
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric synthesis of 1,2-diamines
Francisco
Foubelo
 ab,
Carmen
Nájera
*b,
Ma Gracia
Retamosa
ab,
José M.
Sansano
ab and
Miguel
Yus
b
ab,
Carmen
Nájera
*b,
Ma Gracia
Retamosa
ab,
José M.
Sansano
ab and
Miguel
Yus
b
aDepartamento de Química Orgánica and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, E-03080 Alicante, Spain
bCentro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Alicante, Apdo. 99, E-03080 Alicante, Spain. E-mail: cnajera@ua.es
First published on 11th July 2024
Abstract
The asymmetric catalytic synthesis of 1,2-diamines has received considerable interest, especially in the last ten years, due to their presence in biologically active compounds and their applications for the development of synthetic building blocks, chiral ligands and organocatalysts. Synthetic strategies based on C–N bond-forming reactions involve mainly (a) ring opening of aziridines and azabenzonorbornadienes, (b) hydroamination of allylic amines, (c) hydroamination of enamines and (d) diamination of olefins. In the case of C–C bond-forming reactions are included (a) the aza-Mannich reaction of imino esters, imino nitriles, azlactones, isocyano acetates, and isothiocyanates with imines, (b) the aza-Henry reaction of nitroalkanes with imines, (c) imine–imine coupling reactions, and (d) reductive coupling of enamines with imines, and (e) [3+2] cycloaddition with imines. C–H bond forming reactions include hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds and C–H amination reactions. Other catalytic methods include desymmetrization reactions of meso-diamines.
N bonds and C–H amination reactions. Other catalytic methods include desymmetrization reactions of meso-diamines.
 Francisco Foubelo | Francisco Foubelo was born in 1961 and studied Chemistry at the University of Oviedo where he received BS (1984), MS (1986) and PhD (1989) degrees, under the direction of Professors J. Barluenga, M. Yus and F. J. Fañanás. He then joined the laboratory of Professor M. F. Semmelhack at Princeton University as a Fulbright postdoctoral fellow, and, in 1991, the group of Professor M. Yus at the University of Alicante, where he became an associate professor in 1995, and a full professor in 2002. His current research interests are focused on the development of new synthetic methodologies involving chiral sulfinyl imines, and on metal-promoted functionalization of alkenes and alkynes. |
 Carmen Nájera | Carmen Nájera was born in Nájera (La Rioja, 1951) and received BS (1973) and MS from the University of Zaragoza and PhD (1979) form the University of Oviedo. After postdoctoral stays at the ETH, Oxford, Harvard and Uppsala Universities, she became an associate professor (1985) at the University of Oviedo and a full professor (1993) at the University of Alicante. She is a coauthor of more than 430 papers and supervised 50 PhD students. She has been awarded with several national and international prizes, and in 2012 she was elected as a full member of the Spanish RAC and in 2016–2017 ChemPubSoc Europe Fellow. |
 M Gracia Retamosa | Maria de Gracia Retamosa is a Senior Researcher of the University of Alicante (CIDEGENT program of the G. Valenciana) and received her PhD in 2008 from the University of Alicante (Spain) under the guidance of Prof. Carmen Nájera and José Miguel Sansano. She is a coauthor of 45 articles and 5 patents. She completed several postdoctoral stays [with Prof. Michael Greaney at the University of Edinburgh (UK, 2009), Prof. Jesús M. Sanz at the University Miguel Hernández (Elche, Spain, 2009–2011) and Prof. Fernando P. Cossío at the University of the Basque Country and Donostia International Physics Center (Spain, 2012–2016)]. Recently, she has joined the group of Prof. Rosario Fernández and José M. Lassaletta as a postdoctoral researcher [CSIC (Sevilla, Spain)]. Her current research interests include asymmetric metal, and organocatalysis and synthesis of compounds with pharmacological interest. |
 José M. Sansano | Professor José Miguel Sansano was born in Rojales (Alicante), studied chemistry at the University of Alicante, where he obtained his BSc and PhD degrees in 1988 and 1994, respectively. His thesis was supervised by Prof. C. Nájera and dealt about sulfone chemistry. After spending a two-year postdoctoral stay at the University of Leeds (U.K.) with Prof. R. Grigg, he joined the University of Alicante in 1996, where he was appointed as an Associate Professor in 2001. In 2010, he was promoted to a Full Professor in the same University. He was invited as a visiting Professor at Chuo University in 2014 and in the UFRJ (Brazil). He is a coauthor of more than 140 articles and he has supervised 13 PhD students. |
 Miguel Yus | Miguel Yus was born in Zaragoza (Spain, 1947), and received his BSc (1969), MSc (1971) and PhD (1973) degrees from the University of Zaragoza. After two years as a postdoctoral fellow at Max Planck Institut für Kohlenforschung in Mülheim/Ruhr, he returned to the University of Oviedo, Spain where he became an associate professor (1977) and a full professor (1987). In 1988, he moved to the University of Alicante. He is a co-author of 600+ papers, having 33.000+ citations and h-index 83. Professor Yus has been in the Advisory Board of more than 30 international journals and founded in 2002 the company MEDALCHEMY S.L. |
1. Introduction
Chiral 1,2-diamines are ubiquitous in biologically active compounds, not only in natural products but also synthetic ones including commercial drugs.1 These compounds are widely used in asymmetric synthesis of chiral auxiliaries, ligands and organocatalysts (Table 1).2–14 For these reasons, there is broad interest in the development of catalytic methods for the synthesis of enantioenriched 1,2-diamines mainly based on asymmetric metal catalysis and organocatalysis. Among them are C–N, C–C and C–H bond forming reactions (Scheme 1).
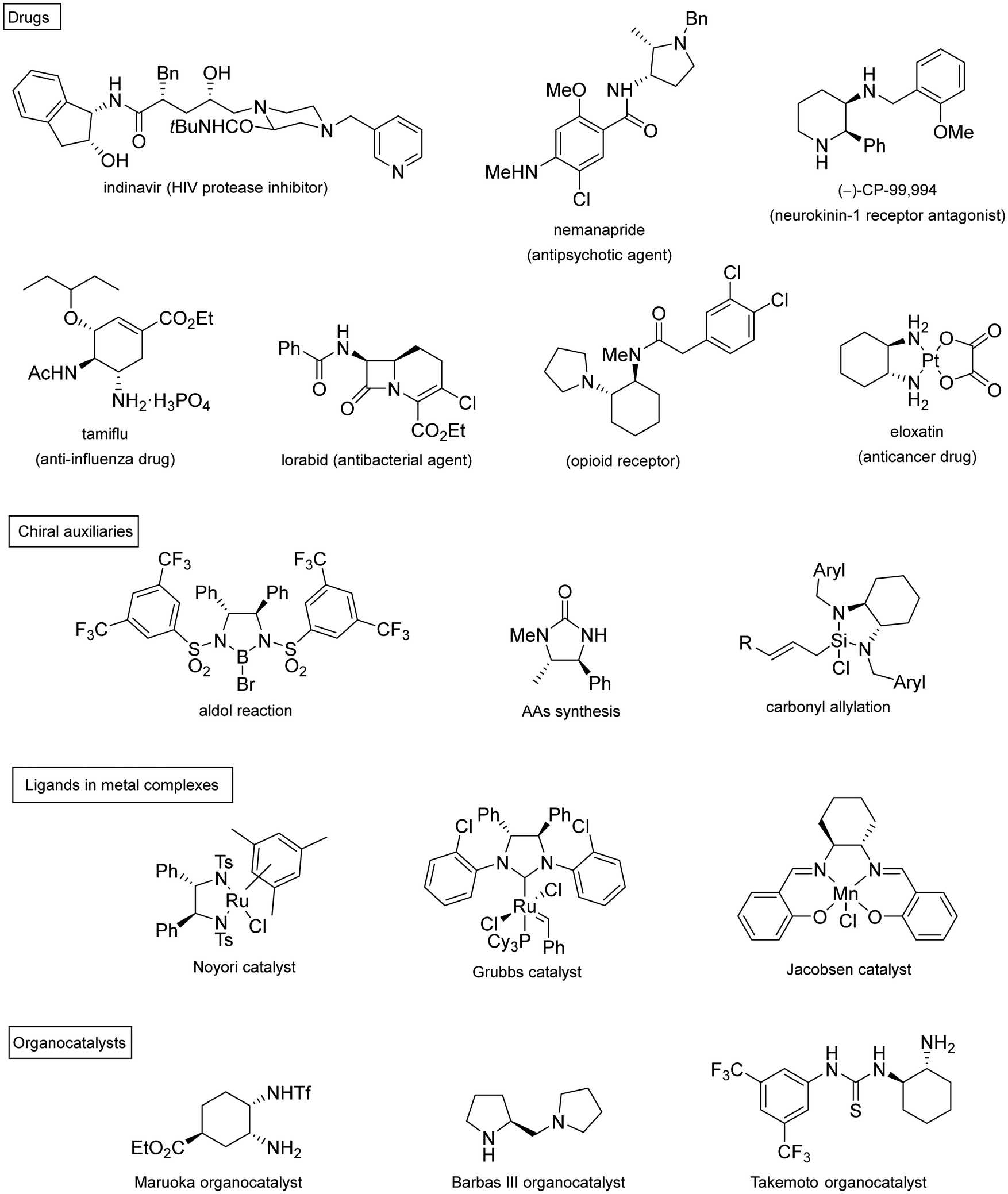
|
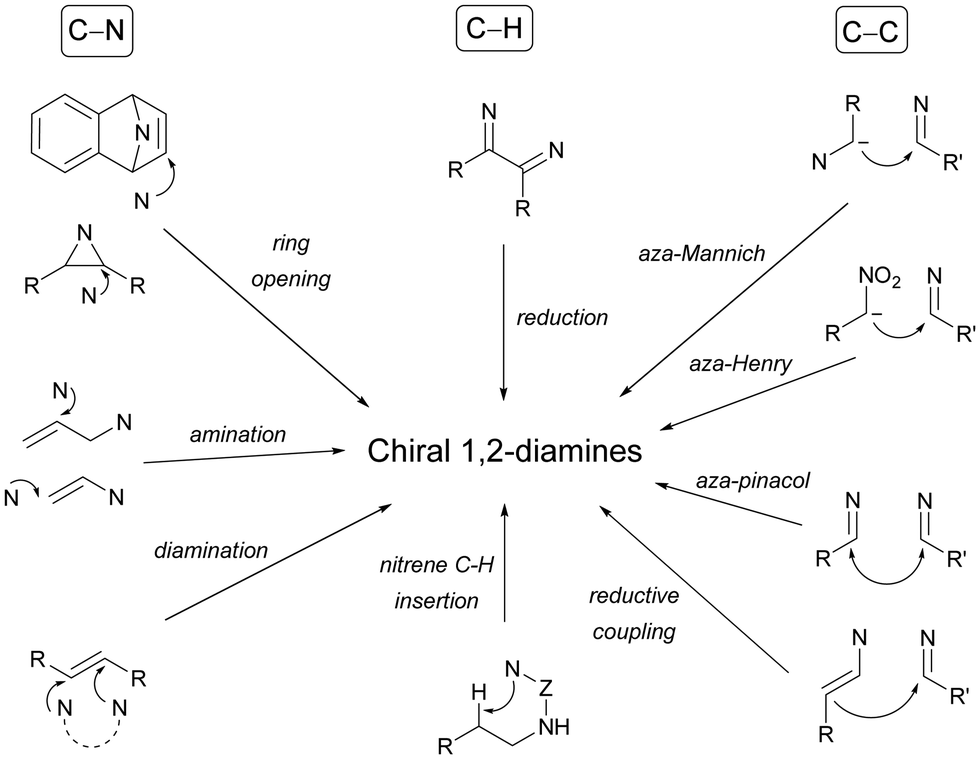 | ||
| Scheme 1 Catalytic methods for the asymmetric synthesis of 1,2-diamines. | ||
In this review article, methods developed mainly in the last 15 years will be considered. For the C–N bond forming reactions, ring-opening of meso-aziridines and azabenzonorbornadienes, amination of allylic amines or enamines, ring opening of azabenzonorbornadienes and diamination of alkenes7–14 will be considered. Classical C–C bond forming reactions such as aza-Mannich-type reactions, aza-Henry reactions followed by the reduction of the nitro group, aza-pinacol reactions, addition of azaallyl anions to imines and 1,3-dipolar cycloaddition of azomethine ylides to imines will be presented. However, due to space limitations, the Strecker reaction, which is mainly applied to the synthesis of α-amino acids, is not included. In the case of C–H forming approaches, reduction of CN bonds by hydrogenation reactions and intramolecular nitrene C–H insertion will be considered. Other catalytic methods will be included in Section 6, such as desymmetrization of meso-diaminocyclopropanes or kinetic resolution of racemic 1,2-diamines. In this catalytic asymmetric methods, metals, organocatalysts and enzymes are extensively used.
2. C–N bond-forming reactions
2.1. Ring-opening of aziridines
The catalytic aminolysis of meso-aziridines 1 is a very efficient strategy to obtain chiral 1,2-diamines with two differently substituted amino groups. This desymmetrization15 methodology can be carried out under chiral Lewis or Brønsted acid catalysis. When trimethylsilyl azide was allowed to react with meso-aziridines 1, the corresponding β-azido amino derivatives 2 were isolated (Scheme 2). Initial pioneering work of Jacobsen and co-workers16 was carried out using a tridentate Schiff base chromium(III) complex 3 derived from 1-amino-2-indanol as a catalyst (Fig. 1). The azido N-2,4-1 products 2 were obtained in 73–95% yields and 83–94% ee and they can be easily transformed into chiral 1,2-diamines (Table 2).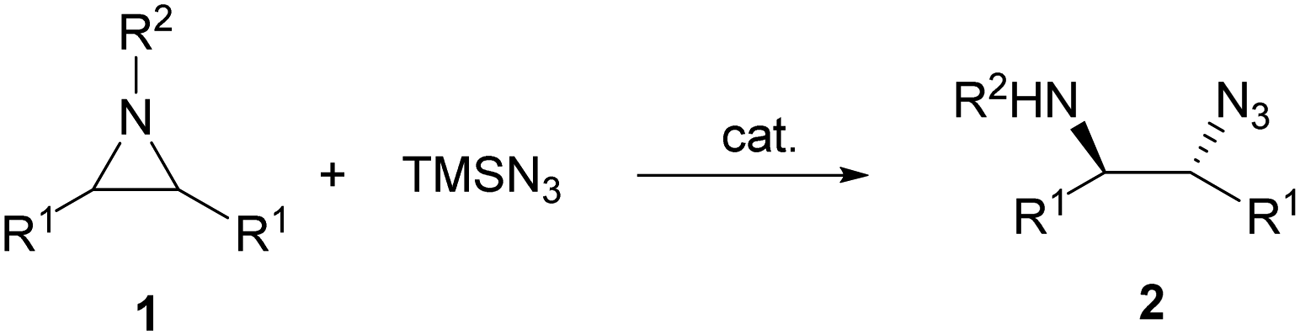 | ||
| Scheme 2 Catalytic ring-opening of meso-aziridines 1 with trimethylsilyl azide. | ||
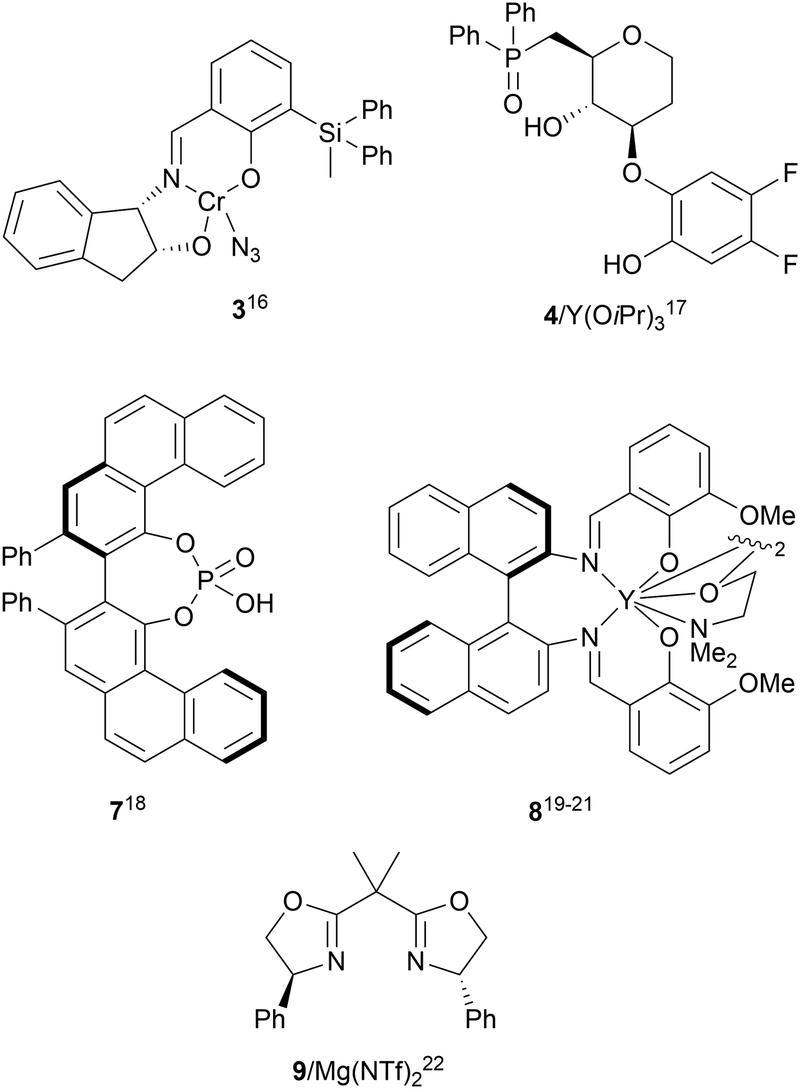 | ||
| Fig. 1 Catalysts for the ring-opening of meso-aziridines with trimethylsilyl azide. | ||
| R1 | R2 | Catalyst | Yield (%) | ee (%) | Ref. |
|---|---|---|---|---|---|
| Me, (CH2)2, (CH2)4, CH2OCH2, 3,5-(NO2)2C6H3CH2, (Z)-CH2CHCHCH2 |
2,4-(NO2)2C6H3CH2 | 3 | 73–95 | 83–94 | 16 |
| Me, Ph, (CH2)3, (CH2)4, (Z)-CH2CHCHCH2, 3,5-(NO2)2C6H3CO, CH2OCH2 |
3,5-(NO2)2C6H3CO | 4 | 94–>99 | 83–96 | 17 |
Me, Ph, (CH2)3, (CH2)4, (Z)-CH2CHCHCH2, 3,5-(NO2)2C6H3CO, CH2OCH2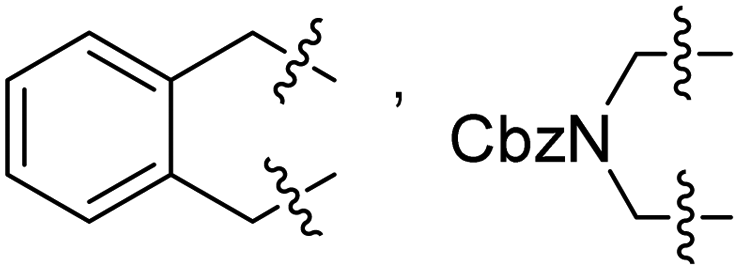 |
3,5-(NO2)2C6H3CO | 8 | 49–97 | 69–95 | 17 |
|
nPr, Ph, (CH2)3, (CH2)4, (Z)-CH2CHCHCH2 |
4-NO2C6H4CO | 11 | 47–99 | 90–99 | 19–21 |
Ph, (CH2)3, (CH2)4, 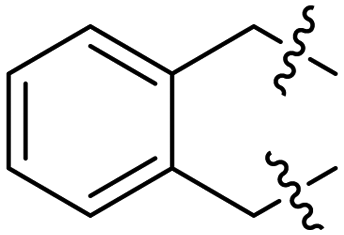 |
2-PySO2, 6-Me, 2-PySO2 | 14 | 51–91 | 64–85 | 22 |
Kanai, Shibasaki and co-workers17 used a yttrium complex formed by a chiral phosphine oxide 4 and Y(OiPr)3 as a catalyst (Fig. 1) for the synthesis of compounds 2 with high yields (94 to >99%) and good enantioselectivities (83–96% ee) (Table 2). Compound 2 [R1–R1(CH2)4] was transformed in 3 steps into trans-1,2-cyclohexanediamine in 96% yield. This reaction was applied to the asymmetric synthesis of Tamiflu starting from aziridine 5, which was transformed into diamine 6 in four steps (Scheme 3).
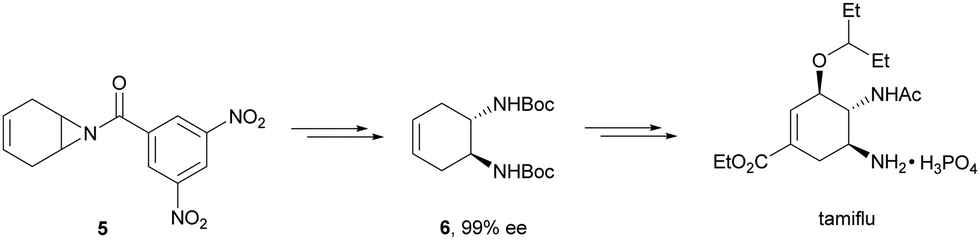 | ||
| Scheme 3 Synthesis of tamiflu by ring-opening of aziridine 5 with trimethylsilyl azide as the key step. | ||
Brønsted acid organocatalysis was performed by Antila and co-workers18 by means of a chiral phosphoric acid (CPA) (S)-Vapol (7) as a catalyst (Fig. 1). This general method was carried out with cyclic and acyclic aziridines 1 to obtain products 2 in good yields and enantioselectivities (Table 2). In the proposed mechanism, the silylation of the CPA firstly took place, which activates the aziridine followed by subsequent nucleophilic attack by the azide. Parquete, RajanBabu and co-workers19–21 employed a dimeric yttrium–salen complex 8 as a chiral catalyst (Fig. 1) for the ring-opening of meso-aziridines 1 with TMSN3. This reaction took place at room temperature to afford products 2 in high yields and enantioselectivities (Table 2). However, monosubstituted aziridines provided a mixture of regioisomeric products.21 Nakamura and co-workers22 used for the first time meso-N-(2-pyridinesulfonyl)aziridines 1 (R2 = 6-Me-2-PySO2), which by reaction with trimethylsilyl azide under Mg(NTf)2/bisoxazoline 9 (Fig. 1) catalysis provided products 2 in good yields and moderate enantioselectivities (Table 2). This procedure was applied to the synthesis of U-50488, which is a highly selective K-opioid agonist, starting from compound 10 which was obtained in 99% ee after recrystallization (Scheme 4).
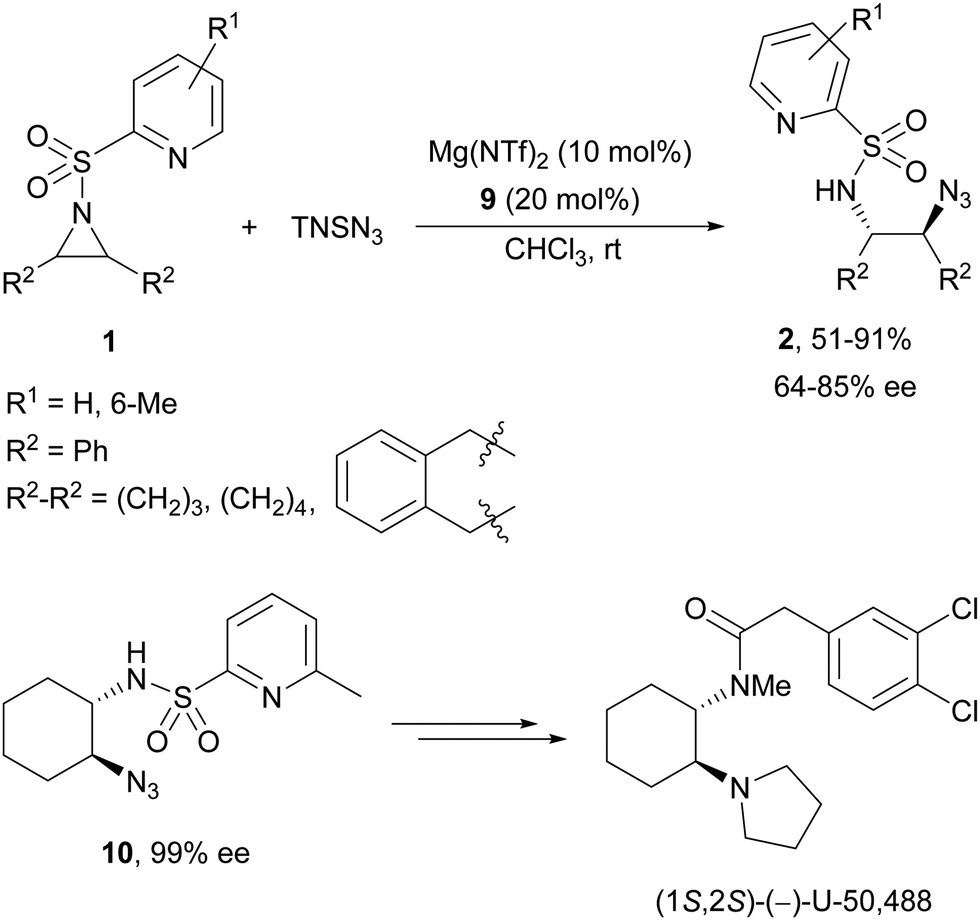 | ||
| Scheme 4 Ring-opening of meso-N-(2-pyridinesulfonyl)aziridines 1 with trimethylsilyl azide under Mg(NTf)2/bisoxazoline 9 catalysis. | ||
Desymmetrization of meso-aziridines with amines was firstly performed under binol derived metal complexes by Kobayashi and co-workers.23–25N-(2-Methoxyphenyl)aziridines 11 were reacted with anilines in the presence of Nb(OiPr)3 and tetradentate binol (R)-12 (Fig. 2) to furnish diamines 13 in good yields and high enantioselectivities (Scheme 5).23
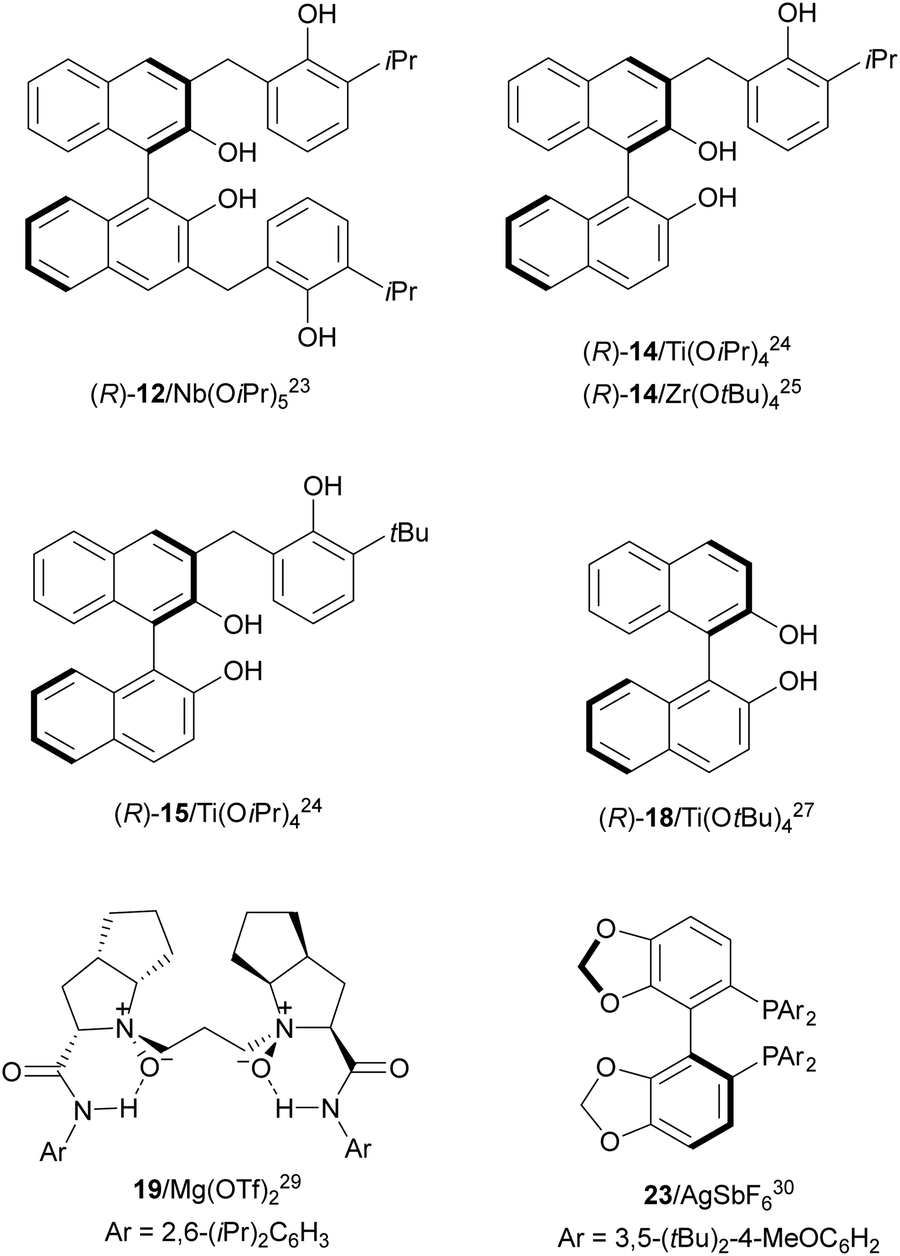 | ||
| Fig. 2 Chiral catalysts for the aminolysis of meso-aziridines. | ||
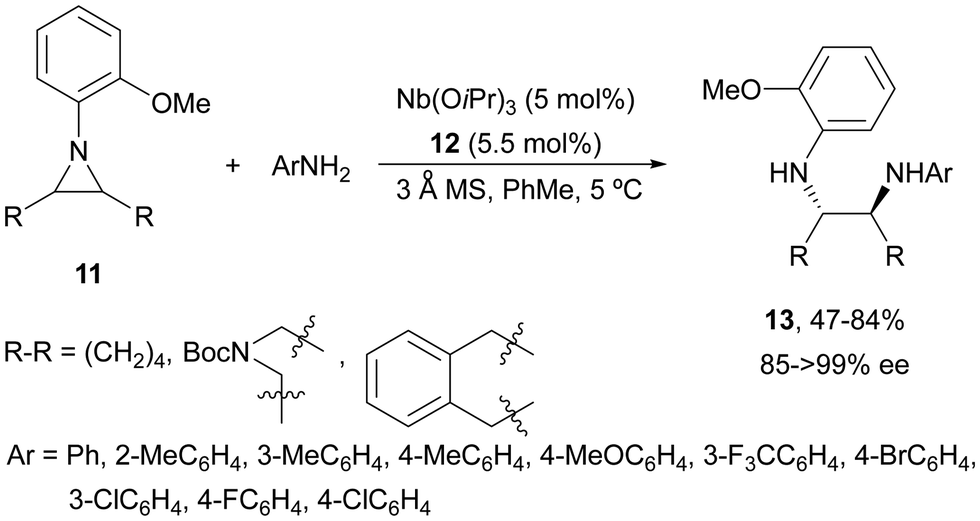 | ||
| Scheme 5 Ring-opening of meso-aziridines 11 with anilines under Nb–binol complex 12 catalysis. | ||
The ring-opening of aziridines 11 with aniline was also studied by the same group24 using Ti(OiPr)4 and tridentate binol derivatives 14 and 15 (Fig. 2) as catalysts. Cyclic meso-aziridines 11 were efficiently transformed into the corresponding N,N′-diaryl 1,2-diamines 13 in good yields (74–94%) and enantioselectivities (61–95%). Ligand 14 and Zr(OtBu)4 were employed as catalysts for the aminolysis of cyclic and acyclic N-benzhydryl aziridines 16 with anilines obtaining products 17 in high yields and enantioselectivities (Scheme 6).25 For the cleavage of the benzhydryl group, hydrogenation on incarcerated Pd26 in EtOH at 70 °C was efficiently performed giving quantitatively the monoamine-free diamine.
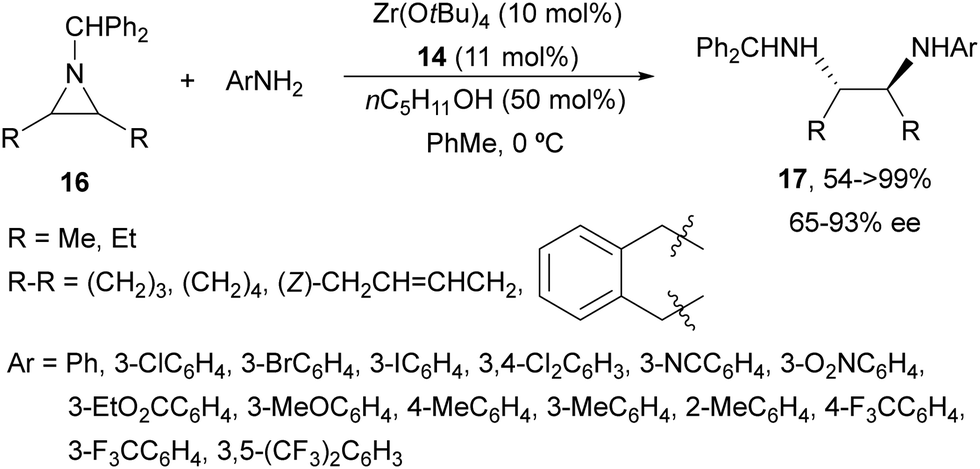 | ||
| Scheme 6 Ring-opening of meso-aziridines 16 with anilines under Zr–binol complex 14 catalysis. | ||
Schneider and co-workers27,28 used Ti(OtBu)4 and (R)-binol (18, Fig. 2) in the aminolysis of cyclic and acyclic N-arylaziridines 11 for the direct preparation of N,N′-diarylated enantioenriched 1,2-diamines 13 (Scheme 7). The chiral catalyst with an oligomeric structure (from ESI-MS experiments) showed, as in the case of Kobayashi's catalyst2415/Ti(OiPr)4, a positive nonlinear effect (NLE). This method is the simplest and in general efficient for the synthesis of these types of diamines.
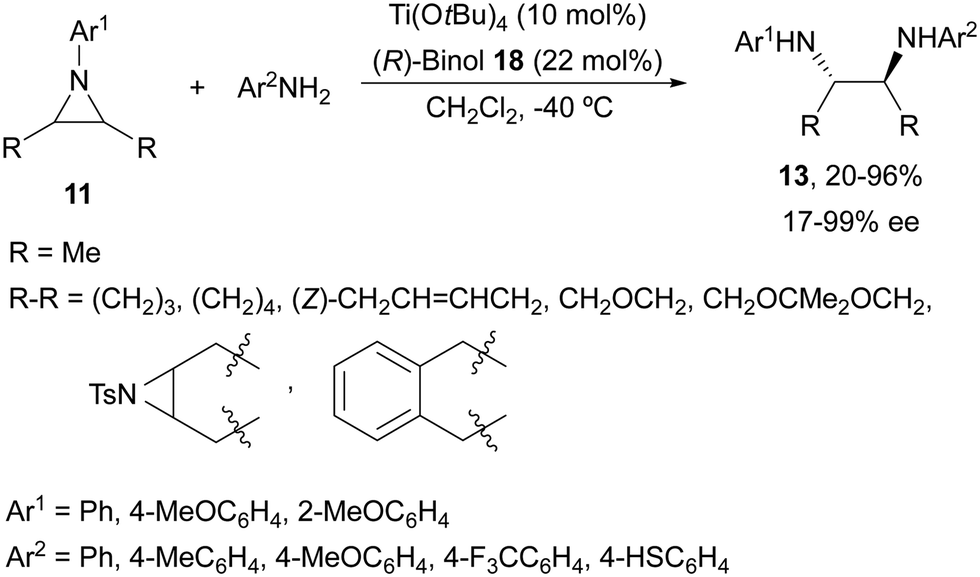 | ||
| Scheme 7 Ring-opening of meso-aziridines 11 with anilines under Ti(OtBu)4/(R)-binol (18) catalysis. | ||
In the studies of meso-aziridines' ring-opening with primary alcohols, Feng and co-workers29 reported a single example with aniline. Chiral Mg(OTf)2/N,N′-dioxide 19 (Fig. 2) acted as a catalyst in the reaction of N-(2-picolinoyl)aziridine 20 with aniline to afford diamine 21 in 97% yield and 95% ee (Scheme 8).
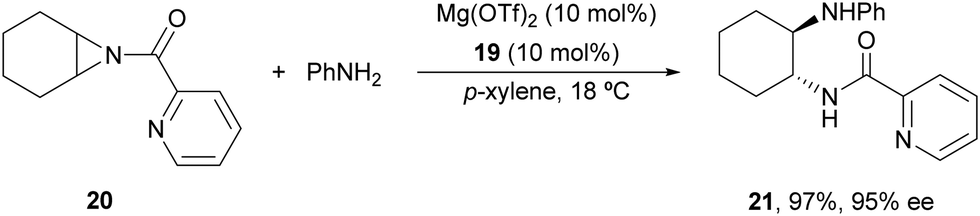 | ||
| Scheme 8 Ring-opening of meso-aziridine 20 with aniline under Mg(OTf)2/N,N′-dioxide 19. | ||
Chai and co-workers30 have reported an Ag(I)-catalyzed enantioselective ring-opening of N-tosylaziridines 22 using (S)-DTBM-Segphos (23) as a chiral bisphosphine (Fig. 2). This desymmetrization was performed with aromatic and aliphatic amines to obtain the corresponding diamines 24 in general with very good yields and enantioselectivities (Scheme 9a). In addition, these reaction conditions have been applied to the kinetic resolution of 2-aryl-N-tosylaziridines 25 with amines to obtain regioselectively diamines 26 with excellent results (Scheme 9b). Gram-scale experiments were performed with meso-cyclohexane aziridine and 4-methoxyaniline, and the reaction of 2-phenyl-1-tosylaziridine with 4-methoxyaniline on 4 and 3 mmol scales, respectively.
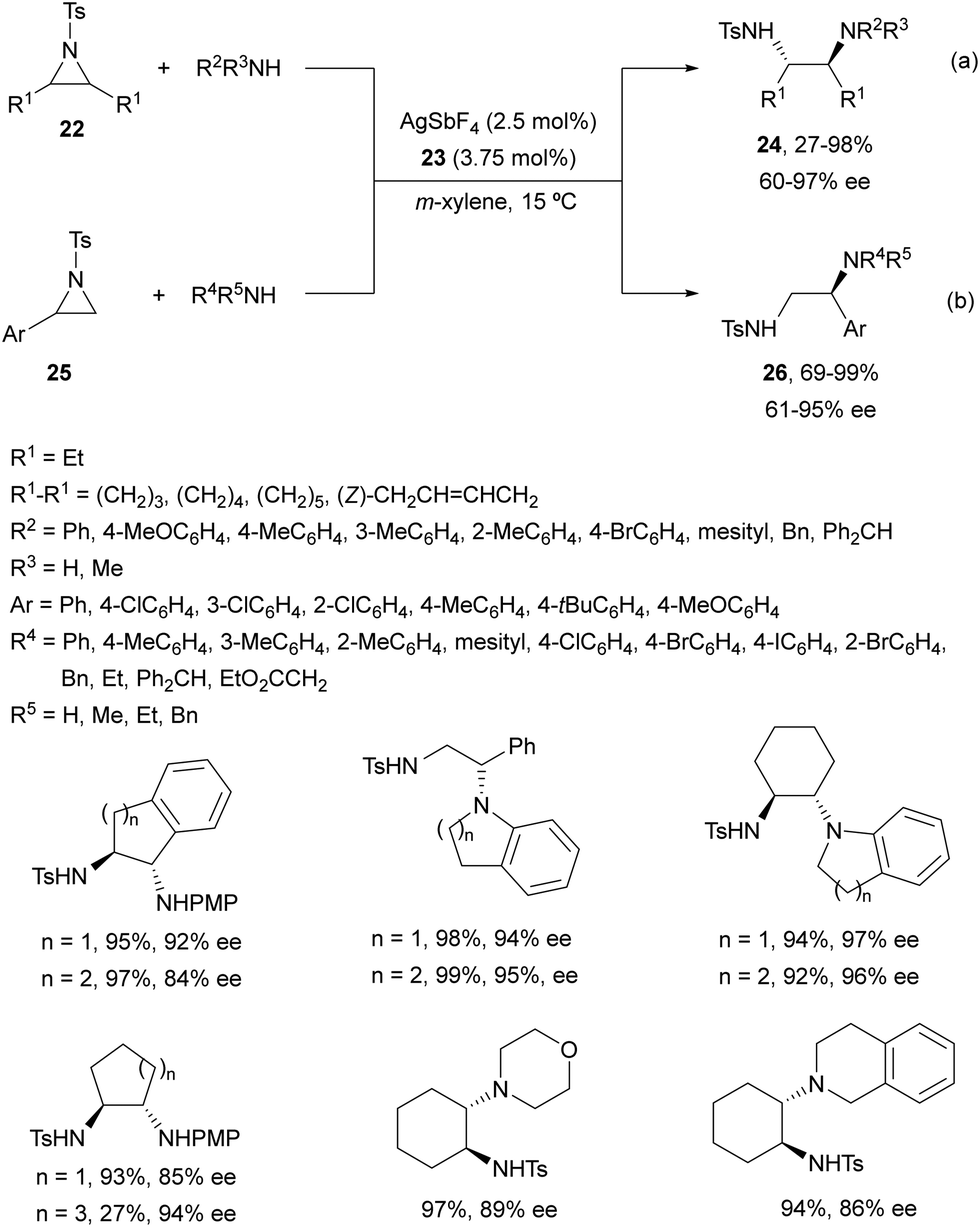 | ||
| Scheme 9 Ring-opening of aziridines 22 and 25 with aromatic and aliphatic amines under AgSbF6/(S)-DTBM-Segphos (23) catalysis. | ||
Other nitrogenated compounds such as substituted hydroxylamines,31 tetrazoles32 and pyrazoles33 have been employed as nucleophiles for desymmetrization of meso-aziridines using Mg(II) complexes as catalysts. Wang and co-workers31 performed the ring-opening of N-(2-picolinoyl)aziridines 20 with different carbonyl protected hydroxylamines 27 to obtain compounds 29 in good yields and enantioselectivities employing n-Bu2Mg and oxazoline 28 as a catalyst (Scheme 10). This Mg-catalyzed desymmetrization can also be carried out on a gram-scale.
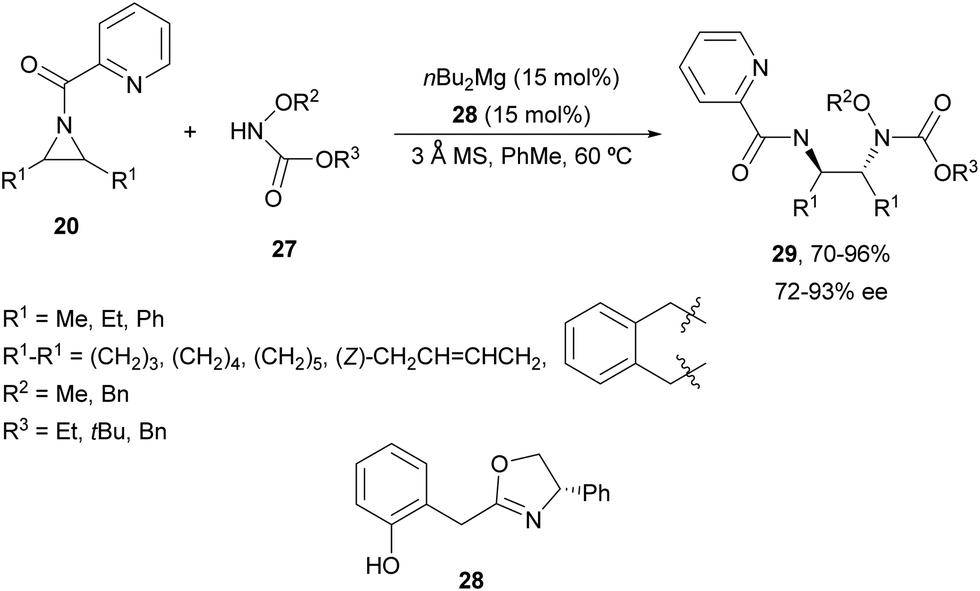 | ||
| Scheme 10 Ring-opening of aziridines 20 with carbonyl-protected hydroxylamines 27 under nBu2Mg/oxazoline 28 catalysis. | ||
In the case of monosubstituted tetrazoles 30, aziridines 20 underwent ring-opening under nBu2Mg and a binolam32 derivative 31 to afford products 32 (Scheme 11).33 These compounds 32 were monodeprotected to the corresponding primary amines with NaOH in EtOH at 90 °C. As a possible mechanism, it has been proposed the formation of chiral catalyst I, which after coordination with the aziridine 20 forms intermediate II. Subsequent coordination of the tetrazole gives intermediate III, which evolves by ring-opening of the aziridine to provide the product and regenerates the catalyst.
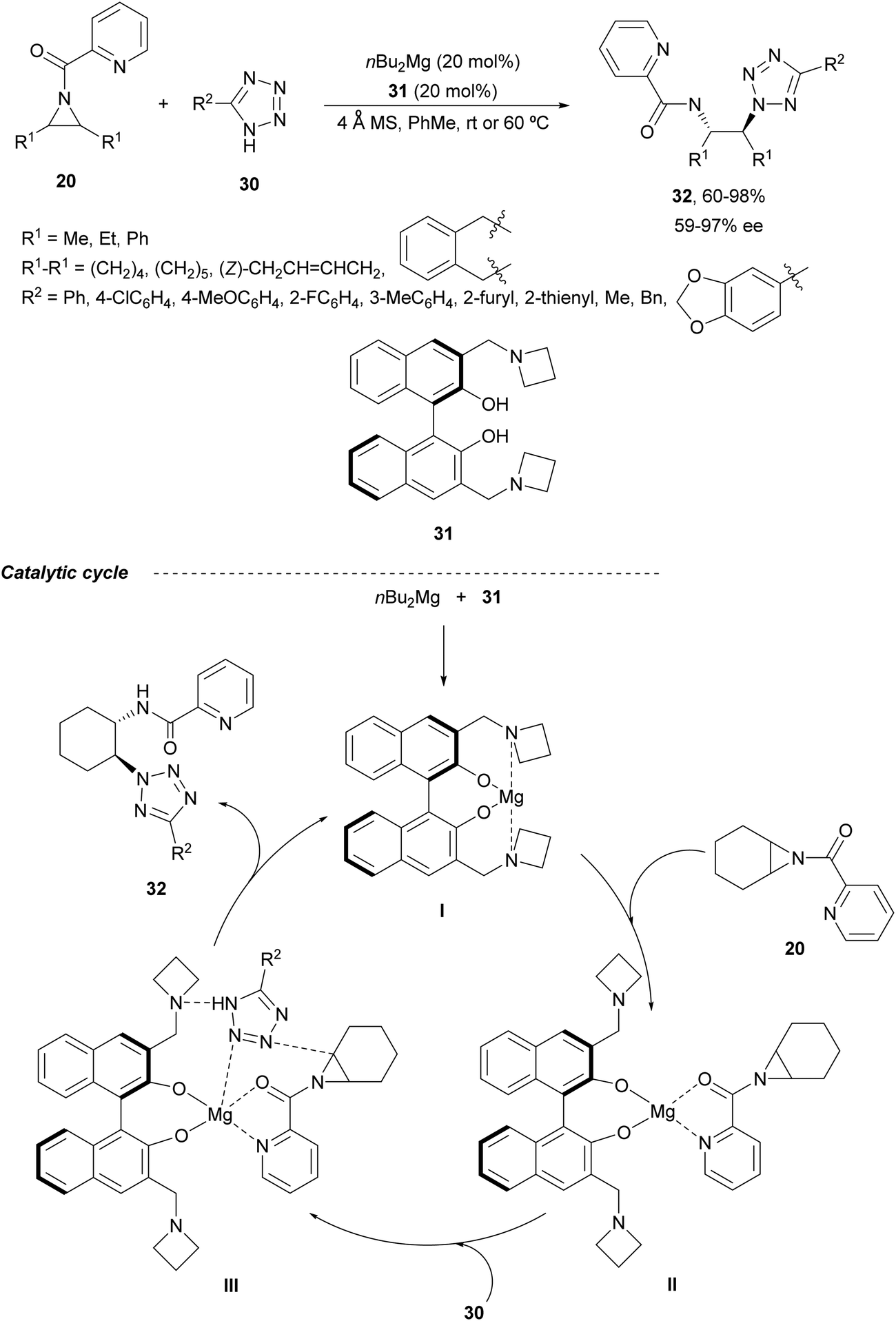 | ||
| Scheme 11 Ring-opening of meso-aziridines 20 with tetrazoles 30 under nBu2Mg/binolam 31 catalysis. | ||
Feng and co-workers34 applied a similar catalyst employed in the ring opening of aziridines 20 with aniline (see Scheme 8) for pyrazoles 33 (Scheme 12). In this case, Mg(OTf)2 and N,N′-dioxide 34 were used as catalysts for the desymmetrization of differently substituted N-(2-picolinoyl)aziridines 20 with pyrazoles 33 to furnish products 35 in good yields (up to 99%) and enantioselectivities (up to 94% ee). Strong positive NLE was observed for this type of desymmetrization. Besides, benzotriazole, 3-phenyltetrazole and trimethylsilyl azide were also applied to this ring-opening of cyclohexane-derived aziridine although with lower enantioselectivities.
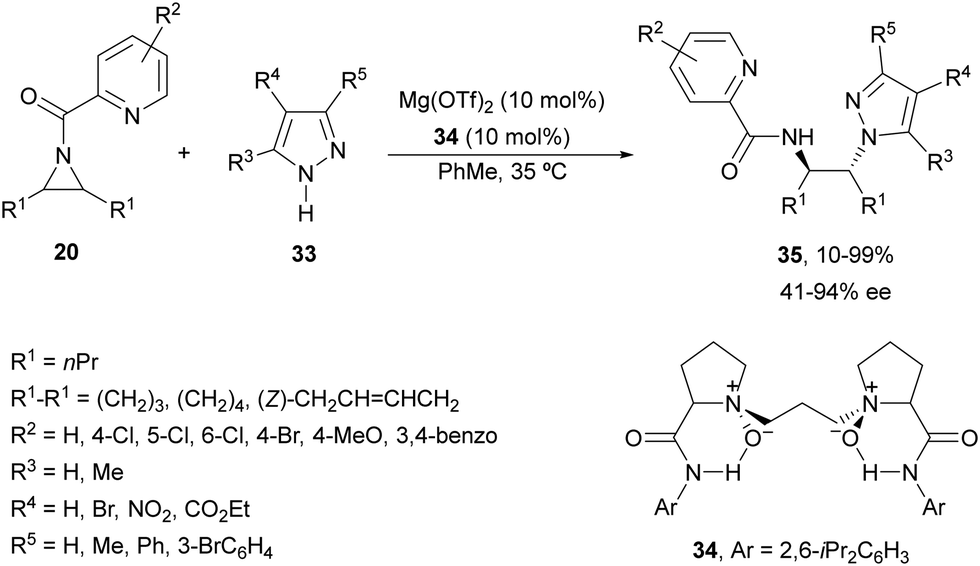 | ||
| Scheme 12 Ring-opening of meso-aziridines 20 with pyrazoles 33 under Mg(OTf)2/N,N′-oxide 34 catalysis. | ||
According to the procedures for the asymmetric ring-opening of aziridines, the initial use of trimethylsilyl azide under metal-catalysis and CPA as an organocatalyst has evolved to aromatic and aliphatic amines under silver catalysis. The nitrogenated nucleophiles such as substituted hydroxylamines, tetrazoles and pyrazoles can be employed under magnesium catalysis.
2.2. Ring-opening of azabenzonorbornadienes
Transition-metal-catalyzed asymmetric ring-opening reaction of azabenzonorbornadienes 36 with amines is a desymmetrization15 strategy for the synthesis of diaminotetralines 37 (Scheme 13). Lautens and co-workers35–37 reported the first example35,36 using the rhodium complex of C2-ferriphos 38 (Fig. 3) and aromatic or aliphatic amines to provide regioselectively diamines 37 in 50–98% yield and 89 to >99% ee. This process was applied to the total synthesis of an analgesic compound 39, a highly selective K-opioid agonist,38 starting from N-Boc-pyrrole. Experimental results36 support the formation of a dimer I, which after exo-binding at the C–C double bond and to the nitrogen atom forms intermediate II. Oxidative insertion of a Rh catalyst to C–N bond gives III and subsequent SN2′ displacement by the amine provides product 37 regenerating the catalyst (Scheme 13).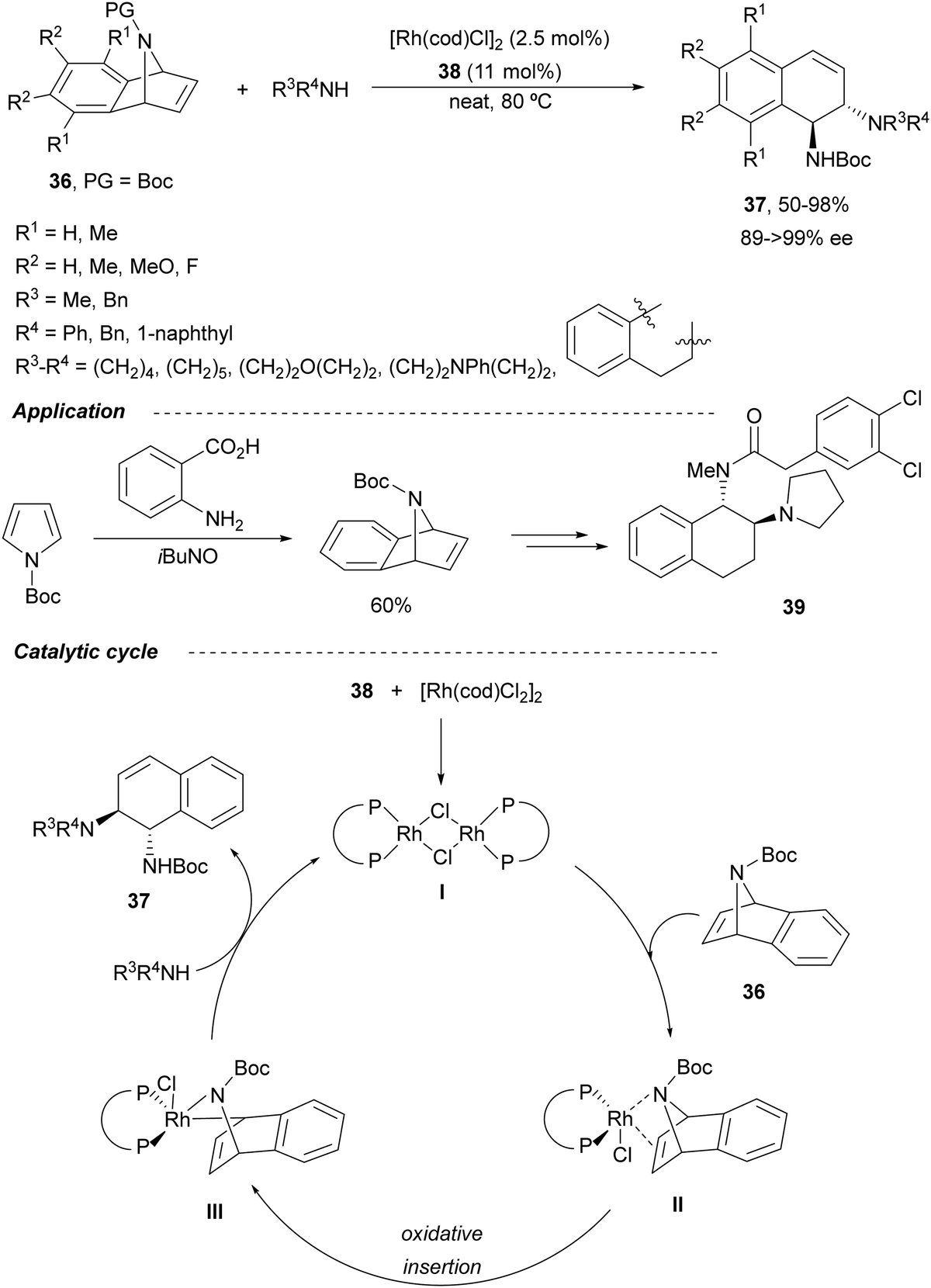 | ||
| Scheme 13 Ring-opening of N-Boc-azabenzonorbornadienes 36 with amines under Rh(I)/C2-ferriphos 38. | ||
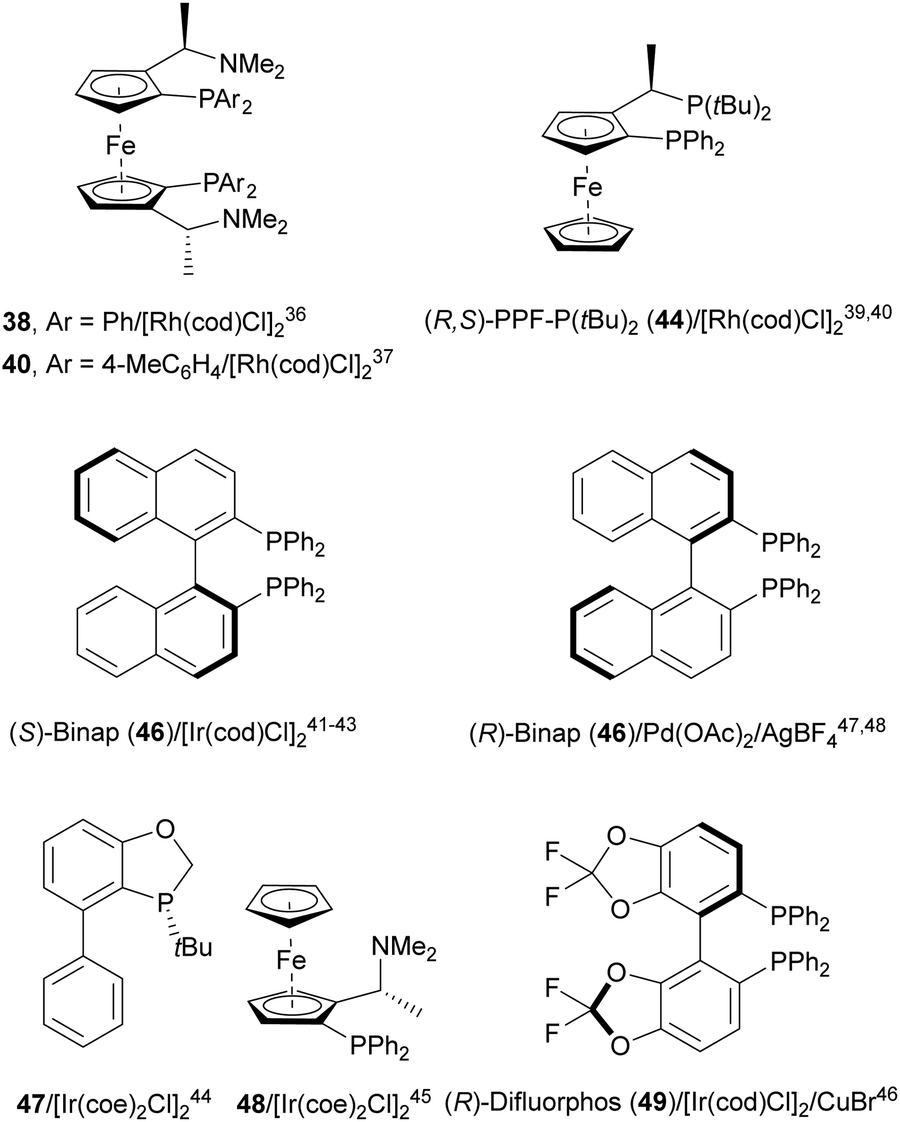 | ||
| Fig. 3 Catalysts for the ring-opening of azabenzonorbornadienes with amines. | ||
Another synthetic application of this Rh-catalyzed ring-opening of azabenzonorbornadienes was the preparation of new chiral ligands.37 In this case, the ring-opening of 36 was carried out with dibenzylamine in the presence of (S,S′)-(R,R′)-C2-ferriphos-tolyl 40 (Fig. 3) at 80 °C to give diamines 37 in 70–99% yield and 97–99% ee. After deprotection of diaminotetralins 37 resulted the corresponding tartrate salts of primary amines 41, and these diamines were transformed into salen-type ligands 42 (Scheme 14).
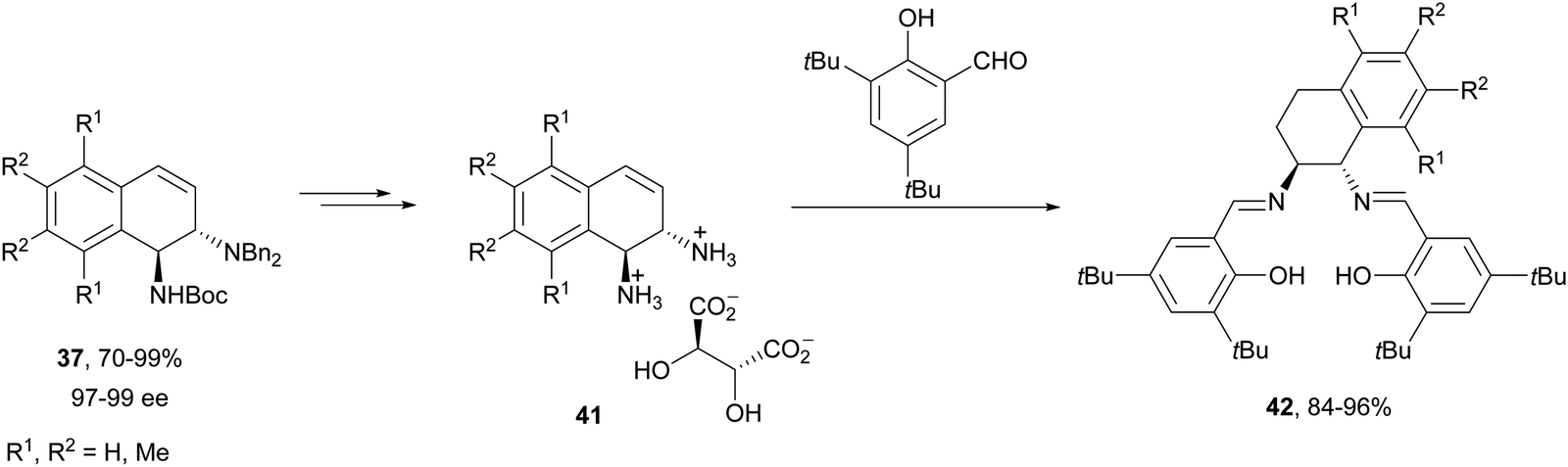 | ||
| Scheme 14 Synthesis of chiral salen-type ligands 42 from diaminotetralins 37. | ||
Yang and co-workers39,40 reported the Rh-catalyzed ring opening of N-Boc-azabenzonorbornadienes 36 with piperazine nucleophiles 43 using (R,S)-PPF-P(tBu)244 as a chiral ligand (Fig. 3). The resulting diaminotetralines 45 were obtained in the presence of ammonium iodide as an additive in tetrahydropyran (THP) at 100 °C with good yields and moderate enantioselectivities (Scheme 15).40
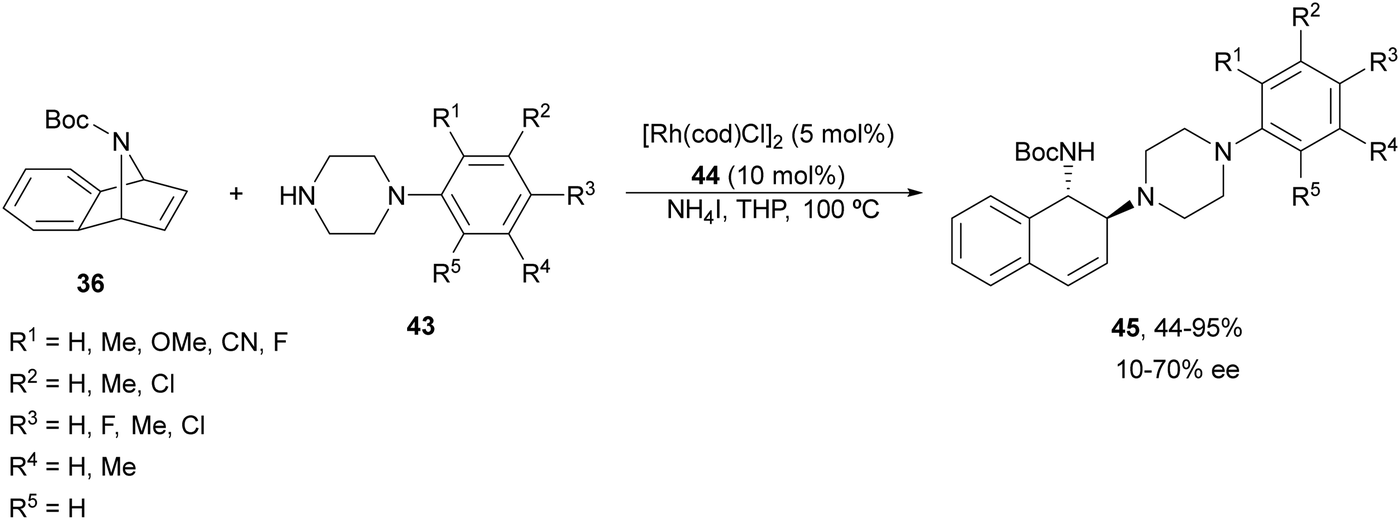 | ||
| Scheme 15 Ring-opening of N-Boc-azabenzonorbornadiene 36 with piperazines 43 under Rh/(R,S)-PPF-P(tBu)2 (44) catalysis. | ||
Iridium-catalyzed desymmetrization of N-Boc-azabenzonorbornadienes 36 with different amines has been described by Yang and co-workers.41–43 Secondary amines41,42 were employed as nucleophiles in the presence of [Ir(cod)Cl]2 and (S)-Binap 46 (Fig. 3) as catalysts in THP at 100 °C. N-Substituted piperazines 43 gave the corresponding diamines with good yields (55–86%) and enantioselectivities (61–87% ee), whereas N-methylanilines provided lower results (24–59% yields and 42–85% ee). The authors proposed the same mechanism as depicted in Scheme 13 for the Rh-catalyzed ring opening of N-Boc-azabenzonorbornadienes. Higher yields and enantioselectivities were obtained in the reaction of different N-substituted azabenzonorbornadienes 36 with primary aromatic amines working on THP at 100 °C (bath temperature) with a lower catalyst loading to furnish diamines 37 in up to 97% yield and up to 97% ee (Scheme 16).43 The best results were obtained with N-tosyl-azabenzonorbornadiene.
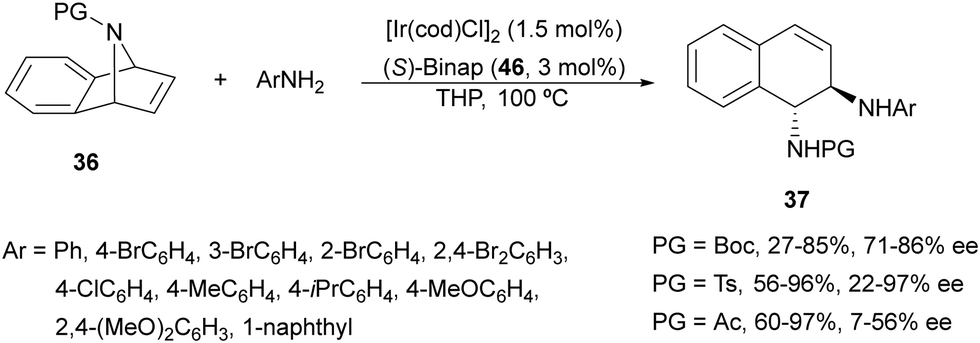 | ||
| Scheme 16 Ring-opening of N-substituted azabenzonorbornadienes 36 with primary aromatic amines under Ir/(S)-Binap 46 catalysis. | ||
A chiral monophosphine 47 (Fig. 3) has been employed by Luo and co-workers44,45 as a ligand for the Ir-catalyzed desymmetrization of oxa- and azabenzonorbornadienes. However, in the case of N-Boc protected compound 36, the reaction with N-phenylpiperazine (43) afforded the corresponding diamine 45 in only 19% yield and 35% ee.44 Lately, they found that [Ir(coe)2Cl]2 and ligand 48 (Fig. 3) were better catalysts for the ring-opening of N-Boc-azabenzonorbornadienes 36 with secondary aliphatic amines in the presence of NaI as an additive (Scheme 17).45 The resulting diamines 45 were obtained in good yields and enantioselectivities and one example was scaled up to ca. 2 g scale.
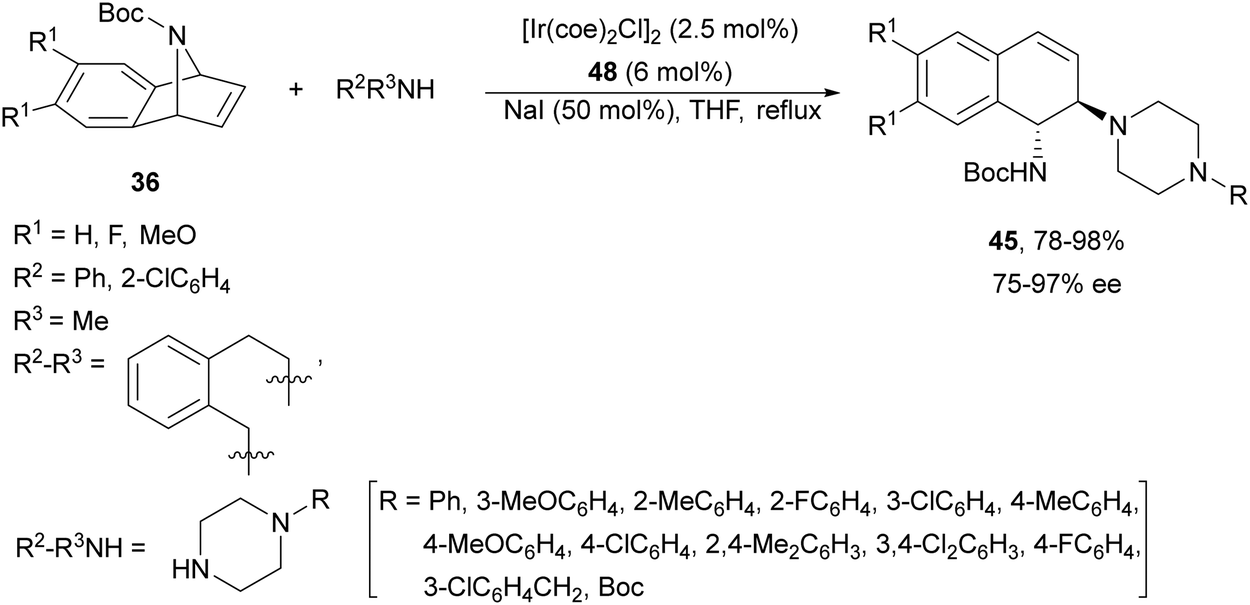 | ||
| Scheme 17 Ring-opening of N-Boc-azabenzonorbornadienes 36 with secondary amines under Ir/48 catalysis. | ||
A combination of Ir/Cu co-catalyzed asymmetric ring-opening of N-substituted azabenzonorbornadienes 36 with amines was reported by Wang, Fan and co-workers.46 Besides [Ir(cod)Cl]2 and (R)-difluorphos 49 (Fig. 3), CuBr (20 mol%) was the best Lewis acid for the desymmetrization affording diamines 37 in very good yields and enantioselectivities (Scheme 18). In this case, for N-Boc derivatives primary and secondary aromatic amines, as well as Bn2NH, worked efficiently. When electro-withdrawing groups at the nitrogen atom of azabenzonorbornadiene were used such as Ts, Ns or CO2Me, instead of a Boc group, the yield decreased, though the enantioselectivity could be maintained. In the proposed mechanism, the catalytic cycle is initiated by coordination of [Ir(cod)Cl]2 with the chiral ligand 49 forming catalyst I. Subsequent coordination with 36, Cu+ and aniline gives intermediate II, which after intramolecular addition reaction generates intermediate III. Final β-elimination reaction provides the copper complex IV, which by cation exchange forms the product 37.
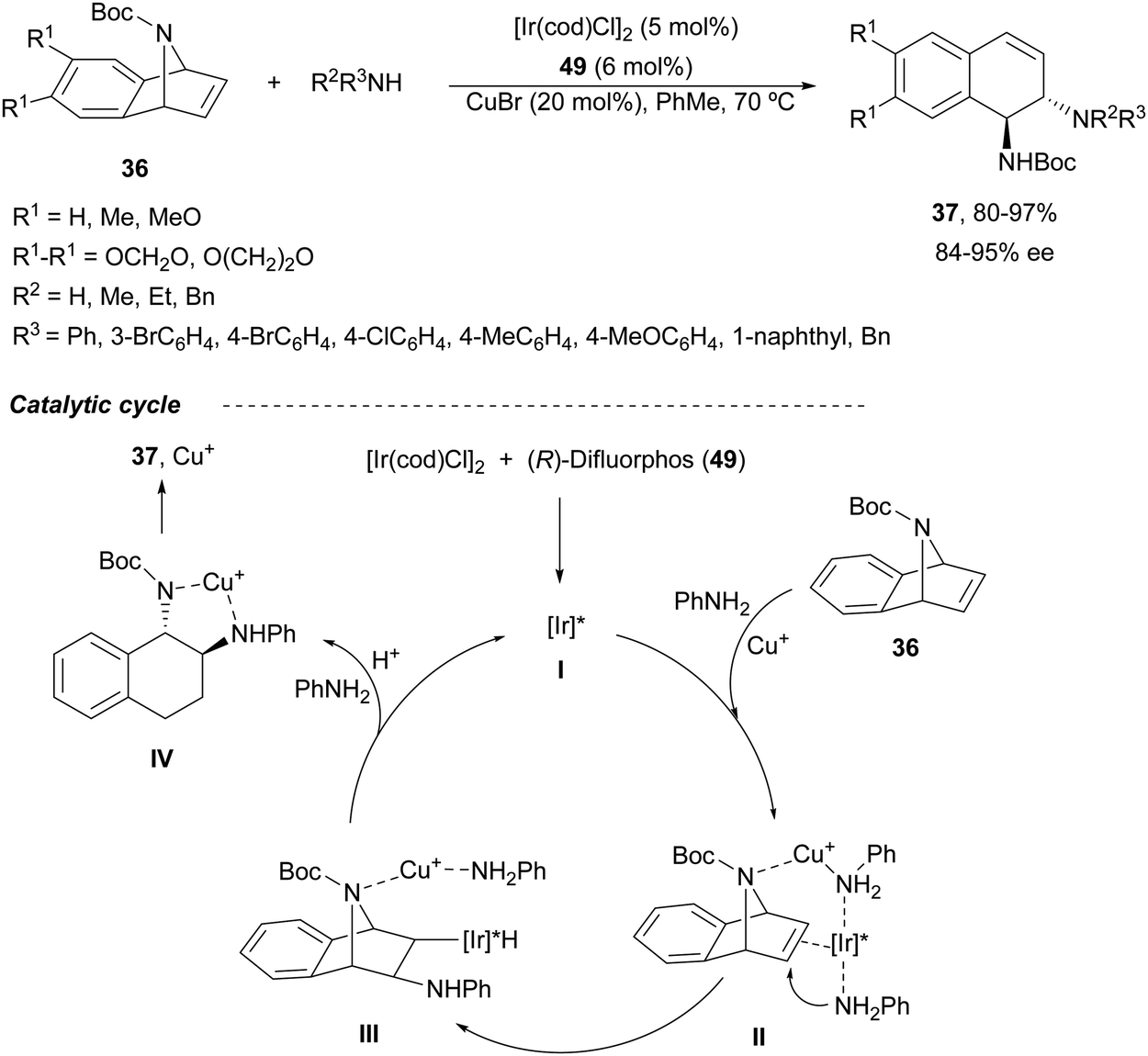 | ||
| Scheme 18 Ring-opening of N-Boc-azabenzonorbornadienes 36 with amines under Ir/49 and CuBr catalysis. | ||
The same group47 employed for the first time Pd(OAc)2, AgBF4 and (R)-Binap (46) (Fig. 3) as catalysts for the ring-opening of N-Boc-azabenzonorbornadienes 36 with primary and secondary aromatic amines (Scheme 19). Resulting diamines 37 were obtained up to 97% yield and up to >99% ee working in toluene at 90 °C.
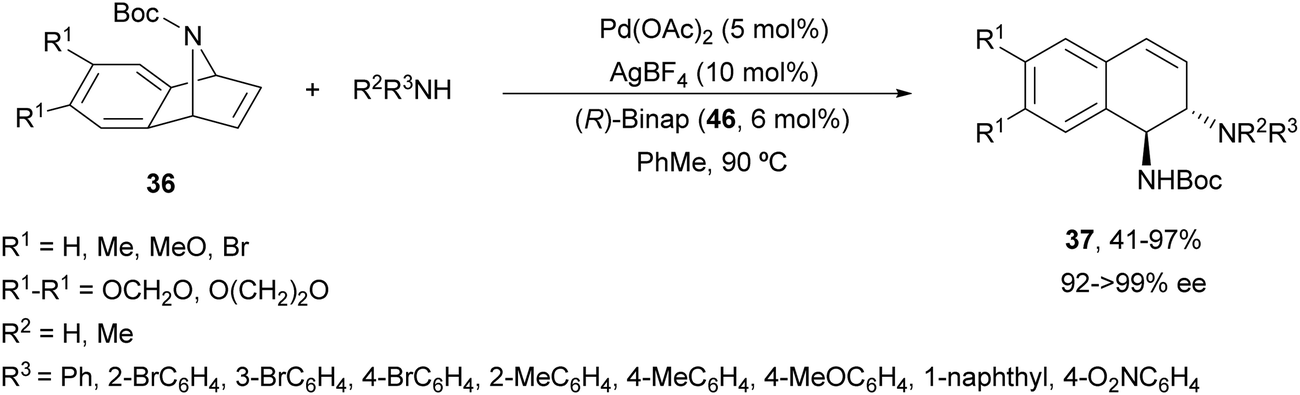 | ||
| Scheme 19 Ring-opening of N-Boc-azabenzonorbornadienes 36 with aromatic amines under Pd/(R)-Binap (46) catalysis. | ||
Under these reaction conditions, N-Boc-azabenzonorbornadienes 36 were reacted with different amides such as sulfonamides, carbamates, carboxamides and phosphoramides to obtain syn-1,2-diamine derivatives 50 (Scheme 20).48 This is the first example of syn-stereoselective ring-opening of compounds 36. Products 50 were obtained in good to high yields (up to 96%) and enantioselectivities (up to 98% ee). In the proposed reaction pathway, the catalytic cycle commenced with the chiral Pd complex I. Subsequent coordination of the catalyst I with benzenesulfonamide forms complex II, which coordinates the silver cation and the azabenzonorbornadiene 36 to provide intermediate III. After nucleophilic addition, intermediate IV is formed, which evolves to give the ring-opening complex V through a β-elimination reaction. Finally, product 50 is formed regenerating the Pd catalyst.
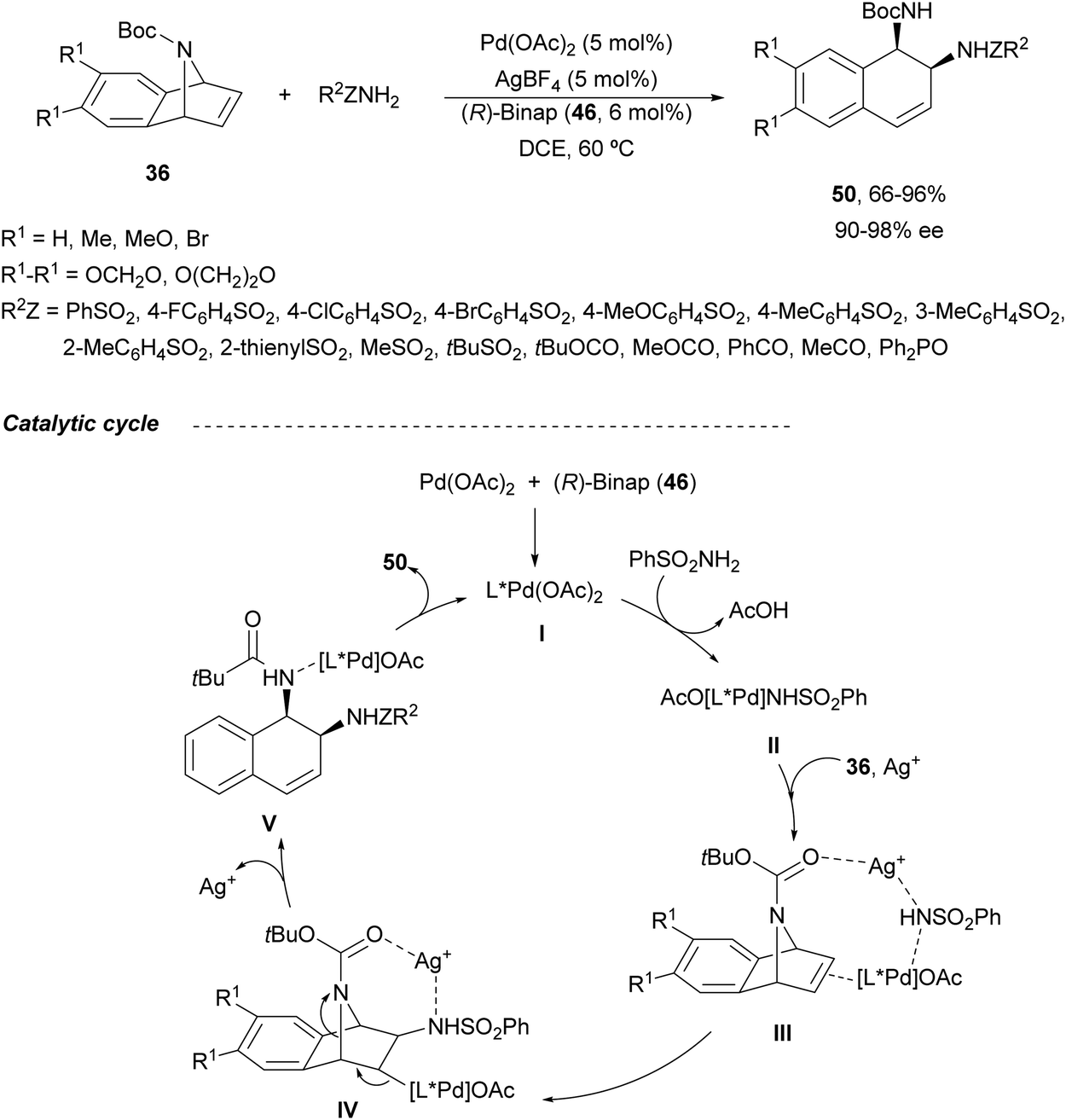 | ||
| Scheme 20 Ring-opening of N-Boc-azabenzonorbornadienes 36 with amides under Pd/(R)-Binap (46) catalysis. | ||
From the results included in this section, it can be concluded that for the asymmetric ring-opening of N-Boc-azabenzonorbornadienes Rh/ferriphos is the best catalyst for both secondary aliphatic and aromatic amines. Besides, Pd/Binap (46) is the simplest catalyst for primary and secondary aromatic amines. anti-Selectivity was observed in all cases except in the case of using amides as nucleophiles, which gave syn-diamine derivatives. The resulting enantioenriched 1,2-diaminotetralines are valuable intermediates for the synthesis of biologically active compounds35 and chiral ligands.37
2.3. Amination of allylic amines
Direct hydroamination of allylamines is an attractive route to 1,2-diamines with 100% atom economy. Intermolecular enantioselective hydroamination (HA) of allylic amines was developed by Beauchemin and co-workers49 with chiral aldehydes as tethering catalysts. Based on previous studies with racemic aldehydes,50,51 they used chiral aldehydes 53 and 54 (20 mol%) for the HA of allylic amines 51 with hydroxylamines 52 (Scheme 21).49 This intermolecular Cope-type HA took place in benzene or hexafluorobenzene under an argon atmosphere and at room temperature for 1 to 3 days reaction time to provide unsymmetrical 1,2-diamine derivatives 55 in good yields and enantioselectivities. Aldehyde 53 derived from (R)-glyceraldehyde gave products (S)-55, whereas bicyclic aldehyde 54 provided access to the corresponding (R)-55 enantiomers in an enantiodivergent manner.52 The proposed mechanism is based on racemic reactions51 involving α-(benzyloxy)acetaldehyde. After condensation of the aldehyde with hydroxylamine 52 a nitrone I is formed, which suffers nucleophilic attack by the allylic amine 51 to furnish intermediate II. This mixed chiral aminal II undergoes, through a bicyclic TS, a Cope-type HA to provide intermediate III with efficient transfer of chirality. Subsequent aminal cleavage forms the iminium intermediate IV, which after condensation with a second molecule of hydroxylamine 52 releases the product 55 and the nitrone I.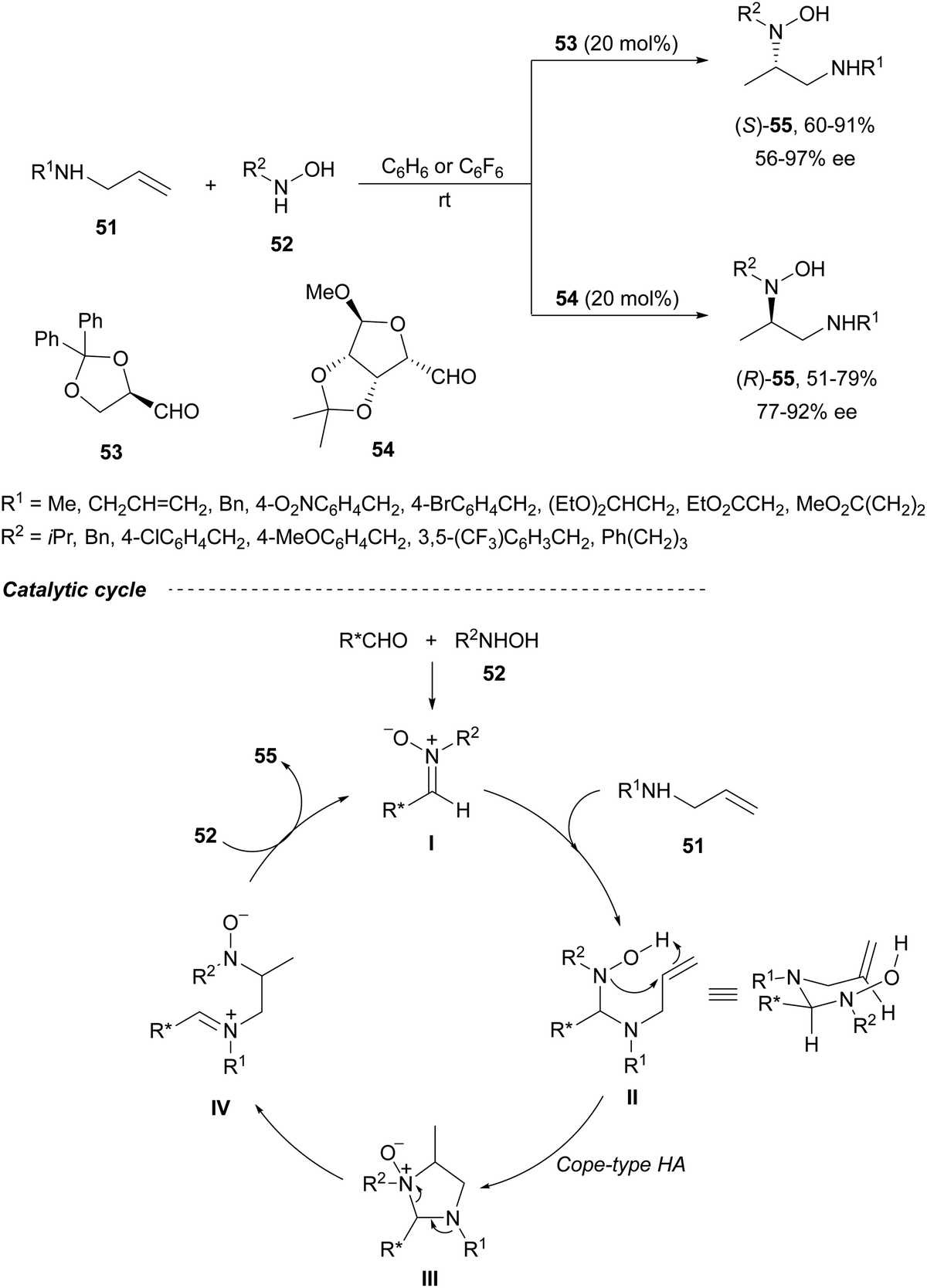 | ||
| Scheme 21 Enantiodivergent hydroamination of allylic amines 51 with chiral aldehydes 53 or 54 as organocatalysts. | ||
The first Rh-catalyzed asymmetric HA of allylic amines 51 was recently described by Hull and co-workers.53 They employed MeO-Biphep 56 as a chiral ligand and secondary aliphatic amines as nucleophiles resulting diamines 57 in moderate to good yields and up to 95% ee (Scheme 22). Diamine 57a was transformed into a methylated antidepressant methyl-moclobemide 58 in 47% overall yield by treatment with phenol and phosphoric acid followed by benzoylation with 4-chlorobenzoyl chloride. According to the previously proposed catalytic cycle for allylic imines,54 the cationic Rh(I) complex coordinates the N and C–C double bond to give intermediate I. This complex I undergoes nucleophilic attack by the amine to form intermediate II, which suffers direct protonation of the [Rh]–C bond or proton transfer to generate a [Rh]–H complex followed by reductive elimination giving intermediate III. Final ligand exchange with the allylic amine provides the product and regenerates the Rh complex I.
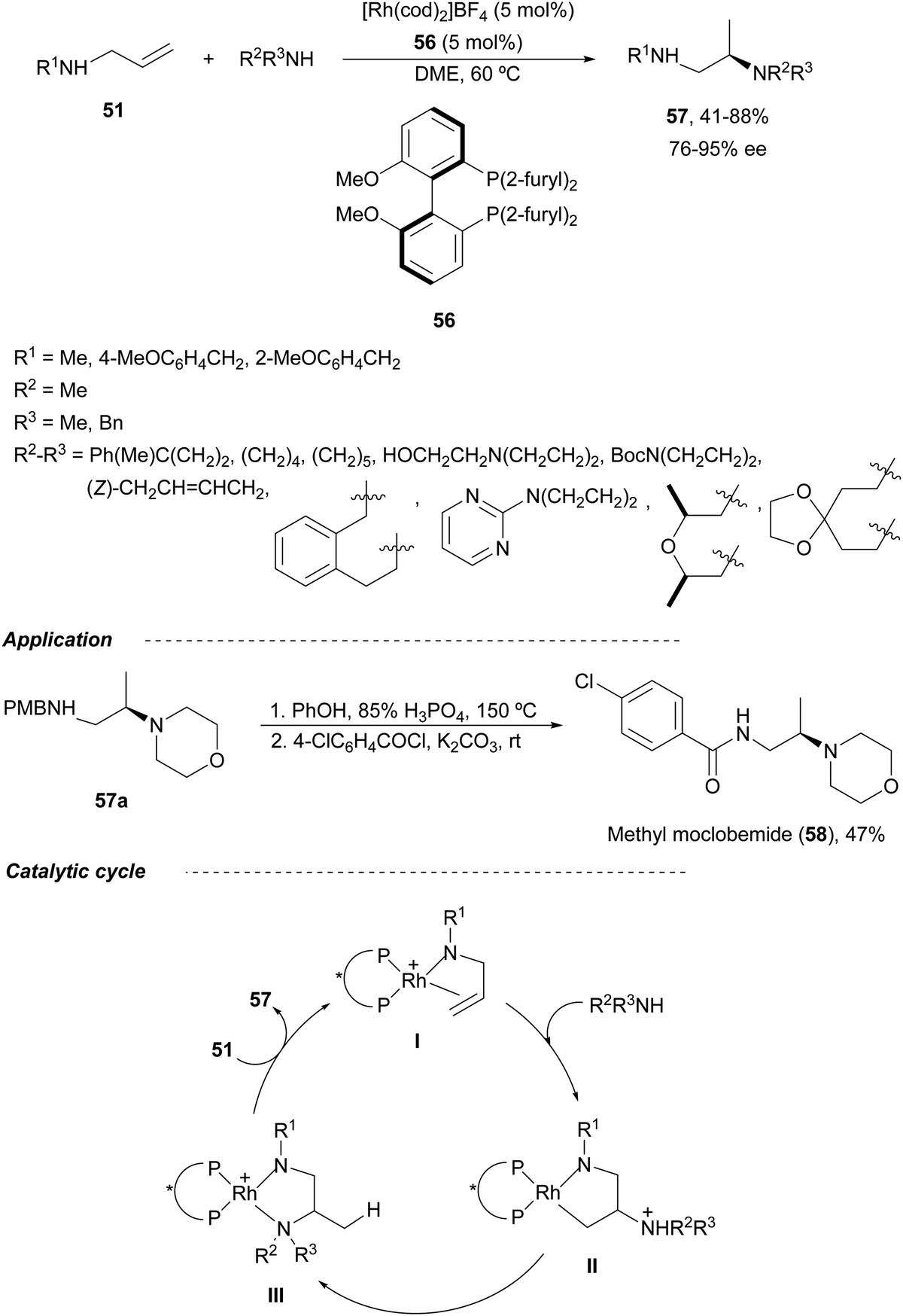 | ||
| Scheme 22 Asymmetric hydroamination of allylic amines 51 with secondary aliphatic amines under Rh/MeO-Biphen (56) catalysis. | ||
Copper-catalyzed asymmetric HA of γ-substituted N-pivaloyl allylic amines 59 has been described by Buchwald and co-workers.55 This type of HA requires as electrophilic amines O-benzoylated hydroxylamines 60 for the C–N bond forming process. The N-pivaloyl group in the allylic amine is essential facilitating the hydrocupration step. Differently protected 1,2-diamine derivatives 61 were obtained using the in situ generated catalyst by mixing Cu(OAc)2, (R)-DTBM-Segphos 23 (Fig. 2) and triphenylphosphine, a mixture known as CuCatMix*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 56 (Scheme 23). In general, this process took place with good regioselectivity (3:1–>20:1) and enantioselectivity (94–98%) and could be performed on a gram-scale. In the proposed mechanism for the CuH-catalyzed HA,57 firstly the allylic amine undergoes hydrocupration to form a chiral alkylcopper intermediate II, which is trapped by hydroxylamine benzoate 60 to provide the product and copper(I) benzoate III. For the generation of the catalyst I, a σ-bond metathesis with the hydrosilane (MeO)2MeSiH occurs. In the undesired β-elimination of intermediate II, the protected group at nitrogen of the allylic amine is crucial, which also controls the regioselectivity of the hydrocupration step.
56 (Scheme 23). In general, this process took place with good regioselectivity (3:1–>20:1) and enantioselectivity (94–98%) and could be performed on a gram-scale. In the proposed mechanism for the CuH-catalyzed HA,57 firstly the allylic amine undergoes hydrocupration to form a chiral alkylcopper intermediate II, which is trapped by hydroxylamine benzoate 60 to provide the product and copper(I) benzoate III. For the generation of the catalyst I, a σ-bond metathesis with the hydrosilane (MeO)2MeSiH occurs. In the undesired β-elimination of intermediate II, the protected group at nitrogen of the allylic amine is crucial, which also controls the regioselectivity of the hydrocupration step.
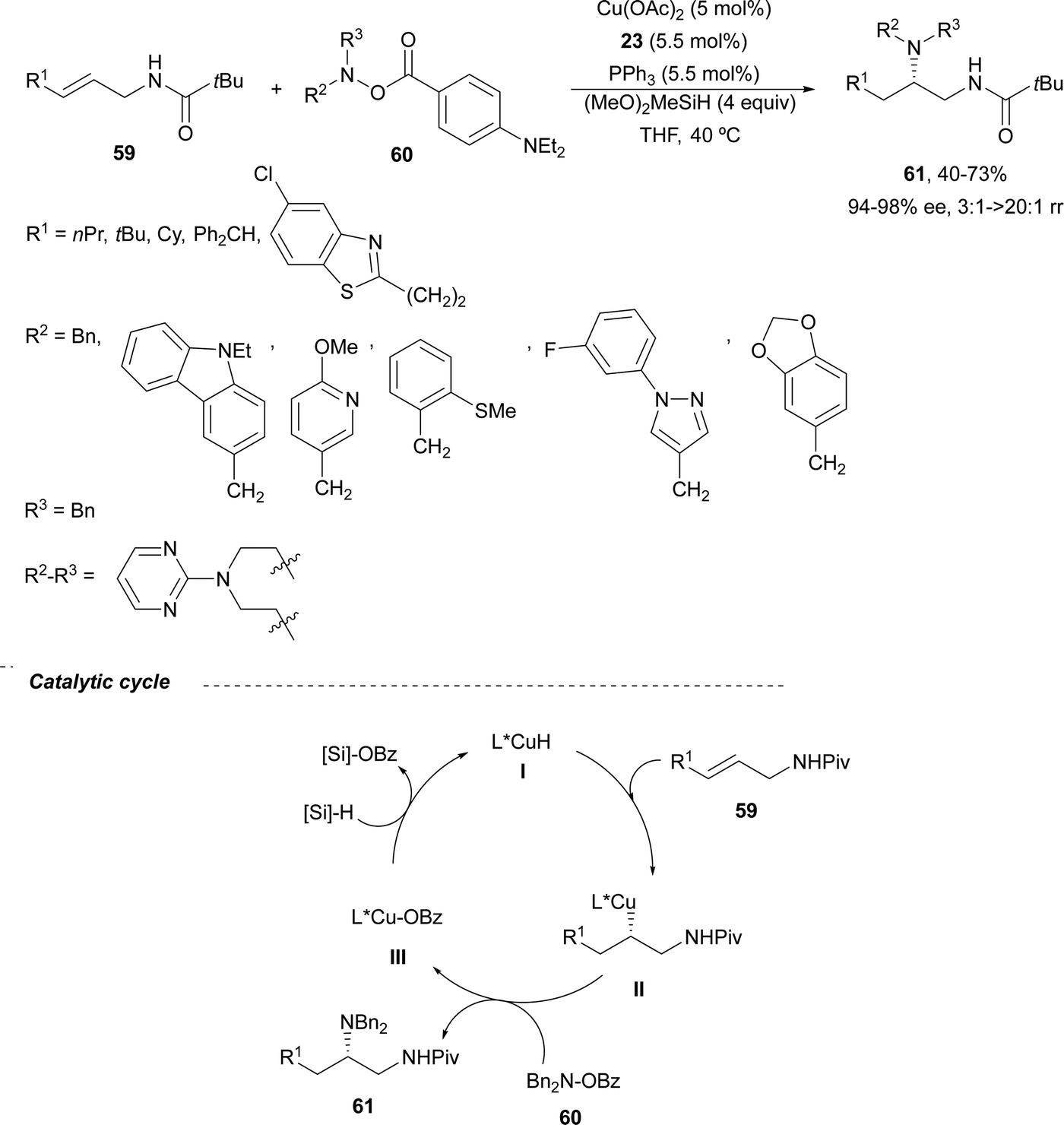 | ||
| Scheme 23 Asymmetric hydroamination of allylic pivalamides 59 with O-benzoylated hydroxylamines 60 by asymmetric hydrocupration. | ||
Intramolecular asymmetric carboamination of allylic ureas58 and sulfamides59 has been extensively studied by Wolfe and co-workers. These cyclizations were carried out under Pd-catalyzed conditions based on carboheterofunctionalization processes.60,61 Eventually, the resulting saturated heterocyclic compounds can be transformed into chiral 1,2-diamines.
Imidazolidin-2-ones 6458 are obtained under Pd-catalysis by carboamination of N-allylureas 62, which can be prepared by a reaction of allylic amines with isocyanates.62 In the presence of Pd2(dba)3 and a chiral phosphoramidite (S)-Siphos-PE 63 as a catalyst, the intramolecular hydroamination followed by C–C bond formation with aryl or alkenyl halides took place (Scheme 24). The resulting 4-aryl or alkenyl methylimidazolidin-2-ones 64 were obtained in good yields and enantioselectivities. According to the experimental results, a plausible catalytic cycle is depicted in Scheme 24. Firstly, oxidative addition to Pd(0) of the aryl/alkenyl halide generates intermediate I, which by reaction with 62 is converted by aminopalladation into Pd-amino complex II. Then, syn-insertion of the alkene into the Pd–N bond yields cyclic intermediate III, which after reductive elimination forms product 64 and regenerates the catalyst. This last process is the enantiodetermining step of the catalytic cycle.
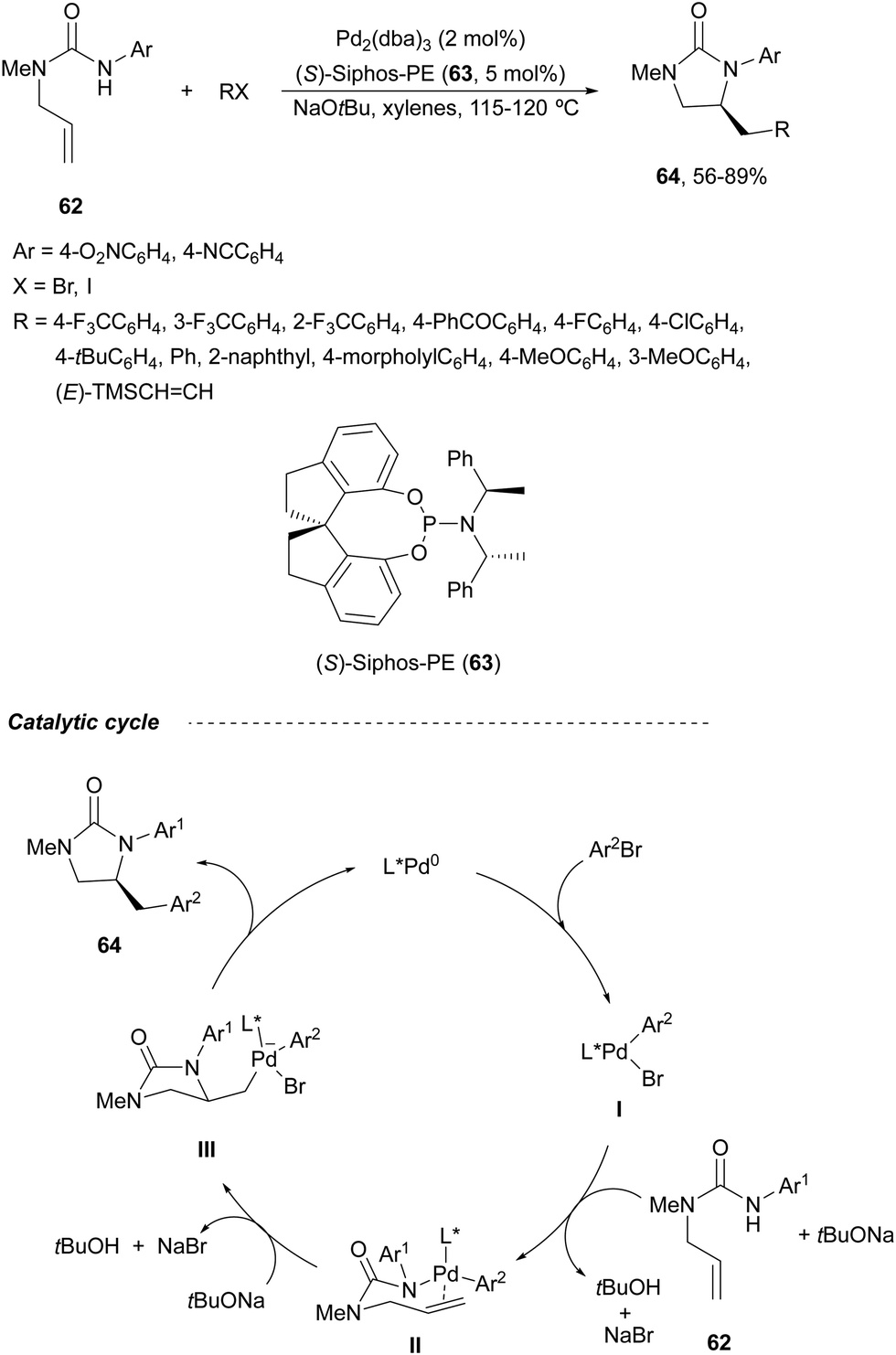 | ||
| Scheme 24 Asymmetric carboamination of allylic ureas 62 with aryl/alkenyl halides under Pd/(S)-Siphos-PE (63) catalysis. | ||
Cyclic sulfamides 6659 have been prepared by asymmetric intramolecular carboamination of N-allylsulfamides 65 using similar reaction conditions to those previously described for allylureas.58 Again, the addition of 2 equivalents of water improved yields and enantioselectivities giving products 66 in up to 96% yield and up to 90% ee (Scheme 25a). Deprotection of the tBu group with TFA at room temperature of 66a (R = Ph) gave 67 in 98% yield and the same 86% ee. In a second step, 67 was treated with HBr in phenol to obtain diamine 68 in 85% yield and 88% ee (Scheme 25b).
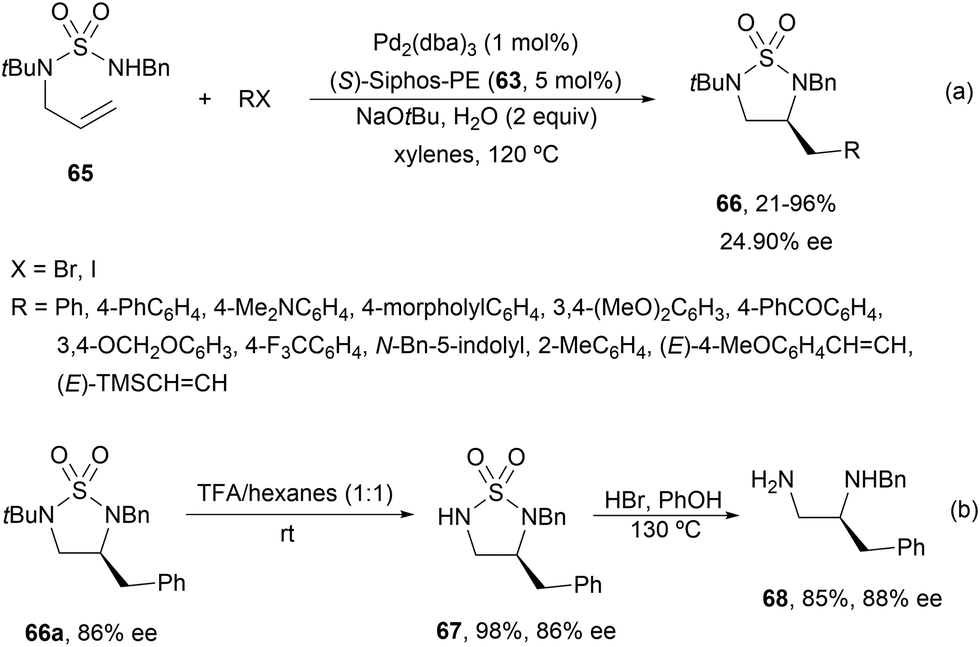 | ||
| Scheme 25 Asymmetric carboamination of N-allylsulfamides 65 with alkyl/alkenyl halides under Pd/(S)-Siphos-PE (63) catalysis. | ||
Buchwald and co-workers63 reported a regioselective intramolecular asymmetric HA of allylic hydroxylamine esters 69 using the same CuCatMix*56 as it was described for the intermolecular HA (Scheme 23). In this case, enantioenriched aziridines 70 were formed in good to excellent yields (Scheme 26a). The aziridine 70a underwent regioselective ring-opening with trimethylsilylazide and acetic acid to provide compound 71 in 92% yield, 98% ee and 9.5:1 rr (Scheme 26b).
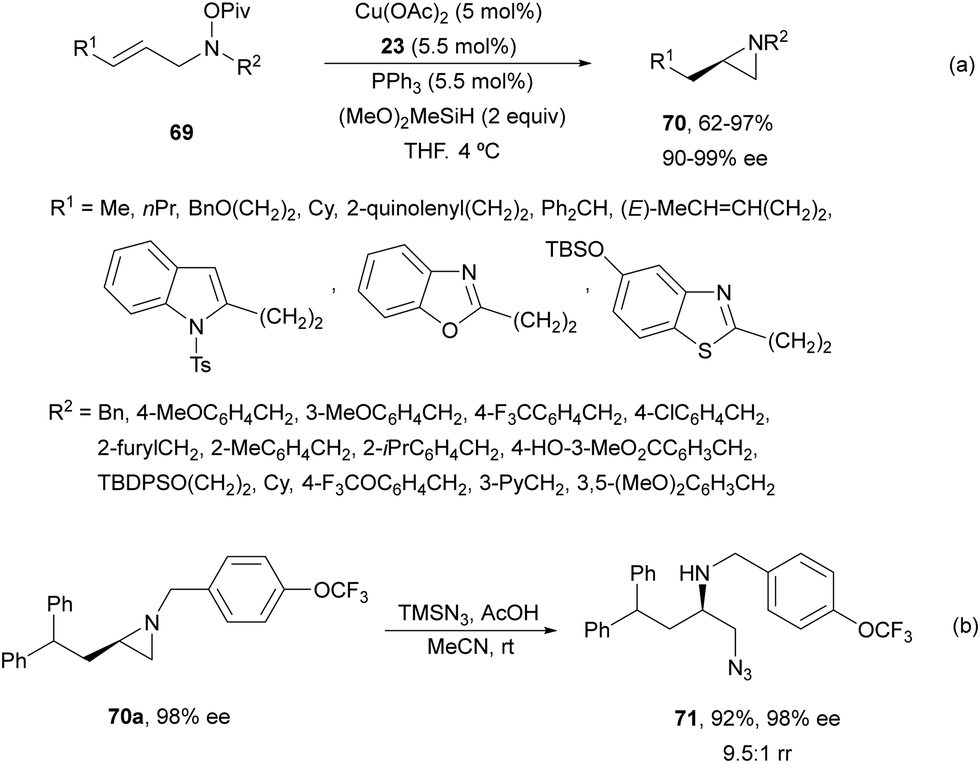 | ||
| Scheme 26 Intramolecular hydroamination of allylic hydroxylamine esters 69 by asymmetric hydrocupration. | ||
The asymmetric intermolecular HA of allylic amines with nucleophilic nitrogenated compounds gives 1,2-diamines either under aldehyde organocatalyzed conditions or under Rh or Cu catalysis. This methodology allows the preparation of propylene diamines, which increases in potency for some drugs called ‘magic methyl effects’.64 Hydrocupration of γ-substituted N-protected allylic amines has to be performed with electrophilic O-benzoylated hydroxylamines. An intramolecular HA of allylic hydroxylamine esters under Cu-catalysis gave enantioenriched aziridines. Besides, asymmetric intramolecular HA of allylic derivatives has been achieved under Pd-catalysis with concomitant coupling with aryl or alkenyl halides to obtain imidazolidinones or cyclic sulfamides precursors of 1,2-diamines.
2.4. Amination of enamines
In this section, hydroamination of enamines by asymmetric hydrocupration, amination of enecarbamates or enamides under metal catalysis, and chiral phosphoric acid catalyzed amination of enecarbamates as well as photoredox processes will be discussed.Based on the hydroamination reaction of allylic amines by asymmetric hydrocupration55 (see, Scheme 23), Somfai and coworkers65,66 reported the asymmetric formal HA of enamines 72 with O-acyl hydroxylamines 60 (Scheme 27). In the presence of Cu(OAc)2/(R)-DTBM-Segphos (23) and with (MeO)2MeSiH as the stoichiometric hydride source, 1,2-diamines 73 were obtained with good yields and regio and enantioselectivities. A one-pot procedure involving the formation of enamine followed by the HA step has been also performed.66 Starting form aldehydes and amines in the presence of anhydrous sodium sulfate (10 equivalents), the corresponding enamines are prepared quantitatively in dichloromethane at room temperature in only one hour. This HA method was applied to the synthesis of orthogonally protected dipiperazine 73a, a precursor of the human melanocortin-4 (MC-4) receptor antagonist 74.67 The MC-4 receptor has been involved in the regulation of feeding, metabolism, sexual functions and emotional states such as anxiety and depression. Based on the previous proposed mechanism for the hydrocupration of alkenes, a catalytic cycle depicted in Scheme 27 was proposed. Hydrocupration of enamine 72 (R1 = Ph; R2 = R3 = Bn) with the in situ generated CuH (I) forms alkylcopper intermediate II in an irreversible and enantiodetermining step. Oxidation of species II with hydroxylamine 60 generates the product 73 and releases the copper benzoate III. Final transmetallation of III with (MeO)2MeSiH regenerates the catalyst I. DFT calculations66 supported that the insertion of the L*CuH catalyst into the enamine is the regio- and enantiodetermining step. In addition, TS(R) explains the stereocontrol of the process and the N–O bond in this TS also determines the enantioselectivity. The energy difference between TS(R) and TS(S) was calculated to be 3.0 kcal mol−1 in accordance with the experimental enantioselectivities.
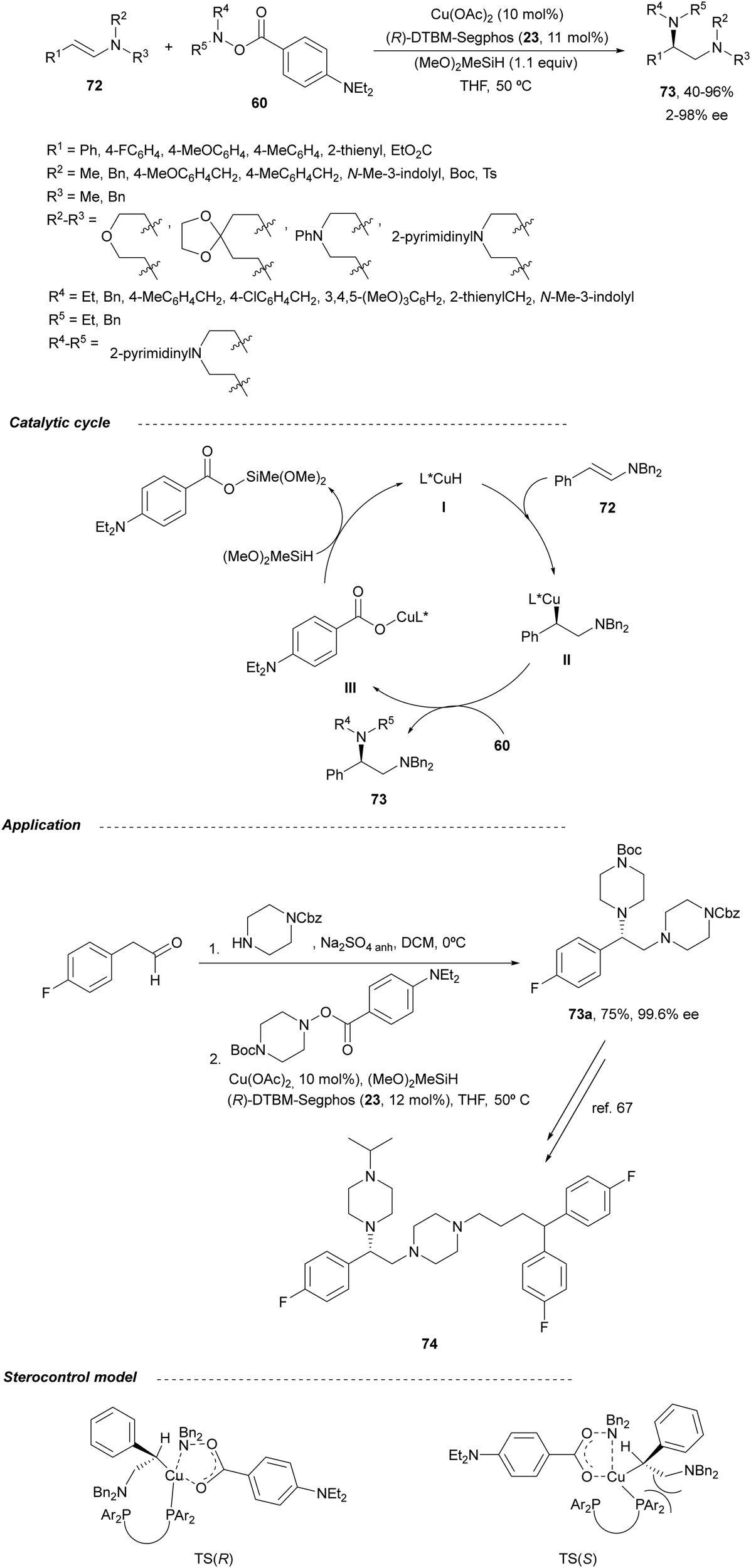 | ||
| Scheme 27 Hydroamination of enamines 72 with O-acylhydroxylamines 60 by asymmetric hydrocupration. | ||
The first catalytic asymmetric amination of enecarbamates 72 with azodicarboxylate 75 using a diamine 76-Cu complex was described by Matsubara and Kobayashi.68 The resulting acylimines 77 were converted into diamine derivatives syn-78 after stereoselective NaBH4 reduction with >95:5 dr (Scheme 28a). syn-1,2-Diamine 79 was obtained by Cbz-deprotection of 78a followed by N–N cleavage with RANEY® Ni and benzoylation in 78% overall yield (Scheme 28b). Both Z and E-enecarbamates gave products with the same configuration. The authors proposed an acyclic concerted TS model in which the Re-face of the azodicarboxylate is shielded by the neighboring benzyl group of the diamine ligand and an enecarbamate attack from the Si-face predominantly. An antiperiplanar TS minimizes steric repulsion between the enecarbamate and the copper catalyst.
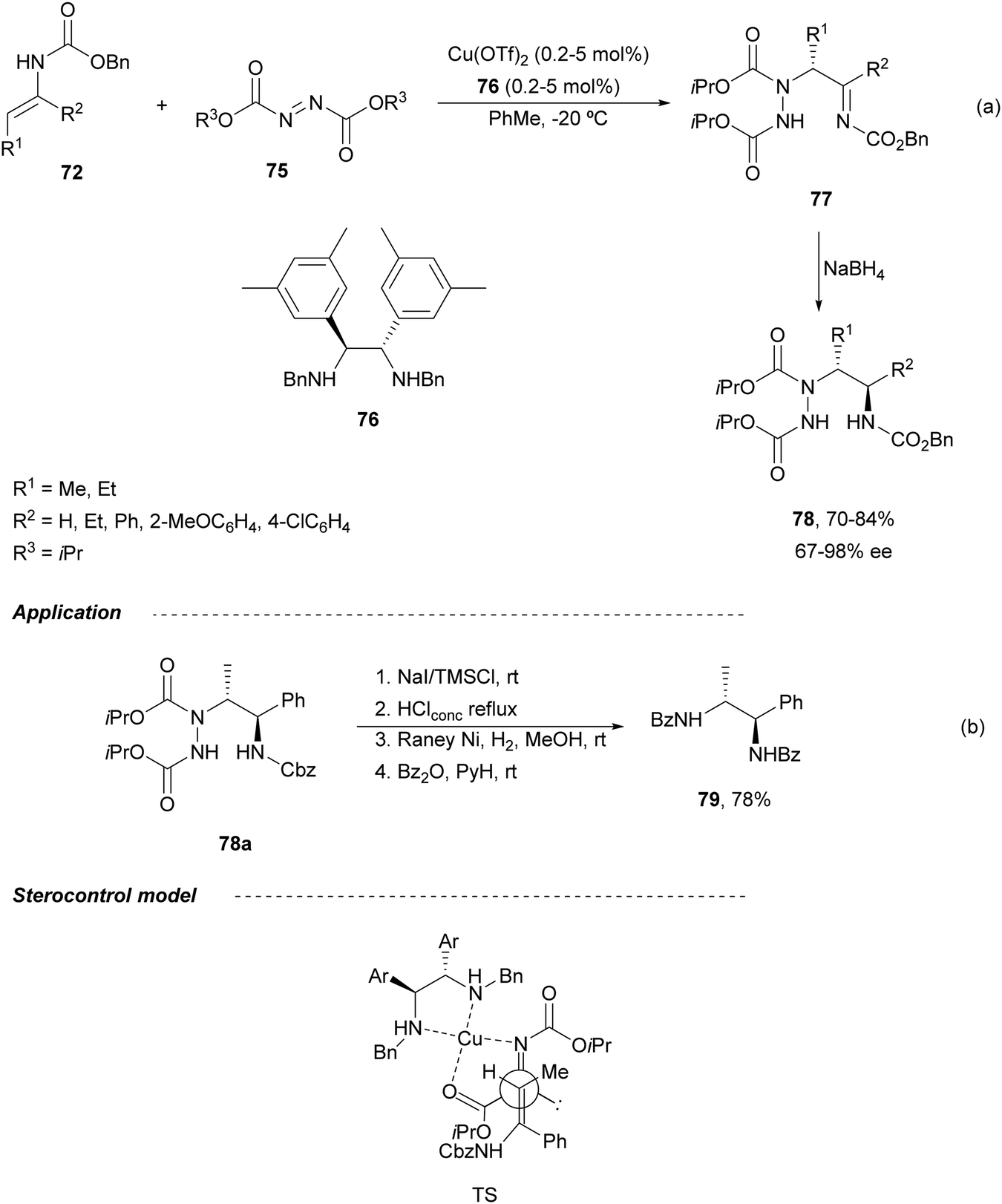 | ||
| Scheme 28 Asymmetric amination of enecarbamates 72 with diisopropyl azodicarboxylate (75) under Cu(OTf)2/diamine 76 catalysis. | ||
Feng and co-workers69 developed later the same amination using (Z)-N-acetyl enamides 80 and dibenzyl azodicarboxylate 75 (R3 = Bn) under Cu(OTf)2 and N,N′-dioxide 19 (Fig. 2) catalysis (Scheme 29). Products 81 were reduced with NaBH4 at −78 to −45 °C to the corresponding precursors of syn-1,2-diamines 82 in good yields and diastereoselectivities (>95:5) and without any loss of enantioselectivity. (E)-Enamides exhibited higher reactivity than the Z counterpart and were converted into the corresponding adducts with the opposite configuration although with lower enantioselectivities.
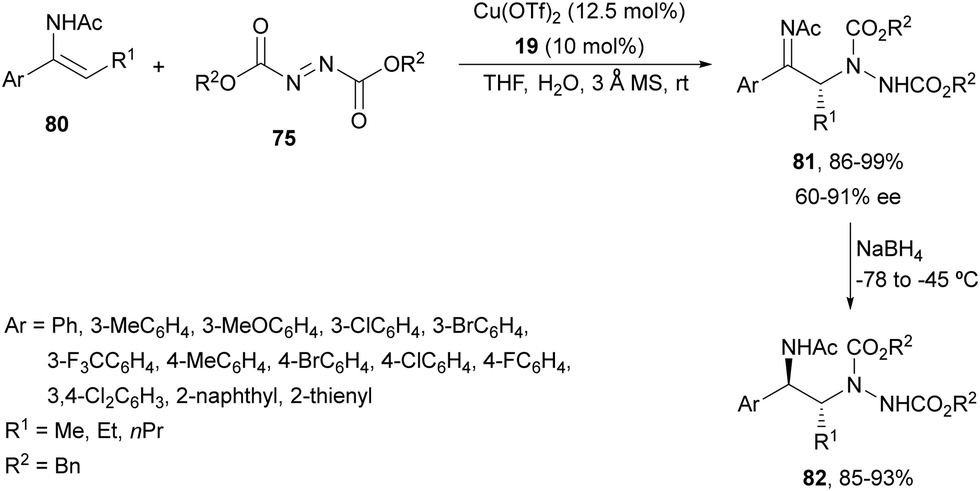 | ||
| Scheme 29 Asymmetric amination of enamides 80 with dibenzyl azodicarboxylate (75) under Cu(OTf)2/N,N′-dioxide 19 catalysis. | ||
Calcium-bis(phosphate) complex 83 was employed as a catalyst by Masson, Zhu and co-workers70,71 for the enantioselective electrophilic amination of (E)-enamides 80 with diisopropyl azodicarboxylate 75 (Scheme 30). This amination took place in dichloromethane at −35 °C followed by NaBH4/MeOH reduction to provide syn-1,2-diamines 82 (R2 = iPr) in >95.5 dr and up to 95% ee. Experimental studies support a catalytic cycle according to nonlinear effects (NLEs) and kinetic measurements as well as DFT calculations.71 After monocoordination of the monomeric complex with azodicarboxylate to form intermediate I, the enamide forms a hydrogen bond providing intermediate II, with the complex acting as a bifunctional catalyst. A Si-face attack of the enamide onto azodicarboxylate gives the zwitterionic species III, which undergoes a proton transfer to generate intermediate IV. Final dissociation and complexation with azodicarboxylate provide α-aminoimine 81 and regenerate intermediate I.
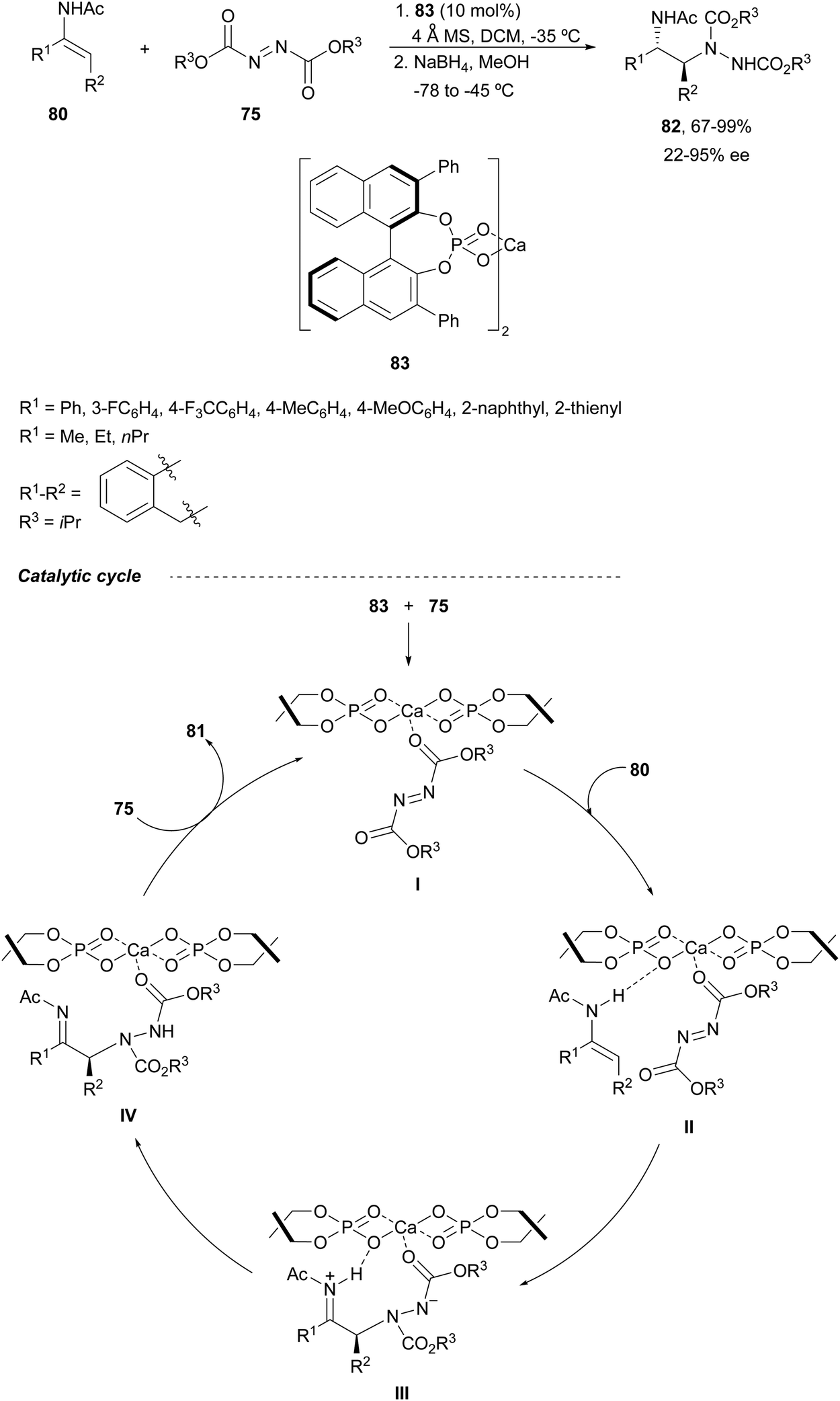 | ||
| Scheme 30 Asymmetric amination of (E)-N-acetyl enamides 80 with diisopropyl azodicarboxylate (75) under calcium-bis(phosphate) complex 83 as a catalyst. | ||
Masson and co-workers have reported in a recent report72 the difunctionalization of enamide derivatives for synthesizing α,β-substituted amines. Concerning the synthesis of enantioenriched 1,2-diamines, in 201573 they performed the amination of (E)-enecarbamates 72 with dibenzyl azodicarboxylate 75 in the presence of EtOH using a catalytic amount of the CPA 84 followed by reduction with triethylsilane (TESH) and trimethylsilyl triflate in a mixture of acetonitrile and methanol at 0 °C (Scheme 31). Under these reaction conditions, intermediates 85 were formed, precursors of α-hydrazinoimines 86, which after reduction afforded diamine derivatives 87 in good yields and excellent enantioselectivities. In the proposed TS for the amination reaction, the CPA may act as a bifunctional catalyst to activate both the enecarbamate and the azodicarboxylate by hydrogen bonding. Accordingly, from this TS, the (R)-adduct 86 will be formed through an intramolecular Si face attack of 72 on 75.
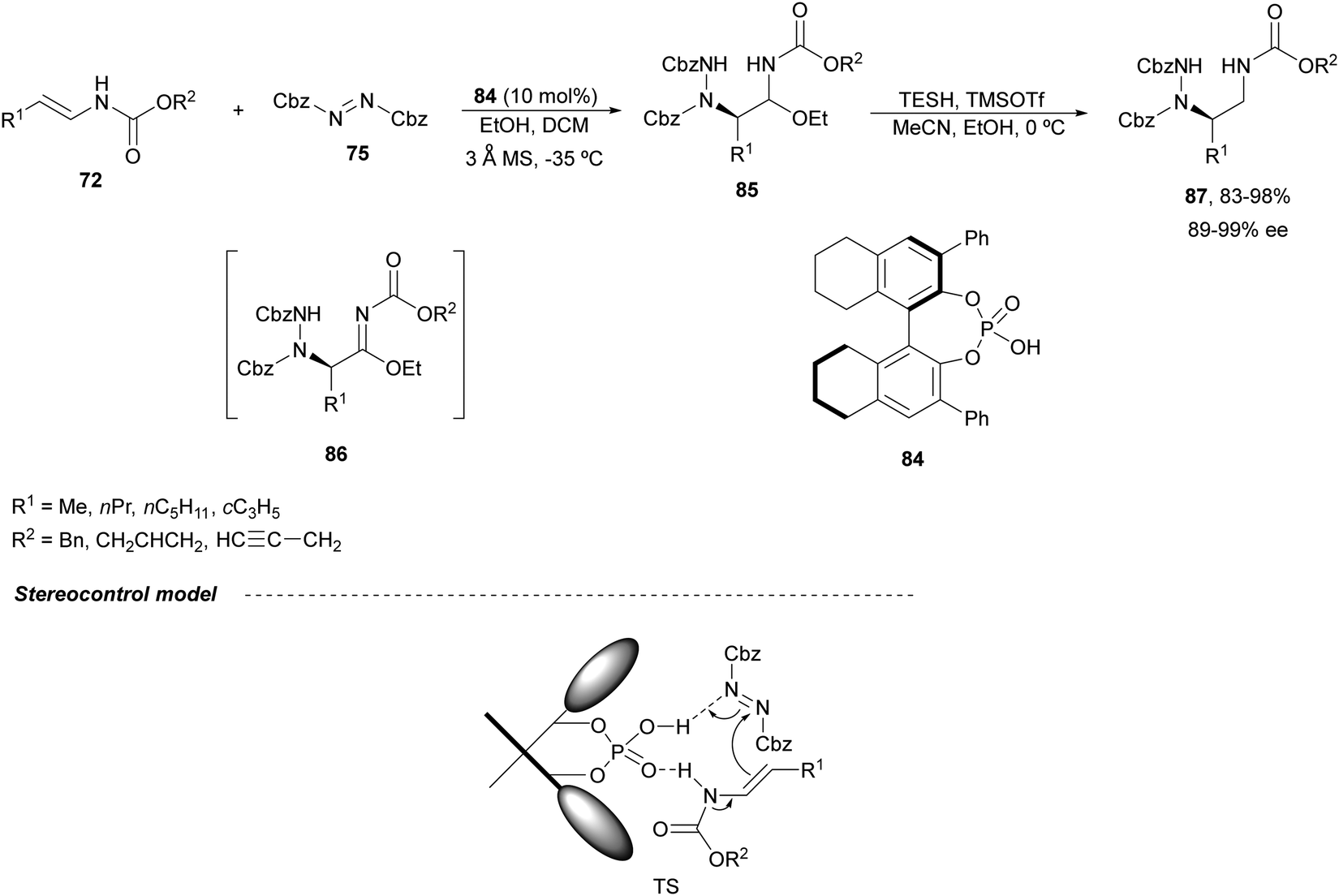 | ||
| Scheme 31 Asymmetric amination of (E)-enecarbamates 72 with dibenzyl azodicarboxylate 75 under CPA 84 catalysis. | ||
Electrophilic amination of enecarbamates using CPA 84 as a catalyst followed by nucleophilic addition to intermediates 86 allowed the synthesis of diamine derivatives.74 When silylated nucleophiles were employed, products 88 were obtained with cyano, azido or allyl groups with 2:1 diastereoselectivity and good enantioselectivity by a two-step sequential procedure (Scheme 32). In this case, BF3·Et2O was used as Lewis acid to promote the formation of compounds 86.
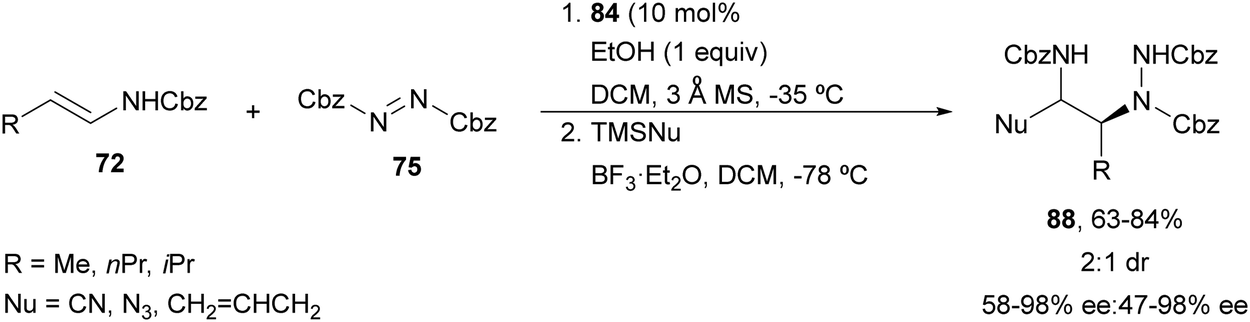 | ||
| Scheme 32 Asymmetric amination of enecarbamates 72 with 75 under CPA 84 catalysis followed by functionalization with silylated nucleophiles. | ||
The former strategy was expanded to the synthesis of N-carbamoylthioethers 89, which were subjected to a N-iodosuccinimide-assisted Friedel–Crafts reaction with 1,3,5-trimethoxybenzene resulting in compounds 90 with >95:5 diastereoselectivity and high enantioselectivity (Scheme 33).74 However, furan gave product 91 in 72% yield and 2:1 dr with 98 and 93% ee. Hydrogenation of the hydrazine group provides diamines 92 in 95:5 dr for 1,3,5-trimethoxyphenyl derivatives and 93 in 2:1 dr for the furan derivative. When indoles were used in the Friedel–Crafts step double addition was observed leading to mainly bisindole 94 in 53–97% yields and 97–99% ee. On the other hand, other heterocyclic systems such as pyrazoles reacted through the nitrogen atom.
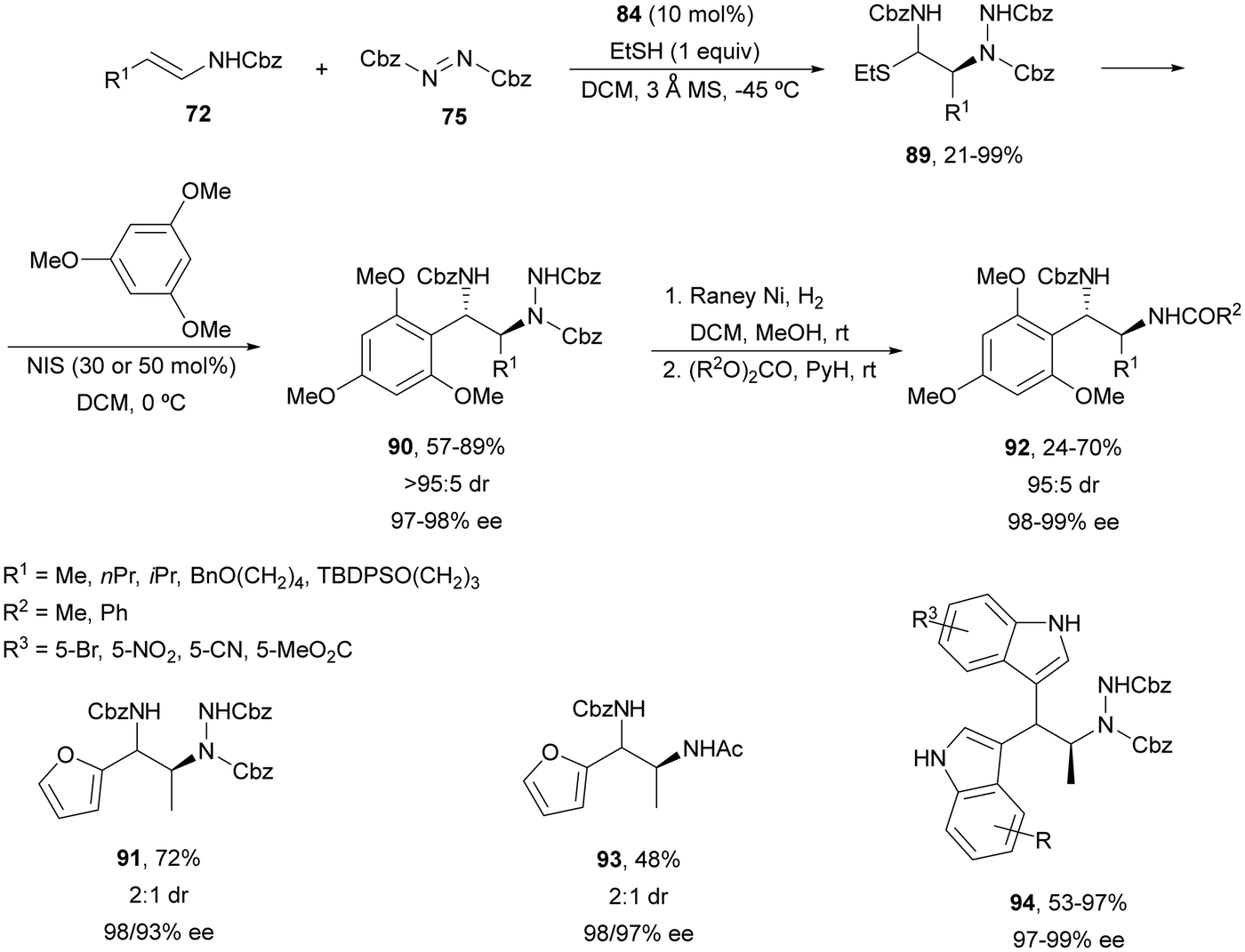 | ||
| Scheme 33 Asymmetric amination of (E)-enecarbamates 72 with 75 under CPA 84 catalysis followed by functionalization by the Friedel–Crafts reaction. | ||
In order to avoid the double addition of indoles to intermediates 89 to form products 94, the same group75 used visible light photoredox catalysis. β-Amino substituted tryptamines 95 were obtained with up to >99:1% ee (Scheme 34). In the catalytic cycles, the Ru(II) photocatalyst reacts with oxygen to form a Ru(III) complex. This species abstracts an electron from the α-amidosulfide 89 (single electron transfer, SET) leading to a radical cation I, which undergoes fragmentation regenerating iminium cation II. After the Friedel–Crafts reaction with indoles, the iminium cation II gives the corresponding tryptamines 95.
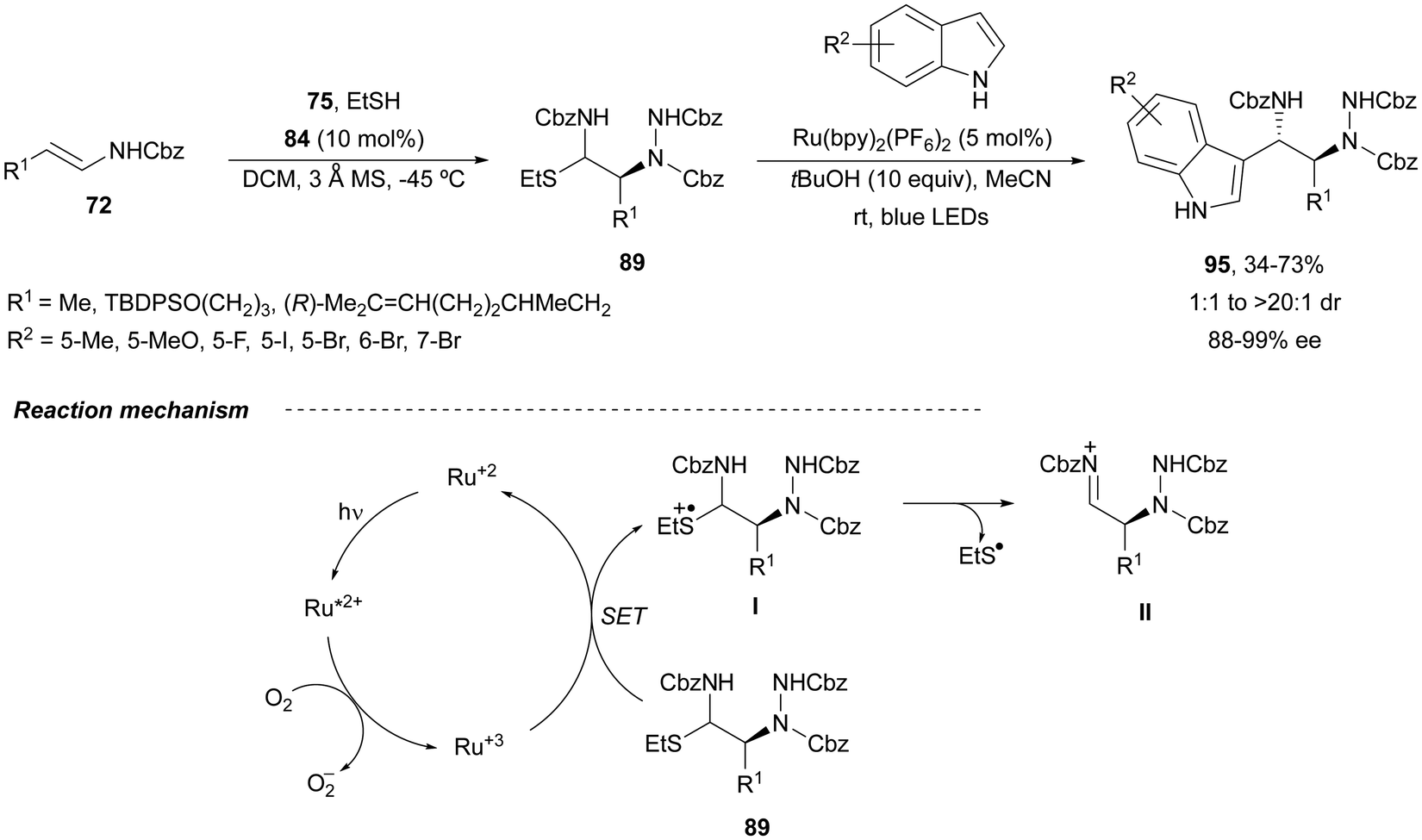 | ||
| Scheme 34 Asymmetric amination of (E)-enecarbamates 72 with dibenzyl azodicarboxylate 75 and EtSH under CPA 84 catalysis followed by Friedel–Crafts reaction with indoles under photoredox catalysis. | ||
Masson and co-workers76 designed a chiral photosensitive organocatalyst 96, which contains a CPA backbone and a visible-light-sensitive thioxanthone unit for the amination of enamides 72 with dibenzyl azodicarboxylate 75 (Scheme 35). This process was carried out in the presence of pyrazoles or 7-bromoindole as nucleophiles to give compounds 97 or 95, respectively, in good yields, excellent enantioselectivity and moderate diastereoselectivity. In the proposed mechanism, intermediate α-carbamoylsulfide 89 would be oxidized by the excited chiral thioxanthone 96* generating the ketal radical anion I and the sulfur radical cation II. A single electron transfer from I to O2 would generate the thioxanthone catalyst 96 and produce the superoxide radical anion O2˙−III. Intermediate II would undergo C–S cleavage to produce imine 86 and thiyl radical IV, which dimerizes to form diethyl disulfide. Finally, the imine 86 suffers addition of the azole and subsequent hydrogen atom abstraction from O2˙− affording products 97 or 95.
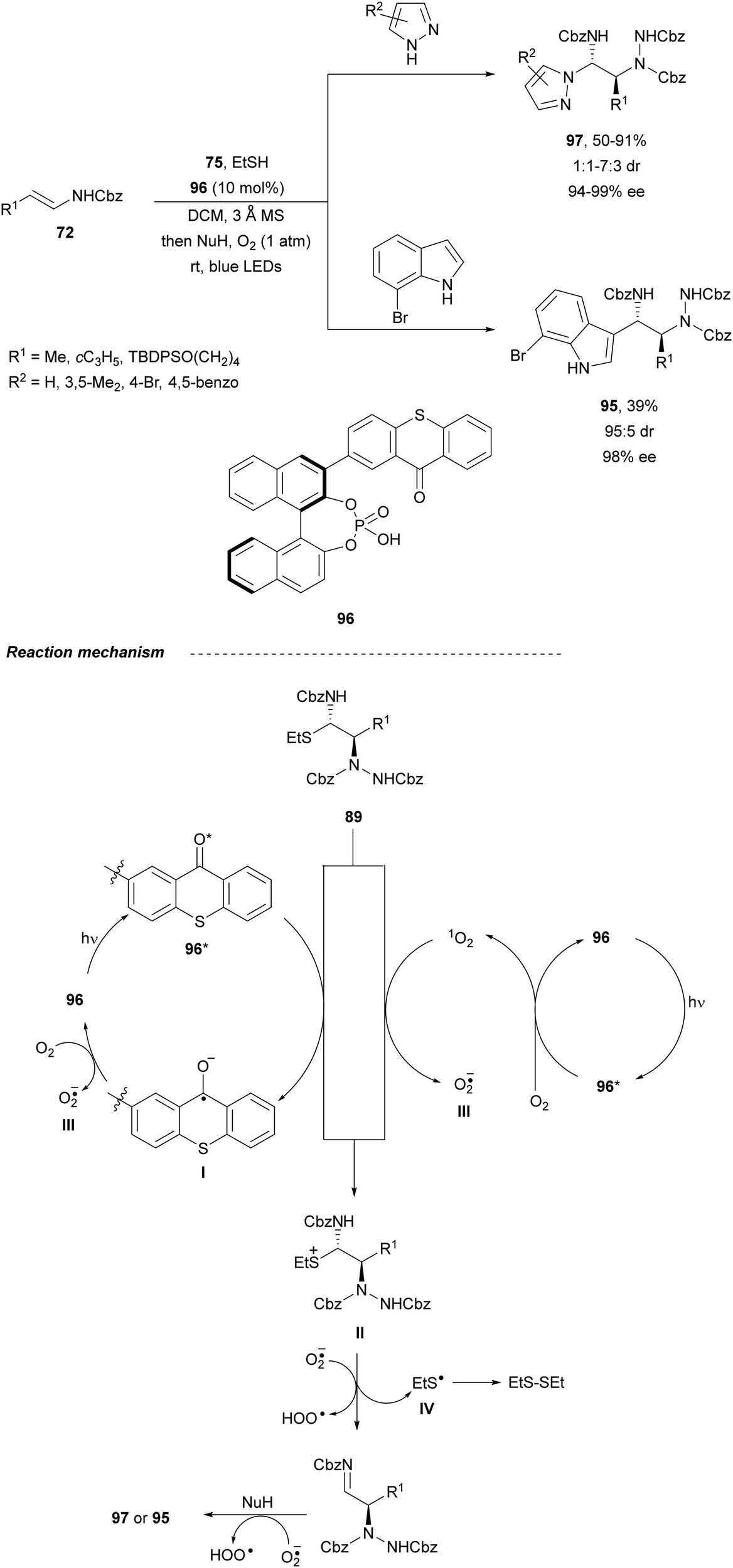 | ||
| Scheme 35 Asymmetric amination of (E)-enecarbamates 72 with dibenzyl azodicarboxylate 75 and EtSH under CPA-thioxanthone 96 photocatalysis and Friedel–Crafts reaction with pyrazoles and 7-bromoindole. | ||
Direct hydroamination of enamines has been performed only under asymmetric hydrocupration conditions using acyl hydroxylamines as nucleophiles. This method has been successfully applied to the synthesis of melanocortin-4 receptor antagonists. For the amination of acetyl enamides dialkyl azodicarboxylates have been used as nucleophiles under copper asymmetric catalysis or with calcium-bis(phosphate) complex as a catalyst. In the case of enecarbamates, this asymmetric amination could be carried out using chiral phosphoric acids as catalysts in the presence of a nucleophile followed by functionalization with silylated nucleophiles or by a Friedel–Crafts reaction under conventional conditions or visible light photoredox catalysis. This amination requires further hydrogenation of the hydrazine group to provide 1,2-diamines, which were obtained mainly as syn-diastereomers.
2.5. Diamination of alkenes
Catalytic asymmetric diamination of C–C double bonds represents one of the most important C–N bond formation strategies to access enantioenriched 1,2-diamines. This subject has been recently reviewed by two groups.13,14 In Scheme 36 are summarized the most efficient synthetic strategies based on (a) the two-electron redox pathway using Pd(0)/Pd(II) or Pd(II)/Pd(IV), I(I)/I(III) and Se(II)/Se(IV) catalytic diaminations, and (b) the one-electron radical mechanism under Cu or Fe catalysis (Scheme 36).14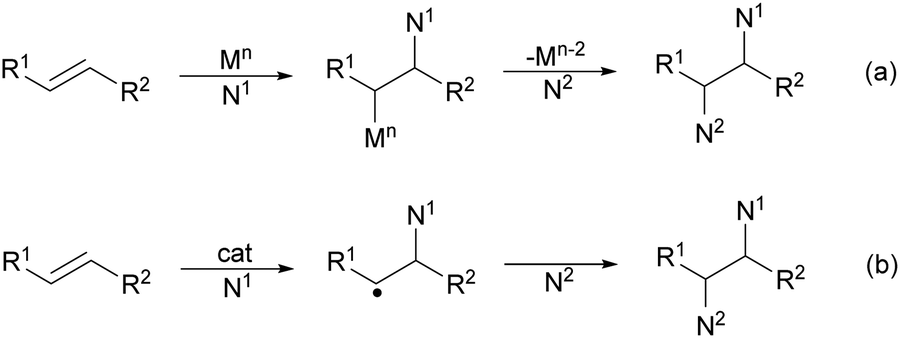 | ||
| Scheme 36 Catalytic mechanisms for CC diamination. | ||
Pd(0)/Pd(II) intermolecular diamination was developed by Shi and co-workers10 using di-tert-butyldiaziridinone or di-tert-butylthiadiaziridine-1,1-dioxide both as a nitrogen source and phosphoramidites 98 as chiral ligands or NHC-Pd(0) 99 as a chiral complex (Fig. 4) to provide trans-imidazolidinones 100 or 101, respectively (Scheme 37a). In 2018, Gong and co-workers77 employed simpler dialkylureas as diamination agents and a Pybox 102 (Fig. 4) as a chiral ligand to give imidazolidinones 103 (Scheme 37b). The regioselectivity occurs at the terminal CC bond of conjugate 1,2-dienes, whereas Shi's method took place at the internal CC bond. Allylpalladium intermediates I and II provided by an intramolecular allylic amination products 100 and 103, respectively.
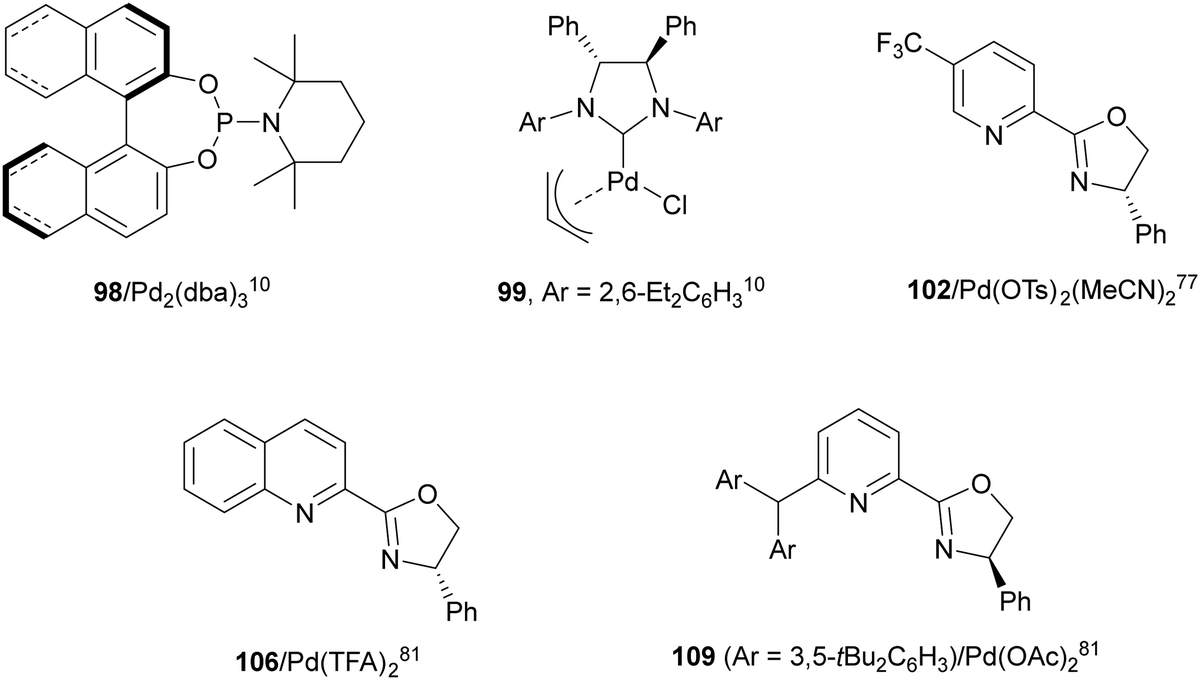 | ||
| Fig. 4 Chiral Pd catalysts for intermolecular and intramolecular asymmetric diaminations. | ||
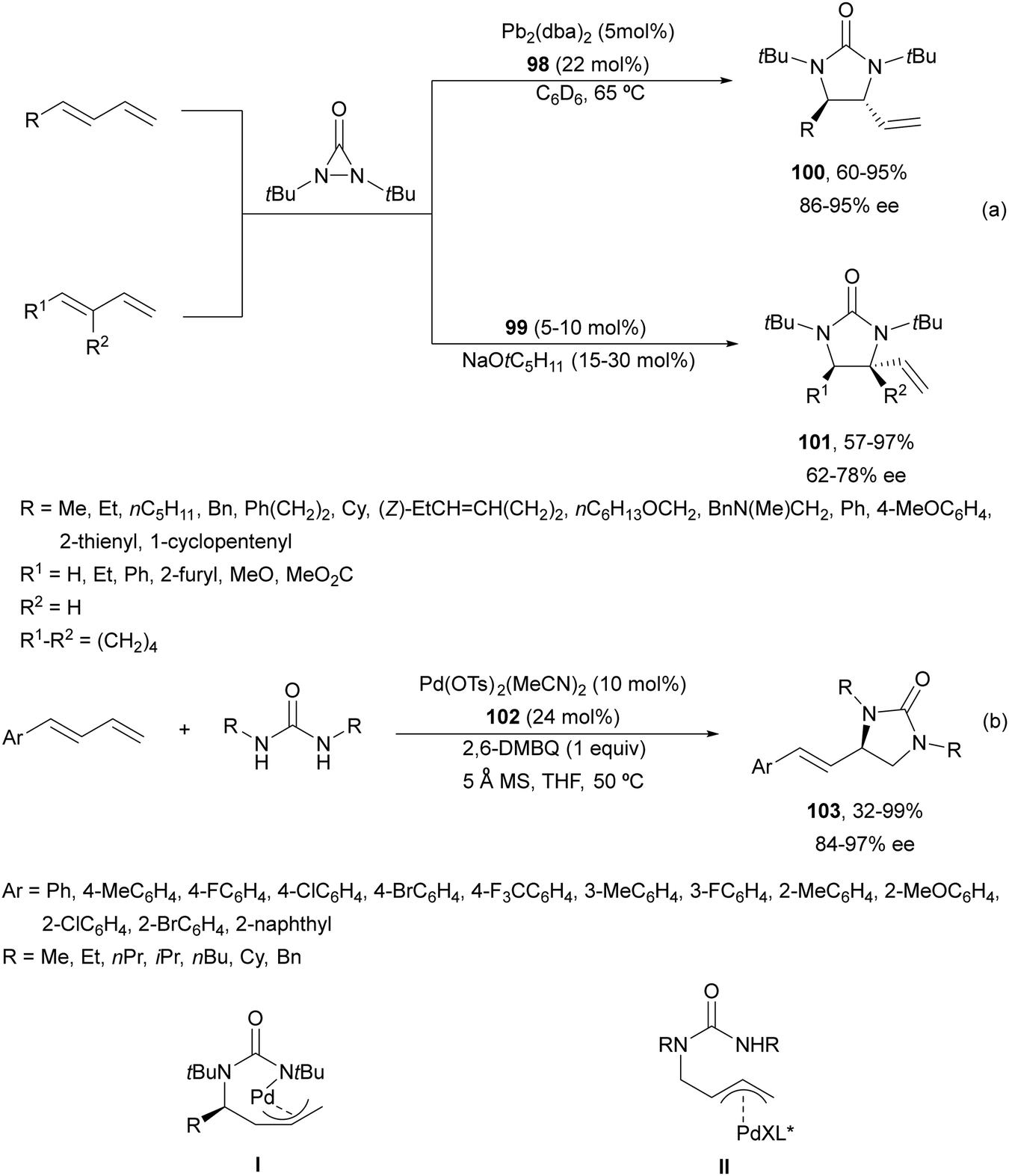 | ||
| Scheme 37 Asymmetric intermolecular diamination of 1,3-dienes under Pd(0)/Pd(II) catalysis. | ||
Recently, Lin and co-workers78 reported an asymmetric sequential diamination of 1,3-enynes using ureas as dinucleophiles (Scheme 38). This process was carried out using Pd(OAc)2 and (S)-Segphos as chiral ligands, and 3,5-bis(tert-butyl)benzoic acid (30 mol%) and Et3N to obtain imidazolidinones of type 104 in good yields and enantioselectivities. Mechanistic studies revealed the initial intermolecular hydroamination with a PdH complex to give an allene intermediate I, which by subsequent asymmetric intramolecular hydroamination furnished the imidazolidinone.
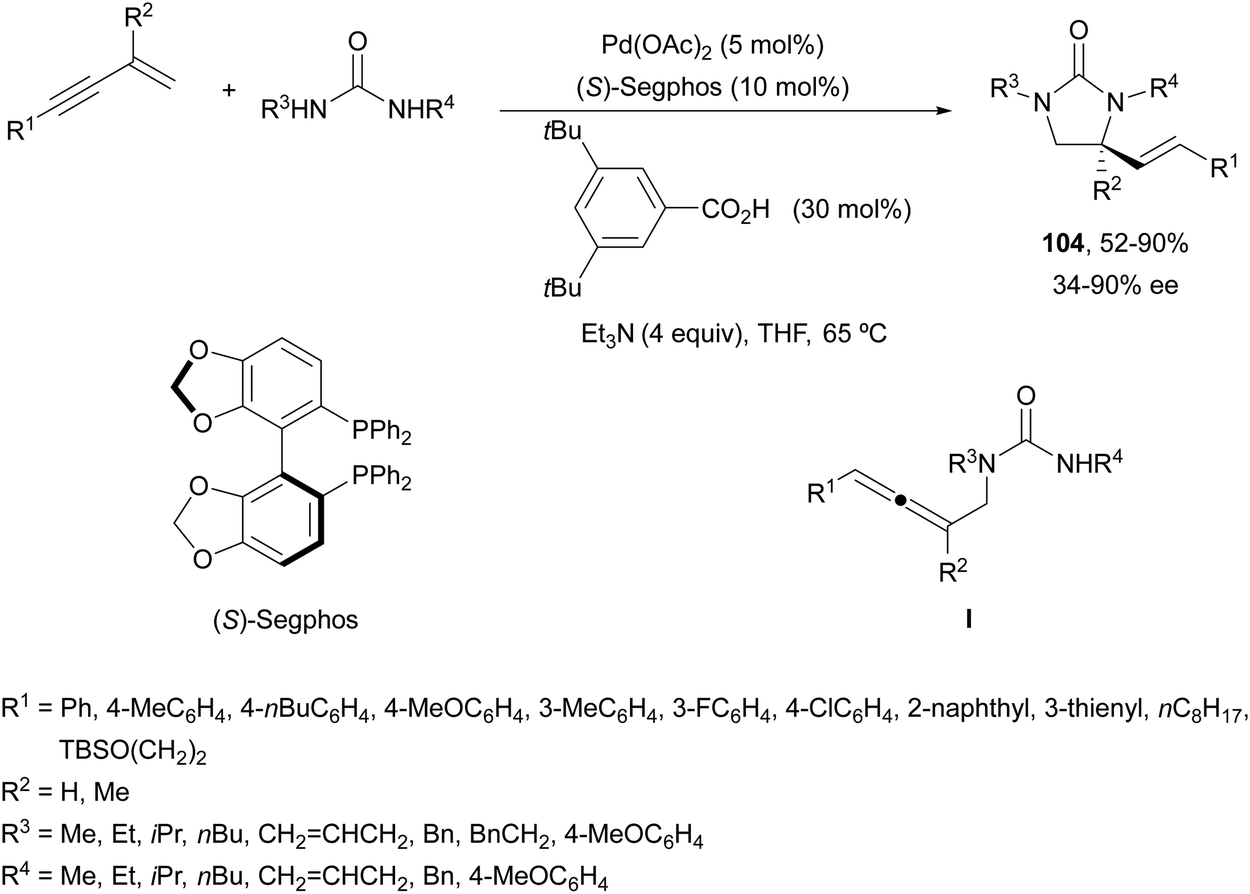 | ||
| Scheme 38 Asymmetric diamination of 1,3-enynes under Pd(0)/Pd(II) catalysis. | ||
Pd(II)/Pd(IV) asymmetric intramolecular diamination was carried out by Michael and co-workers79 on the basis of previous work of Muñiz and co-workers.80 Intramolecular diamination of a tethered double bond in compounds 105 used Ph-quinox 106 (Fig. 4) as a chiral ligand and N-fluorobenzenesulfonimide (NFSI) as a second nitrogen nucleophile and for the oxidation of Pd(II) to Pd(IV) to provide aminomethyl-substituted pyrrolidines 107 (Scheme 39a). Intermediate I is the Pd(II) species which after reductive elimination or displacement furnished products 107. When sulfonamide 108 was used as starting aminoalkene, it was possible to control the cyclization using ligand 106 or 109 (Fig. 4). Thus, Liu and co-workers81 modulated the formation of 6-endo-cyclic piperidine 110 using ligand 106, whereas ligand 109 favored the exo-cyclization to form pyrrolidine 111 (Scheme 39b).
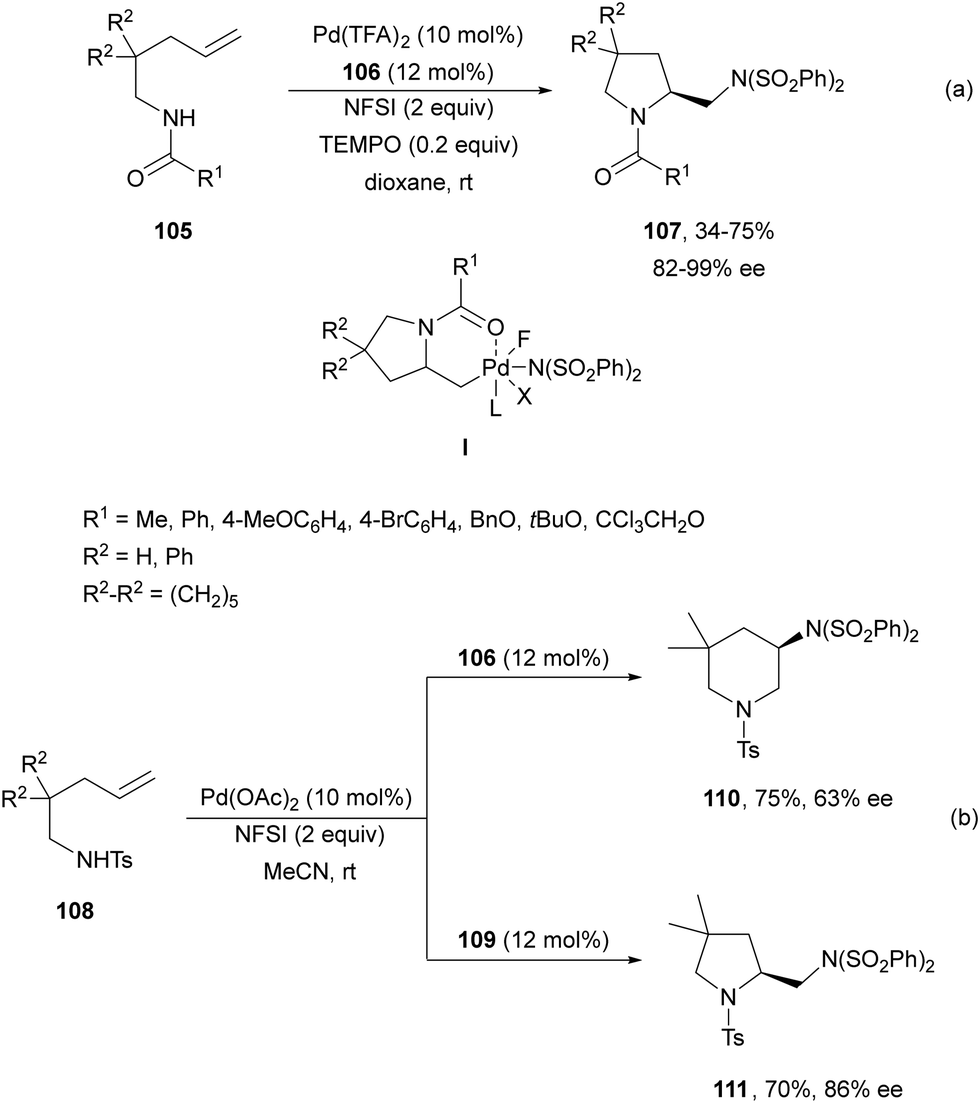 | ||
| Scheme 39 Asymmetric intramolecular diamination under Pd(II)/Pd(IV) catalysis. | ||
Initial studies of intermolecular diamination of styrenes with bismesylimide by Muñiz and co-workers82 employed a chiral λ3-iodane reagent in stoichiometric amounts. In 2014, Wirth and co-workers83 performed an intramolecular diamination using a sulfuryl diamide 112 as a difunctional nucleophile and a chiral λ3-iodane generated from the aryl iodide 113 (20 mol%) as a catalyst with sodium perborate as the oxidant (Scheme 40). In this example, the resulting bicyclic product 114 was obtained in 72% yield and 86% ee.
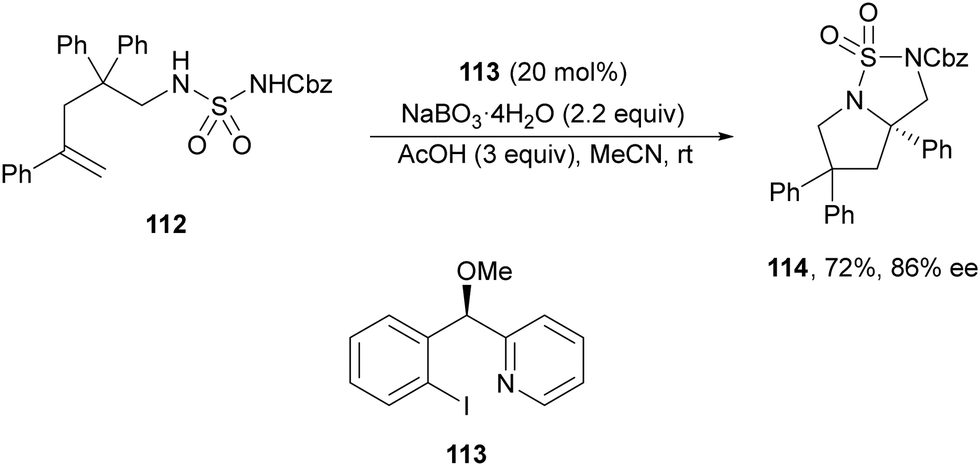 | ||
| Scheme 40 Asymmetric intramolecular diamination under λ3-iodane, from aryl iodide 113, catalysis. | ||
The first intermolecular enantioselective diamination of alkenes using a chiral λ3-iodane was reported by Muñiz and co-workers84 using iodide 115 as a chiral catalyst with MCPBA as an oxidant for the in situ generation of the I(III) compound (Scheme 41). The combination of tert-butyl methyl ether (TBME) and hexafluoroisopropanol (HFIP) as a solvent was crucial to avoid competitive epoxidation. Terminal styrenes gave compounds 116 in good yields and high ee. Lower yields were obtained with internal alkenes providing anti-products 117. Intermediate I would generate by reductive displacement of the intermediate II precursor of diamine derivative 117.
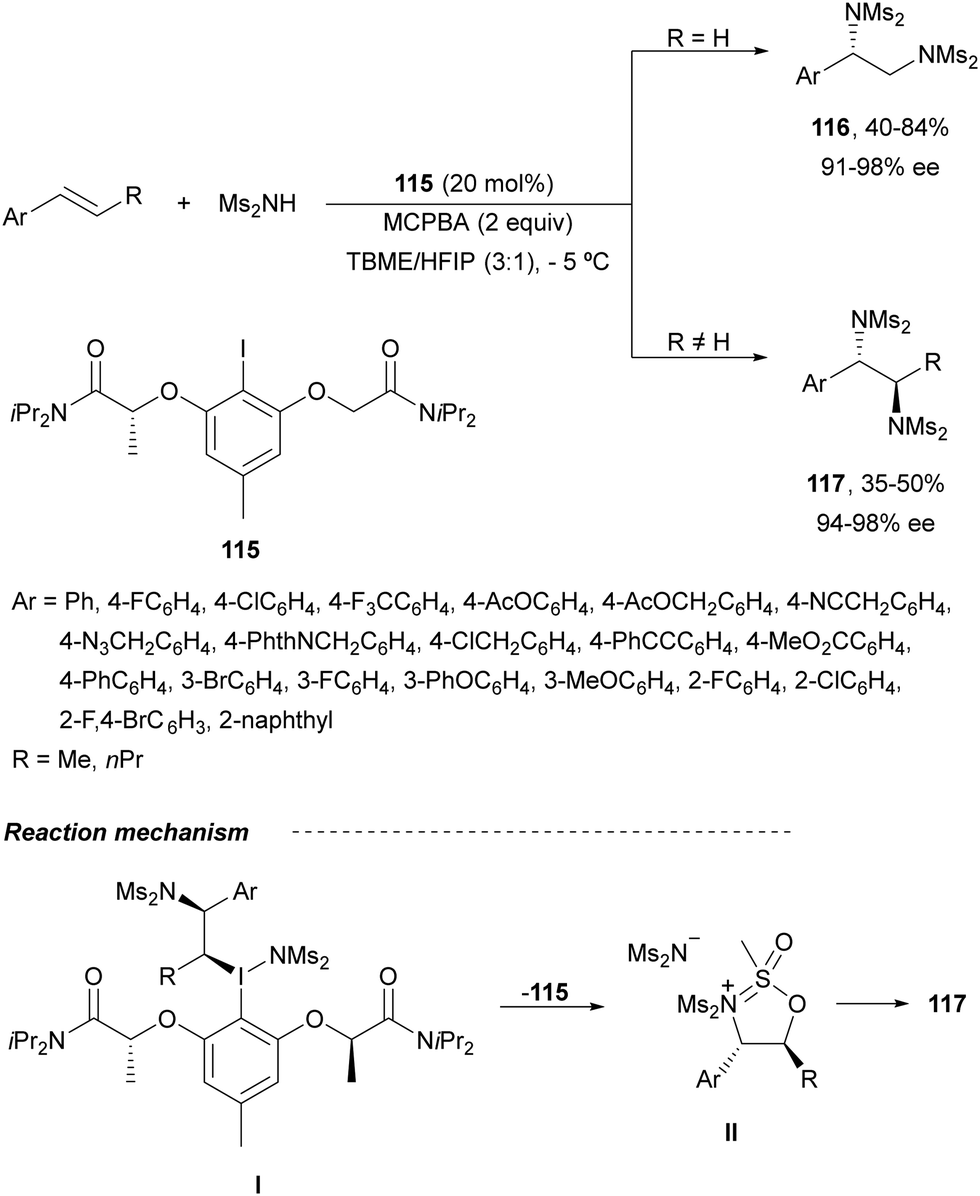 | ||
| Scheme 41 Asymmetric intermolecular anti-diamination under λ3-iodane, from aryl iodide 115, catalysis. | ||
The last methodology based on a two-electron redox pathway (Scheme 36a) is the Se(II)/Se(IV) intermolecular diamination of olefins described by Denmark and co-workers.85 In this case, syn-diamination was achieved with N,N′-bistosylurea using the chiral organoselenium reagent 118 as a catalyst and N-fluorocollidinium tetrafluoroborate 119 as the stoichiometric oxidant (Scheme 42). Diaryl, aryl–alkyl and alkyl–alkyl olefins with a trans-configuration were diaminated to the corresponding trans-imidazolidinones 120 in moderate to good yields and enantioselectivities. This process takes place by formation of a seleniranium ion II by the reaction of the alkene with the species I generated by oxidation of the diaryl diselenide 118. Ring opening of intermediate II by urea provides intermediate III, which after oxidation to Se(IV) forms intermediate IV. Subsequent cyclization gives the final product 120 and regenerates species I.
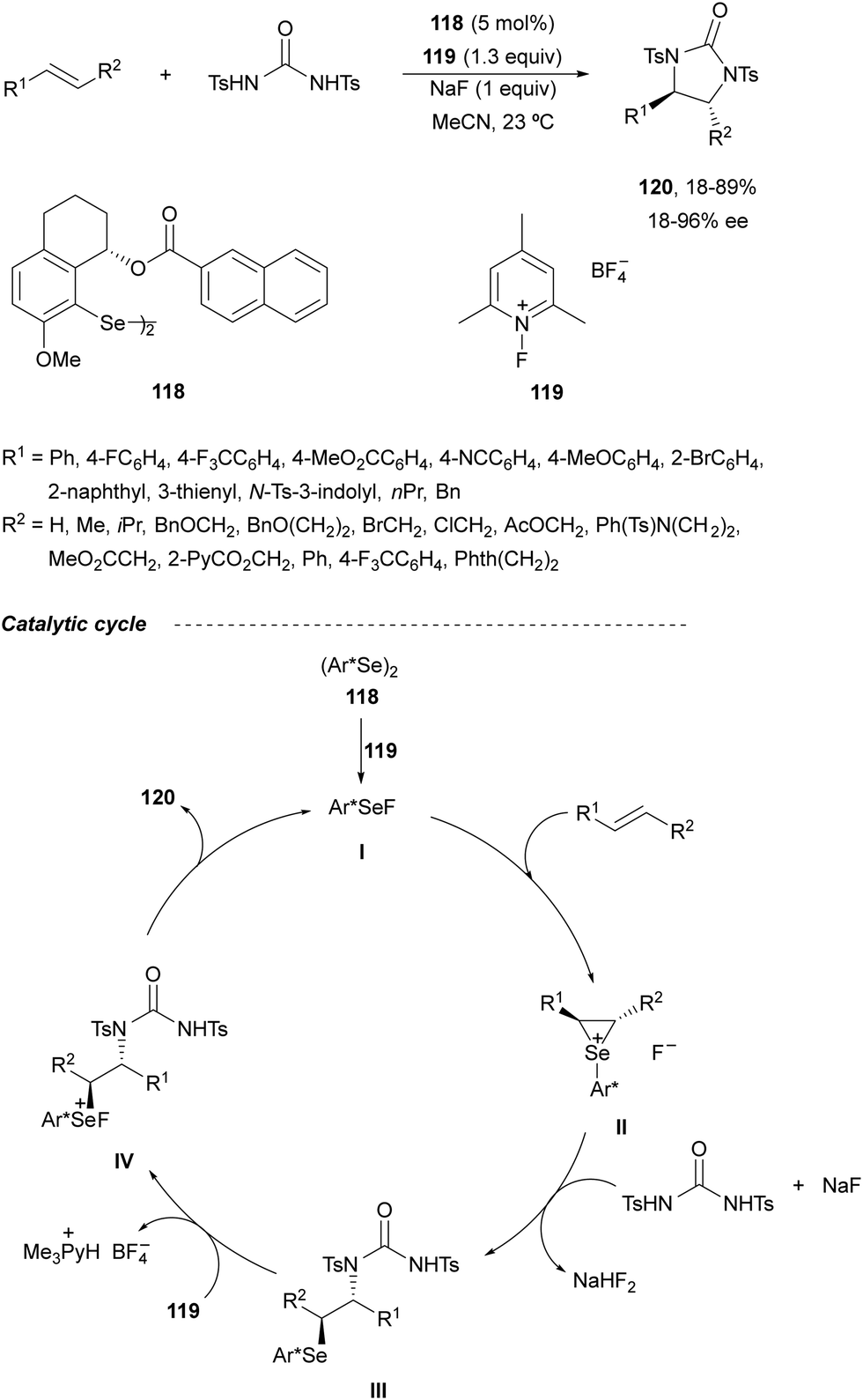 | ||
| Scheme 42 Asymmetric intermolecular syn-diamination of alkenes under Se(II)Se(IV) catalysis. | ||
Enantioselective diaminations by the one-electron radical mechanism have been accomplished under copper or iron catalysis.14 Initial studies of Shi and co-workers10,86 on Cu-catalyzed diamination of dienes were performed with di-tert-butyldiaziridinone and (R)-DTBM-Segphos (23) as a chiral ligand to provide imidazolidinones 121 (Scheme 43a) with different regioselectivity than in the Pd-catalyzed diamination10,87 (Scheme 37). This diamination took place in good yields and moderate enantioselectivities at the terminal double bond (absolute configuration not assigned). In the radical mechanism, intermediate I is formed by N–N bond cleavage of the nucleophile by the Cu(I) catalyst through a SET process. This aminyl radical I adds to the less hindered side of the diene giving radical intermediate II, which reacts with Cu to provide intermediate III. Final reductive elimination of III gives the imidazolidinone 121. Afterwards, the group described the same process using a Cu(I) phosphate 122 as a chiral catalyst (Scheme 43b).88 A positive effect of a chiral phosphate as an anionic counterion was observed in the obtained enantioselectivity (absolute configuration not assigned).
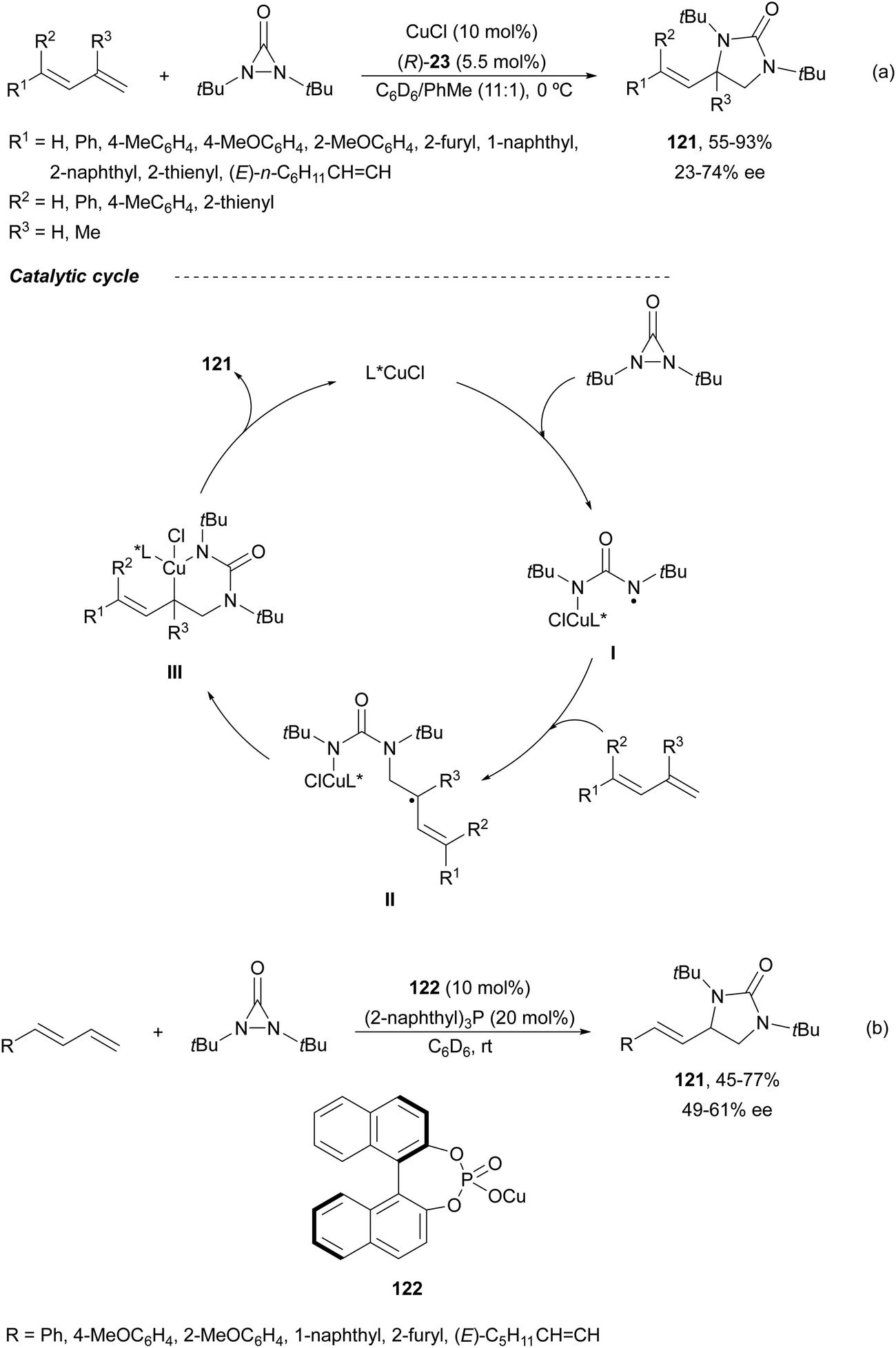 | ||
| Scheme 43 Asymmetric radical intermolecular diamination of conjugated dienes under Cu(I) catalysis. | ||
Intramolecular enantioselective diamination of aminoalkenes has been performed under copper(I) catalysis similarly to the Pd(II)/Pd(IV) redox diamination. Chemler and co-workers89 reported in 2014 the cyclization-amination of γ-alkenyl sulfonamides 123 and 108 in the presence of sulfonamides as external amines using Cu(I)-Phbox 9 as a catalyst and MnO2/KMnO4 as an oxidant (Scheme 44). The corresponding 2-aminomethyl indolines 124 and pyrrolidines 125 were obtained, respectively, in good yields and enantioselectivities. In the proposed radical mechanism, after coordination of the substrate 123 with the catalyst to form intermediate I, syn-aminocupration takes place giving intermediate II. Subsequently, radical III is formed, which after recombination with Cu(II) species generates the Cu(III) intermediate IV. Final reductive elimination forms the second C–N bond to give products 124.
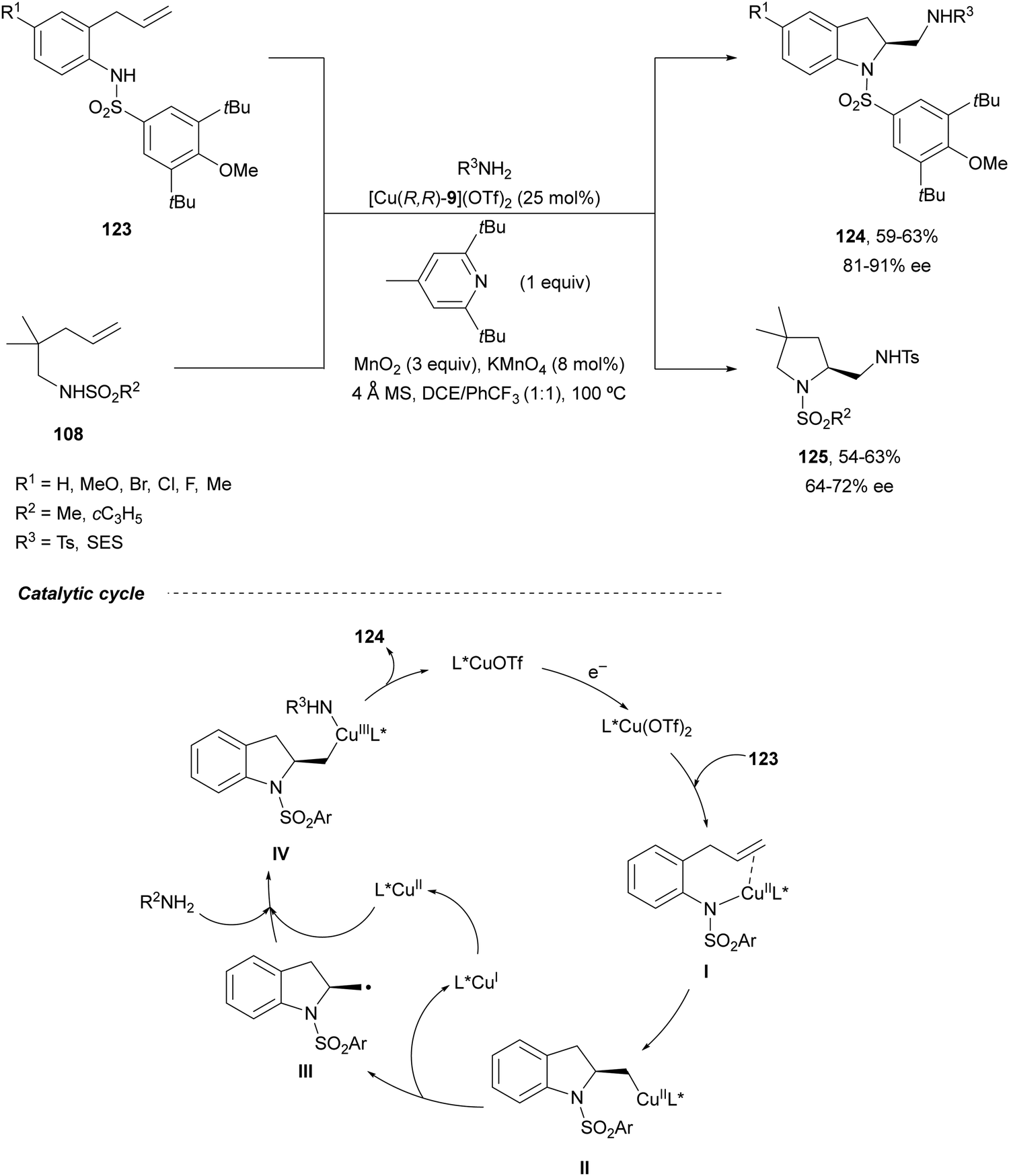 | ||
| Scheme 44 Asymmetric radical intra/intermolecular diamination of γ-alkenyl sulfonamides under Cu(I) catalysis. | ||
In order to avoid the competitive formation of six-membered rings, Zheng and co-workers90 described a double intramolecular diamination of N-alkenylureas 126 (Scheme 45). Under similar reaction conditions to those described by Chemler's group, bicyclic heterocycles 127 were obtained up to 86% yield and up to 98% ee. For one particular example 127 (R1 = R2 = Ph), the treatment with Ba(OH)2 afforded (S)-pyrrolidine 128.
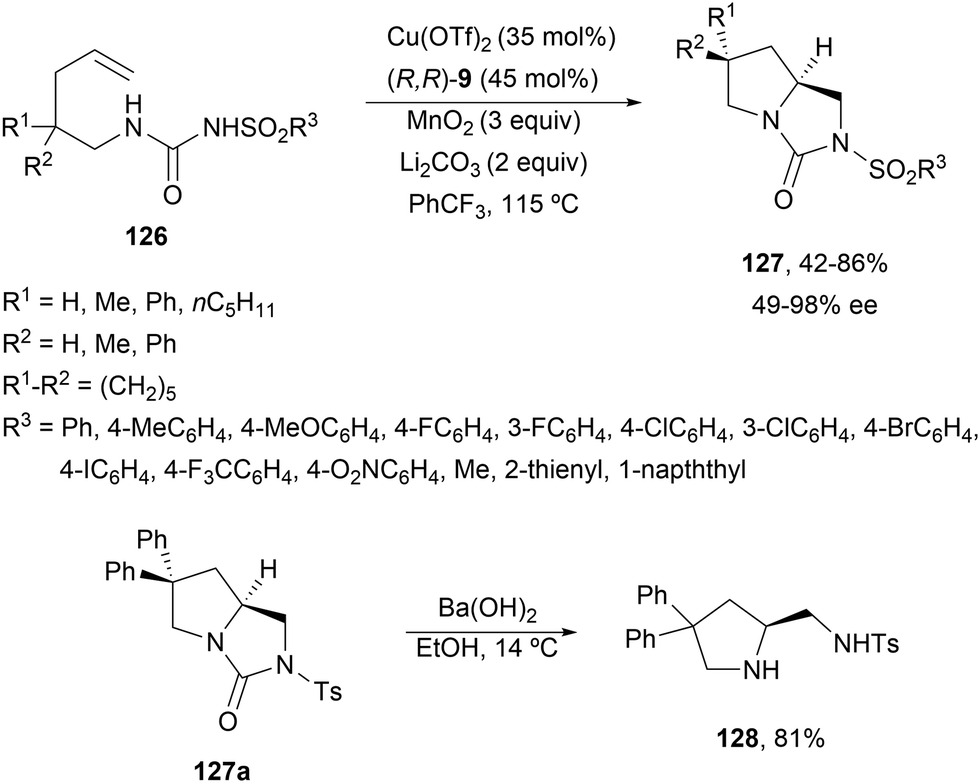 | ||
| Scheme 45 Asymmetric radical intramolecular diamination of γ-alkenylureas 126 under Cu(I) catalysis. | ||
Liu and co-workers91 developed a general method for the asymmetric diamination of γ-alkenylureas 129 in the presence of O-arylhydroxylamines 130 as the external nitrogen source to form enantioenriched β-alkylamino substituted pyrrolidines 131 under mild reaction conditions (Scheme 46a). In this case, Cu(MeCN)4 BF4 and CPA 132 were used as catalysts to give products 131 in good yields and enantioselectivities. A similar chiral copper(I) phosphate derived from the CPA 133 (see Scheme 43) catalyzed the intramolecular amidation followed by intermolecular azidation using a λ3-iodane reagent 134 to provide pyrrolidines 135 (Scheme 46b). In the proposed radical mechanism to explain the formation of products 131, a SET process between the copper phosphate and the O-acylhydroxylamine 130 generates an aminyl radical I. After reaction of I with the starting urea 129, a radical intermediate II is generated by intermolecular addition to the double bond. Subsequent second intramolecular amination followed by reductive elimination forms pyrrolidine 131 and regenerates the catalyst.
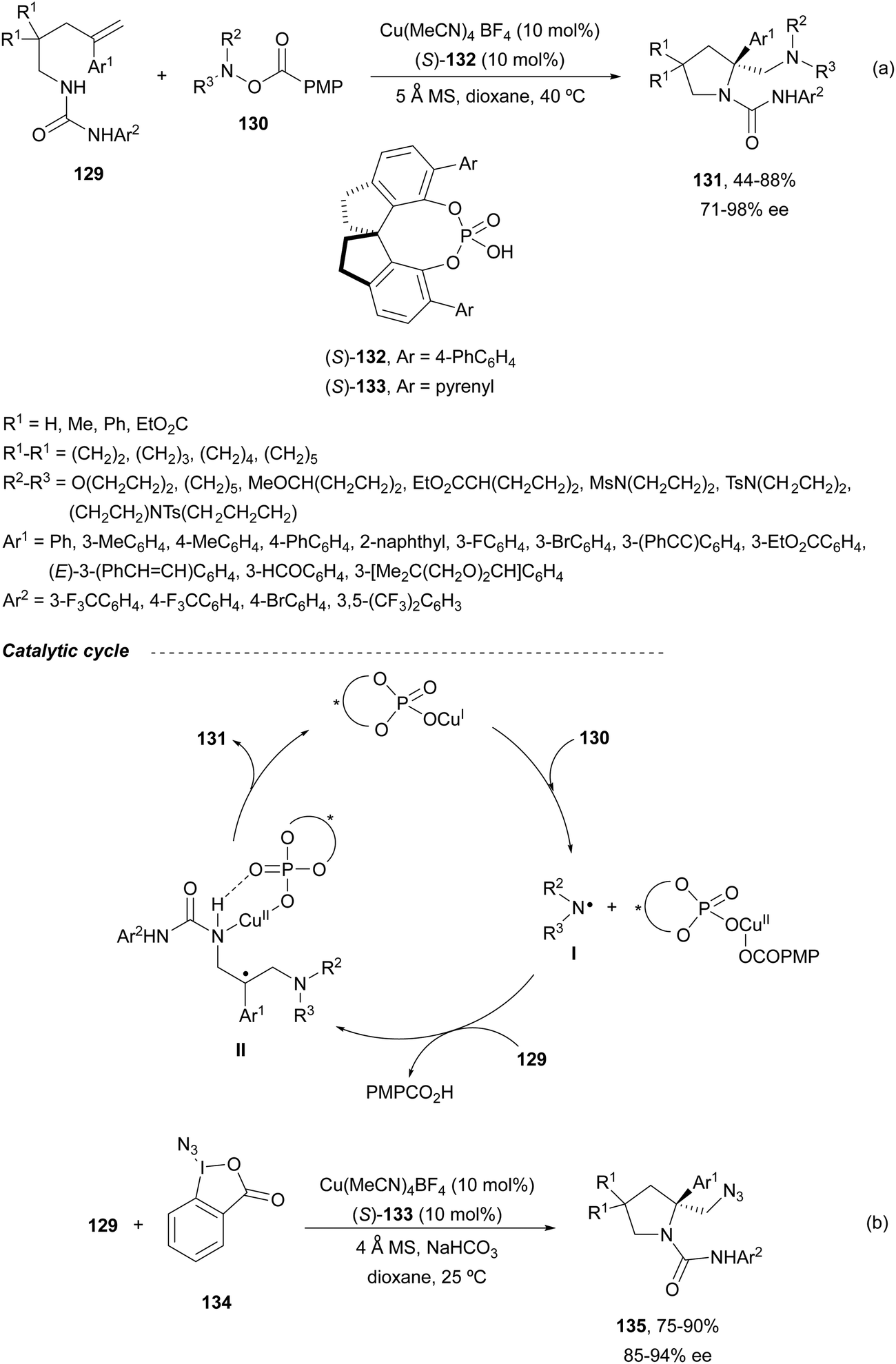 | ||
| Scheme 46 Asymmetric radical inter/intramolecular diamination of γ-alkenylureas 129 under Cu(I) chiral phosphate catalysis. | ||
Recently, Bao and co-workers92 reported the first example of asymmetric intermolecular diamination of styrenes using Fe(OTf)2 and the chiral tridentate ligand 136 (Scheme 47). In the presence of TMSN3 and NFSI, a three-component aminoazidation process took place to provide regioselectively products 137 with very good yields and enantioselectivities. According to experimental studies, a radical mechanism was proposed. The iron complex I reacts with TMSN3 to form the iron(II) azide complex II. A subsequent SET process between NSFI and II forms a Fe(III) azide species III and a bis-sulfonylamidyl radical IV, which reacts with styrene to give a benzyl radical V. This radical V can react with the azide species III to provide the final product. Alternatively, intermediate III can be generated by a SET process between the catalyst I and NSFI to give intermediate VI followed by anion exchange with TMSN3. Products 137 were transformed into different 1,2-diaminated products.
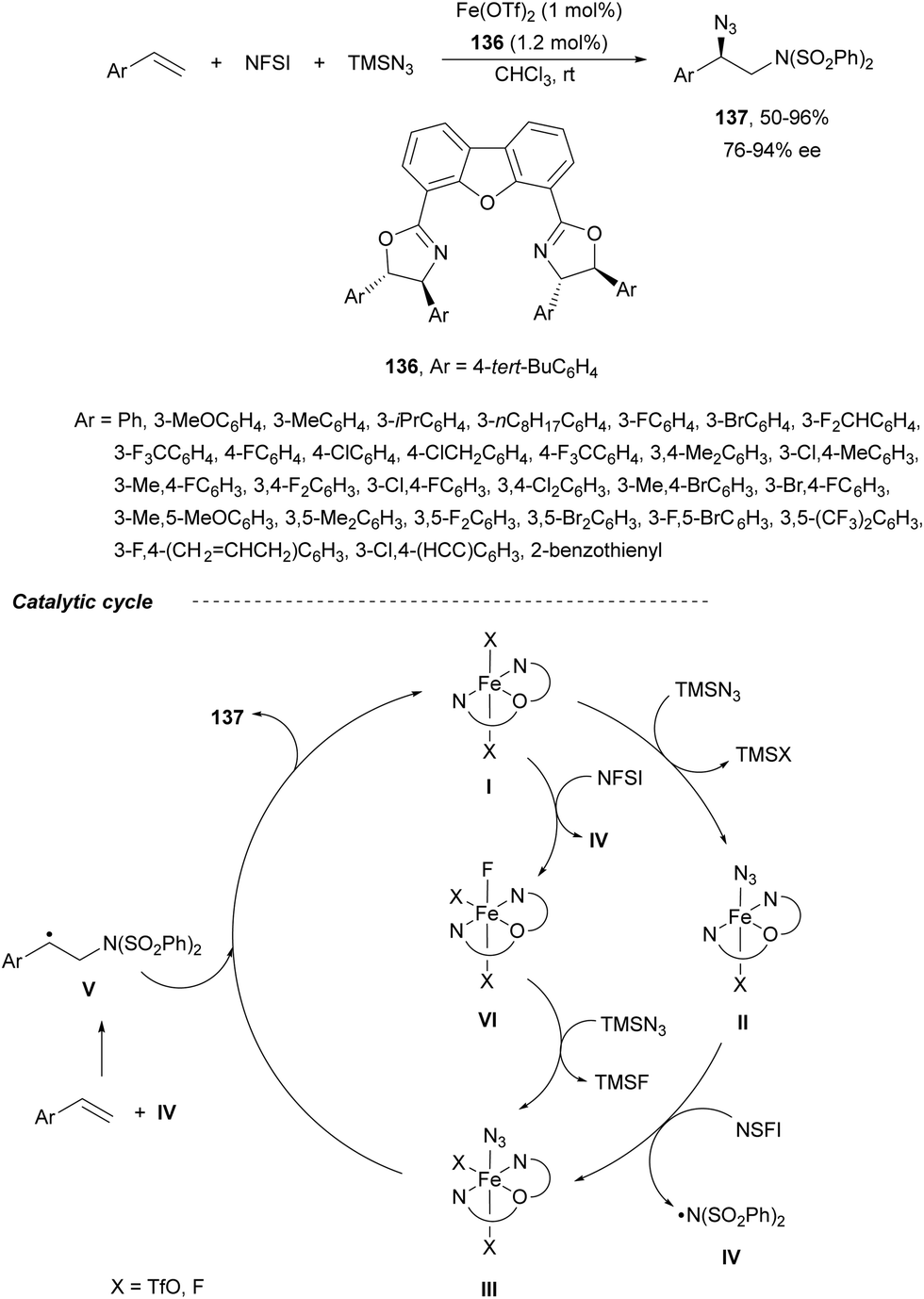 | ||
| Scheme 47 Asymmetric radical aminoazidation of styrenes under Fe(III) catalysis. | ||
Asymmetric diamination processes can be performed intermolecularly with styrenes or dienes, whereas intramolecular diamination must be carried out with amine-tethered alkenes. They are based on a two-electron redox pathway under Pd(0)/Pd(II) or Pd(II)/Pd(IV) catalysis and also with λ3-iodanes or diaryl selenides as chiral catalysts. In the case of the one-electron radical mechanism, Cu(I) and Fe(II) chiral complexes have been used. As nucleophiles, di-tert-butyldiaziridinone and ureas have been efficiently employed for intermolecular processes under Pd(0)/Pd(II) catalysis. However, intramolecular amination has been carried out with amides or sulfonamides and also with λ3-iodanes and diselenides. Cu(I)-catalyzed reactions can be used in inter- and intramolecular processes with di-tert-butyldiaziridinone and with sulfonamides or ureas in the case of intramolecular diaminations. Intermolecular aminoazidation of styrenes can be carried out very efficiently under Fe(II)/bis(oxazoline) catalysis.
2.6. Other amination reactions
Trost and Fandrick93 reported the dynamic kinetic asymmetric transformation (DYKAT) of vinylaziridines 138 to imidazolidinones 139 (Scheme 48a). This cycloaddition of isocyanates to vinylaziridines took place under Pd catalysis by use of the Trost ligand (R,R)-140 in DCM at room temperature to give 4-vinylimidazolidinones 139 up to 95% yield and up to 99% ee. The presence of acetic acid was crucial to improve the enantioselectivity in order to equilibrate the diastereomeric π-allylpalladium intermediates I and II by protonation, which should be faster than cyclization in a DYKAT. Imidazolidinones 139 were converted into diamines firstly by reduction with LiAlH4 affording imidazolidines, followed by hydrolysis with hydroxylamine in diluted HCl. This methodology was applied to the total synthesis of (+)-pseudodistomin D,94 an alkaloid isolated from the Okinawa tunicate Pseudodistamina megalarva.95 These types of alkaloids exhibit calmodulin-antagonist activity and potent cytotoxicity against murine leukemia and human epidermoid carcinoma KB cells.96,97 Starting from vinylaziridine 138a resulted imidazolidinone 139a in 80% yield and 94% ee, which was further transformed into (+)-pseudodistomin D. An alternative method for the asymmetric cycloaddition was simultaneously described by Dong and Alper98 using Binap (46) as a chiral ligand and CeCl3 as Lewis acid (Scheme 48b). Imidazolidinones 139 (R2 = H; absolute configuration not determined) were obtained in good yields and enantioselectivities. In this case, CeCl3 increased the rate of equilibration of π-allylpalladium intermediates.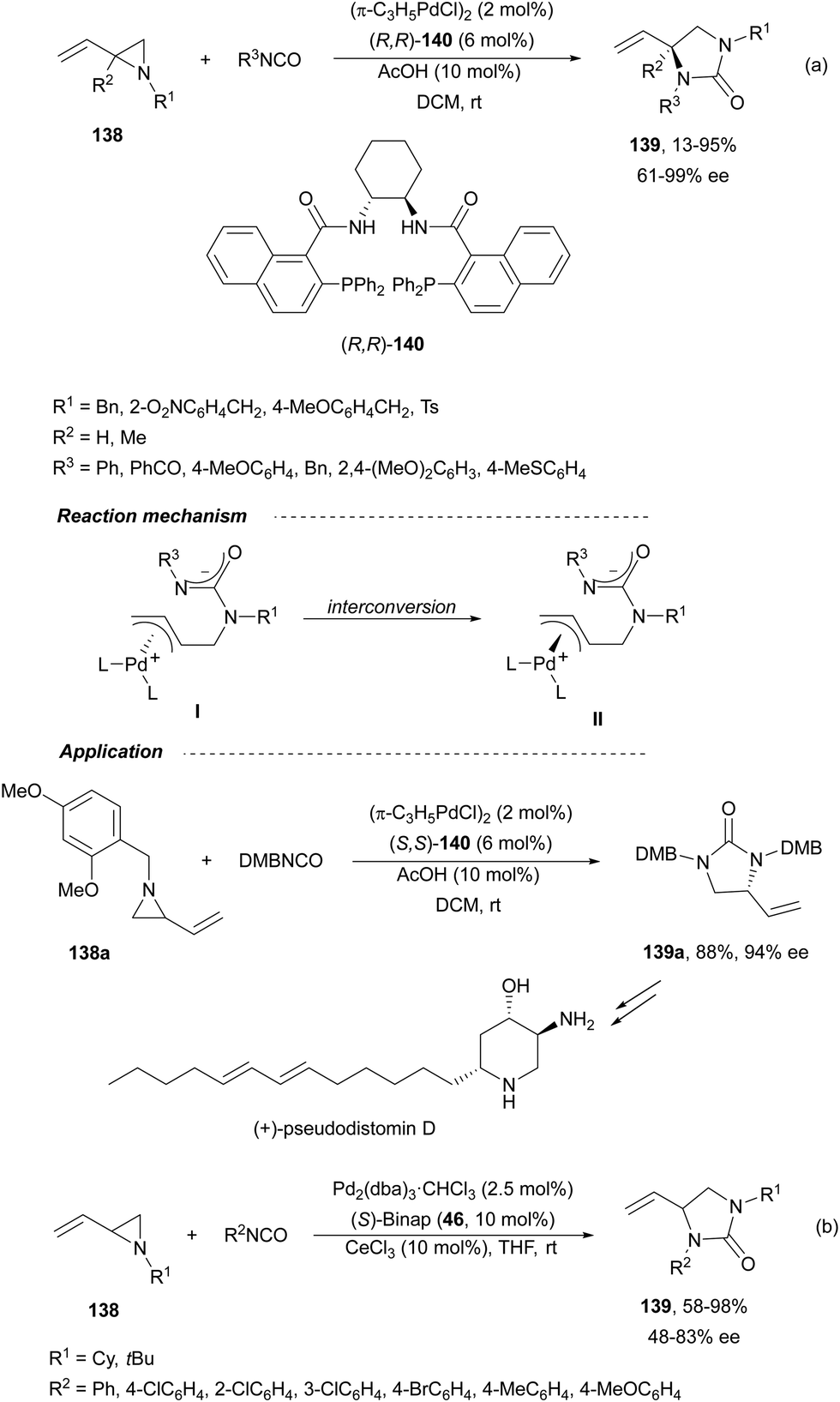 | ||
| Scheme 48 Asymmetric cycloaddition of isocyanates with 2-vinylaziridines 138. | ||
When vinylaziridines 138 were allowed to react with imido carboxylates 141, a dynamic kinetic asymmetric allylic amination and subsequent aryl migration took place through intermediates 142 resulting orthogonally protected enantioenriched 1,2-diamines 143 (Scheme 49).99 In this case, no additives were used and products 143 were obtained in very good yields and enantioselectivities. This dynamic kinetic asymmetric reaction was also carried out with imido dicarboxylates 144 to provide diamines 145 with readily cleavable protecting groups, in lower yields than with benzoyl imido carboxylates 141. This methodology has been applied to the synthesis of a potent protein kinase C (PKC) inhibitor balanol100 starting from diamine 145a.
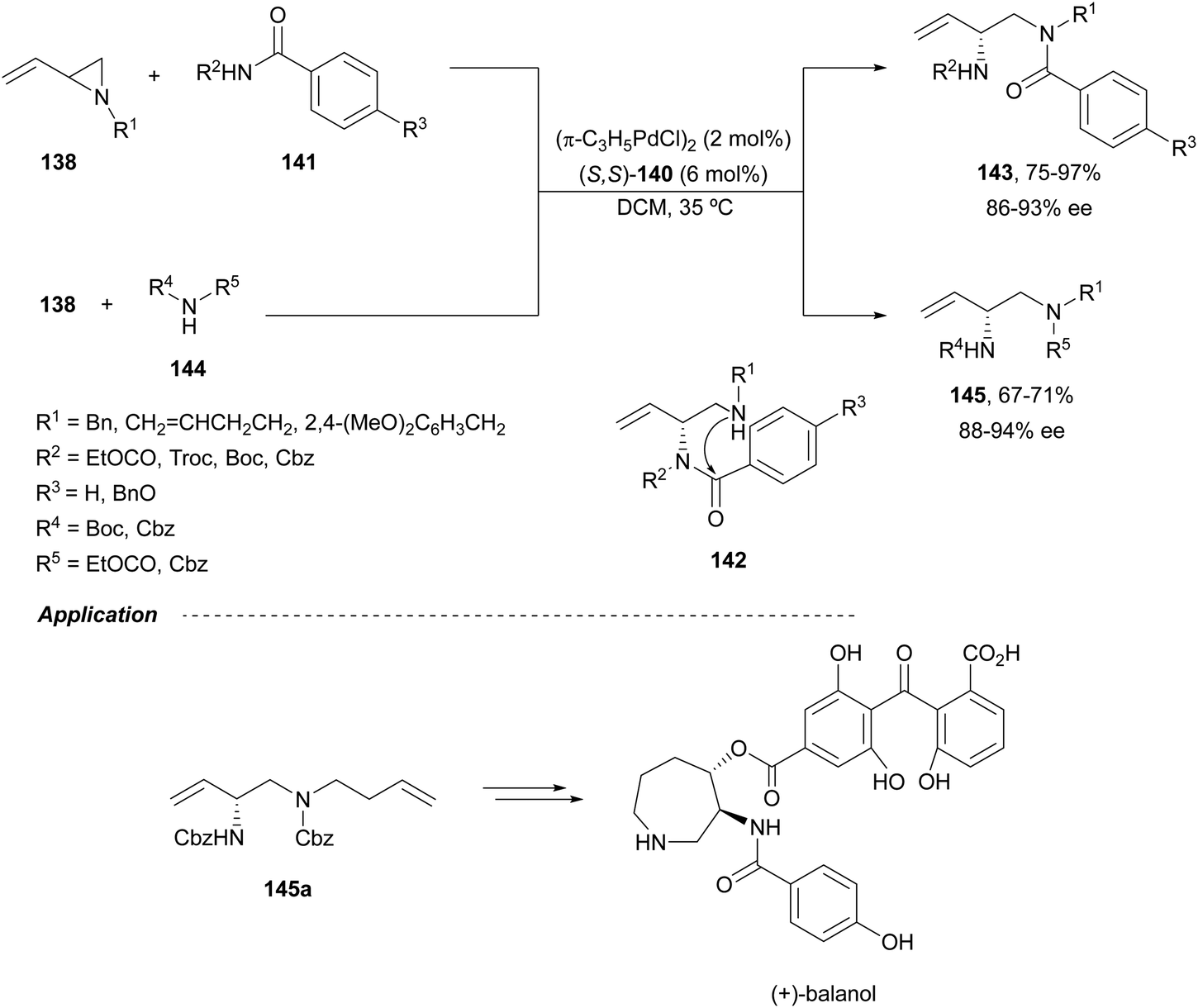 | ||
| Scheme 49 Asymmetric allylic amination and acyl migration of vinylaziridines 138 with imido carboxylates 141. | ||
Johnston and co-workers101 have described an enantioselective synthesis of imidazolidinones from allylic amines and N-tosyl isocyanate using a C2-symmetric bisamidine (BAM) 146 as organocatalyst. This amine-isocyanate capture-aminocyclization was carried out in the presence of N-iodopyrrolidone (NIP) to provide 5-exo-cyclization products 147 in very good yields and enantioselectivities (Scheme 50). However, for E-1,2-disubstituted allylic amines, the endo pathway was favored to produce six-membered urea precursors of 1,3-diamines. The obtained imidazolidinone 147a was transformed into the corresponding diamine 148 by reduction with LiAlH4. This method was also applied to the synthesis of the NK1 antagonist 149 (Schering-Plough).
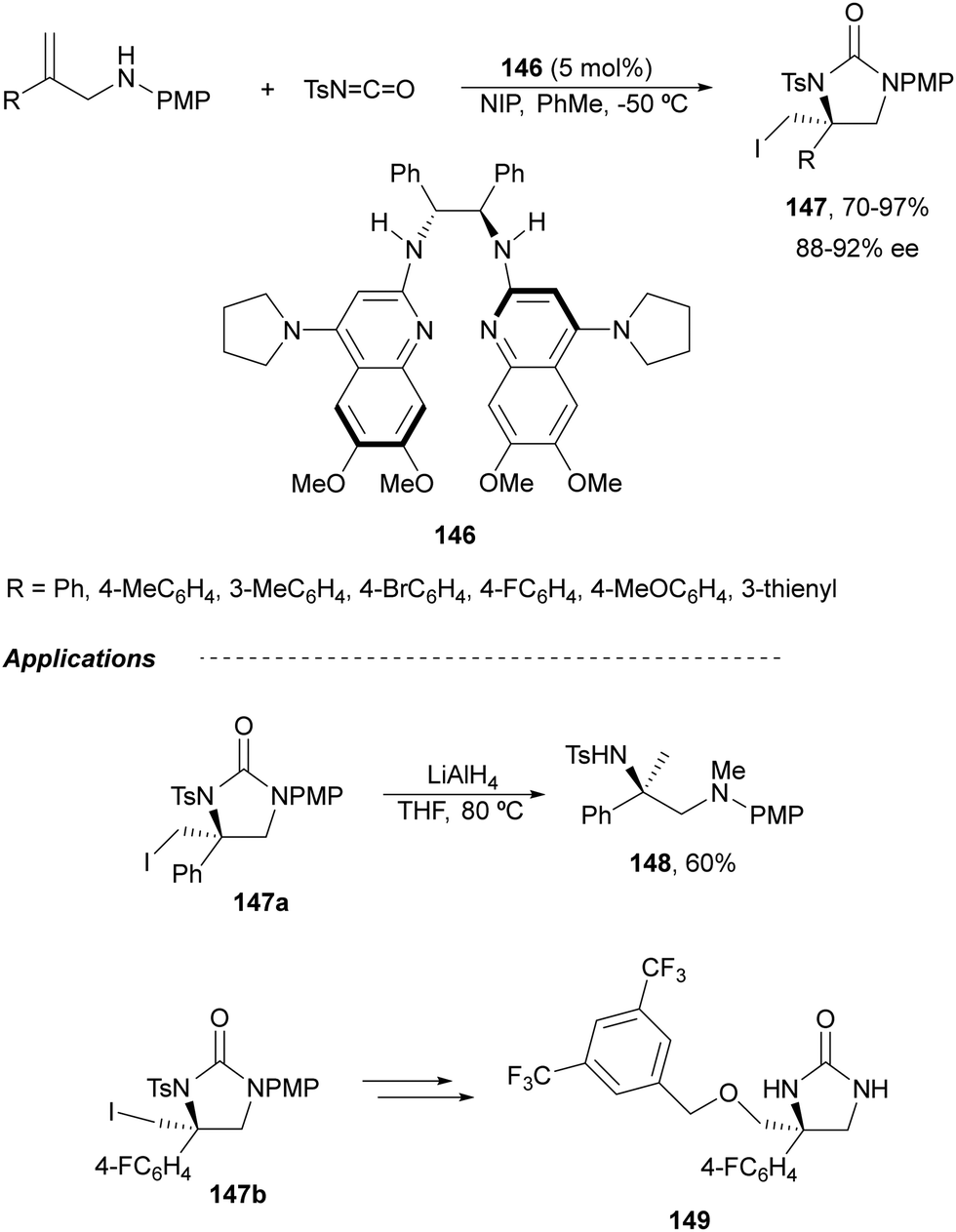 | ||
| Scheme 50 Asymmetric alkene iodoamination by isocyanate capture by allylic amines. | ||
Heterocyclic diamine derivatives 153 have been prepared in an enantioselective manner by a three-component radical cascade reaction of quinolines with N-vinylacetamide and O-acyl hydroxylamines of type 150 (Scheme 51).102 These reactions were performed under photoredox reaction conditions with Ir(ppy)3151 as a catalyst and a (R)-TRIP 152 as chiral Brønsted acid under blue LED irradiation at 10–12 °C. Enantioenriched diamines 153 were obtained in 59–72% yields with high enantioselectivities. In this Minisci-type reaction, an amidyl radical I is formed by a SET reduction of the O-acylhydroxylamine 150 under photoredox reaction conditions, which reacts with the enamine to form radical II. This radical II adds to protonated quinoline by hydrogen bonding formation in intermediate III to provide IV. Probably, IV is formed reversibly as a mixture of diastereomers and one of them is selectively deprotonated to give V and regenerates the CPA 152. Finally, V is oxidized by excited Ir(III)* to provide the product after deprotonation.
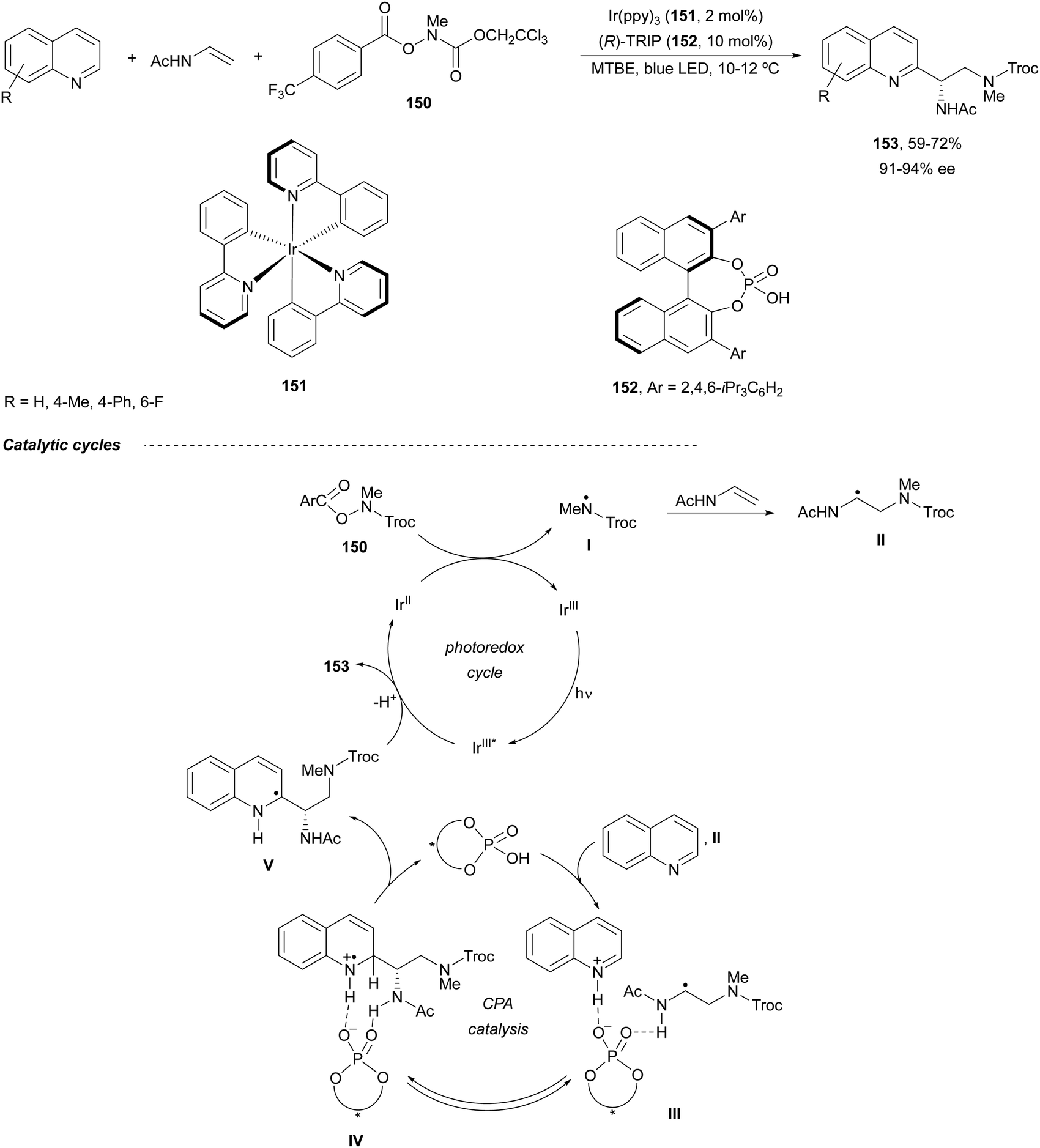 | ||
| Scheme 51 Asymmetric synthesis of diamines 153 by a three-component radical cascade reaction. | ||
3. C–C bond-forming reactions
3.1. Aza-Mannich ractions
In this section, the reaction of α-amino nucleophiles with aldimines or ketimines will be considered. As nucleophiles, imino esters, imino nitriles, azlactones, isocyano acetates and isothiocyanates have been used for the synthesis of α,β-diamino acid derivatives. Other nucleophiles such as α-azido ketones or amides gave α,β-diamino carbonyl compounds. In the case of α-amino acetaldehydes, β,γ-diamino alcohols were obtained. 3-Indoline-2-carboxylates provided β-amino ester derivatives. N-Aryl glycines reacted with hydrazones to provide 1,2-diamines under copper or photoredox catalysis.Coordination of the anion derived from alkyl imino glycinates with a Lewis acid should favor to act as a nucleophile in the aza-Mannich reaction rather than an azomethine ylide resulting in a 1,3-dipolar cycloaddition (see, Section 3.5). Since 2003,105 copper(I) salt with chiral ligand catalysis (154–156) has been widely used for the aza-Mannich reaction of imino glycinates with aldimines (Fig. 5).106,107 Imino glycinates and alaninates were also added to imines under copper(I) and Fesulphos ligand 157 catalysis.108,109 Other ligands such as 158110 and 159111 have also been employed with CuBF4 and Cu(OAc)2 salts, respectively (Fig. 5). The reaction of imino glycinates 160 with N-substituted aldimines 161 gave mainly syn-products 162105,109 or anti-products107–110 depending on electronic effects in the catalyst or in the imine protecting group (Scheme 52).
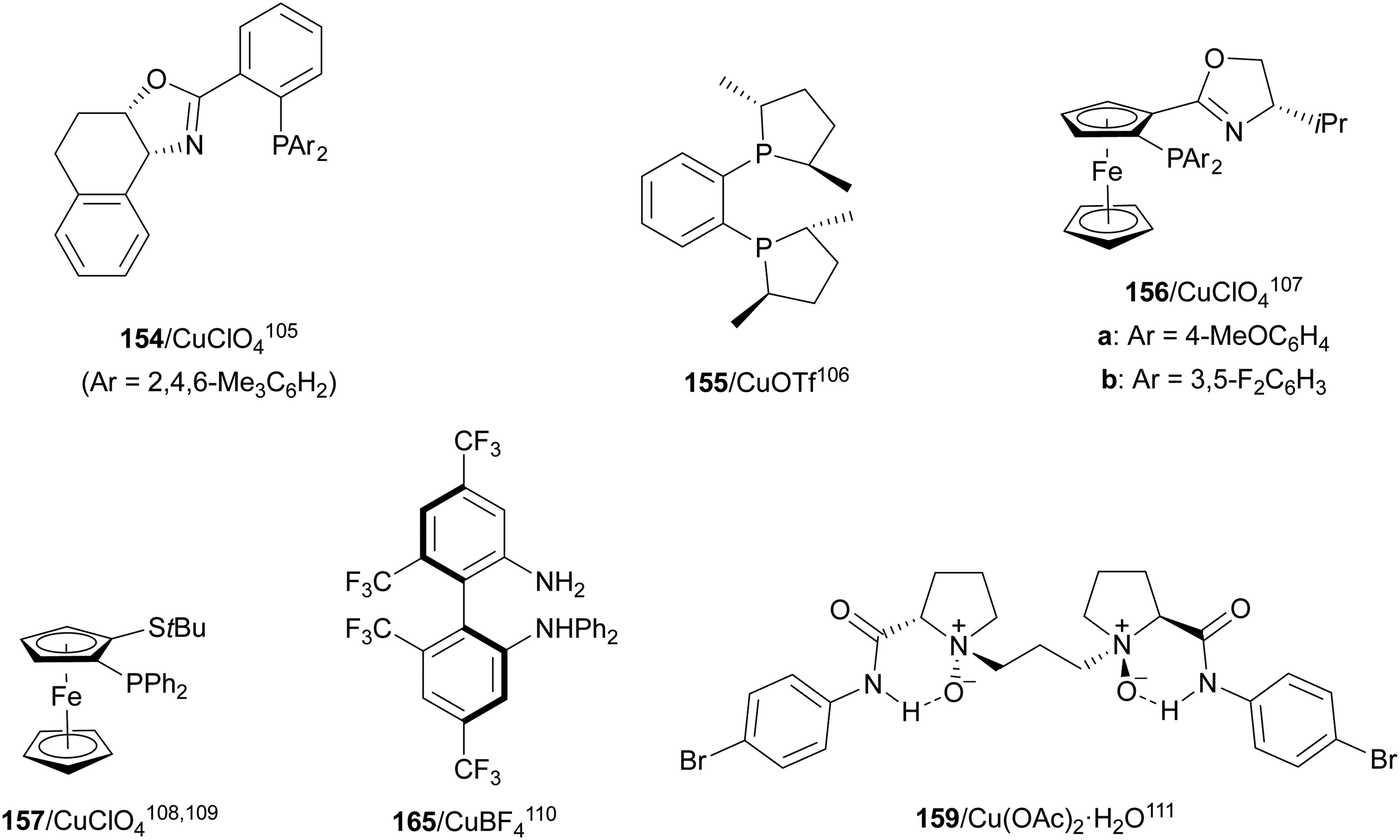 | ||
| Fig. 5 Chiral copper catalysts for the aza-Mannich reaction of imino glycinates with imines. | ||
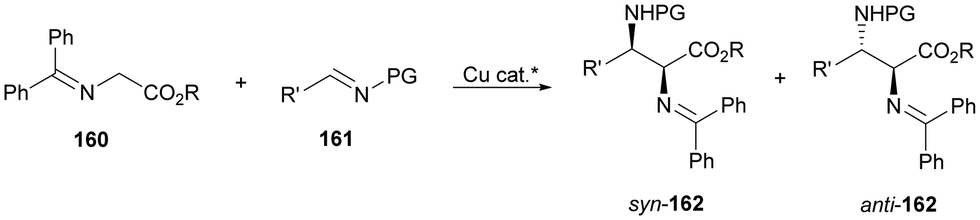 | ||
| Scheme 52 Asymmetric aza-Mannich reaction of benzophenoneimine glycinates with imines. | ||
In 2012, Arai and co-workers112 described a syn-diastereoselective asymmetric Mannich reaction of N-sulfonyl aldimines 161 with imino esters 160 catalyzed by a tridentate ligand PyBidine 163 and Cu(OTf)2 (Scheme 53). α,β-Diamino acid precursors syn-162 were obtained up to 96% yield and up to 99% ee with, in general, high diastereoselectivities. Starting from the N-nosyl product 162a, via two deprotection steps, the α,β-diamino acid methyl ester 164 was obtained with the same diastereo- and enantioselectivities as the starting compound 162a. In the proposed mechanism, the Cu enolate II was initially formed from intermediate I, which reacts with the imine by a syn-approach depicted in the TS to provide the syn-diastereomer 162.
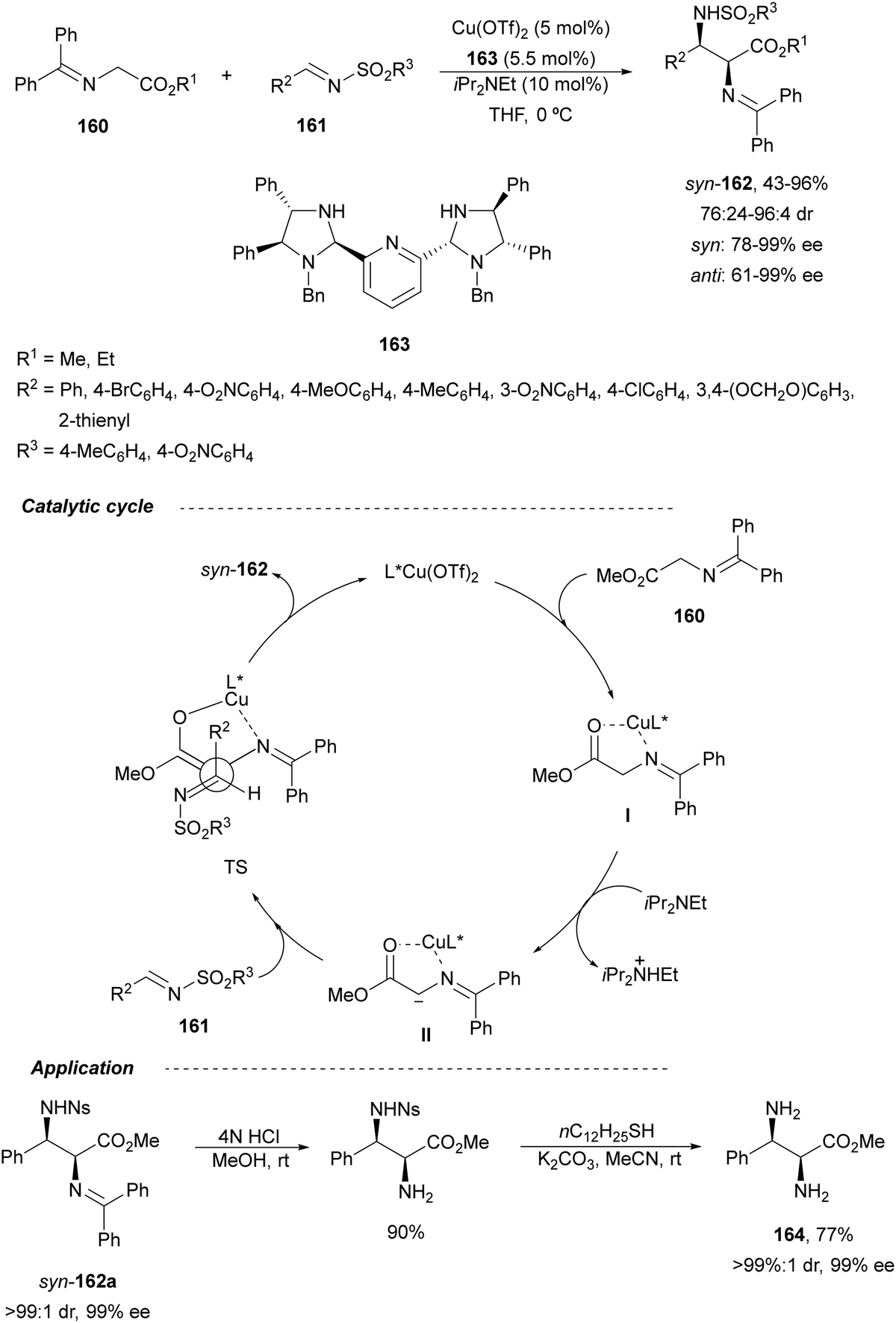 | ||
| Scheme 53 Asymmetric aza-Mannich reaction of imino esters 160 with aldimines 161 under Cu/PyBidine 163 catalysis. | ||
Kobayashi and co-workers113 employed a copper amide CuHMDS, prepared from CuOTf and potassium hexamethyldisilazide (HMDS), and the chiral ligand (R)-DTBM-Segphos (23) for the asymmetric aza-Mannich reaction of glycine imino ester 160 with N-tosylaldimines 161 (R3 = 4-MeC6H4) (Scheme 54). The chiral catalytic complex must be prepared in situ in order to avoid CuHMDS aggregation. Products 162 were obtained in good yields, moderate syn-diastereoselectivity and up to 98% ee.
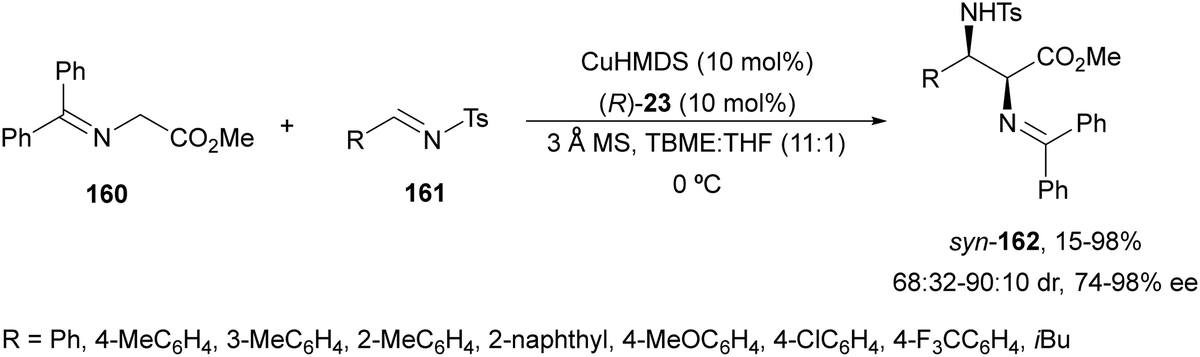 | ||
| Scheme 54 Asymmetric aza-Mannich reaction of imino glycinate methyl ester 160 with N-tosylimines 161 under CuHMDS/(R)-DTBM-Segphos (23) catalysis. | ||
In the case of the Cu(I)/Fesulphos (157)-catalyzed aza-Mannich reaction, a more practical method was described by Carretero's group.114 Easy available and stable α-amido sulfones 165 were used as precursors of unstable aliphatic aldimines based on the previous described asymmetric aza-Mannich reaction of phosphonoglycine imino esters with α-amido sulfones.115 In the presence of Cs2CO3 (1.5 equiv.) as a base, the corresponding N-tosylimines were generated in situ and reacted with glycine imino esters to provide syn-β-alkyl-α,β-diamino acid derivatives 162 with very good yields and high diastereoselectivities and enantioselectivities (Scheme 55). The syn-diastereocontrol was rationalized involving a severe steric repulsion of the bulky N-diarylmethylene group in the ketimine nucleophile with the tosyl group as it is shown in intermediate I. Thereby, the imine approaches from its Si-face via intermediate II, which affords the syn-product.
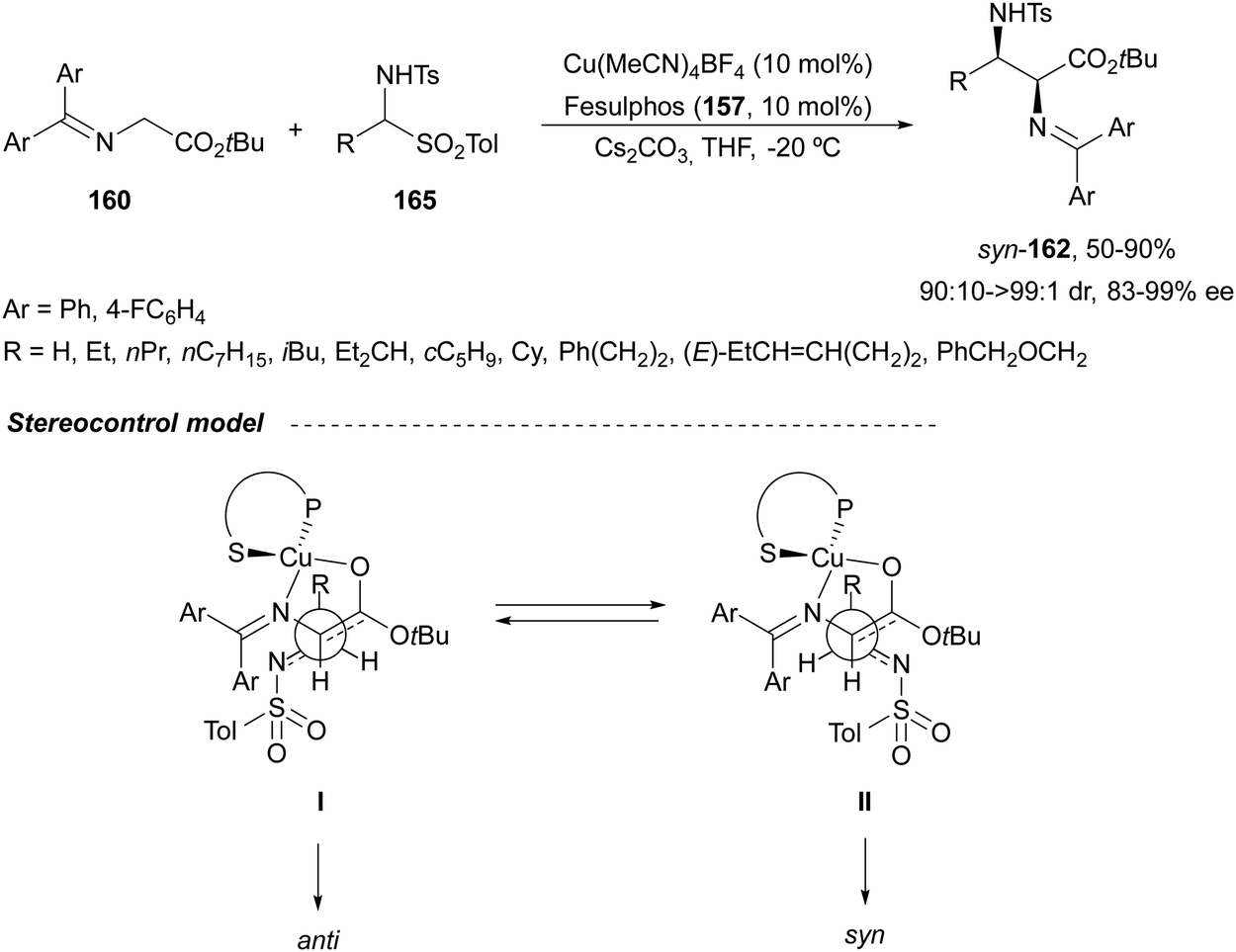 | ||
| Scheme 55 Asymmetric aza-Mannich reaction of glycine imino esters 160 with α-amido sulfones 165 under Cu(I)/Fesulphos 157 catalysis. | ||
Recently, N-diphenylphosphinoyl (DPP) aldimines 167 have been used as electrophiles using a bifunctional Cu(I)/amidophosphine-urea 168 catalyst.116 The reaction of imino esters 166, derived from aldehydes, and imines 167, followed by monodeprotection with aqueous NH2OH·HCl of the crude reaction mixture, provided the aza-Mannich syn-adducts 169 in high yields and up to >99:1 dr and up to 99% ee (Scheme 56). On the basis of the experimental studies and theoretical calculations,117 a plausible TS involving dual activation of the glycine imino ester and N-DPP imine has been proposed. The glycinate enolate is coordinated to the Cu(I) metal center and the N-phosphinoyl imine is activated and oriented by two hydrogen bondings between the urea moiety and the PO unit. A preferential Si-face attack of the imino ester onto the N-phosphinoyl imine gives the (2S,3R)-adduct.
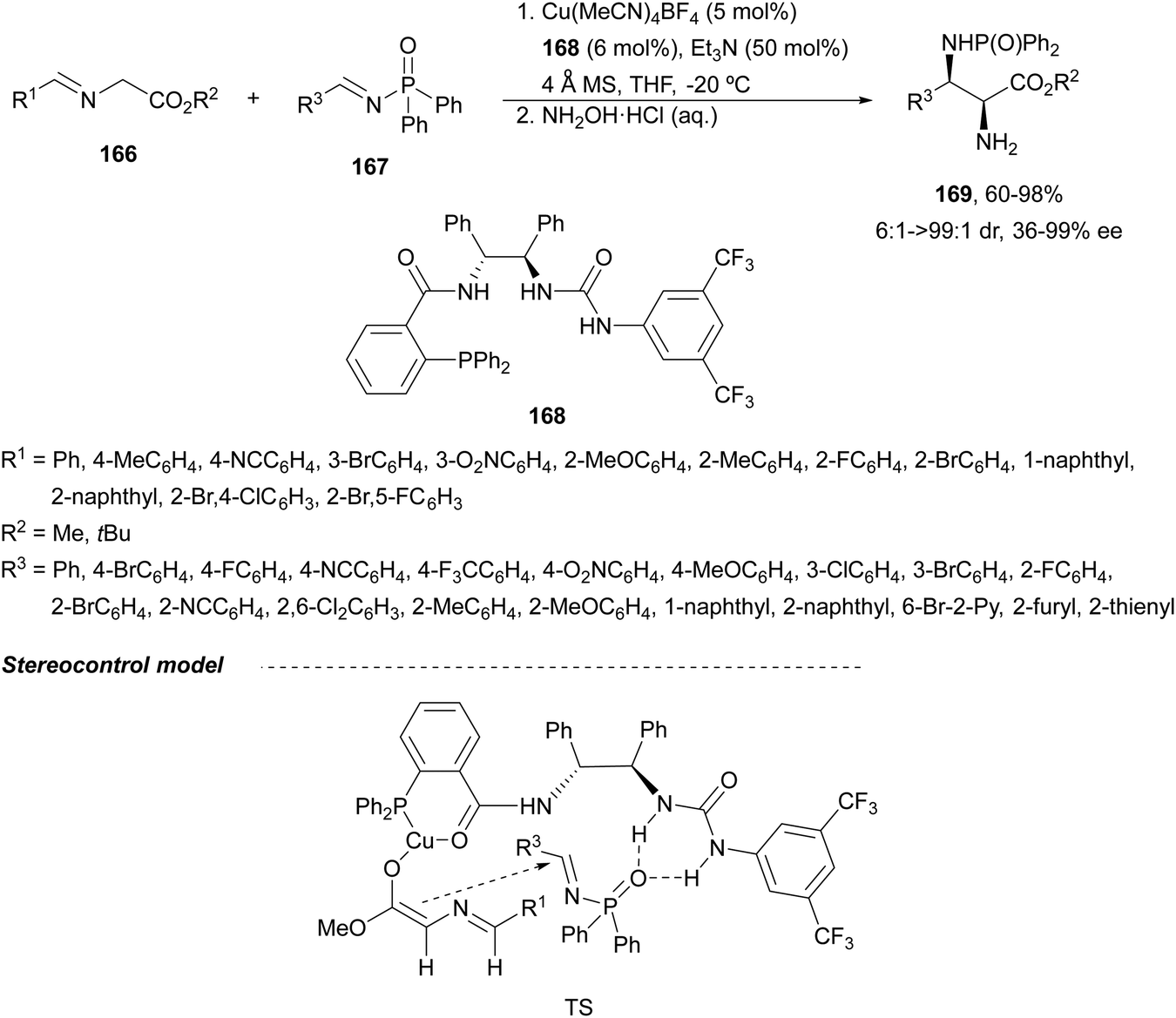 | ||
| Scheme 56 Asymmetric aza-Mannich reaction of glycine imino esters 166 with N-diphenylphosphinoyl imines 167 under Cu(I)-amidophosphine-urea 168 catalysis. | ||
Ketimines 170 derived from isatins have been employed as electrophiles in the aza-Mannich reaction with glycine imino ester 160 under Cu(I)/Ph-Phosferrox 171 catalysis by Yang, Deng and co-workers.118 This method allowed the asymmetric synthesis of 3-aminooxindoles 172 in high yields and enantioselectivities although with moderate to high diastereoselectivities (Scheme 57). The Mannich adduct 172a was transformed into biologically important spirooxindoles119,120173 and 174 in 99% ee.
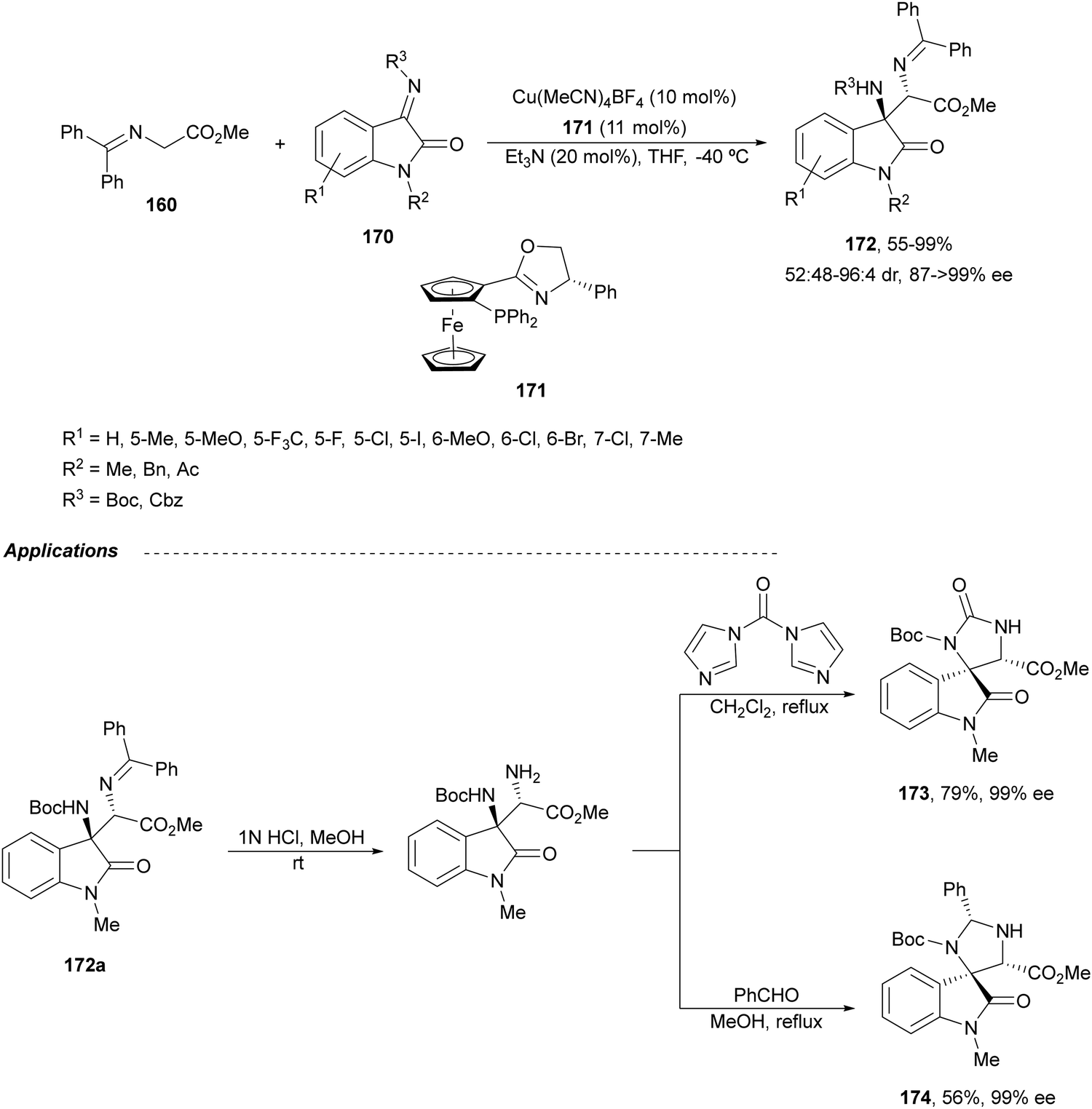 | ||
| Scheme 57 Asymmetric aza-Mannich reaction of glycine imino ester 160 with isatin derived ketimines 170 under Cu(I)/Ph-Phosferrox (171) catalysis. | ||
Cyclic ketimines 175 derived from saccharine have been allowed to react with glycine imino esters 160 under Cu(II)/RuPhox 176 catalysis by Xie and co-workers.121 Mannich-type adducts 177 were obtained up to 99% yield, up to >20:1 dr and 99% ee (Scheme 58). The sterically bulky tert-butyl imino glycinates gave generally better results and the absolute configuration for the (S,S)-diastereomer was determined by X-ray diffraction analysis.
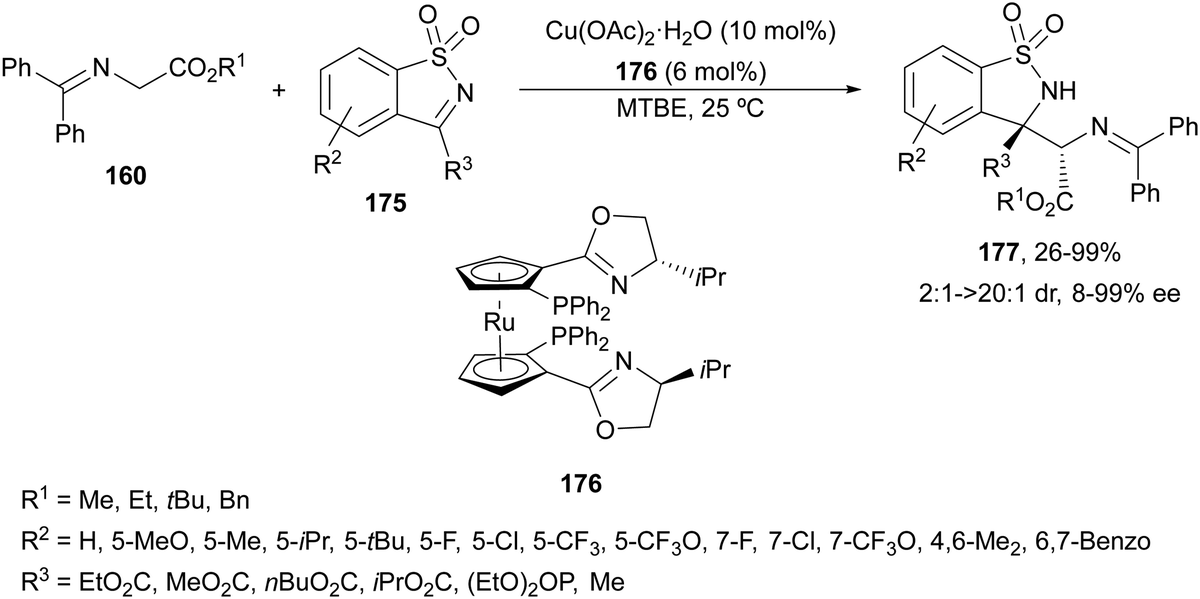 | ||
| Scheme 58 Asymmetric aza-Mannich reaction of glycine imino esters 160 with cyclic ketimines 175 under Cu(II)/Ruphox 176 catalysis. | ||
Silver acetate in combination with chiral ligands such as 178122 and 179123 have been used as catalysts for the aza-Mannich reaction of glycine methyl imino esters 160 and N-tosyl aldimines 161 (Fig. 6).104 These procedures took place with high yields and enantioselectivities but with low diastereoselectivities.
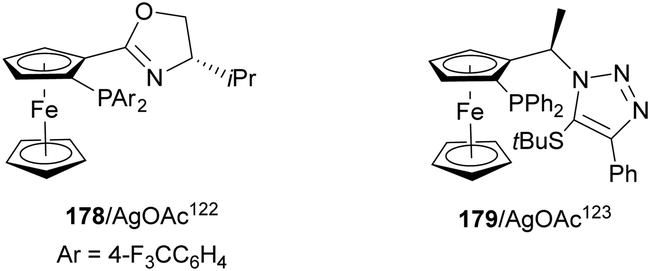 | ||
| Fig. 6 Chiral silver catalysis for the aza-Mannich reaction of methyl iminoglycinate 160 with N-tosyl imines 161. | ||
Sansano and co-workers124 reported this type of asymmetric aza-Mannich reaction using AgOTf and Feringa's phosphoramidite (Sa,R,R)-180 as a catalyst (Scheme 59). Precursors of α,β-diamino acids 162 were obtained up to 70% yield, up to 90:10 dr and 99% ee for the syn-diastereomer. In the proposed TS, the silver enolate attacks the N-tosyl aldimine 161 whose tosyl group is placed far away from the benzylidene moiety of the enolate.
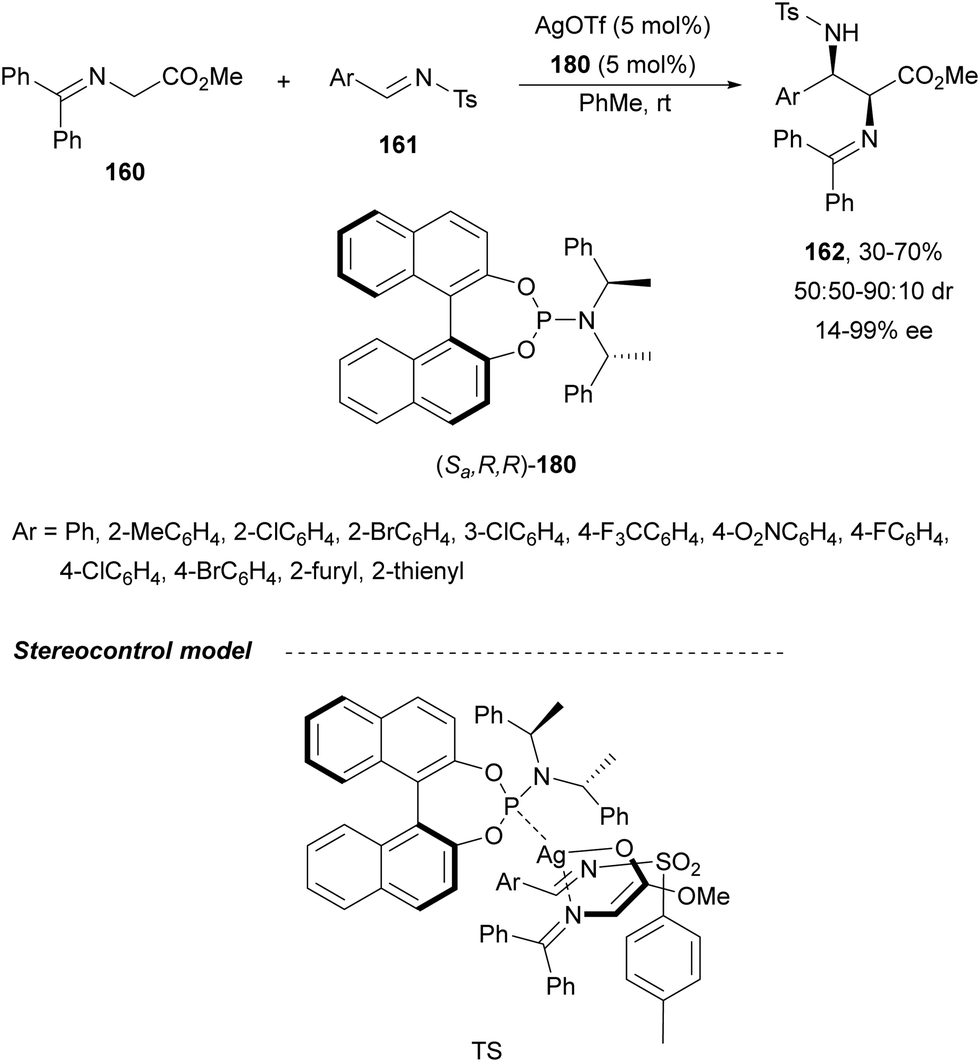 | ||
| Scheme 59 Asymmetric aza-Mannich reaction of glycinate methyl imino ester 160 with N-tosyl aldimines 161 under AgOTf/phosphoramidite 180 catalysis. | ||
Hu and co-workers125 have described that Rh2(OAc)2 and CPA (R)-183 or (R)-184 catalyzed the three-component aza-Mannich reaction of a diazo compound 181, and a carbamate and a N-aryl aldimine 182 furnished both anti- and syn-α,β-diamino acid derivatives 185, respectively (Scheme 60). This diastereodivergent52 trapping of carbamate ammonium ylides with imines takes place by initial formation of the rhodium carbenoid I, which reacts with the carbamate to form the carbamate ammonium ylides II/III. These ylides are trapped by the imine to give products 185, via zwitterion IV. The observed stereoselective control was explained by TSanti and TSsyn formed with the different types of CPAs. In the case of the sterically demanding CPA (R)-183, an open-chain TSanti was suggested. However, with CPA (R)-184 a bifunctional role forms a N–H–OPO–H–N bridge in the TSsyn.
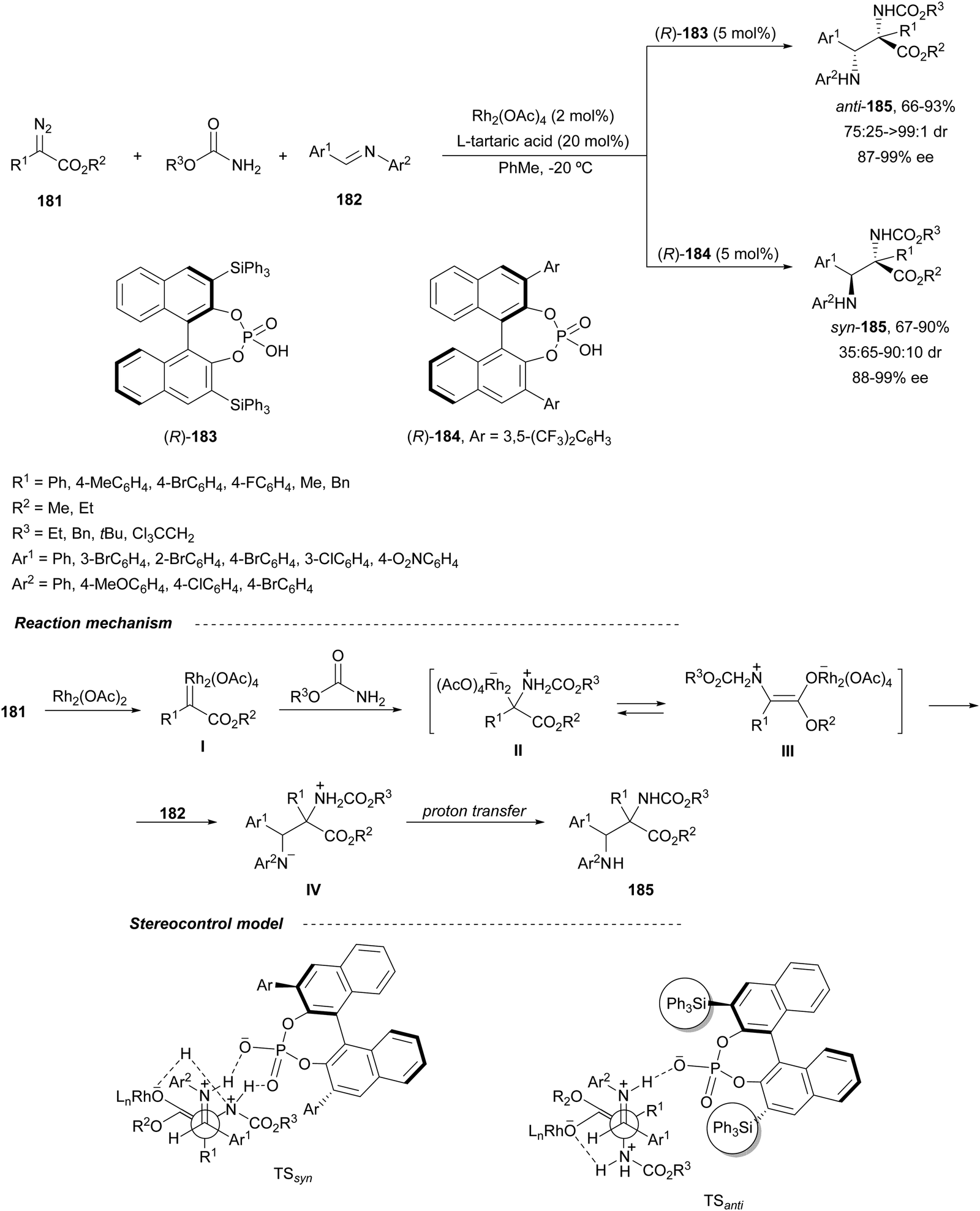 | ||
| Scheme 60 Asymmetric three-component aza-Mannich reaction of diazo compounds 181 with carbamates and N-acyl aldimines 182 under Rh2(OAc)4/CPA (R)-183 and (R)-184 catalysis. | ||
The former asymmetric three-component reaction was also carried out by the same group126 with arylamines instead of carbamates. In this case, (R)-TRIP (152) was used as a CPA to provide anti-α,β-diamino esters 186 up to 90% yield, up to >95:5 dr and 96% ee (Scheme 61).
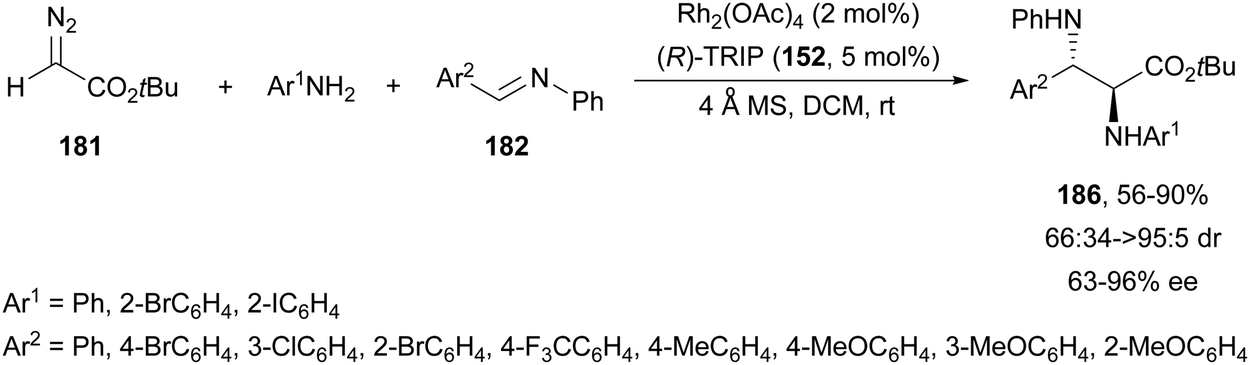 | ||
| Scheme 61 Asymmetric three-component aza-Mannich reaction of tert-butyl diazoacetate 181 with arylamines and N-aryl aldimines 182 under Rh2(OAc)4/(R)-TRIP (152) catalysis. | ||
When phosphoramides 187 and α-imino esters 188 were allowed to react with diazo compounds 181, under Rh/CPA reaction conditions, 2,3-diaminosuccinic acid derivatives 190 were obtained (Scheme 62).127 In this case, the 9-phenantryl (S)-189 was used as Brønsted acid to give products 190 with good yields, moderate to high syn-diastereoselectivities and up to 98% ee. The trapping of protic phosphoramidate ammonium ylides with α-imino esters can be envisaged via intermediate I.
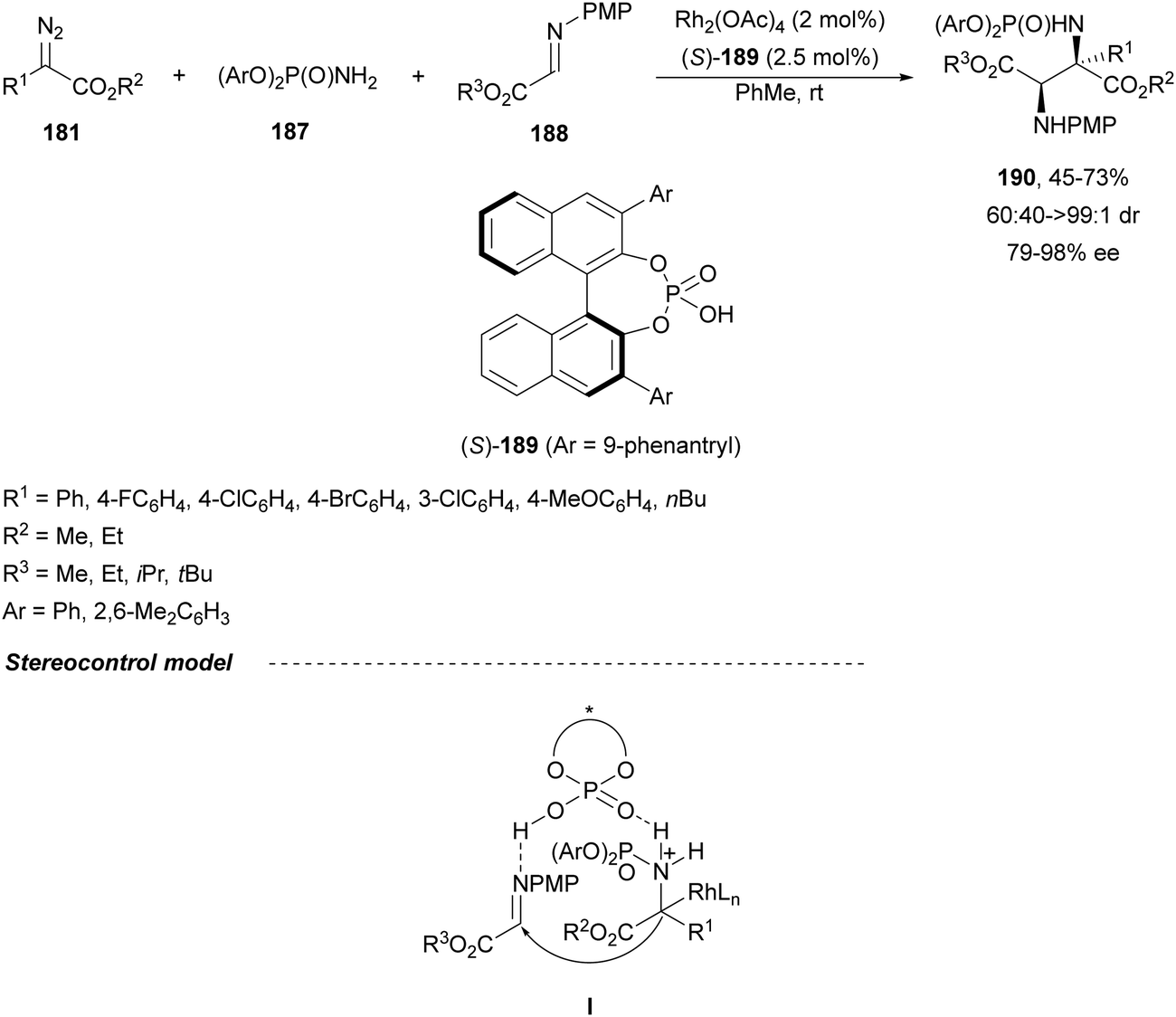 | ||
| Scheme 62 Asymmetric three-component aza-Mannich reaction of diazo compounds 181 with phosphoramides 187 and α-imino esters 188 under Rh/CPA (S)-189 catalysis. | ||
As a summary of the asymmetric metal-catalyzed aza-Mannich reaction, the most studied process between imino esters and imines was carried out under copper catalysis to give mainly syn-α,β-diamino acid derivatives. The three-component reaction of diazo compounds, amines or amides and imines under Rh/CPA catalysis is a versatile strategy which provided mainly the same syn- or anti-products depending on the CPA catalyst.
Organocatalytic asymmetric aza-Mannich reactions have been performed using chiral phase-transfer catalysts (PTC) but also chiral bases. Maruoka and co-workers128 reported the phase-transfer-catalyzed aza-Mannich reaction of benzophenone imine of tert-butyl glycinate ester 160 with N-aryl-α-imino ester 188 using a N-spiro-C2-symmetric chiral quaternary ammonium bromide 191 as a catalyst (Fig. 7). This method enabled the synthesis of a syn-3-aminoaspartate derivative, related to compound 190, in 88% yield, 82:18 dr and 91% ee. This product was converted into a precursor of streptolidine lactam, a constituent of streptothricin antibiotics. A general method for the aza-Mannich reaction of 160 (R = tBu) with N-Boc-aldimines was described by Ohshima, Shibasaki and co-workers129,130 using tartrate-derived diammonium salts 192 as a chiral catalyst (Fig. 7). The corresponding syn-adducts 162 were obtained with high yields (88–96%), diastereoselectivities (97:3–99:1 dr) and good enantiocontrol (70–90% ee). This method was applied to the synthesis of the antipsychotic (+)-nemonapride.
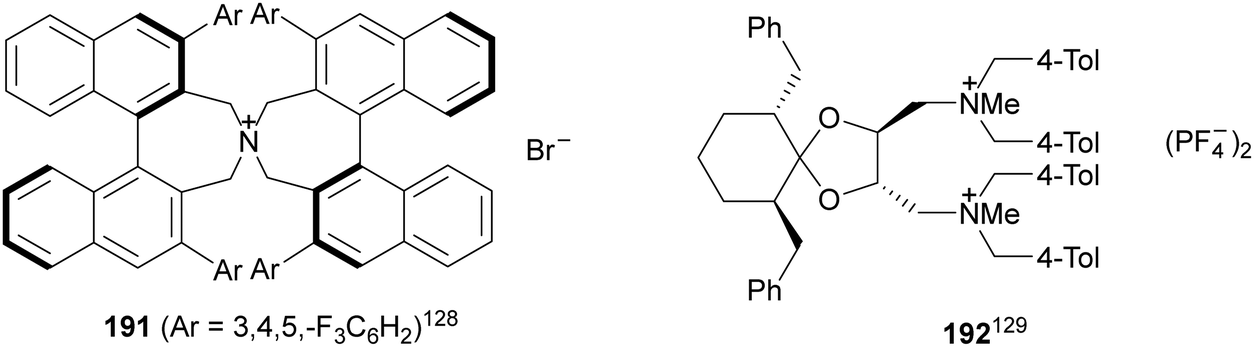 | ||
| Fig. 7 Chiral phase-transfer catalysts for the aza-Mannich reaction of imino glycinates with imines. | ||
Cinchona-Alkaloid derived ammonium salts have been widely used as readily available catalysts in asymmetric PTC in organic synthesis.131 Gong and co-workers132 employed a quininium salt 193 bearing a (R)-binol unit for the asymmetric aza-Mannich reaction of imino ester 160 with N-Boc imines 161 in very high yields, high syn-diastereoselectivity and up to 96% ee (Scheme 63). This reaction was performed in toluene with Cs2CO3 as a base and in the presence of anhydrous Na2SO4 to remove residual water. The additional (R)-axial chirality exerted a great impact on the stereochemical control.
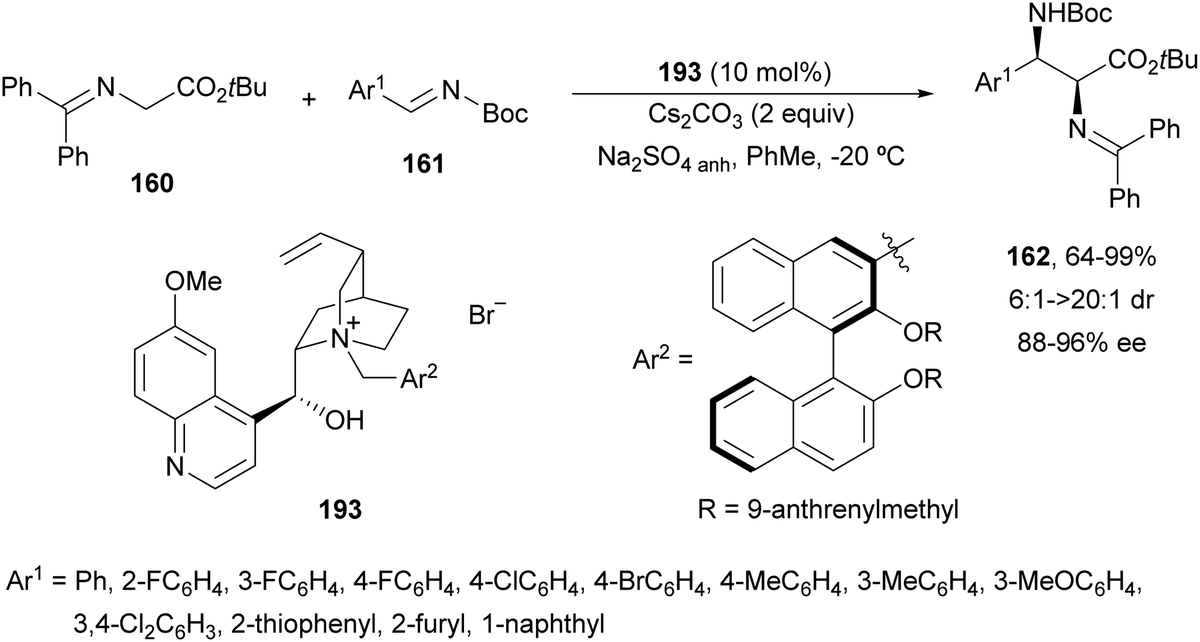 | ||
| Scheme 63 Asymmetric aza-Mannich reaction of imino esters 160 with N-Boc imines 161 using a phase-transfer catalyst 193. | ||
Maruoka and co-workers133 employed in situ generated N-Boc imines from N-Boc aminals 194 under basic conditions. This procedure was very useful for N-Boc imines which cannot be prepared by traditional methods. Imino glycinates 160 reacted with N-Boc-aminals 194 under PTC in the presence of the binaphthyl-based catalyst 195 to furnish after acidic deprotection, products 196 in good yields and diastereo- and enantioselectivities (Scheme 64a). This aza-Mannich reaction was also carried out with the alanine Schiff base 197 and aminals 194 to provide syn-α,β-diamino acid derivatives 198 with a quaternary stereocenter in a diastereomeric ratio of >20:1 (Scheme 64b).
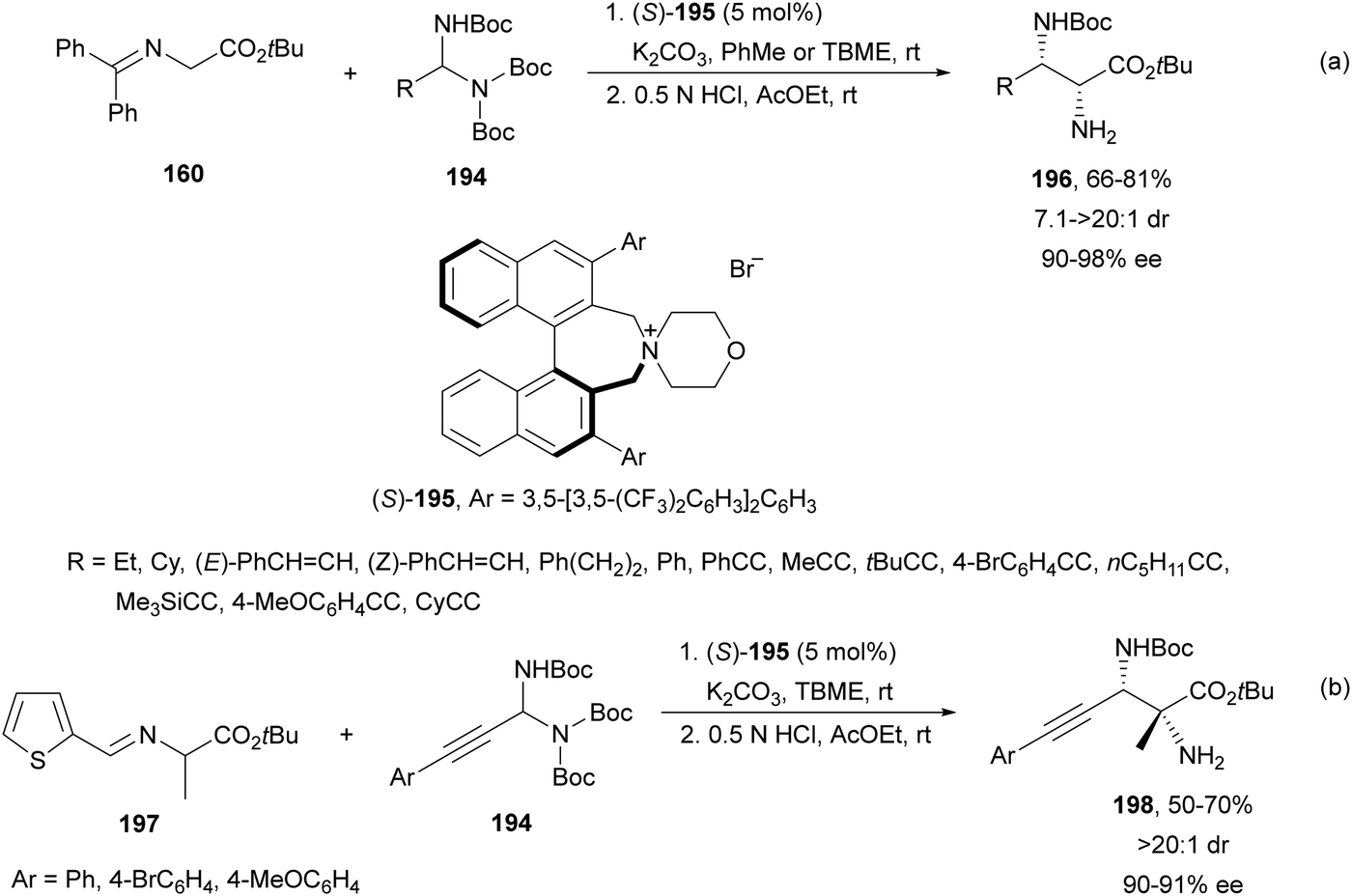 | ||
| Scheme 64 Asymmetric aza-Mannich reaction of imino esters 160 or 197 with N-Boc-aminals 194 using phase-transfer catalyst (S)-195. | ||
α,β-Diaminophosphonic acid derivatives 202 have been synthetized by an aza-Mannich reaction of a phosphoglycine Shiff base 199 with in situ generated N-Boc imines from α-amido sulfones 200 under asymmetric PTC by Bernardi, Ricci and co-workers.134 The corresponding syn-products 202 were obtained in the presence of the quininium salt 201 as a chiral phase-transfer catalyst in good yields, total diastereoselectivity and up to 94% ee (Scheme 65).
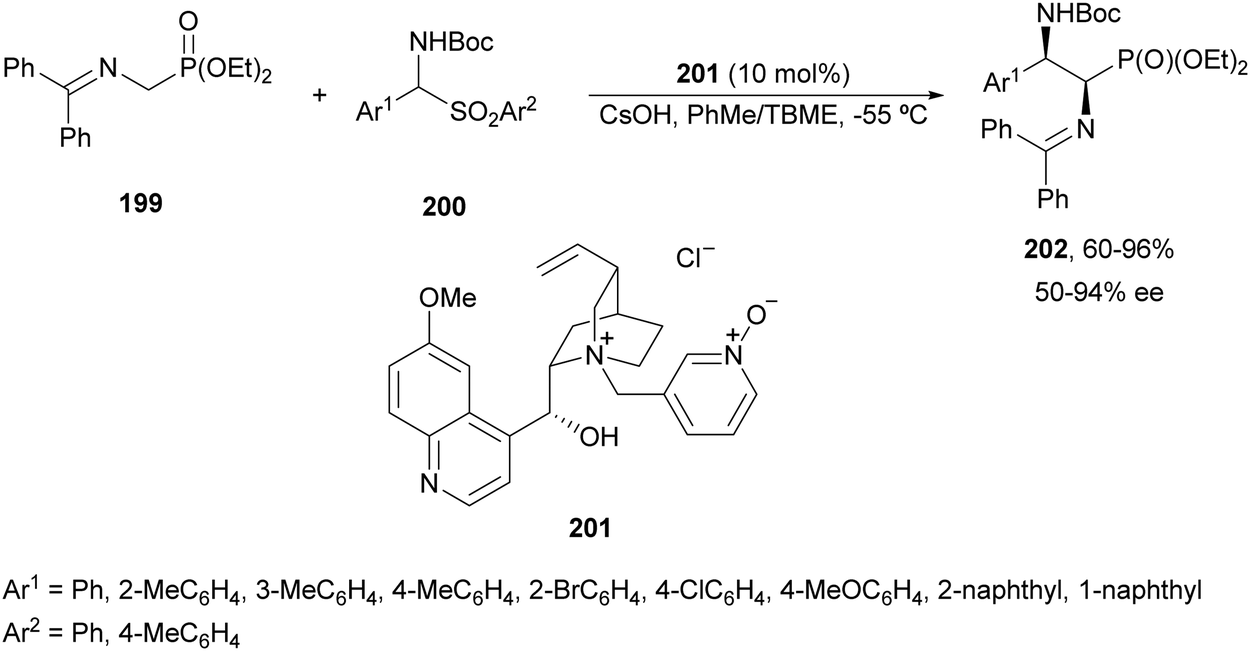 | ||
| Scheme 65 Asymmetric aza-Mannich reaction of phosphoglycine Shiff base 199 with in situ generated N-Boc imines using a phase-transfer catalyst 201. | ||
The first example using a chiral base as a catalyst was described by Kobayashi and co-workers.135 Fluorenone glycine imine 203 has been used for the aza-Mannich reaction with N-Boc imines 161 in the presence of chiral guanidine 204 as an organocatalyst (Scheme 66). α,β-Diamino acid derivatives syn-205 were isolated diastereoselectively in good yields and ee's.
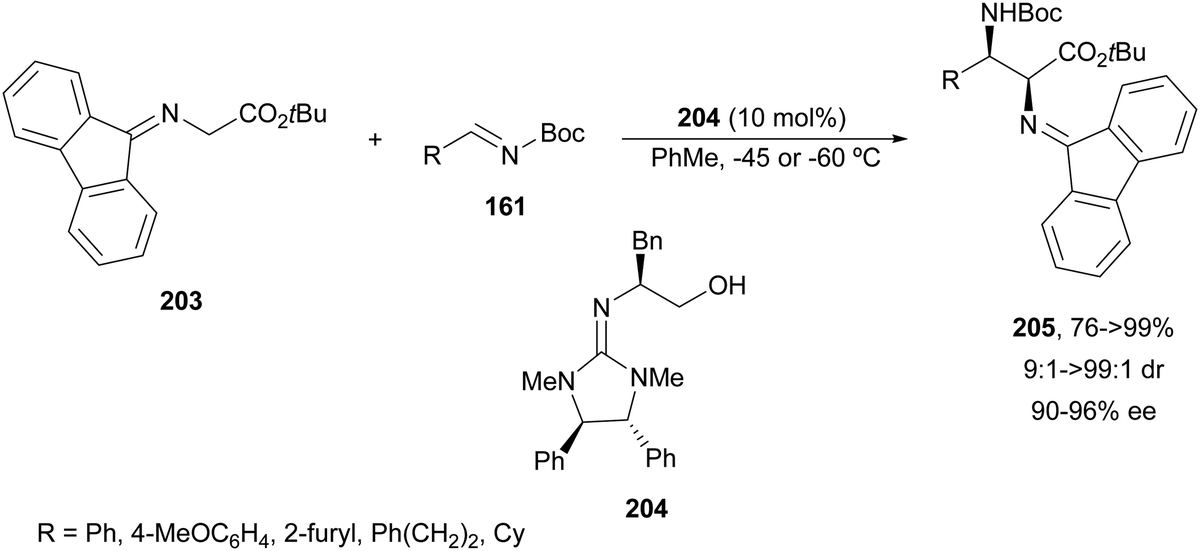 | ||
| Scheme 66 Asymmetric aza-Mannich reaction of fluorenone glycine imino ester 203 with N-Boc imines 161 under base 204 catalysis. | ||
Glycine imino ester 160 (R = Me) has been allowed to react with N-Boc-aldimines generated in situ from amido sulfones 200 using a Cinchona alkaloid thiourea 206 as a chiral organocatalyst (Fig. 8). Barbas III and co-workers136 performed this asymmetric direct aza-Mannich reaction in trifluoromethylbenzene in the presence of a saturated solution of Na2CO3 at 4 °C to provide products syn-162 with high yields (62–98%) and excellent ee (>95–>97%) and dr (>99:1). Bandar and Lambert137 employed a cyclopropenimine 207 as a chiral base for the reaction of glycine imino ester 162 (R = Me, tBu, and Bn) with N-Boc imines from aromatic aldehydes, using NaOtBu as a base in THF at 0 °C (Fig. 8). α,β-Amino acid derivatives syn-162 were obtained in 63–99% yield, 96:4 dr and 38–97% ee.
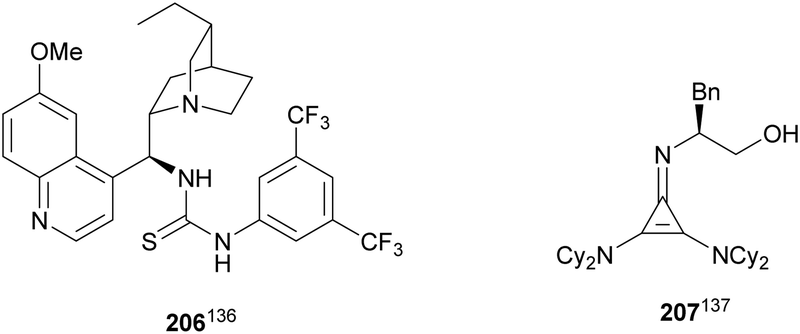 | ||
| Fig. 8 Chiral bases used as organocatalysts for the aza-Mannich reaction of imino glycinates 160 with N-Boc imines. | ||
The chiral guanidine 210 has been used as an organocatalyst for the aza-Mannich reaction of aldimino glycinate 208, derived from 3,5-di-tert-butylsalicylic aldehyde with sterically hindered N-(8-quinolyl)sulfonyl aldimines 209 (Scheme 67).138 Products 211 resulted in very good yields but modest diastereo- and enantioselectivities.
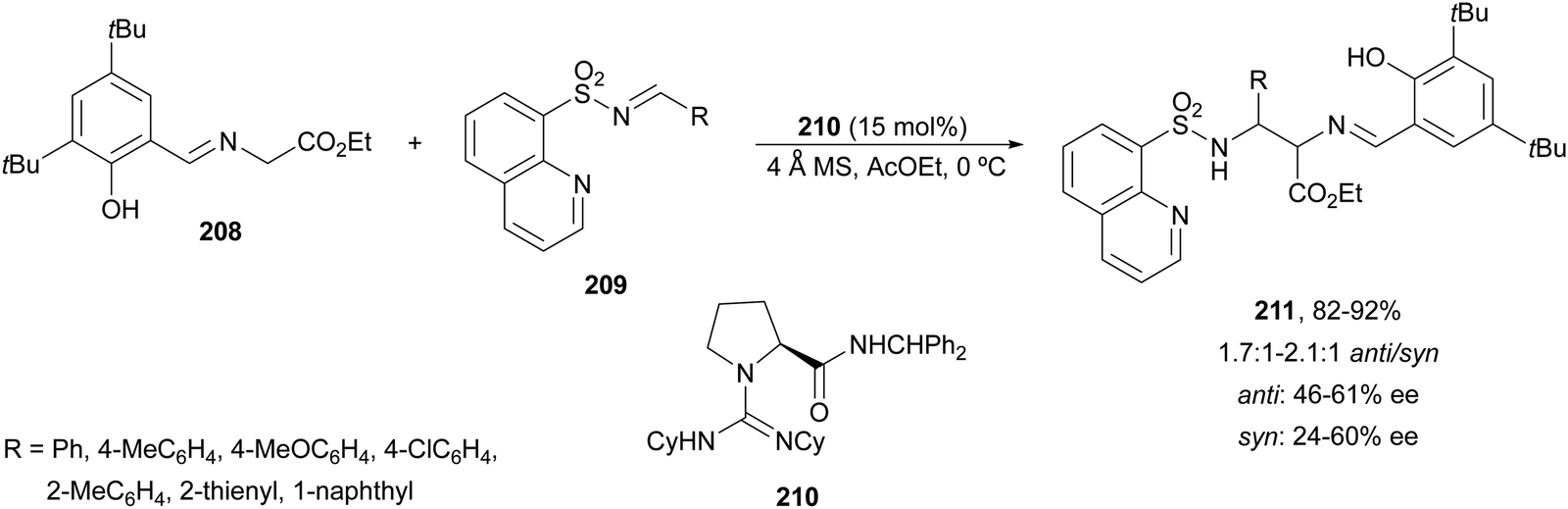 | ||
| Scheme 67 Asymmetric aza-Mannich reaction of glycine imino ester 208 with aldimines 209 under guanidine 210 catalysis. | ||
Yuan, Zhao and co-workers139,140 described a biomimetic asymmetric aza-Mannich reaction using carbonyl catalysis inspired by pyridoxal-depending enzymes. The reaction of unprotected tert-butyl glycinate hydrochloride with N-diphenylphosphinoyl imines 167 was performed in the presence of the chiral pyridoxal (R,S)-212 to furnish mainly anti-products 169 in good yields and excellent diastereo- and enantioselectivities (Scheme 68). In the plausible catalytic mechanism, initial condensation of the catalyst 212 with glycinate forms an imino ester I, which after deprotonation generates carbanion II. Subsequent addition of this enolate to imine 167 through a TS gives intermediate III, which after hydrolysis yields product 169 and regenerates the catalyst.
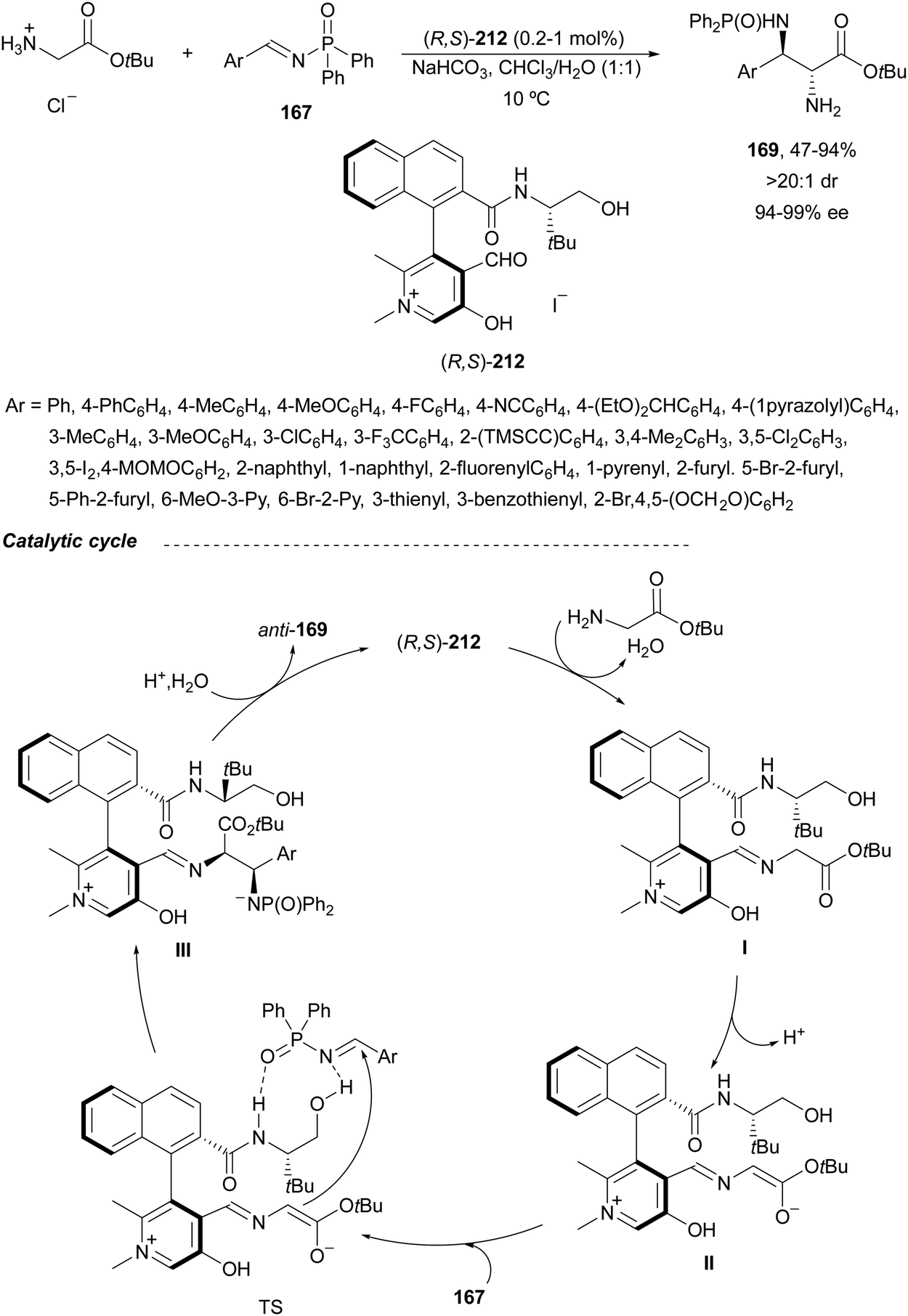 | ||
| Scheme 68 Asymmetric aza-Mannich reaction of tert-butyl glycinate hydrochloride with imines 167 under carbonyl catalysis. | ||
Organocatalytic methods for the asymmetric aza-Mannich reaction of imino glycinates with imines, which are based on chiral phase-transfer catalysis, are better and use more simple reaction conditions than with metal complexes. In the case of using chiral bases, mainly syn-diamino acid derivatives are obtained. However, the biomimetic carbonyl-based catalysis gave very efficient anti-products.
:13 dr and 73% ee.
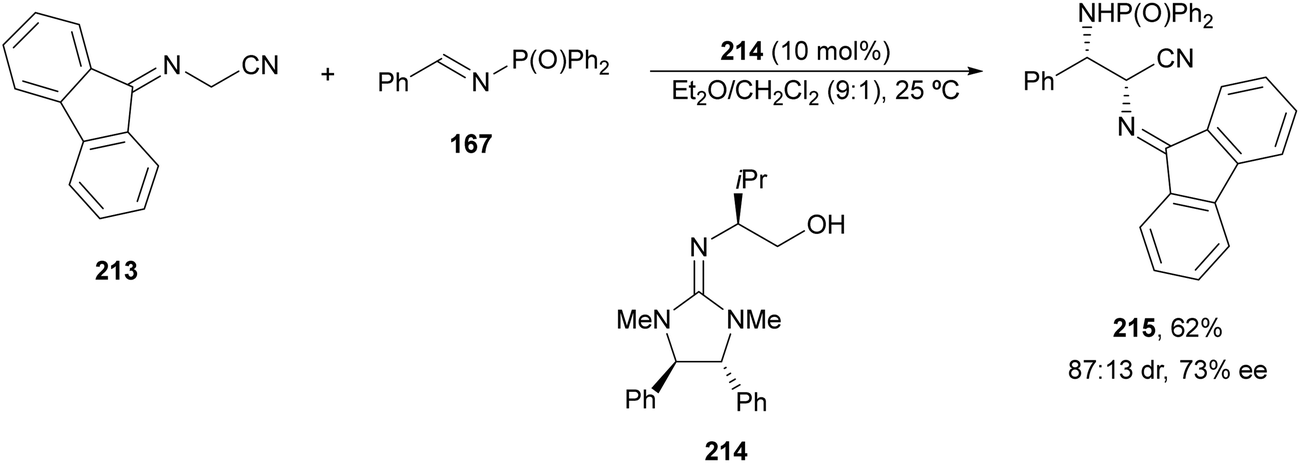 | ||
| Scheme 69 Asymmetric aza-Mannich reaction of imino nitrile 213 with imines 167 under base 214 catalysis. | ||
The former N-fluorenylidene α-amino nitrile 213 was further employed as a nucleophile with ketimines 216 bearing a thiophosphinoyl group by Kumagai, Shibasaki and co-workers.142 The Mannich reaction was carried out under Cu/(R)-DMM-Garphos 217 catalysis using LiOtBu as a base at −78 °C to furnish mainly α,β-diamino alkanenitriles anti-218 with a quaternary stereocenter up to 99% yield, up to 95:5 dr and 95% ee (Scheme 70). The thio-DPP protecting group was transformed into DPP by treatment with H2O2 and subsequently into a NH2 group by hydrolysis with 12 M HCl at 40 °C.
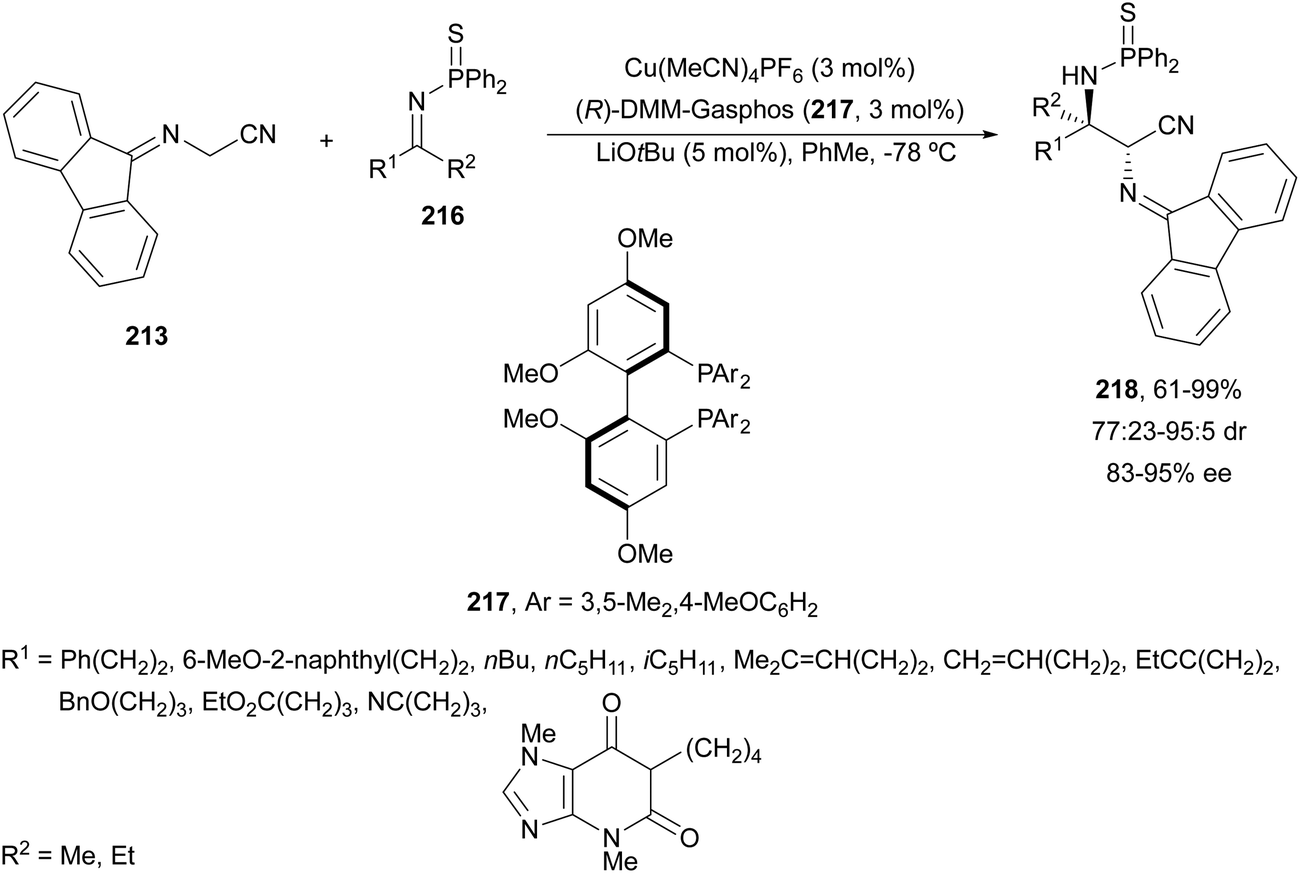 | ||
| Scheme 70 Asymmetric aza-Mannich reaction of imino nitrile 213 with ketimines 216 under Cu/217 catalysis. | ||
Nakamura and co-workers143 reported the same year the benzylidene α-amino nitrile 219 reaction with N-(2-pyridinesulfonyl)imine 220 using a chiral bis(imidazoline)palladium complex 221 and Ag(acac) as a catalyst in THF at −60 °C (Scheme 71). The corresponding products 222 were obtained after deprotection of the benzylidene group with HCl in THF at room temperature. Total deprotection of compound 222a was carried out with Mg in MeOH to provide product 223 in 60% yield. A plausible catalytic cycle was proposed starting from complex I, which reacts with the imino nitrile 219 to give intermediate II by coordination of the cyano group to the Pd atom. After deprotonation of II, the palladium complex III is formed. In the next step, the nucleophilic addition of III to the aldimine 220 gives intermediate IV, which undergoes protonation and decomplexation to provide the product and regenerates the catalyst. On the basis of theoretical calculations, a TS was proposed.
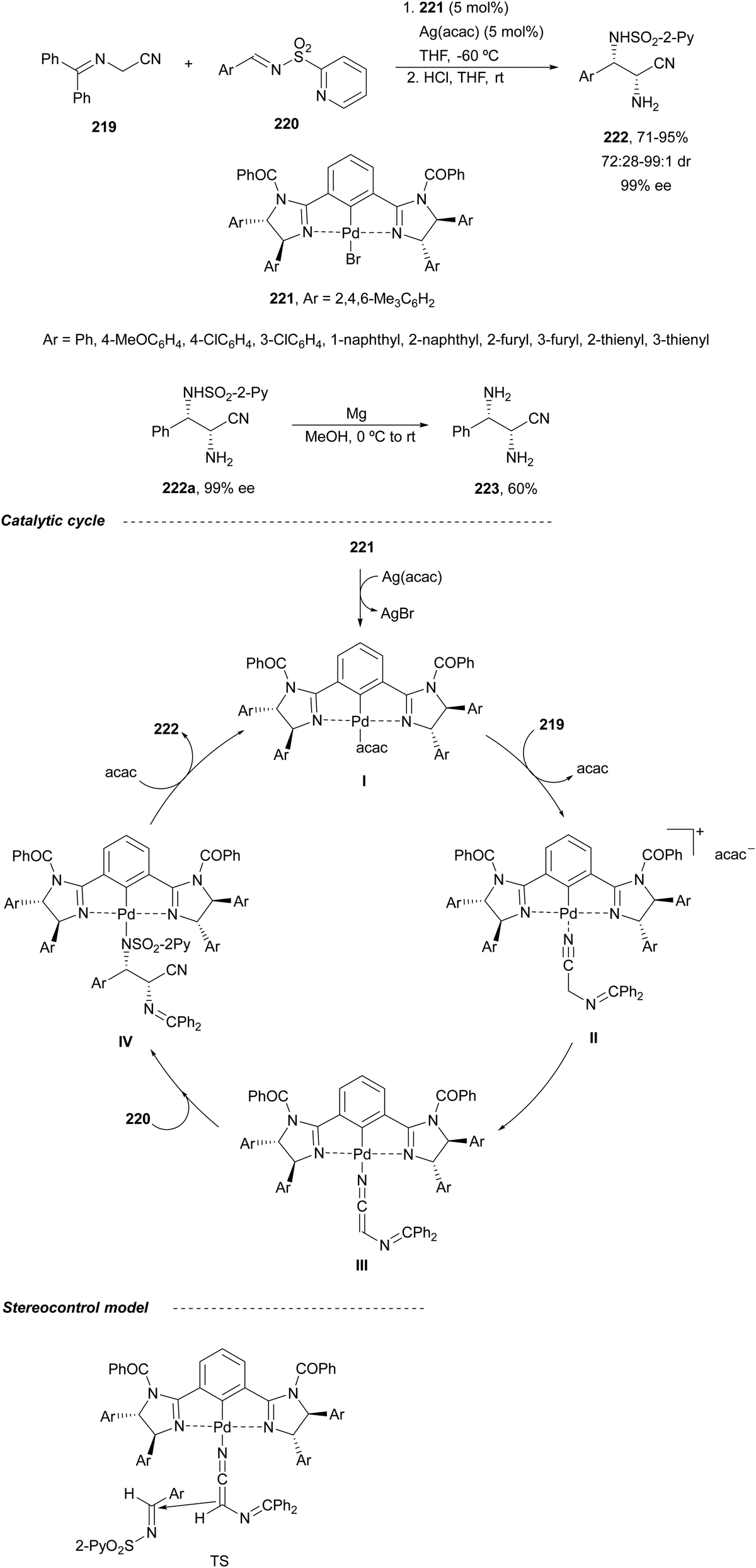 | ||
| Scheme 71 Asymmetric aza-Mannich reaction of imino nitrile 219 with aldimines 220 under Pd complex 221/Ag(acac) catalysis. | ||
Imino nitriles underwent asymmetric aza-Mannich reaction with activated aldimines to give syn-adducts only under metal catalysis. Only ketimines provided anti-adducts under Cu-catalyzed conditions.
 | ||
| Scheme 72 Aza-Mannich reaction of azlactones 224 with N-protected imines 161. | ||
Ooi and co-workers145 described the reaction of azlactones 224 with N-sulfonyl imines 161 using a chiral tetraaminophosphonium carboxylate 226 as a phase-transfer catalyst to give mainly syn-products 225 up to 99% yield, 12:1 dr and 97% ee (Fig. 9). After two-step deprotection with aqueous H2SO4 in THF followed by hydrolysis with concentrated HCl, the corresponding α,β-diamino acid hydrochloride (R1 = Bn, R3 = Me) was obtained in 87% yield maintaining a 92% ee. The same group used a C1 symmetric ammonium betaine 229 as a catalyst for the aza-Mannich reaction of thiazolones 227 with N-Boc imines 161 to give mainly products anti-228 with high yields, up to 15:1 dr and 99% ee (Scheme 73). Compound 228a was transformed in a two-step procedure into α,β-diamino acid ester derivative anti-230 in 60% yield and 99% ee.146
 | ||
| Fig. 9 Chiral catallysts used for the asymmetric aza-Mannich reaction of azlactones 224 with N-arylsulfonyl imines 161. | ||
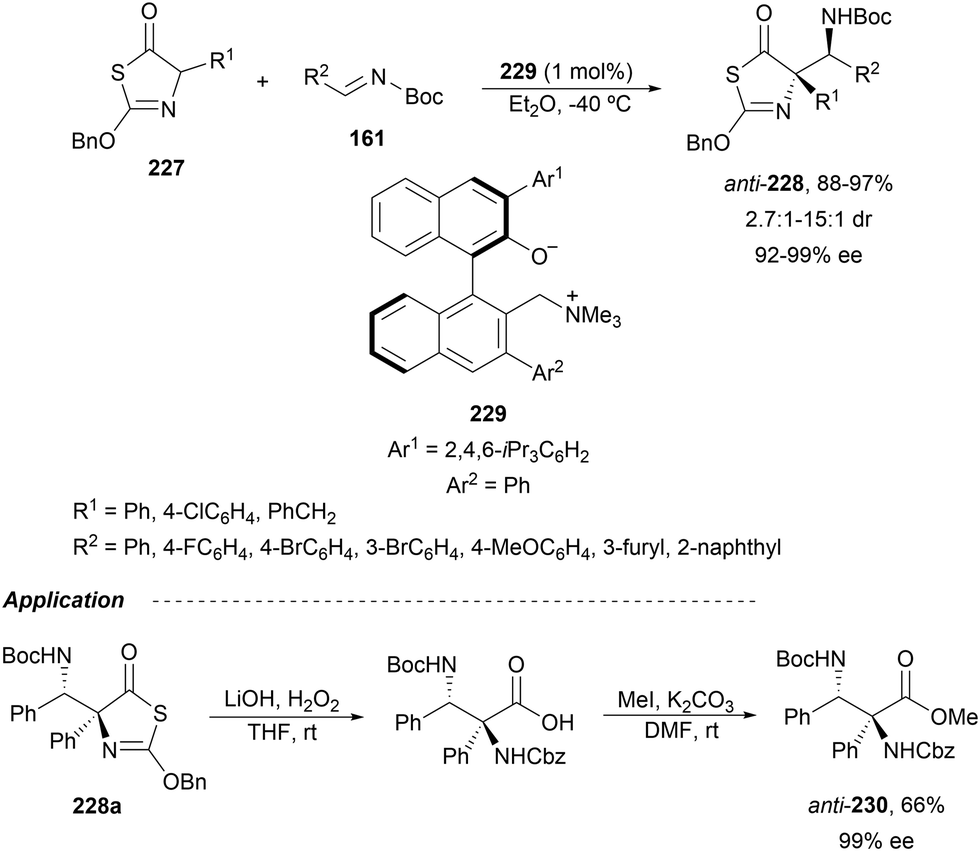 | ||
| Scheme 73 Asymmetric aza-Mannich reaction of thiazolones 227 with N-Boc imines 161 under betaine 229 catalysis. | ||
Wang and co-workers147 employed a chiral Cinchona-derived base 231 for the aza-Mannich reaction of azlactones 224 with N-tosyl imines 161 to furnish syn-products 225 with 49–94% yields, 3:1–>30:1 dr and 80–97% ee (Fig. 9). A chiral bis(betaine) 232 catalyst was employed by Gong and co-workers148 for the asymmetric aza-Mannich reaction of azlactones 224 with aliphatic N-tosyl imines 161 to obtain anti-products 225 with 76–99% yields, 2.1:7.1 dr and 96–99% ee (Fig. 9). The chiral CPA (S)-TRIP (152) was employed by Amarante and co-workers149 for the addition of azlactones 224 to N-sulfonyl imines 161 to afford anti-products 225 up to 74% yield, >19:1 dr and 98% ee. Šebesta and co-workers150 reported the use of a chiral thiourea 233 as a catalyst for the addition of azlactones 224 to N-sulfonyl imines 161 to obtain syn-products 225 up to 82% yield, 12:1 dr and 95% ee (Fig. 9). In 2011, Toste and co-workers151 reported the same aza-Mannich catalyzed by a chiral bisphosphine/(AuOBz)2 complex 234, which activates the azlactones 224 to give anti-products 225 with 50–98% yield, 6:1–>20:1 dr and 83–94% ee (Fig. 9). The combination of a chiral phosphate anion derived from the CPA (S)-183 and silver ion 235 is an excellent ion pair catalyst for this type of aza-Mannich reaction to provide syn-products 225 up to 95% yield, 25:1 dr and 99% ee (Fig. 9).152
In the above mentioned examples, α,β-diamino acid precursors bear a quaternary stereocenter at the α-position. More recently, Ren and co-workers153 reported the synthesis of α,β-diamino acid derivatives containing two consecutive quaternary stereocenters by the aza-Mannich reaction of azlactones 224 with isatin-derived ketimines 170 (Scheme 74). In this case, a chiral bifunctional squaramide 236 was used as an organocatalyst affording Mannich adducts 237 in 40–95% yields, up to >20:1 dr and up to 97% ee. Compound 237a was transformed into the α,β-diamino acid derivative 238 by treatment with K2CO3 in MeOH.
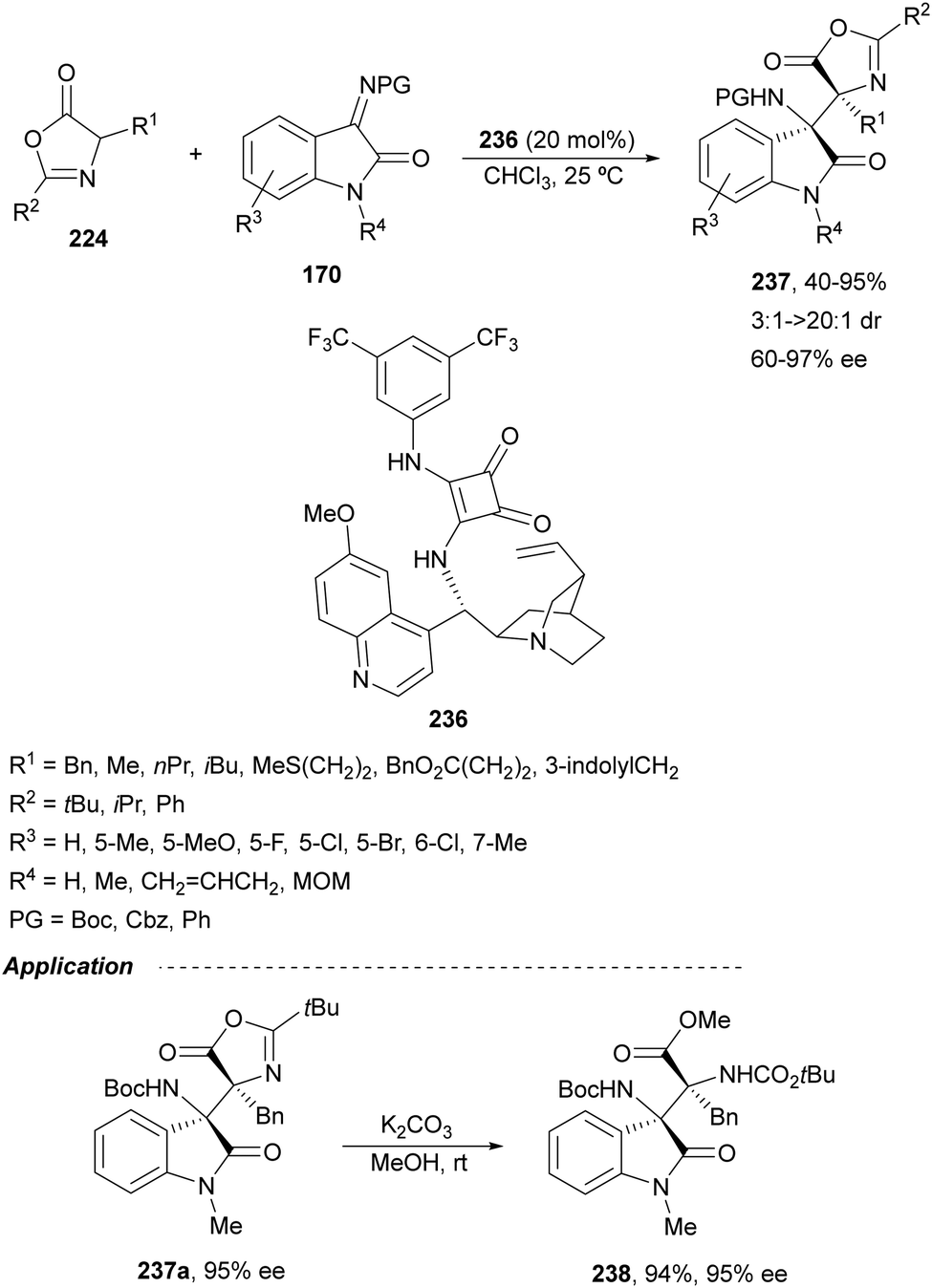 | ||
| Scheme 74 Asymmetric aza-Mannich reaction of azlactones 224 with isatin-derived ketimines 170 under squaramide 236 catalysis. | ||
In the case of asymmetric organocatalytic aza-Mannich reactions, azlactones are the appropriate nucleophiles for the synthesis of α,β-diamino acids with a quaternary stereocenter at the α-position in the case of aldimines and two quaternary stereocenters in the case of ketimines.
Lin and co-workers154–156 reported for the first time the aza-Mannich type reaction of ethyl isocyano acetate 239 with N-tosyl imines 161 in the presence of Au(I)/diphosphine 240 to provide diastereoselectively cis-2-imidazolines 241 in moderated yields and up to 99% ee (Scheme 75). These imidazolines were transformed into anti-α,β-diamino esters 242 by treatment with concentrated HCl in EtOH with 22–56% yields after the two step process.
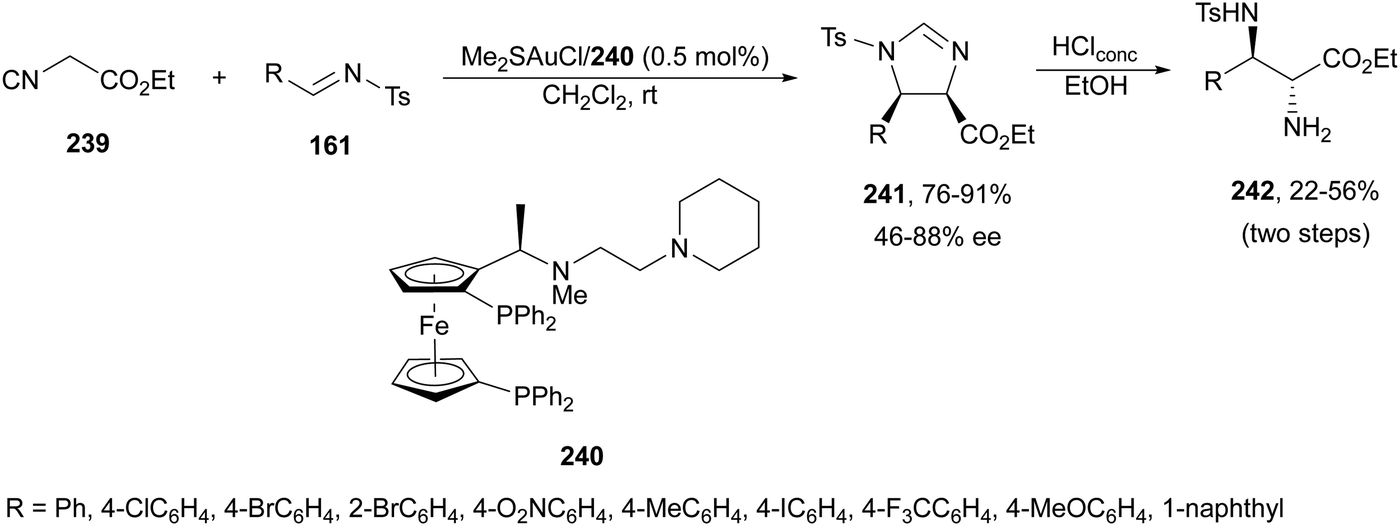 | ||
| Scheme 75 Asymmetric aza-Mannich-cyclization reaction of methyl isocyano acetate 239 with N-tosyl imines 161 under Au(I)/diphosphine 240 catalysis. | ||
The same transformation was carried out by Szabó and co-workers157 using chiral palladium-pincer complexes based on binol or biphenantrol to obtain 2-imidazolines 241 (R = Ph) as a mixture of cis and trans diastereomers (up to 1:4) with high yields (98%) and modest enantioselectivities.
Shi and co-workers158 employed a Cinchona alkaloid squaramide 244 in combination with AgOAc for the reaction of α-substituted isocyano acetates 239 with cyclic trifluoromethyl ketimines 243 to obtain tetrahydroimidazo[1,5-c]quinazoline derivatives 245 in excellent yields and good to excellent diastereo- and enantioselectivities (Scheme 76). In the proposed TS model, the α-proton of isocyano acetate 239a (R1 = Ph, R2 = Me) is deprotonated by the quinuclidine unit of catalyst 244 due to the activation of silver by chelation of the terminal carbon atom of isocyano acetate. This organocatalyst 244 forms a hydrogen bonding of the MeO group of the enolate of 239 and then NH of the squaramide forcing the isocyanate to be delivered through the Re face to its Si face of CN. Subsequent 5-endo–dig cyclization of intermediate I affords the cyclic product 245a.
 | ||
| Scheme 76 Asymmetric aza-Mannich/cyclization of α-substituted isocyano acetates 239 with cyclic trifluoromethyl ketimines 243 under AgOAc/squaramide 244 catalysis. | ||
Oxazole-imidazolines 248 have been prepared by Zhao and co-workers159 using cyclic α-imino esters 246 and 2 equivalents of isocyano acetates 239 (Scheme 77). In this case, Ag2O and the quinine derived phosphine 247 were used as catalysts to furnish products 248 in high yields, total diastereoselectivity and up to 99% ee. Hydrolysis of product 248a with TsOH in CHCl3/H2O (2:1) at room temperature formed the α,β-diamino ester 249 in 90% yield and the same ee.
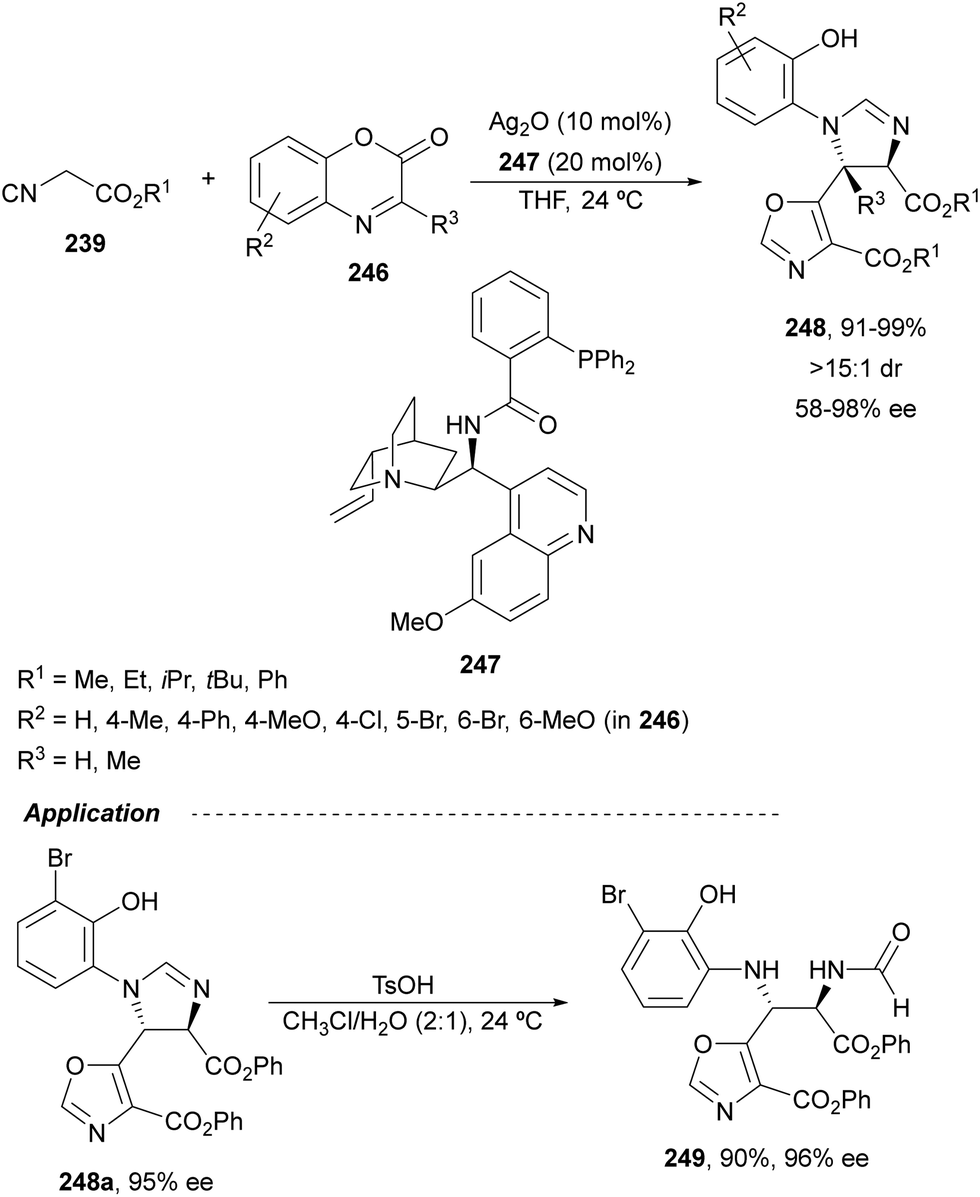 | ||
| Scheme 77 Asymmetric aza-Mannich/cyclization of isocyano acetates 239 with cyclic imino esters 246 under Ag/phosphine 247 catalysis. | ||
Dixon and co-workers160 reported the aza-Mannich/cyclization of isocyano acetates 239 and N-diphenylphosphinoyl (DPP) ketimines 250 using Ag2O and the chinchonine-derived aminophosphine 251 as a catalyst (Scheme 78). trans-2-Imidazolines 252 (R1 = H) were obtained with high yields, up to 99:1 dr and up to 99% ee. The removal of the DPP group was performed with 1 M HCl in DCM at room temperature to provide 2-imidazolines 253 in 59–95% yields. In one example, 253a was chosen to hydrolyze under aqueous KOH to α,β-diamino acid 254 in 45% yield without deterioration of enantiopurity. Later, the same group studied the same process with α-substituted isocyano acetates 239.161 Tetrasubstituted 2-imidazolines 252 (R1 ≠ H) were prepared using AgOAc and the quinine-derived aminophosphine 255 as a catalyst (Scheme 78), which were isolated as unprotected derivatives trans-253 in 28–90% yields and 87–95% ee by treatment of compounds 253 with 1 M HCl in DCM at room temperature. In the proposed TS based on experimental studies, the N atom of the ketimine forms a hydrogen bonding with the N–H of the amide, both of which orientate the electrophile 250 towards the nucleophilic addition of the enolate of the isocyano acetate bonded to the Ag atom. The ester group of 239 is oriented away from the aromatic ring of the ketimine 250.
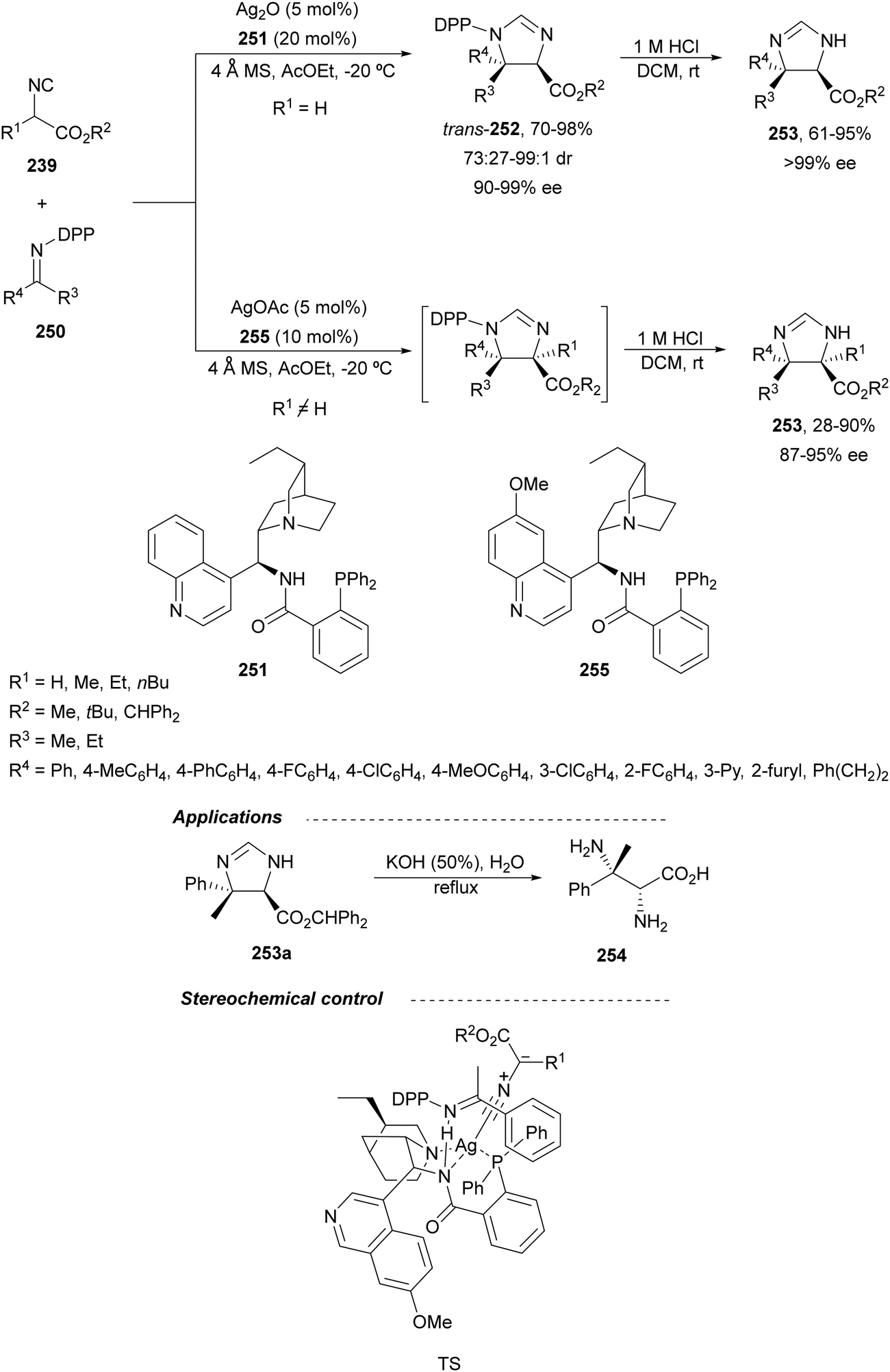 | ||
| Scheme 78 Asymmetric aza-Mannich/cyclization of isocyano acetates 239 with N-diphenylphosphinoyl ketimines 250 under Ag/phosphines 251 and 255 catalysis. | ||
Nakamura's group reported independently similar aza-Mannich/cyclization processes of unsubstituted162 and α-substituted163 isocyano acetates 239 with N-DPP ketimines 250. In the first case,162 they employed Cu(OTf)2 and picolinamide ligand 256, derived from cinchonine, as a catalyst in the presence of Cs2CO3 as a base (Scheme 79). Products cis-252 were not isolated due to thermal instability, instead, the corresponding tosyl imidazolines 257 resulted by a two-step process based on hydrolysis with 4 M HCl in dioxane followed by tosylation with TsCl/Et3N, up to 92:8 dr and 99% ee. Compound 257a was further transformed into the α,β-diamino acid methyl ester derivative 258 quantitatively and in 99% ee. Starting from α-substituted isocyano acetates,163 the same process was carried out with NiCl2 as Lewis acid and 9-amino-9-epi-cinchonidine 259 as catalysts (Scheme 79). In this case, N-DPP imidazolines trans-252 were isolated in high yields and diastereo- and enantioselectivities. Removal of the DPP group was also carried out with 4 M HCl in dioxane for compound 252b (R1 = R2 =Me, R3 = p-tol) to obtain product 253b in 79% yield. Both catalytic cycles with Cu(OTf)2162 and NiCl2163 were proposed. In Scheme 79 is depicted the last case for the synthesis of tetrasubstituted imidazolines trans-252.163 The reaction of isocyano acetate 239 with catalyst 259 and NiCl2 in the presence of Cs2CO3 as a base forms intermediate I. Subsequent coordination of I with ketimine 250 (R4 = Me) forms intermediate II, which undergoes aza-Mannich reaction to give intermediate III. Cyclization of III promotes imidazolidine formation giving intermediate IV, which regenerates I and affords product 252. In the TS for the Mannich reaction, the oxygen of ketimine 250, two nitrogen atoms from the picolinoyl moiety in 259 and the isocyano group coordinate to the nickel atom in a distorted tetrahedral form. In addition, the quinuclidine moiety makes hydrogen bonding with the ketene hemiacetal of α-isocyano acetate, which attacks the ketimine approaching by the Si face in both cases avoiding steric repulsion between the two methyl groups.
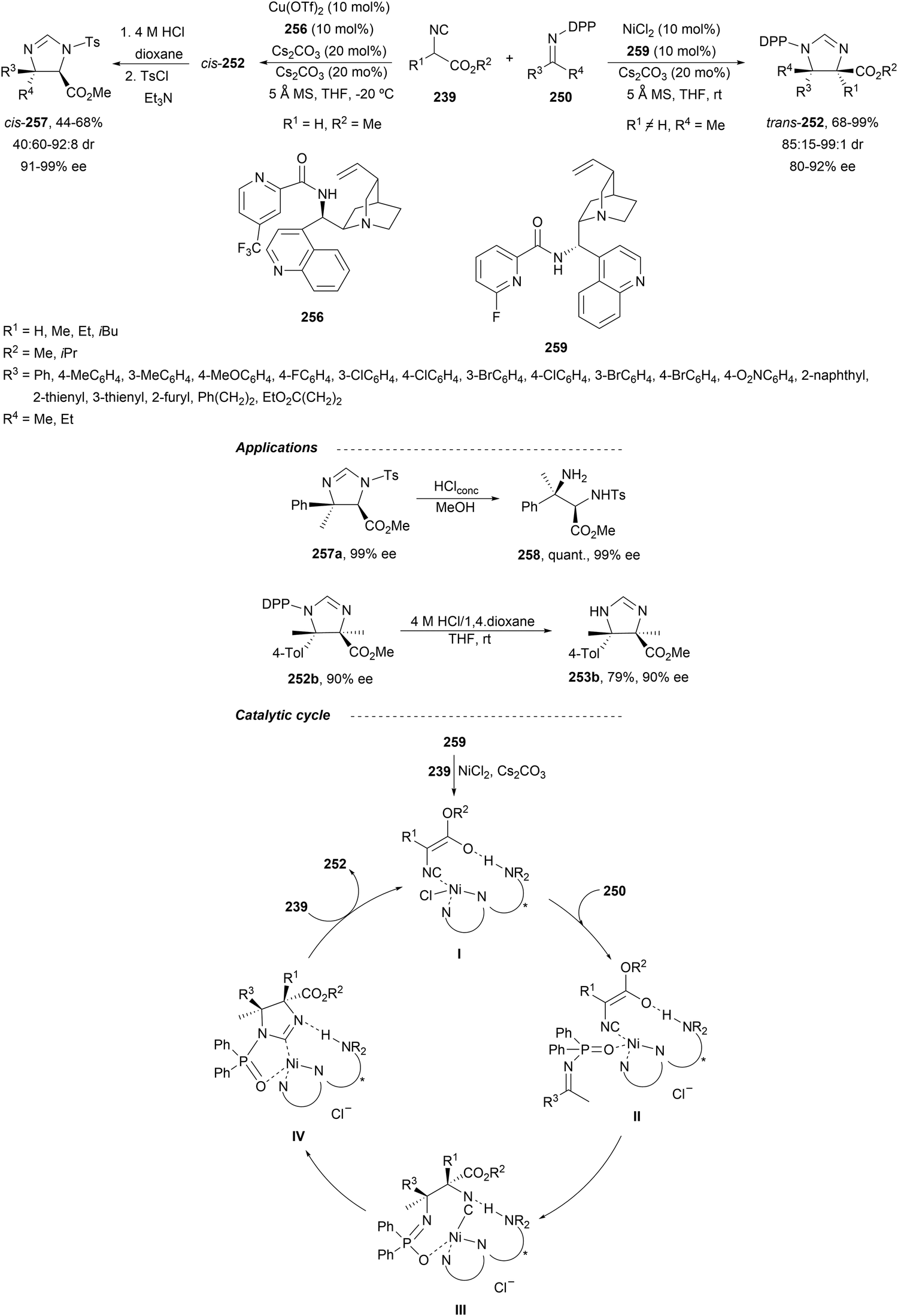 | ||
| Scheme 79 Asymmetric aza-Mannich/cyclization of isocyano acetates 239 with N-DPP ketimines 250 under Cu(OTf)2/cinchonine derivative 256 and NiCl2/cinchonidine derivative 259 catalysis. | ||
The former aza-Mannich/cyclization process has been applied by Zhao, Shi and co-workers164 to cyclic sulfamide ketimines 260 to obtain imidazoline-fused sulfahydantoin derivatives 262 (Scheme 80). Isocyano acetates 239 reacted with 4-aryl-3-carbonyl-1,2,5-thiadiazole-1,1-dioxide type ketimines 260 under cooperatively catalysis by squaramide 261 and AgOAc as Lewis acid to provide compounds 262 in excellent yields and good to excellent stereoselectivities. A plausible TS was proposed to explain the observed stereochemical results. Enolization of isocyano acetate by the quinuclidine nitrogen of catalyst 261 due to activation of Ag chelating to the isocyano group resulted in a hydrogen bonding between the OH group and the tertiary amine. In addition, the squaramide unit directed the ketimine 260 and isocyano acetate 239 through a hydrogen bonding interaction between the NH and the sulfonyl groups, ketimine nitrogen atoms or methoxy groups. These orientation forces the approach of 239 by the Si face to the Re face of the CN giving for 262 the two stereocenters with a (R,S)-configuration.
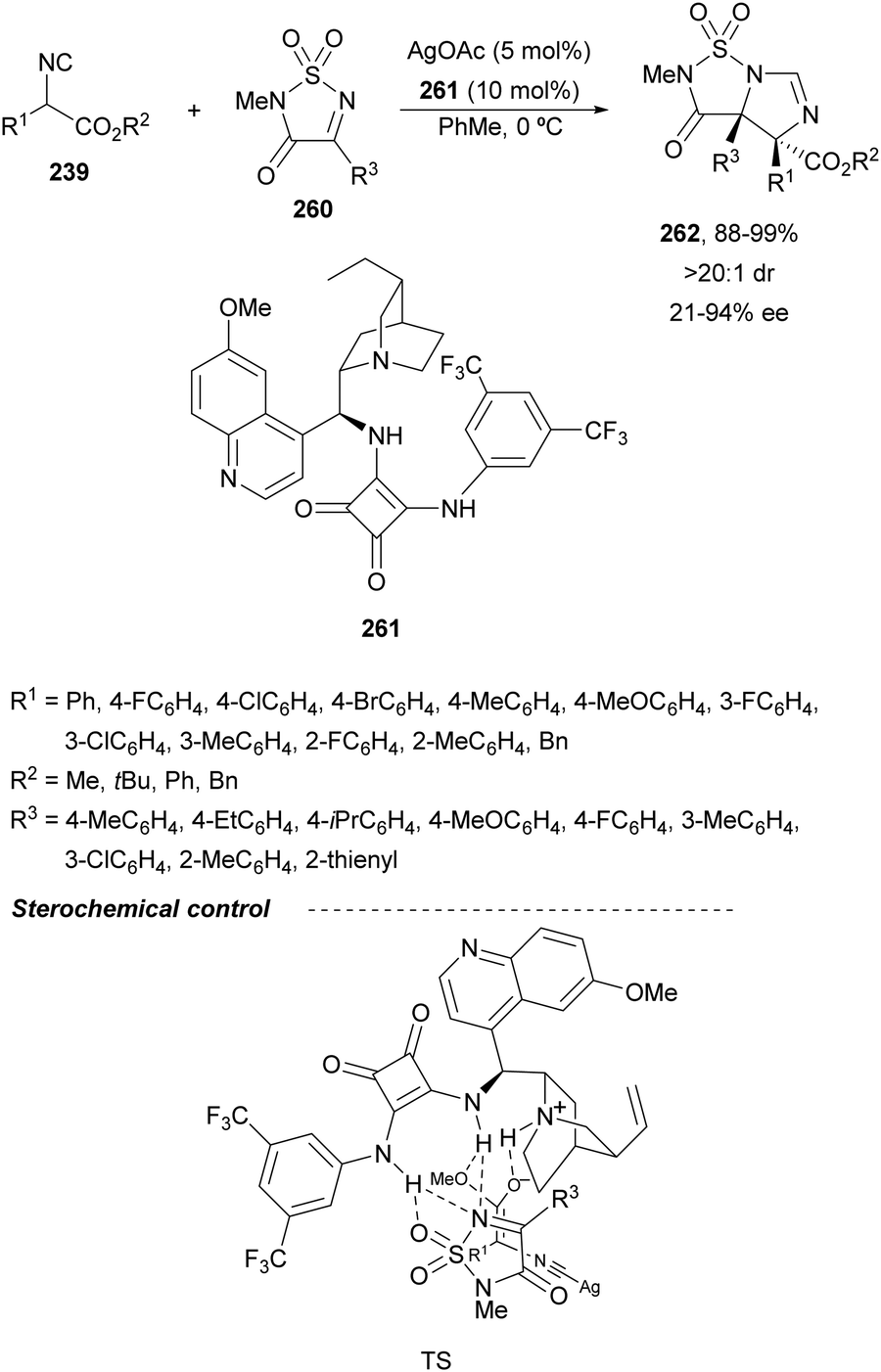 | ||
| Scheme 80 Asymmetric aza-Mannich/cyclization of isocyano acetates 239 with cyclic sulfamide ketimines 260 under AgOAc/squaramide 261 catalysis. | ||
Organocatalyzed aza-Mannich/cyclization processes have been carried out with Cinchona-based tertiary amines as catalysts. Chan and co-workers165 employed O-acetyl quinidine 263 (10 mol%) (Fig. 10) for the reaction of methyl isocyano acetate 239 with N-tosyl imines 161 to furnish mainly trans-2-imidazolines 241 in 35–79% yields, 91:9–>99:1 trans/cis dr and 5–70% ee. Nakamura, Shibata and co-workers166 employed chiral thiourea 264 (10 mol%) as an organocatalyst for the aza-Mannich/cyclization of α-substituted isocyano acetates 239 with 2-pyridylsulfonyl imines 220 (Fig. 10). The resulting trisubstituted (4R,5S)-trans-2-imidazolines were obtained in 71–99% yields, 73:27–99:1 dr and 74–96% ee.
 | ||
| Fig. 10 Organocatalysts used for the asymmetric aza-Mannich/cyclization of isocyano acetates 241 with N-sulfonyl imines. | ||
Chiral thiourea 265 derived from quinidine has been used as an organocatalyst for the aza-Mannich reaction of α-substituted isocyano acetates 239 with isatin-derived ketimines 170 to obtain products 266 up to 93% yield, >20:1 dr and up to 98% ee (Scheme 81). These products 266 were transformed into spirooxindole imidazolines 268 in the presence of thiourea 267 as an organocatalyst.167
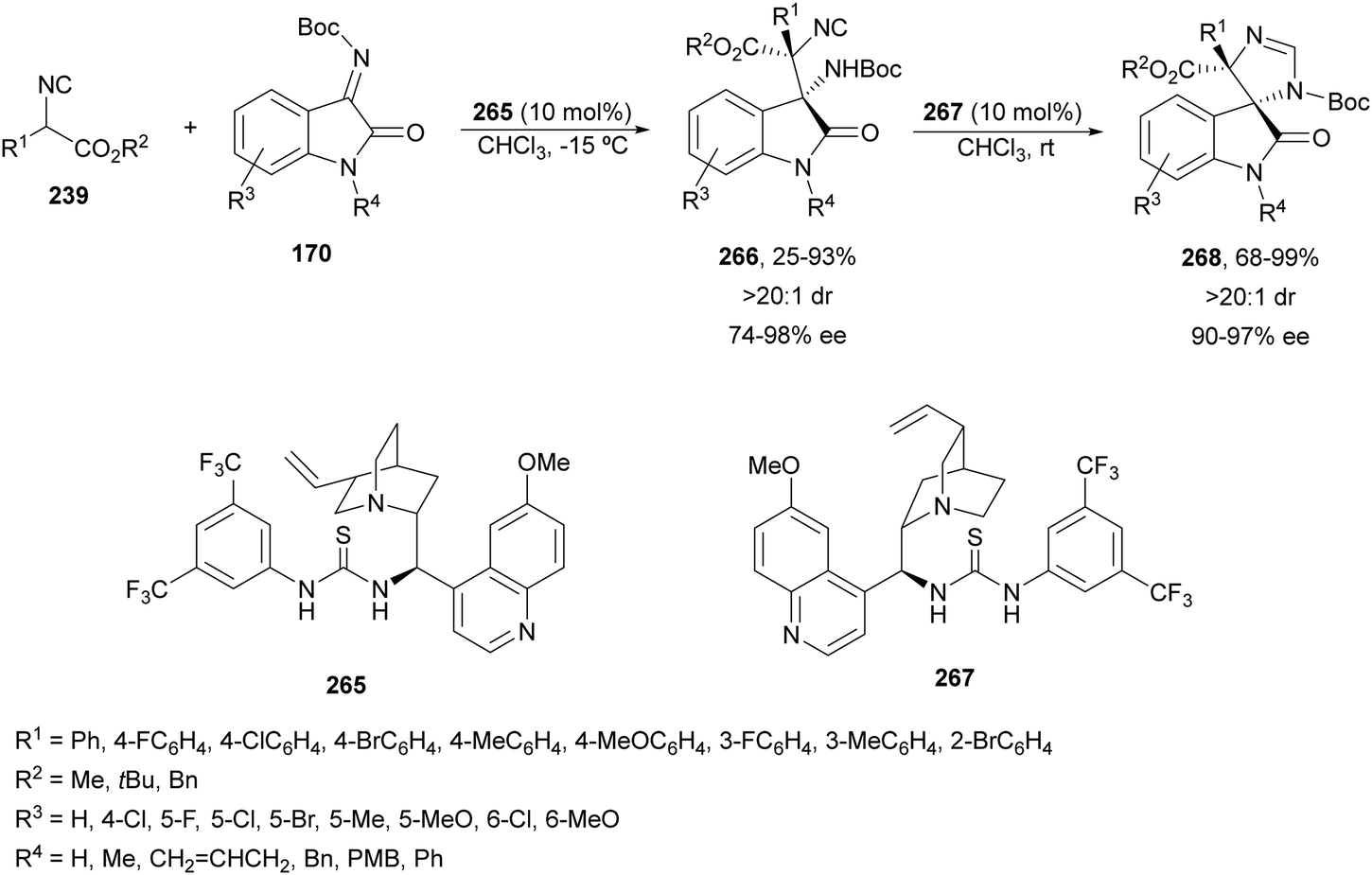 | ||
| Scheme 81 Asymmetric aza-Mannich reaction of isocyano acetates 239 with N-Boc-isatin-derived ketimines 170 under thiourea 265 catalysis. | ||
Recently, Zhao and co-workers168 reported the aza-Mannich/cyclization of α-substituted isocyano acetates 239 with N-(2-benzothiazolyl) imines 269 to obtain benzothiazole-dihydroimidazoles 270 with good to excellent yields, modest dr and excellent ee's (Scheme 82). In this case, the squaramide organocatalyst 261 was used as in the case of cyclic sulfamide ketimines 260.164 Product 270a was treated with NaBH3CN and AcOH in methanol to obtain benzothiazole-imidazolidine 271 as the only diastereomer which has been hydrolyzed using concentrated sulfuric acid to the corresponding N-(2-benzothiazolyl)-α,β-diamino acid 272 in 75% yield and >99% ee.
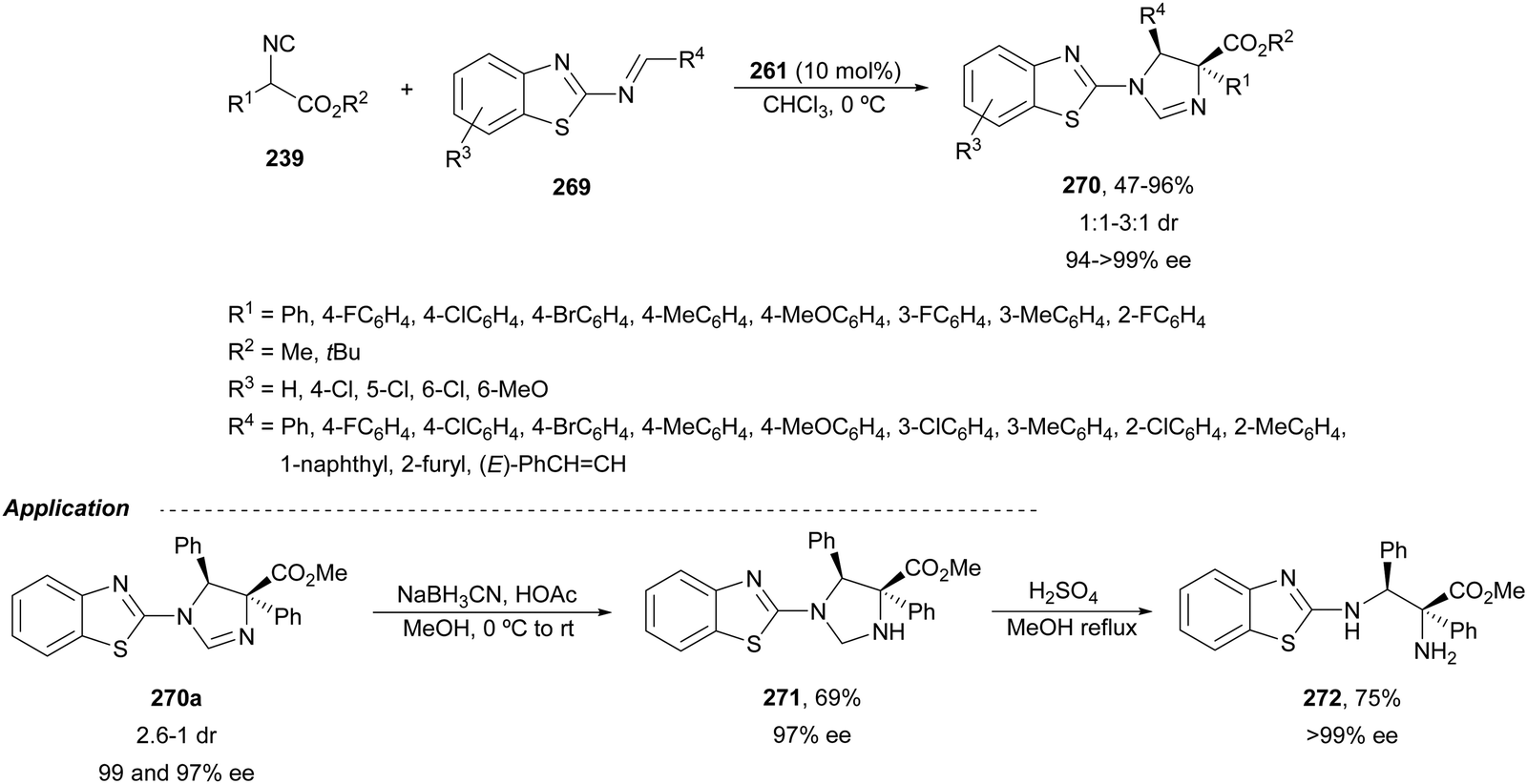 | ||
| Scheme 82 Asymmetric aza-Mannich/cyclization of isocyano acetates 239 and N-(2-benzothiazolyl)imines 269 under squaramide 261 catalysis. | ||
Isocyano acetates react with aldimines and ketimines under metal (Ag, Cu, and Ni) salts and a Cinchona-derived base or under phosphine catalysis to give 2-imidazolines through a aza-Mannich/cyclization process. Organocatalysts derived from Cinchona alkaloids have also been employed for these types of aza-Mannich reactions. These imidazolines are transformed by hydrolysis mainly into anti-α,β-diamino acid derivatives.
In 2007, Willis and co-workers169 reported the enantioselective addition of the isothiocyanate-substituted oxazolidinone 273 to N-tosyl aldimines 161 in the presence of Mg(ClO4)2 and ligand Ph-Dbfox 274 using the Hünig base DIPEA (Scheme 83). cis-Cyclic thioureas 275, precursors of anti-α,β-diamino acid derivatives, resulted from in situ cyclization of the acyclic anti-adducts in high yields, moderate to high diastereoselectivity and high enantioselectivity. Compound 275a was transformed into its iPr ester 276 by reaction with MeMgBr in THF/iPrOH, which by treatment with DBU could be epimerized to the syn-Mannich adduct 277.
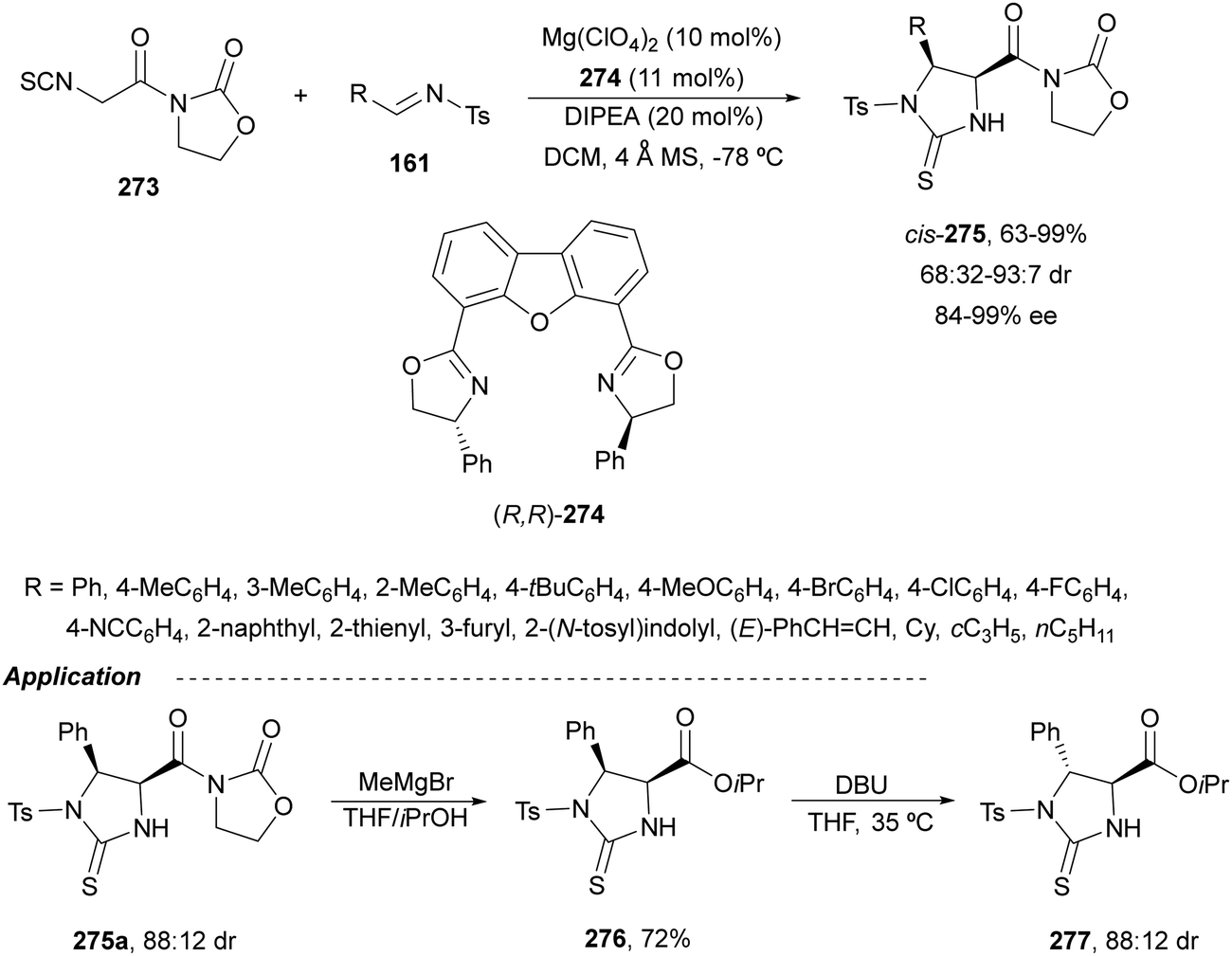 | ||
| Scheme 83 Asymmetric aza-Mannich reaction of α-isothiocyanate N-acyl oxazolidinone 273 with N-tosyl imines 161 under Mg(ClO4)2/Ph-Dbfox 274 catalysis. | ||
Based on the aldol reaction of α-methyl-α-isothiocyanato ester 278,170 Matsunaga, Shibasaki and co-workers171 described the Mannich reaction of this isothiocyanate 278 with N-DPP ketimines 250 under nBu2Mg/Schiff base 279 catalysis (Scheme 84). Cyclic thioureas cis-280 were mainly obtained in good to high yields, dr and good enantioselectivities. However, trans-280 were mainly obtained when Sr(OiPr)2 was used as the metal source. Circular dichroism (CD) spectra of nBu2Mg/279 and Sr(OiPr)2/279 complexes were different suggesting that chiroptically different aggregates were formed. Moreover, the different dihedral angle of the binaphthyl unit in both complexes can play a key role in the sterodiscriminating step and would cause these diastereodivergent52 results.
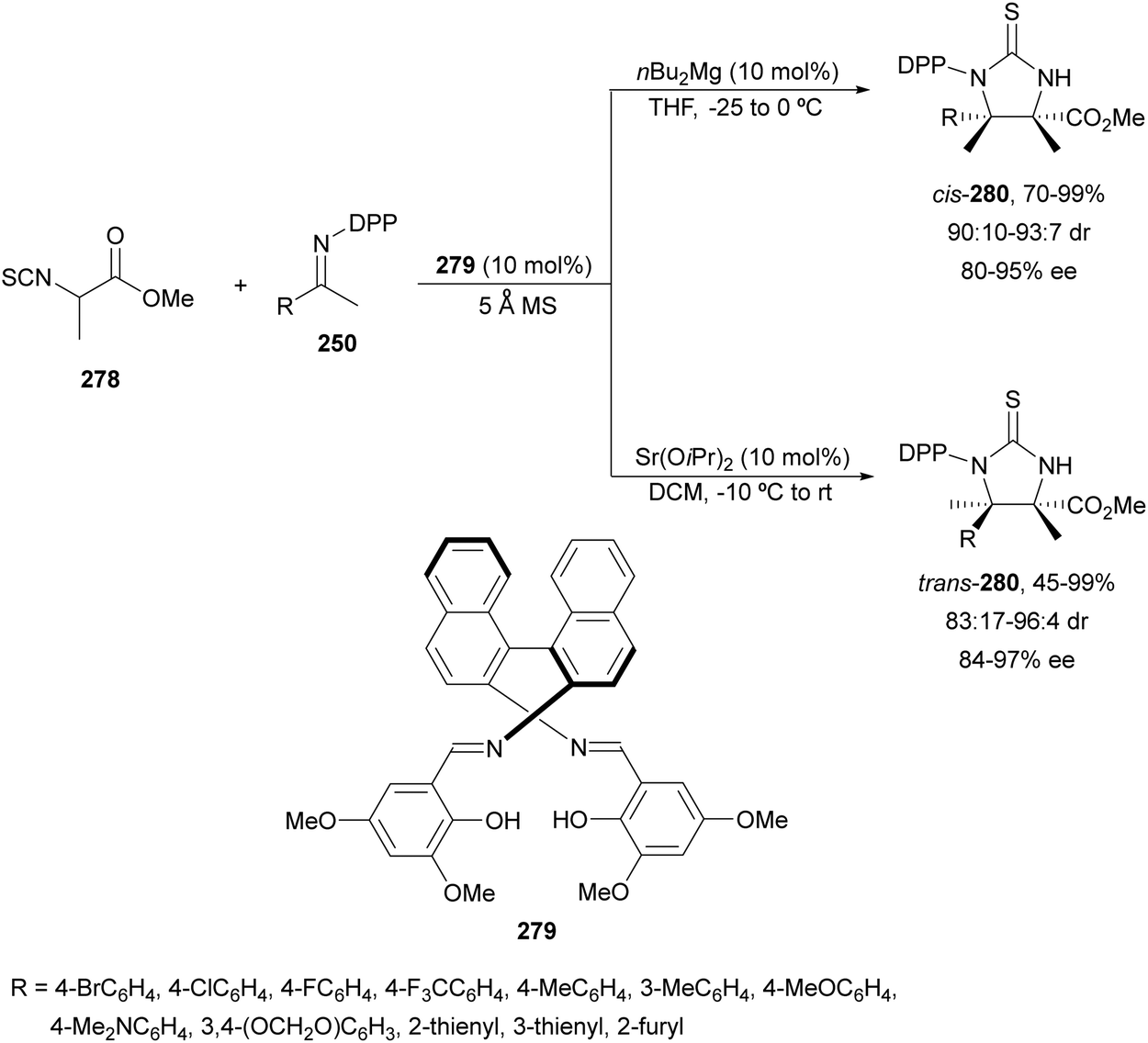 | ||
| Scheme 84 Asymmetric diastereodivergent aza-Mannich reaction of α-mehtyl-α-isothiocyanato ester 278 with ketimines 250 under nBu2Mg or Sr(OiPr)2 and ligand 279 catalysis. | ||
The same group172 performed the asymmetric synthesis of spirooxindoles 283 by an aza-Mannich reaction of isothiocyanato-oxindoles 281 with N-DPP aldimines 167 under Sr(OiPr)2/ligand 282 catalysis (Scheme 85). In this case, the use of nBu2Mg, Ca(OiPr)2, Ba(OiPr)2, Al(OiPr)3 or Ni(OAc)2 gave very poor enantioselectivities. Products 283 were isolated in excellent yields and stereoselectivities and compound 283a was transformed into a spiro[imidazoline-4,3′-oxindole] core. Benzoylation of 283a to give 284 followed by Pd-catalyzed desulfurative cross-coupling with phenylboronic acid, developed by Liebeskind and co-workers,173 afforded product 285, which is related to nutlin,174 an imidazoline-based inhibitor of p53/E3-ubiquitin ligase Mdm2 interaction, and to MI-219,175 a spiro[pyrrolidine-3,3′-oxindole]-based p53/Mdm2 inhibitor.
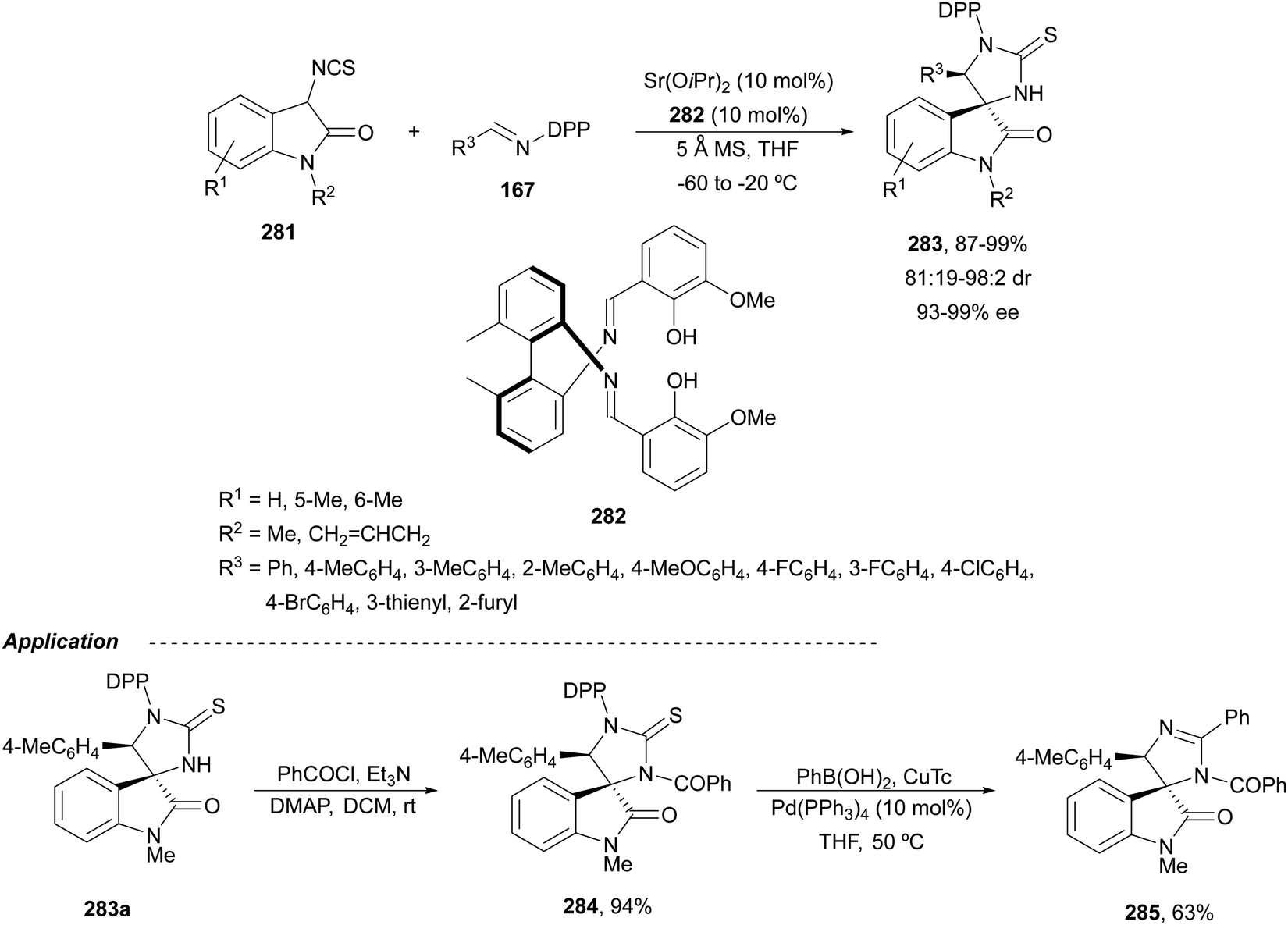 | ||
| Scheme 85 Asymmetric aza-Mannich reaction of isothiocyanato-oxindoles 281 with N-DPP aldimines 167 under Sr(OiPr)2/ligand 282 catalysis. | ||
Seidel and co-workers176 described for the first time the addition of isothiocyanate 273 to N-sulfonyl imines 161 using the quinidine derivative 263 (1 mol%) as a chiral organocatalyst in toluene at room temperature to furnish products trans-275 in 53–99% yields, 72:28–>95:5 dr and 89–99% ee (Scheme 86a). The same process was simultaneously described by Zhong and co-workers177 using a quinine-derived organocatalyst 286. In this case, after the reaction was completed the solvent (m-xylene) was removed and MeMgBr in THF was added as well as EtOH at 0 °C to obtain trans-thiooxazolidines 287 in high yields, up to 97:3 dr and >99% ee (Scheme 86b).
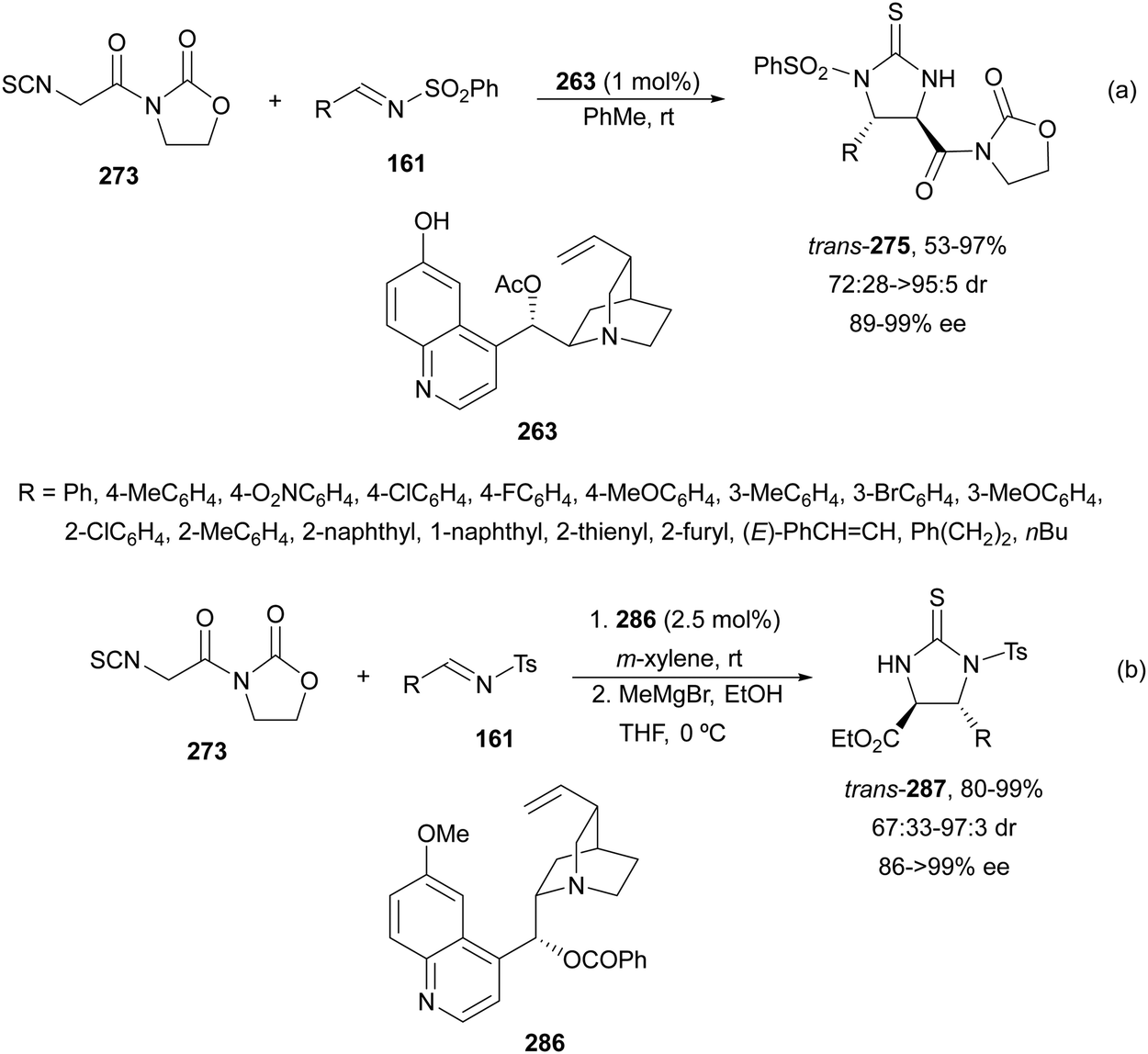 | ||
| Scheme 86 Asymmetric aza-Mannich reaction of isothiocyanato-oxazolidinone 273 with N-sulfonyl imines 161 under quinidine or quinine-derived organocatalysis. | ||
Chiral bisguanidine 288 (5–10 mol%) has been employed by Liu, Feng and co-workers178 for the aza-Mannich reaction of isothiocyanato-oxazolidinone 273 and N-tosyl imines 161 to obtain trans-275 working in the presence of 4-MeC6H4CO2H (7.5 mol%) as an additive in THF/DCM at −12 °C (Fig. 11). Products trans-275 were isolated in 82–99% yields, 80:20–>95:5 dr and 90–>99% ee. The authors proposed a TS in which the catalyst is protonated by the acid to obtain a guanidinium salt. This salt activates the N-Ts imine via hydrogen bonding. The other guanidine unit deprotonates the isothiocyanato-imidazolidine 273, which attacks the Si-face of 161 to form the major (4S,5R)-275 product (Fig. 11).
 | ||
| Fig. 11 Bisguanidine organocatalyst 288 for the asymmetric aza-Mannich reaction of isothiocyanato-oxazolidinone 273 with N-tosyl imines 161 and proposed TS. | ||
Independently, Liu179 and Yuan180 groups have described the reaction of isocyanato-oxindoles 281 with N-tosyl imines 161 to obtain the corresponding spirooxindoles 290. In the first case, thiourea 289 (Takemoto organocatalyst181) was used as a chiral organocatalyst and with or without 4-NCC6H4CO2H as an additive in acetone at −20 °C to room temperature (Scheme 87a). Products 290 were obtained in good yields and excellent enantioselectivities. In the second case, just quinine was used as an organocatalyst (1 mol%) in toluene at 0 °C to provide products ent-290 in moderate to high yields and stereoselectivities (Scheme 87b).
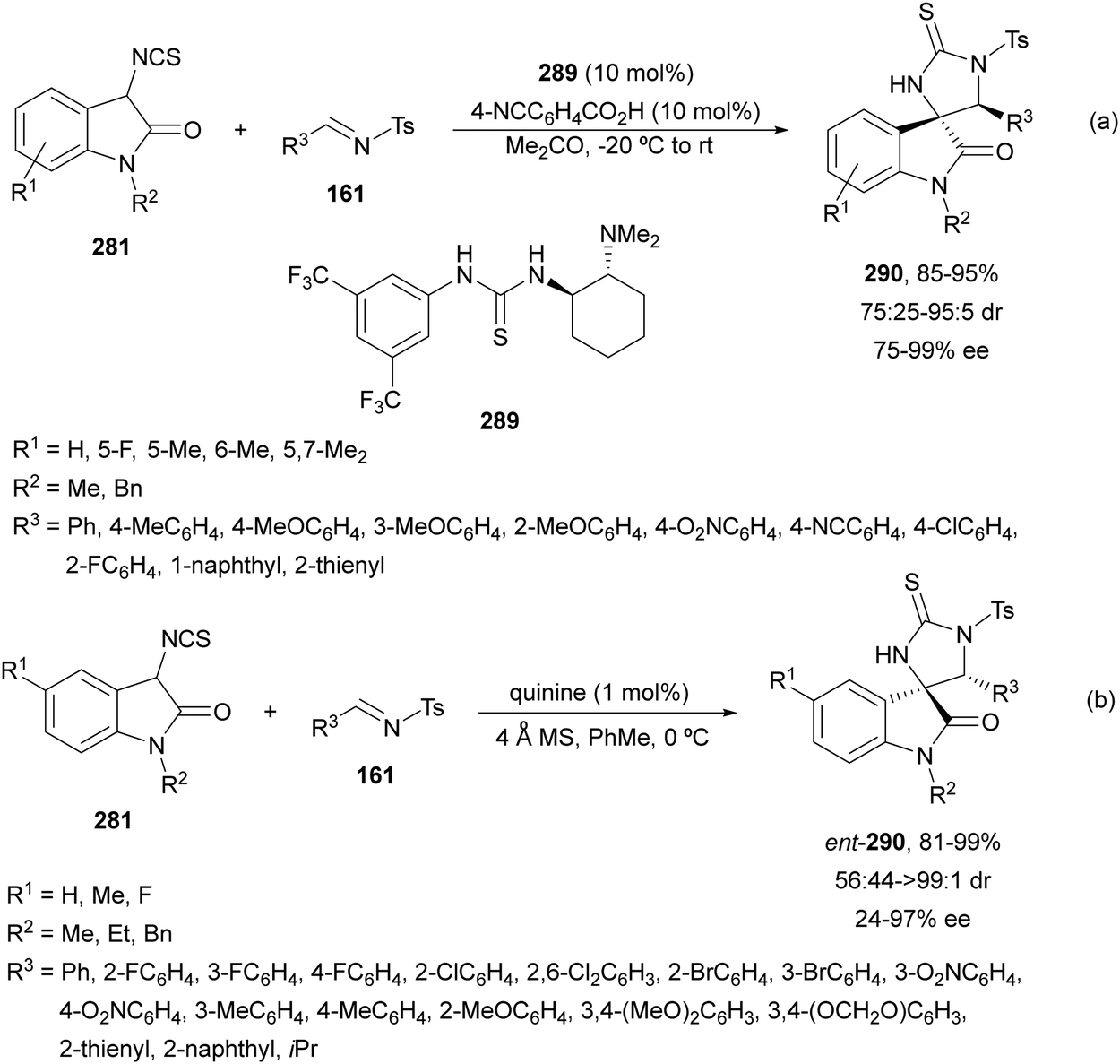 | ||
| Scheme 87 Asymmetric aza-Mannich reaction of isothiocyanato-oxindoles 281 with N-tosyl imines 161 under thiourea 289 or quinine catalysis. | ||
Isothiocyanate derivatives have been mainly used as glycine or other amino acid equivalents for the synthesis of enantioenriched cis- or trans-imidazolidinthiones, precursors of α,β-diamino acids, under Mg or Sr catalysis or under organocatalysis with chiral bases, guanidines and thioureas.
 | ||
| Scheme 88 Asymmetric three-component aza-Mannich reaction of α-azido ketones 291 with aldehydes and p-anisidine under 292 catalysis. | ||
Cyclic α-azido ketones 295 was reacted with N-Boc imines 161 to obtain aza-Mannich adducts 297 possessing a α-quaternary stereocenter with modest to high yields, diastereoselectivities and enantioselectivities (Scheme 89).184 In this case, the H8-CPA (S)-296 was used as an organocatalyst in DCM at 40 °C. Hydrogenation of 297a gave 1,2-diamine 298 in 63% yield without reduction of ee.
 | ||
| Scheme 89 Asymmetric aza-Mannich reaction of α-azido cyclic ketones 295 with N-Boc imines 161 under CPA (S)-296 catalysis. | ||
α-Azido amides 299 of 7-azaindoline have been used as nucleophiles by Kumagai, Shibasaki and co-workers185 in the asymmetric aza-Mannich reaction with N-thiophosphinoyl imines 300 catalyzed by Cu(I)/(R)-xyl-Segphos 301 and Barton's base 302 (Scheme 90). anti-Products 303 were isolated and β-azido-α-amino acid hydrochlorides 304 were obtained by treatment with 6 M HCl at 80 °C with good yields, high diastereoselectivities and enantioselectivities.
 | ||
| Scheme 90 Asymmetric aza-Mannich reaction of α-azido amides 299 with N-thiophosphinoyl imines 300 under Cu(I)/(R)-xyl-Segphos 301 catalysis. | ||
Maruoka and co-workers186 employed N-protected α-amino acetaldehydes 305 in the aza-Mannich reaction with N-Boc imines 161 (Scheme 91). A diastereodivergent52 process was achieved using either L-proline or the axially chiral amino sulfonamide (S)-306 to obtain, after NaBH4 reduction, products 307. In the first case, syn-adducts were mainly obtained according to a (E)-s-trans-enamine TS1, whereas (E)-s-cis-enamine TS2 is mainly involved in the formation of anti-adducts. The former process was applied to the formal synthesis of the marine alkaloid (−)-agelastatin A, which possesses a potent antitumor activity. Mannich product 308a was transformed into the key intermediate 309.187
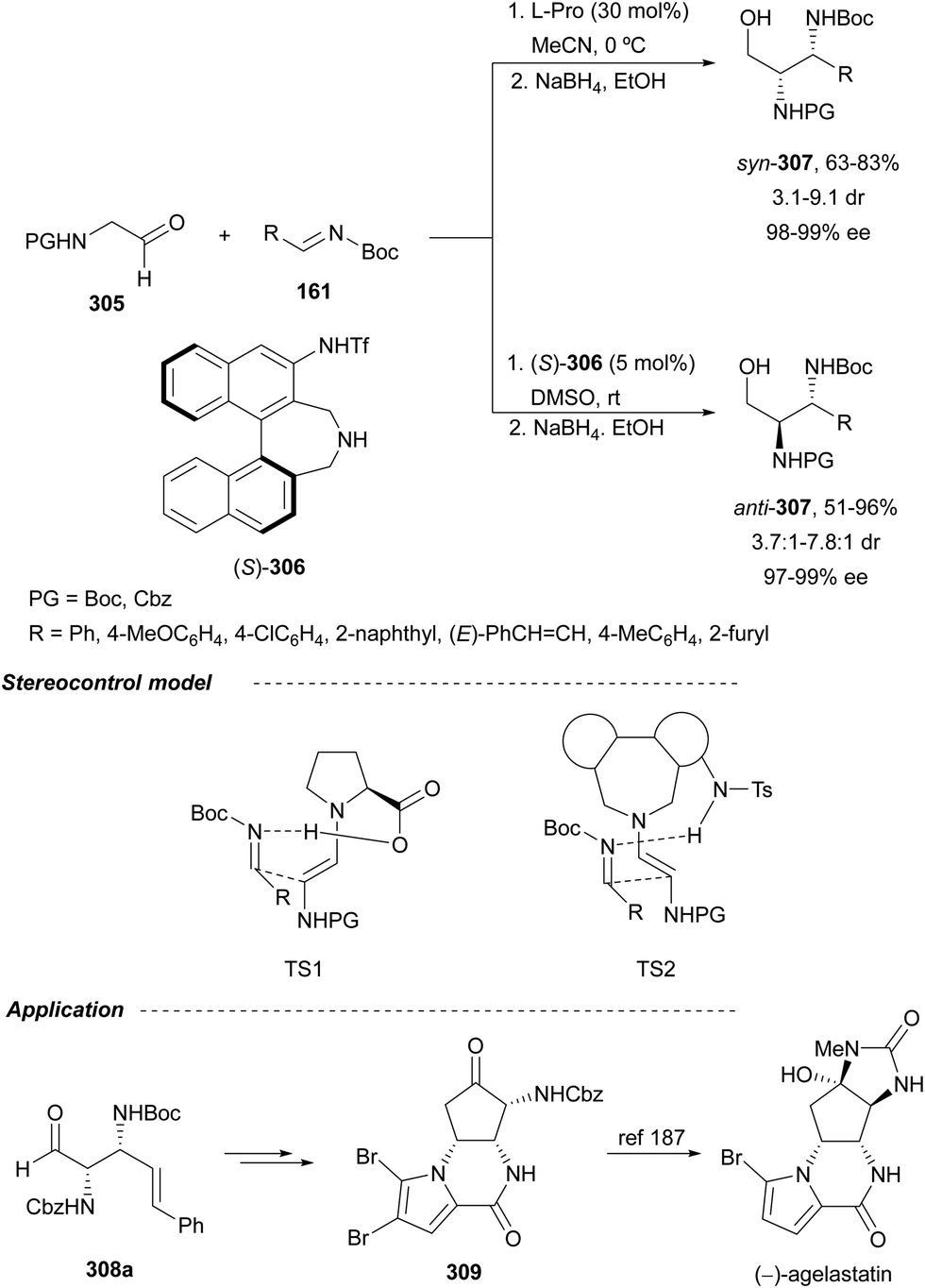 | ||
| Scheme 91 Asymmetric aza-Mannich reaction of α-amino acetaldehydes 305 with N-Boc imines 161 under L-Pro or (S)-306 diastereodivergent catalysis. | ||
Subba Reddy and co-workers188 have employed 3-indolinone-2-carboxylates 310 as nucleophiles in the asymmetric aza-Mannich reaction using thiourea 311 as an organocatalyst (Scheme 92). Chiral β-amino esters 312 were obtained in good yields with ee up to 99% by reaction of compounds 310 with in situ generated N-Boc imines from α-amido sulfones 200. A plausible TS was proposed to explain the mechanism of this organocatalytic process, which involves the formation of hydrogen bonding between the enol of 310 and the tertiary amine. Subsequently, the N-Boc imine is activated by hydrogen bonding with the thiourea unit to form a ternary complex. A preferential Re-face attack of the enolate from 310 onto N-Boc imine would give product 312.
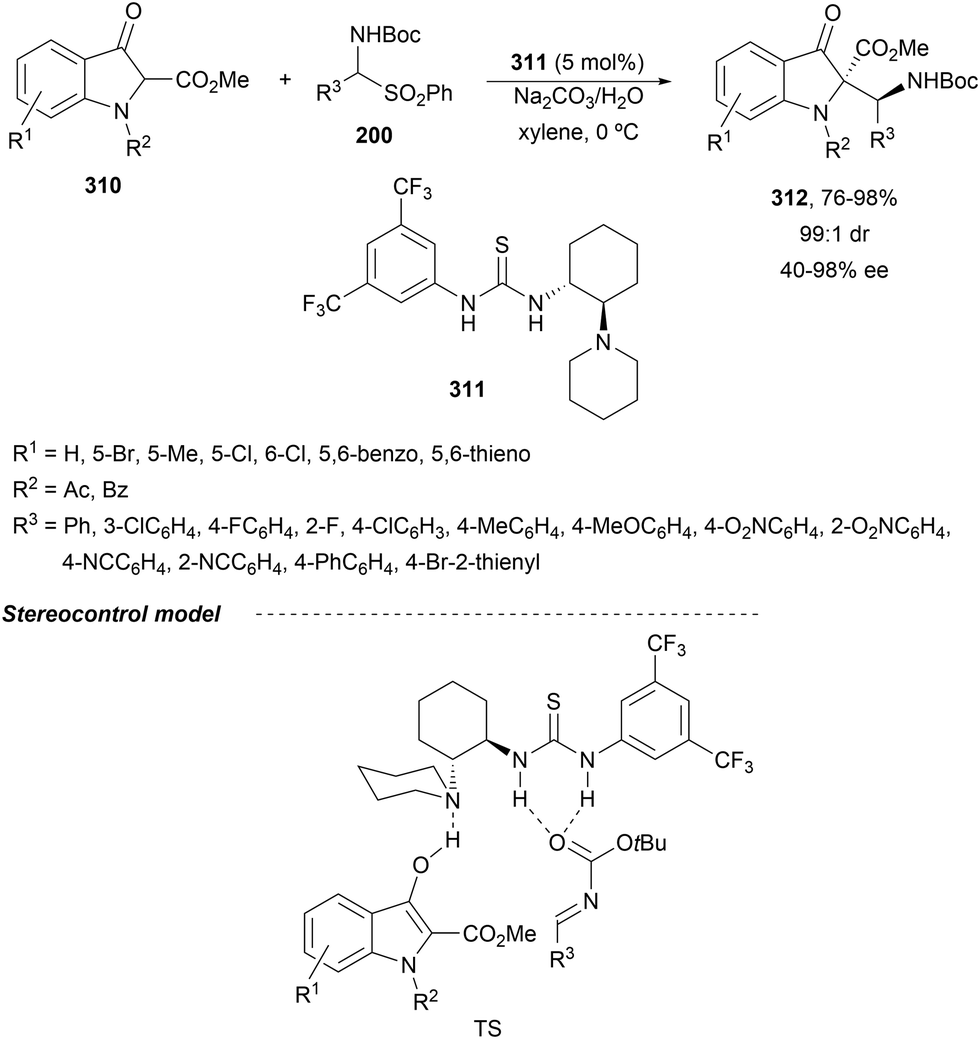 | ||
| Scheme 92 Asymmetric aza-Mannich reaction of methyl 3-indolinone-2-carboxylates 310 with N-Boc imines under thiourea 311 catalysis. | ||
Recently, an aza-Mannich reaction between N-aryl glycines 313 and hydrazones 314 has been carried out by Cu/bisoxazoline 315 and visible light-induced photoredox catalysis.189 The decarboxylative radical coupling/cyclization reaction of glycine derivatives 313, hydrazones 314 and aldehydes gave chiral imidazolidines 318 with high yields and enantioselectivities (Scheme 93). In the absence of paraformaldehyde, diamines 317 could be isolated in good yields and enantioselectivities. Based on electron paramagnetic resonance investigations, a possible reaction mechanism was proposed. The SET between the excited photocatalyst 4CzIPN* (316*) and N-phenylglycine 313 leads to the formation of the radical cation III, which after decarboxylation resulted the radical cation IV. Intermediate I was reduced to intermediate II through a SET pathway and the photocatalyst 316 is generated. Radicals II and IV undergo a radical coupling reaction to provide complex V, which after protonation and ligand exchange generates product 317. Final cyclization of 317 with paraformaldehyde furnishes imidazolidines 318.
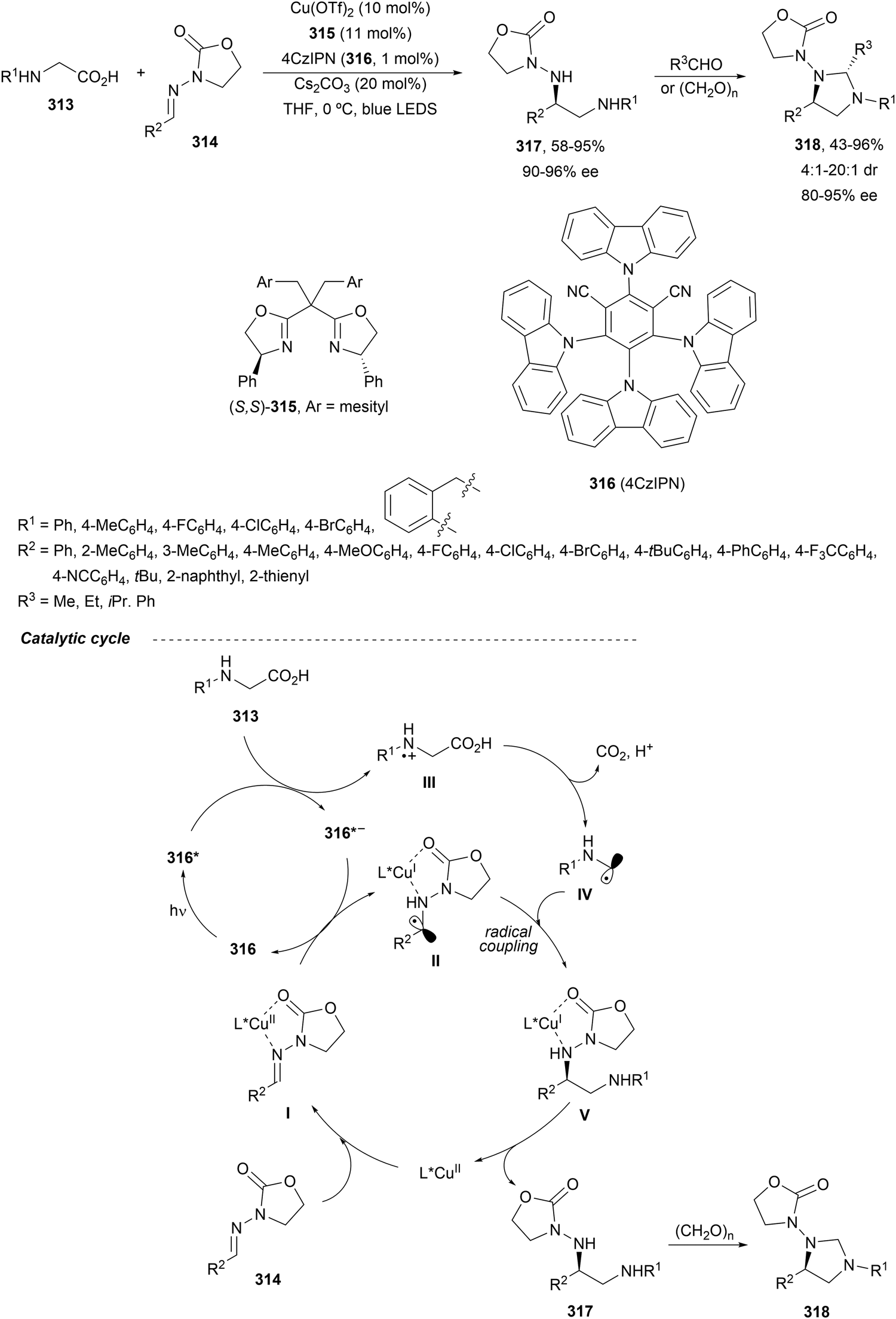 | ||
| Scheme 93 Asymmetric aza-Mannich reaction of N-aryl glycines 313 and hydrazones 314 under Cu/315 and photoredox catalysis. | ||
Different nucleophiles such as α-azido ketones and amides, α-amino acetaldehydes, and 3-indoline-2-carboxylates have been used in the asymmetric aza-Mannich reaction mainly under organocatalysis. Recently, a Cu/Box complex and a photocatalyst have been used to co-catalyze the aza-Mannich reaction of N-aryl glycines and hydrazones to obtain 1,2-diamine derivatives.
3.2. Aza-Henry reaction
The aza-Henry reaction, alternatively the nitro-Mannich reaction, involves the nucleophilic addition of nitroalkanes to imines to provide β-nitroamines, which can be easily reduced to 1,2-diamines. Several revisions have been published based on stereoselective transformations for the synthesis of natural products and biologically active compounds.190,191 Asymmetric reactions are based on the use of chiral metal complexes and mainly with organocatalysts.103,104,192,193 In this section, the most significant recent catalytic methods will be considered.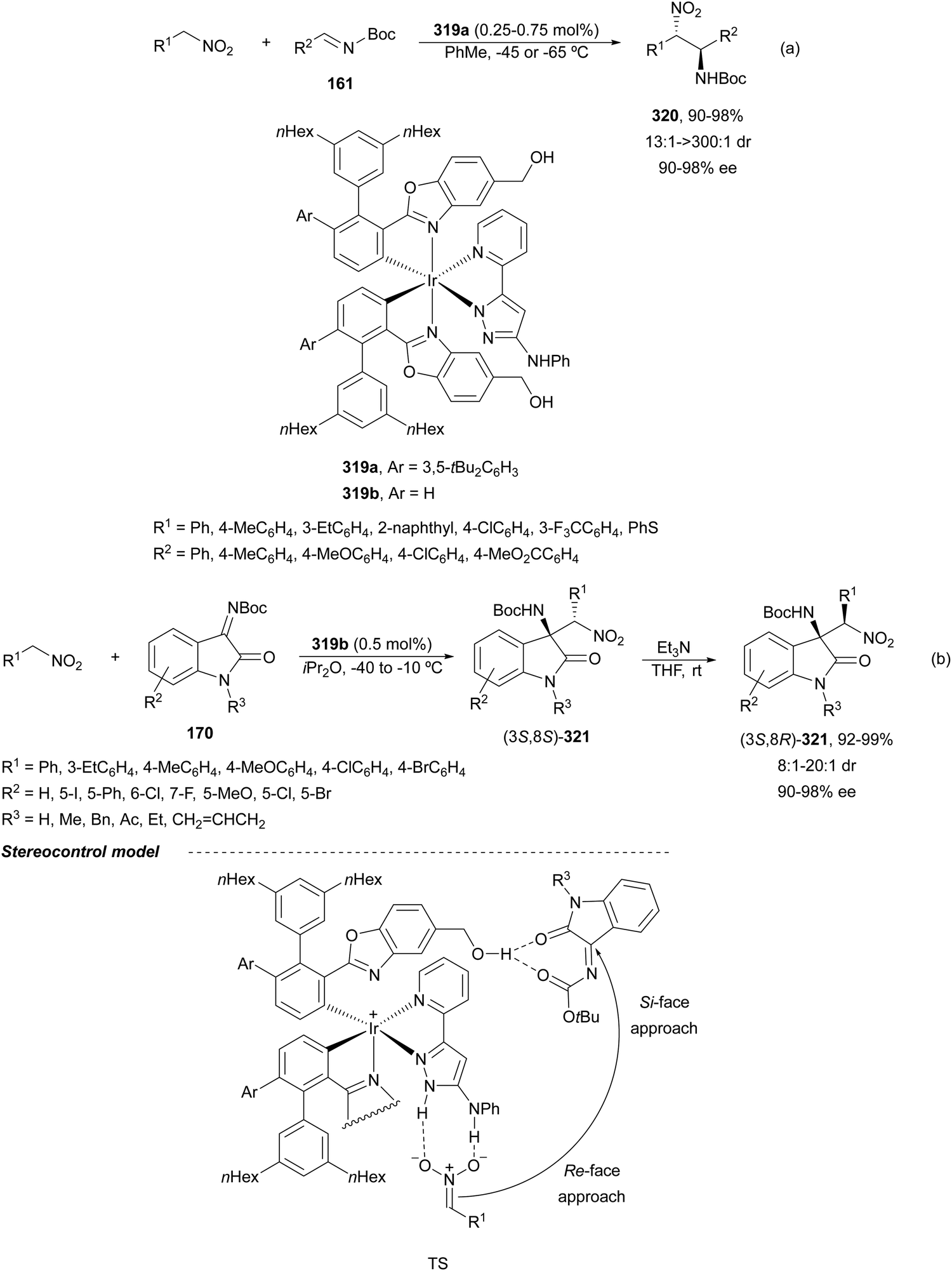 | ||
| Scheme 94 Asymmetric aza-Henry reactions of nitroalkanes with N-Boc imines 161 and isatin ketimines 170 under Ir(III) complex 319 catalysis. | ||
Arai and co-workers198 reported bis(imidazolidine)pyridine (PyBidine) 163 and NiCl2 (5 mol%) catalyzed aza-Henry reaction of isatin N-Boc ketimines 170 with nitromethanes in the presence of 10 mol% of DIPEA. The resulting 3-amino-2-oxindoles (3R)-321 (R1 = H) were obtained in toluene at 30 °C with 19–99% yields and 78–95% ee. For the reduction of the nitro group (3R)-321, (R1 = R2 = H, R3 = Me) was treated with NiCl2·6H2O/NaBH4 in MeOH at 0 °C to give the corresponding diamine in 99% yield and 97% ee. Blay, Pedro and Holmquist199 carried out the same aza-Henry reaction using 10 mol% of Cu(BF4)2 and the bisoxazoline (S,S)-9. Products (3S)-321 (R1 = H) from nitromethane were prepared in the presence of iPr2NH (13 mol%) at room temperature in 82–99% yields and 6.6–99.9% ee.
Chiral dimeric ligand 322 derived from (R)-binol, salen and (1R,2S)-2-aminodiphenylethanol has been used by Kureshy and co-workers200 in the Cu-catalyzed aza-Henry reaction of N-tosyl imines 161 with nitroalkanes (Scheme 95a). This process was performed in toluene at room temperature to give products 323 up to 82% yield and up to >99% ee, mainly as anti-diastereomers in the case of nitroethane up to 98:2 dr. The catalyst Cu(OAc)2/322 was recyclable 5 times in the reaction of 161 (R2 = 2-MeOC6H4) with nitromethane without apparent loss in activity and enantioselectivity. This process was employed to the synthesis of (S)-levamisole, an anthelmintic agent at 1 g scale starting from 323a (Scheme 95b).
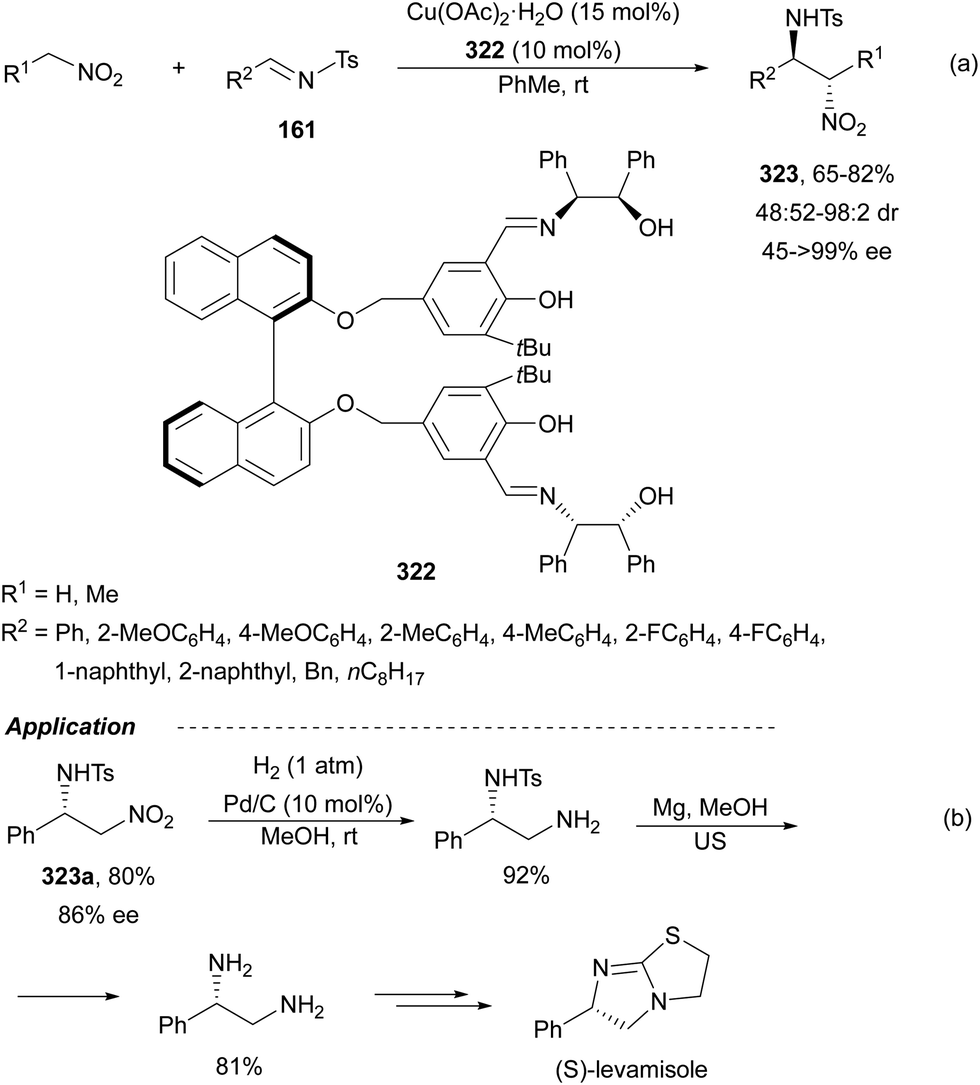 | ||
| Scheme 95 Asymmetric aza-Henry reaction of nitroalkanes with N-tosyl imines 161 under Cu(OAc)2/322 catalysis. | ||
The same group201 reported a chiral binol linked monomeric macrocycle Cu(II)-salen complex as a catalyst (5 mol%) for the asymmetric aza-Henry reaction of N-Boc ketimines derived from isatins 170 with nitromethane at room temperature. The corresponding adducts (3S)-321 (R1 = H) were obtained in 77–80% yields and 46–99% ee.
The first asymmetric aza-Henry reaction mediated by Fe(OTf)2 and (R,R)-TPS-he-Pybox 324 has been reported by Dudek and Mlynarski202 in 2017. Nitromethane reacted with N-diphenylphosphinoyl imines 167 using TEA (0.5 equivalents) as a base in THF at room temperature to give β-nitroamines 325 with good yields and high ee (Scheme 96). However, other secondary nitroalkanes afforded compounds 325 as 1:1 mixture of diastereomers.
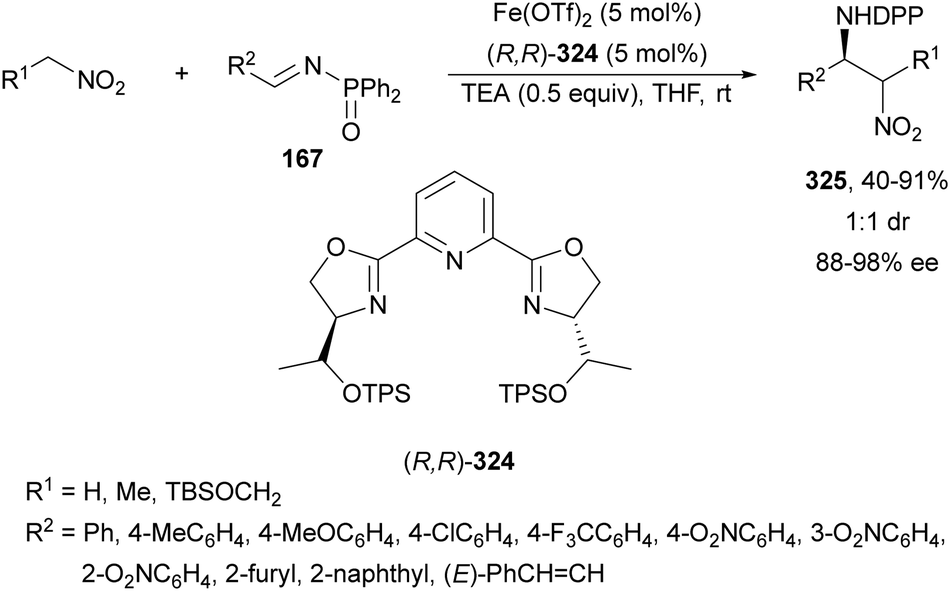 | ||
| Scheme 96 Asymmetric aza-Henry reaction of nitroalkanes with N-diphenylphosphinoyl imines 167 under Fe(OTf)2/(R,R)-324 catalysis. | ||
Recently, Yasukawa, Nakamura and co-workers203 reported the enantioselective reaction of cyclic imino esters 326 with nitroalkanes using Cinchona alkaloid derived sulfonamide 327/zinc complex as a catalyst (Scheme 97). The reaction with nitromethane was also carried out with 6-membered cyclic ketimine 326a and with the benzene fused 5-membered ketimine ester 326b to obtain the corresponding adducts 328a and 328b in 56% yield and 92% ee and in 42% yield and 83% ee, respectively. Products 328 derived from the five-membered ketimines 326 were obtained in 52–86% yields and 70–92% ee with moderate diastereoselectivity.
 | ||
| Scheme 97 Asymmetric aza-Henry reaction of nitroalkanes with cyclic imino esters 326 under Et2Zn/sulfonamide 327 catalysis. | ||
In 2004, Takemoto and co-workers204 reported for the first time a thiourea 289 as a bifunctional organocatalyst for the asymmetric aza-Henry reaction of N-DPP imines 169 with nitromethane to provide compounds 325 (R1 = H) up to 91% yield and 78% ee. Chiral thioureas 289205 and 329206 have been developed for the addition of nitroalkanes to N-Boc imines 161 to obtain anti-320 (Fig. 12). Thiourea 331 bearing 1,2-diamine (DPEN) as a chiral unit showed excellent results for the addition of activated α-nitro esters 330 to N-Boc imines 161 to furnish mainly (2S,3S)-α-nitro-β-amino esters 332 (Scheme 98).207
 | ||
| Fig. 12 Chiral thioureas as catalysts for the aza-Henry reaction of nitroalkanes with N-protected imines. | ||
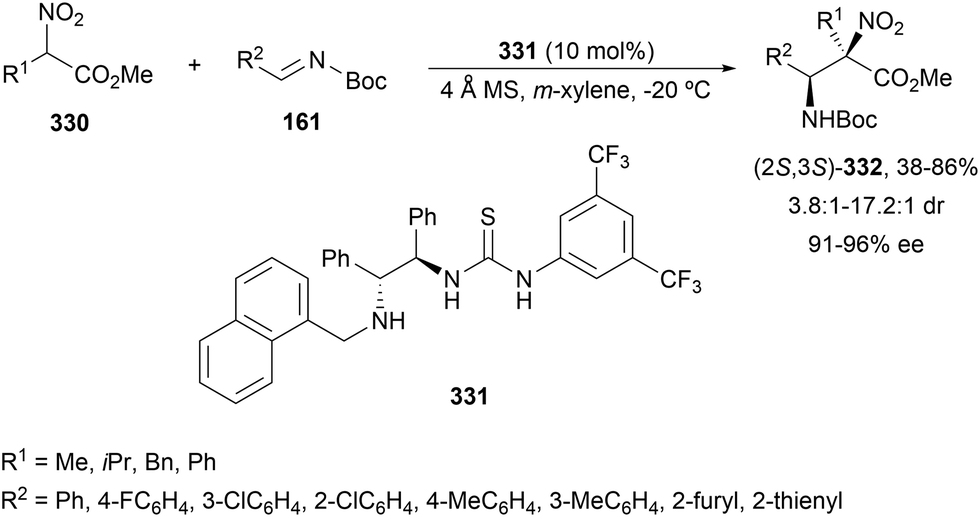 | ||
| Scheme 98 Asymmetric aza-Henry reaction of α-nitro esters 330 with N-Boc imines 161 under thiourea 331 catalysis. | ||
For the asymmetric aza-Henry reaction of N-Boc imines 161 with nitroalkanes, Zhao and co-workers208 employed a combination of a new bifunctional phosphine-thiourea 333 and an acrylate able to generate in situ a zwitterion which acted as a catalyst. This reaction gave the corresponding adducts anti-320 with high yields and diastereo- and enantioselectivities (Scheme 99). According to 31P NMR and mass analysis of 333 and methyl acrylate, the zwitterion intermediate A was detected, which forms an ion-pair with the nitronate. A possible TS has been proposed to explain the observed stereoselectivity.
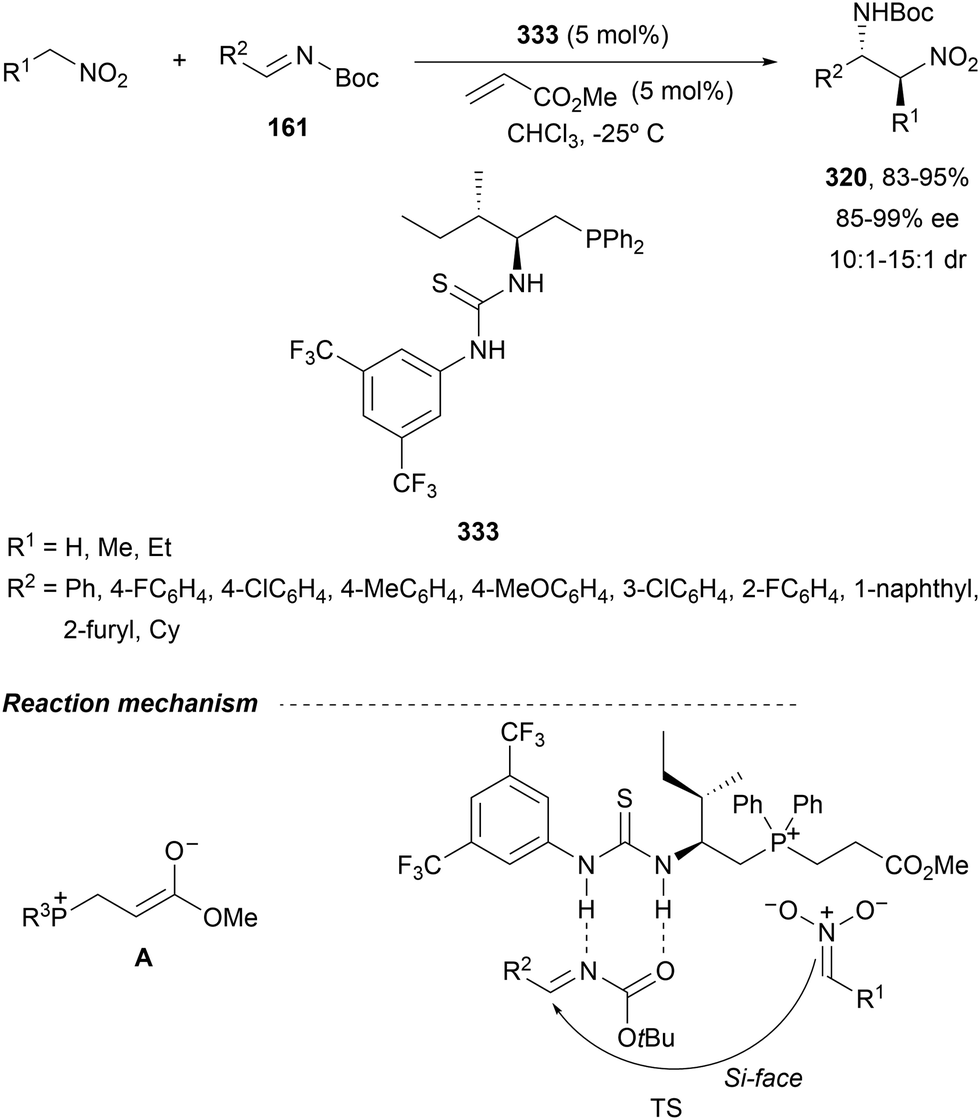 | ||
| Scheme 99 Asymmetric aza-Henry reaction of nitroalkanes with N-Boc imines 161 under ion-pair catalysis formed by phosphine-thiourea 333 and methyl acrylate. | ||
Quinine-derived thiourea 335209 has been employed by Wang and co-workers210 as an organocatalyst for the enantioselective addition of nitroalkanes to cyclic trifluoromethyl ketimines 243 up to 97% yield and up to 98% ee. This method was applied to the synthesis of anti-HIV drug DCP 083 employing 2(1H)-quinazolinone 243a and 1-nitro-2-cyclopropylethane 334 to obtain adduct 336 in 91% yield as a mixture of 1.5:1 of diastereomers in 90% and 70% ee, respectively (Scheme 100).
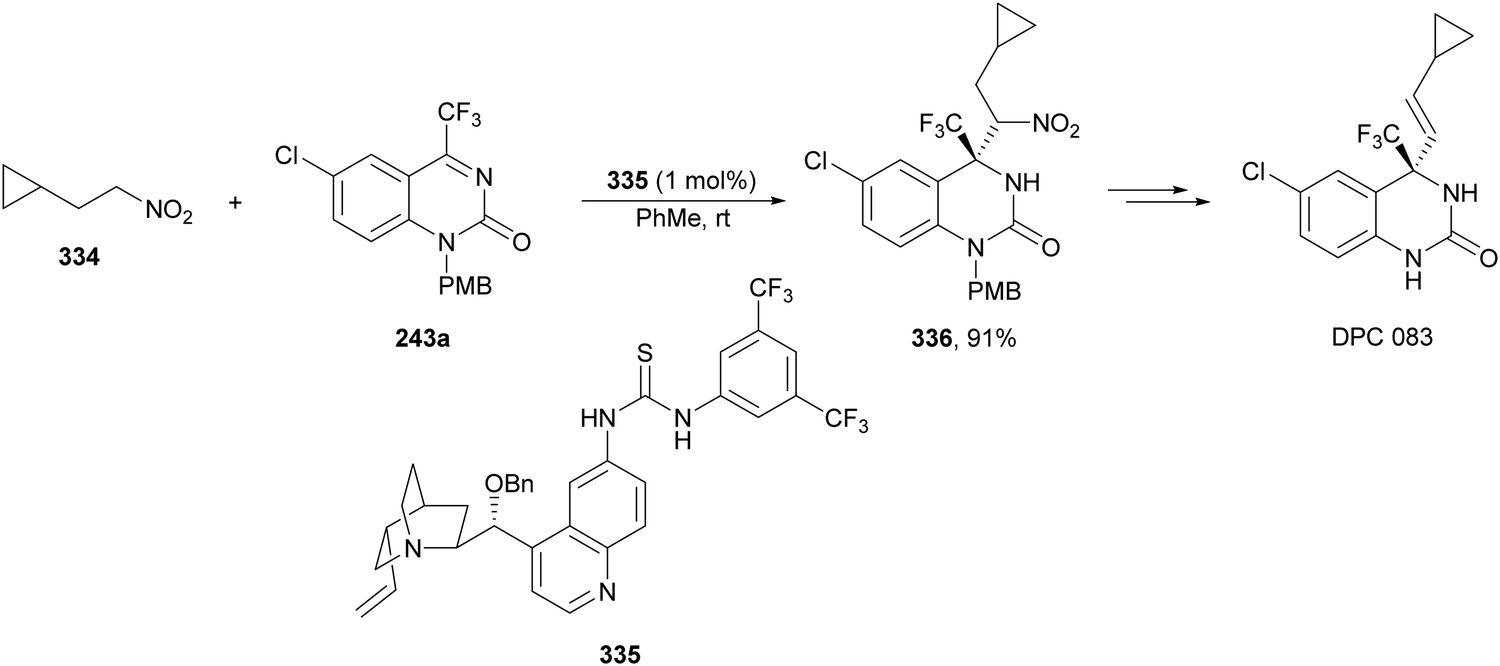 | ||
| Scheme 100 Asymmetric aza-Henry reaction of 1-nitro-2-cyclopropylethane 334 with cyclic trifluoromethyl ketimine 243a towards the synthesis of anti-HIV drug DPC 083. | ||
Alemán and co-workers211 described the asymmetric aza-Henry reaction of cyclic α-carbonyl ketimines 337 with nitromethane catalyzed by the hydroquinine thiourea 338 (Scheme 101). 2-Aryl-3H-indol-3-ones 337 were allowed to react with nitromethane in p-xylene at room temperature to furnish products 339 up to >98% yield and 96% ee. It was proposed that the thiourea unit forms hydrogen bonding with ketimine 337 and the nitronate is coordinated with the catalyst according to the depicted TS. Nitroethane gave product 339 (R = Ph) with low 3:1 dr in 97% yield.
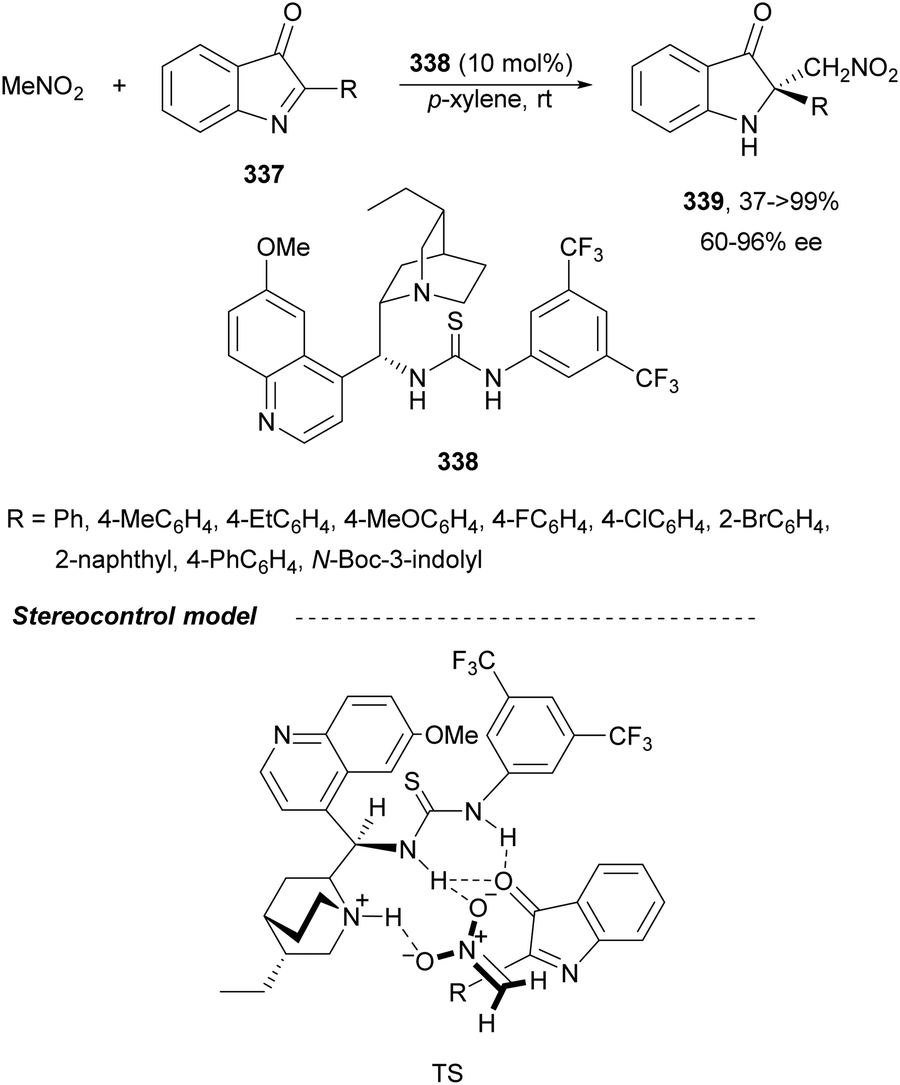 | ||
| Scheme 101 Asymmetric aza-Henry reaction of nitromethane with 2-aryl-3H-indol-3-ones 337 under thiourea 338 catalysis. | ||
Miao and co-workers212 performed the aza-Henry reaction of α-substituted nitroacetates 330 with N-phosphoryl imines 340 under Cinchona-derived thiourea 341 catalysis (Scheme 102). Products 342 were obtained working in toluene at −20 °C in good yields, modest to high anti-diastereoselectivity and up to >99% ee. In the proposed catalytic cycle, the organocatalyst forms a double hydrogen bonding between the two NH groups and the PO group of the imine to give intermediate I. Hydrogen bonding of methyl nitropropanoate with the protonated quinuclidine unit resulted intermediate II. Subsequently, the Si-face attack of the imine through the Si-face of the enolate furnishes intermediate III with (S,S)-configuration. This mechanism was corroborated by 31P NMR spectroscopy.
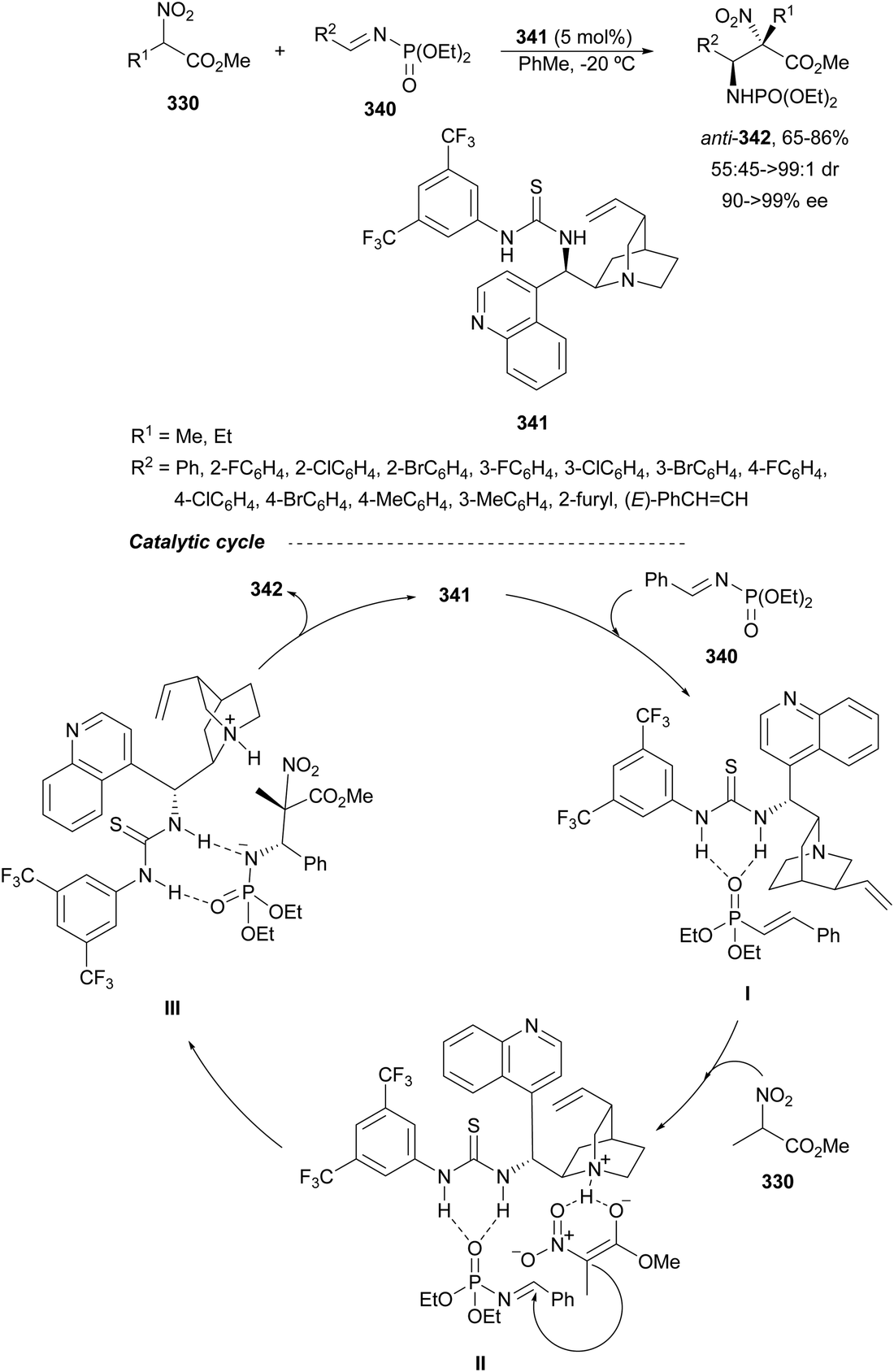 | ||
| Scheme 102 Asymmetric aza-Henry reaction of methyl α-nitro esters 330 with N-phosphoryl imines 340 under thiourea 341 catalysis. | ||
Chiral squaramides behave as thioureas like hydrogen donors in organocatalysis.213,214 Du and co-workers215 reported the asymmetric aza-Henry reaction of N-benzothiazolyl imines 269 with nitromethane in the presence of squaramide 236 as a better organocatalyst than the related thiourea. Compounds 343 were obtained in high yields and enantioselectivities (Scheme 103a). Alternatively, a three-component process was carried out using 2-aminobenzothiazoles 344, aldehydes and nitromethane (Scheme 103b). The corresponding aza-Henry adducts 343 were isolated in modest to good yields and enantioselectivities lower than those of imines 269.
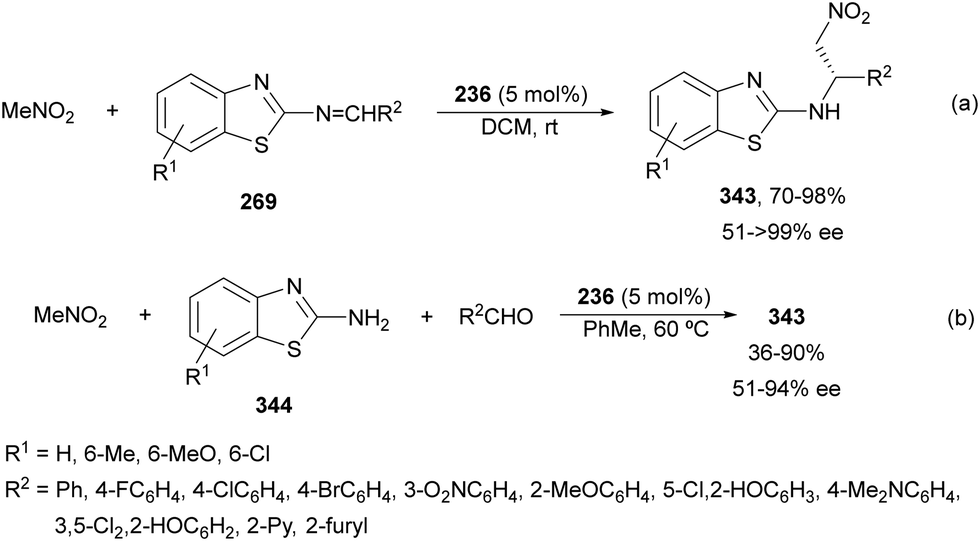 | ||
| Scheme 103 Asymmetric aza-Henry reaction of nitromethane with N-benzothiazolyl imines 269 under squaramide 236 catalysis. | ||
Chiral thiourea 345 bearing a basic iminophosphorane moiety as a Brønsted superbase has been designed by Dixon and co-workers.216 This bifunctional organocatalyst was able to promote the aza-Henry reaction of nitromethane with N-diphenylphosphinoyl ketimines 250 to provide β-nitroamines 346 with high yields and enantioselectivities (Scheme 104). However, when a cinchonine-derived thiourea, such as 341, was used as an organocatalyst no product was detected. This aza-Henry reaction was performed on a multigram scale and product 346a was transformed into 1,2-diamine derivative 347via a nickel boride reduction of the nitro group followed by Cbz protection and final DPP removal.
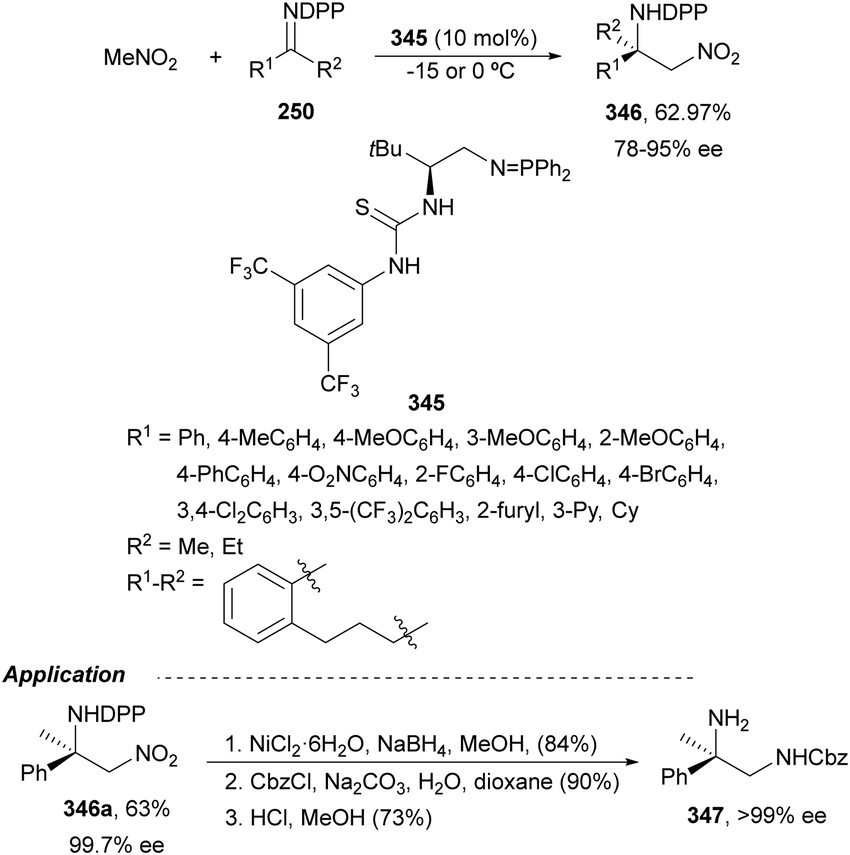 | ||
| Scheme 104 Asymmetric aza-Henry reaction of nitromethane with N-diphenylphosphinoyl ketimines 250 under thiourea-iminophosphorane 345 catalysis. | ||
Phosphorylated ketimines 348 have been transformed into tetrasubstituted α-amino-β-nitrophosphonates 349 by Palacios and co-workers.217 The corresponding asymmetric aza-Henry reaction with nitromethane was carried out under thiourea 341 catalysis (Scheme 105). Products 349 were isolated in good yields and enantioselectivities, and enantiopure compound 349a was transformed into (S)-α,β-diaminophosphonate 350 under hydrogenation conditions in 95% yield.
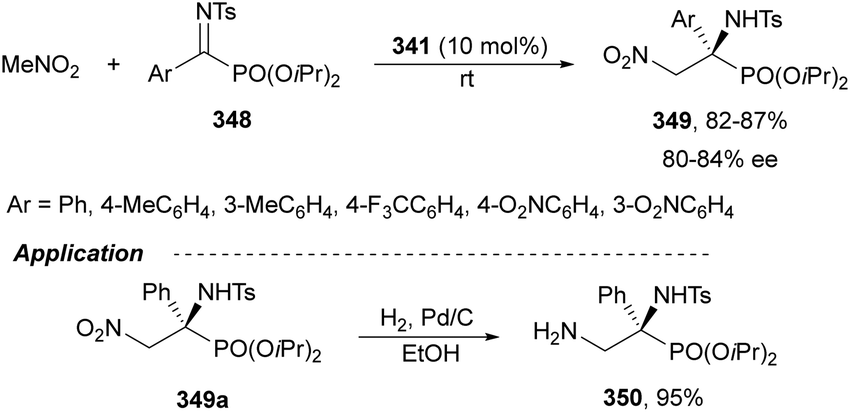 | ||
| Scheme 105 Asymmetric aza-Henry reaction of nitromethane with phosphorylated ketimines 348 under thiourea 341 catalysis. | ||
Aryl α-keto ester-derived N-tosyl ketimines 351 have been subjected to the aza-Henry reaction with nitromethane by Lin, Duan and co-workers218 under thiourea 352 organocatalysis (Scheme 106). This reaction was carried out in fluorobenzene at room temperature to obtain the corresponding products 353 up to 99% yield and 99% ee. A plausible TS was proposed involving the formation of hydrogen bonding between the thiourea and the α-keto ester imine 351 as well as with one of the oxygen atoms of the nitronate. The nitronate also interacts by hydrogen bonding with the nitrogen atom of the quinuclidine unit.
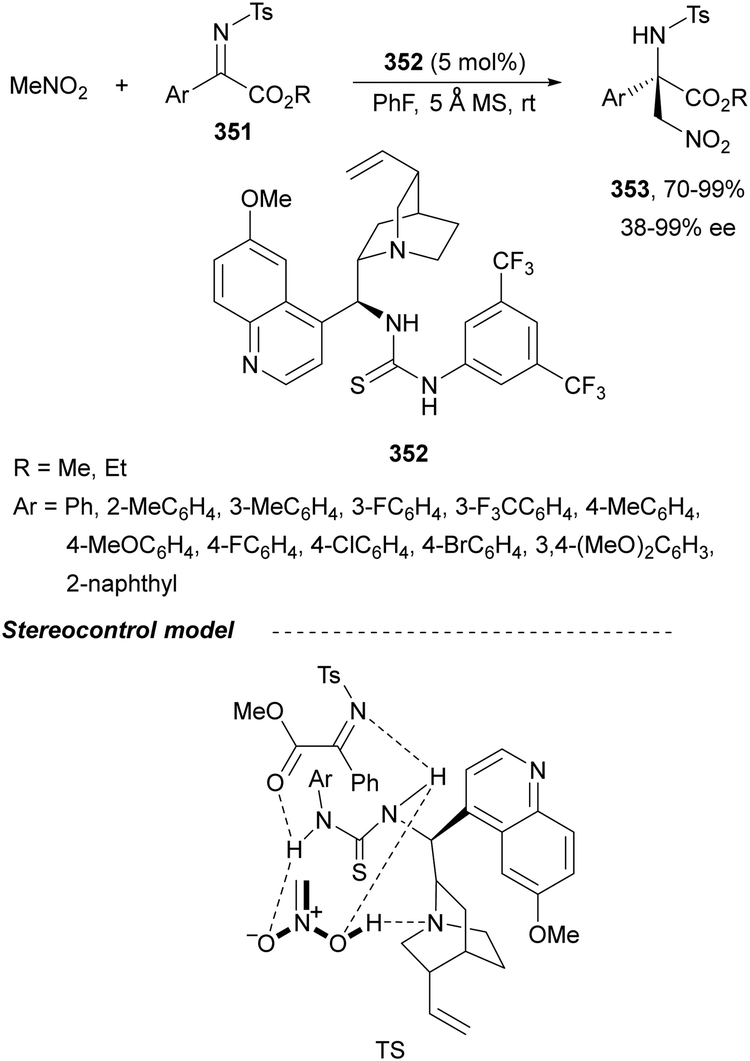 | ||
| Scheme 106 Asymmetric aza-Henry reaction of nitromethane with α-keto ester-derived imines 351 under thiourea 352 catalysis. | ||
The same group219 recently reported that N-Boc ketimines 170 derived from isatins were reacted with nitroalkanes using thiourea 354 derived from hydroquinine and (S)-phenylglycinol as an organocatalyst (Scheme 107). After screening of different hydroquinine-derived thioureas, 354 gave the best results affording products 355 up to 99% yield, 99:1 dr and 99% ee. This process was performed on a gram-scale with isatin-derived imine 170 (R1 = H) and nitroethane obtaining product 355 in 98% yield, 90:10 dr and 98% ee. In the proposed TS, the ketimine is coordinated with the thiourea and the hydroxy units and the nitronate with the protonated nitrogen from the quinuclidine moiety.
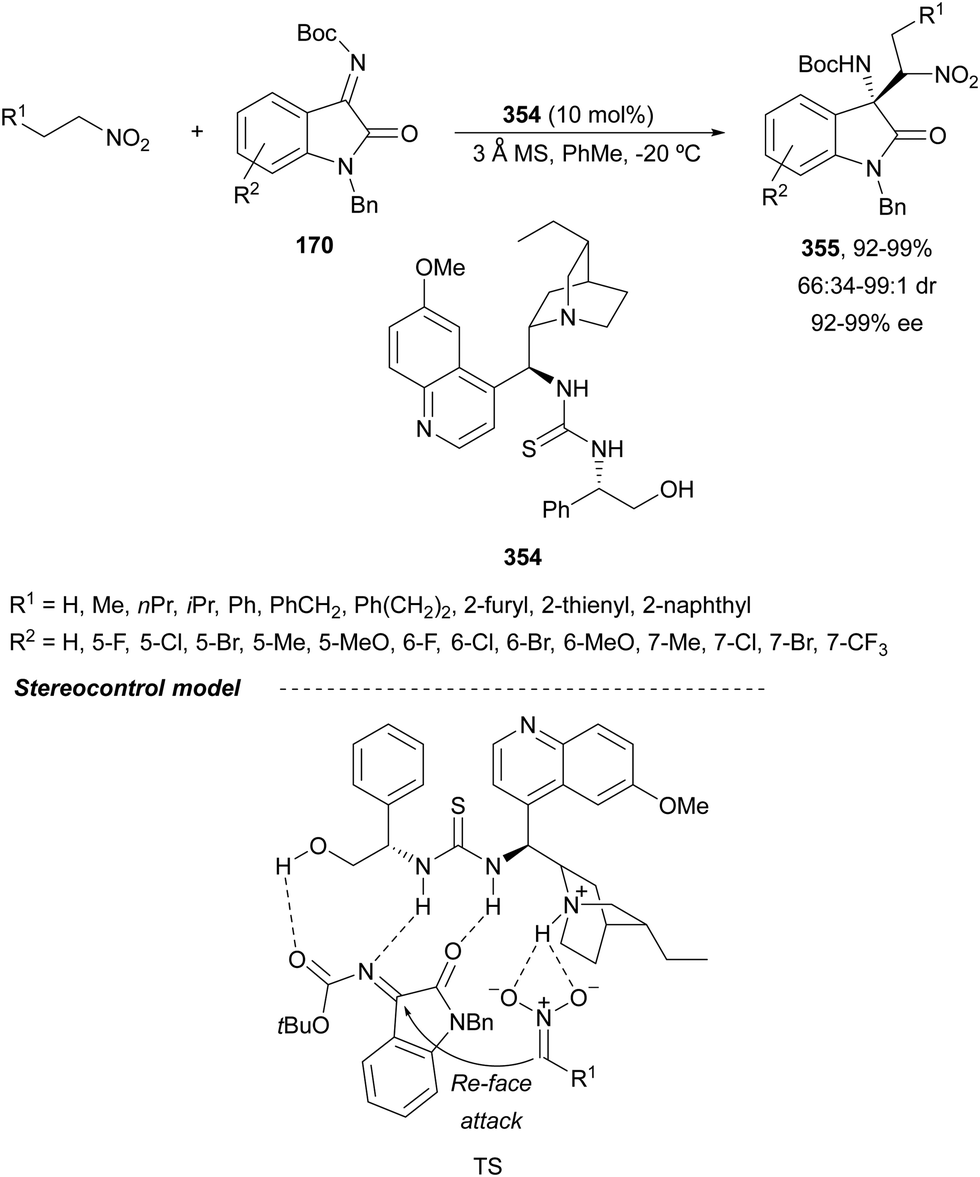 | ||
| Scheme 107 Asymmetric aza-Henry reaction of nitroalkanes with ketimines 170 under thiourea 354 catalysis. | ||
Fluoromethyl imines 356 were reacted with nitromethane under mild reaction conditions using the hydroquinine-derived thiourea 352 to furnish fluoromethylated β-nitroamines 357 in good yields and enantioselectivities (Scheme 108).220 However, other α-alkylated nitroalkanes gave poor results. The trifluoromethyl product 357 (RF = CF3) was reduced to the corresponding diamine via a nickel boride reduction with 68% yield and subsequently transformed into a 4-trifluoromethyl-2-imidazolidone.
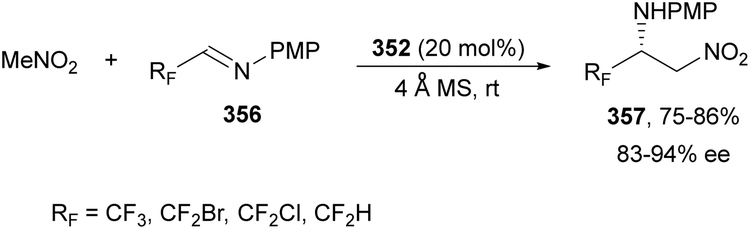 | ||
| Scheme 108 Asymmetric aza-Henry reaction of nitromethane with fluoromethyl imines 356 under thiourea 352 catalysis. | ||
Seven-membered cyclic imines 358 reacted with nitromethane in the presence of quinine-derived thiourea 264 to provide the corresponding adducts 359 in good to excellent yields and enantioselectivities (Scheme 109).221 The inactivated dibenzo[b,f][1,4]oxazepine 358a gave product 359a, which was reduced into a 1,2-diamine derivative by hydrogenation and further tosylated to 360 or underwent reductive amination to product 361 with potential application for the synthesis of bioactive molecules. In the proposed TS, hydrogen bonding interactions between the nitronate group of the thiourea moiety, the protonated quinuclidine and the imine promote the nucleophilic attack on the Si-face of the cyclic imine leading to (R)-enantiomer 359.
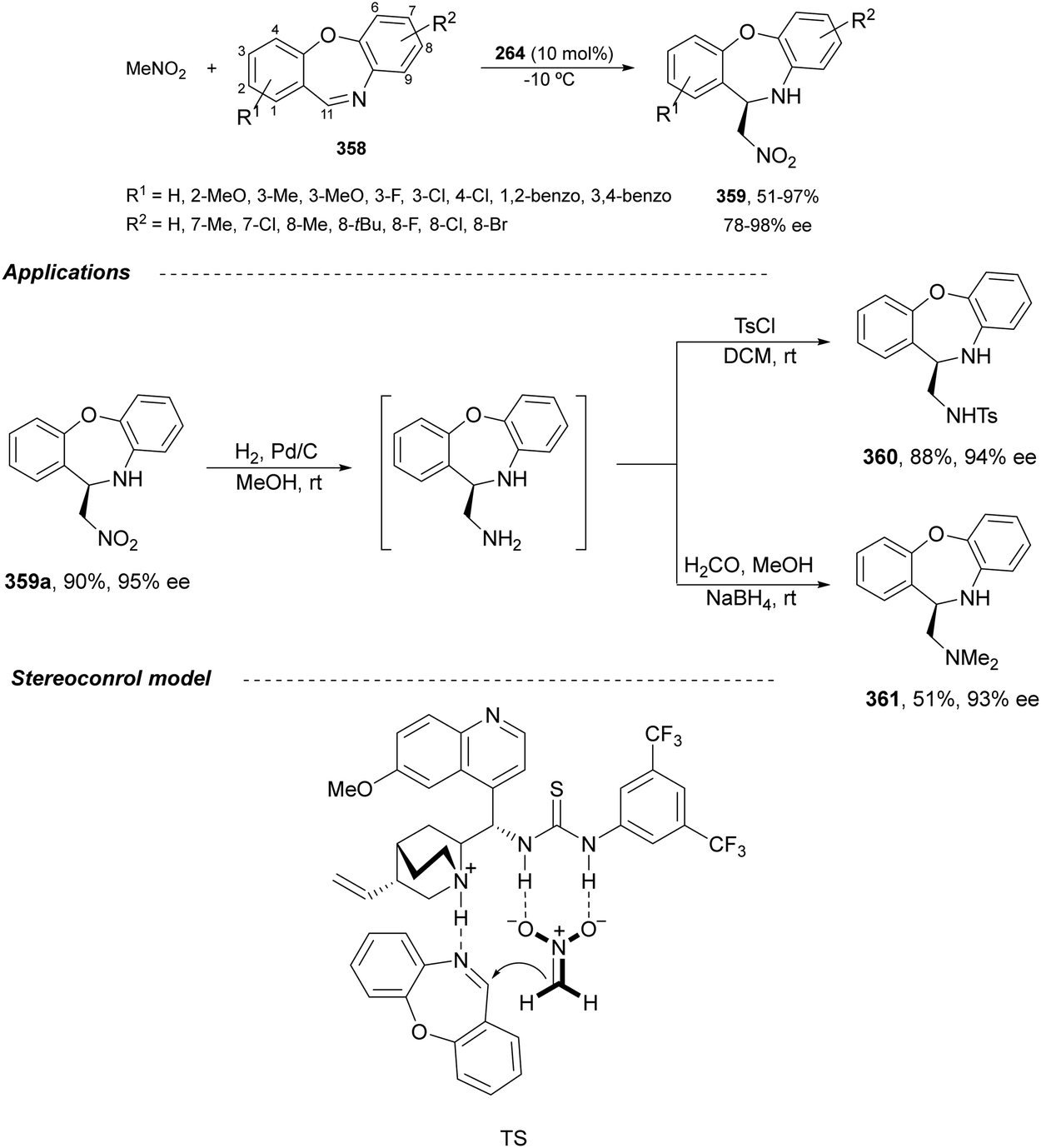 | ||
| Scheme 109 Asymmetric aza-Henry reaction of nitromethane with dibenzo[b,f][1,4]oxazepines 358 under thiourea 264 catalysis. | ||
The same group222 performed the aza-Henry reaction catalyzed by thiourea 264 of nitromethane with indolenines 362 to obtain 2-nitromethyl indolines 363 in good yields and high enantioselectivities (Scheme 110). This protocol was scaled-up to 3 mmol of 3H-indole [R1 = 4,6-Me2, R2–R2 = (CH2)5] 362a giving the corresponding product 363a in 90% yield and 94% ee. The same product 363a was transformed into diamine 364 in 84% yield and ca. 94% ee.
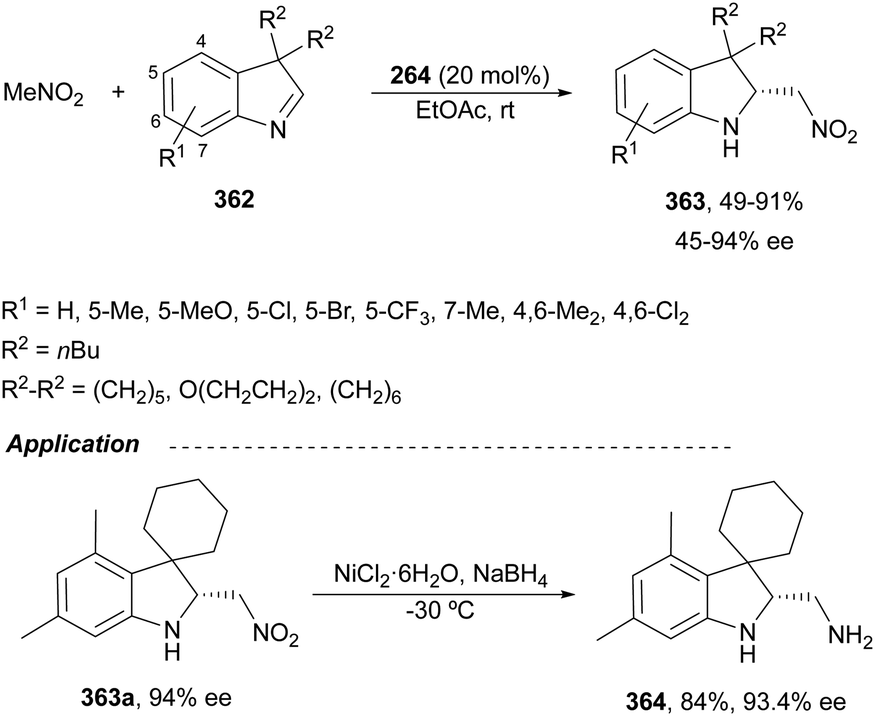 | ||
| Scheme 110 Asymmetric aza-Henry reaction of nitromethane with 3H-indoles 362 under thiourea 264 catalysis. | ||
Recently, Di Mola, Massa and co-workers223 reported an asymmetric cascade aza-Henry/lactamization reaction of α-amido sulfones 365 with nitromethane under Takemoto's thiourea 289 catalysis. α-Amido sulfones 365, prepared from 2-formyl benzoates, generated in situ the corresponding N-protected imines which reacted with nitromethane to form intermediates 366. These compounds underwent in situ cyclization at room temperature to give 3-(nitromethyl)isoindolin-1-ones 367 in good yields and enantioselectivities (Scheme 111). Reduction of the nitro group of product 367a was efficiently carried out with Zn and a small amount of HCl in MeOH at 0 °C to furnish product 368 with 61% yield and the same ee.
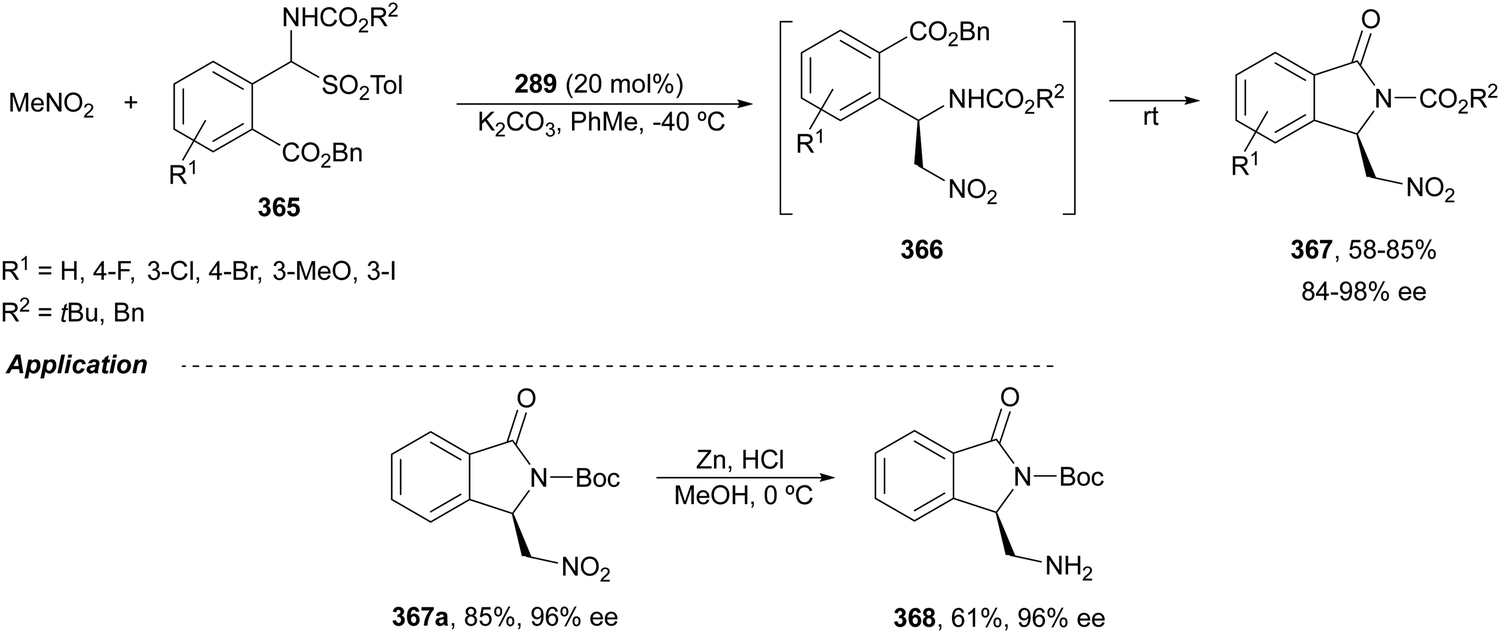 | ||
| Scheme 111 Asymmetric aza-Henry reaction of nitromethane with α-amido sulfones 365 under thiourea 289 catalysis followed by in situ lactamization. | ||
McHardy and co-workers224 recently performed the synthesis of two protein kinase C (PKC)-epsilon inhibitors to treat alcohol use disorder.225 Starting from α-amido sulfone 369, the aza-Henry reaction with nitromethane using Zhao thiourea 333 as an organocataltyst,208 product 370 was obtained with the highest enantioselectivity (95% ee) and with 98% yield (Scheme 112a). Adduct 370 was further transformed in four steps into CIDD-0072424 371. In the case of compound 373, they started from N-acylimine 372 and used a Takemoto catalyst (S,S)-289 for the aza-Henry reaction with nitromethane (Scheme 112b). Product 373 was isolated in 95% yield and 93% ee, and was further converted into compound (S)-374 in 27% overall yield over eight-step sequence.
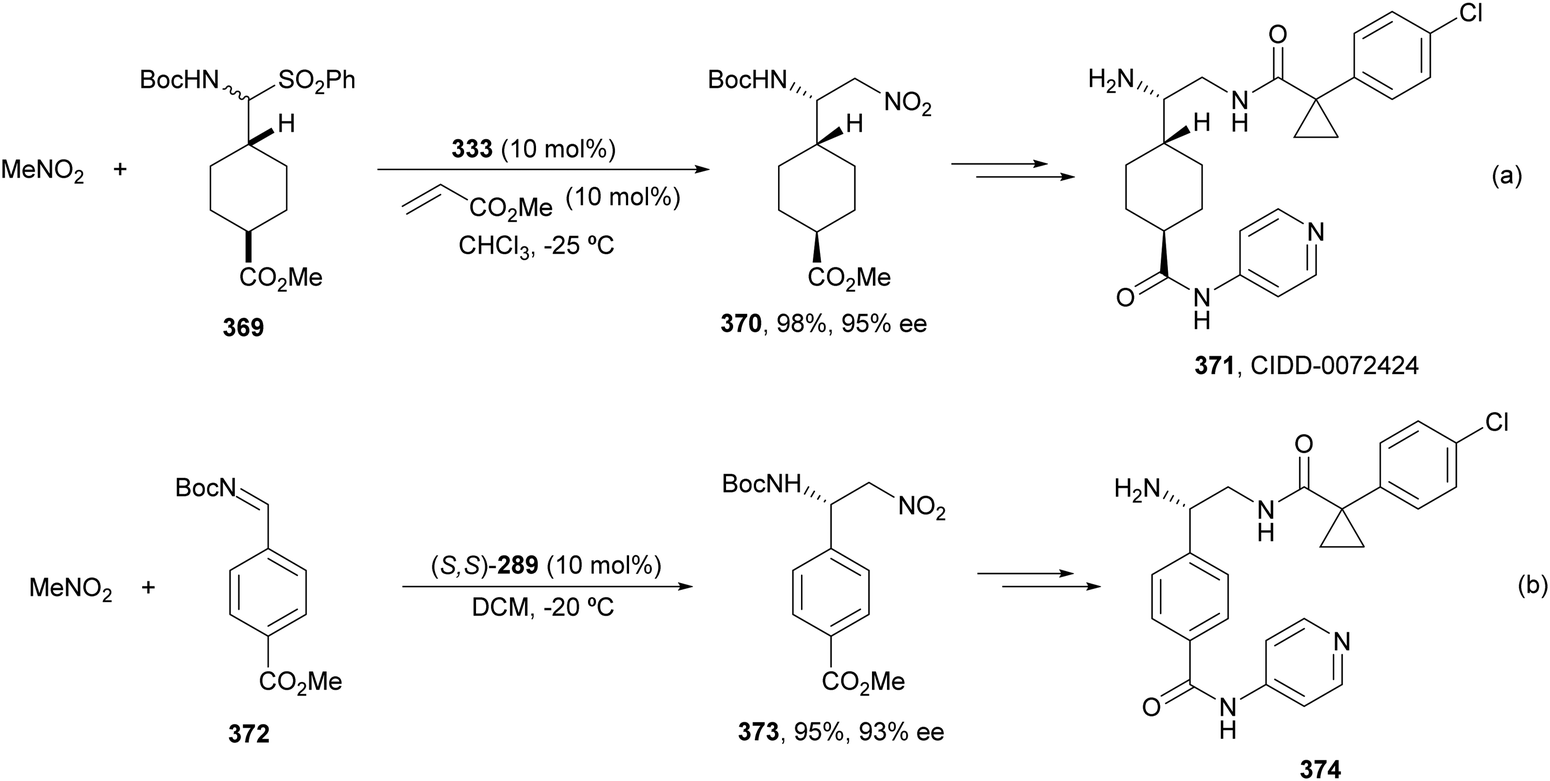 | ||
| Scheme 112 Asymmetric aza-Henry reaction of nitromethane with α-amido sulfone 369 and N-Boc-imine 372 catalyzed by thioureas 333 and 289 towards PKC-epsilon inhibitors 371 and 374, respectively. | ||
It can be generalized that chiral thioureas, especially Cinchona alkaloids derived thioureas, are excellent and versatile organocatalysts for the asymmetric aza-Henry reaction of acyclic and cyclic aldimines as well as ketimines.
In 2005, phase transfer-catalyzed asymmetric aza-Henry reactions were simultaneously reported by Herrera, Bernardi and co-workers226 and by Palomo and co-workers227 using α-amido sulfones of type 165 as precursors of N-carbamoyl imines. N-Benzyl quininium chloride 375 (Fig. 13) was used in both cases as a catalyst in the presence of KOH226 and CsOH·H2O227 as bases in toluene at −45226 and −50 °C,227 respectively. The corresponding (R)-β-nitroamines 320 were obtained in 53–98% yields and 73–98% ee,226 whereas Palomo reported 72–83% yields and 82–98% ee.227 In the last case, the reaction with nitroethane afforded anti-products 320 with 85–88% yields, 75:25–95:5 dr and 91–98% ee. They also studied the reaction mechanism using experimental work and theoretical calculations.228 In the proposed catalytic cycle, the nitronate anion is the base to generate the N-Boc imine from the α-amido sulfone (slow step). The nitronate is also responsible for the addition of the in situ generated imine (fast step) and TS with several hydrogen bonds explains the observed enantioselectivity.
 | ||
| Fig. 13 Phase-transfer catalysis for the asymmetric aza-Henry reaction of N-acyl imines with nitroalkanes. | ||
In Fig. 13, several PTCs such as the N-benzotriazole-quininium salt 376 were used by He and co-workers,229 for the aza-Henry reaction of N-tosyl α-amido sulfones 165 with nitromethane and nitroethane to obtain β-nitro amines 323 with reversal of enantioselectivity than the N-benzyl quininium salt 375. Takada and Nagasawa230 used the guanidinium-cis-thiourea 377 (Fig. 13) for the reaction of N-Boc imines with nitroalkanes obtaining β-nitroamines 320 up to 96% yield, 99:1 dr and 99% ee. Peng, Han and co-workers231 used the same catalyst 377 (Fig. 13) starting from N-Boc α-amido sulfones but with lower diastereoselectivities than N-Boc imines. Dixon and co-workers232 used a quininidium-urea catalyst 378 (Fig. 13) for the aza-Henry reaction of α-amido sulfones and nitroalkanes to obtain products 320 with 83–100% yields, 6:1–24:1 dr and 84–95% ee.
Kumaraswamy and Pitchaiah233 applied Palomo's reaction conditions227 to the synthesis of furyl derived β-nitroamines 323, which were transformed into orthogonally protected (2S)-2,3-diaminopropanoates 380 (Scheme 113). The furan moiety was oxidatively cleaved to obtain the carboxylic acids, which were esterified with diazomethane to provide methyl esters 379. Subsequent reduction of the nitro group provided 2,3-diaminopropanoates 380, which were further protected as Fmoc derivatives.
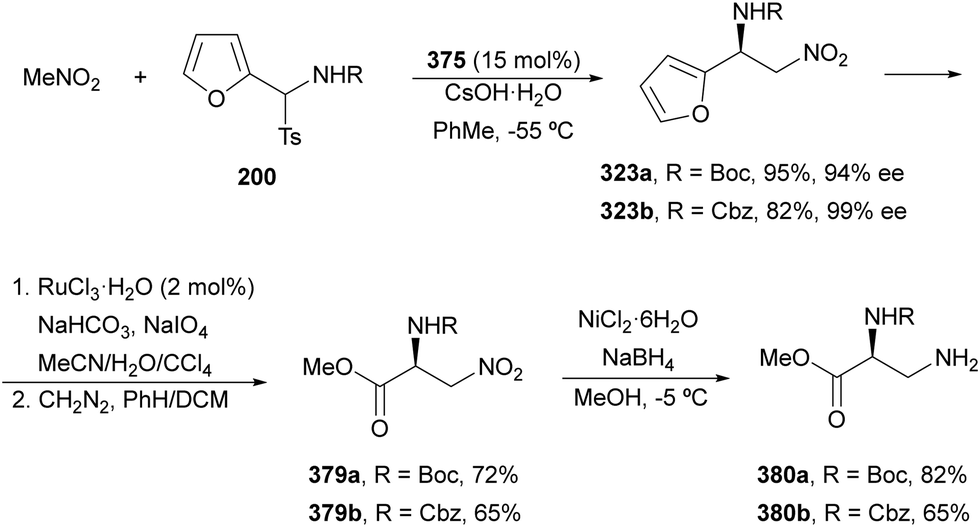 | ||
| Scheme 113 Asymmetric aza-Henry reaction of nitromethane with furfural derived α-amido sulfones 200 using 375 as a phase-transfer catalyst. | ||
The same Indian's group234 applied this aza-Henry reaction to the enantioselective synthesis of (2S,3S)-methyl 3-aminopiperidine-2-carboxylate 383. In this case, the nitro compound 381 was allowed to react with α-amido sulfone 200 (R = Boc) under asymmetric Palomo's PTC conditions227 to afford mainly the corresponding nitro adduct 382, which was further transformed into the pipecolic acid derivative 383 (Scheme 114).
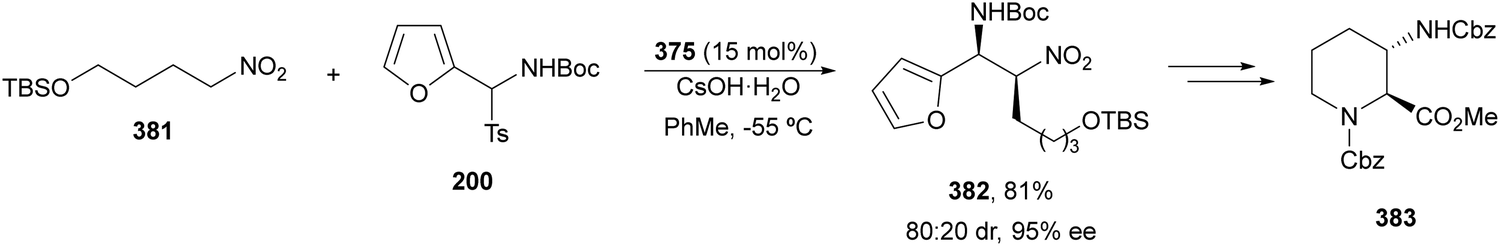 | ||
| Scheme 114 Asymmetric aza-Henry reaction of nitroalkane 381 with α-amido sulfone 200 using 375 as a phase-transfer catalyst. | ||
Cinchona alkaloid-derived bifunctional ammonium salts 384 derived from quinine and 385 derived from quinidine bearing a urea unit with a β-amino alcohol have been used by Lin, Duan and co-workers235 for the enantiodivergent52 aza-Henry reaction of α-amido sulfones 165 with nitroalkanes (Scheme 115). Enantiomeric β-nitroamine derivatives 320 were obtained with good yields and diastereo- and enantioselectivities. Walvoord and Kozlowski236 employed cinchonidinium acetate as a phase-transfer catalyst for the asymmetric synthesis of cis-stilbene diamines. The reaction of α-aryl nitromethanes with N-Boc benzylidene imines 161 using cinchonidine (10 mol%) and HOAc (10 mol%) in dichloromethane at −30 °C provided products anti-320 (R1 = Ar) with 29–99% yields, 97:3–99:1 dr and 26–79% ee. Lin, Duan and co-workers237 performed the same reaction using α-amido sulfones 165 and catalyst 386. Products anti-320 (R1 = Ar) were obtained with excellent yields (93–99%) and diastereo- (96:4–>99:1 dr) and enantioselectivities (91–99% ee).
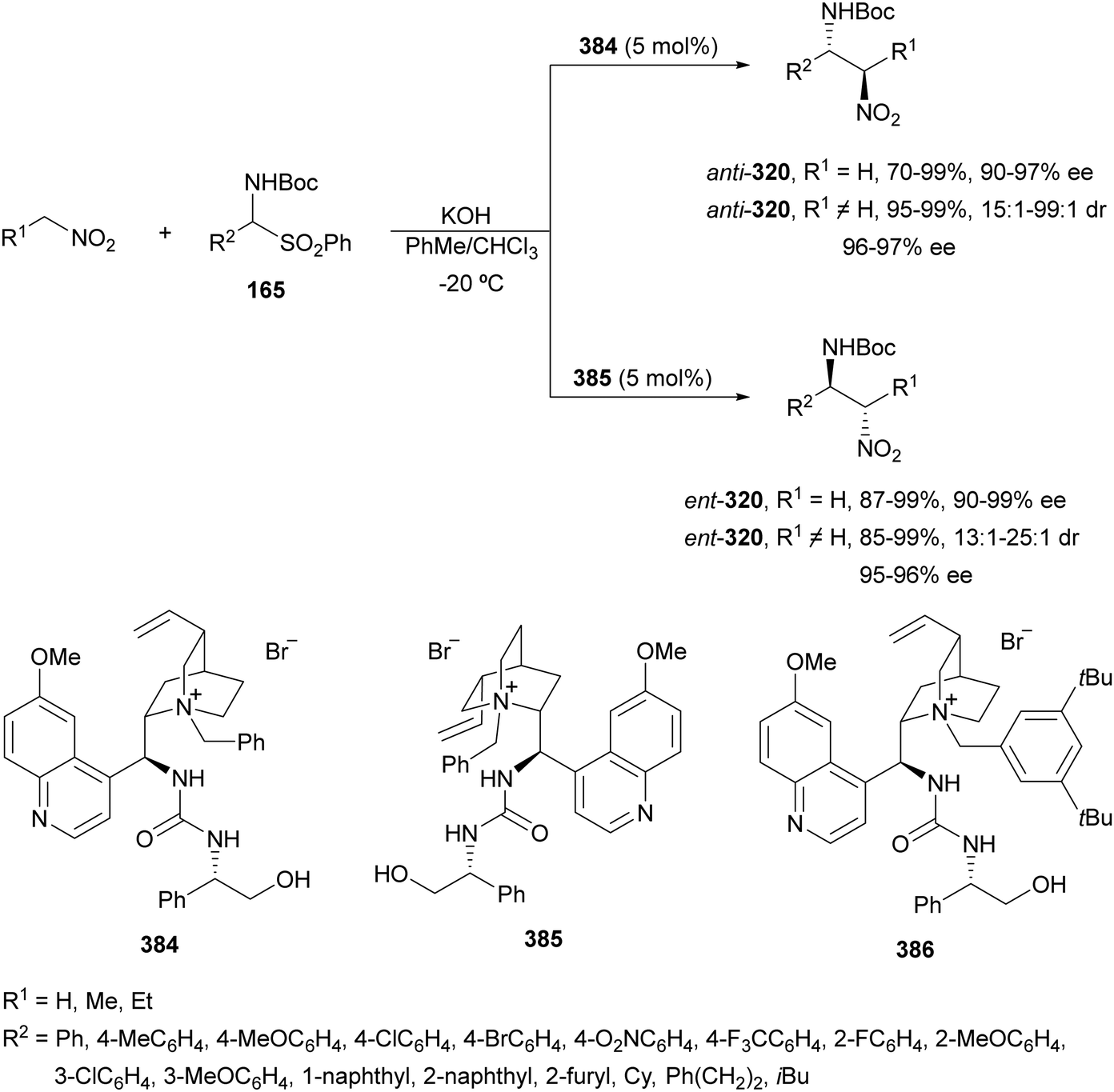 | ||
| Scheme 115 Asymmetric enantiodivergent aza-Henry reaction of nitroalkanes with α-amido sulfones 165 using 384–386 as phase-transfer catalysts. | ||
For the synthesis of the quinolone-fused lactam LP99, the groups of Brennan and Dixon238 developed an aza-Henry reaction/lactamization cascade process. Reaction of the 4-nitrobutanoate 387 with N-Boc imine 388, using the bifunctional quinidinium-urea catalysis 378232 under solid–liquid PTC conditions, provided adducts 389 on a gram scale with 70% yield, 7:1 dr and 90% ee (Scheme 116). Deprotection of N-Boc followed by cyclization and reduction of the NO2 group and subsequent steps gave the corresponding LP99 with a KD value of 99 nM against BRD9. Treatment with LP99 led to displacement of BRD7 and BRD9 from chromatin and down-regulation of the pro-inflammatory cytokine IL-6.
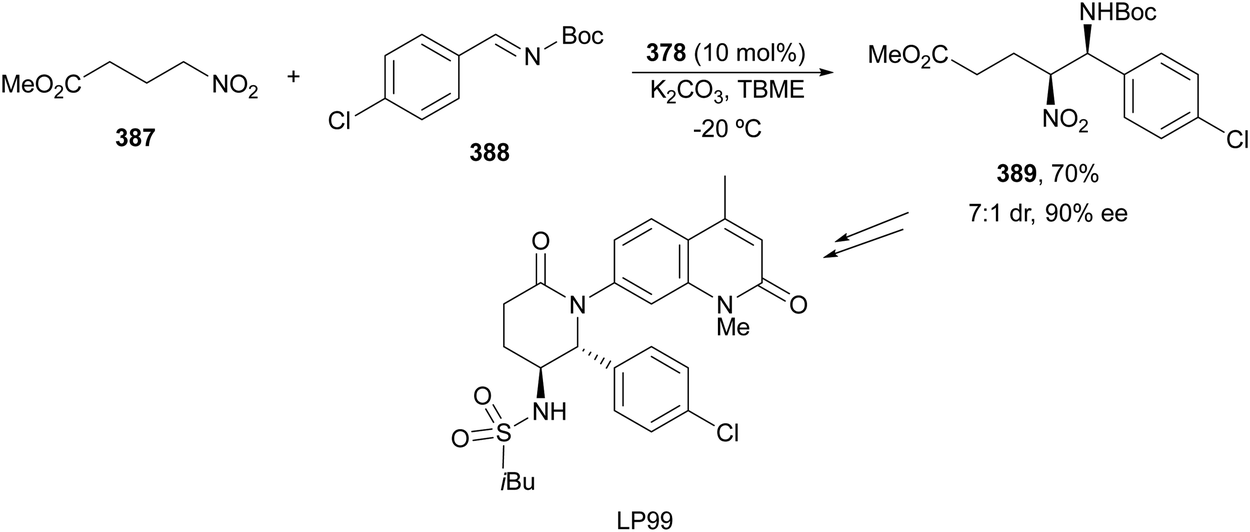 | ||
| Scheme 116 Asymmetric aza-Henry reaction of methyl 4-nitrobutanoate (387) with N-Boc imine 388 using 378 as a phase-transfer catalyst. | ||
Nitroalkenes 390 were reacted with α-amido sulfones 200 under PTC using the quininium salt 386, and LiOH as a base in DCM at −40 °C (Scheme 117).239 This bifunctional catalyst with multiple hydrogen bonding donors provided compounds 391 in excellent yields and diastereo- and enantioselectivities. Based on DFT calculations, a TS has been proposed in which the in situ generated N-Boc imine forms hydrogen bonding with the urea and the hydroxy group of the amino alcohol unit. In addition, the nitronate forms hydrogen bonding with two hydrogen atoms of the quinuclidine moiety.
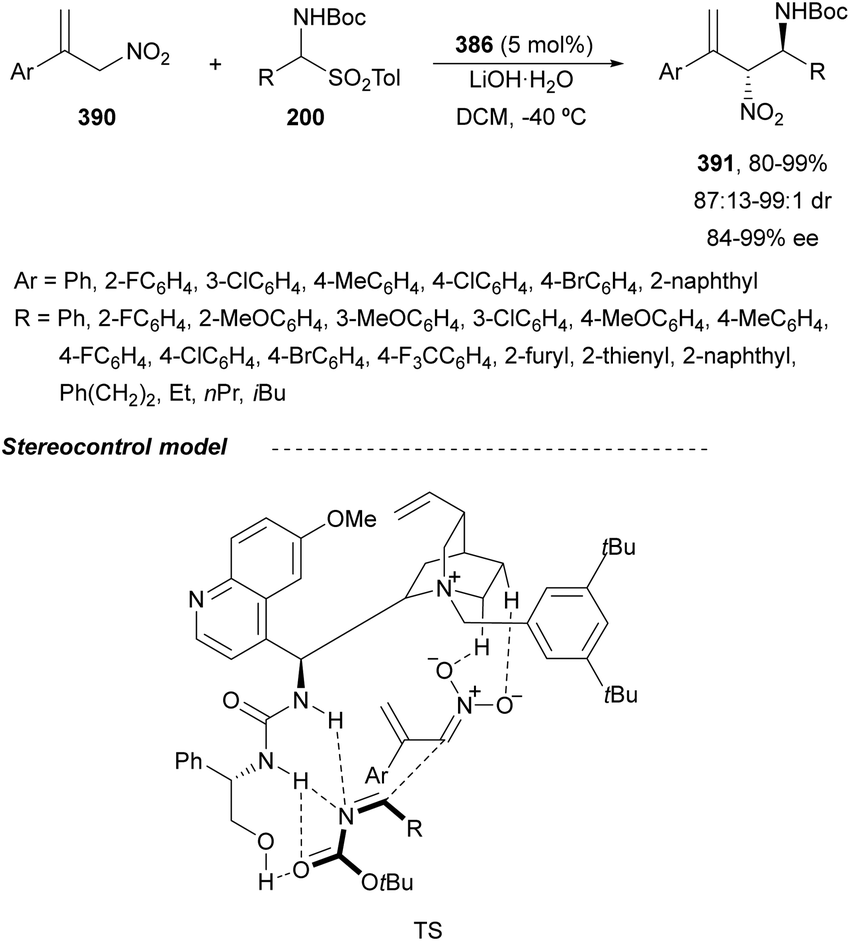 | ||
| Scheme 117 Asymmetric aza-Henry reaction of nitroalkenes 390 with α-amido sulfones 200 using phase-transfer catalyst 386. | ||
Bifunctional thiourea-ammonium salts derived from α-amino acids have been developed by Zhao and co-workers.240 The best phase-transfer catalyst 392 for the aza-Henry reaction of α-amido sulfones 200 with nitroalkanes gave products (S)-320 and (S,S)-320 with 80–99% yields, 6:1–>25:1 dr and 68–99.5% ee. In the proposed TS, the thiourea unit has hydrogen-bonding interactions with the in situ generated N-Boc imine and the nitronate interacts with the ammonium center by Coulomb force (Fig. 14). The same group241 employed for the same aza-Henry reaction thiourea-phosphonium salts also derived from α-amino acids. Catalyst 393 (Fig. 14) and also KOH as a base in toluene at −20 °C provided products (S)-320 and (S,S)-320 in 68–99% yields, moderate diastereoselectivities (syn:anti = 22:78–47:53) and excellent enantioselectivities (86–98% ee). Lin, Duan and co-workers242 performed this type of asymmetric aza-Henry reaction using bifunctional phase-transfer catalysts 394a and 394b derived from α-amino acids (Fig. 14). The corresponding products (S)-330 and (S,S)-320 were prepared with KOH as a base in CHCl3 at −20 °C with excellent yields (93–99%), 90:10–92:8 dr and 90–>99.9% ee. More recently, this group243 performed the reaction of α-aryl nitromethanes with α-amido sulfones in the presence of catalyst 395 in good yields and stereoselectivities (Scheme 118).
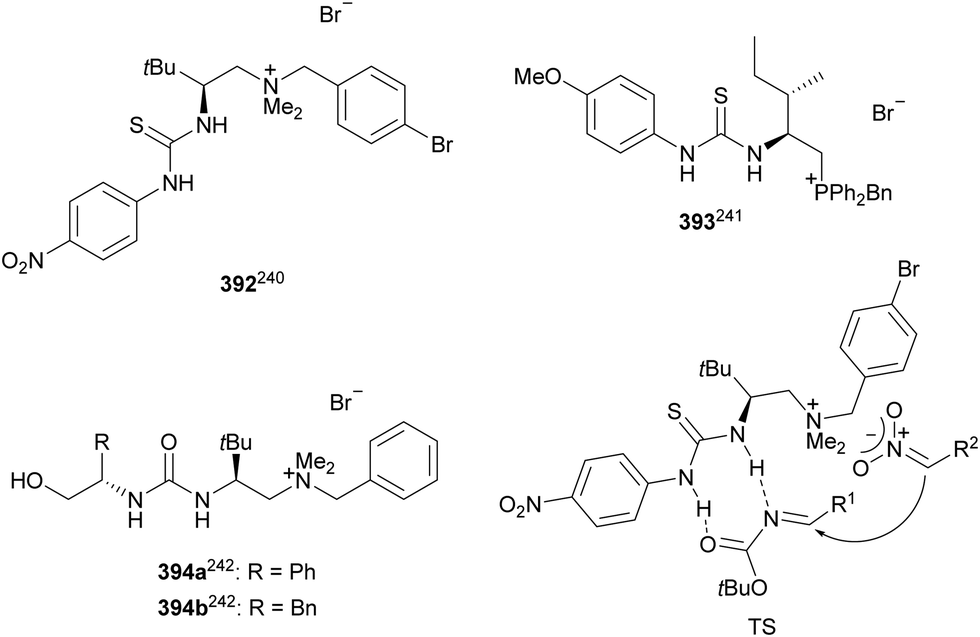 | ||
| Fig. 14 Catalysts used for the asymmetric aza-Henry reaction of nitroalkanes with N-Boc α-amido sulfones 200. | ||
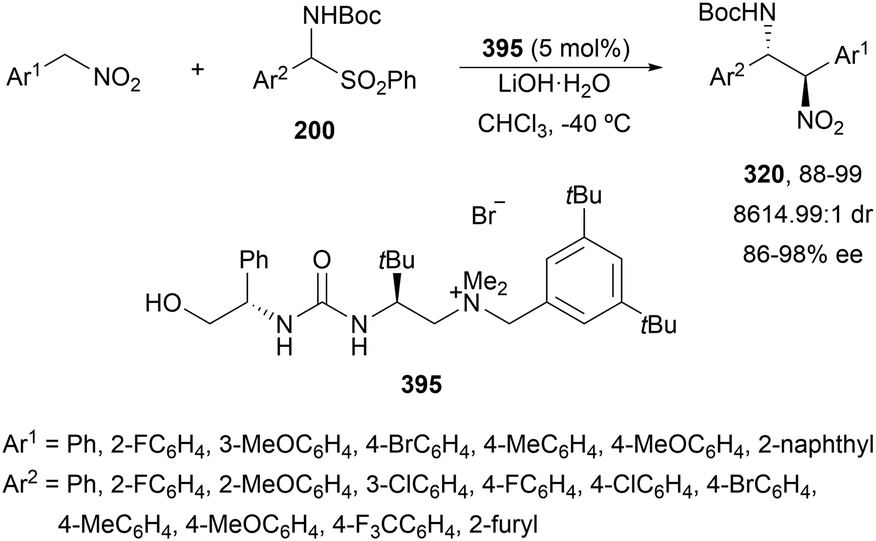 | ||
| Scheme 118 Asymmetric aza-Henry reaction of α-aryl nitromethanes with α-amido sulfones 200 using phase-transfer catalyst 395. | ||
Ketimines 396 bearing a 6-methyl-2-pyridylsulfonyl protecting group were active electrophilic substrates for the asymmetric aza-Henry reaction under PTC conditions. Lin, Duan and co-workers244 used a modified Dixon's catalyst 378 with a 2-methoxyphenyl group instead of a phenyl group at the quinuclidine nitrogen and a thiourea instead of the urea moiety 397. The addition of nitromethane to ketimines 396 took place with good yields and enantioselectivities giving products 398 (Scheme 119). Based on DFT calculations, a TS with a Wynberg ion-pair hydrogen bond type mechanism as well as the formation of hydrogen bonds with the thiourea unit leading to the Si-face addition of ketimine was proposed.
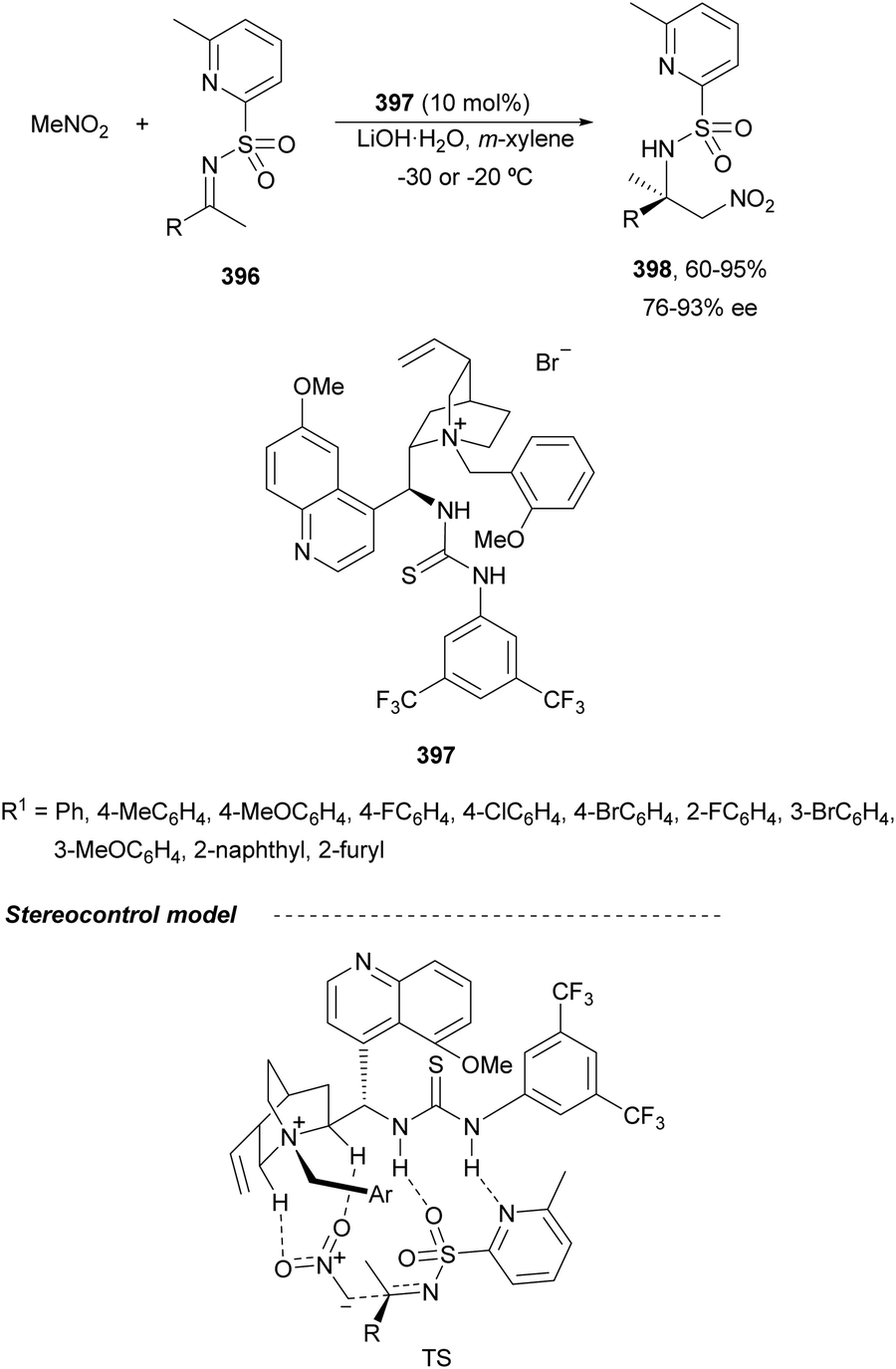 | ||
| Scheme 119 Asymmetric aza-Henry reaction of nitromethane with ketimines 396 using a phase-transfer catalyst 397. | ||
Lin, Duan and co-workers245 modified catalyst 397 for the asymmetric aza-Henry reaction of α-keto esters-derived N-tosyl ketimines 351 with nitromethane. In this case, a 3,5-di-tert-butyl benzyl moiety at the quinuclidine nitrogen was proved to be the best phase-transfer catalyst 399 to give products 353 with very good yields and moderate to high enantioselectivities (Scheme 120). Product 353a was reduced with Zn in HOAc and after acetylation the α,β-diamino ester derivative 400 was obtained in 81% yield and 97% ee.
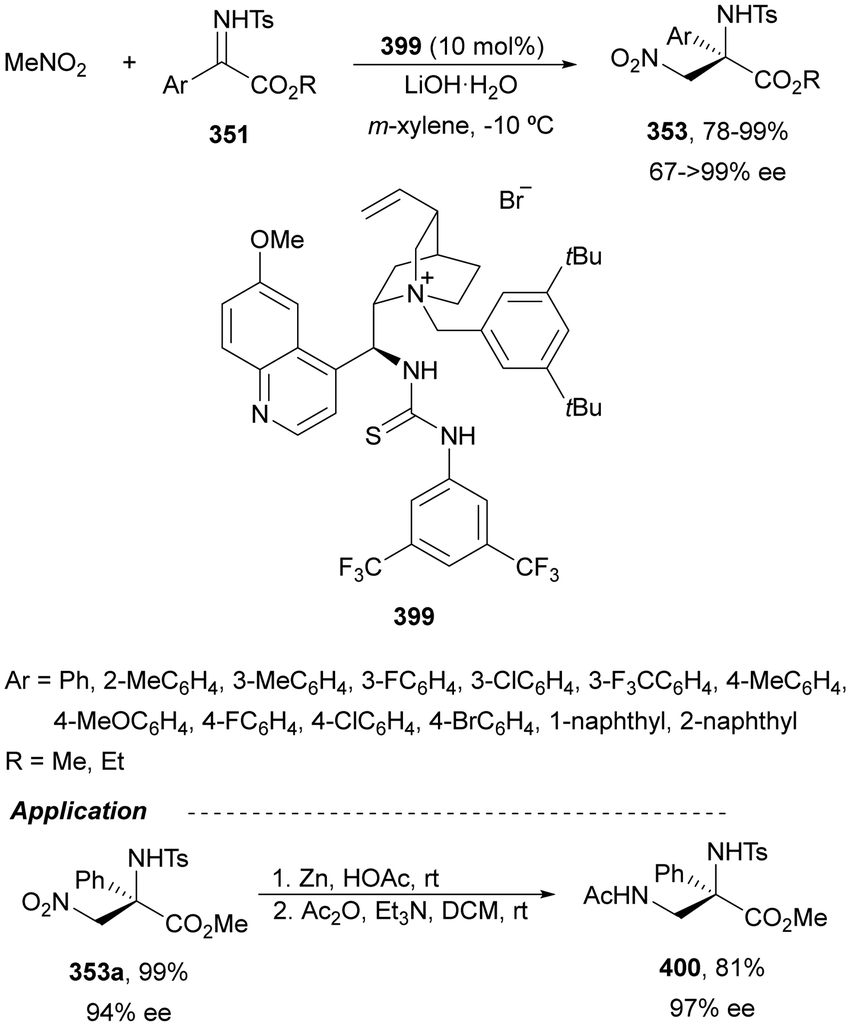 | ||
| Scheme 120 Asymmetric aza-Henry reaction of nitromethane with ketimines 351 using the phase-transfer catalyst 399. | ||
Acyclic trifluoromethyl ketimines 401 have been used for the first time as electrophiles in the asymmetric aza-Henry reaction with nitromethane by Lin, Duan and co-workers.246 After screening of several quininium salts, including Dixon's catalyst 378,232 the catalyst containing (S)-phenylglycinol moiety 386 (Scheme 115) gave the best results (Scheme 121). The resulting fluorinated β-nitroamines 402 were obtained with excellent yields with high enantioselectivities. Product 402a was reduced to N-Boc diamine 403 in 90% yield, using NiCl2/NaBH4, and by further treatment with DBU it was transformed into the imidazolidinone 404 in 90% yield.
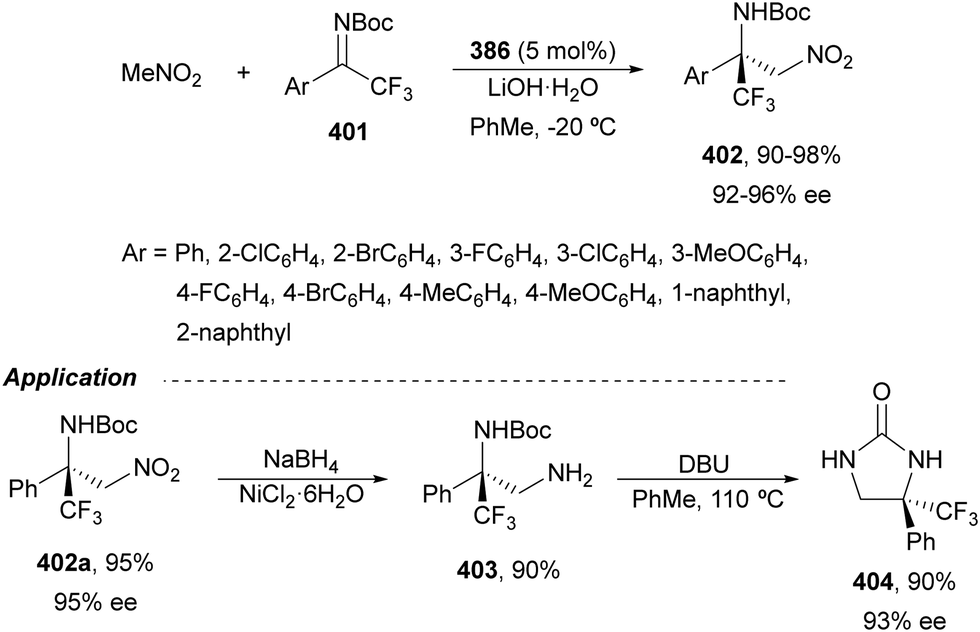 | ||
| Scheme 121 Asymmetric aza-Henry reaction of nitromethane with trifluoromethyl ketimines 401 using a phase-transfer catalyst 386. | ||
The group of Lin and Duan has applied the phase-transfer catalyst 386 to the asymmetric aza-Henry reaction of nitromethane247 and aryl nitromethanes248 to N-Boc ketimines derived from isatins 170. The 3-substituted 3-aminooxindoles 355 were obtained with excellent yields and good enantioselectivities (Scheme 122). In the case of α-aryl nitromethanes, they also employed249 the (S)-tert-leucine-derived urea-ammonium salt 405 as a phase-transfer catalyst to obtain products 355 with similar results (Scheme 122). In the proposed TS for catalyst 405, the urea forms hydrogen-bond interactions with the N-Boc group of the ketimine and the ammonium unit would form an ion pair with the nitronate.
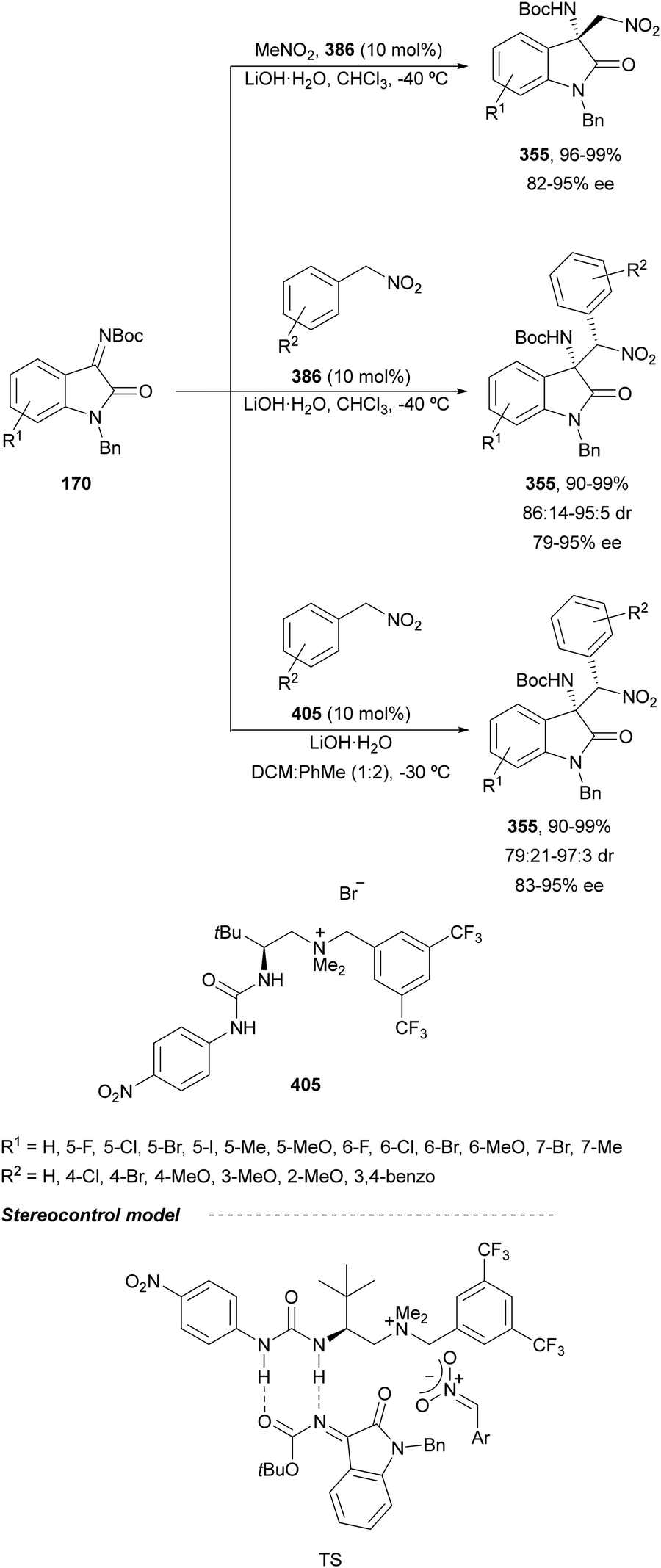 | ||
| Scheme 122 Asymmetric aza-Henry reaction of nitroalkanes with isatin-derived N-Boc ketimines 170 using phase-transfer catalysts 386 and 405. | ||
Phase-transfer catalysis for the asymmetric aza-Henry reaction is based on Cinchona-alkaloids ammonium salts not only for aldimines but also for ketimines. For less reactive ketimines, an extra thiourea unit enhances its reactivity with nitromethane. Under these PTC reaction conditions, α-amido sulfones can be used as precursors of the corresponding aldimines.
Chiral bases are able to deprotonate the nitro compound to form the corresponding nitronate. Ooi and co-workers250 reported in 2008251 a chiral ammonium betaine 406 as a bifunctional base, which catalyzed the aza-Henry reaction of α-nitro esters 330 with N-Boc imines 161, with only 1 mol% catalyst loading, to provide mainly compounds syn-332 (Scheme 123). This process was carried out with only 1 mol% of organocatalyst in toluene at 0 °C giving products 332 in excellent yields, modest diastereoselectivities and excellent enantioselectivities for both diastereomers. The catalytic performance of C1-symmetric chiral ammonium betaine 407 was studied for the same aza-Henry reaction.252 Products syn-332 were obtained with similar yields and diastereo- and enantioselectivities.
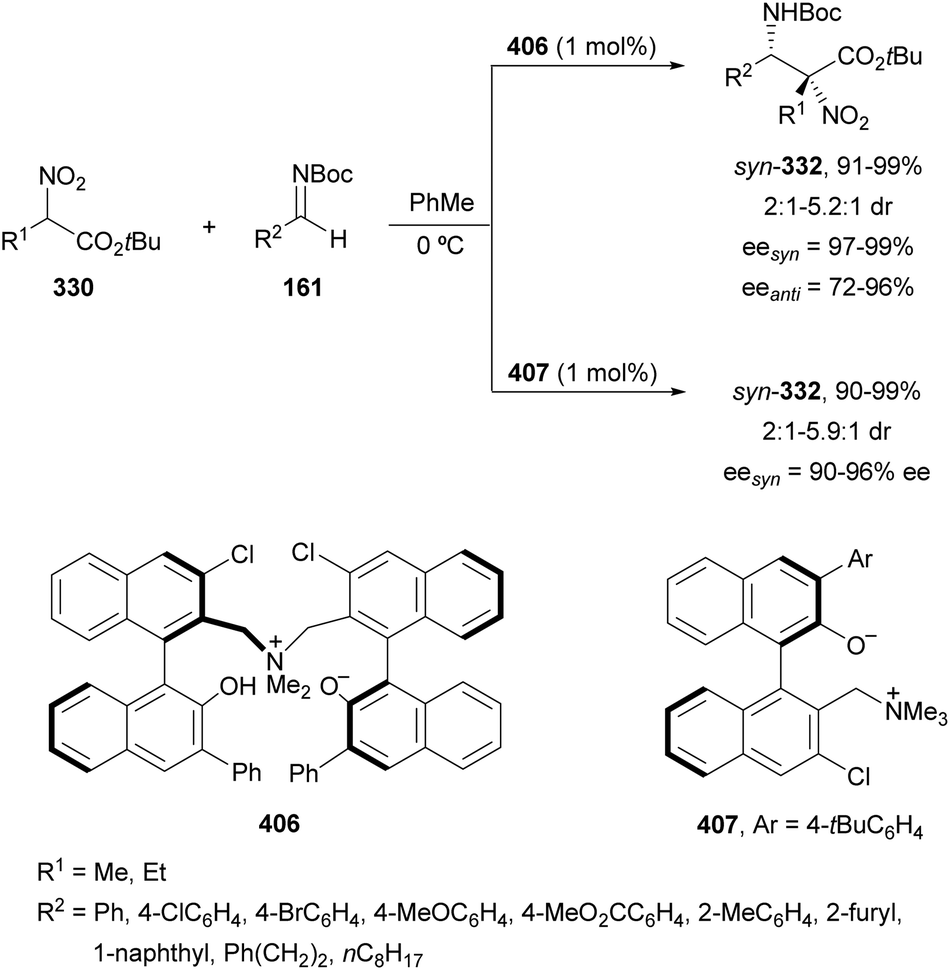 | ||
| Scheme 123 Asymmetric aza-Henry reaction of α-nitro esters 330 with N-Boc imines 161 using betaines 406 and 407 as catalysts. | ||
In the case of β,β-disubstituted nitroolefins 408, the addition of N-Boc aldimines 161 was catalyzed by the axially chiral ammonium betaine 409a (Scheme 124).253 Intermediate vinylogous nitronate I underwent α-addition to the imine to provide mainly β-nitroamines anti-410 in high yields and diastereo- and enantioselectivities. Intermediate II was postulated to be formed by interaction of nitronate I with the catalyst 409a. The reduction of anti-410a (R1 = R3 = Ph, R2 = H) with Zn/HCl in EtOH followed by Boc-protection gave diamine 411, which under hydrogenation conditions gave diastereomers 412, and was also transformed into α,β-diamino ketone 413 by ozonolysis.
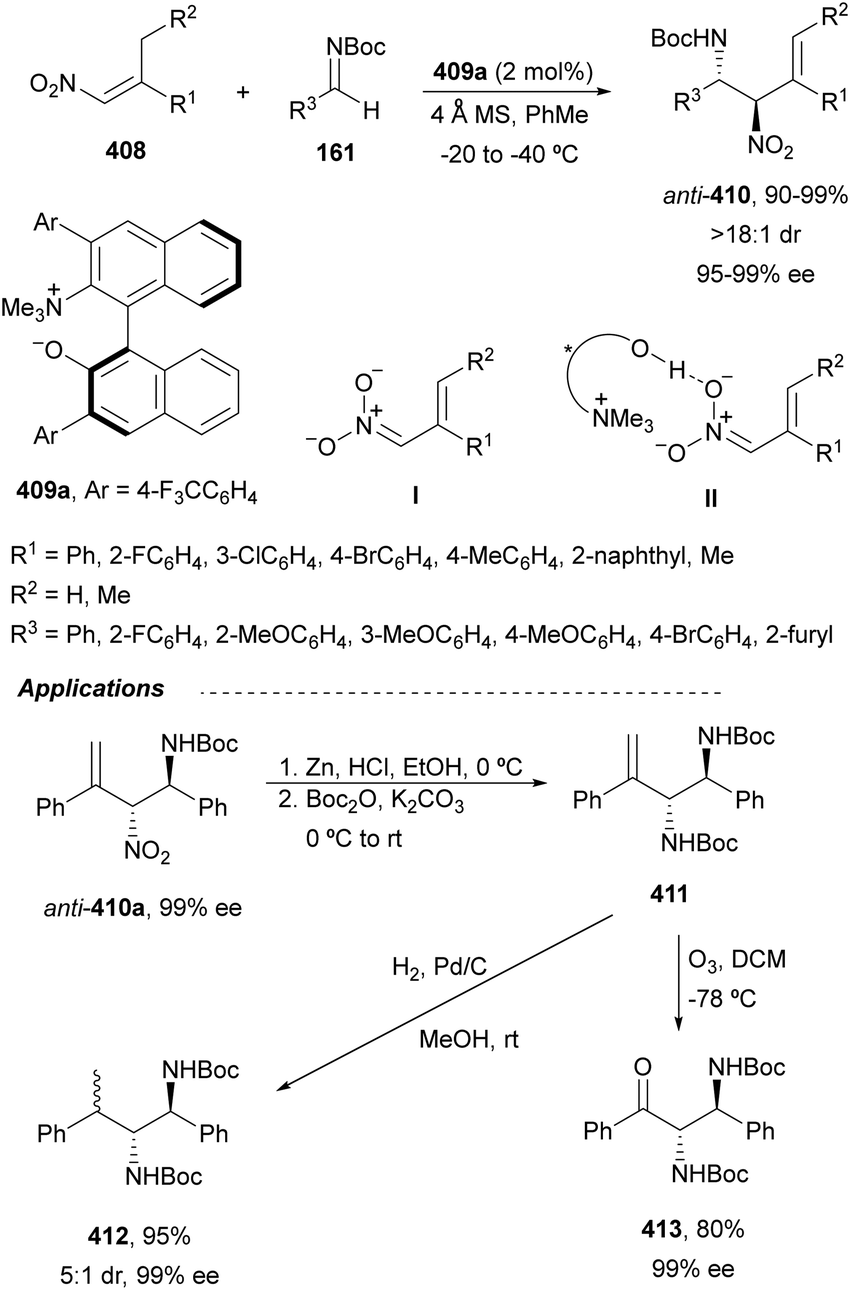 | ||
| Scheme 124 Asymmetric aza-Henry reaction of β,β-disubstituted nitroalkenes 408 with N-Boc imines 161 under betaine 409a catalysis. | ||
The same group reported254 the aza-Henry reaction of α-aryl-β-monosubstituted nitroolefins 414 with N-Boc imines 161 using the betaine 409b as a chiral base and catalyst. This base formed by γ-deprotonation of 414 the vinylogous nitronate I to provide anti-adducts 415 with >55% α-selectivity in good to excellent yields and diastereoselectivities, and high enantioselectivities (Scheme 125). Compound 415a (R1 = H, R2 = Ar = Ph) was reduced with Zn/HCl and protected with CbzCl to the corresponding diamine 416, which was further transformed by ozonolysis into aldehyde 417 and oxidized to the orthogonally protected α,β-diamino acid 418.
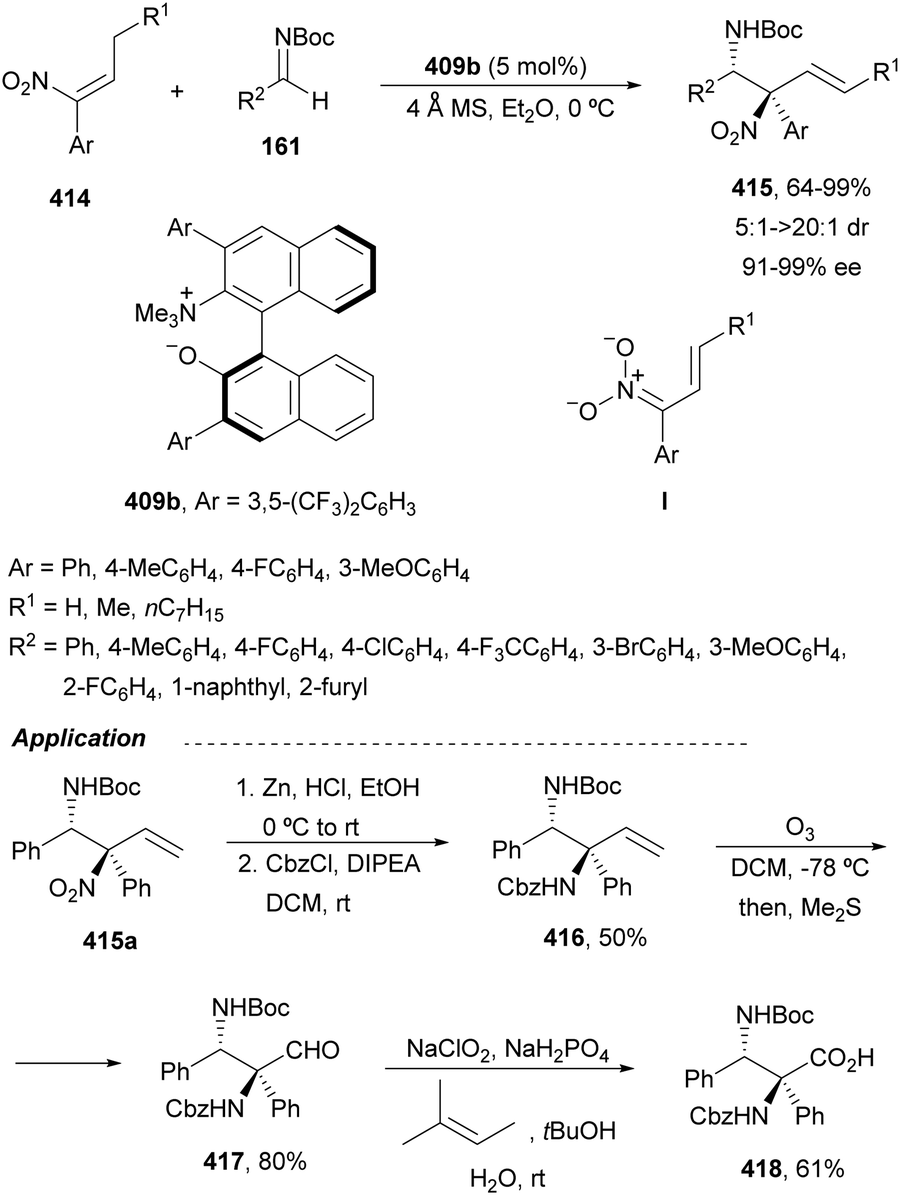 | ||
| Scheme 125 Asymmetric aza-Henry reaction of α-aryl-β-monosubstituted nitroolefins 414 with N-Boc imines 161 under betaine 409b catalysis. | ||
When α-aryl nitromethanes were allowed to react with N-Boc imines 161 in the presence of the base 409a, mainly anti-products 320 were obtained with high yields and enantioselectivities (Scheme 126).255 The β-nitroamines 320a (R = Ar = Ph) and 320b (R = Ph, Ar = 2-FC6H4) were transformed into the corresponding anti-1,2-diamines 419 by reduction with CoCl2/NaBH4. Compound 419b was deprotected by treatment with TFA to obtain diamine 420b, and 419a was tosylated to the orthogonally protected diamine 421a.
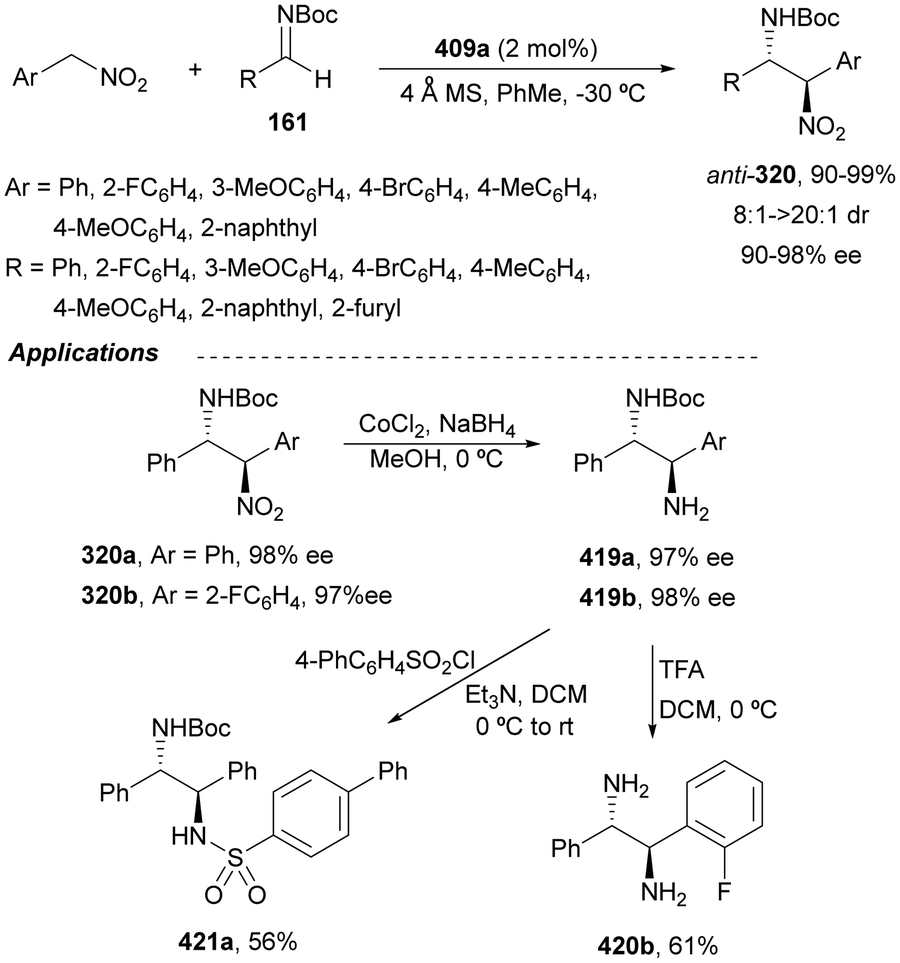 | ||
| Scheme 126 Asymmetric aza-Henry reaction of α-aryl nitromethanes with N-Boc imines 161 under betaine 409a catalysis. | ||
The addition of nitromethane to ketimines derived from α-keto ester 351a or isatin 170a has been performed with quinine-derived bifunctional catalyst 422 (Scheme 127). Zhou and co-workers256 found out that DBU was an appropriate base and catalyst for this addition and as a proof of concept employed 422 for the enantioselective version. Products 353a or 423a were obtained in 68 or 71% ee, respectively.
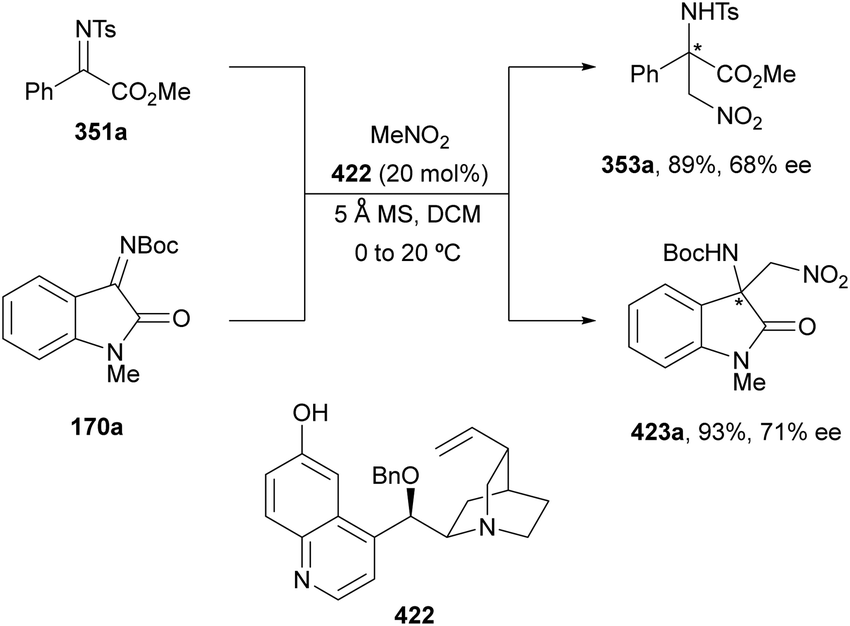 | ||
| Scheme 127 Asymmetric aza-Henry reaction of nitromethane with α-keto ester 351a or isatin-derived ketimine 170a under quinine-derived 422 catalysis. | ||
Chimni and co-workers257 employed the same quinine-derived 6′-OH organocatalyst 422 for the addition of nitroalkanes to isatin-derived N-Boc ketimines 170 to obtain products 423 in 60–80% yields, 54:46–72:28 dr and 56–89% ee (Fig. 15). In the proposed model for the TSI, the quinuclidine tertiary amine can deprotonate the nitroalkane which attacks on the Re-face of the ketimine activated through hydrogen bonding with the C6′–OH of the catalyst. Feng and co-workers258 employed a chiral bifunctional guanidine 424 (Fig. 15) for the aza-Henry reaction of nitromethane with isatin-derived N-Boc ketimines 170. The resulting 3-aminooxindoles 423 were obtained in 81–99% yields and 85–94% ee. In the proposed TSII, the guanidine deprotonates the nitroalkane and forms a dual hydrogen bonding and the amide acts as a Brønsted acid to activate the ketimine by hydrogen bonding.
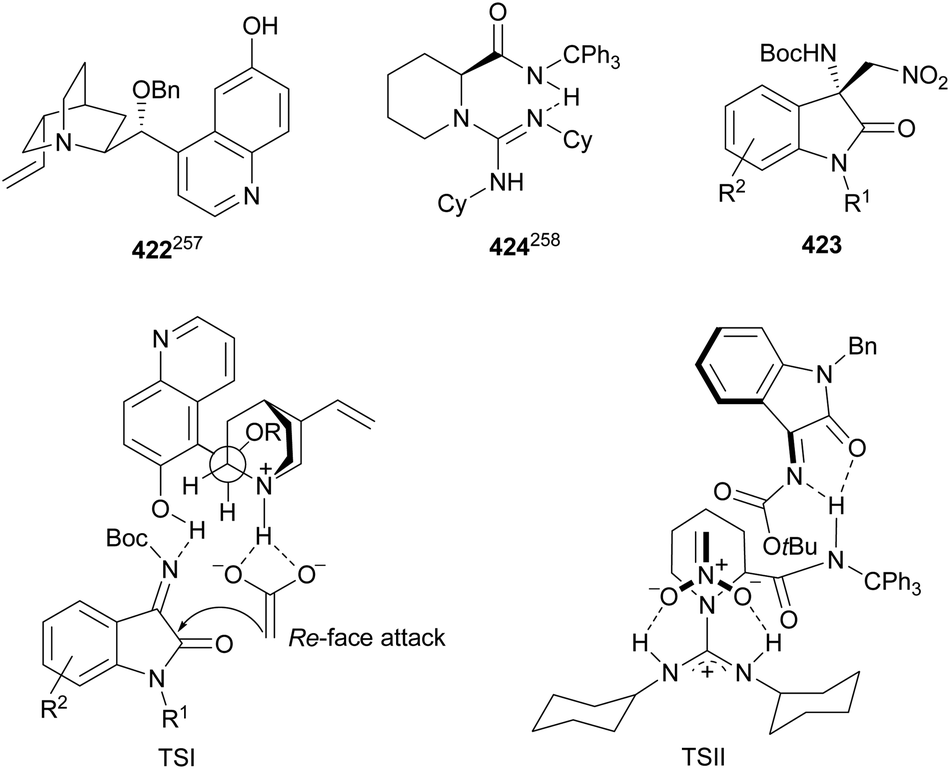 | ||
| Fig. 15 Chiral bases used as catalysts for the addition of nitromethane to isatin-derived N-Boc ketimines 170. | ||
The addition of nitromethane to trifluoromethyl aryl ketimines 401 has been recently carried out by Krstić and co-workers259 using a bifunctional iminophosphorane 425 (Scheme 128). β-Nitroamines 402 were isolated in moderate to good yields and enantioselectivities. A possible TS was proposed in which the thiourea unit interacts by hydrogen bonding with the ketimine and the nitromethane with the iminophosphorane and the thiourea.
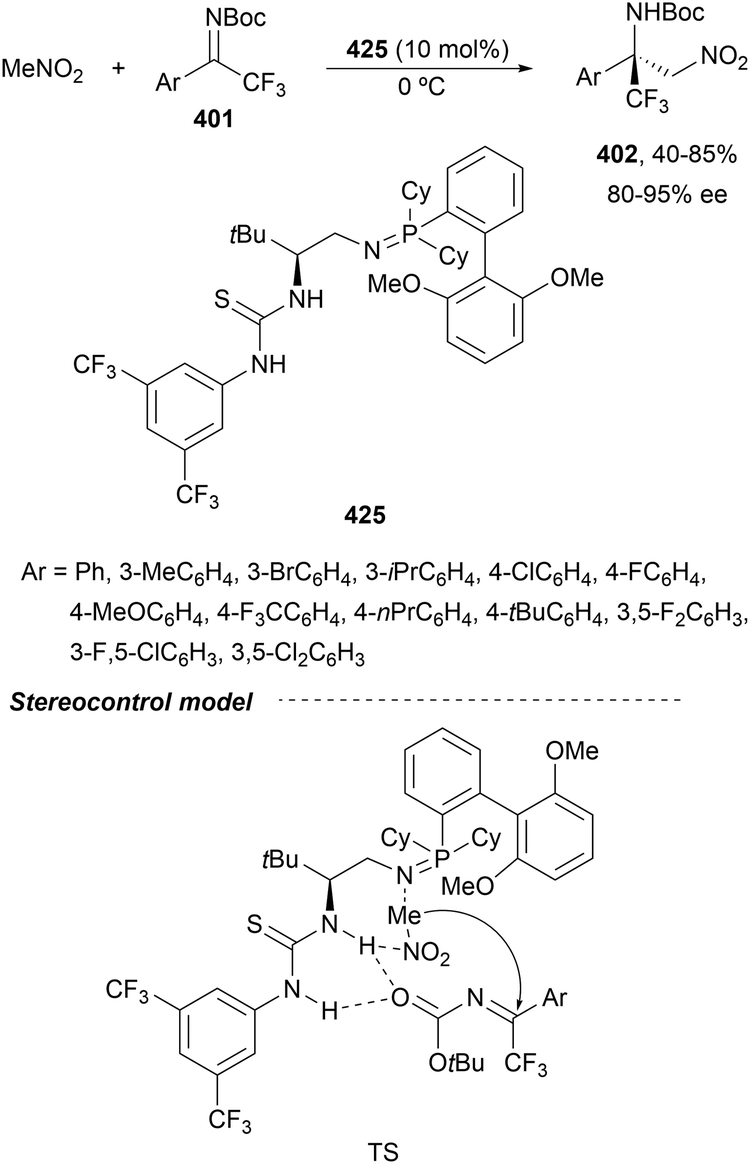 | ||
| Scheme 128 Asymmetric aza-Henry reaction of nitromethane with trifluoromethyl ketimines 401 under phosphorane 425 catalysis. | ||
N-Acyl hydrazones 426 have been used as electrophiles in the asymmetric aza-Henry reaction using Cinchona-derived bases as catalysts.260 Under quinine catalysis, β-nitrohydrazides 427 derived from nitromethane were obtained in moderate to good yields and modest enantioselectivities (Scheme 129). In the case of nitroethane, low diastereoselectivity was observed (up to 2.4:1). According to experimental studies, including kinetics, the catalyst activates both reagents and in the TS the hydrazone protonation and the C–C bond forming reaction occurs through a concerted process.
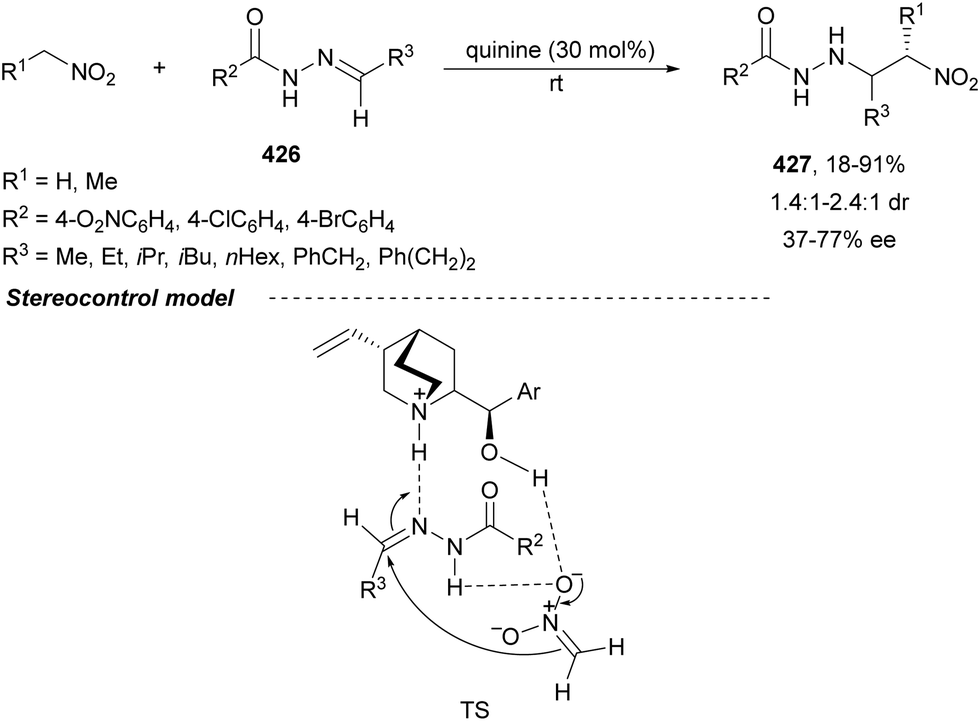 | ||
| Scheme 129 Asymmetric aza-Henry reaction of nitroalkanes with N-acyl hydrazones 426 under quinine catalysis. | ||
In 2004, Johnston and co-workers261 reported an enantioselective aza-Henry reaction of nitromethane and nitroethane with N-Boc imines 161 catalyzed by the Brønsted acid salt 428 (Fig. 16). This process was carried out at −20 °C to obtain anti-β-nitroamines 320 with 50–69% yields, 7:1–19:1 dr and 59–90% ee. A modified chiral bis(amidine) (BAM) Brønsted acid 429 (Fig. 16) was employed as a catalyst for the addition of nitroacetates 330 (R1 = H) to N-Boc imines 161 in toluene at −78 °C to provide mainly anti-products 332, which were further reduced with NaBH4/CoCl2 to the corresponding anti-α,β-diamino acid derivatives 196 in 67–95% overall yield, 1:2–11:1 dr and 69–88 ee.262 In the case of α-substituted nitroacetates 330 (R1 ≠ H) a methoxy substitution in the catalyst 430263 (Fig. 16) improved the results to afford syn-α-nitro-β-amino acid derivatives 332 with 59–88% yield, 5:1–>20:1 dr and 94–99% ee. Pyrrolidine BAM 4312·(HOTf)3264 (Fig. 16) was the most efficient catalyst to improve the enantioselectivity in the reaction of substituted nitroalkanes with N-Boc imines 161 providing nitroamines 320 with 61–100% yield, 7:1–35:1 dr and 71–95% ee working in toluene at −20 °C.
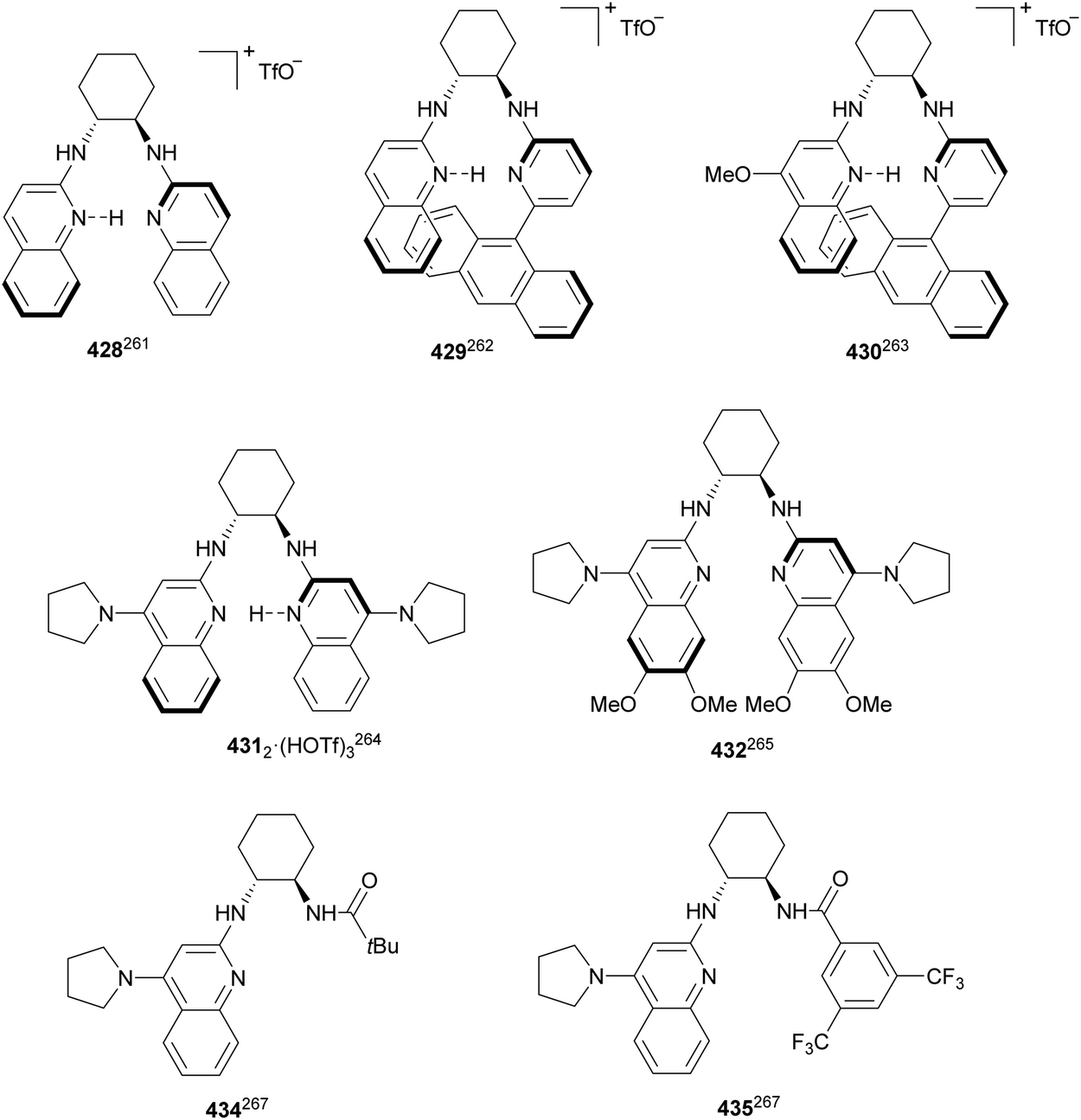 | ||
| Fig. 16 Mono and bis(AMidine)-derived catalysts for the asymmetric aza-Henry reaction. | ||
Subsequent studies of Johnston and co-workers were focused on the synthesis of therapeutics. Based on the asymmetric aza-Henry reaction of α-aryl nitromethanes to N-Boc aldimines 161, anti-products 320 were obtained up to 99% yield, 131:1 dr and 93% ee under modified pyrrolidine BAM 432 catalysis (Fig. 16).265 They found out that the free base was equally effective than the Brønsted acid salt of the BAM compounds. The cis-stilbene 320a was prepared on a large scale using β-MeO PBAM 433 as a catalyst (5 mol%) in toluene at −20 °C (Scheme 130).266 After recrystallization, 15.98 g of product 320a was obtained in 62% yield of diastereo- and enantiomerically pure compound. This compound is the key product for the synthesis of (−)-nutlin-3, a potent cis-imidazoline p53/MDM2 inhibitor discovered by Hoffmann-La Roche.174cis-Imidazolines could disrupt the protein–protein interaction between p53 and MDM2, thereby including apoptosis in cancer cells. Non-symmetric cis-stilbene diamines and cis-imidazolines were accessible by asymmetric aza-Henry reactions of different aryl aldimines with differently substituted α-aryl nitromethanes using different mono(amidine) MAM 434 and 435 (Fig. 16) as an organocatalyst.267 Further development of an intermittent-flow enantioselective aza-Henry reaction for the synthesis of 320a on a multigram scale has been reported by Johnston and co-workers.268
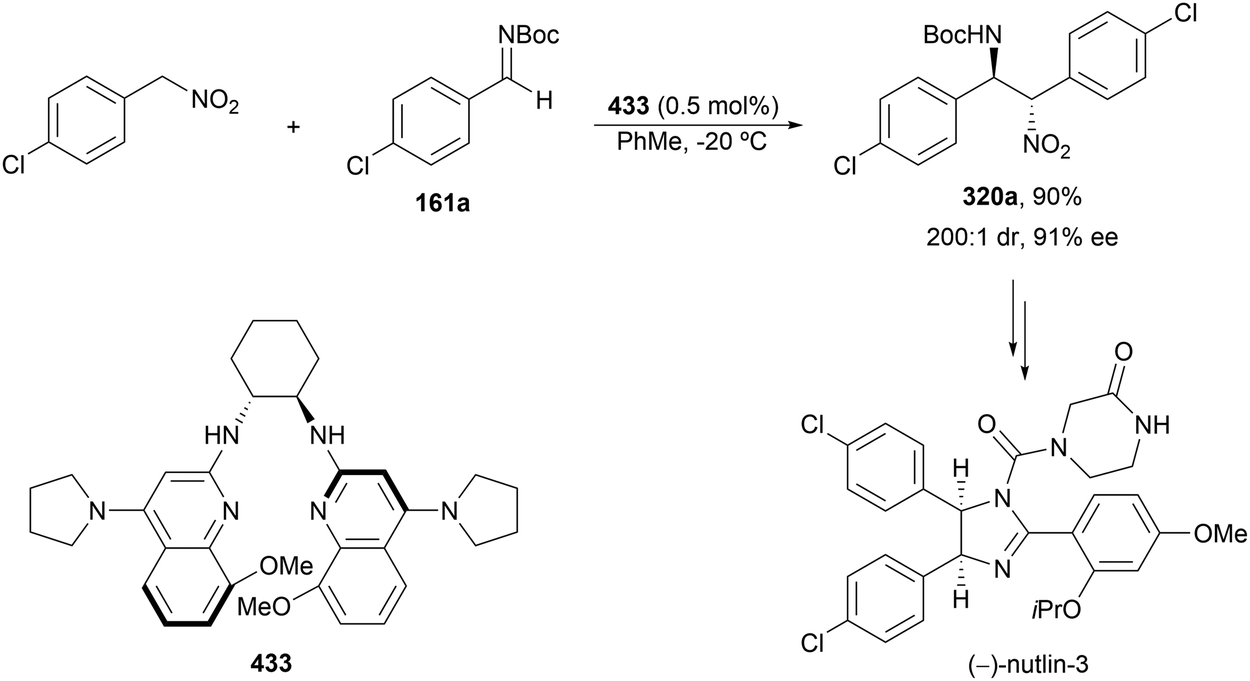 | ||
| Scheme 130 Asymmetric aza-Henry reaction of α-(4-chlorophenyl)nitromethane with N-Boc-4-chlorobenzylidene imine under β-MeO PBAM (433) catalysis and application to (−)-nutlin-3. | ||
The first enantioselective synthesis of a potent GlyT1 inhibitor, which can increase glycine levels and NMDA signaling thereby providing a promising therapeutic target for the treatment of schizophrenia,269 was described by Johnston and co-workers.270 Nitroazetidine 436 reacted with N-Boc benzylidene imine under PBAM (437)·HOTf catalysis in toluene at −20 °C to obtain product 438 in 93% yield and 92% ee, whereas the free amine afforded product 438 with 87% yield and 86% ee (Scheme 131). This intermediate 438 was transformed into the target azetidine (−)-439 through a short reaction sequence.
 | ||
| Scheme 131 Asymmetric aza-Henry reaction of nitroazetidine 436 with N-Boc benzylidene imine 161 under 7(MeO) PBAM (437)·HOTf catalysis and application to (−)-439. | ||
Enantioselective synthesis of VNI, a potent inhibitor of CYP51, which showed a parasitological cure of mice infected with T. cruci (Chagas disease), has been performed by Johnston and co-workers.271 The reaction of α-bromo nitromethane 440 with 2,4-dichlorobenzylidene imine 161 in toluene at −20 °C using 431 as an organocatalyst gave product 441 in >98% yield and 98% ee as a 1:1 mixture of diastereomers on 47 mmol scale (Scheme 132). Treatment of the mixture with cobalt boride provided diamine derivative 442 in 52% yield. However, the aza-Henry reaction with nitromethane as a solvent gave a mixture of mono and diaddition products. Compound 442 was further transformed into (+)-VNI after three more steps. This aza-Henry reaction was performed with N-Boc aliphatic aldimines using the BAM ent-431·HOTf as a catalyst to obtain products of the type ent-441 with 39–94% yield and 76–93% ee, which were transformed into α-amino amides.272
 | ||
| Scheme 132 Asymmetric aza-Henry reaction of bromomethane 440 with 2,4-dichlorobenzylidene imine under BAM 431 catalysis. | ||
DFT calculations by Dudding and co-workers,273,274e.g. for the reaction of nitromethane with N-Boc benzylidene imine using the BAM 428 as a catalyst, supported the cooperative role of this catalyst. In the TS, the imine formed a dual hydrogen bonding as well as the nitronate (Fig. 17). A synclinal alignment of both reagents maximized orbital and electrostatic interactions.
 | ||
| Fig. 17 Proposed TS for the asymmetric aza-Henry reaction catalyzed by BAM 428. | ||
Hindered α-substituted α-nitro esters 330 were used as nucleophiles for the addition of N-Boc imines 161 using 431·HNTf2 as a catalyst.275 In this case, anti-332 were obtained, whereas using catalyst 430263 resulted syn-products. These results are another example of diastereodivergence52 based on the BAM derived Brønsted acid–base organocatalyst (Scheme 133). Compounds anti-332 were obtained with 46–76% yields, 4:1–>20:1 dr and 78–99% ee. Reduction of the nitro group provided α-substituted anti-α,β-diamino esters. Stereochemical-determining arrangements to obtain anti and syn diastereomers are depicted in Scheme 133.
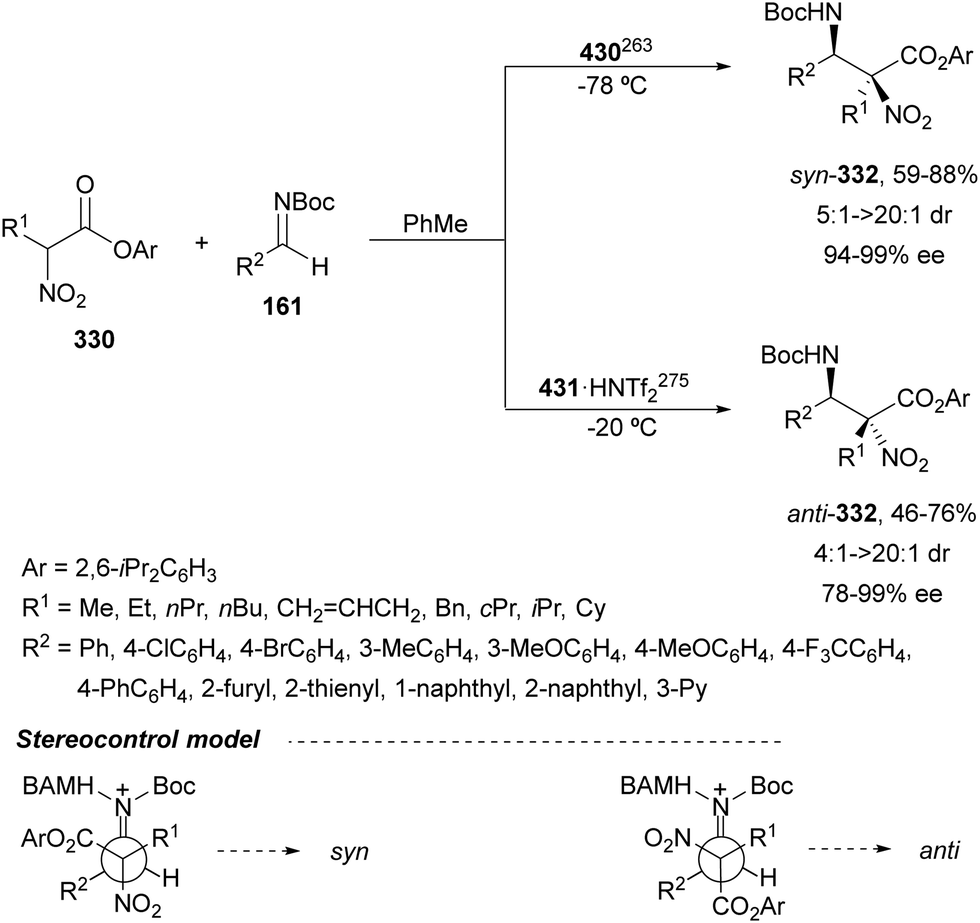 | ||
| Scheme 133 Asymmetric aza-Henry reaction of α-substituted α-nitro esters 330 with N-Boc imines 161 under 430 or 431·HNTf2 catalysis. | ||
In a recent work of Johnston and co-workers,276 α-fluoro nitroalkanes 443 were allowed to react with N-Boc imines 161 using 432 or 432·HNTf2, or N-benzylquininium chloride (375).227 This study was also carried out with non-fluorinated nitroalkanes in order to compare the resulted diastereoselectivity. In the case of α-fluoro nitroalkanes 443, four possible diastereomers 444 can form type I–IV depending on the substituents (Scheme 134). anti-Diastereoselectivity was mainly observed with nitroalkanes (1:1–20:1) and α-aryl-α-fluoro nitromethanes (2.7:1–5.2:1) using the three catalysts, whereas reversal syn-diastereoselectivity was mainly obtained only with type III and type IV α-alkyl-α-fluoro nitromethanes (2.4:1–7.2:1). The stereochemistry-determining arrangements are depicted in Scheme 134. Regardless of the employed catalyst, the imine Si-face is favored and the bifunctional activation of the imine and nitronate favored a synclinal arrangement of the amine nitrogen and NO2 according to Dudding's analysis.273,274
 | ||
| Scheme 134 Asymmetric aza-Henry reaction of α-fluoro nitroalkanes 443 with N-Boc imines under BAM 432 or 432·HNTf2 or 375 catalysis. | ||
Chiral bases such as alkoxides in ammonium betaines can be used for the asymmetric aza-Henry reaction of activated nitro compounds with aldimines. In the case of ketimines, the reaction with nitromethane was efficiently performed with Cinchona-derived alkaloids, guanidines and phosphoranes. Chiral Brønsted acids derived from bis(amidine) and mono bis(amidine) have been employed in the reaction of activated nitro compounds with aldimines. This strategy has been applied to the synthesis of 1,2-diamines, precursors of therapeutics such as (−)-nutlin-3, azetidine (−)-439 and (+)-VNI.
3.3. Imine–imine coupling reactions
Asymmetric imine–imine coupling reactions to provide diastereoselectively and enantiomerically enriched 1,2-diamines under catalytic conditions are some of the most direct and challenging strategies. This type of process has been performed (a) by a homocoupling process of unprotected imines such as an aza-pinacol coupling and (b) by a reaction of N-alkyl imines with aldimines via formation of an azaallyl anion.In 2017, Tang and co-workers277 reported the asymmetric reductive coupling of isoquinolines mediated by chiral diborons (D–B) under mild reaction conditions. Diboron 445 derived from (1S,2S)-1,2-diphenylethane-1,2-diol (0.75 equivalents) reacted with isoquinolines to form intermediates I, which underwent a concerted [3,3]-sigmatropic rearrangement to give products 446. These compounds were treated with acetyl chloride to obtain bisisoquinoline diacetamides 447 in good yields and high enantioselectivities (Scheme 135). Mechanistic investigations278 suggested the activation of the B–B bond via double N–B coordination followed by [3,3]-sigmatropic migration from intermediate I.
 | ||
| Scheme 135 Asymmetric aza-pinacol coupling of isoquinolines mediated by diboron 445. | ||
The former methodology was applied by the same group279 to unsubstituted aldimines prepared in situ by conventional methods using a non-C2-symmetric chiral diboron 448. A broad scope of aromatic, heteroaromatic and aliphatic imines as well as cyclic imines and N-methyl aldimines gave the corresponding syn-1,2-diamines 420, 449 and 450 in high yields and enantioselectivities (Scheme 136a–d). This process was scaled-up to 50 g starting from benzaldehyde and ammonia in MeOH, followed by a reaction with 448 in THF at room temperature with recovery of the diol. Stable aromatic imines such as benzaldimine and N-methylbenzaldimine were transformed into the corresponding 1,2-diamines using a stoichiometric amount of (BNeop)2 and a catalytic amount of chiral diol 451 (30 mol%). In the case of N-methylbenzaldimine, the diamine 450a was obtained in 88% yield and 96% ee, after treatment of N–BNeop diamine with MeOH (Scheme 136e).
 | ||
| Scheme 136 Asymmetric aza-pinacol coupling of aldimines mediated by diboron 448. | ||
The homocoupling of alkyl aryl ketimines 452 provided tetrasubstituted syn-1,2-diamines 453. Tang's group280 performed the reductive coupling of aryl methyl ketimines 452 (R = Me) with diboron 448 to obtain diamines 453 up to 96% yield and 99% ee (Scheme 137). However, for other ketimines 452 with R ≠ Me, the diboron 454 gave better results than 448 affording products 453 (R ≠ Me) up to 92% yield and 99% ee. DFT calculations revealed that the two chiral diborons utilize two different conformational assembling to direct the reductive homocoupling.
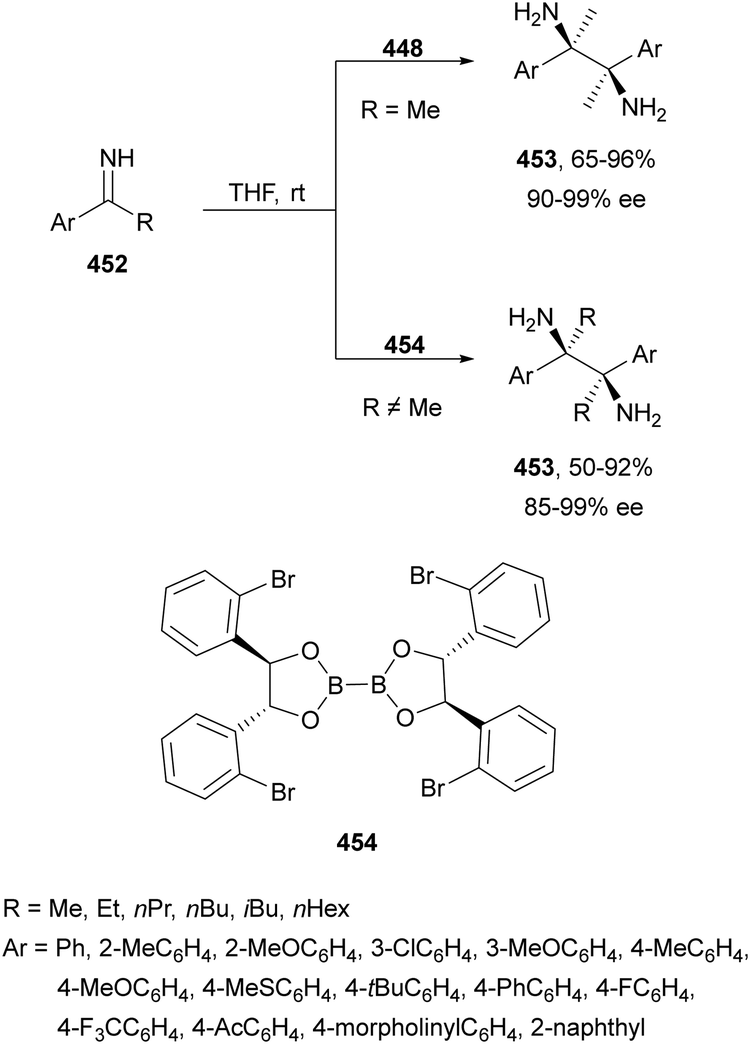 | ||
| Scheme 137 Asymmetric aza-pinacol coupling of ketimines mediated by diborons 448 and 454. | ||
Zhu and co-workers281 have reported the nucleophilic addition of hydrazones 455 to N-Boc aldimines 161 under CPA catalysis. In the presence of (S)-456 working in toluene at −20 °C, N-alkyl hydrazones behaved as α-azo carbanion equivalents to provide 1,2-diamine derivatives anti-457 in excellent yields with high chemo-, diastereo, and enantioselectivity (Scheme 138). Product 457a was transformed into monoprotected diamine 458 in two steps and into unprotected diamine 459 in 43 and 96% yield, respectively. The stereochemical outcome was explained by formation of two hydrogen bonding with bifunctional CPA through the TS depicted in Scheme 138.
 | ||
| Scheme 138 Asymmetric coupling of hydrazones 455 with N-Boc imines 161 under CPA (S)-456 catalysis. | ||
With respect to the second strategy based on the reaction of imines with N-alkyl imines by formation of azaallyl anions,282 Kobayashi and co-workers283 described an imine–imine cross-coupling using imines bearing a 9-fluorenyl moiety at the nitrogen atom and in the presence of a catalytic amount of potassium 2,2,2-trifluoroethoxide as a base. The asymmetric version was performed with a chiral guanidine 204 as a base for the cross-coupling of N-Boc benzaldimine 161 with N-fluorenyl glyoxylateimine 203 to furnish adduct syn-205 in 90% yield, 98:2 dr and 98% ee (Scheme 139). Compound 205 was transformed into a 3-amino-β-lactam by cyclization of the α,β-diamino acid tert-butyl ester. It is assumed that the carbanion I is formed and reacts through the α-position of the ester group.
 | ||
| Scheme 139 Asymmetric imine–imine cross-coupling of N-fluorenyl glyioxylateimine 203 with N-Boc benzaldimine 161 under guanidine 204 catalysis. | ||
The former imine umpolung was recently applied by Luo, Deng and co-workers284 to the imine cross-coupling of ketimines 460 with N-methoxycarbonyl imines 161 under asymmetric PTC conditions (Scheme 140). Newly designed Cinchona-alkaloid ammonium salt 461 or its pseudoenantiomer 462 gave products 463 or ent-463, respectively, and the regioisomer 464 with good yields and diastereo- and enantioselectivities, although with moderate to good regioselectivity. In this procedure, intermediate I is formed by the azaallyl anion and the tetraakyl ammonium cation. On the basis of 1H NMR titration studies, interaction modes A or B between the BArF− ammonium salts 461′ or 462′ with the electrophilic imine were postulated. Computational studies performed by Smith and co-workers285 proposed hydrogen bonding interactions between ammonium α-CH8 with the nitrogen of the imine and H12 and H12′ with the carbonyl group was proposed in activation mode A. Hydrogen bonding interactions between H2, H6 and H12′ of ammonium salts shifted downfield 462′ in the titration studies (mode B).
 | ||
| Scheme 140 Asymmetric imine–imine cross-coupling of ketimines 460 and 161 (PG = CO2Me) under chiral PTC conditions. | ||
Ketimine–ketimine cross-coupling was described by Lin, Lu and co-workers286 using chiral squaramide 236 and thiourea 465 as bifunctional organocatalysts. Isatin-derived N-2,2,2-trifluoromethyl ketimines 466 were used as pronucleophile and N-Boc ketimines 170 as electrophiles to provide, in the presence of squaramide 236, products 467 in high yields and diastereo- and enantioselectivities (Scheme 141). This type of cross-coupling was carried out with ketimines 468 and isatin-derived N-Boc ketimines 170. In this case, the best organocatalyst was thiourea 465 to provide compounds 469 in high yields and enantioselectivities. Both processes were scaled-up to grams maintaining yields and stereoselectivities.
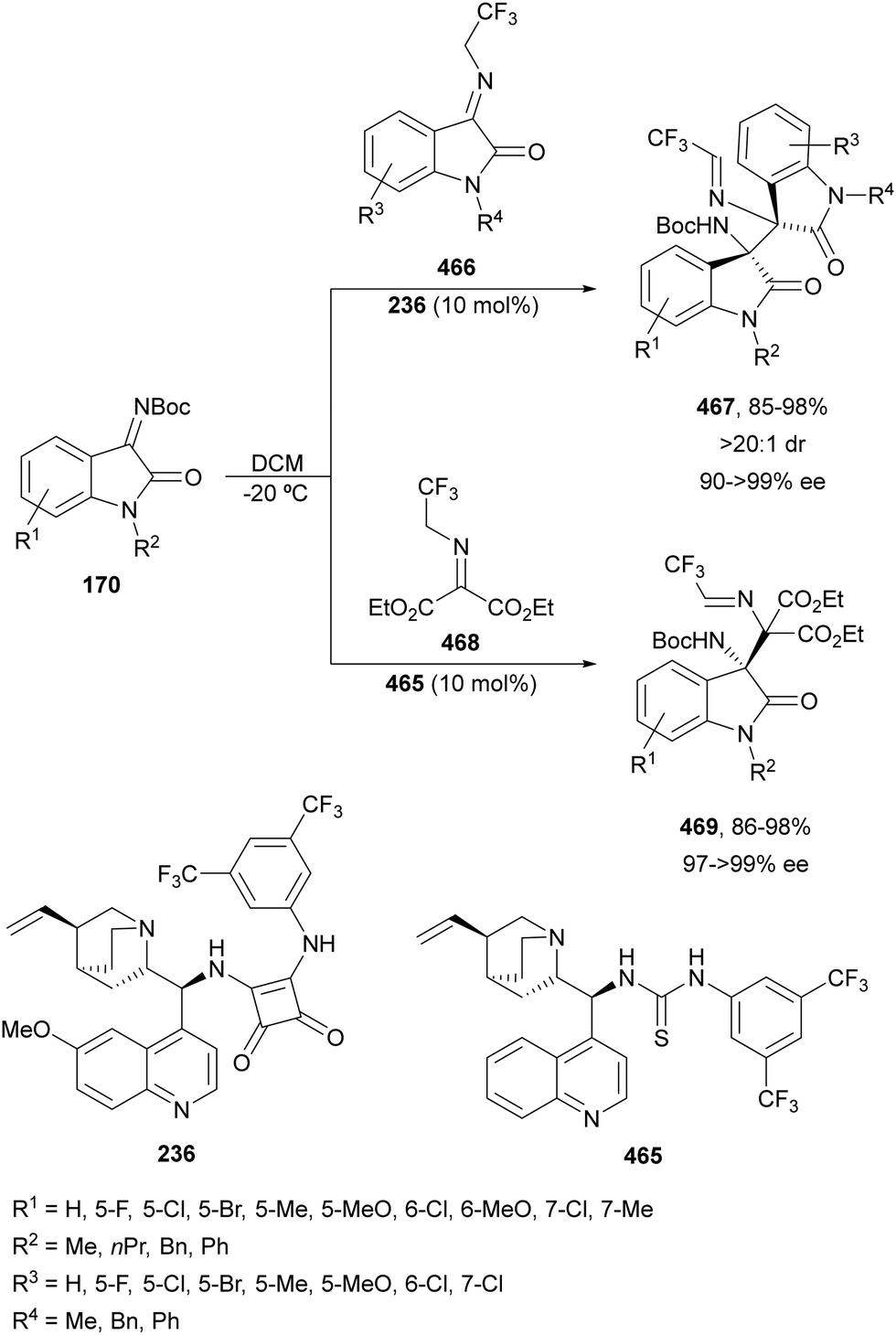 | ||
| Scheme 141 Asymmetric imine–imine cross-coupling of ketimines 466 and 468 with isatin-derived N-Boc ketimines 170 under squaramide 236 and thiourea 465 catalysis. | ||
The copper(I)-catalyzed asymmetric ketimine–imine cross-coupling allows the synthesis of syn-1,2-diamines. Tian, Yin and co-workers287 have performed the addition of ketimines derived from trifluoroacetophenone 470 to N-Boc aldimines 161 using mesitylcopper and (R,RP)-Taniaphos 471 as a chiral catalyst (Scheme 142). A broad scope of products 472 were obtained regioselectively in moderate to good yields and diastereoselectivities, with very good enantioselectivities. A gram-scale reaction was carried out with 4-nitrobenzylamine-derived trifluoroacetophenone imine and N-Boc benzaldimine to obtain syn-472a in 75% yield, >20:1 dr and 96% ee. This product was transformed into N-tosyl diamine 473 by reduction with Zn/HOAc, removal of the ketimine moiety with concentrated HCl and tosylation. Intermediate I has been proposed to explain the formation of the azaallyl anion.
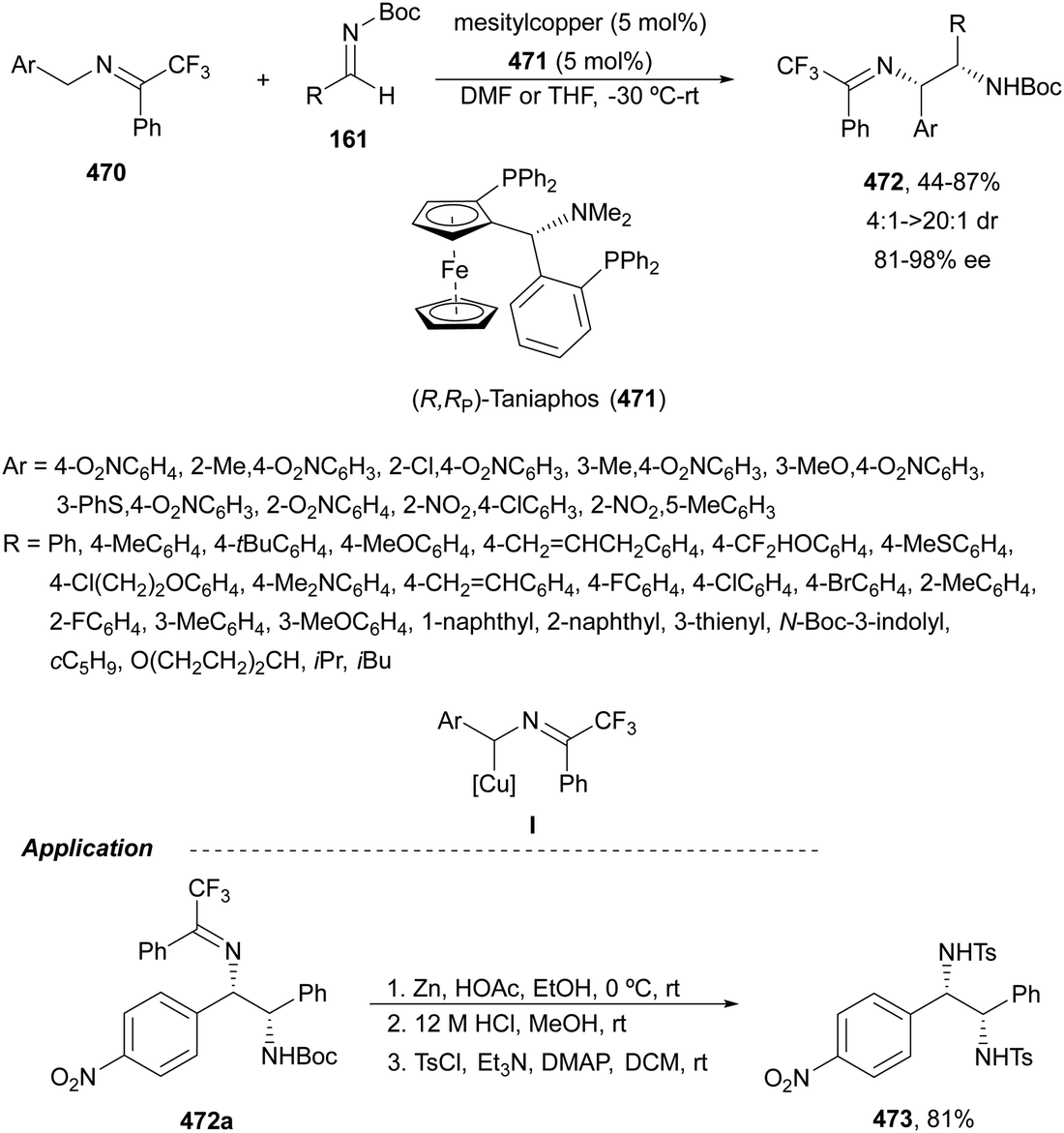 | ||
| Scheme 142 Asymmetric imine–imine cross-coupling of ketimine 470 with N-Boc aldimines 161 under mesitylCu/(R,RP)-Taniaphos (471) catalysis. | ||
Asymmetric homocoupling of imines has been promoted by chiral diborons in stoichiometric amounts to provide syn-1,2-diamines. However, cross-coupling of hydrazones and imines using a CPA formed anti-1,2-diamine derivatives. In the case of a second strategy via formation of an azaallyl anion, N-alkyl imines were added to imines using a chiral organocatalyst (PTC or squaramides and thioureas) to furnish anti-1,2-diamine derivatives. When a Cu/phosphine chiral complex was used as a catalyst, syn-diamines were formed.
3.4. Other addition to imines
Reductive coupling of enamines with imines under asymmetric copper catalysis as well as photocatalytic enantioselective α-amino alkylation of imines will be considered in this section as also other strategies for the synthesis of 1,2-diamines.Asymmetric copper-catalyzed reductive coupling of azadienes 474 with N-diphenylphosphinoyl (DPP) imines 167 or ketimines 250 allowed the synthesis of anti-1,2-diamines. Malcolmson and co-workers288 described this chemoselective transformation by using Cu(OAc)2 and (S,S)-Ph-Bpe (475) as a chiral ligand in the presence of dimethoxymethylsilane (DMMS) as a reducing agent and tBuOH as an additive in THF at 5 °C (Scheme 143). The resulting diamines 476 and 477 were obtained in >20:1 diastereomeric ratio and excellent enantioselectivities. Under these reaction conditions, a Cu-catalyzed imine hydroxylation was carried out to form a azaallyl copper intermediate I which added to the electrophilic imines. Product 476a was transformed into orthogonally protected diamine 478 in two steps in 85% yield and into 479 in 80% yield.
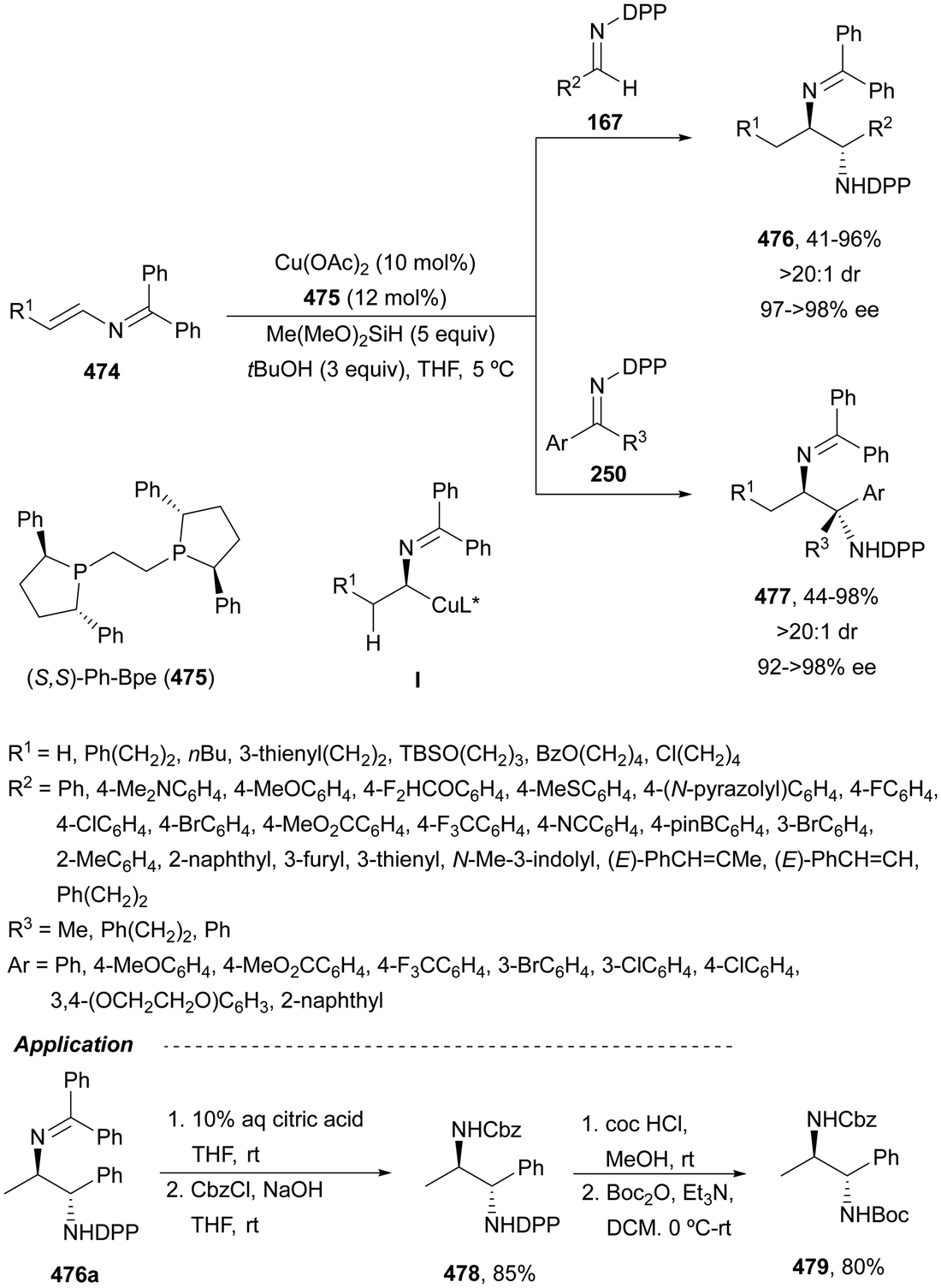 | ||
| Scheme 143 Asymmetric reductive coupling of enamines 474 with aldimines 167 and ketimines 250 under Cu(OAc)2/(S,S)-Ph-Bpe (475) catalysis. | ||
The same group289 recently reported an asymmetric diastereodivergent52 synthesis of syn- and anti-1,2-diamines by a Cu-catalyzed reductive coupling of 2-azatrienes 480 with imines 167. With (S,S)-Ph-Bpe (475) as a chiral ligand, anti-diamines 481 were obtained (up to 97% yield, >20:1 dr, >20:1 rr and 76% ee), whereas with tBu-BDPP (482) resulted syn-diamines 481 (up to 76% yield, >20:1 dr, >20:1 rr and 94% ee) (Scheme 144). Diastereodivergent models I and II were proposed, for Ph-Bpe model I is formed through O-coordination of the imine and for tBu-BDPP in model II the nitrogen atom of the imine was coordinated.
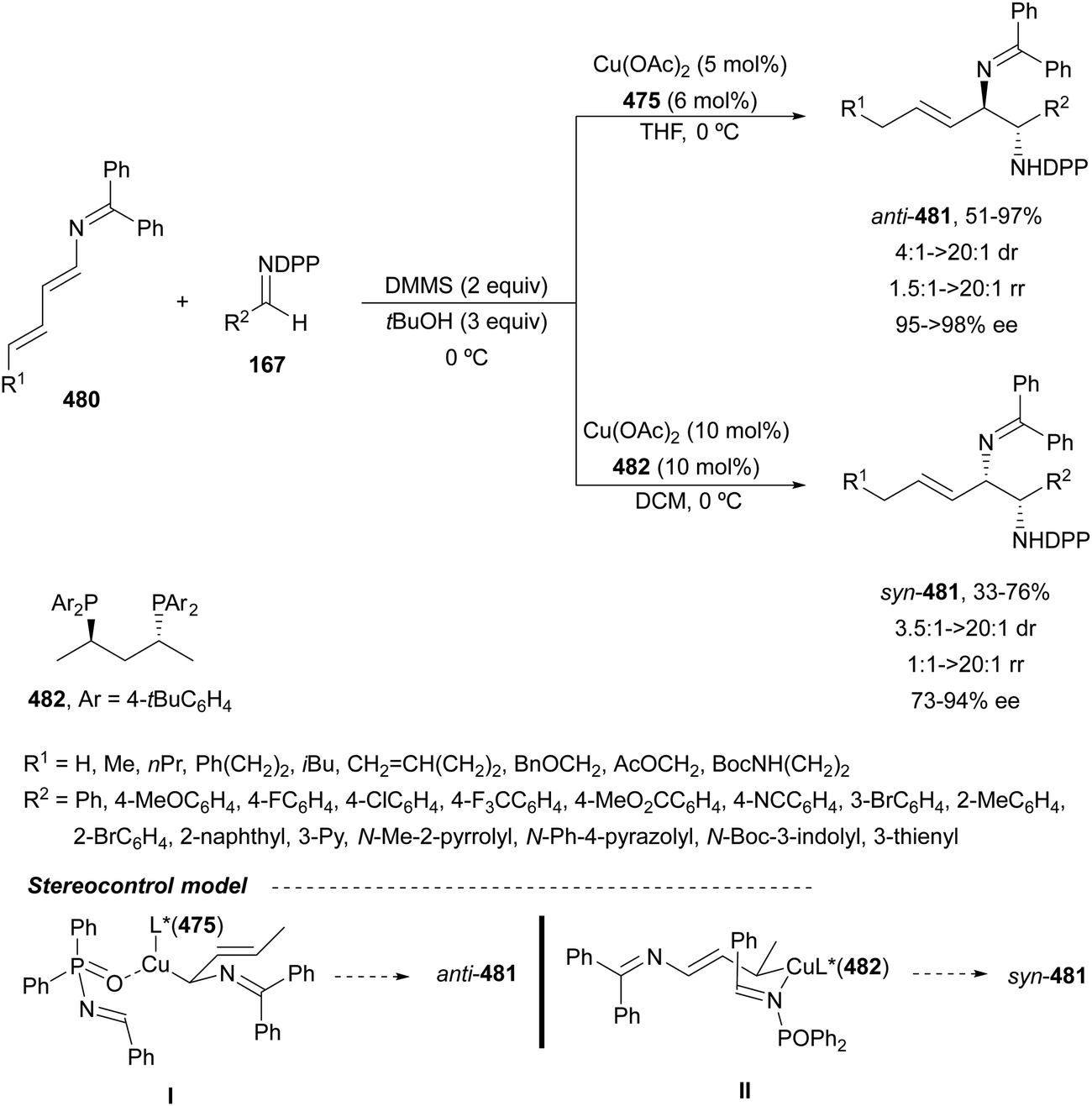 | ||
| Scheme 144 Asymmetric diastereodivergent reductive coupling of 2-azatrienes 480 with imines 167 under Cu(OAc)2/475 and Cu(OAc)2/482 catalysis. | ||
For the enantioselective α-amino alkylation of imines, Ooi and co-workers290 developed a synergistic catalysis based on an ionic Brønsted acid and a photocatalyst. The α-coupling of N,N-disubstituted α-aminomethanes 483 with N-mesyl imines 161 (PG = Ms) gave 1,2-diamine derivatives 486 under visible light irradiation in the presence of the P-spiro chiral arylaminophosphonium barfate 484 as a Brønsted acid and [Ir(ppy)2(Me2phen)]BArF 485 as a photosensitizer (Scheme 145). Diamines 486 were obtained in toluene at room temperature in good yields and enantioselectivities for aromatic aldimines. In contrast, aliphatic imines with a lower reduction potential remained intact. The authors proposed that the imine quenches the photoexcited Ir(III) species with proton loss delivering the α-amino radical I. At the same time, the imine is reduced by the Ir(II) species to give the N-mesyl radical anion which forms an ion-pair with the positive Ir(III) ground state complex. Subsequent ion exchange with the aminophosphonium cation forms the chiral radical ion pair II. After radical–radical coupling with the amino radical, the product is formed enantioselectively. In 2016,291 the authors utilized α-silyl amines as precursors of α-amino radicals which participate in the same catalytic cycle giving diamines 486 in 28–86% yields and 78–97% ee.
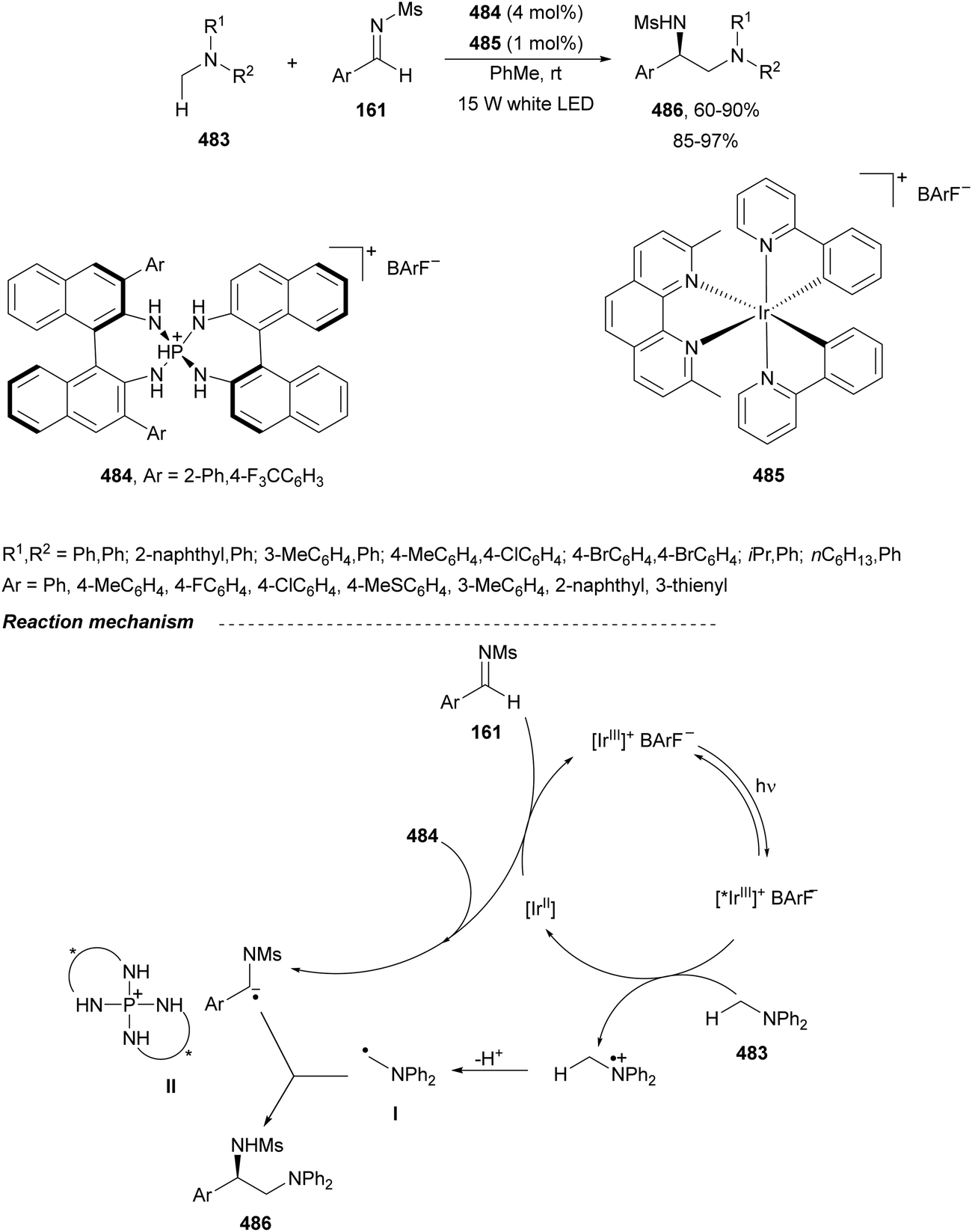 | ||
| Scheme 145 Asymmetric α-amino alkylation of imines 161 with α-aminomethanes 483 under aminophosphonium barfate 484 and Ir complex 485 photocatalysis. | ||
Gong and co-workers292 have described this type of amino alkylation of imines under copper-based asymmetric photocatalysis. α-Silylamines 487 reacted with N-acyl hydrazones 488 with a bis-oxazoline (S,S)-489 copper complex as a catalyst under irradiation with a 24 W blue LED at −40 °C in THF to provide 1,2-diamine derivatives 490 in good yields and enantioselectivities (Scheme 146). In the proposed catalytic cycle, the hydrazone 488 forms complex I with the [L*Cu(II)] catalyst. Meanwhile, a single-electron transfer (SET) between the silylamine 487 and [L*Cu(II)] leads to the formation of the radical cation II and [L*Cu(I)]. Dissociation of the TMS cation from II affords the α-amino alkyl radical III. This nucleophilic radical III adds to the CN of intermediate I to give N-radical species IV. Reduction of IV by the excited [L*Cu(I)] provides cationic complex V, which after protonation and ligand exchange releases the diamine 490 and regenerates intermediate I. Diamine 490a gave protected diamine 491 after reaction with RANEY® nickel in EtOH at 80 °C for one hour in 71% yield and 94% ee.
 | ||
| Scheme 146 Asymmetric α-amino alkylation of hydrazones 488 with α-silylamines 487 under Cu(OTf)2/bisoxazoline (S,S)-489 photocatalysis. | ||
Reductive coupling of azadienes with imines has been carried out with a Cu/diphosphine chiral catalyst to obtain anti-1,2-diamine derivatives by hydrocupration and to obtain an azaallylcopper intermediate. In the case of 2-azatrienes, diastereodivergent52 formation of syn- and anti-1,2-diamines was controlled by the chiral diphosphine ligands. Asymmetric α-aminoalkylation of imines can be performed under photocatalytic conditions using a phosphonium salt or Cu/bisoxazoline as a chiral catalyst.
3.5. Cycloaddition reactions
In this section, asymmetric 1,3-dipolar cycloadditions (1,3-DC) of azomethine ylides and 5-vinyloxazolidinones with imines to obtain imidazolidine precursors of 1,2-diamines will be mainly considered. In addition, [2+2] cycloaddition of allenamides was also included.In 2008, Gong and co-workers293 reported a three-component 1,3-DC of aldehydes, diethyl aminomalonate and anilines catalyzed by CPA (R)-TRIP (152). This process took place in toluene at −10 °C to give chiral imidazolidines 492 up to 99% yield, 91:9 dr and 98% ee (Scheme 147). In the proposed mechanism, the condensation of the aldehyde with aminomalonate is controlled by the CPA to obtain an azomethine ylide coordinate with the CPA I. Meanwhile, condensation of the aldehyde with aniline can be activated by formation of a species II by the CPA. Both intermediates undergo an enantioselective [3+2] cycloaddition to provide imidazolidines 492. Reduction of 492a with NaBH4/LiCl followed by hydrolysis with aqueous phosphoric acid generated a diamine 493 in 50% overall yield.
 | ||
| Scheme 147 Asymmetric three-component 1,3-DC of aldehydes, diethyl aminomalonate and anilines under (R)-TRIP (152) catalysis. | ||
Fluorinated imidazolidines have been prepared by asymmetric CuBF4/(S,RP)-PPFOMe (494) catalysis of azomethine ylides with fluorinated imines. Wang and co-workers294,295 performed the 1,3-DC of N-PMP-trifluoromethyl imines with metallo-dipoles A derived from imino esters 166 (Scheme 148). In the case of aldimines 356, the reaction was carried out with Et3N as a base in ether at −20 °C to obtain 2,5-trans-imidazolidines 495 in good yields and diastereo- and enantioselectivities through TS I, resulting from exo-selective cycloaddition. On the other hand, trifluoromethylated ketimine 496 was reacted with imino esters 166 in the presence of Cs2CO3 through an endo-approach to obtain 2,5-cis-imidazolidines 497via TS II. This mechanistic proposal is based on labelling experiments to discard epimerization processes, and linear effect studies which explain the formation of a monomeric Cu(I) complex. Imidazolidine 495a was transformed into α,β-diamino acid derivative 498 by treatment with p-toluenesulfonic acid in MeOH at room temperature in 80% yield and the same 97% ee.
 | ||
| Scheme 148 Asymmetric 1,3-DC of imino esters 166 with fluoromethylated imines 356 and 496 under CuBF4/(S,RP)-PPFOMe (494) catalysis. | ||
The first asymmetric homo-1,3-DC of azomethine ylides was described by Shi and co-workers296 using a (R)-SPINOL-derived CPA 499. This cycloaddition was carried out starting from an aldehyde and aminomalonate in order to generate in situ the corresponding imino ester working in CHCl3 at −40 °C in the presence of 4 Å MS resulting 2,4-cis-imidazolidines 500 with modest to good yields, high diastereoselectivity and moderate to good enantioselectivities (Scheme 149). In the proposed TS, the catalyst activates both the azomethine ylide and the imine moiety via hydrogen bonding interactions with subsequent endo-[3+2] cycloaddition.
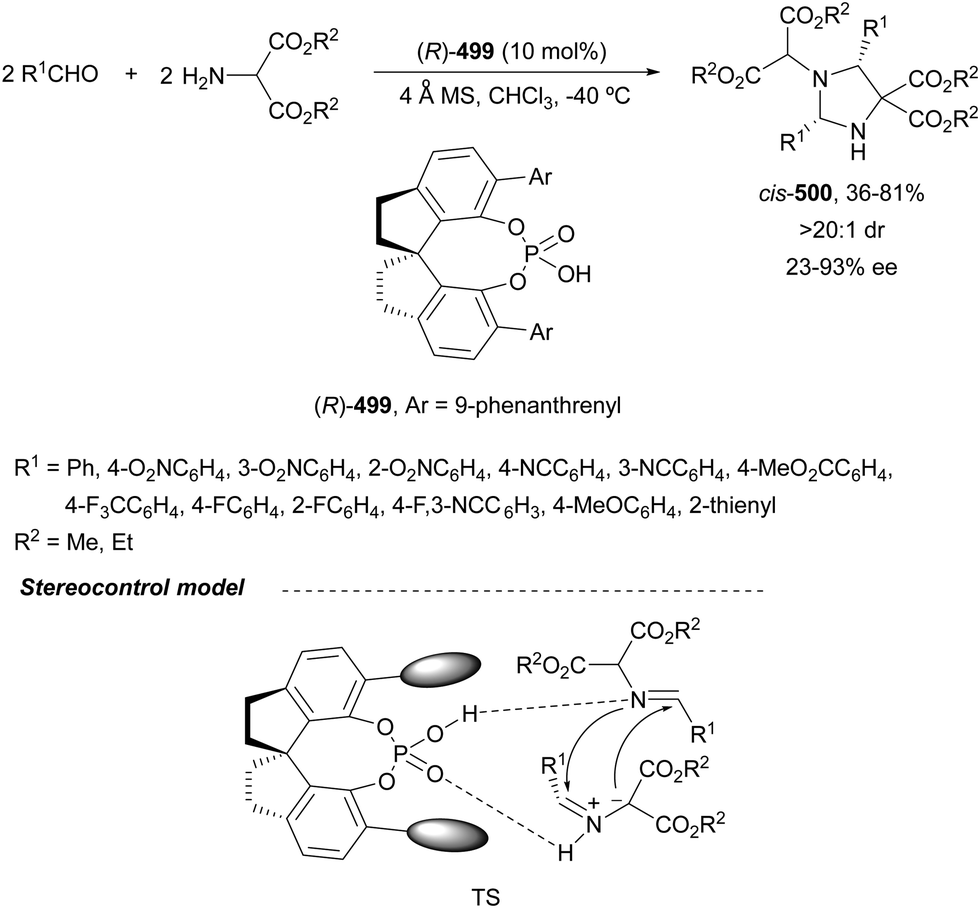 | ||
| Scheme 149 Asymmetric homo-1,3-DC of azomethine ylides with imino esters under CPA (R)-499 catalysis. | ||
Xu and co-workers297 performed the homo-1,3-DC of imino esters 166 by employing AgOAc/Xing-Phos 501 as catalyst to furnish all-cis-imidazolidines 502 with good to excellent diastereo- and enantioselectivities as well as good yields (Scheme 150). The diastereoselectivity of this 1,3-DC was explained by an endo-approach in the TS, which after Mannich addition gave intermediate I. Subsequent intramolecular N-cyclization of I provided the corresponding cis,cis-502. Products cis,trans-502 were obtained as secondary products.
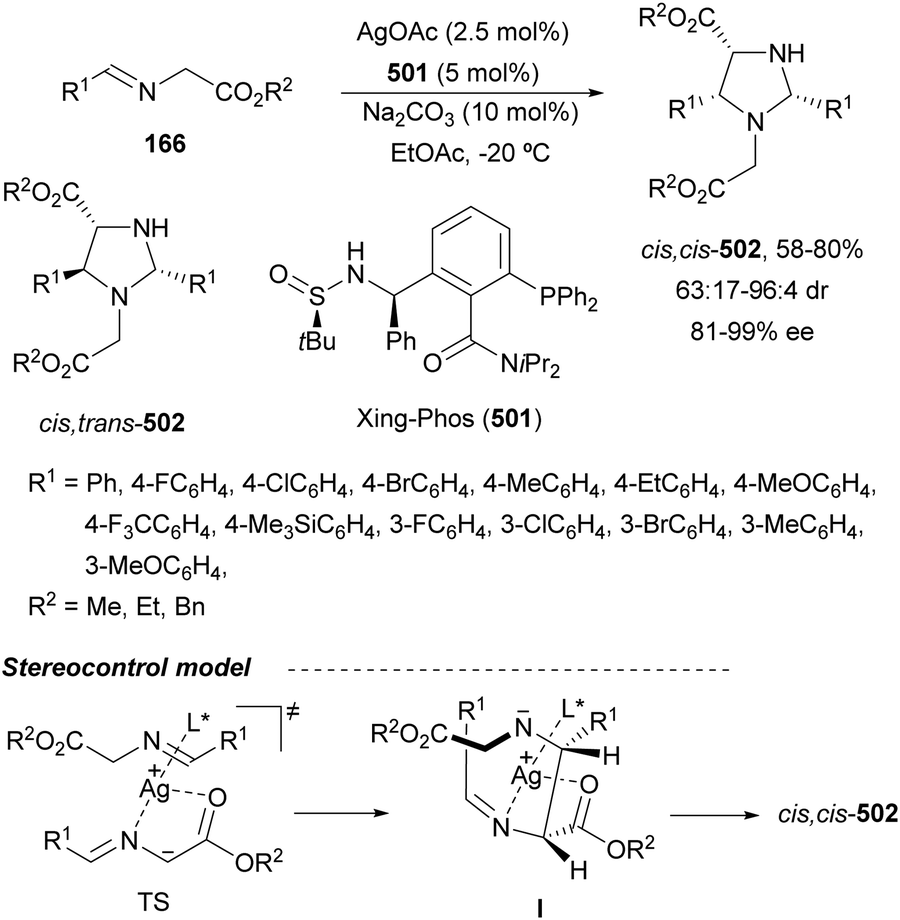 | ||
| Scheme 150 Asymmetric homo-1,3-DC of imino esters 166 under AgOAc/Xing-Phos (501) catalysis. | ||
The same group298 reported a diastereodivergent52 1,3-DC of azomethine ylides with imines. Glycine-derived imino esters 166 reacted with N-alkyl aromatic imines 503 to give under AgOAc/Xing-Phos (501) catalysis all-cis-504 up to 75% yield, 98:2 dr and 99% ee (Scheme 151). On the other hand, 2,3-cis-2,5-trans-imidazolidines 504 were mainly obtained using AgOAc/DTBM-Segphos (23) with 57–74% yields, up to 99:1 dr and 99% ee. The chiral ligand control of the diastereo- and enantioselectivity was explained by steric repulsion in the case of DTBM-Segphos via pathway B and by hydrogen bonding interactions with Xing-Phos (pathway A).
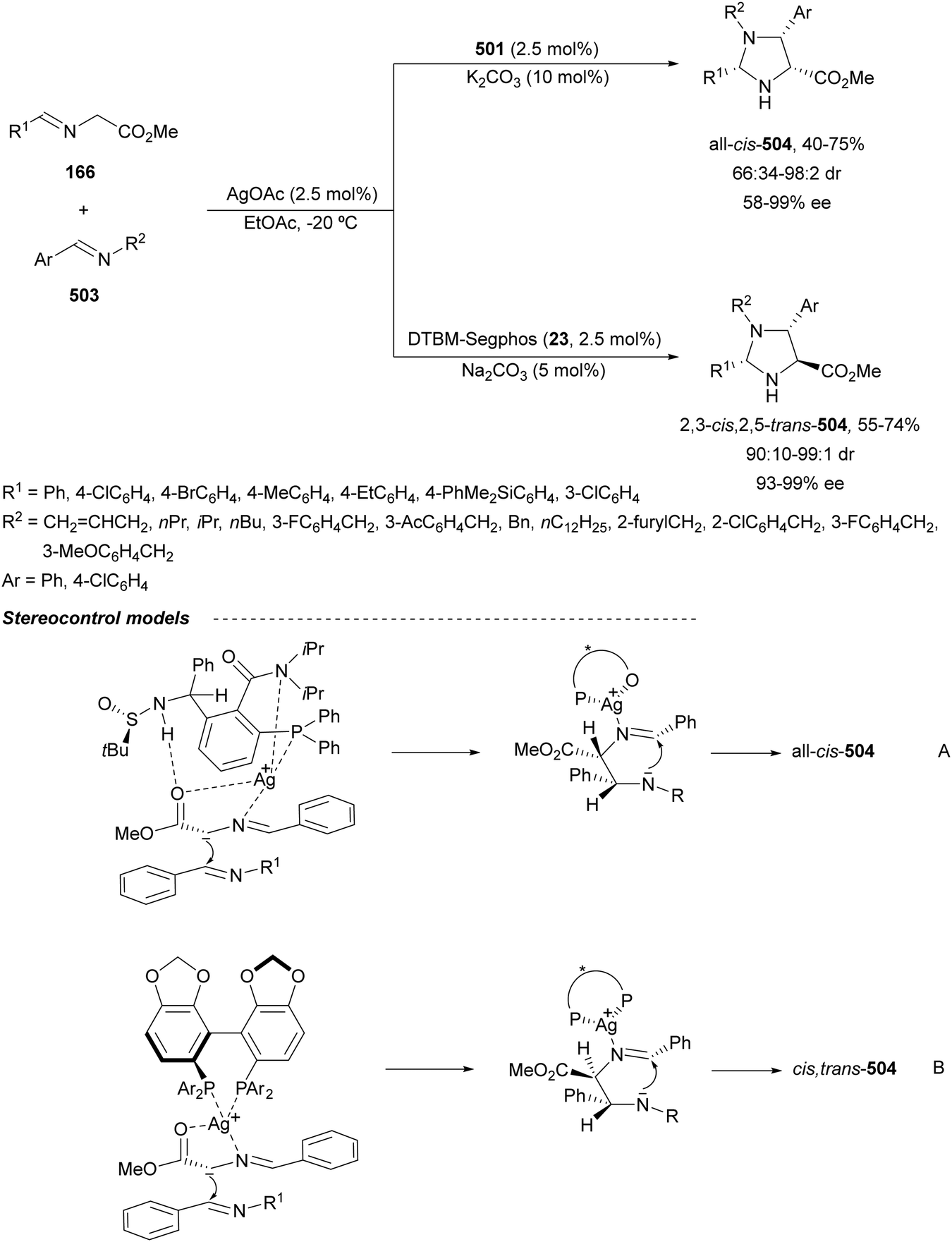 | ||
| Scheme 151 Asymmetric diastereodivergent 1,3-DC of imino ester 166 with N-alkyl aldimines 503 under AgOAc/Xing-Phos (501) or DTBM-Segphos (23) catalysis. | ||
Shi and co-workers299 described a chemoselective 1,3-DC of azomethine ylides with isatin-derived imines 170 using (R)-TRIP (152) as a CPA. This asymmetric [3+2] cycloaddition was performed via a three-component reaction of imines, aldehydes and diethyl aminomalonate in toluene at 0 °C to obtain spiro[imidazolidine-2,3′-oxindole] derivatives 505 up to 76% yield, >95:5 dr and 94% ee (Scheme 152). Based on control experiments, initial homo-1,3-DC of in situ generated imino ester gives product 500, which undergoes a cascade reaction with ketimine 170 promoted by CPA 152 by a dual hydrogen bonding interaction. When racemic 500 was used, product 505 was obtained in 84% ee, whereas 500 was recovered in the racemic form, therefore kinetic resolution during the cascade reaction was discarded.
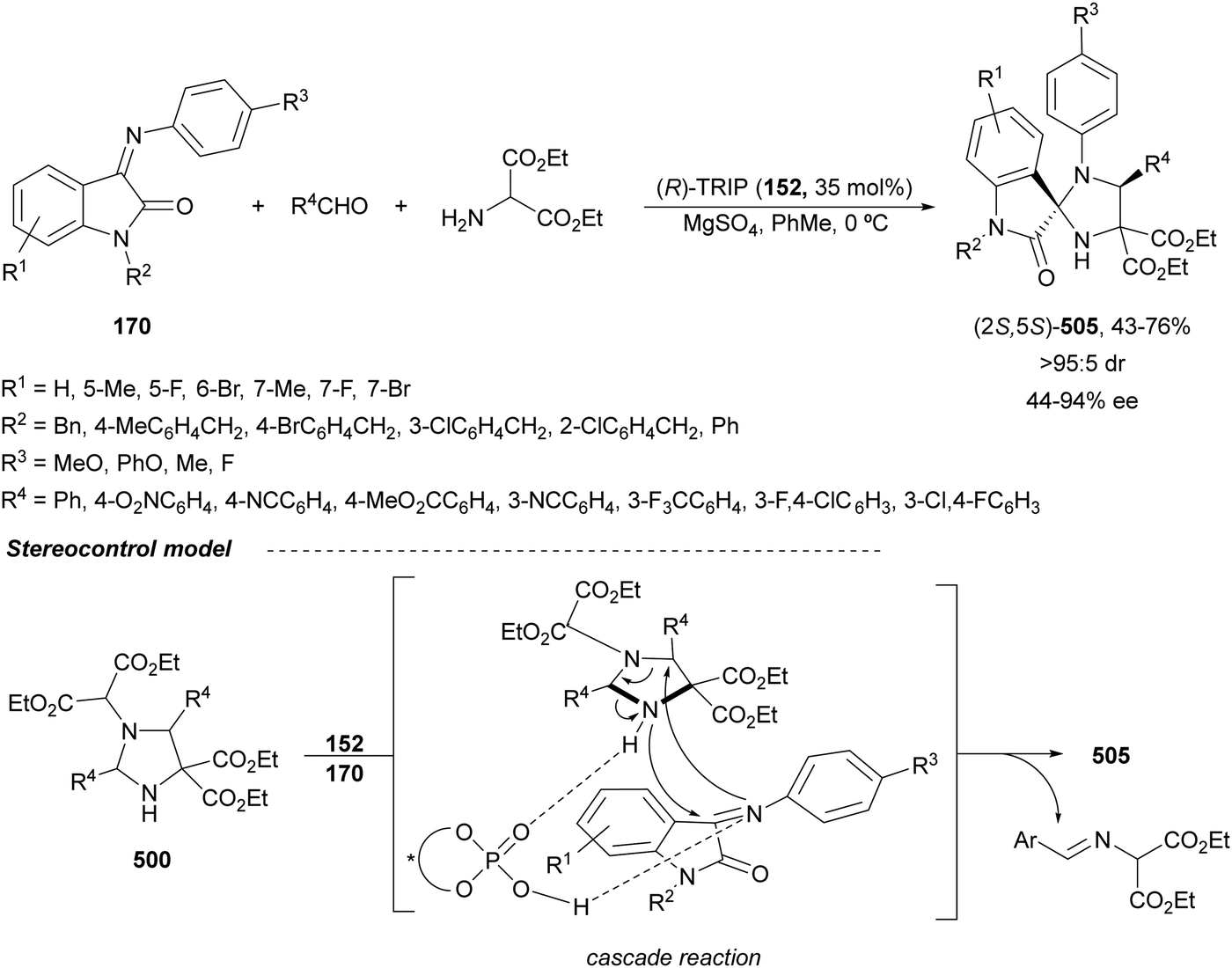 | ||
| Scheme 152 Asymmetric three-component 1,3-DC of aldehydes, diethyl aminomalonate and isatin-derived imines 170 under (R)-TRIP (152) catalysis. | ||
Guo and co-workers300 performed a tandem [3+2] cycloaddition/1,4-addition reaction of azomethine ylides and aza-o-quinone methides to obtain imidazolidines 507. Starting from N-tosyl-o-(chloromethyl)anilines 506 and imino esters 166 in the presence of AgOTs/ferrocenylphosphine 48 as catalyst, with KOH as a base and 18-crown-6 as an additive in dichloromethane at −30 °C, resulted imidazolidines all-cis-507 in good yields and diastereo- and enantioselectivities (Scheme 153). Based on control experiments, a plausible mechanism was proposed involving the homo-1,3-DC of the silver dipole with the imino ester to obtain imidazolidine all-cis-502a, which reacts with the aza-o-quinone methide I to provide product 507a.
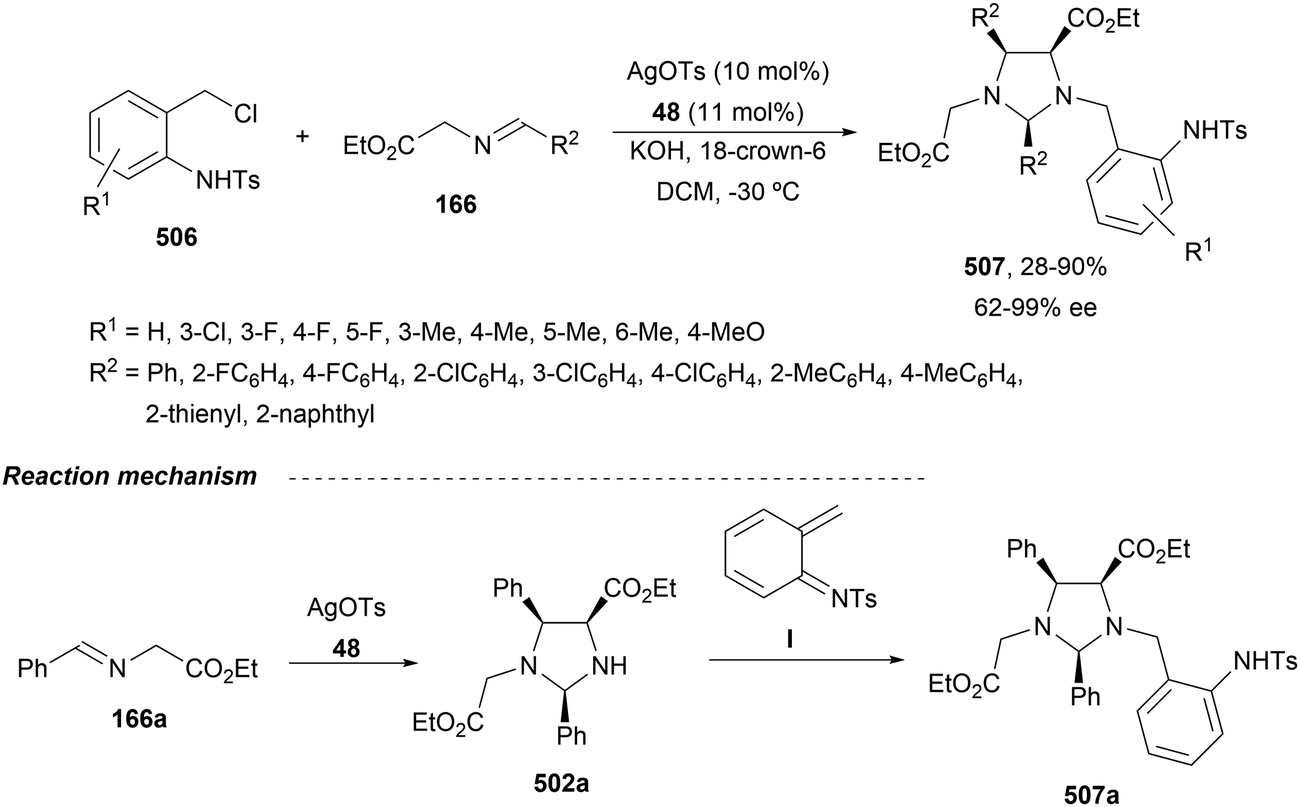 | ||
| Scheme 153 Asymmetric tandem 1,3-DC/1,4-addition of imino esters 166 with N-tosyl-o-(chloromethyl)anilines 506 under AgOTs/ferrocenylphosphine 55 catalysis. | ||
A tandem nucleophilic addition of imino esters 166 to arynes to give an azomethine ylide followed by a 1,3-DC with a metalated α-imino esters afforded the corresponding imidazolidines all-cis-509. This process was described by Shi, Guo and co-workers301 using AgNTf2/ferrocenylphosphine ligand 48 as a catalyst, CsF as a base and 18-crown-6 as an additive in acetonitrile at −10 °C (Scheme 154). O-Silyl aryl triflates 508 were employed as precursors of arynes which by reaction with imino esters 166 derived from aromatic aldehydes provided imidazolidines 509 with a 2S,4S,5S configuration in good yields and enantioselectivities. Two concurrent pathways have been proposed, the [3+2] cycloaddition of aryne-induced ylides with metalated α-imino esters and the metal-catalyzed [3+2] cycloaddition of azomethine ylide with α-imino esters. Imino ester 166a reacts with benzyne, generated from 508 (R1 = H) to form zwitterion I, which after 1,4-hydrogen transfer gives the azomethine ylide II′ and the trans-isomer II. Meanwhile, 166a reacts with the silver catalyst to provide the metalo-dipole III, which undergoes a regioselective endo-[3+2] cycloaddition to give 509a. In the other pathway, azomethine ylide IV reacts with 166a to provide 502a. Subsequent capture of 502a by benzyne generates the product 509a.
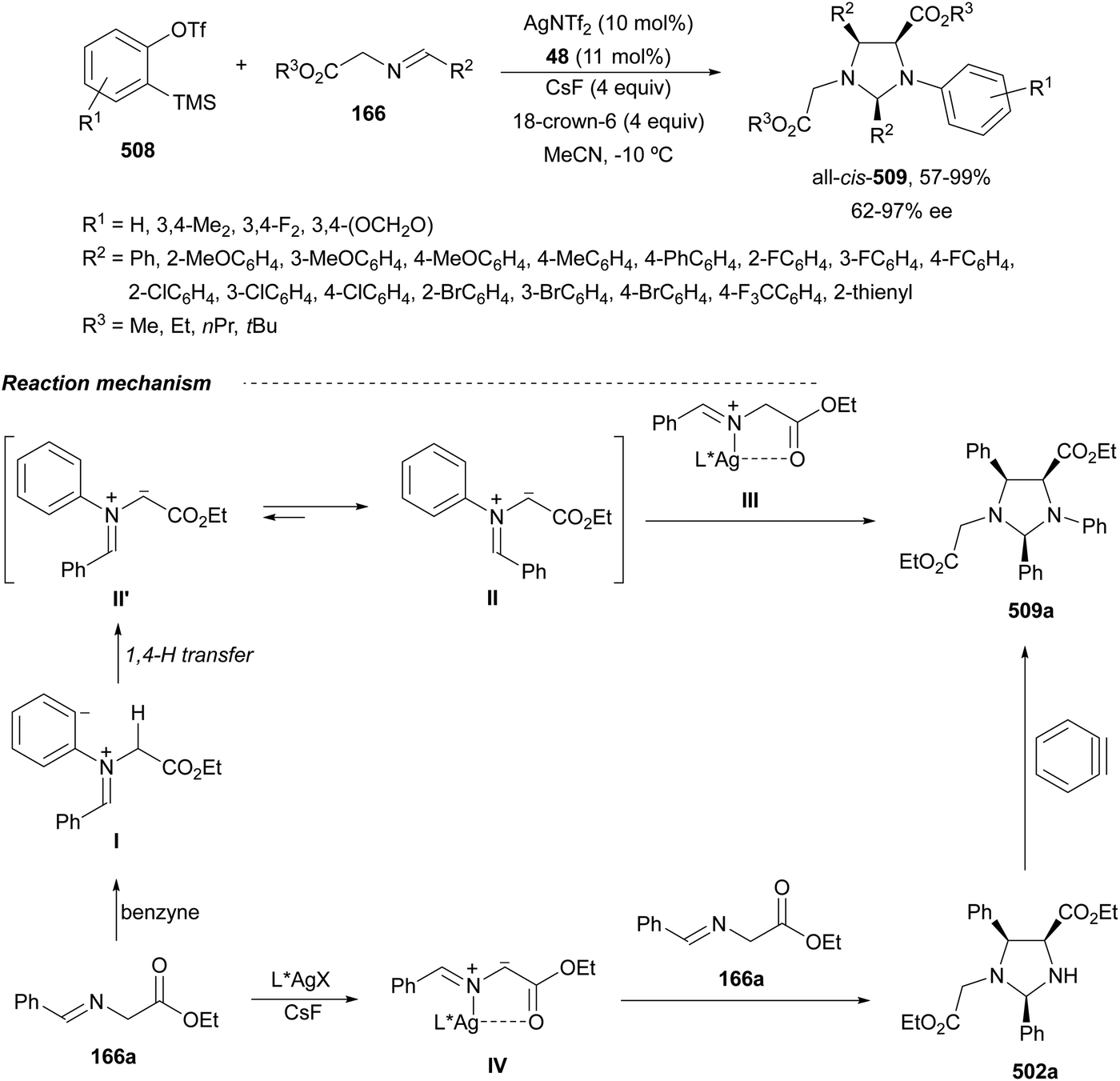 | ||
| Scheme 154 Asymmetric 1,3-DC of imino esters 166 with arynes under AgNTf2/ferrocenylphosphine 48 catalysis. | ||
Ooi and co-workers302 have reported a Pd-catalyzed [3+2] cycloaddition of 5-vinyloxazolidinones 510 with imines 161 to yield imidazolidines 512 (Scheme 155). In the presence of Pd2(dba)3·CHCl3 (Pd 2.5 mol%) and a chiral ammonium-phosphine 511 hybrid ligand, the corresponding imidazolidines cis-512 were obtained in high yields and diastereo- and enantioselectivities. Oxazolidinones 510 undergo ring opening by Pd to provide the π-allylPd intermediate I, which after decarboxylation generates the dipole II. Imidazolidine 512a bearing a quaternary stereocenter was transformed into the 1,2-diamine 513 by deprotection of the mesyl group with dodecanethiol.
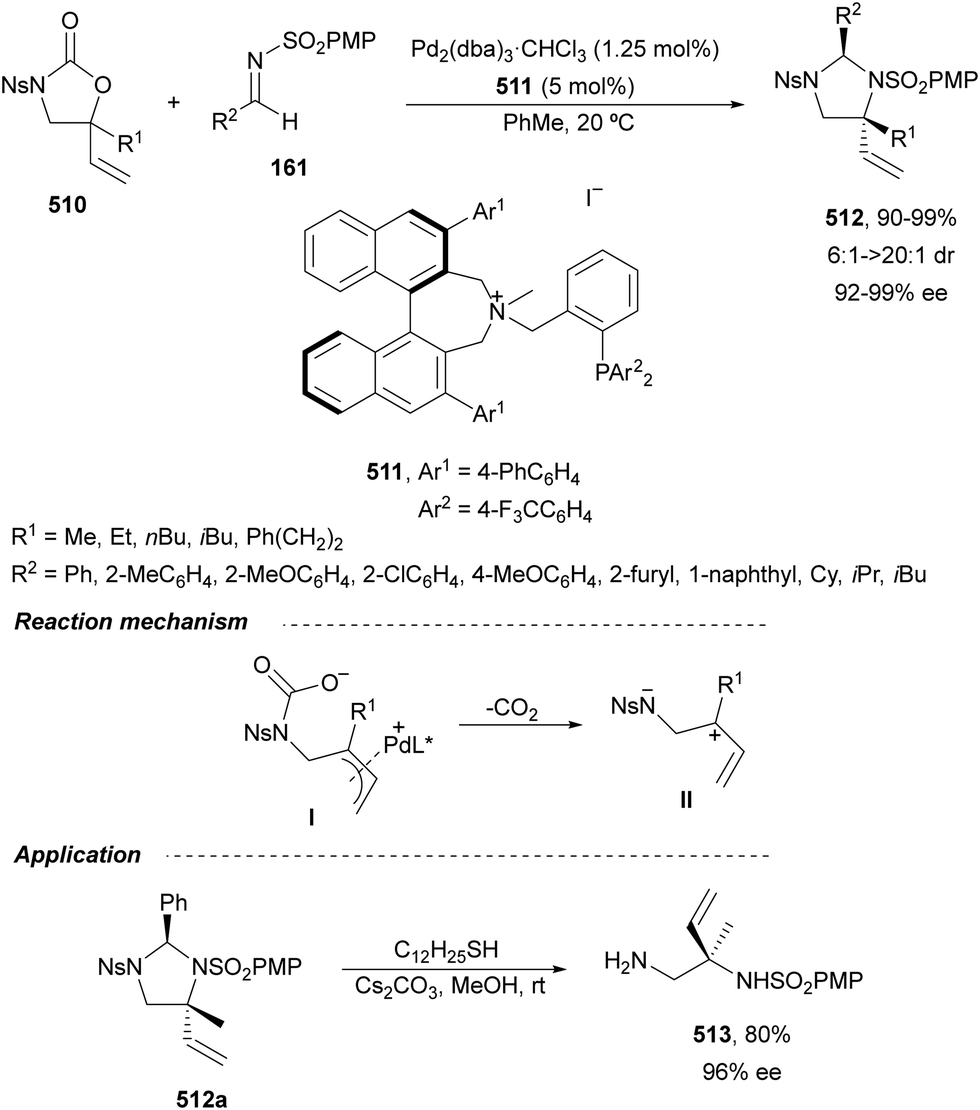 | ||
| Scheme 155 Asymmetric 1,3-DC of 5-vinyloxazolidinone 510 with imines 161 under Pd(0)/511 catalysis. | ||
Rhodium-catalyzed [2+2] cycloaddition of allenamides 514 has been reported by Kang and co-workers.303 This intermolecular head-to-head enantioselective [2+2] cycloaddition was carried out in the presence of 5 mol% of [Rh(cod)Cl]2 and (S)-Binap (46) in 1,2-dichlorobenzene at 40–60 °C to furnish trans-3,4-bis(methylene)cyclobutene-1,2-diamines 515 in good yields and enantioselectivities (Scheme 156). In the proposed mechanism, two allenamide molecules coordinate to the Rh catalyst to produce the rhodiacyclopentane intermediate I. Subsequent enantioselective controlled reductive elimination affords the cyclobutene-1,2-diamine 515.
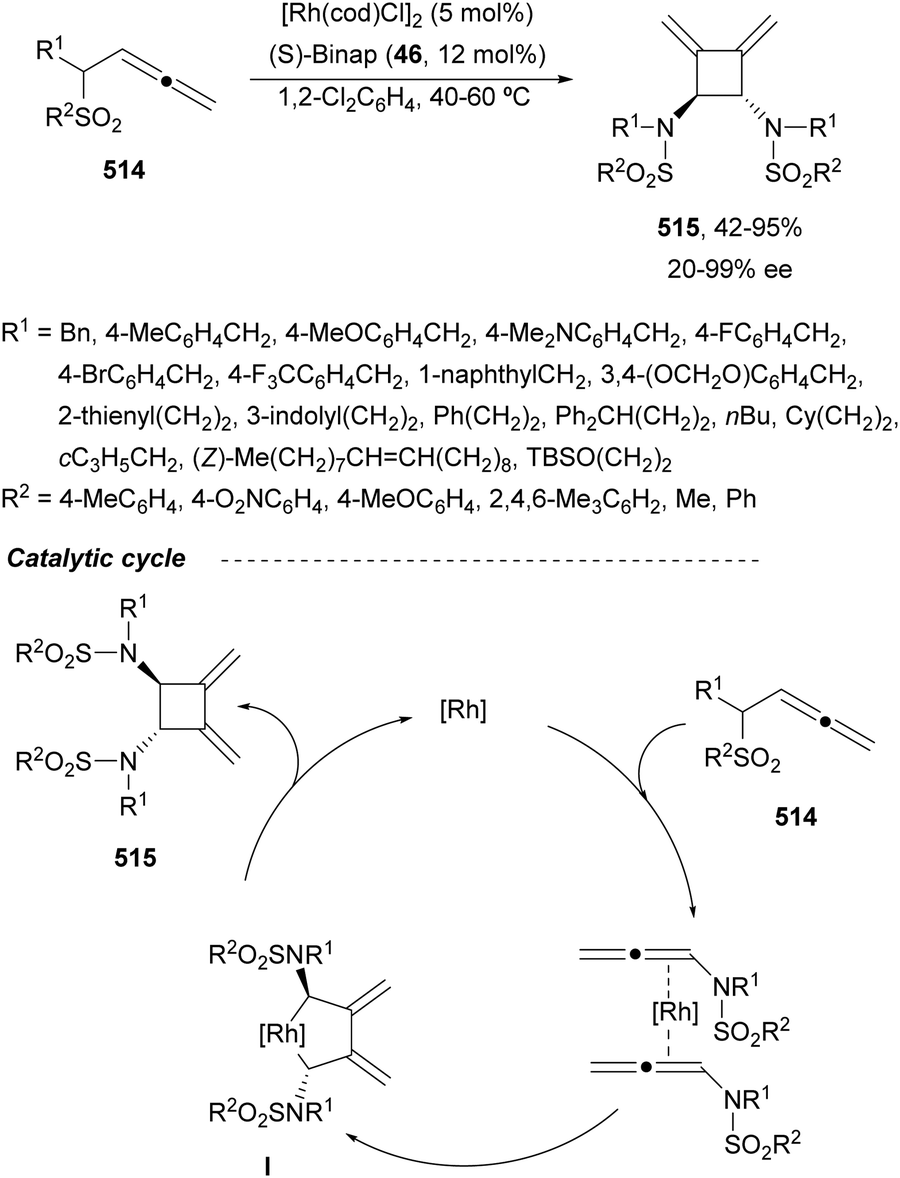 | ||
| Scheme 156 Asymmetric [2+2] cycloaddition of allenamides 514 under Rh(I)/(S)-Binap (46) catalysis. | ||
The asymmetric [3+2] cycloaddition of azomethine ylides with imines gave diastereo- and enantioenriched imidazolidines which are easily transformed into 1,2-diamines. These 1,3-DC are catalyzed by CPA, chiral silver or copper complexes, generally with endo-selectivity to provide cis-imidazolidines in good yields and diastereo- and enantioselectivities. A Rh-catalyzed [2+2] cycloaddition of allenamides provided bis(methylene)-1,2-diamines up to 99% ee.
4. C–H bond-forming reactions
In this section, hydrogenation reactions of CN bonds with molecular hydrogen under metal-catalyzed processes, transfer hydrogenation conditions or hydrogen autotransfer are considered.
The synthesis of chiral piperazines 519–521 has been performed by asymmetric hydrogenation (AH) of N-alkylpyrazinium salts 516–518 under Ir-catalysis, respectively. Zhou and co-workers304 employed (R,SP)-Josiphos (44) as a chiral ligand in the case of 3-substituted pyrazinium salts 516 to obtain chiral piperazines 519 up to 95% yield and 92% ee, whereas (R)-Segphos (23) and (S,S)-522 were the ligands for AH of 3,5- and 2,3-disubstituted pyrazinium salts, 517 and 518, respectively, to provide piperazines 520 and 521 with high yields and enantioselectivities (Scheme 157). This methodology made the pyrazine ring more electron deficient, weakening its coordination ability and facilitating its reduction. In the proposed mechanism, the salt 516a undergoes 1,4-hydride addition to furnish the 1,4-dihydropyrazine I and HBr. In the presence of HBr, intermediate I tautomerizes to II. Subsequent hydrogenation of iminium salt II gives III and then the finally piperazine 519a. This procedure was applied to the synthesis of vestipitant, a potent and selective NK1 receptor antagonist, an antiemetic and anxiolytic drug, in only three steps from piperazine 519b. In the case of mirtazapine, for the treatment of insommia and climacteric symptoms, the precursor piperazine N-Boc-519a was prepared from pyrazinium salt 516a by AH with (R,SP)-44 as a catalyst in 87% yield and 90% ee.
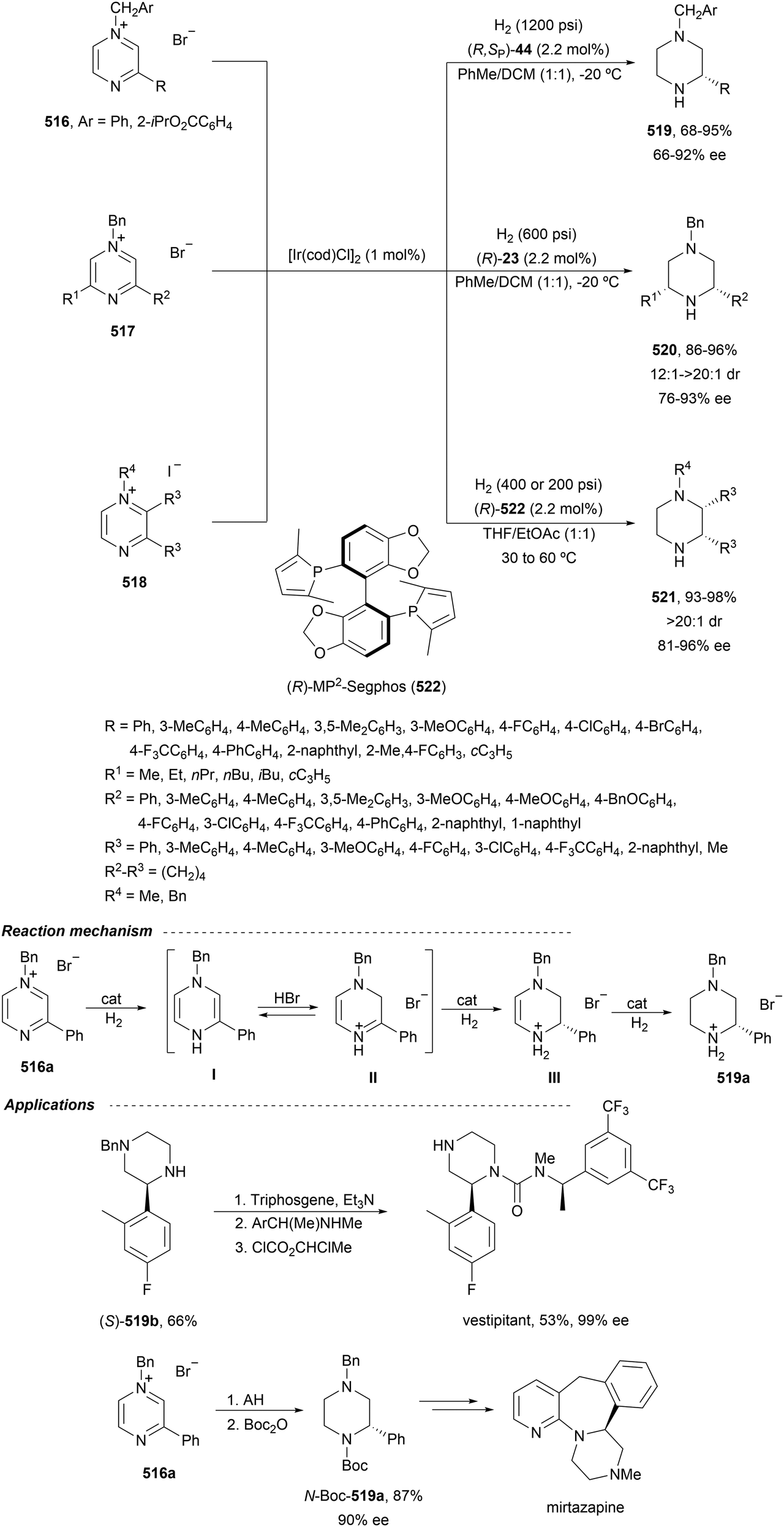 | ||
| Scheme 157 Asymmetric hydrogenation of N-alkyl pyrazinium salts 516–518 to piperazines 519–521 under Ir/ligand 44, 23 and 522 catalysis. | ||
AH of 2,2′-bisquinoline and quinoxaline derivatives 523 has been described by Fan and co-workers305 using chiral cationic Ru(diamine) complexes 524 or 525 under 50 atmospheres. Endocyclic vicinal diamines 526 were obtained in high yields with excellent diastereo- and enantioselectivities (Scheme 158a). This methodology has been previously applied by the same group306 to 2-substituted and 2,9-disubstituted 1,10-phenanthrolines 527 by means of (R,R)-525 to obtain OPhen derivatives 528. In the case of 2,9-disubstituted 527, trans-OPhen 528 were diastereoselectively obtained in high yields and excellent enantioselectivities (Scheme 158b). Products 526 and 528 can be transformed into chiral N-heterocyclic carbene ligands by reaction with neat triethyl orthoformate.
 | ||
| Scheme 158 Asymmetric hydrogenation of 2,2′-bisquinolines or quinoxalines 523 and 1,10-phenanthrolines 527 under Ru(diamine) (R,R)-524 or 525 catalysis. | ||
The same group307 reported a highly enantioselective Ir- or Ru-catalyzed intermolecular reductive amination/asymmetric hydrogenation of 2-quinolinecarbaldehydes 529 and aromatic amines. Chiral vicinal diamines 531 were obtained in good yields and enantioselectivities using the Ir complex (R,R)-530 as a chiral catalyst (Scheme 159). When sterically hindered aromatic amines were used, the Ru(diamine) complex (R,R)-524 in the presence of TfOH was the best catalyst for this transformation. These chiral diamines 531 and their corresponding bulky chiral NHC were used as ligands for the Suzuki–Miyaura cross-coupling reaction and for ring-opening cross-metathesis, respectively.
 | ||
| Scheme 159 Reductive amination/asymmetric hydrogenation of quinoline-2-carbaldehydes 529 with aromatic amines under Ir or Ru complex (R,R)-530 or (R,R)-524 catalysis. | ||
Stoltz and co-workers308 described the AH of 1,3-disubstituted isoquinolines to provide chiral 1,2,3,4-tetrahydroisoquinolines (Scheme 160). In the case of the 1-(N-Boc-aminomethyl)-3-phenylisoquinoline 532, the AH was carried out with [Ir(cod)Cl]2 ligand 533, TBAI as an additive, 60 bar H2, 9:1 THF/AcOH as solvents at 60 °C to obtain cis-diamine derivative 534 in 71% yield, 9:1 dr and 90% ee.
 | ||
| Scheme 160 Asymmetric hydrogenation of 1-(N-Boc-aminomethyl)-3-phenylisoquinoline (532) under Ir/bisphosphine 533 catalysis. | ||
2,3-Disubstituted quinoxalines 535 were transformed by Zhang and Du309 into the corresponding cis-1,2,3,4-tetrahydroquinoxalines 537 by AH using bis(pentafluorophenyl)borane (Piers's borane310) and a chiral diene 536 as a ligand (Scheme 161). This metal-free hydrogenation took place under 20 bar in hexane at room temperature to provide cis-2,3-disubstituted tetrahydroquinoxalines 537 with good yields and enantioselectivities and high diastereoselectivities.
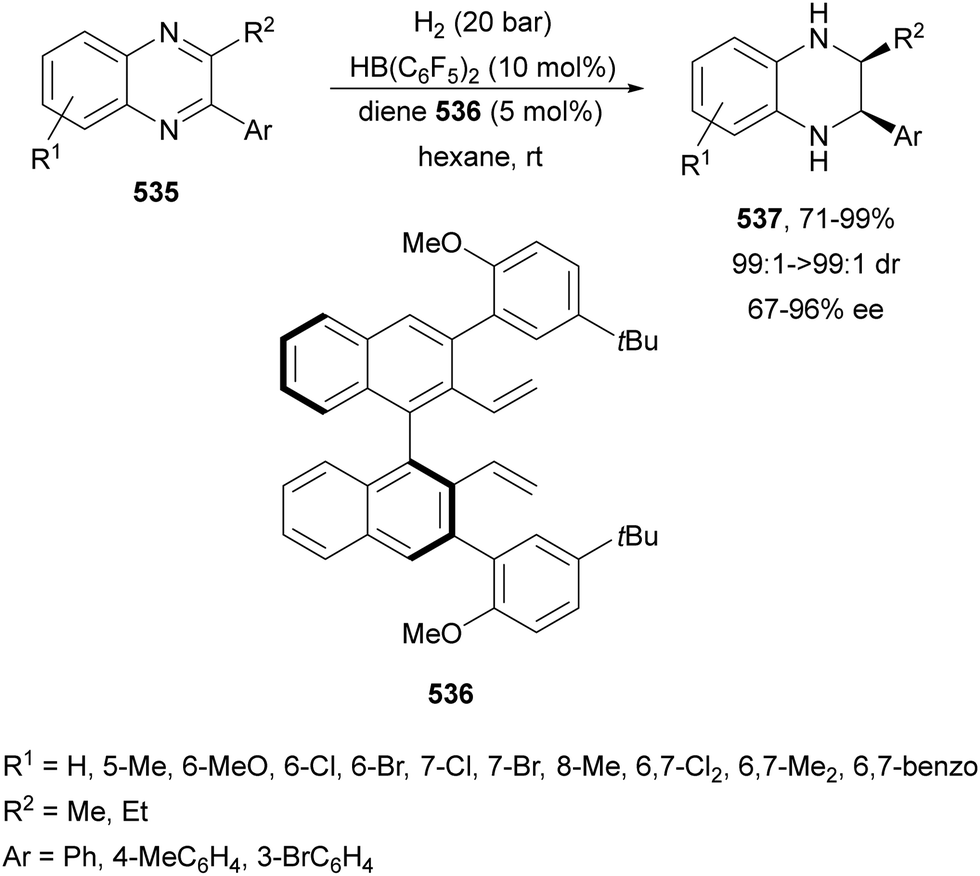 | ||
| Scheme 161 Asymmetric hydrogenation of 2,3-disubstituted quinoxalines 535 under bis(pentafluorophenyl)borane and chiral diene 536 catalysis. | ||
Asymmetric transfer hydrogenation (ATH) has been employed for the enantioselective synthesis of 4-alkylidenethiadiazolines 540 from thiadiazole-1,1-dioxides 538 prepared from 1,2-diketones, by Zezschwitz and co-workers.311 This ATH was carried out with the Noyori catalyst RuCl(TsDpen) 539 working with a 5/2 mixture of formic acid and triethylamine in acetonitrile at −15 °C to provide products 540 up to 97% yield and 98% ee (Scheme 162). Subsequent diastereoselective reduction of compounds 540 with LiBH4 in THF at room temperature afforded cis-1,2,5-thiadiazolidine-1,1-dioxides 541 in high yields and enantioselectivities. These cis-thiadiazolidine derivatives 541 were transformed into the trans-diastereomers by treatment with trifluoroacetic acid or sulfuric acid under preservation of the enantioselectivity. Both cis- and trans-541a were transformed into the anti- and syn-diamines 542, respectively, by refluxing in hydrazine monohydrate. These diamines have been further converted into α,β-diamino acids by oxidation of the PMP group.
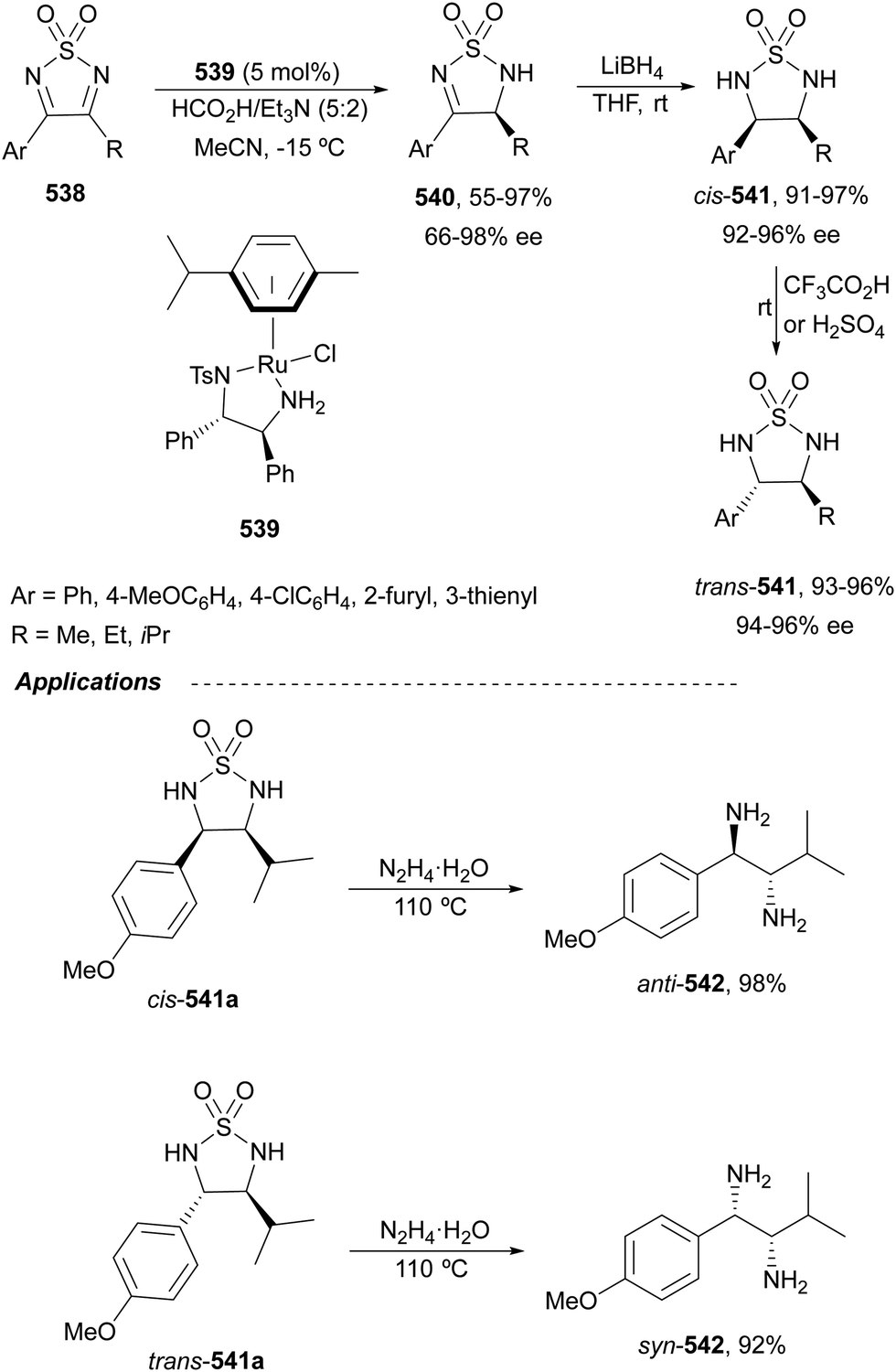 | ||
| Scheme 162 Asymmetric hydrogen transfer of thiadiazoles 538 under RuCl(TsDpen) 539 catalysis. | ||
Yang, Zhao and co-workers312 have recently reported an enantioconvergent313 synthesis of diamines from diols through a catalytic borrowing hydrogen autotransfer process. Readily available secondary-primary diols 543 bearing an aryl substituent were diaminated with amines using [Ir(cod)Cl]2 (2.5 mol%), a chiral bisphosphine 544 (5 mol%) and a CPA (R)-TRIP (152, 10 mol%) as cooperative catalysts to furnish diamines 545 in toluene at 90 °C up to 89% yield and 94% ee (Scheme 163). According to control experiments, a catalytic pathway was proposed. Mono-oxidation of diol by Ir gives the α-hydroxy aldehyde I, which after CPA catalyzed condensation with the amine provides the α-hydroxy imine II. Heyns rearrangement of II forms the α-amino ketone III, which reacts with the amine to give the α-amino imine IV and after reduction of the ketimine with [Ir]H resulted the diamine 545. Alternatively to the enantioconvergent process, a dynamic kinetic asymmetric transformation (DYKAT) can take place by tautomerization of IV to enamine V, and then in the presence of the CPA VI and VI′ are formed. A faster reduction by [Ir]H of VI in preference over VI′ leads to diamine 545.
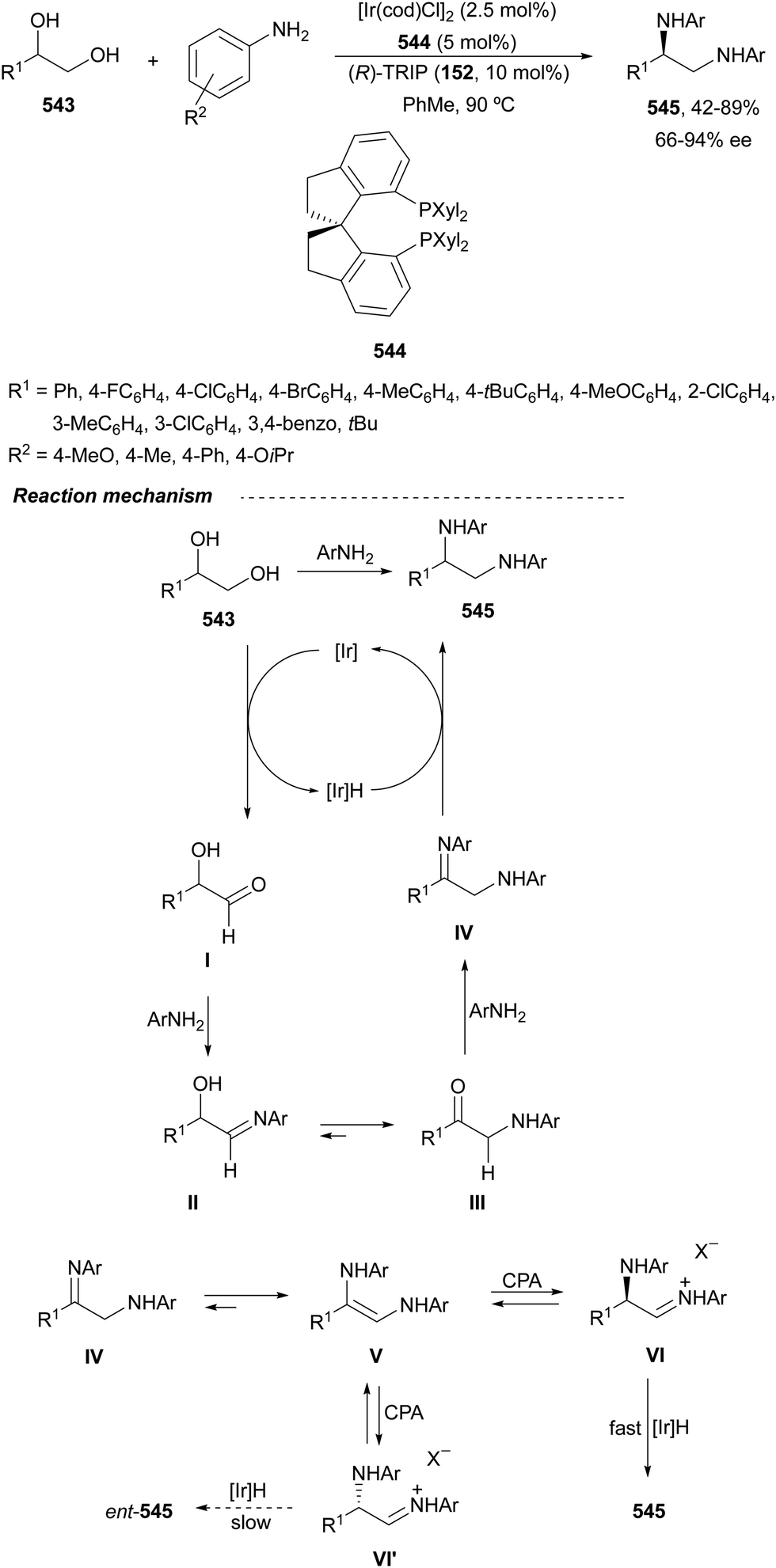 | ||
| Scheme 163 Asymmetric hydrogen autotransfer of diols 543 with amines under [Ir(cod)Cl]2/bisphosphine 544 and (R)-TRIP (152) catalysis. | ||
Asymmetric hydrogenation of N-containing heterocyclic compounds such as N-alkylpyrazinium salts, 2.2′-bisquinoline and quinoxaline derivatives, and isoquinolines can be carried out under Ir/bisphosphine or Ru(diamine) chiral complexes to provide chiral 1,2-diamines. In the case of 2,3-disubstituted quinoxalines, Pier's borane and a chiral diene gave the corresponding chiral cis-2,3-disubstituted tetrahydroquinoxalines. Asymmetric transfer hydrogenation is an efficient reduction for thiadiazoles using RuCl(TsDpen) as a catalyst and formic acid allowed the synthesis of anti- and syn-diamines. Diols have been transformed into chiral diamines under hydrogen autotransfer conditions by means of Ir/bisphosphine and a CPA cooperative catalysis.
5. C–H amination reactions
Intramolecular radical C–H amination by metal nitrenoid species and atroposelective C–H amination will be considered in this section.Zhang and co-workers314 described in 2018 the enantioselective intramolecular C–H amination of sulfamoyl azide compounds under cobalt-amideporphyrin complexes as catalysts to obtain 1,6-aminated products. One year later, the same group315 reported the enantiodivergent52 1,5-C–H amination of sulfamoyl azides 546 using Co-porphyrin complexes 547a (n = 2, Ar = 2,6-(MeO)2C6H3) and 547b (n = 3, Ar = 2,6-tBu2C6H3) as catalysts (Scheme 164). This catalytic radical amination via enantiodifferentiative H-atom abstraction (HAA) and stereoselective radical substitution (RS) afforded cyclic sulfamides 548 in a highly enantioenriched form precursors of chiral 1,2-diamines. Deuterium labelling studies and DFT calculations demonstrated that the cavity size of the cobalt-amideporphyrin affects the enantiodivergence of this process. In the proposed catalytic pathway, a Co(III)-amidyl radical I is formed, which after H-atom abstraction results an alkyl radical II able to undergo RS to give the sulfamide 548 and regenerating the catalyst.
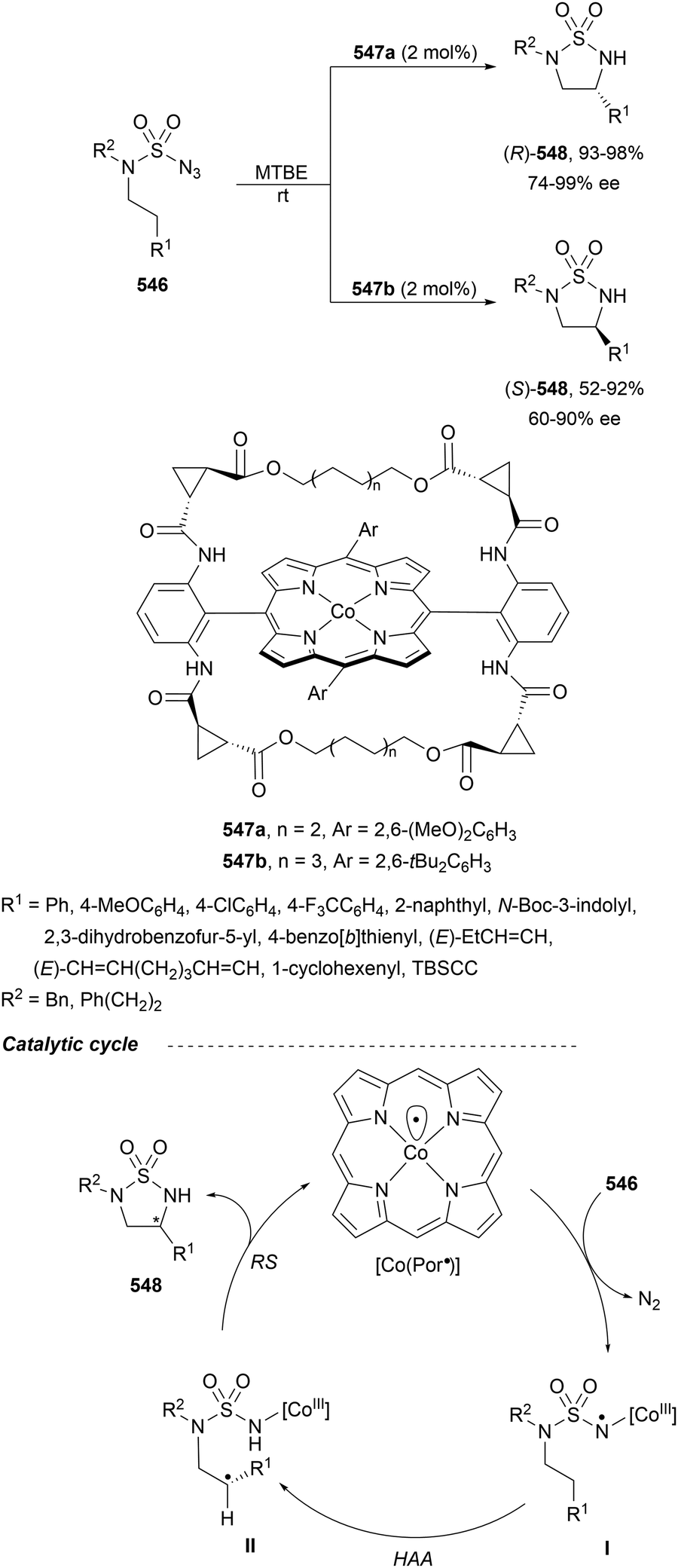 | ||
| Scheme 164 Asymmetric enantiodivergent radical C–H amination of sulfamoyl azides 546 under Co-porphyrin 547 catalysis. | ||
Liu, Arnold and co-workers316 used a mutated variant of cytochromes P441, designed as P441Dianel3, for the intramolecular amination of sulfamoyl azides 546 to cyclic sulfamides (S)-548 in 91–99% ee. Meggers and co-workers317,318 reported the same cyclization using a chiral Ru(II)/pybox complex 549 as a catalyst to obtain cyclic sulfamides (S)-548 up to 98% yield and up to 88% ee (Scheme 165). The reaction of azide 546 (R = Ph) was carried out on a gram-scale to provide (S)-548 (R = Ph) in 95% yield and 95% ee, which was further transformed into the corresponding chiral 1,2-diamine by treatment with hydrazine at 110 °C311 in 95% yield and 95% ee. Mechanistic experiments support a stepwise mechanism in which a Ru-nitrenoid intermediate IS is formed under release of molecular nitrogen. This initially formed nitrenoid in a singlet state is converted to a triplet state IT to form a nitrenoid radical complex, which initiates a 1,5-hydrogen atom transfer (HAT) at the benzylic position to provide diradical II. Subsequent radical–radical rebound forms intermediate III, which releases the product and regenerates the catalyst.
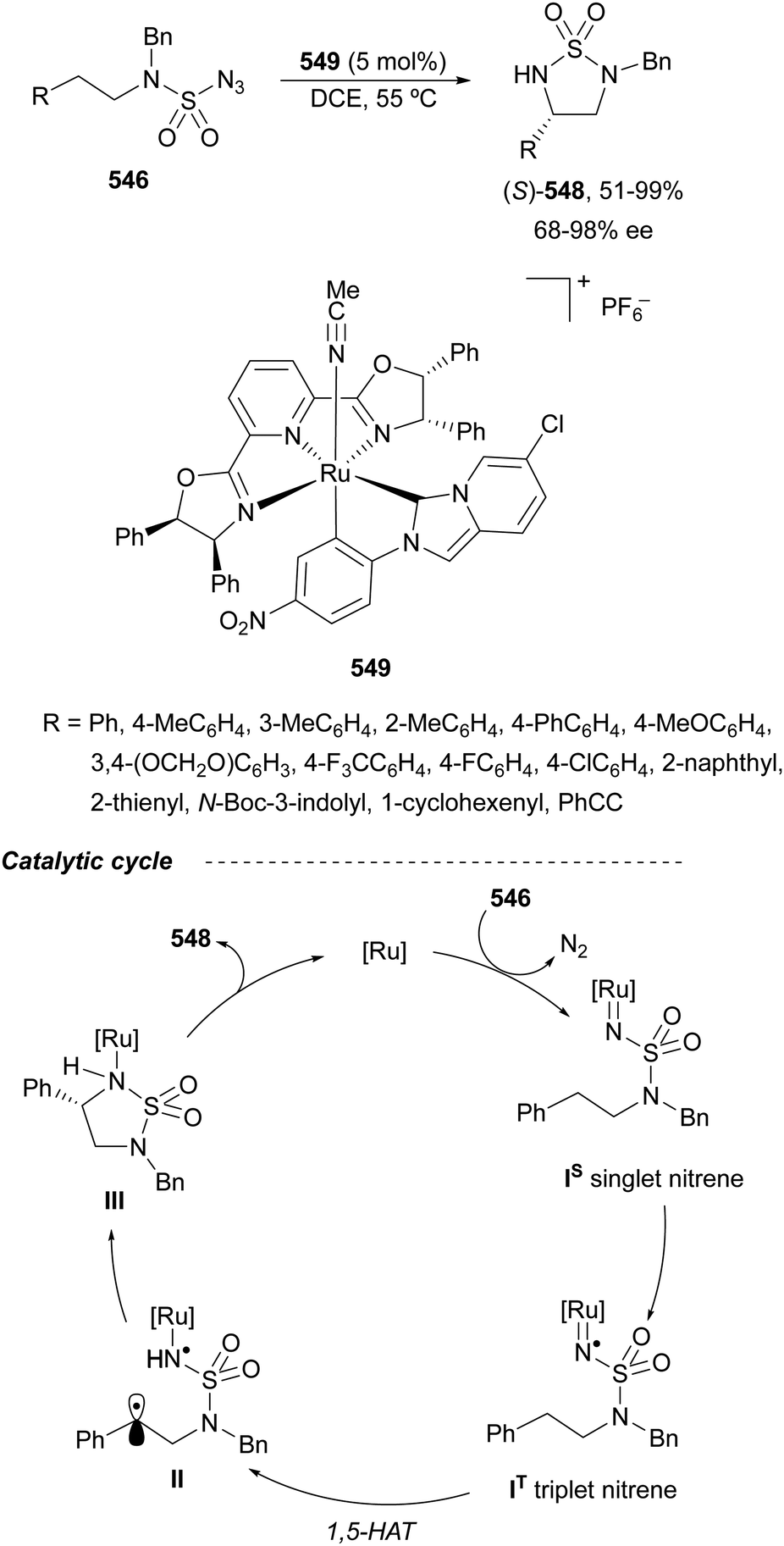 | ||
| Scheme 165 Asymmetric radical C–H amination of sulfamoyl azides 546 under Ru(II) complex 549 catalysis. | ||
The same group319 described the C(sp3)–H amination of urea derivatives 550 by using a chiral-at-metal Ru catalyst 551, providing cyclic ureas 552 up to 99% yield and 99% ee (Scheme 166). This process was carried out with very low catalyst loading down to 0.05 mol% under mild reaction conditions. These products were transformed into chiral 1,2-diamines by treatment with concentrated HCl in AcOH at 85 °C under microwave conditions. In the proposed mechanism, upon release of benzoic acid from N-benzoyloxy urea 550, the Ru catalyst forms a Ru nitrenoid which evolves triplet state I. Subsequent 1,5-HAT provides intermediate II followed by C–N bond formation though radical recombination to give intermediate III. Finally, the release of chiral 2-imidazolidinone 552 and regeneration of the catalyst take place.
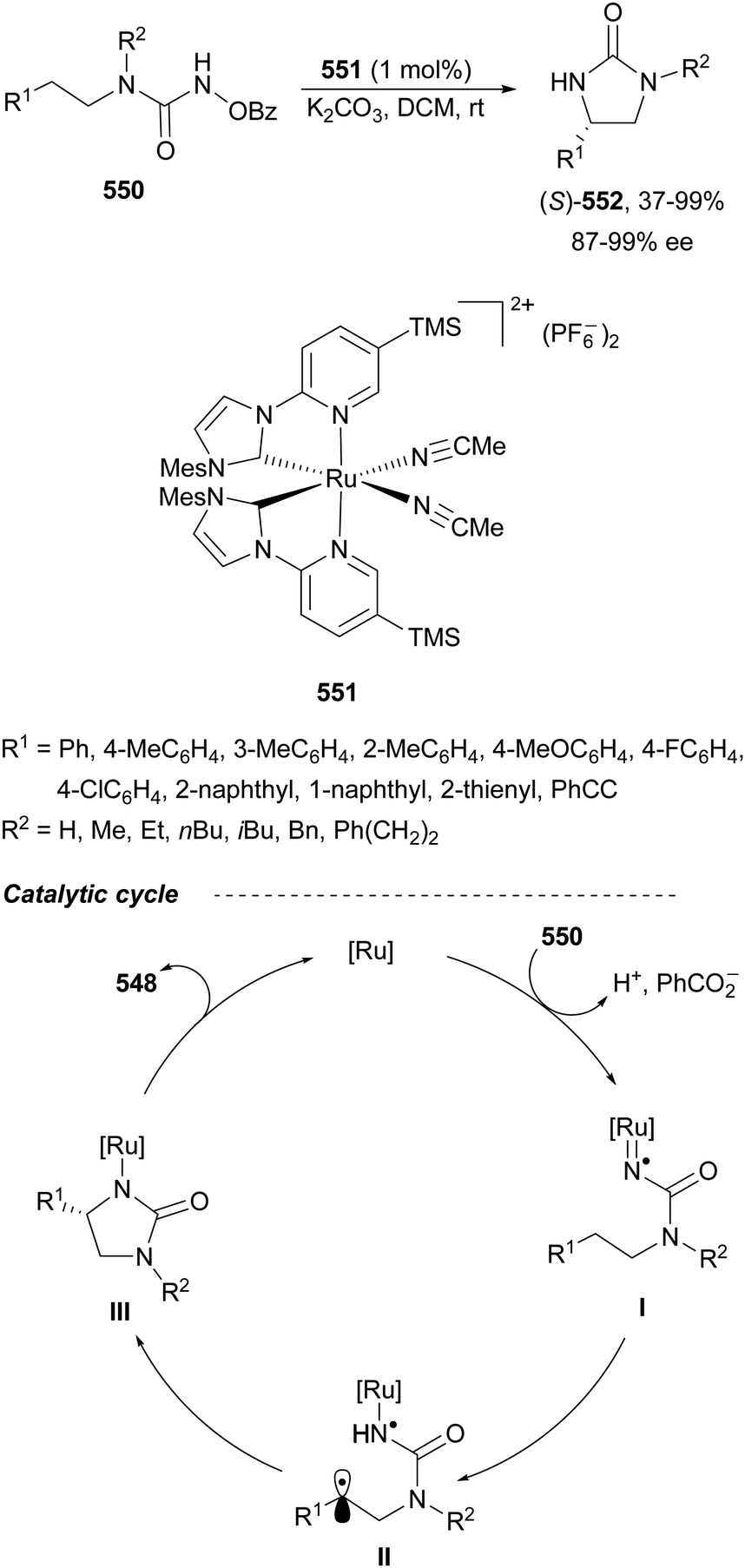 | ||
| Scheme 166 Asymmetric radical C–H amination of N-benzoyloxy ureas 550 under Ru(II) complex 551 catalysis. | ||
Zhang and co-workers320 reported an unusual enantioselective C–H amination of N-aryl-2-naphthylamines 553 with azodicarboxylates under CPA (R)-189 catalysis to provide naphthalene-1,2-diamine derivatives 554 (Scheme 167). This atroposelective direct C–H amination occurred via a concerted control of π–π interactions and dual hydrogen-bonding illustrated in the proposed reaction mechanism. First, the CPA activates the N-phenyl-2-naphthylamine 553a and also the azodicarboxylate to form intermediate I by dual hydrogen-bonding. Then, the π–π interaction assisted by the nucleophilic addition of 553a to N-Boc azodiacarboxylate to form intermediate II followed by rearomatization and stabilization of product 554a by intramolecular hydrogen-bonding.
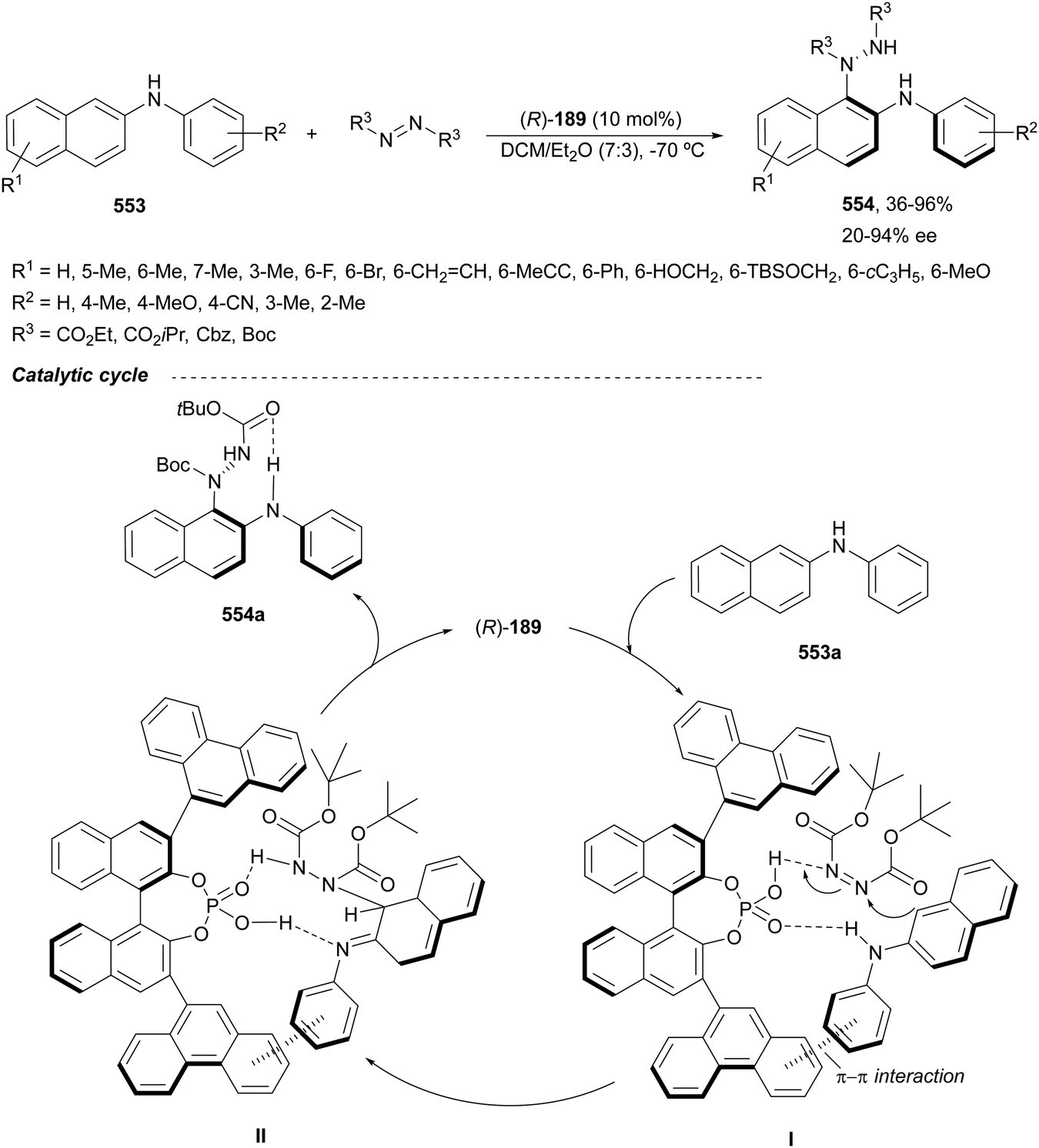 | ||
| Scheme 167 Asymmetric atroposelective C–H amination of N-aryl-2-naphthylamines 553 with azodicarboxylates under CPA (R)-189 catalysis. | ||
Asymmetric intramolecular radical C–H amination of sulfamoyl azides or N-benzoyloxyureas has been carried out efficiently with Ru(II) cationic complexes as catalysts to give enantioenriched cyclic sulfamides or imidazolidinones, respectively, precursors of 1,2-diamines. In the case of N-aryl-2-naphthylamines, an asymmetric atroposelective C–H amination has been performed using CPA as catalyst and azodicarboxylates as electrophiles to obtain naphthalene-1,2-diamine derivatives.
6. Other catalytic methods
Enantioselective desymmetrization15 of meso-diamines 555 has been accomplished by monobenzylation under organocatalysis by De and Seidel.321 The cooperative action of two catalysts, DMAP as an achiral nucleophile and diamide-thiourea 556 as a chiral anion receptor catalyst gave the monoacylated products anti-557 in good yields and good enantioselectivities (Scheme 168). In the proposed mechanism, DMAP reacts with benzoic anhydride to give intermediate I, an achiral ion pair, which interacts with thiourea to form the chiral ion pair II able to carry out the monobenzylation.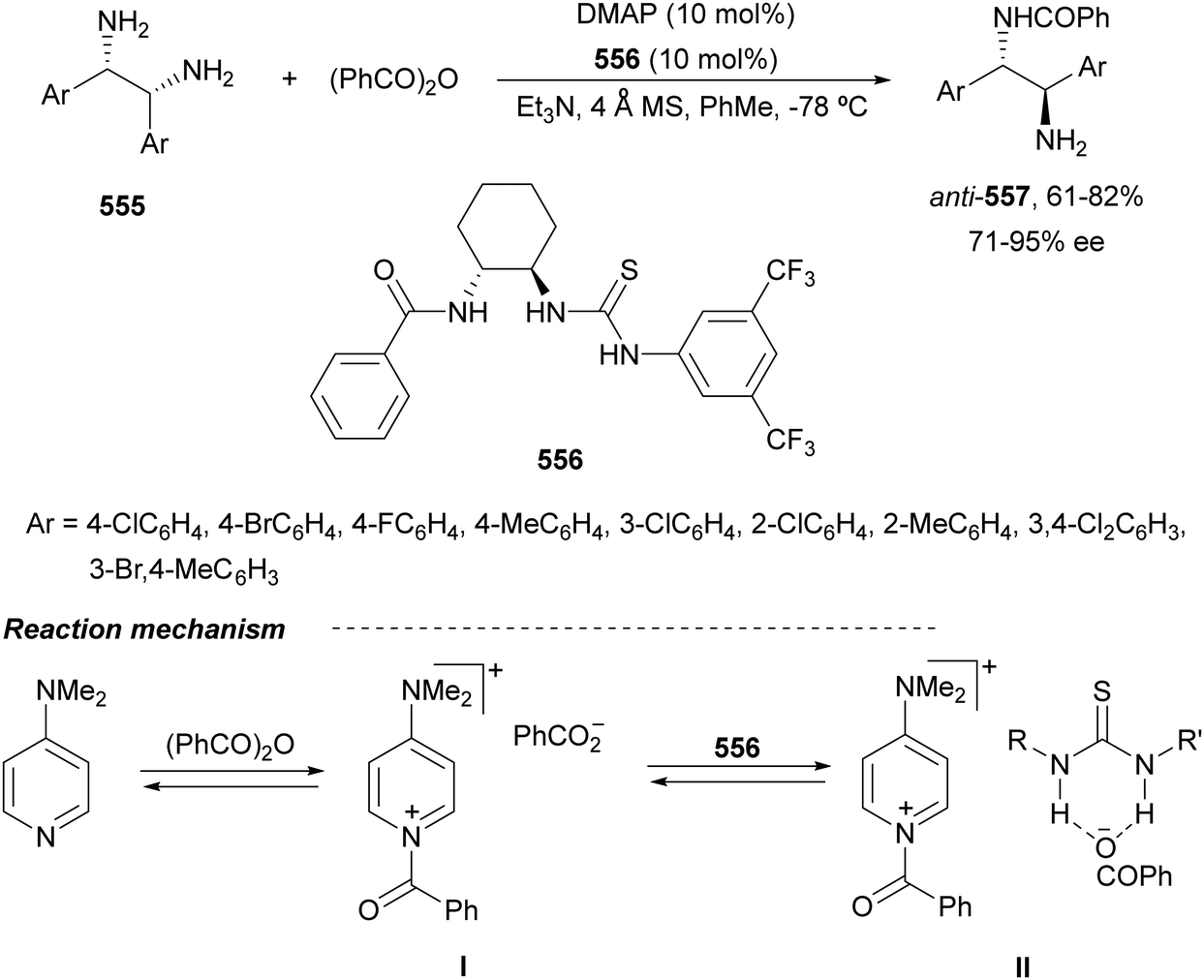 | ||
| Scheme 168 Enantioselective desymmetrization of meso-diamines 555 with benzoic anhydride under cooperative DMPA and thiourea 556 catalysis. | ||
The same group performed322 the kinetic resolution of 1,2-diaryl-1,2-diaminoethanes 555 by monobenzylation using the dual catalysis approach described in Scheme 168. In this case, amide-urea 558 was used as a chiral anion receptor and 4-(di-n-propylamino)pyridine (559) as an achiral nucleophile to form the ion pair II (Scheme 168). The crude reaction mixture was treated with TrocCl to provide a mixture of products 560 and 561 up to 53% conversion and a s-factor up to 30 (Scheme 169).
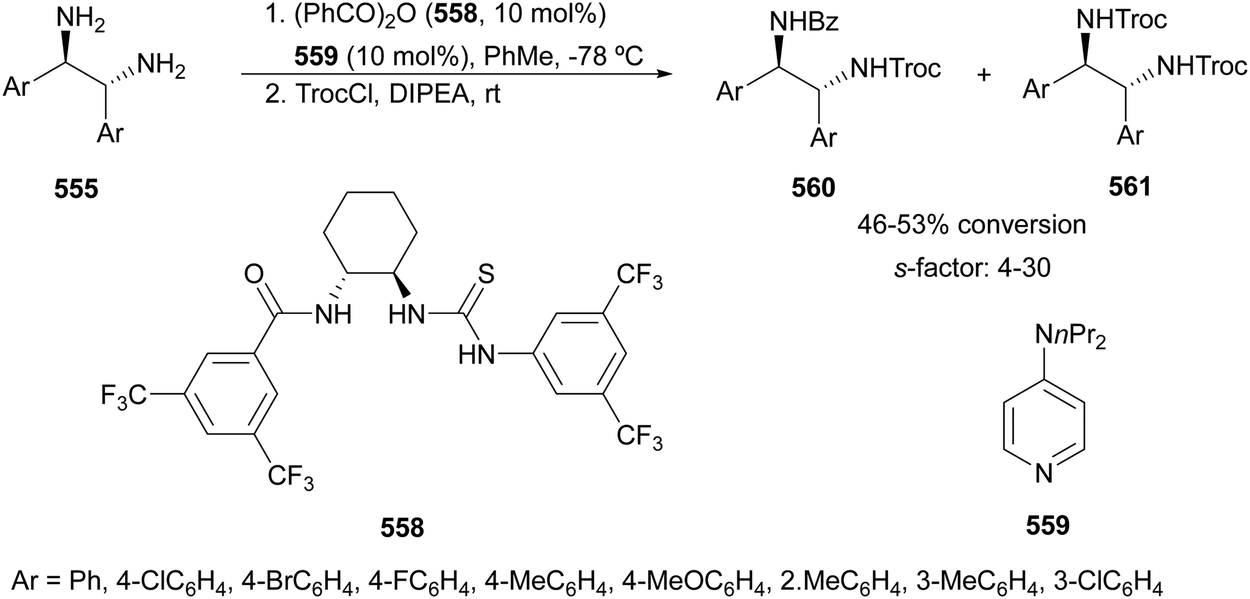 | ||
| Scheme 169 Kinetic resolution of 1,2-diaryl-1,2-diaminoethanes 555 under amide-thiourea 558 and 4-(di-n-propylamino)pyridine (559) dual catalysis. | ||
Kinetic resolution (KR) of 1,2-diamines 562 and 563 has also been performed via organocatalyzed electrophilic amination with dibenzyl azodicarboxylate (75) by He, Yang and co-workers.323 In the case of 1,2-diamines 562 with a α-secondary amine, (R)-456 was used as CPA to obtain (S)-562 (up to >99% ee) and products (R)-564 (up to 94% ee), which were separated by column chromatography (Scheme 170a). For the KR of α-tertiary amines 563, (S)-565 was employed as CPA to provide (R)-563 (up to 97% ee) and compounds (S)-566 (up to 96% ee) (Scheme 170b). Based on control experiments with 562a (R = Ph, R = H) a plausible mechanism was proposed. Under catalyst (S)-456, the dual hydrogen bonding activation of matched (S)-1,2-diamine substrate 563a by the CPA catalyst facilitates the electrophilic addition of the aniline moiety with the diazo group in I to provide dearomatized intermediate II. Subsequent aromatization of II forms (S)-566a. Product (S)-566a (Ar = Ph, R1 = Me, R2 = H) was transformed into (S)-563a by treatment with KOH at 70 °C by removing of the hydrazine moiety. Moreover, oxidative cleavage of the N-aryl group of (S)-566a by using trichloroisocyanuric acid (TCCA) at 80 °C produced the primary amine 567a in 58% yield.
 | ||
| Scheme 170 Kinetic resolution of 1,2-diamines 562 and 563via electrophilic reaction with dibenzyl azodicarboxylate (75) under CPA (R)-456 and (S)-565 catalysis, respectively. | ||
More recently, Waser and co-workers324 reported the enantioselective desymmetrization of meso-diaminocyclopropane 568 under Cu(II)/bis(oxazoline) 569 catalysis. This Friedel–Crafts alkylation of indoles and a pyrrole delivered enantioenriched diastereomerically pure imidazolidinones 570 and 571, respectively (Scheme 171). The trans-relative configuration of products 570 and 571 supports a SN2-like mechanism for the ring-opening of the cyclopropane unit.
 | ||
| Scheme 171 Enantioselective desymmetrization of meso-diaminocyclopropane 568 by Friedel–Crafts alkylation of indoles and a pyrrole under Cu(II)/bis(oxazoline) 569 catalysis. | ||
Rhodium-catalyzed dynamic kinetic asymmetric transformations (DYKAT) of racemic allylic trichloroacetimidates 572 and 573 were employed by Mwenda and Nguyen325 for the enantioselective synthesis of 1,2-diamines 575 and 576, respectively. Chiral diene 574-ligated Rh-catalyst promoted the amination of allylic tetrachloroacetamidates 572 with aromatic amines to provide 1,2-diamines 575 and 576 with tertiary and quaternary stereocenters with good diastereo- and enantioselectivities (Scheme 172). This process occurs by intermediacy of a π-allylrhodium intermediate I, which undergoes fast nucleophilic attack of aniline suppressing vinyl aziridine formation.
 | ||
| Scheme 172 Enantioselective DYKAT of allylic trichloroacetamidates 572 and 573 with aromatic amines under Rh/diene 574 catalysis. | ||
Pyrrolidine-3,4-diamine skeleton is present in some biologically active compounds.326 Enantioselective 1,3-DC of azomethine ylides and (E)-β-naphthalimidonitroethene 577 has been described by Yu, Deng and co-workers327 for the preparation of trans-3,4-diamino derivatives 579 using CuI/N,O-ligands 578 (Scheme 173). Imino esters 166 reacted with dipolarophile 577 to give endo-adducts 579 in high yields and diastereo- and enantioselectivities. The authors propose a TS with the two phenyl groups adjacent to the oxygen atom in the 1,2-dihydroimidazo[1,2-a]quinolone ligand 578 blocking the dipolarophile 577 approaching from the bottom face favoring the formation of the endo-579 through approaching from the top face. Nitro group reduction of cycloadduct 579a with RANEY® Ni and deprotection of phthalyl by methylamine provided trans-3,4-diaminopyrrolidine 580.
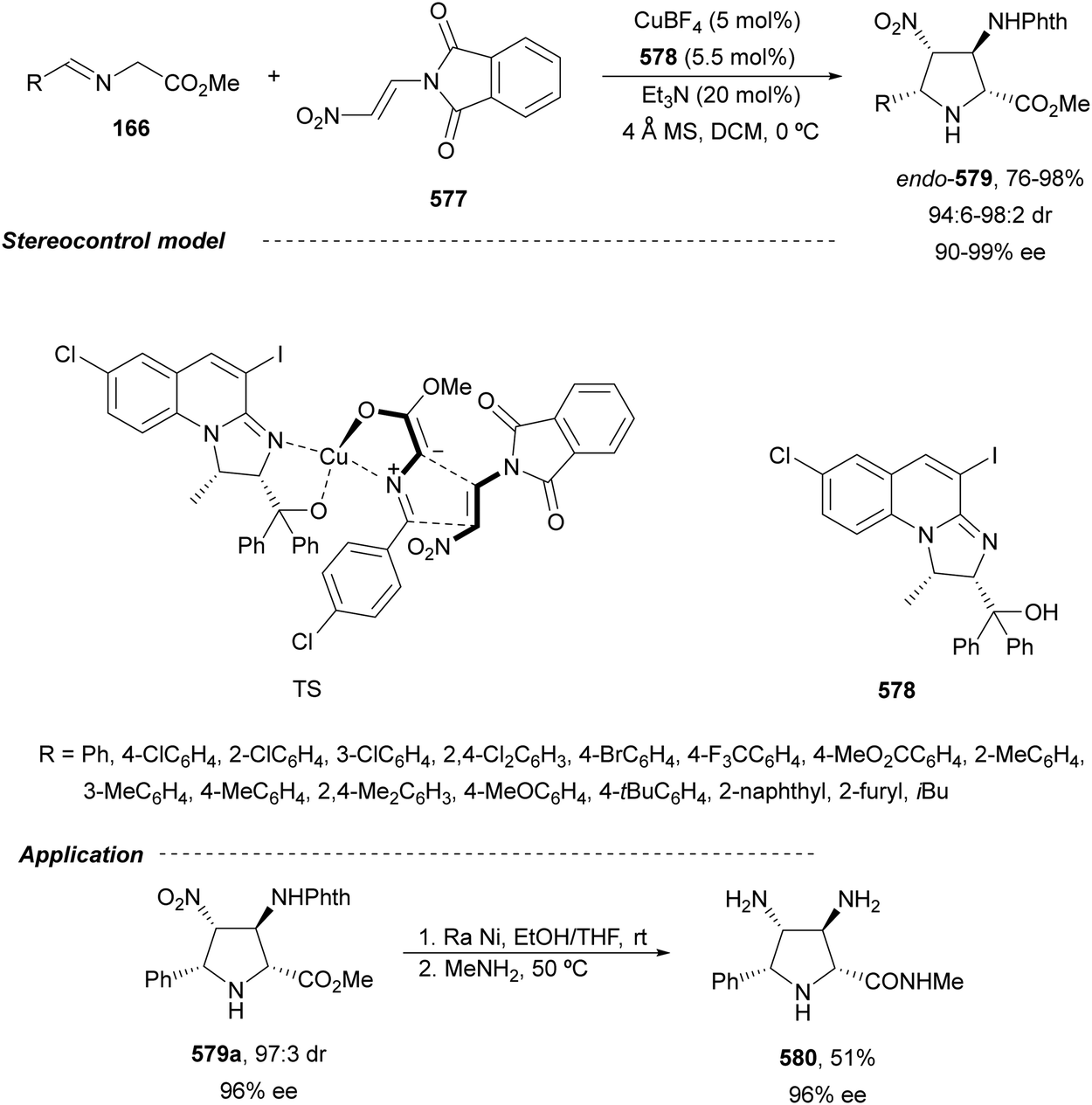 | ||
| Scheme 173 Asymmetric 1,3-DC of imino esters 166 with (E)-β-phthalimidonitroethene 577 under CuBF4/ligand 578 catalysis. | ||
For the enantioselective synthesis of chiral cis-3,4-diaminopyrrolidine derivatives 583, Nájera, Sansano and co-workers328 recently reported the 1,3-DC of azomethine ylides with (Z)-2-amido-1-nitroethenes 581a and 581b.329 Imino esters 166 reacted with (Z)-amidonitroethenes 581a and 581b using AgClO4 or Ag2CO3 and phosphoramidite (Sa,R,R)-180 as a catalyst to furnish pyrrolidines endo-582 (Scheme 174). In the case of cycloadduct endo-582b, the reduction with Zn and concentrated HCl under reflux afforded the cis-3,4-diamine derivative 583 in 90% yield and with the same enantiomeric excess.
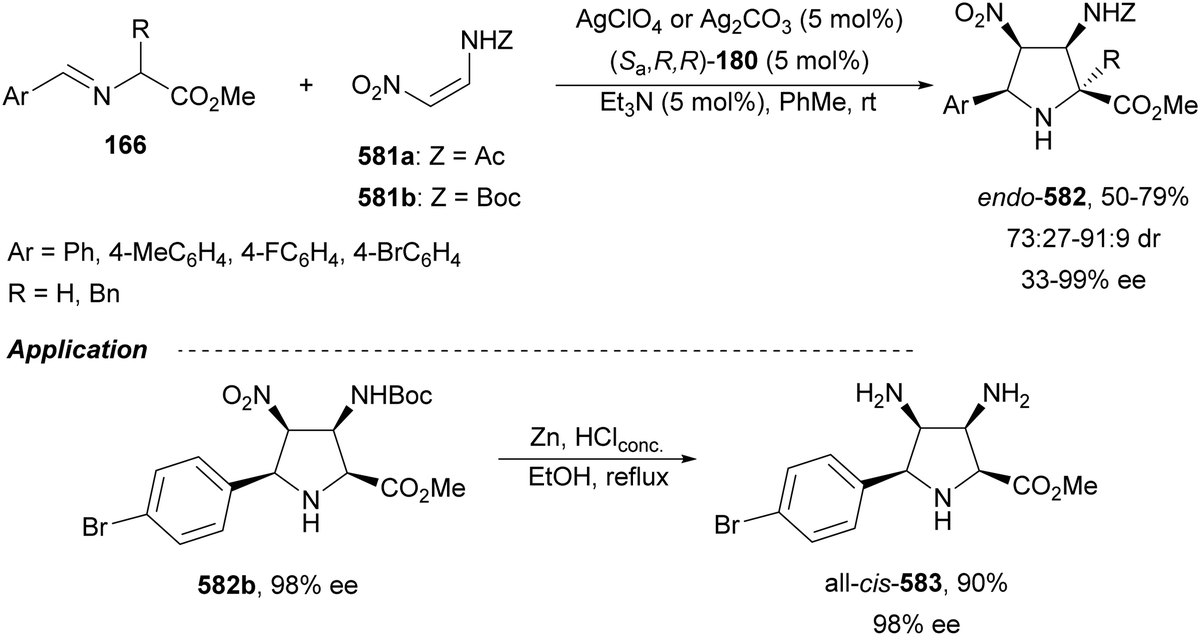 | ||
| Scheme 174 Asymmetric 1,3-DC of imino esters 166 with (Z)-amidonitroethenes 581 under Ag/phosphoramidite 180 catalysis. | ||
Desymmetrization of meso-1,2-diamines by acylation under thiourea catalysis and meso-diaminocyclopropane by Friede-Crafts alkylation of indoles under Cu/bis(oxazoline) catalysis were efficient strategies for the asymmetric synthesis of 1,2-diamines and 4,5-disubstituted imidazolidinones, respectively. Alternatively, KR of 1,2-diamines by acylation under thiourea catalysis has been employed. Electrophilic amination of N-aryl-1,2-diamines with azodicarboxylate under CPA catalysis allowed the KR of these compounds. DYKAT of allylic trichloroacetamidates with aromatic amines has been accomplished under Rh/chiral diene complexes to obtain 1,2-diamines. 1,3-DC of imino esters with β-nitroaminoethenes under Cu or Ag catalysis have been applied to the synthesis of trans- or cis-3,4-diaminoprolinates, respectively.
7. Conclusions
Particular conclusions are included at the end of each section in this review article. We report now general conclusions of this subject. With respect to asymmetric C–N bond-forming reactions detailed in Section 2, classical ring opening of aziridines was accomplished with trimethylsilyl azide using metal-catalyzed methods or Brønsted acids such as CPA, whereas desymmetrization of meso-aziridines with aromatic amines has been mainly accomplished with binol–metal complexes derived from Nb, Ti and Mg, and aliphatic amines were able to open aziridines under Ag/diphosphine catalysis. Other nitrogenated compounds such as hydroxylamines, tetrazoles and pyrazoles were also employed for desymmetrization of meso-aziridines by Mg complexes. Desymmetrization of azabenzonorbornadienes with amines has to be carried out under transition-metal catalysis to obtain anti-diamines, whereas using amides as nucleophiles syn-diamino derivatives were mainly formed.Intermolecular hydroamination of allyl amines has been achieved under Rh catalysis. On the other hand, asymmetric hydrocupration allowed inter and intramolecular hydroaminations. Intramolecular carboamination of allylic ureas and sulfamides has been carried out under Pd catalysis with concomitant cross-coupling with aryl or alkenyl halides at the terminal position. Hydroamination of enamines has been performed by hydrocupration with O-acyl hydroxylamines and by Cu or Ca catalyzed amination by azodicarboxylates, alternatively CPAs can also be used as a catalyst.
Diamination of olefins can be carried out by a two-electron redox pathway using Pd(0)/Pd(II) or Pd(II)/Pd(IV) catalysis under I(I)/I(III) or Se(II)/Se(IV), and by one-electron radical mechanism under Cu or Fe catalysis. 1,3-Dienes and enynes were diaminated with urea under Pd(0)/Pd(II) catalysis, whereas intramolecular diamination of tethered double bonds has been performed under Pd(II)/Pd(IV) catalysis as well as by chiral λ3-iodane reagents. Intermolecular syn-diamination of alkenes was achieved using Se(II) reagents. In the case of one-electron mechanism, intermolecular diamination of 1,3-dienes was accomplished with di-tert-butylaziridinone under Cu catalysis. Intramolecular radical diamination was also achieved under Cu catalysis. Recently, an enantioselective radical aminoazidation of styrenes has been carried out successfully under Fe(II) catalysis. Section 3 discussed C–C bond-forming reactions, such as classical asymmetric aza-Mannich and aza-Henry reactions, which have been widely used for the synthesis of 1,2-diamines.
For the direct aza-Mannich reaction, different nucleophiles such as imino esters, imino nitriles, azlactones, isocyano acetates and isothiocyanates were reacted with imines to obtain α,β-diamino acid derivatives. For imino esters, Cu and Ag catalysis gave mainly syn-α,β-diamino acids. Moreover, organocatalytic methods using asymmetric PTC also gave syn-α,β-diamino acid derivatives. For anti-α,β-diamino acids a biomimetic strategy using a chiral aldehyde as catalyst has been described. Imino nitriles needed activated aldimines and ketimines under Cu or Pd catalysis. Azlactones were specially useful for the synthesis of α,β-diamino acids bearing quaternary stereocenters. Isocyano acetates reacted with aldimines and ketimines under metal salt catalysis and a Cinchona-derived alkaloid as a chiral base as well as thioureas and squaramides to provide imidazolines that can be easily transformed into α,β-diamino acid derivatives. Isothiocyanates reacted with aldimines and activated ketimines under Mg or Sr catalysis but also with Cinchona-derived alkaloids, guanidines and thioureas as chiral organocatalysts. Other nucleophiles such as α-azido carbonyl compounds or amides, as well as α-amino acetaldehydes have been employed under organocatalysis. Recently, N-aryl glycines and hydrazones were reacted under Cu/bis(oxazoline) and visible light-induced photoredox catalysis to obtain 1,2-diamine derivatives.
Asymmetric aza-Henry reactions were considered in Section 3.2 either under metal complexes or mainly under organocatalysis. Recent examples used Ir(III), Cu(II) and Zn(II) complexes to obtain anti-β-nitroamine derivatives. As organocatalysts, chiral thioureas and squaramides derived from Cinchona alkaloids have been efficiently used for acyclic and cyclic aldimines and ketimines. PTC mainly with Cinchona-derived ammonium salts has been widely used in the aza-Henry reaction for aldimines, which can be generated in situ from α-amido sulfones. Ketimines can also be employed with simple nitroalkanes. Recently, chiral bases have been extensively used for the addition of nitroalkanes to aldimines and ketimines, specially mono and bis(amidine)-derived catalysts. This strategy has been employed for the synthesis of 1,2-diamine precursors of (−)-nutlin-3, azetidine (−)-441 and (+)-VNI therapeutics.
In Section 3.3, imine–imine coupling such as aza-pinacol promoted by chiral diboron compounds allowed the synthesis of non-substituted 1,2-diamines. For the coupling of imines with hydrazones, a CPA has been employed. In the second strategy, azaallyl anions acted as nucleophiles with imines and chiral guanidines, thioureas and squaramides were used as catalysts or PTC conditions to obtain syn-diamines. In Section 3.5, 1,3-DC of azomethine ylides or 5-vinyloxazolidinones with imines to obtain imidazolidines were described. CPAs as well as chiral Ag and Cu complexes gave by endo-selectivity cis-imidazolidines. In the case of [2+2] cycloaddition of allenamides under Rh(I)/Binap (53) catalysis, bis(methylene)-1,2-diamines were formed.
Section 4 considered C–H bond-forming reactions by CN asymmetric hydrogenation reactions to obtain saturated N-containing heterocyclic systems such as piperazines, dihydro-2,2′-bisquinolines, quinoxalines and tetrahydroquinolines. Asymmetric transfer hydrogenation with RuCl(TsDpen) has been employed for the reduction of thiadiazole-1,1-dioxides to 4-alkylidenthiadiazolines with cis and trans configurations. Asymmetric hydrogen autotransfer of diols under Ir(I) and (R)-TRIP dual catalysis with anilines allowed general synthesis of N-arylated 1,2-diamines.
Section 5 included C–H amination reactions such as intramolecular 1,5-radical amination under Co and Ru catalysis of sulfamoyl azides or N-benzoyloxyureas to obtain enantioenriched sulfamides or imidazolidinones, respectively. On the other hand, N-aryl-2-naphthylamines underwent an asymmetric atroposelective C–H amination with azodicarboxylates under CPA catalysis to give naphthalene-1,2-diamine derivatives.
Section 6 dealt with other catalytic methods based on desymmetrization reactions for kinetic resolution of 1,2-diamines by acylation under thiourea catalysis. Kinetic resolution of 1,2-diamines via an electrophilic reaction at the N-aryl group by an azodicarboxylate has been performed under CPA catalysis. DYKAT of racemic allyl trichloroacetamidates with anilines was accomplished under Rh/diene catalysis. Finally, [3+2] cycloadditions of imino esters with β-nitroaminoethanes under Cu or Ag catalysis formed trans or cis-3,4-diaminopyrrolidines, respectively.
The great importance of asymmetric 1,2-diamines is and will be related to applications of these chiral building blocks to the synthesis of chiral ligands, organocatalysts, biologically active compounds and pharmaceuticals.
Abbreviations
| Ac | Acetyl |
| acac | Acetylacetate |
| Ad | Adamantyl |
| AH | Asymmetric hydrogenation |
| AHT | Hydrogen atom transfer |
| Am | Amyl (2-methyl-2-butyl) |
| anh | Anhydrous |
| ATH | Asymmetric transfer hydrogenation |
| atm | Atmosphere(s) |
| BAM | Bis(amidine) |
| BArF | Tetrakis(3,5-bis(trifluoromethyl)phenyl)borate |
| Benzhydryl | Diphenylmethyl |
| Binap | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene |
| Binol | 1,1′-Bi(2-naphthol) |
| Binolam | 3,3′-Bis(diethylaminomethyl)-1,1′-bi-2-naphthol |
| Bn | Benzyl |
| (BNeop)2 | Bis(neopentyl glycolato)diboron |
| Boc | tert-Butoxycarbonyl |
| Bpy | 2,2′-Bipirydine |
| Bz | Benzoyl |
| C2-ferriphos-tolyl | 2,2′-Bis[1-(N,N-dimethylamino)ethyl]-1,1′-bis(diphenylphosphino)ferrocene |
| ca. | Circa |
| cat | Catalyst |
| Cbz | Benzyloxycarbonyl |
| CD | Circular dichroism |
| cod | 1,5-Cyclooctadiene |
| coe | Cyclooctene |
| conc | Concentrated |
| CPA | Chiral phosphoric acid |
| cPr | Cyclopropyl |
| CuCatMix* | Mixture of Ph3P, (R)-DTBM-Segphos and Cu(OAc)2 |
| Cy | Cyclohexyl |
| CYP51 | Catalyst for the demethylation of lanosterol |
| 4CzIPN | 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyanobenzene |
| D-B | Diboron |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DC | Dipolar cycloaddition |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DCP 083 | (S,E)-6-Chloro-4-(2-cyclopropylvinyl)-4-(trifluoromethyl)-3,4-dihydroquinazolin-2(1H)-one |
| DFT | Density functional theory |
| DIBAL-H | Diisobutylaluminium hydride |
| Difluorophos | [4-(5-Diphenylphosphanyl-2,2-difluoro-1,3-benzodioxol-4-yl)-2,2-difluoro-1,3-benzodioxol-5-yl]-diphenylphosphine |
| DIPEA | Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| DMB | 2,4-Dimethoxybenzyl |
| DMBQ | 2,6-Dimethoxybenzoquinone |
| DME | Dimethoxyethane |
| DMF | Dimethylformamide |
| DMMGarphos | 2,2′-Bis[bis(4-methoxy-3,5-di-t-butylphenyl)phosphino]-4,4′,6,6′-tetramethoxybiphenyl |
| DMMS | Dimethoxymethylsilane |
| DMSO | Dimethyl sulfoxide |
| DPEN | 1,2-Diphenyethane-1,2-diamine |
| DPP | Diphenylphosphinoyl |
| dr | Diastereomeric ratio |
| DTBM-Segphos | 5,5′-Bis[bis(3,5-di-tert-butyl-4-methoxyphenyl)phosphino]-4,4′-bi-1,3-benzodioxole |
| DYKAT | Dynamic kinetic asymmetric transformation |
| ee | Enantiomeric excess |
| equiv. | Equivalents |
| Fmoc | Fluorenylmethoxycarbonyl |
| Fesulphos | 2-(tert-Butylthio)-1-(diphenylphosphino)ferrocene |
| HA | Hydroamination |
| HAA | H-Atom abstraction |
| Hex | n-Hexyl |
| HFIP | Hexafluoroisopropanol |
| HIV | Human immunodeficiency virus |
| HMDS | Bis(trimethylsilyl)amine |
| Josiphos | [see, PPF-P(tBu)2] |
| KR | Kinetic resolution |
| L | Ligand |
| LED | Light-emitting diode |
| LP99 | N-[(2R,3S)-2-(4-Chlorophenyl)-1-(1,2-dihydro-1,4-dimethyl-2-oxo-7-quinolinyl)-6-oxo-3-piperidinyl]-2-methyl-1-propanesulfonamide |
| M | Metal |
| MAM | Mono(amidine) |
| MC-4 | Melanocortin-4 |
| MCPBA | meta-Chloroperbenzoic acid |
| MeO-Biphen | 2,2′-Bis[di-(2-furyl)phosphino]-6,6′-dimethoxy-1,1′-biphenyl |
| MS | Molecular sieves |
| MTBE | Methyl tert-butyl ether |
| N,Nu | Nucleophile |
| NFSI | N-Fluorobenzenesulfonimide |
| NHC | N-Heterocyclic carbene |
| NIP | N-Iodopyrrolidone |
| NIS | N-Iodosuccinimide |
| NLE | Non-linear effect |
| NMDA | N-Methyl-D-aspartic acid |
| NMR | Nuclear magnetic resonance |
| Ns | 4-Nitrophenylsulfonyl |
| OPhen | 1,2,3,4,7,8,9,10,-1,10-octahydrophenantroline |
| PBAM | H,4PyrrolidineQuin-BAM |
| PG | Protecting group |
| Phbox | 2,2′-Isopropylidenebis[(4S)-4-phenyl-2-oxazoline] |
| Ph-Bpe | 2,5-(Diphenylphospholano)ethane |
| Ph-Dbfox | 4,6-Dibenzofurandiyl-2,2′-bis(4-phenyloxazoline) |
| Ph-Phosferrox | 2-(Diphenylphosphino)ferrocenyl-4-isopropyloxazoline |
| Phth | Phthalimido |
| Piv | Pivaloyl |
| PKC | Protein kinase |
| CPMB | para-Methoxybenzyl |
| PPFOMe | 1-[(SP)-2-(Diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine |
| PPF-P(tBu)2 | (Josiphos): (R)-1-[(SP)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine |
| ppy | 2-(2-Pyridyl)phenyl |
| Pro | Proline |
| psi | Pound-force per square inch |
| PTC | Phase transfer catalysis |
| Py | Pyridyl |
| PyBidine | 2,6-Bis[(2R,4S,5S)-1-benzyl-4,5-diphenylimidazolidin-2-yl]pyridine |
| PYR | 6-Chloro-2,5-diphenylpyrimidin-4-yl |
| Ra | Ni RANEY® nickel |
| rr | Regioisomeric ratio |
| RS | Radical substitution |
| rt | Room temperature |
| RuPhox | Ruthenocenyl phosphino-oxazoline complex |
| Segphos | 5,5′-Bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole |
| SES | 2-(Trimethylsilyl)ethanesulfonyl |
| SET | Single electron transfer |
| Siphos-PE | 10,11,12,13-Tetrahydrodiindeno[7,1-de:1′,7′-fg][1,3,2]dioxaphosphocin-5-bis[(R)-1-phenylethyl]amine, N-Di[(R)-1-phenylethyl]-[(S)-1,1′-spirobiindane-7,7′-diyl]-phosphoramidite |
| SPINOL | 12-Hydroxy-1,10-di(phenanthren-9-yl)-4,5,6,7-tetrahydrodiindeno[7,1-de:1′,7′-fg][1,3,2]dioxaphosphocine 12-oxide |
| TBAI | Tetrabutylammonium iodide |
| TBDPS | tert-Butyldiphenylsilyl |
| TBME | tert-Butyl methyl ether |
| TBS | tert-Butyldimethylsilyl |
| Tc | Thiophene-2-carboxylate |
| TEMPO | 2,2,6,6-(Tetramethylpiperidin-1-yl)oxyl |
| TESH | Triethylsilane |
| Tf | Trifluoromethylsulfonyl |
| TFA | Trifluoroacetic acid, trifluoroacetate |
| THF | Tetrahydrofuran |
| THP | Tetrahydropyrane |
| TMB | 2,4,6-Trimethylbenzyl |
| TMS | Trimethylsilyl |
| tolyl | Methylphenyl |
| tosyl, Ts | 4-Metylphenylsulfonyl |
| TPS | tert-Butyldiphenylsilyl |
| TPS-he-Pybox | 2,6-Bis(tert-butyldiphenylsilyloxyethyloxazolinyl)pyridine |
| TRIP | 3,3′-Bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate |
| Tro | 2,2,2-Trichloroethoxycarbonyl |
| TS | Transition state |
| US | Ultrasounds |
| VNI | Potent inhibitor of CYP51 |
| W | Watts |
| Xing-Phos | 2-[(R)-[[(R)-(1,1-Dimethylethyl)sulfinyl]amino](phenyl)methyl]-6-(diphenylphosphino)-N,N-diisopropylbenzamide |
| Xylene | Dimethylbenzene |
| xyl-Segphos | 5,5′-Bis[di(3,5-xylyl)phosphino]-4,4′-bi-1,3-benzodioxol |
| xylyl | Dimethylphenyl |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Spanish Ministerio de Ciencia, Innovacion y Universidades (project RED2018-102387-T), the Spanish Ministerio de Economía, Industria y Competitividad, Agencia Estatal de Investigacion (AEI) (MCIN/AEI/10.13039/501100011033) and Fondo Europeo de Desarrollo Regional (FEDER, EU) (projects CTQ2017-82935-P, PID2019-107268GB-I00 and PID2021-123079OB-I00), Generalitat Valenciana (IDIFEDER/2021/013, GVA-COVID19/2021/079 and CIDEGENT/2020/058), Medalchemy S. L. (Medalchemy-18T) and the University of Alicante (VIGROB-068, UAUSTI21-05).References
- D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS PubMed.
- S. R. S. Kotti, C. Timmons and G. Li, Chem. Biol. Drug Des., 2006, 67, 101–114 CrossRef CAS PubMed.
- J.-C. Kizirian, Chem. Rev., 2008, 108, 140–205 CrossRef CAS PubMed.
- A. Viso, R. Fernández de la Pradilla, M. Tortosa, A. García and A. Florez, Chem. Rev., 2011, 111, PR1–PR42 CrossRef PubMed.
- O. O. Grygorenko, D. S. Radchenko, D. M. Volochnyuk, A. A. Tolmachev and I. V. Komarov, Chem. Rev., 2011, 111, 5506–5568 CrossRef CAS PubMed.
- S. De Jong, D. G. Nosal and D. J. Wardrop, Tetrahedron, 2012, 68, 4067–4105 CrossRef CAS PubMed.
- F. Cardona and A. Goti, Nat. Chem., 2009, 1, 269–275 CrossRef CAS PubMed.
- R. M. de Figueiredo, Angew. Chem., Int. Ed., 2009, 48, 1190–1193 CrossRef CAS PubMed.
- K. Muñiz and C. Martínez, J. Org. Chem., 2013, 78, 2168–2174 CrossRef PubMed.
- Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Acc. Chem. Res., 2014, 47, 3665–3678 CrossRef CAS PubMed.
- X. Zhang and S. L. You, Chem, 2017, 3, 919–921 CAS.
- J. B. Parry, N. Fu and S. Lin, Synlett, 2018, 257–265 CAS.
- Z. Wu, M. Hu, J. Li, W. Wu and H. Jiang, Org. Biomol. Chem., 2021, 19, 3036–3054 RSC.
- Z.-L. Tao and S. E. Denmark, Synthesis, 2021, 3951–3962 CAS.
- C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Tetrahedron, 2022, 106–107, 132629 CrossRef.
- Z. Li, M. Fernández and E. N. Jacobsen, Org. Lett., 1999, 1, 1611–1613 CrossRef CAS PubMed.
- Y. Fukuta, T. Mita, N. Fukuda, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 6312–6313 CrossRef CAS PubMed.
- E. B. Rowland, G. B. Rowland, E. Ribera-Otero and J. C. Antilla, J. Am. Chem. Soc., 2007, 129, 12084–12085 CrossRef CAS PubMed.
- B. Wu, J. R. Parquette and T. V. RajanBabu, Science, 2009, 326, 1662 CrossRef CAS PubMed.
- B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Angew. Chem., Int. Ed., 2009, 48, 1126–1129 CrossRef CAS PubMed.
- B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Chem. Sci., 2014, 5, 1102–1117 RSC.
- S. Nakamura, M. Hayashi, Y. Kamada, R. Sasaki, Y. Hiramatsu, N. Shibata and T. Toru, Tetrahedron Lett., 2010, 51, 3820–3823 CrossRef CAS.
- K. Arai, S. Lucarini, M. M. Salter, K. Ohta, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2007, 129, 8103–8111 CrossRef CAS PubMed.
- R. Yu, Y. Yamashita and S. Kobayashi, Adv. Synth. Catal., 2009, 351, 147–152 CrossRef CAS.
- K. Seki, R. Yu, Y. Yamazaki, Y. Yamashita and S. Kobayashi, Chem. Commun., 2009, 5722–5724 RSC.
- R. Akiyama and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 3412–3413 CrossRef CAS PubMed.
- S. Peruncheralathan, H. Teller and C. Schneider, Angew. Chem., Int. Ed., 2009, 48, 4849–4852 CrossRef CAS PubMed.
- S. Peruncheralathan, S. Aurich, H. Teller and C. Schneider, Org. Biomol. Chem., 2013, 11, 2787–2803 RSC.
- J. Li, Y. Liao, Y. Zhang, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 6672–6674 RSC.
- Z. Chai, P.-J. Yang, H. Zhang, S. Wang and G. Yung, Angew. Chem., Int. Ed., 2017, 56, 650–654 CrossRef CAS PubMed.
- D. Li, D. Yang, L. Wang, X. Liu, X. Jiang and R. Wang, Chem. – Eur. J., 2016, 22, 17141–17144 CrossRef PubMed.
- C. Nájera, J. M. Sansano and J. M. Saá, Eur. J. Org. Chem., 2009, 2385–2400 CrossRef.
- D. Li, K. Wang, L. Wang, Y. Wang, P. Wang, X. Liu, D. Yang and R. Wang, Org. Lett., 2017, 19, 3211–3214 CrossRef CAS PubMed.
- X. Li, J. Guo, L. Lin, H. Hu, F. Chang, X. Liu and X. Feng, Adv. Synth. Catal., 2017, 359, 3532–3537 CrossRef CAS.
- M. Lautens, K. Fagnou and V. Zunic, Org. Lett., 2002, 4, 3465–3468 CrossRef CAS PubMed.
- Y.-H. Cho, V. Zunic, H. Senboku, M. Ofsen and M. Lautens, J. Am. Chem. Soc., 2006, 128, 6837–6846 CrossRef CAS PubMed.
- Y.-H. Cho, A. Fayol and M. Lautens, Tetrahedron: Asymmetry, 2006, 17, 416–427 CrossRef CAS.
- G. F. Castello, R. James, J. S. Shaw, A. M. Slater and N. C. J. Stutchbury, J. Med. Chem., 1991, 34, 181–189 CrossRef PubMed.
- L. Xie, D. Q. Yang, S. Q. Zhao, H. Wang, L. H. Liang and R. S. Luo, Chin. Chem. Lett., 2007, 18, 127–129 CrossRef CAS.
- Y. Long, D. Yang, H. Zeng, L. Xie, L. Wu, H. Mo and X. Zuo, Chin. J. Chem., 2010, 28, 235–242 CrossRef CAS.
- D. Yang, Y. Long, H. Wang and Z. Zhang, Org. Lett., 2008, 10, 4723–4726 CrossRef CAS PubMed.
- Y. Long, D. Yang, Z. Zhang, Y. Wu, H. Zeng and Y. Chen, J. Org. Chem., 2010, 75, 7291–7299 CrossRef CAS PubMed.
- D. Yang, Y. Long, Y. Wu, X. Zuo, Q. Tu, S. Fang, L. Jiang, S. Wang and C. Li, Organometallics, 2010, 29, 5936–5940 CrossRef CAS.
- R. Luo, J. Liao, L. Xie, W. Tang and A. S. C. Chan, Chem. Commun., 2013, 49, 9959–9961 RSC.
- R. Luo, G. Cheng, Y. Wei, R. Deng, M. Huang and J. Liao, Organometallics, 2018, 37, 1652–1655 CrossRef CAS.
- C. Zeng, F. Yang, J. Chen, J. Wang and B. Fan, Org. Biomol. Chem., 2015, 33, 8425–8428 RSC.
- Z. Lu, J. Wang, B. Han, S. Li, Y. Zhou and B. Fan, Adv. Synth. Catal., 2015, 357, 3121–3125 CrossRef CAS.
- G. Shen, R. Khan, H. Lv, Y. Yang, X. Zhang, Y. Zhan, Y. Zhou and B. Fan, Org. Chem. Front., 2019, 6, 1423–1427 RSC.
- M. J. MacDonald, C. R. Hesp, D. J. Schipper, M. Pesant and A. M. Beauchemin, Chem. – Eur. J., 2013, 19, 2597–2601 CrossRef CAS PubMed.
- M. J. MacDonald, D. J. Schipper, P. J. Ng, J. Moran and A. M. Beauchemin, J. Am. Chem. Soc., 2011, 133, 20100–20103 CrossRef CAS PubMed.
- N. Guimond, M. J. MacDonald, V. Lemieux and A. M. Beauchemin, J. Am. Chem. Soc., 2012, 134, 16571–16577 CrossRef CAS PubMed.
- I. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS PubMed.
- E. P. Vanable, J. L. Kennemur, L. A. Joyce, R. T. Ruck, D. M. Schultz and K. L. Hull, J. Am. Chem. Soc., 2019, 141, 739–742 CrossRef CAS PubMed.
- A. R. Ickes, S. C. Ensign, A. K. Gupta and K. L. Hull, J. Am. Chem. Soc., 2014, 136, 11256–11259 CrossRef CAS PubMed.
- S. Ichikawa, X.-J. Dai and S. L. Buchwald, Org. Lett., 2019, 21, 4370–4373 CrossRef CAS PubMed.
- J. S. Bandar, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 14812–14818 CrossRef CAS PubMed.
- A. A. Thomas, K. Speck, I. Kevlishvili, Z. Lu, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 13976–13984 CrossRef CAS PubMed.
- B. A. Hopkins and J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 9886–9890 CrossRef CAS PubMed.
- Z. J. Garlets, K. R. Parenti and J. P. Wolfe, Chem. – Eur. J., 2016, 22, 5919–5922 CrossRef CAS PubMed.
- D. M. Schultz and J. P. Wolfe, Synthesis, 2012, 351–361 CAS.
- Z. J. Garlets, D. R. White and J. P. Wolfe, Asian J. Org. Chem., 2017, 6, 636–652 CrossRef CAS PubMed.
- J. A. Fritz, J. S. Nakhla and J. P. Wolfe, Org. Lett., 2006, 8, 2531–2534 CrossRef CAS PubMed.
- H. Wang, J. C. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 8428–8431 CrossRef CAS PubMed.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- L. Yu and P. Somfai, Angew. Chem., Int. Ed., 2019, 58, 8551–8555 CrossRef CAS PubMed.
- M. Mohiti, Y. Lu, H. He, S.-F. Ni and P. Somfai, Chem. – Eur. J., 2024, 30, e202303078 CrossRef CAS PubMed.
- D. Nozawa, T. Okubo, T. Ishii, S. Okuyama and A. Nakazato, Chem. Pharm. Bull., 2007, 55, 1044–1050 CrossRef CAS PubMed.
- R. Matsubara and S. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 7993–7995 CrossRef CAS PubMed.
- L. Chang, Y. Kuang, B. Qin, X. Zhou, X. Liu, L. Lin and X. Feng, Org. Lett., 2010, 12, 2214–2217 CrossRef CAS PubMed.
- F. Drouet, C. Lalli, H. Liu, G. Masson and Z. Zhu, Org. Lett., 2011, 13, 94–97 CrossRef CAS PubMed.
- C. Lalli, A. Dumoulin, C. Lebée, F. Drouet, V. Guérineau, D. Touboul, V. Gandon, J. Zhu and G. Masson, Chem. – Eur. J., 2015, 21, 1704–1712 CrossRef CAS PubMed.
- D. Bouchet, T. Varlet and G. Masson, Acc. Chem. Res., 2022, 55, 3265–3283 CrossRef CAS PubMed.
- A. Dumoulin, C. Lalli, P. Retailleau and G. Masson, Chem. Commun., 2015, 51, 5383–5386 RSC.
- A. Dumoulin, G. Bernadat and G. Masson, J. Org. Chem., 2017, 82, 1775–1789 CrossRef CAS PubMed.
- G. Levitre, C. Audubert, A. Dumoulin, N. Goual, P. Petailleau, X. Moreau and G. Masson, ChemCatChem, 2019, 11, 5723–5727 CrossRef CAS.
- J. Lyu, A. Claraz, M. R. Vitale, C. Allain and G. Masson, J. Org. Chem., 2020, 85, 12843–12855 CrossRef CAS PubMed.
- M.-S. Wu, T. Fan, S.-S. Chen, Z.-Y. Han and L.-Z. Gong, Org. Lett., 2018, 20, 2485–2489 CrossRef CAS PubMed.
- Q. Li, X. Fang, R. Pan, H. Yao and A. Lin, J. Am. Chem. Soc., 2022, 144, 11364–11376 CrossRef CAS PubMed.
- E. L. Ingalls, P. A. Sibbald, W. Kaminsky and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 8854–8856 CrossRef CAS PubMed.
- J. Streuff, C. H. Hövelmann, M. Nieger and K. Muñiz, J. Am. Chem. Soc., 2005, 127, 14586–14587 CrossRef CAS PubMed.
- X. Liu, C. Hou, Y. Peng, P. Chen and G. Liu, Org. Lett., 2020, 22, 9371–9375 CrossRef CAS PubMed.
- C. Röben, J. A. Souto, Y. González, A. Lishchynskyi and K. Muñiz, Angew. Chem., Int. Ed., 2011, 50, 9478–9482 CrossRef PubMed.
- P. Mizar, A. Laverny, M. El-Sherbini, U. Farid, M. Brown, F. Malmedy and T. Wirth, Chem. – Eur. J., 2014, 20, 9910–9913 CrossRef CAS PubMed.
- K. Muñiz, L. Barreiro, R. M. Romero and C. Martínez, J. Am. Chem. Soc., 2017, 139, 4354–4357 CrossRef PubMed.
- Z. Tao, B. B. Gilbert and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 19161–19170 CrossRef CAS PubMed.
- H. Du, B. Zhao, W. Yuan and Y. Shi, Org. Lett., 2008, 10, 4231–4234 CrossRef CAS PubMed.
- H. Du, W. Yuan, B. Zhao and Y. Shi, J. Am. Chem. Soc., 2007, 129, 11688–11689 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, J. Org. Chem., 2009, 74, 8392–8395 CrossRef CAS PubMed.
- B. W. Turnpenny and S. R. Chemler, Chem. Sci., 2014, 5, 1786–1793 RSC.
- S. Fu, H. Yang, G. Li, Y. Deng, H. Jiang and W. Zheng, Org. Lett., 2015, 17, 1018–1021 CrossRef CAS PubMed.
- F.-L. Wang, X.-Y. Dong, J.-S. Lin, Y. Zeng, G.-Y. Jiao, Q.-S. Gu, X.-Q. Guo, C.-L. Ma and X.-Y. Liu, Chem, 2017, 3, 979–990 CAS.
- D. Lv, Q. Sun, H. Zhou, L. Ge, Y. Qu, T. Li, X. Ma, Y. Li and H. Bao, Angew. Chem., Int. Ed., 2021, 60, 12455–12460 CrossRef CAS PubMed.
- B. M. Trost and D. R. Fandrick, J. Am. Chem. Soc., 2003, 125, 11836–11837 CrossRef CAS PubMed.
- B. M. Trost and D. R. Fandrick, Org. Lett., 2005, 7, 823–826 CrossRef CAS PubMed.
- A. J. Freyer, A. D. Patil, L. Killmer, N. Troupe, M. Mentzer, B. Carte, L. Faucette and R. K. Johnson, J. Nat. Prod., 1997, 60, 986–990 CrossRef CAS PubMed.
- M. Ishibashi, Y. Ohizumi, T. Sasaki, H. Nakamura, Y. Hirata and J. Kobayashi, J. Org. Chem., 1987, 52, 450–453 CrossRef CAS.
- J. Kobayashi, K. Naitoh, Y. Doi, K. Deki and M. Ishibashi, J. Org. Chem., 1995, 60, 6941–6945 CrossRef CAS.
- C. Dong and H. Alper, Tetrahedron: Asymmetry, 2004, 15, 1537–1540 CrossRef CAS.
- B. M. Trost, D. R. Fandrick, T. Brodmann and D. T. Stiles, Angew. Chem., Int. Ed., 2007, 46, 6123–6125 CrossRef CAS PubMed.
- J. W. Lampe, P. F. Hughes, C. K. Biggers, S. H. Smith and H. Hu, J. Org. Chem., 1996, 61, 4572–4581 CrossRef CAS PubMed.
- T. J. Struble, H. M. Lankswert, M. Pink and J. N. Johnston, ACS Catal., 2018, 8, 11926–11931 CrossRef CAS PubMed.
- D. Zhung and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 15803–15807 CrossRef PubMed.
- R. Gómez Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940–1948 RSC.
- B. Karimi, D. Enders and E. Jafari, Synthesis, 2013, 2769–2812 CrossRef CAS.
- L. Bernardi, A. S. Gothelf, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2003, 68, 2583–2591 CrossRef CAS PubMed.
- M. M. Salter, J. Kobayashi, Y. Shimizu and S. Kobayashi, Org. Lett., 2006, 8, 3533–3536 CrossRef CAS PubMed.
- X.-X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X.-L. Hou and Y.-D. Wu, J. Am. Chem. Soc., 2008, 130, 14362–14363 CrossRef CAS PubMed.
- J. Hernández-Toribio, R. Gómez Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2008, 130, 16150–16151 CrossRef PubMed.
- J. Hernández-Toribio, R. Gómez Arrayás and J. C. Carretero, Chem. – Eur. J., 2010, 16, 1153–1157 CrossRef PubMed.
- G. Liang, M. C. Tong, H. Tao and C. J. Wang, Adv. Synth. Catal., 2010, 352, 1851–1855 CrossRef CAS.
- D. Shang, Y. Liu, X. Zhou, X. Liu and X. Feng, Chem. – Eur. J., 2009, 15, 3678–3681 CrossRef CAS PubMed.
- T. Arai, A. Mishiro, E. Matsumura, A. Awata and M. Shirasugi, Chem. – Eur. J., 2012, 18, 11219–11222 CrossRef CAS PubMed.
- Y. Yamashita, S. Yoshimoto, K. Masuda and S. Kobayashi, Asian J. Org. Chem., 2012, 1, 327–330 CrossRef CAS.
- E. Hernando, R. Gómez Arrayás and J. C. Carretero, Chem. Commun., 2012, 48, 9622–9624 RSC.
- R. D. Momo, F. Fini, L. Bernardi and A. Ricci, Adv. Synth. Catal., 2009, 351, 2283–2287 CrossRef CAS.
- C. Zhang, J. Yang, W. Zhou, Q. Tan, Z. Yang, L. He and M. Zhang, Org. Lett., 2019, 21, 8620–8624 CrossRef CAS PubMed.
- Y. Xiong, Z. Du, H. Zhen, Z. Yang, Q. Tan, C. Zhang, L. Zhu, Y. Lan and M. Zhang, J. Am. Chem. Soc., 2019, 141, 961–971 CrossRef CAS PubMed.
- J.-Y. Zhu, W.-L. Yang, Y.-Z. Liu, S.-J. Shang and W.-P. Deng, Org. Chem. Front., 2018, 5, 70–73 RSC.
- G. S. Sing and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef PubMed.
- M. M. Santos, Tetrahedron, 2014, 70, 9735–9757 CrossRef CAS.
- Q. Shao, L. Wu, J. Chen, I. D. Gridnev, G. Yang, F. Xie and W. Zhang, Adv. Synth. Catal., 2018, 360, 4625–4634 CrossRef CAS.
- Q.-A. Chen, W. Zeng, D.-W. Wang and Y.-G. Zhou, Synlett, 2009, 2236–2241 CAS.
- K. Imae, K. Shimizu, K. Ogata and S. Fukuzawa, J. Org. Chem., 2011, 76, 3604–3608 CrossRef CAS PubMed.
- A. Cayuelas, L. Serrano, C. Nájera and J. M. Sansano, Tetrahedron: Asymmetry, 2014, 25, 1647–1653 CrossRef CAS.
- J. Jiang, H.-D. Xu, J.-B. Xi, B.-Y. Ren, F.-P. Lv, X. Guo, L.-Q. Jiang, Z.-Y. Zhang and W.-H. Hu, J. Am. Chem. Soc., 2011, 133, 8428–8431 CrossRef CAS PubMed.
- L. Jiang, D. Zhang, Z. Wang and W. Hu, Synthesis, 2013, 452–458 CAS.
- J. Jiang, X. Ma, S. Liu, Y. Qian, F. Lv, L. Qiu, X. Wu and W. Hu, Chem. Commun., 2013, 49, 4238–4240 RSC.
- T. Ooi, M. Kameda, J. Fujii and K. Maruoka, Org. Lett., 2004, 6, 2397–2399 CrossRef CAS PubMed.
- A. Okada, T. Shibuguchi, T. Ohshima, H. Masu, K. Yamaguchi and M. Shibasaki, Angew. Chem., Int. Ed., 2005, 44, 4564–4567 CrossRef CAS PubMed.
- T. Shibuguchi, H. Mihara, A. Kuramochi, T. Ohshima and M. Shibasaki, Chem. – Asian J., 2007, 2, 794–801 CrossRef CAS PubMed.
- For a review, see: J. Novacek and M. Waser, Eur. J. Org. Chem., 2013, 637–648 CrossRef CAS.
- Z. Tao, A. Adele, X. Wu and L. Gong, Chin. J. Chem., 2014, 12, 969–973 CrossRef.
- T. Kano, R. Kobayashi and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 8471–8474 CrossRef CAS PubMed.
- R. D. Momo, F. Fini, L. Bernardi and A. Ricci, Adv. Synth. Catal., 2009, 351, 2283–2287 CrossRef CAS.
- S. Kobayashi, R. Yazaki, K. Seki and Y. Yamashita, Angew. Chem., Int. Ed., 2008, 47, 5613–5615 CrossRef CAS PubMed.
- H. Zhang, S. Syed and C. F. Barbas III, Org. Lett., 2010, 12, 708–711 CrossRef CAS PubMed.
- J. S. Bandar and T. H. Lambert, J. Am. Chem. Soc., 2013, 135, 11799–11802 CrossRef CAS PubMed.
- L. Wu, G. Li, M. He, Y. Wang, G. Zhao and Z. Tang, Can. J. Chem., 2016, 94, 769–772 CrossRef CAS.
- J. Chen, X. Gong, J. Li, Y. Li, F. Ma, C. Hou, G. Zhao, W. Yuan and B. Zhao, Science, 2018, 360, 1438–1442 CrossRef CAS PubMed.
- X. Cui, Q. Li, L. Yao, Y. Ma, C. Hou, G. Zhao, W. Yuan and L. Zhao, J. Org. Chem., 2021, 86, 6592–6599 CrossRef CAS PubMed.
- Y. Yamashita, M. Matsumoto, Y.-J. Chen and S. Kobayashi, Tetrahedron, 2012, 68, 7538–7563 CrossRef.
- S. Lin, Y. Kawato, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2015, 54, 5183–5186 CrossRef CAS PubMed.
- M. Kondo, T. Nishi, T. Hatanaka, Y. Funahashi and S. Nakamura, Angew. Chem., Int. Ed., 2015, 54, 8198–8202 CrossRef CAS PubMed.
- P. P. de Castro, A. G. Carpanez and G. W. Amarante, Chem. – Eur. J., 2016, 22, 10294–10318 CrossRef CAS PubMed.
- D. Uraguchi, Y. Ueki and T. Ooi, J. Am. Chem. Soc., 2008, 130, 14088–14089 CrossRef CAS PubMed.
- D. Uraguchi, K. Kashimoto and T. Ooi, Chem. Commun., 2010, 46, 300–302 RSC.
- X. Liu, L. Deng, X. Jiang, W. Fan, C. Liu and R. Wang, Org. Lett., 2010, 12, 876–879 CrossRef CAS PubMed.
- W.-Q. Zhang, L.-F. Cheng, J. Yu and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 4085–4088 CrossRef CAS PubMed.
- E. P. Ávila, R. M. S. Justo, V. P. Gonçalves, A. A. Pereira, R. Diniz and G. W. Amarante, J. Org. Chem., 2015, 80, 590–594 CrossRef PubMed.
- M. Žabka, A. Malastová and R. Šebesta, RSC Adv., 2015, 5, 12890–12893 RSC.
- A. D. Melhado, G. W. Amarante, Z. J. Wang, M. Luparia and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 3517–3527 CrossRef CAS PubMed.
- S.-H. Shi, F.-P. Huang, P. Zhu, Z.-W. Dong and X.-P. Hui, Org. Lett., 2012, 14, 2010–2013 CrossRef CAS PubMed.
- Z. Li, J. Peng, C. He, J. Xu and H. Ren, J. Org. Chem., 2020, 85, 3894–3901 CrossRef CAS PubMed.
- X.-T. Zhou, Y.-R. Lin and L.-X. Dai, Tetrahedron: Asymmetry, 1999, 10, 855–862 CrossRef CAS.
- L.-X. Dai, Y.-R. Lin, X.-L. Hou and Y.-Z. Zhou, Pure Appl. Chem., 1999, 71, 1033–1040 CrossRef CAS.
- X.-T. Zhou, Y.-R. Lin, L.-X. Dai, J. Sun, L.-J. Xia and M.-H. Tang, J. Org. Chem., 1999, 64, 1331–1334 CrossRef CAS.
- J. Aydin, A. Rydén and K. J. Szabó, Tetrahedron: Asymmetry, 2008, 19, 1867–1870 CrossRef CAS.
- M.-X. Zhao, H.-L. Bi, R.-H. Jiang, X.-W. Xu and M. Shi, Org. Lett., 2014, 16, 4566–4569 CrossRef CAS PubMed.
- P.-L. Shao, J.-Y. Liao, Y. A. Ho and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 5435–5439 CrossRef CAS PubMed.
- I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2014, 53, 3462–3465 CrossRef PubMed.
- R. de la Campa, A. D. Gammack Yamagata, I. Ortín, A. Franchino, A. L. Thompson, B. Odell and D. J. Dixon, Chem. Commun., 2016, 52, 10632–10635 RSC.
- M. Hayashi, M. Iwanaga, N. Shiomi, D. Nakane, H. Masuda and S. Nakamura, Angew. Chem., Int. Ed., 2014, 53, 8411–8415 CrossRef CAS PubMed.
- S. Nakamura, R. Yamaji and M. Iwanaga, Chem. Commun., 2016, 52, 7462–7465 RSC.
- M.-X. Zhao, Z.-W. Dong, G.-Y. Zhu, X.-L. Zhao and M. Shi, Org. Biomol. Chem., 2018, 16, 4641–4649 RSC.
- Z.-W. Zhang, G. Lu, M. M. Chen, N. Lin, Y.-B. Li, T. Hayashi and A. S. C. Chan, Tetrahedron: Asymmetry, 2010, 21, 1715–1721 CrossRef CAS.
- S. Nakamura, Y. Maeno, M. Ohara, A. Yamamura, Y. Funahashi and N. Shibata, Org. Lett., 2012, 14, 2960–2963 CrossRef CAS PubMed.
- M.-X. Zhao, L. Jing, H. Zhou and M. Shi, RSC Adv., 2015, 5, 75648–75652 RSC.
- T. Yang, S. Yang, Z.-J. Yu, X.-L. Zhao, M. Shi and M.-X. Zhao, Eur. J. Org. Chem., 2023, e202300275 CrossRef CAS.
- G. A. Cutting, N. E. Stainforth, M. P. John, G. Kociok-Köhn and M. C. Willis, J. Am. Chem. Soc., 2007, 129, 10632–10633 CrossRef CAS PubMed.
- T. Yoshino, H. Morimoto, G. Lu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 17082–17083 CrossRef CAS PubMed.
- G. Lu, T. Yoshino, H. Morimoto, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4382–4385 CrossRef CAS PubMed.
- S. Kato, T. Yoshino, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 7007–7010 CrossRef CAS PubMed.
- C. L. Kusturiu, L. S. Liebeskind and W. L. Neumann, Org. Lett., 2002, 4, 983–985 CrossRef PubMed.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS PubMed.
- L. Li, M. Ganesh and D. Seidel, J. Am. Chem. Soc., 2009, 131, 11648–11649 CrossRef CAS PubMed.
- Z. Shi, P. Yu, P. J. Chua and G. Zhong, Adv. Synth. Catal., 2009, 351, 2797–2800 CrossRef CAS.
- X. Chen, S. Dong, Z. Qiao, Y. Zhu, M. Xie, L. Lin, X. Liu and X. Feng, Chem. – Eur. J., 2011, 17, 2583–2586 CrossRef CAS PubMed.
- H. Cai, Y. Zhou, D. Zhang, J. Xu and H. Liu, Chem. Commun., 2014, 50, 14771–14774 RSC.
- M. Bai, B.-D. Cui, J. Zuo, J.-Q. Zhao, Y. You, Y.-Z. Chen, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2015, 71, 949–955 CrossRef CAS.
- Y. Hoashi, T. Okino and Y. Takemoto, Angew. Chem., Int. Ed., 2005, 44, 4032–4035 CrossRef CAS PubMed.
- N. S. Chowdari, M. Ahmad, K. Albertshofer, F. Tanaka and C. F. Barbas III, Org. Lett., 2006, 8, 2839–2842 CrossRef CAS PubMed.
- S. Saito, H. Nakajima, M. Inaba and T. Moriwake, Tetrahedron Lett., 1989, 30, 837–838 CrossRef CAS.
- X. Ye, Y. Pan and X. Yang, Chem. Commun., 2020, 56, 98–101 RSC.
- Z. Sun, K. Weidner, N. Kumagai and M. Shibasaki, Chem. – Eur. J., 2015, 21, 17574–17577 CrossRef CAS PubMed.
- T. Kano, R. Sakamoto, M. Akakura and K. Maruoka, J. Am. Chem. Soc., 2012, 134, 7516–7520 CrossRef CAS PubMed.
- Y. Ichikawa, T. Yamaoka, K. Nakano and H. Kotsuki, Org. Lett., 2007, 9, 2989–2992 CrossRef CAS PubMed.
- G. Ravi Kumar, B. Ramesh, S. Yarlagadda, B. Sridhar and B. V. Subba Reddy, ACS Omega, 2019, 4, 2168–2177 CrossRef CAS PubMed.
- L. Dai, Q. Zhu, J. Zeng, Y. Liu, G. Zhong, X. Han and X. Zeng, Org. Chem. Front., 2022, 9, 2994–2999 RSC.
- A. Y. Sukhorukov, A. A. Sukhanova and S. G. Zlotin, Tetrahedron, 2016, 72, 6191–6281 CrossRef CAS.
- A. M. F. Phillips, M. F. C. Quedes da Silva and A. J. L. Pombeiro, Front. Chem., 2020, 8, 30 CrossRef CAS PubMed.
- E. Marqués-López, P. Merino, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2009, 2401–2420 CrossRef.
- A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed.
- K.-I. Yamada, S. J. Harwood, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 3504–3506 CrossRef CAS PubMed.
- K. R. Knudsen, T. Risgaard, N. Nishiwaki, K. V. Gothelf and K. A. Jørgensen, J. Am. Chem. Soc., 2001, 123, 5843–5844 CrossRef CAS PubMed.
- J. Ma, X. Ding, Y. Hu, Y. Huang, L. Gong and E. Meggers, Nat. Commun., 2014, 5, 4531 CrossRef CAS PubMed.
- Y. Hu, Z. Zhou, L. Gong and E. Meggers, Org. Chem. Front., 2015, 2, 968–972 RSC.
- T. Arai, E. Matsumura and H. Masu, Org. Lett., 2014, 16, 2768–2771 CrossRef CAS PubMed.
- M. Holmquist, G. Blay and J. R. Pedro, Chem. Commun., 2014, 50, 9309–9312 RSC.
- M. K. Choudhary, A. Das, R. I. Kureshy, M. Kumar, N. H. Khan, S. H. Abdi and C. Bajaj, Cat. Sci. Technol., 2014, 4, 548–565 RSC.
- T. Menapara, R. Tak, S. Saravanan, R. I. Kureshy, N. H. Khan, B. Ganguly and M. K. Si, Tetrahedron, 2018, 74, 7000–7008 CrossRef CAS.
- A. Duded and J. Mlynarski, J. Org. Chem., 2017, 82, 11218–11225 CrossRef PubMed.
- N. Yasukawa, A. Yamanoue, T. Takehara, T. Suzuki and S. Nakamura, Chem. Commun., 2022, 58, 1316–1321 RSC.
- T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625–627 CrossRef CAS PubMed.
- X. Xu, T. Furukawa, T. Okino, H. Miyabe and Y. Takemoto, Chem. – Eur. J., 2006, 12, 466–476 CrossRef PubMed.
- C.-J. Wang, X.-Q. Dong, Z.-H. Zhang, Z.-Y. Xue and H.-L. Teng, J. Am. Chem. Soc., 2008, 130, 8606–8607 CrossRef CAS PubMed.
- B. Han, Q.-P. Liu, R. Li, X. Tian, X.-F. Xiong, J.-G. Deng and Y.-C. Chen, Chem. – Eur. J., 2008, 14, 8094–8097 CrossRef CAS PubMed.
- H.-Y. Wang, K. Zhang, C.-W. Zheng, Z. Chai, D.-D. Cao, J.-X. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2015, 54, 1775–1779 CrossRef CAS PubMed.
- T. Marcelli, R. N. S. van der Haas, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 929–931 CrossRef CAS PubMed.
- H. Xe, Y. Zhang, S. Zhang, X. Chen and W. Wang, Angew. Chem., Int. Ed., 2011, 50, 11773–11776 CrossRef PubMed.
- A. Parra, R. Alfaro, L. Marzo, A. Moreno-Carrasco, J. L. García-Ruano and J. Alemán, Chem. Commun., 2012, 48, 9759–9761 RSC.
- W. Fan, S. Kong, Y. Cai, G. Wu and Z. Miao, Org. Biomol. Chem., 2013, 11, 3223–3229 RSC.
- R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC.
- J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 6890–6899 CrossRef PubMed.
- H.-X. He, W. Yang and D.-M. Du, Adv. Synth. Catal., 2013, 355, 1137–1148 CrossRef CAS.
- M. G. Núñez, A. J. M. Farley and D. J. Dixon, J. Am. Chem. Soc., 2013, 135, 16348–16351 CrossRef PubMed.
- J. Vicario, P. Ortiz, J. M. Ezpeleta and F. Palacios, J. Org. Chem., 2015, 80, 156–164 CrossRef CAS PubMed.
- Y. Fang, N. Lu, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Tetrahedron Lett., 2018, 59, 4371–4375 CrossRef CAS.
- J. Wang, Y. Liu, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, ACS Omega, 2021, 6, 5812–5824 CrossRef CAS PubMed.
- P. Li, D.-W. Sun, M. Jiang and J. T. Liu, Tetrahedron: Asymmetry, 2019, 15, 603–607 CrossRef.
- Y.-D. Shao, D.-D. Han, X.-Y. Yang, D.-D. Zhou, T. Wang and D.-J. Cheng, Eur. J. Org. Chem., 2019, 1957–1962 CrossRef CAS.
- Y.-D. Shao, X.-Y. He, D.-D. Han, X.-R. Yang, H.-B. Yao and D.-J. Cheng, Asian J. Org. Chem., 2019, 8, 2023–2026 CrossRef CAS.
- L. Serusi, L. Palombi, G. Pierri, A. Di Mola and A. Massa, J. Org. Chem., 2022, 87, 8420–8428 CrossRef CAS PubMed.
- H. De Kraker, H.-Y. L. Wang, H. D. Arman, R. N. Renteria, C. N. Fleischer, R. O. Messing and S. F. McHardy, J. Org. Chem., 2024, 89, 5134–5141 CrossRef CAS PubMed.
- A. Blasio, J. Wang, D. Wang, F. P. Varodayan, M. B. Pomrenze, J. Miller, A. M. Lee, T. McMahon, S. Gyawali, H. Wang, M. Roberto, S. F. McHardy, M. A. Pleissaud and R. O. Messing, Biol. Psychiatry, 2018, 84, 193–201 CrossRef CAS PubMed.
- F. Fini, V. Sgarzani, D. Pettersen, R. P. Herrera, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 7975–7978 CrossRef CAS PubMed.
- C. Palomo, M. Oiarbide, A. Laso and R. López, J. Am. Chem. Soc., 2005, 127, 17622–17623 CrossRef CAS PubMed.
- E. Gomez-Bengoa, A. Linden, R. López, I. Múgica-Mendiola, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2008, 130, 7955–7966 CrossRef CAS PubMed.
- Y. Wei, W. He, Y. Liu, P. Liu and S. Zhang, Org. Lett., 2012, 14, 704–707 CrossRef CAS PubMed.
- K. Takada and K. Nagasawa, Adv. Synth. Catal., 2009, 351, 345–347 CrossRef CAS.
- W. Huang, C. Peng, L. Guo, R. Hu and B. Han, Synlett, 2011, 2981–2984 CAS.
- K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nuñez, A. M. Goldys and D. J. Dixon, Org. Lett., 2012, 14, 2492–2495 CrossRef CAS PubMed.
- G. Kumaraswamy and A. Pitchaiah, Helv. Chim. Acta, 2011, 94, 1543–1550 CrossRef CAS.
- G. Kumaraswamy and A. Pitchaiah, Tetrahedron, 2011, 67, 2536–2541 CrossRef CAS.
- B. Wang, Y. Liu, C. Sun, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Org. Lett., 2014, 16, 6432–6435 CrossRef CAS PubMed.
- R. B. Walvoord and M. C. Kozlowski, Tetrahedron Lett., 2015, 56, 3037–3074 CrossRef PubMed.
- N. Lu, R. Li, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, J. Org. Chem., 2017, 82, 4668–4676 CrossRef CAS PubMed.
- P. G. K. Clark, L. C. C. Vieira, C. Tallant, O. Fedorov, D. C. Singleton, C. M. Rogers, O. P. Monteiro, J. M. Bennett, R. Baronio, S. Müller, D. L. Daniels, J. Méndez, S. Knapp, P. E. Brennan and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 6217–6221 CrossRef CAS PubMed.
- N. Lu, F. Bai, Y. Fang, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Adv. Synth. Catal., 2017, 359, 4111–4116 CrossRef CAS.
- H.-Y. Wang, Z. Chai and G. Zhao, Tetrahedron, 2013, 69, 5104–5111 CrossRef CAS.
- D. Cao, Z. Chai, J. Zhang, Z. Ye, H. Xiao, H. Wang, J. Chen, X. Wu and G. Zhao, Chem. Commun., 2013, 49, 5972–5974 RSC.
- Y. Liu, Z. Wei, Y. Liu, J. Cao, D. Liang, Y. Lin and H. Duan, Org. Biomol. Chem., 2017, 15, 9234–9242 RSC.
- Y. Liu, Z. Wei, Y. Liu, J. Wang, J. Cao, D. Liang, H. Duan and Y. Lin, Chem. Res. Chin. Univ., 2018, 34, 333–337 CrossRef CAS.
- B. Wang, T. Xu, L. Zhu, Y. Lan, J. Wang, N. Lu, Z. Wei, Y. Lin and H. Duan, Org. Chem. Front., 2017, 4, 1266–1271 RSC.
- N. Lu, Y. Fang, Y. Gao, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, J. Org. Chem., 2018, 83, 1486–1493 CrossRef CAS PubMed.
- X. Wang, Y. Gao, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Org. Chem. Front., 2019, 6, 3269–3273 RSC.
- Y. Liu, Y. Liu, J. Wang, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Tetrahedron Lett., 2017, 58, 2400–2403 CrossRef CAS.
- Y. Liu, J. Wang, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, New J. Chem., 2018, 42, 1608–1611 RSC.
- J. Wang, Y. Liu, Y. Liu, Z. Wei., J. Cao, D. Liang, Y. Lin and H. Duan, Tetrahedron, 2019, 75, 2883–2892 CrossRef CAS.
- D. Uraguchi and T. Ooi, J. Synth. Org. Chem., Jpn., 2018, 76, 1144–1153 CrossRef CAS.
- D. Uraguchi, K. Koshimoto and T. Ooi, J. Am. Chem. Soc., 2008, 130, 10878–10879 CrossRef CAS PubMed.
- D. Uragachi, K. Koshimoto, C. Sanada and T. Ooi, Tetrahedron: Asymmetry, 2010, 21, 1189–1190 CrossRef.
- D. Uraguchi, K. Oyaizu and T. Ooi, Chem. – Eur. J., 2012, 18, 8306–8309 CrossRef CAS PubMed.
- K. Oyaizu, D. Uraguchi and T. Ooi, Chem. Commun., 2015, 51, 4437–4440 RSC.
- D. Uraguchi, K. Oyaizu, H. Noguchi and T. Ooi, Chem. – Asian J., 2015, 10, 344–347 CrossRef PubMed.
- Y.-H. Wang, Y.-L. Liu, Z.-Y. Cao and J. Zhou, Asian J. Org. Chem., 2014, 3, 429–432 CrossRef CAS.
- A. Kumar, J. Kaur, S. S. Chimni and A. K. Jassal, RSC Adv., 2014, 4, 24816–24819 RSC.
- B. Fang, X. Liu, J. Zhao, Y. Tang, L. Lin and X. Feng, J. Org. Chem., 2015, 80, 3332–3338 CrossRef CAS PubMed.
- M. Kristić, M. Benaglia, M. Gazzotti, E. Colombo and M. Sanz, Adv. Synth. Catal., 2023, 365, 1093–1098 CrossRef.
- I. G. Sonsona, J. V. Alegre-Requena, E. Marqués-López, M. C. Gimeno and R. P. Herrera, Chem. – Eur. J., 2020, 26, 5469–5478 CrossRef CAS PubMed.
- B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418–3419 CrossRef CAS PubMed.
- A. Sing, R. A. Yoder, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2007, 129, 3466–3467 CrossRef PubMed.
- A. Sing and J. N. Johnston, J. Am. Chem. Soc., 2008, 130, 5866–5867 CrossRef PubMed.
- T. A. Davis, J. C. Wilt and J. N. Johnston, J. Am. Chem. Soc., 2010, 132, 2880–2882 CrossRef CAS PubMed.
- T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076–1079 RSC.
- T. A. Davis, A. E. Vilgelm, A. Richmond and J. N. Johnston, J. Org. Chem., 2013, 78, 10605–10616 CrossRef CAS PubMed.
- B. A. Vara, A. Mayasundari, J. C. Tellis, M. W. Danneman, V. Arredondo, T. A. Davis, J. Min, K. Finch, R. K. Guy and J. N. Johnston, J. Org. Chem., 2014, 79, 6913–6938 CrossRef CAS PubMed.
- S. V. Tsukanov, M. D. Johnson, S. A. May, M. Rosemeyer, M. A. Watkins, S. P. Kolis, M. H. Yates and J. N. Johnston, Org. Process Res. Dev., 2016, 20, 215–226 CrossRef CAS PubMed.
- C. R. Hopkins, ACS Chem. Neurosci., 2011, 2, 685–686 CrossRef CAS PubMed.
- T. A. Davis, M. A. Danneman and J. N. Johnston, Chem. Commun., 2012, 48, 5578–5580 RSC.
- M. C. Dobish, F. Villalta, M. R. Waterman, G. I. Lepesheva and J. N. Johnston, Org. Lett., 2012, 14, 6322–6325 CrossRef CAS PubMed.
- K. E. Schwieter and J. N. Johnston, ACS Catal., 2015, 5, 6559–6562 CrossRef CAS PubMed.
- L. Belding, S. M. Taimoory and T. Dudding, ACS Catal., 2015, 5, 343–349 CrossRef CAS.
- S. M. Taimoory and T. Dudding, J. Org. Chem., 2016, 81, 3286–3295 CrossRef CAS PubMed.
- D. J. Sprague, A. Sing and J. N. Johnston, Chem. Sci., 2018, 9, 2336–2339 RSC.
- J. A. Bing, N. D. Schley and J. N. Johnston, Chem. Sci., 2022, 13, 2614–2623 RSC.
- D. Chen, G. Xu, Q. Zhou, L. W. Chung and W. Tang, J. Am. Chem. Soc., 2017, 139, 9767–9770 CrossRef CAS PubMed.
- Q. Zhou, W. Tang and L. W. Chung, J. Organomet. Chem., 2018, 864, 97–104 CrossRef CAS.
- M. Zhou, K. Li, D. Chen, R. Xu, G. Xu and W. Tang, J. Am. Chem. Soc., 2020, 42, 10337–10342 CrossRef PubMed.
- M. Zhou, Y. Lin, X.-X. Chen, G. Xu, L. W. Chung and W. Tang, Angew. Chem., Int. Ed., 2023, 62, e202300334 CrossRef CAS PubMed.
- Y. Wang, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5612–5615 CrossRef CAS PubMed.
- S. Tang, X. Zhang, J. Sun, D. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393–10457 CrossRef CAS PubMed.
- M. Matsumoto, M. Harada, Y. Yamashita and S. Kobayashi, Chem. Commun., 2014, 50, 13041–13044 RSC.
- X.-L. Han, B. Hu, C. Fei, Z. Li, Y. Yu, C. Cheng, B. Foxman, J. Luo and L. Deng, J. Am. Chem. Soc., 2023, 145, 4400–4407 CrossRef CAS PubMed.
- C. P. Jonston, A. Kothari, T. Sergeieva, S. L. Okovytyy, K. E. Jackson, R. S. Paton and M. D. Smith, Nat. Chem., 2015, 7, 171–177 CrossRef PubMed.
- W.-R. Zhu, K. Liu, J. Weng, W.-H. Huang, W.-J. Huang, Q. Chen, N. Lin and G. Lu, Org. Lett., 2020, 22, 5014–5019 CrossRef CAS PubMed.
- X.-C. Gan, C.-Y. Zhang, F. Zhong, P. Tian and Y. Yin, Nat. Commun., 2020, 11, 4973 CrossRef PubMed.
- X. Shao, K. Li and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 7083–7087 CrossRef CAS PubMed.
- P. Zhou, X. Shao and S. J. Malcolmson, J. Am. Chem. Soc., 2021, 143, 13999–14008 CrossRef CAS PubMed.
- D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768–13771 CrossRef CAS PubMed.
- T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953–6958 CrossRef CAS PubMed.
- B. Han, Y. Li, Y. Yu and L. Gong, Nat. Commun., 2019, 10, 3804 CrossRef PubMed.
- W.-J. Liu, X.-H. Chen and L.-Z. Gong, Org. Lett., 2008, 10, 5357–5360 CrossRef CAS PubMed.
- Q.-H. Li, L. Wei, X. Chen and C.-J. Wang, Chem. Commun., 2013, 49, 6277–6279 RSC.
- L. Wei, Q.-H. Li and C.-J. Wang, J. Org. Chem., 2018, 83, 11814–11824 CrossRef CAS PubMed.
- R.-Y. Zhu, C.-S. Wang, F. Jiang, F. Shi and S.-J. Tu, Tetrahedron: Asymmetry, 2014, 25, 617–624 CrossRef CAS.
- B. Yu, X.-F. Bai, J.-Y. Lv, Y. Yuan, J. Cao, Z.-J. Zheng, Z. Xu, Y.-M. Cui, K. F. Yang and L.-W. Xu, Adv. Synth. Catal., 2017, 359, 3577–3584 CrossRef CAS.
- B. Yu, K.-F. Yang, X.-F. Bai, J. Cao, Z.-J. Zheng, Y.-M. Cui, Z. Xu, L. Li and L.-W. Xu, Org. Lett., 2018, 20, 2551–2554 CrossRef CAS PubMed.
- Y.-M. Wang, H.-H. Zhang, C. Li, T. Fan and F. Shi, Chem. Commun., 2016, 52, 1804–1807 RSC.
- H. Jia, H. Liu, Z. Guo, J. Huang and H. Guo, Org. Lett., 2017, 19, 5236–5239 CrossRef CAS PubMed.
- H. Jia, Z. Guo, H. Liu, B. Mao, X. Shi and H. Guo, Chem. Commun., 2018, 54, 7050–7053 RSC.
- K. Ohmatsu, S. Kawai, N. Imagawa and T. Ooi, ACS Catal., 2014, 4, 4304–4306 CrossRef CAS.
- W.-F. Zheng, G.-J. Sun, L. Chen and Q. Kang, Adv. Synth. Catal., 2018, 360, 1790–1794 CrossRef CAS.
- W.-X. Huang, L.-J. Liu, B. Wu, G.-S. Feng, B. Wang and Y.-G. Zhou, Org. Lett., 2016, 18, 3082–3085 CrossRef CAS PubMed.
- W. Ma, J. Zhang, C. Xu, F. Chen, Y.-M. He and Q.-H. Fan, Angew. Chem., Int. Ed., 2016, 55, 12891–12894 CrossRef CAS PubMed.
- T. Wang, F. Chen, J. Qin, Y.-M. He and Q.-H. Fan, Angew. Chem., Int. Ed., 2013, 52, 7172–7176 CrossRef CAS PubMed.
- Y. Chen, Y. Pan, Y.-M. He and Q.-H. Fan, Angew. Chem., Int. Ed., 2019, 58, 16831–16834 CrossRef CAS PubMed.
- A. N. Kim, A. Ngamnithipon, E. R. Welin, M. T. Daiger, C. U. Grünanger, M. D. Bartberger, S. C. Virgil and B. M. Stoltz, ACS Catal., 2020, 10, 3241–3248 CrossRef CAS PubMed.
- Z. Zhang and H. Du, Angew. Chem., Int. Ed., 2015, 54, 623–626 CrossRef CAS PubMed.
- D. J. Parks, R. E. von, H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809–811 CrossRef CAS.
- C. Schüttler, Z. Li-Böhmer, K. Harms and P. von Zezschwitz, Org. Lett., 2013, 15, 800–803 CrossRef PubMed.
- H.-J. Pan, Y. Lin, T. Gao, K. K. Lau, W. Feng, B. Yang and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 18599–18604 CrossRef CAS PubMed.
- M. Yus, C. Nájera, F. Foubelo and J. M. Sansano, Chem. Rev., 2023, 123, 11817–11893 CrossRef CAS PubMed.
- C. Li, K. Lang, H. Lu, Y. Hu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16837–16841 CrossRef CAS PubMed.
- K. Lang, S. Torker, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2019, 141, 12388–12396 CrossRef CAS PubMed.
- Y. Yang, I. Cho, X. Qi, P. Liu and F. H. Arnold, Nat. Chem., 2019, 11, 987–993 CrossRef CAS PubMed.
- L. Li, F. Han, X. Nie, Y. Hong, S. Ivlev and E. Meggers, Angew. Chem., Int. Ed., 2020, 59, 12392–12395 CrossRef CAS PubMed.
- X. Nie, Z. Yan, S. Ivlev and E. Meggers, J. Org. Chem., 2021, 86, 750–761 CrossRef CAS PubMed.
- Z. Zhou, Y. Tan, T. Yamahira, S. Ivlev, X. Xie, R. Riedel, M. Hemming, M. Kimura and E. Meggers, Chem, 2020, 6, 2024–2034 CAS.
- H.-Y. Bai, F.-X. Tan, T.-Q. Liu, G.-D. Zhu, J.-M. Tian, T.-M. Ding, Z.-M. Chen and S.-Y. Zhang, Nat. Commun., 2019, 10, 3063 CrossRef PubMed.
- C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 14538–14541 CrossRef CAS PubMed.
- C. Min, N. Mittal, C. K. De and D. Seidel, Chem. Commun., 2012, 48, 10853–10855 RSC.
- J. Xie, Z. Guo, W. Liu, D. Zhang, Y.-P. He and X. Yang, Chin. J. Chem., 2022, 40, 1674–1680 CrossRef CAS.
- D. Perrota, M.-M. Wang and J. Waser, Angew. Chem., Int. Ed., 2018, 57, 5120–5123 CrossRef PubMed.
- E. T. Mwenda and H. M. Nguyen, Org. Lett., 2017, 19, 4814–4817 CrossRef CAS PubMed.
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 139–165 RSC.
- F. S. He, H. Zhu, Z. Wang, M. Gao, X. Yu and W.-P. Deng, Org. Lett., 2015, 17, 4988–4991 CrossRef CAS PubMed.
- E. García-Mingüens, M. Ferrándiz-Saperas, M. de, G. Retamosa, C. Nájera, M. Yus and J. M. Sansano, Molecules, 2022, 27, 4579 CrossRef PubMed.
- S. Zhu, S. Yu, Y. Wang and D. Ma, Angew. Chem., Int. Ed., 2010, 49, 4656–4660 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |