneous | 218 |
| Red blood cell | Blood | Drug delivery | Tumor cells | Activate RIG-I pathway | Metastatic | 219 |
| Embryonic kidney cell | HEK293T cell line | Antigen-presenting | Tumor cells | Increase T lymphocyte infiltrations | Orthotopic | 220 |
| Mesenchymal stem cell | Bone marrow | RNA cargo | Tumor cells | Inhibit CD38 enzyme activity | Subcutaneous | 268 |
| Mesenchymal stem cell | Bone marrow | Drug delivery | Tumor cells | Activate dendritic cells | Subcutaneous | 269 |
| Mesenchymal stem cell | Bone marrow | Antigen-presenting | Tumor cells | Induce ICD | Orthotopic | 271 |
| Mesenchymal stem cell | Bone marrow | Antigen-presenting | Tumor cells | Promotes immune escape | Subcutaneous | 270 |
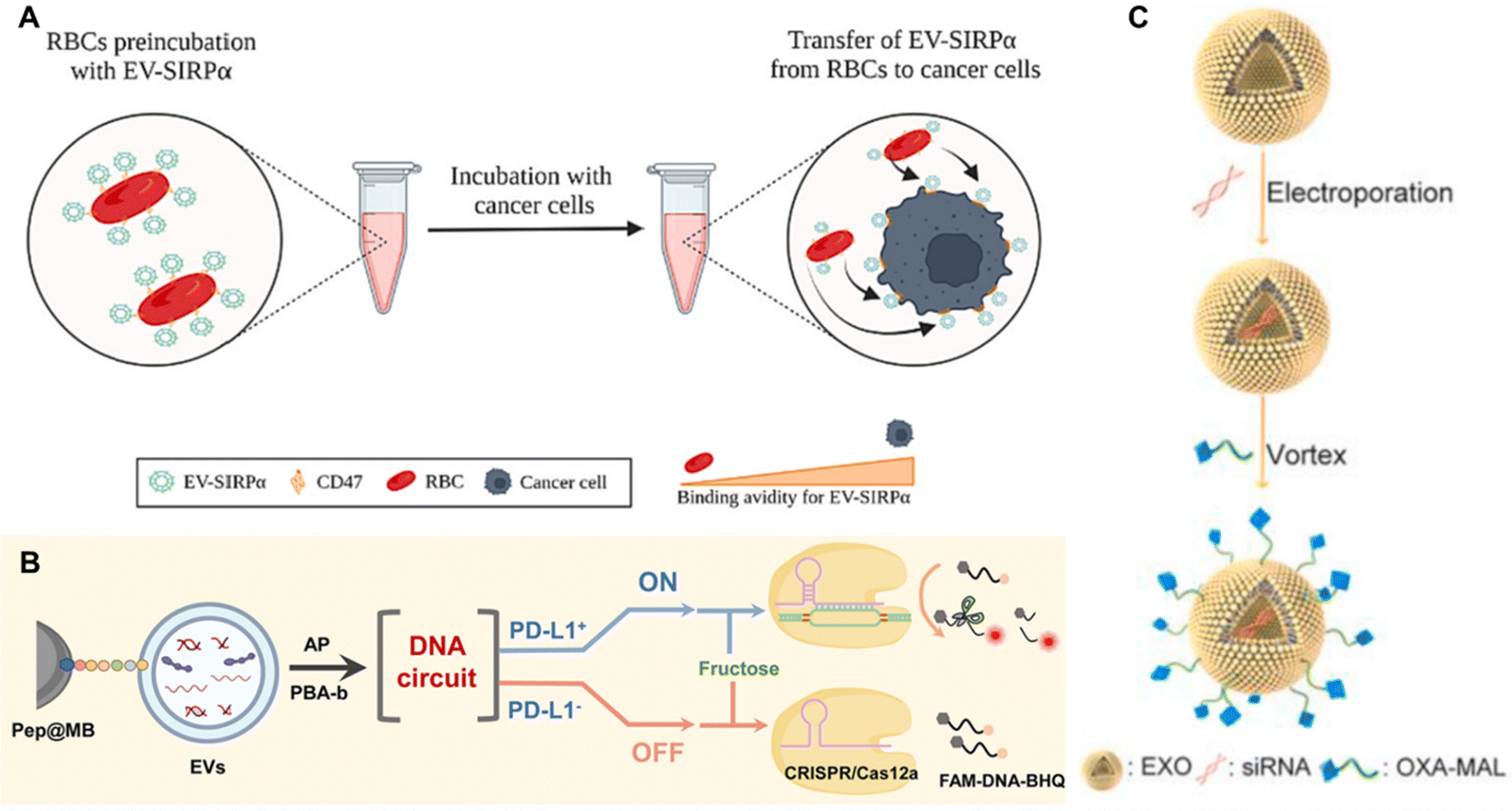 | ||
| Fig. 20 Exosomes derived from other various sources. (A) EVs that express SIPRα, EV-SIRPα, is a RBC-derived exosomes that can hinder the CD47 “don’t eat me” signaling present on tumor for cancer immunotherapy. Reproduced from ref. 220 Copyright 2022 with permission from Elsevier. (B) Circulating exosomes for lung cancer diagnosis and immunotherapy response prediction. A programmable DNA circuit will translate the presence of PD-L1 into the appearance of duplex DNA probes on the surface of the EVs to activate he trans-cleavage activity of CRISPR/Cas12a system, which finally produces a significant fluorescence signal. Reproduced from ref. 265 Copyright 2023 with permission from American Chemical Society. (C) Bone marrow mesenchymal stem cell (MSC)-derived exosomes for immunotherapy of pancreatic ductal carcinoma (PDAC). The exosomes from MSCs were isolated by gradient centrifugation and loaded with galectin 9 siRNA via electroporation. The surface of the exosome was modified with oxaliplatin (OXA) to trigger immunogenic cell death. Reproduced from ref. Copyright 2021 with permission from Elsevier. | ||
Stem cell-derived exosomes represent a potent avenue in cancer immunotherapy, showcasing their ability to deliver therapeutic agents, reshape the tumor microenvironment, and enhance immunotherapeutic efficacy (Table 5). Recent studies underscore their potential applications. CD38 siRNA-loaded exosomes from bone marrow mesenchymal stem cells (BM-MSCs) were harnessed to counter immunosuppression in hepatocellular carcinoma, inhibiting tumor growth and metastasis.268 Similarly, nanovesicles derived from BM-MSCs incorporated anti-PD-L1 antibodies for targeted drug delivery, resulting in robust immune activation and tumor ablation in photoimmunotherapy.269 A dual delivery system from BM-MSCs was also demonstrated, triggering immunogenic cell death and enhancing therapeutic efficacy in cancer cell aggressiveness and immune evasion.270 Zhou et al. fabricated OXA-MAL, a MSC-based exosomes functionalized with oxaliplatin prodrug on its surface to trigger immunogenic cell death. Additionally, the construct was loaded with galectin 9 siRNA to target the M2 phenotype mediated immunosuppression via the galectin 9/dectin 1 axis (Fig. 20C). In vivo efficacy testing showed an immune response centred on increased M1/M2 phenotype ratio in the tumor as well as Treg cell decrease and infiltration of CD8+ T-cell in the tumor lesions.271
4.3. Immunotherapy via multifunctional delivery using versatile albumin nanoparticles
Albumin is the most abundant protein in the blood and plays many roles in the body, including modulating the plasma oncotic pressure and transporting various endogenous and exogenous substances, such as hormones and drugs.275 The protein has an intrinsic ability to target and accumulate in tumors due to enhanced passive uptake via the EPR effect. Albumin can also bind to specific surface receptors such as the secreted protein acidic and rich in cysteine (SPARC) and the 60-kDa glycoprotein (gp60) receptor. Uptake of albumin via these receptors can enhance the delivery of drugs to tumor cells. For example, the transport of nab-paclitaxel (nab-PTX, abraxane), an FDA-approved albumin-bound paclitaxel, was more efficient by 4.2 folds compared to standard paclitaxel solution.276 Albumin possesses many functional groups that can be used for functionalization and for drug loading.277 Due to its intrinsic role in transport biology, albumin-mediated delivery is stable and protects the cargo load from degradation. Research groups have spent efforts in formulating albumin-based compounds for immunotherapy due to its status as an FDA-approved bionanomaterial.Albumin can be extracted from different sources such as ovalbumin from chicken egg and rat serum albumin. This section will focus on human (HSA) and bovine serum albumin (BSA), the most used protein bionanoparticle platforms, to assess the strategies used for endowing the albumin nanoparticle complexes with immunomodulatory properties. Albumin on its own is non- or mildly immunogenic exhibiting no cytotoxicity. Hence, it serves as a blank canvas for loading compounds that can elicit an immune response. The most applied strategy for immunomodulation with albumin is via the induction of ROS to trigger oxidative stress resulting in cell death and the use of checkpoint inhibition. ROS induced cell death is known to recruit and stimulate the immune system. Albumin nanoparticles were used as a photosensitizer delivery vehicle or assembled with zinc sulfide (ZnS) to trigger the formation of ROS. In most of these studies, phototherapy is a central treatment method to induce ROS buildup.
A formulation comprising human serum albumin HSA holding IR780 (a dye photosensitizer) and zinc sulfide (ZnS) was formulated by Yang et al. via self-assembly to generate IR780-ZnH@HSA complexes (Fig. 21A).278 The zinc ions from ZnS can induce ROS of facilitated by generation of hydrogen sulfide (H2S). IR780-ZnH@HSA was used as a photothermal therapy (PTT) and photodynamic therapy (PTT) effector that induces cells death via pyroptosis by activating the caspase-3-GSDME signaling pathway combined with anti PD-L1 therapy (aPD-L1). In vivo efficacy testing against a subcutaneous 4T1 mammary carcinoma model in mice showed that three doses of PDT/PTT and aPD-L1 combination therapy with IR780-ZnH@HSA controlled both primary tumor and distant metastases, indicating an immune response which was amplified systematically (Fig. 21B and C). Mice in the PDT/PTT with and without aPD-L1 exhibited better survival over 60 days. Most importantly, the treatment also induced increased CD8+T-cell and decreased Foxp3 Tregs populations in the primary tumor. Elevated serum cytokine levels indicated a systemically activated immune response (Fig. 21D and E). A similar immune activation effect is observed by Cen et al. with their ZnS@BSA formulation designed to result in a ROS induced cell death via the release of Zn ions. The albumin nanoparticle combined with aPD-L1 treatment also promoted distant tumor growth inhibition and CD8+ T-cell recruitment and activation in the primary tumor in a subcutaneous model of HCC with Hepa1-6 cells.279
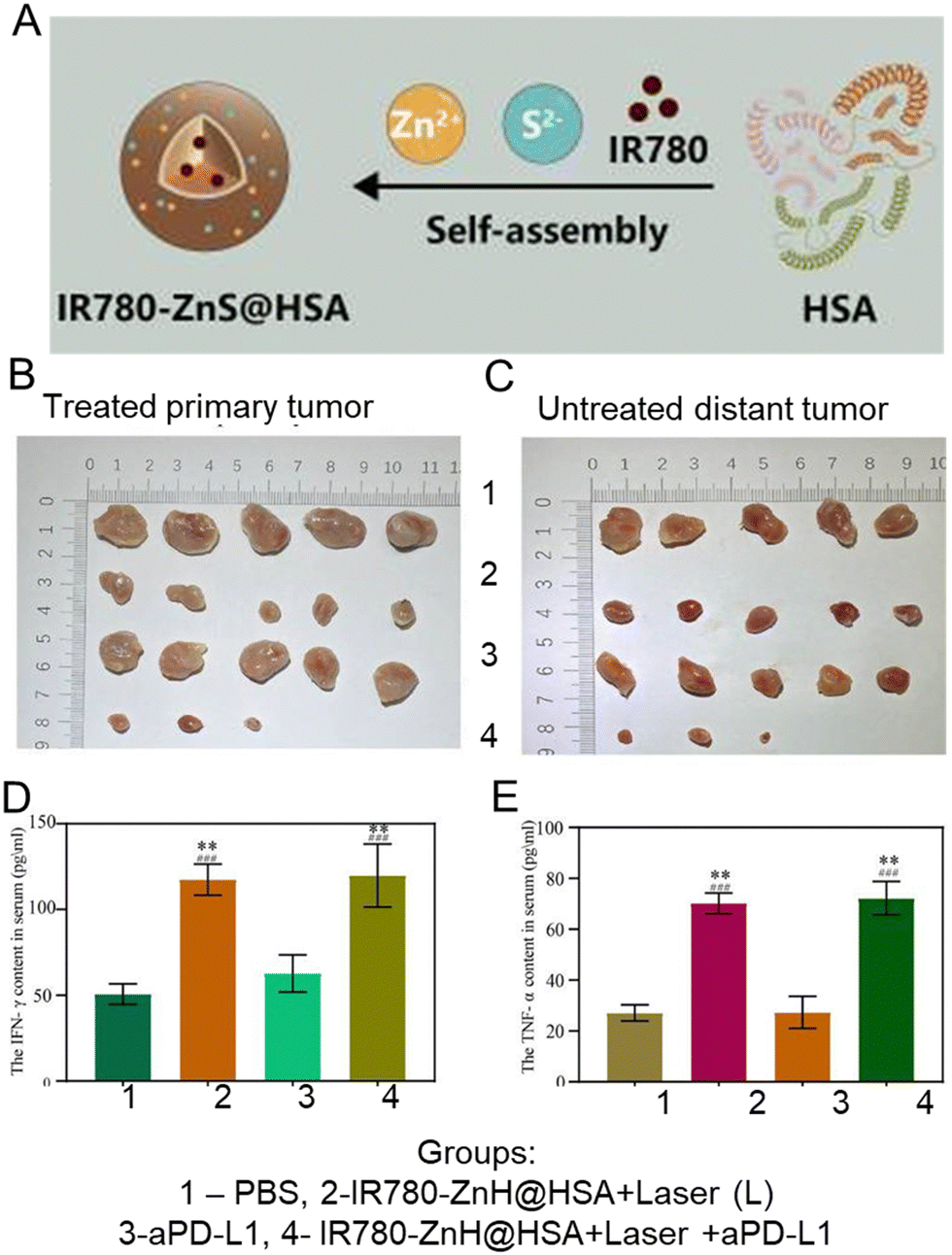 | ||
| Fig. 21 The antitumor and immunomodulatory effects of IR780-ZnS@HSA nanoparticles (NPs). (A) A schematic of the fabrication strategy of IR780-ZnS@HSA nanoparticles (NPs). Photographs of (B) treated primary tumor, and (C) distant untreated tumors of 4T1 subcutaneous lesions after three phototherapy doses with IR780-ZnS@HSA nanoparticles and aPD-L1. Analysis of serum (D) IFN-γ and (E) TNF-α three days after various treatments. Data is represented as mean ± SD. **P < 0.01. (n = 3). Reproduced from ref. 278 Copyright 2023 originally published by and used with permission from frontiers. | ||
Various BSA-based formulations carrying photosensitizers for PDT, such as MHI148 dye, chlorin e6, and TPA-Erdn, exhibited immunomodulatory mechanisms via increased CD8+ or CD4+ T-cell accrual to the tumor sites.280–282 Hence, it is noteworthy that without checkpoint inhibition, ROS induced cell death that occurs sufficiently at a threshold above cellular tolerance point can edit the TME to a pro-inflammatory state via the release of dead tumor cell debris, essentially converting a cold tumor into a hot tumor state. Combining phototherapy with checkpoint inhibition enhances the treatment outcomes in vivo, adding another layer of immunomodulation guarantee in the package. Coating of albumin with gold nanoparticles (AuNPs) for as a photothermal agent is another viable strategy for generating antitumor immunity. Zhang et al. fabricated HSA@AuNPs that also deliver human melanoma peptide antigen gp10025–33 (hgp100) as a PTT plus vaccination combination strategy, taking full advantage of the versatile binding sites of the HSA molecule.283 Another Au–albumin complex, mPEG-GNRs@BSA/R837, was formed by preparation of mPEG (polyethylene glycol monomethyl ether with sulfhydryl end group)-Au nanorods (NRs) core followed by coating with BSA and loading with an immunoadjuvant imiquimod (R837) through electrostatic binding. PTT with mPEG-GNRs@BSA/R837 combined with anti-PD1 inhibitor provided long-term antitumor immunity (100 days surveillance) and inhibited tumor metastases in mice inoculated subcutaneously with metastatic melanoma cell line, B16F10. The release of inflammatory cytokines, TNF-α, IL-6, and IL-12 was significantly elevated in the serum three days after PTT and CD8+ T-cells were detected in B16F10 tumor sections.284 A similar complex of albumin-modified AuNRs for PTT triggered the activation of immature DCs in a contact-dependant manner between DCs and 4T1-Luc mammary tumor cells.285 In summary, oxidative and heat stress are efficient methods to induce cellular death that can activate an antitumor immune response but such methods must be used with caution because mild levels of stress exposure can promote tumor progression.286 Thus, it is necessary to utilize an exposure of the stress at levels beyond the cellular repair threshold, thereby directing the cellular fate towards death.
Checkpoint inhibition therapy has been gaining traction in the treatment of various advanced malignancies. Hence, efforts were focused on fabricating a single platform albumin-based nanomedicine bound with checkpoint inhibition molecules. Lai et al. fabricated a fexofenadine-loaded albumin nanoparticles functionalized with PD-L1 aptamers (PDL1-NP-FEXO) for the treatment of a subcutaneous colon carcinoma model in mice using CT26 cells. Fexofenadine is a H1-antihistamine and is known to stimulate the immune response by reducing M2 phenotype macrophages in the TME. The PDL1-NP-FEXO bind to PD-L1 positive MDA-MB-231 cells in vitro and significantly inhibited CT26 tumor growth in vivo without generating systemic toxicity.287 A similar formulation was fabricated by Yao et al. with CTLA-4 aptamers (CTLA-4-NP-FEXO) which could bind CTLA-4 positive cells and improved antitumor immunity in vivo via lymphocyte activation.288 Combination therapy was explored with HSA-PTX generated via albumin bound technology and then pooled with anti-PD-L1 monoclonal antibody through a pH-sensitive linker (Fig. 22A). The complex was termed PD-L1/PTX@HSA and was tested in a mouse subcutaneous model of mammary carcinoma with EMT-6 cells. Intravenous injection of the albumin complex was found to be distributed predominantly in the liver and kidneys with tumor site accumulation after 24 hours post intravenous injection. No organ toxicity was observed upon treatment completion. The treatment comprises five doses of the albumin complex, and mice were sacrificed on day 15 of the study, three days after the last treatment dose. Successful tumor growth inhibition was observed along with the infiltration of CD4+ and CD8+ T-cells in the tumor, especially when combined with CTLA-4 inhibition (Fig. 22B-E). The infiltration of immunosuppressive Foxp3+ and PD-L1+ cell-types was significantly abrogated (Fig. 22F and G) signifying immunoediting towards an inflammatory antitumor environment.289
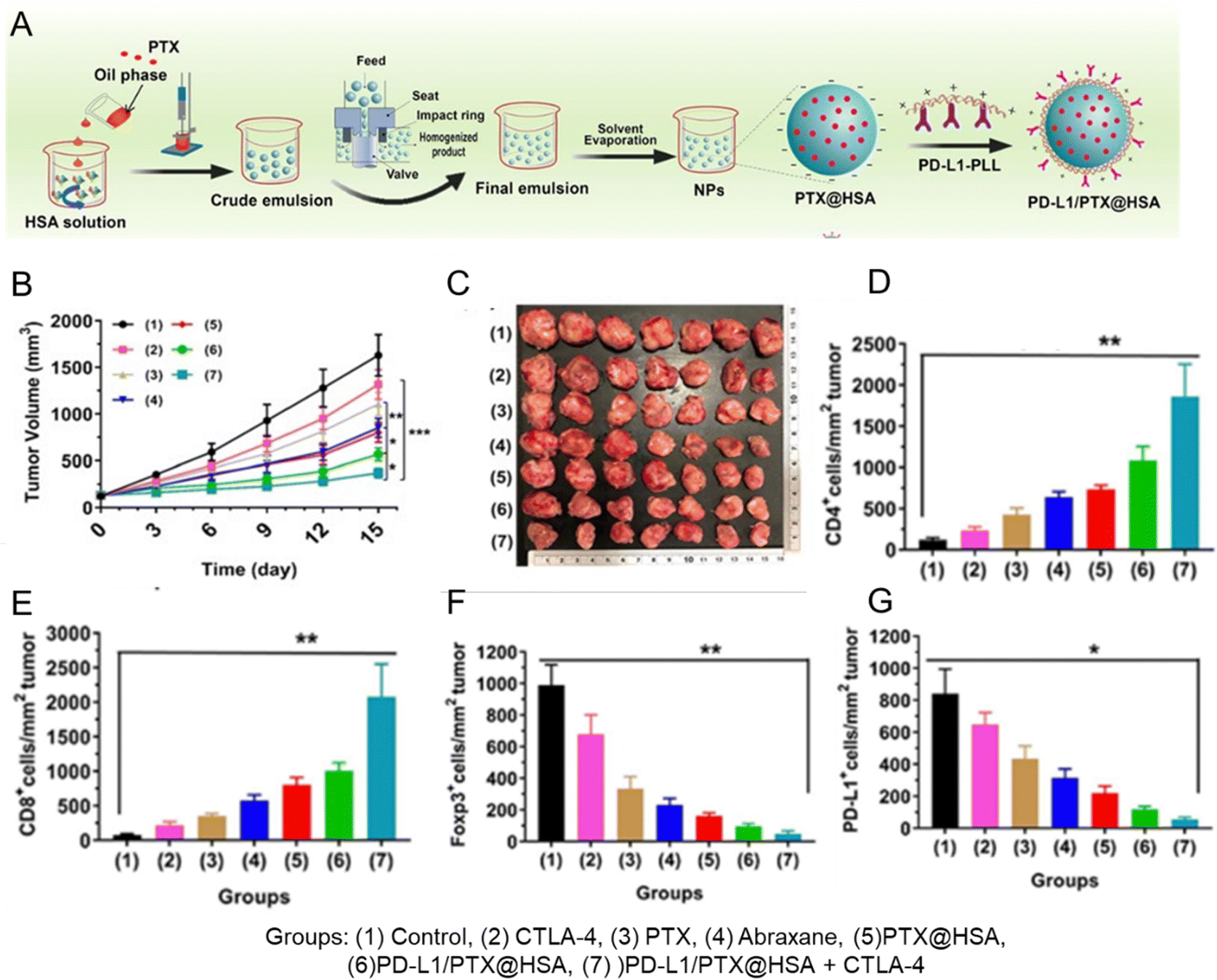 | ||
| Fig. 22 Combination of chemotherapy and immunotherapy PDL1/PTX@HSA. (A) Schematic of the PDL1/PTX@HSA synthesis procedure. (B) 15-day examination of tumor volumes in each group. (C) Representative tumors excised after termination. Data are represented as means ± SD (n = 7). *P < 0.05, **P < 0.01, ***P < 0.001. Percentage of (D) CD4+ T-cells, (E) CD8+ T-cells, (F) Foxp3+ Tregs, and (G) PD-L1+ cells from immunohistochemistry analysis of the excised tumors after study period data are represented as means ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001. Reproduced from ref. 289 Copyright 2021 with permission from Elsevier. | ||
The versatility of the albumin macromolecule facilitates binding to various drugs and molecules. Ai et al. used BSA molecules as a nanocarrier for the delivery of CpG oligodeoxynucleotides (ODNs) to macrophages targeted via mannosylation of cationic BSA (ODN@MCBSA).290 The mannose moiety functionalization of the BSA endows macrophage targeting through its interaction with the mannose receptor on the macrophages. The complex facilitated the polarization of macrophages to the M1 phenotype in vitro. Furthermore, the team demonstrated the enhanced secretion of pro-inflammatory cytokines such as IL-12, IL-6, and TNF-α after macrophage treatment with ODN@MCBSA in vitro. However, their formulation was used solely as a macrophage targeting agent to induce activation and polarization towards the M1 phenotype and did not exhibit any toxicity towards HeLa cells in vitro.290 Another formulation aimed at overcoming the blood brain barrier (BBB) by functionalizing the albumin with a brain-targeting peptide sequence, DCDX (cgreirtgraerwsekf, D-form sequence), that can target the nicotininc acetylcholine receptors (nAChRs) expressed highly in the endothelial cells of the brain.291 The functionalized albumin was used to co-encapsulate celastrol (CELA), and mTOR inhibitor, and LY2157299, a TGF-β receptor I (TGFβRI) inhibitor. CELA was reported to promote M2 to M1 macrophage polarization while the TGFβRI inhibitor can downregulate the effects of TGF-β in the tumor microenvironment, alleviating the tumor-induced immunosuppression. The DCDX-BSA NPs demonstrated efficient in vivo targeting to implanted orthotopic brain tumors in mice, revealing intracranial accumulation at 4.2 times higher than that of BSA-NPs group. Analysis of the immune environment in the glioma revealed a significant increase in the percentage of mature DCs and a decrease in the overall TAMs population.291 Efforts to deliver inhibitors to metabolic actors responsible in tumor-induced immunosuppression, such as IDO and PI3Kγ, have also demonstrated success in modulating the TME in vivo.292–294
5. Limitations
The section discourse delves into the challenges of utilizing bio-nanoparticles for tumor immunomodulation, focusing on nuanced aspects crucial for therapeutic development. The choice of cell membrane source plays a pivotal role in dictating the system's efficacy, given the diverse interactions of various cell types with the immune system.168,169 A paramount concern arises from the immunogenicity of cell membrane-based platforms, as these structures may be recognized and eliminated by the host immune system, potentially hampering their therapeutic effectiveness. Studies have explored innovative strategies, such as employing hybrid membranes like erythrocyte-cancer cell hybrids, to mitigate immunogenicity and amplify therapeutic outcomes.167,205 Furthermore, the immunosuppressive milieu within tumor microenvironments poses formidable challenges. Tumor cells employ immune tolerance mechanisms, such as upregulating immune checkpoint molecules, impeding the effectiveness of cell membrane-based immunotherapies.178,205 Despite efforts to bolster immune responses, these approaches encounter hurdles in overcoming the immunosuppressive microenvironment orchestrated by cancer cells. Combining cell membrane-based immunotherapy with immune checkpoint inhibitors exhibits promise in preclinical studies; however, further optimization is requisite for clinical translation.In the realm of exosome-mediated immunomodulation, a fundamental obstacle lies in the incomplete understanding of the mechanisms underpinning this process. While existing literature underscores the immunomodulatory effects of exosomes derived from diverse cell types, the intricate mechanisms governing their interactions with immune cells and regulation of immune responses remain insufficiently elucidated.208,209,232 It is imperative to delve deeper into these interactions, delineating the key molecular constituents orchestrating exosome-mediated immunomodulation. Moreover, researchers must consider the intricate immune microenvironment and the influence of diverse factors in the context of cancer immunomodulation, necessitating meticulous investigations. The precise biodistribution and targeted delivery of exosomes to tumor sites present significant challenges.229,230,236 Despite some studies demonstrating the tumor-targeting capabilities of exosomes, enhancing strategies are indispensable to augment their accumulation and persistence within tumor tissues. This optimization is critical for ensuring the efficient delivery of therapeutic payloads, thereby maximizing the inherent therapeutic efficacy of exosomes. Sustained efforts in this domain are pivotal for advancing the potential applications of exosome-mediated immunomodulation in cancer therapy. Careful considerations must also be observed when using exosomes derived from tumor cells due to their involvement in cancer progression and therapy resistance.295
Tumor antigen heterogeneity poses a formidable obstacle for personalized cancer vaccines, as acquiring patient-specific antigens proves impractical. Exosome heterogeneity introduces challenges in treatment consistency, necessitating standardized approaches. The stability of cell membrane coatings emerges as a critical concern for long-term efficacy, urging further research efforts. Scalability issues plague both exosome and cell membrane production, as their production relies heavily on top-down approaches, demanding the development of efficient methods to meet the demands of therapeutic applications.296,297 Safety concerns encompass potential autoimmune reactions, immune related adverse events, and off-target effects, mandating comprehensive studies to evaluate the safety profiles of these therapies.134
Albumin nanoparticles enjoy varying degrees of success as a drug delivery vehicle, transporting and releasing chemotherapeutic drugs in cancer therapy. Their low toxicity and biocompatibility also define the success of albumin as a carrier, as the carrier protein shows no toxicity to cancer or healthy cells. However, albumin is limited by its cargo loading ability, with issues in payload capacity and conjugation efficiency.298 Without intrinsic immune potential, albumin is restricted in its role as a carrier and is limited in the selection of agents and pharmaceuticals that can be loaded into the structure. However, the albumin nanoparticle platform may offer the best chance of clinical translation due to their regulatory precedent. The prevalence of small sample sizes in existing research restricts the generalizability of findings. Undertaking larger-scale studies is imperative to validate the predictive and therapeutic potential of bionanoparticles in cancer immunomodulation. Addressing these multifaceted challenges through standardization, rigorous evaluation, and expansive studies is indispensable for propelling the field of cancer immunotherapy toward enhanced efficacy and broader applicability.
6. Conclusion and outlook
In this review, the authors comprehensively summarized the tumor-induced immune aberrations to enable a broad and general understanding of the challenges in tackling tumor-induced immune suppression using nanotechnology approaches. Furthermore, recent advances in fabricating membrane biomimetic bionanoparticles were discussed and organized according to membrane source. Fabricating bio-nanoparticles for immunotherapeutic purposes is still in a nascent stage, but developing rapidly, with huge potential to impact clinical practice. The addition of immunomodulation potential ups the ante by tipping the balance of a developed immunosuppressive environment, a hallmark of cancer, into an activated state equipped to recognize and mount a tumor-specific response. Various efforts on the bench provide a positive outlook with an excellent opportunity for growth and expansion. Currently, membrane coating and exosomes are the predominant strategies utilized to generate immunomodulating platforms for cancer therapy, with some focus on developing the FDA-approved albumin nanoparticle. Efforts in other areas can complement the progress in this field, such as developing computational and AI-based methods for modeling tumor-induced immune aberrations and using the model to predict outcomes of exploratory treatments. Other biological and bionanoparticle platforms that can be exploited for immunomodulation are listed below. Suggestions for combination approaches are also included for each platform in a point-by-point manner:• Caged protein nanocarriers. These nanoparticle complexes possess hollow structures with nearly monodispersed-sized distribution.299 Some of the members of this family are naturally immunogenic, such as virus-based proteins, and they can be combined with checkpoint blockade to stimulate the immune response and subdue the immunosuppressive mechanisms induced by their structure.300 They can also be functionalized with membrane proteins for tumor targeting.
• Cytokines and peptides. These proteins are potent stimuli that can recruit and activate innate and adaptive immune components. Customization and encapsulation of protein cocktails can be done to achieve the desired immune effect, such as the polarization of M2 to M1 phenotype macrophages or proliferation and clonal expansion of T-cells.301 However, cytokines are generally pleiotropic, and the release of cytokines should be designed with stimuli responsiveness and spatially controlled in the TME.
• Lipid rewiring. The formation of lipid rafts and the accumulation of lipids are recently discovered mechanisms inducing dysfunction in antigen presentation and cytokine secretion. Lipid rewiring nanoparticles and blockage of lipid uptake using composite hydrogels have demonstrated the reactivation of immune cells and inhibition of immunosuppressive immune cells.302,303
• Artificial antigen-presenting cells. Synthetic artificial antigen-presenting cells were formulated for T-cell activation.304 A biological-based platform can be fabricated with the MHC-peptide complex subunits and an immunostimulatory agent, such as viral-based nanoparticles or exosomes to generate a tumor-specific response and an inflammatory reaction conducive to tumor eradication.
• RNA interference, mRNA technology, and gene editing techniques. The structure-to-function relationship of biological molecules is well studied. Hence, bionanoparticles are amenable to modifications for assembly and functionalization via the genetic code, nucleotide, and protein chemistry. The fine-tuning of such modifications is achievable via genetic engineering techniques and mRNA technology. The encapsulation of messenger RNA (mRNA) in lipid nanoparticles is one of the most advancing strategies in this field, spurred by the success of the COVID-19 mRNA vaccine.305 Similarly, RNA interference can be used to silence unfavourable phenotypes. Such approaches can also be attempted to generate exosomes with desirable cargo protein load at increased concentrations.
Nanotechnology applications in medicine have improved clinical management, from diagnostics to treatment and prognosis. Advances in bionanoparticle design for cancer therapy can draw inspiration and lessons from the journey of its synthetic counterparts. In a heavily regulated environment, the risk-to-benefit ratio of exploratory treatments is weighted carefully before approval for use in human cancer patients. Hence, most synthetic nanoplatforms fail due to biodistribution and toxicity issues. This risk is especially avoided to protect cancer patients whose systems are already compromised by the malignant disease. Better safety profiles are anticipated for bionanoparticle formulations. However, rendering immunomodulatory functions can be a double-edged sword, as the immune system is a powerful entity with a far-reaching influence on human physiology. As an example, debilitating and even fatal side effects were reported with the breakthrough CAR-T cell therapy despite its biological origin. Hence, careful considerations need to be made about the spatiotemporal manner of immune activation.
Certain cancers will benefit from a tissue-targeted treatment concept. Hence, adding tumor-targeting moiety and stimuli-responsiveness will help confine the activation in the TME, increasing treatment safety. Others, such as hematological malignancies, will require activation in the circulatory system, which presents more risks. Regardless, advanced nanotherapy design must consider the biological barriers to tissue-specific delivery and implement controls to circumvent the barriers, e.g., endothelial barriers such as leaky tumor vasculature and extrinsic barriers such as high interstitial fluid pressure in tumors. Most importantly, rigorous pre-clinical studies done using orthotopic animal models that recapitulate the malignant disease in humans are pertinent for determining the efficacy, safety, toxicity, and long-term effects of the bionanoparticles before transitioning to clinical trials. Finally, multidisciplinary collaborative efforts and perspective input from material scientists, immunologists, pharmacologists, and oncologists are vital in advancing the development of bionanoparticles in the clinic. This development will be a boon for cancer patients for whom the standard of care and alternative therapies fail to provide a lasting cure.
Author contributions
Juwita N. Rahmat conceptualisation, writing – original draft, writing – review & editing; Jiayi Liu writing – original draft; Taili Chen writing – original draft; ZhiHong Li supervision; Yong Zhang conceptualisation, writing – review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge financial support from Singapore's National Medical Research Council (NMRC, MOH-000640, MOH-001114-00) and the City University of Hong Kong (project number 9380160).References
- D. T. Debela, S. G. Muzazu, K. D. Heraro, M. T. Ndalama, B. W. Mesele, D. C. Haile, S. K. Kitui and T. Manyazewal, SAGE Open Med., 2021, 9, 20503121211034366 CrossRef PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 CrossRef PubMed.
- S. Sim and N. K. Wong, Biomed. Rep., 2021, 14(5), 42, DOI:10.3892/br.2021.1418.
- L. A. Dykman and N. G. Khlebtsov, Acta Naturae, 2011, 3, 34–55 CrossRef CAS.
- S. Medici, M. Peana, V. M. Nurchi and M. A. Zoroddu, J. Med. Chem., 2019, 62, 5923–5943 CrossRef CAS PubMed.
- T. Alberti, D. Coelho, A. P. L. Voytena, H. D. S. Pitz, M. D. Prá, L. Mazzarino, S. Kuhnen, R. M. Ribeiro-do-Valle, M. Maraschin and B. Veleirinho, Curr. Pharm. Des., 2017, 23(24), 3515–3528, DOI:10.2174/1381612823666170503152550.
- P. N. Navya, A. Kaphle, S. P. Srinivas, S. K. Bhargava, V. M. Rotello and H. K. Daima, Nano Converge., 2019, 6(1), 23, DOI:10.1186/s40580-019-0193-2.
- A. Karabasz, M. Bzowska and K. Szczepanowicz, Int. J. Nanomed., 2020, 15, 8673–8696, DOI:10.2147/ijn.s231477.
- S. Svenson, Chem. Soc. Rev., 2015, 44(12), 4131–4134, 10.1039/c5cs00288e.
- R. A. Alshehri, A. M. Ilyas, A. Hasan, A. Arnaout, F. Ahmed and A. Memić, J. Med. Chem., 2016, 59(18), 8149–8167, DOI:10.1021/acs.jmedchem.5b01770.
- Y. Luo, X. Cai, H. Li, Y. Lin and D. Du, ACS Appl. Mater. Interfaces, 2016, 8(6), 4048–4055, DOI:10.1021/acsami.5b11471.
- P. C. Lippert and J. C. Zachos, Paleoceanography, 2007, 22(4), PA4104, DOI:10.1029/2007PA001471.
- S. Negrini, V. G. Gorgoulis and T. D. Halazonetis, Nat. Rev. Mol. Cell Biol., 2010, 11(3), 220–228, DOI:10.1038/nrm2858.
- K. Kunimasa and T. Goto, Int. J. Mol. Sci., 2020, 21(2), 597, DOI:10.3390/ijms21020597.
- B. V. Kumar, T. J. Connors and D. L. Farber, Immunity, 2018, 48, 202–213 CrossRef CAS PubMed.
- Y. Ohue and H. Nishikawa, Cancer Sci., 2019, 110, 2080–2089 CrossRef CAS PubMed.
- T. Saito, H. Nishikawa, H. Wada, Y. Nagano, D. Sugiyama, K. Atarashi, Y. Maeda, M. Hamaguchi, N. Ohkura, E. Sato, H. Nagase, J. Nishimura, H. Yamamoto, S. Takiguchi, T. Tanoue, W. Suda, H. Morita, M. Hattori, K. Honda, M. Mori, Y. Doki and S. Sakaguchi, Nat. Med., 2016, 22, 679–684 CrossRef CAS PubMed.
- C. A. Janeway Jr, P. Travers and M. Walport, Immunobiology: The Immune System in Health and Disease, Garland Science, New York, 5th edn, 2001 Search PubMed.
- T. Sarkar, S. Dhar and G. Sa, Curr. Res. Immunol., 2021, 2, 132–141 CrossRef CAS PubMed.
- M. Kondo, S. Kumagai and H. Nishikawa, Int. Immunol., 2023, 36, 75–86 CrossRef PubMed.
- T. Hofer, O. Krichevsky and G. Altan-Bonnet, Front. Immunol., 2012, 3, 268 Search PubMed.
- B. Mirlekar, SAGE Open Med., 2022, 10, 20503121211069012 Search PubMed.
- C. Alsinet, M. N. Primo, V. Lorenzi, E. Bello, I. Kelava, C. P. Jones, R. Vilarrasa-Blasi, C. Sancho-Serra, A. Knights, J.-E. Park, B. S. Wyspianska, G. Trynka, D. F. Tough, A. Bassett, D. J. Gaffney, D. Álvarez-Errico and R. Vento-Tormo, Nat. Commun., 2022, 13, 2885, DOI:10.1038/s41467-022-30557-4.
- T. Matsuguchi, S. Okamura, C. Kawasaki, K. Shimoda, F. Omori, S. Hayashi, N. Kimura and Y. Niho, Eur. J. Haematol., 1991, 47, 128–133 CrossRef CAS PubMed.
- T. Condamine, J. Mastio and D. I. Gabrilovich, J. Leukoc Biol., 2015, 98, 913–922 CrossRef CAS PubMed.
- S. Ostrand-Rosenberg, D. W. Beury, K. H. Parker and L. A. Horn, Cancer Immunol. Immunother., 2020, 69, 215–221 CrossRef CAS PubMed.
- F. Veglia, E. Sanseviero and D. I. Gabrilovich, Nat. Rev. Immunol., 2021, 21, 485–498 CrossRef CAS PubMed.
- D. Raman, P. J. Baugher, Y. M. Thu and A. Richmond, Cancer Lett., 2007, 256, 137–165 CrossRef CAS PubMed.
- P. Li, M. Lu, J. Shi, Z. Gong, L. Hua, Q. Li, B. Lim, X. H. Zhang, X. Chen, S. Li, L. D. Shultz and G. Ren, Nat. Immunol., 2020, 21, 1444–1455 CrossRef PubMed.
- L. Yang, Q. Liu, X. Zhang, X. Liu, B. Zhou, J. Chen, D. Huang, J. Li, H. Li, F. Chen, J. Liu, Y. Xing, X. Chen, S. Su and E. Song, Nature, 2020, 583, 133–138 CrossRef CAS PubMed.
- W. Zheng, Y. Zhu, X. Chen and J. Q. Zhao, Annals Transl. Med., 2021, 9(14), 1148, DOI:10.21037/atm-21-2589.
- H. Kuroda, S. Mabuchi, E. Yokoi, N. Komura, K. Kozasa, M. Kawano, R. Takahashi, T. Sasano, K. Shimura, M. Kodama, K. Hashimoto, K. Sawada, E. Morii and T. Kimura, Oncotarget, 2018, 9(91), 36317–36330, DOI:10.18632/oncotarget.26347.
- M. F. Chen, P. T. Chen, F. C. Kuan and W. C. Chen, Annals Surgical Oncology, 2018, 26(1), 190–199, DOI:10.1245/s10434-018-6944-1.
- U. K. Scarlett, M. R. Rutkowski, A. M. Rauwerdink, J. Fields, X. Escovar-Fadul, J. Baird, J. R. Cubillos-Ruiz, A. C. Jacobs, J. L. Gonzalez, J. Weaver, S. Fiering and J. R. Conejo-Garcia, J. Exp. Med., 2012, 209, 495–506 CrossRef CAS PubMed.
- P. L. Kuo, M. S. Huang, D.-E. Cheng, J. Y. Hung and S. H. Chou, J. Biol. Chem., 2012, 287(13), 9753–9764, DOI:10.1074/jbc.m111.321190.
- D. L. Herber, W. Cao, Y. Nefedova, S. V. Novitskiy, S. Nagaraj, V. A. Tyurin, A. Corzo, H. I. Cho, E. Celis, B. Lennox, S. C. Knight, T. Padhya, T. V. McCaffrey, J. C. McCaffrey, S. Antonia, M. Fishman, R. L. Ferris, V. E. Kagan and D. I. Gabrilovich, Nat. Med., 2010, 16, 880–886 CrossRef CAS PubMed.
- Y. Hong, I. Manoharan, A. Suryawanshi, T. Majumdar, M. L. Angus-Hill, P. A. Koni, B. Manicassamy, A. L. Mellor, D. H. Munn and S. Manicassamy, Cancer Res., 2015, 75, 656–665 CrossRef CAS PubMed.
- M. Martínez-Blanco, D. Lozano-Ojalvo, L. Pérez-Rodríguez, S. Benedé, E. Molina and R. López-Fandiño, Front. Immunol., 2021, 12, 675733, DOI:10.3389/fimmu.2021.675733.
- D. H. Munn and A. L. Mellor, Trends Immunol., 2016, 37, 193–207 CrossRef CAS PubMed.
- H. Yun, Z. Chen, Y. Yang, Z. Jiang, Y. Gu, Y. Liu, C.-K. Lin, Z. Y. Pan, Y. Yu, M. Jiang, W. Zhou and X. Cao, Hepatology, 2013, 59(2), 567–579, DOI:10.1002/hep.26694.
- Q. Liu, C. Zhang, A. Sun, Y. Zheng, L. Wang and X. Cao, J. Immunol., 2009, 182, 6207–6216 CrossRef CAS PubMed.
- W. Niedbala, B. Cai and F. Y. Liew, Ann. Rheum. Dis., 2006, 65(Suppl 3), iii37–40 Search PubMed.
- K. Zhong, W. Song, Q. Wang, C. Wang, X. Liu, D. Chen, Z. Zhu, Y. Wu, W. Zhang and M. Zhang, PLoS One, 2012, 7, e49378 CrossRef CAS PubMed.
- P. Kaliński, P. L. Vieira, J. H. Schuitemaker, E. C. de Jong and M. L. Kapsenberg, Blood, 2001, 97, 3466–3469 CrossRef PubMed.
- M. S. von Bergwelt-Baildon, A. Popov, T. Saric, J. Chemnitz, S. Classen, M. S. Stoffel, F. Fiore, U. Roth, M. Beyer, S. Debey, C. Wickenhauser, F. G. Hanisch and J. L. Schultze, Blood, 2006, 108, 228–237 CrossRef CAS PubMed.
- X. Yu, J. Qian, L. Ding, S. Yin, L. Zhou and S. Zheng, Int. J. Mol. Sci., 2023, 24(7), 6501, DOI:10.3390/ijms24076501.
- J. M. Ilarregui, D. O. Croci, G. A. Bianco, M. A. Toscano, M. Salatino, M. E. Vermeulen, J. R. Geffner and G. A. Rabinovich, Nat. Immunol., 2009, 10, 981–991 CrossRef CAS PubMed.
- T. Dalotto-Moreno, D. O. Croci, J. P. Cerliani, V. C. Martinez-Allo, S. Dergan-Dylon, S. P. Méndez-Huergo, J. C. Stupirski, D. Mazal, E. Osinaga, M. A. Toscano, V. Sundblad, G. A. Rabinovich and M. Salatino, Cancer Res., 2013, 73, 1107–1117 CrossRef CAS PubMed.
- J. Krempski, L. Karyampudi, M. D. Behrens, C. L. Erskine, L. Hartmann, H. Dong, E. L. Goode, K. R. Kalli and K. L. Knutson, J. Immunol., 2011, 186, 6905–6913 CrossRef CAS PubMed.
- P. W. Kantoff, C. S. Higano, N. D. Shore, E. R. Berger, E. J. Small, D. F. Penson, C. H. Redfern, A. C. Ferrari, R. Dreicer, R. B. Sims, Y. Xu, M. W. Frohlich and P. F. Schellhammer, N. Engl. J. Med., 2010, 363, 411–422 CrossRef CAS PubMed.
- B. De Keersmaecker, S. Claerhout, J. Carrasco, I. Bar, J. Corthals, S. Wilgenhof, B. Neyns and K. Thielemans, J. Immunother. Cancer, 2020, 8(1), e000329, DOI:10.1136/jitc-2019-000329.
- M. Ogasawara, M. Miyashita, Y. Yamagishi and S. Ota, Ther. Apher. Dial., 2019, 23, 279–288 CrossRef CAS PubMed.
- S. Cozzi, M. Najafi, M. Gomar, P. Ciammella, C. Iotti, C. Iaccarino, M. Dominici, G. Pavesi, C. Chiavelli, A. Kazemian and A. Jahanbakhshi, Curr. Oncol., 2022, 29, 881–891 CrossRef PubMed.
- J. Cao, F. H. Kong, X. Liu and X. B. Wang, World J. Gastroenterol., 2019, 25, 3649–3663 CrossRef CAS PubMed.
- E. Uribe-Querol and C. Rosales, Front. Immunol., 2020, 11, 1066, DOI:10.3389/fimmu.2020.01066.
- C. Yunna, H. Mengru, W. Lei and C. Weidong, Eur. J. Pharmacol., 2020, 877, 173090 CrossRef PubMed.
- C. Zhu, J. M. Kros, C. Cheng and D. Mustafa, Neuro-Oncology, 2017, 19, 1435–1446 CrossRef CAS PubMed.
- N. Wang, H. Liang and K. Zen, Front. Immunol., 2014, 5, 614, DOI:10.3389/fimmu.2014.00614.
- F. A. W. Verreck, T. d Boer, D. M. L. Langenberg, M. A. Hoeve, M. Kramer, E. Vaisberg, R. A. Kastelein, A. Kolk, R. D. W. Malefyt and T. H. M. Ottenhoff, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(13), 4560–4565, DOI:10.1073/pnas.0400983101.
- C. Ngambenjawong, H. H. Gustafson and S. H. Pun, Adv. Drug Delivery Rev., 2017, 114, 206–221 CrossRef CAS PubMed.
- K. Kawamura, Y. Komohara, K. Takaishi, H. Katabuchi and M. Takeya, Pathol. Int., 2009, 59(5), 300–305, DOI:10.1111/j.1440-1827.2009.02369.x.
- H. Zhao, Q. Chen, A. Alam, J. Cui, K. C. Suen, A. P. Soo, S. Eguchi, J. Gu and D. Ma, Cell Death Dis., 2018, 9, 356 CrossRef PubMed.
- Y. Tan, L. Zhao, Y. G. Yang and W. Liu, Front. Oncol., 2022, 12, 953283 CrossRef CAS PubMed.
- S. Gampala and J. Y. Yang, Cells, 2021, 10(11), 3135, DOI:10.3390/cells10113135.
- M. Yin, X. Li, S. Tan, H. J. Zhou, W. Ji, S. Bellone, X. Xu, H. Zhang, A. D. Santin, G. Lou and W. Min, J. Clin. Invest., 2016, 126, 4157–4173 CrossRef PubMed.
- R. T. Annamalai, P. A. Turner, W. F. Carson, B. Levi, S. Kunkel and J. P. Stegemann, Biomaterials, 2018, 161, 216–227 CrossRef CAS PubMed.
- M. N. Hasan, O. Capuk, S. M. Patel and D. Sun, Cancers, 2022, 14(14), 3331, DOI:10.3390/cancers14143331.
- S. H. Aliyah, Y. N. Ardiyan, I. Mardhiyah, C. Herdini, E. K. Dwianingsih, S. Aning, N. S. N. Handayani, W. Asmara, J. Fachiroh and D. K. Paramita, Asian Pac. J. Cancer Prev., 2021, 22, 3447–3453 CrossRef CAS PubMed.
- Q. Lou, M. Zhao, Q. Xu, S. Xie, Y. Liang, J. Chen, L. Yuan, L. Wang, L. Jiang, L. Mou, D. Lin and M. Zhao, Front. Cell Dev. Biol., 2021, 9, 658757, DOI:10.3389/fcell.2021.658757.
- T. Fujimura and S. Aiba, Biomolecules, 2020, 10(8), 1087, DOI:10.3390/biom10081087.
- K. H. Susek, M. Karvouni, E. Alici and A. Lundqvist, Front. Immunol., 2018, 9, 2159 CrossRef PubMed.
- C. Bernsmeier, S. van der Merwe and A. Périanin, J. Hepatol., 2020, 73, 186–201 CrossRef CAS PubMed.
- S. Fan, H. G. Fehr and D. Adams, Cell. Immunol., 1991, 135, 78–87 CrossRef CAS PubMed.
- C. Zhao, Q. Han, H.-Y. Ying, X.-X. Gu, N. Yang, L. Li and Q.-Z. Zhang, Oncoimmunology, 2022, 11(1), 2032918, DOI:10.1080/2162402x.2022.2032918.
- T. Cavalleri, L. Greco, F. Rubbino, T. Hamada, M. Quaranta, F. Grizzi, E. Sauta, V. Craviotto, P. Bossi, S. Vetrano, L. Rimassa, V. Torri, R. Bellazzi, A. Mantovani, S. Ogino, A. Malesci and L. Laghi, J. Pathology Clin. Res., 2022, 8(4), 307–312, DOI:10.1002/cjp2.267.
- Y. M. Yeh, S. J. Hsu, P. Lin, K. F. Hsu, P. Y. Wu, W. C. Su, J. Y. Chang and M. R. Shen, Clin. Cancer Res., 2017, 23(20), 6021–6030, DOI:10.1158/1078-0432.ccr-17-1007.
- M. H. Fadhil, O. F. Abdul-Rasheed and M. A. Al-Naqqash, Acta Biochim. Pol., 2019, 66(4), 437–443, DOI:10.18388/abp.2019_2855.
- L. E. Hudson and R. L. Allen, Front. Immunol., 2016, 7, 281, DOI:10.3389/fimmu.2016.00281.
- A. A. Barkal, K. Weiskopf, K. S. Kao, S. R. Gordon, B. Rosental, Y. Y. Yiu, B. M. George, M. Markovic, N. G. Ring, J. M. Tsai, K. M. McKenna, P. Y. Ho, R. Z. Cheng, J. Y. Chen, L. J. Barkal, A. M. Ring, I. L. Weissman and R. L. Maute, Nat. Immunol., 2018, 19, 76–84 CrossRef CAS PubMed.
- W. Wang, J. M. Marinis, A. M. Beal, S. Savadkar, Y. Wu, M. Khan, P. S. Taunk, N. Wu, W. Su, J. Wu, A. Ahsan, E. Kurz, T. Chen, I. Yaboh, F. Li, J. Gutierrez, B. Diskin, M. Hundeyin, M. Reilly, J. D. Lich, P. A. Harris, M. K. Mahajan, J. H. Thorpe, P. Nassau, J. E. Mosley, J. Leinwand, J. A. Kochen Rossi, A. Mishra, B. Aykut, M. Glacken, A. Ochi, N. Verma, J. I. Kim, V. Vasudevaraja, D. Adeegbe, C. Almonte, E. Bagdatlioglu, D. J. Cohen, K.-K. Wong, J. Bertin and G. Miller, Cancer Cell, 2018, 34, 757–774.e757 CrossRef CAS PubMed.
- J. M. Jaynes, R. Sable, M. Ronzetti, W. Bautista, Z. Knotts, A. Abisoye-Ogunniyan, D. Li, R. Calvo, M. Dashnyam, A. Singh, T. Guerin, J. White, S. Ravichandran, P. Kumar, K. Talsania, V. Chen, A. Ghebremedhin, B. Karanam, A. Bin Salam, R. Amin, T. Odzorig, T. Aiken, V. Nguyen, Y. Bian, J. C. Zarif, A. E. de Groot, M. Mehta, L. Fan, X. Hu, A. Simeonov, N. Pate, M. Abu-Asab, M. Ferrer, N. Southall, C.-Y. Ock, Y. Zhao, H. Lopez, S. Kozlov, N. de Val, C. C. Yates, B. Baljinnyam, J. Marugan and U. Rudloff, Sci. Transl. Med., 2020, 12, eaax6337 CrossRef CAS PubMed.
- M. Lauss, M. Donia, I. M. Svane and G. Jonsson, Clin. Cancer Res., 2022, 28, 1751–1758 CrossRef CAS PubMed.
- N. M. de Gruijter, B. Jebson and E. C. Rosser, Clin. Exp. Immunol., 2022, 210, 253–262 CrossRef PubMed.
- L. Jerby-Arnon, P. Shah, M. S. Cuoco, C. Rodman, M. J. Su, J. C. Melms, R. Leeson, A. Kanodia, S. Mei, J. R. Lin, S. Wang, B. Rabasha, D. Liu, G. Zhang, C. Margolais, O. Ashenberg, P. A. Ott, E. I. Buchbinder, R. Haq, F. S. Hodi, G. M. Boland, R. J. Sullivan, D. T. Frederick, B. Miao, T. Moll, K. T. Flaherty, M. Herlyn, R. W. Jenkins, R. Thummalapalli, M. S. Kowalczyk, I. Canadas, B. Schilling, A. N. R. Cartwright, A. M. Luoma, S. Malu, P. Hwu, C. Bernatchez, M. A. Forget, D. A. Barbie, A. K. Shalek, I. Tirosh, P. K. Sorger, K. Wucherpfennig, E. M. Van Allen, D. Schadendorf, B. E. Johnson, A. Rotem, O. Rozenblatt-Rosen, L. A. Garraway, C. H. Yoon, B. Izar and A. Regev, Cell, 2018, 175, 984–997e924 CrossRef CAS PubMed.
- D. Michaud, C. R. Steward, B. Mirlekar and Y. Pylayeva-Gupta, Immunol. Rev., 2021, 299, 74–92 CrossRef CAS PubMed.
- C. M. Laumont, A. C. Banville, M. Gilardi, D. P. Hollern and B. H. Nelson, Nat. Rev. Cancer, 2022, 22, 414–430 CrossRef CAS PubMed.
- X. Zhou, Y. X. Su, X. M. Lao, Y. J. Liang and G. Q. Liao, Oral Oncol., 2016, 53, 27–35 CrossRef CAS PubMed.
- W. W. Wang, X. L. Yuan, H. Chen, G. H. Xie, Y. H. Ma, Y. X. Zheng, Y. L. Zhou and L. S. Shen, Oncotarget, 2015, 6, 33486–33499 CrossRef PubMed.
- Y. Yamamoto, H. Kasashima, Y. Fukui, G. Tsujio, M. Yashiro and K. Maeda, Cancer Sci., 2023, 114, 16–24 CrossRef CAS PubMed.
- G. Biffi and D. A. Tuveson, Physiol. Rev., 2021, 101, 147–176 CrossRef CAS PubMed.
- M. Bartoschek, N. Oskolkov, M. Bocci, J. Lovrot, C. Larsson, M. Sommarin, C. D. Madsen, D. Lindgren, G. Pekar, G. Karlsson, M. Ringner, J. Bergh, A. Bjorklund and K. Pietras, Nat. Commun., 2018, 9, 5150 CrossRef PubMed.
- V. Bernard, A. Semaan, J. Huang, F. A. San Lucas, F. C. Mulu, B. M. Stephens, P. A. Guerrero, Y. Huang, J. Zhao, N. Kamyabi, S. Sen, P. A. Scheet, C. M. Taniguchi, M. P. Kim, C. W. Tzeng, M. H. Katz, A. D. Singhi, A. Maitra and H. A. Alvarez, Clin. Cancer Res., 2019, 25, 2194–2205 CrossRef CAS PubMed.
- H. Li, E. T. Courtois, D. Sengupta, Y. Tan, K. H. Chen, J. J. L. Goh, S. L. Kong, C. Chua, L. K. Hon, W. S. Tan, M. Wong, P. J. Choi, L. J. K. Wee, A. M. Hillmer, I. B. Tan, P. Robson and S. Prabhakar, Nat. Genet., 2017, 49, 708–718 CrossRef CAS PubMed.
- D. Ohlund, A. Handly-Santana, G. Biffi, E. Elyada, A. S. Almeida, M. Ponz-Sarvise, V. Corbo, T. E. Oni, S. A. Hearn, E. J. Lee, I. I. C Chio, C. I. Hwang, H. Tiriac, L. A. Baker, D. D. Engle, C. Feig, A. Kultti, M. Egeblad, D. T. Fearon, J. M. Crawford, H. Clevers, Y. Park and D. A. Tuveson, J. Exp. Med., 2017, 214(3), 579–596, DOI:10.1084/jem.20162024.
- G. Comito, E. Giannoni, C. P. Segura, P. Barcellos-de-Souza, M. R. Raspollini, G. Baroni, M. Lanciotti, S. Serni and P. Chiarugi, Oncogene, 2014, 33, 2423–2431 CrossRef CAS PubMed.
- B. C. Ozdemir, T. Pentcheva-Hoang, J. L. Carstens, X. Zheng, C. C. Wu, T. R. Simpson, H. Laklai, H. Sugimoto, C. Kahlert, S. V. Novitskiy, A. De Jesus-Acosta, P. Sharma, P. Heidari, U. Mahmood, L. Chin, H. L. Moses, V. M. Weaver, A. Maitra, J. P. Allison, V. S. LeBleu and R. Kalluri, Cancer Cell, 2014, 25, 719–734 CrossRef CAS PubMed.
- E. J. Kim, V. Sahai, E. V. Abel, K. A. Griffith, J. K. Greenson, N. Takebe, G. N. Khan, J. L. Blau, R. Craig, U. G. Balis, M. M. Zalupski and D. M. Simeone, Clin. Cancer Res., 2014, 20, 5937–5945 CrossRef CAS PubMed.
- D. Ganguly, R. Chandra, J. Karalis, M. Teke, T. Aguilera, R. Maddipati, M. B. Wachsmann, D. Ghersi, G. Siravegna, H. J. Zeh, 3rd, R. Brekken, D. T. Ting and M. Ligorio, Cancers, 2020, 12(9), 2652, DOI:10.3390/cancers12092652.
- B. G. Neel, H. Gu and L. Pao, Trends Biochem. Sci., 2003, 28, 284–293 CrossRef CAS PubMed.
- M. Ahmadzadeh, L. A. Johnson, B. Heemskerk, J. R. Wunderlich, M. E. Dudley, D. E. White and S. A. Rosenberg, Blood, 2009, 114, 1537–1544 CrossRef CAS PubMed.
- M. E. Keir, M. J. Butte, G. J. Freeman and A. H. Sharpe, Annu. Rev. Immunol., 2008, 26, 677–704 CrossRef CAS PubMed.
- T. Yamazaki, H. Akiba, H. Iwai, H. Matsuda, M. Aoki, Y. Tanno, T. Shin, H. Tsuchiya, D. M. Pardoll, K. Okumura, M. Azuma and H. Yagita, J. Immunol., 2002, 169, 5538–5545 CrossRef CAS PubMed.
- J. Cai, D. Wang, G. Zhang and X. Guo, OncoTargets Therapy, 2019, 12, 8437–8445 CrossRef CAS PubMed.
- P. Yadollahi, Y.-K. Jeon, W. L. Ng and I. Choi, BMB Rep., 2020, 54(1), 12–20, DOI:10.5483/BMBRep.2021.54.1.241.
- W. Qin, L. Hu, X. Zhang, S. Jiang, J. Li, Z. Zhang and X. Wang, Front. Immunol., 2019, 10, 2298, DOI:10.3389/fimmu.2019.02298.
- M. Yi, M. Niu, L. Xu, S. Luo and K. Wu, J. Hematol. Oncol., 2021, 14, 10 CrossRef CAS PubMed.
- N. Patsoukis, Q. Wang, L. Strauss and V. A. Boussiotis, Sci. Adv., 2020, 6, eabd2712 CrossRef CAS PubMed.
- Q. Wang, K. Bardhan, V. A. Boussiotis and N. Patsoukis, Adv. Biol., 2021, 5, 2100758 CrossRef CAS PubMed.
- Y. Zhao, D. L. Harrison, Y. Song, J. Ji, J. Huang and E. Hui, Cell Rep., 2018, 24, 379–390.e376 CrossRef CAS PubMed.
- Y. Han, D. Liu and L. Li, Am. J. Cancer Res., 2020, 10, 727–742 CAS.
- Y. Li, M. He, Y. Zhou, C. Yang, S. Wei, X. Bian, O. Christopher and L. Xie, Front. Pharmacol., 2019, 10, 139 CrossRef CAS PubMed.
- Y. Qu, Q. Li, T. Ren, W. Xia, Y. Peng, G. L. Liu, H. Luo, Y. Yang, X. Dai, S. F. Zhou and D. Wang, Drug Des. Dev. Therapy, 2015, 9, 901–909, DOI:10.2147/dddt.s75152.
- S. Jiang, X. Li, L. Huang, X. Zhang and J. Lin, Front. Immunol., 2022, 13, 988416, DOI:10.3389/fimmu.2022.988416.
- M. B. Cirqueira, C. R. Mendonça, L. R. Soares, M. A. D. P. C. Cysneiros, R. R. Paulinelli, M. A. R. Moreira and R. Freitas-Júnior, Cancers, 2021, 13(23), 6090, DOI:10.3390/cancers13236090.
- R. Sabatier, P. Finetti, É. Mamessier, J. Adélaïde, M. Chaffanet, H. R. Ali, P. Viens, C. Caldas, D. Birnbaum and F. Bertucci, Oncotarget, 2014, 6(7), 5449–5464, DOI:10.18632/oncotarget.3216.
- X. Wang, F. Teng, L. Kong and J. Yu, Oncotargets Therapy, 2016, 9, 5023–5039, DOI:10.2147/ott.s105862.
- Q. Tang, S. Zhao, N. Zhou, J. He, L. Zu, T. Liu, Z. Song, L. Peng and S. Xu, Int. J. Oncol., 2023, 62(4), 49, DOI:10.3892/ijo.2023.5497.
- B. Rowshanravan, N. Halliday and D. M. Sansom, Blood, 2018, 131, 58–67 CrossRef CAS PubMed.
- C. A. Chambers, M. Krummel, B. Boitel, A. Hurwitz, T. Sullivan, S. Fournier, D. Cassell, M. Brunner and J. P. Allison, Immunol. Rev., 1996, 153, 27–46, DOI:10.1111/j.1600-065x.1996.tb00919.x.
- H. M. Gibson, C. J. Hedgcock, B. M. Aufiero, A. J. Wilson, M. S. Hafner, G. C. Tsokos and H. K. Wong, J. Immunol., 2007, 179, 3831–3840 CrossRef CAS PubMed.
- U. A. Ramagopal, W. Liu, S. C. Garrett-Thomson, J. B. Bonanno, Q. Yan, M. S. Srinivasan, S. Wong, A. Bell, S. Mankikar, V. S. Rangan, S. Deshpande, A. J. Korman and S. C. Almo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(21), E4223–E4232, DOI:10.1073/pnas.1617941114.
- A. DeMaio, S. Mehrotra, K. Sambamurti and S. Husain, J. Neuroinflammation, 2022, 19, 251 CrossRef PubMed.
- B. Salik, M. J. Smyth and K. Nakamura, J. Hematol. Oncol., 2020, 13, 111 CrossRef CAS PubMed.
- L. Lisi, P. M. Lacal, M. Martire, P. Navarra and G. Graziani, Pharmacol. Res., 2022, 175, 105997 CrossRef CAS PubMed.
- Y. Zhao, W. Yang, Y. Huang, R. Cui, X. Li and B. Li, Cell. Physiol. Biochem., 2018, 47, 721–734 CrossRef CAS PubMed.
- H. Schneider, E. Valk, R. Leung and C. E. Rudd, PLoS One, 2008, 3(12), e3842, DOI:10.1371/journal.pone.0003842.
- S. Kubsch, E. Graulich, J. Knop and K. Steinbrink, Eur. J. Immunol., 2003, 33, 1988–1997 CrossRef CAS PubMed.
- C. Olsson, K. Riesbeck, M. Dohlsten and E. Michaëlsson, J. Biol. Chem., 1999, 274, 14400–14405 CrossRef CAS PubMed.
- L. Ciszak, I. Frydecka, D. Wołowiec, A. Szteblich and A. Kosmaczewska, Clin. Exp. Med., 2015, 16, 317–332, DOI:10.1007/s10238-015-0360-7.
- S. Chikuma and J. A. Bluestone, Eur. J. Immunol., 2007, 37(5), 1285–1289, DOI:10.1002/eji.200737159.
- P. Hu, Q. Liu, G. Deng, J. Zhang, N. Liang, J. Xie and J. Zhang, Sci. Rep., 2017, 7, 42913, DOI:10.1038/srep42913.
- E. J. Lipson and C. G. Drake, Clin. Cancer Res., 2011, 17(22), 6958–6962, DOI:10.1158/1078-0432.ccr-11-1595.
- J. C. Yang, M. Hughes, U. Kammula, R. Royal, R. M. Sherry, S. L. Topalian, K. B. Suri, C. Levy, T. Allen, S. Mavroukakis, I. Lowy, D. E. White and S. A. Rosenberg, J. Immunother., 2007, 30, 825–830 CrossRef CAS PubMed.
- A. Winer, J. N. Bodor and H. Borghaei, J. Thorac. Dis., 2018, 10, S480–s489 CrossRef PubMed.
- L. Paz-Ares, T. E. Ciuleanu, M. Cobo, M. Schenker, B. Zurawski, J. Menezes, E. Richardet, J. Bennouna, E. Felip, O. Juan-Vidal, A. Alexandru, H. Sakai, A. Lingua, P. Salman, P. J. Souquet, P. De Marchi, C. Martin, M. Pérol, A. Scherpereel, S. Lu, T. John, D. P. Carbone, S. Meadows-Shropshire, S. Agrawal, A. Oukessou, J. Yan and M. Reck, Lancet Oncol., 2021, 22, 198–211 CrossRef CAS PubMed.
- M. Reck, T. E. Ciuleanu, M. Cobo, M. Schenker, B. Zurawski, J. Menezes, E. Richardet, J. Bennouna, E. Felip, O. Juan-Vidal, A. Alexandru, H. Sakai, A. Lingua, F. Reyes, P. J. Souquet, P. De Marchi, C. Martin, M. Pérol, A. Scherpereel, S. Lu, L. Paz-Ares, D. P. Carbone, A. Memaj, S. Marimuthu, X. Zhang, P. Tran and T. John, ESMO Open, 2021, 6, 100273 CrossRef CAS PubMed.
- L. Monney, C. A. Sabatos, J. L. Gaglia, A. Ryu, H. Waldner, T. Chernova, S. Manning, E. A. Greenfield, A. J. Coyle, R. A. Sobel, G. J. Freeman and V. K. Kuchroo, Nature, 2002, 415, 536–541 CrossRef CAS PubMed.
- J. H. Meyers, C. A. Sabatos, S. Chakravarti and V. K. Kuchroo, Trends Mol. Med., 2005, 11, 362–369 CrossRef CAS PubMed.
- L. Cai, Y. Li, J. Tan, L. Xu and Y. Li, J. Hematol. Oncol., 2023, 16, 101 CrossRef CAS PubMed.
- X. Lu, L. Yang, D. Yao, X. Wu, J. Li, X. Liu, L. Deng, C. Huang, Y. Wang, D. Li and J. Liu, Cell. Immunol., 2017, 313, 43–51 CrossRef CAS PubMed.
- G. Curigliano, H. Gelderblom, N. Mach, T. Doi, D. Tai, P. M. Forde, J. Sarantopoulos, P. L. Bedard, C. C. Lin, F. S. Hodi, S. Wilgenhof, A. Santoro, C. A. Sabatos-Peyton, T. A. Longmire, A. Xyrafas, H. Sun, S. Gutzwiller, L. Manenti and A. Naing, Clin. Cancer Res., 2021, 27, 3620–3629 CrossRef CAS PubMed.
- F. Triebel, S. Jitsukawa, E. Baixeras, S. Roman-Roman, C. Genevee, E. Viegas-Pequignot and T. Hercend, J. Exp. Med., 1990, 171, 1393–1405 CrossRef CAS PubMed.
- S. Hannier, M. Tournier, G. Bismuth and F. Triebel, J. Immunol., 1998, 161, 4058–4065 CrossRef CAS.
- H. A. Tawbi, D. Schadendorf, E. J. Lipson, P. A. Ascierto, L. Matamala, E. Castillo Gutierrez, P. Rutkowski, H. J. Gogas, C. D. Lao, J. J. De Menezes, S. Dalle, A. Arance, J. J. Grob, S. Srivastava, M. Abaskharoun, M. Hamilton, S. Keidel, K. L. Simonsen, A. M. Sobiesk, B. Li, F. S. Hodi, G. V. Long and R. Investigators, N. Engl. J. Med., 2022, 386, 24–34 CrossRef CAS PubMed.
- X. Yu, K. Harden, L. C. Gonzalez, M. Francesco, E. Chiang, B. Irving, I. Tom, S. Ivelja, C. J. Refino, H. Clark, D. Eaton and J. L. Grogan, Nat. Immunol., 2009, 10, 48–57 CrossRef CAS PubMed.
- D. Qiu, X. Liu, W. Wang, X. Jiang, X. Wu, J. Zheng, K. Zhou, X. Kong, X. Wu and Z. Jin, Clin. Exp. Med., 2023, 23, 165–174 CrossRef CAS PubMed.
- A. A. Badawy, Neuropharmacology, 2017, 112, 248–263 CrossRef CAS PubMed.
- M. Platten, M. Weller and W. Wick, CNS Oncol, 2012, 1, 99–106 CrossRef CAS PubMed.
- O. Warburg, F. Wind and E. Negelein, J. Gen. Physiol., 1927, 8, 519–530 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhai, J. Duan, X. Wang, J. Zhong, L. Wu, A. Li, M. Cao, Y. Wu, H. Shi, J. Zhong and Z. Guo, Front. Endocrinol., 2022, 13, 901495 CrossRef PubMed.
- A. Carracedo, L. C. Cantley and P. P. Pandolfi, Nat. Rev. Cancer, 2013, 13, 227–232 CrossRef CAS PubMed.
- F. Hossain, A. A. Al-Khami, D. Wyczechowska, C. Hernandez, L. Zheng, K. Reiss, L. D. Valle, J. Trillo-Tinoco, T. Maj, W. Zou, P. C. Rodriguez and A. C. Ochoa, Cancer Immunol. Res., 2015, 3, 1236–1247 CrossRef CAS PubMed.
- S. Zhang, K. Lv, Z. Liu, R. Zhao and F. Li, Cell Death Discovery, 2024, 10, 39 CrossRef CAS PubMed.
- A. J. de Jong, M. Kloppenburg, R. E. Toes and A. Ioan-Facsinay, Front. Immunol., 2014, 5, 483 Search PubMed.
- G. T. Wurz, C. Y. Kao, M. Wolf and M. W. DeGregorio, Hum. Vaccines Immunother., 2014, 10(11), 3383–3393, DOI:10.4161/hv.29836.
- U. Şahin, P. Oehm, E. Derhovanessian, R. A. Jabulowsky, M. Vormehr, M. Gold, D. Maurus, D. Schwarck-Kokarakis, A. N. Kuhn, T. Omokoko, L. M. Kranz, M. Diken, S. Kreiter, H. Haas, S. Attig, R. Rae, K. Ćuk, A. Kemmer-Brück, A. Breitkreuz, C. Tolliver, J. Caspar, J. Quinkhardt, L. Hebich, M. Stein, A. Hohberger, I. Vogler, I. Liebig, S. Renken, J. Sikorski, M. Leierer, V. Müller, H. Mitzel-Rink, M. Miederer, C. Huber, S. Grabbe, J. Utikal, A. Pinter, R. Kaufmann, J. C. Hassel, C. Loquai and Ö. Türeci, Nature, 2020, 585(7823), 107–112, DOI:10.1038/s41586-020-2537-9.
- T. Gargett, M. N. Abbas, P. Rolan, J. F. Price, K. M. Gosling, A. Fèrrante, A. Ruszkiewicz, I. I. Atmosukarto, J. G. Altin, C. R. Parish and M. P. Brown, Cancer Immunol., Immunother., 2018, 21, 593–602, DOI:10.1007/s00262-018-2207-z.
- P. Zhao, Y. Xu, W. Ji, L. Li, L. Qiu, S. Zhou, Z. Qian and H. Zhang, Int. J. Nanomed., 2022, 17, 73–89 CrossRef CAS PubMed.
- D. T. Johnson, J. Zhou, A. V. Kroll, R. H. Fang, M. Yan, C. Xiao, X. Chen, J. Kline, L. Zhang and D. E. Zhang, Leukemia, 2022, 36, 994–1005 CrossRef CAS PubMed.
- D. Huang, T. Wu, S. Lan, C. Liu, Z. Guo and W. Zhang, Biomaterials, 2022, 289, 121808 CrossRef CAS PubMed.
- Y. Tang, H. K. Bisoyi, X. M. Chen, Z. Liu, X. Chen, S. Zhang and Q. Li, Adv. Mater., 2023, 35, e2300232 CrossRef PubMed.
- Y. Li, H. Zhang, R. Wang, Y. Wang, R. Li, M. Zhu, X. Zhang, Z. Zhao, Y. Wan, J. Zhuang, H. Zhang and X. Huang, Adv. Mater., 2023, 35, e2208923 CrossRef PubMed.
- Y. Chen, S. Zhi, J. Ou, J. Gao, L. Zheng, M. Huang, S. Du, L. Shi, Y. Tu and K. Cheng, ACS Nano, 2023, 17, 16620–16632 CrossRef CAS PubMed.
- X. Wen, X. Xiong, G. Yang, W. Xiao, J. Hou, T. Pan, Y. Hu and S. Zhou, J. Controlled Release, 2023, 353, 535–548 CrossRef CAS PubMed.
- J. Li, Y. Wu, J. Wang, X. Xu, A. Zhang, Y. Li and Z. Zhang, ACS Nano, 2023, 17, 322–336 CrossRef CAS PubMed.
- L. Hou, X. Gong, J. Yang, H. Zhang, W. Yang and X. Chen, Adv. Mater., 2022, 34, e2200389 CrossRef PubMed.
- W. Ma, D. Zhu, J. Li, X. Chen, W. Xie, X. Jiang, L. Wu, G. Wang, Y. Xiao, Z. Liu, F. Wang, A. Li, D. Shao, W. Dong, W. Liu and Y. Yuan, Theranostics, 2020, 10, 1281–1295 CrossRef CAS PubMed.
- M. Lin, Y. Li, H. Long, Y. Lin, Z. Zhang, F. Zhan, M. Li, C. Wu and Z. Liu, Int. J. Biol. Macromol., 2023, 225, 873–885 CrossRef CAS PubMed.
- J. Xiong, M. Wu, J. Chen, Y. Liu, Y. Chen, G. Fan, Y. Liu, J. Cheng, Z. Wang, S. Wang, Y. Liu and W. Zhang, ACS Nano, 2021, 15, 19756–19770 CrossRef CAS PubMed.
- Q. Jiang, K. Wang, X. Zhang, B. Ouyang, H. Liu, Z. Pang and W. Yang, Small, 2020, 16, e2001704 CrossRef PubMed.
- X. Liang, X. Ye, C. Wang, C. Xing, Q. Miao, Z. Xie, X. Chen, X. Zhang, H. Zhang and L. Mei, J. Controlled Release, 2019, 296, 150–161 CrossRef CAS PubMed.
- L. E. Munoz, L. Monterroza, R. Bommireddy, Y. Shafizadeh, C. D. Pack, S. Ramachandiran, S. J. C. Reddy and P. Selvaraj, Int. J. Mol. Sci., 2021, 22(16), 8377, DOI:10.3390/ijms22168377.
- B. Liu, Y. Yang, Y. Chao, Z. Xiao, J. Xu, C. Wang, Z. Dong, L. Hou, Q. Li and Z. Liu, Nano Lett., 2021, 21, 9410–9418 CrossRef CAS PubMed.
- C. D. Pack, R. Bommireddy, L. E. Munoz, J. M. Patel, E. N. Bozeman, P. Dey, V. Radhakrishnan, V. F. Vartabedian, K. Venkat, S. Ramachandiran, S. J. C. Reddy and P. Selvaraj, Hum. Vaccines Immunother., 2020, 16, 3184–3193 CrossRef CAS PubMed.
- Y. N. Tan, J. D. Huang, Y. P. Li, S. S. Li, M. Luo, J. Luo, A. W. Lee, L. Fu, F. Q. Hu and X. Y. Guan, Adv. Healthcare Mater., 2022, 11, e2101496 CrossRef PubMed.
- W. Xie, W. W. Deng, M. Zan, L. Rao, G. T. Yu, D. M. Zhu, W. T. Wu, B. Chen, L. W. Ji, L. Chen, K. Liu, S. S. Guo, H. M. Huang, W. F. Zhang, X. Zhao, Y. Yuan, W. Dong, Z. J. Sun and W. Liu, ACS Nano, 2019, 13, 2849–2857 CrossRef CAS PubMed.
- L. Shi and H. Gu, Nanomedicine, 2023, 48, 102648 CrossRef CAS PubMed.
- J. Wang, B. Sun, L. Sun, X. Niu, L. Li and Z. P. Xu, Biomater. Sci., 2023, 11, 2020–2032 RSC.
- J. Gan, G. Du, C. He, M. Jiang, X. Mou, J. Xue and X. Sun, J. Controlled Release, 2020, 326, 297–309 CrossRef CAS PubMed.
- D. Wu, X. Shou, Y. Zhang, Z. Li, G. Wu, D. Wu, J. Wu, S. Shi and S. Wang, Nanomedicine, 2021, 32, 102333 CrossRef CAS PubMed.
- C. Hu, R. Man, H. Li, M. Xia, Z. Yu and B. Tang, Anal. Chem., 2023, 95, 13575–13585 CrossRef CAS PubMed.
- J. Luo, X. Wang, Z. Shi, Y. Zeng, L. He, J. Cao, Y. Sun, T. Zhang and P. Huang, J. Nanobiotechnol., 2022, 20, 228 CrossRef CAS PubMed.
- D. Wang, J. Liu, J. Duan, Y. Ma, H. Gao, Z. Zhang, J. Liu, J. Shi and K. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 44183–44198 CrossRef CAS PubMed.
- S. Li, S. Jiang, M. S. U. Rahman, J. Mei, X. Wang, J. Jiang, Y. Chen, S. Xu and Y. Liu, Small Methods, 2023, 7, e2201569 CrossRef PubMed.
- T. Du, Y. Wang, Z. Luan, C. Zhao and K. Yang, Int. J. Pharm., 2022, 624, 121911 CrossRef CAS PubMed.
- Y. Zhai, J. Wang, T. Lang, Y. Kong, R. Rong, Y. Cai, W. Ran, F. Xiong, C. Zheng, Y. Wang, Y. Yu, H. H. Zhu, P. Zhang and Y. Li, Nat. Nanotechnol., 2021, 16, 1271–1280 CrossRef CAS PubMed.
- X. Li, W. Zhang, J. Lin, H. Wu, Y. Yao, J. Zhang and C. Yang, Biomater. Sci., 2021, 9, 3453–3464 RSC.
- Q. Kou, Y. Huang, Y. Su, L. Lu, X. Li, H. Jiang, R. Huang, J. Li and X. Nie, Nanoscale, 2023, 15, 9457–9476 RSC.
- Y. Wang, W. Li, Z. Li, F. Mo, Y. Chen, M. Iida, D. L. Wheeler and Q. Hu, Sci. Adv., 2023, 9, eadf6854 CrossRef CAS PubMed.
- H. Wang, C. Wu, X. Tong and S. Chen, J. Controlled Release, 2023, 353, 727–737 CrossRef CAS PubMed.
- B. Bahmani, H. Gong, B. T. Luk, K. J. Haushalter, E. DeTeresa, M. Previti, J. Zhou, W. Gao, J. D. Bui, L. Zhang, R. H. Fang and J. Zhang, Nat. Commun., 2021, 12, 1999 CrossRef CAS PubMed.
- Y. Zhang, Z. Huang, J. Cheng, H. Pan, T. Lin, X. Shen, W. Chen, Q. Chen, C. Gu, Q. Mao and Y. Liang, Front. Endocrinol., 2022, 13, 865655 CrossRef PubMed.
- H. Yan, Y. Zhang, Y. Zhang, Y. Li, X. Kong, D. Liu, J. Li, Y. Xi, J. Ji, L. Ye and G. Zhai, Biomater. Sci., 2022, 10, 6583–6600 RSC.
- M. Chen, Y. Qiao, J. Cao, L. Ta, T. Ci and X. Ke, J. Nanobiotechnol., 2022, 20, 273 CrossRef CAS PubMed.
- Q. Chen, L. Zhang, L. Li, M. Tan, W. Liu, S. Liu, Z. Xie, W. Zhang, Z. Wang, Y. Cao, T. Shang and H. Ran, J. Nanobiotechnol., 2021, 19, 449 CrossRef CAS PubMed.
- Y. Wang, Z. Zhao, C. Liu, M. Hao, C. Kong, X. Zhao, Y. Gao, Y. Zhang, W. Cui, C. Zhang and J. Jiang, Int. J. Nanomed., 2022, 17, 855–868 CrossRef CAS PubMed.
- X. Meng, J. Wang, J. Zhou, Q. Tian, B. Qie, G. Zhou, W. Duan and Y. Zhu, Acta Biomater., 2021, 127, 266–275 CrossRef CAS PubMed.
- S. Li, S. Dong, J. Wu, X. Lv, N. Yang, Q. Wei, C. Wang and J. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 7878–7886 CrossRef CAS PubMed.
- C. Hu, T. Lei, Y. Wang, J. Cao, X. Yang, L. Qin, R. Liu, Y. Zhou, F. Tong, C. S. Umeshappa and H. Gao, Biomaterials, 2020, 255, 120159 CrossRef CAS PubMed.
- J. Zhang, L. Wei, X. Ma, J. Wang, S. Liang, K. Chen, M. Wu, L. Niu and Y. Zhang, Acta Biomater., 2023, 166, 470–484 CrossRef CAS PubMed.
- W. L. Liu, M. Z. Zou, T. Liu, J. Y. Zeng, X. Li, W. Y. Yu, C. X. Li, J. J. Ye, W. Song, J. Feng and X. Z. Zhang, Nat. Commun., 2019, 10, 3199 CrossRef PubMed.
- W. L. Liu, M. Z. Zou, T. Liu, J. Y. Zeng, X. Li, W. Y. Yu, C. X. Li, J. J. Ye, W. Song, J. Feng and X. Z. Zhang, Adv. Mater., 2019, 31, e1900499 CrossRef PubMed.
- P. Pan, X. Dong, Y. Chen, J. J. Ye, Y. X. Sun and X. Z. Zhang, Biomaterials, 2022, 289, 121810 CrossRef CAS PubMed.
- L. Rao, L. Wu, Z. Liu, R. Tian, G. Yu, Z. Zhou, K. Yang, H. G. Xiong, A. Zhang, G. T. Yu, W. Sun, H. Xu, J. Guo, A. Li, H. Chen, Z. J. Sun, Y. X. Fu and X. Chen, Nat. Commun., 2020, 11, 4909 CrossRef PubMed.
- Y. Wang, Z. Luan, C. Zhao, C. Bai and K. Yang, Eur. J. Pharm. Sci., 2020, 142, 105136 CrossRef CAS PubMed.
- C. Gong, X. Yu, W. Zhang, L. Han, R. Wang, Y. Wang, S. Gao and Y. Yuan, J. Nanobiotechnol., 2021, 19, 58 CrossRef CAS PubMed.
- W. Zhong, Z. Xiao, Z. Qin, J. Yang, Y. Wen, Z. Yu, Y. Li, N. C. Sheppard, S. Y. Fuchs, X. Xu, M. Herlyn, C. H. June, E. Puré and W. Guo, Cancer Res., 2023, 83, 2790–2806 CrossRef CAS PubMed.
- W. Zhou, X. Chen, Y. Zhou, S. Shi, C. Liang, X. Yu, H. Chen, Q. Guo, Y. Zhang, P. Liu, C. Li, Y. Chu, Y. Luo, Y. Wang, Z. Zhou, Z. Zhao, Q. Chen, T. Sun and C. Jiang, Biomaterials, 2022, 280, 121306 CrossRef CAS PubMed.
- Y. Guo, S. Z. Wang, X. Zhang, H. R. Jia, Y. X. Zhu, X. Zhang, G. Gao, Y. W. Jiang, C. Li, X. Chen, S. Y. Wu, Y. Liu and F. G. Wu, Nat. Commun., 2022, 13, 6534 CrossRef CAS PubMed.
- Y. Jang, H. Kim, S. Yoon, H. Lee, J. Hwang, J. Jung, J. H. Chang, J. Choi and H. Kim, J. Controlled Release, 2021, 330, 293–304 CrossRef CAS PubMed.
- L. Tang, Y. Yin, Y. Cao, C. Fu, H. Liu, J. Feng, W. Wang and X. J. Liang, Adv. Mater., 2023, 35, e2303835 CrossRef PubMed.
- J. Lin, N. Huang, M. Li, M. Zheng, Z. Wang, X. Zhang, H. Gao, Y. Lao, J. Zhang and B. Ding, Drug Des., Dev. Ther., 2023, 17, 2087–2106 CrossRef CAS PubMed.
- J. Li, J. Li, Y. Peng, Y. Du, Z. Yang and X. Qi, J. Controlled Release, 2023, 353, 423–433 CrossRef CAS PubMed.
- X. Xiong, X. Ke, L. Wang, Y. Lin, S. Wang, Z. Yao, K. Li, Y. Luo, F. Liu, Y. Pan, S. J. Yeung, W. Helfrich and H. Zhang, J. Extracell. Vesicles, 2022, 11, e12243 CrossRef CAS PubMed.
- S. Won, C. Lee, S. Bae, J. Lee, D. Choi, M. G. Kim, S. Song, J. Lee, E. Kim, H. Shin, A. Basukala, T. R. Lee, D. S. Lee and Y. S. Gho, J. Extracell. Vesicles, 2023, 12, e12357 CrossRef PubMed.
- X. Z. Liu, Z. J. Wen, Y. M. Li, W. R. Sun, X. Q. Hu, J. Z. Zhu, X. Y. Li, P. Y. Wang, J. L. Pedraz, J. H. Lee, H. W. Kim, M. Ramalingam, S. Xie and R. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 3744–3759 CrossRef CAS PubMed.
- Y. Li, K. Zhang, Y. Wu, Y. Yue, K. Cheng, Q. Feng, X. Ma, J. Liang, N. Ma, G. Liu, G. Nie, L. Ren and X. Zhao, Small, 2022, 18, e2107461 CrossRef PubMed.
- C. Zhang, X. Chong, F. Jiang, J. Gao, Y. Chen, K. Jia, M. Fan, X. Liu, J. An, J. Li, X. Zhang and L. Shen, J. Extracell. Vesicles, 2022, 11, e12209 CrossRef CAS PubMed.
- B. Peng, T. M. Nguyen, M. K. Jayasinghe, C. Gao, T. T. Pham, L. T. Vu, E. Y. M. Yeo, G. Yap, L. Wang, B. C. Goh, W. L. Tam, D. Luo and M. T. Le, J. Extracell. Vesicles, 2022, 11, e12187 CrossRef CAS PubMed.
- Y. K. Kim, Y. Hong, Y. R. Bae, J. Goo, S. A. Kim, Y. Choi, G. H. Nam, M. Kwon, S. G. Yun, G. Lee, C. Jeong and I. S. Kim, J. Controlled Release, 2022, 351, 727–738 CrossRef CAS PubMed.
- X. Zhu, H. Hu, Y. Xiao, Q. Li, Z. Zhong, J. Yang, P. Zou, Y. Cao, F. Meng, W. Li, Y. You, A. Y. Guo and X. Zhu, Cancer Lett., 2022, 536, 215668 CrossRef CAS PubMed.
- V. M. Ukrainskaya, O. E. Musatova, D. V. Volkov, D. S. Osipova, D. S. Pershin, A. M. Moysenovich, E. G. Evtushenko, E. A. Kulakovskaya, E. G. Maksimov, H. Zhang, Y. P. Rubtsov, M. A. Maschan, A. V. Stepanov and A. G. Gabibov, Sci. Rep., 2023, 13, 463 CrossRef CAS PubMed.
- P. Gupta, I. P. Kadamberi, S. Mittal, S. W. Tsaih, J. George, S. Kumar, D. K. Vijayan, A. Geethadevi, D. Parashar, P. Topchyan, L. McAlarnen, B. F. Volkman, W. Cui, K. Y. J. Zhang, D. Di Vizio, P. Chaluvally-Raghavan and S. Pradeep, Adv. Sci., 2022, 9, e2104452 CrossRef PubMed.
- M. Böttcher, R. Böttcher-Loschinski, S. Kahlfuss, M. Aigner, A. Gießl, A. Mackensen, U. Schlötzer-Schrehardt, T. Tüting, H. Bruns and D. Mougiakakos, Cells, 2022, 11(14), 2176, DOI:10.3390/cells11142176.
- J. Wu, D. Zeng, S. Zhi, Z. Ye, W. Qiu, N. Huang, L. Sun, C. Wang, Z. Wu, J. Bin, Y. Liao, M. Shi and W. Liao, J. Transl. Med., 2021, 19, 381 CrossRef CAS PubMed.
- Z. Jiang, Y. Zhang, Y. Zhang, Z. Jia, Z. Zhang and J. Yang, Cell Commun. Signaling: CCS, 2021, 19, 93 CrossRef CAS PubMed.
- L. Jiang, H. Fei, A. Yang, J. Zhu, J. Sun, X. Liu, W. Xu, J. Yang and S. Zhang, Cancer Lett., 2021, 520, 332–343 CrossRef CAS PubMed.
- M. Tan, Y. Chen, Y. Guo, C. Yang, M. Liu, D. Guo, Z. Wang, Y. Cao and H. Ran, Biomater. Sci., 2020, 8, 6703–6717 RSC.
- Y. Zhang, T. Ge, M. Huang, Y. Qin, T. Liu, W. Mu, G. Wang, L. Jiang, T. Li, L. Zhao and J. Wang, Int. J. Nanomed., 2023, 18, 49–63 CrossRef CAS PubMed.
- B. Lin, Y. Wang, K. Zhao, W. D. Lü, X. Hui, Y. Ma and R. Lv, Biomater. Sci., 2022, 10, 744–752 RSC.
- D. Wang, Z. Wan, Q. Yang, J. Chen, Y. Liu, F. Lu and J. Tang, Drug Delivery, 2022, 29, 702–713 CrossRef CAS PubMed.
- M. Hu, J. Zhang, L. Kong, Y. Yu, Q. Hu, T. Yang, Y. Wang, K. Tu, Q. Qiao, X. Qin and Z. Zhang, ACS Nano, 2021, 15, 3123–3138 CrossRef CAS PubMed.
- J. Chen, J. Yang, W. Wang, D. Guo, C. Zhang, S. Wang, X. Lu, X. Huang, P. Wang, G. Zhang, J. Zhang, J. Wang and Z. Cai, Cellular Mol. Immunol., 2022, 19, 1290–1301 CrossRef CAS PubMed.
- J. Chen, Y. Song, F. Miao, G. Chen, Y. Zhu, N. Wu, L. Pang, Z. Chen and X. Chen, Cancer Sci., 2021, 112, 3437–3454 CrossRef CAS PubMed.
- N. Pakravan, A. Abbasi and Z. M. Hassan, Oxid. Med. Cell. Longevity, 2021, 2021, 5529484 Search PubMed.
- S. Heidegger, F. Stritzke, S. Dahl, J. Daßler-Plenker, L. Joachim, D. Buschmann, K. Fan, C. M. Sauer, N. Ludwig, C. Winter, S. Enssle, S. Li, M. Perl, A. Görgens, T. Haas, E. T. Orberg, S. Göttert, C. Wölfel, T. Engleitner, I. Cortés-Ciriano, R. Rad, W. Herr, B. Giebel, J. Ruland, F. Bassermann, C. Coch, G. Hartmann and H. Poeck, Cell Rep. Med., 2023, 4, 101171 CrossRef CAS PubMed.
- X. Wu, H. Zhang, G. Jiang, M. Peng, C. Li, J. Lu, S. Jiang, X. Yang and Y. Jiang, Clin. Exp. Immunol., 2022, 210, 309–320 CrossRef PubMed.
- A. Asadirad, K. Baghaei, S. M. Hashemi, S. Dehnavi, H. Ghanbarian, E. Mortaz, A. Anissian, H. Asadzadeh Aghdaei and D. Amani, Int. Immunopharmacol., 2022, 104, 108493 CrossRef CAS PubMed.
- Q. Zhou, S. Wei, H. Wang, Y. Li, S. Fan, Y. Cao and C. Wang, Front. Immunol., 2023, 14, 1130033 CrossRef CAS PubMed.
- M. R. Hussain, M. Baig, H. S. Mohamoud, Z. Ulhaq, D. C. Hoessli, G. S. Khogeer, R. R. Al-Sayed and J. Y. Al-Aama, Saudi J. Biol. Sci., 2015, 22, 359–373 CrossRef CAS PubMed.
- D. Wang, G. Qiu, X. Zhu, Q. Wang, C. Zhu, C. Fang, J. Liu, K. Zhang and Y. Liu, J. Immunother. Cancer, 2023, 11(5), 6516, DOI:10.1136/jitc-2022-006516.
- J. Hong, M. Kang, M. Jung, Y. Y. Lee, Y. Cho, C. Kim, S. Y. Song, C. G. Park, J. Doh and B. S. Kim, Adv. Mater., 2021, 33, e2101110 CrossRef PubMed.
- M. Fan, H. Liu, H. Yan, R. Che, Y. Jin, X. Yang, X. Zhou, H. Yang, K. Ge, X. J. Liang, J. Zhang and Z. Li, Biomaterials, 2022, 282, 121424 CrossRef CAS PubMed.
- A. Aharon, G. Horn, T. H. Bar-Lev, E. Zagagi Yohay, T. Waks, M. Levin, N. Deshet Unger, I. Avivi and A. Globerson Levin, Hum. Gene Ther., 2021, 32, 1224–1241 CrossRef CAS PubMed.
- B. Zuo, Y. Zhang, K. Zhao, L. Wu, H. Qi, R. Yang, X. Gao, M. Geng, Y. Wu, R. Jing, Q. Zhou, Y. Seow and H. Yin, J. Hematol. Oncol., 2022, 15, 46 CrossRef CAS PubMed.
- Y. Li, L. Tian, T. Zhao and J. Zhang, Cancer Immunol., Immunother.: CII, 2023, 72, 1673–1683 CrossRef CAS PubMed.
- Y. Zhao, Y. Zheng, Y. Zhu, H. Li, H. Zhu and T. Liu, J. Nanobiotechnol., 2022, 20, 359 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, X. Mu, C. R. Tu, Y. Chung, S. W. Tsao, G. C. Chan, W. H. Leung, Y. L. Lau, Y. Liu and W. Tu, J. Immunother. Cancer, 2022, 10(2), 32–38, DOI:10.1136/jitc-2021-003832.
- M. Lu, H. Xing, W. Shao, T. Zhang, M. Zhang, Y. Wang, F. Li, Y. Weng, A. Zheng, Y. Huang and X. J. Liang, Adv. Mater., 2022, 34, e2204765 CrossRef PubMed.
- M. Wang, J. Jia, Y. Cui, Y. Peng and Y. Jiang, Cell Death Dis., 2021, 12, 1065 CrossRef CAS PubMed.
- T. Lu, Z. Zhang, J. Zhang, X. Pan, X. Zhu, X. Wang, Z. Li, M. Ruan, H. Li, W. Chen and M. Yan, J. Extracell. Vesicles, 2022, 11, e12218 CrossRef CAS PubMed.
- H. M. Binder, N. Maeding, M. Wolf, A. Cronemberger Andrade, B. Vari, L. Krisch, F. G. Gomes, C. Blöchl, K. Muigg, R. Poupardin, A. M. Raninger, T. Heuser, A. Obermayer, P. Ebner-Peking, L. Pleyer, R. Greil, C. G. Huber, K. Schallmoser and D. Strunk, Cells, 2021, 10(12), 21–33, DOI:10.3390/cells10123321.
- X. Liu, C. A. Wills, L. Chen, J. Zhang, Y. Zhao, M. Zhou, J. M. Sundstrom, T. Schell, V. S. Spiegelman, M. M. Young and H. G. Wang, J. Immunother. Cancer, 2022, 10(4), 43–99, DOI:10.1136/jitc-2021-004399.
- Z. L. Yu, X. C. Liu, M. Wu, S. Shi, Q. Y. Fu, J. Jia and G. Chen, J. Immunother. Cancer, 2022, 11, e12214 CAS.
- R. E. Veerman, G. G. Akpinar, A. Offens, L. Steiner, P. Larssen, A. Lundqvist, M. C. I. Karlsson and S. Gabrielsson, Cancer Immunol., Immunother. Res., 2023, 11, 217–227 CrossRef CAS PubMed.
- M. Jung, M. Kang, B. S. Kim, J. Hong, C. Kim, C. H. Koh, G. Choi, Y. Chung and B. S. Kim, Adv. Mater., 2022, 34, e2106516 CrossRef PubMed.
- A. S. Hansen, L. S. Jensen, K. R. Gammelgaard, K. G. Ryttersgaard, C. Krapp, J. Just, K. L. Jønsson, P. B. Jensen, T. Boesen, M. Johansen, A. Etzerodt, B. W. Deleuran and M. R. Jakobsen, J. Extracell. Vesicles, 2023, 12, e12350 CrossRef PubMed.
- P. Yang, X. Cao, H. Cai, P. Feng, X. Chen, Y. Zhu, Y. Yang, W. An, Y. Yang and J. Jie, Cell. Immunol., 2021, 360, 104262 CrossRef CAS PubMed.
- K. Svennerholm, K. S. Park, J. Wikström, C. Lässer, R. Crescitelli, G. V. Shelke, S. C. Jang, S. Suzuki, E. Bandeira, C. S. Olofsson and J. Lötvall, Sci. Rep., 2017, 7, 17434 CrossRef PubMed.
- J. Klimentova and J. Stulik, Microbiol. Res., 2015, 170, 1–9 CrossRef CAS PubMed.
- K. S. Park, K. Svennerholm, R. Crescitelli, C. Lässer, I. Gribonika and J. Lötvall, J. Extracell. Vesicles, 2021, 10, e12120 CrossRef CAS PubMed.
- Z. W. Luo, K. Xia, Y. W. Liu, J. H. Liu, S. S. Rao, X. K. Hu, C. Y. Chen, R. Xu, Z. X. Wang and H. Xie, Int. J. Nanomed., 2021, 16, 2949–2963 CrossRef PubMed.
- R. Rezaei Adriani, S. L. Mousavi Gargari, H. Bakherad and J. Amani, Sci. Rep., 2023, 13, 16403 CrossRef CAS PubMed.
- W. R. Zhuang, Y. Wang, Y. Lei, L. Zuo, A. Jiang, G. Wu, W. Nie, L. L. Huang and H. Y. Xie, Nano Lett., 2022, 22, 4491–4500 CrossRef CAS PubMed.
- B. Bo, W. Li, J. Li, C. Han, Q. Fang, M. Yang, J. Ni and C. Zhou, ACS Appl. Mater. Interfaces, 2023, 15, 17696–17704 CrossRef CAS PubMed.
- J. Zhang, Y. Zhu, M. Guan, Y. Liu, M. Lv, C. Zhang, H. Zhang and Z. Zhang, Nanoscale, 2022, 14, 8995–9003 RSC.
- D. de Miguel-Perez, A. Russo, M. Gunasekaran, F. Buemi, L. Hester, X. Fan, B. A. Carter-Cooper, R. G. Lapidus, A. Peleg, M. Arroyo-Hernández, A. F. Cardona, A. Naing, F. R. Hirsch, P. C. Mack, S. Kaushal, M. J. Serrano, V. Adamo, O. Arrieta and C. Rolfo, Cancer, 2023, 129, 521–530 CrossRef CAS PubMed.
- J. Deng and H. Ke, Oncoimmunology, 2023, 12, 2152635 CrossRef PubMed.
- H. Chen, P. Zhang, Y. Shi, C. Liu, Q. Zhou, Y. Zeng, H. Cheng, Q. Dai, X. Gao, X. Wang and G. Liu, J. Nanobiotechnol., 2022, 20, 61 CrossRef CAS PubMed.
- S. Li, G. Yan, M. Yue and L. Wang, BMC Cancer, 2021, 21, 349 CrossRef CAS PubMed.
- W. Zhou, Y. Zhou, X. Chen, T. Ning, H. Chen, Q. Guo, Y. Zhang, P. Liu, Y. Zhang, C. Li, Y. Chu, T. Sun and C. Jiang, Biomaterials, 2021, 268, 120546 CrossRef CAS PubMed.
- P. Gellings, M. Galeas-Pena and L. A. Morici, Clin. Exp. Med., 2023, 23, 519–527 CrossRef CAS PubMed.
- F. Meng, L. Li, Z. Zhang, Z. Lin, J. Zhang, X. Song, T. Xue, C. Xing, X. Liang and X. Zhang, J. Extracell. Vesicles, 2022, 11, e12289 CrossRef PubMed.
- Y. Zhai, Y. Ma, B. Pang, J. Zhang, Y. Li, Y. Rui, T. Xu, Y. Zhao, Z. Qian, Y. Gu and S. Li, J. Nanobiotechnol., 2021, 19, 434 CrossRef CAS PubMed.
- R. N. Moman, N. Gupta and M. Varacallo, StatPearls [Internet], StatPearls Publishing, Treasure Island, Florida, 2023 Search PubMed.
- I. Hassanin and A. Elzoghby, Cancer Drug Resist., 2020, 3, 930–946 CAS.
- F. F. An and X. H. Zhang, Theranostics, 2017, 7, 3667–3689 CrossRef CAS PubMed.
- J. Yang, W. Guo, R. Huang, J. Bian, S. Zhang, T. Wei, C. He, Z. Hu, J. Li, C. Zhou and M. Lu, Front. Immunol., 2023, 14, 1173487 CrossRef CAS PubMed.
- D. Cen, Q. Ge, C. Xie, Q. Zheng, J. Guo, Y. Zhang, Y. Wang, X. Li, Z. Gu and X. Cai, Adv. Mater., 2021, 33, e2104037 CrossRef PubMed.
- Z. Zhou, J. Chen, Y. Liu, C. Zheng, W. Luo, L. Chen, S. Zhou, Z. Li and J. Shen, Acta Pharm. Sin. B, 2022, 12, 4204–4223 CrossRef CAS PubMed.
- Z. Liu, J. Zhang, H. Liu, H. Shen, N. Meng, X. Qi, K. Ding, J. Song, R. Fu, D. Ding and G. Feng, Adv. Mater., 2023, 35, e2208692 CrossRef PubMed.
- Y. Zhu, J. Xue, W. Chen, S. Bai, T. Zheng, C. He, Z. Guo, M. Jiang, G. Du and X. Sun, J. Controlled Release, 2020, 322, 300–311 CrossRef CAS PubMed.
- D. Zhang, P. Liu, X. Qin, L. Cheng, F. Wang, X. Xiong, C. Huang and Z. Zhang, J. Mater. Chem. B, 2022, 10, 8750–8759 RSC.
- B. Zhou, J. Song, M. Wang, X. Wang, J. Wang, E. W. Howard, F. Zhou, J. Qu and W. R. Chen, Nanoscale, 2018, 10, 21640–21647 RSC.
- X. Wang, J. Li, N. Kawazoe and G. Chen, Materials, 2018, 12(1), 31, DOI:10.3390/ma12010031.
- M. Maghsudlu and E. Farashahi Yazd, J. Dig. Dis., 2017, 18, 431–444 CrossRef CAS PubMed.
- X. Lai, F. Yao, Y. An, X. Li and X. D. Yang, Molecules, 2023, 28(6), 25–56, DOI:10.3390/molecules28062556.
- F. Yao, Y. An, X. Lai, X. Li, Z. Yu and X. D. Yang, J. Cancer Res. Clin. Oncol., 2023, 149, 7515–7527 CrossRef CAS PubMed.
- L. M. Pham, K. Poudel, W. Ou, C. D. Phung, H. T. Nguyen, B. L. Nguyen, P. Karmacharya, M. Pandit, J. H. Chang, J. H. Jeong, S. K. Ku, C. S. Yong, H. G. Choi and J. O. Kim, Int. J. Pharm., 2021, 605, 120816 CrossRef CAS PubMed.
- S. L. Ai, X. Y. He, B. Y. Liu, R. X. Zhuo and S. X. Cheng, Mol. Pharm., 2019, 16, 2616–2625 CrossRef CAS PubMed.
- S. Zhu, F. Sun, P. Zhao, G. Liang, X. Sun, L. Zeng and Y. Huang, Int. J. Pharm., 2022, 619, 121709 CrossRef CAS PubMed.
- Z. Hu, B. Zheng, J. Xu, S. Gao and W. Lu, Nanotechnology, 2020, 31, 295101 CrossRef CAS PubMed.
- Y. Hu, X. Chen, Y. Xu, X. Han, M. Wang, T. Gong, Z. R. Zhang, W. John Kao and Y. Fu, Nanoscale, 2019, 11, 16476–16487 RSC.
- Y. Song, L. Bugada, R. Li, H. Hu, L. Zhang, C. Li, H. Yuan, K. K. Rajanayake, N. A. Truchan, F. Wen, W. Gao and D. Sun, Sci. Transl. Med., 2022, 14, eabl3649 CrossRef CAS PubMed.
- I. Li and B. Y. Nabet, Mol. Cancer, 2019, 18(1), 32, DOI:10.1186/s12943-019-0975-5.
- Z. Zhou, R. Wang, J. Wang, Y. Hao, Q. Xie, L. Wang and X. Wang, Front. Immunol., 2022, 13(9), 33–36, DOI:10.3389/fimmu.2022.933736.
- J. M. J. M. Ravasco, A. C. Paiva-Santos and J. Conde, Nat. Rev. Bioeng., 2023, 1(1), 156–158, DOI:10.1038/s44222-022-00008-2.
- F. Kratz, A. Warnecke, K. Scheuermann, C. Stockmar, J. Schwab, P. Lázár, P. Drückes, N. Esser, J. Drevs, D. Rognan, C. Bissantz, C. Hinderling, G. Folkers, I. Fichtner and C. Unger, J. Med. Chem., 2002, 45(25), 5523–5533, DOI:10.1021/jm020276c.
- M. Neek, T. I. Kim and S. W. Wang, Nanomedicine, 2019, 15, 164–174 CrossRef CAS PubMed.
- K. B. Narayanan and S. S. Han, Virus Genes, 2018, 54(5), 623–637, DOI:10.1007/s11262-018-1583-y.
- W. Sun, W. Ji, Q. Hu, J. Yu, C. Wang, C. Qian, G. Hochu and Z. Gu, Biomaterials, 2016, 96, 1–10, DOI:10.1016/j.biomaterials.2016.04.011.
- C. Xu, X. Ji, Y. Zhou, C. Yu-chun, D. Guo, Q. Li, N. Chen and C. Fan, Adv. Mater., 2023, 35(30), 11–15, DOI:10.1002/adma.202211415.
- J. Ma, D. Guo, X. Ji, Y. Zhou, C. Liu, Q. Li, J. Zhang and C. Fan, Adv. Mater., 2023, 14, 15–79, DOI:10.1002/adma.202211579.
- A. C. Wauters, J. F. Scheerstra, I. G. Vermeijlen, R. Hammink, M. Schluck, L. Woythe, H. Wu, L. Albertazzi, C. G. Figdor, J. Tel, L. K. E. A. Abdelmohsen and J. C. M. V. Hest, ACS Nano, 2022, 16(9), 15072–15085, DOI:10.1021/acsnano.2c06211.
- H.-L. Wang, Z.-G. Wang and S.-L. Liu, Molecules, 2022, 27, 5607 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |