
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGel polymer electrolytes for rechargeable batteries toward wide-temperature applications
Xiaoyan
Zhou†
ab,
Yifang
Zhou†
a,
Le
Yu†
a,
Luhe
Qi
a,
Kyeong-Seok
Oh
c,
Pei
Hu
*b,
Sang-Young
Lee
 *c and
Chaoji
Chen
*a
*c and
Chaoji
Chen
*a
aSchool of Resource and Environmental Sciences, Hubei Biomass-Resource Chemistry and Environmental Biotechnology Key Laboratory, Wuhan University, Wuhan 430079, P. R. China. E-mail: chenchaojili@whu.edu.cn
bSchool of Science, Hubei University of Technology, Wuhan 430070, P. R. China. E-mail: hupei@hbut.edu.cn
cDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul, Republic of Korea. E-mail: syleek@yonsei.ac.kr
First published on 18th April 2024
Abstract
Rechargeable batteries, typically represented by lithium-ion batteries, have taken a huge leap in energy density over the last two decades. However, they still face material/chemical challenges in ensuring safety and long service life at temperatures beyond the optimum range, primarily due to the chemical/electrochemical instabilities of conventional liquid electrolytes against aggressive electrode reactions and temperature variation. In this regard, a gel polymer electrolyte (GPE) with its liquid components immobilized and stabilized by a solid matrix, capable of retaining almost all the advantageous natures of the liquid electrolytes and circumventing the interfacial issues that exist in the all-solid-state electrolytes, is of great significance to realize rechargeable batteries with extended working temperature range. We begin this review with the main challenges faced in the development of GPEs, based on extensive literature research and our practical experience. Then, a significant section is dedicated to the requirements and design principles of GPEs for wide-temperature applications, with special attention paid to the feasibility, cost, and environmental impact. Next, the research progress of GPEs is thoroughly reviewed according to the strategies applied. In the end, we outline some prospects of GPEs related to innovations in material sciences, advanced characterizations, artificial intelligence, and environmental impact analysis, hoping to spark new research activities that ultimately bring us a step closer to realizing wide-temperature rechargeable batteries.
 Xiaoyan Zhou | Xiaoyan Zhou is currently a lecturer at the School of Science at the Hubei University of Technology and a visiting scholar at the laboratory of Prof. Chaoji Chen at Wuhan University. She received her PhD (2022) in Materials Science and Engineering from Huazhong University of Science and Technology (HUST) under the supervision of Prof. Guo Xin, where she worked on solid electrolytes and solid-state batteries. Her research interests are biomass-based functional gels and their applications in energy storage. |
 Le Yu | Le Yu is currently a postdoctoral fellow at the laboratory of Prof. Chaoji Chen at Wuhan University. Prior to joining the Chen Lab, he received his PhD degree (2021) in materials science and engineering from Huazhong University of Science and Technology under the supervision of Prof. Xianluo Hu, where he worked on ionic liquids and ionogel electrolytes for high-temperature electrochemical energy storage devices. His current interests focus on the development of biomass-based ionic conductors and their application in energy storage. |
 Pei Hu | Pei Hu is an associate professor at the School of Science at the Hubei University of Technology. He received his BS and PhD degrees in Materials Science and Engineering from Huazhong University of Science and Technology (HUST) under the supervision of Prof. Yunhui Huang. He then joined the School of Electrical and Electronic Engineering at HUST as a post-doctoral fellow. His research interests focus on energy storage and conversion through nanomaterial synthesis and electrode/electrolyte design, and the industrialization of battery materials and systems. |
 Sang-Young Lee | Sang-Young Lee, Professor of Chemical and Biomolecular Engineering at Yonsei University, Korea, received his BA in Chemical Engineering from Seoul National University (1991) and MS/PhD in Chemical Engineering from KAIST (1993, 1997). He served as a postdoctoral fellow at Max-Planck Institute and later led the development of ceramic-coated separators (SRS®) as a principal research scientist at Batteries R&D, LG Chem. He is a fellow of the Korean Academy of Science and Technology and a fellow of the National Academy of Engineering of Korea. His research focuses on liquid/solid electrolytes, high-mass-loading electrodes, cellulose-based batteries, and flexible/wearable power sources. |
 Chaoji Chen | Chaoji Chen is a full professor at the School of Resource and Environmental Sciences at Wuhan University. He received his PhD degree (2015) in Materials Science and Engineering from Huazhong University of Science and Technology (HUST). He then joined Prof. Liangbing Hu's Lab at the University of Maryland College Park as a post-doctoral fellow. He is the academic editor of The Innovation Materials and youth editor of The Innovation, Research, SusMat, Environmental Science & Ecotechnology, and Green Carbon. His research focuses on the multiscale engineering and sustainable utilization of bio-sourced materials such as wood, bamboo, cellulose toward addressing global material-energy-environment challenges. |
1. Introduction
Global energy consumption has risen greatly with the development of human society. To replace traditional fossil fuels, developing clean and renewable energy sources such as solar, wind and tide has become an urgent demand to an unprecedented level. However, these energy sources are intermittent and random, requiring reliable energy storage and conversion devices to balance the electricity supply and flexible deployment of the electrical grid system and to fill the gap between the installed capacities and the actual amount of electricity.1–3Among the various types of energy storage and conversion devices, rechargeable (or secondary) batteries are among the most successful proven technologies that can repeatedly convert chemical energy into clean electricity and have achieved a giant leap in energy density over the past two decades. Meanwhile, diversifying rechargeable batteries with different design configurations (e.g., coin/cylindrical/prismatic shape, blade/magazine stack) and functionalities were developed, and novel battery chemistries were proposed to meet the requirements of extensive application scenarios including portable electronic consumer devices, electric vehicles (EVs) and large-scale electricity storage in smart or intelligent grids.4–7 Though energy/power density, safety, and stability of rechargeable batteries at room temperature have been greatly improved, the demand for batteries that can survive and operate under extreme temperatures is soaring.
In this century, severe summers and winters occurred frequently over the world. As displayed in Fig. 1(a), the record temperatures of capital cities around the world have been frequently broken: the high temperatures in some countries were over 40 °C, e.g., India (45.6 °C in 2020), Egypt (45 °C in 2019), and Australia (46 °C in 2020), and low temperatures approximately approach −30 °C, e.g., Russia (−33 °C in 2012), Canada (−36 °C in 2013), and Ukraine (−30 °C in 2012). It is worth noting that these are only the data for capital cities rather than the whole countries, taking the United States as an example, the extreme temperatures can hit down to −62.2 °C and up to 56.7 °C.8 Given the diversity and variability of climate on a global scale, EVs are expected to operate stably and safely under a wide temperature range. Extensive research has demonstrated that the driving range of EVs heavily depends on the environmental temperatures (Fig. 1(b)). According to the test results on thousands of EVs performed by Seattle-based Recurrent, some vehicles saw a 30% drop in their driving ranges at near subzero temperatures.9,10 Moreover, some special application scenarios, such as military, high-flying drones, and space exploration, urgently need batteries that can be operated under more challenging temperature conditions, for example, −40 to 70 °C for military applications.11–13
 | ||
| Fig. 1 Schematic showing the driving range anxiety and safety issues of EVs raised by global extreme temperatures. (a) Global extreme low (blue) and high (red) temperatures of capital cities of different countries in the last 10 years (from Jan. 2012 to July 2023), data collected from the Climate Change Institute, USA. (b) Change in driving range of EVs with the environmental temperature. | ||
Until now, however, the state-of-the-art rechargeable batteries face formidable challenges of serious electrochemical performance degradation and safety issues at both low and high temperatures due to the chemical/electrochemical limitations of conventional electrolytes, thus severely limiting their applications at harsh temperature conditions:14–16 (i) at low temperatures, the increased viscosity or even solidification of electrolytes causes sluggish ion-transfer kinetics both in bulk electrolytes and electrolyte/electrode interfaces as well as the slow ion solid-state diffusion within the active electrode materials, lowering the practical energy densities; (ii) at high temperatures, the thermodynamic instability and increased reactivity of electrolytes toward the electrodes would lead to serious safety hazards of rechargeable batteries. Considering these demands, rechargeable batteries capable of operating at wide temperature ranges have attracted tremendous research attention in recent years.
To date, most research on wide-temperature rechargeable batteries has been largely devoted to the discovery of novel electrolytes due to their crucial bridge role in electrochemical battery systems (Fig. 2(a)). However, it remains challenging to explore a kind of electrolyte that can operate well in extremely cold and hot environments. Standing out among various kinds of electrolyte systems, gel polymer electrolyte (GPE) combines the high ionic conductivity and excellent interfacial compatibility of liquid electrolytes as well as the high safety of all-solid-state electrolytes, promising to realize rechargeable batteries with wide-temperature operability (Fig. 2(b)) and has become a research hot spot in both academic and industrial world.17–19 Although great progress on GPEs has been made, the investigation on GPEs and batteries operating at extreme temperatures is still in their early stage.20–22 To date, reviews on GPEs have been mainly focused on the synthetic strategies, properties, and their battery performance at room temperature, with limited coverage of extreme temperature conditions. To the best of our knowledge, there has been no comprehensive review discussing the design and engineering strategies of GPEs toward wide-temperature rechargeable batteries. Also, the structure–property relationships of GPEs and their effects on the wide-temperature performance of rechargeable batteries have not been comprehensively and thoroughly summarized. Given these facts, it is crucial and timely to review on the progress and advances and to discuss the design principles, engineering strategies, challenges, opportunities, and prospects of wide-temperature adaptable GPEs.
 | ||
| Fig. 2 Overview of the research trend of wide-temperature rechargeable batteries based on various electrolyte systems. (a) Number of publications on “wide-temperature/all-climate/high-temperature/elevated-temperature/low-temperature/extreme-temperature/extreme-condition/all-weather/extreme-weather/environment-adaptable/all-temperature”, up to December 2023. Database: Web of Science. (b) Comparison of wide-temperature performance between liquid, all-solid electrolytes and GPE. | ||
In this review, we first overview the major challenges encountered in the development of wide-temperature adaptable GPEs. Next, the key requirements and design principles of GPEs for wide-temperature applications are discussed. Then, we thoroughly discuss the engineering strategies of GPEs in terms of salts, polymer matrices, solvents, additives, and hosts to enable their wide-temperature operability. Finally, we offer our perspectives on the future research directions and opportunities of wide-temperature adaptable GPEs through innovations in material sciences, advanced characterizations, artificial intelligence, and environmental impact analysis for addressing the global energy challenges using gel polymer-based materials.
2. Main challenges of wide-temperature rechargeable batteries
The electrolyte, which physically interacts with all other components, stands out as the most distinctive component in a rechargeable battery.23,24 Temperature significantly affects the electrolyte and its interfaces with other components whether it is an organic nonaqueous or aqueous system. At both high and low temperatures, the electrolyte experiences substantial degradation, leading to increased interfacial polarization and a decline in the performance of rechargeable batteries. Given the wide range of physical and electrochemical events that occur in rechargeable batteries, particularly under extreme temperatures, there are numerous complex challenges to overcome in developing suitable electrolytes.2.1 Challenges at low temperature
During the charging process of rechargeable batteries, the cations experience several steps: (i) solvation in bulk electrolyte, (ii) migration of solvated cations across the electrolyte to the surface of active material, (iii) desolvation of solvated cations at the electrolyte/electrode interphase, (iv) cations passing through the electrolyte/electrode interphase layer and intercalation into/plating onto the anode. At low temperatures, however, the electrolyte freezing and the intrinsic kinetic constraints of charge carriers/mass-transfer can induce the reduction of ionic conductivity of GPE, and high desolvation barrier, SEI thickening and dendrite growth in the GPE/electrode interfaces, significantly deteriorating the discharge capacity and causing excessive overpotentials (Fig. 3(a)). | ||
| Fig. 3 Main challenges for rechargeable batteries at low temperatures. (a) Schematic representation of the main challenges for rechargeable batteries at low temperatures. The arrows represent the main ion transport processes during the charging process. (b) Schematic illustration of the temperature-dependent ionic conductivity based on the Arrhenius and VTF equations. (c) Variation of the dendrite growth rate ratio it/if with temperature and Li plating current density, data extracted from ref. 45 with permission from Elsevier, Copyright 2013. (d) Schematic plot of the dependence of the critical Li nuclei radius and areal nuclei density on the Li deposition overpotential, data extracted from ref. 51 with permission from American Chemical Society, Copyright 2017. | ||
The conductivity of an electrolyte is a measure of its ability to transport ions. Researchers often use two empirical equations, namely, the Arrhenius equation (eqn (1)) and Vogel–Tammann–Fulcher (VTF) equation (eqn (2)), to describe the temperature dependence of ionic conductivity in the electrolyte (Fig. 3(b)).25,26
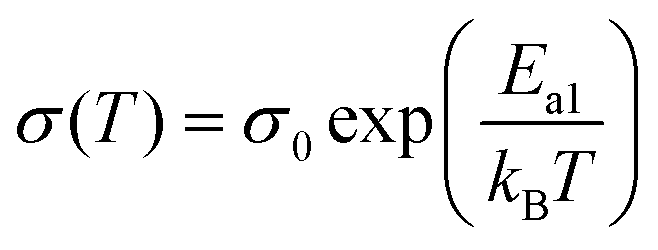 | (1) |
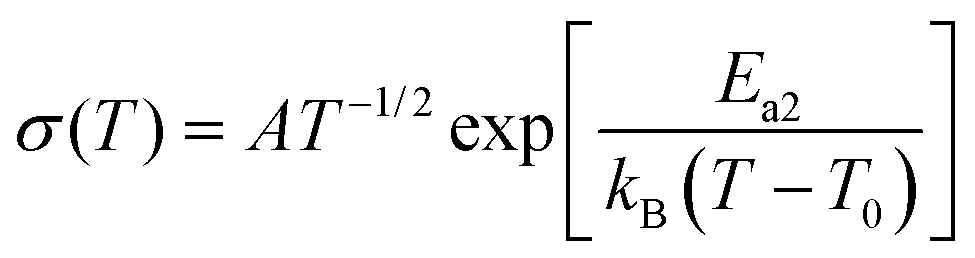 | (2) |
The ionic conductivity of conventional carbonate-based liquid electrolytes can be determined by eqn (3)
 | (3) |
 | (4) |
Early research suggested that the ion diffusion within the anode material (such as graphite) was the main obstacle to battery performance.28,29 However, recent studies have recognized that the process of ion migration at the electrolyte/electrode interface, involving the desolvation of cations from the solvent sheath and subsequent ion diffusion across the solid interfacial interphase into the active electrode material, is the primary factor affecting battery performance, especially at low temperatures.30,31 The ion-transfer kinetics at low temperatures are reflected by charge transfer resistance (Rct, the mid-frequency semicircle in EIS spectra). A high Rct value indicates a slow charge transfer kinetics. Zhang et al. found that at temperatures below −20 °C, the value of Rct became dominant, almost equaling the total resistance (RCell), whereas at 20 °C, it only accounted for approximately 50% of the RCell.32 According to the Arrhenius equation and the Butler–Volmer equation, Rct is related to the activation energy (Ea3) of the charge transfer process.20,33
 | (5) |
Besides the desolvation process, the ion migration across the SEI layer is also a key challenge at low temperatures, which depends on the nature and composition of the SEI layer. Thermodynamically, lowering the temperature not only slows down ion transport but also alters the decomposition reaction paths of electrolyte components. Wang et al. proposed that the SEI layer formed at low temperatures was mainly composed of intermediate products-rich organic species due to the incomplete decomposition of salts and solvents, which were metastable and easily dissolve in liquid electrolytes upon cell cycling, inhibiting the ion transport and inducing undesired side reactions.34,35 Furthermore, the lithium plating phenomenon of lithium-ion batteries was reported to occur at the anodic side at low temperature; it subsequently reacted with the solvent and lithium salt in electrolytes and the reaction products on the anodic side, increasing the thickness of the SEI layer. The increased SEI thickness lengthens the diffusion distance, causing a large interfacial impedance and polarization.36,37 It has been reported that an inorganic-rich SEI layer, particularly one containing fluorine, with a low thickness can lower the energy barrier for desolvation and facilitate rapid ion diffusion through the SEI layer.38–40 Given this understanding, it is crucial to develop suitable electrolyte compositions and SEI structures to enhance the performance of rechargeable batteries at low temperatures.
Kinetically, the decreased temperature reduces the mass transport limitation at the electrolyte/electrode interface and increases the reaction kinetic energy barrier of metal-ion deposition, leading to the slow and uneven plating of metallic species. This causes the formation and accumulation of dendrites near the anode.41–43 Love et al. found that the temperature directly affected the dendrite morphology and the dendrite formation rate (vd) and thus tuned the short-circuit time (tsc),44 as shown in eqn (6).
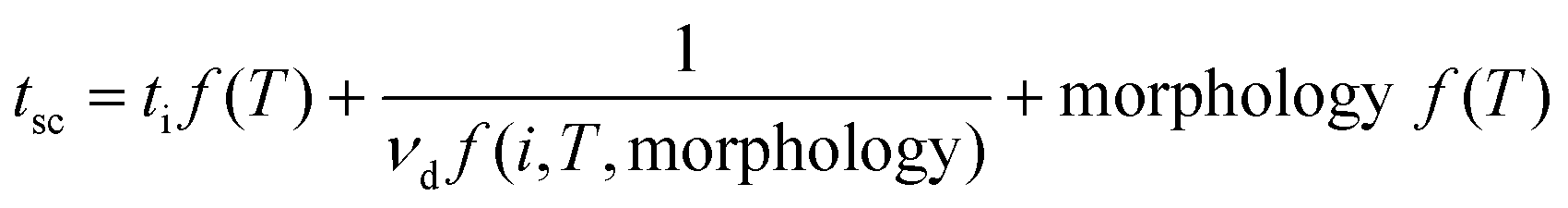 | (6) |
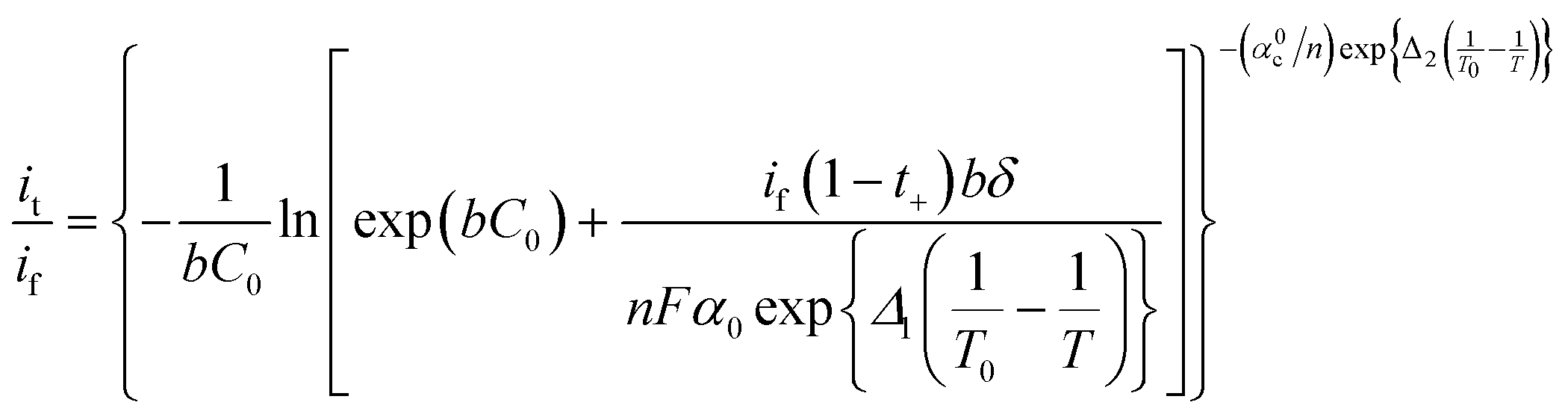 | (7) |
 | (8) |
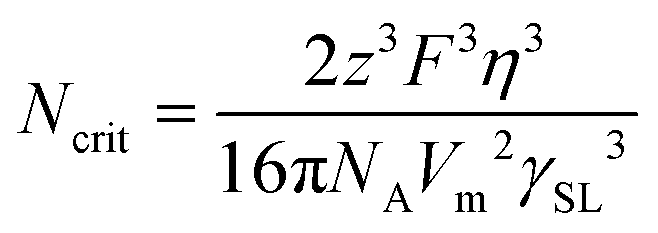 | (9) |
2.2 Challenges at high temperature
Batteries often perform better at high temperatures due to increased ionic conductivity and speedier chemical reactions, this is only somewhat reliable within a −20 to 55 °C temperature range, based on numerous past studies. However, the performance of most documented rechargeable batteries at temperatures exceeding 55 °C remains questionable. This is mainly attributed to the electrolyte evaporation and decomposition, pronounced solubility and reconstruction of SEI, and uncontrollable dendrite growth. These factors trigger dynamic and intricate structural alterations during the mass and charge transfer process within the batteries, eventually leading to battery performance degradation and even causing safety hazards (Fig. 4(a)).56 Physically, elevated temperatures would increase the vapor pressure of the liquid components in the electrolytes, raising the risk of evaporation or leakage, electrochemical reactions instability, and even safety hazards. Thermodynamically, the instability and decomposition of electrolytes at high temperatures can be attributed to the fact that these batteries operate at voltages beyond the thermodynamic stability range of the electrolytes, which is determined by the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO). The chemical potential of cathode materials (μc) and anode materials (μa) was reported to shift with the increase in temperature. This process can be evaluated according to the Nernst equation57 | (10) |
 | ||
| Fig. 4 Main challenges of rechargeable batteries at high temperatures. (a) Schematic illustrations of the main challenges of rechargeable batteries at high temperatures. (b) Change of chemical potential of cathode materials (μc) and anode materials (μa) with the increase in temperature, data extracted from ref. 16 with permission from RSC publishing, Copyright 2022. (c) Schematic illustration of the SEI/CEI energy level shift to reach a new steady state as the chemical potential of the cathode (anode) shifts with temperature. (d) Change of the thickness of the anode particles with temperature, data deduced from ref. 60 with permission from Elsevier, Copyright 2014. (e) Relationship between SEI thickness and Young's modulus at different temperatures, data extracted from ref. 65 with permission from RSC publishing, Copyright 2014. (f) Change in the dendrite length and width with the temperature, data extracted form ref. 67 with permission from Elsevier, Copyright 2018. Insets: The morphology of Li dendrites at the temperature of 0 and 50 °C. | ||
In addition to unfavorable interfacial reactions, dendrite growth is also a main obstacle at high temperatures, especially in rechargeable metal batteries. The uncontrollable growth of these metallic protrusions can cause internal short-circuits. Theoretically, high temperatures have an inhibitory effect on the growth of dendrite.66 As displayed in eqn (8), (9) and Fig. 3(d), a high temperature can facilitate ion diffusion, leading to the decrease of η and contributing to the metal deposits with larger nuclei size and low nucleation density. The integrated temperature-dependent phase-field models of Li-dendrite presented by Chen et al. and the Zn-dendrite model presented by Wang et al. both demonstrate that high temperature leads to a shorter dendrite length and larger width than those at low temperatures (Fig. 4(f)), thus weakening the dendrite growth.67,68
However, in fact the experimental results of dendrite formation at high temperatures have been widely reported.69,70 In these cases, it is commonly assumed that the incompatible kinetics and uneven charge distribution at the electrolyte/electrode interfaces are responsible for dendritic growth at elevated temperatures. On the one hand, the ionic conductivity of the SEI is several orders of magnitude lower than that of the bulk electrolyte, causing faster ion diffusion in the electrolyte than in the SEI and leading to the formation of ionic depletion layer and strong concentration gradient, which aggravates dendrite growth. On the other hand, the unevenly distributed component and morphology of SEI accumulated by the degradation products induces an uneven impedance and current density distribution, promoting dendrite formation.70,71 Additionally, the irregular SEI breakdown (solubility or mechanical instability, voids and/or cracks formation, etc.) at the electrode vicinity due to the large volume change of the anode and the porous SEI augmentation by the electrolyte decomposition during repetitive plating/stripping at high temperature results in variations of ion concentration and mobility across the electrolyte/electrode interfaces, further exacerbating the dendrite formation.57,72
3. Key requirements of wide-temperature rechargeable batteries
As discussed above, both high and low environmental temperatures would have an impact on the ion-transport kinetics and stabilities of electrolytes and electrolyte/electrode interfaces in rechargeable batteries. Rational electrolyte design is required for both aqueous and non-aqueous battery systems to enhance the electrochemical performance and avoid severe degradation and safety risks at extreme temperature conditions. In order to meet the requirements of rechargeable batteries to withstand wide-temperature applications, advanced electrolytes are required to overcome the above-mentioned drawbacks and achieve performance including rapid ionic charges and mass transport, excellent chemical/electrochemical stability against electrodes, and satisfactory thermal/mechanical tolerance. The detailed requirements are shown in Fig. 5 and comprehensively discussed in the following sections. | ||
| Fig. 5 Key requirements of wide-temperature rechargeable batteries. The temperature-dependent electrochemical/physicochemical/mechanical properties, cost, and environmental impact should be comprehensively considered. | ||
3.1 High ionic conductivity
Ionic conductivity is considered the most important parameter in all electrolyte research works as it primarily determines whether the electrolyte could be operated to deliver the theoretical energy density of the designed battery system. It is timely to establish a temperature-dependent threshold value of ionic conductivity, applicable to various electrolyte systems, to guide the electrolyte optimization towards wide temperature operability.73–76 Due to that the ionic conductivity is not the limiting factor for high temperature battery operation, the ionic conductivity requirements in this section refer specifically to conditions of low temperature. In contrast to the relatively short research history of GPEs toward wide temperature applications, research progress on the more developed liquid electrolytes may provide some reference for evaluating the ionic conductivity requirements, it is demonstrated that the rationally designed electrolyte with an ionic conductivity of ∼2.7 mS cm−1 at −20 °C could allow a NCM811//Li battery with a high areal capacity of 3.5 mA h cm−2 to operate efficiently and stably at −20 °C and even below.77 Another groundbreaking example is the recent demonstration of a remarkable energy density of 110 W h kg−1 at extremely low temperature of −40 °C for Ah-level sodium-ion batteries using a low-temperature specific liquid electrolyte.78 Summarizing these achievements, it is found that at the very low environmental temperature of −20 °C (approximately the lowest temperature that most regions of the earth could reach), an ionic conductive value of 3–10 mS cm−1 is required for the efficient operation of these practical-level batteries. Considering that the ionic conductivities of liquid electrolytes would retain only one tenth to one sixth of their initial values when being impregnated in the commercial polyolefin separator, 1 mS cm−1 at −20 °C could be preliminarily considered as the threshold requirement to guide the exploration of GPEs applicable at low temperatures.79–81 Furthermore, a low activation energy (i.e., Ea) is desired for easy ion conduction and to minimize the impact of temperature on ionic conductivity.82,83 This can be achieved by improving cation concentration, enhancing cation migration rate and providing excess cation conduction paths. Since GPEs possesses cohesive properties of solids and diffusive properties of liquids simultaneously, proper tuning of GPE components, including polymer matrix and enclosed liquids, is necessary to achieve high ionic conductivity and low activation energy over a wide temperature range.84,853.2 High target-ion transference number
Although necessary, high ionic conductivity alone does not guarantee excellent performance of wide-temperature rechargeable batteries, given the fact that ion conductivity is the result of the collective movement of all ionic species, whereas the faradaic current is predominantly balanced by the cations rather than their counterions.86 In practical electrolyte systems, cations and anions move in opposite directions. However, interactions (e.g., coordination bonds and hydrogen bonds) between cations and polar molecules such as solvents cause anions to move faster than cations, leading to the anion accumulation and the buildup of a concentration overpotential near the electrode, which can seriously lower the energy efficiency, particularly at extreme temperatures.87,88 In this case, the importance of target-ion transference number (t+) is highly emphasized since it reflects the ionic conductivity contribution of specific ions (t+ = Nv,sDs/∑Nv,sDs, where Nv,s and Ds are the number density and diffusion coefficient of individual ions, respectively), a high t+ would represent a relative higher target-ion mobility than other ionic species in electrolyte.89,90 According to the classical Newman model, an electrolyte with a lithium-ion transference number (tLi+) close to unity can warrant a considerably higher discharge capacity compared to the traditional liquid electrolytes with tLi+ of approximately 0.2, primarily attributed to the negligible concentration polarization.91,92 Following the theoretical Chazalviel model and experimental measurements, it was found that the increase in dendrite growth is favored in electrolytes with low t+ due to the cation depletion and formation of space charge layer.93,94 Conversely, improved t+ promotes the cation distribution homogeneity and inhibits dendrite growth.95,96 It is suggested that modest improvements in t+, to 0.5 to 0.7, would be beneficial, particularly allowing more uniform electric field distribution on the electrode surfaces, which reduces the risk of hotspots and thus extends the life of the battery.88 This suggests that optimizing the transference number along with the ionic conductivity could be a key strategy in the development of advanced electrolytes, especially for low temperature applications where the total ionic conductivities of electrolytes have been significantly lowered.3.3 Wide electrochemical stability window
For achieving stable and reversible redox reactions in batteries, a desirable electrolyte must have a sufficient electrochemical stability window against electroreduction and electrooxidation over a wide range of operating temperatures and working voltages without causing detrimental side reactions.97–99 The electrochemical stability window is typically defined as the gap between the HOMO and LUMO of the electrolyte. However, recent research suggests that the widening of electrochemical stability window requires the consideration of chemical/electrochemical interfacial compatibility beyond just the HOMO and LUMO of electrolyte components.100,101 The traditional methods of predicting electrochemical stability windows, such as HOMO/LUMO, overlook the electron transfer between reactants (neutral molecules) and products (charged molecules), as well as the structural evolution in the redox reaction, can lead to large prediction errors of up to ∼3.25 V.100 It can be expected that the extreme temperatures can further increase the error due to the temperature-dependent nature of the electron transfer behaviors. Additionally, temperature can affect the thermodynamic reactivity of electrolyte components and the kinetic behavior of the SEI/CEI layer, which determines the electrochemical stability window. Higher temperatures can push the potential of the cathode or reduce the potential of the anode into the unstable zone of the electrolytes. Constructing electrochemically stable SEI/CEI layers can effectively broaden the electrochemical stability window, which can prevent direct contact between the electrodes and electrolytes consistently even when the electrolyte operates at extremely high temperatures.102–104 Therefore, choosing thermodynamically stable components for the electrolyte and constructing reliable SEI/CEI layers are two major pathways to widen the electrochemical stability window of the electrolyte over a wide temperature range.Regulating the ionic solvation structure is a practical strategy to customize SEI/CEI and improve the electrochemical stability window of electrolytes, in which the size of ions, the coordination number of polar solvent/water molecules in the solvation sheath structure, and the interaction strength between ion and polar molecules affects the properties of SEI/CEI.105,106 For example, modifying the solvation structure using additives or cosolvents (e.g., dimethyl sulfoxide (DMSO), dimethyl carbonate (DMC), and triethyl phosphate (TEP)) that are easier to coordinate with cations to reduce the solvent/water molecules in the primary solvation structure or by forming an anion-derived interphase has been reported to weaken the activity of the solvent and inhibit the active solvent-related decomposition and parasitic reactions and enlarge the electrochemical stability window.107–109 In some studied GPEs, the polar functional groups in their polymer chains have been reported to interact with the electroactive metal ions (such as Li+ in lithium-ion batteries) and thus participate in the regulation of the solvation structure.84,85 Although there is a preliminary understanding of some polymers, such as polynitrile, and their ability to extend the electrochemical stability window when are they used as a polymer matrix in GPEs, it remains unclear how and to what extent they affect the electrochemical stability windows, there is an urgent need for quantitative studies and internal guidelines.14,110 Of note, the influence of properties (e.g., components, morphologies, and structures) and electrochemical reaction thermodynamics (e.g., the formation mechanism and ion transport across SEI/CEI) of SEI/CEI on the electrochemical stability window of the electrolyte and the electrochemical performance of rechargeable batteries remains controversial, calling for deeper exploration in the future.
3.4 High thermal stability
High thermal stability is crucial for wide-temperature operability of GPEs as it encompasses thermodynamic, kinetic, and process stability related to heat generation and material stability upon exposure to extreme temperatures.Several factors affect the thermostability of electrolytes. Firstly, the inherent electrolyte components affect the thermal behaviors of the overall bulk electrolyte system. The most used electrolytes for rechargeable batteries at large scale are carbonate solvents in non-aqueous electrolyte systems and water in aqueous electrolyte systems; they generally possess low boiling temperatures and are easy to volatilize or burn at elevated temperatures. Meanwhile, most salts with low electrostatic energy suffer from decomposition at elevated temperatures, indicating low thermal stability. Na salts showed better stability compared to the Li counterparts (e.g., LiPF6 (∼175 °C) < NaPF6 (∼325 °C)).111 In addition, the disproportionation and the reactions with charged electrodes of hexafluorophosphate are more likely to occur at high temperatures (above 50 °C), even in the presence of traces of protic impurities, to produce toxic and corrosive HF.112,113 Therefore, it is required to choose salts with resistance to heat for wide-temperature electrolytes.
Secondly, the interplays among the electrolyte components critically affect the freezing and boiling points of electrolytes and influence the resulting thermal stability. It was reported that in some electrolyte systems such as localized high concentration electrolyte systems, the solvation structure possessed enhanced anion–cation interactions to reduce the reduction of the un-coordinated “free” solvent molecules, resulting in reduced solvent evaporation and improved thermal stability of the electrolyte.114,115 According to Chen et al., the enhancement of interactions between the polymer matrix and solvents can improve the thermal stability of GPEs by modifying the polymer matrix with similar functional groups to those found in the solvent based on the similarity-intermiscibility theory.116
Thirdly, when the electrolytes are used in rechargeable batteries, the thermal stability of electrolytes at the electrolyte/electrode interfaces must be taken into account. Side reactions between the electrolyte and the charged electrodes are one of the main factors contributing to the safety hazard of rechargeable batteries. In particular, the chemical transformations and parasitic reactions of electrolytes were susceptible to accelerate at elevated temperatures. Taking commercial lithium-ion battery as an example, the previous study presented that the SEI/CEI layers had the lowest decomposition onset temperature (60–120 °C) prior to the melting of the PE separator (∼135 °C) and the decomposition of the cathode (∼170 °C), electrolyte (>200 °C) and binder (>260 °C). Thus, the decomposition of the SEI/CEI layers determines the thermal stability of the overall rechargeable battery system.117,118 Due to the strong dependence of SEI/CEI stability on the solvent structure of the electrolyte, rational solvation structure engineering to construct thermally stable SEI/CEI is further highlighted.119,120 Recently, several methods have been successfully applied to facilitate the formation of a thermally stable SEI/CEI, including adjusting the participation and contribution of ion species, solvent/water molecules, and polymer dipoles in the coordination of metal-ion solvation structure by component regulation.116,121,122 Still, the effects of salts, solvents, polymers, additives, and concentrations of salt on the thermally stable SEI/CEI are unclear, and further research is needed.
3.5 High dimensional/mechanical stability
Existing GPE materials generally have limitations in weak mechanical properties at room temperature, which would be further magnified due to their vulnerability to freezing at low temperatures and matrix structure failure at high temperatures.123,124 The freezing of electrolytes at low temperatures could induce hardening issues such as salt precipitation, volume contraction of the GPEs. At high temperatures, structure failure induced by the evaporation and leakage of solvent, as well as the softening, shrinkage and even melting of the polymer matrix may lead to damage to the structural integrity and cause many dire consequences such as battery performance degradation, short-circuit between cathode and anode and even hazardous thermal runaway.125–127 Targeting high reliability in extreme temperature conditions, the ideal electrolyte should possess both high mechanical strength and good flexibility across a wide temperature range. Moreover, considering the external stress that the GPEs would bear during battery manufacturing, certain safety margins should be included in setting the mechanical performance parameters.Due to the shortcomings discussed above, previously, one could only increase the thickness of the GPE or incorporate a reinforcing matrix, both of which inevitably come at the expense of ionic conductance. Therefore, what the high mechanical strength nature of a GPE can additionally promote is the dramatic reduction of the thickness towards high mass and volume energy density of the assembled batteries.128,129 According to the development trend of solid polymer electrolyte, if the tensile strength of GPEs could be improved to the level of 10 MPa, ultrathin GPEs with a thickness of 5–20 μm, even thinner than state-of-the-art separators, are expected without experiencing damage during the battery assembly and operation process.130,131
3.6 Facile processability, low cost and low environmental/health impact
The most widely reported preparation methods of GPEs include ex situ solution impregnation/casting and in situ polymerization. The solution impregnation method is widely used to fabricate the GPE membranes with linear polymer matrices, such as polyethylene oxide (PEO), polydimethylsiloxane (PDMS), polyvinylidene fluoride (PVDF), and poly(acrylic acid) (PAA). It typically involves dissolving the polymer and forming dry membranes through casting and solvent evaporation, followed by a liquid swelling (soaking) step to produce GPEs. This approach is well-suited to current lithium-ion battery production if the dry membranes are used as the separator before solvent infiltration. In this case, these dry membranes need to meet similar general requirements for separators to overcome the damage caused by subsequent processing in battery production, such as curling, stacking, and packaging during, battery assembly.132 According to the USABC stipulations, the puncture strength of the separator needs to be greater than 100 kgf mil−1 (1 kgf = 9.8 N, 1 mil = 25.4 μm) and the tensile strain should be less than 2% when an external force of 1000 psi (1 psi = 0.006895 MPa) is applied.133 At the same time, the thickness of GPEs must be well-controlled to be comparable to commercial polyolefin separators (15 to 25 μm) and be competitive. The solution casting method produces self-standing GPE membranes without requiring solvent evaporation, streamlining the manufacturing process. However, it presents challenges concerning the mechanical properties of the resultant GPE membrane. For instance, mechanical brittleness can be induced by the osmotic swelling of liquid solutions. Likewise, molecular treatments aimed at enhancing the wide temperature operability can result in weakened mechanical strength. All these issues should be considered in the context of the reel-to-reel production process in battery manufacturing. Furthermore, the inadequate interfacial contacts between the prepared GPEs and the porous electrodes with high mass loading could be a tricky question that needs to be addressed prior to practical applications.134,135 The in situ polymerization method offers an alternative by generating GPE directly within the battery assembly.136–138 The process involves injecting a homogeneous precursor solution containing low-viscosity monomers or oligomers, salts, solvents, and initiators into the battery's electrodes and inducing polymerization via UV or thermal energy to form the GPE, ensuring good electrolyte-to-electrode contact. This technique is particularly useful for fabricating cross-linked GPEs, such as polyacrylate, polycarbonate, and polyether. A noteworthy advancement in this field was made by Lee and colleagues, who achieved a uniform distribution of components in the through-thickness direction of an NCM811 cathode with high areal mass loading of 60 mg cm−2 by the in situ introduction of a UV-cured GPE in the cathode.139 Although this result offers a practical solution, its universality in other practical battery systems needs further exploration. Future investigations should also consider the limited penetration of UV light and the light-absorbing characteristics of carbon materials in electrodes. Furthermore, the enclosed environments of the battery system limit the use of UV-induced in situ polymerization, making thermally-induced free radical or ionic in situ polymerization a more practical approach. The use of appropriate monomers and initiators that avoid gas or by-product formation is essential for quality GPE production. Typically, a separator is included to prevent short-circuits during in situ polymerization, but it often obstructs the ion transport, inevitably adding unnecessary weight and reducing ion conductivity. Research is therefore aimed at developing thin or even separator-free electrolytes and designing ion-conducting separators or hosts for in situ polymerization to improve the overall battery performance.The development and application of GPEs on a large scale necessitates a significant consideration of material and production costs. Economically viable alternatives or commercially proven materials can be used as polymer matrices and solvents within GPE systems, which are generally not prohibitive and can even be cost-effective. Certain additives, such as fluorine-containing species, despite being relatively expensive, can improve the performance of GPEs with a small dosage, thus minimizing the overall material cost. Therefore, material costs may not be the primary concern at this stage. The focus should rather be on production costs as the intricate preparation, production line, and sealing technologies associated with GPE represent significant challenges that need to be circumvented. Innovative techniques, such as rapid room-temperature in situ polymerization, can help reduce production cycle time and enable continuous production. Additionally, the economic and environmental implications of GPE production, including the consumption and recycling of organic solvents such as NMP, AN, and acetone, should be considered. The feasible and scalable processability of GPEs is highly demanded in pursuing the practical applications of wide-temperature rechargeable batteries, especially when applied in large-scale energy storage scenarios.
The requirements for GPEs have extended beyond their physical and electrochemical properties, delving into the realm of environmental and health impacts. It is of paramount importance to ensure that the synthesis, use, and disposal of these materials do not pose possible threats to the environment and human health. A primary concern is the heavy use of organic solvents in the production of GPEs as these solvents generally present potential hazards to both the environment and human health due to their volatility and toxicity. Therefore, more benign alternatives need to be pursued. Some GPE systems based on green solvents, such as IL and water, have been proposed. Moreover, the incorporation of biomass materials or other biodegradable polymers, including polylactic acid, cellulose, chitosan, and xanthan gum, into the electrolyte matrix could offer a promising solution. In particular, cellulose and cellulose derivatives as ion-conductors have offered an interesting opportunity for replacing fossil oil-based polymers in GPEs.140,141 Not only would this approach reduce the reliance on non-renewable resources but it would also mitigate the environmental burden associated with polymer waste. Additionally, rigorous safety testing must be conducted to minimize any potential health risks associated with the use of GPEs. This includes examining the potential for harmful emissions during operation, assessing the risk of fire or explosion, and evaluating the potential toxicity of the materials to humans.
4. Design and engineering strategies of GPEs for wide-temperature rechargeable batteries
As can be expected from the above section, it is not easy to find an electrolyte that fulfills all the requirements for wide-temperature rechargeable batteries. To guarantee the workability of the rechargeable batteries over a wide temperature range, many strategies have shown the effectiveness of modifying the GPE chemistry by adjusting the relative composition of the electrolyte and species of salts, polymer matrices, solvents, additives, etc.4.1 Salts
Salt is the key determinant of electrolyte performance. With respect to wide-temperature operability, the addition of salt reduces the freezing point of the electrolyte according to the colligative property, and the anionic species largely determine the properties of the SEI/CEI layers.105,142 For achieving wide-temperature applications, an ideal salt is expected to have superior solubility to completely dissolve and dissociate in electrolyte and the solvated cations should have high mobility to achieve high ionic conductivity of the bulk electrolyte. Meanwhile, it should have high thermal and electro-/chemical stability.Over the past few decades, significant efforts have been dedicated to designing, synthesizing, and characterizing novel salts as potential alternatives both in liquid electrolytes and GPEs. The most intensively investigated salts are thermally and chemically stable borate salts (e.g., lithium bis(oxalate)borate (LiBOB), sodium bis(oxalate)borate (NaBOB) and lithium difluoro(oxalate)borate (LiDFOB)) and imide salts (e.g., lithium bis(fluorosulfonyl)imide (LiFSl), sodium bis(fluorosulfonyl)imide (NaFSl), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and sodium bis(trifluoromethanesulfonyl)imide (NaTFSI)). However, borate salts have low solubility in carbonate solvents at room temperature, which worsens at low temperatures. While imide salts have shown good performance, especially at low temperatures, the significant corrosion toward aluminium current collector at relatively high voltages limits their application scenarios. Additionally, the high cost of these salts remains a major obstacle. Though most of the newly-developed salts demonstrated outstanding performance at room temperature, their use at low and high temperatures is still to be studied.145,146 Therefore, the strategy of using blended salts containing at least two main salts (usually binary/ternary main salts) offers a feasible and cost-effective approach to enhance the practical applications of salts and extend the operating temperature ranges of GPEs.
As shown in Table 1, the successful application of blended salts in electrolytes has been demonstrated at both low and high temperatures.147–151 The reasons for the substantial improvement in the low- and high-temperature performance using blended salts in electrolytes are summarized in Fig. 6. At low temperatures, the functions of blended salts are summarized as follows: (i) improving the ionic conductivity of the electrolytes. It was found that the dissociation ability of a single salt could be improved by introducing other salts, further influencing the dissociation balance reaction of a single salt, inducing the forward reactions, and improving the ionic conductivity.152,153 For example, the addition of LiDFOB in the LiBF4-based electrolyte enhances its ionic conductivity by reducing salt association (see Fig. 6(a)) thanks to the larger size and electron-withdrawing nature of the ODFB− anions compared to BF4− anions.154 (ii) Facilitating the desolvation process of target ions. Recent studies have shown that anionic coordination with target ions can regulate the solvation structure by reducing the interaction strength between the target ions and the solvents, which facilitates easier desolvation at the electrolyte/electrode interfaces. In a study by Hu et al., both experimental results and DFT calculations showed that with the addition of LiFSI in the LiTFSI-based electrolyte, the inclusion of FSI− in the Li+ solvation sheath decreases the Li+ desolvation energy from 63.9 kJ mol−1 to 56.7 kJ mol−1, thereby enhancing the Li+ desolvation kinetics at low temperatures (Fig. 6(b)).155 (iii) Promoting the formation of SEI/CEI with low ion transport barrier. The salts with good film-formability can preferentially decompose to form low ion-resistive SEI/CEI layers on the surface of electrodes, allowing fast ion-migration across the interfaces.
| Electrolyte formulation | Cathode//anode | Working temperature (°C) | Battery performance (initial capacity/capacity retention/cycles) | Ref. |
|---|---|---|---|---|
| 0.05 M LiPO2F2 + 1 M LiTFSI0.6–LiTFPFB0.4 in PC:EC:EMC | NCM532//Li | −40 to 90 | 154.4 mA h g−1/69.6%/300 cycles@60 °C | 147 |
| 108.1 mA h g−1/93.7%/50 cycles@−20 °C | ||||
| 1 M LiTFSI0.6–LiTFPFB0.4 in PC:EC:EMC | 153 mA h g−1/39.2%/300 cycles@60 °C | |||
| 105.9 mA h g−1/91.4%/50 cycles@−20 °C | ||||
| 1 M LiPF6 in PC:EC:EMC | Failed after 300 cycles@60 °C | |||
| 0.8 M LiTFSI-0.2 M LiDFOB-0.01 M LiPF6 in EC/PC | LiCoO2//Li | RT to 80 | 2.4 mA h cm−2/900%/100 cycles@80 °C | 148 |
| 1 M LiPF6 in EC/PC | —/4%/100 cycles@80 °C | |||
| 1 M LiTFSI0.5–LiBOB0.5 in EC/EMC | LFP//Li | RT to 60 | 55%/1000 cycles@60 °C | 149 |
| 0.5 M LiPF6–0.5 M LiBOB in EC/EMC | Unstable at 60 °C | |||
| 0.8 M Zn(OTf)2-0.2 M Zn(ClO4)2 in 2-MeTHF/H2O | PANI@V2O5//Zn | −20 to 60 | —/—(78.80 mA h g−1)/250 cycles@−20 °C | 169 |
| 216 mA h g−1/250 cycles@60 °C | ||||
| Cross-linked PEGDA-ETPTA + LiTFSI–LiPF6 in EC:DMC:DEC | LFP//Li | 25 | 116.9 mA h g−1/87.93%/300 cycles@25 °C | 165 |
| poly-DOL + 0.3 M LiDFOB-2 M LiTFSI | LiCoO2//Li | RT to 40 | 138.3 mA h g−1/∼58%/60 cycles@40 °C | 170 |
| PVDF-HFP + 0.8 M LiBF4-0.8 M LiTFSI-1 wt% LiBOB in EC | LMO + LMFP//graphite | −18 to 45 | 84%/100 cycle@45 °C | 171 |
| ∼63% of RT capacity/—/—@−18 °C | ||||
| PVDF-HFP + 0.8 M LiBF4-0.8 M LiTFSI in EC | 51% over 100 cycles@45 °C | |||
| ∼60% of RT capacity/—/—@−18 °C | ||||
| PTFE + PVDF-HFP in FEC/adiponitrile | NMC811//Li | 143 mA h g−1/78.3%/1000 cycles@RT | 166 | |
| PVDF-1 M LiTFSI-1 M LiBOB in PC/EC/FEC | LFP//Li | −10 to 80 | 165 mA h g−1/92.9%/300 cycles@70 °C | |
| —/—(79 mA h g−1)/300 cycles@−10 °C | ||||
| Poly-DOL-LiPF6-LiDFOB in DOL | LiCoO2//Li | 5 to 100 | 118 mA h g−1/87.2%/1200 cycles@15 °C | 172 |
| —/86.5%/520 cycles@80 °C | ||||
| Polysaccharide κ-carrageenan/PAAm 1 M ZnCl2-7 M LiCl in H2O | LFP//Li | −40 to RT | —/78.3%/1500 cycles@RT | 168 |
| —/80.5%/2000 cycles@−20 °C | ||||
| —/95.2%/2000 cycles@−40 °C |
 | ||
| Fig. 6 Fundamental electrochemical aspects of the blended salt strategy towards wide-temperature batteries. Effects or proposed mechanisms of this strategy in regulating/improving (a) ionic conductivity, data extracted from ref. 154 with permission from Springer Nature, Copyright 2018; (b) desolvation kinetics, data extracted from ref. 155 with permission from Elsevier, Copyright 2023; (c) thermal stability, data extracted from ref. 158 with permission from IOP publishing, Copyright 2004; (d) formation of ion-conductive and thermostable SEI layer; (e) TEM images of cycled Li anode disassembled from single- and blended-salt systems. Reproduced from ref. 167 with permission from Springer Nature, Copyright 2023. | ||
At high temperatures, the blended salt strategy can improve the battery performance in two ways: (i) improving the thermal stability of electrolytes. The thermal stability of electrolyte systems can be improved by blending salts that are more thermally stable.156,157 For example, due to the higher thermal stability of LiBF4, the exothermic reaction of the LiPF6–LiBF4 dual salt carbonate electrolyte system has a higher onset temperature compared to the pure LiPF6 carbonate electrolyte system (Fig. 6(c)).158 (ii) Forming thermostable SEI/CEI layers to prevent electrolyte decomposition.159,160 Some salts in blended salt systems can serve as sacrificial agents. When working at elevated temperatures, these salts can decompose to form thermally and chemically stable SEI/CEI layers on the surface of charged electrodes, protecting electrolytes from further decomposing and ensuring the stability of the interfaces (Fig. 6(d)).161,162 Thus, the effectiveness of these electrolytes is mainly attributed to the blended salts used, which synergistically modify the interfacial chemistry on the surface of the cathode and anode. As a typical example, Luo et al. reported a dual-salt electrolyte based on sulfite solvent (0.4 M LiDFOB + 0.6 M LiFSI in dimethyl sulfite (DMS)) for the wide-temperature operation of lithium-ion batteries. LiFSI has a strong dissociation ability in DMS, while LiDFOB has a strong affinity for Li+. At the low-temperature end, LiFSI provides ultrahigh ionic conductivity, and the weakly dissociated LiDFOB promotes Li+ desolvation by adjusting the solvation structure. At the high-temperature end, the interface stability of both the cathode and anode is improved by the preferential self-limiting decomposition of DFOB− anions to generate inorganic-rich SEI/CEI layers, largely suppressing the decomposition of DMS solvent molecules. This synergistic effect of LiFSI and LiDFOB allows the LCO//graphite full cells to withstand a wide temperature range from −78 to 60 °C.163
The above advantages make blended-salt electrolytes a shining star for wide-temperature GPEs.164,165 For example, Guo et al. designed a poly(ethylene glycol)diacrylate (PEGDA)-based GPE using a LiTFSI–LiPF6 dual-salt. They demonstrated that the introduction of dual salts improved the ionic conductivity to 5.6 × 10−4 S cm−1@ 25 °C, which is higher than that for single LiTFSI and LiPF6 salt in GPEs.165 In a recent study, Cuicci and co-workers used a LiTFSI–LiBOB dual-salt in a PVDF-based GPE. The synergic effect of LiBOB and LiTFSI helped to achieve high ionic conductivities at both low and high temperatures (0.16 mS cm−1@−20 °C, 1.93 mS cm−1@100 °C). In addition, LiBOB has a lower LUMO energy level than LiTFSI, making it more likely to be preferentially reduced on the surface of the anode to form a stable SEI layer enriched with ion-conductive and thermostable LiF and Li–B–O-based species. Therefore, the LiTFSI–LiBOB dual-salt in GPEs enables fast ion migration at low temperatures and high interfacial stability at high temperatures, resulting in the extremely stable cycling performance of LFP//Li batteries over a wide operating temperature range (between −10 and 80 °C).166 Very recently, from the aspect of salt, Wagemaker et al. raised the entropy of electrolytes by introducing multiple salts (e.g., LiFSI, LiTFSI, LiDFOB, and LiNO3) in dimethoxyethane (DME). Due to the participation of more anionic groups in the solvation structures, the solvation strength of Li+–DME is largely weakened, and the formation of a thin (∼6 nm) SEI layer enriched with ion-conductive inorganics is promoted. In contrast, the electrolyte with LiFSI as the single salt forms a thick organic-rich SEI layer (Fig. 6(e)).167 Although this study does not specifically address the low- and high-temperature performance of the electrolyte, we believe that this strategy has great potential for wide-temperature rechargeable batteries as the fast desolvation and migration of Li+ ions across the interfaces are widely recognized as necessary for improving the low-temperature performance, while a stable SEI layer holds promise for enhancing the high-temperature performance. However, although the positive effects of high-entropy salts on liquid electrolytes have been proven, there is a lack of knowledge regarding the properties of this class of liquid solutions in GPEs as well as their wide-temperature applications.
In addition to the abovementioned nonaqueous GPEs, the blended-salt GPEs with water as solvent have been also reported to achieve stable wide-temperature performance. For instance, Li et al. designed a polysaccharide κ-carrageenan/PAAm-based hydrogel electrolyte containing ZnCl2–LiCl dual-salt. LiCl provides Li+ ions for combining H2O molecules (competing against Zn2+ ions) and Cl− ions for forming more Zn–Cl super halide anions, suppressing the formation of insoluble hydrolysis byproducts of Zn(OH)2/ZnO and the freezing of the GPE. As a result, the LFP//Zn battery displays outstanding cycling stability at a low temperature of −40 °C.168 In another study, Wang et al. reported an aqueous electrolyte containing Zn(OTf)2–Zn(ClO4)2 dual-salt. The ClO4− anions in Zn(ClO4)2 act as hydrogen bonding acceptors to change the solvation structure and disturb the hydrogen bonding network, lowering the electrolyte solidification point and improving the Zn2+ transport kinetics. At the same time, Zn(OTf)2 facilitates the formation of ZnF2-rich SEI layer on the zinc anode surface, effectively inhibiting the growth of zinc dendrites and the parasitic reaction of H2O molecules. As a result, the PANI@V2O5//Zn cells delivered improved cycling stability across a wide temperature range from −20 to 60 °C.169
Overall, the blended-salt strategy opens up a wide range of opportunities for designing GPEs with improved thermal stability, desirably high ionic conductivity and the ability to form more durable electrolyte/electrode interfaces in practical batteries. Due to the very limited research experience yet high chemical/structural complexity of blended salt systems, previous efforts in this direction have mainly focused on the characterizations of the GPEs’ basic properties and demonstrations of battery performance improvements, lacking mechanistic investigation on how different salts can work collaboratively to realize these achievements. Continual research should focus on the precise detection of electrolyte/electrode interface evolution behaviors and the reconstruction of interactions among various ionic and molecular species within the blended-salt systems, this could involve the use of cutting-edge spectroscopic and imaging techniques to observe the dynamic processes at the molecular level, potentially leading to the discovery of novel blended-salt formulations for wide-temperature GPEs.
| Electrolyte | σ | Cathode//anode | Working temperature (°C) | Battery performance | Ref. |
|---|---|---|---|---|---|
| —/5 M LiTFSI in G4 | 9 × 10−5 S cm−1@25 °C | S//Li | 25 to 60 | 666 mA h g−1 over 100 cycles@60 °C | 179 |
| —/2.0 M LiDFOB in EC/DMC | — | LCO/graphite | 25 to 90 | 99.6% after 160 cycles@90 °C | 180 |
—/1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M LiTFSI MDFA/MDFSA-TTE M LiTFSI MDFA/MDFSA-TTE |
0.6mScm−1@20 °C |
NMC811//graphite | −60 to 60 | 160.2mA hg−1/80% after 360cycles@−30 °C |
108 |
| PVDF-HFP/6 M LiTFSI/DMSO | 0.89 mS cm−1@50 °C; | LFP//Li | −10 to 100 | 102.2 mA h g−1/50C@100 °C; | 188 |
| 0.031 mS cm−1@0 °C | 158.7 mA h g−1/2C@−10 °C | ||||
| —/19 M CH3COONH4/H2O | — | NiHCF@CNTs//poly(1,5-NAPD) | −40 to 80 | 0.05 A g−1, 130 mA h g−1@−40 °C; | 197 |
| 4 A g−1, 84 mA h g−1@25 °C | |||||
| 4 A g−1, 102 mA h g−1@80 °C | |||||
| Sodium polyacrylate (PVNa)/6 M KOH in H2O | 5.7 S m−1@−20 °C; | NiCo//Zn | −20 to 25 | 87%/10000 cycles@RT |
198 |
| 12.6 S m−1@24 °C; | |||||
| 16.3 S m−1@50 °C | |||||
| Polyelectrolyte poly(2-acrylamido-2-methylpropanesulfonic acid potassium salt) and methyl cellulose/5 M KOH in H2O | 0.18 S cm−1@−20 °C; | Co3O4/carbon black/air cathode//Zn | −20 to 50 | 764.7 mA h g−1@25 °C; 758.5 mA h g−1@0 °C; 754.2 mA h g−1@−20 °C | 199 |
| 1.05 S cm−1@25 °C | |||||
| PVA/33 M LiNO3 in H2O | 2.51 × 10−2 S cm−1 at RT | LiMn2O4//V2O5 | RT to 80 | 64.7 mA h g−1 –/–@RT | 192 |
| 54.5 mA h g−1 76.6%/176 cycles@60 °C | |||||
| PAM/3 m Zn(BF4)2 in H2O | 2.38 mS cm−1@−70 °C | PANI@SWCNTs | −70 to 25 | —/≈100%/100 cycles@−70 °C | 189 |
| ∼106 mA h g−1/94%/1000 cycles@25 °C | |||||
| Polyacrylamide (PAM)/7.5 M ZnCl2 in H2O | 6 mS cm−1@RT | WO3//Zn | −30 to 30 | —/—(278.3 mA h m−2)/9200 cycles@RT | 200 |
| 1.28 mS cm−1@ −30 °C | |||||
| Xanthan gum/12 M ZnCl2 in H2O | 13.8 mS cm−1@20 °C | NH4V3O8·1.9H2O//Zn | −20 to 20 | At 1.5 A g−1, 166 mA h g−1@20 °C; 97 mA h g−1@0 °C | 201 |
| 5.0 mS cm−1@−20 °C | 185 mA h g−1/90.3%/800 cycles@20 °C; 129 mA h g−1/90.7%/800 cycles | ||||
| PAM/3 M Zn(OTf)2 in H2O | 1.9 mS cm−1@−30 °C | MgVO//Zn | −30 to 80 | 484 mA h g−1/without any capacity sacrifice/100 cycles@60 °C | 191 |
| 27.1 mS cm−1@30 °C | —/almost unchanged/100 cycles@−30 °C | ||||
| Poly(2-acrylamide-2-methyl-1-propanesulfonic acid co-acrylamide)/3 M Zn(ClO4)2 in H2O | 59.9 mScm−1@25 °C |
PANI//Zn | −35 to 25 | —/great stability for >30000 cycles@25°C |
202 |
| 14.2 mScm−1@−35 °C |
—/great stability for >70000 cycles@−35 °C |
||||
| PVDF-HFP/6 M LiTFSI in DMSO | 0.27 mS m−1@20 °C | LFP//Li | −10 to 100 | 163.5 mA h g−1/89.6%/800 cycles @25 °C | 188 |
| 0.89 mS m−1@50 °C | —/stable for over 100 cycles@100 °C | ||||
| 2C, 133.5 mA h g−1@−10 °C | |||||
| PVA/33 M LiNO3 in H2O | 2.51 × 10−2 S cm−1@RT | LiMn2O4//VO2 | RT to 80 | 73.0 mA h g−1/—/—@80 °C | 192 |
| 71.1 mA h g−1/76.6%/176 cycles@60 °C | |||||
| PAA/1 M Zn(OAc)2 and 40 M KOAc in H2O | 3.74 × 10−3 S cm−1@RT | MnO2//Zn | 25 to 80 | 299.0 mA h g−1/90.6%/300 cycles@60 °C | 194 |
In conventional liquid electrolyte recipes, the ionic conductivity increases drastically as the salt concentrates until it peaks at an optimum value, in which the optimum ionic conductivity can be achieved by the trade-off between the ionic carrier number proportional to the dissolution and dissociation of salts and the ionic mobility that is inversely proportional to the viscosity of the solution. After that, the increase in the concentration will lead to a reduction in the ionic mobility due to an increase in viscosity (Fig. 7(a)–(c)).173–175 Another critical feature brought by concentration alternation involves ion-solvation structure and the interfacial chemistry (Fig. 7(d)). In low-concentration electrolyte (typically, <1 M), the number of solvent molecules is much greater than that of salts, in which metal ions are completely coordinated with solvent molecules in the solvation structure, and free solvent molecules are the dominant species (Fig. 7(d1)).106,176 Conventional medium-concentration electrolytes (usually 1 to 2 M) exhibit a balanced number of anions and solvents; the solvent separated ion pairs (SSIPs) and free solvent molecules are the main species in solvation structure (Fig. 7(d2)). Such an electrolyte is desired because only a small amount of salt is required, which reduces the cost of electrolyte. However, the free solvent molecules are prone to suffer from freezing, and the strong binding of solvent molecules to the ions in SSIPs may limit the desolvation dynamics at low temperatures. Moreover, free solvent molecules and loosely-coordinated anions in SSIP can intensely decompose near the electrolyte/electrode interface at high temperatures. All these effects will limit the wide-temperature use of low- and medium-concentration electrolytes. With ascending salt concentration to above a threshold (usually >2 M, depending on the salt-solvent combinations), the interactions between cations and anions are enhanced, and the solvation sheath of the cations is increasingly dominated by salt anions, leading to the structural transition from SSIPs to contact ion pairs (CIPs) and ionic aggregates (AGGs) (Fig. 7(d3)). The new solution structure makes the location of the LUMO shift from the solvent towards the salt, resulting in the reductive predominant decomposition of the anions before the solvent to form an anion-derived SEI at low potential. This anion-derived passivation was revealed to be inorganic-rich, providing low interfacial charge-transfer resistance and high stability, which is beneficial for wide-temperature performance.167,177 Moreover, the dramatical drop of free solvent molecules in the liquid solution shows less decomposition and passivation in the electrolyte–electrode interfaces, improving the thermodynamic stability and electrochemical stability of the electrolyte.178 In addition, as anions are confined kinetically to CIPs and AGGs, the cationic transference number will increase in high-concentration electrolytes, which favors the improvement of rate capability. Therefore, increasing the salt concentration of the electrolyte provides a potential solution for widening the operating temperature window.
 | ||
| Fig. 7 Extending the operating temperature of GPEs via salt concentration adjustment. (a) Dependence of ionic conductivity on the salt concentration; (b) Variations of viscosity on the salt concentration at different temperatures, data extracted from ref. 174. Reproduced from ref. 174 with permission from American Chemical Society, Copyright 2020; (c) the comparison of high- and low-concentration electrolytes.196 Reproduced from ref. 196 with permission from CC BY 4.0; (d) the solvation structure and SEI evolution of (d1) low, (d2) medium-concentration, (d3) high-concentration and (d4) localized high-concentration electrolytes; the schematic illustrations of (e) the intermolecular interactions and solvation structures in a solidified localized high-concentration electrolyte. | ||
High-concentration salt electrolytes have been demonstrated to boost the cycling stability of batteries at high temperatures. For example, Sun et al. developed an electrolyte for Li–S battery with 5 M LiFSI dissolved in tetraethylene glycol dimethyl ether (G4). They suggested that the SEI layer formed in this high-concentration electrolyte consisted mainly of LiF, which was derived from the LiTFSI salt rather than the G4 solvent. The LiF-rich SEI layer helps to suppress the unwanted side reactions and dendrite growth on the Li anode, enabling the safe and stable operation of Li–S batteries at 60 °C.179 Similarly, a concentrated electrolyte of 2.0 M LiDFOB in EC/DMC was proved to boost the high-temperature cycling stability of a graphite//LCO cell at 90 °C. They attributed this to the formation of a compact LiF-rich CEI layer in the high-concentration electrolyte, which could effectively inhibit the solvent decomposition and the dissolution of Co2+ in the LCO cathode.180
In spite of the improved high-temperature electrochemical performance enabled by high-concentration electrolytes, their applications below room temperature are rarely reported. The main reason is their dramatically increased viscosity at low temperatures, which leads to low ionic conductivity and inferior wetting property.174 The novel concept of localized high concentration electrolytes (LHCE), as proposed in recent studies, is particularly interesting and is attracting increasing attention.181–183 By adding a small amount of non-polar or low-polar solvents such as 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE), bis(2,2,2-trifluoroethyl)ether (BTFE), and methyl difluoroacetate (MDFA), the concentrated electrolyte solution can be diluted while maintaining the originally favorable solvation structures (highly concentrated coordination clusters) (Fig. 7(d4)).184–187 For example, Huang et al. revealed that the fluorinated ether cosolvent with low dielectricity and weak binding strength with Li+ intrinsically facilitated the anion–cation interactions by affording a specific low-dielectric environment, thus promoting the generation of AGGs and favoring the formation of a dense and smooth SEI layer on the Li metal anode.184 In a more recent study, Wang et al. developed an LiFSI-based LHCE diluted with TTE and MDFA. They revealed that the addition of TTE and MDFA minimized the Li+–solvent binding energy and promoted the formation of CIPs and AGGs, enabling the improvement of ionic conductivity and the formation of TFSI− anion-derived LiF-rich SEI/CEI layers. As a result, the NMC811//graphite full cells achieved stable cycling performance over a wide temperature range from −60 to 60 °C.108 However, developing solvent systems that can concurrently show good salt solubility and high compatibility with polymer matrices remains challenging. Very recently, a “solidified localized high-concentration electrolyte” was proposed through the gelation of a concentrated solution containing 11.92 M LiTFSI/DMSO within the PVDF-HFP membrane, in which the non-solvating PVDF-HFP framework participated in the regulation of the solvation structure. By interacting with Li+ ions and DMSO molecules, the PVDF-HFP framework can confine solvent molecules and promote the formation of ion aggregates (donated as CIP-2 and AGG-2) in an ultrahigh salt concentration regime (Fig. 7(e)). The unique solvation structure enables a high ionic conductivity (0.031 mS cm−1 at 0 °C) as well as the formation of an LiF-rich SEI layer, resulting in the stable operation of LMBs over a wide temperature range from −10 to 100 °C.188 Although only very limited progress has been achieved, we anticipate that studies on GPEs with (localized) high salt concentration will increase in the years to come.
In aqueous electrolyte systems, high-concentration salts are thought to help reduce hydrogen bonding density between free water molecules, inhibiting the formation of ice crystals and lower the freezing point of aqueous electrolytes.189 The wide-temperature effectiveness of high-concentration (“salt-in-water”) electrolytes has been demonstrated for both liquid electrolytes and GPEs.157,190 A concentrated Zn(OTf)2-based polyacrylamide (PAM) GPE, for example, was reported to show satisfactory low temperature ionic conductivity of 1.9 mS cm−1@−30 °C and thus boosting the wide-temperature performance of the Zn–MgxV2O5·nH2O (MgVO) battery from −30 to 80 °C. This is because the strong coordination between water and ions in the concentrated electrolyte reduces the intermolecular hydrogen bonds, thus preventing the electrolyte from freezing at low temperatures and suppressing the electrochemical activity of water from side reactions on both the surfaces of the MgVO cathode and Zn anode at high temperatures.191 Even when increasing the salt concentration to an oversaturated state, GPEs still showed an expanded operating temperature range.192–194 A recent work by Tao et al. demonstrated that a poly(acrylic acid) (PAA)-based GPE with oversaturated salts (1 M Zn(OAc)2 + 40 M KOAc) and configuration merely composed of inactive “frozen-like” solvent and contacted cation solvation sheath helped to improve the electrochemical stability and suppress Zn dendrites formation at an elevated temperature of 60 °C.194
Therefore, constructing salt-concentrated GPEs may be a promising direction in developing wide-temperature adaptable batteries. However, the lack of a general principle for guiding the construction of salt-concentrated GPEs poses many uncertainties to researchers in this field. Laborious trial and error experiments are needed for the optimization of salt type and concentration. In addition, developing low-cost salt alternatives that do not compromise the electrochemical performance of the GPEs is a critical yet thorny issue. It should also be noted that increasing the salt concentration may negatively affect the GPEs’ mechanical properties.195 Therefore, the rational design of polymer matrices becomes crucial in balancing their electrochemical performance and mechanical stability, especially under extreme temperature conditions.
4.2 Polymer matrices
In addition to conditioning salts, the modification of polymer matrices in GPEs is another commendable option to achieve stable performance under wide-temperature conditions (Table 3). The most frequently used homopolymer and copolymer matrix materials in GPEs include synthetic and natural polymers. Different polymers possess different polymer chain structures, functional groups, properties and costs, as presented in Fig. 8. Synthetic polymers, such as PEO, PVDF, polyacrylates (polymethyl methacrylate) (PMMA), poly(acrylamide) (PAM), polyacrylonitrile (PAN), and PAA, possessing exceptional ionic conduction properties and excellent thermal stability, are widely used as polymer matrices of wide-temperature adaptable GPEs. However, these synthetic polymer matrices generally come with high costs due to their complex synthesis processes and raw material costs. This can significantly impact the overall cost of the GPEs and, consequently, the final battery products. Therefore, natural polymers derived from biomass, such as cellulose, chitin, agar, sodium alginate, starch, and silk fibroin, are emerging as sustainable and cost-effective alternatives. In addition, these renewable materials offer decent mechanical properties for GPEs.141,203,204 However, their performance, particularly their ionic conduction properties, might not match those of synthetic polymers to satisfy the demand for long cyclability over a wide temperature range. Therefore, engineering the polymers to balance these factors—ionic conduction, thermal stability, and cost—is crucial for developing wide-temperature adaptable GPEs.| Polymer matrices | Functional groups | Liquid solution | Ionic conductivity | Working temperature (°C) | Cathode//anode | Battery performance | Ref. |
|---|---|---|---|---|---|---|---|
| PAM-SC | –COO− | 6 M KOH in H2O | 324.68 mS cm−1@20 °C | −40 to 40 | Air//Zn | —/—/700 cycles@−40 °C | 250 |
| 73.96 mS cm−1@−40 °C | —/—/400 cycles@0 °C | ||||||
| —/—/170 cycles@40 °C | |||||||
| G-CyBA/PAAm double network | –NH2 | 6 M KOH in H2O | 300.1 mS cm−1@−80 °C | −50 to 100 | Air//Zn | —/—/42 cycles@100 °C | 225 |
| –OH | 431.7 mS cm−1@100 °C | —/—/192 cycles@70 °C | |||||
| —/—/600 cycles@RT | |||||||
| —/—/>240 cycles@−50 °C | |||||||
| Poly(TMPTA) | –C–O–C– | 1 M NaPF6 in EC:DEC:FEC | 1.13 × 10−4 S cm−1@0 °C | 0 to 60 | NMT//Zn | 71.6 mA h g−1/98%/100 cycles@0 °C | 233 |
–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
7.16 × 10−4 S cm−1@30 °C | 83.8 mA h g−1/80%/1000 cycles@60 °C | |||||
| 1.13 × 10−3 S cm−1@60 °C | |||||||
| Polymeric acid | –SO3 | 3 M Zn(ClO4)2 in H2O | 14.2 mScm−1@−35 °C |
−35 to 25 | PANI//Zn | 79.6 mA hg−1/—/70000 cycles@−35 °C |
202 |
| PAAm | OC– |
Zn(CF3SO3)2 in DMSO/H2O | — | −40 to 60 | Zn3V2O8//Zn | —/95.27%/3000 cycles@RT; | 249 |
| ∼175 mA h g−1/—/75000 cycles@−40 °C |
 | ||
| Fig. 8 Functions of polymer matrices in extending the operating temperature. Molecular structures, thermal properties and estimated costs of various synthetic and natural polymers having been widely used or showing great application potential. | ||
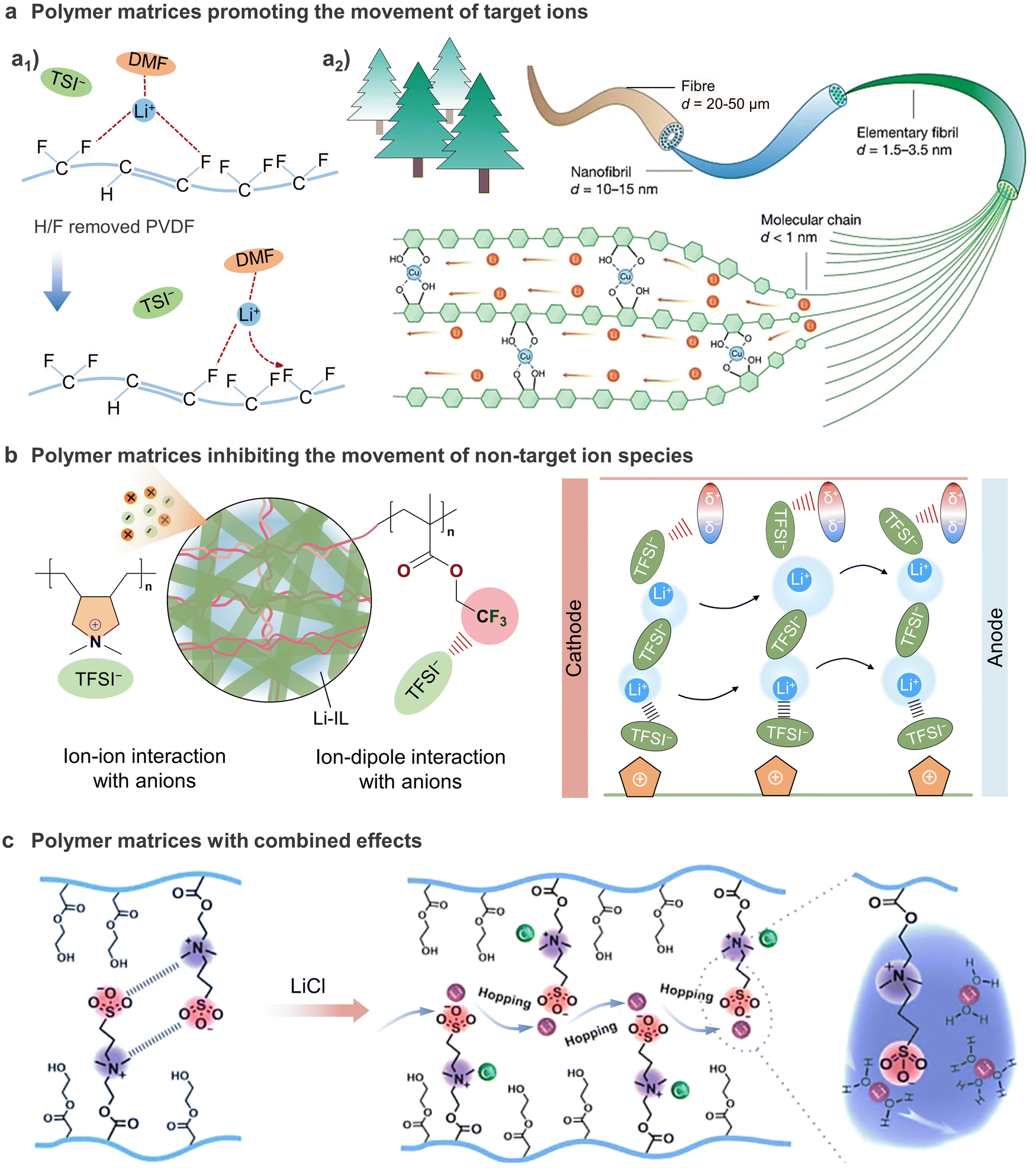
Apart from interaction with cations, some polar functional groups can also interact with anions to affect ion conduction.211,212 Yu et al. reported an ionogel membrane (IGEM) composing polymerized ionic liquid (PIL) of poly(diallyldimethylammonium) bis(trifluoromethanesulfonyl)imide (PDADMATFSI) nanofibers and cross-linked poly(2,2,2-trifluoroethyl methacrylate) (PTFEMA), in which the positively charged PIL and functional groups (–CH2CF3) with strong electronegativity on PTFEMA were able to balance the ion–dipole interactions and selectively anchor TFSI−.122 By regulating the environment surrounding the Li+ ions, the ion transport within the IGEM shifts from a sluggish vehicular Li+ transport, where Li+ migrates together with its coordination shell, to fast structural Li+ diffusion, where Li+ migrates by anion exchange in the first coordination shell, thus boosting the Li+ mobility (Fig. 9(b)). Benefiting from the enhanced Li+ conductivity, the LFP//Li cells with the IGEM allows low interfacial polarization, reduces the side reactions and achieves reasonably high rate capability and stable cycling stability in a wide temperature range from 0 to 90 °C. In another study, Coskun's group introduced a Lewis-acidic fluorinated alkyl side chain to functionalize the GPE matrix. The Lewis acidic sites immobilize TFSI− anions through noncovalent interactions, decreasing the affinity of Li+ towards oxygen atoms of glycol chains and promoting the Li+ mobility, thus realizing a high room-temperature Li+ conductivity (9.16 mScm−1) and high t+ value of 0.69 simultaneously.213
 | ||
| Fig. 9 Three proposed mechanisms possibly to facilitate the ion transport in GPEs. (a1) Schematic illustration for the interactions and lithium transport in the “dried” PVDF-DMF-LiFSI electrolyte. Reproduced from ref. 207 with permission from Elsevier, Copyright 2022. (a2) Schematic illustration of the structure and ion-transport channels in cellulose-based GPE. Reproduced from ref. 209 with permission from Springer Nature, Copyright 2021. (b) Schematic illustration of the Li+ transport mechanism in IGEM. Reproduced from ref.122 with permission from John Wiley & Sons, Copyright 2021. (c) Proposed Li+ migration mechanism in polySH electrolyte. Reproduced from ref. 215 with permission from John Wiley & Sons, Copyright 2021. | ||
Beyond the individual interactions with either cations or anions, some polymer matrices can interact with both anions and cations in GPEs. One such example is the zwitterionic polymer, in which the cation and anion are covalently bonded to form the side chain. This unique structure allows the dissociation of salts and increases the abundance of free ions through electrostatic interactions. Additionally, the zwitterionic polymer can also serve as migration pathways for both cations and anions under an applied electric field. These properties have made zwitterionic polymers highly attractive as matrices for constructing high-performance GPEs.214–216 Liu's group designed a zwitterionic polymer by randomly copolymerizing zwitterionic monomer (SBMA) and 2-hydroxyethyl acrylate (HEA) in the presence of LiCl salt. They proposed a direct hopping migration mechanism, wherein the interactions between anionic and cationic counterions on zwitterionic chains and Li+ ions facilitates the dissociation of lithium salt, allowing the formed hydrated lithium-ion (Li+(H2O)n) to hop and migrate through the channel of zwitterionic groups, as depicted in Fig. 9(c). This unique migration mechanism greatly improves the ionic conductivity of the GPE. As a result, the GPE exhibited an impressive ionic conductivity of 146 mS cm−1 at room temperature and 12.6 mS cm−1 at −40 °C, which is much higher than that of the conventional PVA-based GPEs.215
In these cases, apart from providing mechanically stable networks for the liquid solution, the polymers serve as the ion transport medium. GPE with rationally tailored polymer chain chemistry is therefore promising to deliver desirable ionic conductive ability. This would be further highlighted under the development trend that liquid-lean GPEs are nowadays actively pursued:217,218 as the liquid fraction is reduced, the ion transport may not depend largely on the liquid solvent molecules, and the role of the polymer matrix in ion conduction can become very prominent. This leads to a new sub-class of GPEs with improved stability and safety and may imply new thoughts for designing batteries with an extended operating temperature range.
% KOH).221 Liu et al. confined DMSO within a 4,4′-bis(stearoylamino)diphenyl ether-based polymer matrix to fabricate GPE. The strong interactions between the N–H bonds in BSDE and the SO bonds in DMSO (N–H⋯OS) broke the original weak hydrogen bonds and ordered molecular arrangement of DMSO, damaging the frozen structure of DMSO solvent at sub-zero temperatures.222 Wu's group reported that the abundant functional groups in modified polysaccharides carboxymethyl chitosan–PAM hydrogel could form ternary and weak hydrogen bonding with chaotropic ClO4− anions and water molecules, which enabled the polymer chains to break the hydrogen bonds between water molecules to remarkably decrease the electrolyte freezing point.223
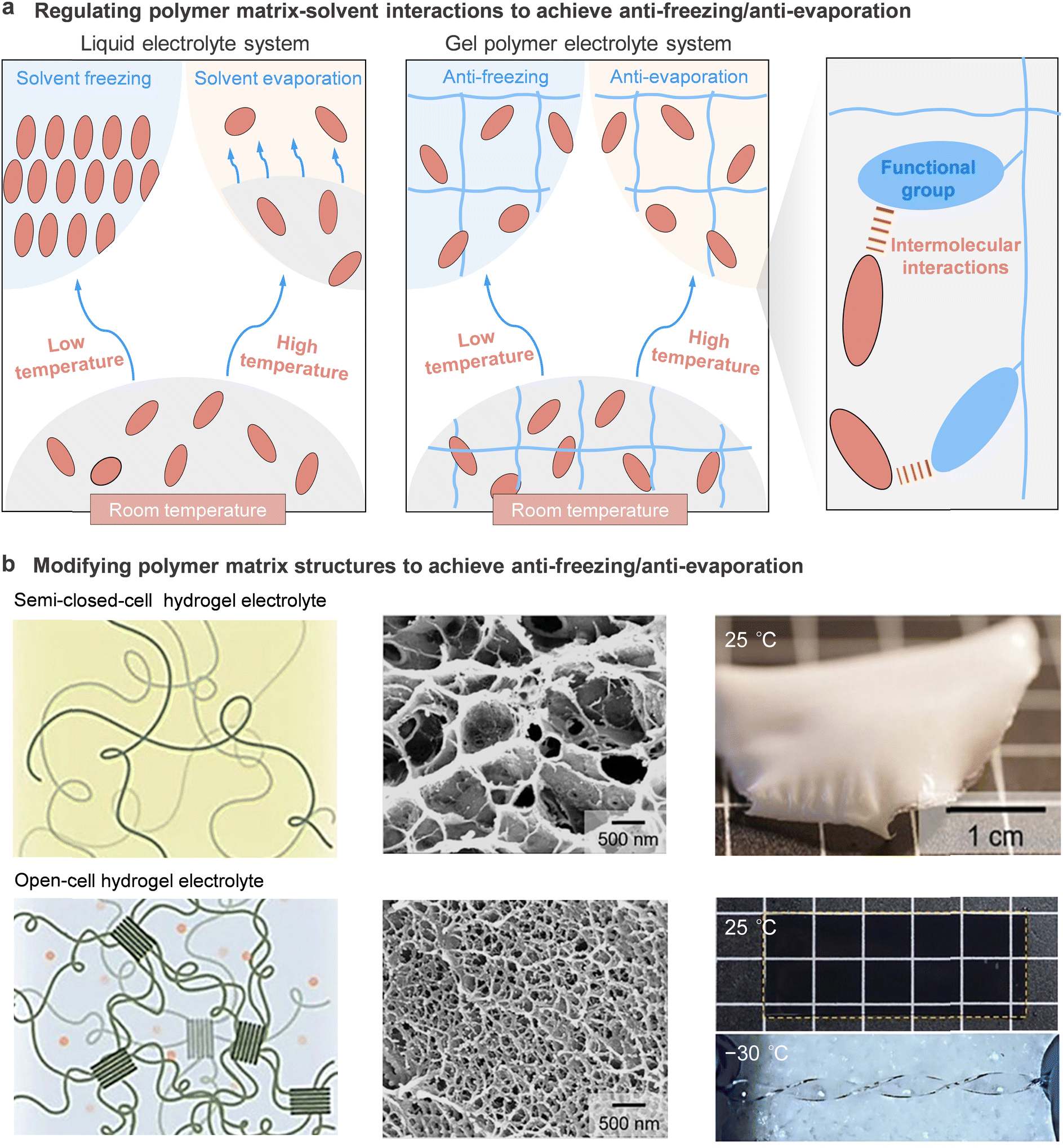 | ||
| Fig. 10 Beneficial effects of polymer matrices on suppressing the freezing and evaporation of confined solvents. (a) Illustrations of the mechanisms involved in the suppression of freezing and evaporation. (b) Schematics, SEM images and optical photos of GPEs featuring polymer matrices with open-cell and semi-close-cell structures. Reproduced from ref. 224 with permission from John Wiley & Sons, Copyright 2023. | ||
In addition, the structure of polymer matrices influences the temperature tolerance, mechanical and ion transporting properties of GPEs. As shown in Fig. 10(b), He's group fabricated a PVA-based GPEs with unique open-cell porous structures via a strategy combining co-nonsolvency and “salting-out”. They used “salting-out” K+ and acetate to toughen hydrogels via ion-promoted chain aggregation and added DMSO cosolvent to the precursor, synergistically promoting the formation of open-cell porous structures with a densified polymer network. The open-cell porous structure allows the fast diffusion of the ions and achieves enhanced mass transport (10× lower overpotential) compared to the PVA hydrogel with a semi-closed-cell structure. Moreover, the hydrogel electrolyte contains strongly aggregated polymer chains and disrupted hydrogen bonds among free water molecules, contributing to a high mechanical strength (tensile strength of 15.6 MPa) and excellent freezing tolerance (<−77 °C).224 Gu et al. designed a supramolecular double-network hydrogel electrolyte with a hierarchical porous structure by interpenetrating guanosine–cyclohexylboronic acid (G-CyBA) with a polyacrylamide (PAAm) network in the presence of concentrated aqueous KOH. The O atom in the B–O bonds of G-CyBA nanowires intensively interact with –NH2 groups in PAAm via multiple N–H⋯O hydrogen bonds. Also, the additional hydrogen bonding sites in the –OH groups of the G-quadruplex nanowires and –NH2 groups of the PAAm chain were supposed to further interact with water molecules to immobilize the solvents. The obtained hydrogels exhibit high mechanical robustness and stretchability, ultra-high conductivity, as well as superior anti-freezing (−196 °C) and anti-dehydration (100 °C) properties.225
Of note, the use of nonflammable polymer matrices is effective to improve the safety of GPE at high temperature. For example, Liu et al. fabricated the succinonitrile-based PIL GPE. With the thermally stable PIL as the polymer matrix, the thermal stability of GPE was improved up to 300 °C, 70 °C higher than that of GPE without the PIL matrix.226 In another study, Zhang et al. synthesized nonflammable or less-flammable poly(phosphonate/phosphates)-containing polymer matrices. These GPEs show good thermal stability and efficient flame retardance even filled with 90wt% of traditional liquid electrolytes.227,228
Overall, by fine-tuning the polymer–solvent interactions and the structure of the polymer matrix, it is possible to extend the operating temperature range of GPEs, making them more versatile for a wide range of applications.
First, the optimization of polymer matrices of GPEs can enhance the interfacial compatibility by introducing adhesive functional groups or creating “conformal” interfacial contacts (Fig. 11(a)).233,234 For instance, Li et al. reported a adhesive hydrogel electrolyte prepared through a mild free radical copolymerization with sodium lignosulfonate and acrylamide as the monomers. The catechol groups from sodium lignosulfonate strongly interact and adhere well with both the cathode and anode materials, which significantly reduces the contact resistances and improves the affinity between the electrolyte and electrode (Fig. 11(a1)).235 In another study, Cao et al. demonstrated a cellulose nanofibril-based morphing GPE to improve the interfacial contact, it is mechano-adaptive and beneficial to the formation of interlocked structure on the surface of electrodes, which largely reduces the interfacial contact resistance and promotes uniform nucleation.236 In addition, chemical bonds form between polar groups in polymer matrices of GPE and electrodes (especially when employing organic electrodes), which enhances the affinity of electrolyte and electrodes. Recently, Shim et al. fabricated a GPE for PANI//Zn batteries using methanesulfonic acid and PVA. The intermolecular interactions between the electrolyte and the PANI cathode enable the close contact between the electrolyte and the polyaniline cathode, thus increasing the adhesion of the electrolyte and the electrode. The increased adhesion not only reduces the interfacial impedance but also enhances the interfacial charge transfer performance by increasing the localized sites for ion hopping.237 Additionally, the enhancement of the interfacial compatibility can be achieved by in situ synthesis methods such as UV/thermal polymerization (Fig. 11(a2)).238–240 The polymer electrolyte with compact interfacial contact developed by the in situ polymerization of trihydroxymethylpropyl triacrylate (TMPTA) not only provides an integrated 3D network throughout the whole interior of battery with well-connected Na+ ion migration pathways but also forms a favorable NaF-rich SEI layer to prevent the sodium metal anode from forming dendrites, endowing the Na//Na2/3Ni1/3Mn1/3Ti1/3O2 (NMT) batteries with excellent cycling stability in the temperature range from 0 to 60 °C.233
 | ||
| Fig. 11 Influence of polymer matrix on the interfacial chemistry. (a) Enhancing interfacial compatibility by: (a1) introducing adhesive functional groups; (a2) creating “conformal” contact. (b) Accelerating interfacial ion transport by: (b1) regulating the ion solvation structure; (b2) guiding uniform ion transport. | ||
Second, the molecularly designed polar groups in the polymer chain were reported to coordinate with target-ions and delicately modulate the solvation structure, thus accelerating the target-ion desolvation and regulating the SEI layer on the surface of the electrode (see Fig. 11(b1)).84,206,229,241 For example, Luo and co-workers developed a GPE composed of 0.5 M NaPF6/diglyme in polymerized 1,3-dioxolane (poly-DOL). They indicated that the poly-DOL segments in the GPE help to weaken the interaction between Na+ ions and diglyme solvents and confine the PF6− anions. This leads to a local high concentration of Na+ ions and significantly speeds up the Na+ desolvation process, promoting the formation of a thin inorganic SEI like Na2O and NaF. These inorganic components in SEI restrain side reactions and provide faster channels for interfacial Na+ migration. As a result, the NVP//Na cells presented excellent performance after 1000 cycles with a capacity retention of ∼99% at −20 °C, better than the regular liquid electrolyte.241 The participation of the polymer matrix in the solvation structure can expel partial active solvent molecules from the solvent sheath, thus reducing the formation of undesirable substances in the SEI layer.84,242 In a recent work, Zhou et al. found that the abundant functional carbonyl groups (OC) groups in the polymer chains of the PEGDA-based GPE weakened the interactions between Na+ and propylene carbonate (PC) solvent molecules, greatly decreasing the activity of organic PC and inhibiting the parasitic reactions.84
Additionally, the abundant charged groups and the unique backbone structure of polymer matrices can provide additional driving force and specific channels to accelerate ion transport (Fig. 11(b2)).202,243,244 Recent studies have demonstrated that some polymer networks possess metal-ionophilic properties to uniformize metal ion flux distributions and constrain two-dimensional diffusion over the surface of anode, thus achieving highly efficient ion migration.245–247 For instance, Hu's group developed a negatively charged carboxylate hydrogel electrolyte (gelatin/sodium alginate–acetate), in which the zincophilic carboxyl functional groups and acetate ions guided the directional migration of hydrated Zn2+ ions along polymer chains, stabilizing the interfacial electrochemistry.248 A recent study showed that the abundant negatively charged carbonyl groups in the PAAm chain of a cross-linked PAAm-based GPE could coordinate with Zn ions to improve their distribution, which could restrict the cusp effect and attract more accumulation of Zn salts. As a result, the dendrite growth was suppressed and the operation temperature of the Zn3V2O8//Zn full-cell was extended to −40 to 60 °C.249
As seen in this research direction, previous interest has mainly been focused on the role of polymer matrices on the electrolyte/electrode interfacial compatibility. Future efforts such as molecular and structural engineering of polymer matrices to tailor the solvation/desolvation dynamics of electroactive ions towards better electrochemical performance of batteries are of great significance. It is expected that a thorough understanding and thus control of the interactions between the polymer matrices and ionic/molecular species of GPEs could lead to further breakthroughs in all-temperature batteries.
4.3 (Co-)solvents
For GPEs, the solvent, both water and organic ones, primarily functioned as the liquid plasticizer to reduce the polymer matrix crystallinity, thus enabling good mechanical reliance and significantly improved ionic conductivity. Additionally, it also plays an important role in determining wide-temperature operability, mainly referring to high-temperature thermal/electrochemical stability, freezing tolerance, and SEI stability at both high and low temperatures, by regulating the microstructure (especially solvation structure of target ion) of liquid components confined in the matrix.For achieving wide-temperature applications, the ideal solvent should satisfy the requirements as follows: (i) good solvation ability to salts; (ii) chemical inertness toward battery components; (iii) wide electrochemical widow; and (iv) low viscosity and freezing point to favor ion mobility at low temperature, high boiling point to improve the safety at high temperature. However, as displayed in Fig. 12, the properties of single-solvent electrolytes are limited and do not meet the requirements of wide-temperature or even room-temperature electrolytes. In addition, the complex interfacial chemistry at extreme temperatures further complicates the solvent selection. A common strategy to overcome these challenges and extend the operating temperature range is the addition of cosolvents.
 | ||
| Fig. 12 Summary of freezing points and boiling points of popular solvents used to prepare GPEs. This data could roughly guide the design of GPEs with the desired working temperature range. For example, sulfones and ILs may be applied to stabilize the GPEs toward high temperature applications. | ||
Different from reducing the freezing point of the electrolyte directly, some cosolvents were found to participate in the solvation structure, helping to improve the ion transport kinetics of batteries at low temperatures.256,263 In commercial electrolytes, EC has been established as the main constituent in the primary solvation sheath of Li+, making EC the major chemical source for the desolvation and formation of SEI. However, the strong affinity toward Li+, together with the organic-rich EC-derived SEI, unavoidably hinders Li+ desolvation kinetics and slows the migration rate of Li+ across the SEI, especially at low temperatures.264 It was reported that the addition of linear DMS or DES as the cosolvent is capable of simultaneously decreasing the freezing point of electrolytes and forming a robust SEI layer on the anode, thus keeping the low irreversible charge losses at low temperatures. Guo et al. very recently found that the single-electron reduction of DMS in baseline electrolyte (1M LiPF6 in EC/EMC) facilitated the formation of ionic conductive SEI layer, leading to a low Rct and RSEI in graphite//Li half-cell and retarding the capacity decay of LiNi0.5Co0.2Mn0.3O2//graphite batteries at −20 °C.263 Fluorinated solvents, such as fluoroethylene carbonate (FEC), 2,2,2-trifluoro-N,N-dimethylacetamide (FDMA) and methyl-3,3,3-trifluoropropanoate (MTFP), have been widely used as low-temperature cosolvents due to their low freezing points and favorable effects of electron-withdrawing fluorine on desolvation and interfacial chemistry.182,265,266 A recent study by Xiong et al. showed that the Li+–dipole interaction strength decreased from −1.90 to −1.66 and −1.44 eV with the increased fluorination degree from EC to FEC and difluoroethylene carbonate (DFEC), respectively. Owing to the lower interaction strength, FEC and DFEC can desolvate from the Li+ solvation sheath more easily than EC molecules to produce a LiF-rich SEI layer. As a result, the NCM811//Li batteries with FEC- and DFEC-based electrolytes retain 45% and 51% of room-temperature capacity at −30 °C, respectively, superior to the nearly zero capacity in the EC-based electrolyte.267 He et al. fabricated a PEGDA-based GPE containing 1 M LiPF6 in ethyl propanoate (EP)/EMC/FEC cosolvent. Benefiting from the high ionic conductivity of 1.2 mS cm−1 at −40 °C and LiF-rich CEI and SEI, the LFP//Li batteries show a high discharge capacity (72.1% of the room temperature capacity) even at a low temperature of −40 °C.268,269 Guo et al. demonstrated a NCM811//Li battery with a low-temperature adaptable GPE consisting of FDMA and FEC as the cosolvent, LiDFOB as the salt, and poly(1,3,5-trioxane) as the polymer matrix.17 The implementation of this design allows the stable cycling of NCM811//Li batteries at −30 °C. This impressive behavior is mainly attributed to the formation of a LixBOyFz/LiF-rich and Li2CO3-poor SEI with a low energy barrier for Li+ diffusion. At the same time, the formed amorphous CEI helps to stabilize the NCM811 cathode from the dissolution of transition metal and structural cracking.
The cosolvent strategy is also found useful in improving the freezing tolerance and ionic conductivity of hydrogel electrolytes. Organic cosolvents commonly used in hydrogel electrolytes include nitriles, esters, and amides.270,271 For example, using the low-freezing point DMF (−60.5 °C) as a cosolvent, the freezing point of the ZnSO4/DMF/H2O electrolyte drops down to −32 °C, lower than that of the ZnSO4/H2O electrolyte (−11 °C).272 Similarly, Chen et al. prepared a PVA-based hydrogel using an IL/water (EMImAc/H2O) binary solvent. The freezing points of the prepared hydrogels (−65 °C) were much lower than those of pure water and EMImAc (−20 °C) due to the strong interactions between EMImAc and H2O.273 Unlike aprotic organic solvents, water molecules can be used as both electron acceptors and electron donors. Therefore, another strategy to weaken the water–water interaction and improve the low-temperature performance of aqueous electrolytes is to add cosolvents such as dimethyl sulfoxide (DMSO) and polyol solvents.274–276 When polyol cosolvents are added to the electrolyte, the strong interactions of –OH groups of polyol solvents with water molecules through hydrogen bonding severely impair the interactions among water molecules, thus inhibiting the ice crystallization and lowering the freezing temperature. At the same time, the attraction of polyol molecules to H2O could reduce the solvation degree of Zn2+–H2O, which is beneficial to ion migration in the electrolytes. For example, Li et al. added a low freezing point EG cosolvent to water (EG:H2O = 3:2 by volume), reducing the freezing point of the electrolyte to −33 °C.277 DFT calculation results showed stronger hydrogen bonding interactions of EG–H2O (10.40 kcal mol−1) than H2O–H2O (5.78 kcal mol−1), indicating the capability of EG in locking free water and inhibiting freezing.278 In GPE systems, the polyol cosolvents can assist the PAM polymer network to break the hydrogen bond networks of water, endowing PAM-based hydrogel electrolyte with excellent anti-freezing ability down to −40 °C.279 In addition to polyols, DMSO as a cosolvent has also been extensively evaluated in hydrogel electrolytes to improve their low-temperature operability. For example, Li et al. designed a DMSO-containing PAAm/agar gel electrolyte that could operate at −30 °C.280 A recent study by Liu et al. demonstrated that DMSO in GPE could stabilize the Zn2+ solvation structure and reduce the proportion of networked water, inhibiting the freezing of the hydrogel electrolyte and lowering the operating temperature of the Zn battery to −20 °C.281
Recently, liquid electrolytes using high-entropy solvents have been recognized as a potential solution for low-temperature electrolytes. By increasing the solvent molecular diversity to modulate the solvation entropy and tune the mesoscopic solvation structure, the operating temperature of the liquid electrolyte could be extended to extremely low temperatures.282 For example, Chen et al. configured a series of high-entropy solvent-based electrolytes. They indicated that the freezing point of electrolytes decreased greatly with an increase in the number of solvents from binary to decimal. The high entropy of solvents endows the high disorder degree of mixtures, making the EC solvent molecules to nearly completely separate from each other and largely reducing the viscosity of the electrolyte. As a result, the decimal high-entropy EC-based electrolyte (EC/DEC/PC/EMC/EP/EA/MB/BA/MP/PB) showed an ultralow freezing temperature of −130 °C. Moreover, the electrolyte achieved a remarkably high ionic conductivity of 0.62 mS cm−1 at −60 °C and excellent electrochemical performance of the LMO//LTO batteries at −40 °C, which was far superior to that of commercial counterparts.283
Obviously, the development of high-entropy electrolytes with more than five organic solvents is of great research value for both low-temperature liquid electrolytes and GPEs. However, the precise selection and optimization of solvent combinations from the vast number of possible solvents remains challenging. Moreover, the complexity of high-entropy solvents increases the difficulty of understanding the physical and chemical interactions within the electrolyte system, not to mention at extreme temperatures. In addition, the introduction of multiple solvents into the GPE systems may lead to potential compatibility issues or unwanted side reactions. It is expected that these issues will be well addressed prior to the low temperature applications of GPEs with high-entropy solvents.
Despite their less pronounced stability, molecular-type solvents of sulfites and dinitriles have also been widely applied.290 For example, sulfolane, one of the most common sulfites, was successfully applied to prepare a PVDF/PEO-based GPE, which remarkably enhanced the cycling and safety performances of LMBs. When used in the Li//LCO battery, the sulfolane-containing electrolyte enables long-term cycling stability at high temperatures of up to 90 °C. Moreover, the full-charged Li//LCO pouch cells exhibited a higher onset temperature of thermal runaway at 190 °C, 60 °C higher than their commercial counterpart of LiPF6//EC/DMC/EMC.291 Succinonitrile (boiling point: 266 °C), a representative dinitrile, has a similar function to sulfolane. It has been reported that the PEO-based GPE plasticized with succinonitrile achieved a superior high-temperature-tolerance characteristic up to 170 °C.292 At a high temperature of 55 °C, the LCO//Li batteries using succinonitrile/LiTFSI-plasticized polyurethane acrylate GPE demonstrated improved cycling stability, compared to the one plasticized with liquid electrolyte (LiPF6 in EC/DMC).293
Despite the high boiling points of these solvents, their limited salt solubility and low reductive stability pose challenges in exceeding the upper temperature limits for batteries using them as plasticizers in GPEs. The addition of cosolvents, such as carbonates, and carboxylates, can help to solve these issues. Laskovic reported a PVDF-HFP-based GPEs gelled by liquids of LiTFSI in PC/isobutyronitrile/trimethyl acetonitrile/ethylene sulfate (ES)/vinylene carbonate (VC). Using ES and VC as SEI forming agents and employing EC and PC to dissolve LiTFSI, along with isobutyronitrile and trimethyl acetonitrile to enhance the thermal stability at high temperatures, PVDF-HFP-based GPEs demonstrated high thermal stability and improved cycling performance in NMC622//graphite full cells, even at 60 °C.294 In particular, fluorinated solvents have been extensively utilized as high-temperature cosolvents due to their ability to promote the formation of thermally stable LiF-rich SEI/CEI layers. Deng et al. introduced a fluorinated ether co-solvent (1,1,1-trifluoro-2-[(2,2,2-trifluoroethoxy) methoxy]ethane (TTME)) to the conventional ether electrolyte system comprising DME and LiFSI to form a fluorinated ether-based electrolyte. TTME not only helps to generate a stable SEI on the surface of graphite and to inhibit the electrolyte decomposition but also improves the overall oxidation stability of the electrolyte to prevent the degradation of the electrolyte on the surface of the LCO cathode. As a result, the LCO//graphite batteries performed much better in terms of cycling performance than their counterparts using carbonate electrolyte at elevated temperatures.295
Compared to non-aqueous electrolytes, achieving high-temperature applications with aqueous electrolytes poses more challenges. The lower boiling point of water, in comparison to most non-aqueous solvents, leads to the evaporation of water solvent and increased water activity in aqueous electrolyte systems operating at elevated temperatures. This can result in significant electrolyte loss and decomposition at the electrolyte/electrode interfaces. The addition of high boiling point cosolvents in the plasticizer of hydrogel electrolytes can improve their thermal stability and inhibit their interfacial side reactions. The main cosolvents of high-temperature aqueous electrolytes include amides, sulphones, and polyols. These cosolvents not only possess high boiling point values but can also manipulate the solvation structure and interfacial chemistry, inhibiting the evaporation of water inside the electrolyte and suppressing the interfacial side reactions, thus improving the high-temperature performance of electrolytes and batteries.296–298 Xiong et al. modified the ZnSO4 aqueous electrolyte with DMF (boiling point: 153 °C). They demonstrated that the DMF can intensify the hydrogen bonds of DMF–H2O and weaken the solvation interaction of Zn2+ with H2O, facilitating the in situ formation of a Zn2+-conducting SEI layer (Zn5(CO3)2(OH)6) on the electrode surface during the cycling process. The formed SEI layer, featuring a high thermal decomposition temperature of 250 °C, enables the electrolyte to achieve outstanding performances and stabilities at high temperatures of 70 °C.272 The sulphones (e.g., DMSO) and polyols (e.g., glycol, glycerol, etc.) have been proven to be able to form hydrogen bonding with water molecules and suppress the water activity, mitigating the electrolyte decomposition at the electrolyte/electrode interfaces at high temperatures.249 It is foreseeable that the high temperature performance of aqueous electrolytes can be improved by increasing the content of organic co-solvents to water. However, this approach would defeat the original purpose of aqueous batteries as low cost, safe and environmentally friendly; thus, the ratio of organic co-solvents to water should be carefully adjusted.
For this consideration, the incorporation of ILs into hydrogel electrolytes appears to be a promising approach for high-temperature aqueous batteries as it combines the benefits of the high-temperature stability of ILs while preserving the advantages of aqueous electrolytes. However, the use of ILs in aqueous battery systems has been limited due to their thermodynamic immiscibility with water and poor salt solubility. Nevertheless, a recent study by Yu et al. has demonstrated a breakthrough in this regard. They developed an electrolyte consisting of a water-immiscible ionic liquid diluent, specifically 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)amide (EmimFSI), in water. Remarkably, this electrolyte enabled the operation of Zn0.25V2O5·nH2O//Zn cells at a temperature as high as 100 °C.299 This work opens new possibilities for the high-temperature utilization of ILs in aqueous battery systems regardless of liquid electrolytes or GPEs.
4.4 Additives
Unlike the salts and co-solvents, which are the dominant constituents of the electrolytes, additives are generally characterized by their small dosage but “quick-effect” nature. Due to their unique roles in regulating the ion transport behaviors and in improving the stability of electrolytes and the electrolyte/electrode interfaces, numerous types of additives are employed to improve the low- and/or high-temperature performance of GPEs and thus the batteries. In accordance with their respective roles under different temperature ranges, the functions of the additives are summarized in Fig. 13. In the case of low-temperature operation scenario, additives are strategically incorporated to (i) increase ionic conductivity as solubilizers; (ii) accelerate the rate-determining desolvation process as solvation tailors; and (iii) assist in the formation of thin and ion-conducting SEI as interface modifiers. Under high-temperature conditions, the additive engineering of GPEs shifts mainly to (i) improving the thermal stability as a thermal tolerance enhancer; (ii) protecting the GPE from flame as fire preventers; and (iii) helping to form thermostable SEI at the GPE/electrode interfaces as interfacial protectors.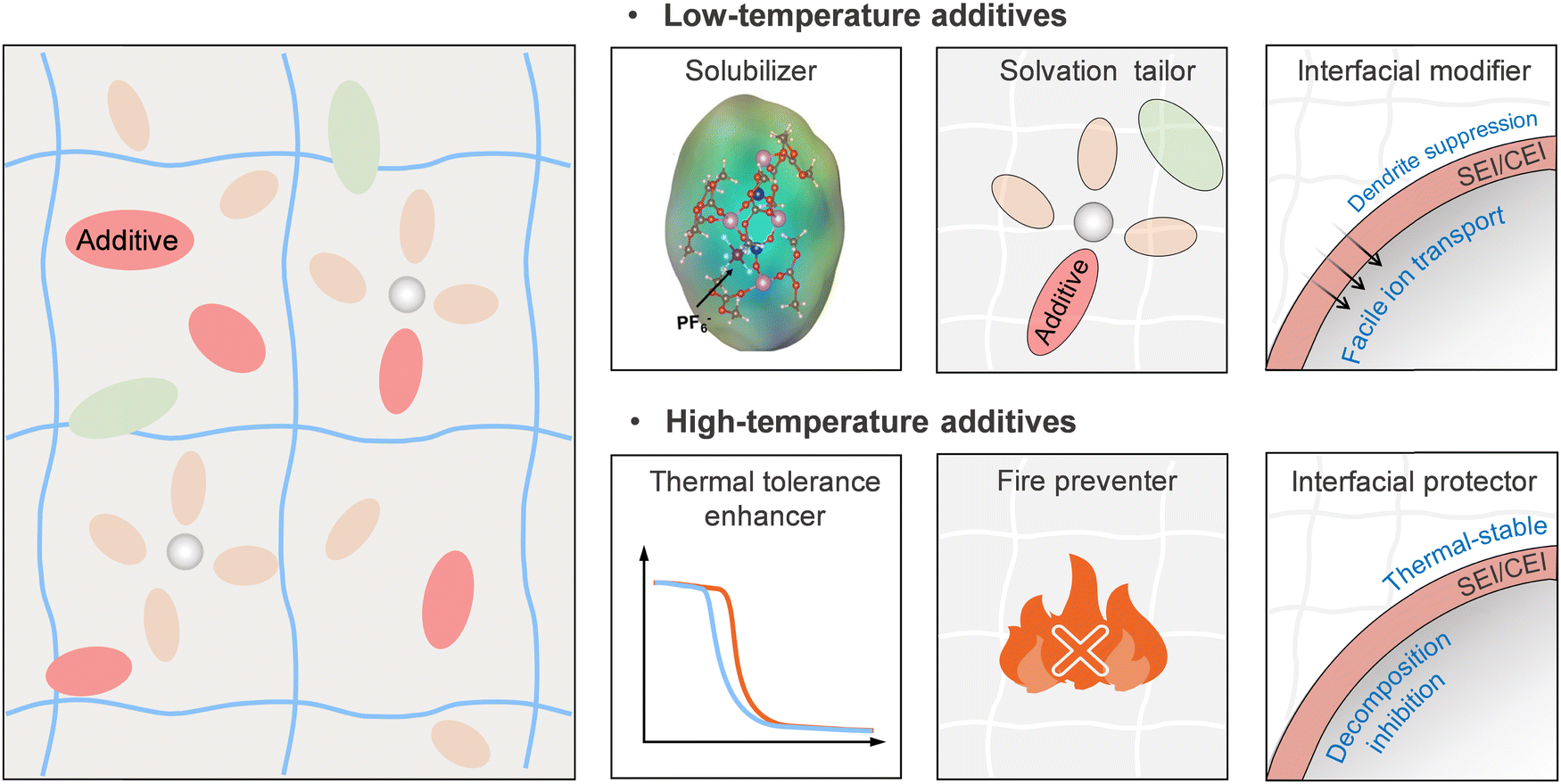 | ||
| Fig. 13 Effects of additives on extending the operating temperature of GPEs. Additives can function as solubilizer, solvation tailor, and interfacial modifier to ensure the low-temperature battery performance, while in the cases aiming at high-temperature operability, they usually act to improve the thermal stability, fire resistance and structural stability of the solid electrolyte interphase. Solubilizer image adapted from ref. 300 with permission from John Wiley & Sons, Copyright 2022. | ||
At the low-temperature end, although some organic additives with low freezing point and low viscosity can inhibit the solvent solidification or freezing of liquid solution and improve the ionic conductivity of GPE to a certain extent, considering their small dosages in the whole GPE systems, researchers are currently focusing more on their functions in regulating the interfacial chemistry of batteries.304 The organic additive can participate in the regulation of the ion solvation structure and formation of interfaces and interphases on the surface of electrodes, reducing the barrier of the desolvation process and across the SEI/CEI layers and eventually facilitating the ion-transport kinetics.305–307 Some organic solvents, such as VC, ES, AN, EC, and FEC, can function as film-forming additives to create interfacial layers with low ion transport barriers between the electrolyte and electrode.302,308,309 For example, the co-addition of 2 wt% ES and 2 wt% VC in a PVDF-HFP-based GPE with PC as the solvent contributes to the formation of a stable SEI, making the charge transfer and SEI impedances independent of the cycling process. This formulation achieved a remarkable cycle life for the NMC532/graphite cells.310 At low temperatures, however, VC leads to a thicker SEI with a low lithium ion conductivity, decreasing the discharge capacity.311 FEC, as an alternative, has been investigated in detail as a film-forming additive to create a LiF-rich SEI with superior low-temperature performance and improve the ionic conductivity of the electrolytes at low temperatures.307,312,313 In a recent study, Liao et al. found that the addition of 2 wt% FEC in the liquid electrolyte could increase the discharge capacity and CE by forming SEI layers on the graphite anode and LFP cathode at −40 °C.314,315 Moreover, Ciucci et al. demonstrated that FEC additive in a poly-DOL-based GPE participated in the first Li+ solvation sheath and reduced the binding energy of MP solvents with Li+, resulting in easier Li+ transport. At the same time, the presence of FEC induced the formation of fluorine-rich interphases, contributing to enhanced interfacial stability. Li//LFP and high-voltage Li//NCM811 batteries demonstrate excellent long-term stability (>2000 cycles at room temperature) and can operate effectively at a low temperature of −30 °C.316 Interestingly, the introduction of FEC is reported to be able to regulate the Li deposition morphologies on the Li anode from porous loosened deposits with dendritic structure into large particles with a dense structure at a low temperature of −40 °C,317 which provides a valuable solution for dendrite suppression of low-temperature adaptable alkali-metal batteries. These findings certainly motivate the exploration of more novel F-containing compounds.
At the high-temperature end, organic additives can also enhance the battery's thermal tolerance by forming a protective yet ion-conductive layer on the surfaces of both the cathode and anode. For example, the unsaturated organic compound of VC can be readily reduced to form a SEI layer rich in polymeric components via a ring-opening reaction at high temperatures. Its effectiveness has been proven in several battery systems including lithium-ion, lithium–sulfur, sodium-ion, and potassium-ion batteries.318,319 In addition, fluorides, such as FEC, can form LiF components on the anode and polyether species on the cathode, protecting the electrolyte from degradation, thus improving the reversibility of the battery at high temperature.320 Although some electrolytes discussed above could be operated at a high temperature, most of them contain flammable components, which may induce safety issues at abnormal temperatures. Flame retardants, such as organic phosphorus (e.g., triethyl phosphate (TEP), triphenyl phosphate (TPP), and trimethyl phosphate (TMP)) and fluorinated compounds (e.g., tris(pentafluorophenyl)borane, TB), can reduce the flammability of the electrolyte and passivate the electrodes by forming thermally stable SEI/CEI layers simultaneously, preventing possible hazards resulting from the thermal runaway of batteries.121,262,321–323 For example, TMP can release phosphorus-containing free radicals PO˙ and PO2˙ to prevent the chain reaction in batteries by capturing the H˙ and HO˙ radicals released from the burning composition.318,324 In an attempt to develop a safe and wide-temperature operable quasi-solid-state Li–S battery, TB additive was introduced to improve the flame retardance via decomposing and generating fluorine radicals to trap the highly reactive radical released by the GPE. At the same time, the TB with high fluoride content contributes to the formation of a highly stable and resilient LiF-rich SEI layer. With these merits, Li–S batteries assembled with this electrolyte demonstrated stable cycling over a wide temperature range from −20 to 50 °C.323
Similarly, for hydrogel electrolytes, the low-temperature performance was reported to be improved by adding organic additives such as polyols, sorbitol, and ILs.325,326 These additives feature high solubility in water, and the ability to strongly interact with water molecules via hydrogen bonding can improve the low-temperature ionic conductivities of hydrogel electrolytes. For instance, Chen et al. reported that the DMSO additive in PAAM-based GPEs could form abundant hydrogen bonds with both the functional groups of the polymer matrix and water molecules, enabling the formation of optimized GPEs with a high ionic conductivity of 0.82 S m−1 at −20 °C, comparable to that at room temperature.327 Moreover, at the electrolyte/electrode interfaces, the organic additive molecules are able to participate in the solvation structure and affect the interfacial chemistry through the interactions with target ions.281,304 A study by Hou et al. demonstrated that the DMSO additive in a PAM-based hydrogel electrolyte could coordinate with Zn2+ in the solvation structure to reduce the Zn2+–O(H2O) pair, lowering the energy barrier for Zn2+desolvation and improving the reversibility of Zn plating/stripping at low temperatures.281,319,328 At elevated temperatures, the organic additives were demonstrated to be able to inhibit the water decomposition via hydrogen evolution reaction and oxygen evolution reaction at the surface of discharged electrodes.329,330 A study by Hu et al. proposed that the addition of glycol and AN additive could adjust the affinity between H2O and oxygen-containing groups in the PAM-based GPE, enabling high ionic conductivity at low temperatures and reduced H2O reactivity at high temperatures. The GPE exhibited good anti-freezing properties at −20 °C and a slow water evaporating rate at 60 °C, enabling a high-performance Zn//V2O5 full cell that can cycle stably in a wide temperature range from −20 to 60 °C.274
Compared to the relatively limited choice of solvents/liquid plasticizers, it remains highly challenging to select suitable additives from the vast chemical space of possible compounds. Despite many successful additives for various battery chemistries, traditional trial and error experiments for additive searching is still far from efficient. Fortunately, the fast development of data-driven quantitative structure–property relationship models can substantially accelerate the findings of additives, promoting more advancements in the development of GPE-based batteries with wide temperature operability.
Many salts, such as LiBOB, LiDFOB, and LiPO2F2, have been investigated as auxiliary additives due to their film-forming ability.145,333,334 These salts can be sacrificially reduced or oxidized at the anode or cathode surface prior to solvent decomposition, thereby forming low-resistive and stable SEI/CEI layers composed of inorganic-rich components. For example, LiBOB can be electrochemically reduced to form a low-resistive SEI film at low temperatures. When cooperated with 4-vinyl-1,3-dioxolan-2-one (VEC) additive in a PVDF-HFP-based GPE, a robust and highly ionic-conductive SEI film can be built up to enhance the electrolyte/electrode interfacial stability, enabling the superior cycling performances of LMO+LiMn0.7Fe0.3PO4 (LMFP)//graphite battery at temperatures ranging from −18 to 45 °C.171 In addition to LiBOB, LiDFOB was also recognized as an effective auxiliary additive. With the contribution of LiDFOB decomposition to form low-resistance and stable F-rich and B-containing SEI film, the PVA-based GPE with LiDFOB presented lower capacity fading at low temperatures.335 F-containing metal-ion salt additives are especially attractive for constructing stable electrolyte/electrode interfaces/interphases. Their decomposition products, such as LiF and NaF, are believed to be able to reduce the interfacial resistance and improve the toughness of the SEI/CEI layers, therefore facilitating the interfacial ion transport and stability and boosting the wide-temperature performance.336–338 In a recent work by Cui and co-workers, 0.05 M LiPO2F2 was added to LiTFSI/lithium trifluoro(perfluoro-tert-butyloxyl)borate (LiTFPFB) dual salt-based GPE, dramatically improving the cyclability and rate capability of an LiNi0.5Mn0.3Co0.2O2//Li battery over an impressively wide temperature range from −40 to 90 °C. LiPO2F2 plays a dominant role in forming an F-rich SEI with both low impedance and high-temperature durability. In particular, the inorganic Li-containing species (especially LiF and Li2O, ionic conductive) and P–O species at the outer shell of the SEI layer can suppress Li dendrite growth and their reactions with carbonate solvents.147 These mechanisms have also been proved via the construction of N-rich or S-rich or B-containing SEI/CEI layers.335,339–341
Recently, nitrate salts (e.g., LiNO3 and NaNO3) have also been used as auxiliary additives to improve the wide-temperature tolerance of GPEs. Archer et al., for example, reported that the addition of LiNO3 in PEG-based GPEs showed a high ionic conductivity of >1mS cm−1 even at −30 °C, making it suitable for low-temperature batteries without any compromises in power density.342 Furthermore, they presented that the physical interactions between the electron-lacking N-atom in LiNO3 additive and DOL solvents could distort bonds in DOL and coupled motions of individual molecules, thus arresting the liquid to crystalline solid thermal transition and making the electrolyte preserve abnormally high bulk and interfacial ionic conductivities at temperatures down to −50 °C.343 Furthermore, the LiNO3-containing electrolyte can facilitate the formation of dendrite-free Li deposition.344 Additionally, it was reported that the addition of a small dosage of Lewis acidic salts (e.g., Al(OTf)3, Mg(OTf)2, LiPF6, and LiDFOB) or halides (e.g., AlI3 or AlF3) to a baseline electrolyte can initiate the in situ polymerization of monomers inside a battery cell without the addition of initiators, enabling the facile preparation of GPEs with high thermal, mechanical, and electrochemical stability. Because the formed GPEs originated from liquids are able to wet all components of the cell, “conformal” interfacial contact could be realized and thus the low interfacial resistance and homogeneous interfacial charge transfer, which represents a promising approach for constructing GPEs and battery systems that are adaptable over a wide range of temperatures.164,238,345–347
For aqueous GPEs, the introduction of soluble ions into water can lower the freezing point of electrolytes according to the colligative properties observed in dilute solutions. For example, the freezing point of water can be significantly lowered by the dissolution of inorganic salts such as CaCl2, LiCl, ZnSO4, or organic zwitterions.250 Chen's group introduced sodium citrate (SC) into PAM-based hydrogel electrolyte. The intensive interactions between the hydrophilic –COO− functional groups of SC molecules with H2O molecules facilitate the localization of water molecules in hydrogels, preventing water from freezing and evaporation. The electrolyte delivers a high ionic conductivity of 73.96 mS cm−1 even at a low temperature of −40 °C as well as high water retention of 96.85% after 96 h exposure, both of which are substantially higher than those of the control samples. As a result, high-performance zinc–air batteries with PAM-SC hydrogel electrolyte can be achieved over a wide temperature range from −40 to 40 °C.250
At low temperatures, inorganic fillers are reported to interact with ion species and ease the diffusion barrier.350,351 Zhang et al. incorporated an –NH2-functionalized vermiculite (Vr-NH2) as fillers into GPE, in which IL was confined in the regular interlayer nanochannels of the lamellar Vr-NH2 framework. The fine-tuning of the microstructure can trigger the rearrangement and crystallinity of ILs in nanochannels, leading to the formation of continuous and densely-aligned nanocrystalline IL layers and fast lithium-ion hopping transport along the 2D channels (Fig. 14(a)–(c)). With such unique structural merits, the developed electrolyte delivered a high ionic conductivity of 0.09 to 1.35 mS cm−1 over a wide temperature range from −40 to 100 °C. As a result, the LiFePO4//Li pouch cells presented an excellent wide-temperature electrochemical performance (93.8 and 45.0 mA h g−1 at 60 and −20 °C with capacity retention of 97% and 98% after 50 cycles, respectively).352 Recently, Ren and coworkers utilized the graphene oxide quantum dots (GOQDs) as an additive of aqueous electrolyte. They found that GOQDs strongly adsorb on the ice crystal surface to inhibit their growth, thus effectively inhibiting the growth of ice crystals and expanding the interconnected liquid regions to offer more spacious ion-transport channels, resulting in improved cell capacity and cycling stability at low temperatures.
 | ||
| Fig. 14 Compositing GPEs with inorganic additives to extend the operating temperature limits. (a) Schematic illustration of the Li+ transport mechanism along the 2D interlayer channel of the ionogel electrolyte with Vr-NH2 as the host; cross-sectional SEM and HR-TEM images (insets) of (b) the Vr-NH2 framework and (c) L-ILCE-NH2.352 Reproduced from ref. 352 with permission from John Wiley & Sons, Copyright 2023. (d) Schematic illustration of the function of MOF filler at high temperature; TGA curves of (e) the typical liquid electrolyte (1 M LiTFSI-PC) and (f) GPE with MOF as the filler.353 Reproduced from ref. 353 with permission from Springer Nature, Copyright 2022. | ||
At high temperatures, the addition of inorganic fillers can improve the thermal stability of GPEs by confining the liquid components. For instance, Zhou and coworkers used MOF (CuBTC-PSS, 6.5Å) that possesses a nanoporous (sub-nanoporous) structure as the host to fabricate the gel electrolyte. Due to the physical sub-nanoconfinement and chemical coordination with the MOF host (Fig. 14(d)), the solvents in GPE are unlikely to evaporate and decompose at the electrolyte/electrode interfaces. As a result, the decomposition temperature (highlighted in yellow) of the liquid solvent within the GPE began to decompose at nearly 200 °C, over 100 °C higher than that of the LE (Fig. 14(e) and (f)). Furthermore, the high-voltage NCM811//Li pouch cells using this GPE with a high NCM811 mass loading of 20 mgcm−2 delivered highly stable electrochemical performances with a high capacity of 171 mA hg−1 and 89% capacity retention after 300 cycles, even at a high working temperature of 90 °C.353
4.5 Hosts
Usually, the pursuit of mechanical strength and structural/dimensional stability of GPE is realized by increasing the mass ratio of the polymer matrix, which would unavoidably sacrifice the ion transport properties. Moreover, even with this strategy, the mechanical performance cannot always satisfy the requirements of GPE towards practical applications due to the theoretically limited strength and thermal stability of the molecularly dispersed polymer network, especially at extreme temperature conditions. With this consideration, the incorporation of host materials that are swelling-resistant against water/organic solvent has been considered an effective approach of counter-balancing the plasticizing effect. Familiar hosts include cellulose membrane, glass fiber filter, silica aerogel, and polymer nonwovens, which are typically in the form of interwoven structured or 3D porous membranes.354,355 In addition to good swelling resistance, they share similar advantageous properties of high mechanical strength reaching the 102 MPa level and excellent thermal stability higher than 300 °C. Therefore, their use can dramatically improve the mechanical performance of GPE mainly in terms of tensile strength, Young's modulus and thermal/dimensional stability, preventing short-circuit caused by dendrite growth and thermal shrinkage/cold contraction.356–358Thanks to the integrated 3D hosts, which offer continuous paths for ion transport, the GPEs can deliver enhanced ionic conductivity across a broad temperature spectrum.372–374 Lu's group fabricated a PVDF-HFP-based GPE with a uniformly cross-linked β′′-Al2O3 nanowire as the host (ANs–GPE) (Fig. 15(a)). They revealed that the electrolyte can suppress the side reactions between the sodium metal and liquid electrolyte, as well as sodium dendrite formation even at an elevated temperature of 60 °C. They attributed this to the effective immobilization of the liquid electrolyte by the β′′-Al2O3 host to construct highly efficient, uniform, and continuous solid–liquid hybrid Na-ion transportation channels through and along the β′′-Al2O3 nanowires (Fig. 15(b)), promoting uniform sodium deposition and formation of a stable and flat solid electrolyte interface on the sodium metal anode (Fig. 15(c)). As a result, the NVP//Na batteries using this electrolyte presented excellent cycling performance in a wide temperature range from 25 to 60 °C, with 95.3% and 78.8% capacity retentions after 1000 cycles at 1C at 25 and 60 °C, respectively.375
 | ||
| Fig. 15 Regulating ion transport behaviors in GPEs by incorporating functional hosts. (a) Planar SEM image of GPE with β′′-Al2O3 nanowires as the host; (b) schematic diagram of the Na+ transportation path along the nanowire; (c) working mechanism of NVP//Na batteries with the ANs-GPE.375 Reproduced from ref. 375, Springer Nature, Copyright 2019. (d) Cross-sectional SEM image of the VTMMT membrane; illustrations of the Li+ diffusion pathways (e) along the VTMMT and (f) within the GPE.376 Reproduced from ref. 376 with permission from Springer Nature, Copyright 2022. | ||
To further improve the ion transportation efficiency in GPE, aligned networks have been proposed.376,377 Ding et al. incorporated poly(ethylene glycol) diacrylate-based precursor into the vertically-aligned MMT scaffold (VTMMT, as shown in Fig. 15(d)) to form a GPE via in situ polymerization. The uniform and fast Li+ diffusion pathways along the continuous interface/interlayer of the low-tortuosity VTMMT provided benefits to the GPE by improving the Li+ transport capacity and reducing the occurrence of side reactions (Fig. 1(5e) and (f)). As a result, the LFP//Li full cell using this GPE exhibited outstanding rate performance and cycling stability with high CE and high-capacity retention at a wide temperature range from 0 to 60 °C.376 The development of GPEs with aligned host provides a promising approach for improving the ion conduction via novel nano-architecture designs.
Despite these advantages, there are still challenges associated with the use of inorganic hosts in wide-temperature GPEs. One challenge is the limited compatibility between the inorganic host and the polymer matrix. The incorporation of inorganic particles into the polymer matrix can disrupt the molecular order and hinder ion mobility, resulting in decreased ionic conductivity and deteriorated electrochemical performance. Therefore, the careful selection and optimization of both the inorganic host and the polymer matrix are essential to achieve optimal compatibility and maintain good ion transport properties.
4.6 Multiple combined strategies
The multiple combined strategies that can integrate the advantageous effects of various strategies are proposed to circumvent the limitation that most of the “single” strategies discussed above can only prioritize either low- or high-temperature performance. For instance, some solvents having low melting points can favor the low-temperature operability of batteries but are susceptible to volatilization and decomposition at slightly higher temperatures; IL plasticizers are particularly advantageous in ensuring excellent high-temperature oxidation stability, but the resulting demerit of low target-ion transference number would severely limit the rate performance of batteries. Obviously, to battery chemists, electrolyte design/engineering has always been the game of overcoming trade-offs between various property/performance indicators, while a single tuning strategy often appears to be less competent. In fact, the multiple combined strategies have been widely adopted by many works discussed in previous sections: strategies of co-solvent incorporation and salt concentration adjustment are simultaneously involved in the design of the organohydrogel with enhanced freezing tolerance and thermal stability; the ionogel electrolyte with its polymer matrix rationally designed to assist the ion transport is exactly a multiple strategy combining both solvent optimization and matrix design. Nevertheless, this part is written separately to highlight the great significance of the multiple combined strategies.In the effort to address the issue of low Li+ transference number of ionogel electrolytes for Li battery use, Song et al. designed a composite ionogel electrolyte showing superior comprehensive performance in terms of concurrent high ionic conductivity, oxidation stability up to 5.5 V, and, most importantly, a high tLi+ of 0.89, by taking the advantages of the IL solvent of EmimTFSI, high-concentration Li salt of 3 M LiTFSI, and inorganic filler (host) of LLZO, in which the cooperation of high concentration Li salt and inorganic filler with tLi+ approaching unity is believed to mainly responsible for the desired high tLi+ value. With these advantages, LMBs using the as-obtained ionogels as GPE and high-voltage NCM materials as the cathode deliver a high discharge capacity, which is hardly achievable for conventional ionogel electrolytes.378
Additionally, from our perspective, the multiple combined strategies may also refer to the preparation of hybrid electrolytes that feature an A/B double-layer structure (or A/B/A and A/B/C three-layer structures in some cases), of which process A and B layers are prepared based on different strategies. For example, one can construct an ultrathin inorganic layer with high Young's modulus on the side of a conventional GPE interfaced with the metallic anode to physically inhibit the dendrite growth, without significantly sacrificing the ion transport properties; distinct chemical/electrochemical requirements of anodes and cathodes could be elegantly addressed by designing hybrid GPEs consisting of two thermodynamically immiscible functional layers.
As a notable example, Lv et al. designed a hierarchically-structured polymerized tetrahydrofuran (poly-THF) GPE with one layer doped with the LixGa86In14 alloy, in which the THF solvent with low viscosity and melting point ensured high ion conductivity at low temperatures, while the LixGa86In14-doped layer helped to homogenize the Li+ flux and formed a stable SEI layer, thus enabling high discharge areal capacity and good cycling stability of LMBs over a wide temperature range of −20 to 55 °C.379 Despite being in an early stage, these results suggest the great potential of the multiple combined strategies, namely, rationally integrating two or more strategies, in sparking unprecedented breakthroughs in the design and synthesis of advanced GPEs toward wide temperature applications.
5. Summary and perspectives
Advancing the electrolytes play a critical role in enabling rechargeable batteries to operate reliably in extreme conditions. The emergence of high- and low-temperature batteries highlights the potential of GPEs in facilitating wide-temperature adaptable batteries. This review focuses on the GPEs and their associated wide-temperature applications, emphasizes the interest of the scientific community in both (i) the factors leading to capacity loss of batteries at low and high temperatures, and (ii) the latest strategies in GPE engineering and related interface chemistry to improve low-temperature kinetics and high-temperature stability. On this basis, new insights into viable solutions for wide-temperature GPEs are proposed, as displayed in Fig. 16, and discussed in more detail below. | ||
| Fig. 16 Research and industrialization opportunities of wide-temperature GPEs. Opportunities stemming from the gain of deeper mechanism insights, AI-assisted GPE design, advanced characterizations, and balancing the environmental impact and battery performance are discussed. Adapted with permission from: ref. 104 (cryo-TEM image), John Wiley & Sons, Copyright 2022; ref. 380 (Raman image), Springer Nature, Copyright 2022; ref. 381, (time lapse SEM), John Wiley & Sons, Copyright 2017; ref. 382 (Micro-CT), Springer Nature, Copyright 2022. | ||
5.1 In-depth study of battery performance degradation mechanisms at extreme temperatures
The practical application of GPE with wide-temperature operability hinges on continued research of the deterioration mechanism of the electrolyte itself and battery performance under thermal extremes. Such investigations must be devoted to the deep understanding of the intermolecular interactions between the polymer matrices and liquid plasticizers in response to temperature variation in a wide range. Then, the cause-to-effect relationship between the nanoscale interaction, the macroscopic electrochemical properties of GPEs, and the battery working and failure mechanisms should be studied. Hence, from our perspective, the leading issue is to quantify the contribution of temperature-dependent polymer-ion interaction to the ionic conductivity of GPE with different mass ratios of the liquid plasticizer. Previously, most reports on GPEs with high ionic conductivity are realized by increasing the proportion of liquid plasticizers, the role of polymer matrices in ion transport is barely illustrated. Considering the fact that the mobility of chain segments increases with temperature, it is crucial not to disregard the influence of polymer matrices on ion transport within GPEs, especially at high temperatures. This effect has not been thoroughly elucidated thus far and warrants further investigation.The in-depth investigations of the electrolyte/electrode interface chemistry of GPEs at extreme temperatures should be more underscored. It is widely accepted that the retarded ion kinetics and accelerated electrolyte decomposition at the electrolyte/electrode interfaces are the main factors causing battery performance degradation at low and high temperatures, respectively. However, the most fundamental questions such as the effect of operating temperature on the GPE/electrode interfacial chemistry and charge carrier/mass transport kinetics, remain unanswered. Special attention should be given to the impact of temperature on the interactions among various components and ion solvation structure. Though it has been reported that the ion–dipole interactions between solvent and salt increase with temperature,383,384 these are only hypothesis, and concrete characterizations are lacking. Recent studies have suggested that the interactions between polymer matrices and ionic species or solvents in GPE greatly affect the ion solvation structure and interfacial stability of batteries; yet, their dynamic changes and the effects on electrochemical kinetics under different temperatures should be thoroughly investigated.
At the battery level, further in-depth research on the influence of temperature on the electrochemical performance remains indispensable and urgent. Some research directions can be highlighted in future endeavors, such as the interfacial stress evolution induced by the low-temperature freezing and high-temperature melting of GPEs. In addition, the structural reversibility of GPE and GPE/electrode interfaces with the temperature changes, if any, remains ambiguous, which still requires in-depth research both theoretically and experimentally.
A thorough understanding of these mechanisms can guide the design of GPEs with efficient charge transfer, minimized side reactions, and enhanced overall battery performance across a wide temperature range.
5.2 Developing novel electrolyte systems with wide-temperature operability
Precisely controlled components with tailorable structures are also desired for wide-temperature GPEs and rechargeable batteries. As a prerequisite, highly thermally stable electrolyte components should be designed to accommodate temperature variations. On this basis, the high ionic conductivity and the component formation of the ion-conductive and thermostable SEI layer should be highlighted when exploiting novel electrolyte materials. Recent advancements in GPEs with wide temperature adaptability have shown that the introduction of ionophilic/solvent–philic polymer matrices can enhance ion diffusion in the bulk electrolyte and improve the interfacial stability, thereby enhancing the performance of GPEs at high and low temperatures. However, further research is needed to systematically evaluate the impact of these advancements on the wide-temperature tolerance of GPE and battery systems.It is likewise important to focus more on the self-protection design of novel GPE to prevent the hazards of thermal runaway of batteries when operating in extreme temperature conditions. Thermoresponsive materials show promise in this regard through several mechanisms: (i) inhibiting ion transport at high temperature to reduce the likelihood of thermal runaway; (ii) absorbing or dispersing heat generated to prevent the temperature from escalating to dangerous levels during thermal runaway events; and (iii) acting as thermal insulators, reduce the heat transfer from the surroundings to the battery or between different components within the battery, preventing overheating or excessive cooling in extreme temperature conditions. For instance, Qian et al. integrated a thermoresponsive phase-change material, polyethylene glycol (PEG), into agarose-based GPEs.385 Once the operating temperature reaches the phase-change temperature, the PEG can absorb heat and inhibit ion transport by undergoing a phase change from crystalline to amorphous, alleviating heat generation inside the batteries under thermal shock. Zhang et al. introduced thermal-polymerization VC as a thermoresponsive solvent that could boost the thermal stability of both the SEI and electrolyte, raising the critical temperature for thermal safety (the beginning temperature of obvious self-heating) from 71.5 to 137.4 °C.386 Leveraging the unique properties of the thermoresponsive materials, the safety and performance of GPEs and batteries could be collectively enhanced in extreme temperature conditions, making them potential candidates for novel wide-temperature GPE design as polymer matrices or functional components.386–388
Further work is needed to develop unscalable manufacturing methods and achieve mass production and industrial scale-up of laboratory GPEs. This work should focus on using safe and cost-effective reactants and synthetic processes. The manufacturing of ionogels based on the biomass polymer matrices and ILs solvents shows promise and should therefore be given a higher priority. Considering the possible interfacial compatibility of ex situ production processes of GPEs, the facile synthesis technology based on the in situ thermo-polymerization seems like a prompt pathway as it enables the creation of an integrated battery structure with seamless interfacial contact, satisfactory electrochemical performance, as well as the good compatibility with existing lithium-ion battery production processes.239,389,390 Additionally, the balance between electrochemical properties and mechanical properties, such as strength, flexibility, and stretchability, should be carefully regulated through polymer engineering and optimizing the polymer-to-liquid plasticizer ratio. Furthermore, as the electrolyte thickness decreases, the importance of ionic conductance (G = σA/l) in evaluating ionic conduction should be emphasized.391,392
Overall, the physical, chemical and electrochemical properties of GPEs in a wide temperature range should be focused on for meeting the criteria for designing usable and safe rechargeable batteries. Since the wide-temperature battery is a complicated topic, in addition to the material challenges and solutions presented in this review, the technical innovations in battery configuration and the full operational simulation in different temperature environments before stepping into the practical manufacturing market are also important research directions and should therefore be paid attention to. All of these will help to ensure the reliability and functionality of wide-temperature GPE-based rechargeable batteries.
5.3 Wide-temperature adaptable non-destructive and in-operando tools
The complex nature of GPEs makes it difficult to obtain detailed information about their bulk and molecular structures as well as their evolution upon the battery life cycle and under different temperature conditions. This complexity spans multiple scales, including atomic and ionic chemistry, microscopic molecular structures, mesoscopic electrolyte materials and interfaces, and macroscopic battery systems. Conventional characterization techniques, such as X-ray diffraction (XRD), SEM and X-ray photoelectron spectroscopy (XPS), are usually performed ex situ and require battery disassembly, hampering the mechanistic examination and continuous research of GPEs and rechargeable batteries, thereby obscuring some of the fundamental principles essential for the design of wide-temperature batteries.393Recent emphasis has been applied to in situ/operando techniques to study batteries in operation. Such tools can avoid the negative effects and uncertainty from the post-treatment of samples and thus provide an accurate information of the electrochemical processes.394–396 Examples include the in situ/operando X-ray computed tomography (CT) for non-invasive information about a battery's morphology and internal structure, the non-destructive ultrasonic scanning and imaging technique for examining the battery state of charge and health, and time-lapse SEM for observing the detailed lithium deposition processes.381,397,398 Advanced techniques like time-of-flight secondary ion mass spectroscopy (TOF-SIMS) are suitable for interfacial composition characterization because of their high mass sensitivity simultaneously and ultrahigh spatial resolution. The in situ XRD techniques has been utilized for investigating the structure and interfacial evolution of graphite anode in Al-ion batteries assembled with GPE.399 At the microscale, Raman and FTIR spectra are important techniques to study the presence of intermolecular interactions between the constituents of polymer electrolytes, such as ion–ion, ion–solvent, ion–nanofiller, polymer–ion, polymer–solvent, and polymer–ion–solvent. It provides in-hand information about the complex formation and examines by identifying the shift in the peak position of the characteristic functional groups. Mao and coworkers elucidated the SEI formation and evolution pathways during the operation of LMBs using a depth-sensitive plasmon-enhanced Raman spectroscopy for the monitoring of dynamic nature of SEI formation and evolution.400 A deeper insight into the atomic/ionic structure can be provided by aberration-corrected high angle annular dark field scanning TEM (HAADF-STEM), annular bright field STEM, neutron diffraction, X-ray absorption near edge structure (XANES), etc.
Building upon the progress in in situ/operando characterizations, there are several considerations worth investigating. Firstly, the liquid–solid duality of GPEs suggest that these in situ characterizations and analyses of both liquid and solid electrolyte-based batteries can be extended to GPEs-based battery systems. Secondly, combining various in situ/operando characterization techniques can provide diverse information at different scales for optimizing wide-temperature adaptable GPEs and rechargeable batteries. Thirdly, in situ sensing techniques show great promise for the in situ/operando evaluation of battery electrochemical behaviour by correlating them with parameters (e.g., stress, temperature, and gas) of interest.401,402 As a notable example, Tarascon et al. monitored electrolyte evolution in 18650 Na-ion and Li-ion cells under real working conditions by in operando IR fiber evanescent wave spectroscopy (FBWS). The FBWS helps to reveal changes in the dynamics of the solvents and additives in electrolytes and identify the nature of the electrolyte decomposition species without any latency as well as changes in the ion solvation dynamics as a function of voltage and current.403 These efforts can contribute to a deeper understanding of the reaction processes, degradation mechanisms, and thermal decomposition mechanisms of GPEs under various temperature conditions, guiding rational multiscale materials design for optimized properties and performance.
5.4 Artificial intelligence (AI)-assisted GPE design
The rechargeable battery with GPE is a complex electrochemical system governed by electro-/chemical reactions, charge, and ion transport in solid and liquid phases, making it challenging to describe with a simple physics formula, particularly under extreme temperature conditions. Additionally, the properties of GPEs, including ionic conductivity, cationic transference number and temperature tolerance, significantly impact the battery's performance in terms of charging and discharging rates, cycle life, and energy density at extreme temperatures. Although researchers have made significant progress in the experimental modification of electrolyte formulations and theoretical mole including DFT modeling and molecular dynamics, accurate and versatile prediction tools are urgently needed to explore optimal battery performance and corresponding gel electrolyte formulations under various temperature scenarios, crossing large time scale and under practical application conditions. Machine learning and deep learning algorithms offer a widely applicable and automated tool that have been carried out to assist the electrode and electrolyte design.404 For example, deep learning algorithms can be used to improve the accuracy and efficiency of numerical simulation. By implementing the neural network to learn the atomic polarizabilities and charges, the molecular dynamics prediction on the polarization terms in the liquid electrolyte can be effectively accelerated, and dependencies on the 3D conformation can be avoided.405 Moreover, by informing the neural networks about the surrounding environment of the atoms or molecules, the accuracy and speed of the prediction could be further improved.406,407 In addition, machine learning has been demonstrated to facilitate the guidance of empirical data on electrolyte synthesis. Cui's group used the elemental composition of electrolytes as model features of a machine learning model to identify key features for predicting CE and created fluorine-free solvent-based electrolyte formulations, helping to achieve a remarkable CE of 99.70% according to the as-trained machine learning model.408 These advancements have demonstrated the potential of AI-assisted models and algorithms in efficiently designing GPE and rechargeable batteries that are capable of operating in a wide temperature range. By harnessing AI-assisted models and algorithms, researchers are expected to efficiently analyze large amounts of data, identify patterns, and establish correlations between composition and electrolyte and electrolyte/electrode interfacial evolution, as well as the corresponding battery performance at different temperatures, which can help to screen and optimize gel-polymer electrolyte candidates. Additionally, AI is able to predict material properties and behaviors, which can lead to a reduction in time-consuming and expensive experimental tests. As AI algorithms continue to evolve and improve, their integration into the design of wide-temperature GPEs is expected to revolutionize the development of advanced energy storage systems, leading to more efficient and durable devices for a sustainable future.5.5 Balancing the environmental sustainability and battery performance
Balancing environmental sustainability and battery performance is an increasingly important consideration in the future design of GPEs and rechargeable batteries. A forward-looking approach involves the development of quantitative approaches, such as life cycle assessment (LCA), to provide a comprehensive analysis of the entire life cycle of the batteries, from raw material extraction through manufacturing, use, and disposal, and to evaluate the environmental and economic impacts and resource risks of GPEs and rechargeable batteries in a dispersive temperature regime. In recent years, there have been several LCA studies on lithium-ion and lithium-beyond batteries with liquid electrolytes based on different life cycle impact assessment methodologies, life cycle inventory databases, and different assumptions about the energy intensity of battery cell production.409,410 However, there are very few studies available on the environmental assessment of GPEs and corresponding rechargeable batteries. Moreover, most LCA studies focus on the carbon emissions and environmental indicators of the battery manufacturing phase rather than on the whole battery cycling lifetime, and, in particular, neglect the stages of material recycling and remanufacturing phases of spent batteries.411 In addition, there is still a lack of a consistent definition, and guidelines for functional units and sufficient data (original and current) for LCA assessment are still lacking.412,413 For example, several functional units such as “battery pack”, “1 kg of battery”, “1 kW h of storage capacity”, and “1 km (the distance traveled by the electric vehicle during its lifetime)” have been proposed in previous works, making it difficult to compare different LCA studies.383,414 Therefore, there is a need to promote reproducible, comparable, and verifiable LCA studies on different GPEs and rechargeable systems.Exploring more biodegradable GPEs and battery systems is a promising approach to address the environmental concerns associated with traditional GPEs made from fossil oil-based polymers, such as PEO, PMMA, and PVDF and their derivatives. These species are non-biodegradable and easily result in energy waste and environmental “white pollution”, making it hard to meet the ever-growing demands for both comprehensive performance and environmental protection of rechargeable batteries. In this respect, biomass materials, including polylactic acid, cellulose, chitosan, and xanthan gum, can offer an interesting opportunity for replacing synthetic polymers and developing GPEs with reduced environmental impact.415–417 A wide variety of cellulose and derivatives have gained considerable attention due to their abundant biomass sources (e.g., bacteria, wood, bamboo, and grass), high ionic conductivity, satisfactory mechanical strength and good freezing tolerance, creating a range of possibilities for developing functional, inexpensive, safe, and environment-friendly GPEs.230,418–420 We expect that their applications could be extended to wide-temperature scenarios and more rechargeable battery systems.
In addition, the innovation of recycling strategies that align with the principles of green chemistry is also an integral part of the future development of wide-temperature GPEs and rechargeable batteries. Efficient and sustainable recycling practices will minimize the waste and environmental pollution associated with the disposal of GPEs and rechargeable batteries.421 The traditional linear model of sending raw materials to landfills or partial recycling causes significant pollution and waste.422,423 Existing battery recycling strategies, such as classical pyrometallurgy, hydrometallurgy and emerging bio-metallurgy, and direct regeneration techniques focus on the extraction of valuable elements or materials from the cathode, which typically requires expensive reagents or complex processes and often neglects the recovery of low-value components such as electrolytes and graphite.424–426 In the future, researchers should focus on exploring more environment-friendly recycling processes, including the separation of GPEs from batteries, and the recovery and reuse of GPEs and other valuable materials from rechargeable batteries. All the above efforts will contribute to the creation of more environment-friendly and sustainable battery technologies, promoting a greener future for energy storage.
Author contributions
Conceptualization, X. Zhou, Y. Zhou, L. Yu, P. Hu, and C. Chen; data curation, X. Zhou and Y. Zhou; visualization, X. Zhou, L. Yu, L. Qi, and Y. Zhou; writing – original draft, X. Zhou and L. Yu; writing – review & editing, X. Zhou, L. Yu, Y. Zhou, K. Oh, S.-Y. Lee, P. Hu, and C. Chen; resources, C. Chen; supervision, P. Hu, S.-Y. Lee, C. Chen.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 52273091 and 21776225), the Fundamental Research Funds for the Central Universities (grant no. 2042022kf1177), the start-up fund of Wuhan University (grant no. 691000003), and the Technology Innovation Program (20012216) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).References
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- Z. Wang, H. Ji, J. Zhou, Y. Zheng, J. Liu, T. Qian and C. Yan, Nat. Commun., 2023, 14, 2267 CrossRef CAS PubMed.
- C. Zhang, Y. Yang, X. Liu, M. Mao, K. Li, Q. Li, G. Zhang and C. Wang, Innovation, 2023, 4, 100518 Search PubMed.
- J. Yang, G. Liu, M. Avdeev, H. Wan, F. Han, L. Shen, Z. Zou, S. Shi, Y. S. Hu, C. Wang and X. Yao, ACS Energy Lett., 2020, 5, 2835–2841 CrossRef CAS.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- J. Huang, K. Wu, G. Xu, M. Wu, S. Dou and C. Wu, Chem. Soc. Rev., 2023, 52, 4933–4995 RSC.
- S. Chen, X. Wei, G. Zhang, X. Wang, J. Zhu, X. Feng, H. Dai and M. Ouyang, Innovation, 2023, 4, 100465 CAS.
- W. contributors, List of weather records, (accessed 21 December, 2023).
- A. Kalmowitz, It's Too Hot For EVs To Work Right, (accessed 21 July 2023).
- S. Rivers, Excessive Heat Can Axe Almost A Third Of Range In Some Electric Vehicles, (accessed 20 July 2023).
- C.-Y. Wang, G. Zhang, S. Ge, T. Xu, Y. Ji, X.-G. Yang and Y. Leng, Nature, 2016, 529, 515–518 CrossRef CAS PubMed.
- S. Liu, R. Zhang, J. Mao, Y. Zhao, Q. Cai and Z. Guo, Sci. Adv., 2022, 8, eabn5097 CrossRef CAS PubMed.
- A. Gupta and A. Manthiram, Adv. Energy Mater., 2020, 10, 2001972 CrossRef CAS PubMed.
- M.-T. F. Rodrigues, G. Babu, H. Gullapalli, K. Kalaga, F. N. Sayed, K. Kato, J. Joyner and P. M. Ajayan, Nat. Energy, 2017, 2, 17108 CrossRef CAS.
- Z. Chen, K. Wang, P. Pei, Y. Zuo, M. Wei, H. Wang, P. Zhang and N. Shang, Nano Res., 2023, 16, 2311–2324 CrossRef.
- Y. Feng, L. Zhou, H. Ma, Z. Wu, Q. Zhao, H. Li, K. Zhang and J. Chen, Energy Environ. Sci., 2022, 15, 1711–1759 RSC.
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 482 CrossRef PubMed.
- X. Zhou, J. Fu, Z. Li, R. Yu, S. Liu, Z. Li, L. Wei and X. Guo, Chin. Sci. Bull., 2022, 67, 842–859 CrossRef.
- F. Gebert, J. Knott, R. Gorkin, S.-L. Chou and S.-X. Dou, Energy Storage Mater., 2021, 36, 10–30 CrossRef.
- X. Su, Y. Xu, Y. Wu, H. Li, J. Yang, Y. Liao, R. Qu and Z. Zhang, Energy Storage Mater., 2023, 56, 642–663 CrossRef.
- F. Han, Z. Chang, X. Liu, A. Li, J. Wang, H. Ding and S. Lu, J. Phys.: Conf. Ser., 2021, 2009, 012069 CrossRef.
- Q. Nian, T. Sun, S. Liu, H. Du, X. Ren and Z. Tao, Chem. Eng. J., 2021, 423, 130253 CrossRef CAS.
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, eabq3750 CrossRef CAS PubMed.
- H. Zhang, L. Qiao, H. Kühnle, E. Figgemeier, M. Armand and G. G. Eshetu, Energy Environ. Sci., 2023, 16, 11–52 RSC.
- D. Baril, C. Michot and M. Armand, Solid State Ionics, 1997, 94, 35–47 CrossRef CAS.
- M. Nookala, B. Kumar and S. Rodrigues, J. Power Sources, 2002, 111, 165–172 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- C. K. Huang, J. S. Sakamoto, J. Wolfenstine and S. Surampudi, J. Electrochem. Soc., 2000, 147, 2893 CrossRef CAS.
- Y.-B. He, F. Ning, B. Li, Q.-S. Song, W. Lv, H. Du, D. Zhai, F. Su, Q.-H. Yang and F. Kang, J. Power Sources, 2012, 202, 253–261 CrossRef CAS.
- Q. Li, D. Lu, J. Zheng, S. Jiao, L. Luo, C.-M. Wang, K. Xu, J.-G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 42761–42768 CrossRef CAS PubMed.
- L. Wu, Y. Wang, X. Guo, P. Ding, Z. Lin and H. Yu, SusMat, 2022, 2, 264–292 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, Electrochim. Acta, 2004, 49, 1057–1061 CrossRef CAS.
- P. Suresh, A. K. Shukla and N. Munichandraiah, J. Appl. Electrochem., 2002, 32, 267–273 CrossRef CAS.
- S. Weng, X. Zhang, G. Yang, S. Zhang, B. Ma, Q. Liu, Y. Liu, C. Peng, H. Chen, H. Yu, X. Fan, T. Cheng, L. Chen, Y. Li, Z. Wang and X. Wang, Nat. Commun., 2023, 14, 4474 CrossRef PubMed.
- B. Ramasubramanian, M. V. Reddy, K. Zaghib, M. Armand and S. Ramakrishna, Nanomaterials, 2021, 11, 2476 CrossRef CAS PubMed.
- V. Agubra and J. Fergus, Materials, 2013, 6, 1310–1325 CrossRef CAS PubMed.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- Q. Zhao, S. Stalin and L. A. Archer, Joule, 2021, 5, 1119–1142 CrossRef CAS.
- H. Zhang, Y. Chen, C. Li and M. Armand, SusMat, 2021, 1, 24–37 CrossRef CAS.
- D. Luo, L. Zheng, Z. Zhang, M. Li, Z. Chen, R. Cui, Y. Shen, G. Li, R. Feng, S. Zhang, G. Jiang, L. Chen, A. Yu and X. Wang, Nat. Commun., 2021, 12, 186 CrossRef CAS PubMed.
- X. Xu, Y. Liu, J.-Y. Hwang, O. O. Kapitanova, Z. Song, Y.-K. Sun, A. Matic and S. Xiong, Adv. Energy Mater., 2020, 10, 2002390 CrossRef CAS.
- Y. Liu, Y. Shi, C. Gao, Z. Shi, H. Ding, Y. Feng, Y. He, J. Sha, J. Zhou and B. Lu, Angew. Chem., Int. Ed., 2023, 62, e202300016 CrossRef CAS PubMed.
- C. T. Love, O. A. Baturina and K. E. Swider-Lyons, ECS Electrochem. Lett., 2015, 4, A24 CrossRef CAS.
- R. Akolkar, J. Power Sources, 2014, 246, 84–89 CrossRef CAS.
- R. Akolkar, J. Power Sources, 2013, 232, 23–28 CrossRef CAS.
- Z. Li, J. Huang, B. Yann Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- B. Thirumalraj, T. T. Hagos, C.-J. Huang, M. A. Teshager, J.-H. Cheng, W.-N. Su and B.-J. Hwang, J. Am. Chem. Soc., 2019, 141, 18612–18623 CrossRef CAS PubMed.
- K. Yan, J. Wang, S. Zhao, D. Zhou, B. Sun, Y. Cui and G. Wang, Angew. Chem., Int. Ed., 2019, 58, 11364–11368 CrossRef CAS PubMed.
- A. C. Thenuwara, P. P. Shetty and M. T. McDowell, Nano Lett., 2019, 19, 8664–8672 CrossRef CAS PubMed.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- H. Wang, E. Matios, J. Luo and W. Li, Chem. Soc. Rev., 2020, 49, 3783–3805 RSC.
- X. Zhou, Q. Zhang, Z. Hao, Y. Ma, O. A. Drozhzhin and F. Li, ACS Appl. Mater. Interfaces, 2021, 13, 53227–53234 CrossRef CAS PubMed.
- J. Sun, J. Peng, T. Ring, L. Whittaker-Brooks, J. Zhu, D. Fraggedakis, J. Niu, T. Gao and F. Wang, Energy Environ. Sci., 2022, 15, 5284–5299 RSC.
- F.-N. Jiang, S.-J. Yang, H. Liu, X.-B. Cheng, L. Liu, R. Xiang, Q. Zhang, S. Kaskel and J.-Q. Huang, SusMat, 2021, 1, 506–536 CrossRef CAS.
- G. Yan, K. Reeves, D. Foix, Z. Li, C. Cometto, S. Mariyappan, M. Salanne and J.-M. Tarascon, Adv. Energy Mater., 2019, 9, 1901431 CrossRef CAS.
- Y. Zhu, W. Li, L. Zhang, W. Fang, Q. Ruan, J. Li, F. Zhang, H. Zhang, T. Quan and S. Zhang, Energy Environ. Sci., 2023, 16, 2825–2855 RSC.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. Hou, M. Yang, D. Wang and J. Zhang, Adv. Energy Mater., 2020, 10, 1904152 CrossRef CAS.
- T. Waldmann, M. Wilka, M. Kasper, M. Fleischhammer and M. Wohlfahrt-Mehrens, J. Power Sources, 2014, 262, 129–135 CrossRef CAS.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS PubMed.
- X. Lin, M. Salari, L. M. R. Arava, P. M. Ajayan and M. W. Grinstaff, Chem. Soc. Rev., 2016, 45, 5848–5887 RSC.
- H. Wang, L. Sheng, G. Yasin, L. Wang, H. Xu and X. He, Energy Storage Mater., 2020, 33, 188–215 CrossRef.
- R. Fang, H. Xu, B. Xu, X. Li, Y. Li and J. B. Goodenough, Adv. Funct. Mater., 2021, 31, 2001812 CrossRef CAS.
- J. Zheng, H. Zheng, R. Wang, L. Ben, W. Lu, L. Chen, L. Chen and H. Li, Phys. Chem. Chem. Phys., 2014, 16, 13229–13238 RSC.
- H. Yang and Z. Wang, J. Solid State Electrochem., 2023, 27, 2607–2618 CrossRef CAS.
- H. H. Yan, Y. H. Bie, X. Y. Cui, G. P. Xiong and L. Chen, Energy Convers. Manage., 2018, 161, 193–204 CrossRef CAS.
- K. Wang, Y. Xiao, P. Pei, X. Liu and Y. Wang, J. Electrochem. Soc., 2019, 166, D389 CrossRef CAS.
- J. Jeon, G. H. Yoon, T. Vegge and J. H. Chang, ACS Appl. Mater. Interfaces, 2022, 14, 15275–15286 CrossRef CAS PubMed.
- J. Wang, W. Huang, A. Pei, Y. Li, F. Shi, X. Yu and Y. Cui, Nat. Energy, 2019, 4, 664–670 CrossRef CAS.
- J. Su, X. Yin, H. Zhao, H. Yang, D. Yang, L. He, M. Wang, S. Jin, K. Zhao, Y. Wang and Y. Wei, Nano Lett., 2022, 22, 1549–1556 CrossRef CAS PubMed.
- J. Kasemchainan, S. Zekoll, D. Spencer Jolly, Z. Ning, G. O. Hartley, J. Marrow and P. G. Bruce, Nat. Mater., 2019, 18, 1105–1111 CrossRef CAS PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- E. Metwalli, M. V. Kaeppel, S. J. Schaper, A. Kriele, R. Gilles, K. N. Raftopoulos and P. Müller-Buschbaum, ACS Appl. Energy Mater., 2018, 1, 666–675 CrossRef CAS.
- L. Qin, L. Schkeryantz, J. Zheng, N. Xiao and Y. Wu, J. Am. Chem. Soc., 2020, 142, 11629–11640 CrossRef CAS PubMed.
- H. Zhang, Y. Geng, J. Huang, Z. Wang, K. Du and H. Li, Energy Environ. Sci., 2023, 16, 889–951 RSC.
- G. Zhang, J. Chang, L. Wang, J. Li, C. Wang, R. Wang, G. Shi, K. Yu, W. Huang, H. Zheng, T. Wu, Y. Deng and J. Lu, Nat. Commun., 2023, 14, 1081 CrossRef CAS PubMed.
- Q. Zhu, D. Yu, J. Chen, L. Cheng, M. Tang, Y. Wang, Y. Li, J. Yang and H. Wang, Joule, 2024, 8, 1–14 CrossRef.
- P. Lu, Y. Xia, G. Sun, D. Wu, S. Wu, W. Yan, X. Zhu, J. Lu, Q. Niu, S. Shi, Z. Sha, L. Chen, H. Li and F. Wu, Nat. Commun., 2023, 14, 4077 CrossRef CAS PubMed.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- D. Zhou, R. Liu, Y.-B. He, F. Li, M. Liu, B. Li, Q.-H. Yang, Q. Cai and F. Kang, Adv. Energy Mater., 2016, 6, 1502214 CrossRef.
- R. Collongues, A. Kahn and D. Michel, Annu. Rev. Mater. Res., 1979, 9, 123–150 CrossRef CAS.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- X. Zhou, Z. Li, W. Li, X. Li, J. Fu, L. Wei, H. Yang and X. Guo, Adv. Funct. Mater., 2022, 33, 2212866 CrossRef.
- W. Zhang, J. Zhang, X. Liu, H. Li, Y. Guo, C. Geng, Y. Tao and Q.-H. Yang, Adv. Funct. Mater., 2022, 32, 2201205 CrossRef CAS.
- Y. Xu, X. Zhou, Z. Chen, Y. Hou, Y. You and J. Lu, Mater. Today, 2023, 66, 339–347 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- H. Liu, X.-B. Cheng, J.-Q. Huang, H. Yuan, Y. Lu, C. Yan, G.-L. Zhu, R. Xu, C.-Z. Zhao, L.-P. Hou, C. He, S. Kaskel and Q. Zhang, ACS Energy Lett., 2020, 5, 833–843 CrossRef CAS.
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16 CrossRef CAS.
- J. Newman, K. E. Thomas, H. Hafezi and D. R. Wheeler, J. Power Sources, 2003, 119–121, 838–843 CrossRef CAS.
- J. N. Chazalviel, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 7355–7367 CrossRef CAS PubMed.
- C. Brissot, M. Rosso, J. N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81–82, 925–929 CrossRef CAS.
- C. Z. Zhao, X. Q. Zhang, X. B. Cheng, R. Zhang, R. Xu, P. Y. Chen, H. J. Peng, J. Q. Huang and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11069–11074 CrossRef CAS PubMed.
- X. Zhou, X. Li, Z. Li, H. Xie, J. Fu, L. Wei, H. Yang and X. Guo, J. Mater. Chem. A, 2021, 9, 18239–18246 RSC.
- S. Huo, L. Sheng, W. Xue, L. Wang, H. Xu, H. Zhang and X. He, InfoMat, 2023, 5, e12394 CrossRef CAS.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- X. Xie, Z. Wang, S. He, K. Chen, Q. Huang, P. Zhang, S.-M. Hao, J. Wang and W. Zhou, Angew. Chem., Int. Ed., 2023, 135, e202218229 CrossRef.
- D. Wang, T. He, A. Wang, K. Guo, M. Avdeev, C. Ouyang, L. Chen and S. Shi, Adv. Funct. Mater., 2023, 33, 2212342 CrossRef CAS.
- Y. Zou, G. Liu, Y. Wang, Q. Li, Z. Ma, D. Yin, Y. Liang, Z. Cao, L. Cavallo, H. Kim, L. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, Adv. Energy Mater., 2023, 13, 2300443 CrossRef CAS.
- X. Wang, X. Li, H. Fan and L. Ma, Nano-Micro Lett., 2022, 14, 205 CrossRef CAS PubMed.
- C.-Z. Zhao, B.-C. Zhao, C. Yan, X.-Q. Zhang, J.-Q. Huang, Y. Mo, X. Xu, H. Li and Q. Zhang, Energy Storage Mater., 2020, 24, 75–84 CrossRef.
- C. B. Jin, N. Yao, Y. Xiao, J. Xie, Z. Li, X. Chen, B. Q. Li, X. Q. Zhang, J. Q. Huang and Q. Zhang, Adv. Mater., 2023, 35, 2208340 CrossRef CAS PubMed.
- Z. Huang, X. Li, Z. Chen, P. Li, X. Ji and C. Zhi, Nat. Rev. Chem., 2023, 7, 616–631 CrossRef PubMed.
- L. Zhou, Z. Cao, W. Wahyudi, J. Zhang, J. Y. Hwang, Y. Cheng, L. Wang, L. Cavallo, T. Anthopoulos, Y. K. Sun, H. N. Alshareef and J. Ming, ACS Energy Lett., 2020, 5, 766–776 CrossRef CAS.
- X. Hou, R. Wang, X. He, T. P. Pollard, X. Ju, L. Du, E. Paillard, H. Frielinghaus, L. C. Barnsley, O. Borodin, K. Xu, M. Winter and J. Li, Angew. Chem., Int. Ed., 2021, 60, 22812–22817 CrossRef CAS PubMed.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X. Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- X. Li, X. Wang, L. Ma and W. Huang, Adv. Energy Mater., 2022, 12, 2202068 CrossRef CAS.
- X. Fan, C. Zhong, J. Liu, J. Ding, Y. Deng, X. Han, L. Zhang, W. Hu, D. P. Wilkinson and J. Zhang, Chem. Rev., 2022, 122, 17155–17239 CrossRef CAS PubMed.
- X. Zhu and L. Wang, EcoMat, 2020, 2, e12043 CrossRef CAS.
- Y. Yao, X. Wang, C. Dong, J. Chen, D. Wang and Z. Mao, J. Power Sources, 2022, 523, 231034 CrossRef CAS.
- D. S. Hall, R. Gauthier, A. Eldesoky, V. S. Murray and J. R. Dahn, ACS Appl. Mater. Interfaces, 2019, 11, 14095–14100 CrossRef CAS PubMed.
- Q. Liu and L. Wang, Adv. Energy Mater., 2023, 13, 2301742 CrossRef CAS.
- Q.-K. Zhang, X.-Q. Zhang, L.-P. Hou, S.-Y. Sun, Y.-X. Zhan, J.-L. Liang, F.-S. Zhang, X.-N. Feng, B.-Q. Li and J.-Q. Huang, Adv. Energy Mater., 2022, 12, 2200139 CrossRef CAS.
- X. He, Y. Ni, Y. Hou, Y. Lu, S. Jin, H. Li, Z. Yan, K. Zhang and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 22672–22677 CrossRef CAS PubMed.
- S. Kundu and Y. Ein-Eli, J. Power Sources, 2023, 553, 232267 CrossRef CAS.
- P. Jindal and J. Bhattacharya, J. Electrochem. Soc., 2019, 166, A2165 CrossRef CAS.
- X. Wang, E. Yasukawa and S. Kasuya, J. Electrochem. Soc., 2000, 147, 2421 CrossRef CAS.
- L. Dong, S. Zhong, B. Yuan, Y. Ji, J. Liu, Y. Liu, C. Yang, J. Han and W. He, Research, 2022, 2022, 1–52 CrossRef PubMed.
- Z. Li, S. Weng, J. Fu, X. Wang, X. Zhou, Q. Zhang, X. Wang, L. Wei and X. Guo, Energy Storage Mater., 2022, 47, 542–550 CrossRef.
- L. Yu, L. Yu, Q. Liu, T. Meng, S. Wang and X. Hu, Adv. Funct. Mater., 2021, 32, 2110653 CrossRef.
- Q. Rong, W. Lei, J. Huang and M. Liu, Adv. Energy Mater., 2018, 8, 1801967 CrossRef.
- C. Deng, Y. Li and J. Huang, Small Methods, 2023, 2300832 CrossRef PubMed.
- D. Yu, X. Pan, J. E. Bostwick, C. J. Zanelotti, L. Mu, R. H. Colby, F. Lin and L. A. Madsen, Adv. Energy Mater., 2021, 11, 2003559 CrossRef CAS.
- Y. H. Li, X. L. Wu, J. H. Kim, S. Xin, J. Su, Y. Yan, J. S. Lee and Y. G. Guo, J. Power Sources, 2013, 244, 234–239 CrossRef CAS.
- X.-Q. Xu, X.-B. Cheng, F.-N. Jiang, S.-J. Yang, D. Ren, P. Shi, H. Hsu, H. Yuan, J.-Q. Huang, M. Ouyang and Q. Zhang, SusMat, 2022, 2, 435–444 CrossRef CAS.
- J. Chen, Z. Yang, G. Liu, C. Li, J. Yi, M. Fan, H. Tan, Z. Lu and C. Yang, Energy Storage Mater., 2020, 25, 305–312 CrossRef.
- S. Wen, C. Luo, Q. Wang, Z. Wei, Y. Zeng, Y. Jiang, G. Zhang, H. Xu, J. Wang, C. Wang, J. Chang and Y. Deng, Energy Storage Mater., 2022, 47, 453–461 CrossRef.
- Z. Wang, L. Shen, S. Deng, P. Cui and X. Yao, Adv. Mater., 2021, 33, 2100353 CrossRef CAS PubMed.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L. Q. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS PubMed.
- H. Hao, T. Hutter, B. L. Boyce, J. Watt, P. Liu and D. Mitlin, Chem. Rev., 2022, 122, 8053–8125 CrossRef CAS PubMed.
- W. Lin, F. Wang, H. Wang, H. Li, Y. Fan, D. Chan, S. Chen, Y. Tang and Y. Zhang, ChemSusChem, 2022, 15, e202201464 CrossRef CAS PubMed.
- P. Wen, P. Lu, X. Shi, Y. Yao, H. Shi, H. Liu, Y. Yu and Z.-S. Wu, Adv. Energy Mater., 2020, 11, 2002930 CrossRef.
- Y. Kuang, C. Chen, D. Kirsch and L. Hu, Adv. Energy Mater., 2019, 9, 1901457 CrossRef.
- V. Vijayakumar, B. Anothumakkool, S. Kurungot, M. Winter and J. R. Nair, Energy Environ. Sci., 2021, 14, 2708–2788 RSC.
- G. Xiao, H. Xu, C. Bai, M. Liu and Y.-B. He, Interdiscip. Mater., 2023, 2, 609–634 CrossRef CAS.
- C. Wang, H. Zhang, S. Dong, Z. Hu, R. Hu, Z. Guo, T. Wang, G. Cui and L. Chen, Chem. Mater., 2020, 32, 9167–9175 CrossRef CAS.
- J.-H. Kim, J.-M. Kim, S.-K. Cho, N.-Y. Kim and S.-Y. Lee, Nat. Commun., 2022, 13, 2541 CrossRef CAS PubMed.
- X. Sun, Z. Pang, Y. Zhu, Z. Yu, P. Yang, L. Liu, S. Renneckar, T. Li and F. Jiang, Innovations Mater., 2023, 1, 100040 CrossRef.
- Y. Ye, L. Yu, E. Lizundia, Y. Zhu, C. Chen and F. Jiang, Chem. Rev., 2023, 123, 9204–9264 CrossRef CAS PubMed.
- Z. Li, Y.-X. Yao, S. Sun, C.-B. Jin, N. Yao, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303888 CrossRef CAS PubMed.
- F. Cheng, M. Cao, Q. Li, C. Fang, J. Han and Y. Huang, ACS Nano, 2023, 17, 18608–18615 CrossRef CAS PubMed.
- J. Liu, Y. Zhang, J. Zhou, Z. Wang, P. Zhu, Y. Cao, Y. Zheng, X. Zhou, C. Yan and T. Qian, Adv. Funct. Mater., 2023, 33, 2302055 CrossRef CAS.
- L. Li, W. Lv, J. Chen, C. Zhu, S. Dmytro, Q. Zhang and S. Zhong, ACS Appl. Energy Mater., 2022, 5, 11900–11914 CrossRef CAS.
- L. Zheng, H. Zhang, P. Cheng, Q. Ma, J. Liu, J. Nie, W. Feng and Z. Zhou, Electrochim. Acta, 2016, 196, 169–188 CrossRef CAS.
- X. Shangguan, G. Xu, Z. Cui, Q. Wang, X. Du, K. Chen, S. Huang, G. Jia, F. Li, X. Wang, D. Lu, S. Dong and G. Cui, Small, 2019, 15, e1900269 CrossRef PubMed.
- Z. Geng, J. Lu, Q. Li, J. Qiu, Y. Wang, J. Peng, J. Huang, W. Li, X. Yu and H. Li, Energy Storage Mater., 2019, 23, 646–652 CrossRef.
- X. Chen, W. Xu, M. H. Engelhard, J. Zheng, Y. Zhang, F. Ding, J. Qian and J.-G. Zhang, J. Mater. Chem. A, 2014, 2, 2346–2352 RSC.
- H. M. Law, J. Yu, S. C. T. Kwok, G. Zhou, M. J. Robson, J. Wu and F. Ciucci, Energy Storage Mater., 2022, 46, 182–191 CrossRef.
- T. Zheng, J. Xiong, B. Zhu, X. Shi, Y.-J. Cheng, H. Zhao and Y. Xia, J. Mater. Chem. A, 2021, 9, 9307–9318 RSC.
- S. Chereddy, P. R. Chinnam, V. Chatare, S. P. diLuzio, M. P. Gobet, S. G. Greenbaum and S. L. Wunder, Mater. Horiz., 2018, 5, 461–473 RSC.
- X. Li, J. Zheng, M. H. Engelhard, D. Mei, Q. Li, S. Jiao, N. Liu, W. Zhao, J.-G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 2469–2479 CrossRef CAS PubMed.
- L. Zhang, Y. Sun, Y. Zhou, C. Hai, S. Hu, J. Zeng, Y. Shen, S. Dong, G. Qi and F. Li, Ionics, 2018, 24, 2995–3004 CrossRef CAS.
- H. Cheng, X. Jin, S. Liu, T. Zhang, Z. Song, L. Wang and F. Hu, J. Power Sources, 2023, 567, 232975 CrossRef CAS.
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed.
- Y.-P. Yang, A.-C. Huang, Y. Tang, Y.-C. Liu, Z.-H. Wu, H.-L. Zhou, Z.-P. Li, C.-M. Shu, J.-C. Jiang and Z.-X. Xing, Polymers, 2021, 13, 707 CrossRef CAS PubMed.
- E. S. Hong, S. Okada, T. Sonoda, S. Gopukumar and J.-I. Yamaki, J. Electrochem. Soc., 2004, 151, A1836 CrossRef CAS.
- G. Xu, X. Shangguan, S. Dong, X. Zhou and G. Cui, Angew. Chem., Int. Ed., 2020, 59, 3400–3415 CrossRef CAS PubMed.
- S. Chen, M. Zhang, P. Zou, B. Sun and S. Tao, Energy Environ. Sci., 2022, 15, 1805–1839 RSC.
- S. Li, Y.-M. Chen, W. Liang, Y. Shao, K. Liu, Z. Nikolov and Y. Zhu, Joule, 2018, 2, 1838–1856 CrossRef CAS.
- L. Xia, Y. Jiang, Y. Pan, S. Li, J. Wang, Y. He, Y. Xia, Z. Liu and G. Z. Chen, ChemistrySelect, 2018, 3, 1954–1960 CrossRef CAS.
- Y. Zhao, Z. Hu, Z. Zhao, X. Chen, S. Zhang, J. Gao and J. Luo, J. Am. Chem. Soc., 2023, 145, 22184–22193 CrossRef CAS PubMed.
- F. Q. Liu, W. P. Wang, Y. X. Yin, S. F. Zhang, J. L. Shi, L. Wang, X. D. Zhang, Y. Zheng, J. J. Zhou, L. Li and Y. G. Guo, Sci. Adv., 2018, 4, eaat5383 CrossRef CAS PubMed.
- W. Fan, N. W. Li, X. Zhang, S. Zhao, R. Cao, Y. Yin, Y. Xing, J. Wang, Y. G. Guo and C. Li, Adv. Sci., 2018, 5, 1800559 CrossRef PubMed.
- X. Lin, J. Yu, M. B. Effat, G. Zhou, M. J. Robson, S. C. T. Kwok, H. Li, S. Zhan, Y. Shang and F. Ciucci, Adv. Funct. Mater., 2021, 31, 2010261 CrossRef CAS.
- Q. Wang, C. Zhao, J. Wang, Z. Yao, S. Wang, S. G. H. Kumar, S. Ganapathy, S. Eustace, X. Bai, B. Li and M. Wagemaker, Nat. Commun., 2023, 14, 440 CrossRef CAS PubMed.
- Z. Zhou, H. Wang, W. Yi, S. Wu, X. Sun and J. Li, J. Power Sources, 2023, 570, 233066 CrossRef CAS.
- J. Zhou, D. Zhang, H. Yuan, Y. Ding, H. Li, R. Wang, Y. Cao and H. Wang, Appl. Surf. Sci., 2023, 638, 158087 CrossRef CAS.
- Q. Liu, B. Cai, S. Li, Q. Yu, F. Lv, F. Kang, Q. Wang and B. Li, J. Mater. Chem. A, 2020, 8, 7197–7204 RSC.
- L. Quan, Q. Su, H. Wu, W. Huang, M. Liu, Y. Lu, Z. Li, H. Liu, L. Xing and W. Li, Chem. Eng. J., 2023, 454, 140086 CrossRef CAS.
- Y. Chen, F. Huo, S. Chen, W. Cai and S. Zhang, Adv. Funct. Mater., 2021, 31, 2102347 CrossRef CAS.
- C. Wang, D. Li, N. Liu, G. Bai, W. He, X. Zhou, J. Wang, J. Zhang and X. Liu, Polym. Chem., 2023, 14, 1094–1102 RSC.
- Y. Li, Y. Yang, Y. Lu, Q. Zhou, X. Qi, Q. Meng, X. Rong, L. Chen and Y. S. Hu, ACS Energy Lett., 2020, 5, 1156–1158 CrossRef CAS.
- G. A. Giffin, Nat. Commun., 2022, 13, 5250 CrossRef CAS PubMed.
- Z. Tian, Y. Zou, G. Liu, Y. Wang, J. Yin, J. Ming and H. N. Alshareef, Adv. Sci., 2022, 9, 2201207 CrossRef CAS PubMed.
- Y. Chen, M. Li, Y. Liu, Y. Jie, W. Li, F. Huang, X. Li, Z. He, X. Ren, Y. Chen, X. Meng, T. Cheng, M. Gu, S. Jiao and R. Cao, Nat. Commun., 2023, 14, 2655 CrossRef CAS PubMed.
- X. Zheng, L. Huang, X. Ye, J. Zhang, F. Min, W. Luo and Y. Huang, Chem, 2021, 7, 2312–2346 CAS.
- M. He, X. Li, N. G. Holmes, R. Li, J. Wang, G. Yin, P. Zuo and X. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 38296–38304 CrossRef CAS PubMed.
- M. Zhang, J. Zhang, J. Yang, J. Yao, Z. Chen, C. Lu, X. Du, Z. Zhang, H. Zhang and G. Cui, Chem. Commun., 2019, 55, 9785–9788 RSC.
- Z. Yu, J. Zhang, T. Liu, B. Tang, X. Yang, X. Zhou and G. Cui, Acta Chim. Sin., 2020, 78, 114–124 CrossRef CAS.
- X. Cao, P. Gao, X. Ren, L. Zou, M. H. Engelhard, B. E. Matthews, J. Hu, C. Niu, D. Liu, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020357118 CrossRef CAS PubMed.
- Y. Zhao, T. Zhou, L. P. H. Jeurgens, X. Kong, J. W. Choi and A. Coskun, Chem, 2023, 9, 682–697 CAS.
- J.-F. Ding, R. Xu, N. Yao, X. Chen, Y. Xiao, Y.-X. Yao, C. Yan, J. Xie and J.-Q. Huang, Angew. Chem., Int. Ed., 2021, 60, 11442–11447 CrossRef CAS PubMed.
- J. He, A. Bhargav, W. Shin and A. Manthiram, J. Am. Chem. Soc., 2021, 143, 20241–20248 CrossRef CAS PubMed.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- J. He, A. Bhargav, W. Shin and A. Manthiram, J. Am. Chem. Soc., 2021, 143, 20241–20248 CrossRef CAS PubMed.
- S. Xu, R. Xu, T. Yu, K. Chen, C. Sun, G. Hu, S. Bai, H.-M. Cheng, Z. Sun and F. Li, Energy Environ. Sci., 2022, 15, 3379–3387 RSC.
- Y. Shi, R. Wang, S. Bi, M. Yang, L. Liu and Z. Niu, Adv. Funct. Mater., 2023, 33, 2214546 CrossRef CAS.
- D.-T. Shieh, P.-H. Hsieh and M.-H. Yang, J. Power Sources, 2007, 174, 663–667 CrossRef CAS.
- W. Deng, Z. Zhou, Y. Li, M. Zhang, X. Yuan, J. Hu, Z. Li, C. Li and R. Li, ACS Nano, 2020, 14, 15776–15785 CrossRef CAS PubMed.
- S. Chen, P. Sun, B. Sun, J. Humphreys, P. Zou, K. Xie and S. Tao, Energy Storage Mater., 2021, 37, 598–608 CrossRef.
- J. Wei, G. Wei, Y. Shang, J. Zhou, C. Wu and Q. Wang, Adv. Mater., 2019, 31, 1900248 CrossRef PubMed.
- S. Chen, P. Sun, J. Humphreys, P. Zou, M. Zhang, G. Jeerh and S. Tao, Energy Storage Mater., 2021, 42, 240–251 CrossRef.
- R. Tamate, Y. Peng, Y. Kamiyama and K. Nishikawa, Adv. Mater., 2023, 35, 2211679 CrossRef CAS PubMed.
- C. Tian, K. Qin and L. Suo, Mater. Futures, 2023, 2, 012101 CrossRef.
- L. Yan, Y.-e Qi, X. Dong, Y. Wang and Y. Xia, eScience, 2021, 1, 212–218 CrossRef.
- H. Wang, J. Liu, J. Wang, M. Hu, Y. Feng, P. Wang, Y. Wang, N. Nie, J. Zhang, H. Chen, Q. Yuan, J. Wu and Y. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 49–55 CrossRef CAS PubMed.
- N. Sun, F. Lu, Y. Yu, L. Su, X. Gao and L. Zheng, ACS Appl. Mater. Interfaces, 2020, 12, 11778–11788 CrossRef CAS PubMed.
- X. Ai, Q. Zhao, Y. Duan, Z. Chen, Z. Zhang, Y. Liu and Y. Gao, Cell Rep. Phys. Sci., 2022, 3, 101148 CrossRef CAS.
- Y. Chen, J. Zhao and Y. Wang, ACS Appl. Energy Mater., 2020, 3, 9058–9065 CrossRef CAS.
- D. Feng, Y. Jiao and P. Wu, Angew. Chem., Int. Ed., 2023, 62, e202215060 CrossRef CAS PubMed.
- D. Zhao, Y. Zhu, W. Cheng, G. Xu, Q. Wang, S. Liu, J. Li, C. Chen, H. Yu and L. Hu, Matter, 2020, 2, 390–403 CrossRef CAS.
- C. Chen and L. Hu, Adv. Mater., 2021, 33, 2002890 CrossRef CAS PubMed.
- X. Zhou, Z. Li, W. Li, X. Li, J. Fu, L. Wei, H. Yang and X. Guo, Adv. Funct. Mater., 2023, 33, 2212866 CrossRef CAS.
- G. Guo, C. Ji, H. Mi, C. Yang, M. Li, C. Sun and L. Sun, Adv. Funct. Mater., 2023, 34, 2308405 CrossRef.
- Q. Liu, G. Yang, X. Li, S. Zhang, R. Chen, X. Wang, Y. Gao, Z. Wang and L. Chen, Energy Storage Mater., 2022, 51, 443–452 CrossRef.
- S.-Y. Lee, Nat. Energy, 2021, 6, 949–950 CrossRef.
- C. Yang, Q. Wu, W. Xie, X. Zhang, A. Brozena, J. Zheng, M. N. Garaga, B. H. Ko, Y. Mao, S. He, Y. Gao, P. Wang, M. Tyagi, F. Jiao, R. Briber, P. Albertus, C. Wang, S. Greenbaum, Y.-Y. Hu, A. Isogai, M. Winter, K. Xu, Y. Qi and L. Hu, Nature, 2021, 598, 590–596 CrossRef PubMed.
- D. Wang, H. Xiea, Q. Liu, K. Mu, Z. Song, W. Xu, L. Tian, C. Zhu and J. Xu, Angew. Chem., Int. Ed., 2023, 62, e202302767 CrossRef CAS PubMed.
- F. Ackermann and M. Schönhoff, J. Phys. Chem. C, 2021, 125, 266–274 CrossRef CAS.
- L. Wan, H. Zhang, M. Qu, M. Feng, Z. Shang, R. Wang, D. Lei and Y. Cui, Energy Storage Mater., 2023, 63, 102982 CrossRef.
- T. Zhou, Y. Zhao, J. W. Choi and A. Coskun, Angew. Chem., Int. Ed., 2021, 60, 22791–22796 CrossRef CAS PubMed.
- S. Liu, H. Cheng, R. Mao, W. Jiang, L. Wang, Z. Song, M. Pei, T. Zhang and F. Hu, Adv. Energy Mater., 2023, 13, 2300068 CrossRef CAS.
- J. Yang, Z. Xu, J. Wang, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Funct. Mater., 2021, 31, 2009438 CrossRef CAS.
- X. Sui, H. Guo, P. Chen, Y. Zhu, C. Wen, Y. Gao, J. Yang, X. Zhang and L. Zhang, Adv. Funct. Mater., 2020, 30, 1907986 CrossRef CAS.
- L. Xu, Y. Lu, C.-Z. Zhao, H. Yuan, G.-L. Zhu, L.-P. Hou, Q. Zhang and J.-Q. Huang, Adv. Energy Mater., 2021, 11, 2002360 CrossRef CAS.
- Y. Wang, Q. Li, H. Hong, S. Yang, R. Zhang, X. Wang, X. Jin, B. Xiong, S. Bai and C. Zhi, Nat. Commun., 2023, 14, 3890 CrossRef CAS PubMed.
- D. Dong, T. Wang, Y. Sun, J. Fan and Y.-C. Lu, Nat. Sustain., 2023, 6, 1474–1484 CrossRef.
- J. Xu, X. Cai, S. Cai, Y. Shao, C. Hu, S. Lu and S. Ding, Energy Environ. Mater., 2023, 6, e12450 CrossRef CAS.
- Z. Pei, Z. Yuan, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 4793–4799 CrossRef CAS PubMed.
- W. Cui, C. Ma, X. Lei, Y. Lv, Q. Zhang, W. Guan and X. Liu, J. Power Sources, 2023, 577, 233264 CrossRef CAS.
- S. Huang, L. Hou, T. Li, Y. Jiao and P. Wu, Adv. Mater., 2022, 34, 2110140 CrossRef CAS PubMed.
- Y. Yan, S. Duan, B. Liu, S. Wu, Y. Alsaid, B. Yao, S. Nandi, Y. Du, T.-W. Wang, Y. Li and X. He, Adv. Mater., 2023, 35, 2211673 CrossRef CAS PubMed.
- C. Gu, X.-Q. Xie, Y. Liang, J. Li, H. Wang, K. Wang, J. Liu, M. Wang, Y. Zhang, M. Li, H. Kong and C.-S. Liu, Energy Environ. Sci., 2021, 14, 4451–4462 RSC.
- K. Liu, Q. Zhang, B. P. Thapaliya, X.-G. Sun, F. Ding, X. Liu, J. Zhang and S. Dai, Solid State Ionics, 2020, 345, 115159 CrossRef CAS.
- J. Zheng, Y. Yang, W. Li, X. Feng, W. Chen and Y. Zhao, J. Mater. Chem. A, 2020, 8, 22962–22968 RSC.
- J. Zheng, Y. Sun, W. Li, X. Feng, W. Chen and Y. Zhao, ACS Appl. Mater. Interfaces, 2021, 13, 25024–25035 CrossRef CAS PubMed.
- Y. Hao, D. Feng, L. Hou, T. Li, Y. Jiao and P. Wu, Adv. Sci., 2022, 9, 2104832 CrossRef CAS PubMed.
- J. Huang, L. Yu, S. Wang, L. Qi, Z. Lu, L. Chen, D. Xu, H. Deng and C. Chen, Innovations Mater., 2023, 1, 100029 CrossRef.
- Y.-N. Liu, Z. Xiao, W.-K. Zhang, J. Zhang, H. Huang, Y.-P. Gan, X.-P. He, G. G. Kumar and Y. Xia, Rare Met., 2022, 41, 3762–3773 CrossRef CAS.
- Y. Zhu, Y. Guo, K. Cao, S. Zeng, G. Jiang, Y. Liu, W. Cheng, W. Bai, X. Weng, W. Chen, D. Zhao, H. Yu and G. Yu, Nat. Synth., 2023, 2, 864–872 CrossRef.
- Y. N. Zhou, Z. Xiao, D. Han, L. Yang, J. Zhang, W. Tang, C. Shu, C. Peng and D. Zhou, Adv. Funct. Mater., 2022, 32, 2111314 CrossRef CAS.
- P. Jaumaux, Q. Liu, D. Zhou, X. Xu, T. Wang, Y. Wang, F. Kang, B. Li and G. Wang, Angew. Chem., Int. Ed., 2020, 59, 9134–9142 CrossRef CAS PubMed.
- J. Li, P. Yu, S. Zhang, Z. Wen, Y. Wen, W. Zhou, X. Dong, Y. Liu and Y. Liang, J. Colloid Interface Sci., 2021, 600, 586–593 CrossRef CAS PubMed.
- F. Cao, B. Wu, T. Li, S. Sun, Y. Jiao and P. Wu, Nano Res., 2022, 15, 2030–2039 CrossRef CAS.
- G. Shim, M. X. Tran, G. Liu, D. Byun and J. K. Lee, Energy Storage Mater., 2021, 35, 739–749 CrossRef.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- Q. Liu, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2300798 CrossRef CAS.
- H.-X. Xie, Q.-G. Fu, Z. Li, S. Chen, J.-M. Wu, L. Wei and X. Guo, Chem. – Eur. J., 2021, 27, 7773–7780 CrossRef CAS PubMed.
- T. Zhao, X. Zheng, D. Wang, L. Huang, B. Li, X. Liu, H. Yang, Y. Dai, Y. Huang and W. Luo, Adv. Funct. Mater., 2023, 33, 2304928 CrossRef CAS.
- M. Sun, G. Ji and J. Zheng, Chem. Eng. J., 2023, 463, 142535 CrossRef CAS.
- Y. Ren, M. Yang, Z. Shi, J. Guo, D. Chu, F. Feng, H. Li, Z.-F. Ma, S. Chen and T. Liu, Energy Storage Mater., 2023, 61, 102909 CrossRef.
- M. Peng, X. Tang, K. Xiao, T. Hu, K. Yuan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202302701 CrossRef CAS PubMed.
- H. Du, R. Zhao, Y. Yang, Z. Liu, L. Qie and Y. Huang, Angew. Chem., Int. Ed., 2022, 61, e202114789 CrossRef CAS PubMed.
- Z. Huang, Z. Li, Y. Wang, J. Cong, X. Wu, X. Song, Y. Ma, H. Xiang and Y. Huang, ACS Energy Lett., 2023, 8, 372–380 CrossRef CAS.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- C. Tian, J. Wang, R. Sun, T. Ali, H. Wang, B.-B. Xie, Y. Zhong and Y. Hu, Angew. Chem., Int. Ed., 2023, 62, e202310970 CrossRef CAS PubMed.
- H. Lu, J. Hu, L. Wang, J. Li, X. Ma, Z. Zhu, H. Li, Y. Zhao, Y. Li, J. Zhao and B. Xu, Adv. Funct. Mater., 2022, 32, 2112540 CrossRef CAS.
- M. Jiao, L. Dai, H.-R. Ren, M. Zhang, X. Xiao, B. Wang, J. Yang, B. Liu, G. Zhou and H.-M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202301114 CrossRef CAS PubMed.
- N. Zhang, T. Deng, S. Zhang, C. Wang, L. Chen, C. Wang and X. Fan, Adv. Mater., 2022, 34, 2107899 CrossRef CAS PubMed.
- M. S. Ding, K. Xu, S. Zhang and T. R. Jow, J. Electrochem. Soc., 2001, 148, A299 CrossRef CAS.
- M. S. Ding, K. Xu and T. R. Jow, J. Electrochem. Soc., 2000, 147, 1688 CrossRef CAS.
- M. C. Smart, B. V. Ratnakumar and S. Surampudi, J. Electrochem. Soc., 1999, 146, 486 CrossRef CAS.
- L. Chen, H. Wu, X. Ai, Y. Cao and Z. Chen, Battery Energy, 2022, 1, 20210006 CrossRef CAS.
- G. H. Wrodnigg, J. O. Besenhard and M. Winter, J. Power Sources, 2001, 97–98, 592–594 CrossRef CAS.
- B. T. Yu, W. H. Qiu, F. S. Li and L. Cheng, J. Power Sources, 2006, 158, 1373–1378 CrossRef CAS.
- Y. Zhang, Y. Zhong, Z. Wu, B. Wang, S. Liang and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 7797–7802 CrossRef CAS PubMed.
- Y. Zhu, S. He, J. Ding, G. Zhao and F. Lian, Nano Res., 2023, 16, 3855–3863 CrossRef CAS.
- M. C. Smart, B. L. Lucht, S. Dalavi, F. C. Krause and B. V. Ratnakumar, J. Electrochem. Soc., 2012, 159, A739 CrossRef CAS.
- M. C. Smart, B. V. Ratnakumar, K. B. Chin and L. D. Whitcanack, J. Electrochem. Soc., 2010, 157, A1361 CrossRef CAS.
- J. Zhao, M. Li, H. Su, Y. Liu, P. Bai, H. Liu, L. Ma, W. Li, J. Sun and Y. Xu, Small Methods, 2023, 7, 2300228 CrossRef CAS PubMed.
- R. Guo, Y. Che, G. Lan, J. Lan, J. Li, L. Xing, K. Xu, W. Fan, L. Yu and W. Li, ACS Appl. Mater. Interfaces, 2019, 11, 38285–38293 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- O. Lavi, S. Luski, N. Shpigel, C. Menachem, Z. Pomerantz, Y. Elias and D. Aurbach, ACS Appl. Energy Mater., 2020, 3, 7485–7499 CrossRef CAS.
- Y. Wang, Z. Li, Y. Hou, Z. Hao, Q. Zhang, Y. Ni, Y. Lu, Z. Yan, K. Zhang, Q. Zhao, F. Li and J. Chen, Chem. Soc. Rev., 2023, 52, 2713–2763 RSC.
- A. Hu, F. Li, W. Chen, T. Lei, Y. Li, Y. Fan, M. He, F. Wang, M. Zhou, Y. Hu, Y. Yan, B. Chen, J. Zhu, J. Long, X. Wang and J. Xiong, Adv. Energy Mater., 2022, 12, 2202432 CrossRef CAS.
- H. He, Y. Wang, M. Li, J. Qiu, Y. Wen and J. Chen, Chem. Eng. J., 2023, 467, 143311 CrossRef CAS.
- H. He, Y. Wang, M. Li, J. Qiu, Y. Wen and J. Chen, J. Electroanal. Chem., 2022, 925, 116870 CrossRef CAS.
- X. Gao, J. Yang, Z. Xu, Y. Nuli and J. Wang, Energy Storage Mater., 2023, 54, 382–402 CrossRef.
- Y. Sun, Y. Wang, L. Liu, B. Liu, Q. Zhang, D. Wu, H. Zhang and X. Yan, J. Mater. Chem. A, 2020, 8, 17998–18006 RSC.
- P. Xiong, Y. Kang, N. Yao, X. Chen, H. Mao, W.-S. Jang, D. M. Halat, Z.-H. Fu, M.-H. Jung, H. Y. Jeong, Y.-M. Kim, J. A. Reimer, Q. Zhang and H. S. Park, ACS Energy Lett., 2023, 8, 1613–1625 CrossRef CAS.
- Y. Liu, W. Wang, K. Gu, J. Yao, Z. Shao and X. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 29008–29020 CrossRef CAS PubMed.
- T. Wei, Y. Ren, Z. Li, X. Zhang, D. Ji and L. Hu, Chem. Eng. J., 2022, 434, 134646 CrossRef CAS.
- M. Chen, W. Zhou, A. Wang, A. Huang, J. Chen, J. Xu and C.-P. Wong, J. Mater. Chem. A, 2020, 8, 6828–6841 RSC.
- F. Mo, G. Liang, Q. Meng, Z. Liu, H. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC.
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC.
- Z. Liu, J. Zhang, J. Liu, Y. Long, L. Fang, Q. Wang and T. Liu, J. Mater. Chem. A, 2020, 8, 6219–6228 RSC.
- R. Wang, M. Yao, S. Huang, J. Tian and Z. Niu, Sci. China Mater., 2022, 65, 2189–2196 CrossRef CAS.
- B. Li, M. Ullah Khan, C. Qv, L. Chen, Y. Xiong, L. Zhang and M. Wu, Chem. Phys. Lett., 2023, 832, 140883 CrossRef CAS.
- M. Li, C. Xi, X. Wang, L. Li, Y. Xiao, Y. Chao, X. Zheng, Z. Liu, Y. Yu and C. Yang, Small, 2023, 19, 2301569 CrossRef CAS PubMed.
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Y. Yang, Z. Yu, S. T. Oyakhire, W. Zhang, L. C. Greenburg, M. S. Kim, D. T. Boyle, P. Sayavong, Y. Ye, J. Qin, Z. Bao and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef CAS.
- W. Zhang, H. Xia, Z. Zhu, Z. Lv, S. Cao, J. Wei, Y. Luo, Y. Xiao, L. Liu and X. Chen, CCS Chem., 2021, 3, 1245–1255 CrossRef CAS.
- C. Xu and C. Chen, Innovations Mater., 2023, 1, 100033 CrossRef.
- L. Yu, S. Guo, Y. Lu, Y. Li, X. Lan, D. Wu, R. Li, S. Wu and X. Hu, Adv. Energy Mater., 2019, 9, 1900257 CrossRef.
- R. Jamil and D. S. Silvester, Curr. Opin. Electrochem., 2022, 35, 101046 CrossRef CAS.
- Q. Yang, Z. Zhang, X.-G. Sun, Y.-S. Hu, H. Xing and S. Dai, Chem. Soc. Rev., 2018, 47, 2020–2064 RSC.
- S. Zheng, Z.-S. Wu, F. Zhou, X. Wang, J. Ma, C. Liu, Y.-B. He and X. Bao, Nano Energy, 2018, 51, 613–620 CrossRef CAS.
- R. S. Kühnel, N. Böckenfeld, S. Passerini, M. Winter and A. Balducci, Electrochim. Acta, 2011, 56, 4092–4099 CrossRef.
- H. Moon, S.-J. Cho, D.-E. Yu and S.-Y. Lee, Energy Environ. Mater., 2023, 6, e12383 CrossRef CAS.
- X. Yu and X. Hu, Sci. China Mater., 2022, 65, 2967–2974 CrossRef CAS.
- Y.-H. Li, X.-L. Wu, J.-H. Kim, S. Xin, J. Su, Y. Yan, J.-S. Lee and Y.-G. Guo, J. Power Sources, 2013, 244, 234–239 CrossRef CAS.
- P. Lv, Y. Li, Y. Wu, G. Liu, H. Liu, S. Li, C. Tang, J. Mei and Y. Li, ACS Appl. Mater. Interfaces, 2018, 10, 25384–25392 CrossRef CAS PubMed.
- C. H. Krause, P. Röring, H. Onishi, D. Diddens, J. H. Thienenkamp, G. Brunklaus, M. Winter and I. Cekic-Laskovic, J. Power Sources, 2020, 478, 229047 CrossRef CAS.
- W. Ruo, W. Haonan, Z. Huajun, Y. Mingman, L. Zhongbo, Z. Guangzhao, Z. Tong, Q. Yunxian, W. Jun, L. Iseult and D. Yonghong, Energy Mater., 2023, 3, 300040 Search PubMed.
- M. Li, X. Wang, J. Meng, C. Zuo, B. Wu, C. Li, W. Sun and L. Mai, Adv. Mater., 2023, 36, 2308628 CrossRef PubMed.
- H. Wang, Z. Chen, Z. Ji, P. Wang, J. Wang, W. Ling and Y. Huang, Mater. Today Energy, 2021, 19, 100577 CrossRef CAS.
- J.-Q. Huang, X. Guo, X. Lin, Y. Zhu and B. Zhang, Research, 2019, 2019, 2635310 CAS.
- L. Yu, J. Huang, S. Wang, L. Qi, S. Wang and C. Chen, Adv. Mater., 2023, 35, 2210789 CrossRef CAS PubMed.
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS PubMed.
- Q. Nian, T. Sun, Y. Li, S. Jin, S. Liu, X. Luo, Z. Wang, B.-Q. Xiong, Z. Cui, D. Ruan, H. Ji, Z. Tao and X. Ren, Angew. Chem., Int. Ed., 2023, 62, e202217671 CrossRef CAS PubMed.
- H. Chen, M. Zheng, S. Qian, H. Y. Ling, Z. Wu, X. Liu, C. Yan and S. Zhang, Carbon Energy, 2021, 3, 929–956 CrossRef CAS.
- R. Poiana, E. Lufrano, A. Tsurumaki, C. Simari, I. Nicotera and M. A. Navarra, Electrochim. Acta, 2022, 401, 139470 CrossRef CAS.
- Q. Nian, J. Wang, S. Liu, T. Sun, S. Zheng, Y. Zhang, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 16994–16999 CrossRef CAS PubMed.
- Y. Cui, R. Zhang, S. Yang, L. Liu and S. Chen, Mater. Futures, 2024, 3, 012102 CrossRef.
- J. Liu, B. Yuan, N. He, L. Dong, D. Chen, S. Zhong, Y. Ji, J. Han, C. Yang, Y. Liu and W. He, Energy Environ. Sci., 2022, 16, 1024–1034 RSC.
- W. Ren, Y. Zhang, R. Lv, S. Guo, W. Wu, Y. Liu and J. Wang, J. Power Sources, 2022, 542, 231773 CrossRef CAS.
- Y. Liao, G. Li, N. Xu, T. Chen, X. Wang and W. Li, Solid State Ionics, 2019, 329, 31–39 CrossRef CAS.
- J. Castillo, A. Santiago, X. Judez, I. Garbayo, J. A. Coca Clemente, M. C. Morant-Miñana, A. Villaverde, J. A. González-Marcos, H. Zhang, M. Armand and C. Li, Chem. Mater., 2021, 33, 8812–8821 CrossRef CAS.
- H. Jia, H. Onishi, N. von Aspern, U. Rodehorst, K. Rudolf, B. Billmann, R. Wagner, M. Winter and I. Cekic-Laskovic, J. Power Sources, 2018, 397, 343–351 CrossRef CAS.
- H. Ota, K. Shima, M. Ue and J.-I. Yamaki, Electrochim. Acta, 2004, 49, 565–572 CrossRef CAS.
- L. Liu, S. Wang, Z. Zhang, J. Fan, W. Qi and S. Chen, Ionics, 2019, 25, 1035–1043 CrossRef CAS.
- S. J. Cho, D. E. Yu, T. P. Pollard, H. Moon and S. Y. Lee, iScience, 2020, 23, 100844 CrossRef CAS PubMed.
- L. Liao, X. Cheng, Y. Ma, P. Zuo, W. Fang, G. Yin and Y. Gao, Electrochim. Acta, 2013, 87, 466–472 CrossRef CAS.
- L. Liao, P. Zuo, Y. Ma, Y. An, G. Yin and Y. Gao, Electrochim. Acta, 2012, 74, 260–266 CrossRef CAS.
- J. Yu, X. Lin, J. Liu, J. T. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2022, 12, 2102932 CrossRef CAS.
- A. C. Thenuwara, P. P. Shetty, N. Kondekar, S. E. Sandoval, K. Cavallaro, R. May, C.-T. Yang, L. E. Marbella, Y. Qi and M. T. McDowell, ACS Energy Lett., 2020, 5, 2411–2420 CrossRef CAS.
- X. Lin, G. Zhou, J. Liu, J. Yu, M. B. Effat, J. Wu and F. Ciucci, Adv. Energy Mater., 2020, 10, 2001235 CrossRef CAS.
- C. Zu, H. Yu and H. Li, InfoMat, 2021, 3, 648–661 CrossRef CAS.
- J. Liu, X. Shen, J. Zhou, M. Wang, C. Niu, T. Qian and C. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 45048–45056 CrossRef CAS PubMed.
- L. Han, L. Wang, Z. Chen, Y. Kan, Y. Hu, H. Zhang and X. He, Adv. Funct. Mater., 2023, 33, 2300892 CrossRef CAS.
- T.-H. Park, M.-S. Park, A. H. Ban, Y.-S. Lee and D.-W. Kim, ACS Appl. Energy Mater., 2021, 4, 10153–10162 CrossRef CAS.
- J. Xiang, Y. Zhang, B. Zhang, L. Yuan, X. Liu, Z. Cheng, Y. Yang, X. Zhang, Z. Li, Y. Shen, J. Jiang and Y. Huang, Energy Environ. Sci., 2021, 14, 3510–3521 RSC.
- B. Qiu, K. Liang, W. Huang, G. Zhang, C. He, P. Zhang and H. Mi, Adv. Energy Mater., 2023, 13, 2301193 CrossRef CAS.
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- F. Chen, D. Zhou, J. Wang, T. Li, X. Zhou, T. Gan, S. Handschuh-Wang and X. Zhou, Angew. Chem., Int. Ed., 2018, 130, 6678–6681 CrossRef.
- Y. Liu, H. Li, X. Wang, T. Lv, K. Dong, Z. Chen, Y. Yang, S. Cao and T. Chen, J. Mater. Chem. A, 2021, 9, 12051–12059 RSC.
- S. Huang, S. He, Y. Li, S. Wang and X. Hou, Chem. Eng. J., 2023, 464, 142607 CrossRef CAS.
- R. Wang, Q. Ma, L. Zhang, Z. Liu, J. Wan, J. Mao, H. Li, S. Zhang, J. Hao, L. Zhang and C. Zhang, Adv. Energy Mater., 2023, 13, 2302543 CrossRef CAS.
- Y. Deng, H. Wang, M. Fan, B. Zhan, L.-J. Zuo, C. Chen and L. Yan, J. Am. Chem. Soc., 2023, 145, 20109–20120 CrossRef CAS PubMed.
- X. Yu, Z. Jiang, R. Yuan and H. Song, Nanomaterials, 2023, 13, 1789 CrossRef CAS PubMed.
- N. Badi, A. M. Theodore, S. A. Alghamdi, H. A. Al-Aoh, A. Lakhouit, P. K. Singh, M. N. F. Norrrahim and G. Nath, Polymers, 2022, 14, 3101 CrossRef CAS PubMed.
- Q. Li, G. Liu, H. Cheng, Q. Sun, J. Zhang and J. Ming, Chem. – Eur. J., 2021, 27, 15842–15865 CrossRef CAS PubMed.
- F. Meng, X. Xiong, L. Tan, B. Yuan and R. Hu, Energy Storage Mater., 2022, 44, 390–407 CrossRef.
- E. Zhao, S. Luo, Y. Gu, L. Yang and S.-I. Hirano, ACS Appl. Mater. Interfaces, 2021, 13, 59843–59854 CrossRef CAS PubMed.
- X. Lan, S. Yang, T. Meng, C. Zhang and X. Hu, Adv. Energy Mater., 2023, 13, 2203449 CrossRef CAS.
- Y. He, C. Zhen, M. Li, X. Wei, C. Li, Y. Zhu, X. Yang and M. D. Gu, ACS Nano, 2023, 17, 21730–21738 CrossRef PubMed.
- M. Baek, J. Kim, J. Jin and J. W. Choi, Nat. Commun., 2021, 12, 6807 CrossRef CAS PubMed.
- Q. Wang, P. Zhang, W. Zhu, Z. Li, D. Zhang, H. Wang, H. Sun, B. Wang and S.-S. Chi, J. Power Sources, 2023, 564, 232898 CrossRef CAS.
- Z. Wang, Z. Sun, J. Li, Y. Shi, C. Sun, B. An, H. M. Cheng and F. Li, Chem. Soc. Rev., 2021, 50, 3178–3210 RSC.
- R. Fang, B. Xu, N. S. Grundish, Y. Xia, Y. Li, C. Lu, Y. Liu, N. Wu and J. B. Goodenough, Angew. Chem., Int. Ed., 2021, 133, 17842–17847 CrossRef.
- S. Choudhury, Z. Tu, A. Nijamudheen, M. J. Zachman, S. Stalin, Y. Deng, Q. Zhao, D. Vu, L. F. Kourkoutis, J. L. Mendoza-Cortes and L. A. Archer, Nat. Commun., 2019, 10, 3091 CrossRef PubMed.
- Q. Zhao, X. Liu, J. Zheng, Y. Deng, A. Warren, Q. Zhang and L. Archer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26053–26060 CrossRef CAS PubMed.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y.-M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- Z. Yu, Y. Cui and Z. Bao, Cell Rep. Phys. Sci., 2020, 1, 100119 CrossRef.
- S. Zhang, F. Sun, X. Du, X. Zhang, L. Huang, J. Ma, S. Dong, A. Hilger, I. Manke, L. Li, B. Xie, J. Li, Z. Hu, A. C. Komarek, H.-J. Lin, C.-Y. Kuo, C.-T. Chen, P. Han, G. Xu, Z. Cui and G. Cui, Energy Environ. Sci., 2023, 16, 2591–2602 RSC.
- Z. Li, W. Tang, Y. Deng, M. Zhou, X. Wang, R. Liu and C.-A. Wang, J. Mater. Chem. A, 2022, 10, 23047–23057 RSC.
- W. Li, S. Zhang, B. Wang, S. Gu, D. Xu, J. Wang, C. Chen and Z. Wen, ACS Appl. Mater. Interfaces, 2018, 10, 23874–23882 CrossRef CAS PubMed.
- M. Yang, F. Feng, Y. Ren, S. Chen, F. Chen, D. Chu, J. Guo, Z. Shi, T. Cai, W. Zhang, Z.-F. Ma, S. Chen and T. Liu, Adv. Funct. Mater., 2023, 33, 2305383 CrossRef CAS.
- X. Lu, H. Wu, D. Kong, X. Li, L. Shen and Y. Lu, ACS Mater. Lett., 2020, 2, 1435–1441 CrossRef CAS.
- X. Bian, J. Liang, X. Tang, R. Li, L. Kang, A. Su, X. Su and Y. Wei, J. Alloys Compd., 2019, 803, 1075–1081 CrossRef CAS.
- Y. Zhang, J. Huang, H. Liu, W. Kou, Y. Dai, W. Dang, W. Wu, J. Wang, Y. Fu and Z. Jiang, Adv. Energy Mater., 2023, 13, 2300156 CrossRef CAS.
- Z. Chang, H. Yang, X. Zhu, P. He and H. Zhou, Nat. Commun., 2022, 13, 1510 CrossRef CAS PubMed.
- Y. Xia, Q. Wang, Y. Liu, J. Zhang, X. Xia, H. Huang, Y. Gan, X. He, Z. Xiao and W. Zhang, J. Colloid Interface Sci., 2023, 638, 908–917 CrossRef CAS PubMed.
- L. Yu, T. Gao, R. Mi, J. Huang, W. Kong, D. Liu, Z. Liang, D. Ye and C. Chen, Nano Res., 2023, 16, 7609–7617 CrossRef CAS.
- J. Hu, P. He, B. Zhang, B. Wang and L.-Z. Fan, Energy Storage Mater., 2020, 26, 283–289 CrossRef.
- Z. Shen, J. Zhong, S. Jiang, W. Xie, S. Zhan, K. Lin, L. Zeng, H. Hu, G. Lin, Y. Lin, S. Sun and Z. Shi, ACS Appl. Mater. Interfaces, 2022, 14, 41022–41036 CrossRef CAS PubMed.
- A. A. AbdelHamid, J. L. Cheong and J. Y. Ying, Nano Energy, 2020, 71, 104633 CrossRef CAS.
- W. Ling, N. Fu, J. Yue, X. X. Zeng, Q. Ma, Q. Deng, Y. Xiao, L. J. Wan, Y. G. Guo and X. W. Wu, Adv. Energy Mater., 2020, 10, 1903966 CrossRef CAS.
- H. Duan, M. Fan, W. P. Chen, J. Y. Li, P. F. Wang, W. P. Wang, J. L. Shi, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Mater., 2019, 31, e1807789 CrossRef PubMed.
- F. He, W. Tang, X. Zhang, L. Deng and J. Luo, Adv. Mater., 2021, 33, 2105129 Search PubMed.
- H. X. Yang, Z. K. Liu, Y. Wang, N. W. Li and L. Yu, Adv. Funct. Mater., 2023, 33, 2209837 CrossRef CAS.
- X. Zhou, X. Li, Z. Li, J. Fu, S. Xu, W. Zhou, S. Gui, L. Wei, H. Yang, J. Wu and X. Guo, Mater. Today Energy, 2022, 26, 100990 CrossRef CAS.
- Q. Zhou, J. Ma, S. Dong, X. Li and G. Cui, Adv. Mater., 2019, 31, e1902029 CrossRef PubMed.
- J.-F. Wu and X. Guo, Small, 2019, 15, 1804413 CrossRef PubMed.
- P. Zhou, X. Zhang, Y. Xiang and K. Liu, Nano Res., 2023, 16, 8055–8071 CrossRef CAS.
- Z. Xu, T. Yang, X. Chu, H. Su, Z. Wang, N. Chen, B. Gu, H. Zhang, W. Deng, H. Zhang and W. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 10341–10349 CrossRef CAS PubMed.
- C. Ding, Y. Liu, L. K. Ono, G. Tong, C. Zhang, J. Zhang, J. Lan, Y. Yu, B. Chen and Y. B. Qi, Energy Storage Mater., 2022, 50, 417–425 CrossRef.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- M. Li, T. Chen, S. Song, Y. Li and J. Bae, Nanomaterials, 2021, 11, 736 CrossRef CAS PubMed.
- K. Li, W. Shen, T. Xu, L. Yang, X. Xu, F. Yang, L. Zhang, Y. Wang, Y. Zhou, M. Zhong and D. Wei, Carbon Energy, 2021, 3, 916–928 CrossRef CAS.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 2096–2100 CrossRef CAS PubMed.
- K. Aruchamy, S. Ramasundaram, S. Divya, M. Chandran, K. Yun and T. H. Oh, Gels, 2023, 9, 585 CrossRef CAS PubMed.
- J. Zheng, W. Li, X. Liu, J. Zhang, X. Feng and W. Chen, Energy Environ. Mater., 2023, 6, e12422 CrossRef CAS.
- D. Lei, Y. B. He, H. Huang, Y. Yuan, G. Zhong, Q. Zhao, X. Hao, D. Zhang, C. Lai, S. Zhang, J. Ma, Y. Wei, Q. Yu, W. Lv, Y. Yu, B. Li, Q. H. Yang, Y. Yang, J. Lu and F. Kang, Nat. Commun., 2019, 10, 4244 CrossRef PubMed.
- X. Li, Y. Wang, K. Xi, W. Yu, J. Feng, G. Gao, H. Wu, Q. Jiang, A. Abdelkader, W. Hua, G. Zhong and S. Ding, Nano-Micro Lett., 2022, 14, 210 CrossRef CAS PubMed.
- H. Joukhdar, A. Seifert, T. Jüngst, J. Groll, M. S. Lord and J. Rnjak-Kovacina, Adv. Mater., 2021, 33, 2100091 CrossRef CAS PubMed.
- Y. Zhai, W. Hou, M. Tao, Z. Wang, Z. Chen, Z. Zeng, X. Liang, P. Paoprasert, Y. Yang, N. Hu and S. Song, Adv. Mater., 2022, 34, 2205560 CrossRef CAS PubMed.
- C. Ma, X. Liu, H. Geng, X. Qu, W. Lv and Y. Ding, Fundam. Res., 2022, 2, 611–618 CrossRef CAS.
- E. Miele, W. M. Dose, I. Manyakin, M. H. Frosz, Z. Ruff, M. F. L. De Volder, C. P. Grey, J. J. Baumberg and T. G. Euser, Nat. Commun., 2022, 13, 1651 CrossRef CAS PubMed.
- G. Rong, X. Zhang, W. Zhao, Y. Qiu, M. Liu, F. Ye, Y. Xu, J. Chen, Y. Hou, W. Li, W. Duan and Y. Zhang, Adv. Mater., 2017, 29, 1606187 CrossRef PubMed.
- J. Scharf, M. Chouchane, D. P. Finegan, B. Lu, C. Redquest, M.-C. Kim, W. Yao, A. A. Franco, D. Gostovic, Z. Liu, M. Riccio, F. Zelenka, J.-M. Doux and Y. S. Meng, Nat. Nanotechnol., 2022, 17, 446–459 CrossRef CAS PubMed.
- J. Chen, Y. Peng, Y. Yin, Z. Fang, Y. Cao, Y. Wang, X. Dong and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 23858–23862 CrossRef CAS PubMed.
- B. S. Krumgal’z, J. Struct. Chem., 1973, 13, 727–731 CrossRef.
- Y. Meng, L. Zhang, M. Peng, D. Shen, C. Zhu, S. Qian, J. Liu, Y. Cao, C. Yan, J. Zhou and T. Qian, Adv. Funct. Mater., 2022, 32, 2206653 CrossRef CAS.
- F. N. Jiang, X. B. Cheng, S. J. Yang, J. Xie, H. Yuan, L. Liu, J. Q. Huang and Q. Zhang, Adv. Mater., 2023, 35, 2209114 CrossRef CAS PubMed.
- S. Chen, Y. Li, Y. Feng and W. Feng, Mater. Chem. Front., 2023, 7, 1562–1590 RSC.
- J. Zhu, M. Yao, S. Huang, J. Tian and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 16480–16484 CrossRef CAS PubMed.
- T. Liu, J. Zhang, W. Han, J. Zhang, G. Ding, S. Dong and G. Cui, J. Electrochem. Soc., 2020, 167, 070527 CrossRef CAS.
- Z. Bi, W. Huang, S. Mu, W. Sun, N. Zhao and X. Guo, Nano Energy, 2021, 90, 106498 CrossRef CAS.
- J. Wu, Z. Rao, Z. Cheng, L. Yuan, Z. Li and Y. Huang, Adv. Energy Mater., 2019, 9, 1902767 CrossRef CAS.
- D. Zhou, Y.-B. He, R. Liu, M. Liu, H. Du, B. Li, Q. Cai, Q.-H. Yang and F. Kang, Adv. Energy Mater., 2015, 5, 1500353 CrossRef.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q.-H. Yang, M. Wagemaker, F. Kang, X.-Q. Yang and B. Li, Adv. Mater., 2019, 31, 1806620 CrossRef PubMed.
- S. Luo, X. Liu, X. Zhang, X. Wang, Z. Wang, Y. Zhang, H. Wang, W. Ma, L. Zhu and X. Zhang, ACS Energy Lett., 2022, 7, 3064–3071 CrossRef CAS.
- D. T. Boyle, W. Huang, H. Wang, Y. Li, H. Chen, Z. Yu, W. Zhang, Z. Bao and Y. Cui, Nat. Energy, 2021, 6, 487–494 CrossRef CAS.
- S. Tan, Z. Shadike, J. Li, X. Wang, Y. Yang, R. Lin, A. Cresce, J. Hu, A. Hunt, I. Waluyo, L. Ma, F. Monaco, P. Cloetens, J. Xiao, Y. Liu, X.-Q. Yang, K. Xu and E. Hu, Nat. Energy, 2022, 7, 484–494 CrossRef CAS.
- Z. Deng, Z. Huang, Y. Shen, Y. Huang, H. Ding, A. Luscombe, M. Johnson, J. E. Harlow, R. Gauthier and J. R. Dahn, Joule, 2020, 4, 2017–2029 CrossRef CAS.
- G. Qian, Y. Li, H. Chen, L. Xie, T. Liu, N. Yang, Y. Song, C. Lin, J. Cheng, N. Nakashima, M. Zhang, Z. Li, W. Zhao, X. Yang, H. Lin, X. Lu, L. Yang, H. Li, K. Amine, L. Chen and F. Pan, Nat. Commun., 2023, 14, 6048 CrossRef CAS PubMed.
- Z. Yu, S. Jiao, S. Li, X. Chen, W.-L. Song, T. Teng, J. Tu, H.-S. Chen, G. Zhang and D.-N. Fang, Adv. Funct. Mater., 2019, 29, 1806799 CrossRef.
- Y. Gu, E.-M. You, J.-D. Lin, J.-H. Wang, S.-H. Luo, R.-Y. Zhou, C.-J. Zhang, J.-L. Yao, H.-Y. Li, G. Li, W.-W. Wang, Y. Qiao, J.-W. Yan, D.-Y. Wu, G.-K. Liu, L. Zhang, J.-F. Li, R. Xu, Z.-Q. Tian, Y. Cui and B.-W. Mao, Nat. Commun., 2023, 14, 3536 CrossRef CAS PubMed.
- Y. Jin, Z. Zheng, D. Wei, X. Jiang, H. Lu, L. Sun, F. Tao, D. Guo, Y. Liu, J. Gao and Y. Cui, Joule, 2020, 4, 1714–1729 CrossRef CAS.
- P. Liu, L. Yang, B. Xiao, H. Wang, L. Li, S. Ye, Y. Li, X. Ren, X. Ouyang, J. Hu, F. Pan, Q. Zhang and J. Liu, Adv. Funct. Mater., 2022, 32, 2208586 CrossRef CAS.
- C. Gervillié-Mouravieff, C. Boussard-Plédel, J. Huang, C. Leau, L. A. Blanquer, M. B. Yahia, M. L. Doublet, S. T. Boles, X. H. Zhang, J. L. Adam and J. M. Tarascon, Nat. Energy, 2022, 7, 1157–1169 CrossRef.
- C. Sui, Z. Jiang, G. Higueros, D. Carlson and P.-C. Hsu, Nano Res. Energy, 2024, 3, e9120102 CrossRef.
- E. Heid, M. Fleck, P. Chatterjee, C. Schröder and A. D. MacKerell, Jr., J. Chem. Theory Comput., 2019, 15, 2460–2469 CrossRef CAS PubMed.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, J. Chem. Phys., 2018, 148, 241722 CrossRef PubMed.
- T. Xie, A. France-Lanord, Y. Wang, Y. Shao-Horn and J. C. Grossman, Nat. Commun., 2019, 10, 2667 CrossRef CAS PubMed.
- S. C. Kim, S. T. Oyakhire, C. Athanitis, J. Wang, Z. Zhang, W. Zhang, D. T. Boyle, M. S. Kim, Z. Yu, X. Gao, T. Sogade, E. Wu, J. Qin, Z. Bao, S. F. Bent and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2214357120 CrossRef CAS PubMed.
- X. Lai, Q. Chen, X. Tang, Y. Zhou, F. Gao, Y. Guo, R. Bhagat and Y. Zheng, eTransportation, 2022, 12, 100169 CrossRef.
- A. Bouter and X. Guichet, J. Cleaner Prod., 2022, 344, 130994 CrossRef CAS.
- P. Mandade, M. Weil, M. Baumann and Z. Wei, Chem. Eng. J. Adv., 2023, 13, 100439 CrossRef CAS.
- F. Arshad, J. Lin, N. Manurkar, E. Fan, A. Ahmad, M.-u-N. Tariq, F. Wu, R. Chen and L. Li, Resour., Conserv. Recycl., 2022, 180, 106164 CrossRef CAS.
- M. Dieterle, P. Fischer, M.-N. Pons, N. Blume, C. Minke and A. Bischi, Sustainable Energy Technol., 2022, 53, 102457 Search PubMed.
- J. Porzio and C. D. Scown, Adv. Energy Mater., 2021, 11, 2100771 CrossRef CAS.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- J. Song, C. Chen, S. Zhu, M. Zhu, J. Dai, U. Ray, Y. Li, Y. Kuang, Y. Li, N. Quispe, Y. Yao, A. Gong, U. H. Leiste, H. A. Bruck, J. Y. Zhu, A. Vellore, H. Li, M. L. Minus, Z. Jia, A. Martini, T. Li and L. Hu, Nature, 2018, 554, 224–228 CrossRef CAS PubMed.
- S. Wang, L. Yu, S. Wang, L. Zhang, L. Chen, X. Xu, Z. Song, H. Liu and C. Chen, Nat. Commun., 2022, 13, 3408 CrossRef CAS PubMed.
- P. Gu, W. Liu, Q. Hou and Y. Ni, J. Mater. Chem. A, 2021, 9, 14233–14264 RSC.
- S. Jing, L. Wu, A. P. Siciliano, C. Chen, T. Li and L. Hu, ACS Nano, 2023, 17, 22196–22226 CrossRef CAS PubMed.
- Y. Zhou, C. Chen, X. Zhang, D. Liu, L. Xu, J. Dai, S.-C. Liou, Y. Wang, C. Li, H. Xie, Q. Wu, B. Foster, T. Li, R. M. Briber and L. Hu, J. Am. Chem. Soc., 2019, 141, 17830–17837 CrossRef CAS PubMed.
- Z. Chen, R. Feng, W. Wang, S. Tu, Y. Hu, X. Wang, R. Zhan, J. Wang, J. Zhao, S. Liu, L. Fu and Y. Sun, Nat. Commun., 2023, 14, 4648 CrossRef CAS PubMed.
- D. Song, J. Yu, M. Wang, Q. Tan, K. Liu and J. Li, Energy Storage Mater., 2023, 61, 102870 CrossRef.
- H. Bae and Y. Kim, Mater. Adv., 2021, 2, 3234–3250 RSC.
- X. Zhang, L. Li, E. Fan, Q. Xue, Y. Bian, F. Wu and R. Chen, Chem. Soc. Rev., 2018, 47, 7239–7302 RSC.
- K. Du, E. H. Ang, X. Wu and Y. Liu, Energy Environ. Mater., 2022, 5, 1012–1036 CrossRef CAS.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |