
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot chemo- and photo-enzymatic linear cascade processes
J. M.
Carceller
 ,
K. S.
Arias
,
M. J.
Climent
,
S.
Iborra
* and
A.
Corma
*
,
K. S.
Arias
,
M. J.
Climent
,
S.
Iborra
* and
A.
Corma
*
Instituto de Tecnología Química (Universitat Politècnica de València-Agencia Estatal Consejo Superior de Investigaciones Científicas), Avda dels Tarongers s/n, 46022, Valencia, Spain. E-mail: acorma@itq.upv.es; siborra@itq.upv.es; Fax: +34 963877809
First published on 5th July 2024
Abstract
The combination of chemo- and photocatalyses with biocatalysis, which couples the flexible reactivity of the photo- and chemocatalysts with the highly selective and environmentally friendly nature of enzymes in one-pot linear cascades, represents a powerful tool in organic synthesis. However, the combination of photo-, chemo- and biocatalysts in one-pot is challenging because the optimal operating conditions of the involved catalyst types may be rather different, and the different stabilities of catalysts and their mutual deactivation are additional problems often encountered in one-pot cascade processes. This review explores a large number of transformations and approaches adopted for combining enzymes and chemo- and photocatalytic processes in a successful way to achieve valuable chemicals and valorisation of biomass. Moreover, the strategies for solving incompatibility issues in chemo-enzymatic reactions are analysed, introducing recent examples of the application of non-conventional solvents, enzyme–metal hybrid catalysts, and spatial compartmentalization strategies to implement chemo-enzymatic cascade processes.
 J. M. Carceller | Jose M. Carceller was born in Valencia, Spain, in 1991. He studied Chemistry at the Universidad de Valencia and completed his PhD in the research group of Prof. Avelino Corma under the supervision of Prof S. Iborra and Prof. M. José Climent at the Institute of Chemical Technology at the Universitat Politècnica de València (UPV-CSIC). Then he received the Margarita Salas grant and carried out his postdoctoral studies with Prof. Todd Hyster at the Department of Chemistry at Princeton University. Then, he came back to Spain and continued his postdoctoral studies at the Institute of Chemical Technology (UPV-CSIC) in the research group of Prof. Corma. His research interests include the development of chemo-enzymatic processes using heterogeneous catalysts and immobilized enzymes for the synthesis of fine chemicals and biologically active compounds, and the development of new enzyme activities using directed evolution and photocatalysis. |
 K. S. Arias | Karen S. Arias received her BS in Chemistry from the University of Quindío (Colombia) and her MS in Sustainable Chemistry from the Polytechnique University of Valencia (UPV) (Spain). After completing her PhD study in 2016 under the supervision of Prof. Maria J. Climent at the Institute of Chemical Technology (ITQ, UPV), she joined the group of Prof. Avelino Corma at the ITQ as a researcher associate. Since then, her research has mainly focused on converting biomass-derived platform molecules into high valued chemicals and fuels through enzymatic and heterogeneous catalyses. |
 M. J. Climent | Maria Jose Climent Olmedo was born in Alginet (Spain). She is a Full Professor at the Chemistry Department of the Technical University of Valencia. She obtained her PhD in 1991 from the University of Valencia and in 1993 she became a member of the Institute of Chemical Technology (ITQ) at the Technical University of Valencia working in the group of Prof. Avelino Corma. Her main research focuses on the application of heterogeneous catalysts and enzymes to the synthesis of fine chemicals and valorization of biomass-derived platform molecules into chemicals and biofuels through one-pot cascade strategies. |
 S. Iborra | Sara Iborra (Carlet, Valencia) is a Full Professor at the Chemistry Department of the Polytechnical University of Valencia (UPV). She received her PhD in 1987 from the Universidad de Valencia and in the same year she joined the Chemistry Department of the UPV as an Assistant Professor. She has been a member of the Institute of Chemical Technology (ITQ) at the UPV since 1991, where she works in the research group of Prof. Avelino Corma. The main focus of her current research involves the application of heterogeneous catalysts (acid–base and redox) to the synthesis of fine chemicals and valorization of biomass. |
 A. Corma | Avelino Corma is a Professor and founder of the Instituto de Tecnología Química (CSIC-UPV) in Valencia (Spain). He has been carrying out research on heterogeneous catalysis in academia and in collaboration with companies. He has worked on fundamental aspects of acid–base and redox catalyses with the aim of understanding the nature of the active sites and reaction mechanisms. With this basis, he has developed catalysts that are being used commercially in several industrial processes. He is an internationally recognized expert in solid acid and bifunctional catalysts for oil refining, petrochemistry and chemical processes. He has published more than 1000 research papers and is an inventor of more than 130 patents. |
1. Introduction
Nature has developed over the years the most complex and optimized multistep catalytic processes in the form of cascade reactions catalysed by enzymes, which are involved in the metabolic networks needed for life.1 The ingenious strategies and highly efficient cascade reactions in the microenvironment of a cell, basically in a one-pot system, have inspired scientists to perform organic synthesis through cascade reactions using mono- or multifunctional catalytic systems. A cascade reaction can be defined as the reaction with at least two consecutive events, where the product of the first step serves as the substrate for the subsequent step. It can be performed in a sequential mode or in a concurrent (or simultaneous) mode. While in the concurrent mode all the catalysts and reactants are present from the beginning and the reaction conditions are unchanged, in the sequential mode (also called relay or temporal compartmentalization), only after the completion of the first step, an additional component (catalyst, co-factor, solvent, etc.) is added, and supplementary operations (changes in temperature, concentration, pH, etc) can also be performed.Cascade processes in a one-pot mode offer a variety of advantages over the conventional step-by-step approach, such as the avoidance of purification of intermediates, reducing therefore the operation time, waste production and cost of the process. Additionally, atom economy, stereochemical control and substrate scope can be improved, while the cooperative effect between the different catalytic sites can increase reactivity and selectivity by allowing equilibrium reaction steps to proceed to practically full conversion. Therefore, process intensification through cascade catalysed processes is currently an area of increasing interest.2–5
On the other hand, biocatalysis has generated even more interest in industry and academia over the past few years and has emerged as a sustainable and economically viable approach to produce chemicals that require high chemo-, regio- and stereoselectivities. The combination of chemocatalysis with biocatalysis, which couples the flexible reactivity of the chemocatalysts with the highly selective and environmentally friendly nature of biocatalysts, is, therefore, of most interest. Traditionally the chemo-enzymatic processes have been carried out as multi-pot multi-step processes, where an intermediate separation/isolation stage after each reaction step is required.6 However, the design of chemo-enzymatic cascade processes in a one-pot mode, mimicking the nature, which occur in a concurrent or sequential mode, is a research area of increasing interest.
While multi-enzymatic cascade reactions are often successfully achieved as they take place under similar reaction conditions,7 the combination of chemo- and biocatalysts in one-pot is challenging because the optimal operating conditions of both catalysts may be rather different. In fact, the majority of chemocatalysts are active under relatively severe reaction conditions (temperature, pH, organic solvents, etc.) that are not compatible with enzymes, which usually operate under very mild reaction conditions (room temperature, neutral pH, aqueous solutions, etc.).8,9 Furthermore, the different catalyst stabilities and the mutual deactivation of the chemo- and/or biocatalysts are additional problems often encountered in one-pot processes. A typical example is the inactivation or inhibition of enzymes by metal ions.10 Thus, the search for a compatibility window in which both catalysts coexist and operate with reasonable activity is key for the success of a chemo-enzymatic one-pot process. It is not surprising that the number of examples where both classes of catalysts work concurrently in an efficient way is limited when compared with processes performed in a sequential mode, where there is more freedom to operate. Therefore, there are no standard procedures for the development and optimization of chemo-enzymatic cascades. As a result, researchers have developed ingenious strategies to overcome the barriers between both catalytic toolboxes such as the use of bio-conjugated nanohybrids or spatial compartmentalization, which includes use of biphasic systems, membrane filtration, catalyst encapsulation, and performing reactions in a flow mode.11,12
The potential of chemo-enzymatic processes was first highlighted in the early 1980s by the van Bekkum group for the production of mannitol from glucose. The authors combined an enzyme (glucose isomerase) with a heterogeneous metal catalyst (Pt/C). In this process glucose is first in situ transformed into fructose using an immobilized glucose isomerase, which is subsequently hydrogenated to mannitol in the presence of Pt supported on carbon.13 Further works addressing catalyst and process development were performed by both the Steward and van Bekkum groups.14,15
However, after these pioneering studies of combining metals and biocatalysts, there was for more than a decade a lack of significant studies on chemo-enzymatic reactions up to the pioneering contributions on dynamic kinetic resolutions (DKRs). These processes are mainly applied to the preparation of enantiomerically pure amines and alcohols from racemic mixtures, which combine chemocatalytic racemization and hydrolase-catalyzed resolution in water16–19 or lipase-catalyzed resolution in organic solvents.20–23 In fact, in the last 30 years, most of the chemo-enzymatic processes that have been reported correspond to just two groups: dynamic kinetic resolutions and enzymatic processes with concomitant regeneration of co-factors.5,24,25
As DKR has been extensively reviewed,26–30 recent reviews on chemo-enzymatic cascade processes have focused on different particular aspects, such as the synthesis of chiral compounds by combining metal chemocatalysts with enzymes,31–33 chemo-enzymatic cascades in aqueous media34 and the prevailing methods to overcome compatibility issues.12,35,36
The aim of this review is to give the readers a wide overview of the variety of transformations and strategies adopted for combining enzymes and chemo- and photocatalytic processes in a successful way to achieve valuable chemicals and bring out the vast potential that the combination of both catalytic systems has for organic synthesis in the field of sustainable chemistry. Thus, we have focused on prominent and up-to-date examples of linear chemo-enzymatic cascades, where the starting substrate is first transformed by the chemocatalyst prior to the biocatalyzed reaction or vice versa in one-pot. In the first section a series of selected examples of chemo-enzymatic cascades have been classified according to the type of compound synthesized and the different approaches adopted have been discussed. In the second section, different strategies for solving incompatibility issues in chemo-enzymatic reactions will be discussed, introducing recent examples of the application of enzyme–metal hybrid catalysts as well as examples of spatial compartmentalization strategies to implement chemo-enzymatic cascade processes. Finally, the combination of photocatalytic and biocatalytic transformations in linear cascades will also be discussed.
2. Synthesis of organic compounds through chemo-enzymatic cascade strategies
2.1. Synthesis of alcohols and amines
A variety of chemo-enzymatic approaches, particularly focused on the synthesis of chiral compounds, have been reported. In this section, we will discuss the different strategies adopted for the synthesis of these compounds.Alcohol dehydrogenases (ADHs) (also termed ketoreductases, KREDs) are an important family of enzymes that are able to catalyse the complete reduction of carbonyl groups (aldehydes or ketones) to the respective chiral alcohols. These biocatalysts have been successfully used for the production of chiral alcohols, amino alcohols, etc.49,50 Compared with hydrolytic enzymes, ADHs have been much less used for the production of optically pure alcohols since ADHs require stoichiometric amounts, with respect to the substrate, of co-factors such as nicotinamide adenine dinucleotide (NADH) or nicotinamide adenine dinucleotide phosphate (NADPH). However, recently, the development of highly efficient methods for the regeneration of the co-factors, such as the use of formate dehydrogenase or isopropanol as a co-substrate of ADH, has allowed the use of catalytic amounts of co-factors efficiently.51–53
In the case of amines, enzymes such as ω-transaminases (ω-TAs) have emerged as powerful alternatives to hydrolases and proteases for chiral amine preparation.54
Transaminases catalyse the transfer of an amino group from an amino donor on a carbonyl group in the presence of the co-factor, pyridoxal-5′-phosphate (PLP). Thus, the increased accessibility of ω-TAs and the development of engineered enzymes have converted the biotransamination process into a viable and attractive production route for enantiopure amines in the pharmaceutical industry.55
In this section, we will discuss several chemo-enzymatic strategies for the synthesis of chiral alcohols and amines starting from different substrates that involve the formation of a prochiral ketone as the first step.
Based on the chemo-enzymatic oxidation of racemic alcohols combined with the bioreduction approach, Lavandera et al.56 developed an efficient one-pot process in a sequential mode for deracemization of secondary alcohols (Scheme 1). The authors combined a TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl) based oxidation system with alcohol dehydrogenase (ADH) catalyzed enantioselective bioreduction. A key point for this study was the optimization of the oxidative step in a buffer medium, which is necessary for the ADH-catalyzed reduction.
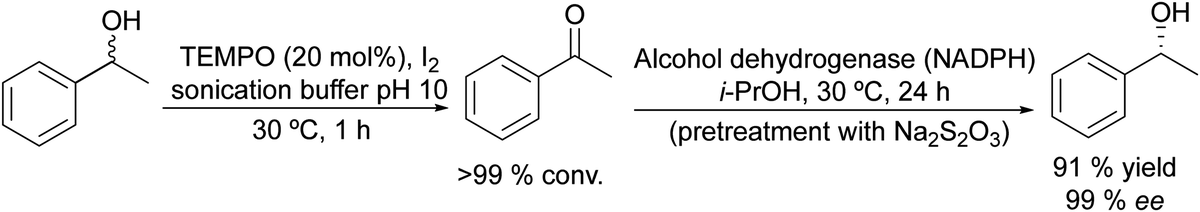 | ||
| Scheme 1 Synthesis of chiral alcohols from racemic mixtures by combining chemocatalyzed oxidation and bioreduction. | ||
Selecting as a model reaction the oxidation of (rac)-phenylethanol to acetophenone, TEMPO combined with different oxidants (that allow the reoxidation of TEMPO) was tested. The study of the compatibility of the ADH in the presence of the different oxidation systems showed that TEMPO combined with I2 was the most effective. Moreover, it was fully compatible with the ADH-catalyzed bioreduction of the resulting ketones when the excess of iodine (that affects the labile co-factor (NADPH) and the enzyme) is quenched with an aqueous Na2S2O3 solution. The protocol was applied to a wide variety of secondary alcohols using Prelog and antiPrelog alcohol dehydrogenases achieving enantiopure (S)- and (R)-alcohols with conversions over 90%.
Following the same strategy González-Sabín et al.57 reported the chemo-enzymatic deracemization of secondary alcohols using the organic radical 2-azaadamantane N-oxyl (AZADO)/sodium hypochlorite as the oxidation system combined with ketoreductases in a sequential mode. It was shown that the presence of AZADO did not affect the enzyme activity, but the excess NaClO used in the first step is detrimental for the subsequent biotransformation. In this case, the problem was easily solved because the isopropyl alcohol used in excess as the hydrogen donor in the enzymatic step quenches the remaining NaOCl in the reaction medium. Following this protocol, a variety of racemic secondary alcohols were converted into enantiomerically pure alcohols in overall yields of up to 98%. Moreover, the strategy was also applied efficiently to the conversion of rac/mesodiols into only one enantiomer. This strategy has advantages over the multienzymatic deracemization, providing higher enantiomeric excess of the chiral alcohol and working at higher concentrations of substrates. The authors applied this approach to the synthesis of chiral amines by coupling the oxidation step with the bioamination of the intermediate prochiral ketone catalyzed by ω-transaminases (ω-TA)58 (Scheme 2). To combine both steps, first, the authors focused on the exploration of an efficient method to perform the oxidation step in aqueous media, compatible with transaminases. It was shown that 2-azaadamantano-N-oxyl (AZADO) combined with NaOCl resulted in an excellent oxidation system able to work in aqueous media at basic pH to perform the oxidation of a wide variety of secondary alcohols into the corresponding prochiral ketones. Then, the amination process of the different prochiral ketones was optimized by screening the activity of a variety of commercial ω-transaminases at basic pH. Optimization of the bioamination step leads to the formation of a variety of valuable chiral amines with high diastereo- and enantioselectivities. The possibility of coupling both steps in a concurrent mode was studied using 1-phenylpropanol as a model substrate. However, several important incompatibilities exist, such as the deactivation of the co-factor of the ω-TA, pyridoxal-5′-phosphate (PLP), in the medium, the inhibition of AZADO by DMSO, the co-solvent required for the bioamination, and the inhibition of alcohol oxidation at the low concentration typically required for ω-TAs (10–20 mM), precluding the concurrent mode. However, the cascade process was successfully achieved in a sequential mode by performing the alcohol oxidation first at the required concentration (250 mM) in an alkaline buffer solution. Once the oxidation was completed, the reaction mixture was diluted to 10 mM with an alkaline buffer solution containing the (R) or (S) ω-transaminase, isopropylamine (as the amino group donor), PLP (co-factor), and DMSO as a co-solvent. The corresponding chiral amine, 1-phenylpropanamine, was obtained in 87% yield and with >99% ee (Scheme 2). The method showed broad substrate scope and was successfully applied to a variety of substrates including sterically hindered β-substituted cycloalkanols, giving excellent results in terms of conversion (up to 97%) and stereoselectivity.
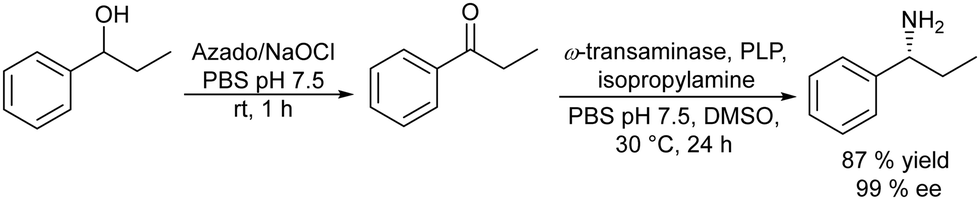 | ||
| Scheme 2 Synthesis of chiral amines from racemic alcohols by combining chemocatalyzed oxidation and bioamination. | ||
On the other hand, the synthesis of chiral secondary aromatic alcohols at laboratory and industrial scales is an important topic in chemistry. The alcohols are usually achieved by the asymmetric reduction of the corresponding aryl ketones (obtained through a Friedel–Crafts acylation) or through enzymatic kinetic resolutions.59,60 However, non-activated alkenes, such as styrene, could also be a source of chiral secondary alcohols through a direct asymmetric hydration process of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, although efficient methodologies have not been sufficiently developed.61 Therefore, Gröger et al.62 developed a chemo-enzymatic process in a sequential mode to produce chiral secondary benzylic alcohols from styrene derivatives. This formal hydration process is based on the Wacker–Tsuji oxidation of the alkene (catalyzed by PdCl2 in the presence of benzoquinone) into a ketone intermediate, which is subsequently reduced to the chiral alcohol in the presence of an alcohol dehydrogenase (ADH) (Scheme 3). Using styrene as a model substrate, the authors showed that Pd species formed during the Wacker–Tsuji oxidation step have a strong inhibitory effect on the ADH activity, leading to a very low yield of the target chiral alcohol (14%). Then, the strategy used to suppress the inhibitory effect of Pd(II) species was the addition of different Pd(II) complexing agents such as thiourea, EDTA, or 2,2′-bipyridine after the oxidation step. Using this protocol, high yield of the corresponding chiral benzyl alcohol could be obtained and the process was extended to a variety of styrene derivatives with good success. Therefore, considering the lack of a generally applicable and efficient chiral catalyst suitable for the direct transformation of non-activated alkenes into chiral aromatic alcohols this chemo-enzymatic process represents an important tool to synthesize these types of compounds.
C bond, although efficient methodologies have not been sufficiently developed.61 Therefore, Gröger et al.62 developed a chemo-enzymatic process in a sequential mode to produce chiral secondary benzylic alcohols from styrene derivatives. This formal hydration process is based on the Wacker–Tsuji oxidation of the alkene (catalyzed by PdCl2 in the presence of benzoquinone) into a ketone intermediate, which is subsequently reduced to the chiral alcohol in the presence of an alcohol dehydrogenase (ADH) (Scheme 3). Using styrene as a model substrate, the authors showed that Pd species formed during the Wacker–Tsuji oxidation step have a strong inhibitory effect on the ADH activity, leading to a very low yield of the target chiral alcohol (14%). Then, the strategy used to suppress the inhibitory effect of Pd(II) species was the addition of different Pd(II) complexing agents such as thiourea, EDTA, or 2,2′-bipyridine after the oxidation step. Using this protocol, high yield of the corresponding chiral benzyl alcohol could be obtained and the process was extended to a variety of styrene derivatives with good success. Therefore, considering the lack of a generally applicable and efficient chiral catalyst suitable for the direct transformation of non-activated alkenes into chiral aromatic alcohols this chemo-enzymatic process represents an important tool to synthesize these types of compounds.
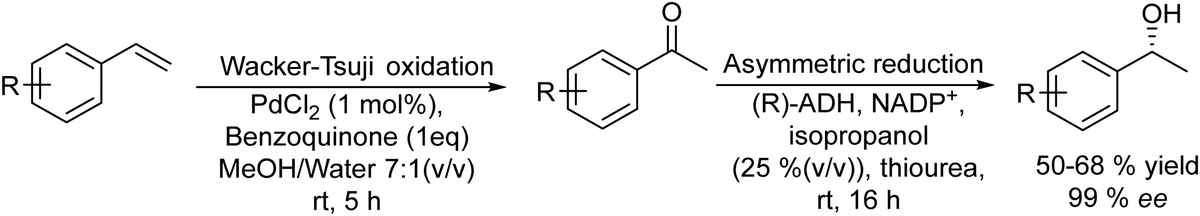 | ||
| Scheme 3 Synthesis of chiral aromatic alcohols from non-activated alkenes by combining oxidation and bioreduction. | ||
Chiral amines were also successfully obtained using a similar strategy. For instance, Gröger et al.63 combined the Wacker oxidation of non-activated alkenes using PdCl2/CuCl as the catalyst with transamination using transaminases64 or reductive amination using an amine dehydrogenase enzyme that uses ammonia as a nitrogen source. In both cases, excellent results in terms of yield and enantioselectivity were obtained in these one-pot two-step processes.
Gotor-Fernández et al.65 synthesized enantiopure 1-arylpropan-2-amines through the Wacker oxidation of allylbenzene derivatives catalysed by palladium(II) Pd(TFA)2 and iron(III) as the oxidant, followed by biotransamination. The authors showed for the first time the possibility to combine Pd(II)/Fe(III) (chemo-oxidative system) with an enzyme catalysed step without affecting the enzymatic activity. The target chiral amines were obtained in 72–92% yield with 99% ee.
Besides alkenes, alkynes can be also used as starting substrates for the synthesis of prochiral ketones by means of the acid catalysed nucleophilic water addition to the alkyne. The use of gold salts as catalysts for alkyne hydration represents a more environmentally friendly method compared with the traditional oxymercuration or the use of large amounts of mineral acids.66,67
The synthesis of chiral alcohols from alkynes was reported by Mihovilovic et al.68 The chemo-enzymatic method involves the Au-catalyzed (AuCl3) alkyne hydration followed by ADH-catalyzed bioreduction in a sequential mode, where the solvent used for the chemocatalyzed step (isopropanol) is used as the secondary substrate for co-factor regeneration in the biocatalyzed step. A variety of S- and R-chiral alcohols were successfully obtained using alcohol dehydrogenases derived from Rhodococcus ruber and Lactobacillus kefir respectively (Scheme 4).
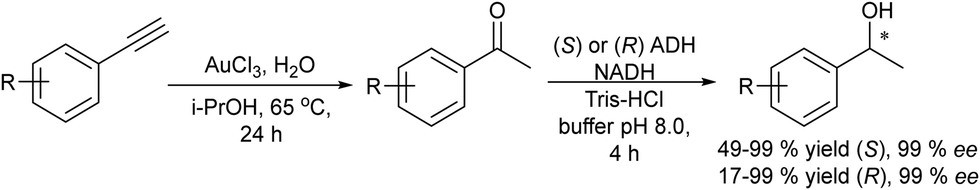 | ||
| Scheme 4 Synthesis of chiral alcohols from alkynes by combining alkyne hydration and bioreduction. | ||
A related transformation was reported by González-Sabín et al.69 by using KAuCl4 as the catalyst and pentynoic acid as the substrate. Thus, the alkyne was first hydrated into a keto group, which was subsequently reduced by a ketoreductase into the corresponding alcohol, which cyclized spontaneously to the chiral lactone (Scheme 5).
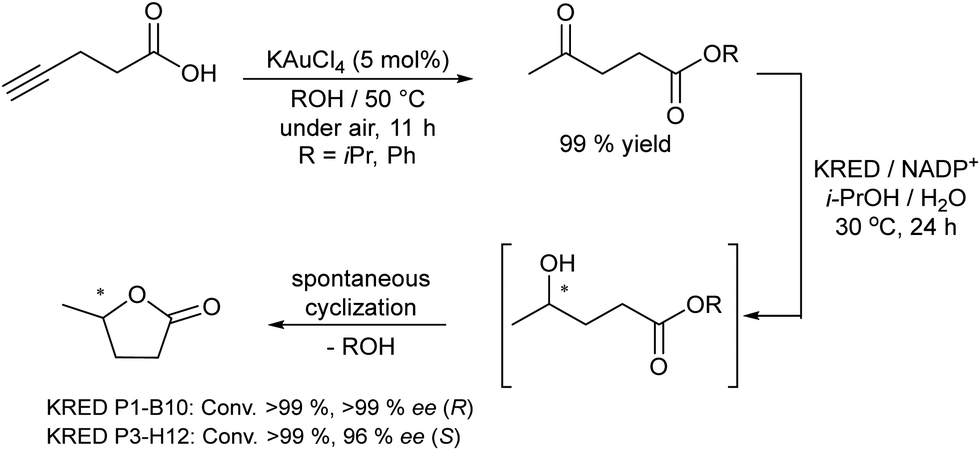 | ||
| Scheme 5 Synthesis of chiral lactones by combining chemocatalyzed hydration of alkynes and bioreduction. | ||
A similar strategy involving gold-catalyzed alkyne hydration and transamination of the intermediate ketone with transaminases was employed by Rueping et al.70 for the synthesis of chiral amines with excellent success.
Recently, Gotor-Fernandez et al.71 reported a chemo-enzymatic process by combining a gold(I) catalyst and enzymes to transform propargylic derivatives into a variety of nor(pseudo)ephedrine derivatives in a regio- and stereoselective way. Ephedrine-type alkaloids are an important family of nitrogen-containing compounds with multiple applications in medicinal chemistry as mimics of adrenaline. Due to the presence of two stereogenic centers, highly stereoselective multienzymatic processes are the preferred synthetic methods.72–74
The chemo-enzymatic cascade process involves as the first step the highly regioselective hydration of racemic or enantiopure propargyl esters or amides into the corresponding ketone ester or keto amide intermediates catalyzed by a gold(I) N-heterocyclic carbene (NHC) complex (IPrAuNTf2) followed by (dynamic) asymmetric biotransamination or bioreduction of the corresponding keto ester or keto amide intermediate (Scheme 6). Under optimized reaction conditions, a series of protected diamine, diol, amino-alcohol derivatives were obtained from propargyl esters or amines in variable yields (57–86% isolated yield), while the (stereo)selectivity of the overall cascade process was determined by the type of biocatalyst, achieving diastereomeric excess and enantiomeric excess in the range of 70–99% and >98% respectively. The advantage of the strategy is that it allows access to a variety of nor(pseudo)ephedrine derivatives and analogues bearing two chiral centers in a direct manner, avoiding multienzymatic methods that usually require low substrate concentration and suffer from mutual deactivation of enzymes.
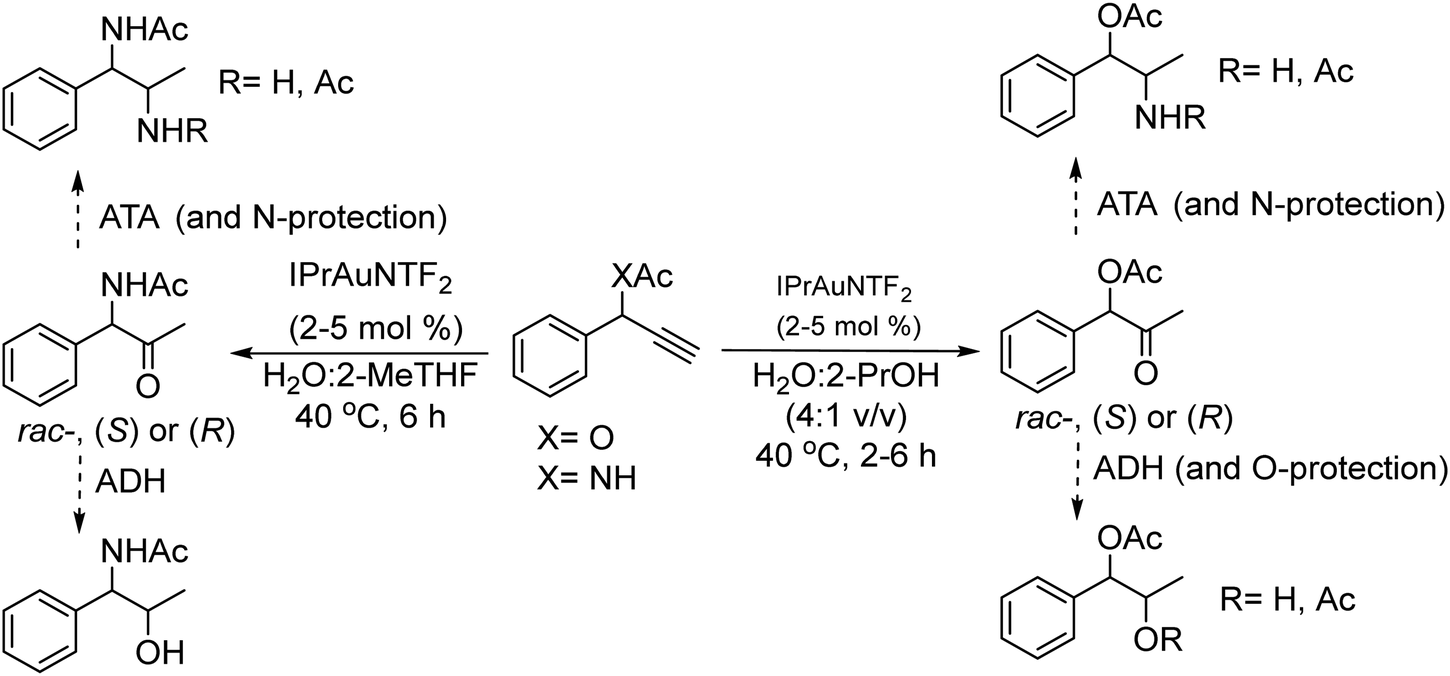 | ||
| Scheme 6 Gold(I)-catalyzed hydration of propargylic ester and amide and subsequent asymmetric enzymatic reactions. | ||
Another class of substrates that allow access to prochiral ketones are allylic alcohols, which can be isomerized to ketones using transition metal complexes (Ru, Rh, and Ni) as catalysts.75,76
González-Sabín et al.77 coupled, for the first time, the ruthenium-catalysed isomerisation of allylic alcohols with asymmetric bioamination catalyzed by ω-transaminases in an aqueous medium in a sequential mode. The authors evaluated three highly efficient ruthenium(IV) catalysts (A–C) (see Scheme 7) in the allylic isomerization of α-vinylbenzyl alcohol, using the reaction conditions required for the enzymatic step, i.e. a phosphate buffer solution containing isopropyl amine as the amino donor. Interestingly, while most of the Ru catalysts previously reported did not show activity for the isomerisation reaction when working in pure water as a solvent and below 80–100 °C, the three complexes were active and selective catalysts for obtaining the desired ketones, working at 50 °C, with complex C being the most efficient catalyst. The authors tried to perform the cascade process in a concurrent mode; however, the deactivation of the Ru catalyst by both the enzyme and pyridoxal-5′-phosphate (co-factor) was observed. Therefore, the cascade reaction was performed in a sequential mode by adding the enzyme and co-factor after the first step before diluting the reaction media to the required concentration of ketone. The cascade process was applied to the synthesis of a variety of chiral amines with high yields and enantiomeric excesses (Scheme 8).
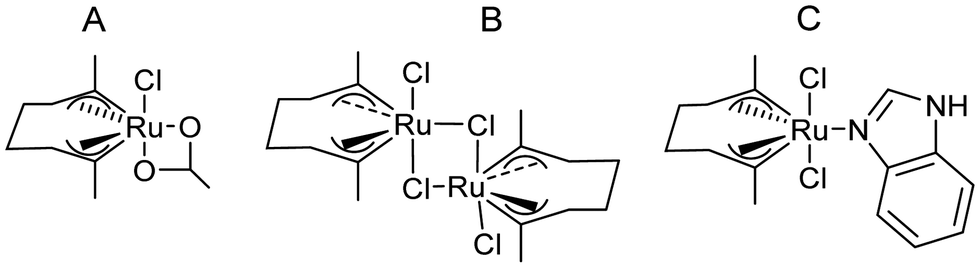 | ||
| Scheme 7 Ru based chemocatalyst complexes. | ||
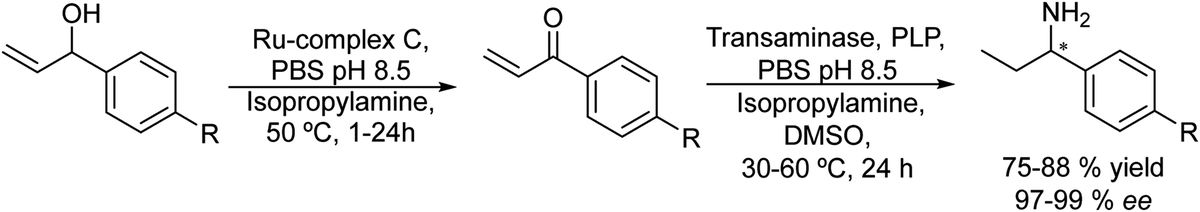 | ||
| Scheme 8 Synthesis of chiral amines from allylic alcohols in a sequential mode by combining chemocatalyzed isomerization and bioamination. | ||
Applying a similar protocol and using a ketoreductase in the enzymatic step, the same authors also synthesized chiral alcohols from racemic allylic alcohols.78 The study of compatibility of the Ru-catalysts (A, B and C) with the enzyme and the required additives (NADPH and the co-factor regenerating agent isopropanol) showed that the Ru-complex B could maintain its activity in the presence of the additives. Then, with optimum concentrations of the different components and temperature, the reaction could be performed in a concurrent mode achieving yields of the desired chiral alcohols between 60 and 86% and ee >99%, (Scheme 9). This case constitutes the first example of a concurrent metal-catalyzed and biocatalyzed reaction in water.
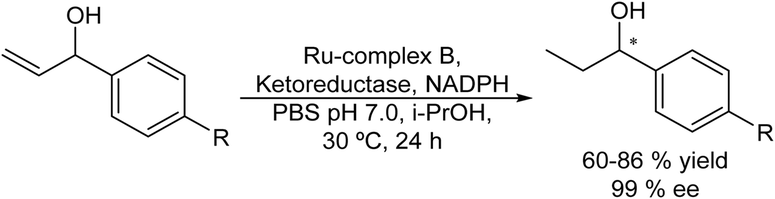 | ||
| Scheme 9 Synthesis of chiral amines from allylic alcohols in a concurrent mode by combining chemocatalyzed isomerization and bioreduction. | ||
Another type of isomerization that gives access to prochiral ketones is for instance the Meyer–Schuster rearrangement of propargylic alcohols. The process comprises the metal catalysed (Cu, Ag, Au, In…) transformation of propargylic (acetylated) alcohols into the corresponding α,β-unsaturated carbonyl compounds, which occurs via a formal 1,3-hydroxyl shift and tautomerisation usually under harsh reaction conditions. The chemo-enzymatic approach combining the Meyer–Schuster rearrangement with enantioselective ketone bioreduction was reported for the first time by Gotor-Fernández et al.79 to obtain chiral β,β-disubstituted allylic alcohols. These compounds are important synthetic intermediates and have been used for instance as precursors of aroma compounds.80 The process combines in a concurrent mode the rearrangement of propargylic alcohols catalysed by a gold carbene complex and bioreduction with ADH of the produced ketone to afford the desired chiral allylic alcohol. Therefore, the authors performed an exhaustive optimization of the reaction conditions of the gold catalyst, to find a compatible reaction window where the gold catalyst and enzyme (ADH) can work in an aqueous medium. Thus, the combination of an active carbene gold(I) catalyst [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene][bis(trifluoromethanesulfonyl)-imide]gold(I) (IPrAuNTf2) with alcohol dehydrogenases (ADHs derived from Rhodococcus ruber) allowed the synthesis of a broad panel of optically active (E)-4-arylpent-3-en-2-ols in good isolated yields (65–86%) (Scheme 10). The chemo-enzymatic cascade was also effectively expanded to a variety of 2-hetarylpent-3-yn-2-ol, hexynol and butynol derivatives. Moreover, the use of alcohol dehydrogenases of opposite enantioselectivity allowed the direct synthesis of both allyl alcohol enantiomers (93–>99% ee) under very mild reaction conditions. Using a similar strategy, the authors combined the Meyer–Schuster rearrangement of propargylic alcohols catalysed by IPrAuNTf2 with transaminases in aqueous media. In this case, in order to avoid the deactivation of the gold catalyst in the presence of the nucleophilic amine derivatives, the reaction was performed in a sequential mode, achieving the corresponding chiral allylic amines in good conversion (67–95%) and excellent selectivities (>97% ee).81
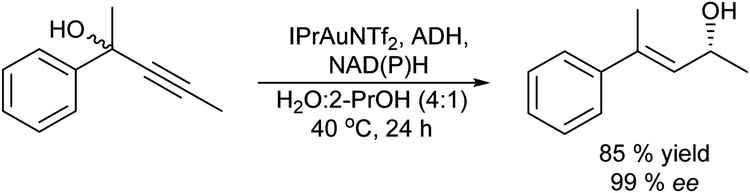 | ||
| Scheme 10 Synthesis of chiral β,β-disubstituted allylic alcohols from propargylic alcohols in a concurrent mode by combining a metal catalyzed Meyer–Schuster rearrangement with bioreduction. | ||
Finally, the selective C–H oxidation of saturated hydrocarbons is another route to obtain alcohols and ketones. However, this type of oxidation is highly challenging, since many catalysts are less selective and overoxidation of products is usually observed. Hutchings et al.82 presented an elegant chemo-enzymatic process to produce alcohols (including chiral alcohols) from hydrocarbons, which involves the in situ generation of H2O2via oxygen reduction catalyzed by gold–palladium nanoparticles supported on TiO2 combined with an unspecific peroxygenase PaDa-I that allows the efficient and selective oxidation of C–H bonds. The authors selected the oxidation of cyclohexane to cyclohexanol as the model reaction and after an exhaustive optimization of the reaction parameters the one-pot chemo-enzymatic process could be achieved in a concurrent mode (Scheme 11). Thus, the substrate, the heterogeneous catalyst (0.5 mol% Au and 0.5 mol%Pd@TiO2) and the biocatalyst were added in a batch reactor under 2 bar (80/20 H2/air), which generates H2O2 at a concentration of 10–20 ppm. Working at room temperature and after 16 h, 87% yield of the desired cyclohexanol with a negligible amount of cyclohexanone was obtained. Under the optimized conditions the authors studied the formation of the chiral R-1 phenylethanol and excellent results in terms of yield and enantioselectivity were obtained (78% yield, 98% ee).
 | ||
| Scheme 11 Synthesis of alcohols by the oxidation of saturated hydrocarbons in a concurrent mode. | ||
The Heck reaction is a C–C cross-coupling process between aryl halides or vinyl halides and activated alkenes catalyzed by palladium catalysts in the presence of a base. By combining the Heck cross-coupling reaction with bioreduction, enantiomerically pure allylic alcohols have been efficiently obtained. These compounds are useful building blocks in organic synthesis for the preparation of a variety of biologically active molecules.83–85 The most practical procedures to produce chiral allylic alcohols are based on kinetic resolution via asymmetric enzymatic acylation and on dynamic kinetic resolution methods of racemic allylic alcohols.86–90 While these procedures are based on the conversion of the preformed carbon structure into the desired chiral allylic alcohol, the chemo-enzymatic strategies allow constructing directly chiral allylic alcohols with a desired carbon structure. Cacchi et al.91 developed for the first time a sequential chemo-enzymatic process for the preparation of chiral allylic alcohols by combining the Heck reaction of aryl iodide derivatives with butenone in the presence of palladium acetoacetonate (Pd(OAc)2) as the catalyst with the enantioselective reduction of the α,β-unsaturated ketone intermediate using an alcohol dehydrogenase (ADH). The Heck reaction conducted selectively with the α,β-unsaturated ketone intermediate was performed by a method developed by the authors under solvent-free conditions in the presence of a high affinity proton acceptor (proton sponge) (1,8-bis(dimethylamino)naphthalene (1,8-BDAN) and 2,4,6-trimethoxyphenyl)phosphine (tmpp) and using an excess of butenone. After reaction completion, the excess of butanone was removed by evaporation, and the ADH and the required additives (see Scheme 12) were added to the reaction mixture. Following the optimized protocol, a series of aryl iodides provided the corresponding (R)- (with LB-ADH) and (S)- (with TADH) allylic alcohols in general in excellent isolated yields and >99% ee. Interestingly, no enzyme inhibition was observed in the biocatalytic step due to the presence of Pd or other reactants in the reaction medium. The method proved to be superior in terms of yield and enantioselectivity respect to conventional dynamic kinetic resolution methods.16,92,93
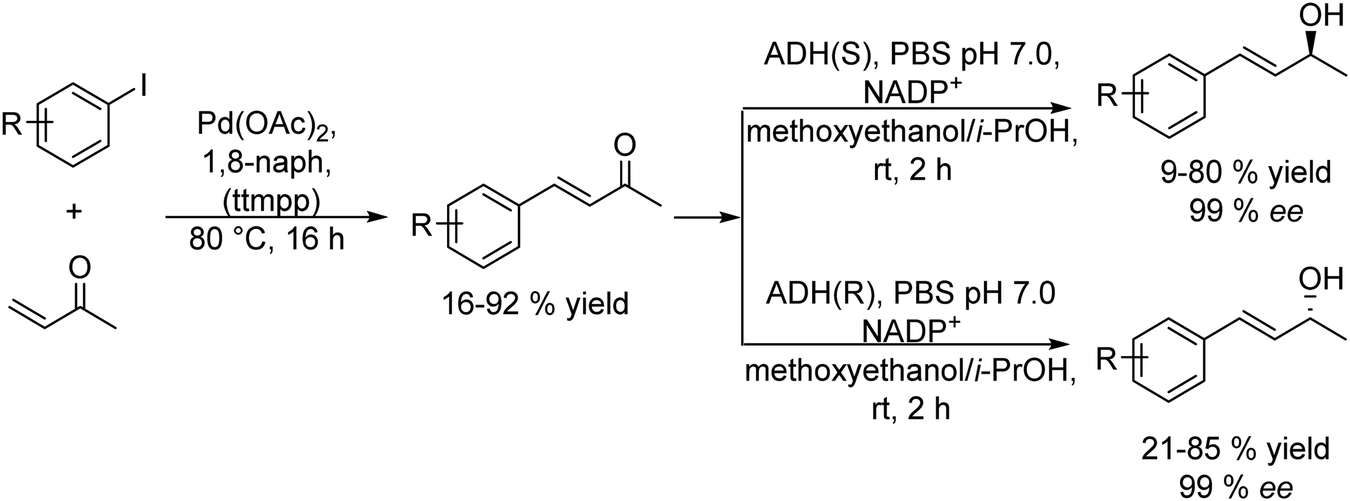 | ||
| Scheme 12 Synthesis of chiral allylic alcohols by combining a metal catalyzed Heck reaction with bioreduction. | ||
Based on the combination of Heck reaction and bioreduction, the same authors performed the synthesis of R-(−)-rhododendrol.94 This compound is an aglycone intermediate used for the synthesis of rhododendrin,95–97 a natural product widely used for its analgesic and anti-inflammatory properties.98,99
In this study, the authors prepared two different Pd chemocatalysts immobilized on a fluorous silica gel, both through fluorous–fluorous interactions (Pdnp-A/FSG) and covalent bonding (Pdnp-B) (Scheme 13), which were tested in the Heck reaction in aqueous media. After optimizing the buffer solution and pH, the Pd catalysts were successfully applied to the Heck reaction of a variety of aryl iodides with 3-buten-2-ol, showing the high stability of the Pd catalysts that could be recycled several times retaining their activity and selectivity. Then, synthesis of (R)-(−)-rhododendrol was selected as a probe for evaluating the feasibility of the chemo-enzymatic process. For doing that, the Heck reaction between 4-iodophenol and 3-buten-2-ol was performed under optimized conditions (see Scheme 14). Then, after cooling, alcohol dehydrogenase [(R)-ADH], the co-factor (NADPH), and isopropanol (as the co-factor regenerating agent) were added to the reaction mixture, and the enzymatic step was completed at room temperature in 24 h. After that, (R)-(−)-rhododendrol was isolated in 90% yield and >99% ee (Scheme 14).
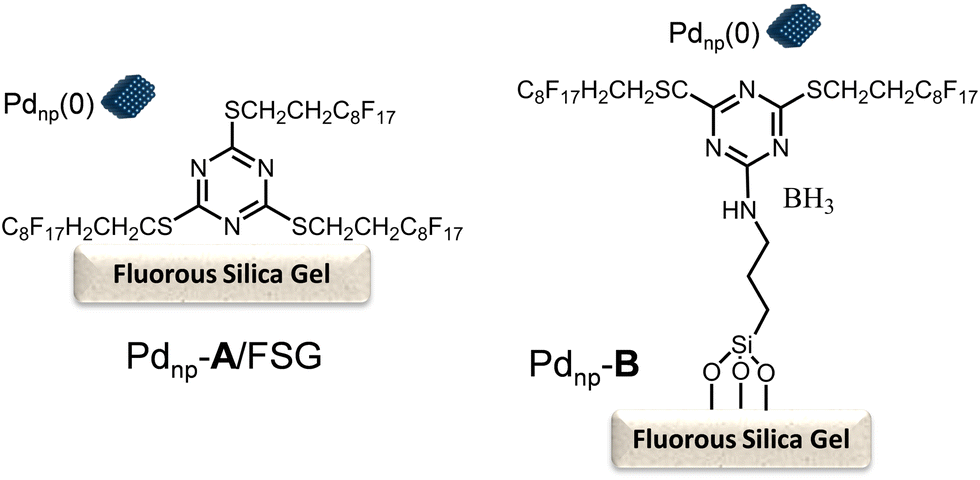 | ||
| Scheme 13 Perfluoro-tagged palladium nanoparticles immobilized on a fluorous silica gel through fluorous–fluorous interaction (Pdnp-A/FSG) and covalent bonding (Pdnp-B). | ||
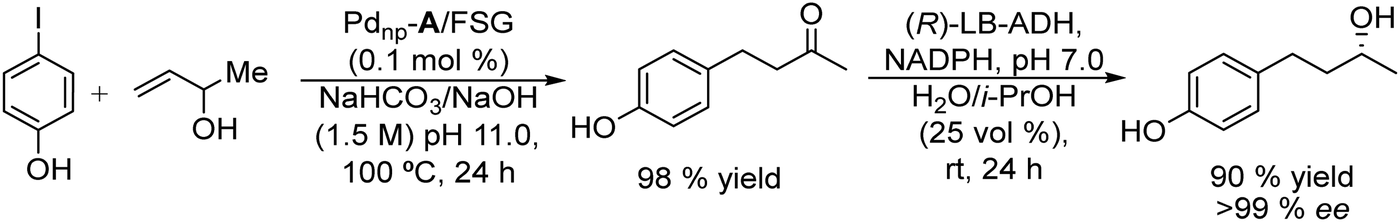 | ||
| Scheme 14 Synthesis of (R)-(−)-rhododendrol by combining a Pd catalyzed Heck reaction with bioreduction. | ||
The process was satisfactorily extended to prepare a variety of other chiral alcohols and represents an alternative to the use of air-sensitive phosphine Pd complexes.
Metal catalyzed Suzuki–Miyaura cross-coupling between aryl halides and phenyl boronic acid derivatives is another C–C bond forming reaction that has been successfully combined with ketoreductases and transaminases to obtain chiral biaryl alcohols and amines in high enantioselectivity.
Thus, chiral biaryl alcohols, which are of great interest in the synthesis of polymers, fluorescent brighteners and chiral ligands100 were prepared by Gröger et al.101 for the first time through a one-pot process by combining a Suzuki cross-coupling reaction catalyzed by a Pd complex with a subsequent asymmetric enzymatic reduction in the presence of the enzyme ADH in aqueous reaction media (Scheme 15).
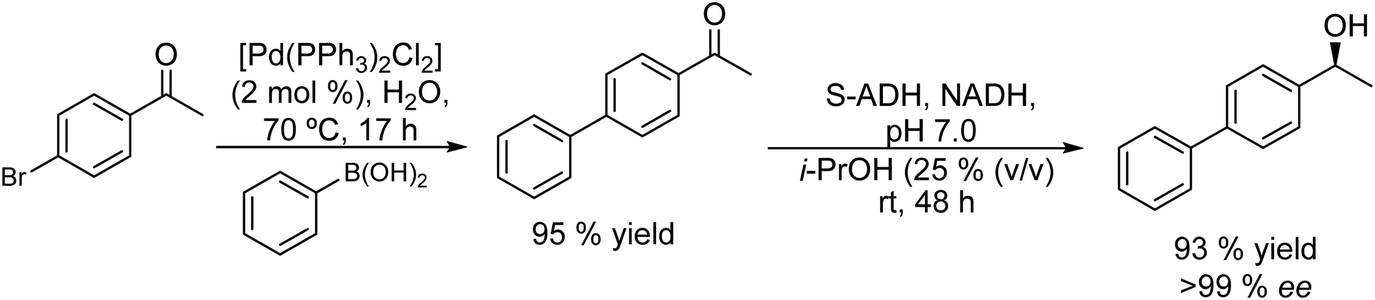 | ||
| Scheme 15 Synthesis of chiral biaryl alcohols by combining a Pd catalysed Suzuki cross-coupling and bioreduction. | ||
Taking as a model the reaction between 4-bromoacetophenone and the boronic acid, the authors studied the possible incompatibilities between the chemical and enzymatic steps. They found that while the Pd complex had a minor negative impact on the enzyme activity, an excess of boronic acid and the use of triphenylphosphine as a base in the Suzuki step strongly inhibited the ADH activity. Then, triphenyl phosphine was substituted by sodium carbonate, and boronic acid was added in stoichiometric amounts. Under these reaction conditions the cascade process was performed in a sequential mode and then to achieve the Suzuki reaction towards practical completion (>95%), the pH was adjusted (pH 7) and the ADH, the corresponding co-factor and isopropanol as the co-factor regenerating agent were added to the reaction media. The corresponding chiral biaryl alcohol was achieved in excellent yield (91%) and >99% ee. The process was applied to a variety of substituted bromoacetophenones and boronic acids, affording the corresponding chiral biaryl alcohols in excellent yields. Based on this pioneering work and to avoid the use of air-sensitive phosphine ligands, Cacchi et al.102 prepared water-soluble palladium nanoparticles, stabilized primarily within a protein cavity (Pdnp/Te-Dps), to synthesize chiral biaryl alcohols in aqueous media using the same strategy. Thus, the Suzuki–Miyaura cross-coupling step followed by the asymmetric enzymatic reduction catalysed by an alcohol dehydrogenase derived from Lactobacillus brevis was performed efficiently. However, it was not possible to reuse the aqueous solution containing the catalyst system (Scheme 16).
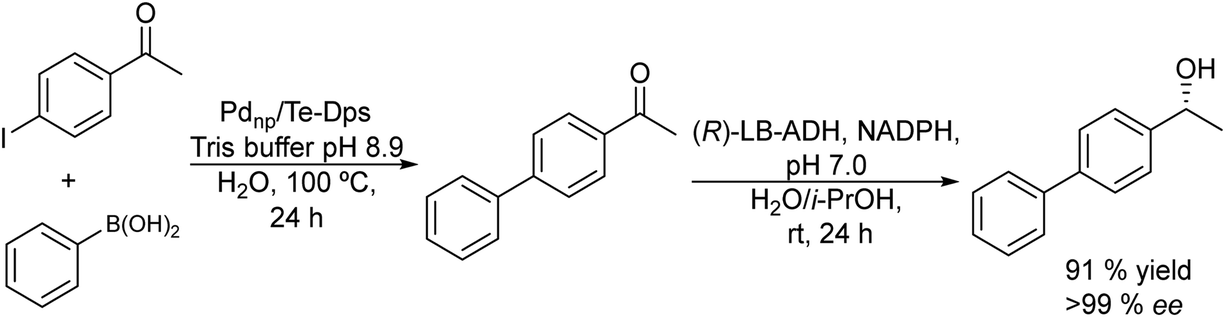 | ||
| Scheme 16 Synthesis of chiral biaryl alcohols by combining Pd catalysed Suzuki cross-coupling and bioreduction. | ||
Symmetrical chiral biaryl diols (Scheme 17) were also synthesized by Gröger et al.103 following a similar strategy. Symmetrical chiral diols are of interest as chiral ligands in asymmetric synthesis and also as monomers for the synthesis of enantiomerically pure polymers,104 such as polyesters.
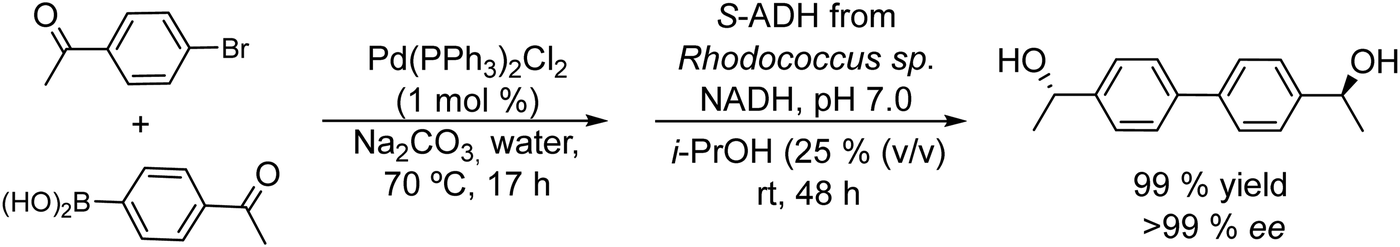 | ||
| Scheme 17 Synthesis of symmetrical chiral biaryl alcohols by combining Pd catalyzed Suzuki cross-coupling and bioreduction. | ||
The authors performed the Suzuki cross-coupling reaction between the halogenated ketone and the phenylboronic substituted ketone, using a triphenylphosphine Pd complex [Pd(PPh3)2Cl2] under aqueous conditions at moderate temperatures (70 °C). Excellent results were obtained in terms of yields of the desired ketones (92–99%) in moderate time (17 h). After pH adjustment to 7 the enzyme ADH derived from Rhodococcus sp. and the required additives (isopropanol and NADP+) were added to the reaction medium. Under these conditions excellent yields and enantioselectivities of the desired biaryl diols were obtained (yield >99% and ee >99%) (Scheme 17).
Gotor-Fernández et al.105 applied this strategy to synthesize a chiral building block of the drug odanacatib (Scheme 18), which is used in the treatment of osteoporosis. The desired chiral intermediate was obtained with excellent yield (97%) and enantioselectivity (99%) (Scheme 19).
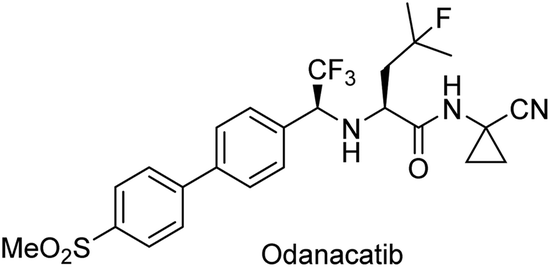 | ||
| Scheme 18 Odanacatib used for treatment of the osteoporosis disease. | ||
 | ||
| Scheme 19 Synthesis of the chiral building block of odanacatib by combining Suzuki cross-coupling and bioreduction. | ||
These strategies have important advantages with respect to conventional synthesis of biaryl alcohols, which requires stoichiometric amounts of a chiral auxiliary for the reduction of the intermediate ketone or is based on enzymatic resolutions,106,107 which in general give poor yields of the target chiral alcohol.
An example of the combination of the Suzuki–Miyaura cross-coupling reaction with a biocatalyzed transamination step to produce chiral biaryl amines was reported by Bornscheuer et al.108 The authors performed the Suzuki–Miyaura coupling reaction of aryl bromides such as 5-bromo-3-acetyl-pyridine with boronic acid in the presence of PdCl2/Na2CO3 as a catalytic system, dissolved in a 50% aqueous solution of N,N-dimethylformamide (DMF). When the corresponding biaryl ketone was formed in quantitative yield, an engineered transaminase derived from Aspergillus fumigatus along with the different required additives was added to the reaction media (see Scheme 20). Interestingly, a conveniently modified transaminase showed excellent compatibility with the reaction conditions of the Suzuki–Miyaura step, achieving an 84% yield of the chiral biaryl amine with 99% ee. The robust feature of the enzyme, allowed the design of a transamination step in a flow reactor using the transaminase immobilized on resins (EziGTM carriers). Thus, after the Suzuki–Miyaura step, the resulting reaction medium was directly used as feed of the continuous flow reactor after adjusting the different required additives for the transamination; however, the yield of the target biaryl amine was moderate (43%). This approach has an important advantage with respect to methods that require amine group protection strategies since free amines are known to be Pd-chelating functional groups. Moreover, it is known that the efficiency of the Suzuki–Miyaura reaction is low when N-protected substrates are involved.109
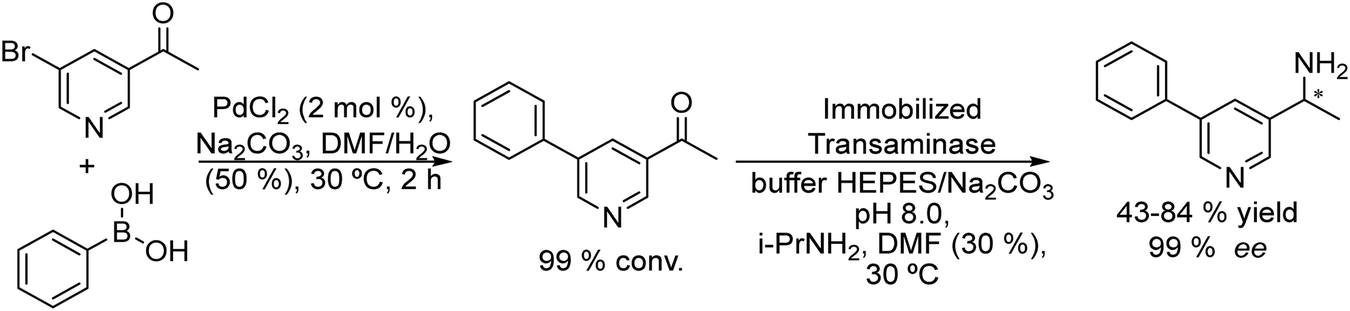 | ||
| Scheme 20 Synthesis of chiral biaryl amines by combining a Suzuki cross-coupling reaction in a batch mode with bioamination in a flow reactor. | ||
Chiral diarylmethanols useful as intermediates for the synthesis of antihistaminics such as (R)-orphenadrine, were synthesized by Garg et al.110 The process combines a Suzuki–Miyaura coupling reaction of arylamides with phenylboronic acid pinacol ester derivatives catalysed by nickel and an asymmetric reduction catalysed by an engineered ketoreductase (KRED P1-B12) (Scheme 21). The process was performed in a sequential mode in water, and the elimination of the acetone produced during the enzymatic step allowed driving the reaction to completion. An important finding for the success of the process was that the Suzuki–Miyaura coupling can be successfully performed in aqueous medium and that the engineered KRED P1-B12 can work in the stereoselective reduction step using i-PrOH–NADPH as the co-factor recycling system.
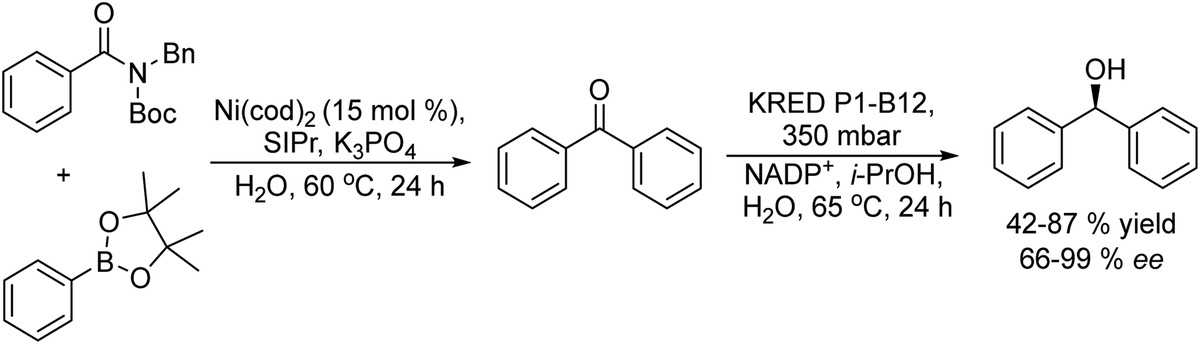 | ||
| Scheme 21 Synthesis of chiral diarylmethanols by combining a Suzuki cross-coupling reaction with bioreduction (SIPr: 1,3-bis(2,6-diisopropylphenyl)imidazolidine). | ||
On the other hand, recent developments in asymmetric organocatalysis in homogeneous media have established that these catalysts are broadly applicable and constitute efficient synthetic tools for the preparation of many types of enantiomerically enriched and enantiomerically pure molecules.111–113 Considering that the reaction diversity of asymmetric organo- and biocatalysis is complementary, the combination of both catalysts in chemo-enzymatic one-pot processes can enable a wide range of exclusive synthetic cascades. The potential of such chiral organocatalysts to form C–C bonds through an aldol condensation when combined with enzymes was demonstrated by Gröger et al.114,115 for the synthesis of valuable 1,3-diols with two stereogenic centers. The chemo-enzymatic process couples the sequential stereoselective formation of both stereogenic centers by combining, in a sequential mode, an asymmetric aldol condensation using a chiral organocatalyst and a bioreduction using an alcohol dehydrogenase (ADH).
As a model reaction of the chemo-enzymatic process, the authors performed the enantioselective aldol condensation reaction of p-chlorobenzaldehyde and acetone in the presence of a proline derived asymmetric organocatalyst (S,S or R,R, Scheme 22), obtaining the corresponding enantiomerically enriched (R) or (S) β-hydroxyketones respectively. Then, bioreduction employing (R or S) ADH was performed at room temperature by addition of a phosphate buffer solution containing the co-factor (NAD+ and NADP+) and 2-propanol (as a co-factor regenerator), achieving successfully the corresponding stereoisomer with high ee (see Scheme 22). Interestingly, it was found that the generation of the second stereocenter was controlled only by the biocatalyst, while the undesired influence of the first generated stereogenic center in the aldol adduct was not observed. Then, it is possible to define the enantioselectivity of each stereocenter by modulating only the organo- and the biocatalyst. Furthermore, the authors expanded the chemo-enzymatic process to other substituted aldehydes and the aldol step was optimized in saturated aqueous NaCl solution.115 The study showed that the organocatalyst concentration was key for controlling the enantioselectivity of the β-hydroxyketone intermediate. Thus, working in aqueous media and at low organocatalyst concentration (0.5 mol%), the aldol condensation yielded the chiral β-hydroxyketone with excellent success (99% ee), which was coupled with the enzymatic reduction. Following the same protocol, similar results in terms of yield and selectivity of chiral 1,3-diols were reported by Aoki et al.116 by using chiral Zn+2 complexes for the aldol reactions between acetone and substituted benzaldehydes followed by bioreduction of the β-hydroxyketone intermediate with ADH. Interestingly, Gröger et al.117 developed later the chemo-enzymatic process in a spatial compartmentalized mode in a continuous flow system. As proof of principle, they performed the reaction between 3-chorobenzaldehyde and acetone by combining two continuous flow reactors. In the first reactor containing the S,S organocatalyst immobilized over acrylic beads, the asymmetric aldol condensation was performed in organic media (cyclohexane) at 3 °C for 24 h, obtaining similar yields to those obtained with the free catalyst in aqueous media.115 Then, after evaporation of the excess of acetone, the reaction mixture was used as a feed (along with isopropanol) of the second reactor, where ADH(S) and its co-factor NAD+ were co-immobilized on the superabsorbent polymer Favor SXM 9155. The target chiral 1,3-diol was obtained in high conversion (89%) and excellent diastereo- and enantioselectivities (diastereomer ratio (dr) >35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and >99% ee) after 24 h at room temperature.
:1 and >99% ee) after 24 h at room temperature.
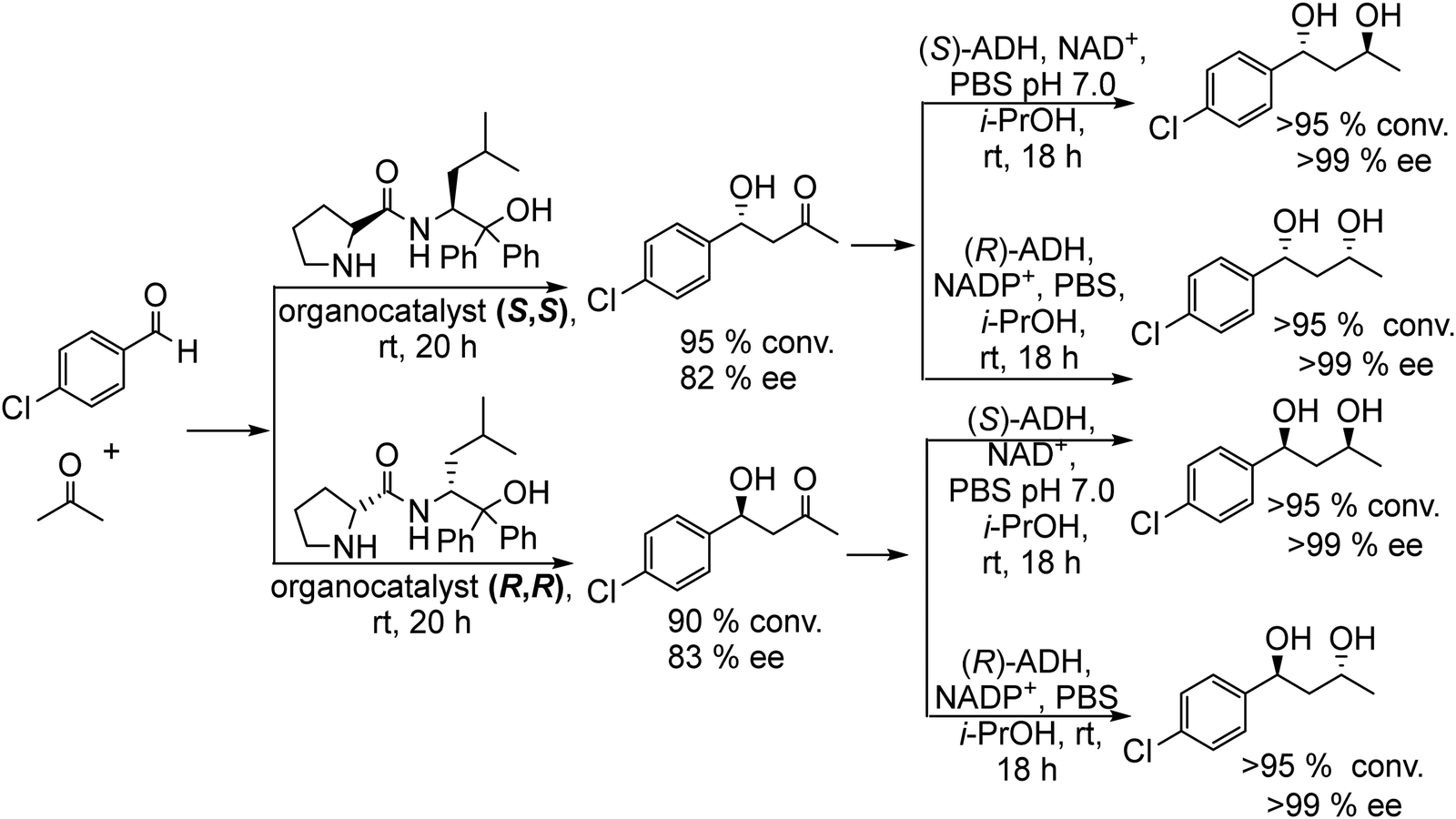 | ||
| Scheme 22 Synthesis of chiral 1,3-diols by combining organocatalyzed asymmetric aldol condensation with bioreduction. | ||
More recently, the authors reported the diastereoselective and enantioselective synthesis of 1,3-diols by combining the S,S organocatalyst and ADH (S) in aqueous media in a concurrent mode, where organo- and biocatalysts are introduced together at the beginning of the reaction.118 First, the compatibility of the organocatalyst, the enzyme and the different required additives was shown. Then, a crucial task was to identify the appropriated reaction condition window under which both reaction steps could proceed efficiently, while side reactions (e.g., reduction of the aldehyde used in the aldol step by the enzyme involved in the second step) were suppressed. Thus, by adjusting the reaction time and the amount of the different components (organo- and biocatalysts, aldehyde/acetone ratio and 2-propanol), 60% of the target chiral 1,3-diol was obtained (see Scheme 23). Interestingly, the process was further successfully implemented in a flow mode.119
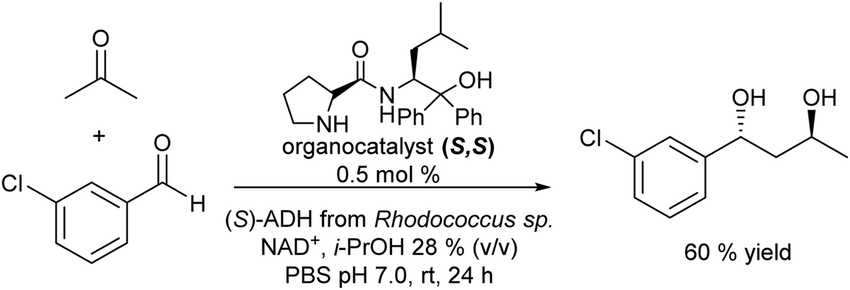 | ||
| Scheme 23 Synthesis of chiral 1,3-diols in a concurrent mode by combining organocatalyzed asymmetric aldol condensation with bioreduction. | ||
Another example of combination of asymmetric organocatalysts with enzymes to generate two stereogenic centers was reported by Mangas-Sánchez and co-workers120 for synthesizing chiral γ-nitroalcohols. Chiral γ-nitro alcohols (and ketones) are essential molecules in the synthesis of pharmaceutical compounds.121 They can be produced via iminium-catalysed asymmetric Michael addition of nitromethane to α,β-unsaturated carbonyl compounds.122,123
The authors120 reported a three-step one-pot process to produce γ-chiral nitroalcohols starting from commercially available benzaldehyde derivatives by combining a Wittig reaction, chiral thiourea-mediated asymmetric conjugate addition of nitromethane, and bioreduction (Scheme 24).
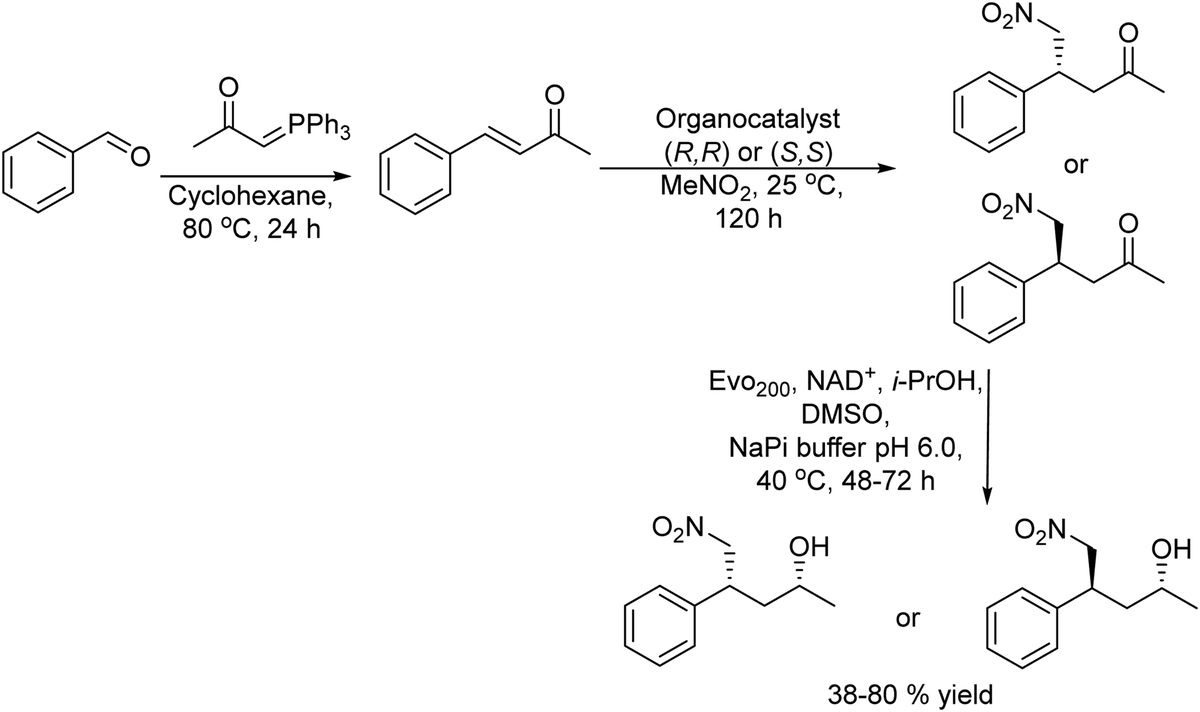 | ||
| Scheme 24 Synthesis of γ-chiral nitro alcohols by combining a Wittig reaction, chiral-thiourea-mediated asymmetric conjugate addition of nitromethane, and bioreduction. | ||
Under optimized reaction conditions chiral γ-nitroalcohols were synthesized by reacting the benzaldehyde derivative with the preformed phosphonium ylide compound followed by the asymmetric 1,4-addition of nitromethane using cyclohexanediamine-based thiourea (R,R) or (S,S) as the asymmetric catalyst to form the intermediate chiral γ-nitro ketone in a one-pot sequential process. After reaction completion, the ketoreductase (Evo200) in a buffer solution and the co-factor and isopropanol as the co-factor regenerating agent were added (Scheme 24). Interestingly, the selection of a ketoreductase resistant to deactivation by nitromethane was a key point for the success of the process. Using this one-pot cascade process in a sequential mode and without significant adjustment of the reaction conditions, a variety of γ-nitro alcohols were obtained in 38–80% yield after three steps, while high diastereomeric and enantiomeric ratios were also obtained (up to ≥97:3 dr and er). The process represents the first case of the coupling of an asymmetric organocatalyzed step via iminium catalysis with a bioreduction step, in one-pot, to form two new stereogenic centers from a prochiral substrate.
2.1.3.1. Synthesis of homoallylic alcohols. Homoallylic alcohols are important building blocks, and are precursors, for instance, for the synthesis of chiral chromanes, lactones or urea derivatives.124–127 Fuchs et al.128 reported a chemo-enzymatic procedure for the synthesis of homoallylic alcohols, which involves the biooxidation of primary alcohols to the corresponding aldehydes in the presence of oxygen and the subsequent chemocatalyzed allylation to the target homoallylic alcohols (Scheme 25). Starting from phenylmethanol as the model substrate, the biooxidation was performed in phosphate buffer solution (PBS) using galactose oxidase, in the presence of a Cu(II) salt as a co-factor, and horseradish peroxidase (HRP) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) for the removal hydrogen peroxide that was produced during the reaction. To achieve complete conversion to the benzaldehyde intermediate, optimization of the reaction conditions, such as oxygen pressure and concentrations of the Cu(II) salt and substrate, was carried out. The second step, the allylation process, was performed following two different methods: (A) a Barbier-type reaction that involves the nucleophilic addition between an alkyl halide and a carbonyl group catalysed by indium(0) and (B) metal-free addition of allylboronic esters.129,130 It was shown that the combination of both steps in a concurrent mode was not possible because the biooxidation was completely inhibited in the presence of the allylating reagents (route A or B), due to the allylation of the active sites of the enzyme.
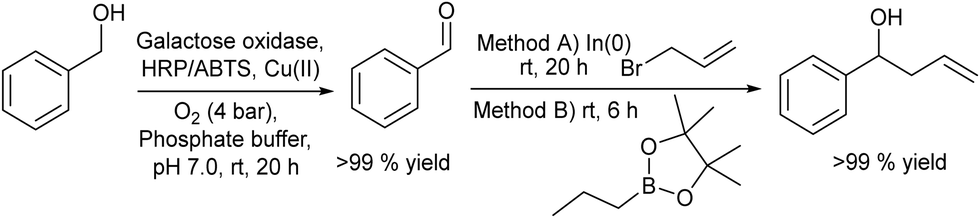 | ||
| Scheme 25 Synthesis of homoallylic alcohols from benzylic alcohols by combining bio-oxidation with chemocatalyzed C–C coupling. | ||
However, the cascade process could be successfully achieved when In(0) and allyl bromide or the allylboronic ester were added to the reaction medium after the completion of biooxidation.
The process was extended to a variety of benzylic alcohols that were transformed into the corresponding homoallylic alcohols with good to excellent conversion. A remarkable feature of the process is the functional group tolerance, which includes unprotected phenolic hydroxyl groups and basic heteroaromatic moieties.
2.1.3.2. Synthesis of catechols. Catechols (1,2-dihydroxybenzenes) are widespread in nature and are widely used in the pharmaceutical and chemical industries. For instance, catechols and their derivatives are used in a variety of processes, such as the manufacture of inks, insecticides, plastics, photographic developers, rubber, etc.131 Moreover, in cells, catechols work as antioxidants, preventing diseases produced by free radicals, such as heart disease, cancer and immune system decline. The synthesis of catechol derivatives is usually difficult and sometimes requires harsh reaction conditions, leading to the formation of isomeric mixtures of 3- and 4-catechols in low yields.132
The potential of combining a dioxygenase-catalysed cis-dihydroxylation of monosubstituted benzenes with a dehydrogenation step catalysed by Pd/C in a one-pot process to obtain catechols in aqueous media was reported by Hardacre et al.133 As an example (Scheme 26) fluorobenzene is transformed into the corresponding cis-diol using the enzyme dioxygenase present in whole cells of P. putida. Subsequently, the cis-diol is transformed into the desired catechol (3-fluorocatechol) using Pd/C as the heterogeneous catalyst.
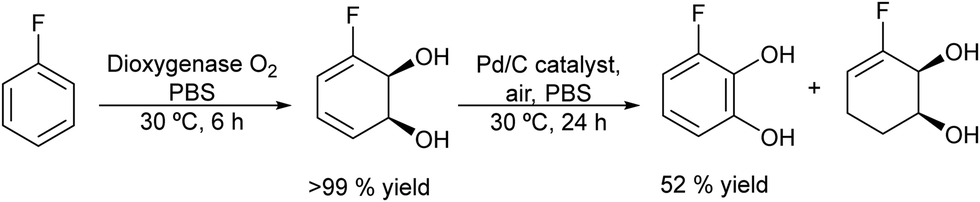 | ||
| Scheme 26 Synthesis of 3-substituted catechols from monosubstituted aromatics by combining cis-hydroxylation with a Pd catalyzed dehydrogenation step. | ||
Optimization of the dehydrogenation step was performed initially using the Pd/C catalyst under different reaction conditions in order to minimize the formation of the cis-dihydrodiol byproduct. Starting from different monosubstituted aromatics, yields between 40 and 66% at 100% conversion were achieved. Moreover, the Pd/C catalyst was found to be stable and could be reused for 4 cycles without loss of activity and selectivity.
Finally, the one-pot process starting from fluorobenzene in aqueous phosphate buffer could be achieved by performing the biotransformation using a large volume of culture medium. Then, after concentration of the reaction mixture containing the target cis-diol and bacterial cells, the former is dehydrogenated in the presence of the Pd/C catalyst under an air atmosphere, achieving 52% yield of 3-fluorocatechol. In addition, the chemo-enzymatic process was compared with a biocatalyzed cascade method using whole cells of P. putida for cis-dihydroxylation combined with E. coli nar B for dehydrogenation, achieving similar yields of the target catechol.
2.1.3.3. Synthesis of para-quinols. The p-quinol is a key structural moiety found in numerous natural products,134 pharmaceuticals,135 and synthetic building blocks.136,137 The main synthetic methods of quinols involve the oxidation of 4-alkylphenols and the direct alkylation of quinones using alkyllithium and Grignard reagents. These procedures require corrosive, harsh and water-free conditions or the utilization of toxic transition metal catalysts, achieving in most cases modest yield of quinol.138–140 Recently, Koszelewski et al.141 reported the synthesis of p-quinols through the copper catalysed addition of boronic acid to quinone under aqueous conditions. However, the method suffers from a low atom economy and is limited to benzoquinone derivatives. To overcome these drawbacks, the authors envisioned142 a chemo-enzymatic approach that combines laccase-catalysed in situ oxidation of hydroquinones to the corresponding quinones with subsequent CuI-catalysed C–C coupling with arylboronic acids in a concurrent mode (Scheme 27). After optimization of the reaction conditions, the synthetic applicability was shown for a wide spectrum of p-quinol derivatives that were obtained in yields higher than or similar to those obtained from quinones. Interestingly, the authors found that the enzyme not only participates in the oxidation reaction of the hydroquinone, but also increases the activity of the copper catalyst in the carbon–carbon bond formation of the second step by forming Cu complexes with the enzyme without key changes in the geometry of the active sites of the biocatalyst. The wide applicability of this chemo-enzymatic cascade strategy allows access to quinol structures difficult to obtain by traditional methods, representing an efficient synthetic alternative to existing procedures.
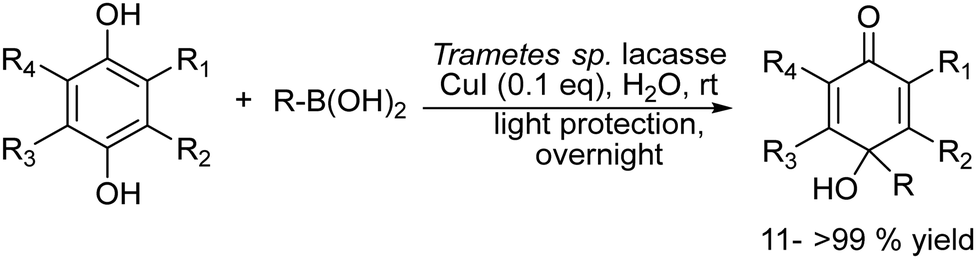 | ||
| Scheme 27 Synthesis of p-quinols by combining laccase-catalyzed in situ oxidation of hydroquinones with the corresponding quinones. | ||
2.1.3.4. Synthesis of chiral vicinal diols. Enantioenriched vicinal diols are important intermediates in the pharmaceutical industry.143 Monti et al.144 developed for the first time a chemo-enzymatic one-pot two-step sequential procedure to obtain highly enantioenriched limonene 1,2-diols by combining the epoxidation of (+)- and (−)-limonene in the presence of heterogeneous titanium-grafted silica catalysts with a biocatalyzed hydrolytic epoxide ring-opening in the presence of limonene-1,2-epoxide hydrolase derived from Rhodococcus erythropolis. For the first step an excess of tert-butyl hydroperoxide (TBHP) in the presence of a titanium on silica catalyst was selected since it directs the epoxidation preferentially towards the endocyclic double bond of limonene (C1–C2).145
For instance, using (−)-limonene as the substrate and Ti/SiO2 as the catalyst (Scheme 28), excellent selectivity of monoepoxides (85% at 93% conversion), as a mixture of cis-endo, trans-endo, and exo isomers (an endo/exo ratio of 6:1), was obtained after 24 h at reflux temperature with trifluorotoluene (TFT) as a solvent. For the hydrolysis step, the hydrolase in a buffer solution was added to the reaction mixture and the reaction proceeded in the biphasic TFT–water medium at room temperature for 24 h. Under these conditions, both endo-epoxides were completely hydrolyzed and an overall yield of 72% of the enantiomerically and diastereomerically enriched (R,R,S)-diol was achieved by this simple and straightforward procedure, while the exo-epoxides were not hydrolyzed. Interestingly the enzymatic activity was not influenced by the presence of the titanium catalyst and the residual amounts of TBHP oxidant.
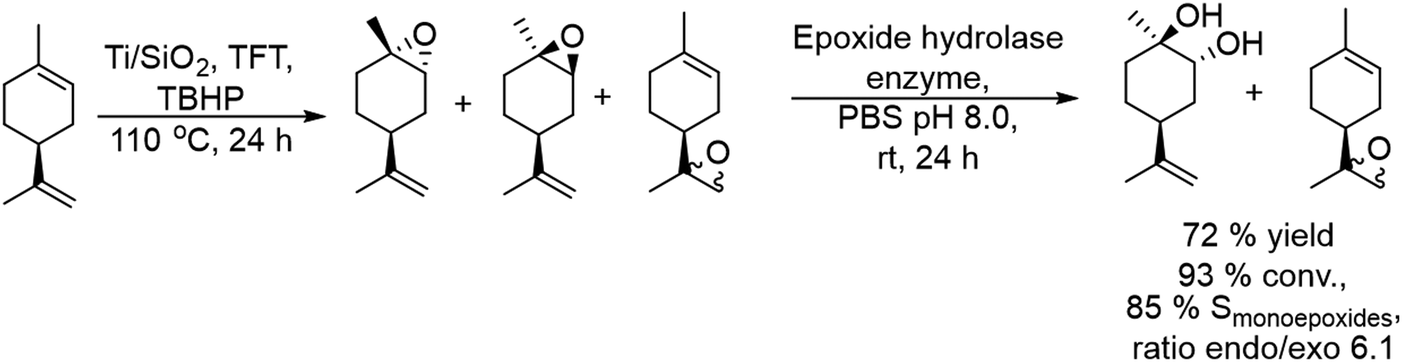 | ||
| Scheme 28 Synthesis of chiral vicinal diols by combining a chemocatalyzed epoxidation reaction with biocatalyzed epoxide hydrolysis. | ||
In summary, chemo-enzymatic cascade processes can allow access to a variety of valuable chiral alcohols and amines. In general, main advantages of these cascade processes compared with conventional methods, involving kinetic resolutions or asymmetric catalysis, are associated with the possibility to start from very structurally different substrates, while higher yields and enantioselectivities of the desired product can be achieved under mild reaction conditions in aqueous media. In addition, the combination of chemocatalyzed C–C-bond forming reactions with enzymes allows access to a variety of more complex chiral structures, difficult to achieve by conventional asymmetric methods, including the easy generation of various stereogenic centers with high stereoselectivity by combining asymmetric organocatalysts with biocatalysts.
2.2. Synthesis of aldehydes
The selective synthesis of aldehydes by the partial reduction of esters is an important reaction in organic chemistry.146 However, the reaction is usually performed through non-environmentally friendly protocols that require special reducing agents such as sodium diisobutyl-t-butoxyaluminum hydride and very low reaction temperature (between 0 °C and −78 °C) and that, in general, lead to a low yield of the corresponding aldehyde.147 Recently, an environmentally friendly chemo-enzymatic method has been reported by Weberskirch et al.148, which involves the lipase catalyzed hydrolysis of the ester followed by the selective oxidation of the intermediate alcohol into the aldehyde in aqueous medium. A highly active and selective Cu(I)/bipyridine/N-oxyl catalytic system, which can operate under mild reaction conditions, was selected for the aerobic oxidation of the alcohol intermediate into the aldehyde.149 However, an important drawback of the Cu(I)/bipyridine catalyst is its water-sensitive character. Therefore, to produce a water-resistant Cu(I)/bipyridine catalyst a core-crosslinked nanoparticle with a hydrophilic shell and a hydrophobic core was prepared by a microemulsion process using hexanediol dimethacrylate as a cross-linking monomer. The polymeric nanoparticles that provide a hydrophobic nano-environment for the Cu/bipyridine system were highly active and stable catalysts for performing the oxidation of a variety of aromatic and allylic alcohols in aqueous media using TEMPO or ABNO (9-azabicyclo[3.3.1]nonane N-oxyl) as the N-oxyl source. Interestingly, the polymeric shell of the nanoparticles also avoids the interaction of the chemocatalyst with the enzyme, preventing their mutual inhibition. Thus, the chemo-enzymatic process was efficiently performed in a sequential mode, where the total hydrolysis of the acetyl ester was first achieved in the presence of the enzyme CAL-B at 40 °C for 20 min. Then, after neutralization of the acetic acid produced during the hydrolysis, the Cu/bipyridine catalyst was added to the reaction media to perform the aerobic oxidation of the produced alcohol intermediates. The corresponding aldehydes were obtained in high yields (73–93%) (Scheme 29).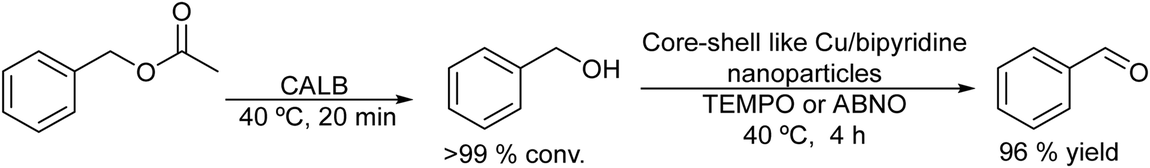 | ||
| Scheme 29 Synthesis of aldehydes from esters by combining enzymatic hydrolysis with oxidation. | ||
This chemo-enzymatic process represents a direct and eco-friendly route for the synthesis of aldehydes with high selectivity from the corresponding esters, avoiding conventional methodologies that use metal hydride salts involving high generation of wastes.
Another synthetic strategy to obtain chiral aldehydes, which can be used as fragrances and flavors, has been studied by Rudroff et al.150 The authors described a chemo-enzymatic cascade process that involves as the first step the palladium-catalyzed Heck-coupling between aryl trifluoroborate derivatives and acrylic acids giving cinnamic acid derivatives, followed by two biocatalysed reduction steps where the acid group is first reduced to the corresponding alpha-unsaturated aldehyde (catalyzed by Carboxylate Reductase (NcCAR) whole cell) and finally, the CC bond is asymmetrically reduced to the final chiral aldehyde by an enereductase (OYE1 whole cell) (Scheme 30).
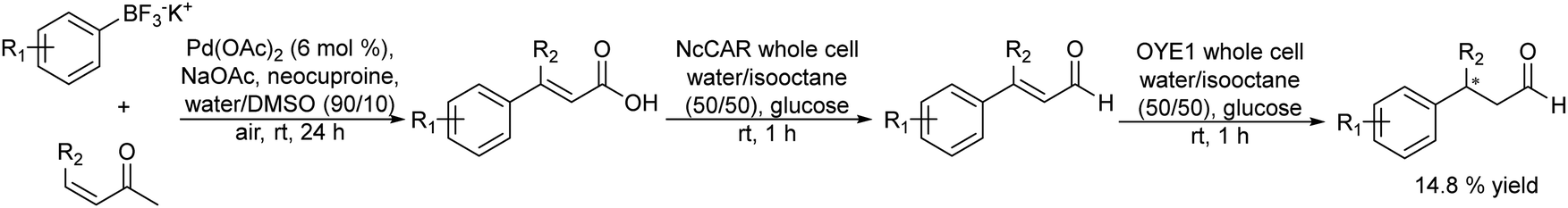 | ||
| Scheme 30 Synthesis of chiral aldehydes in a concurrent mode by combining C–C Heck coupling with biocatalysed carboxylic acid reduction into the aldehyde and asymmetric alkene reduction. | ||
A key point of this study was to find an enzyme with high activity to perform the selective bioreduction of the carboxylic acid into the aldehyde while avoiding the over-reduction of the aldehyde to the undesired alcohol. For this, the authors combined the random mutagenesis biotechnological strategy151 with an in situ product removal (ISPR) strategy by working in a biphasic system (isooctane–water, 50/50 v/v), where the aldehyde is extracted from the aqueous phase, achieving complete conversion to the desired unsaturated aldehyde. The fusion of the two proteins necessary for the two biocatalysed steps, the NcCAR and OYE1, combined with the biphasic strategy, enabled both biocatalyzed steps to occur simultaneously, affording the chiral aldehyde in excellent yield (80%) when starting from the pure carboxylic acid derivative.
However, when the whole chemo-enzymatic cascade process was performed in a concurrent mode the yield of the desired chiral aldehyde was only 14.8% due to the incompatibility between the CAR enzyme and the acrylic acid substrate necessary for the Heck reaction.
With interest in synthesizing fragrant aldehydes the same authors proposed two different catalytic routes starting from substituted phenyl propenes.152 Route A (Scheme 31(a)) involves catalytic isomerization of the allylic alkene group followed by the alkene group cleavage performed using an aromatic dioxygenase enzyme (ADO). Route A was performed in a sequential mode by using PdCl2 as the isomerization catalyst, followed by the addition of ADO (whole cells) after dilution of the reaction media with phosphate buffer and EtOH. However, due to the substrate specificity of the ADO enzyme, the chemo-enzymatic process only worked with substrates bearing a hydroxy group at the para position of the aromatic ring. Vanillin could be obtained through this approach in 53% yield (Scheme 31(a)).
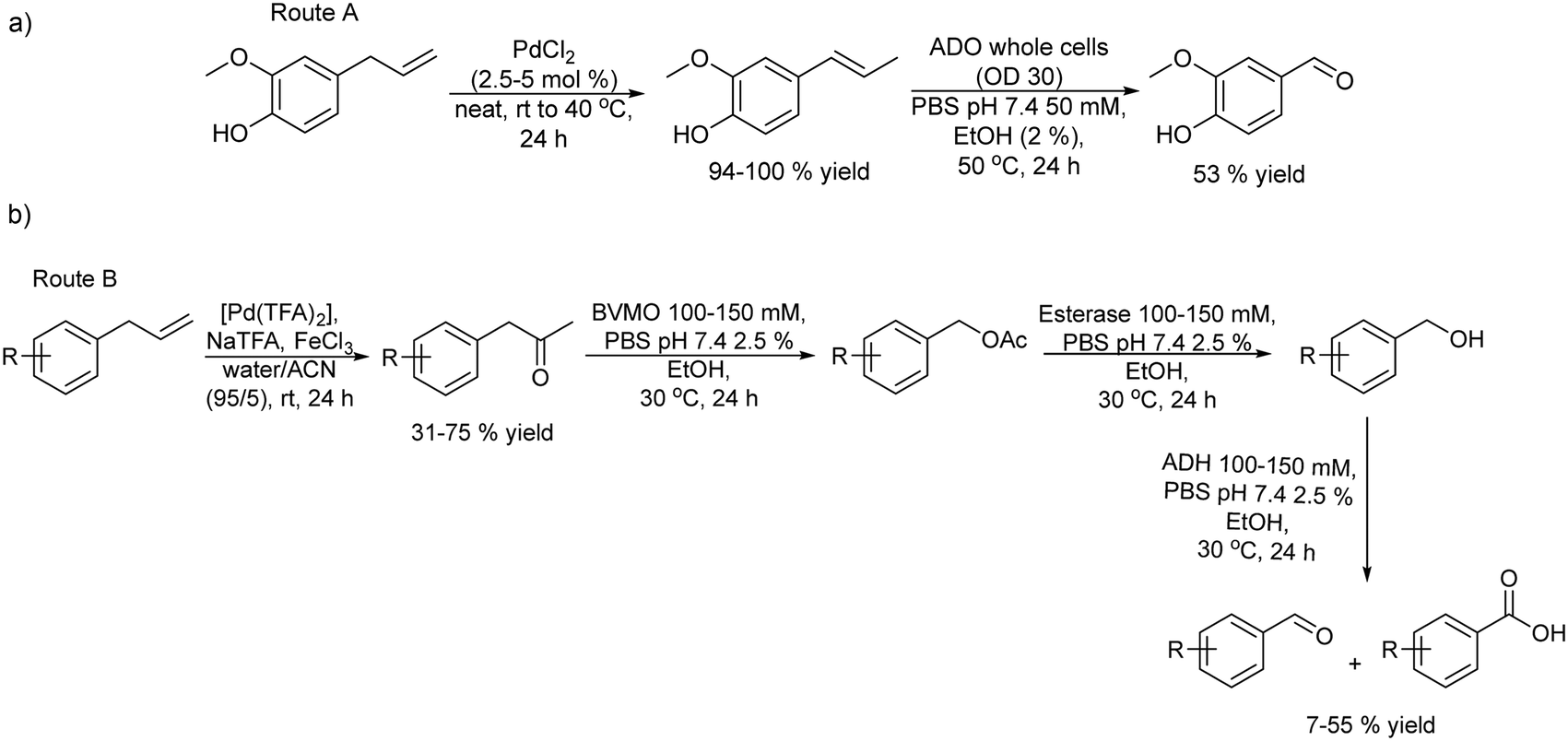 | ||
| Scheme 31 Synthesis of aldehydes from aryl propenes. Route (A): palladium-catalyzed isomerization followed by alkene cleavage. Route (B): Wacker oxidation followed by three consecutive bioreactions involving Baeyer–Villiger oxidation, ester hydrolysis, and oxidation. | ||
Therefore, the authors explored another route (route B) in a sequential mode where the initial step is Wacker oxidation of the phenylpropene substrate to the ketone followed by three biocatalysed steps that involve Baeyer–Villiger monooxygenase oxidation of the ketone to the ester, hydrolysis of the ester performed by an esterase giving an alcohol that is finally biooxidized to the aldehyde (Scheme 31(b)). An extensive study of a series of potential enzymes for this cascade process showed that phenylacetone monooxygenase (PAMO) as Baeyer–Villiger–monooxygenase, AlkJ derived from Pseudomonas putida as alcohol dehydrogenase and an esterase derived from Pseudomonas fluorescens (PfeI) were the optimum enzymes. The authors formed a mixed-culture with the three biocatalysts and studied the conversion of the ketone to the desired aldehyde. A crucial point in this study was to avoid the over-oxidation of the desired aldehyde into the undesired acid in the reaction. It was shown that the control of the buffer concentration and the addition of a small amount of EtOH in the reaction medium were key to achieving this goal. Thus, in the chemo-enzymatic process, the Wacker oxidation step was performed using palladium(II) trifluoroacetate [Pd(TFA)2] and iron(III) in water/acetonitrile (95/5% v/v). Then the reaction mixture was diluted with phosphate buffer, the pH was adjusted to 7.4 and the mixed-culture with the three biocatalysts was added. Moderate yields (7–55%) of the desired aldehydes were obtained after 24 h.
2.3. Synthesis of aldoximes
Aldoximes are important molecules with a wide range of applications in medicine, industry and analytical chemistry. In addition, they are very useful and versatile intermediates in synthetic organic chemistry to prepare oximes, nitriles, amides, etc.153 Preparation of aldoximes by a one-pot process from primary alcohols is of special interest since habitually aldehydes exhibit low stability, making it challenging to prepare aldoximes in high yields. Molinari et al.154 developed a chemo-enzymatic cascade process in a concurrent mode for the synthesis of aldoximes from primary alcohols in aqueous media. The process combines the oxidation of primary alcohols by acetic acid bacteria dehydrogenases with the subsequent condensation of the aldehyde formed with hydroxylamine chlorohydrate (Scheme 32). In the enzymatic oxidation, the reaction kinetics has to be considered since the possible over oxidation of the aldehyde formed to the corresponding carboxylic acid can also occur. Using 2-phenylethanol as the model substrate three strains of acetic acid bacteria (Acetobacter sp. MIM 2000/61, Gluconobacter oxydans DSM 2343 and Asaia bogorensis SF2.1.) were tested in the oxidation step, showing that only Gluconobacter oxydans was mainly selective towards the corresponding aldehyde, while Acetobacter sp. and Asia bogorensis were fully selective towards the corresponding carboxylic acid. However, when the chemo-enzymatic process was performed in a concurrent mode (i.e. the biooxidation was performed in the presence of hydroxylamine) the three strains gave as the main product the aldoxime. These results suggest that the intermediate aldehyde (phenylacetaldehyde) reacts faster with the hydroxylamine than the oxidation to the acid form. It was found that high concentrations of hydroxylamine inhibit the enzymatic oxidation, therefore, by optimizing the reaction conditions, the chemo-enzymatic process could be applied to the synthesis of a variety of aldoximes from phenyl-alcohol derivatives in moderate to high yields (50–96%).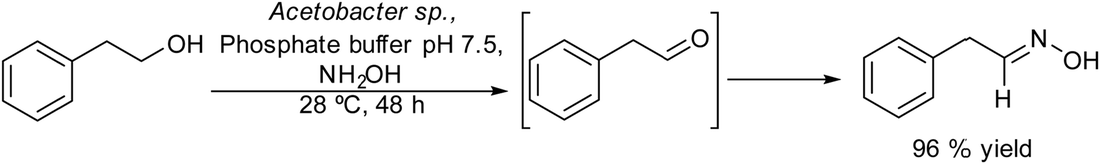 | ||
| Scheme 32 Synthesis of aldoximes from alcohols by combining an enzymatic oxidation and a reaction with hydroxylamine in a concurrent mode. | ||
2.4. Synthesis of nitriles
Nitriles are important chemicals, and particularly, long-chain alkyl nitriles have attracted interest as solvents and as intermediates for amines in the surfactant field.155 The conventional methods to synthesize nitriles are based on substitution or addition reactions with hydrogen cyanide or cyanides; however, an important drawback is the need for stoichiometric amounts of highly toxic cyanide.156 Other alternative methods are based on ammoxidation157 and dehydration of amides;155 however, the main drawback of these processes is the high temperature required, which affects the selectivity of the process and economic cost.Recently, Gröger et al.158 developed an alternative chemo-enzymatic approach to produce nitriles by dehydration of aldoximes using aldoxime dehydratase. The starting aldehyde, nonanal, was first obtained through a hydroformylation step of octene in aqueous media using a rhodium complex as a catalyst. Then, the required amount of the aldehyde was taken from the formylation reactor and subsequently condensed with hydroxylamine in aqueous media to produce quantitatively the corresponding aldoxime. To avoid the inhibition effect of the residual hydroxylamine on the enzyme, the former was subjected to in situ thermal decomposition (100 °C, 16 h) and the recombinant whole-cell expressing aldoxime dehydratase was added to the reaction media (Scheme 33), achieving the corresponding nitrile in 67% yield.
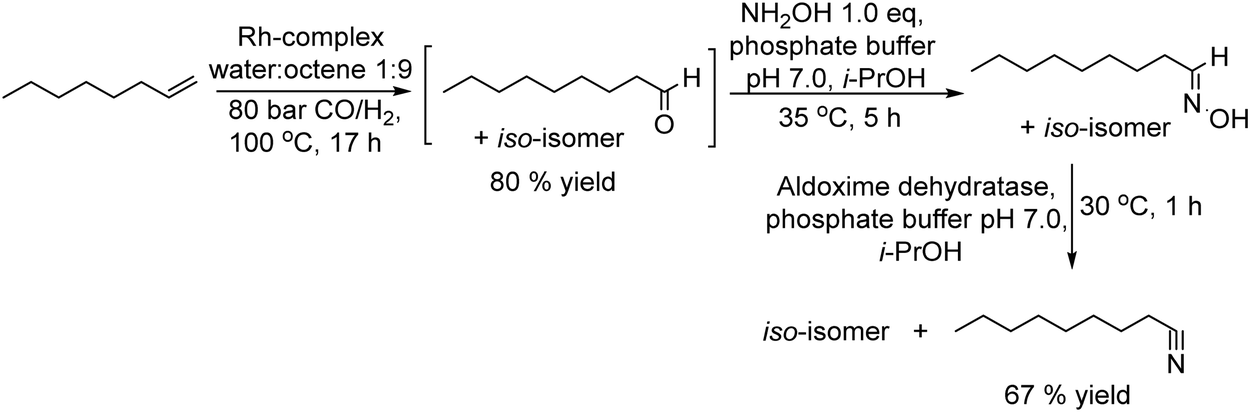 | ||
| Scheme 33 Synthesis of nitriles from 1-alkenes by combining a hydroformylation step, a reaction with hydroxylamine and enzymatic dehydration of the aldoxime intermediate. | ||
A related chemo-enzymatic cascade process to synthesize aliphatic nitriles from alcohols was reported by the same group.159 In this case, the aliphatic alcohol was converted into the aldehyde through a TEMPO-catalysed oxidation using hypochlorite as the oxidation agent and an aliphatic nitrile as an organic solvent. After oxidation completion, a hydroxylamine hydrochloride solution in water was added and stirred at room temperature up to the complete conversion of the aldehyde to the aldoxime. After phase separation, the organic phase bearing the aldoxime was directly used for the biocatalytic step using a supported aldoxime dehydratase, leading to the nitriles.
These chemo-enzymatic processes, which correspond to a formal hydrocyanation of alkenes, allow access to nitriles in moderate yields and avoid the use of harmful cyanide derivatives, while occurring under mild reaction conditions.
Halogenation of organic substrates using halogenases that use benign inorganic halide salts as the halogen sources represents an environmentally friendly alternative to conventional methods that often require the use of hazardous chemicals and harsh reaction conditions. Taking advantage of these enzymes, Micklefield et al.160,161 developed a chemo-enzymatic method for the C–H functionalization of aromatic moieties of aryl and heteroaryl derivatives to nitrile groups, in a highly regioselective manner, by integrating a flavin adenine dinucleotide (FAD)-dependent halogenase (Fl-Hal) with Pd-catalysed cyanation. For instance, starting from indole scaffolds, the chemo-enzymatic cascade performed in a sequential mode involves as a first step the regioselective halogenation of the aromatic ring catalysed by the halogenase followed by palladium chemocatalyzed CN insertion using potassium ferrocyanide (K4[Fe(CN)6]) as a non-toxic cyanide source (see Scheme 34). The enzyme was immobilized in the form of cross-linked enzyme aggregates (CLEAs) that showed higher halogenation efficacy, while improving biocatalyst stability and tolerance to higher substrate loadings. Moreover, enzyme directed evolution enabled the access to the C5, C6 and C7 positions of the indole scaffold regioselectively. Additionally, the encapsulation of the Fl-Hal–CLEA in a molecular weight cut-off (MWCO) membrane (12–14 kDa) enabled the easy separation of the enzyme from the reaction media before the addition of cyanation components and recycling the enzyme. Moreover, to take advantage of the versatility of the nitrile group, the cascade was extended to the synthesis of amides and carboxylic acids through the enzymatic hydration and hydrolysis of nitriles generated in situ, using nitrile hydratase and NITR enzymes within E. coli cells, respectively. This chemo-enzymatic process represents a valuable strategy to introduce nitrile groups in a regioselective manner, giving access through further transformation to a variety of important functionalities such as amines, oximes, amides, etc.
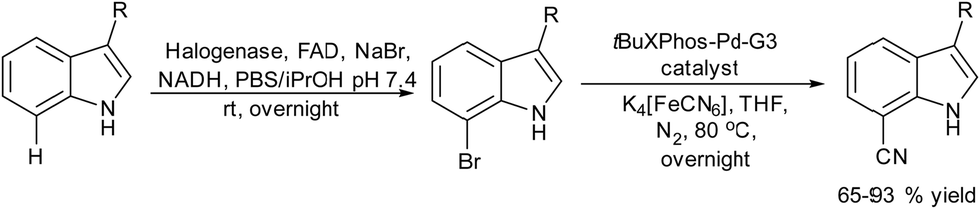 | ||
| Scheme 34 Synthesis of cyano-substituted indole derivatives by combining enzymatic halogenation and cyano group insertion. | ||
2.5. Synthesis of chiral epoxides
Epoxides are attractive synthons used in organic synthesis due to their chemical versatility.162 Conventional methods for the synthesis of chiral epoxides are based on kinetic resolutions of racemic epoxides via epoxide-hydrolase catalysed enantioselective hydrolysis and chemical asymmetric epoxidation of alkenes, using the Sharpless, Jacobsen and Yian Shi methods.163 However, these methods require expensive catalysts that work at low substrate/catalyst ratios, with limited efficiency and variable degrees of enantioselectivities. To overcome these drawbacks direct biocatalyzed stereospecific epoxidation of alkenes by monooxygenases (e.g., cytochrome P450s) represents an interesting alternative.164 Taking advantage of monooxygenases, Zhao et al.165 reported the synthesis of chiral aryl epoxides by combining Ru-complex catalyzed cross-alkene metathesis of stilbene and symmetric alkenes with biocatalyzed stereoselective epoxidation (see Scheme 35). Optimization of reaction conditions (Ru catalyst, substrate loading, organic cosolvent, reaction volume, buffer solution concentration, etc.) and enzyme engineering were required to perform the reaction in a concurrent mode in the aqueous phase to achieve moderate yield and high enantioselectivity of the aryl epoxide that can be regenerated by the glucose dehydrogenase (GDH)/glucose enzymatic system. In this study, the authors used a glucose dehydrogenase (GDH) to recycle NADPH continuously by reduction of NADP+ with concomitant conversion of glucose to gluconic acid. The cascade process gives the E form of the corresponding epoxide with an enantiomeric excess of 94% and moderate yield (50%). The strategy allows constructing a variety of structurally different chiral epoxides in a direct way and under very mild reaction conditions.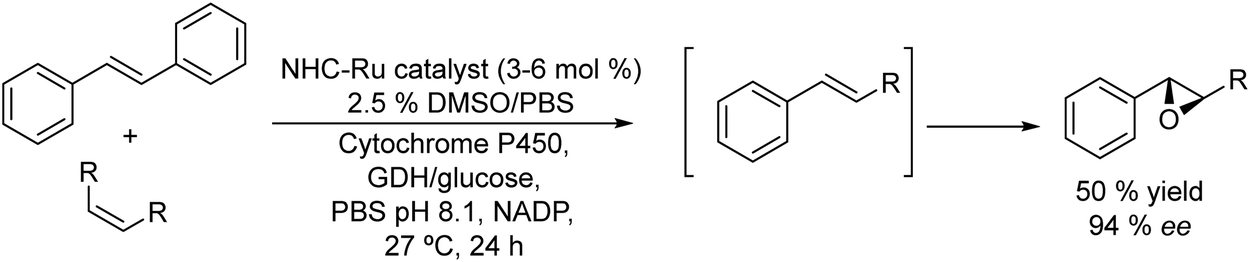 | ||
| Scheme 35 Synthesis of chiral aryl epoxides from alkenes in a concurrent mode by combining a metathesis reaction with biooxidation. | ||
Another efficient chemo-enzymatic method for the synthesis of enantiopure epoxides starts from alpha-chloroketones.166 In this study, the bioreduction of alpha-chloroketones using a newly developed ketoreductase (KcDH) (overexpressed in Escherichia coli) was combined with the base catalysed ring closure of the intermediate halohydrin (see Scheme 36). After the biocatalyzed reduction of chloroacetophenone and chloro-1-phenoxy-2-propanone into the enantiomerically pure halohydrin intermediates, the corresponding chiral epoxides were easily obtained by adding an excess of NaOH while maintaining their ee. For instance, the (R)-styrene oxide was obtained in nearly enantiopure form (98% ee) and 86% yield. As a byproduct, the corresponding vicinal diol resulting from the epoxide opening was obtained in yield lower than 20%.
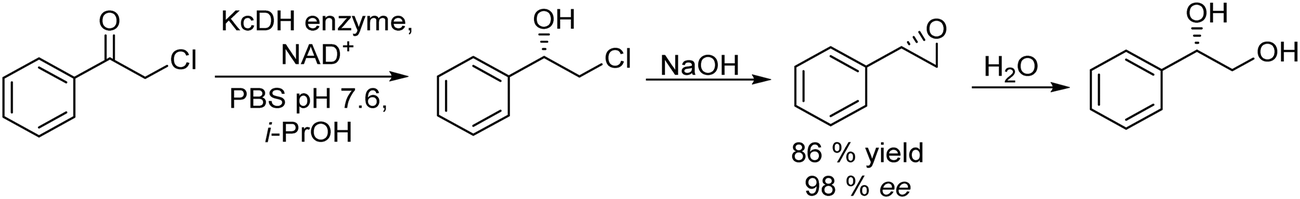 | ||
| Scheme 36 Synthesis of chiral epoxides from α-haloketones by combining bioreduction and ring closure. | ||
Another efficient chemo-enzymatic method to produce chiral epoxides and a variety of versatile chiral halohydrins has been recently reported by Gotor-Fernandez et al.167 Taking advantage of the recently reported compatibility of ADHs with the N-heterocyclic carbene gold(I) complex (1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene][bis(trifluoromethanesulfonyl)imide]gold-(I)) (IPrAuNTf2),79 the authors designed a cascade in a concurrent mode to synthesize chiral halohydrins, which involves the gold-catalyzed regioselective hydration of haloalkynes, followed by the stereoselective bioreduction of the corresponding α-halomethyl ketone intermediates into chiral halohydrins using alcohol dehydrogenases (ADHs). The hydration step was thoroughly investigated in order to conduct the chemo-enzymatic process in a concurrent mode, and under optimized reaction conditions, a variety of chiral halohydrins in yields ranging from 65–86% (isolated yields) and >98% ee were obtained. The success of the process was due to the thermodynamically favored reduction of the α-haloketone intermediate, and therefore a small excess of isopropanol (acting as a co-factor recycling agent and as a co-solvent) was required, thus limiting the formation of byproducts, which are produced when using larger quantities of isopropanol. Finally, the chemo-enzymatic synthesis of the chiral halohydrin was integrated with cyclization into a chiral styrene oxide that was successfully accomplished by the addition of NaOH to the reaction medium (Scheme 37).
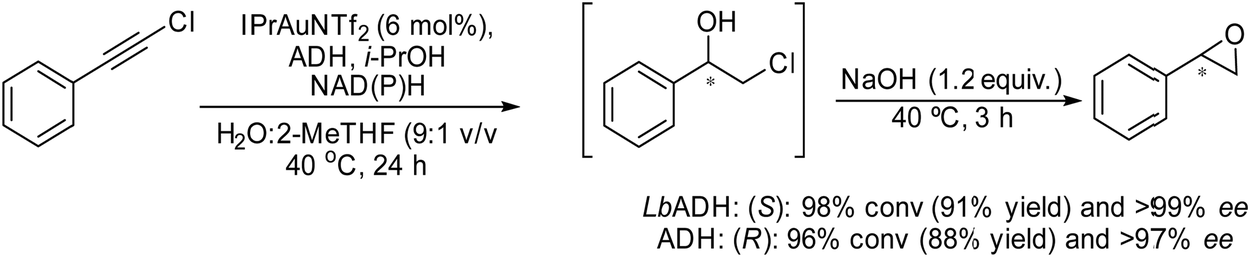 | ||
| Scheme 37 Synthesis of chiral epoxides from haloalkynes by combining a chemocatalyzed hydration step and bioreduction in a concurrent mode followed by cyclization of the chiral halohydrins. | ||
2.6. Synthesis of carboxylic acids and esters
Carboxylic acids and their derivatives, particularly chiral compounds, are important scaffolds in organic synthesis for the preparation of a wide variety of pharmaceuticals, agrochemicals, flavors, etc. For instance, the chiral monoester, (1S,2R)-2-(methoxycarbonyl)-4-cyclohexenecarboxylic acid, is a strategic intermediate for the synthesis of drug candidates for the modulation of chemokine receptor activity.168,169 Classically, the monoester is produced in two separate steps that involve first the esterification of (1R,2S)-4-cyclohexene-1,2-dicarboxylic acid (Scheme 38) with methanol in the presence of a strong acid as a catalyst. The dimethyl ester formed (meso-diester) is subsequently isolated and purified and then selectively hydrolysed by an esterase into the desired monoester in ≥99.5% enantiomeric excess.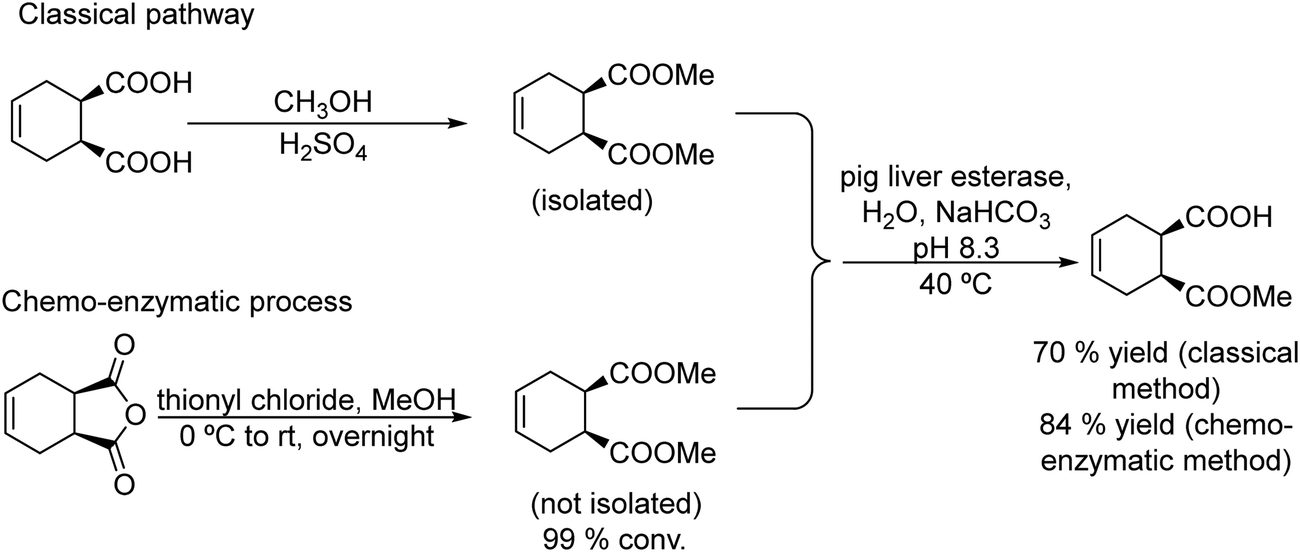 | ||
| Scheme 38 Synthesis of (1S,2R)-2-(methoxycarbonyl)-4-cyclohexenecarboxylic acid through the classical and chemo-enzymatic processes. | ||
An improved chemo-enzymatic process for the synthesis of this high value monoester was developed by Wardenga et al.170 The method involves first a chemical esterification starting from meso-phthalic anhydride to produce the meso-diester followed by the selective enzymatic hydrolysis of one ester group catalyzed by a pig liver esterase. Optimization of the esterification step was performed, showing that the esterification of the meso-anhydride with methanol in the presence of thionyl chloride, instead of sulphuric acid, as the catalyst, was fully selective towards the meso-diester. Then, the reaction conditions were adjusted and esterase was added to perform the biocatalytic step (see Scheme 38). Under these conditions, the desired monoester was obtained in 84% yield (instead of 70% obtained using the classical procedure) and ≥99.5% ee. Several advantages are associated with the one-pot protocol, such as the use of the meso-anhydride as the starting reagent (cheaper than the diacid) and the overall yield improvement of the desired product, while the time-consuming workup steps and waste production can be reduced.
Enantiopure 2-methyl-substituted carboxylic acids are commonly used as pharmaceutical compounds, building blocks, flavors and fragrances.171,172
Recently, Kourist et al.173 reported a chemo-enzymatic cascade synthesis to produce optically pure short-chain 2-methylalkanoic acids.171,172 The chemo-enzymatic strategy combined the enzymatic asymmetric decarboxylation of methylvinylmalonic acid derivatives and the chemical reduction of the non-activated CC double bond (Scheme 39). The decarboxylation step of methylvinylmalonic acid derivatives was performed using engineered arylmalonate decarboxylase (AMDase) variants IPLL and CLGIPL derived from Bordetella bronchiseptica as biocatalysts and tris-HCl buffer at 30 °C. After full substrate conversion into monoacids with high optical purities, the second step, the CC double bond reduction, was performed through the in situ generated diimide. Diimide is a hydrogen donor that can selectively reduce non-activated unsaturated bonds by a concerted hydrogen transfer without producing racemization of the stereocenter. Due to its instability, the diimide has to be generated in situ from its precursor hydrazine by using an oxidation catalyst such a copper salt. The chemo-enzymatic sequential cascade using R- and S-selective AMDase variants produced chiral short-chain 2-methylalkanoic acids in excellent yield and ee (up to 83% isolated yield and 98% ee).
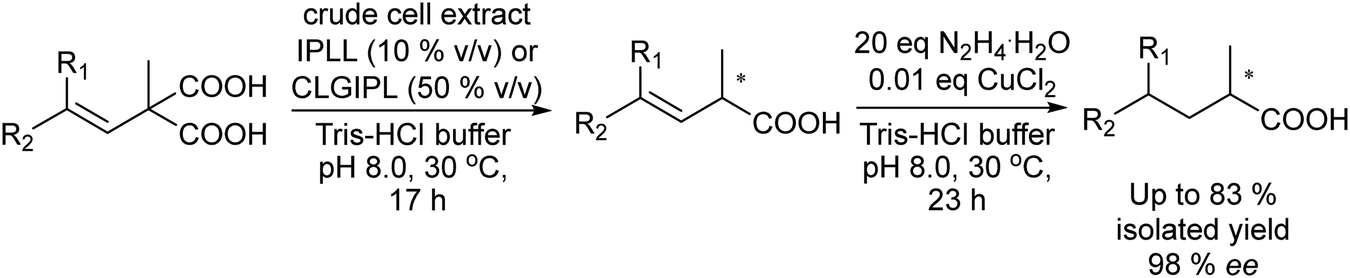 | ||
| Scheme 39 Synthesis of chiral 2-methyl substituted carboxylic acids from methylvinylmalonic acid derivatives by combining biodecarboxylation with CC bond reduction. | ||
This strategy represents an important advancement compared to the conventional methods to prepare these challenging compounds, which is mainly based on kinetic resolutions (inherently limited to 50% maximum yield) or asymmetric hydrogenation of α,β-carboxylic acids, leading to moderate enantioselectivities and lower yields.174,175 Interesting chemo-enzymatic processes performed by combining C–C bond forming reactions with enzymes have also been reported for the synthesis of carboxylic acids. Thus, Gröger et al.176 reported a new synthetic process for the preparation of cyclic malonic acid monoesters, which are of interest as intermediates for the preparation of non-natural amino acids bearing a quaternary carbon center. The authors combined a metathesis reaction of unsaturated malonic acid ester derivatives with ester hydrolysis catalyzed by esterases in a cascade process in a sequential mode. Commercially available Ru carbene Grubbs’ II metathesis catalyst, which is able to act efficiently in water,177 was selected for the first step. For instance, excellent conversion of the substrate diethyl 2,2-diallylmalonate into the corresponding cyclopentene diester derivative was obtained under optimized reaction conditions in aqueous media (see Scheme 40). After completion of the metathesis step, the pig liver esterase (PLE) enzyme was added to the reaction media. Interestingly, it was found that the enzyme was compatible with the metal catalyst, and the enzymatic hydrolysis proceeds with high conversion. However, the selectivity towards the desired monoester was moderate due to the subsequent decarboxylation of the monoester formed. Optimization of the solvent composition for the enzymatic step (i.e. using a mixture of water and t-butanol) allowed the suppression of the decarboxylation process, achieving high selectivity towards the monoester. The process was also successfully applied to the synthesis of cyclohexene monoester derivatives (see Scheme 40b).
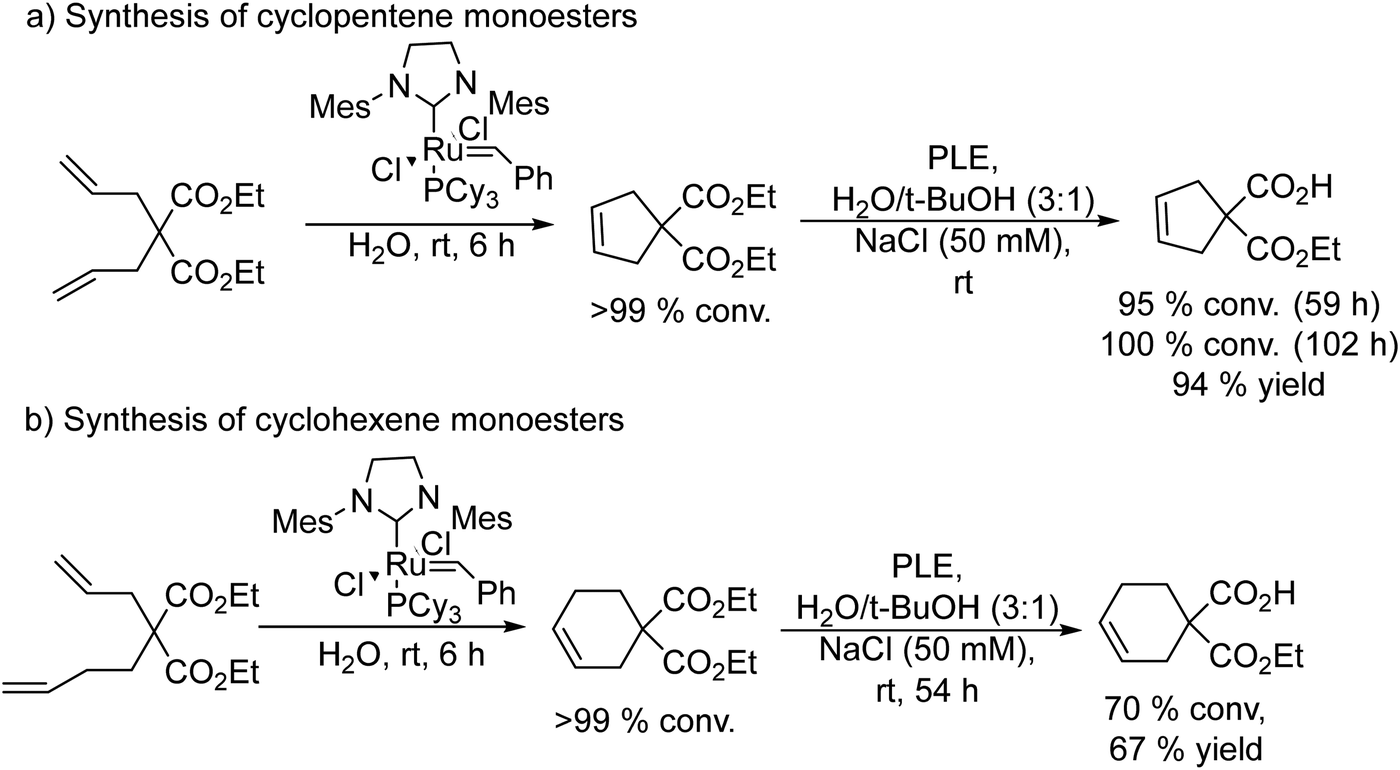 | ||
| Scheme 40 Synthesis of cyclic malonic acid monoesters from unsaturated malonic acid ester derivatives by combining a metathesis reaction with enzymatic hydrolysis. | ||
The process constitutes an interesting straight-forward route to access these types of compounds, avoiding the conventional, multistep and low-selective route starting from dihalogenate alkenes and malonic acids.
Chiral unsymmetrical diesters such as 2-substituted succinate derivatives are important intermediates178–181 in the synthesis of biologically active compounds such as γ-butyrolactone derivatives.182,183 Zhao et al.184 developed a one-pot chemo-enzymatic procedure in a sequential fashion to prepare chiral 2-substituted succinate derivatives. The process comprises rhodium-catalyzed heterocoupling of two diazoesters, which provides the alkene derivative, which is subsequently reduced by an ene-reductase (ER), which occurs with up to 99% enantiomeric excess (Scheme 41).
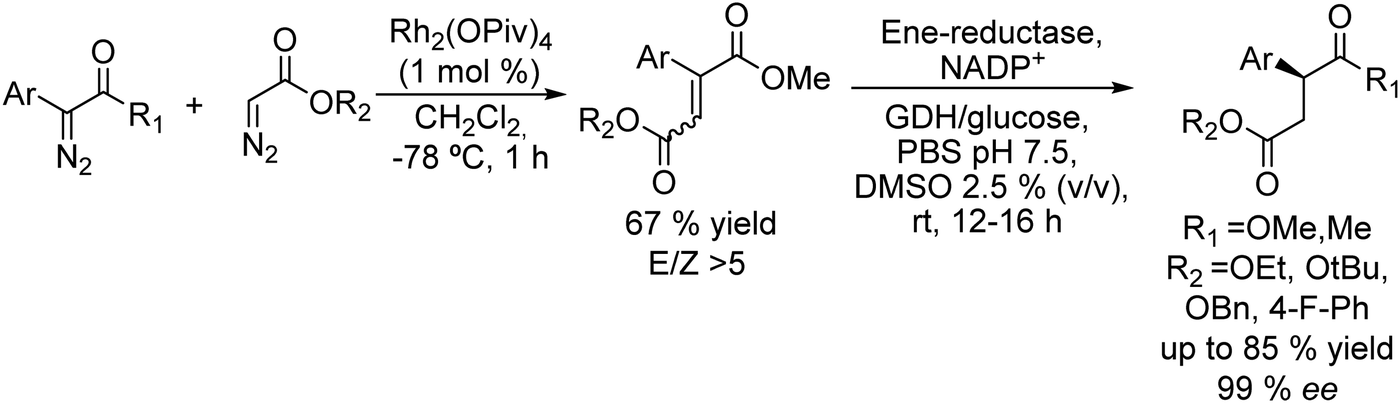 | ||
| Scheme 41 Synthesis of chiral 2-aryl substituted succinate derivatives from diazoesters by combining a diazocoupling reaction and bioreduction. | ||
The authors selected dirhodium(II) tetrakispivaloate [Rh2(OPiv)4] as the catalyst for the diazo coupling of aryldiazo compounds with diazoacetate derivatives, due to its demonstrated high chemo- and stereoselective activity for diazo coupling reactions,185 promoting preferentially the formation of the E-alkene. On the other hand, ene-reductases (ERs) have been shown to be efficient and cost-effective catalysts for the enantioselective reduction of 2-substituted fumaric acid derivatives.186 ERs containing flavin mononucleotides (FMN) require NADPH or NADH as a co-factor to catalyze asymmetric reduction. Interestingly, the enzymatic process only promotes the enantioselective reduction of (E)-2-aryl-substituted dicarbonyl alkenes among a mixture of the E and Z isomers, while occurring efficiently in the presence of the Rh-complex catalyst, being possible to couple both steps in one-pot. Thus, the diazo coupling step was carried out in the presence of [Rh2(OPiv)4] at −78 °C using CH2Cl2 as a solvent to give the corresponding E,Z-alkene. Then, after removing CH2Cl2, the enzymatic system in a buffer solution using DMSO as a co-solvent was added to the reaction mixture and the reduction proceeded at room temperature. Under these conditions, excellent results for the corresponding chiral 2-aryl-substituted succinate derivatives (up to 85% isolated yield and 99% ee) were achieved. The success of the process in terms of high yield and enantioselectivity is attributed to the preferential production of the (E)-alkene in the diazo coupling step along with the selective reduction promoted by the ER of the (E)-alkene in a mixture of (E) and (Z) isomers. Thus, the process allows access to chiral 2-substituted succinate derivatives in higher yields and enantioselectivities than other synthesis methods based on organocatalytic and transition metal-catalyzed asymmetric hydrogenation of prochiral aryl-substituted fumaric (E) acid derivatives.187,188
2.7. Synthesis of amides
Amides represent another class of important scaffolds in organic synthesis, and a variety of structurally different amides can be obtained through chemo-enzymatic processes. For instance, the preparation of primary amines from aldehydes was reported by Sugai et al.189 (Scheme 42). In this process the authors combine in a sequential mode the conversion of aldehydes into nitriles following Fang's procedure190 using NH3 (aq), I2 and DMSO as a solvent (step (i), Scheme 42) with biological action to hydrate the intermediate nitrile (step (ii), Scheme 42) catalyzed by whole cells of Rhodococcus rhodochrous IFO 15564. It is interesting to notice that the biocatalyst used possesses two different activities, the nitrile hydratase that catalyzes the hydration of nitriles into the corresponding amides and amidase that catalyzes the hydrolysis of amides into carboxylic acids. The authors showed that by using DMSO as solvent the amidase activity could be suppressed. Then, a variety of primary amides were obtained in excellent yields starting from the corresponding aldehydes (see Scheme 43) through a one-pot process involving 3 steps: (1) chemical reaction using Fang's procedure in DMSO; (2) neutralization of the excess of I2 by adding Na2S2O3 and adjusting to pH 8 and (3) adding the whole cells of harvested cells of Rhodococcus bacteria in phosphate buffer.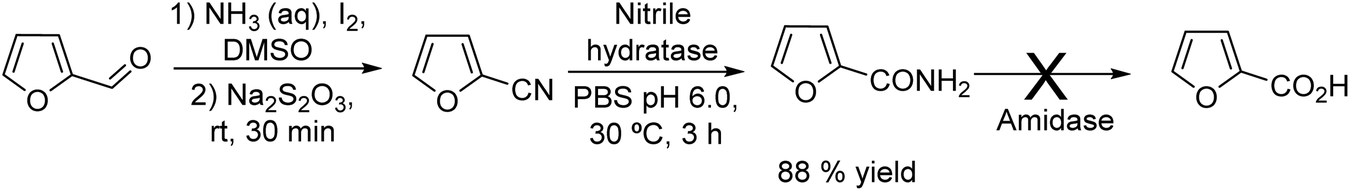 | ||
| Scheme 42 Synthesis of furan-2-carboxamide from furfural by combining the generation of a cyano group with biocatalyzed hydration. | ||
 | ||
| Scheme 43 Carboxamides obtained through the chemo-enzymatic process. | ||
Very recently, Micklefield et al.161 have developed a chemo-enzymatic method for the synthesis of amides starting from nitriles. The process involves as a first step the hydration of nitriles catalyzed by a nitrile hydratase followed by an Ullmann-type N-arylation with aryl iodide catalyzed by copper using trans-N,N′-dimethylcyclohexane-1,2-diamine as the ligand (Scheme 44).
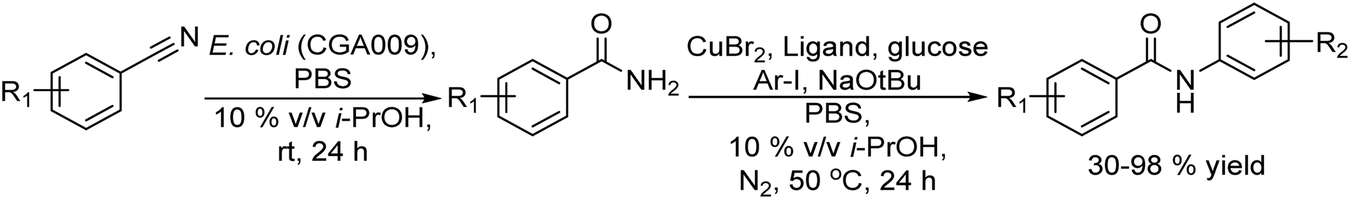 | ||
| Scheme 44 Synthesis of amides by combining biocatalyzed hydration of nitriles with Ullman type arylation. | ||
The authors explored a wide range of reaction conditions and several parameters were optimized such as the enzyme, the form that the enzyme should be present (purified or directly inside the cells), ligand, temperature, amount of catalyst, etc. Under optimized reaction conditions (see Scheme 44) and working in a sequential mode in aqueous buffer supplemented with isopropanol, a variety of aromatic, heterocyclic and aliphatic amides (more than 50 examples), including drug molecules such as periciazine and probenecid, were obtained in good yields. Moreover, the authors showed that the integrated reactions are easily scalable, and the addition of a surfactant (TPGS-750-M), which provides micellar organo-compartments for the chemocatalyzed step, significantly increases the productivity of the process at very high substrate concentrations (up to 1200 mM), which are beyond of the scope of the enzyme. Compared with conventional amide synthesis, this integrated one-pot hydration/arylation cascade presents important advantages such as improvements in space–time yields, does not require protecting groups and circumvents harmful reagents. β-Hydroxyamides are an essential class of compounds in organic synthesis, specifically useful for the preparation of a variety of heterocycles such as oxazolidinones, azetidines, 1,4-diazepanes, or β-lactams,191 and the access to these types of compounds is usually accomplished by hydration of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of β-hydroxynitriles. The chemo-enzymatic synthesis of enantiopure β-hydroxyamides from β-ketonitriles was reported for the first time by González-Sabín et al.192 through a hydration/bioreduction cascade process in aqueous medium occurring in a concurrent mode. The nitrile hydration performed using a commercially available Ru(IV) complex and the bioreduction of the keto group with a ketoreductase (KRED-P1-A0) took place simultaneously under mild reaction conditions, providing the corresponding (R)-β-hydroxyamides in high yields and enantioselectivity (Scheme 45). The key to the success is attributed to the use of ruthenium(IV) catalysts, which exhibit higher compatibility with enzymes than ruthenium(II) complexes. Interestingly, the chemo-enzymatic process could be applied to prepare in very high yields the enantiopure alkaloids (S)-(+)-tembamide and (R)-(−)-aegeline through a simple synthetic sequence (Scheme 46). Interestingly, an alternative method to produce enantiopure β-hydroxyamides from β-ketonitriles is based on two-enzymatic steps catalyzed by a double enzymatic catalytic system composed of a ketoreductase (KRED) and a nitrile hydratase derived from whole cells of R. rhodochrous.193 However, although the ee is very high (>99%) the final yield of the target compound is considerably lower than that achieved through the chemo-enzymatic process.
N bond of β-hydroxynitriles. The chemo-enzymatic synthesis of enantiopure β-hydroxyamides from β-ketonitriles was reported for the first time by González-Sabín et al.192 through a hydration/bioreduction cascade process in aqueous medium occurring in a concurrent mode. The nitrile hydration performed using a commercially available Ru(IV) complex and the bioreduction of the keto group with a ketoreductase (KRED-P1-A0) took place simultaneously under mild reaction conditions, providing the corresponding (R)-β-hydroxyamides in high yields and enantioselectivity (Scheme 45). The key to the success is attributed to the use of ruthenium(IV) catalysts, which exhibit higher compatibility with enzymes than ruthenium(II) complexes. Interestingly, the chemo-enzymatic process could be applied to prepare in very high yields the enantiopure alkaloids (S)-(+)-tembamide and (R)-(−)-aegeline through a simple synthetic sequence (Scheme 46). Interestingly, an alternative method to produce enantiopure β-hydroxyamides from β-ketonitriles is based on two-enzymatic steps catalyzed by a double enzymatic catalytic system composed of a ketoreductase (KRED) and a nitrile hydratase derived from whole cells of R. rhodochrous.193 However, although the ee is very high (>99%) the final yield of the target compound is considerably lower than that achieved through the chemo-enzymatic process.
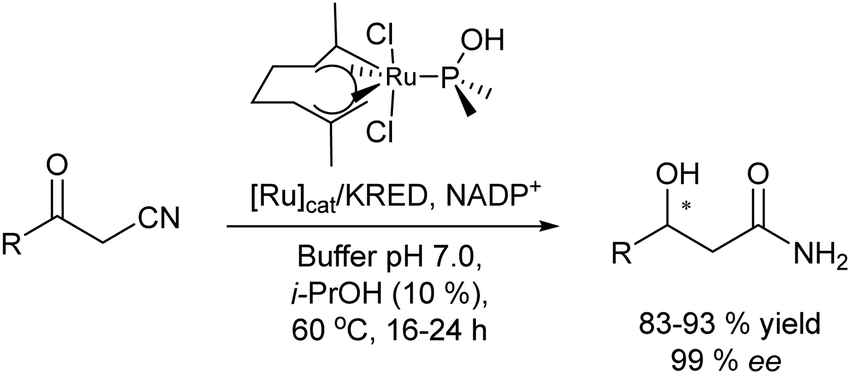 | ||
| Scheme 45 Synthesis of chiral β-hydroxyamides from β-ketonitriles in a concurrent mode by combining bioreduction with chemocatalyzed nitrile hydration. | ||
 | ||
| Scheme 46 Enantiopure alkaloids obtained through the chemo-enzymatic process. | ||
Cordova et al.194 reported the integration of heterogeneous metal catalysis with heterogeneous enzymatic catalysis for the one-pot sequential synthesis of optically active and biologically important amines and amides (such as nonivamide and other capsaicinoid derivatives) starting from aldehydes, ketones or alcohols. An example of the chemo-enzymatic process for the synthesis of nonivamide (a food additive) is presented in Scheme 47. The process involves the Pd catalyzed oxidation of 4-(hydroxymethyl)-2-methoxyphenol into the aldehyde derivative followed by the reductive amination with ammonium formate affording a primary amine. In the last step, a lipase (CALB) catalyzes the coupling of the amine with the corresponding carboxylic acid into amide. Interestingly, all steps were performed in the same solvent (toluene) and at the same temperature (80 °C), taking advantage of the lipophilicity of the selected lipase. The authors developed two heterogeneous chemocatalysts based on Pd immobilized on aminopropyl-controlled pore glass (Pd0-AmP-CPG) and over aminopropyl-mesocellular foam (Pd0-AmP-MFC), which were highly selective for the alcohol oxidation and reductive amination respectively. Interestingly, the Pd catalyst accelerates the decomposition of formate/formic acid, which inhibits the catalytic activity of the lipase, offering the possibility to perform suitably the chemo-enzymatic process.
 | ||
| Scheme 47 Synthesis of nonivamide by combining alcohol oxidation, reductive amination and bioamidation. | ||
Combination of C–N bond forming reactions with enzymes to produce amides was reported by Reek et al.195 The cascade process in a concurrent mode involves Pd catalyzed coupling between an aryl halide and an amine (Buchwald–Hartwig coupling) and lipase catalyzed amidation (Scheme 48).
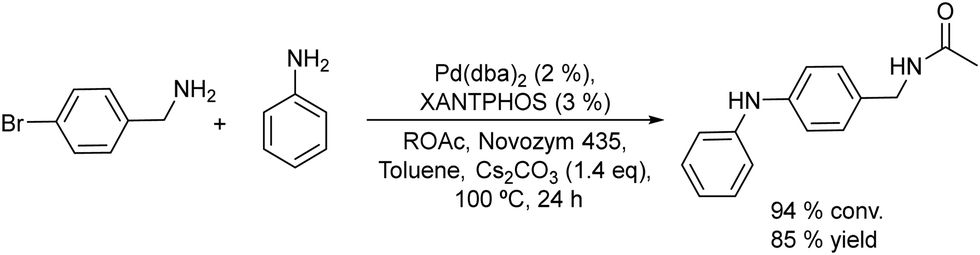 | ||
| Scheme 48 Synthesis of amides in a concurrent mode by combining a C–N bond forming reaction (Buchwald–Hartwig coupling) with bioamidation. | ||
The authors showed that the amidation reaction of 4-bromobenzylamine with ethyl or methyl acetate as the acyl donor proceeded satisfactorily (100% conversion) using the resin bound lipase (Novozym 435®) as a catalyst in the presence of the reagents required for Pd-catalyzed coupling (Pd salts, phosphorus ligands, and Cs2CO3 as a base) in toluene at 100 °C, giving the corresponding 4-bromobenzyl acetamide. Due to the robustness of the lipase under these reaction conditions, the cascade process could be optimized using different Pd ligands and acyl donors. It was found that using methyl acetate as the acyl donor and by performing the cascade process in a concurrent mode there was a synergistic effect between both catalytic processes, leading to higher performances than those achieved when the process was performed in separate steps. This behavior was explained by the presence of small amounts of CsOMe coming from the methanol released in the amidation step. The generation of the methoxide species, which are more basic and soluble in toluene than Cs2CO3, increases the amination rate, thus increasing the performance of the process.
Interestingly, the concept was extended to two other Pd-catalyzed coupling reactions such as the Suzuki–Miyaura (Scheme 49a) and Sonogashira (Scheme 49b) reactions. In both cases the conversion of 4-bromobenzylamine was completed after 20 h, achieving high selectivity towards the final cascade products (70–89%).
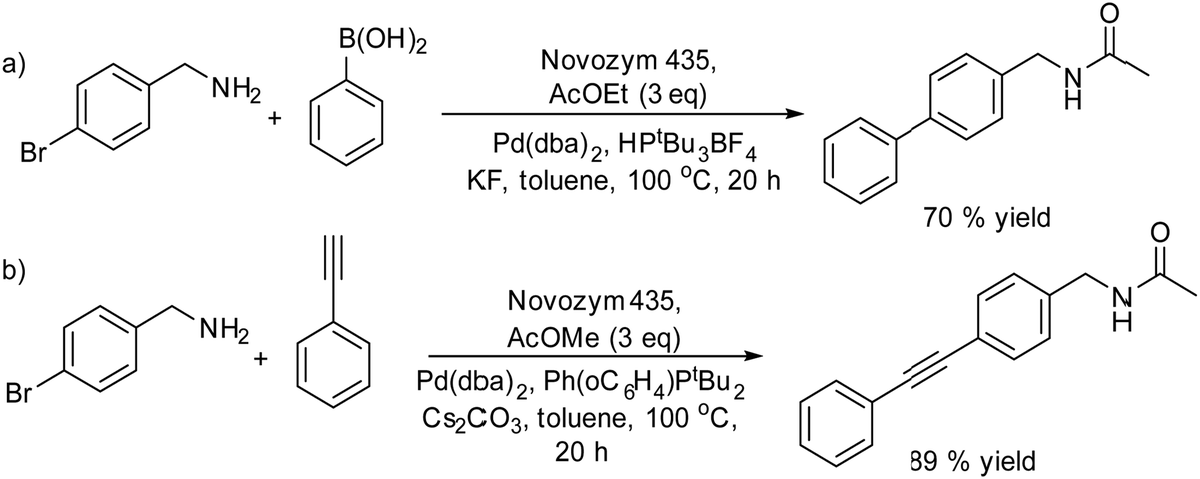 | ||
| Scheme 49 Synthesis of amides by combining a Suzuki reaction (a) or a Sonogashira coupling reaction (b) with biocatalyzed amidation in a concurrent mode. | ||
2.8. Synthesis of amino acids
The enantiopure amino acids (AAs) are useful intermediates in the synthesis of medicines, plant protection products and fine chemicals; for example, tryptophan is an essential chiral AA that plays a key role in the metabolic processes of the human body and can only be produced by microorganisms or plants.196,197Enantiopure amino acids are generally produced using asymmetric reactions that produce high amounts of wastes. To overcome this drawback, Sheldon et al.198 studied a chemo-enzymatic one-pot procedure taking as the model reaction the synthesis of L-alanine, starting from methyl 2-acetamidoacrylate. The reaction involves the asymmetric hydrogenation of 2-acetamidoacrylate using an immobilised Rh-complex followed by hydrolysis of the chiral amidoester intermediate by using an aminoacylase (AMA) (Scheme 50).
 | ||
| Scheme 50 Synthesis of L-alanine from methyl 2-acetoamidoacrylate by combining organocatalyzed asymmetric hydrogenation with enzymatic hydrolysis. | ||
The chemocatalyzed step was first optimized by testing different Rh-complexes anchored on a high area mesoporous aluminosilicate (ALTUD-1) under different reaction conditions and in different solvents. Results showed that the supported Rh-complex (3) (Scheme 51) was a very active, selective and stable chemocatalyst for the hydrogenation of 2-acetamidoacrylate in aqueous solution, achieving 100% conversion with 95% ee, after 1 h reaction time. The high stability against the leaching of the supported Rh-complex (3) in highly polar solvents such as water was attributed to the presence of more hydrophobic ligands compared with the Rh-complexes 1 and 2. For the biocatalyzed step two different acylases derived from Aspergillus melleus (AM) and the porcine kidney (PK) were selected. Both acylases are active for hydrolysing the amide group after the ester group is removed, since they require a terminal carboxylate group for binding with N-acyl-L-amino acids. Advantageously, both acylases were able to hydrolyse the ester group affording N-acetylalanine, which is subsequently converted into the desired L-alanine. In the chemo-enzymatic process, after hydrogenation completion, the acylase was added into a concentrated phosphate buffer solution and the reaction proceeded for 24 h. Interestingly, the acylase was highly active in the presence of the Rh-catalyst, while the presence of the chemocatalyst was detrimental for the PK acylase, and filtration of the chemocatalyst prior to the enzymatic process was required to maintain the enzyme activity. Additionally, the authors intended to perform the cascade process in a concurrent mode by adding all reactants and catalysts from the beginning; however, under these reaction conditions a strong inhibition of the process was observed mainly attributed to the detrimental influence of the buffer on the hydrogenation step and the adsorption of the enzyme on the chemocatalyst.
 | ||
| Scheme 51 Rh-complexes tested for reduction of methyl 2-acetamidoacrylate. | ||
2.9. Synthesis of heterocyclic compounds
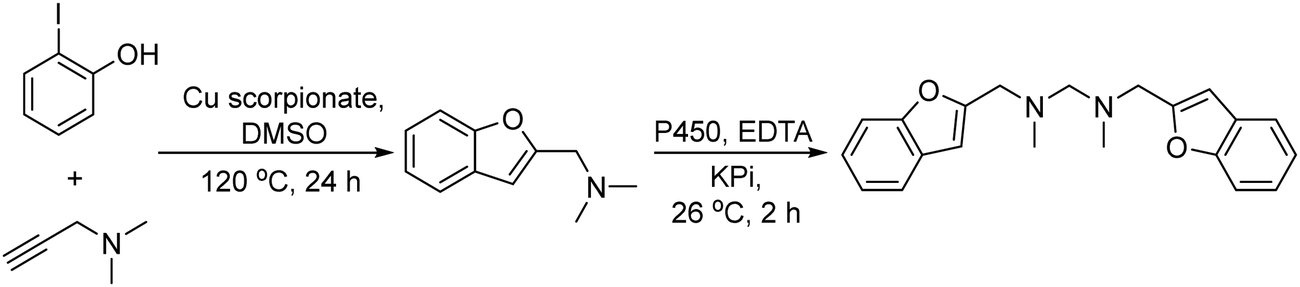 | ||
| Scheme 52 Synthesis of bis(2-substituted benzofuran) derivatives by combining Sonogashira coupling with bio oxidation. | ||
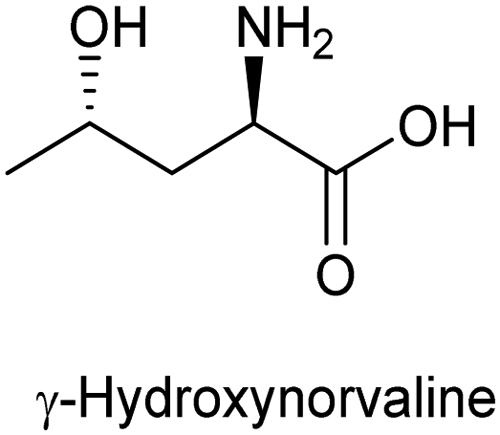 | ||
| Scheme 53 γ-Hydroxynorvaline. | ||
Stereoselective syntheses of α-amino-γ-butyrolactones are mainly based on chemo-catalyzed processes,204 which require auxiliary-assisted multiple steps and allow access to only one or two of four possible stereoisomers.205,206 Faber et al.207 developed a protocol that allows access to the four possible γ-butyrolactone diastereoisomers by sequential construction of the stereogenic centers. This was accomplished by combining an asymmetric Mannich-type reaction catalyzed by (D), or (L)-proline followed by a stereoselective biocatalytic reduction with (S, R) ADH (see Scheme 54). For instance, in the synthesis of the α-amino-γ-butyrolactone precursor of γ-hydroxynorvaline, the process involves as the first step the reaction between ethyl 2-oxoetanoate and 4-methoxyaniline to give the corresponding 4-methoxyphenyl (PMP) aldimine. The PMP-protected aldimine reacts with acetone (used also as a solvent) through a Mannich type reaction catalysed by (L)-proline giving the corresponding (S)-aminoketoester (ethyl (S) 2-amino-PMP-4-oxo-pentanoate) with 99% conversion and in optically pure form (ee 99%). After evaporation of acetone, the crude product was dissolved in buffer solution (PBS) and reduced by (S)-ADH in the presence of the co-factor NAD+ and isopropanol as the co-factor regenerating agent, giving the ethyl (2S,4R) 4-hydroxy-2-aminoester. Spontaneous lactonization of this compound was not complete, and in order to achieve the total lactonization of the 4-hydroxy-2-aminoester its transesterification with methanol was required. After this, the diastereomerically pure (3R,5S) amino lactone was obtained in 51% yield with an excellent diastereoselective yield (a dr of 99:1). Starting from this protected γ-butyrolactone, the γ-hydroxynorvaline was obtained in 58% yield after deprotection and lactone ring-opening (Scheme 54).
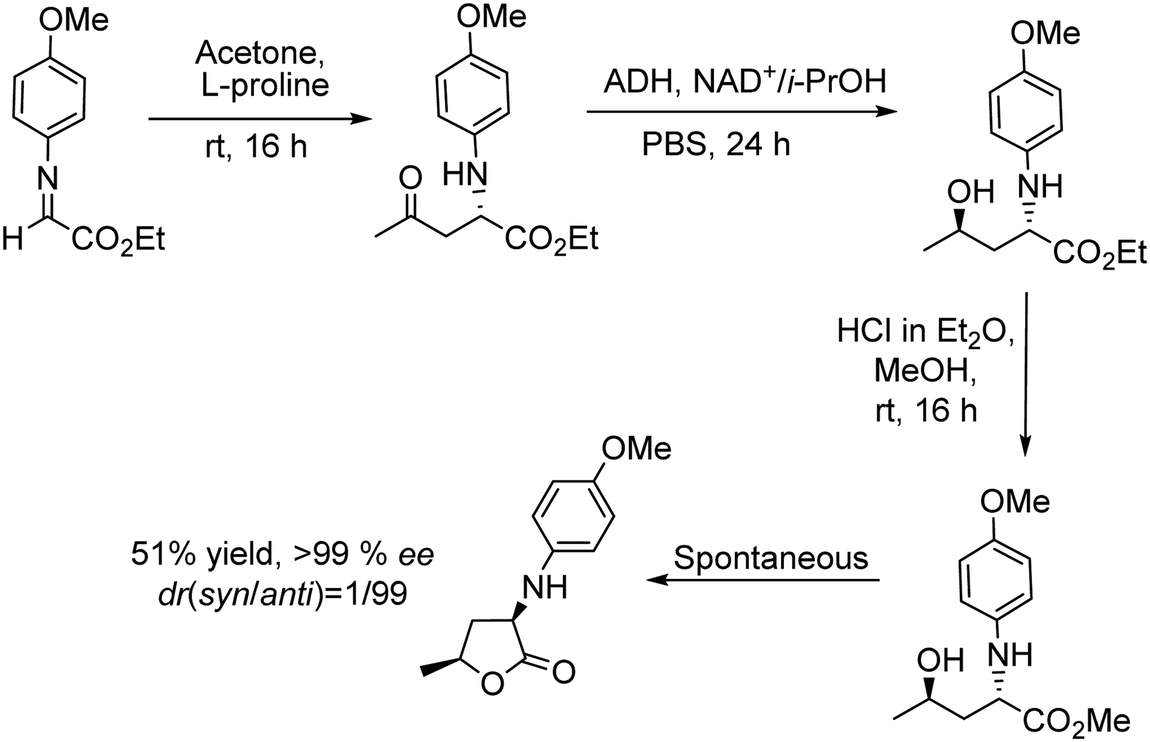 | ||
| Scheme 54 Synthesis of the α-amino-γ-butyrolactone precursor of γ-hydroxynorvaline by combining an organocatalyzed asymmetric Mannich reaction with subsequent transesterification and bioreduction. | ||
The framework of chiral oxy-functionalized indole derivatives can be built through the asymmetric Friedel–Crafts type conjugate addition of α,β-unsaturated aldehydes to an indole followed by oxygenation at the α-position of the carbonyl group. The synthesis is usually performed through two separate steps, resulting in limited yields and enantioselectivities and generation of a high amount of wastes. To overcome these drawbacks Kudo et al.211 developed for the first time the synthesis of oxy-functionalized indole derivatives through a one-pot chemo-enzymatic process. They combined the asymmetric Friedel–Crafts type alkylation of the indole with the bio-oxyamination at the α-position of the aldehyde group of the adduct intermediate. The asymmetric Friedel–Crafts type reaction is performed using a resin supported peptide as a catalyst affording the chiral aldehyde intermediate (see Scheme 55). In the second step, the laccase enzyme was added for the α-oxyamination with TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl). Both steps proceeded smoothly in aqueous media at room temperature and could be applied to the synthesis of several substituted indoles and pyrroles with high enantioselectivities.
 | ||
| Scheme 55 Synthesis of chiral oxy-functionalized indole derivatives by combining symmetric Friedel–Crafts-type alkylation and bio-α-oxyamination. | ||
Chiral hydroxy triazoles have been synthesized by Feringa et al.222 using a smart one-pot chemo-enzymatic process performed in a concurrent mode, where all additives and catalysts were added at the beginning of the reaction. The process involves as the first step the enantioselective azidolysis of aromatic epoxides to 1,2 azidoalcohols catalyzed by the enzyme halohydrin dehalogenase (developed by the authors)223 followed by the [3+2] cycloaddition of the hydroxyazide intermediate to alkynes catalyzed by copper salts (Scheme 56). To develop the process in a concurrent mode the compatibility of the different additives required in both steps was thoroughly investigated. It was shown that the selectivity of the enzyme was preserved in the presence of the different additives required for the click reaction (CuSO4, the reducing agent sodium ascorbate and the MonoPhos ligand), and the chemo-enzymatic process could be performed successfully under optimized conditions. For instance, 2-(4-nitrophenyl)oxirane was 43% converted after 24 h at room temperature into the corresponding triazole with 99% ee, along with the remaining epoxide with 75% ee (Scheme 56). However, the authors showed that the concentration of substrate and the structures of the epoxide and alkyne have a strong influence on the enzyme activity and selectivity.
 | ||
| Scheme 56 Synthesis of chiral β-hydroxytriazoles from aryl epoxides in a concurrent mode by combining enzymatic azidolysis with [3+2] cycloaddition of alkynes. | ||
Better results in the synthesis of chiral β-hydroxytriazoles through a different chemo-enzymatic cascade were achieved by Orden and coworkers.224 In this case, the authors started from α-haloketones, which underwent azidation and subsequent copper-catalyzed azide–alkyne cycloaddition with acetylene derivatives giving the corresponding β-ketotriazoles that are enantioselectively reduced by Rhodotorula mucilaginosa cells into the corresponding (R)-β-hydroxytriazole (Scheme 57). The authors showed that starting from α-chloroacetophenones, and working in a sequential mode, the isolated yields of (R)-β-hydroxytriazoles were 55–65%. However, in a concurrent mode a competitive bioreduction of the starting α-chloroacetophenone into the corresponding chlorohydrine was observed leading to low conversion of the target product. To overcome this drawback, α-bromoketones, which are not prone to be bioreduced into the bromohydrine, were selected as starting reagents. Thus, the cascade process could be effectively accomplished in a concurrent mode, achieving 67–80% isolated yields of the (R)-β-hydroxytriazole derivatives. The fast and complete replacement of the bromine by the azide and the low capability of the enzyme to reduce bromoketones and azidoketones giving undesired products were the main factors responsible for the high yields of (R)-β-hydroxytriazoles achieved.
 | ||
| Scheme 57 Synthesis of chiral β-hydroxytriazoles in a concurrent mode from α-haloketones by combining azidation and subsequent copper-catalyzed azide–alkyne cycloaddition followed by bioreduction. | ||
Structurally different chiral hydroxytriazoles were synthesized by Omori et al.225 following a chemo-enzymatic process in a sequential mode. In this case, the cascade process involves as the first step the enantioselective reduction of azidoacetophenone into the enantiomerically pure azidoalcohol intermediate that is subsequently coupled with alkynes through a click reaction catalyzed by copper salts. The authors performed the enantioselective reduction of azidoacetophenone using carrot bits as a biocatalyst, whose activity is attributed to the endophytic microorganism (Daucus carota) existing in the carrot roots and that previously had shown high levels of enantioselectivity for reducing a variety of prochiral ketones.225 Thus, the reduction of azidoacetophenone was coupled with the click reaction using a variety of alkynes giving the target triazoles in moderate yields with an ee >99%; however, long reaction times were required (Scheme 58).
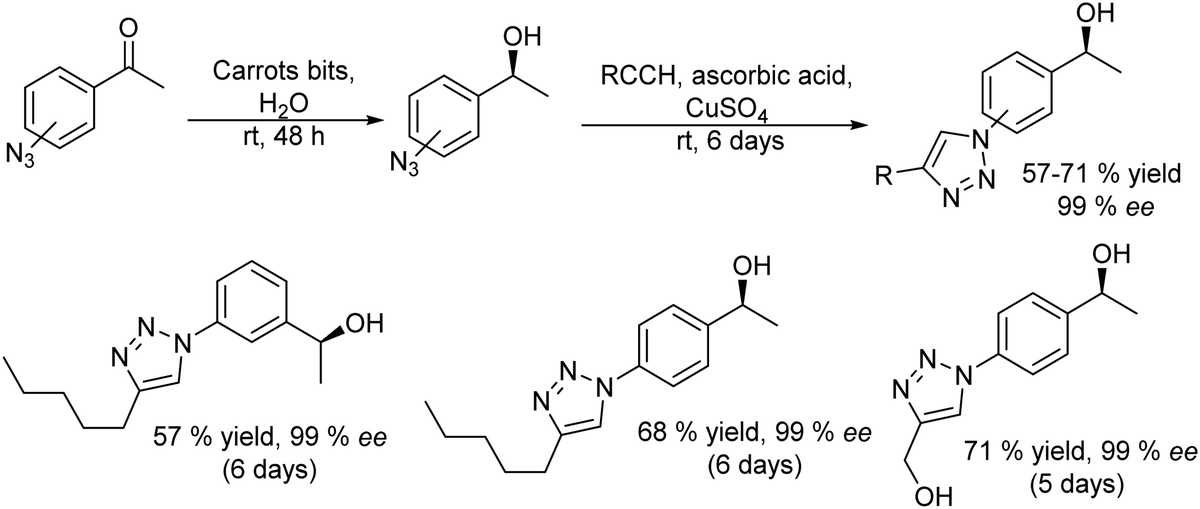 | ||
| Scheme 58 Synthesis of chiral hydroxytriazoles from azidoacetophenone by combining bioreduction with a click reaction. | ||
 | ||
| Scheme 59 Synthesis of iminocyclitols from azidoaldehyde derivatives and dihydroxyacetone by combining biocatalyzed aldol condensation with reductive amination. | ||
Norcoclaurine synthase (NCS) catalyzes the Pictect–Spengler reaction between 3-hydroxyphenethylamines (e.g. dopamine) and a carbonyl compound to form (1S)-THIQs with high stereoselectivity. Moreover, the enzyme has shown a remarkably wide carbonyl compound tolerance, accepting a variety of aldehydes and ketones.238,239 Hailes et al.240 developed a sustainable asymmetric route to produce tetrahydroisoquinoline alkaloids through a one-pot chemo-enzymatic process in a sequential mode, using the enzyme norcoclaurine synthase (NCS), followed by a cyclisation process, to obtain alkaloids with two new heterocyclic rings (Scheme 60). The process combines the Pictect–Spengler reaction that involves a Schiff reaction between a linear methyl carboxylate bearing an aldehyde group and the amine group of the dopamine, followed by a Mannich-type reaction and an electrophilic aromatic substitution giving a tetrahydroisoquinoline intermediate, which after addition of Na2CO3 is cyclized to the target compound. The authors showed that starting from dopamine and methyl 4-oxobutanoate, 74% yield and ee = 95% for (S)-trolline could be obtained in the chemo-enzymatic process. Moreover, the process was successfully extended to other dopamine derivatives and aldehyde derivatives with different chain lengths giving the corresponding THIQs.
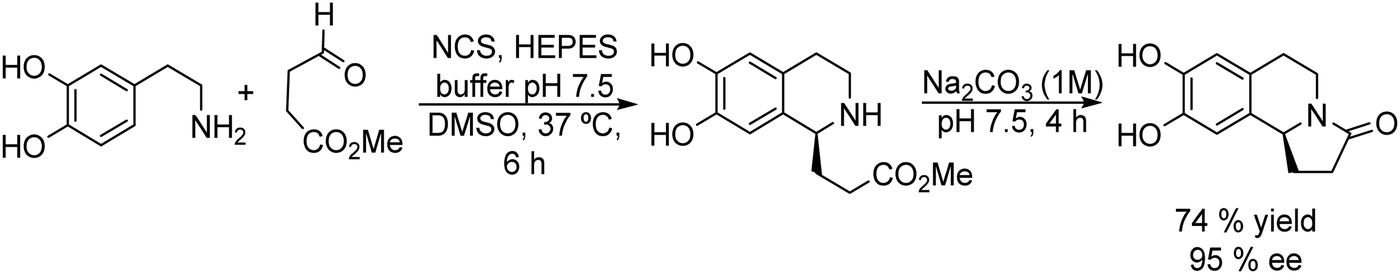 | ||
| Scheme 60 Synthesis of the alkaloid S-trolline from dopamine by combining a biocatalyzed Pictet–Spengler reaction and cyclization. | ||
Also, complex 1,3,4-trisubstituted tetrahydroisoquinolines (THIQs) with three chiral centers were synthesized in a selective manner without intermediate purification by Rother et al.241 through a chemo-enzymatic sequential process in the aqueous phase. These compounds are interesting for the pharmaceutical industry due to the wide scope of activities, such as antitumoral,242 antiparasitic,243 and anticholinergic244 properties. The cascade process involves two biocatalyzed steps combined with a chemocatalyzed path. In the first step the carboligation between 3-hydroxybenzaldehyde and sodium pyruvate in a buffer system was performed using the carboligase EcAHAS-1 derived from Escherichia coli (produced by the authors) as a biocatalyst (Scheme 61a). The produced chiral α-hydroxy ketone intermediate was subjected in the second step to a transamination process with a transaminase (CV2025) (using isopropylamine as the amine donor and PLP as the enzyme co-factor) yielding 3-((1R,2S)-2-amino-1-hydroxypropyl)phenol. Then, after removing the enzymes by ultrafiltration, the 3-((1R,2S)-2-amino-1-hydroxypropyl)phenol is reacted with 2-Br-benzaldehyde through a chemocatalyzed Pictect–Spengler reaction catalyzed by potassium phosphate, yielding the 1,3,4-trisubstituted tetrahydroisoquinoline derivative with three chiral centers. To show the stereoselectivity of the phosphate-catalyzed Pictect–Spengler reaction, different aldehydes were screened, giving the corresponding chiral THIQs with variable success. Interestingly, when the Pictect–Spengler reaction step was performed using norcoclaurine synthase (NCS), instead of potassium phosphate, the THIQs obtained resulted in opposite stereoselectivities at the C1 position, providing access to both possible orientations of the C1 substituent of the THIQs (Scheme 61b).
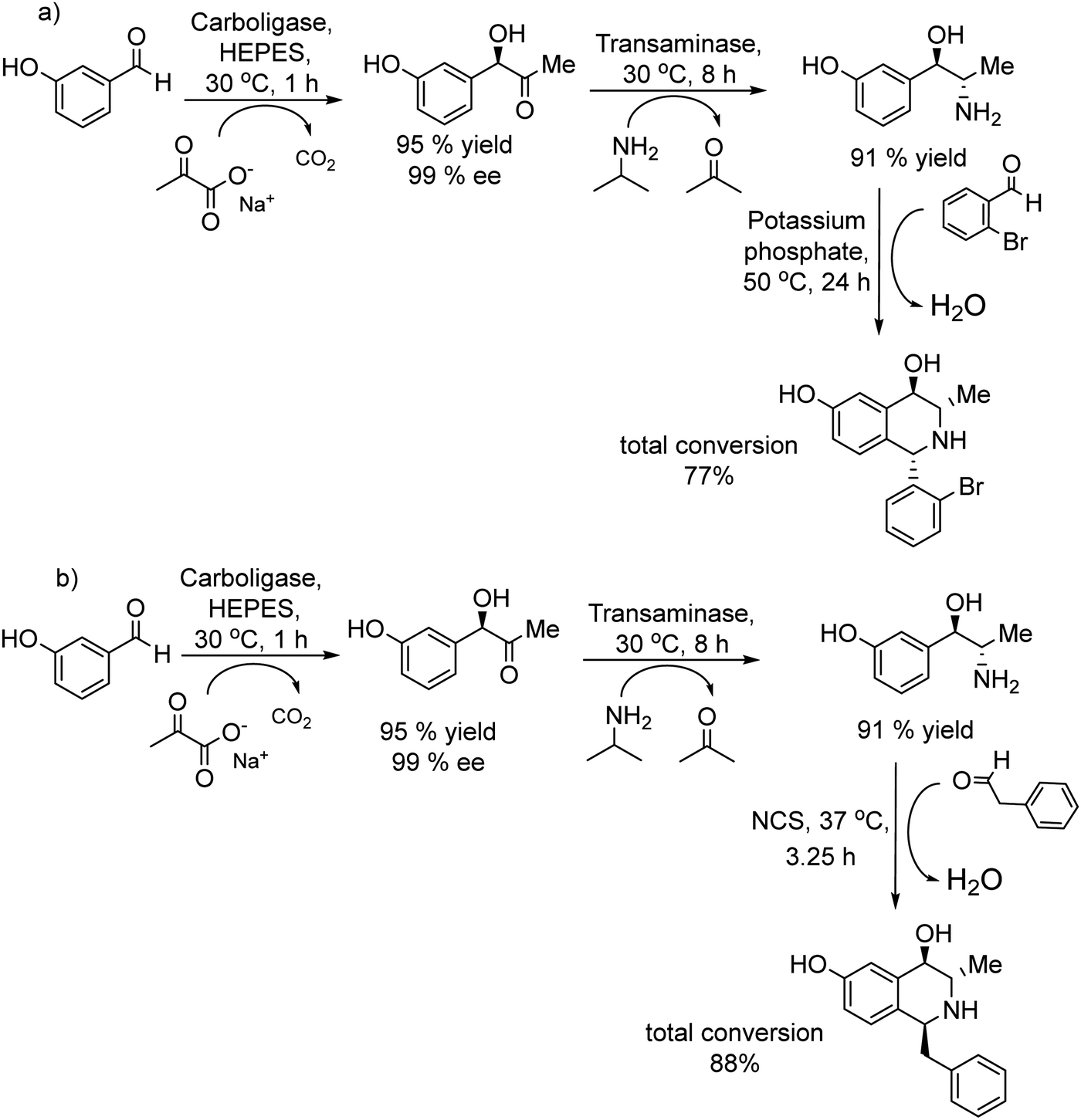 | ||
| Scheme 61 Synthesis of 1,3,4-trisubstituted tetrahydroisoquinolines (THIQs) with three chiral centers: (a) chemo-enzymatic and (b) three step biocatalyzed cascade. | ||
An interesting chemo-enzymatic process giving access to C-1 alkynyl substituted N-alkyltetrahydroisoquinolines was developed by Turner et al.245 by integrating in a concurrent mode biocatalytic oxidation of the N-alkyltetrahydroisoquinolines with gold catalyzed C–C coupling with acetylene derivatives in phosphate buffer. In this process, the enzyme monoamine oxidase (MAO) oxidizes the tetrahydroisoquinoline derivative, which subsequently couples with the alkyne. Under optimized reaction conditions, a variety of alkynyl substituted tetrahydroisoquinoline derivatives were obtained in variable yields under very mild reaction conditions in aqueous media (Scheme 62).
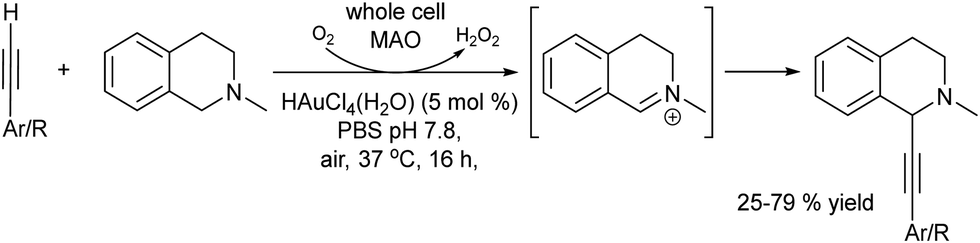 | ||
| Scheme 62 Synthesis of alkynyl substituted tetrahydroisoquinoline from tetraisoquinolines and alkynes by combining biocatalytic oxidation and coupling with alkynes. | ||
Compared with multistep asymmetric strategies to synthesize THIQs, where lower yields are obtained and strong bases and toxic solvents are required,237,246,247 the chemo-enzymatic processes described above represent much more efficient, sustainable and rapid routes to achieve these important compounds.
Other related important scaffolds present in many drugs are 2-quinolones. The challenge in the conventional synthetic methods of these N-heterocycles is their functionalization due to low reactivity of their π-electron deficient skeleton.248 Recently, Castagnolo et al.249 have reported for the first time the synthesis of 2-quinolones through a chemo-enzymatic process in a sequential mode. The process uses N-cyclopropyl-N-alkylaniline derivatives as starting compounds that are transformed in the first step into quinolinium ions through a cyclation/aromatization sequence biocatalyzed by horseradish peroxidase (HRP). The quinolinium ions are further carbonylated into 2-quinolone derivatives using K3Fe(CN)6 in the second step (Scheme 63). A variety of 2-quinolone derivatives could be obtained in good isolated yields.
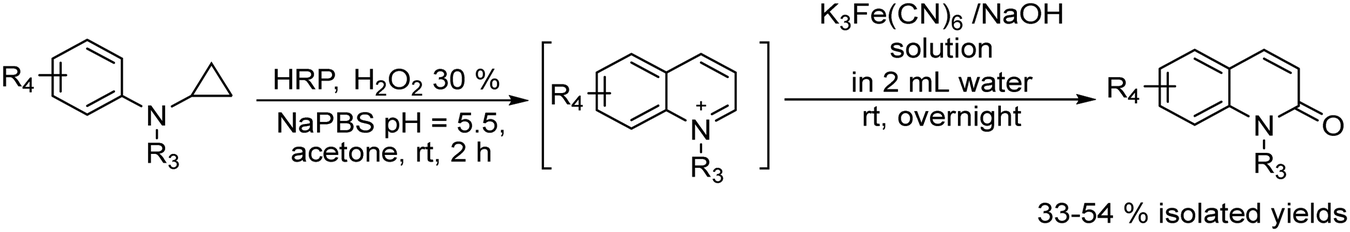 | ||
| Scheme 63 Synthesis of 2-quinolones starting from N-cyclopropyl-N-alkylaniline derivatives through a biocatalyzed cyclation/aromatization sequence followed by carbonylation. | ||
3. Chemo-enzymatic processes for biomass valorization
3.1. Synthesis of HMF from glucose
In the last few decades, the valorization of biomass resources into valuable chemicals has gained increased interest in academic and industrial areas.250 Chemo-enzymatic approaches have high potential to upgrade easily available and renewable bio-based compounds in a minimal number of steps in a selective and productive fashion. An interesting example of a chemo-enzymatic strategy for biomass valorization is the conversion of glucose into 5-hydroxymethylfurfural (HMF). This compound is a versatile platform molecule produced by dehydration of hexoses in acid media and can be converted into valuable derivatives such as 2,5-dimethylfuran (2,5-DMF), monomers like 2,5-furandicarboxylic acid (FDCA) and a variety of fine chemicals,251 and biofuels.252–256 The simplest and efficient way for producing HMF is by the acid-catalyzed dehydration of fructose. However, its production by isomerization of the most abundant and cheapest glucose is highly desired. However, the chemical or enzymatically catalyzed glucose–fructose isomerization is controlled by the thermodynamic equilibrium, which limits the conversion to almost isoquantities of both sugars. Therefore, a key issue in the synthesis of HMF from glucose is to shift the equilibrium from glucose to fructose. The strategy to shift the equilibrium towards the formation of fructose involves the use of sugar-complexing agents such as boronic acid derivatives, which exhibit higher affinity to fructose than to glucose allowing the shift of the equilibrium toward the fructose side.257 Qi et al.258 developed a sequential chemo-enzymatic process involving as the first step the borate-assisted isomerization of glucose into fructose catalyzed by a commercial immobilized glucose isomerase (Sweetzyme IT), which is a cross-linked enzyme with glutaraldehyde (CLEA) (Scheme 64). It was found that at a borate-to-glucose ratio of 0.5% the isomerization equilibrium of glucose to fructose shifted from 53% to 88% yield. After the isomerization step, the glucose isomerase was separated by vacuum filtration and the acid catalyst (HCl) and an organic solvent (1-butanol), along with NaCl to improve the partitioning of HMF into the organic phase, were added. The dehydration step of the resulting sugar mixtures was performed at 190 °C achieving 63.3% HMF yield.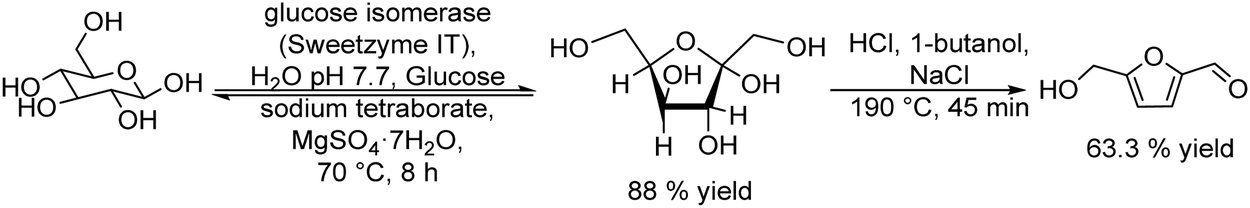 | ||
| Scheme 64 Synthesis of HMF from glucose by combining biocatalyzed isomerization and chemical dehydration. | ||
Zhao et al.259 combined a thermophilic glucose isomerase electrostatically immobilized on mesoporous silica functionalized with amino groups for glucose to fructose isomerization with mesoporous silica functionalized with sulfonic groups (–SO3H) for fructose dehydration (Scheme 65). A suitable monophasic reaction medium (THF:H2O) was chosen to accommodate both reactions in the tandem catalytic sequence in one pot using a combination of both heterogeneous catalysts under separately optimized conditions. Thus, by keeping the temperature first at 90 °C for 1 h the enzyme catalyzed glucose isomerization occurred, giving up to 61% fructose, and then the temperature was increased to 130 °C to accelerate fructose dehydration. Under these reaction conditions, 30% HMF with 64% of selectivity after 24 h was obtained. Unfortunately, the biocatalyst could not be recycled because of its short half-life (t1/2 = 15.8 min) in the organic solvent. Moreover, it was observed that the efficiency of the tandem process is significantly limited by the large difference in rates for the two catalysts.
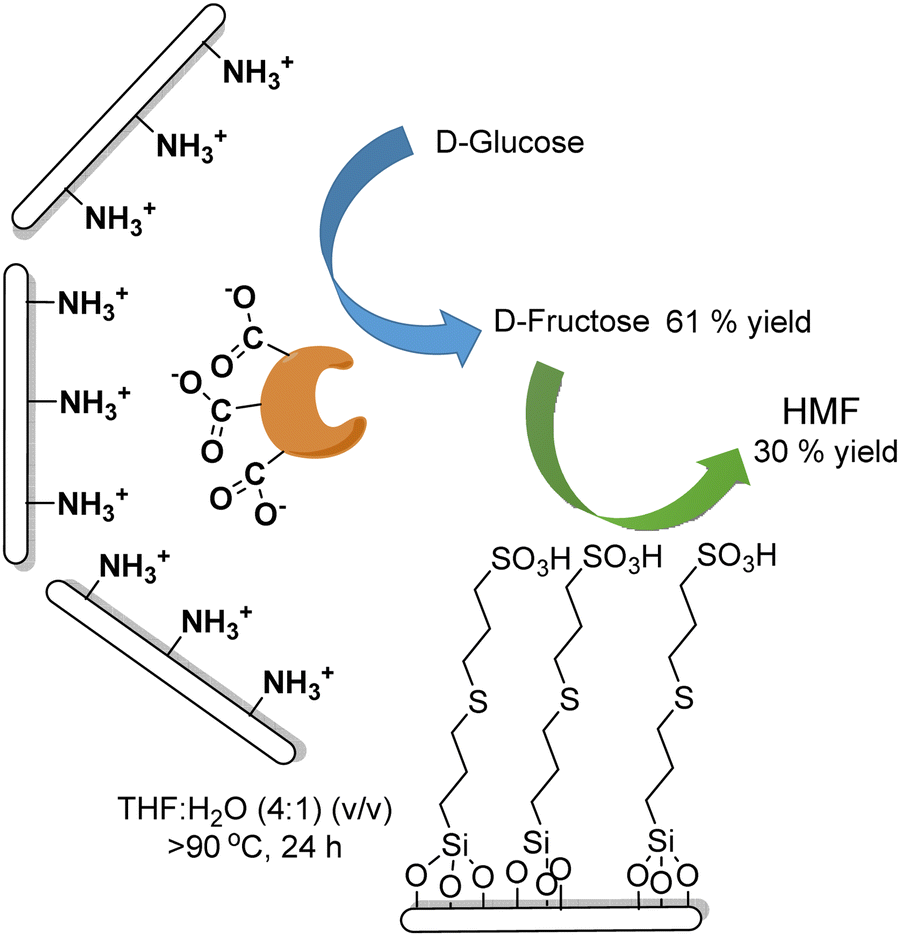 | ||
| Scheme 65 Biocatalyzed isomerization of glucose to fructose followed by chemical dehydration over heterogeneous catalysts to obtain HMF. | ||
These chemo-enzymatic strategies allow access to HMF in moderate yields; however, it is interesting to notice that recently different heterogeneous chemocatalytic systems have been developed containing Lewis acid sites (for glucose isomerization) and Brønsted acid sites (for fructose dehydration) that allow obtaining HMF from glucose with similar HMF yields.260 For instance, Davis et al.261 combined HCl as the Brønsted acid and the Sn-beta catalyst bearing Lewis acid sites in a biphasic THF:water medium. The synergistic effects of this catalytic system allowed obtaining 79% glucose conversion with 70% selectivity for HMF at 180 °C.
3.2. Synthesis of furfuryl alcohol and furfuryl amine from xylose
Furfuryl alcohol (FOL) is an important biomass derived alcohol used as an intermediate for the production of a variety of fine chemicals such as lysine amino acid, vitamin C, lubricants and fragrances.250,262–264 Currently, FOL is produced at the industrial scale by liquid- or vapor-phase hydrogenation of furfural (FAL), which is obtained by the acid catalyzed dehydration of pentoses, such as xylose. However, for FAL hydrogenation, high pressure and/or noble metals along with high energy consumption are required.265,266 Therefore bioreduction of FAL into FOL under mild reaction conditions represents an interesting alternative due to the product specificity and its high efficiency.267–269 On the other hand, furfuryl amine (FLA) is an important platform molecule derived from furfural that is used for producing polymers, agrochemicals, additives, fragrances and pharmaceuticals.270–274 Furfuryl amine is usually synthesized through the reductive amination of furfural with ammonia, using metal catalysts under harsh conditions.275,276 Thus, bioamination of furfural represents an attractive alternative method to biosynthesize FLA under environmentally friendly reaction conditions.272He et al. have performed extensive research on converting xylose and biomass-rich xylose into furfuryl alcohol and furfuryl amine through a sequential chemo-enzymatic process involving as a first step the acid catalyzed dehydration of xylose into furfural using tin-based heterogeneous catalysts. In the second step, bioreduction or bioamination is performed using recombinant Escherichia coli whole cells harboring reductase or ω-transaminase.273,277–282
For instance, xylose dehydration was performed using an optimized heterogeneous acid catalyst based on a modified montmorillonite (SO42−/SnO2-MMT), using water as a solvent at 170 °C, achieving 42% yield of furfural. After this, temperature was decreased to 30 °C, the recombinant Escherichia coli CCZU-K14 whole cells harboring a NADH-dependent reductase were added to the reaction media, pH was adjusted and the required co-factor (glucose) regenerating agent was added. The biocatalytic step yielded 100% FOL after 24 h reaction time (Scheme 66).278
 | ||
| Scheme 66 Synthesis of furfuryl alcohol from xylose by combining chemical dehydration with bioreduction. | ||
The same chemo-enzymatic process was also performed using SO42−/SnO2-attapulgite as the acid catalyst for the dehydration of xylose-rich hydrolysate into furfural, while the recombinant Escherichia coli CCZU-A13 harboring a NADH-dependent reductase immobilized on carrageenan was used for the bioreduction to furfuryl alcohol. Similar results for the furfuryl alcohol production were obtained, but in this case the carrageenan immobilized whole-cells and solid acid catalyst could be recycled up to 5 runs, retaining their activity.280 More recently, and following the same protocol, the authors reported the direct conversion of the raw bamboo shoot shell and corncob into furfuryl alcohol or furfuryl amine.273,281,282 In all cases, although the furfural yield (40–50% yield) was dependent on the type of solid tin-catalyst and the starting raw material, the selectivity towards the target product was 100%.
3.3. Synthesis of furan amino acid derivatives
Ni and co-workers285 reported the one-pot chemo-enzymatic synthesis of a chiral furan-based amino acid from corncob by integrating a magnetic core–shell solid acid catalyst, Fe3O4@MCM-41/SO42−, with PLP-dependent threonine aldolase derived from P. putida (PpThA) (Scheme 67) to catalyze the aldol condensation between furfural and glycine. After process optimization, furfural was obtained from corncob in 63.6% yield using Fe3O4@MCM-41/SO42− as a magnetic solid acid catalyst at 180 °C in 40 min. Subsequently, the biomass-derived furfural was freely transformed into the aldol-adduct β-(2-furyl)-L-serine in 73.6% yield, 99% ee, and 20% de using immobilized cells expressing PpThA, and without significantly reducing the product yields, the magnetic and biocatalysts could be efficiently recovered and reused for five cycles.
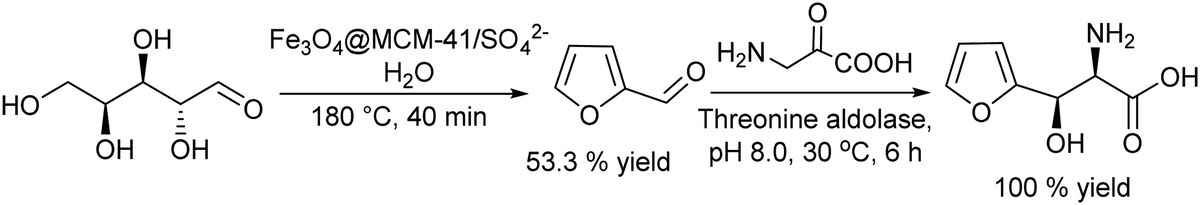 | ||
| Scheme 67 Synthesis of β-(2-furyl) serine from xylose by combining dehydration with biocatalyzed aldol condensation. | ||
Compared with the conventional synthesis of β-(2-furyl)serine involving the chemocatalyzed aldol condensation between furfural and glycine, which leads to a racemic mixture, this chemo-enzymatic approach represents a straight-forward method to convert raw biomass into valuable chiral furyl amino acids.
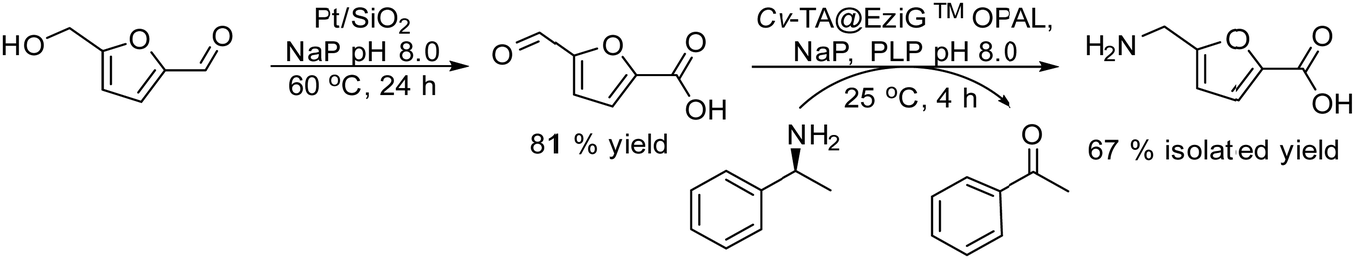 | ||
| Scheme 68 Synthesis of 5-aminomethyl-2-carboxylic acid from 5-hydroxymethyl furfural by combining oxidation with biocatalyzed reductive amination. | ||
3.4. Synthesis of 3-acetamido-5(1-hydroxyethyl)furan from N-acetyl glucosamine
Besides lignocellulose, chitin is also a non-food type biomass, and after cellulose, it is the second most abundant biopolymer on earth. Chitin is widely found in the shells of crustaceans and insects and its annual production is about 100 billion tons.292 It is constituted of units of N-acetyl glucosamine linked by β (1–4) linkages. The hydrolysis of chitin leads to N-acetyl glucosamine (NAG), which is a promising platform molecule to produce highly valuable N-containing chemicals. Thus, dehydration of NAG leads to 3-acetamido-5-acetylfuran (3A5AF), which is a versatile N-containing furan based building block. For instance, the asymmetric reduction of the keto group can afford the corresponding chiral furfuryl alcohol, (3-acetamido-5(1-hydroxyethyl)furan) (3A5HEF), which is a key intermediate for the synthesis of a variety of drugs.293–295 Recently, Li et al. designed a chemo-enzymatic one-pot strategy to synthesize the chiral 3A5HEF starting from N-acetyl glucosamine in a sequential mode. The process involves as the first step the dehydration of NAG using tyrosine hydrochloride/CaCl2 as an enzyme-friendly chemocatalyst in N,N-dimethylformamide (DMA) followed by the asymmetric reduction using a carbonyl reductase derived from Streptomyces coelicolor (ScCR) (see Scheme 69). To improve the reductase activity and stability the authors performed the structure-guided engineering of the enzyme, and two robust mutants were obtained, which showed 6-fold higher catalytic efficiency than the parent enzyme and improved thermal and organic solvent stability. Thus, 53% yield (99% ee) of the target chiral alcohol could be obtained in the one-pot chemo-enzymatic approach, which represents an important advantage compared with other reported methods where the isolation of the intermediate 3A5AF was required, which reduced the overall yield, while producing increased wastes.296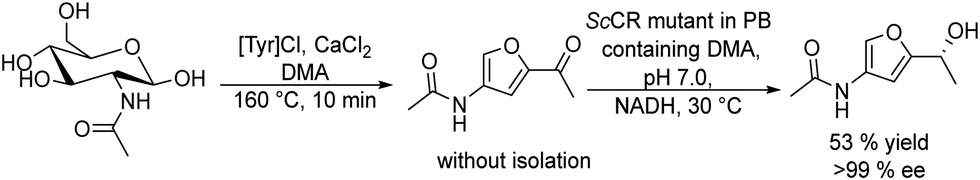 | ||
| Scheme 69 Synthesis of 3-acetamido-5-(1-hydroxyethyl)furan from N-acetyl glucosamine in a sequential mode by combining dehydration with biocatalyzed asymmetric reduction. | ||
Considering that the main drawbacks encountered when dealing with biomass derived molecules are their poor stability under harsh reaction conditions and limited solubility in organic solvents, these examples show that chemo-enzymatic processes occurring in aqueous media and under mild reaction conditions can be an interesting alternative to transform biomass platform molecules into valuable compounds with high conversion and selectivity.
4. Reaction engineering strategies for solving incompatibility issues in chemo-enzymatic reactions
As we showed in the previous section, the synthesis of organic compounds through chemo-enzymatic cascade reactions suffers in general from a lack of compatibility between the chemo- and biocatalytic systems. Then most chemo-enzymatic methodologies are based on sequential processes where reaction conditions (catalysts, temperature, solvent, pH, substrate concentration, required additives) can be adjusted for each reaction step. However, researchers have developed different strategies to overcome these incompatibilities by performing, in many cases, the chemo-enzymatic processes in a concurrent mode. In this section, we will discuss the different approaches adopted for this purpose. In this way, adjusting the solvent compatibility, for instance, increasing the solubility of organic compounds (including biomass raw materials), is an important approach, which can be achieved by using biocompatible neoteric solvents such as ionic liquids (ILs) and deep eutectic solvents (DESs). Also, the mutual inactivation of the chemo- and biocatalysts is an important drawback, which can be particularly significant when metal-based catalysts and enzymes are involved in the cascade process. This has been solved by designing catalytic systems bearing spatial separation between the catalytic sites, such as the enzyme–metal hybrid catalysts, designed by co-immobilization of both catalysts on the same carrier or by bio-conjugation. Moreover, additional advantages of these systems can be their recyclability and the increase in activity by reducing the diffusion distance between catalytic sites of the involved intermediates. Particularly, they have been shown to be very effective in enzymatic oxidation where H2O2 is released, and its accumulation results in strong inhibition of the enzymatic activity poisoning of the enzyme. Thus, the fast migration of H2O2 toward the metal site nearby allows its rapid decomposition, preventing enzyme deactivation. As we will show, other strategies are based on spatial compartmentalization where enzymes, chemocatalysts, and required additives can be confined and can work effectively without interaction. Examples of these strategies are the use of porous hydrophobic membranes and biphasic and micellar systems (using biocompatible surfactants). They are of special interest for enzymes that do not work in organic media, while the solubility of organic substrates in the organic/lipophilic phase is increased. Encapsulation of enzymes and metal catalysts inside a cover has been particularly useful for organometallic complexes and represents another strategy to avoid mutual catalyst deactivation. Moreover, in some cases, the metal complexes are stabilized by encapsulation while increasing their water solubility and the turnover number of the metal catalyst. Finally, the combination of continuous flow reactors to perform compartmentalized chemo-enzymatic reactions will be discussed. This strategy, which requires the use of solid chemocatalysts and supported enzymes, represents a versatile approach where each catalytic system can efficiently work under very different reaction conditions, resulting in completely automated processes with high performances.4.1. Chemo-enzymatic processes in non-conventional media
In the last few decades, neoteric solvents, such as ionic liquids (ILs) and deep eutectic solvents (DESs), have emerged as environmentally friendly solvents, which are exciting alternatives to traditional solvents for chemo- and biocatalytic processes due to their wide range of physicochemical properties.297–306 Deep eutectic solvents are formed by a mixture of salts, for instance quaternary ammonium salts, such as choline chloride, which act as hydrogen bond acceptors (HBAs), and uncharged hydrogen-bond donors (HBDs) (such as urea, carboxylic acids or polyols) (Scheme 70). The mixture forms a wide H-bond network that stabilizes liquid configurations and results in lower melting points than those of their individual components. DESs are attractive because of their easy and inexpensive preparation, tunable properties, biodegradability, and non-volatility. Moreover, they are strongly water-miscible and hygroscopic.307 Their lower cost and diminished environmental impact make them an effective alternative to ILs in multiple applications including organic synthesis.308 Moreover, DESs have been applied as solvents in biocatalysis, where it has been shown that they can improve the efficiency of these reactions via increasing the substrate solubility and the activity and stability of enzymes.309–313 The low solubility of most organic substrates in water limits the maximum substrate concentration; however, the unique properties of DESs allow reaching not only high substrate concentration, but also higher enantioselectivities than that can be achieved in water solutions in enzymatic processes.314,315 These characteristics have stimulated their utilization in chemo-enzymatic cascades, although these systems also suffer from the typical drawbacks such as the incompatibility of the involved catalysts with reaction conditions or undesired catalyst cross-deactivation. | ||
| Scheme 70 Some examples of DESs possessing HBAs (quaternary ammonium salts) and HBDs (alcohols, carboxylic acids and amines). | ||
The first application of DESs in chemo-enzymatic cascade processes was reported by González-Sabín et al. in 2018.316 The process was based on ruthenium-catalyzed isomerization of allylic alcohols to α,β-saturated ketones followed by asymmetric enzymatic reduction promoted by ketoreductases (KREDs) to produce chiral alcohols. (Scheme 71).
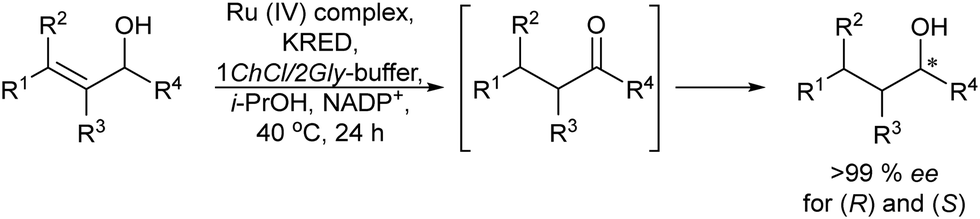 | ||
| Scheme 71 Synthesis of optically active alcohols by combining Ru-(IV)-catalyzed isomerization with enzymatic reduction in DES–buffer medium in a concurrent mode. | ||
An optimization study of different DES systems for the bioreduction of prochiral aromatic ketones performed in DES–water mixtures showed that the DES based on sorbitol or glycerol as a HBD and choline chloride as a hydrogen bond acceptor (HBA) gave the best results at 50% w/w DES. Then, the cascade process was performed in a sequential mode, where metal-catalysed isomerization of allylic alcohols was performed using a ruthenium(IV) complex as a catalyst. When the isomerization was complete, the KRED and co-factor were added and the reaction proceeded at 30 °C for 24 h. Thus, various chiral alcohols were obtained in isolated yields between 60 and 95% and ee (93–99%) using this protocol. The results showed that KRED was compatible with the reaction medium of the metal-catalyzed step, with the effect of the Ru(IV) catalyst on enzyme performance being insignificant. This behavior enabled the process to occur successfully in a concurrent mode, and a variety of allylic alcohols were transformed into the enantiopure saturated alcohols with good to excellent conversions (68–96%), improving previous results obtained in aqueous buffer media.317,318
In a parallel work, the same group319 reported the use of mixtures of DES–buffer media in cascade reactions involving the palladium-catalyzed Suzuki cross-coupling followed by enzymatic reduction mediated by KRED to produce chiral biaryl alcohols (Scheme 72).
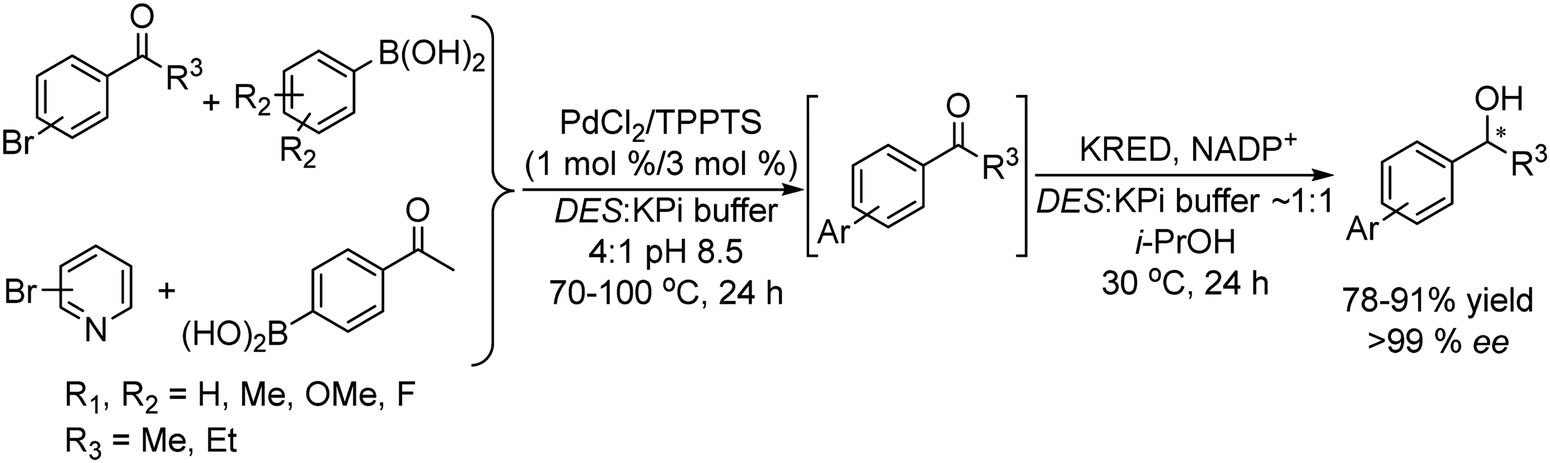 | ||
| Scheme 72 Synthesis of biaryl alcohols by combining palladium-catalyzed Suzuki cross-coupling with enzymatic reduction. TPPTS: triphenylphosphine trisulfonate; KPi: potassium phosphate. | ||
The optimization of the required parameters, such as the nature of the DES, DES–water ratio, temperature and substrate concentration, allowed the efficient coupling of both steps in a sequential mode, achieving the target chiral alcohols in 78–91% yield and >99% ee. Interestingly, the use of DES–water mixtures allowed increasing the substrate concentration up to 75 mM in the biocatalyzed step, which is considerably superior to those used in water solution (33–44 mM).
A similar protocol was applied to the synthesis of chiral biaryl amines,320 by sequentially coupling a Suzuki reaction with asymmetric bioamination catalyzed by an amino transaminase (ATA) (Scheme 73). In this case, and owing to the limitations in the use of amine transaminases to convert bulky ketones, such as biaryl ketones, the authors used an (R)-selective amine transaminase derived from Exophiala xenobiotica, which was identified, using data mining, as an efficient biocatalyst to convert biaryl ketones into the corresponding chiral amines with excellent enantioselectivity.321 Optimization of the reaction conditions in both steps allowed finding a compatible window to perform the chemo-enzymatic process in a sequential mode. A critical point in the biotransformation step was to adjust the concentration of DES in the DES–buffer mixture, since the activity of the ATA was considerably reduced at DES concentrations higher than 15%. Therefore, after the Suzuki step (performed at 100 °C and 200 mM substrate concentration in a DES:water 4:1 mixture) the resultant reaction mixture was diluted to 25 mM (which results in a DES concentration of 10%) prior to the biotransformation step. The protocol was applied successfully to a variety of substrates, where the metal-catalyzed step proceeded quantitatively in all cases, and the resulting biaryl ketones were converted by the ATA quantitatively (>95%) into the corresponding (R)-biaryl amines with >99% ee.
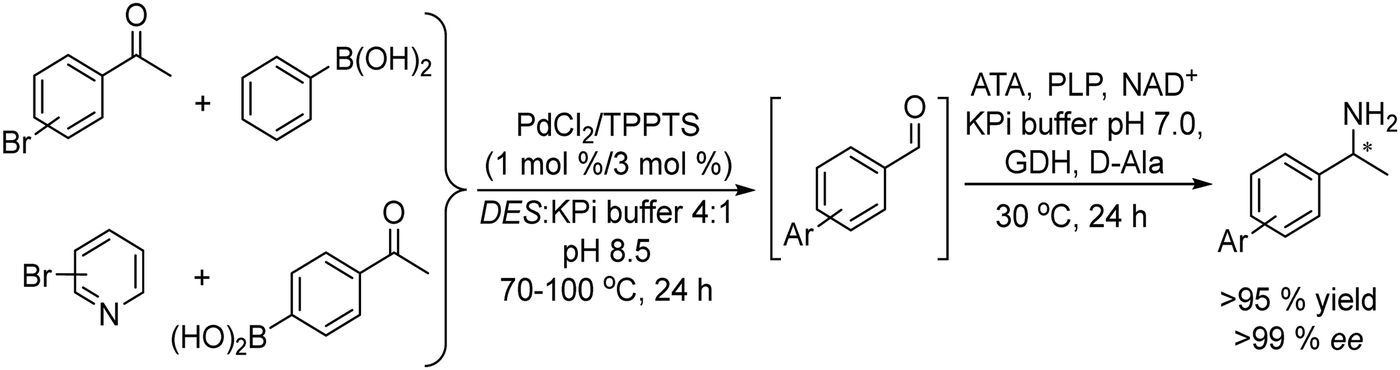 | ||
| Scheme 73 Synthesis of biaryl amines by combining palladium-catalyzed Suzuki cross-coupling with enzymatic transamination. | ||
Very recently, He and co-workers have reported a series of studies on the valorization of biomass in DES–water systems. For instance, the production of furfuryl alcohol and furfuryl amine (FLA) from biomass rich-xylose322,323 was achieved through a sequential chemo-enzymatic cascade reaction, by combining dehydration of xylose into furfural with enzyme catalyzed reduction or transamination. In the chemo-enzymatic process to produce furfuryl amine (Scheme 74) for the chemocatalyzed step, shrimp shell (SS) waste was used as the biobased support to prepare a sulfonated tin-based solid acid (Sn–SS) catalyst for converting sugarcane bagasse (SB) into furfural in DES–water (choline chloride/ethylene glycol–water). After optimization of reaction conditions (ChCl/EG loading, Sn–SS amount, temperature, and reaction time), it was possible to achieve 62.3% conversion of SB to furfural after 20 min of reaction time in ChCl/EG–water (20:80 v/v), which was 1.37 times greater than that of the aqueous system. As an explanation for this enhanced selectivity, the authors proposed that ChCl in the DES could increase proton reactivity and availability by influencing the stability of protons. The bioamination of furfural into furfuryl amine was carried out using newly formed recombinant Eschericia coli pRSFDuet-CV-AlaDH cells expressing ω-transaminase and L-alanine dehydrogenase. The chemo-enzymatic cascade process was performed in a sequential mode, where first the SB was converted into furfural using the Sn–SS catalyst in ChCl/EG–water (20:80 v/v) at 170 °C for 20 min. After this, the solution was adjusted to pH 8.0 and diluted with a buffer solution and then pRSFDuet-CV-AlaDH whole-cells and NH4Cl as the amine donor were added. The transamination proceeded at 35 °C for 1 h and the SB-derived FAL could be entirely converted into FLA with a productivity of 0.458 g FLA per g xylan in SB. Similar results concerning FLA productivity (0.43 g per g xylan) were obtained when the process was performed starting from concorb in EaCl:Gy–water and using a sulfonated tin based hydroxyapatite combined with oxalic acid as the dehydrating agent.324
 | ||
| Scheme 74 Production of furfuryl amine (FLA) from xylose-rich biomass by combining xylose dehydration with enzymatic transamination. | ||
To increase thermostability and catalytic activity in the presence of high substrate concentration, the authors developed mutant transaminase strains using side-directed mutagenesis.325–327 For instance, the authors showed that a triple mutant Aspergillus terreus ω-transaminase (HNILQE) could aminate furfural into furfuryl amine in high yield at 500 mM furfural concentration, which is considerably superior to the usual concentrations (100–300 mM) used for the wild-type strain transaminases. The chemo-enzymatic process proceeded in a three-component DES (choline chloride–malonic acid–lactic acid) that acted as a solvent and as an acid catalyst. Starting from D-xylose the dehydration step was performed at 190 °C for 45 min, giving furfural (70.6% yield), which was aminated by the enzyme mutant after adjusting the pH to 8 and the temperature (35 °C), affording the furfuryl amine in 97.6% yield after 24 h.325
Following a similar strategy, the authors developed chemo-enzymatic cascades in a DES–water system to produce efficiently furfuryl amine and furoic acid starting from different xylose sources such as corncob, bagasse, bamboo shoot shell, corn stalk, etc.328–330 Tin based catalysts supported on biological macromolecules were used as chemocatalysts for the first step. By using a shrimp shell-supported solid acid catalyst (Sn–DAT–SS) in DES (ChCl:EG)−water (10:90, v/v) after 30 min at 170 °C, corncob afforded the highest furfural yield (52.4%). The obtained furfural could be completely aminated to FLA by E. coli CCZU-XLS160 cells harboring ω-transaminase after 24–72 h. On the other hand, furfural derived from different sources of biomass could be fully transformed into furoic acid using Escherichia coli HMFOMUT cells containing dehydrogenase.330
The authors also developed efficient chemo-enzymatic processes for the valorization of D-fructose and bread waste into 2,5-bis(hydroxymethyl)furan (BHMF) in acidic DES in a sequential mode.331,332 Using lactic acid:betaine (LA:B)–water (15:85, v-v) as a solvent and acid catalysts, fructose was dehydrated into 5-hydroxymethylfurfural (HMF) in 92% yield at 150 °C in 1.5 h. HMF derived from fructose was then converted into BHMF (98.4% yield) using Pseudomonas putida S12 within 24 h at 30 °C and pH 8.0 (Scheme 75). Starting from bread waste, the HMF yield reached 44.2 mol% (based on the bread waste) and it could be transformed into 2,5-bis(hydroxymethyl) furan (using E. Coli HMFOMUT harbouring reductase activity) in 84.5% yield after 1 day at 37 °C, which represents a yield of 0.230 kg of BHMF per kg of bread.
 | ||
| Scheme 75 Synthesis of 2,5-bis(hydroxymethyl)furan (BHMF) from fructose by combining fructose dehydration with enzymatic reduction. | ||
Conversion of fructose and bread waste into 5-hydroxymethyl-2-furfurylamine has been recently developed in acidic DES using mutant biocatalysts.333–335 For instance, in the valorization of bread waste, the cascade reaction was performed using betaine:malonic acid–water as the reaction medium. Starting from bread, in the first step, the DES catalyzed the hydrolysis, isomerization and dehydration steps achieving 30% yield of HMF at 190 °C in 45 min. Then HMF was subsequently aminated using E. coli cells expressing L-alanine dehydrogenase and the ω-transaminase mutant as the biocatalyst and using D-alanine as the amino donor, achieving 92.1% yield of the amino-alcohol at 35 °C in 12 h. Interestingly, the use of the mutant biocatalyst not only reduced the amount of the amino donor but also allowed increasing the substrate tolerance (up to 500 mM HMF) of the whole cells, while enhancing the transamination activity of the enzyme.
The authors also developed a chemo-enzymatic cascade to produce 2,5-diformylfuran (DFF), an important building block for furan-based chemicals, from fructose and from waste bread in the acidic lactic acid:betaine–water.336 In this protocol, the fructose is efficiently dehydrated into HMF in the first step, and the HMF is biologically oxidized into DFF (Scheme 76) by E.coli GOase. A productivity of 0.631 g DFF g−1 fructose and 0.323 g DFF g−1 bread could be respectively achieved.
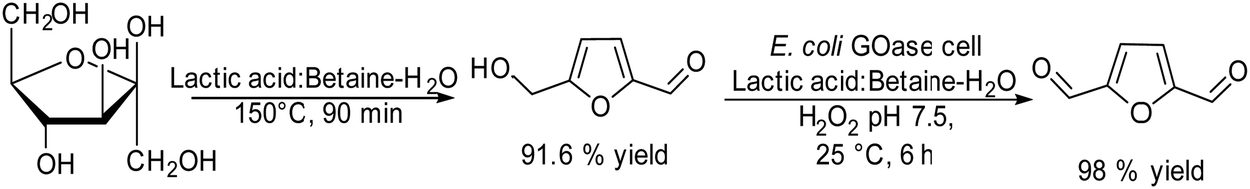 | ||
| Scheme 76 Synthesis of 2,5-diformylfuran (DFF) from fructose by combining fructose dehydration with enzymatic oxidation. | ||
4.2. Enzyme–metal hybrid catalyst
As shown for the different examples summarized in this work, the direct use of enzymes and metal catalysts in chemo-enzymatic one-pot reactions commonly leads to mutual inactivation of the catalysts. Therefore, the spatial separation of catalytic sites is a straightforward method to overcome incompatibility problems between catalytic species involved in chemo-enzymatic processes. An effective strategy is through the rational design of enzyme–metal hybrid catalysts.337 The generation of incompatible metal and enzyme active sites on the same carrier not only provides catalyst stability, but moreover enzyme and metal active sites in close proximity exhibit improvement in activity (by reducing the diffusion distance of involved intermediates), which along with catalyst recyclability are important advantages of this strategy. The enzyme–metal hybrid catalysts can be synthesized through co-immobilization and bio-conjugation methods. In the following sections, relevant and updated examples of both strategies will be presented. | ||
| Scheme 77 Cascade reaction in a concurrent mode involving benzaldehyde reduction followed by esterification using a co-immobilized CalB–Pd hybrid catalyst. | ||
Prasad et al.341 prepared a bifunctional catalyst combining gold nanoparticles and glucosidase. The as-synthesized Au NPs were coated with a silica shell using a sol–gel method, resulting in a core–shell architecture (Au@mSiO2). Then, the shell surface of Au@mSiO2 was functionalized with epoxy groups, which were later used to attach the glucosidase through covalent binding. The resulting catalyst (Au@mSiO2@glucosidase) was used in a cascade reaction for converting 4-nitrophenyl-β-glucopyranoside into 4-aminophenol in a sequential mode. Thus, 4-nitrophenyl-β-glucopyranoside was first hydrolyzed by the enzyme into p-nitrophenol in aqueous solution up to completion. Then an excess of NaBH4 was added to reduce the nitrophenol intermediate into aminophenol (Scheme 78). In this catalytic system, the presence of pores on the surface of silica lets both 4-nitrophenol and NaBH4 to diffuse to the surface of the Au nanoparticles, where the reduction occurs.
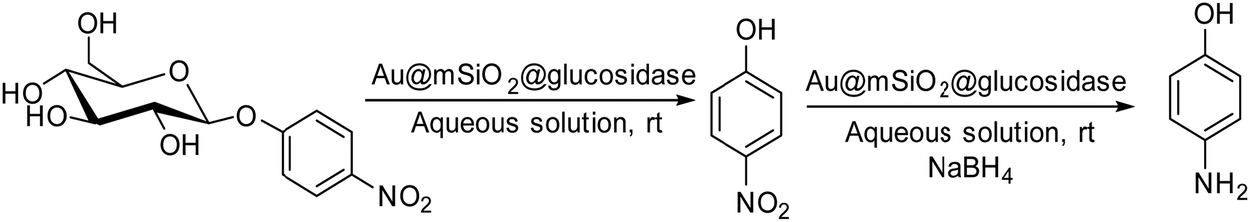 | ||
| Scheme 78 A cascade process involving hydrolysis and reduction sequences catalyzed by a co-immobilized Au–glucosidase hybrid catalyst. | ||
However, the bifunctional catalyst becomes rapidly deactivated after consecutive reaction cycles, probably due to the denaturalization of the enzyme caused by the very high pH generated by the hydrolysis of NaBH4.
Recently, dendritic organosilica nanoparticles (DONs) have been used as excellent supports for co-immobilization of metals and enzymes, without extra hydrophobic modification.342 DONs are a class of mesoporous materials with intrinsic hydrophobicity, bearing ordered center-radial mesoporous channels and high accessible internal surface areas.343 The DONS were prepared by a continuous phase microemulsion method and were subsequently used to stepwise immobilize Pd nanoparticles and the enzyme. Finally, the DON@Pd–enzyme obtained was covered by a polydopamine shell (PDA) by self-polymerization of dopamine, thus obtaining the DON@Pd-Enzyme@PDA catalyst (Scheme 79).
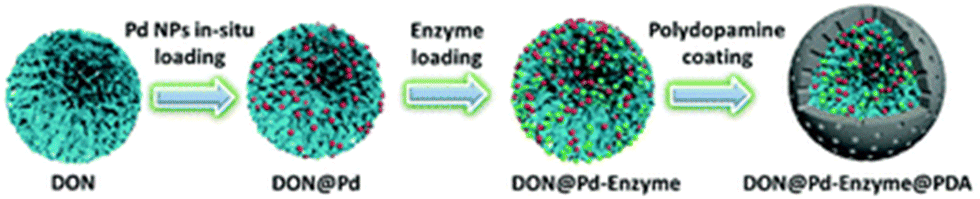 | ||
| Scheme 79 Formation mechanism and process of the amphiphilic chemo-enzymatic nanocatalyst. Reproduced with permission: Copyright 2020, The Royal Society of Chemistry (adapted from ref. 342). | ||
The novel core–shell catalyst showed enhanced catalytic performance in chemo-enzymatic asymmetric synthesis of chiral benzyl alcohols through a two-step concurrent cascade reaction. The process combines a Pd/Cu-catalyzed Liebeskind–Srogl (L–S) cross-coupling reaction between thioesters and boronic acids with asymmetric reduction catalyzed by alcohol dehydrogenase (ADH) derived from Rhodococcus ruber (Scheme 80). A variety of enantiomerically pure benzyl alcohol derivatives were obtained in high yields (60–86%). Additionally, the catalyst was also used for the DKR of racemic amines to obtain chiral amines in high yields. The authors showed that the hydrophobic microenvironment is responsible for increased stability, activity and cascade efficiency of the catalyst, while the hydrophilic PDA shell improves the dispersibility of the catalyst in water and acts as a protective barrier for the inner core. In fact, when the reaction was performed using DON@Pd–ADH without the PDA shell, an important decrease of the yield was observed because the exposed ADH was severely inactivated by copper ions and boronic acid.
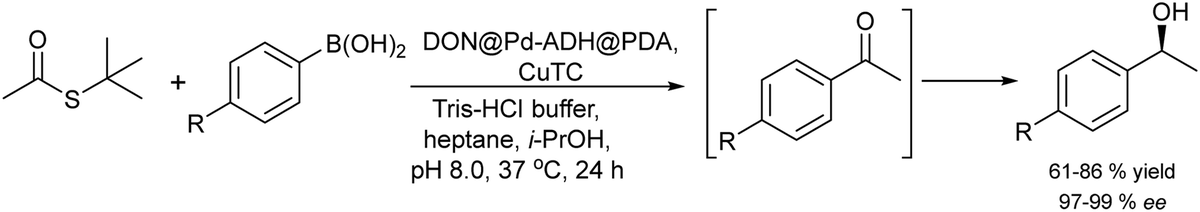 | ||
| Scheme 80 Synthesis of chiral alcohols in a concurrent mode using metals and enzymes co-immobilized on dendritic organosilica nanoparticles (DON) as a hybrid catalyst. (CuTC: (copper(I) thiophene-2-carboxylate). | ||
A core–shell structured magnetic wrinkled organosilica-based metal–enzyme integrated catalyst was developed by Jiang et al.344 The wrinkled organosilica nanoparticles (WOS) were prepared starting from an optimized ratio of tetraethylorthosilicate (TEOS) as a silicon source, bis(triethoxysilyl)ethane, (BTSE) organosilica, Fe3O4 nanoparticles, and urea. The urea was used to promote the hydrolysis and condensation of the silicon shell over the Fe3O4 nanoparticles, and it was shown that the urea concentration had a significant influence on the wrinkled structures. Subsequent functionalization of the silica with amino groups allowed, on the one hand, the high dispersion of Pd ions (from a Pd salt), which were in situ reduced with NaBH4, and on the other hand, the amino groups serve as sites to anchor the enzymes through covalent bonds. The authors showed that by anchoring lipases the optimized hybrid catalyst was highly active in the dynamic kinetic resolution of chiral amines in an organic solvent. Moreover, by anchoring an alcohol dehydrogenase (ADH&Pd@WOS) chemo-enzymatic synthesis of chiral alcohols in water from α,β-unsatured ketones could be performed in high yield and enantiomeric excess (Scheme 81).
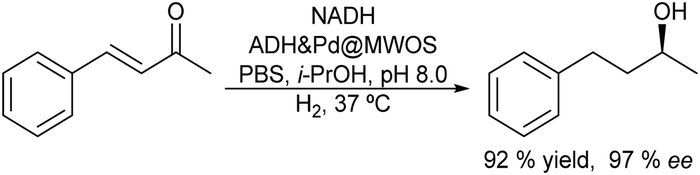 | ||
| Scheme 81 Synthesis of chiral alcohols from α-β-unsaturated ketones using a co-immobilized Pd@ADH hybrid catalyst. | ||
Recently, Tyagi et al.345 developed a biohybrid catalyst [Pd(0)-CALB@SiO2] by immobilizing Candida Antarctica lipaseB and Pd(PPh3)4 within a silica framework. The hybrid catalyst was prepared starting from an emulsion of oleic acid in a mixture of EtOH:water, which acted as a template for the growth of spherical silica particles, while avoiding the contraction of the particles during drying. Then, after the addition of the lipase and Pd(PPh3)4, (3-aminopropyl)triethoxysilane (APTES) and tetraethylorthosilicate (TEOS) were added generating the biohybrid catalyst with a silica framework and amine functionality.
The [Pd(0)–CALB@SiO2] biohybrid was tested in a chemo-enzymatic cascade in a sequential mode involving as the first step the lipase catalyzed Groebke–Blackburn–Bienayme (GBB) multicomponent reaction, followed by the Pd catalyzed Suzuki–Miyaura coupling with boronic acids (Scheme 82). The process was exploited to produce a variety of clinically important imidazo[1,2-a]pyridine derivatives in isolated yields ranging from 49 to 87%. Interestingly, when Pd(PPh3)4 and CALB-enzyme were used in the pure form as a catalyst in a one-pot process, the reaction afforded the target product only in trace amounts, suggesting the mutual deactivation of both catalysts. The biohybrid was not stable over several reuses and a decrease of activity was observed after the second run. This was attributed to the leaching of the enzyme and/or palladium from the catalysts.
 | ||
| Scheme 82 Chemo-enzymatic synthesis of aryl substituted imidazo[1,2-a]pyridines. | ||
Metal–organic frameworks (MOFs) are crystalline structures with high versatility for the design of pore geometries, porosity, and functionality since they can be finely adjusted by the rational design of the metallic centers and organic linkers or by post-synthetic methods.346–348
Preliminary studies performed by Chen et al.349 showed that the specific interactions between the organic components of the MOFs and the supported enzyme prevent enzyme leaching and deactivation. Therefore, MOFs with large pores are considered to be promising candidates for the immobilization of enzymes. Recently, these features of MOFs have been successfully exploited for the preparation of enzyme–metal nanohybrids as catalysts for chemo-enzymatic cascades. For instance, Dutta et al.350 performed the synthesis of a cobalt-based zeolitic imidazolate framework (ZIF67) in the presence of Pd nanoparticles and polyvinylpyrrolidone, to generate defect-rich Co(II) nodes and to produce large mesopores (∼30 nm). This material, bearing stable Pd nanoparticles encapsulated inside the mesopores of the MOF, was incubated with Candida antartica lipase A (CalA), which was also placed inside the mesopores, generating the multifunctional catalysts. The authors applied the hybrid catalysts in a cascade reaction that couples in a concurrent mode a nitroaldol condensation (catalyzed by Lewis acid sites, associated with Co(II) unsaturated nodes) with the Pd catalysed racemization of the nitroaldol adduct intermediate, followed by their dynamic kinetic resolution catalyzed by the lipase (Scheme 83). The reactions performed in a mixture of THF/toluene (4:1) at room temperature allowed a variety of chiral acetylated nitroalcohols to be obtained in high yields and ee >99%.
 | ||
| Scheme 83 Combination of nitroalcohol condensation with DKR in a concurrent mode on a MOF-based Pd–lipase nanohybrid catalyst. | ||
A zirconium-based UiO-66 MOF, bearing amino groups (UiO-66-NH2), was used as a carrier to confine Pd nanoparticles in the inner pores of the MOF, while a lipase CALB was physically adsorbed on the surface.351 Moreover, after Pd immobilization, the hydrophobicity of the MOF could be tuned by ligand exchange reaction with lauric acid. The increase in hydrophobicity allowed enhancing the amount of immobilized enzyme and the dispersibility of the catalyst in organic solvents. The lipase–Pd–UiO catalyst was used in a chemo-enzymatic cascade to convert benzaldehyde into benzyl alcohol, which was subsequently esterified to benzyl hexanoate through a two-step sequential process (Scheme 84). Interestingly, the yield of the ester was higher with the hybrid catalysts than in a control experiment with a separate Pd-MOF and free-lipase, possibly due to the aggregation of free CalB in toluene. The reuse of the catalyst showed gradual deactivation, which was attributed to the denaturation of the lipase during the cascade process.
 | ||
| Scheme 84 Combination of aldehyde reduction with transesterification on a MOF-based Pd-lipase nanohybrid catalyst. | ||
Biohybrids have been especially operative for enzymatic oxidation reactions, where H2O2 is evolved, the accumulation of which in the reaction media not only deactivates the enzyme, but also generates byproducts. To overcome the negative effects of H2O2, the oxidation reactions are usually performed in the presence of catalase for H2O2 decomposition. However, these multienzymatic approaches in general have limited stability, poor recyclability and high cost. To overcome these drawbacks Wu et al.352 prepared a zirconium-based UiO-66 MOF with confined Pt nanoparticles inside the crystals and adsorbed L-amino acid oxidase (LAAO) on the external surface. The catalyst was applied to the synthesis of indole-3-pyruvic acid from L-tryptophan in buffer solution (see Scheme 85). In the biohybrid catalytic system based on UiO-66 MOF/Pt, the angstrom-scale pore size of UiO-66 MOF ensured the rapid diffusion of H2O2 produced to Pt sites, where the fast decomposition suppressed enzyme deactivation and the decarboxylation of the target α-keto acid into indole-3-acetic acid. Moreover, with this catalytic system, the deactivation of the enzyme observed when brought into direct contact with Pt nanoparticles was suppressed due to the spatial separation of the metal nanoparticles and the enzyme leading to a 99.7% yield of α-keto acid, exceeding the yield of α-keto acid achieved using the free enzyme (41.2%).
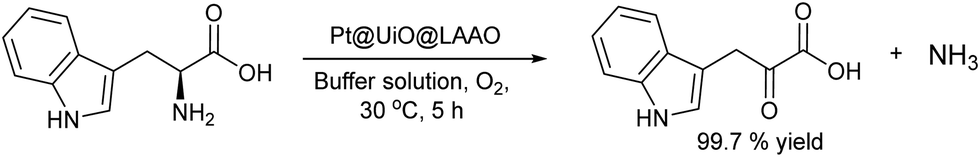 | ||
| Scheme 85 Chemo-enzymatic synthesis of indole-3-pyruvic acid from L-tryptophan on a MOF-based Pt-amino acid oxidase nanohybrid catalyst. | ||
The same tandem reaction for the synthesis of indole-3-pyruvic acid from L-tryptophan was studied by Sun et al.353 using as a bifunctional catalyst a nanohybrid of L-aminooxidase supported/encapsulated on cobalt phosphate nanocrystals (LAAO@CoPs). The catalyst was prepared by a one-pot biomimetic mineralization method under mild conditions, starting from Co salts and L-amino acid oxidase (LAAO). The nanohybrid exhibited a hierarchical nanoflower structure, where the enzyme was sporadically distributed in the inner part of the nanoflower. Thus, during the enzymatic oxidation of L-tryptophan, the nanobiohybrid structure maximizes the in situ decomposition of the H2O2 intermediate through the channelling effect towards the Co sites, which are in direct contact with catalytic sites of LAAO, avoiding H2O2 accumulation and by-product generation. The catalytic system achieved 100% conversion of L-tryptophan with the highest selectivity towards 3-indole-3-pyruvic acid reported up to now. Moreover, the catalyst was reused for several cycles maintaining 75% of its initial activity, while showing high stability against heat and proteolytic treatments.
Using a similar strategy, recently, Du et al.354 reported a new bio-hybrid catalyst (GLOX@Mn3O4) by combining glutamate oxidase (GLOX) and trimanganese tetroxide (Mn3O4) nanocrystals through a one-step biomineralization method for chemo-enzymatic synthesis of α-ketoglutaric acid from L-glutamate (Scheme 86).
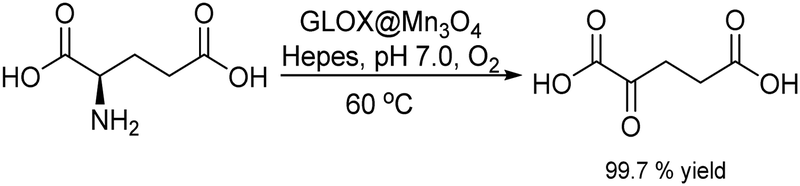 | ||
| Scheme 86 Synthesis of α-ketoglutaric acid from L-glutamate by combining glutamate oxidase (GLOX) and Mn3O4 nanocrystals. | ||
The biohybrid catalyst maximizes the substrate channelling effects for in situ rapid decomposition of H2O2, formed during the enzymatic oxidation of sodium glutamate, diminishing the inhibition of GLOX by H2O2 accumulation. The GLOX@Mn3O4 catalyst exhibited excellent stability and reusability, and practically 100% yield of α-ketoglutaric acid was achieved, which is 4.7 times higher than that obtained using the free GLOX system.
Another interesting example was reported by Sun et al.355 for the production of gluconic acid from starch. In this case the authors immobilized Au nanoparticles onto sulfhydrylated mesoporous silica followed by the co-immobilization of glucoamylase (GA) and glucose oxidase (GOx). The biohybrid (GA&GOx@Au–SiO2) catalyst was highly efficient achieving a gluconic acid yield of 98% within 30 min, which is considerably superior than that obtained using the partly integrated system and free system. The high performance is attributed to the proximity effect of Au nanoparticles, GOx, and GA allowing the rapid transport of the reaction intermediates, glucose and H2O2, the latter being decomposed on the Au sites. Moreover, the GA&GOx@Au–SiO2 catalyst conserved 84% of its initial activity after 10 consecutive runs, which was higher than that without immobilized Au nanoparticles. These examples show that these catalytic systems are interesting alternatives to the use of multienzymatic approaches in terms of performance and stability.
Carbon nitride (C3N4) was used for the first time as a support by Wu et al.356 to construct a biohybrid catalyst through stepwise immobilizing Pd NPs and CalB. The 2D structure of C3N4 providing an abundant amount of N atoms facilitated the immobilization of the Pd NPs. Then, CalB was covalently immobilized on the surface of C3N4 using glutaraldialdehyde as the crosslinker. The bifunctional catalyst showed excellent activity in the hydrogenation of benzaldehyde with H2 at room temperature, while the immobilized CalB showed higher activity in the esterification of the benzyl alcohol than the free enzyme. The cascade process for obtaining benzyl hexanoate from benzaldehyde and ethyl hexanoate was successfully achieved in toluene as solvent in a concurrent mode. The study of the recyclability of the catalyst showed that while the activity of the Pd in the reduction of benzaldehyde was maintained, a strong deactivation of the lipase was observed along consecutive reaction cycles.
Palomo et al.358,359 reported the synthesis of novel nanobiohybrids based on enzyme–metal nanoparticles, which were generated in situ from an aqueous noble metal salt solution and a lipase (CALB). In this protocol, the enzyme acted at the same time as the supporting agent of metal ions and the reducing agent for metal nanoparticle formation, which in addition is stable against aggregation. Different Ag, Au, and Pd nanohybrids (CALB–PdNPs) composed of enzyme aggregates, where the metal nanoparticles are dispersed, were tested in typical metal catalysed reactions such as Heck and Suzuki–Miyaura cross coupling reactions. Taking advantage of the biocatalytic activity of the nanohybrid, which conserved its intrinsic enzymatic activity, and the stability of the metal nanoparticles in aqueous solutions, the authors applied the nanohybrid as a catalyst in a DKR process and in a chemo-enzymatic cascade model reaction. The cascade process, performed in a sequential mode, involves the enzymatic hydrolysis of 4-nitrophenylbutyrate to 4-nitrophenol, followed by the Pd catalyzed chemical reduction of the nitro group to 4-aminophenol using NaBH4 as a reducing agent (Scheme 87). The process was successfully performed using these Pd-nanohybrids as chemo-enzymatic catalysts, and the TOF (defined as the moles of 4-aminophenol per mole of noble metal atoms in the nanocatalyst per minute) was in the range that was previously reported for the metal.
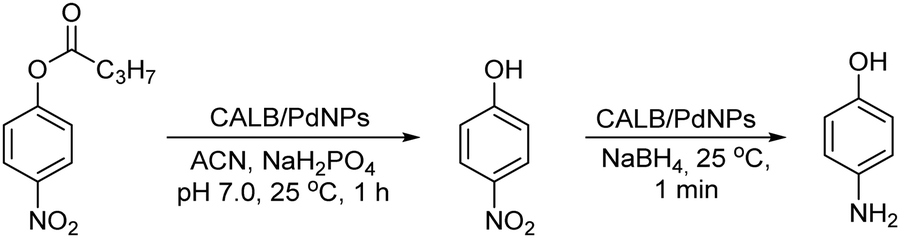 | ||
| Scheme 87 One-pot chemo-enzymatic reaction to obtain 4-aminophenol from 4-nitrophenylbutanoate combining ester hydrolysis with reduction on a lipase–Pd bioconjugated nanohybrid catalyst. | ||
Porcine pancreatic lipase (PPL) encapsulated into a copper ion coordinated nucleotide composite (PPL@CuNC) was prepared through a simple in situ assembly approach followed by the metal reduction with NaBH4.360 The biohybrid catalyst was tested in the same cascade reaction presented in Scheme 87 in a concurrent mode, achieving a 100% yield of 4-aminophenol. However, deactivation of the catalyst was observed, mainly due to the leaching of Cu metal nanoparticles, and after seven consecutive runs only 40% of its initial activity was retained. Recently, Palomo et al.361,362 reported a novel strategy for the preparation of silver and gold nanoparticles–enzyme–polymer conjugate hybrids as bifunctional catalysts for cascade reactions. The authors prepared dextran–aspartic acid polymers that were used for encapsulating CALB through the ion exchange between the amino groups of the enzyme and the carboxylic groups of the polymer to obtain polymer–CALB conjugates, where Au and Ag ions were supported. It was observed that the enzyme–polymer conjugates directly caused the formation of smaller nanoparticles in both metal systems, without the necessity of an external reducing agent. The bifunctional catalyst was effectively applied in the stereoselective hydrolysis/cyclization cascade process starting from an allenic acetate to produce a 2,5-dihydrofuran in exceptional yield (88–95%) and enantiopurity (Scheme 88).
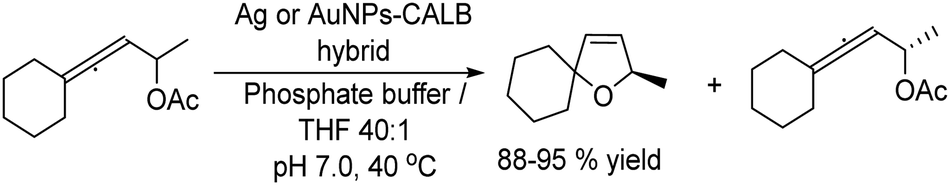 | ||
| Scheme 88 Lipase–silver and gold bioconjugated nanohybrids as catalysts for the hydrolysis/cyclization of allenic acetate. | ||
Interestingly, the authors also reported the synthesis of magnetic Fe nanoparticles–enzyme nanobiohybrids consisting of iron nanoparticles homogeneously dispersed in a lipase matrix that confers high stability against aggregation and oxidation of Fe. These novel magnetic nanostructures were used as heterogeneous catalysts for C–C coupling reactions, chemoselective hydrogenation and degradation of pollutants.363
Bäckvall's group364 developed a bifunctional catalyst based on cross-linked enzyme aggregates (CLEAs) of a lipase where Pd nanoparticles were introduced.
The CLEA was obtained by cross-linking with glutaraldehyde the lipase CALB and subsequently it was treated with palladium acetate that was reduced with Na(CN)BH3 to generate the Pd(0)–CALB CLEA. The amorphous structure of the nanohybrid suggested that the Pd NPs were integrated and bound inside the CLEA matrix. These particles were used as catalysts in the Pd catalysed cycloisomerization of 4-pentynoic acid to form a lactone that acted as an acyl donor for the lipase catalyzed kinetic resolution of a variety of benzyl alcohols (Scheme 89). Interestingly, the bifunctional catalyst was stable along six consecutive reaction cycles, retaining its activity and selectivity.
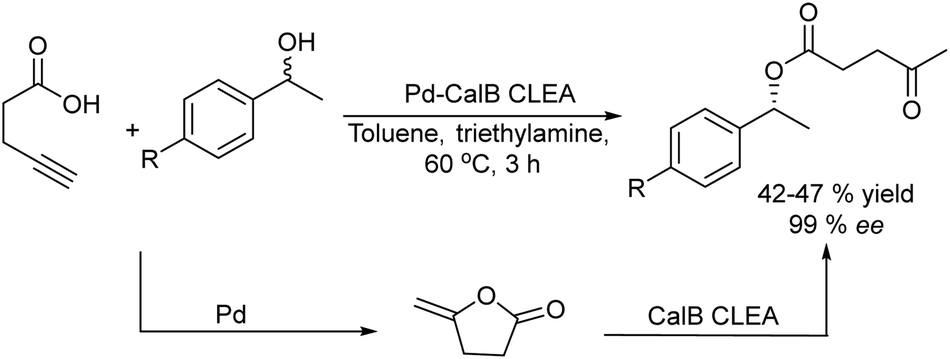 | ||
| Scheme 89 Kinetic resolution of benzyl alcohols using a the Pd–CalB–CLEA bioconjugated nanohybrid as the catalyst. | ||
An advantage of the CLEA–metal catalytic system with respect to the biocatalyst supported on inert carriers is the increase of the catalytic activity per mass unit of the catalytic material. However, a drop in the CLEA enzymatic activity with respect to free enzymes is commonly observed.
In a similar way, Ge et al.365 prepared an enzyme-bimetallic hybrid catalyst by in situ reduction of PdCu nanoclusters on cross-linked lipase aggregates (CALB CLEAs). The catalytic activity of PdCu/CALB CLEAs was evaluated in the chemo-enzymatic synthesis of (R)-N-[1-(4-(phenylethynyl)phenyl)ethyl]-acetamide (Scheme 90), which is an important building block of chiral compounds. In this one-pot cascade reaction that occurs in a concurrent mode, lipase catalyzes the acylation of the (R) enantiomer of amine and the bimetallic nanoclusters have two different functions: they catalyze the Sonogashira reaction and the racemization of the (S) enantiomer of amine, simultaneously (Scheme 90). The strong bimetallic synergistic effect existing between Pd and Cu within PdCu/CALB CLEAs provided excellent activity in the cascade process, achieving 90% yield with 99% ee of the target product. In addition, the catalyst retains 70% of its initial activity after ten consecutive runs.
 | ||
| Scheme 90 Chemo-enzymatic synthesis of (R)-N-[1-(4-(phenylethynyl)phenyl)ethyl]-acetamide in a concurrent mode using the PdCu/CALB CLEA catalyst. | ||
4.3. Spatial compartmentalization
The generation of different microenvironments in cellular processes enabling the compatibility of specific tasks in enzymatic reactions in a one-pot mode has inspired researchers to develop different strategies to combine chemo- and biocatalysts by compartmentalizing the catalytic systems. In this section, a variety of existing methods for spatial compartmentalization of chemo- and enzymatic catalysts applied to relevant chemo-enzymatic transformations will be discussed.The combination of cross-alkene metathesis with biocatalyzed stereoselective epoxidation in a concurrent mode was first reported by Hartwig et al.366 using a Ru carbene catalyst and cytochrome P450 to perform the metathesis and epoxidation respectively in a biphasic system (Scheme 91). The cascade reaction that involves the metathesis between terminal unsaturated carboxylic acids and symmetrical alkenes followed by epoxidation with P450 was performed in a biphasic isooctane–buffer medium. An air and water stable second-generation Hoveyda–Grubbs’ catalyst was selected for the metathesis step, while isooctane was selected as the organic phase due to its suitability as a solvent for olefin metathesis and compatibility with P450 enzymes. In this process, the equilibrium-controlled metathesis process that leads to a mixture of alkenes occurs in the organic phase, while the P450 enzyme in the aqueous phase transforms selectively one alkene from the mixture of alkenes into epoxide. Owing to the irreversible enzymatic step that shifted the metathesis equilibrium, the target product could be obtained in higher yield than those achieved in a stepwise process.
 | ||
| Scheme 91 Chemo-enzymatic cross-metathesis/epoxidation in a biphasic system. | ||
Using a similar strategy, Castagnolo and co-workers reported the direct synthesis of pyrroles and furans from diallyl amines and diallyl ethers respectively in a biphasic isooctane-buffer medium. Pyrroles were obtained through one-pot cascade ring-closing metathesis/aromatization by a combination of ruthenium Grubbs’ catalyst and monoamine oxidase derived from Aspergillus niger (MAO-N) (Scheme 92a).367 Furans were obtained by combining the ruthenium Grubbs’-catalyzed ring-closing metathesis of diallyl ethers and Trametes versicolor laccase/TEMPO for aromatization (Scheme 92b).368 The strategy was effectively applied to synthesise a variety of pyrroles and furans in moderate to good yields.
 | ||
| Scheme 92 Chemo-enzymatic ring-closing metathesis/aromatization cascade in a biphasic system to obtain pyrroles from diallyl amines (a) and furans from diallyl ethers (b). | ||
Chemo-enzymatic systems in biphasic media have also been utilized for valorization of biomass. For instance, and as discussed above, the integration of glucose to fructose isomerization and fructose dehydration into a one-pot system to produce HMF is of relevant importance. Dumeignil et al.369 developed a strategy for the synthesis of HMF, combining the enzymatic isomerization of glucose into fructose using the glucose isomerase followed by the chemical dehydration of fructose. The strategy was based on the compartmentalization of both catalysts (the enzyme and the chemocatalyst) in two isolated aqueous phases separated by an organic–liquid interphase. Thus, glucose is first enzymatically isomerized to fructose in the aqueous phase. Fructose is then preferentially complexed with aryl boronic acid at the interphase of the organic phase, and the fructose complex is transported through the organic phase to the interphase of the second aqueous receiving phase containing citrate buffer (pH 3), where the complex is hydrolyzed and subsequently fructose dehydrates to HMF. The fructose transport allows overcoming the limitation of thermodynamic equilibrium of glucose–fructose isomerization, achieving 70% glucose conversion instead of 46% without fructose transport. However, the reversibility process between the two aqueous phases limited the final yield of HMF (4%), although selectivity was high (70%) (Scheme 93).
 | ||
| Scheme 93 Steps for the synthesis of HMF from glucose in a biphasic system. | ||
More recently, He and co-workers reported two chemo-enzymatic one-pot processes to valorize corncob into furfuryl amine and furoic acid370 in a biphasic medium. In these processes, performed in a sequential mode, biomass is first converted to furfural in the presence of a heterogeneous acid catalyst and subsequently biotransformed into furoic acid or furfuryl amine. For instance, corncob was transformed into furfural using an optimized heterogeneous acid catalyst based on tin-based solid acid Sn-bentonite (Sn-BTN) and methyl isobutyl ketone (MIBK)–H2O as the optimized biphasic medium at 170 °C, yielding 53.3% furfural. After this, the temperature was decreased to 30 °C, pH was adjusted, and Brevibacterium lutescens whole cells were added to the reaction medium. The enzymatic step achieved 100% furoic acid within 18 h (Scheme 94a). In a similar way, for the production of furfuryl amine, corncob was converted into furfural using sulfonated aluminium-based alkaline-treated graphite (Al-AG) as a catalyst in a γ-valerolactone–water biphasic medium (1:4) at 180 °C, for 40 min, and a maximum furfural yield of 56.8% was obtained. Then, the enzymatic step was performed at 35 °C by adding Aspergillus terreus whole cells expressing ω-transaminase and isopropyl amine as the amine donor. After 2 h, furfuryl amine was obtained in 79.0% yield (Scheme 94b). Recently these results were improved by using a robust mutant of Aspergillus terreus whole cells achieving 90–93% yield of furfuryl amine, at higher substrate concentration (500 mM).327,371
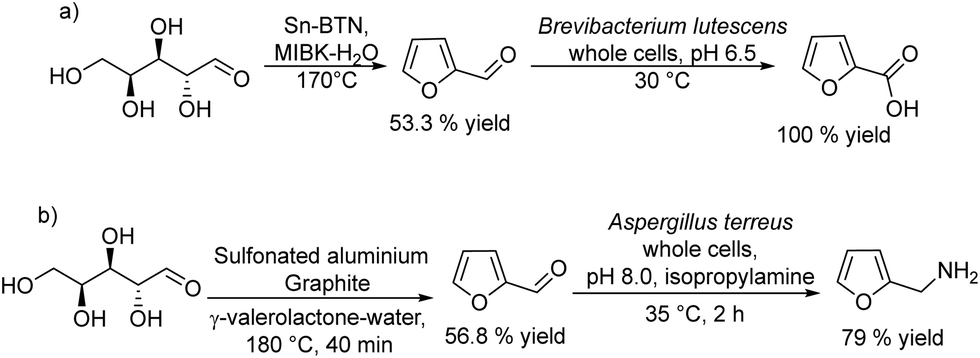 | ||
| Scheme 94 Chemo-enzymatic one-pot processes to valorize corncob into (a) furoic acid and (b) furfuryl amine in a biphasic medium. | ||
Recently, it has been reported that carboxylic acids, which are easily available from biomass resources, can be converted into nitriles through a new chemo-enzymatic cascade process in a biphasic system.372 The strategy combines the aldehyde synthesis from carboxylic acids using a carboxylic acid reductase (CAR) followed by chemical in situ aldoxime formation and its subsequent dehydration to nitrile catalyzed by an aldoxime dehydratase (Scheme 95). The process was performed in a biphasic buffer/heptane system in a sequential mode, where first CAR harbouring cells were incubated with hydroxylamine for 4 h. Then, aldoxime dehydratase (OxdBr1) harbouring cells were added and reaction proceeded for 24 h. The cytotoxic activity of the aldehyde intermediate on CAR was avoided by removing the aldehyde as aldoxime, while a key point was to select an aldoxime dehydratase able to work under the aerobic conditions required for the CAR enzyme. Interestingly the presence of the organic phase improved product recovery of aliphatic and volatile nitriles with respect to the single phase buffer media.
 | ||
| Scheme 95 Chemo-enzymatic conversion of carboxylic acids into nitriles in biphasic media. | ||
A biphasic system composed of an ionic liquid (IL) and an aqueous buffer was used by Schmitzer et al.373 to synthesize chiral biaryl alcohols by combining the chemocatalyzed Suzuki cross-coupling reaction and the enantioselective enzymatic bioreduction. Optimization of the Suzuki step showed that the water-immiscible (1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide) [bmim][NTf2] was optimal for the cross-coupling reaction between 4-iodoacetophenone and boronic acids. Additionally, for the enzymatic step, lyophilized cells of E. coli containing an over-expressed organic solvent tolerant ADH were used. In good agreement with the results reported by Gröger et al.374 in aqueous media, it was found that the Pd catalyst did not have a negative impact on the catalytic activity of ADH, while the base and the boronic acid had an important detrimental effect on the enzymatic activity. By changing the nature of the base (triethylamine instead of K2CO3) and controlling the equivalents of boronic acid during the first step, the one pot reaction was performed successfully in a sequential mode. Thus, when the Suzuki reaction, performed at 110 °C, was over, the system was cooled at room temperature and the E. coli cells, along with the required additives, were added to the reaction media. The cascade sequence proceeded (Scheme 96) successfully with a global conversion higher than 94% and the chiral alcohol was obtained in >99% ee. The main advantage of this one-pot biphasic reaction is that both phases containing the different catalysts can be easily separated and reused (the cells in the aqueous phase and the Pd complex in IL). Additionally, the biphasic system was successfully applied to the synthesis of a variety of biaryl chiral alcohols.
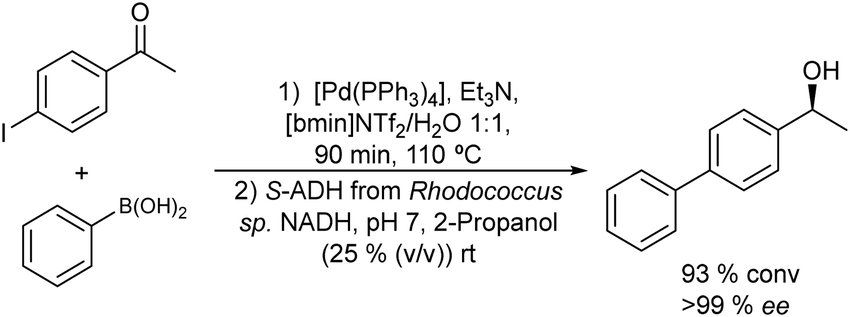 | ||
| Scheme 96 Synthesis of biaryl chiral alcohols by combining the chemocatalyzed Suzuki cross-coupling reaction with enantioselective enzymatic bioreduction in a sequential mode and in biphasic media. | ||
More recently Jiang et al.375 used the same biphasic buffer/[bmim][NTf2] media to perform the synthesis of chiral alpha arylcycloketones in a sequential mode. The process involves as the first step the palladium catalyzed Suzuki cross-coupling of 2-iodocyclohexenones with boronic acids, followed by the bioreduction of the alkene group using E. Coli whole cells expressing ene-reductase (Scheme 97).
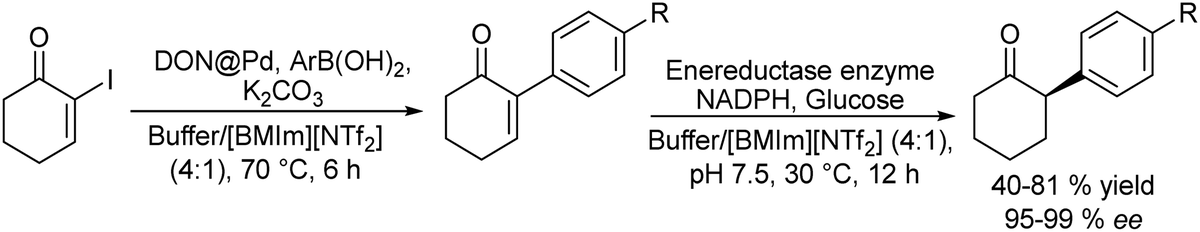 | ||
| Scheme 97 Synthesis of chiral α-aryl cycloketones by combining the chemocatalyzed Suzuki cross-coupling with enzymatic bioreduction in biphasic media. | ||
To overcome the inhibitory effect on the enzyme activity of the triphosphine ligands of the Pd homogeneous catalyst, the first step was performed using Pd-immobilized on dendritic organosilica nanoparticles (DON@Pd) as a heterogeneous catalyst. Under optimized reaction conditions of the chemo-enzymatic process, Pd-catalyzed C–C formation took place in the IL phase, and the enzymatic reduction in the buffer phase was satisfactorily achieved and applied efficiently to the synthesis of a variety of chiral alpha arylcycloketones. Moreover, to access enantiocomplementary products site-directed mutation of the ene-reductase was performed.
Cycloalkenes are important products of the petrochemical industry and are employed in several processes like solvents, building blocks for cyclic compounds, etc. Ward et al. developed a chemo-enzymatic process that allows access to cycloalkenes (C5–C7) from renewable sources.376 The authors performed a cascade process, where a diacid substrate is bis-decarboxylated via a decarboxylase enzyme (UndB), followed by a ring-closing metathesis (RCM) catalyzed by ruthenium Grubbs’ catalyst(I) (Scheme 98).
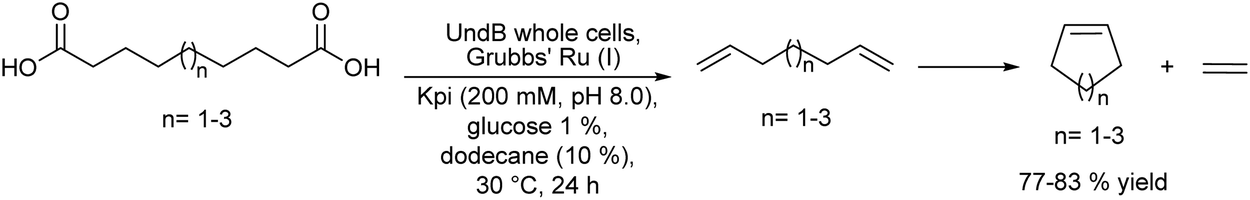 | ||
| Scheme 98 Chemo-enzymatic conversion of diacids into cycloalkenes in biphasic media and in a concurrent mode. | ||
The decarboxylase UndB expressed in E. Coli whole cells was identified among different decarboxylases, as the most active, while a biocompatible commercial Grubbs’ ruthenium(I) catalyst was selected for the RCM step. The low solubility of the substrate in water limited the yield of the cycloalkene, and therefore to increase substrate solubility different biphasic and micellar systems were studied. Thus, using a biphasic medium composed of water/n-dodecane (10%) the chemo-enzymatic process could be successfully performed in sequential and concurrent modes achieving the corresponding cycloalkenes in 87–90% yield. Interestingly, the authors developed engineered E. coli cells, to convert oleic acid into diacids, which when combined with engineered E. coli cells expressing lipase, the enzyme-optimized decarboxylase and the Ru catalyst were able to transform olive oil into different cycloalkenes in moderate yields (29–33%).
The use of aqueous micellar systems as a new and efficient alternative to traditional organic solvents for metal-catalyzed processes such as Heck, Suzuki, Sonogashira C–C couplings has been recently demonstrated by Lipshutz et al.377 With this idea, micelles formed by a tailor-made TPGS-750-M surfactant were applied by the authors378 in some chemo-enzymatic one-pot processes by combining metal-catalyzed reactions with bioreduction using alcohol dehydrogenase (ADH) to produce chiral secondary alcohols. The TPGS-750-M surfactant, containing racemic vitamin E as the hydrophobic tail (Scheme 99), is able to form non-anionic micelles of 50 nm size fully compatible with the ADH. In this system, the metal-catalyzed reaction takes place inside the micelle, while the bioreduction occurs in the aqueous phase. Moreover, the authors showed that micelles are responsible for increasing the enzymatic activity towards highly lipophilic substrates, acting as reservoirs for substrates, catalysts, and products of enzymatic reduction, minimizing enzymatic inhibition and moderating the degree of enzyme saturation.
 | ||
| Scheme 99 Structure of the TPGS-750-M surfactant. | ||
Different chemo-enzymatic processes involving Pd-catalyzed Sonogashira and Heck couplings, Au-Ag-catalyzed alkyne hydration and asymmetric Rh-catalyzed 1,4-addition were successfully coupled after adjusting substrate concentration and pH with the ADH catalysed reduction in a sequential mode. An example of the combination of Sonogashira cross-coupling with bioreduction in the micellar system is presented in Scheme 100.
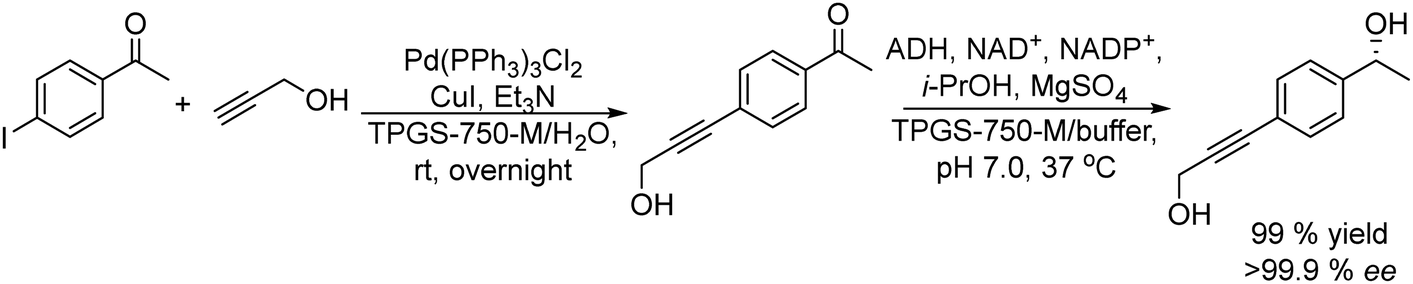 | ||
| Scheme 100 Combination of Pd-catalyzed Sonogashira cross-coupling with bioreduction in a micellar system. | ||
The biocompatibility of TPGS-750-M allowed the efficient application of the aqueous micellar approach in a variety of two-step and multistep chemo-enzymatic sequences involving ene-reductases379 and alcohol dehydrogenases.380
Turner et al.381 also used this surfactant to combine biocatalyzed chiral amine synthesis with the Pd-mediated Buchwald–Hartwig amination to obtain a variety of chiral N-aryl amines, which are key scaffolds of many active pharmaceutical components. Amine dehydrogenases (AmDHs) were used in the first step to produce chiral amines from prochiral ketones that were coupled in the second step with aryl bromides in a sequential mode. The addition of the surfactant in the second step allowed retaining the reactants and catalyst inside the micelles, thus preventing the deactivation of the Pd catalyst by the species present in the aqueous media (Scheme 101).
 | ||
| Scheme 101 Synthesis of chiral aniline derivatives by combining ketone amination with Buchwald–Hartwig amination in a micellar system. | ||
A similar strategy was adopted by Paradisi and Heckman382 by using ω-transaminases in the first step, while in the second step, the coupling of the chiral amine with aryl bromides was performed in a biphasic toluene–buffer medium, instead of micelles, to avoid the deactivation of the Pd catalyst. A variety of chiral N-arylamines in excellent enantioselectivities (>99.5% ee) and high yields (up to 89%) were obtained.
Hastings et al.383 also used the TPGS-750-M surfactant to combine efficiently two transition metal-catalyzed steps (Heck and metathesis reactions) with enzymatic hydrolysis in a concurrent mode. The reactions proceeded with high yields under mild conditions and at ambient temperature.
Gonzalez-Sabin et al.384 developed a chemo-enzymatic one-pot process in a sequential mode to obtain stilbene derivatives from carboxylic acids. The process combines enzymatic decarboxylation with Pd catalyzed Heck coupling in aqueous media in the presence of different surfactants. Particularly, the Cremophor EL® surfactant, a polyethoxylated castor oil classically used in the formulation of low water soluble drugs, resulted in the highly efficient decarboxylation of p-hydroxycinnamic acid into 4-vinylphenol using phenolic acid decarboxylase (PAD), which was coupled with the Pd-catalyzed Heck reaction with iodobenze to obtain (E)-4-styrylphenol (Scheme 102).
 | ||
| Scheme 102 Combination of a biocatalyzed decarboxylation step with a Heck reaction in a micellar system to synthesize stilbene derivatives from carboxylic acids. | ||
Interestingly, the micellar compartmentalization avoids the inhibitory effect of the enzyme on the Pd catalyst, allowing superior performances of the chemo-enzymatic process when compared with the process performed using a mixture of water–eutectic solvent (choline chloride/glycerol (1ChCl/2Gly:H2O) as the reaction medium with free or immobilized PAD.385
An interesting study recently reported by Kunz et al.386 on the impact of the solvent when combining transition metals, biocatalysts and hydrophobic substrates in one-pot chemo-enzymatic reactions concluded that, while using micellar solutions can improve the process, the formation of micelles protecting the enzyme is not the key factor for the success of the process. In fact, the authors showed that when a chemo-enzymatic process combining Heck cross-coupling and ADH bioreduction of ketone was performed in a binary mixture of water/isopropanol (67/33 (w/w)), the enzyme remained partially undissolved and folded and the transition metal catalyst did not have a negative impact on its activity, while the yield of the target compound increased with respect to the process performed in the presence of a surfactant (TPGS-750-M). Moreover, further improvement in performance was achieved in a surfactant-free microemulsion composed of water/isopropanol/benzyl alcohol, concluding that the solubility and stability of the components in the reaction media are the key parameters controlling the performance of the cascade process.
The use of particle-assisted emulsions, also referred to as Pickering emulsions, is an alternative to the use of molecular surfactants to confine the catalytic species. Recently, Wu et al.387 reported the Pickering chemo-enzymatic cascade for the one-pot synthesis of chiral esters from ketones (Scheme 103). The authors prepared a silica nanoparticle catalyst containing anchored chiral Ru(II) complexes that act not only as heterogeneous metal catalysts to reduce the ketone into the alcohol, but also as active emulsifiers that form Pickering emulsions to encapsulate the lipase CALB, which catalyzes the subsequent enantioselective esterification of the alcohol. Thus, the first step was performed through an asymmetric transfer hydrogenation promoted by an optimized Ru(II) catalyst using water as a solvent, achieving 100% conversion and ee >90%. Then, the Pickering emulsion was formed by adding hexane directly to the aqueous solution of the first-step reaction by vortex, and then CALB along with the substrate vinyl butyrate was added to the reaction system for the second step, and the reaction proceeded at room temperature for over 12 h, affording 38% of the chiral ester (ee 99%). Interestingly, the particle-stabilized emulsion gave a much better conversion than the classical two-phase systems, which can be attributed to the large-interface area in the Pickering emulsion and the high local concentration of the chiral Ru complex on the particles.
 | ||
| Scheme 103 Combination of symmetric transfer hydrogenation of ketone with bioesterification in Pickering emulsion. | ||
 | ||
| Scheme 104 Combination of biodecarboxylation with metathesis using the encapsulated enzyme. | ||
Encapsulation of chemical catalysts within protein frameworks, which are known as artificial metallo enzymes,389 is another strategy to separate biocatalysts from chemo-catalysts in cascade processes. This strategy has been mainly applied to catalysts based on organometallic complexes, where the mutual deactivation of the enzyme and the metal complex is commonly observed. Thus, Ward et al.390 incorporated a biotinylated iridium complex [Cp*Ir(Biot-p-L)Cl] within streptavidin (Sav) creating a novel artificial transfer-hydrogenase (ATHase), where the iridium complex is protected by the protein from other catalytic species outside the Sav shell (Fig. 1).
 | ||
| Fig. 1 Reaction cascades resulting from combining an ATHase with a biocatalyst. Reproduced with permission: Copyright 2013, Springer Nature (adapted from ref. 390). | ||
The authors performed a variety of concurrent cascades by combining the ATHase with several co-factor-dependent redox enzymes using sodium formate as a hydride source. As an example, in Scheme 105 is presented the synthesis of L-pipecolic acid from L-lysine.
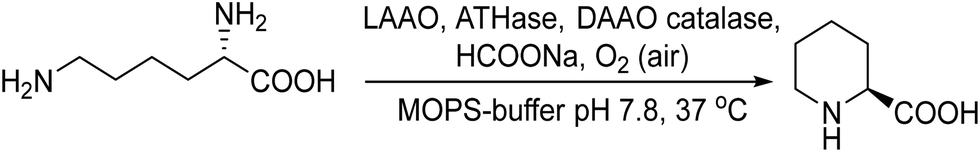 | ||
| Scheme 105 Combination of ATHase with L-amino acid oxidase (LAAO) and D-amino acid oxidase (DAAO) in a concurrent mode to produce L-pipecolic acid from L-lysine. MOPS-buffer: 3-morpholinopropane-1-sulfonic acid. | ||
Following a similar strategy, the authors also developed an NAD(P)H-dependent artificial transfer hydrogenase (ATHase), which enabled imine reduction with NADPH as the hydride source and which was concurrently regenerated by a glucose dehydrogenase (GDH) using glucose as the substrate. The integration of this catalytic system with a catalase and a monoamine oxidase enables the production of enantiopure amines in a four-enzyme cascade process under physiological conditions.391 The benefit of compartmentalizing the Ir complex inside the protein was the improvement of the Ir turnover number (TON) of at least 30 (6% yield) for the free metal complex to TON > 445 (89% yield) with the ATHase. Moreover, the suppression of formate as the hydride source, which may cause deactivation of natural enzymes, is another important advantage of this catalytic system. Additionally, and based on this approach, the authors modified ATHase for regeneration of NADH mimics using formate as the hydride source. The combination of the ATHase with a variety of ene-reductases was used for the synthesis of chiral maleimides in a concurrent mode.392
More recently, Schwaneberg et al.393 developed an artificial metallo enzyme composed of a covalently anchored Grubbs’–Hoveyda type catalyst on an engineered variant of the outer membrane protein (FhuA) of Escherichia coli. The metallo enzyme was successfully combined with a decarboxylase to produce symmetric stilbenes from cinnamic acid. The chemo-enzymatic process involving cinnamic acid decarboxylation followed by metathesis reaction was performed in aqueous media in a sequential mode, yielding the stilbene derivatives in 64–74% yields (Scheme 106). In contrast, when a commercial and water-soluble Grubbs’–Hoveyda catalyst (AquaMet) was used in the second step, only traces of the corresponding stilbene derivatives were detected in the reaction media. This finding is attributed to deactivation of the metal catalyst due to its unspecific coordination with the enzyme, which does not occur when the metallo enzyme is used as the catalyst for the metathesis step.
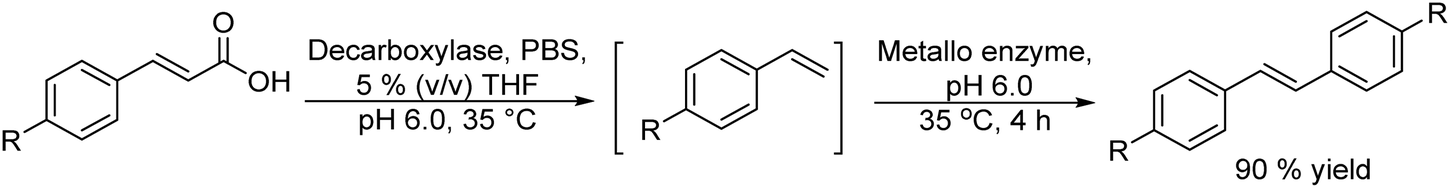 | ||
| Scheme 106 Synthesis of symmetric stilbenes from cinnamic acid by combining a decarboxylase with a metallo enzyme. | ||
Encapsulation of transition-metal complexes inside organic-metal supramolecular clusters to compartmentalize metal catalysts was performed in a seminal work conducted by Toste et al.394 In this remarkable work, the authors encapsulated both Au(I) and Ru(II) within Ga4L6 (L = N,N′-bis(2,3-dihydroxy-benzoyl)-1,5-diaminonapthalene) tetrahedral clusters (see Fig. 2). The authors showed that the encapsulation stabilizes the metal complex, enhances its water solubility and prevents the interaction of the metal with amino acid groups of the enzyme. Then, they performed different concurrent chemo-enzymatic cascade processes by coupling the metal complex activity with esterases, lipases and alcohol dehydrogenases. For instance, by coupling esterase catalyzed hydrolysis of acetate allene derivatives with Au(I)-catalyzed cyclization of the alcohol intermediate, substituted tetrahydrofurans were obtained in 100% selectivity (see Scheme 107). In another example an encapsulated Ru complex was used to isomerize 2-propen-1-ol into propanal, which was subsequently reduced to propanol by ADH using formate as the hydride source. This study showed the high potential of supramolecular clusters as hosts of metal complexes for developing chemo-enzymatic cascades in a concurrent mode.
 | ||
| Fig. 2 Schematic view of the Ga4L6 tetrahedral supramolecular assembly in which each edge of the tetrahedron represents a bisbidentate ligand and each vertex represents a gallium center. | ||
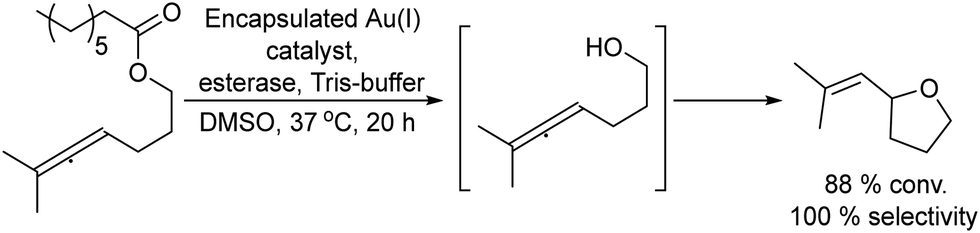 | ||
| Scheme 107 Chemo-enzymatic cascade in a concurrent mode involving acetate allene hydrolysis followed by cyclization using an encapsulated Au(I) catalyst. | ||
Recently, Patel et al.395 reported a chemo-enzymatic process in a sequential mode, involving Ru-catalyzed metathesis of unsaturated malonic acid esters followed by ester hydrolysis catalysed by esterases, to obtain cyclic malonic acid monoesters (see Scheme 108), using encapsulated chemo- and biocatalysts. Ru Grubbs’ catalyst and the esterase were separately encapsulated in hydrogel beads derived from the biopolymer alginate. The encapsulation of both catalysts avoided the contact between them, increasing the enzyme activity, but a decrease of the metal catalyst activity was observed upon encapsulation. However, the advantage of this strategy with respect to the use of the free catalysts previously reported176 was that the encapsulated catalysts could be recycled for seven consecutive runs showing good performances, and thus a better use of the catalysts was achieved. Interestingly, later, the reaction could be performed in ac concurrent mode by co-encapsulating both catalysts in different compartments of a core–shell cross-linked chitosan bead.396
 | ||
| Scheme 108 A chemo-enzymatic process to obtain cyclic malonic acid monoesters using encapsulated chemo- and biocatalysts. | ||
Guohua et al.397 developed a chemo-enzymatic process to produce chiral amines from propargylethers (Scheme 109). The authors combined an Au/carbene complex encapsulated in hollow-shell-structured mesoporous silica with an engineered thermostable phenylalanine dehydrogenase (from Geobacillus kaustophilus) (GkAmDH). The process, which comprises an initial gold-catalyzed hydration of propargylethers into ketones followed by the reductive amination of the in situ generated ketones, can be performed in a concurrent mode, owing to the encapsulation of the gold catalyst preventing enzyme deactivation in the aqueous buffer reaction media. The process provides access to a wide range of chiral amines (25 examples) in 82–97% yield with 96–99% ee. Moreover, the enzyme and chemocatalyst could also be recovered and reused for five consecutive runs, representing a useful method for the preparation of chiral amines.
 | ||
| Scheme 109 Synthesis of chiral amines from propargylethers by combining alkyne hydration with an encapsulated Au complex with bioamination in a concurrent mode. | ||
Recently, Yang et al.398 have reported the simultaneous encapsulation of a lipase CALB and Grubbs’ catalyst, in a multi-compartmental MOF microreactor. The bifunctional catalyst was synthesized through a Pickering double emulsion-directed interfacial synthesis method, by adding both catalysts during the MOF synthesis, specifically MOF-74. The catalytic performance of the prepared Grubbs’/CALB@MOF was tested in the ring-closing metathesis/transesterification cascade process of 1,6-heptadien-4-ol as a model substrate to form cyclopentenyl acetate in a concurrent mode (Scheme 110). After optimization of the lipase/Grubbs’ catalyst ratio a conversion of 97.6% and 98% selectivity to the target compound could be achieved.
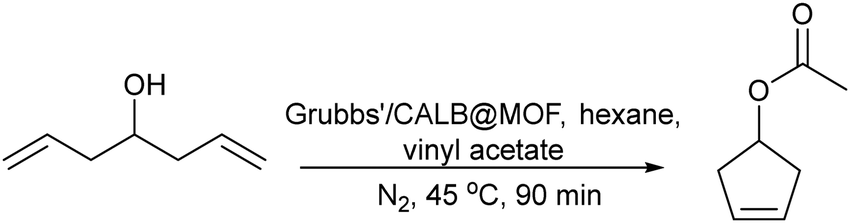 | ||
| Scheme 110 One-pot ring-closing metathesis/transesterification cascade reaction of 1,6-heptadien-4-ol to form 3-cyclopentenyl acetate in a concurrent mode. | ||
Interestingly, the activity of the Grubbs’/CALB@MOF catalyst was considerably superior to those exhibited by the homogeneous counterparts (Grubbs’/CALB), a physical mixture of the catalysts (Grubbs’@MOF/CALB@MOF), and traditionally immobilized catalysts on MOF (Grubbs’@MOF/CALB@MOF) and on mesoporous SBA-15 (Grubbs’/CALB@SBA-15). The higher activity exhibited by the Grubbs’/CALB@MOF catalyst is attributed to the restrained mutual inactivation and substrate channeling effects. Thus, the close confinement of the catalytic sites and the dense packing compartments of the MOF facilitate a high local concentration and the efficient transportation of the intermediates to the enzyme sites. Moreover, the catalyst was found to be stable, retaining its activity over five consecutive runs. The combination of encapsulated solid acid catalysts with enzymatic reactions in a one pot linear cascade has been recently reported by Bornscheuer et al.399 The authors performed the synthesis of homovanillyl butyrate (HVB), a derivative of homovanillyl alcohol (HVA) with antioxidant properties,400 through a chemo-enzymatic cascade in a concurrent mode involving the deacetalization, reduction, and acylation of the lignin depolymerization product G-C2 dioxolane phenol (DOX) (Scheme 111). In this strategy, the acid catalyzed deacetalization of DOX to homovanillin (HV) in aqueous media is achieved by Amberlyst-15 encapsulated in the hydrophobic polydimethylsiloxane (Amberlyst15@PDMS). Then, the HV formed is reduced to HVA by horse liver alcohol dehydrogenase (HLADH) recombinantly produced in E. coli, preventing the accumulation and polymerization of HV. Then, through an acyl transferase reaction (using an acyltransferase/hydrolase variant optimized by the authors (PestE)) the HVA is transformed into butyl ester by transesterification with butyl butyrate, shifting the equilibrium of the HLADH reduction step. The encapsulation of the solid acid catalyst not only prevents the enzyme deactivation by the acid sites, but also allows two incompatible reactions in water media such as deacetalization and transesterification to take place in the same reaction vessel. This example represents an interesting approach to valorize lignin derivatives such as DOX, which is accessible through diol-assisted lignocellulose acid catalyzed depolymerization giving lignin derived acetals.
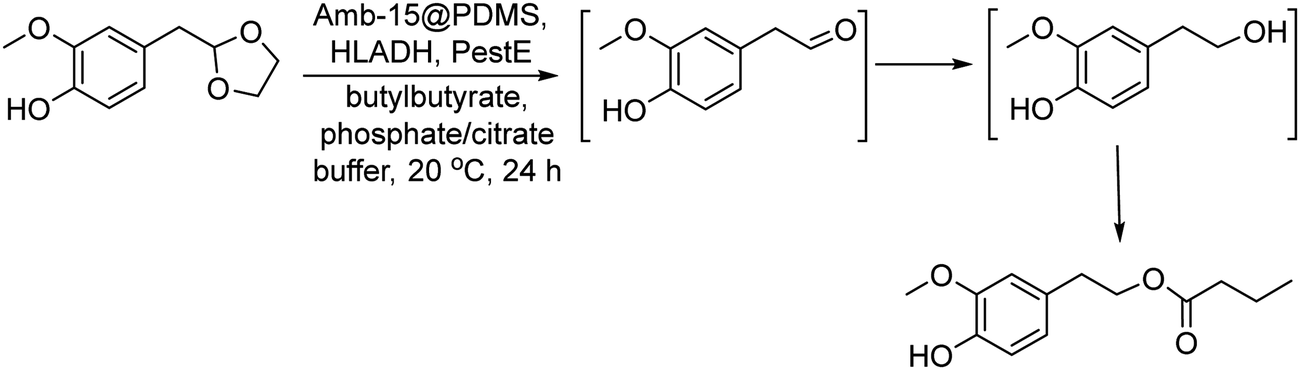 | ||
| Scheme 111 Synthesis of homovanillyl butyrate by combining acetal hydrolysis with bioreduction followed by bioesterification. | ||
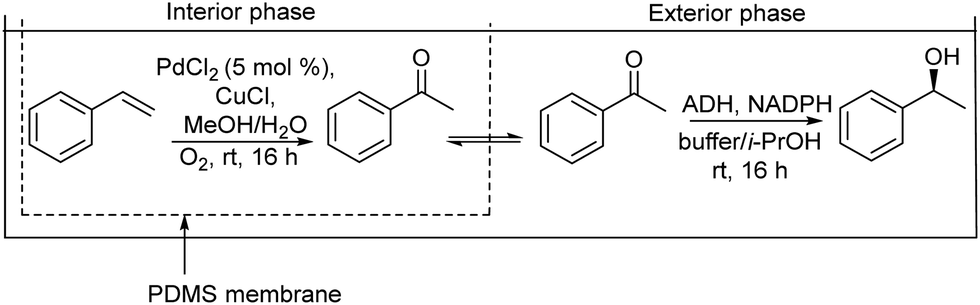 | ||
| Scheme 112 Combination of Wacker oxidation and enzymatic reduction using a PDMS thimble as the compartmentalization strategy. | ||
Preliminary studies showed that Cu species had a strong negative effect on the enzyme activity. Therefore, to prevent the catalyst deactivation the metals and enzymes were separated using the PDMS thimble. Thus, the Wacker oxidation occurs inside the PDMS membrane, and due to its hydrophobic properties, it allows the intermediate acetophenone formed inside to pass through the membrane to the exterior where the stereoselective enzymatic reduction takes place. Moreover, the membrane retains the metals inside, and the enzyme and co-factor outside, achieving an effective compartmentalization. Following this strategy, the process was performed in a concurrent mode; however, the oxidation in the interior leads to only 20% conversion, as a consequence of the extraction of the methanol (the solvent component required for an efficient Wacker oxidation) into the exterior. To suppress the methanol leaching, the cascade process (Wacker oxidation and enzymatic reduction) was performed in a sequential mode. Thus, ADH and the co-factor were added in a buffer/isopropanol solution to the exterior after completion of the initial Wacker oxidation in the interior. Following this method, the accumulated acetophenone was converted in high yield into the chiral 1-phenylpropanol. The process was successfully extended to a variety of substituted styrenes that were converted into the corresponding chiral 1-phenylpropanol derivatives in high yield and 94–99% ee. Interestingly, the Pd catalyst could be easily recovered and recycled over 15 consecutive cycles and remained active for at least 60 days. A similar strategy was used for deracemization of achiral 1-phenylethanol derivatives into (R)-phenyl ethanol derivatives.402 The racemic mixture of alcohols is oxidized into acetophenone in the interior chamber of PDMS using MnO2 as the oxidant, which is detrimental for the enzyme (ADH). The acetophenone passes through the membrane to the exterior chamber, and after oxidation completion, the ADH is added into the exterior chamber where the ketone is enantioselectively reduced. The process performed in a sequential mode could be applied to the deracemization of a wide range of methyl- and chloro-substituted 1-phenylethanols achieving up to 93% yield and >99% ee.
Following the PDMS thimble compartmentalization method, Latham et al.403 combined the regioselective flavin-dependent halogenase (Fl-Hal) enzyme with palladium-catalysed Suzuki–Miyaura cross-coupling to perform the regio-controlled arylation of a variety of aromatic compounds. Preliminary optimization studies showed that the enzyme inhibits the metal catalyst due mainly to metal complexation. The PDMS thimble approach overcame this drawback and improved the synthetic efficiency. All reagents were placed in the reaction system from the beginning of the reaction in an aqueous buffer. First, the enzymatic halogenation takes place in the exterior of the thimble at room temperature and after completion, temperature is adjusted to 80 °C to perform the Pd catalysed cross-coupling inside the thimble (see Scheme 113). Moreover, the authors combined the use of halogenase crosslinked aggregates (CLEAs) with membrane compartmentalization, which allowed decreasing the Pd loading from 50 mol % to 2 mol %. The chemo-enzymatic cascade process allowed the regio-controlled arylation of a variety of inactivated aromatic compounds such as benzamides, isoquinolines and indoles, which usually cannot be accomplished through biocatalyzed or chemocatalyzed processes alone.
 | ||
| Scheme 113 Regio-controlled arylation of aromatic compounds in a PDMS thimble system. | ||
Rudroff et al.404 performed a chemo-enzymatic cascade reaction in a concurrent mode by combining the Cu/Pd catalyzed C–C cross-coupling of thioesters with boronic acids (Liebeskind and Srogl (L–S) coupling) and the subsequent ADH catalyzed reduction of the ketone intermediate in a reactor with two compartments separated by a PDMS membrane (see Fig. 3). Although L–S coupling generally works in organic solvents, previous studies showed that t-butyl thioester is stable to the hydrolysis when the L–S reaction is performed in aqueous media. Thus, in the cascade process, this substrate could be successfully coupled with boronic acids in a water-isopropanol solvent, yielding the corresponding ketone intermediate, which is transported to the enzymatic compartment where the ADH along with the co-factor (NADP+) in a buffer solution at pH 8 are retained, giving the chiral secondary alcohol. The control of the pH was key for the enzymatic step since the acidic pH caused mainly by the excess of boronic acid used deactivated the enzyme. The compartmentalized chemo-enzymatic process in a concurrent mode was extended to a variety of halogenated phenylboronic acids and the corresponding chiral secondary alcohols were obtained in up to 99% yield and high enantioselectivity (>99% ee) over two steps.
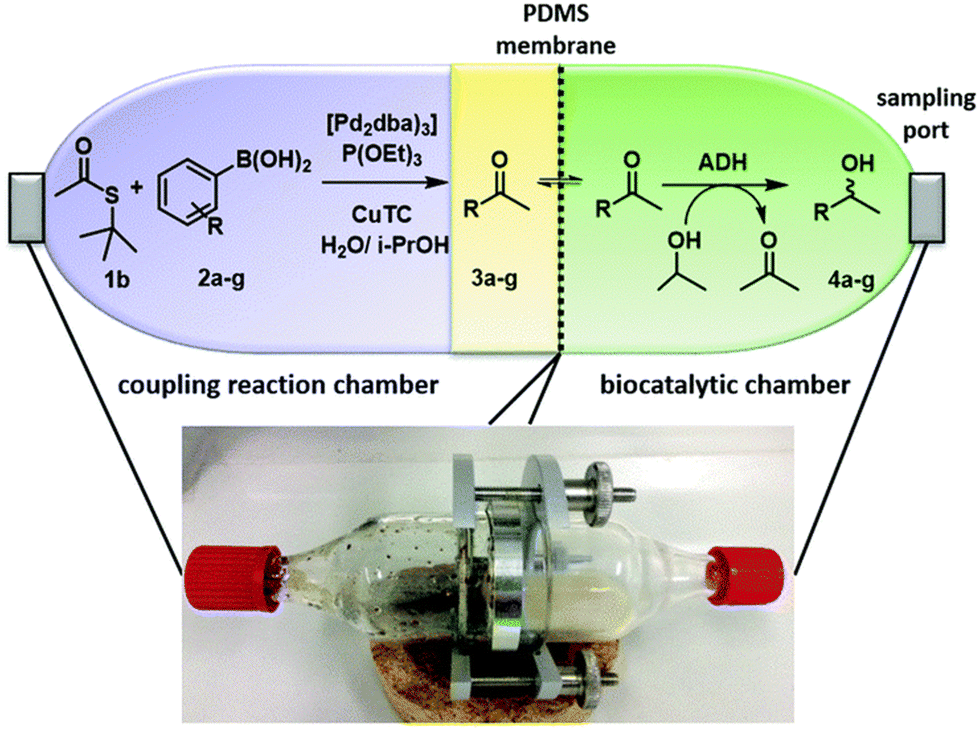 | ||
| Fig. 3 Reactor device for a chemo-enzymatic reaction sequence in a concurrent fashion. Pd2(dba)3: tris(dibenzylideneacetone)-dipalladium (0), P(OEt)3: triethyl phosphite, CuTC: copper-(I)-thiophene-2-carboxylate. Reproduced with permission: Copyright 2018, The Royal Society of Chemistry (adapted from ref. 404). | ||
The high versatility of the flow systems towards various reaction conditions allows their combination to perform reaction sequences providing very different reaction conditions for the catalysts, but moreover the flow reactors can be combined with other technologies that can result in the development of entirely automated process with an enlarged throughput.
While immobilized enzymes have been used for a long time in flow systems,408 the combination of chemo-enzymatic reactions in separate flow reactors has recently emerged as an approach for effective compartmentalization of catalysts and reaction conditions that avoid incompatibility problems. For instance, Gröger et al.409 developed a solvent-free sequential chemo-enzymatic continuous process for the production of chiral β-amino acid esters. Thus, ethyl (S)-3-(benzylamino)butanoate) (Scheme 114) was obtained by combining the thermal aza-Michael addition of benzylamine to trans-ethyl crotonate with a subsequent lipase-catalyzed kinetic resolution via enantioselective aminolysis of the racemic intermediate product. The reactor set-up combined a plug-flow reactor for the thermal aza-Michael reaction and a packed-bed reactor for the lipase Novozym 435 (which is commercially available Candida antarctica lipase B (CALB)) physically immobilized on a mesoporous poly(methyl methacrylate) support.
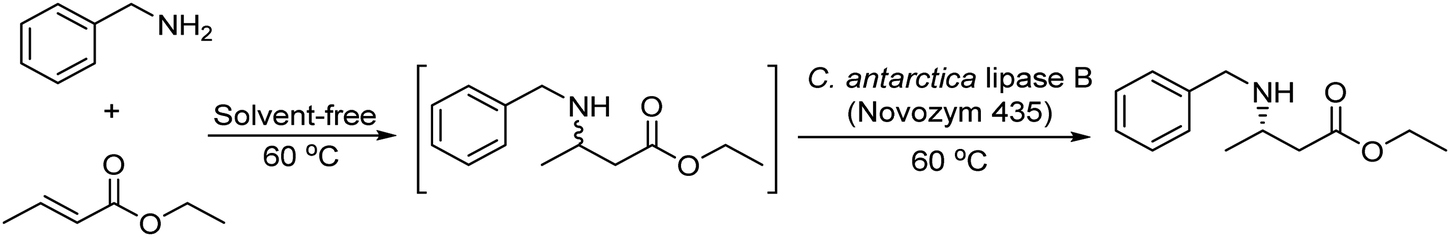 | ||
| Scheme 114 Solvent-free chemo-enzymatic reaction sequence for the synthesis of β-amino acid ester in flow reactors. | ||
The combined reactors worked continuously for 80 h without significant loss of enzyme activity. The target β-amino ester was obtained with 52% yield and enantiomeric excess of >98%, which is superior to that obtained in a batch mode. The combined flow system allows for higher substrate concentrations, while the productivity of the process is comparable to established industrial processes.
Corma et al.410 developed a continuous flow system to deracemize valuable chiral alcohols through a chemo-enzymatic process by combining two-packed bed reactors (Scheme 115). In a first reactor, the racemic alcohol was oxidized to the corresponding prochiral ketone via Oppenauer oxidation using a Zr-beta zeolite as a catalyst at 60 °C and using acetone as the hydrogen acceptor. Then, after removal of the excess of acetone and further dilution with buffer solution, the ketone intermediate was pumped into a second packed-bed reactor where an ADH supported on a 2D zeolite (ITQ-2) performed the stereoselective reduction at 25 °C. A variety of alcohols were deracemized by this procedure in high yields and enantiomeric excess (>99%), independently of the ADH stereo-preference. Moreover, the system could be operated, at least, for 16 days. An interesting feature of this chemo-enzymatic process is that the system meets the principle of 100% atom economy because the isopropanol co-produced in the first step acts as a co-factor regenerator in the bioreduction step, while the acetone accompanying the target chiral alcohol is reincorporated at the outset of the process.
 | ||
| Scheme 115 Chemo-enzymatic synthesis of chiral alcohol from racemic mixtures using two fixed bed reactors. | ||
The same authors411 developed a chemo-enzymatic process to prepare diesters of 2,5- bis(hydroxymethyl)furan (BHMF), useful as biobased plasticizers that can be substitutes of phthalates derived from petroleum.412 Starting from 5-hydroxymethylfurfural (HMF), the process involves as a first step the hydrogenation of HMF into 2,5-bis(hydroxymethyl)furan (BHMF) using monodispersed metallic Co nanoparticles with a thin carbon shell (Co@C) as a catalyst followed by the BHMF bioesterification with vinyl esters using a supported commercial lipase (Novozym 435) (Scheme 116). The process was performed in an integrated two-step continuous flow reactor by combining two fixed bed reactors. In the first reactor the hydrogenation of HMF to BHMF was performed at 90 °C and 10 bar H2, using methanol as a solvent. After methanol evaporation, a solution containing the vinyl ester in 2-methyltetrahydrofuran (2-MTHF) was used as the feed for the second fixed bed reactor containing the lipase and heated at 35 °C. The combination of two flow reactors working under optimized reaction conditions (for reduction and subsequent esterification) allowed achieving ∼90% overall yield of the diesters of BHMF, while the activity was retained at least for 60 h of operation.
 | ||
| Scheme 116 Synthesis of diesters of 2,5-bis(hydroxymethyl)furan from HMF by combining reduction with bioesterification in two continuous flow reactors. | ||
Recently, Kourist et al.8 reported the synthesis of stilbene derivatives of interest in the pharmaceutical industry due to their anti-oxidant and anti-inflammatory activities. For instance, the synthesis of (E)-4-hydroxy-stilbene was performed by a two-step continuous flow process involving the enzyme catalyzed decarboxylation of para-coumaric acid and subsequent Pd-catalyzed Heck cross-coupling with an aryl halide (Scheme 117). Thus, in the first fixed bed reactor the decarboxylation of para-coumaric acid catalyzed by an immobilized phenolic acid decarboxylase (PAD) takes place. The resulting 4-hydroxystyrene is pumped along with the aryl halide into the second reactor where the heterogeneous Pd catalyst (Sn0.79Ce0.2Pd0.01O2-d) is placed, giving the target compound. The process was successfully operated for more than 16 h. In this system the use of a choline chloride-based deep eutectic solvent (DES) played a key role to overcome the problem of solvent compatibility while improving substrate concentration (from 5 mM in buffer to 20 mM in DES). Optimization of the reaction conditions allowed broadening the concept for the synthesis of pterostilbene and resveratrol.413
 | ||
| Scheme 117 Synthesis of (E)-4-hydroxy-stilbene from p-coumaric acid by combining decarboxylation with Heck reaction in a two-step continuous flow reactor. | ||
Recently, the synthesis of hordenine (4-(2-dimethylaminoethyl)phenol), a valuable phenolic phytochemical, has been performed through a novel chemo-enzymatic strategy in a fully-automated continuous flow system, starting from the broadly available L-tyrosine.414 The two-step cascade involves as the first step the decarboxylation of L-tyrosine into tyramine in a buffer solution catalyzed by an immobilized tyrosine decarboxylase derived from Lactobacillus brevis (LbTDC). The N,N-dimethylation of the resulting tyramine is performed with formaldehyde (reductive amination) in a second reactor where the water insoluble pic-BH3 (picoline borane) as a reducing agent suspended on Celite is placed. The complete conversion to hordenine is achieved in less than 5 minutes residence time, and the produced hordenine is extracted from the aqueous phase with ethyl acetate in 77% yield (Scheme 118). Compared to the metal-catalyzed N,N-dimethylation of tyramine, the chemo-enzymatic strategy reduces the environmental impact of the process and considerably improves its productivity.
 | ||
| Scheme 118 Chemo-enzymatic synthesis of hordenine from L-tyrosine in two-step continuous flow reactors. | ||
Besides the above-presented continuous processes performed entirely by combining flow reactors, there are interesting compartmentalized chemo-enzymatic processes that combine flow reactors with batch reactors and with biphasic systems.
For instance, Mihovilovic et al.415 developed a chemo-enzymatic process for the synthesis of the natural (S,S)-aerangis lactone, a compound of interest in the fragrance industry. The process combines the syn-hydrogenation of dihydrojasmone in a flow reactor filled with Rh/C and Cs2CO3, followed by the Baeyer–Villiger oxidation catalysed by cyclododecanone monooxygenase (CDMO) in a batch reactor (Scheme 119). The hydrogenation in the flow reactor was performed using heptane as a solvent, and the efflux of the flow reactor containing the cis-cyclopentanone derivative intermediate was pumped to the batch reactor containing the enzyme and the required additives in an aqueous buffer solution. To facilitate material transfer between the organic and aqueous phases the use of a non-ionic, non-denaturing surfactant (Triton X-100) was required. The cis-aerangis lactone was obtained as the sole product (>99% de).
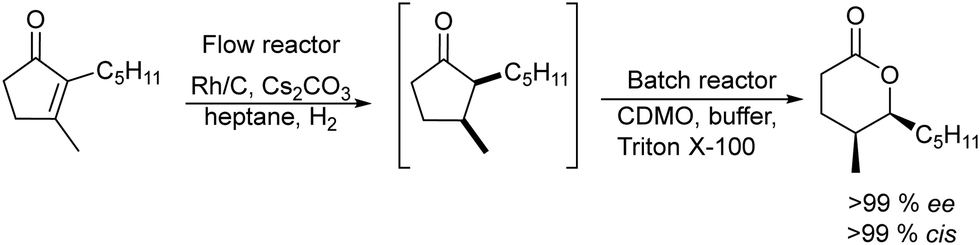 | ||
| Scheme 119 Stereoselective synthesis of natural (S,S)-aerangis lactone from dihydrojasmone through a chemo-enzymatic process combining flow and batch reactors. | ||
Souza et al.416 developed a chemo-enzymatic continuous flow process for the synthesis of a biaryl intermediate of odanacatib, a drug for the treatment of postmenopausal osteoporosis. The process was performed by combining asymmetric biocatalyzed reduction of 4-bromo-2,2,2-trifluoroacetophenone with ADH in isopropanol as a solvent in a batch mode with a Pd-catalyzed Suzuki–Miyaura cross coupling in a continuous reactor. Thus, the chiral alcohol intermediate obtained in the first step and the Pd catalyst in an isopropanol solution along with an aqueous solution of the boronic acid and the required base were pumped from separate feeds through a PFA-coil reactor at 110 °C under microwave heating. The desired Odanacatib biaryl intermediate was obtained in 73% overall yield, without isolation of the chiral alcohol intermediate (Scheme 120).
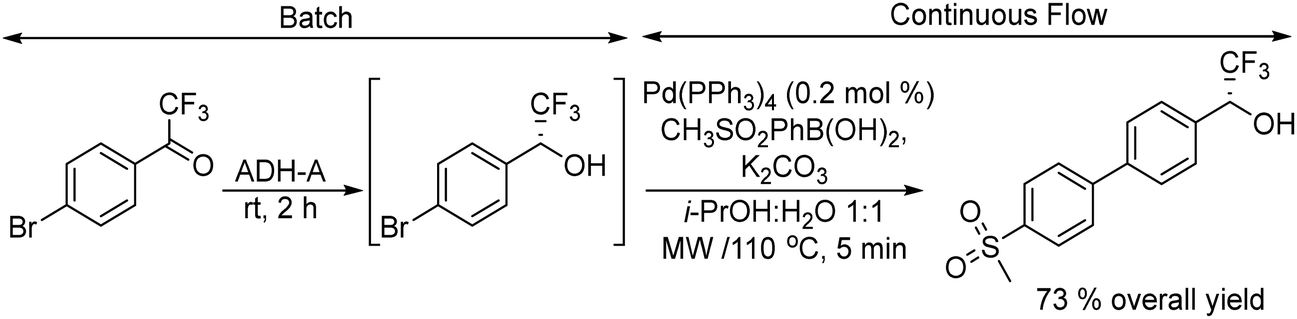 | ||
| Scheme 120 Chemo-enzymatic continuous flow process for the synthesis of a biaryl intermediate of odanacatib by combining batch and flow reactors. | ||
Sieber et al.9 developed a cascade process for the direct synthesis of 2-keto-3-deoxy sugar acids, as important biobased building blocks, starting from the corresponding sugars. The process involves as the first step the chemical oxidation of the sugar, using Au supported on Al2O3, which is subsequently dehydrated using a dihydroxyacid dehydratase, namely, DHAD derived from Sulfolobus solfataricus (SsDHAD) (which has been described as a thermostable enzyme with activity for a broad range of sugar acids) to give the desired 2-keto-3-deoxy sugar acid (Scheme 121). Preliminary studies showed the mutual deactivation of the Au catalyst and the enzyme. To overcome this problem the catalysts were compartmentalized using a fed-batch continuous flow system. So, for instance, in the first step, the L-arabinose was oxidized to L-arabonate in a semicontinuous batch reactor (SCBR) with O2 flow in the presence of Au/Al2O3 and using water as a solvent yielding L-arabonate (90% yield). Then, the solid catalyst was filtered and a catalase enzyme was added to the mixture to eliminate the H2O2 produced during the oxidation step that deactivates the DHAD enzyme. Subsequently the reaction mixture was introduced in a fixed bed flow reactor, which contains the ssDHAD enzyme immobilized over a Histrap FF crude column. Under these conditions, the desired 2-keto-3-deoxy sugar acid was obtained in good yields (58%). Other aldoses such as xylose, galactose and glucose were transformed into the corresponding keto-deoxy sugar acids in a yield range of 69–91%.
 | ||
| Scheme 121 Chemo-enzymatic synthesis of 2-keto-3-deoxy sugar acids from aldoses by combining a semicontinuous batch reactor with a fixed bed flow reactor. | ||
Very recently, Corma et al.417 reported the synthesis of chiral 4-phenyl-2-butanamine derivatives, which are important chiral inductors and building blocks for the synthesis of drugs, starting from prochiral ketones sourced from biomass through a very high atom-economy process. The strategy involves a one-pot aldol condensation-reduction step between benzaldehyde derivatives and acetone using a bifunctional base/metal catalyst (Pd/MgO) to perform the aldol condensation and CC hydrogenation respectively. The prochiral 4-phenyl-2-butanone obtained is aminated into the target amine using an electrostatically immobilized transaminase (ATA) on a 2D ITQ-2 zeolite. The first step was performed in a semicontinuous batch reactor (SCBR) at 100 °C, using acetone as the reactant and solvent. After reaction completion, the excess of acetone was eliminated and the obtained 4-phenyl-2-butanone (after adjusting concentration and addition of the required additives) was pumped into a fixed bed reactor where the ATA immobilized NITQ-2 was placed. After the amination step (performed at 37 °C), the corresponding chiral amines were obtained in yields >95% and ∼100% ee (Scheme 122).
 | ||
| Scheme 122 Chemo-enzymatic synthesis of 4-phenyl-2-butanamine derivatives by combining a semicontinuous batch reactor with a continuous fixed bed flow reactor. | ||
A sophisticated continuous process to produce HMF from glucose was reported by Alipour418 by combining a packed-bed reactor and a biphasic system as the compartmentalization strategy (Scheme 123). The continuous process has three main steps that are shown in Scheme 123. In the first step, a glucose aqueous solution was isomerized into fructose using an immobilized glucose isomerase in a packed-bed reactor. Then, the resulting glucose/fructose solution was allowed to come into contact with an immiscible organic phase (octanol) containing a boronic acid derivative (ABA) for preferentially complexing fructose and driving the equilibrium towards more fructose formation. This simultaneous isomerization and reactive extraction (SIRE) are coupled with a second step where the back-extraction of fructose from the organic phase into an immiscible acidic ionic liquid (IL) occurs achieving 90% yield of fructose. Finally, fructose dehydration to HMF is conducted at 50 °C in the biphasic IL/organic solvent reaction medium in the presence of an acid catalyst (HCl) resulting in high HMF yield (>80%).
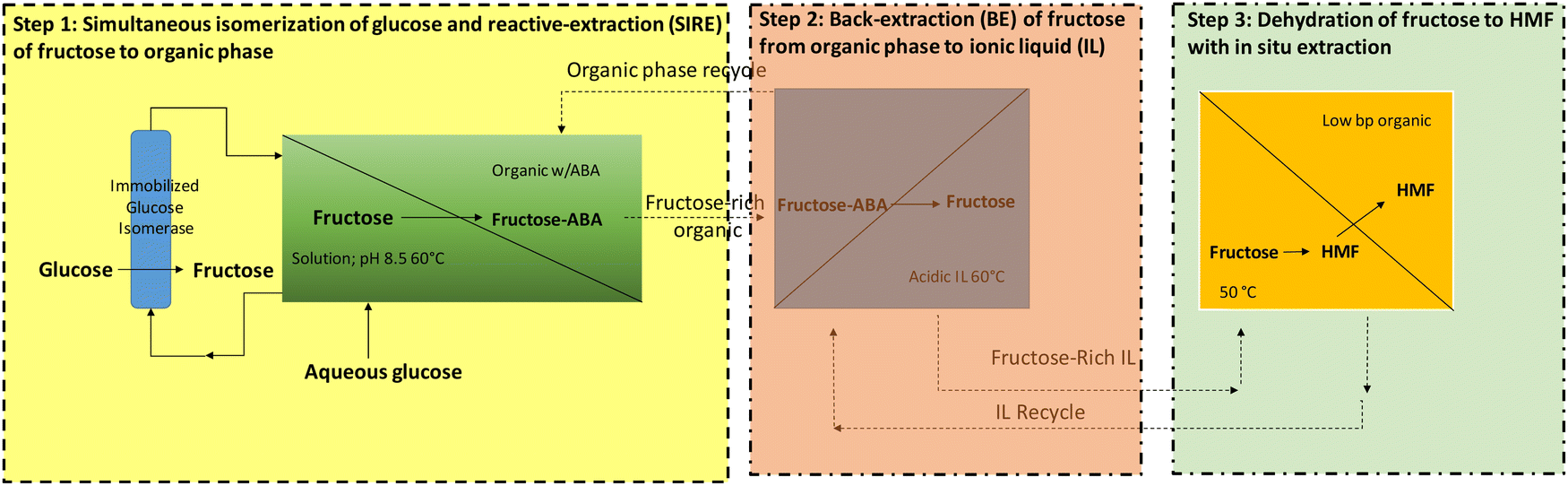 | ||
| Scheme 123 Continuous process for production of HMF from glucose by combining a continuous flow reactor with biphasic systems. | ||
Then, the in situ extraction of HMF with a low boiling point organic solvent (such as THF or methyl isobutyl ketone) is performed. Interesting features of this compartmentalized process are the mild reaction conditions under which all steps (at <70 °C and at ambient pressure) are operated along with the possibility to recycle the organic phase, the IL, and THF; furthermore, this process has also been implemented using a hydrolysate from corn stover as the substrate.
5. Photo-biocatalytic cascades
In the last few decades, photocatalysis using visible light as the energy source has emerged as an important tool in organic synthesis. The wide substrate scope and mild reaction conditions required for photocatalytic processes provide an interesting opportunity to be combined with biocatalysis in cascade processes. Artificial photosynthesis systems (APSs) that mimic natural photo-synthesis by combining photocatalysts and enzymes have been widely described in several reviews419–424 mainly focused on the design of cascade reaction systems, such as the photoactivation of redox enzymes and photoregeneration of co-factors. In contrast, the combination of photocatalytic and biocatalytic transformations in linear cascades in which both steps contribute to the formation of the final product has been scarcely reported. The main reason may be attributed to the biocatalyst inhibition in the presence of highly reactive oxygen species or radical species generated in the photocatalytic step.425 The first one-pot sequential cascade example was reported by Lauder et al.426 for the synthesis of enantiopure 1,3-mercaptoalkanols. 1,3-Mercaptoalkanols belong to the volatile sulphur compounds (VSCs), which are responsible for the aroma of a variety of foods and beverages.In this example the authors coupled in a concurrent mode a photocatalyzed thio-Michael addition between terminal α,β-unsaturated ketones and thiols using [Ru(bpy)3]Cl2 as a photocatalyst under visible light irradiation with subsequent biocatalytic reduction of the ketone group using two highly selective complementary ketone reductases (KREDs) (Scheme 124). The first step was accomplished in only 5 min, while the bioreduction of the ketone group required 24 h. Following this protocol, a variety of chiral 1,3-mercaptotoalkanols were obtained in moderate to good yields (38–73%) with excellent optical purity (99%).
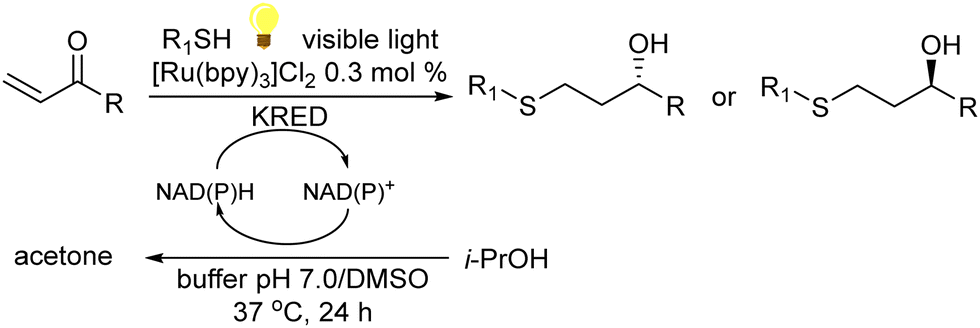 | ||
| Scheme 124 Photo-enzymatic synthesis of enantiopure 1,3-mercaptoalkanols in a concurrent mode. | ||
In another example, Wenger et al.427 developed a photoredox biocatalytic cascade in a concurrent mode for the enantioselective synthesis of amines from imines by a cycling reaction network strategy (Scheme 125). In this approach cyclic imines were reduced into the racemic amines in the first step of the cascade by employing the water soluble Na3[Ir(ppy)3] as a photocatalyst, under visible light irradiation. In the enzymatic step, a monoamine oxidase (MAO-N-9) is used to oxidize enantioselectively one of the enantiomers of the amine to the corresponding imine at the expense of molecular oxygen. The cascade required the presence of hydrogen atomic transfer donors such as ascorbic acid (AscH2) or thiols that capture the highly reactive α-amino alkyl radical intermediates formed in the photocatalyzed step. The continuous cycling of the imine reduction and enantioselective amine oxidation leads to chiral cyclic amines in high yields (95%) and enantiomeric excess (99%).
 | ||
| Scheme 125 Photoredox biocatalytic cascade in a concurrent mode for the enantioselective synthesis of amines from imines. | ||
Visible-light photocatalyzed isomerization of alkenes and the subsequent CC bond stereoselective reduction by ene-reductases were reported by Litman et al.428 In the first step, the photoreduction of E- and Z-alkenes into a E/Z mixture was performed efficiently using flavin mononucleotide (FMN) or a cationic Ir complex as a photocatalyst. In the second step, ene-reductases, which exclusively reduce the E- or Z-isomer, were employed to obtain the desired chiral compound (Scheme 126). The selection of an adequate photocatalyst for the first step that does not inhibit the reductase or glucose dehydrogenase (used as a co-factor regenerating agent) was a critical point. A variety of 2-phenylbut-2-enedioic acid dimethyl ester derivatives and cyanoacrylates were used as substrates obtaining the target enantiomers with moderate to high yields and high enantioselectivity (99%). Interestingly, the authors showed how the enantioenriched compounds obtained through the photo-enzymatic cascade could be transformed into a variety of biologically active compounds and valuable synthetic intermediates.
 | ||
| Scheme 126 Visible-light photocatalyzed isomerization of alkenes and the subsequent double-bond reduction by an ene-reductase in a concurrent mode. GDH/glucose (glucose dehydrogenase/glucose as a co-factor regeneration system). | ||
Schmidt et al.425 reported a photo-enzymatic cascade process by combining photooxidation of alkanes (particularly toluene, aryl alkanes or cycloalkane derivatives) with the corresponding aldehydes or ketones followed by an asymmetric biocatalytic transformation (Scheme 127). Thus, from eleven starting alkanes, and using sodium anthraquinone sulfate (SAS) as the photocatalyst combined with 8 different enzymes, 26 products (chiral hydroxynitriles, amines, acyloins and α- chiral ketones) were obtained with up to 99% ee. The degradation of substrates and inhibition of the biocatalysts due to reactive oxygen species generated in the photocatalytic step as limiting factors affecting the compatibility of both catalytic systems were resolved by using a two-phase system or by temporal and spatial separation of the catalysts.
 | ||
| Scheme 127 SAS-photocatalysed oxyfunctionalisation of alkenes followed by an enzymatic transformation. | ||
This photo-enzymatic cascade approach was also used to obtain chiral amines from alcohols by coupling the photooxidation of several alcohols followed by the reductive amination of the corresponding carbonyl compound intermediate using a variety of ω-transaminases (Scheme 128).73 The system worked in a concurrent mode; however, the productivity was significantly enhanced when the process was performed in a sequential mode.
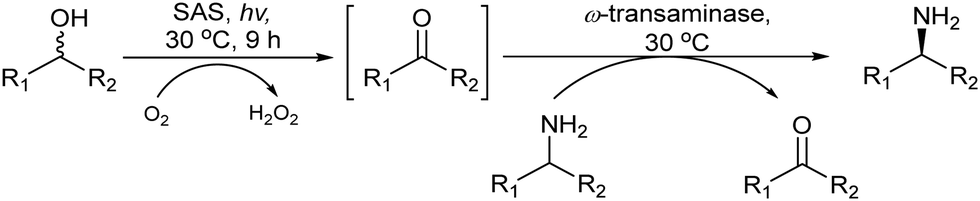 | ||
| Scheme 128 Photo-enzymatic cascade reaction to produce chiral amines from alcohols. | ||
Recently, photooxidation of cyclohexane into cyclohexanone has been combined with the Baeyer–Villiger oxidation using cyclohexanone monooxygenase (CHMO) whole-cells to obtain ε-caprolactone (Scheme 129).429 Au-doped TiO2 (Au-TiO2) and graphitic carbonitride (g-C3N4) were evaluated as photocatalysts under visible-light irradiation. The results showed that when the reaction was performed in a concurrent mode no product could be detected, mainly due to the fast photooxidation of the co-factor NADPH. Therefore, the process was performed in a sequential mode, and after the first step, the biocatalyst was added to the reaction mixture and the reaction proceeded for 24 h in the dark. The highest yield of ε-caprolactone from cyclohexane was achieved using g-C3N4 as the photocatalyst (41%), which was more stable against poisoning than Au–TiO2.
 | ||
| Scheme 129 Photo-biocatalytic cascade oxidation of cyclohexane to ε-caprolactone. | ||
The synthesis of asymmetric 2,2-disubstituted indol-3-ones from 2-arylindoles was reported by He et al.430 by coupling photocatalytic oxidation of 2-arylindoles with the subsequent enantioselective alkylation catalyzed by a lipase in a photo-enzymatic cascade process in a concurrent mode. The photooxidation step was performed using Ru (bpy)3Cl2 as a photocatalyst to obtain the intermediate indolone, which was subsequently asymmetrically alkylated by a wheat germ lipase (WGL) (Scheme 130). A variety of substituted aryl indoles were combined with linear and cyclic ketones as well as dicarbonyl compounds giving the corresponding asymmetric indoles with yields of up to 70% and enantioselectivities of up to 93:7 enantiomeric ratio.
 | ||
| Scheme 130 Photo-enzymatic cascade for the synthesis of asymmetric 2,2-disubstituted indol-3-ones from 2-arylindoles in a concurrent mode. | ||
Recently, Yu et al.431 reported for the first time a photo-enzymatic cascade process for the synthesis of 4H-pyrimido[2,1-b]benzothiazole derivatives from alcohols. In the first step, Rose Bengal (rB) was employed as photocatalyst to oxidize alcohols to aldehydes under visible-light irradiation. Then, the aldehyde intermediate was coupled with ethyl acetoacetate and 2-aminobenzothiazole through the Biginelli reaction biocatalyzed by trypsin to achieve the target product. Intent to do the reaction in a concurrent mode was unsuccessful, probably due to the inactivation of the enzyme by the H2O2 produced in the photocatalytic step. Then, the reaction was optimized in a sequential mode in such a way that after the photocatalytic step, and turning off the light source, the enzyme and substrates (ethyl acetoacetate and 2-aminobenzothiazole) were added and the biocatalyzed multicomponent reaction proceeded (see Scheme 131). Optimization of the solvent showed that a solvent-free procedure was the most favourable approach. Employing a variety of substituted benzyl alcohols fourteen 4H-pyrimido[2,1-b]benzothiazole derivatives were prepared by a simple operation in high yields (up to 98%) under mild reaction conditions, providing a more efficient and eco-friendly strategy, when compared with conventional methods.
 | ||
| Scheme 131 A sequential photo-enzymatic cascade process for obtaining 4H-pyrimidine [2,1-b] benzothiazole derivatives. | ||
A light-driven Wacker oxidation of allyl (hetero)arene derivatives into ketones was coupled with a biotransamination or bioreduction step to obtain enantioenriched amines and alcohols respectively.432 The Wacker oxidation was performed in aqueous media using a Pd(II) complex as a catalyst under aerobic conditions in the presence of 9-mesityl-10-methylacridinium perchlorate ([Acr-Mes]ClO4) as a photosensitizer, under blue light irradiation. After oxidation completion, the enzyme (transaminase or alcohol dehydrogenase) and the required reagents and co-factors were added. A wide panel of chiral amines and alcohols were achieved in good isolated yields, up to 83% (99% ee), under mild reaction conditions in aqueous media (Scheme 132).
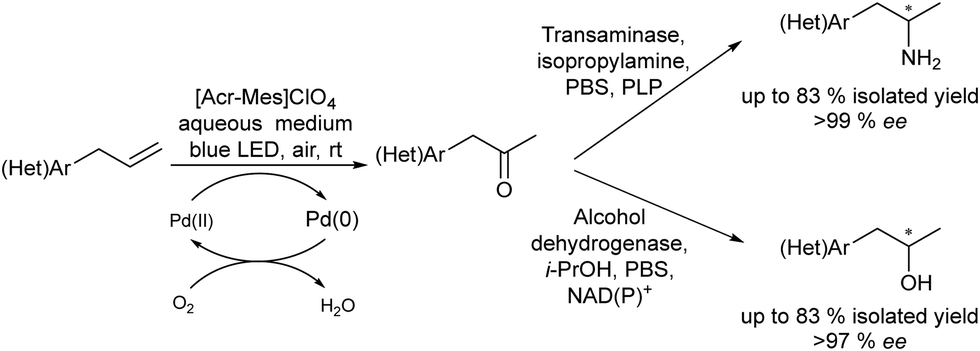 | ||
| Scheme 132 Photo-enzymatic cascade for the synthesis of chiral amines and alcohols. | ||
In a different approach to obtain optically active 1-arylpropan-2-ols, the authors combined the photocatalytic Meerwein arylation between aromatic diazonium salts and isopropenyl acetate under blue LED light irradiation with the bioreduction of the corresponding ketone intermediates.433 The first step was performed in aqueous medium using ([Acr-Mes]ClO4) as a photocatalyst, and the resulting ketone was reduced to the corresponding chiral alcohol in a sequential step using an alcohol dehydrogenase. Using ADHs of opposite stereopreference a total of 19 pairs of 1-arylpropan-2-ol enantiomers were obtained with global yields of up to 76% and high to excellent stereoselectivity (90 to >99% ee) (Scheme 133). The process allows access to an important chiral scaffold from easily accessible reactants and under mild reaction conditions. Interestingly, the authors also showed that it was possible to obtain both 1-phenylpropan-2-ol antipodes, starting from aniline and generating in situ the diazonium salt through a one-pot three-step sequential photobiocatalytic protocol.
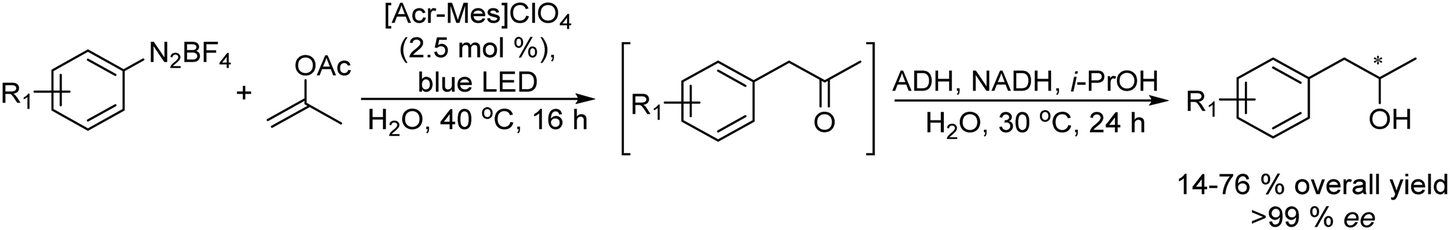 | ||
| Scheme 133 Photo-enzymatic cascade for the synthesis of chiral aryl propanols. | ||
6. Conclusions
In this review, we have presented recent examples of the synthesis of valuable organic compounds (including chiral molecules) through linear cascade processes involving the participation of chemocatalysts (homogeneous and heterogeneous) and enzymes.In the first part of the review, we have shown that one-pot multistep methodologies in a sequential mode, where the different components are added consecutively and the reaction conditions (pH, temperature, substrate concentration, co-solvent, etc) can be adjusted for each reaction step, are the preferred approaches. This is a consequence of the difficulty in finding a compatible operational window where both catalysts can perform efficiently. However, in the last few years the pool of chemo-catalysts (for instance metallic catalysts) able to operate in water has extended enormously, providing new opportunities to combine with biocatalysts, which work in water as their natural world. For instance, the combination of the Wacker oxidation, Suzuki and Heck cross-coupling, and olefin metathesis reactions with enzymatic transformations has been successfully achieved. In addition, while enzyme inhibition by heavy metals is a known phenomenon, catalysts based on transition metals such as Au, Ru, Pd, or Fe have displayed great compatibility with enzymes allowing some cascade processes to proceed in a concurrent manner, as shown by several examples discussed herein. Therefore, searching for compatible catalysts and reaction conditions is one of the main challenges in the future. In this sense, the design of new enzymes based on computational modelling and protein engineering provides enzymes with novel activity, reactivity and stability, which can be substantially improved by the advancements in machine learning and enzyme directed evolution strategies. This fact, along with the development of chemocatalysts able to work efficiently under mild and even physiological reaction conditions, will extend the operational window to combine both catalytic systems, increasing the synthetic applications of chemo-enzymatic cascade reactions.
Furthermore, reaction engineering strategies for solving incompatibility issues in chemo-enzymatic reactions have also been developed, for instance, the use of deep eutectic solvents to solve substrate solubility problems or the spatial separation of catalytic sites through the rational design of enzyme-metal hybrid catalysts to overcome incompatibility problems between catalytic species. However, spatial compartmentalization of the catalytic systems where chemo- and biocatalysts can work effectively without interaction represents a more promising strategy. Thus, the use of biphasic systems and aqueous micellar systems, catalyst encapsulation where the chemo- or the biocatalyst is shielded from external incompatible catalytic species, the use of the porous membranes to confine chemo- and biocatalysts and the use of continuous flow-reactors are reported strategies to generate separate compartments. However, among them, the most promising compartmentalization strategy from the point of view of scalability relies on the continuous flow processes, where incompatible chemo- and biocatalyzed steps can run completely separately under optimized reaction conditions. Although the number of examples of chemo-enzymatic cascades in flow reactors is still very limited, their high potential to manufacture specific compounds on a large scale with high productivity will drive their development in the coming years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Spanish Ministry of Science and Innovation through the “Severo Ochoa Program” (CEX-2021-001230-S) and the PID2021-125897OB-I00 (MCIU/AEI/) 10.13039/501100011033 by “ERDF A way of making Europe” project. J. M. C. thanks the Margarita Salas grant from the Ministerio de Universidades, Universitat Politécnica de Valencia, Spain, funded by the European Union-Next Generation EU (2021–2023).References
- M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. Van Hest, Curr. Opin. Biotechnol, 2014, 28, 10–16 CrossRef CAS PubMed.
- X. Ma and W. Zhang, iScience, 2022, 25, 105005 CrossRef CAS PubMed.
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- M. Mifsud, S. Gargiulo, S. Iborra, I. W. C. E. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- F. Dumeignil, M. Guehl, A. Gimbernat, M. Capron, N. L. Ferreira, R. Froidevaux, J. S. Girardon, R. Wojcieszak, P. Dhulster and D. Delcroix, Catal. Sci. Technol., 2018, 8, 5708–5734 RSC.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- B. Grabner, A. K. Schweiger, K. Gavric, R. Kourist and H. Gruber-Woelfler, React. Chem. Eng., 2020, 5, 263–269 RSC.
- J. M. Sperl, J. M. Carsten, J. K. Guterl, P. Lommes and V. Sieber, ACS Catal., 2016, 6, 6329–6334 CrossRef CAS.
- J. Enoki, J. Meisborn, A. C. Müller and R. Kourist, Front. Microbiol., 2016, 7, 1–8 CrossRef PubMed.
- D. Kracher and R. Kourist, Curr. Opin. Green Sustainable Chem., 2021, 32, 100538 CrossRef CAS.
- Y. Liu, P. Liu, S. Gao, Z. Wang, P. Luan, J. González-Sabín and Y. Jiang, Chem. Eng. J., 2021, 420, 127659 CrossRef CAS.
- M. Makkee, A. P. G. Kieboom, H. Van Bekkum and J. A. Roels, J. Chem. Soc., Chem. Commun., 1980, 19, 930–931 RSC.
- J. F. Ruddlesden and A. Stewart, J. Chem. Res., 1981, 13, 378–379 Search PubMed.
- M. Makkee, A. P. G. Kieboom and H. van Bekkum, Carbohydr. Res., 1985, 138, 237–245 CrossRef CAS.
- J. V. Allen and J. M. J. Williams, Tetrahedron Lett., 1996, 37, 1859–1862 CrossRef CAS.
- P. M. Dinh, J. A. Howarth, A. R. Hudnott, J. M. J. Williams and W. Harris, Tetrahedron Lett., 1996, 37, 7623–7626 CrossRef CAS.
- S. T. Chen, W. H. Huang and K. T. Wang, J. Org. Chem., 1994, 59, 7580–7581 CrossRef CAS.
- T. Riermeier, U. Dingerdissen, P. Groß, W. Holla, M. Beller and D. A. Schichl, DE19955283, 2001.
- A. L. E. Larsson, B. A. Person and J. Backvall, Angew. Chem., Int. Ed. Engl., 1997, 36, 1211–1212 CrossRef CAS.
- J. H. Choi, Y. H. Kim, S. H. Nam, S. T. Shin, M. J. Kim and J. Park, Angew. Chem., Int. Ed., 2002, 41, 2373–2376 CrossRef CAS PubMed.
- O. Pàmies and J. E. Bäckvall, Chem. Rev., 2003, 103, 3247–3261 CrossRef PubMed.
- A. Berkessel, M. L. Sebastian-Ibarz and T. N. Müller, Angew. Chem., Int. Ed., 2006, 45, 6567–6570 CrossRef CAS PubMed.
- V. Köhler and N. J. Turner, Chem. Commun., 2014, 51, 450–464 RSC.
- H. Gröger, in Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, ed. R. Peters, Wiley, 2015, pp. 325–350 Search PubMed.
- P. Hoyos, V. Pace and A. R. Alcántara, Adv. Synth. Catal., 2012, 354, 2585–2611 CrossRef CAS.
- O. Verho and J. E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- P. Hoyos, V. Pace, M. J. Hernáiz and A. R. Alcántara, in Dynamic Kinetic Resolution via Hydrolase–Metal Combo Catalysis, ed. R. N. Patel, John Wiley & Sons, 2016, pp. 373–396 Search PubMed.
- B. Martin-Matute and J.-E. Bäckvall, in Asymmetric Organic Synthesis with Enzymes, ed. V. Gotor, I. Alfonso and E. García-Urdiales, Wiley, 2008, pp. 89–113 Search PubMed.
- C. Ascaso-Alegre and J. Mangas-Sanchez, Eur. J. Org. Chem., 2022, e202200093 CrossRef CAS.
- S. González-Granda, L. Escot, I. Lavandera and V. Gotor-Fernández, Angew. Chem., Int. Ed., 2023, 62, e202217713 CrossRef PubMed.
- S. González-Granda, J. Albarrán-Velo, I. Lavandera and V. Gotor-Fernández, Chem. Rev., 2023, 123, 5297–5346 CrossRef PubMed.
- H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- D. Kracher and R. Kourist, Curr. Opin. Green Sustainable Chem., 2021, 32, 100538 CrossRef CAS.
- H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367–1376 CrossRef CAS PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- S. K. Talapatra, B. Talapatra and K. C. Nicolaou, Chemistry of plant natural products: Stereochemistry, conformation, synthesis, biology, and medicine, Springer, Berlin Heidelberg, 2015 Search PubMed.
- T. Wright, Pharmaceuticals: classes, therapeutic agents, areas of application., Wiley–VCH, Weinheim, 2001 Search PubMed.
- J. H. Lee, K. Han, M. J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999–1015 CrossRef CAS.
- A. Parvulescu, J. Janssens, J. Vanderleyden and D. De Vos, Top. Catal., 2010, 53, 931–941 CrossRef CAS.
- C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 CrossRef CAS.
- C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed.
- A. Díaz-Rodríguez, N. Ríos-Lombardía, J. H. Sattler, I. Lavandera, V. Gotor-Fernández, W. Kroutil and V. Gotor, Catal. Sci. Technol., 2015, 5, 1443–1446 RSC.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- W. Hussain, D. J. Pollard, M. Truppo and G. J. Lye, J. Mol. Catal. B: Enzym., 2008, 55, 19–29 CrossRef CAS.
- H. Kohls, M. Anderson, J. Dickerhoff, K. Weisz, A. Cõrdova, P. Berglund, H. Brundiek, U. T. Bornscheuer and M. Höhne, Adv. Synth. Catal., 2015, 357, 1808–1814 CrossRef CAS.
- E. Keinan, K. K. Seth and R. Lamed, Ann. N. Y. Acad. Sci., 1987, 501, 130–149 CrossRef.
- W. Hummel, Appl. Microbiol. Biotechnol., 1990, 34, 15–19 CrossRef CAS.
- D. Metrangolo-Ruiz de Temiño, W. Hartmeier and M. B. Ansorge-Schumacher, Enzyme Microb. Technol., 2005, 36, 3–9 CrossRef.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- D. Méndez-Sánchez, J. Mangas-Sánchez, I. Lavandera, V. Gotor and V. Gotor-Fernández, ChemCatChem, 2015, 7, 4016–4020 CrossRef.
- E. Liardo, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. Rebolledo, Eur. J. Org. Chem., 2018, 3031–3035 CrossRef CAS.
- E. Liardo, N. Ríos-Lombardía, F. Morís, F. Rebolledo and J. González-Sabín, ACS Catal., 2017, 7, 4768–4774 CrossRef CAS.
- X. Li, L. Xu, G. Wang, H. Zhang and Y. Yan, Process Biochem., 2013, 48, 1905–1913 CrossRef CAS.
- R. A. Sheldon,Chirotechnology: Industrial synthesis of optically active compounds, Marcel Dekker, Inc., New York, 1993 Search PubMed.
- L. Hintermann, Top. Organomet. Chem., 2010, 31, 123–155 CrossRef CAS PubMed.
- I. Schnapperelle, W. Hummel and H. Gröger, Chem. – Eur. J., 2012, 18, 1073–1076 CrossRef CAS PubMed.
- F. Uthoff and H. Gröger, J. Org. Chem., 2018, 83, 9517–9521 CrossRef CAS PubMed.
- F. Thoff, H. Sato and H. Röger, ChemCatChem, 2017, 9, 555–558 CrossRef.
- D. González-Martínez, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2019, 361, 2582–2593 CrossRef.
- L. Hintermann and A. Labonne, Synthesis, 2007, 1121–1150 CrossRef CAS.
- G. A. Olah and D. Meidar, Synthesis, 1978, 671–672 CrossRef CAS.
- P. Schaaf, V. Gojic, T. Bayer, F. Rudroff, M. Schnürch and M. D. Mihovilovic, ChemCatChem, 2018, 10, 920–924 CrossRef CAS.
- M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, S. Schumacher, D. Pérez-Iglesias, F. Morís, V. Cadierno, J. García-Álvarez and J. González-Sabín, ACS Catal., 2017, 7, 7753–7759 CrossRef.
- S. Mathew, A. Sagadevan, D. Renn and M. Rueping, ACS Catal., 2021, 11, 12565–12569 CrossRef CAS.
- S. González-Granda, G. Steinkellner, K. Gruber, I. Lavandera and V. Gotor-Fernández, Adv. Synth. Catal., 2023, 365, 1036–1047 CrossRef.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. Von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- J. Gacs, W. Zhang, T. Knaus, F. G. Mutti, I. W. C. E. Arends and F. Hollmann, Catalysts, 2019, 9, 305 CrossRef.
- M. L. Corrado, T. Knaus and F. G. Mutti, ChemBioChem, 2021, 22, 2345–2350 CrossRef CAS PubMed.
- N. Ahlsten, A. Bartoszewicz and B. Martín-Matute, Dalton Trans., 2012, 41, 1660–1670 RSC.
- P. Lorenzo-Luis, A. Romerosa and M. Serrano-Ruiz, ACS Catal., 2012, 2, 1079–1086 CrossRef CAS.
- N. Ríos-Lombardía, C. Vidal, M. Cocina, F. Morís, J. García-Álvarez and J. González-Sabín, Chem. Commun., 2015, 51, 10937–10940 RSC.
- N. Ríos-Lombardía, C. Vidal, E. Liardo, F. Morís, J. García-Álvarez and J. González-Sabín, Angew. Chem., Int. Ed., 2016, 55, 8691–8695 CrossRef PubMed.
- S. González-Granda, I. Lavandera and V. Gotor-Fernández, Angew. Chem., 2021, 133, 14064–14070 CrossRef.
- A. Abate, E. Brenna, G. Fronza, C. Fuganti, F. G. Gatti, S. Serra and E. Zardoni, Helv. Chim. Acta, 2004, 87, 765–780 CrossRef CAS.
- S. González-Granda, N. V. Tzouras, S. P. Nolan, I. Lavandera and V. Gotor-Fernández, Adv. Synth. Catal., 2022, 364, 3856–3866 CrossRef.
- S. J. Freakley, S. Kochius, J. van Marwijk, C. Fenner, R. J. Lewis, K. Baldenius, S. S. Marais, D. J. Opperman, S. T. L. Harrison, M. Alcalde, M. S. Smit and G. J. Hutchings, Nat. Commun., 2019, 10, 4178 CrossRef PubMed.
- J. Bach, R. Berenguer, J. Garcia and J. Vilarrasa, Tetrahedron Lett., 1995, 36, 3425–3428 CrossRef CAS.
- J. Aleu, B. Bergamo, E. Brenna, C. Fuganti and S. Serra, Eur. J. Org. Chem., 2000, 3031–3038 CrossRef CAS.
- E. Brenna, C. Fuganti, P. Grasselli and S. Serra, Eur. J. Org. Chem., 2001, 1349–1357 CrossRef CAS.
- K. Burgess and L. D. Jennings, J. Am. Chem. Soc., 1991, 113, 6129–6139 CrossRef CAS.
- T. Itoh, E. Akasaki and Y. Nishimura, Chem. Lett., 2002, 154–155 CrossRef CAS.
- A. Ghanem and V. Schurig, Tetrahedron: Asymmetry, 2003, 14, 57–62 CrossRef CAS.
- E. Lindner, A. Ghanem, I. Warad, K. Eichele, H. A. Mayer and V. Schurig, Tetrahedron: Asymmetry, 2003, 14, 1045–1053 CrossRef CAS.
- A. Kamal, M. Sandbhor, A. A. Shaik and V. Sravanthi, Tetrahedron: Asymmetry, 2003, 14, 2839–2844 CrossRef CAS.
- S. Sgalla, G. Fabrizi, R. Cirilli, A. Macone, A. Bonamore, A. Boffi and S. Cacchi, Tetrahedron: Asymmetry, 2007, 18, 2791–2796 CrossRef CAS.
- D. Lee, E. A. Huh, M. J. Kim, H. M. Jung, J. H. Koh and J. Park, Org. Lett., 2000, 2, 2377–2379 CrossRef CAS PubMed.
- Y. K. Choi, J. H. Suh, D. Lee, I. T. Lim, J. Y. Jung and M. J. Kim, J. Org. Chem., 1999, 64, 8423–8424 CrossRef CAS PubMed.
- A. Boffi, S. Cacchi, P. Ceci, R. Cirilli, G. Fabrizi, A. Prastaro, S. Niembro, A. Shafir and A. Vallribera, ChemCatChem, 2011, 3, 347–353 CrossRef CAS.
- S. Fushiya, Y. Kabe, Y. Ikegaya and F. Takano, Planta Med., 1998, 64, 598–602 CrossRef CAS PubMed.
- I. Gill and R. Valivety, Angew. Chem., 2000, 39, 3804–3808 CrossRef CAS.
- G. Sabitha, B. Thirupathaiah and J. S. Yadav, Synth. Commun., 2007, 37, 1683–1688 CrossRef CAS.
- S. Barradas, M. C. Carreño, M. González-López, A. Latorre and A. Urbano, Org. Lett., 2007, 9, 5019–5022 CrossRef CAS PubMed.
- S. Barradas, A. Urbano and M. C. Carreño, Chem. – Eur. J., 2009, 15, 9286–9289 CrossRef CAS PubMed.
- J. M. Longmire, Z. Guoxin and X. Zhang, Tetrahedron Lett., 1997, 38, 375–378 CrossRef CAS.
- E. Burda, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2008, 47, 9551–9554 CrossRef CAS PubMed.
- A. Prastaro, P. Ceci, E. Chiancone, A. Boffi, R. Cirilli, M. Colone, G. Fabrizi, A. Stringaro and S. Cacchi, Green Chem., 2009, 11, 1929–1932 RSC.
- E. Burda, W. Bauer, W. Hummel and H. Gröger, ChemCatChem, 2010, 2, 67–72 CrossRef CAS.
- S. Itsuno, Prog. Polym. Sci., 2005, 30, 540–558 CrossRef CAS.
- D. González-Martínez, V. Gotor and V. Gotor-Fernández, ChemCatChem, 2019, 11, 5800–5807 CrossRef.
- J. M. Longmire, G. Zhu and X. Zhang, Tetrahedron Lett., 1997, 38, 375–378 CrossRef CAS.
- P. V. Ramachandran, G. M. Chen, Z. H. Lu and H. C. Brown, Tetrahedron Lett., 1996, 37, 3795–3798 CrossRef CAS.
- A. W. H. Dawood, J. Bassut, R. O. M. A. de Souza and U. T. Bornscheuer, Chem. – Eur. J., 2018, 24, 16009–16013 CrossRef CAS PubMed.
- C. A. Fleckenstein and H. Plenio, Chem. – Eur. J., 2008, 14, 4267–4279 CrossRef CAS PubMed.
- J. E. Dander, M. Giroud, S. Racine, E. R. Darzi, O. Alvizo, D. Entwistle and N. K. Garg, Commun. Chem., 2019, 2, 82 CrossRef PubMed.
- A. Berkessel and H. Gröger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- B. List and J. W. Yang, Science, 2006, 313, 1584–1586 CrossRef CAS PubMed.
- P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS PubMed.
- K. Baer, M. Kraußer, E. Burda, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2009, 48, 9355–9358 CrossRef CAS PubMed.
- G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944–7947 CrossRef CAS PubMed.
- S. Sonoike, T. Itakura, M. Kitamura and S. Aoki, Chem. – Asian J., 2012, 7, 64–74 CrossRef CAS PubMed.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099–1103 CrossRef CAS.
- G. Rulli, N. Duangdee, W. Hummel, A. Berkessel and H. Gröger, Eur. J. Org. Chem., 2017, 812–817 CrossRef CAS.
- L. Schober, F. Tonin, U. Hanefeld and H. Gröger, Eur. J. Org. Chem., 2022, e202101035 CrossRef CAS.
- C. Ascaso-Alegre, R. P. Herrera and J. Mangas-Sánchez, Angew. Chem., Int. Ed., 2022, 61, e202209159 CrossRef CAS PubMed.
- J. Tian, J. Zhong, Y. Li and D. Ma, Angew. Chem., Int. Ed., 2014, 53, 13885–13888 CrossRef CAS PubMed.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- D. Roca-Lopez, D. Sadaba, I. Delso, R. P. Herrera, T. Tejero and P. Merino, Tetrahedron: Asymmetry, 2010, 21, 2561–2601 CrossRef CAS.
- L. F. Tietze, F. Stecker, J. Zinngrebe and K. M. Sommer, Chem. – Eur. J., 2006, 12, 8770–8776 CrossRef CAS PubMed.
- L. F. Tietze, K. M. Sommer, J. Zinngrebe and F. Stecker, Angew. Chem., Int. Ed., 2004, 44, 257–259 CrossRef PubMed.
- C. J. Li and T. H. Chan, Organometallics, 1991, 10, 2548–2549 CrossRef CAS.
- B. Zhao, X. Peng, S. Cui and Y. Shi, J. Am. Chem. Soc., 2010, 132, 11009–11011 CrossRef CAS PubMed.
- M. Fuchs, M. Schober, J. Pfeffer, W. Kroutil, R. Birner-Gruenberger and K. Faber, Adv. Synth. Catal., 2011, 353, 2354–2358 CrossRef CAS.
- C. J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed.
- T. Ishiyama, T. A. Ahiko and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 12414–12415 CrossRef CAS PubMed.
- N. Schweigert, A. J. B. Zehnder and R. I. L. Eggen, Environ. Microbiol., 2001, 3, 81–91 CrossRef CAS PubMed.
- T. V. Hansen and L. Skattebøl, Tetrahedron Lett., 2005, 46, 3357–3358 CrossRef CAS.
- V. Berberian, C. C. R. Allen, N. D. Sharma, D. R. Boyd and C. Hardacre, Adv. Synth. Catal., 2007, 349, 727–739 CrossRef CAS.
- Y. X. Li, W. J. Zuo, W. L. Mei, H. Q. Chen and H. F. Dai, Chin. J. Nat. Med., 2014, 12, 297–299 CAS.
- S. P. B. Ovenden, M. Cobbe, R. Kissell, G. W. Birrell, M. Chavchich and M. D. Edstein, J. Nat. Prod., 2011, 74, 74–78 CrossRef CAS PubMed.
- J. M. Berry, T. D. Bradshaw, I. Fichtner, R. Ren, C. H. Schwalbe, G. Wells, E. H. Chew, M. F. G. Stevens and A. D. Westwell, J. Med. Chem., 2005, 48, 639–644 CrossRef CAS PubMed.
- D. Mal and S. Ray, Eur. J. Org. Chem., 2008, 3014–3020 CrossRef CAS.
- D. Pan, S. K. Mal, G. K. Kar and J. K. Ray, Tetrahedron, 2002, 58, 2847–2852 CrossRef CAS.
- T. Ramnial, S. A. Taylor, J. A. C. Clyburne and C. J. Walsby, Chem. Commun., 2007, 2066–2068 RSC.
- M. Ghandi and M. Shahidzadeh, J. Organomet. Chem., 2006, 691, 4918–4925 CrossRef CAS.
- D. Koszelewski, D. Paprocki, A. Brodzka, A. Kęciek, M. Wilk and R. Ostaszewski, Sustainable Chem. Pharm., 2022, 25, 100576 CrossRef CAS.
- J. Samsonowicz-Górski, A. Hrunyk, A. Brodzka, R. Ostaszewski and D. Koszelewski, Green Chem., 2023, 25, 6306–6314 RSC.
- R. Kourist, Biocatalysis in Organic Synthesis. Science of Synthesis, Thieme, Stuttgart, 2015, vol. 1–3 Search PubMed.
- C. Palumbo, E. E. Ferrandi, C. Marchesi, D. Monti, S. Riva, R. Psaro and M. Guidotti, ChemistrySelect, 2016, 1, 1795–1798 CrossRef CAS.
- C. Cativiela, J. M. Fraile, J. I. García and J. A. Mayoral, J. Mol. Catal. A: Chem., 1996, 112, 259–267 CrossRef CAS.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- J. I. Song and D. K. An, Chem. Lett., 2007, 36, 886–887 CrossRef CAS.
- H. Sand and R. Weberskirch, RSC Adv., 2017, 7, 33614–33626 RSC.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed.
- D. Schwendenwein, A. K. Ressmann, M. Entner, V. Savic, M. Winkler and F. Rudroff, Catalysts, 2021, 11, 932 CrossRef CAS.
- D. Schwendenwein, A. K. Ressmann, M. Doerr, M. Höhne, U. T. Bornscheuer, M. D. Mihovilovic, F. Rudroff and M. Winkler, Adv. Synth. Catal., 2019, 361, 2544–2549 CrossRef CAS.
- S. Giparakis, M. Winkler and F. Rudroff, Green Chem., 2023, 26, 1338–1344 RSC.
- A. Rapeyko, M. J. Climent, A. Corma, P. Concepción and S. Iborra, ChemSusChem, 2015, 8, 3270–3282 CrossRef CAS PubMed.
- P. Zambelli, A. Pinto, D. Romano, E. Crotti, P. Conti, L. Tamborini, R. Villa and F. Molinari, Green Chem., 2012, 14, 2158–2161 RSC.
- R. A. Reck, J. Am. Oil Chem. Soc., 1985, 62, 355–365 CrossRef CAS.
- T. V. RajanBabu and A. L. Casalnuovo, in Comprehensive asymmetric catalysis I–III, ed. E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer, Berlin, 1999, p. 367 Search PubMed.
- M. A and N. V. Kalevaru, in Industrial Catalysis and Separations Innovations for Process Intensification, ed. K. V. Raghavan and B. M. Reddy, Apple Academic Press, 2014, pp. 61–103 Search PubMed.
- C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke and H. Gröger, ACS Catal., 2019, 9, 5198–5203 CrossRef CAS.
- A. Hinzmann, M. Stricker and H. Gröger, ACS Sustainable Chem. Eng., 2020, 8, 17088–17096 CrossRef CAS.
- E. J. Craven, J. Latham, S. A. Shepherd, I. Khan, A. Diaz-Rodriguez, M. F. Greaney and J. Micklefield, Nat. Catal., 2021, 4, 385–394 CrossRef CAS.
- L. Bering, E. J. Craven, S. A. Sowerby Thomas, S. A. Shepherd and J. Micklefield, Nat. Commun., 2022, 13, 380 CrossRef CAS PubMed.
- J. Gorzynski Smith, Synthesis, 1984, 629–656 CrossRef CAS.
- W. J. Choi and C.-Y. Choi, Biotechnol. Bioprocess Eng., 2005, 10, 167–179 CrossRef CAS.
- A. Archelas and R. Furstoss, Top. Curr. Chem., 1999, 159–191 CrossRef CAS.
- C. A. Denard, M. J. Bartlett, Y. Wang, L. Lu, J. F. Hartwig and H. Zhao, ACS Catal., 2015, 5, 3817–3822 CrossRef CAS.
- K. Wu, L. Chen, H. Fan, Z. Zhao, H. Wang and D. Wei, Tetrahedron Lett., 2016, 57, 899–904 CrossRef CAS.
- S. González-Granda, L. Escot, I. Lavandera and V. Gotor-Fernández, ACS Catal., 2022, 12, 2552–2560 CrossRef.
- R. J. Cherney, P. Carter, J. V. Duncia, D. S. Gardner and J. B. Santella, WO2004071460A2, 2004.
- P. H. Carter, R. J. Cherney, D. G. Batt, J. V. Duncia, D. S. Gardner, S. Ko, A. S. Srivastava and M. G. Yang, WO2005021500A1, 2005.
- P. Süss, S. Borchert, J. Hinze, S. Illner, J. Von Langermann, U. Kragl, U. T. Bornscheuer and R. Wardenga, Org. Process Res. Dev., 2015, 19, 2034–2038 CrossRef.
- R. Kourist, P. Domínguez de María and K. Miyamoto, Green Chem., 2011, 13, 2607–2618 RSC.
- S. Barth and F. Effenberger, Tetrahedron: Asymmetry, 1993, 4, 823–833 CrossRef CAS.
- J. Enoki, C. Mügge, D. Tischler, K. Miyamoto and R. Kourist, Chem. – Eur. J., 2019, 25, 5071–5076 CrossRef CAS PubMed.
- Y. S. Lin, P. Y. Wang, A. C. Wu and S. W. Tsai, J. Mol. Catal. B: Enzym., 2011, 68, 245–249 CrossRef CAS.
- T. Uemura, X. Zhang, K. Matsumura, N. Sayo, H. Kumobayashi, T. Ohta, K. Nozaki and H. Takaya, J. Org. Chem., 1996, 61, 5510–5516 CrossRef CAS.
- K. Tenbrink, M. Seßler, J. Schatz and H. Gröger, Adv. Synth. Catal., 2011, 353, 2363–2367 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed.
- G. Moroy, C. Denhez, H. El Mourabit, A. Toribio, A. Dassonville, M. Decarme, J. H. Renault, C. Mirand, G. Bellon, J. Sapi, A. J. P. Alix, W. Hornebeck and E. Bourguet, Bioorg. Med. Chem., 2007, 15, 4753–4766 CrossRef CAS PubMed.
- J. J. Chen, Y. Zhang, S. Hammond, N. Dewdney, T. Ho, X. Lin, M. F. Browner and A. L. Castelhano, Bioorg. Med. Chem. Lett., 1996, 6, 1601–1606 CrossRef CAS.
- P. Galatsis, B. Caprathe, J. Gilmore, A. Thomas, K. Linn, S. Sheehan, W. Harter, C. Kostlan, E. Lunney, C. Stankovic, J. Rubin, K. Brady, H. Allen and R. Talanian, Bioorg. Med. Chem. Lett., 2010, 20, 5184–5190 CrossRef CAS PubMed.
- J. Liu, Y. Yang and R. Ji, Helv. Chim. Acta, 2004, 87, 1935–1939 CrossRef CAS.
- M. Korpak and J. Pietruszka, Adv. Synth. Catal., 2011, 353, 1420–1424 CrossRef CAS.
- M. T. Barros, C. D. Maycock and M. R. Ventura, Org. Lett., 2003, 5, 4097–4099 CrossRef CAS PubMed.
- Y. Wang, M. J. Bartlett, C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2017, 7, 2548–2552 CrossRef CAS.
- J. H. Hansen, B. T. Parr, P. Pelphrey, Q. Jin, J. Autschbach and H. M. L. Davies, Angew. Chem., 2011, 123, 2592–2596 CrossRef.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892–914 CrossRef CAS.
- Y. C. Chung, D. Janmanchi and H. L. Wu, Org. Lett., 2012, 14, 2766–2769 CrossRef CAS PubMed.
- M. H. Wang, D. T. Cohen, C. B. Schwamb, R. K. Mishra and K. A. Scheidt, J. Am. Chem. Soc., 2015, 137, 5891–5894 CrossRef CAS PubMed.
- M. Kashiwagi, K. I. Fuhshuku and T. Sugai, J. Mol. Catal. B: Enzym., 2004, 29, 249–258 CrossRef CAS.
- S. Talukdar, J. L. Hsu, T. C. Chou and J. M. Fang, Tetrahedron Lett., 2001, 42, 1103–1105 CrossRef CAS.
- M. M. Meloni and M. Taddei, Org. Lett., 2001, 3, 337–340 CrossRef CAS PubMed.
- E. Liardo, R. González-Fernández, N. Ríos-Lombardía, F. Morís, J. García-Álvarez, V. Cadierno, P. Crochet, F. Rebolledo and J. González-Sabín, ChemCatChem, 2018, 10, 4676–4682 CrossRef CAS.
- E. Liardo, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. Rebolledo, Org. Lett., 2016, 18, 3366–3369 CrossRef CAS PubMed.
- C. Palo-Nieto, S. Afewerki, M. Anderson, C. W. Tai, P. Berglund and A. Córdova, ACS Catal., 2016, 6, 3932–3940 CrossRef CAS.
- A. Caiazzo, P. M. L. Garcia, R. Wever, J. C. M. Van Hest, A. E. Rowan and J. N. H. Reek, Org. Biomol. Chem., 2009, 7, 2926–2932 RSC.
- H. Salminen, H. Jaakkola and M. Heinonen, J. Agric. Food Chem., 2008, 56, 11178–11186 CrossRef CAS PubMed.
- M. C. Bohin, J. P. Vincken, H. T. W. M. Van Der Hijden and H. Gruppen, J. Agric. Food Chem., 2012, 60, 4136–4143 CrossRef CAS PubMed.
- C. Simons, U. Hanefeld, I. W. C. E. Arends, T. Maschmeyer and R. A. Sheldon, Top. Catal., 2006, 40, 35–44 CrossRef CAS.
- J. A. Karlowsky, N. M. Laing, T. Baudry, N. Kaplan, D. Vaughan, D. J. Hoban and G. G. Zhanel, Antimicrob. Agents Chemother., 2007, 51, 1580–1581 CrossRef CAS PubMed.
- L. De Luca, G. Nieddu, A. Porcheddu and G. Giacomelli, Curr. Med. Chem., 2008, 16, 1–20 CrossRef PubMed.
- M. A. S. Mertens, F. Thomas, M. Nöth, J. Moegling, I. El-Awaad, D. F. Sauer, G. V. Dhoke, W. Xu, A. Pich, S. Herres-Pawlis and U. Schwaneberg, Eur. J. Org. Chem., 2019, 6341–6346 CrossRef CAS.
- C. Broca, M. Manteghetti, R. Gross, Y. Baissac, M. Jacob, P. Petit, Y. Sauvaire and G. Ribes, Eur. J. Pharmacol., 2000, 390, 339–345 CrossRef CAS PubMed.
- G. Ribes, C. Broca, P. Petit, M. Jacob, Y. Baissac, M. Manteghatti, M. Roye and Y. Sauvaire, Diabetologia, 1996, 39, A234 Search PubMed.
- M. Seitz and O. Reiser, Curr. Opin. Chem. Biol., 2005, 9, 285–292 CrossRef CAS PubMed.
- M. Sendzik, W. Guarnieri and D. Hoppe, Synthesis, 1998, 1287–1297 CrossRef CAS.
- M. D. Swift and A. Sutherland, Org. Biomol. Chem., 2006, 4, 3889–3891 RSC.
- R. C. Simon, E. Busto, J. H. Schrittwieser, J. H. Sattler, J. Pietruszka, K. Faber and W. Kroutil, Chem. Commun., 2014, 50, 15669–15672 RSC.
- J. B. Routien, J. Bacteriol., 1966, 91, 1663 CrossRef CAS PubMed.
- R. G. Werner, L. F. Thorpe, W. Reuter and K. H. Nierhaus, Eur. J. Biochem., 1976, 68, 1–3 CrossRef CAS PubMed.
- N. Tabata, H. Tomoda, Y. Takahashi, K. Haneda, Y. Iwai, H. B. Woodruff and S. Omura, J. Antibiot., 1993, 46, 756–761 CrossRef CAS PubMed.
- K. Akagawa, R. Umezawa and K. Kudo, Beilstein J. Org. Chem., 2012, 8, 1333–1337 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- R. Breinbauer and M. Köhn, ChemBioChem, 2003, 4, 1147–1149 CrossRef CAS PubMed.
- V. D. Bock, H. Hiemstra and J. H. Van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS.
- K. B. Sharpless and R. Manetsch, Expert Opin. Drug Discovery, 2006, 1, 525–538 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- K. C. Hartmuth and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef PubMed.
- M. Meldal and C. W. Tomøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- R. Huisgen, 1,3-Dipolar cycloaddition chemistry, Wiley, New York, 1984 Search PubMed.
- S. Su, J. R. Giguere, S. E. Schaus and J. A. Porco, Tetrahedron, 2004, 60, 8645–8657 CrossRef CAS.
- L. S. Campbell-Verduyn, W. Szymański, C. P. Postema, R. A. Dierckx, P. H. Elsinga, D. B. Janssen and B. L. Feringa, Chem. Commun., 2010, 46, 898–900 RSC.
- J. E. T. Van Hylckama Vlieg, L. Tang, J. H. Lutje Spelberg, T. Smilda, G. J. Poelarends, T. Bosma, A. E. J. Van Merode, M. W. Fraaije and D. B. Janssen, J. Bacteriol., 2001, 183, 5058–5066 CrossRef CAS PubMed.
- C. Aguirre-Pranzoni, R. D. Tosso, F. R. Bisogno, M. Kurina-Sanz and A. A. Orden, Process Biochem., 2019, 79, 114–117 CrossRef CAS.
- C. De Souza De Oliveira, K. T. De Andrade and A. T. Omori, J. Mol. Catal. B: Enzym., 2013, 91, 93–97 CrossRef CAS.
- N. Asano, Glycobiology, 2003, 13, 93–104 CrossRef PubMed.
- R. A. Dwek, T. D. Butters, F. M. Platt and N. Zitzmann, Nat. Rev. Drug Discovery, 2002, 1, 65–75 CrossRef CAS PubMed.
- G. S. Jacob, Curr. Opin. Struct. Biol., 1995, 5, 605–611 CrossRef CAS PubMed.
- R. M. Moriarty, C. I. Mitan, N. Branzǎ-Nichita, K. R. Phares and D. Parrish, Org. Lett., 2006, 8, 3465–3467 CrossRef CAS PubMed.
- A. K. Samland and G. A. Sprenger, Appl. Microbiol. Biotechnol., 2006, 71, 253–264 CrossRef CAS PubMed.
- L. J. Whalen and C.-H. Wong, Aldrichimica Acta, 2006, 39, 63–71 Search PubMed.
- M. Sugiyama, Z. Hong, P. H. Liang, S. M. Dean, L. J. Whalen, W. A. Greenberg and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 14811–14817 CrossRef CAS PubMed.
- E. Sverrisdóttir, T. M. Lund, A. E. Olesen, A. M. Drewes, L. L. Christrup and M. Kreilgaard, Eur. J. Pharm. Sci., 2015, 74, 45–62 CrossRef PubMed.
- M. A. Rashid, K. R. Gustafson, Y. Kashman, J. H. Cardellina II, J. B. Mc Mohan and M. R. Boyd, Nat. Prod. Lett., 1995, 6, 153–156 CrossRef CAS.
- J. D. Guzman, T. Pesnot, D. A. Barrera, H. M. Davies, E. McMahon, D. Evangelopoulos, P. N. Mortazavi, T. Munshi, A. Maitra, E. D. Lamming, R. Angell, M. C. Gershater, J. M. Redmond, D. Needham, J. M. Ward, L. E. Cuca, H. C. Hailes and S. Bhakta, J. Antimicrob. Chemother., 2014, 70, 1691–1703 CrossRef PubMed.
- L. Moreno, J. Párraga, A. Galán, N. Cabedo, J. Primo and D. Cortes, Bioorg. Med. Chem., 2012, 20, 6589–6597 CrossRef CAS PubMed.
- W. Lin and S. Ma, Org. Chem. Front., 2017, 4, 958–966 RSC.
- T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2012, 354, 2997–3008 CrossRef CAS.
- A. Bonamore, L. Calisti, A. Calcaterra, O. H. Ismail, M. Gargano, I. D’Acquarica, B. Botta, A. Boffi and A. Macone, ChemistrySelect, 2016, 1, 1525–1528 CrossRef CAS.
- J. Zhao, B. R. Lichman, J. M. Ward and H. C. Hailes, Chem. Commun., 2018, 54, 1323–1326 RSC.
- V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS PubMed.
- K. Ye, Y. Ke, N. Keshava, J. Shanks, J. A. Kapp, R. R. Tekmal, J. Petros and H. C. Joshi, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1601–1606 CrossRef CAS PubMed.
- G. François, G. Timperman, W. Eling, L. Ake, J. Holenz and G. Bringmann, Antimicrob. Agents Chemother., 2000, 41, 2533–2539 CrossRef PubMed.
- C. R. Chapple, L. Cardozo, W. D. Steers and F. E. Govier, Int. J. Clin. Pract., 2006, 60, 959–966 CrossRef CAS PubMed.
- M. Odachowski, M. F. Greaney and N. J. Turner, ACS Catal., 2018, 8, 10032–10035 CrossRef CAS.
- T. Kanemitsu, Y. Yamashita, K. Nagata and T. Itoh, Heterocycles, 2007, 74, 199–203 CrossRef CAS PubMed.
- W. Liu, S. Liu, R. Jin, H. Guo and J. Zhao, Org. Chem. Front., 2015, 2, 288–299 RSC.
- Y. Jin, L. Ou, H. Yang and H. Fu, J. Am. Chem. Soc., 2017, 139, 14237–14243 CrossRef CAS PubMed.
- H. Xiang, S. Ferla, C. Varricchio, A. Brancale, N. L. Brown, G. W. Black, N. J. Turner and D. Castagnolo, ACS Catal., 2023, 13, 3370–3378 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- F. Chacón-Huete, C. Messina, B. Cigana and P. Forgione, ChemSusChem, 2022, 15, e202200328 CrossRef PubMed.
- A. Velty, S. Iborra and A. Corma, ChemSusChem, 2022, 15, e202200181 CrossRef CAS PubMed.
- H. Son Le, Z. Said, M. Tuan Pham, T. Hieu Le, I. Veza, V. Nhanh Nguyen, B. Deepanraj and L. Huong Nguyen, Fuel, 2022, 324, 124474 CrossRef CAS.
- B. Hurtado, K. S. Arias, M. J. Climent, P. Concepción, A. Corma and S. Iborra, ChemSusChem, 2022, 15, e202200194 CrossRef CAS PubMed.
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS.
- R. van den Berg, J. A. Peters and H. van Bekkum, Carbohydr. Res., 1994, 253, 1–12 CrossRef CAS PubMed.
- R. Huang, W. Qi, R. Su and Z. He, Chem. Commun., 2010, 46, 1115–1117 RSC.
- H. Huang, C. A. Denard, R. Alamillo, A. J. Crisci, Y. Miao, J. A. Dumesic, S. L. Scott and H. Zhao, ACS Catal., 2014, 4, 2165–2168 CrossRef CAS.
- C. Megías-Sayago, S. Navarro-Jaén, F. Drault and S. Ivanova, Catalysts, 2021, 11, 1395 CrossRef.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- X. Chen, L. Zhang, B. Zhang, X. Guo and X. Mu, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
- M. J. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580–585 CrossRef CAS.
- K. Yan, J. Liao, X. Wu and X. Xie, RSC Adv., 2013, 3, 3853–3856 RSC.
- M. Audemar, C. Ciotonea, K. De Oliveira Vigier, S. Royer, A. Ungureanu, B. Dragoi, E. Dumitriu and F. Jérôme, ChemSusChem, 2015, 8, 1885–1891 CrossRef CAS PubMed.
- T. Ema, S. Ide, N. Okita and T. Sakai, Adv. Synth. Catal., 2008, 350, 2039–2044 CrossRef CAS.
- Y. Ni, Y. Su, H. Li, J. Zhou and Z. Sun, J. Biotechnol., 2013, 168, 493–498 CrossRef CAS PubMed.
- Y. C. He, Z. C. Tao, X. Zhang, Z. X. Yang and J. H. Xu, Bioresour. Technol., 2014, 161, 461–464 CrossRef CAS PubMed.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC.
- Z.-Y. Yang, Y.-C. Hao, S.-Q. Hu, M.-H. Zong, Q. Chen and N. Li, Adv. Synth. Catal., 2021, 363, 1033–1037 CrossRef CAS.
- A. Dunbabin, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, Green Chem., 2017, 19, 397–404 RSC.
- P. Zhang, X. Liao, C. Ma, Q. Li, A. Li and Y. He, ACS Sustainable Chem. Eng., 2019, 7, 17636–17642 CrossRef CAS.
- X.-L. Liao, Q. Li, D. Yang, C.-L. Ma, Z.-B. Jiang and Y.-C. He, Appl. Biochem. Biotechnol., 2020, 192, 794–811 CrossRef CAS PubMed.
- M. Jin, L. Da Costa Sousa, C. Schwartz, Y. He, C. Sarks, C. Gunawan, V. Balan and B. E. Dale, Green Chem., 2016, 18, 957–966 RSC.
- X. Liu, Y. Wang, S. Jin, X. Li and Z. Zhang, Arab. J. Chem., 2020, 13, 4916–4925 CrossRef CAS.
- Q. Yang, Z. Tang, J. Xiong and Y. He, Catalysts, 2023, 13, 37 CrossRef CAS.
- Y. C. He, C. X. Jiang, J. W. Jiang, J. H. Di, F. Liu, Y. Ding, Q. Qing and C. L. Ma, Bioresour. Technol., 2017, 238, 698–705 CrossRef CAS PubMed.
- X. X. Xue, C. L. Ma, J. H. Di, X. Y. Huo and Y. C. He, Bioresour. Technol., 2018, 268, 292–299 CrossRef CAS PubMed.
- Y. He, Y. Ding, C. Ma, J. Di, C. Jiang and A. Li, Green Chem., 2017, 19, 3844–3850 RSC.
- X. Q. Feng, Y. Y. Li, C. L. Ma, Y. Xia and Y. C. He, RSC Adv., 2020, 10, 40365–40372 RSC.
- Y. Y. Li, Q. Li, P. Q. Zhang, C. L. Ma, J. H. Xu and Y. C. He, Bioresour. Technol., 2021, 320, 124267 CrossRef CAS PubMed.
- R. M. Williams, Aldrichimica Acta, 1992, 25, 11–25 CAS.
- S. Servi, D. Tessaro and G. Pedrocchi-Fantoni, Coord. Chem. Rev., 2008, 252, 715–726 CrossRef CAS.
- L. Gong, Y. Xiu, J. Dong, R. Han, G. Xu and Y. Ni, Bioresour. Technol., 2021, 337, 125344 CrossRef CAS PubMed.
- T. K. Chakraborty, S. Tapadar and S. Kiran Kumar, Tetrahedron Lett., 2002, 43, 1317–1320 CrossRef CAS.
- C. P. Ferraz, M. Zieliński, M. Pietrowski, S. Heyte, F. Dumeignil, L. M. Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- C. P. Ferraz, N. J. S. Costa, E. Teixeira-Neto, Â. A. Teixeira-Neto, C. W. Liria, J. Thuriot-Roukos, M. T. Machini, R. Froidevaux, F. Dumeignil, L. M. Rossi and R. Wojcieszak, Catalysts, 2020, 10, 75 CrossRef CAS.
- L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS.
- M. M. Cajnko, U. Novak, M. Grilc and B. Likozar, Biotechnol. Biofuels, 2020, 13, 1–11 CrossRef PubMed.
- A. Lancien, R. Wojcieszak, E. Cuvelier, M. Duban, P. Dhulster, S. Paul, F. Dumeignil, R. Froidevaux and E. Heuson, ChemCatChem, 2021, 13, 247–259 CrossRef CAS.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- J. Dai, F. Li and X. Fu, ChemSusChem, 2020, 13, 6498–6508 CrossRef CAS PubMed.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- Y. C. Hao, M. H. Zong, Z. L. Wang and N. Li, Bioresour. Bioprocess., 2021, 8, 1–9 CrossRef PubMed.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS PubMed.
- M. Moniruzzaman, K. Nakashima, N. Kamiya and M. Goto, Biochem. Eng. J., 2010, 48, 295–314 CrossRef CAS.
- A. A. M. Elgharbawy, M. Moniruzzaman and M. Goto, Biochem. Eng. J., 2020, 154, 107426 CrossRef CAS.
- P. Xu, S. Liang, M.-H. Zong and W.-Y. Lou, Biotechnol. Adv., 2021, 51, 107702 CrossRef CAS PubMed.
- A. Schindl, M. L. Hagen, S. Muzammal, H. A. D. Gunasekera and A. K. Croft, Front. Chem., 2019, 7, 347 CrossRef CAS PubMed.
- H. T. Imam, V. Krasňan, M. Rebroš and A. C. Marr, Molecules, 2021, 26, 4791 CrossRef CAS PubMed.
- M. K. Potdar, G. F. Kelso, L. Schwarz, C. Zhang and M. T. W. Hearn, Molecules, 2015, 20, 16788–16816 CrossRef CAS PubMed.
- Y. Dai, J. van Spronsen, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- M. Shaibuna, L. V. Theresa and K. Sreekumar, Soft Matter, 2022, 18, 2695–2721 RSC.
- B. Nian and X. Li, Int. J. Biol. Macromol., 2022, 217, 255–269 CrossRef CAS PubMed.
- M. Pätzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk and D. Holtmann, Trends Biotechnol., 2019, 37, 943–959 CrossRef PubMed.
- P. Xu, G.-W. Zheng, M.-H. Zong, N. Li and W.-Y. Lou, Bioresour. Bioprocess., 2017, 4, 34 CrossRef PubMed.
- L. Cicco, G. Dilauro, F. M. Perna, P. Vitale and V. Capriati, Org. Biomol. Chem., 2021, 19, 2558–2577 RSC.
- V. Gotor-Fernández and C. E. Paul, J. Biotechnol., 2019, 293, 24–35 CrossRef PubMed.
- Z. Maugeri and P. Domínguez de María, ChemCatChem, 2014, 6, 1535–1537 CrossRef CAS.
- P. Vitale, V. M. Abbinante, F. M. Perna, A. Salomone, C. Cardellicchio and V. Capriati, Adv. Synth. Catal., 2017, 359, 1049–1057 CrossRef CAS.
- L. Cicco, N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, F. M. Perna, V. Capriati, J. García-Álvarez and J. González-Sabín, Green Chem., 2018, 20, 3468–3475 RSC.
- E. Liardo, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. Rebolledo, Org. Lett., 2016, 18, 3366–3369 CrossRef CAS PubMed.
- N. Ríos-Lombardía, C. Vidal, E. Liardo, F. Morís, J. García-Álvarez and J. González-Sabín, Angew. Chem. Int. Ed., 2016, 55, 8691–8695 CrossRef PubMed.
- J. Paris, N. Ríos-Lombardía, F. Morís, H. Gröger and J. González-Sabín, ChemCatChem, 2018, 10, 4417–4423 CrossRef CAS.
- J. Paris, A. Telzerow, N. Ríos-Lombardía, K. Steiner, H. Schwab, F. Morís, H. Gröger and J. González-Sabín, ACS Sustainable Chem. Eng., 2019, 7, 5486–5493 CrossRef CAS.
- A. Telzerow, J. Paris, M. Håkansson, J. González-Sabín, N. Ríos-Lombardía, M. Schürmann, H. Gröger, F. Morís, R. Kourist, H. Schwab and K. Steiner, ACS Catal., 2019, 9, 1140–1148 CrossRef CAS.
- D. Xu, W. Tang, Z. Tang and Y. He, Catalysts, 2023, 13, 467 CrossRef CAS.
- Q. Li, J. Di, X. Liao, J. Ni, Q. Li, Y. C. He and C. Ma, Green Chem., 2021, 23, 8154–8168 RSC.
- W. He, Y. C. He and J. Ye, Front. Bioeng. Biotechnol., 2023, 11, 1–10 Search PubMed.
- L. Li, Q. Li, J. Di, Y. He and C. Ma, ACS Sustainable Chem. Eng., 2023, 11, 7515–7525 CrossRef CAS.
- Y. Liu, L. Li, C. Ma and Y. C. He, Bioresour. Technol., 2023, 387, 129638 CrossRef CAS PubMed.
- J. Di, Q. Li, C. Ma and Y. C. He, Bioresour. Technol., 2023, 369, 128425 CrossRef CAS PubMed.
- J. Ni, Q. Li, L. Gong, X. L. Liao, Z. J. Zhang, C. Ma and Y. He, ACS Sustainable Chem. Eng., 2021, 9, 13084–13095 CrossRef CAS.
- W. He, J. Ni, Y. C. He and J. Ye, Int. J. Biol. Macromol., 2022, 222, 1201–1210 CrossRef CAS PubMed.
- Y. Liu, Y. Wu, Y. C. He and C. Ma, Ind. Crops Prod., 2023, 202, 117033 CrossRef CAS.
- S. Zhang, C. Ma, Q. Li, Q. Li and Y. C. He, Bioresour. Technol., 2022, 344, 126299 CrossRef CAS PubMed.
- S. Zhang, C. Wu, C. Ma, L. Li and Y. C. He, Bioresour. Technol., 2023, 371, 128579 CrossRef CAS PubMed.
- C. Wu, Q. Li, J. Di, Y. C. He and C. Ma, Fuel, 2023, 343, 127830 CrossRef CAS.
- R. Gao, Q. Li, J. Di, Q. Li, Y. C. He and C. Ma, Ind. Crops Prod., 2023, 193, 116199 CrossRef CAS.
- C. Wu, C. Ma, Q. Li, H. Chai and Y. C. He, Bioresour. Technol., 2023, 385, 129454 CrossRef CAS PubMed.
- Q. Li, C. L. Ma and Y. C. He, Bioresour. Technol., 2023, 378, 128965 CrossRef CAS PubMed.
- X. Li, X. Cao, J. Xiong and J. Ge, Small, 2020, 16, 1902751 CrossRef CAS PubMed.
- K. E. Metzger, M. M. Moyer and B. G. Trewyn, ACS Catal., 2021, 11, 110–122 CrossRef CAS.
- K. Engström, E. V. Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C.-W. Tai and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006–14010 CrossRef PubMed.
- N. Zhang, R. Hübner, Y. Wang, E. Zhang, Y. Zhou, S. Dong and C. Wu, ACS Appl. Nano Mater., 2018, 1, 6378–6386 CrossRef CAS.
- A. K. Ganai, P. Shinde, B. B. Dhar, S. Sen Gupta and B. L. V. Prasad, RSC Adv., 2013, 3, 2186–2191 RSC.
- L. Gao, Z. Wang, Y. Liu, P. Liu, S. Gao, J. Gao and Y. Jiang, Chem. Commun., 2020, 56, 13547–13550 RSC.
- M. Kalantari, Y. Liu, E. Strounina, Y. Yang, H. Song and C. Yu, J. Mater. Chem. A, 2018, 6, 17579–17586 RSC.
- Y. Liu, N. Guo, W. Kong, S. Gao, G. Liu, L. Zhou, J. Gao and Y. Jiang, Green Synth. Catal., 2023, In press Search PubMed.
- M. Budhiraja, A. Ali and V. Tyagi, Eur. J. Org. Chem., 2023, e202201426 CrossRef CAS.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- M. Viciano-Chumillas, M. Mon, J. Ferrando-Soria, A. Corma, A. Leyva-Pérez, D. Armentano and E. Pardo, Acc. Chem. Res., 2020, 53, 520–531 CrossRef CAS PubMed.
- Y. Chen, S. Han, X. Li, Z. Zhang and S. Ma, Inorg. Chem., 2014, 53, 10006–10008 CrossRef CAS PubMed.
- S. Dutta, N. Kumari, S. Dubbu, S. W. Jang, A. Kumar, H. Ohtsu, J. Kim, S. H. Cho, M. Kawano and I. S. Lee, Angew. Chem., Int. Ed., 2020, 59, 3416–3422 CrossRef CAS PubMed.
- Y. Wang, N. Zhang, E. Zhang, Y. Han, Z. Qi, M. B. Ansorge-Schumacher, Y. Ge and C. Wu, Chem. – Eur. J., 2019, 25, 1716–1721 CrossRef CAS PubMed.
- Y. Wu, J. Shi, S. Mei, H. A. Katimba, Y. Sun, X. Wang, K. Liang and Z. Jiang, ACS Catal., 2020, 10, 9664–9673 CrossRef CAS.
- Z. Wang, Y. Liu, X. Dong and Y. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 49974–49981 CrossRef CAS PubMed.
- H. Hu, Y. Chang, Z. Wang, J. Cui, S. Jia and Y. Du, J. Colloid Interface Sci., 2023, 650, 1833–1841 CrossRef CAS PubMed.
- J. Gao, Z. Wang, R. Guo, Y. Hu, X. Dong, Q. Shi and Y. Sun, Catal. Sci. Technol., 2022, 13, 991–999 RSC.
- Y. Wang, N. Zhang, R. Hübner, D. Tan, M. Löffler, S. Facsko, E. Zhang, Y. Ge, Z. Qi and C. Wu, Adv. Mater. Interfaces, 2019, 6, 1801664 CrossRef.
- J. M. Palomo, Chem. Commun., 2019, 55, 9583–9589 RSC.
- M. Filice, M. Marciello, M. D. P. Morales and J. M. Palomo, Chem. Commun., 2013, 49, 6876–6878 RSC.
- M. Filice, N. Losada-Garcia, C. Perez-Rizquez, M. Marciello, M. D. Morales and J. M. Palomo, Appl. Nano, 2021, 2, 1–13 CrossRef.
- X. Wu, S. Liu, J. Xiong, B. Chen, M.-H. Zong, J.-G. Yang and W.-Y. Lou, Appl. Catal., A, 2020, 608, 117899 CrossRef CAS.
- J. M. Naapuri, N. Losada-Garcia, J. Deska and J. M. Palomo, Nanoscale, 2022, 14, 5701–5715 RSC.
- J. M. Naapuri, N. Losada-Garcia, R. A. Rothemann, M. C. Pichardo, M. H. G. Prechtl, J. M. Palomo and J. Deska, ChemCatChem, 2022, 14, e202200362 CrossRef CAS PubMed.
- R. Benavente, D. Lopez-Tejedor, M. del Puerto Morales, C. Perez-Rizquez and J. M. Palomo, Nanoscale, 2020, 12, 12917–12927 RSC.
- T. Görbe, K. P. J. Gustafson, O. Verho, G. Kervefors, H. Zheng, X. Zou, E. V. Johnston and J.-E. Bäckvall, ACS Catal., 2017, 7, 1601–1605 CrossRef.
- X. Li, X. Hu, Y. Qiao, T. Lu, Y. Bai, J. Xiong, X. Li, Q. Gou and J. Ge, Chem. Eng. J., 2023, 452, 139356 CrossRef CAS.
- C. A. Denard, H. Huang, M. J. Bartlett, L. Lu, Y. Tan, H. Zhao and J. F. Hartwig, Angew. Chem., 2014, 126, 475–479 CrossRef.
- N. Scalacci, G. W. Black, G. Mattedi, N. L. Brown, N. J. Turner and D. Castagnolo, ACS Catal., 2017, 7, 1295–1300 CrossRef CAS.
- C. Risi, F. Zhao and D. Castagnolo, ACS Catal., 2019, 9, 7264–7269 CrossRef CAS.
- A. Gimbernat, M. Guehl, M. Capron, N. Lopes Ferreira, R. Froidevaux, J. S. Girardon, P. Dhulster, D. Delcroix and F. Dumeignil, ChemCatChem, 2017, 9, 2080–2084 CrossRef CAS.
- R. Q. Zhang, C. L. Ma, Y. F. Shen, J. F. Sun, K. Jiang, Z. B. Jiang, Y. J. Dai and Y. C. He, Catal. Lett., 2020, 150, 2220–2227 CrossRef CAS.
- L. Zhu, J. Di, Q. Li, Y. C. He and C. Ma, J. Mol. Liq., 2023, 380, 121741 CrossRef CAS.
- M. Horvat, V. Weilch, R. Rädisch, S. Hecko, A. Schiefer, F. Rudroff, B. Wilding, N. Klempier, M. Pátek, L. Martínková and M. Winkler, Catal. Sci. Technol., 2022, 12, 62–66 RSC.
- V. Gauchot, W. Kroutil and A. R. Schmitzer, Chem. – Eur. J., 2010, 16, 6748–6751 CrossRef CAS PubMed.
- E. Burda, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2008, 47, 9551–9554 CrossRef CAS PubMed.
- P. Luan, Y. Liu, Y. Li, R. Chen, C. Huang, J. Gao, F. Hollmann and Y. Jiang, Green Chem., 2021, 23, 1960–1964 RSC.
- S. Wu, Y. Zhou, D. Gerngross, M. Jeschek and T. R. Ward, Nat. Commun., 2019, 10, 5060 CrossRef PubMed.
- B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672–6695 CrossRef CAS PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J. Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- N. Akporji, V. Singhania, J. Dussart-Gautheret, F. Gallou and B. H. Lipshutz, Chem. Commun., 2021, 57, 11847–11850 RSC.
- A. B. Wood, J. R. A. Kincaid and B. H. Lipshutz, Green Chem., 2022, 24, 2853–2858 RSC.
- S. C. Cosgrove, M. P. Thompson, S. T. Ahmed, F. Parmeggiani and N. J. Turner, Angew. Chem., Int. Ed., 2020, 59, 18156–18160 CrossRef CAS PubMed.
- C. M. Heckmann and F. Paradisi, Chem. – Eur. J., 2021, 27, 16616–16620 CrossRef CAS PubMed.
- C. J. Hastings, N. P. Adams, J. Bushi and S. J. Kolb, Green Chem., 2020, 22, 6187–6193 RSC.
- N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, R. Kourist, N. Comino, F. López-Gallego, J. González-Sabín and J. García-Álvarez, Front. Chem., 2020, 8, 1–11 CrossRef PubMed.
- N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, R. Kourist, N. Comino, F. López-Gallego, J. González-Sabín and J. García-Álvarez, Front. Chem., 2020, 8, 139 CrossRef PubMed.
- E. Hofmann, L. Schmauser, J. Neugebauer, D. Touraud, F. Gallou and W. Kunz, Chem. Eng. J., 2023, 472, 144599 CrossRef CAS.
- S. Wang, L. Scandurra, R. Hübner, U. G. Nielsen and C. Wu, ChemCatChem, 2023, 15, e202201229 CrossRef CAS.
- Á. Gómez Baraibar, D. Reichert, C. Mügge, S. Seger, H. Gröger and R. Kourist, Angew. Chem., Int. Ed., 2016, 55, 14823–14827 CrossRef PubMed.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS PubMed.
- V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2012, 5, 93–99 CrossRef PubMed.
- Y. Okamoto, V. Köhler and T. R. Ward, J. Am. Chem. Soc., 2016, 138, 5781–5784 CrossRef CAS PubMed.
- Y. Okamoto, V. Köhler, C. E. Paul, F. Hollmann and T. R. Ward, ACS Catal., 2016, 6, 3553–3557 CrossRef CAS.
- M. A. S. Mertens, D. F. Sauer, U. Markel, J. Schiffels, J. Okuda and U. Schwaneberg, Catal. Sci. Technol., 2019, 9, 5572–5576 RSC.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 CrossRef CAS PubMed.
- J. Pauly, H. Gröger and A. V. Patel, ChemCatChem, 2019, 11, 1503–1509 CrossRef.
- J. Pauly, H. Gröger and A. V. Patel, Catalysts, 2019, 9, 547 CrossRef CAS.
- F. Chang, C. Wang, Q. Chen, Y. Zhang and G. Liu, Angew. Chem. Int. Ed., 2022, 61, e202114809 CrossRef CAS PubMed.
- D. Tian, R. Hao, X. Zhang, H. Shi, Y. Wang, L. Liang, H. Liu and H. Yang, Nat. Commun., 2023, 14, 1–14 Search PubMed.
- H. Terholsen, J. R. H. Meyer, Z. Zhang, P. J. Deuss and U. T. Bornscheuer, ChemSusChem, 2023, 16, e202300168 CrossRef CAS PubMed.
- S. Grasso, L. Siracusa, C. Spatafora, M. Renis and C. Tringali, Bioorg. Chem., 2007, 35, 137–152 CrossRef CAS PubMed.
- H. Sato, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2015, 54, 4488–4492 CrossRef CAS PubMed.
- H. Sato, R. Yamada, Y. Watanabe, T. Kiryu, S. Kawano, M. Shizuma and H. Kawasaki, RSC Adv., 2022, 12, 10619–10624 RSC.
- J. Latham, J. M. Henry, H. H. Sharif, B. R. K. Menon, S. A. Shepherd, M. F. Greaney and J. Micklefield, Nat. Commun., 2016, 7, 1–8 CrossRef PubMed.
- P. Schaaf, T. Bayer, M. Koley, M. Schnürch, U. T. Bornscheuer, F. Rudroff and M. D. Mihovilovic, Chem. Commun., 2018, 54, 12978–12981 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- P. De Santis, L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- C. Yang, R. Li, K. A. I. Zhang, W. Lin, K. Landfester and X. Wang, Nat. Commun., 2020, 11, 1239 CrossRef CAS PubMed.
- R. A. Messing and A. M. Filbert, J. Agric. Food Chem., 1975, 23, 920–923 CrossRef CAS PubMed.
- S. Strompen, M. Weiß, H. Gröger, L. Hilterhaus and A. Liese, Adv. Synth. Catal., 2013, 355, 2391–2399 CrossRef CAS.
- J. M. Carceller, M. Mifsud, M. J. Climent, S. Iborra and A. Corma, Green Chem., 2020, 22, 2767–2777 RSC.
- K. S. Arias, J. M. Carceller, M. J. Climent, A. Corma and S. Iborra, ChemSusChem, 2020, 13, 1864–1875 CrossRef CAS PubMed.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Renew. Sustainable Energy Rev., 2017, 74, 230–257 CrossRef CAS.
- F. Lackner, K. Hiebler, B. Grabner and H. Gruber-Woelfler, Catalysts, 2020, 10, 1–13 CrossRef.
- S. Gianolio, D. Roura Padrosa and F. Paradisi, Green Chem., 2022, 24, 8434–8440 RSC.
- M. J. Fink, M. Schön, F. Rudroff, M. Schnürch and M. D. Mihovilovic, ChemCatChem, 2013, 5, 724–727 CrossRef CAS.
- R. de Oliveira Lopes, A. S. de Miranda, B. Reichart, T. Glasnov, C. O. Kappe, R. C. Simon, W. Kroutil, L. S. M. Miranda, I. C. R. Leal and R. O. M. A. de Souza, J. Mol. Catal. B: Enzym., 2014, 104, 101–107 CrossRef.
- J. M. Carceller, K. S. Arias, M. J. Climent, S. Iborra and A. Corma, Natl. Sci. Rev., 2022, 9, nwac135 CrossRef CAS PubMed.
- S. Alipour, Green Chem., 2016, 18, 4990–4998 RSC.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS PubMed.
- J. A. Maciá-Agulló, A. Corma and H. Garcia, Chem. – Eur. J., 2015, 21, 10940–10959 CrossRef PubMed.
- J. Kim and C. B. Park, Curr. Opin. Chem. Biol., 2019, 49, 122–129 CrossRef CAS PubMed.
- L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 CrossRef CAS.
- F. F. Özgen, M. E. Runda and S. Schmidt, ChemBioChem, 2021, 22, 790–806 CrossRef PubMed.
- M. A. Emmanuel, S. G. Bender, C. Bilodeau, J. M. Carceller, J. S. DeHovitz, H. Fu, Y. Liu, B. T. Nicholls, Y. Ouyang, C. G. Page, T. Qiao, F. C. Raps, D. R. Sorigué, S. Z. Sun, J. Turek-Herman, Y. Ye, A. Rivas-Souchet, J. Cao and T. K. Hyster, Chem. Rev., 2023, 123, 5459–5520 CrossRef CAS PubMed.
- W. Zhang, E. F. Fueyo, F. Hollmann, L. L. Martin, M. Pesic, R. Wardenga, M. Höhne and S. Schmidt, Eur. J. Org. Chem., 2019, 80–84 CrossRef PubMed.
- K. Lauder, A. Toscani, Y. Qi, J. Lim, S. J. Charnock, K. Korah and D. Castagnolo, Angew. Chem., Int. Ed., 2018, 57, 5803–5807 CrossRef CAS PubMed.
- X. Guo, Y. Okamoto, M. R. Schreier, T. R. Ward and O. S. Wenger, Chem. Sci., 2018, 9, 5052–5056 RSC.
- Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 CrossRef CAS PubMed.
- P. Li, Y. Ma, Y. Li, X. Zhang and Y. Wang, ChemBioChem, 2020, 21, 1852–1855 CrossRef CAS PubMed.
- X. Ding, C.-L. Dong, Z. Guan and Y.-H. He, Angew. Chem., Int. Ed., 2019, 58, 118–124 CrossRef CAS PubMed.
- Y. Yu, W.-F. Lu, Z.-J. Yang, N. Wang and X.-Q. Yu, Bioorg. Chem., 2021, 107, 104534 CrossRef CAS PubMed.
- J. Albarrán-Velo, V. Gotor-Fernández and I. Lavandera, Adv. Synth. Catal., 2021, 363, 4096–4108 CrossRef.
- L. Rodríguez-Fernández, J. Albarrán-Velo, I. Lavandera and V. Gotor-Fernández, Adv. Synth. Catal., 2023, 365, 1883–1892 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |