
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of molecules with multiple stereogenic elements
Arthur
Gaucherand
 ,
Expédite
Yen-Pon
,
Antoine
Domain
,
Alix
Bourhis
,
Jean
Rodriguez
and
Damien
Bonne
*
,
Expédite
Yen-Pon
,
Antoine
Domain
,
Alix
Bourhis
,
Jean
Rodriguez
and
Damien
Bonne
*
Aix Marseille Univ, CNRS, Centrale Med, Marseille, ISM2, France
First published on 30th September 2024
Abstract
This review explores the fascinating world of molecules featuring multiple stereogenic elements, unraveling the different strategies designed over the years for their enantioselective synthesis. Specifically, (dynamic) kinetic resolutions, desymmetrisations and simultaneous installation of stereogenic elements exploiting either metal- or organo-catalysis are the principal approaches to efficiently create and control the three-dimensional shapes of these attractive molecules. Although most molecules presented in this review possess a stereogenic carbon atom in combination with a stereogenic axis, other combinations with helices or planes of chirality have started to emerge, as well as molecules displaying more than two different stereogenic elements.
 Arthur Gaucherand | Arthur Gaucherand studied general chemistry at CPE Lyon Engineering School (France). After spending a year as an exchange student at the Université de Montréal (Canada), where he specialised in organic chemistry, he obtained his master's degree in 2020. He then pursued a PhD at Aix-Marseille University (France), focusing on organocatalytic enantiocontrol of multiple stereogenic elements under the supervision of Prof. Damien Bonne and Prof. Jean Rodriguez. Upon completing his PhD in 2023, he joined Prof. Jérôme Lacour's group at the University of Geneva (Switzerland) for postdoctoral training, where he is currently based. His research interests centre on the stereocontrol of novel chiral molecules. |
 Expédite Yen-Pon | Expédite Yen-Pon obtained her PhD in chemistry in 2016 working on kinase inhibitors with Dr Huixiong Chen (University Paris Cité). After her first postdoctoral experience in medicinal chemistry with Dr Samir Messaoudi (University Paris Saclay), she joined Dr Davide Audisio’ group (CEA Saclay) to work on helicene synthesis. She then worked with Prof. Gary A. Molander (University of Pennsylvania) on photoredox chemistry. In 2022, she joined Aix-Marseille University to work with Prof. Damien Bonne and Prof. Jean Rodriguez on helicene enantioselective synthesis and, afterward, with Dr Yoann Coquerel on PAH preparation. Since 2023, she has been appointed as a CNRS researcher in the same team. |
 Antoine Domain | Antoine Domain studied chemistry for his bachelor's degree at the University of Rennes and a master's degree in organic chemistry at Nantes-Université. After completing his studies, he began his PhD at Aix-Marseille Université under the direction of Pr. Damien Bonne and Pr. Jean Rodriguez. His research focuses on the development of new synthetic methodologies for the control of multiple stereogenic elements. |
 Alix Bourhis | Alix Bourhis studied general chemistry at ECPM Strasbourg Engineering School (France). During her final year at ECPM, she had the opportunity to participate in an exchange program at the Université de Sherbrooke (Canada), where she obtained a master's degree in 2022, specialising in organic and pharmaceutical chemistry. She is currently pursuing her academic studies as a PhD student at Aix-Marseille University (France), within the Stereo team of the ISM2 under Prof. Damien Bonne and Prof. Jean Rodriguez's supervision. Her research focuses on the atroposelective synthesis of benzopyrans. |
 Jean Rodriguez | Jean Rodriguez was born in Cieza (Spain) in 1958 and studied chemistry at the University of Aix-Marseille (France), he completed his PhD in 1987, and his Habilitation in 1992. He is currently Professor and was appointed Director of the UMR-CNRS-7313-iSm2 until December 2023. His research interests include the development of multiple bond-forming transformations and their application in stereoselective organocatalysed synthesis. In 2021 he was awarded the “Grand Prix Emile Jungfleisch” from the French Academy of Sciences. |
 Damien Bonne | Damien Bonne obtained his PhD in 2006 from the ‘Institut de Chimie des Substance Naturelles’ (ICSN) under the direction of Prof. Jieping Zhu working on multicomponent reactions. After a postdoc in the group of Prof. Varinder Aggarwal (Bristol), he was appointed Lecturer at Aix-Marseille University in 2007. He was promoted to Full Professor in 2022 and in 2023, he became “Junior Distinguished Member” of the French Chemical Society. His research focuses on the development of enantioselective methodologies for the control of axial and helical chiralities. |
Introduction
In the complex field of three-dimensional molecular architectures, the presence of multiple stereogenic elements in a molecule significantly influences its biological and physical properties. Molecules adorned with multiple stereogenic elements have attracted attention from chemists for exploiting their specific three-dimensional form in several domains. A broad range of natural products bearing multiple stereogenic elements,1 such as diconophylline A,2 vescalin,3 cordypyridone A,4 chaetochromin A,5 and TMC-95 D,6 often possess a stereogenic C–C bond and one or several stereogenic carbon atoms (Fig. 1). This structural feature is also found in synthetic pharmaceutical ingredients, for example, BI 224436, whose antiviral activity was greatly enhanced by the introduction of two stereogenic elements7 or diazocanone 1 exhibiting excellent NK1 antagonistic activities.8 Sotorasib represents the first FDA-approved monotherapy to be manufactured and marketed as a configurationally stable C–N atropisomerically pure compound,9 which demonstrated a ten-fold difference in potency against non-small-cell lung cancer.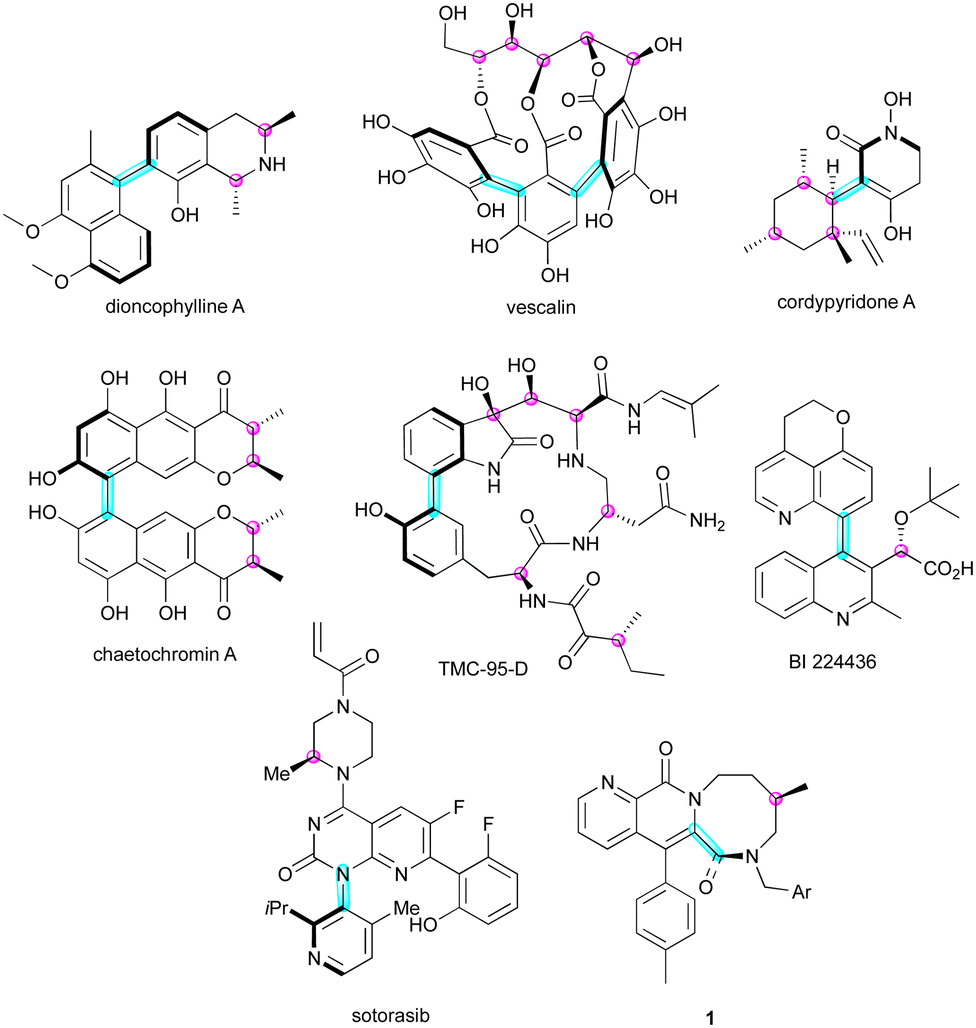 | ||
| Fig. 1 Natural and non-natural products bearing multiple stereogenic elements. | ||
These intriguing molecules are not only found in nature, but have also been designed for other applications such as ligands10 and organocatalysts11 for enantioselective catalysis or in materials science (Fig. 2).12 In these applications, other combinations of stereogenic elements are found, notably with the incorporation of planar or helical systems.
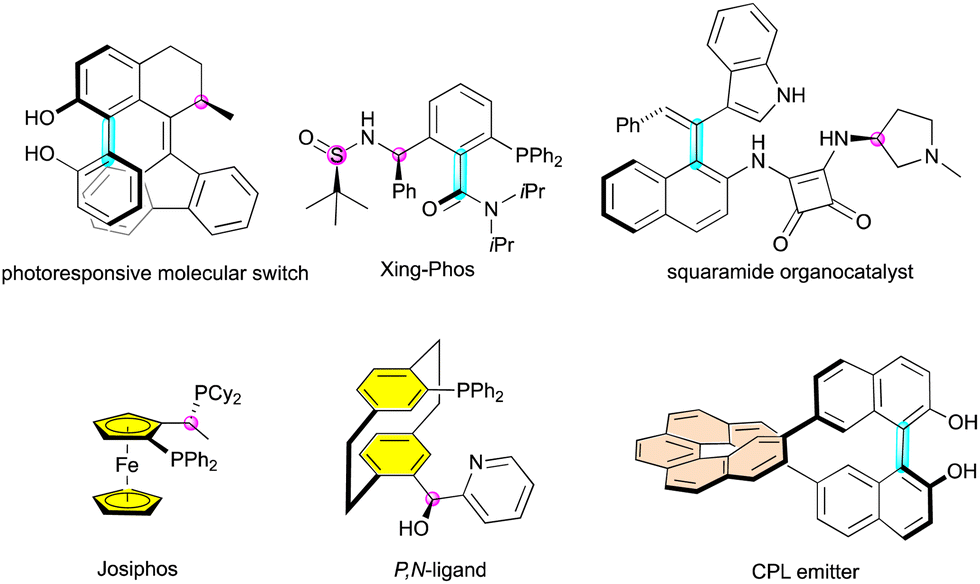 | ||
| Fig. 2 Applications of multiple stereogenic element-containing molecules. | ||
For years, the most common methods for accessing these attractive molecules bearing multiple stereogenic elements have relied on diastereoselective synthesis, including cyclisation,13 coupling,14 rearrangement,15 and C–H functionalisation,16 but alternative methods have also been developed,17 as well as post-functionalisations of optically active substrates.18 The enantioselective synthesis of these molecules has remained a formidable challenge for several years with sporadic examples,19 but renewed interest has been witnessed in the last five years, which justifies this review.20,21 Hence, we present the history of the development in this field based on the classification of strategies for the direct catalytic enantioselective construction of molecules containing multiple stereogenic elements in the atropoisomeric, planar, helicoidal, and allenic series. To access these attractive systems, various complementary strategies have been developed to date and are presented in five sections: (1) kinetic resolution, (2) dynamic kinetic resolution, (3) desymmetrisation, (4) simultaneous installation of stereogenic elements, and (5) enantioselective synthesis of axially chiral allenes bearing one additional stereogenic centre. Each section is organised based on the nature of the stereogenic elements present in the molecule and the catalytic activation involved in stereocontrol.
Kinetic resolution
Planar/central stereogenic elements
Among the possible strategies enabling the enantioselective synthesis of molecules bearing different stereogenic elements, kinetic resolution (KR) was the first to be used. Indeed, this technique allows for efficient deracemisation22 and sometimes results in the formation of an additional stereogenic element with good diastereo- and enantioselectivities.Interestingly, one of the first reported enantiocontrol of multiple stereogenic elements was done on a planar chiral paracyclophane using a kinetic resolution strategy. In 2001, Kagan and colleagues applied the Corey–Bakshi–Shibata reduction with C1 as the catalyst to a racemic sample of 4-acetyl[2.2]paracyclophane (±)-2, leading to the formation of the secondary benzylic alcohol (R,Rp)-3 with good stereoselectivity (9.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 78% ee), together with unreacted substrate (S)-2 in very high enantiopurity (Scheme 1).23
:1 dr and 78% ee), together with unreacted substrate (S)-2 in very high enantiopurity (Scheme 1).23
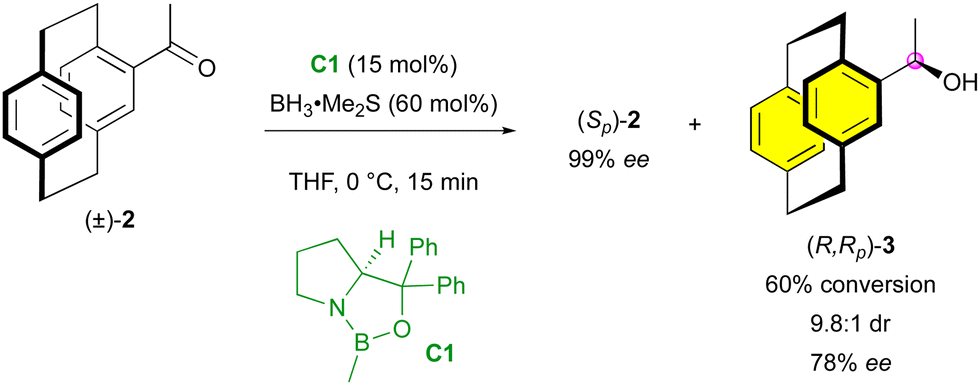 | ||
| Scheme 1 KR of racemic 4-acetyl[2.2]paracyclophane. | ||
Fourteen years later, the Kudo group proposed the use of an artificial peptide for the enantioselective addition of nitromethane to conjugated ketone (±)-4 (Scheme 2).24 The formation of a chiral iminium intermediate (Rp)-6 after the condensation of the catalyst on the ketone guided the addition of nitromethane via a hydrogen-bonding network. The stereo-defined structure of peptide catalyst C2 resulted in the faster formation of (S,Rp)-5 with very good diastereoselectivity (11.5:1 dr) and high enantiomeric excess (90% ee).
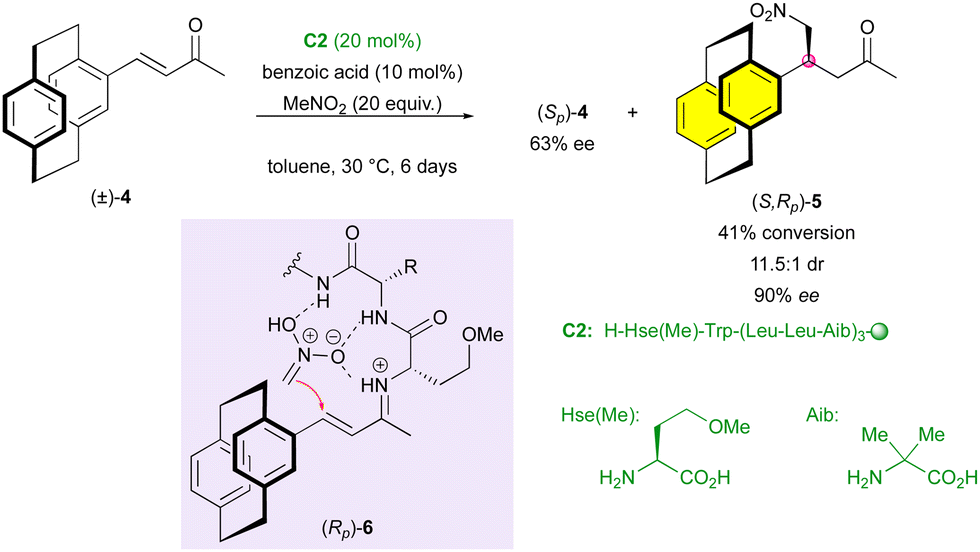 | ||
| Scheme 2 KR via enantioselective Michael addition. | ||
Likewise, enantioselective addition has also been used recently by Zhou and collaborators for the KR of paracyclophanes. In 2019, they developed the palladium-catalysed addition of arylboronic acids onto N-sulfonyl imines (±)-7 bearing planar chirality (Scheme 3).25 The use of the planarly and centrally chiral ligand L1 allows the synthesis of (S,Sp)-8 with good to high diastereoselectivity (7:1 to >20:1 dr) and excellent enantiocontrol (96% to 99% ee). Two years later, they applied their method to cyclic N-sulfonyl imines (±)-9 using a similar ligand L2.26 Interestingly, when applied to these substrates, the reaction led to the formation of (R,Sp)-10 with always perfect diastereoselectivity (>20:1 dr), which is probably due to the rigid cyclic structure of the imine function, although the enantiomeric excess was slightly impacted (76% to 97% ee).
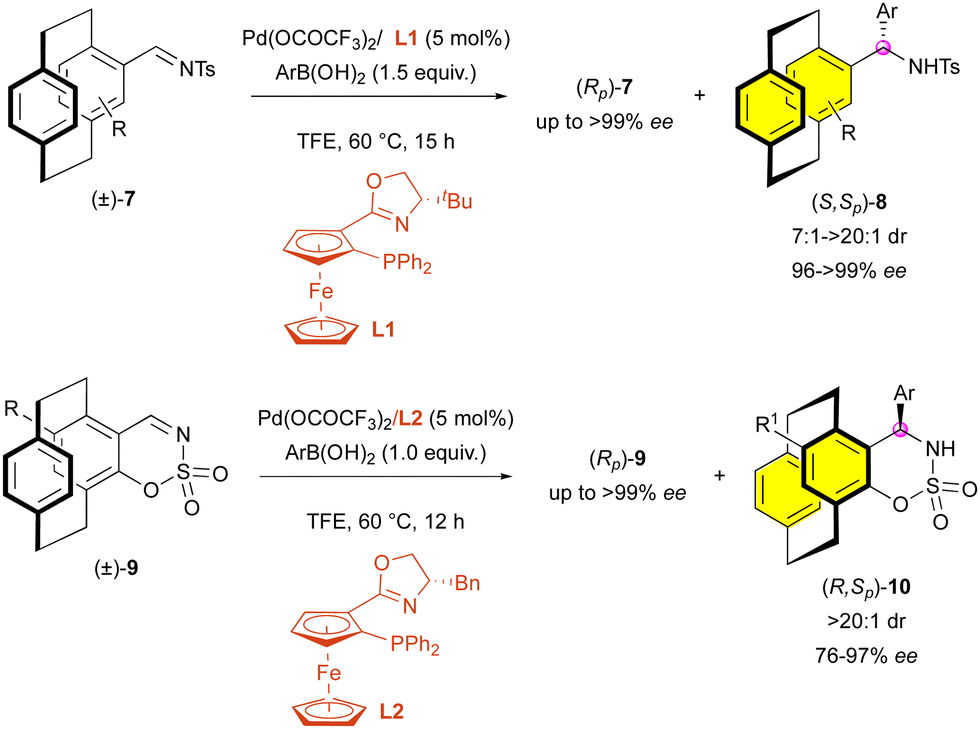 | ||
| Scheme 3 Pd-Catalysed addition of arylboronic acids on planarly chiral N-sulfonyl imines. | ||
Although less examples exist, the enantioselective formation of a stereocentre for the KR of planar-chiral compounds has been applied to metallocenes as well. As early as 2009, Rios, Moyano and coworkers performed the KR of ferrocenecarbaldehyde (±)-11 bearing a pyrimidine substituent (Scheme 4).27 The L-proline-catalysed aldolisation with acetone resulted in the isolation of three different products, enantioenriched unreacted substrate (Rp)-11, target aldol adduct (R,Sp)-12 featuring planar and central chirality, and the corresponding dehydrated compound (Rp)-13. Although only moderate enantiomeric excess and low yield (up to 57% ee with 15% yield and down to 38% ee with 26% yield) could be obtained, this simple approach constitutes the first remarkable example of this stereochemical combination.
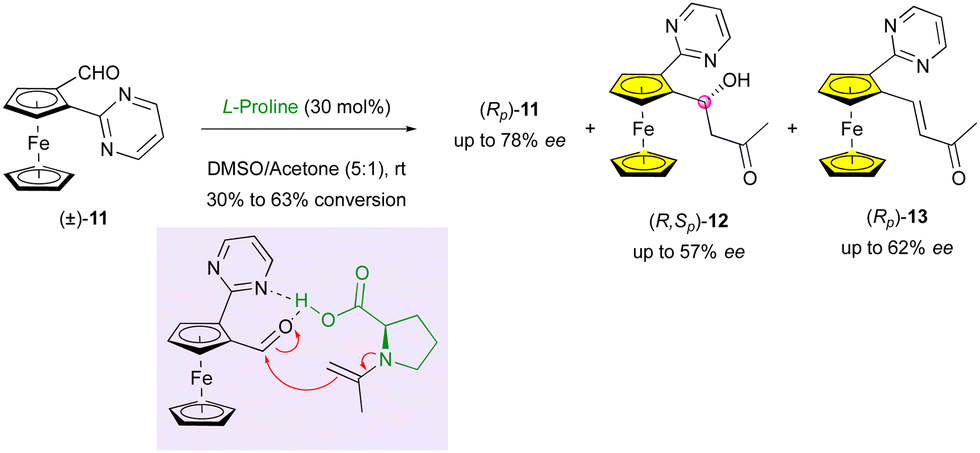 | ||
| Scheme 4 KR of ferrocenecarbaldehyde via aldolisation. | ||
In 2014, the Kudo group applied their peptide-catalysed enantioselective addition of nitromethane to planar-chiral metallocenes (±)-14, achieving efficient KR (Scheme 5).28 Products (S,Rp)-15 displayed good to perfect diastereoselectivity (4.6:1 to >99:1 dr) and modest to excellent enantiomeric excess (59% to 93% ee). As expected, the stereoselectivity was closely related to the conversion, where higher yields led to a decrease in the diastereo- and enantioselectivity. Interestingly, ruthenocene reacted faster than its ferrocene analogue, leading to a lower stereocontrol (49:1 dr, 92% ee and 36% yield for M = Fe, R = I vs. 4.6:1 dr, 66% ee and 38% yield for M = Ru, R = I).
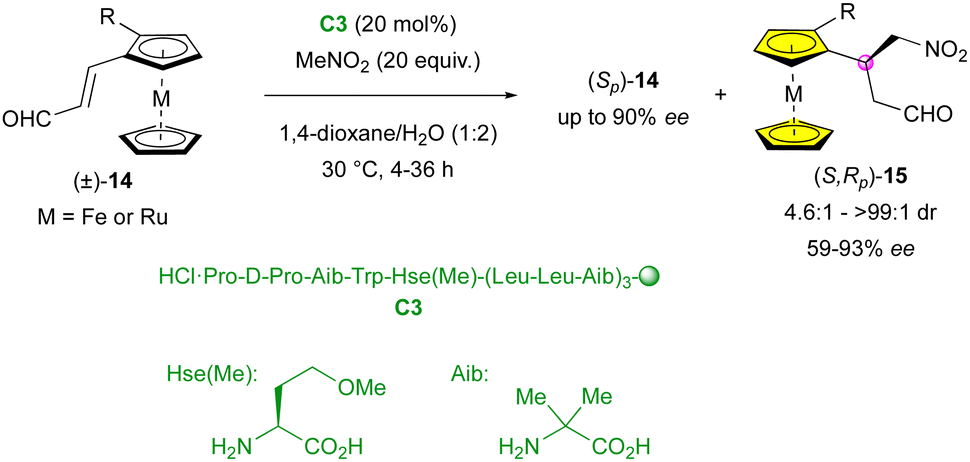 | ||
| Scheme 5 KR of metallocenes via organocatalysed enantioselective Michael addition. | ||
Planar/axial stereogenic elements
Besides paracyclophanes and metallocenes, planar chirality can be observed in macrocycles. Thus, very recently, the Wang group proposed the original KR of axially chiral indoles (±)-16via NHC-catalysed oxidative macrocyclisation with pre-catalyst C4 (Scheme 6).29 Various sizes of macrocycles 17 could be obtained, always with excellent diastereoselectivity (>19:1 dr) and enantioselectivity (88% to 98% ee), endowing (aR)-16 with moderate enantioselectivities. The planar configuration of the macrocycle was confirmed by X-ray diffraction analysis of a similar non-axially chiral compound, but the absolute configuration of the stereogenic axis was not determined.
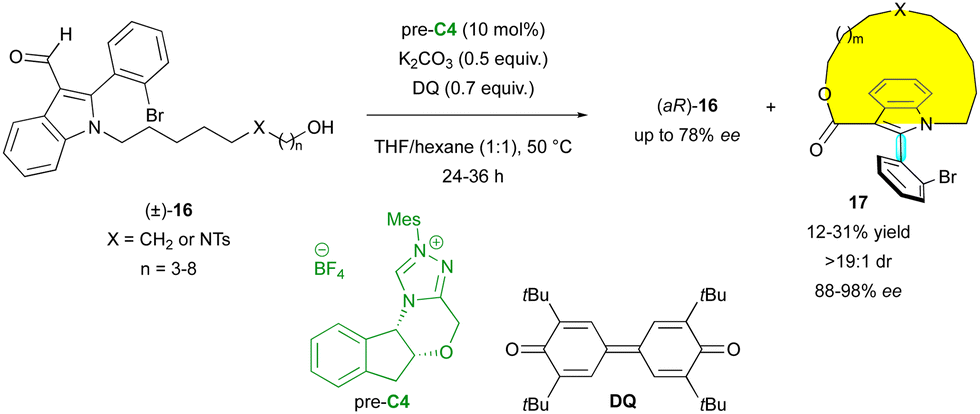 | ||
| Scheme 6 KR of axially chiral indoles via NHC-catalysed macrocyclisation. | ||
Central/axial stereogenic elements
Among the methodologies developed for the enantioselective synthesis of molecules containing multiple stereogenic elements, the combination of a stereogenic centre with an axis is the most common. In this regard, in 2018, the Wang group described the catalytic KR of racemic anilide hemiaminals (±)-18 by an enantioselective NHC-catalysed acylation with propargylic aldehydes 19 involving pre-catalyst C5 (Scheme 7).30 The resulting catalytic C–O bond formation provided C–N axially chiral isoindolinones 20 and allowed the separation of the two enantiomers of anilides giving (S)-18 efficiently in high yield with excellent enantioselectivity.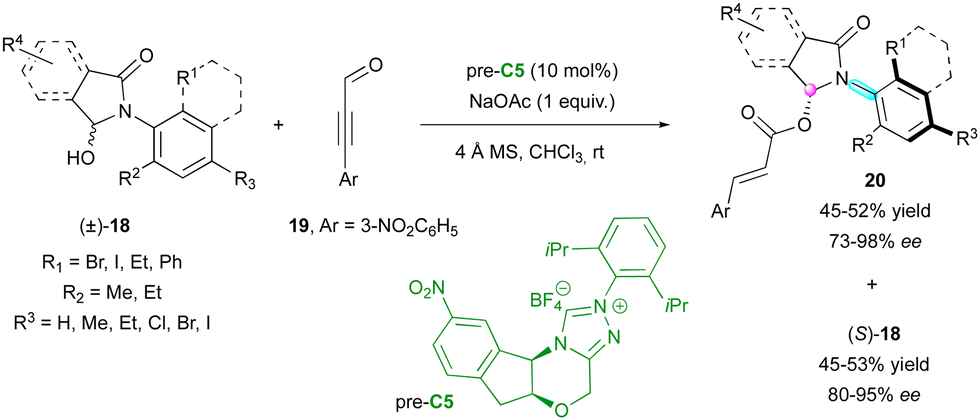 | ||
| Scheme 7 KR of axially chiral anilides via enantioselective NHC-catalysed acylation. | ||
More recently, chiral NHC-catalysed (3+3) annulation was proposed by the Biju group for the KR of racemic N-aryl aminomaleimides 21 with 2-bromoenals 22 to furnish C–N axially chiral fused-dihydropyridinones (M,S)-23 bearing a stereogenic centre and leaving the enantioenriched starting material (P)-21 (Scheme 8).31 The observed remote chirality induction provided by pre-catalyst C6 was governed by the crucial bulky t-Bu group and the aminoindanol moiety of the α,β-acylazolium intermediate 24, both imposing good facial diastereoselectivity, which resulted in good to excellent enantioselectivities but moderate diastereoselectivities.
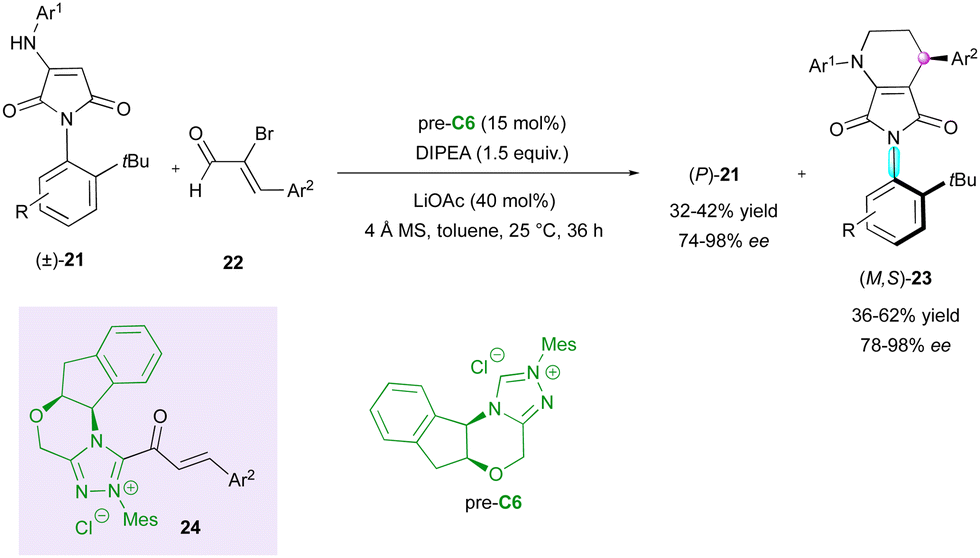 | ||
| Scheme 8 NHC-Catalysed (3+3) annulation. | ||
Very recently, the Li group exploited the rhodium-catalysed C–H activation of pyridone rings for the KR of C–N axially chiral isoquinolines (±)-25. In this process, N-protected O-allylhydroxyamines 26, with Rh-catalyst C7, underwent enantio- and diastereoselective C–H carboamidation directed by the large 8-substituted isoquinolyl motif (Scheme 9).32 The starting 2-pyridones 25 were efficiently resolved and the centrally and axially chiral amino alcohols (aR,R)-27 were obtained as one diastereomer with excellent enantiomeric excess (93% to 99% ee) and s-factor of up to >600, allowing the recovery of (aS)-25 with high enantiomeric excesses. Several sulfonyl substituents were tolerated; however, a bulky enough R2 substituent was necessary to prevent free rotation around the C–N axis (ΔG‡ = 118 kJ mol−1 for R1 = H, R2 = Cl).
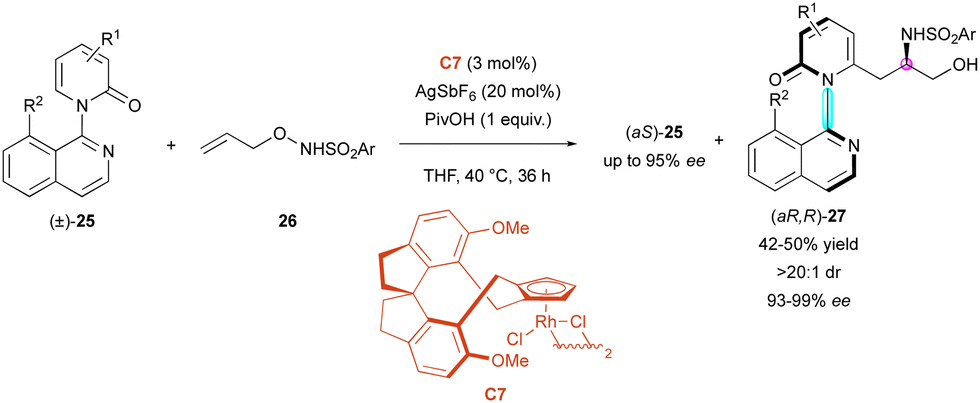 | ||
| Scheme 9 Access to chiral amino alcohols via enantioselective carboamidation. | ||
Concerning access to C–C axially chiral biaryls bearing a stereogenic centre, in 2019, the Li and Zhou group described the catalytic KR of axially chiral biaryl nitroalkenes (±)-28via an enantioselective Michael addition upon enamine activation of acetone (Scheme 10).33 This method relies on the hydrogen bond network established between the substrate and bifunctional thiophosphinamide catalyst C8 bearing a primary amine, which guides the addition of the chiral enamine intermediate. The reaction delivered molecules (S,aR)- (ok) with very high enantiomeric excess but poor diastereomeric ratio in most cases and enantioenriched starting material (aS)-28 was recovered with good selectivity. However, the diastereoselectivity of (S,aR)-29 could be enhanced in the presence of bulky R2 substituents in the ortho position.
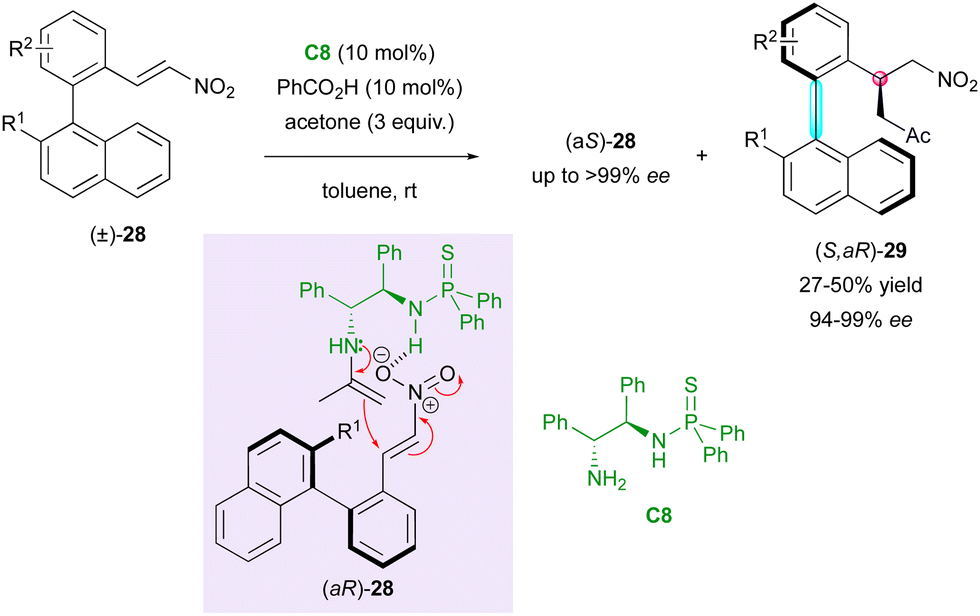 | ||
| Scheme 10 Organocatalysed KR of axially chiral 2-nitrovinyl biaryls. | ||
One year later, Zhang, Tan and Shi attempted the KR of the original non-biaryl atropisomers using chiral phosphoric acid catalyst (CPA) C9 (Scheme 11).34 The CPA-catalysed enantioselective nucleophilic substitution of axially chiral anilines (±)-30 on centrally chiral azalactones (±)-31 resulted in the formation of rare tetrasubstituted axially chiral styrene-type atropisomers (aS,S)-32 bearing an additional stereocentre with good diastereoselectivity (5.3:1 to 15.7:1 dr) and high enantiomeric excess (76% to 95% ee). Besides leaving highly enantioenriched staring anilines (aR)-30, the method resulted in the concomitant dynamic kinetic resolution of centrally chiral azalactones (±)-31, whose racemisation was fast under these reaction conditions.
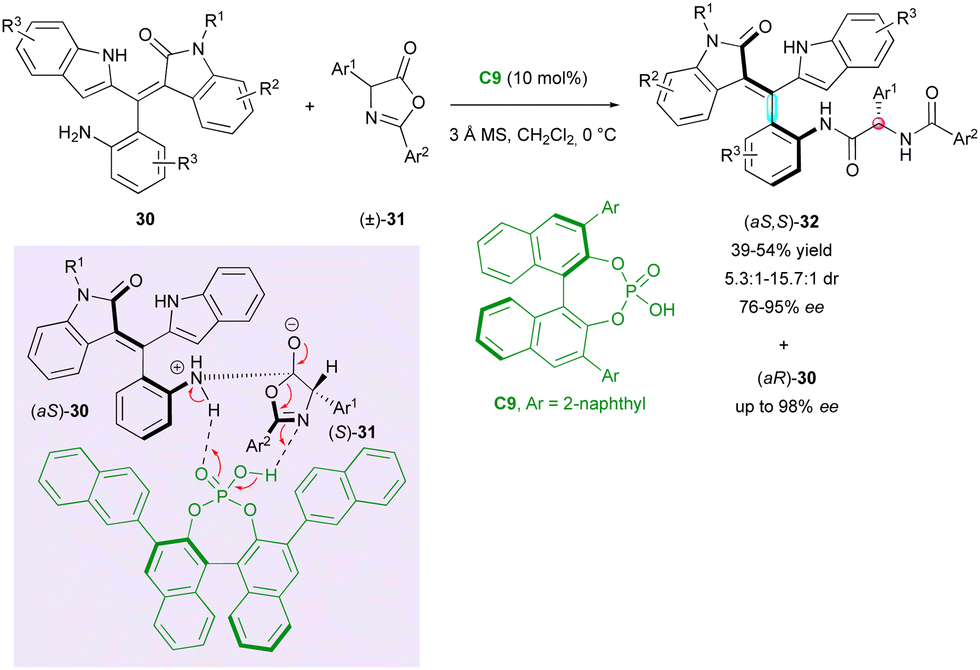 | ||
| Scheme 11 Atroposelective access to oxindole-based chiral styrenes via KR. | ||
Dynamic kinetic resolution
Organocatalysed activation
The inherent conformational flexibility of atropisomers makes them ideal substrates for dynamic kinetic resolution (DKR).35 This was first exploited by Clayden and co-workers in 1999 for the synthesis of atropisomeric amides 36 by the dynamic resolution of ortho-formyl naphthamides 3336 using enantiopure proline-derived diamine 34 as a resolving agent via thermodynamic control during the formation of aminal intermediates 35 in a 9:1 diastereomeric ratio (Scheme 12).
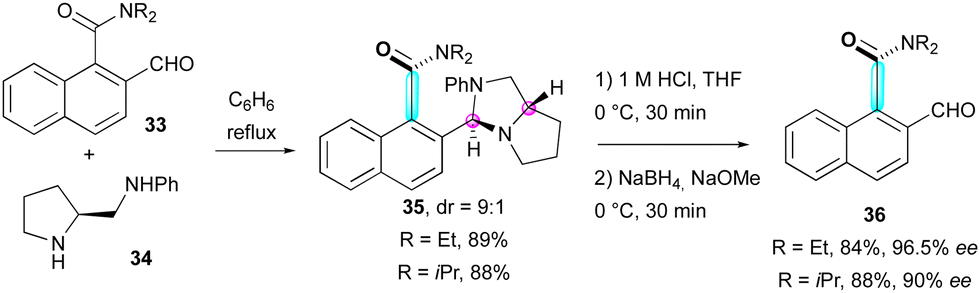 | ||
| Scheme 12 Dynamic resolution of atropisomeric amides. | ||
Following this pioneer result, in 2004, the Walsh group reported the first example of the DKR strategy using an organocatalyst for the control of two different stereogenic elements (Scheme 13).37 They proposed an efficient way to afford molecules 38 bearing both a stereodefined secondary alcohol and a stereogenic axis starting from 2-formylbenzamides 37 and using L-proline as the catalyst. Initially conducted in DMSO/acetone as the solvent, the reaction gave higher enantio- and diastereoselectivities using neat acetone. This strategy gave very good yields (up to >99%) and high enantiomeric excesses (up to 95%) but moderate diastereoselectivity (1.6:1 to 8:1). To account for the observed stereochemistry, transition state 40 should be less favorable than 39 as a result of the unfavorable interaction between the amide moiety and the axial hydrogen atom in the Zimmerman–Traxler six-membered chair-like transition state model.
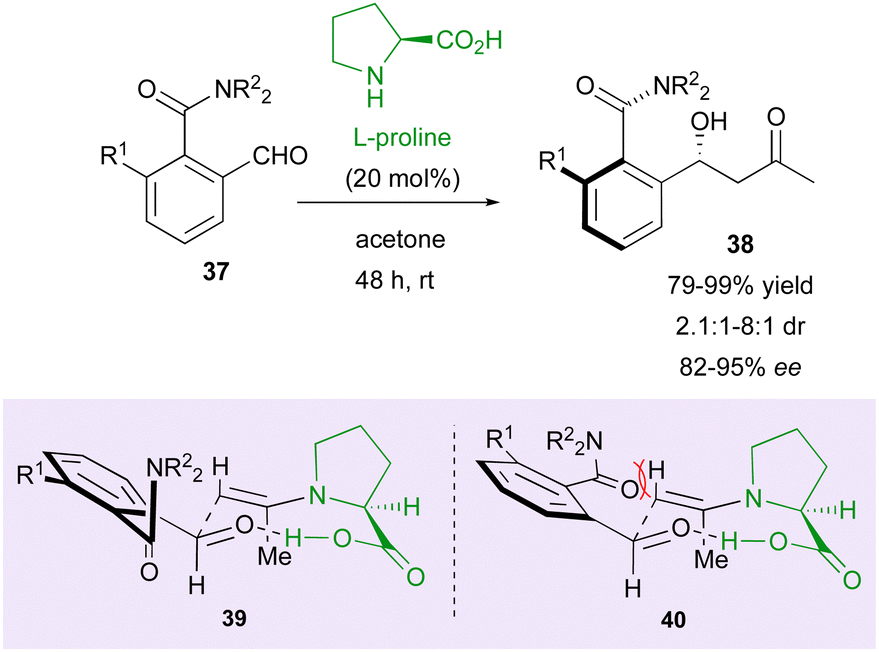 | ||
| Scheme 13 DKR of atropisomeric amides via enantioselective aldolisation. | ||
In 2020, capitalising on Clayden's aminal formation,35 Hang, Zhang and Jiang performed an elegant enantioselective CPA-(C10)-catalysed aminocyclisation involving ortho-formyl 1-naphthamides 41 and pyrrolylanilines 42 (Scheme 14).38 The control experiments showed that the catalyst plays a crucial role in the chiral recognition for each step of the sequence to organise a postulated transition state, which reasonably explains the observed stereoselectivity after the addition of the pyrrole to the intermediate imine. This putative synergy results in the efficient formation of axially and centrally chiral pyrrolopyrazines 43 in very good yield, excellent diastereoselectivity and enantiomeric excess in most cases.
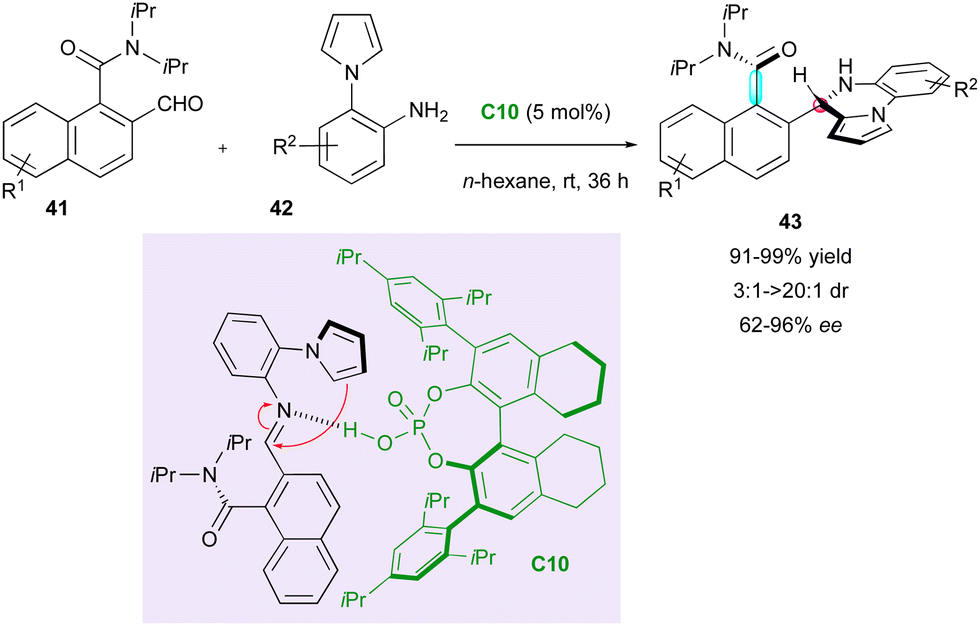 | ||
| Scheme 14 CPA-Catalysed DKR of atropisomeric o-formyl naphthamides. | ||
Shortly after, Li's group was interested in the atroposelective O-allylation of related ortho-hydroxy naphthamide 44 with Morita–Baylis–Hillman (MBH) adduct 45, employing hydroquinine as the catalyst and reported a single example of DKR with modest yield and diastereoselectivity with a decent enantiomeric excess of 90% for product 46 (Scheme 15).39 The catalyst interacts with the deprotonated naphthol through hydrogen bonding interactions and controls the stereoselectivity of the reaction.
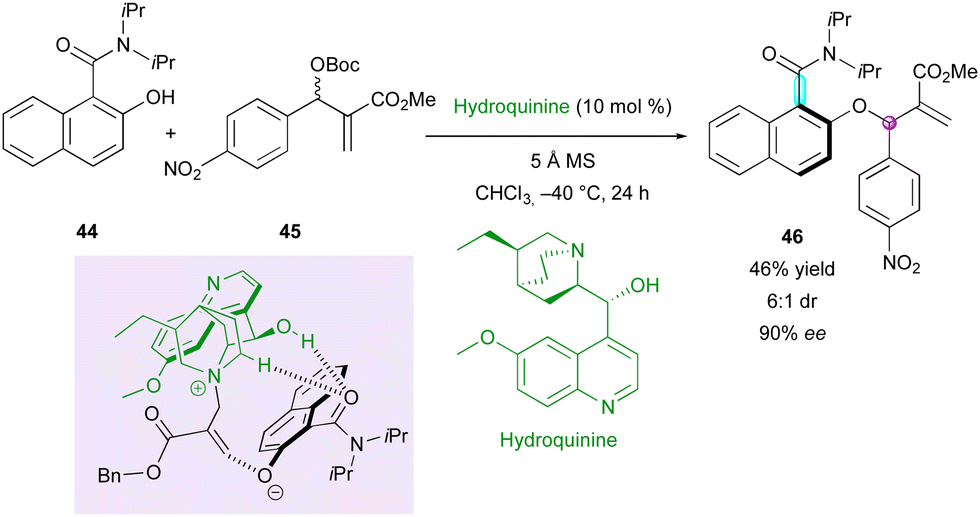 | ||
| Scheme 15 DKR of o-hydroxy 1-naphthamide via atroposelective allylation. | ||
The incorporation of a stereogenic atom other than carbon in the final structure is not a common feature and still constitutes a real synthetic challenge. In 2021, the same group proposed the related N-allylation of N-aryl sulfinamides 47 with MBH-adducts 48, employing quinine as the catalyst (Scheme 16).40 They showed an unusual simultaneous control of a stereogenic sulfur atom and a C–N stereogenic axis. Although most of the examples exhibited moderate diastereoselectivity (4:1 to 9:1 dr), a few allylated sulfinamides 49 were obtained with very good diastereomeric ratio (dr > 19:1), always with excellent enantiocontrol.
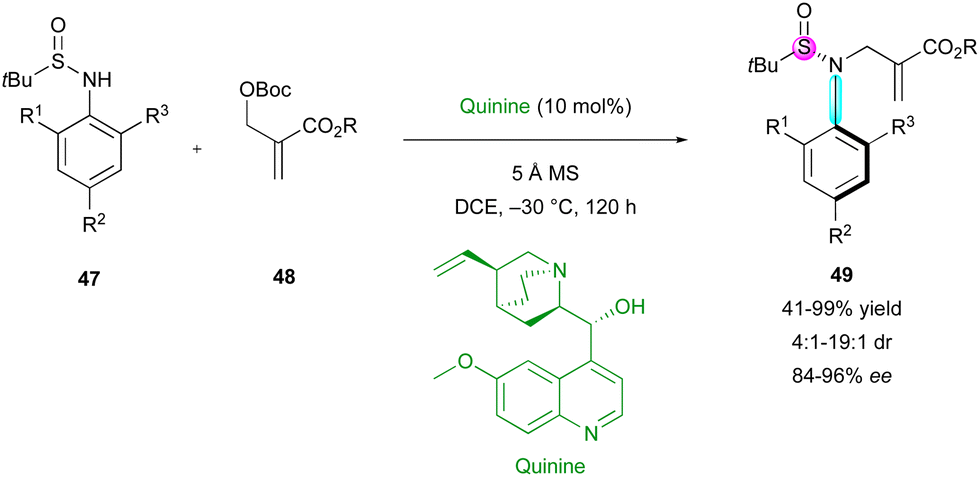 | ||
| Scheme 16 DKR of sulfinamides via atroposelective N-allylation. | ||
The functionalisation of 3-substituted arylindoles has allowed the design of several organocatalysed DKR for the enantioselective synthesis of axially and centrally chiral indole-based heterocycles. The pioneering work was disclosed by Shi's group in 2019 who proposed the interesting enantioselective synthesis of naphthyl-indole atropisomers bearing a stereogenic centre (Scheme 17).41 They achieved control of both stereogenic elements employing SPINOL-derivative catalyst C11. Initially, phosphoric acid reacts with the phenol derivatives 51, forming ortho-quinone methide (o-QM) intermediate 53. Subsequently, the CPA efficiently controls the stereoselectivity of the nucleophilic addition through hydrogen bond interactions with naphthyl-indole substrates 50 and delivers centrally chiral indole atropisomers 52 in high efficiency. Recently, this strategy was applied by the same group for the enantioselective diastereodivergent synthesis of 2-alkenylindoles bearing both axial and central stereogenic elements.42
 | ||
| Scheme 17 DKR of naphthyl-indoles via atroposelective functionalisation with o-QM. | ||
Following the pioneer work by Shi, Fu and co-workers applied the same strategy with 2-aryl-3H-indol-3-ones 54 as electrophiles for the direct synthesis of arylindolyl indolin-3-ones 55 with the creation and control of both a stereogenic axis and a quaternary stereogenic centre with good to excellent enantiomeric excess (Scheme 18).43 The experimental evidence indicated the slight influence of the naphthyl-indole hydroxyl group of the starting 3-substituted arylindoles 50 on the enantioselectivity.
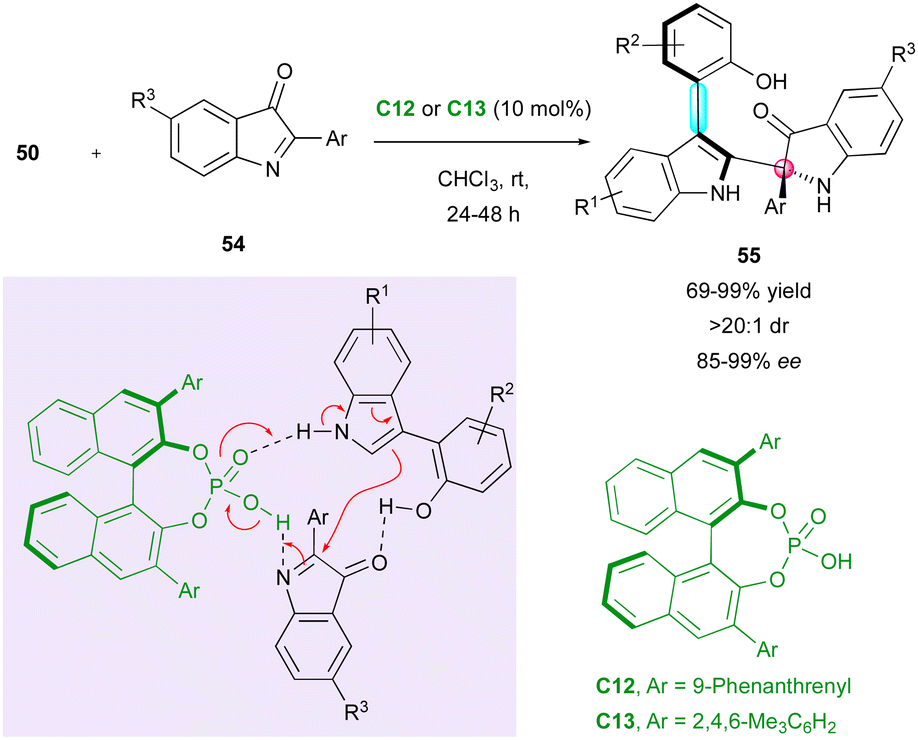 | ||
| Scheme 18 Synthesis of arylindolyl indolin-3-ones with axial/central chiralities. | ||
Concurrently, Shi's research group developed the first example of simultaneous control of a stereogenic axis between two indole nuclei and a stereogenic quaternary centre through the DKR strategy of 2-substituted 3,3′-bisindoles 56 (Scheme 19).44 They selected BINOL-derivative C14 as the catalyst, enabling interaction with both substrates 56 and 57, while governing the reactivity and selectivity of the reaction. The diastereo- and enantioselectivity were excellent, affording complex tris-indole scaffolds 58, representing a rare example of two five-membered ring atropisomers. In 2020, the same group demonstrated the feasibility of the reaction with isatin-derived imines as electrophiles utilising SPINOL-derivative catalyst C15, which led to related atropisomeric bis-indoles 60 featuring a C–N stereogenic centre with very high stereoselectivities.45
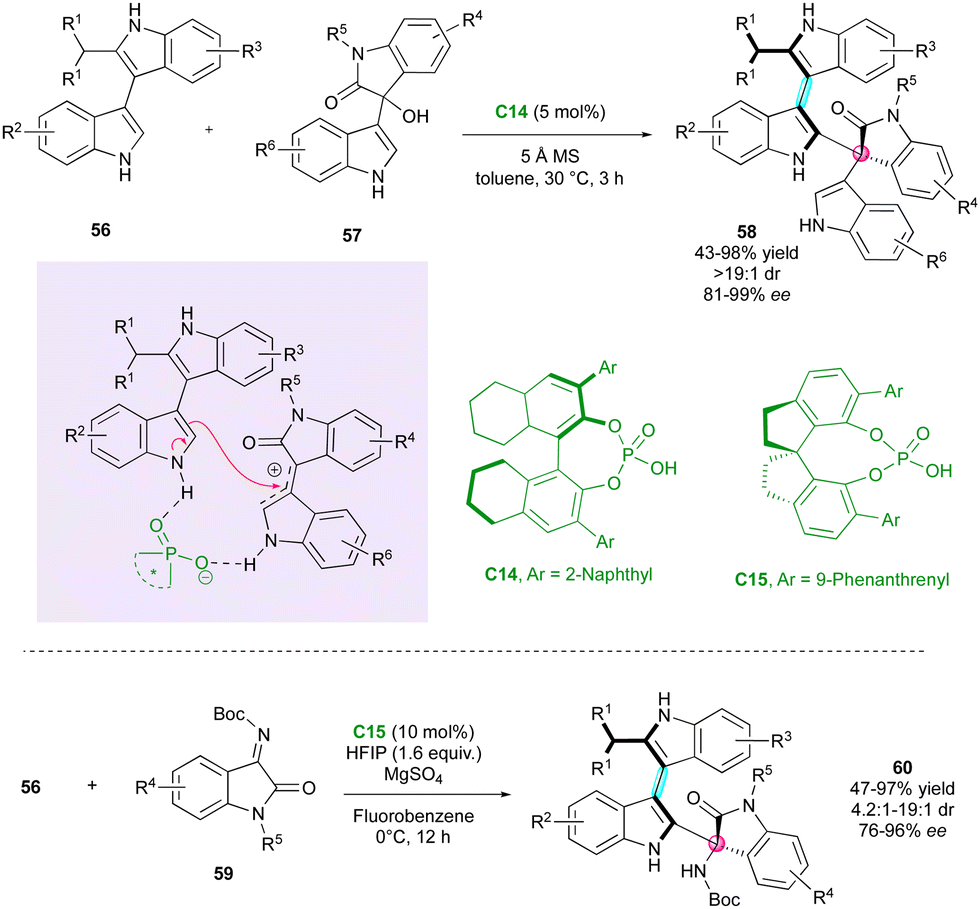 | ||
| Scheme 19 Enantioselective construction of axially chiral 3,3′-bisindole scaffolds. | ||
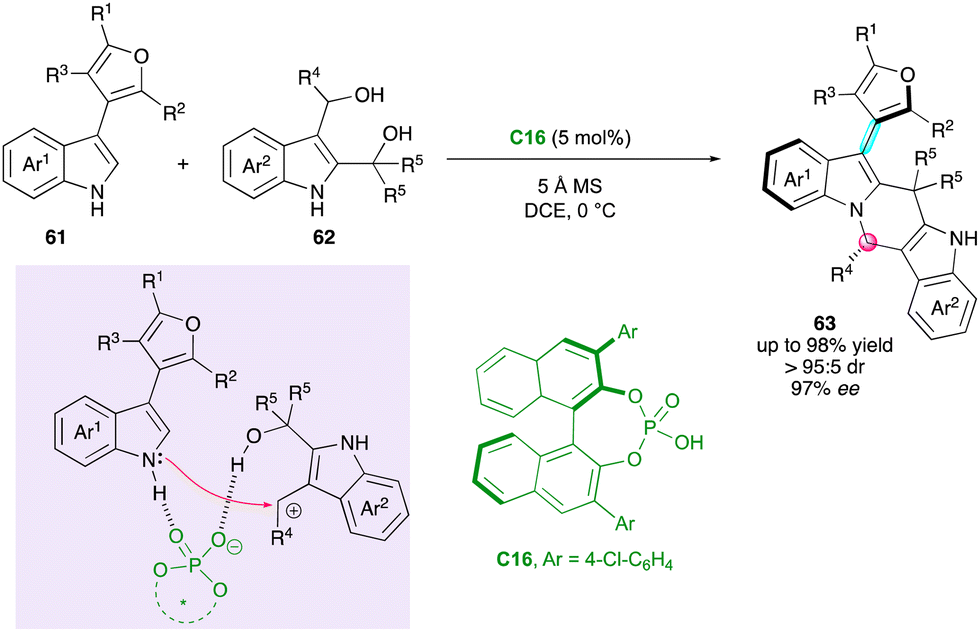
Shi, Tan and Ni applied their previous methodology for the enantioselective and diastereoselective synthesis of furan atropisomers 63 bearing an additional stereogenic carbon atom via the formal (4+2) cycloaddition between pro-atropisomeric 3-furan-indole scaffolds 61 and 2,3-indolyldimethanols 62 (Scheme 20).46 Under CPA organocatalysis with C16, the regioselective functionalisation of both nitrogen and C2 atoms of the indole moiety resulted in a significant increase in the rotational barrier around the C–C bond, giving rise to desired products 63 with high barriers to diastereomerisation via the DKR process. The synthesis of furan atropisomers is a great challenge, as attested by the availability of only a few methods,47 which makes the present work significant. Many examples have been realised with constant good enantio- and diastereoselectivities. In addition, several useful post-functionalisations and deep study on the reaction mechanism were provided, allowing a clear explanation of the enantioselectivity as well as the uncommon regioselectivity observed for this type of cycloaddition.
 | ||
| Scheme 20 Enantioselective synthesis of 3-furan-indoles with central and axial chirality. | ||
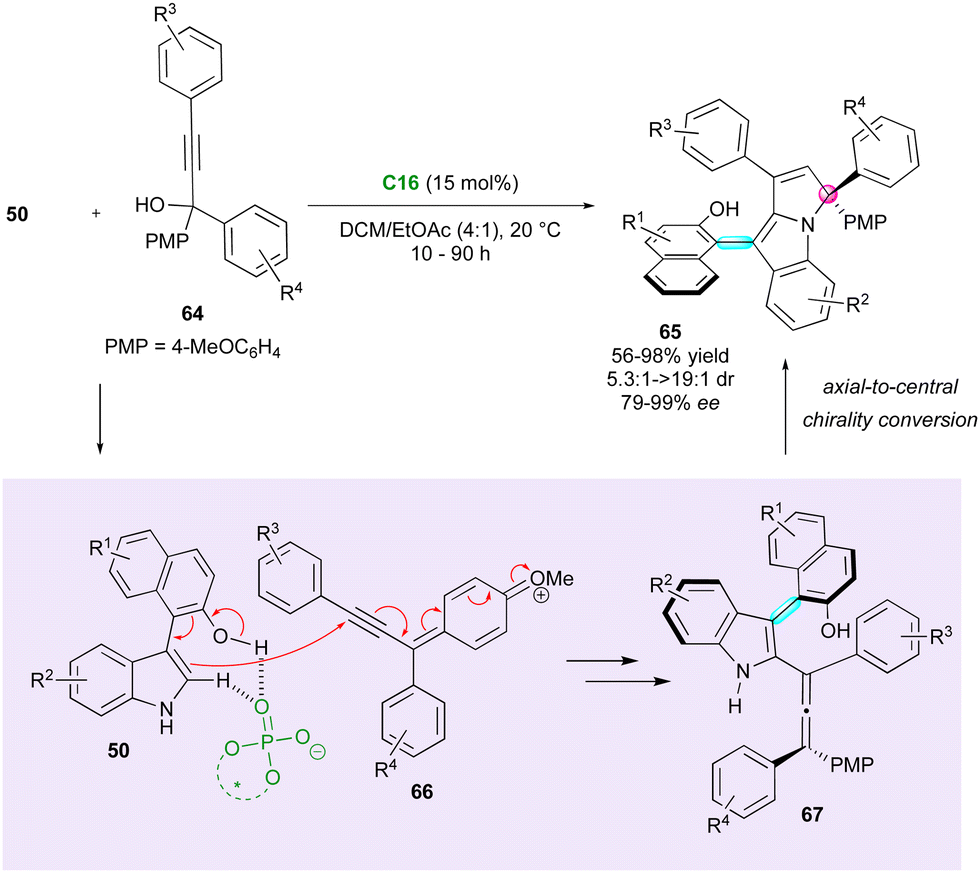
Axially chiral allenes can be considered as crucial intermediates controlling the diastereo- and enantioselectivity during the formation of centrally and axially chiral compounds. In 2023, Jiao, Tan and Shi presented a notable example of this strategy, wherein an elegant DKR approach was employed to create arylpyrroloindoles 65 bearing both a stereogenic axis and a quaternary stereocentre (Scheme 21).48 CPA C16 involved in the reaction governs both the stereochemistry and regioselectivity. The catalyst activates the OH and NH groups of arylindoles 50 and triggers the formation of key p-quinone methide cations 66, resulting in the generation of reactive chiral allene intermediates 67. Subsequent protonation of the allene function by the CPA and trapping by the nitrogen atom of indole, with axial-to-central conversion of chirality, finalised the overall (2+3) cycloaddition to fused tricyclic atropisomers 65. This methodology afforded good to excellent diastereoselectivity and moderate to excellent enantioselectivity. However, a slight limitation reported is the required presence of a p-methoxyphenyl substituent (PMP) to generate the key p-quinone methide cation intermediate.
 | ||
| Scheme 21 Organocatalytic enantioselective (2+3) cyclisation for the synthesis of axially chiral arylpyrroloindoles. | ||
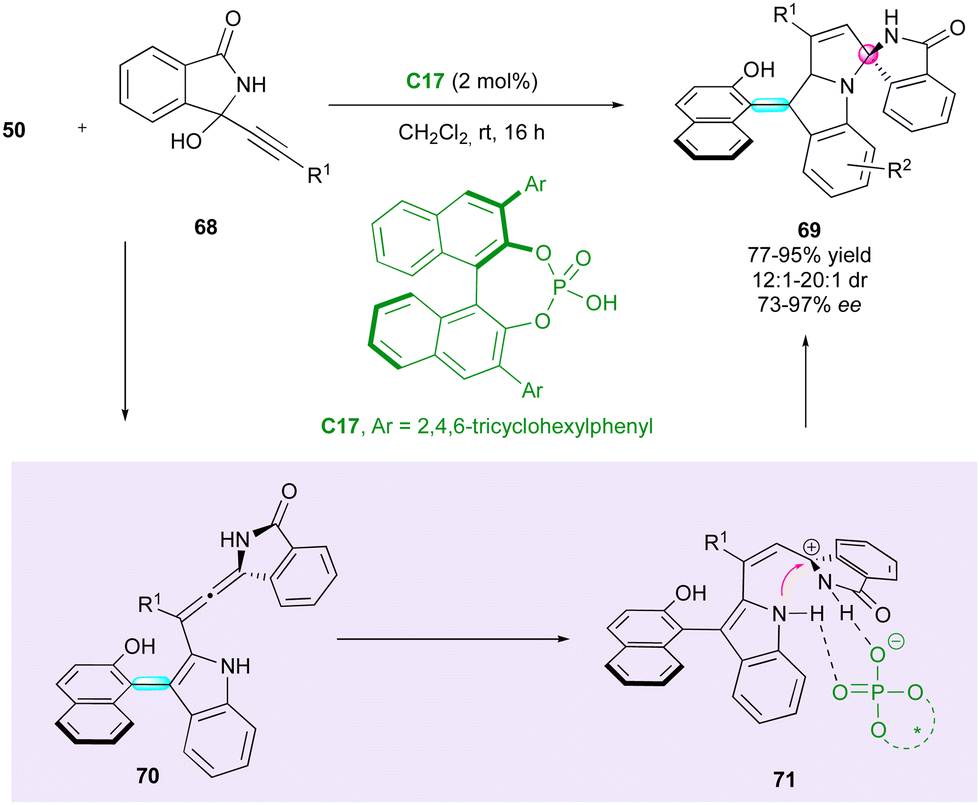
Shortly after, Li, Dong and Li reported a related approach for enantioselective access to indolyl-pyrroloindoles 69 bearing both axial and spiro-central chirality (Scheme 22).49 This (3+2) cyclisation between arylindoles 50 and propargylic alcohols 68 was catalysed by CPA C17, albeit requiring a lower catalytic loading (2 mol%). Notably, the diastereoselectivity exhibited moderate improvements for specific products compared with previous work. It is imperative to note that the presence of an R1 aryl group plays a crucial role in governing the enantioselectivity. Indeed, the introduction of a cyclopropyl group resulted in the formation of racemic product 69. From a mechanistic point of view, dehydration of 68 gave a N-acyl propargylic imine, which underwent enantioselective 1,4-addition with 50, giving rise to enantioenriched chiral allene intermediate 70. Protonation of the latter led to cationic intermediate 71 and subsequent diastereoselective intramolecular annulation furnished the desired product 69.
 | ||
| Scheme 22 Enantioselective synthesis of axially/centrally chiral indolyl-pyrroloindoles. | ||
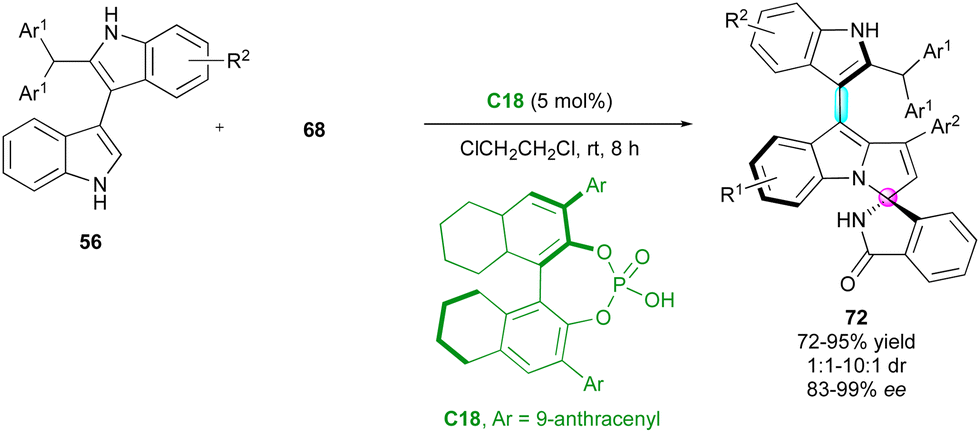
The last closely related example of this spiroannulation strategy was published by Zhang, Shi and co-workers (Scheme 23).50 This challenging regio-, diastereo- and enantioselective cycloaddition involved 2-substituted 3,3′-bisindoles 56 and isoindolinone-based propargylic alcohols 68 catalysed by CPA C18. In comparison with the precedent examples, the final bisindolyl-pyrroloindoles 72 exhibited lower diastereomeric ratios but with consistently excellent enantiomeric excesses.
 | ||
| Scheme 23 Enantioselective synthesis of bisindolyl-pyrroloindoles. | ||
Axially chiral C–N atropisomers featuring a five-membered ring are not commonly encountered in the literature and still constitute a challenging synthetic target. In 2021, Kwon's group published the first enantio- and atroposelective Pictet–Spengler reaction of pro-atropisomeric amino-functionalised N-aryl indoles 73 and benzaldehydes 74 involving a CPA-(C17)-organocatalysed DKR strategy (Scheme 24).51 Although the diastereoselectivity was moderate to good, the enantiomeric excesses were excellent despite the high temperature of the condensation, highlighting the strong configurational stability of the obtained axially and centrally chiral N-aryl-tetra-hydro-β-carbolines 75. The presence of a secondary amine in the ortho position of the N-aryl substituent of 73 was crucial for the stereocontrol due to the hydrogen-bond interactions with the CPA catalyst. Moreover, reactivity was only observed when electron-withdrawing groups were introduced on benzaldehydes 74 (R2 = CN, CF3, NO2). The same group exploited this approach using related indoles bearing an o-amidoaryl group on the N-1, C-2 or C-3 positions, which underwent efficient DKR under CPA catalysis with bulky electrophiles, yielding valuable C–N indole atropisomers with central chirality.52
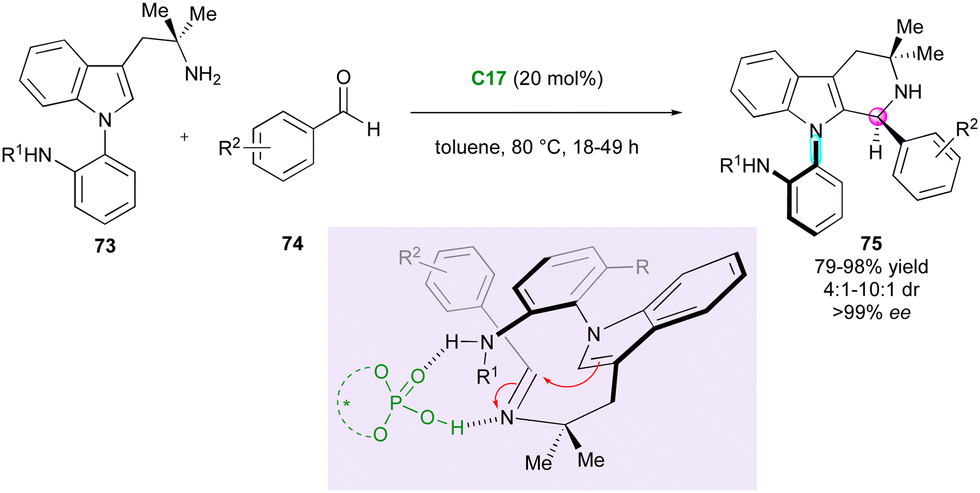 | ||
| Scheme 24 CPA-Catalysed atroposelective Pictet–Spengler reaction. | ||
To date, all the DKR strategies reported in this review were centred on the resolution of pro-axially chiral substrates with low barriers to enantiomerisation. However, in 2020, Chen and Chen demonstrated an interesting complementary extension to racemic centrally chiral compounds via remote vinylogous DKR. They reported an original cascade catalysed by Takemoto thiourea C19 between chiral indenylidene malononitrile (±)-76 and ortho-substituted β-nitrostyrenes 77, simultaneously governing the configuration of a stereogenic axis and a stereocentre (Scheme 25).53 The racemisation of (±)-76 relies on a fast reversible deprotonation-protonation process at the benzylic position, and both the diastereomeric ratio and the enantiomeric excess are controlled during the Michael addition of chiral vinylogous nucleophilic intermediate 79. Subsequently, the stereogenic axis is formed in the final oxidative aromatisation under an oxygen atmosphere through a central-to-axial chirality conversion. However, although this strategy is efficient in terms of diastereo- and enantiocontrol and elegant in the construction of diverse stereogenic elements, the presence of the strongly electron-withdrawing α,α-dicyanoolefin motif is required to perform the DKR strategy.
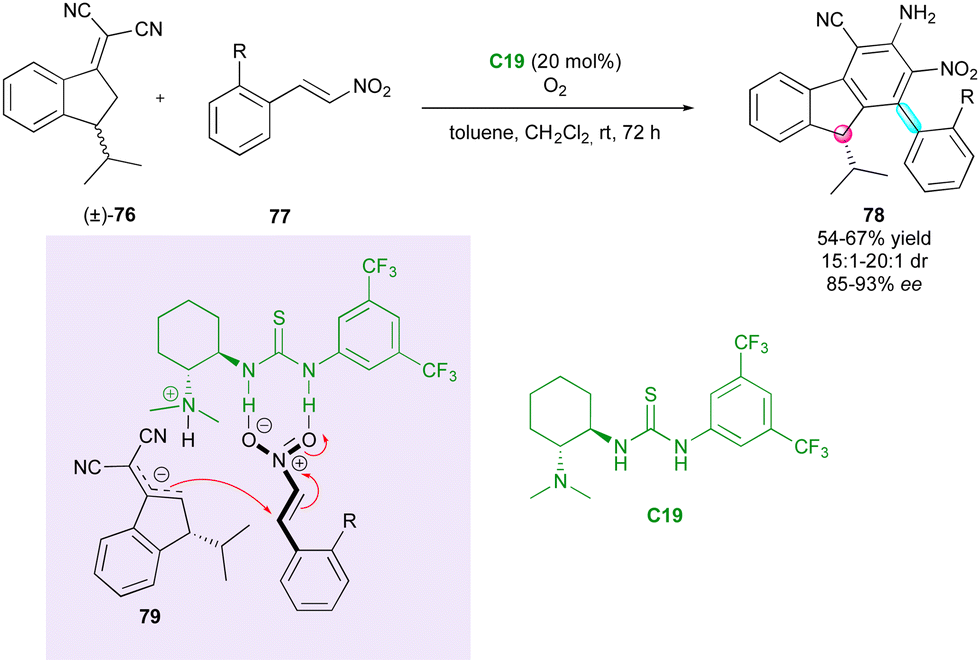 | ||
| Scheme 25 Synthesis of enantioenriched 9H-fluorenes scaffolds via vinylogous DKR. | ||
Recently, chiral bridged biaryls have attracted interest from synthetic chemists due to their numerous applications54 but their enantioselective synthesis is a still significant challenge. An initial contribution in this direction was reported by the Yeung group in 2017, describing access to a rare family of axially and centrally chiral six-membered ring fused biaryls via an organocatalytic enantioselective DKR-semipinacol rearrangement (Scheme 26).55 Racemic fluorenols 80 underwent ring expansion to form brominated axially and centrally chiral 9,10-dihydrophenanthrenones 81 with a central-to-axial conversion of chirality triggered by N-bromophthalimide (NBP) in the presence of (DHQD)2PHAL C20 as a chiral catalyst. The addition of racemic camphor sulfonic acid provided an appreciable improvement in the enantioselectivity and excellent diastereoselectivity.
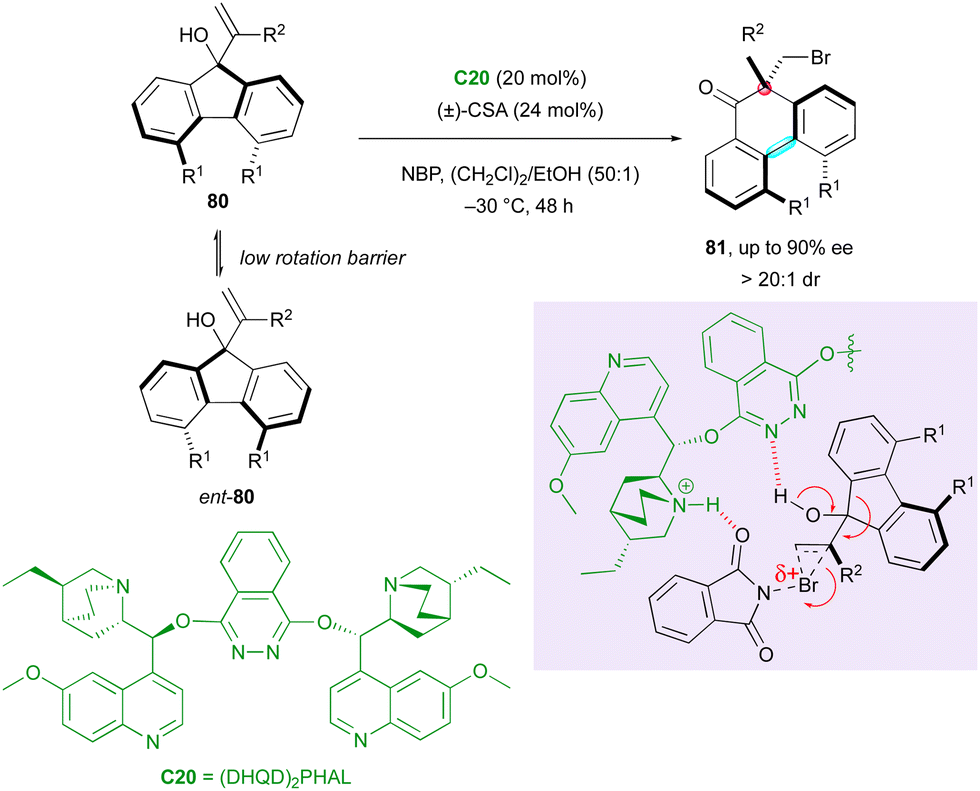 | ||
| Scheme 26 Semipinacol rearrangement to centrally and axially chiral biaryls. | ||
The plausible mechanism based on the configurationally labile starting fluorenols with a low barrier to racemisation relies on the possible activation of NBP by the protonated quinuclidine core during the formation of the bromonium intermediate, while the basic nitrogen of the phthalazine can deprotonate the tertiary alcohol of the substrate, triggering the semipinacol rearrangement.
Another pioneering contribution was reported by the Luan group in 2018, with the first example of the catalytic enantioselective tautomerisation of structurally labile but isolable enamines to access to chiral imines (Scheme 27).56 Kinetically stable pro-axially chiral enamine-based dibenzo[b,d]azepines 82 were tautomerised by simple chiral BINOL–phosphoric acid C21, which offered a range of seven-membered imine products 83 featuring both central and axial stereogenic elements, achieving good yields (up to 96%) with excellent enantioselectivity and diastereoselectivity (up to 97% ee, >20:1 dr). This highly atom-economical process is a rare example of converting one class of kinetically stable enols or enols equivalents into their enantioenriched keto tautomers through an enantioselective tautomerisation strategy.
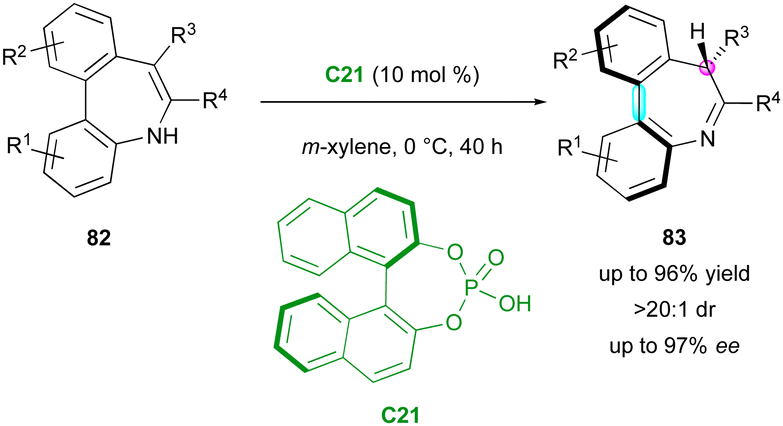 | ||
| Scheme 27 Catalytic enantioselective tautomerisation of enamines. | ||
More recently, the Luo and Zhao group reported an example of enantioselective domino thia-Michael/aldol reaction, simultaneously controlling the formation of three stereogenic centres and one stereogenic axis starting from pro-axially chiral seven-membered ring bridged biaryl ketones 84 as Michael acceptors and 2-mercaptoacetophenones 85 as thia-nucleophiles (Scheme 28).57 Among the different catalysts, the optimal conditions were found with CPA C22 in cyclohexane, directing both the enantio-determining thia-Michael addition and the following diastereoselective aldol reaction by its dual ability to be a hydrogen bond donor and acceptor. The resulting multi-chiral thiochromene-fused dibenzoannulenone derivatives 86 were obtained with excellent yield, enantio- and diastereoselectivity (up to 95% yield, up to 99% ee, >20:1 dr). DFT calculations gave an energy difference of 9.1 kcal mol−1 between the two possible atropodiastereomers, in perfect agreement with the constantly high diastereoselectivity observed.
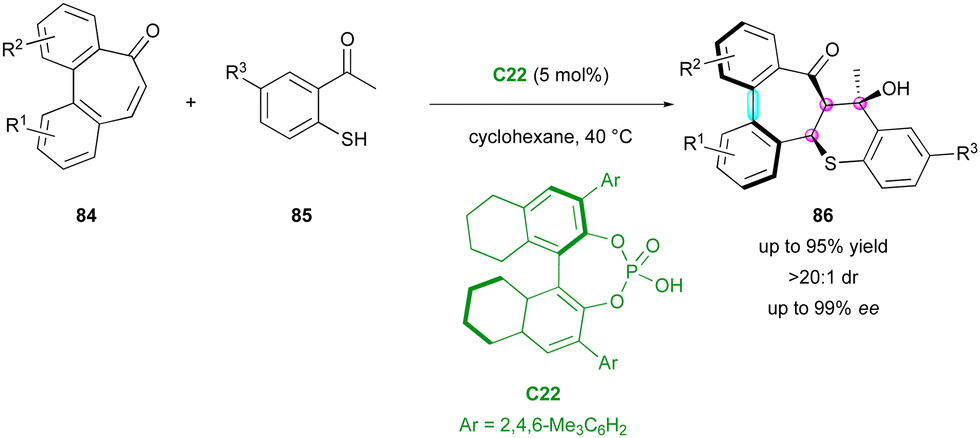 | ||
| Scheme 28 Enantioselective thia-Michael/aldol for simultaneous stereogenic centres and chiral axis formation. | ||
Smith and coworkers reported the unique and elegant enantioconvergent counterion-directed cyclisation of racemic biaryl anilides 87 under phase-transfer catalysis (PTC), allowing the formation of multi-chiral eight-membered ring lactams 88 (Scheme 29).58 Although starting anilides 87 existed as a complex mixture of enantiomers and diastereomeric conformers due to the presence of multiple axes of restricted rotation and a stereogenic centre, chiral ammonium salt catalyst C23 could differentiate between a complex and rapidly equilibrating in situ-formed mixture of enolates and rotational isomers. The overall dynamic and enantioconvergent process allowed the highly enantioselective alkylative ring closure from favored chiral enolates 89, with efficient control of three stereogenic elements including an all-carbon quaternary centre and a two-axis (C–C and C–N) system by virtue of the N-aryl and biaryl bonds.
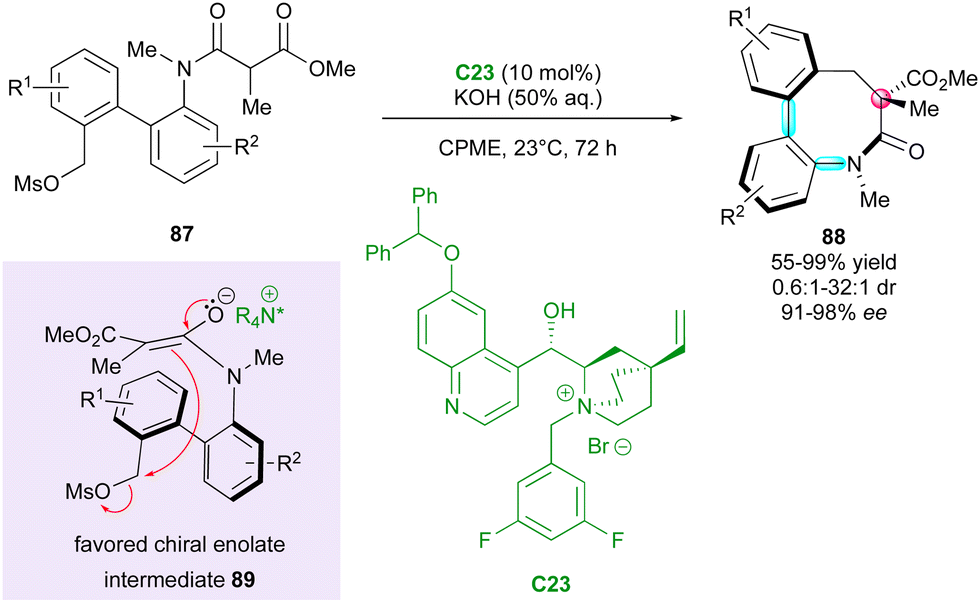 | ||
| Scheme 29 PTC for enantioconvergent synthesis of bis-axially and centrally chiral medium-sized lactams. | ||
Interestingly, the fused biaryl lactam products 88 adopt a deep bowl-like arrangement with a torsion angle of about 60° and a measured barrier to diastereomerisation (bowl inversion) of 124.1 kJ mol−1, meaning that the bowl inversion does not occur at room temperature.
In complement, the Chauhan group simultaneously reported an enantioselective organocatalytic process to obtain various seven-membered ring bridged biaryls, containing one chiral axis and up to four stereogenic centres (Scheme 30).59 This challenging process is based on a stereoselective domino 1,4/1,2 addition or 1,2/oxa-Michael addition, depending on the nucleophile class. Due to squaramide catalysts C24–C26, simple pro-stereogenic biaryl substrates 91 could react with different nucleophiles, such as tert-butylhydroperoxyde, 3-substituted 2-oxindoles and substituted 1,3-dicarbonyls, giving the corresponding products 90, 92 and 93, respectively. After the optimisation of the conditions for these three classes of nucleophiles, the reaction scope was investigated. Only rare examples did not provide the corresponding product with good stereoselectivity, while a broad range of substrates could be employed, giving good yield (39–98%) with high enantio- and diastereoselectivity (1:1 to 20:1 dr, up to >99% ee).
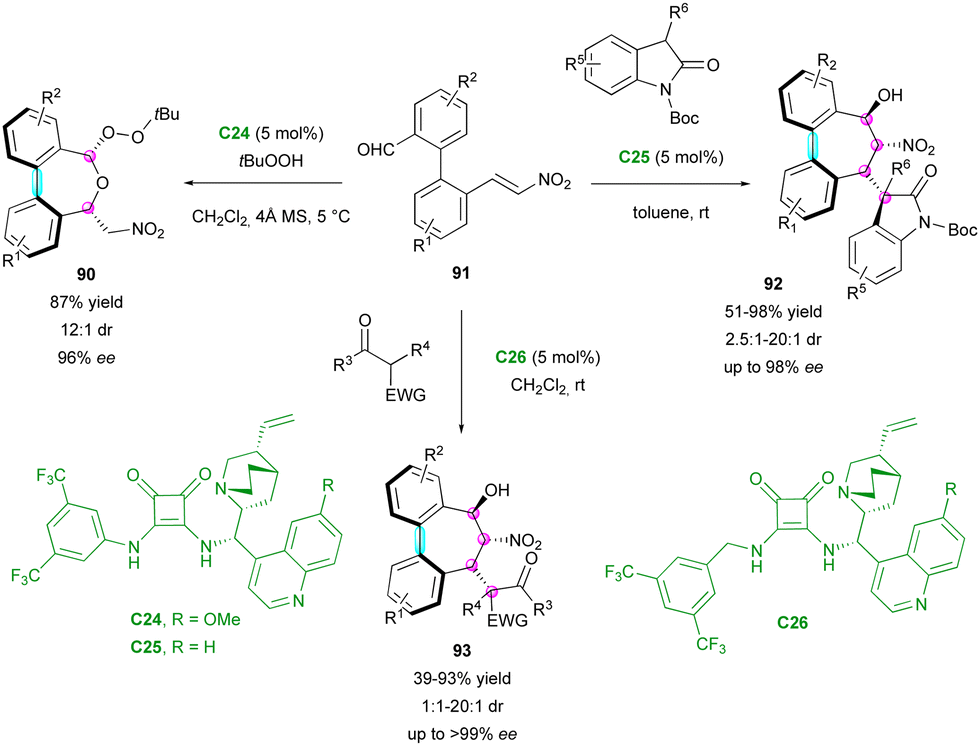 | ||
| Scheme 30 Enantioselective synthesis of seven-membered ring bridged biaryls via nucleophile-dependent switchable domino process. | ||
Very recently, pro-stereogenic biaryl aldehydes 95 were ingeniously used by Wang and co-workers in combination with amino oxindoles 94 for the atropo- and highly diastereoselective synthesis of eight-membered bridged (hetero)biaryl lactones fused to spiro pyrrolidine-oxindole derivatives 96 and 97 (Scheme 31).60 Depending on the structure of staring substrate 95 (X = C or N), these complex molecular architectures displayed either a C–C and C–N stereogenic axis and four contiguous stereocentres. The high degree of diastereocontrol was due to the stereospecificity and stereoselectivity of intramolecular (3+2) cycloaddition involving azomethine intermediate 98 together with the tendency of eight-membered bridged biaryls to form a stable boat conformation. In addition, CPA catalyst C12 or C18 activated both the ester moiety and the 1,3-dipole through hydrogen-bonds, delivering the final products 96 or 97, respectively, with high levels of stereocontrol.
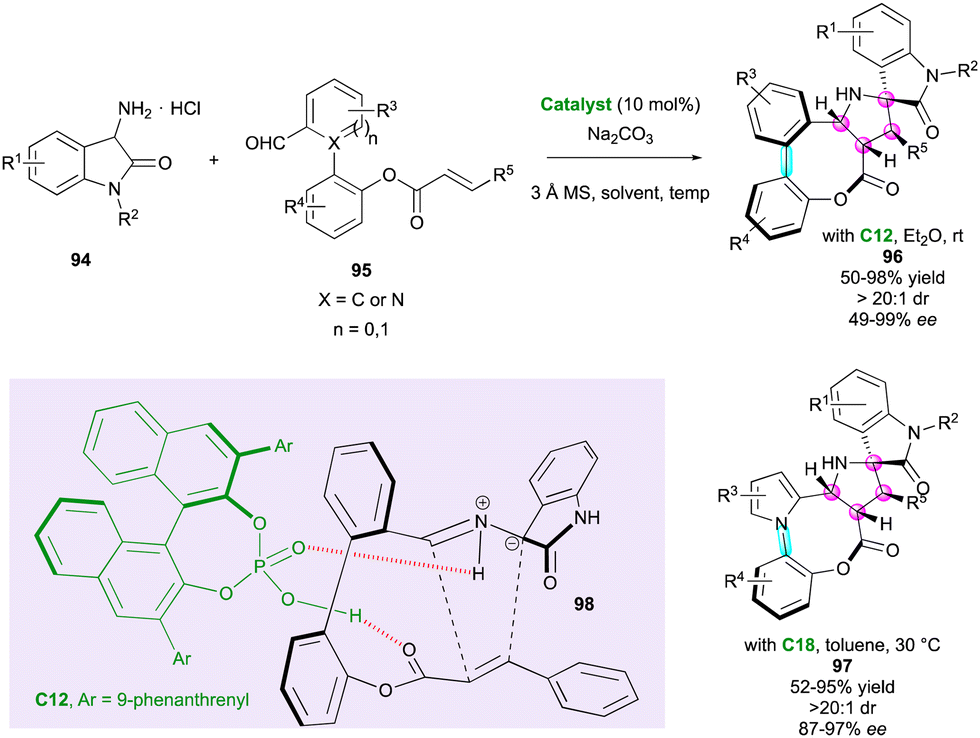 | ||
| Scheme 31 Atroposelective intramolecular (3+2) cycloaddition to eight-membered bridged (hetero)biaryls. | ||
Finally, in 2023, Wang's group published an interesting approach for the simultaneous control of central and helicoidal stereogenic elements through a DKR strategy involving conformationally labile oxa[5]nitro helicenes (±)-100 as Michael acceptors towards various phosphine oxides 99 (Scheme 32).61 The phospha-Michael addition was catalysed by peptide phosphonium salt C27 interacting with both substrates through a hydrogen bonding network and ionic interactions. This innovative methodology enabled the synthesis of various phosphorus-containing hetero helicenoid structures 101 with excellent diastereoselectivity and good to excellent enantiomeric excess.
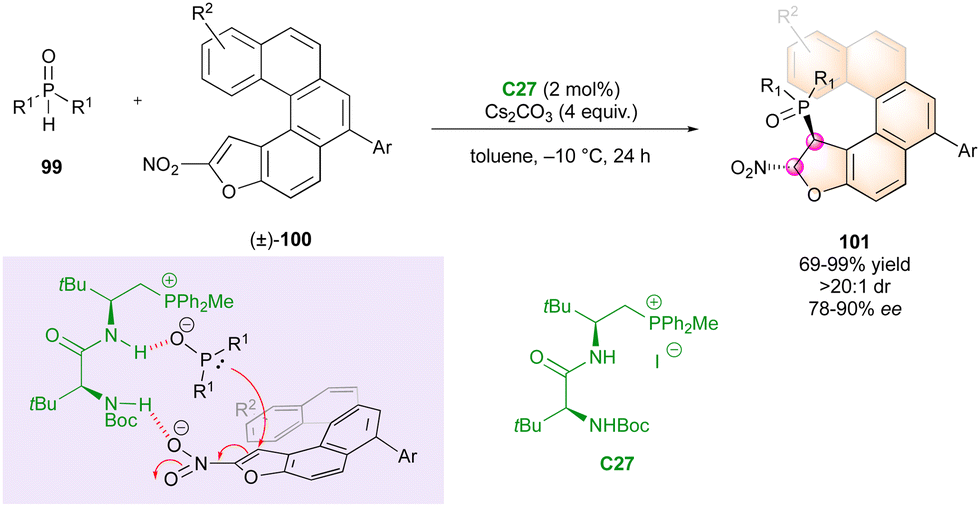 | ||
| Scheme 32 DKR for the enantioselective synthesis of P-containing helicenoids. | ||
Metal-catalysed activation
Besides organocatalysed methodologies, the DKR strategy for accessing molecules bearing multiple stereogenic elements can also be efficient using transition metal catalysis. In this regard, in 2018, the Fernández and Lassaletta group published a series of works on the DKR of N-heterobiaryls. Their first proposal dealt with a palladium-catalysed atroposelective Heck reaction with chiral BINAP-type ligand L3 of a variety of quinoline, quinazoline, phthalazine, and picoline sulfonate derivatives 102 with 2,3-dihydrofuran 103 (Z = O) and N-Boc protected 2,3-dihydropyrrole 103 (Z = NBoc) (Scheme 33).62 From a mechanistic point of view corroborated by experimental observations and DFT studies, after oxidative addition, the formation of five-membered cationic palladacycle 106 is essential for reactivity. Then, the following insertion/β-elimination sequence leads to the major axially and centrally chiral olefins 104 together with the corresponding isomerised olefins 105 with good selectivity ratios, ranging from 6:1 to >20:1, for dihydrofurans 104/105 (Z = O), while dihydropyrroles 104/105 (Z = NBoc) were obtained with slightly lower selectivity from 3:1 to 6:1.
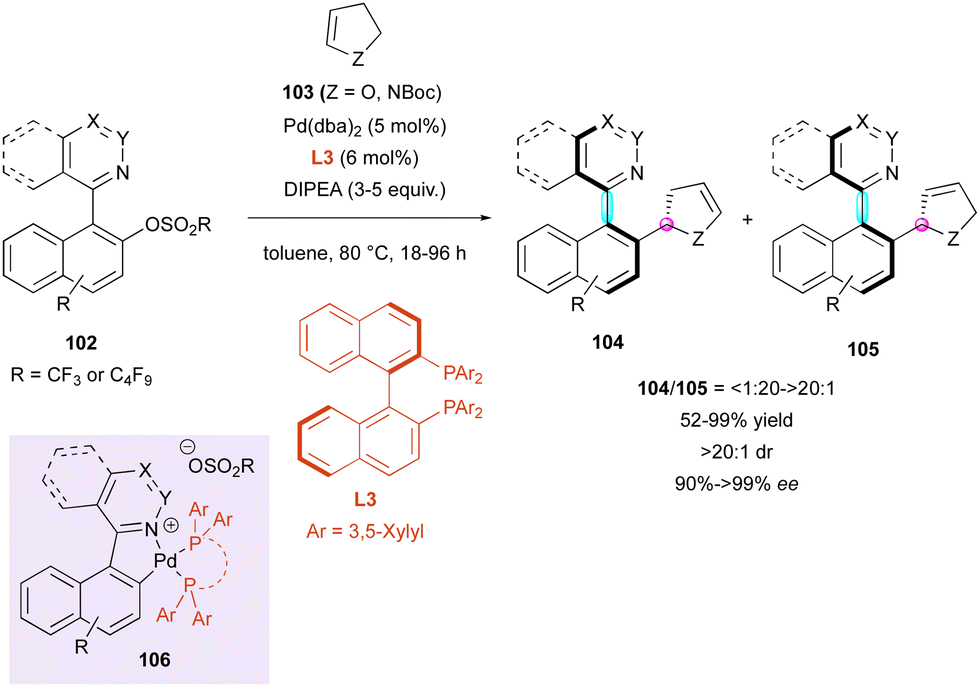 | ||
| Scheme 33 DKR via Pd-catalysed atroposelective Heck reaction. | ||
Another complementary DKR approach by the same group concerns the enantioselective reduction of racemic configurationally labile heterobiaryl ketones (±)-107 (Scheme 34). Thus, they developed a zinc-catalysed enantioselective hydrosilylation with chiral diamine ligand L4, leading to axially and centrally chiral N-heterocycles 108 with moderate to excellent diastereoselectivity (2:1 to >20:1 dr) and very good enantioselectivity (80% to 98% ee).63 This strategy is based on racemisation by a ring-closing/ring-opening event through a transient intramolecular Lewis acid–base interaction, resulting in five-membered ring cyclic intermediate 109. Intriguingly, perfect diastereoselectivity was observed in the case of the sterically challenging ketone (dr >20:1 for R = CH2tBu), albeit with lower enantioselectivity (80% ee) and yield (52%). Unfortunately, beside this example, the diastereoselectivities were moderate (up to 8.5:1 dr), but with consistently high enantioselectivities (>90% ee).
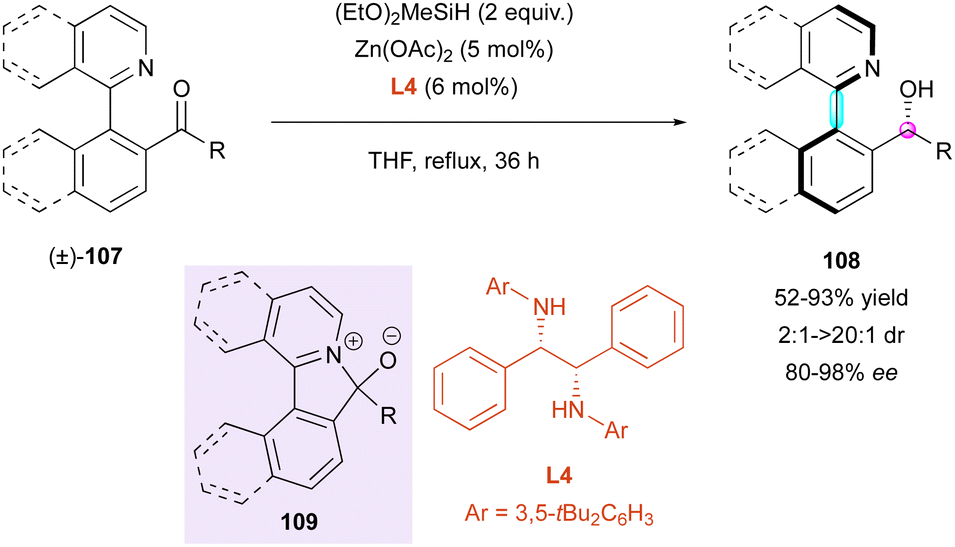 | ||
| Scheme 34 Zn-Catalysed enantioselective hydrosilylation. | ||
Recently, this group applied their precedent ring-closing/ring-opening strategy61 through transient Lewis acid–base interaction to very similar substrates (±)-110, forming six-membered ring intermediate 112 (Scheme 35). In that respect, aldehydes 110 could undergo enantioselective iridium-catalysed transfer hydrogenative allylation, leading to the formation of centrally and axially chiral homoallylic alcohols 111 with good diastereoselectivity (4.2:1 to 10:1 dr) and excellent enantioselectivity (88% to 98% ee).64 Although no value was given, supposedly high rotational barrier of products 111 allowed the reaction to be performed at high temperature (100 °C, 20 h) without any reduction in the stereoselectivity.
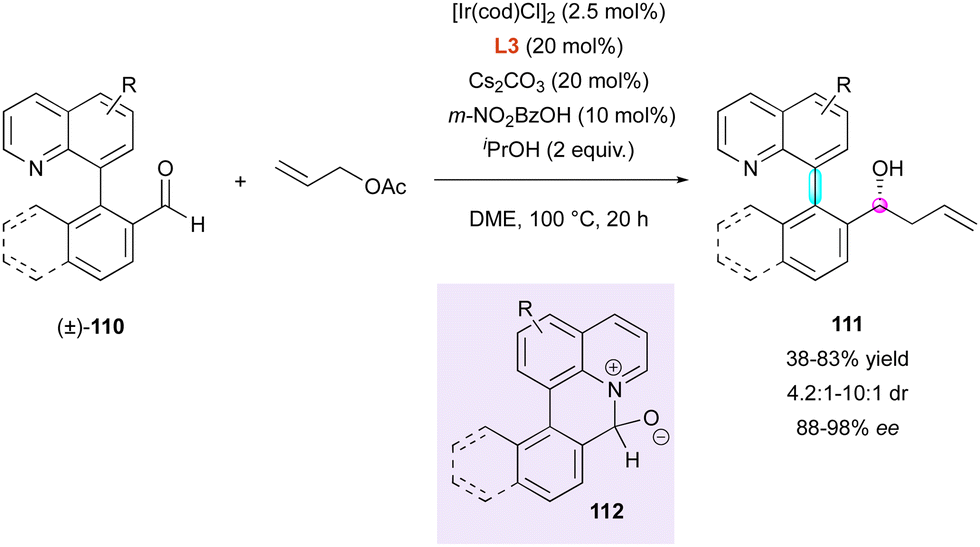 | ||
| Scheme 35 Ir-Catalysed transfer hydrogenative allylation. | ||
Simultaneously, the same group proposed a closely related transient Lewis acid–base interaction for the DKR of configurationally labile 3-thioaryl indole-2-carbaldehydes (±)-113 (Scheme 36).65 This method exploited the reactivity of chiral bis-oxazoline rhodium-complex C29 as the catalyst in a reductive aldol reaction with simple acrylates, simultaneously creating two adjacent stereocentres together with the stereogenic axis on products 114 with uniformly very high enantiomeric excesses (87% to 99% ee). In this case, the DFT computational studies were consistent with a racemisation process involving 2H-thiopyran transition-state 115 with a barrier to enantiomerisation of 100.7 kJ mol−1, in total agreement with a possible DKR at ambient temperature and above. It is worth noting that good diastereoselectivity was observed for R1 = Me (3:1 to >20:1 dr), whereas perfect diastereoselectivity was achieved for R1 = Et or Bn (>20:1 dr). Interestingly, n-butyl acrylate could be used instead of tert-butyl acrylate, albeit with moderate enantioselectivity (69% ee for R1 = Me, R2 = H, R3 = Bn).
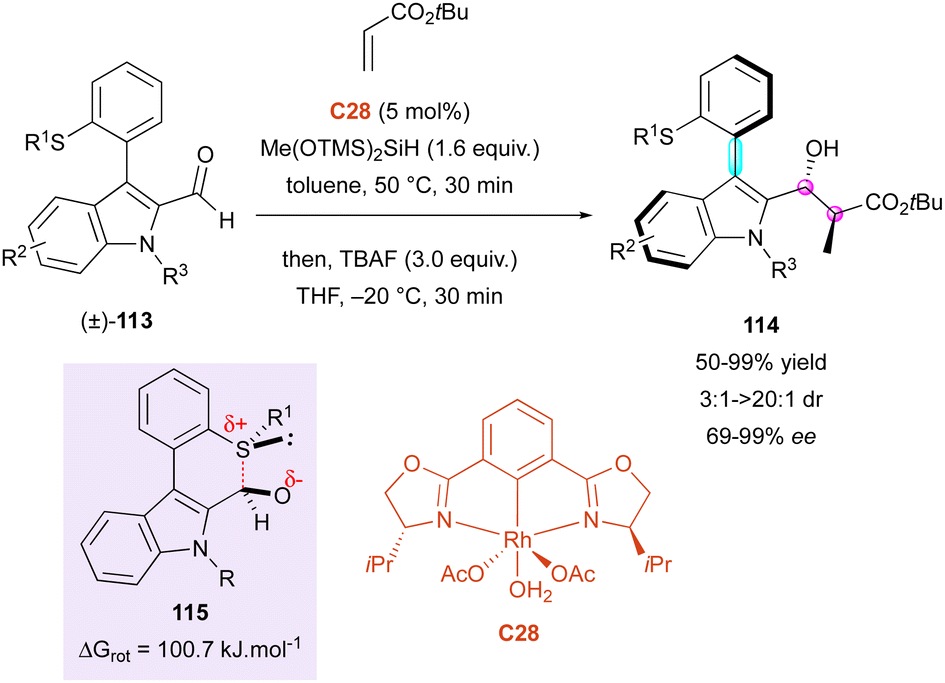 | ||
| Scheme 36 Rh-Catalysed reductive aldol reaction. | ||
Another strategy to functionalize pro-axially chiral substrates is to develop C–H functionalisation close to the stereogenic axis, which delivers atropostable products via DKR. In this case, the Fernández and Lassaletta group developed the Ir-catalysed atroposelective C–H functionalisation of pro-axially chiral heterobiaryls 116via the hydroarylation of acyclic vinyl ethers and bicycloalkenes 119 (Scheme 37).66 In the presence of chiral diphosphine ligand L5, simple vinyl ethers gave complete branched/linear selectivity, leading to axially and centrally chiral heterobiaryls 117 with excellent diastereomeric ratio and high enantioselectivity. In contrast, 2,3-dihydrofuran gave the opposite configuration and the corresponding hydroarylated derivatives 118 were obtained with high enantioselectivity but moderate diastereoselectivity (dr from 2:1 to 3.5:1). In fact, the diastereoselectivity seemed to be closely related to the steric hindrance of the ether, as also shown by the relatively lower diastereomeric ratio of ethyl vinyl ether (dr <15:1 for R2 = Et) compared to all others (dr > 20:1). Alternatively, the use of prochiral norbornene substrates 119 led to a double desymmetrisation process, leading to hydroarylated heterocyclic systems 120 with perfect control of up to five stereogenic carbon atoms and a stereogenic axis in high yields with excellent enantioselectivity.
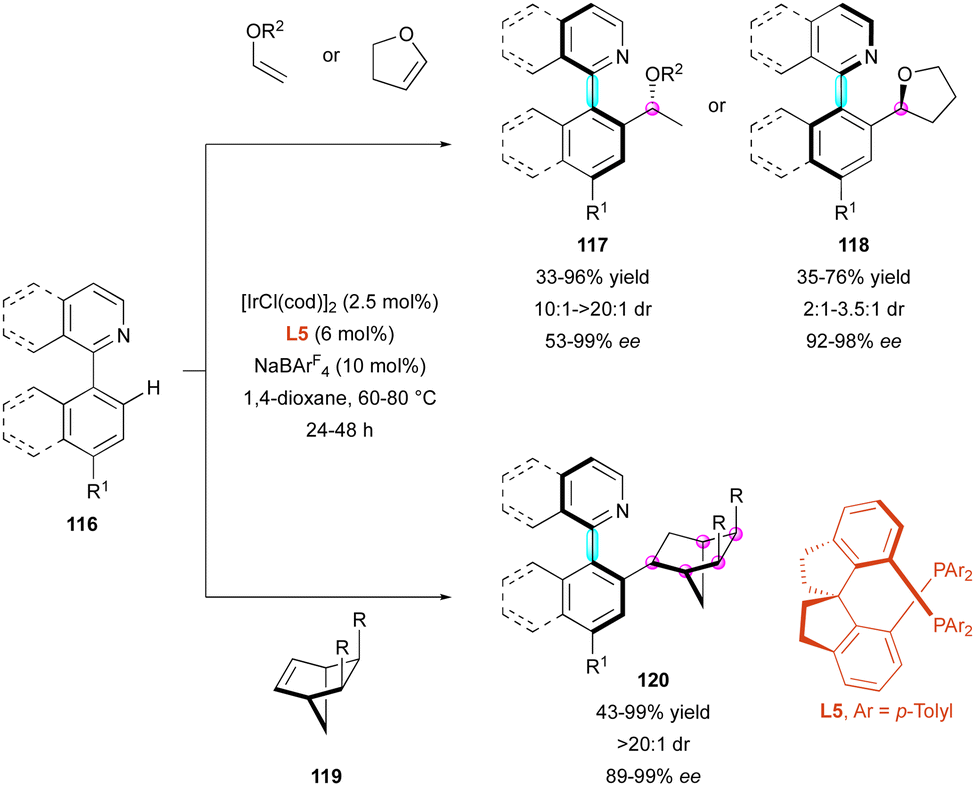 | ||
| Scheme 37 DKR of heterobiaryls via Ir-catalysed atroposelective hydroarylation. | ||
Similarly, the Xing group exploited the aforementioned Ir-catalysed atroposelective C–H functionalisation to broaden the DKR of pro-axially chiral 121 using aliphatic alkenes (Scheme 38).67 Slight modifications of the reaction conditions involving diphosphine ligand L6 allowed the construction of nine new examples of heterobiaryls 122 with perfect diastereo- and enantioselectivities.
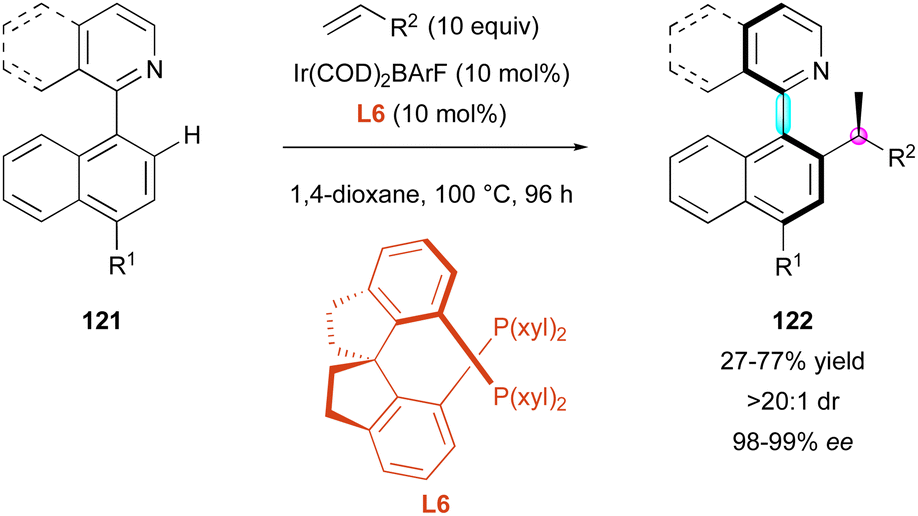 | ||
| Scheme 38 Ir-Catalysed atroposelective hydroarylation with aliphatic alkenes. | ||
Recently, the Ackermann group also exploited ruthenium catalysis for the enantioselective C–H activation of pro-axially chiral N-isoquinoline-indoles 123 with functionalised terminal alkenes 124 in the presence of chiral carboxylic acid ligand L7 (Scheme 39).68 In this case, the authors could isolate a wide range of branched products 124 bearing a C–N stereogenic axis and a stereocentre as single diastereomers (>20:1 dr). The large number of examples highlighted the beneficial effect of an aromatic ring on the olefin partners given that alkyl-substituted alkenes (R3 = Alk) proceeded with lower enantioselectivity compared to vinylarenes (R3 = Ar). These results are consistent with a probable π-chelating interaction between ligand L7 and alkenes, playing a pivotal role in the enantiocontrol. Also, vinyl silanes (R3 = SiR3) were well supported by this method to produce valuable chiral organosilanes 124 (R3 = SiR3) with good enantioselectivities (68% to 95% ee), albeit with a small drop in the yield (35% to 75% yield).
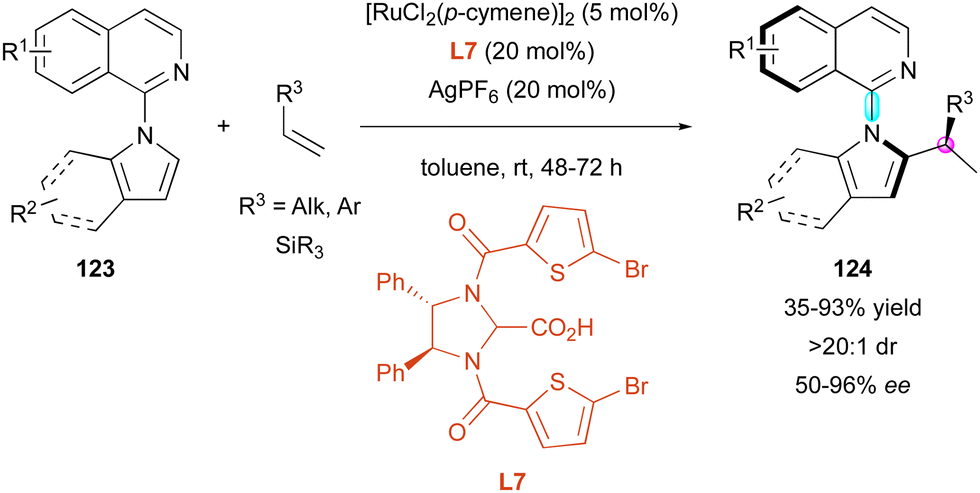 | ||
| Scheme 39 DKR of N-isoquinoline-indoles via Ru-catalysed C–H activation. | ||
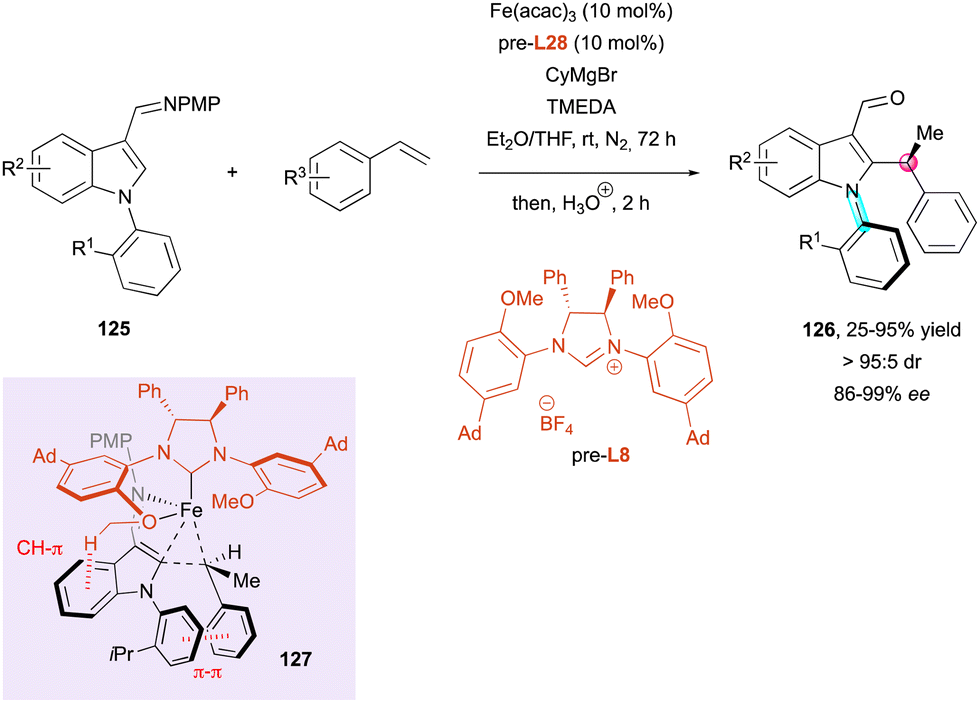
An impressive iron-catalysed C–H alkylation of indoles 125 with alkenes was recently disclosed by Neidig, Wencel-Delord and Ackermann for the simultaneous introduction and control of central and axial chirality in substituted indoles 126 (Scheme 40).69 The careful design of NHC ligand pre-L8 allowed excellent diastereocontrol and very high enantioselectivities to be achieved in final products 126. Deuterium-labelling experiments and DFT calculations shed light on the reaction mechanism, which occurs by ligand-to-ligand hydrogen transfer (LLHT) rather than the classical C–H bond oxidative addition. In addition, the stereocontrol of both stereogenic elements is supposed to occur via transition state 127, which displays favorable CH–π and π–π interactions and determines both the configuration of the stereogenic axis and the carbon atom. These findings highlight the crucial role of the methoxy group of the NHC ligand in the stereochemical outcome of the reaction.
 | ||
| Scheme 40 Fe-Catalysed C–H alkylation of indoles. | ||
In their work on Ni/Al-catalysed atroposelective C–H alkylation using heteroatom-substituted secondary phosphine oxide (HASPO) preligands, Hong and Ackermann described two examples of benzimidazole C–N atropisomers bearing an additional stereocentre with very high diastereo- and enantioselectivities.70
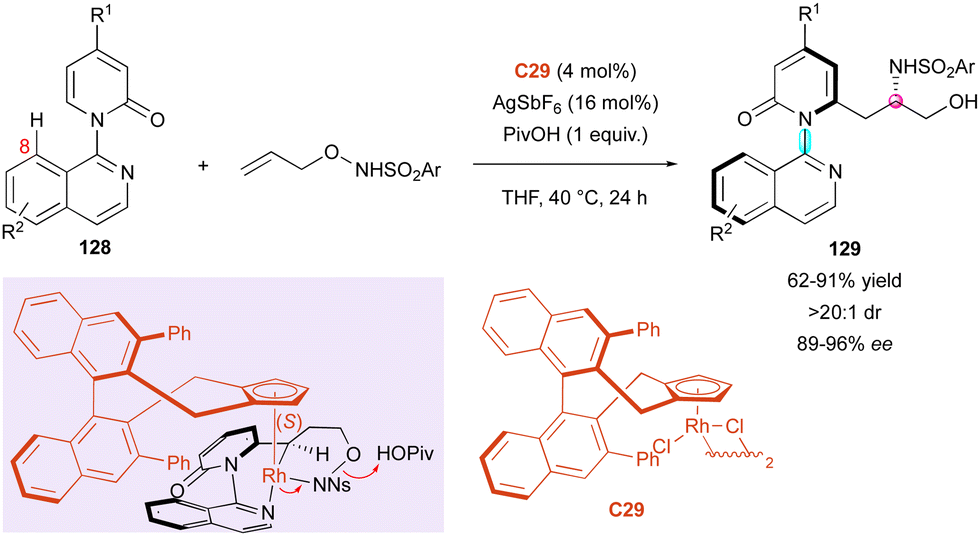
The Li group capitalised on their work on rhodium-catalysed C–H activation for the kinetic resolution of racemic axially chiral pyridone rings (±)-25 (Scheme 9).31 In continuation, the authors proposed a complementary DKR process involving pro-axially chiral pyridones 128 lacking substitution at the 8 position (Scheme 41). The resulting carboamidation with N-protected O-allylhydroxyamines proceeded with perfect diastereoselectivities (>20:1 dr) and high enantiomeric excesses (89% to 96% ee). Nevertheless, modification of the substrates mostly concerned the N-protected O-allylhydroxyamine substrate with a large panel of sulfonyl groups but only one variation of R1 in 128 (R1 = H or R1 = CO2Me). The proposed transition state with Rh-complex C29 coordinated with the pyridine ring and the sulfonyl amide accounts for the excellent enantioselectivity. Due to the relatively free rotation around the C–N axis, which allows substrate 128 to react in its aS configuration, atropisomeric amino alcohols (aS,S)-129 are selectively obtained.
 | ||
| Scheme 41 Rh-Catalysed C–H activation of pyridone rings towards amino alcohol atropisomers. | ||
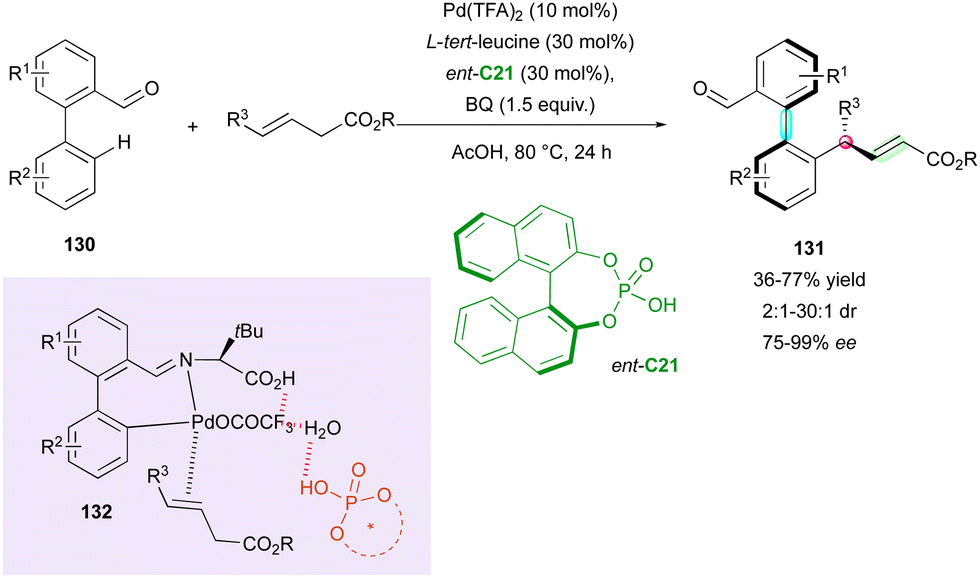
The aldehyde function can also be used as a directing group for the C–H functionalisation of biaryls. Recently, the Ge, Sunoj and Maiti groups proposed the palladium-catalysed C–H allylation of pro-axially chiral biaryl aldehydes 130 with trans-3-alkeneoates proceeding via a DKR process with control of three stereogenic elements, i.e., one C–C axis, one central chirality and the alkene configuration (Scheme 42).71 Cooperative assistance by CPA ent-C21 and L-tert-leucine provided a highly stereoselective transformation with good enantioselectivity and perfect regio- and (E)-selectivity. The DKR pathway was supported by the low barrier to interconversion of the starting racemic aryl aldehydes 130, calculated at 80.6 kJ mol−1, in agreement with rapid racemisation and indicating the faster reactivity of (Ra)-130. In-depth DFT calculations unveiled the mechanistic pathway, indicating that the axial chirality is controlled by the formation of chiral transient imine 132 as a directing function, while the central chirality and stereochemistry of the alkene are controlled by the migratory insertion of [Pd]. The Re prochiral face is energetically favored, leading to (Sa,R) products 131.
 | ||
| Scheme 42 Pd-Catalysed DKR C–H allylation. | ||
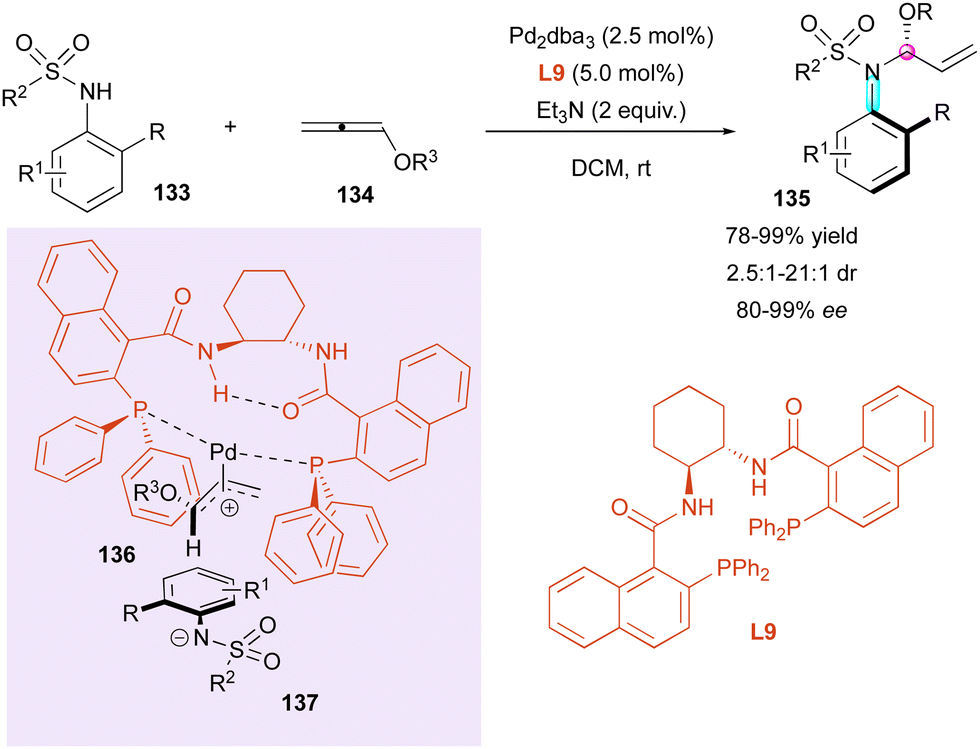
Inspired by the work reported by Rhee on the hydroamination of allenes 134 to construct N,O-acetals with one stereogenic centre,72 in 2021, Zhou and Zhang developed an hydroamination of allenes with pro-axially chiral sulfonamides to implement a C–N axis as an additional stereogenic element (Scheme 43).73 Using bidentate chiral ligand L9 with Pd2dba3 as the catalyst, high enantio- and diastereoselectivities were obtained with aryl sulfonamides 133 as the starting material, while alkyl sulfonamides led to a reduction in the diastereomeric ratio (dr = 2.5:1). The origin of the stereoselectivity was rationalised by DFT calculations, with a more favorable transition state involving palladium π-allyl intermediate 136, which underwent nucleophilic addition of sulfonamide anion 137.
 | ||
| Scheme 43 Pd-Catalysed hydroamination of allenes. | ||
The formation of axially chiral bridged molecules via DKR of pro-stereogenic substrates can be an efficient approach for the introduction and control of multiple stereogenic elements. In 2020, Zhang and Yin reported a reductive amination/ring closing cascade to afford enantioenriched bridged atropisomers 139 of the dibenzo[c,e]azepin-5-one family, catalysed by a ruthenium complex (Scheme 44).74 The reaction was optimised starting from biaryl keto esters 138 using Ru(L10)(OAc)2 as the catalyst, excess of NH4OAc as the ammonia source under a high-pressure hydrogen atmosphere (50 bar) in trifluoromethanol at high temperature (100 °C). These very harsh conditions gave the best results in terms of yield and enantioselectivity (up to 99% ee). Methyl or ethyl ketones performed the best, while phenyl ketones required the stoichiometric use of Ti(OiPr)4 and only afforded the products in low yield and lower enantioselectivity (45% yield, 81% ee). They proposed that the configuration of the stereogenic axis is induced by the configuration of the stereogenic centre, which is created and controlled in the initial reductive amination step.
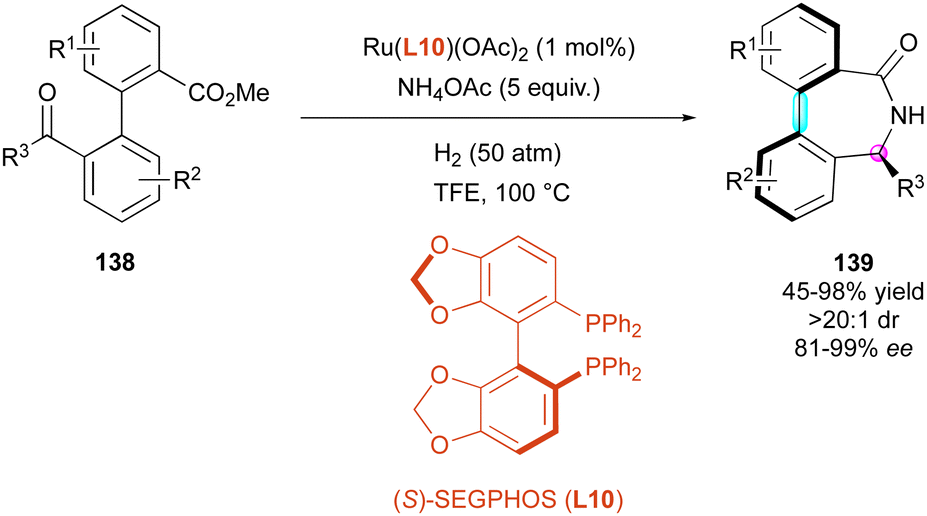 | ||
| Scheme 44 Ru-Catalysed reductive amination/ring closing cascade. | ||
In 2021, the Luo and Zhu groups reported the enantioselective synthesis of related dibenzo[b,d]azepin-6-ones 141via a carbopalladation/carbonylation cascade (Scheme 45).75 The prepared heterocyclic seven-membered ring lactams contain both central and axial stereogenic elements. This palladium-catalysed reaction was optimised with 2′-iodo-[1,1′-biphenyl]-2-yl-N-methylacrylamides 140 under a CO atmosphere and various nucleophiles. As predicted by DFT calculations, excellent diastereo- and enantioselectivity were obtained using (S)-AntPhos L11 as a chiral ligand after 10 h of reaction in toluene. Although a high ligand loading is required {[Pd]/L = 1:9.6}, different nucleophiles such as anilines and alcohols were successfully engaged in this reaction. The observed configuration was explained by the strong steric repulsion after the oxidative addition of the aryl iodide to the [Pd(0)] species, which forces the terminal alkene to approach [Pd(II)] away from the ligand. As suggested by 1H NMR, where only one product was detected, the atropisomerism is thermodynamically controlled by the newly formed stereogenic centre.
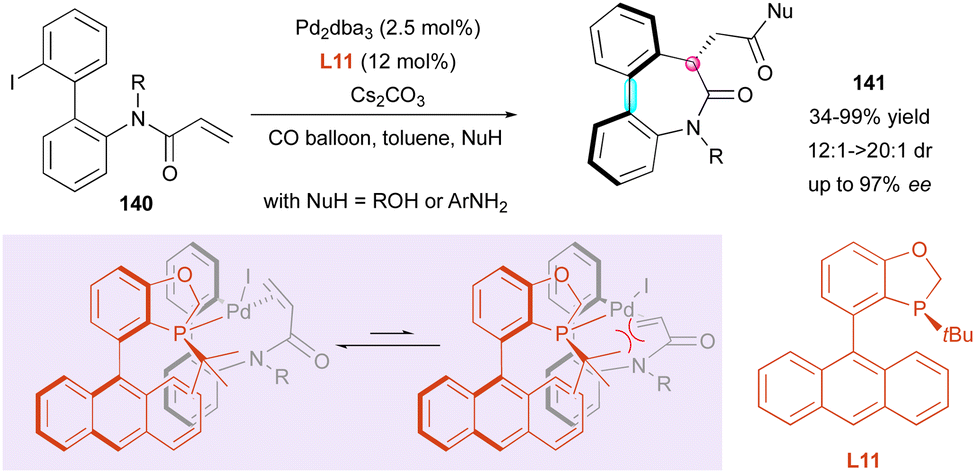 | ||
| Scheme 45 Pd-Catalysed carbopalladation/carbonylation cascade. | ||
Finally, Wang and Li reported the original atroposelective hydrophosphination of 1-alkynyl indoles 142 with simple non-symmetrical configurationally labile diarylphosphines (±)-143 as nucleophiles catalysed by a chiral palladium complex for the synthesis of P-stereogenic substituted alkenes 144 (Scheme 46).76 Their initial efforts at optimising the chiral ligand allowed (S)-Me-Bophoz L12 to be selected as optimal to give, after in situ hydrogen peroxide oxidation, configurationally stable vinylphosphine oxides 144 featuring a C–N axially chiral trisubstituted olefin and a P-stereogenic centre with perfect regio- and (E)-selectivity, moderate to good diastereocontrol (5.2:1 to 10:1 dr) and enantioselectivity above 81%. Supported by DFT calculations, only the observed axial chirality was rationalised, and the migratory insertion was identified as the enantio-determining step with two essential arguments, where firstly, the more hindered indole moiety tends to be placed downward with minimal repulsion between R1 and PPhR2 groups as the depicted intermediate. Secondly, the π-acidic alkyne is trans to the more donating arylphosphino group, favoring the (S) configuration of the product.
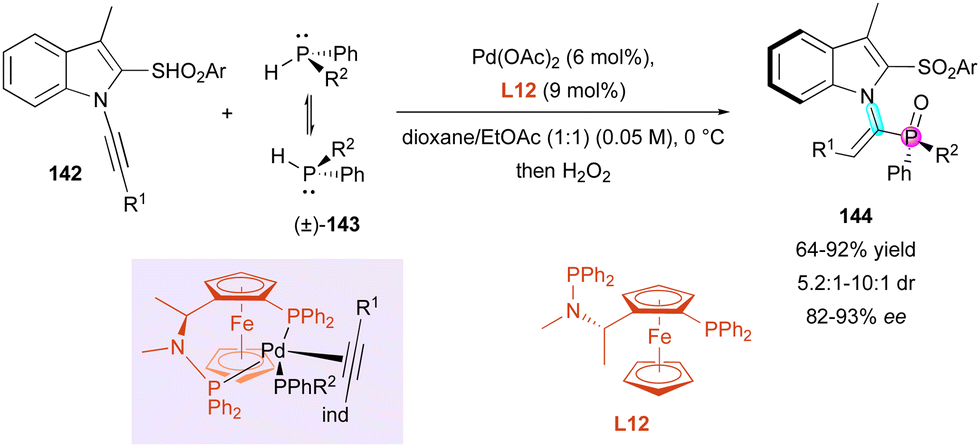 | ||
| Scheme 46 Pd-Catalysed hydrophosphination of 1-alkynyl indoles. | ||
Another recent DKR for the atroposelective synthesis of C–N axially chiral compounds featuring a stereogenic centre was proposed by Wang, Ren and colleagues.77 They envisaged a unique Rh(II)-catalysed carbene N–H insertion strategy of diazonaphthoquinones 146 to racemic 3-indolinone spiroacetals (±)-145 in the presence of chiral amino acid L13 as the ligand (Scheme 47). Although the mechanism was not explained, ring-opening/ring-closing of spiro-acetal may occur to enable the observed diastereoselective DKR, leading to a new family of C–N atropisomers 147 with excellent enantioselectivity (72% to 99% ee). Although the diastereoselectivity was generally moderate (mostly 1.5:1 to 2:1 dr), a better result was observed for R3 = 3-OnPr (8:1 dr). Interestingly, increased yields were obtained in the case of electron-donating substituents as R1 and R3. Remarkably, the authors proposed an efficient one-pot protocol for the formation of 147 (R1 = R2 = R3 = H) directly from isatin 148, triggering both N–H and C–O carbene insertion with concomitant N-arylation and spirocyclisation with the same efficient creation of the two different stereogenic elements (45% yield, 3:1 dr, 96% ee).
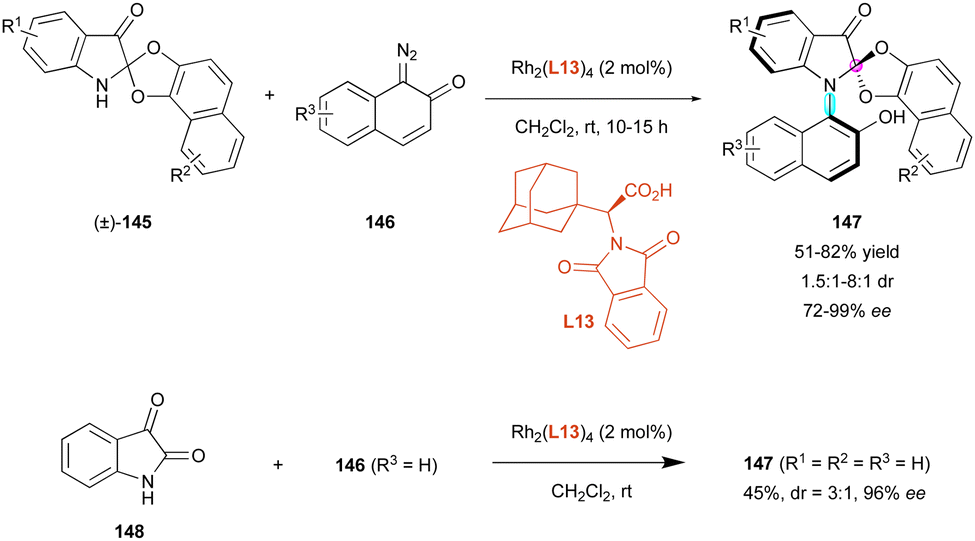 | ||
| Scheme 47 Rh-Catalysed enantioselective N–H bond insertion of indolinone-spiroacetals. | ||
Desymmetrisation
Maleimide/succinimide substrates
Enantioselective desymmetrisation is a well-established and powerful method to reveal multiple stereogenic elements from simple achiral, prochiral or meso substrates by breaking one or more elements of symmetry involving either enzymatic78 or non-enzymatic79 catalytic transformations. In this context, the last two decades have witnessed the emergence of new strategies for the synthesis of compounds featuring both central and axial chirality.The initial contribution in this direction was from Simpkings and collaborators,80 who proposed the desymmetrisation of N-(2-tert-butylphenyl)succinimide 149 involving chiral lithium amide 150 (Scheme 48). The enantiotopic deprotonation of 149 by mono-lithiated base 150 generated atropisomeric enolate intermediate 152, which reacted diastereoselectively with various electrophiles, leading to enantioenriched succinimides 151 featuring both central and axial chirality. The reaction proceeded with moderate yields (40–56%), good to high diastereoselectivity (6:1–25:1 dr) and high enantiomeric excess (85–95% ee).
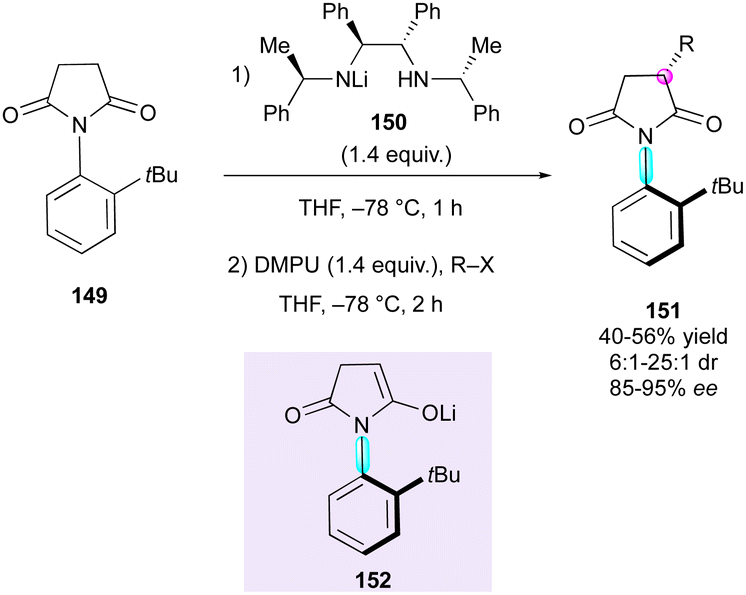 | ||
| Scheme 48 Enantiotopic desymmetrisation of N-(2-tert-butylphenyl). | ||
Another stereoselective approach to centrally and axially chiral succinimides 154 relies on the desymmetrisation of maleimides 153 triggered by stereoselective 1,4-additions largely inspired by the pioneer Curran's diastereoselective Giese reaction.81 The first report by the Hayashi and Shintani group deals with the rhodium-catalysed enantio- and diastereoselective conjugate addition of arylboronic acids controlled by the commercially available chiral 2,5-diphenyl bicyclo[2.2.2]octadiene ligand L14 (Scheme 49).82 Square planar Rh-coordinated transition state 155 minimising the steric repulsion between the tBu-substituent of maleimide and the phenyl groups of the ligand was proposed to support the very high stereoselectivities observed.
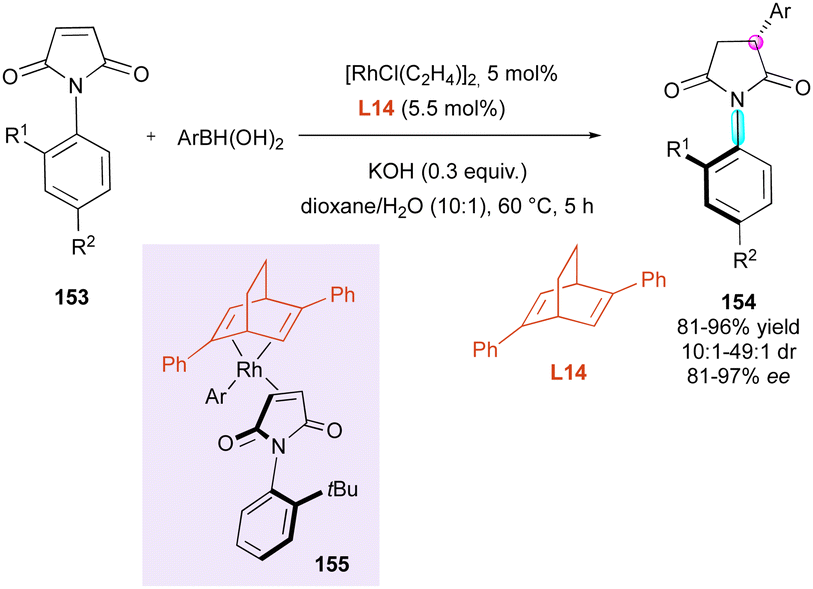 | ||
| Scheme 49 Rh-Catalysed conjugate addition of arylboronic acids. | ||
After these seminal contributions, the powerful Michael acceptor reactivity of maleimides for desymmetrisation leading to multi-chiral succinimides was advanced in 2014 by the Bencivenni group.83 Their methodology deals with the amino-catalytic desymmetrisation of N-arylmaleimides 160via the vinylogous Michael addition of 3-substituted cyclohexenones 158 to N-(2-tert-butylphenyl) succinimides 159 with remote control of the axial chirality (Scheme 50, up right).84 The salt made of 9-amino(9-deoxy)epi-quinine C30 and N-Boc phenylglycine catalysed the enantioselective desymmetrisation via dienamine activation, leading to atropisomeric succinimides 159 with two adjacent stereocentres. Primary amine catalysis was crucial for achieving enantioselective desymmetrisation, enabling simultaneous and exclusive remote control of both the stereogenic axis and newly formed stereocentres. This process resulted in moderate diastereoselectivity but consistently high enantioselectivity. Following these promising results, the same group developed complementary organocatalysed desymmetrisations of N-(2-tert-butylphenyl)maleimides 160via either Michael addition of α-activated carbonyl compounds 162–16485 catalysed by C31 and C26 (Scheme 50, bottom left, centre and right) or formal Diels–Alder cycloaddition with α,β-unsaturated ketones 15786 promoted by catalyst C30 (Scheme 50, up left). In the first case, activated carbonyl compounds, such as cyanomalonates 162 or 3-aryloxindoles 164, allowed the control of two stereogenic centres and one C–N axis,87 while with the last approach, control of three stereogenic centres and one C–N axis in 156 could be reached via dienamine activation.
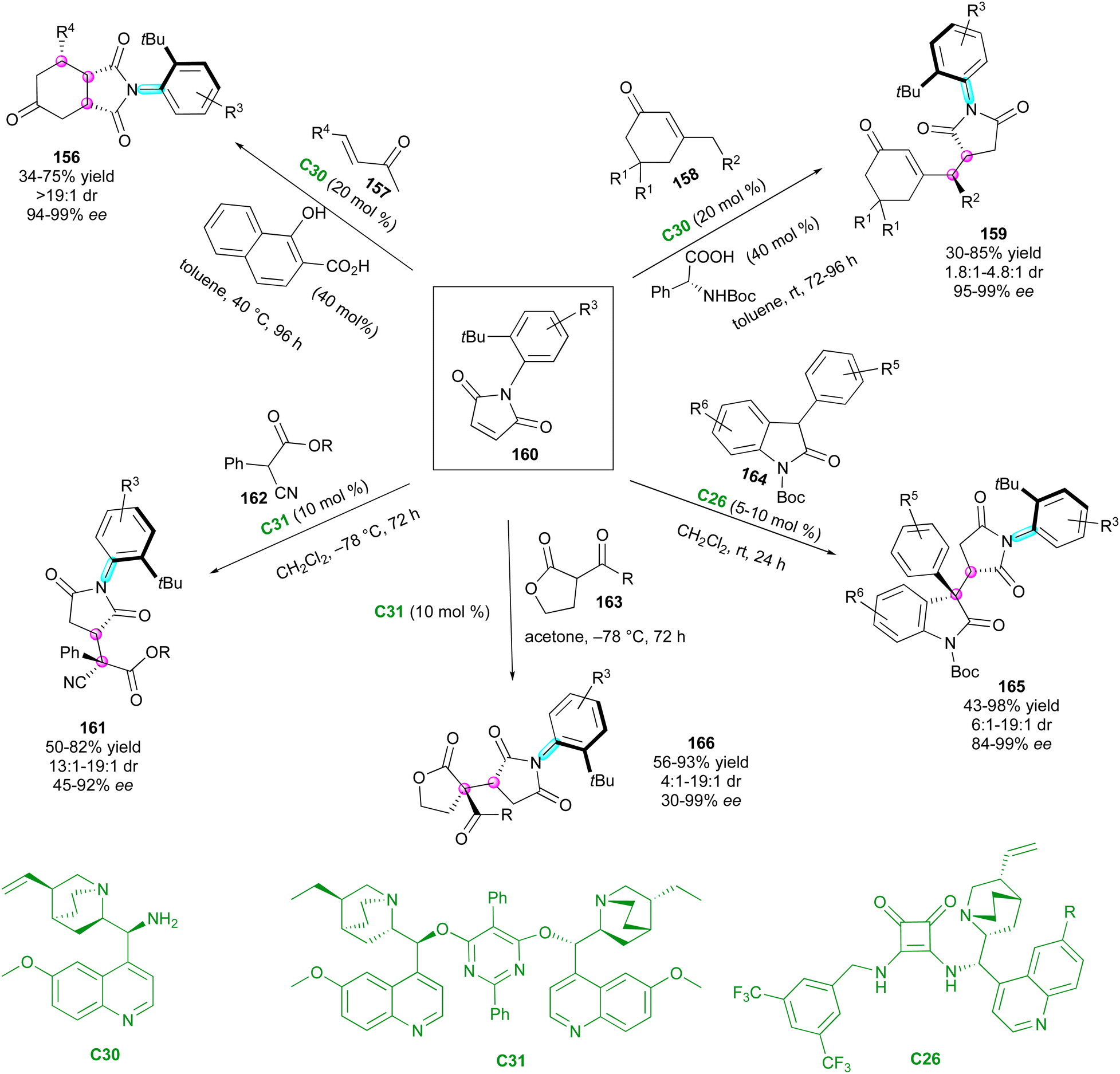 | ||
| Scheme 50 Amino-catalytic desymmetrisation for remote control of axial chirality. | ||
Contemporaneously, Feng and collaborators88 applied their chiral N,N′-dioxide metal complexes for the desymmetrisation of N-(2-tert-butylphenyl)maleimide 160 (R3 = H) by Michael addition with NH-free 3-benzyl-2-oxindoles 167 (Scheme 51). Among the various combinations, with the L14-Sc(OTf)3 complex, the yields and stereoselectivities of 168 remained consistently high, tolerating a wide array of 3-benzyl groups, while 3-phenyl- or 3-methyl analogues gave poorer results with 89% ee, 9:1 dr and 88% ee, 3:1 dr, respectively. A tetradentate coordination of the Sc(III) by chiral ligand L15 was proposed as the active catalytic species, allowing to organise an hexacoordinated transition state accounting for the observed stereoselectivities.
 | ||
| Scheme 51 Sc(III)-Catalysed desymmetrisation of N-(2-tert-butylphenyl) maleimide. | ||
Correspondingly, six years later, Li and collaborators89 proposed the elegant C–H alkylation of benzamides 170 with Cramer's chiral CpxRh-(III)-catalysts C32 for the efficient desymmetrisation of a large series of N-aryl maleimides 169 as electrophilic Michael acceptor partners (Scheme 52). The coupling system features a broad substrate scope, including N-naphthylmaleimides, and proceeds with excellent enantio- and diastereoselectivity for simultaneous control of the axial and central chirality in 171. The experimental mechanistic studies clearly indicated the reversibility of the C–H bond activation, whose cleavage significantly contributes to the overall kinetic barrier. Moreover, the excellent stereoselectivities were rationalised based on Cramer's stereocontrol model via the endo coordination of the substrates to Rh with the relatively bulky tertiary amide directing group pointing frontward to minimize the steric repulsions.
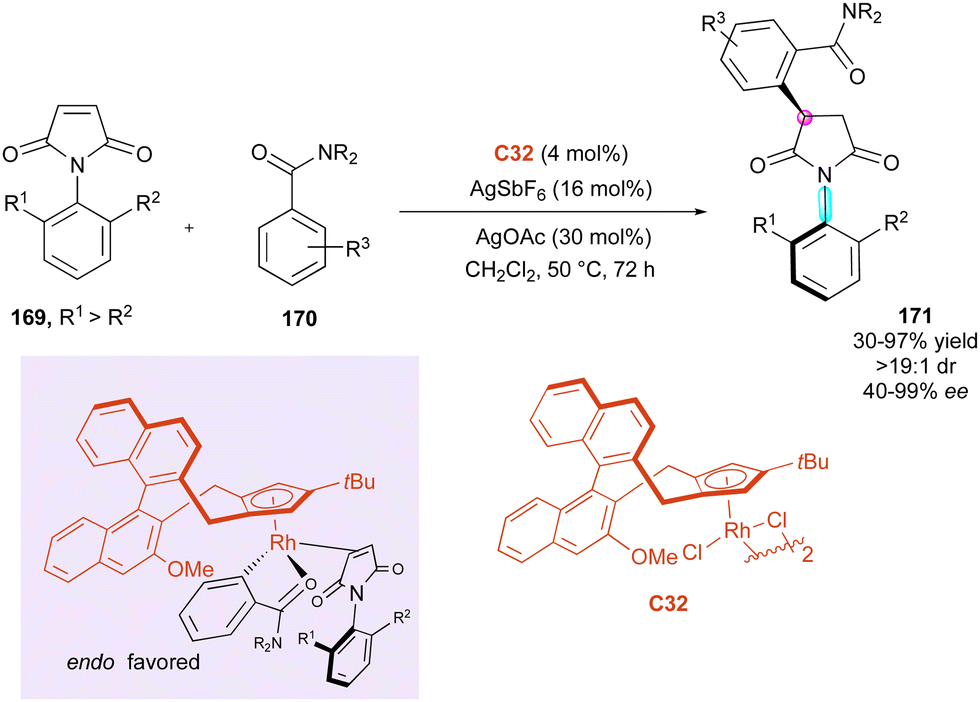 | ||
| Scheme 52 Rh-Catalysed C–H enantioselective alkylation of benzamides. | ||
Enantioselective metal-catalysed hydrofunctionalisation of olefins has attracted significant attention for the control of central chirality,90 and more recently found some interesting applications for the desymmetrisation of functionalised maleimides. In 2020, the Xu91 group developed a stereospecific Si–C coupling with remote control of the axial chirality upon the enantioselective palladium-catalysed hydrosilylation of maleimides 172 (Scheme 53). Among the various chiral ligands, TADDOL-derived phosphoramidites of type L16 bearing aromatic bulky groups were revealed to be the most efficient, inhibiting both the competitive O-hydrosilylation and reductive hydrosilylation. Therefore, the chemo- and enantioselective desymmetrisation tolerated many functional variations including different silanes 173 and proceeded with generally high yields and excellent stereoselectivities for the simultaneous remote control of the stereogenic axis and the newly formed silylated stereocentre in products 174.
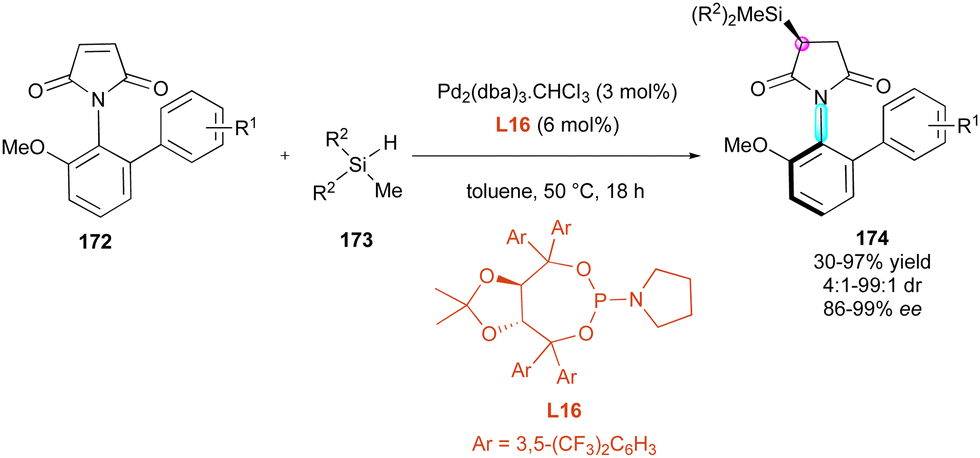 | ||
| Scheme 53 Pd-Catalysed enantioselective hydrosilylation of maleimides. | ||
Similarly, the following year, the Fang and Chen group92 reported the efficient enantioselective nickel-catalysed hydrocyanative desymmetrisation of N-aryl-norbornene-2,3-dicarboximide derivatives 175 (Scheme 54). Acetone cyanohydrin as the in situ source of HCN and Ni(cod)2 as the catalyst in the presence of SegPhos bidentate ligand L17 were selected as the benchmark combination, giving high yields and excellent stereoselectivities even at relatively elevated temperature (60 °C to 100 °C). Consequently, a large series of functionalised succinimides 176 featuring three stereogenic centres and a C–N axially chiral bond was easily available by a remarkable remote control. The complementary experiments combined with DFT calculations clearly indicated that the rigid structure of the cyclic imide was essential to control the enantioselectivity determined during the reductive elimination step through a more favorable transition state (ΔΔG0 = 2.3 kcal mol−1), which is consistent with the high ee values obtained experimentally.
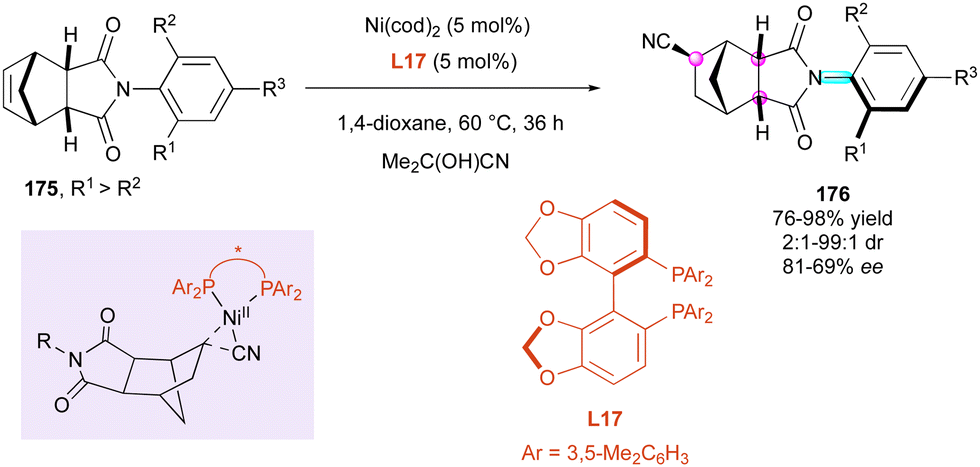 | ||
| Scheme 54 Ni-Catalysed enantioselective hydrocyanation of norbornenes. | ||
More recently, the Michael acceptor character of maleimides was exploited by the Jindal, Mukherjee and Biju group93 in an efficient N-heterocyclic carbene-(NHC)-catalysed Stetter-aldol domino sequence involving aromatic dialdehydes 177 and prochiral N-(2-tert-butylphenyl)maleimide 160 (R3 = H), followed by subsequent oxidation (Scheme 55). This two-step approach afforded atropisomeric N-aryl succinimides 178 combining an axially chiral C–N bond and a spirocyclic stereogenic centre in good yields and ee values. The reaction is mostly limited to o-tBu-N-aryl derivatives and proceeds with 10 mol% loading of the N-heterocyclic carbene generated in situ by deprotonation of chiral triazolium salt pre-C6 with DIPEA. DFT calculation indicated that the Michael addition of transient Breslow intermediate 179 is the enantio-determining step.
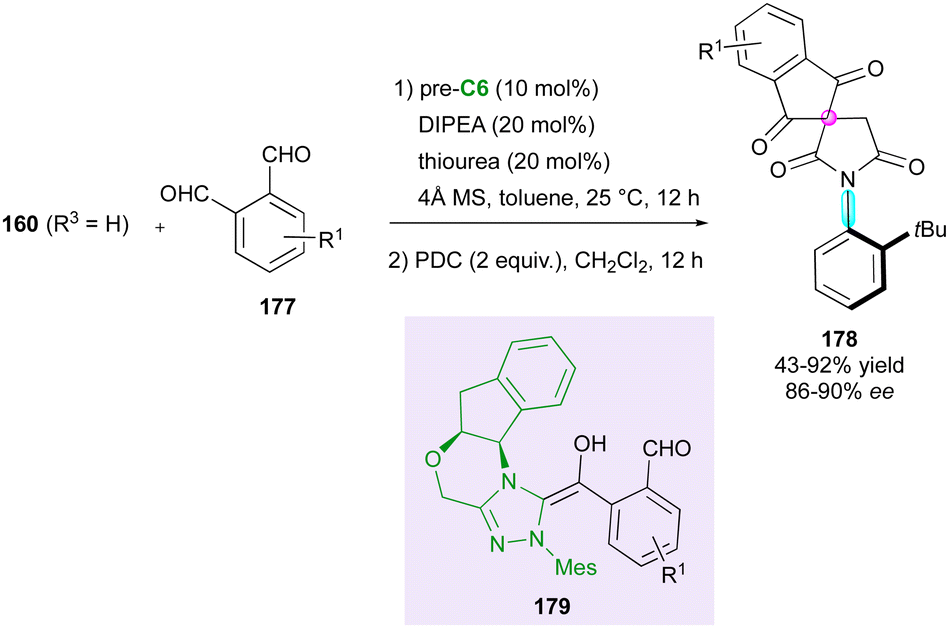 | ||
| Scheme 55 NHC-Catalysed enantioselective spiro-desymmetrisation of maleimides. | ||
Maleimides have also been known for a long time as excellent dipolarophiles and dienophiles in enantioselective cycloadditions, and recently have emerged as an interesting tool to control both central and axial chirality by desymmetrisation of prochiral maleimides. One of the first contributions was by Wang and collaborators in 2016,94 who proposed the 1,3-dipolar cycloaddition of azomethine ylides to N-(2-tert-butylphenyl)maleimide 160 (R3 = H) as a dipolarophile (Scheme 56). The reaction proceeded smoothly with only 3 mol% loading of silver(I)/L18 catalytic system formed in situ and various imino esters 180. Interestingly, no additional base was required to generate the reactive azomethine ylides, affording enantioenriched octahydropyrrolo[3,4-c]pyrrole derivatives 181 bearing four adjacent stereocentres and one C–N stereogenic axis with very high yields and stereoselectivities even when a quaternary stereogenic centre is created (R2 = Me, Et, tBu…). Following these pioneer results, Zhang and collaborators95 proposed alternative trifluoromethylated sulfonamide phosphine ligand L19, providing similar results but in the presence of the mineral base Cs2CO3.
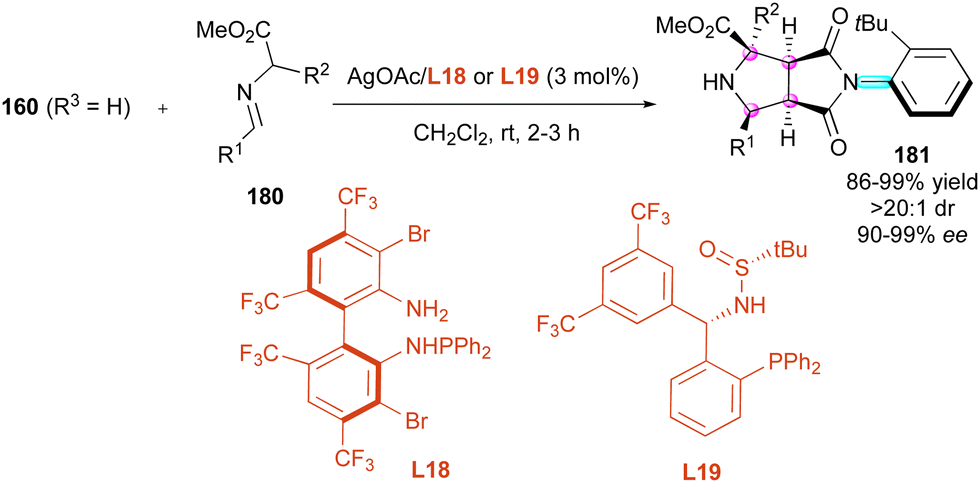 | ||
| Scheme 56 Ag(I)-Catalysed enantioselective (3+2) cycloaddition of azomethine ylides. | ||
More recently, two complementary approaches for the simultaneous control of C–N axial and central chirality via the (3+2) cycloaddition of prochiral maleimides 182 were reported (Scheme 57). Based on the silver-catalysed 1,3-dipolar cycloaddition, the Qian and Liao group96 reported the first utilisation of activated isocyanides 183 as 1,3-dipolarophiles for the synthesis of C–N atropisomers in the presence of chiral diamine ligand L20 (Scheme 57, up). Simultaneously, Wang, Lin and collaborators97 disclosed the new atroposelective (3+2) annulation of Morita–Baylis–Hillman carbonates 185 catalysed by chiral phosphine C33. The first approach gave fused bicyclic 1-pyrrolines 184 featuring a distal C–N stereogenic axis and three adjacent stereogenic centres in high yields and stereoselectivities, while C–N axially chiral cyclopentannulated N-aryl succinimides 186 having two stereogenic centres were obtained by the alternative method with comparable efficiencies. In addition, a closely related protocol of silver-catalysed 1,3-dipolar cycloaddition with isocyanides 183 was applied by Qian and Liao's group for the desymmetrisation of quinazolinone maleimides 187, delivering bicyclic heterocycles 188 displaying a stereogenic N–N axis and three contiguous stereogenic carbon atoms (Scheme 57, bottom).98 Interestingly, a simple ligand switch allowed the development of a diastereodivergent approach with facile and efficient access to both endo and exo diastereomers of 188 in excellent dia- and enantioselectivities. This protocol was also applicable to N-indole maleimide starting materials.
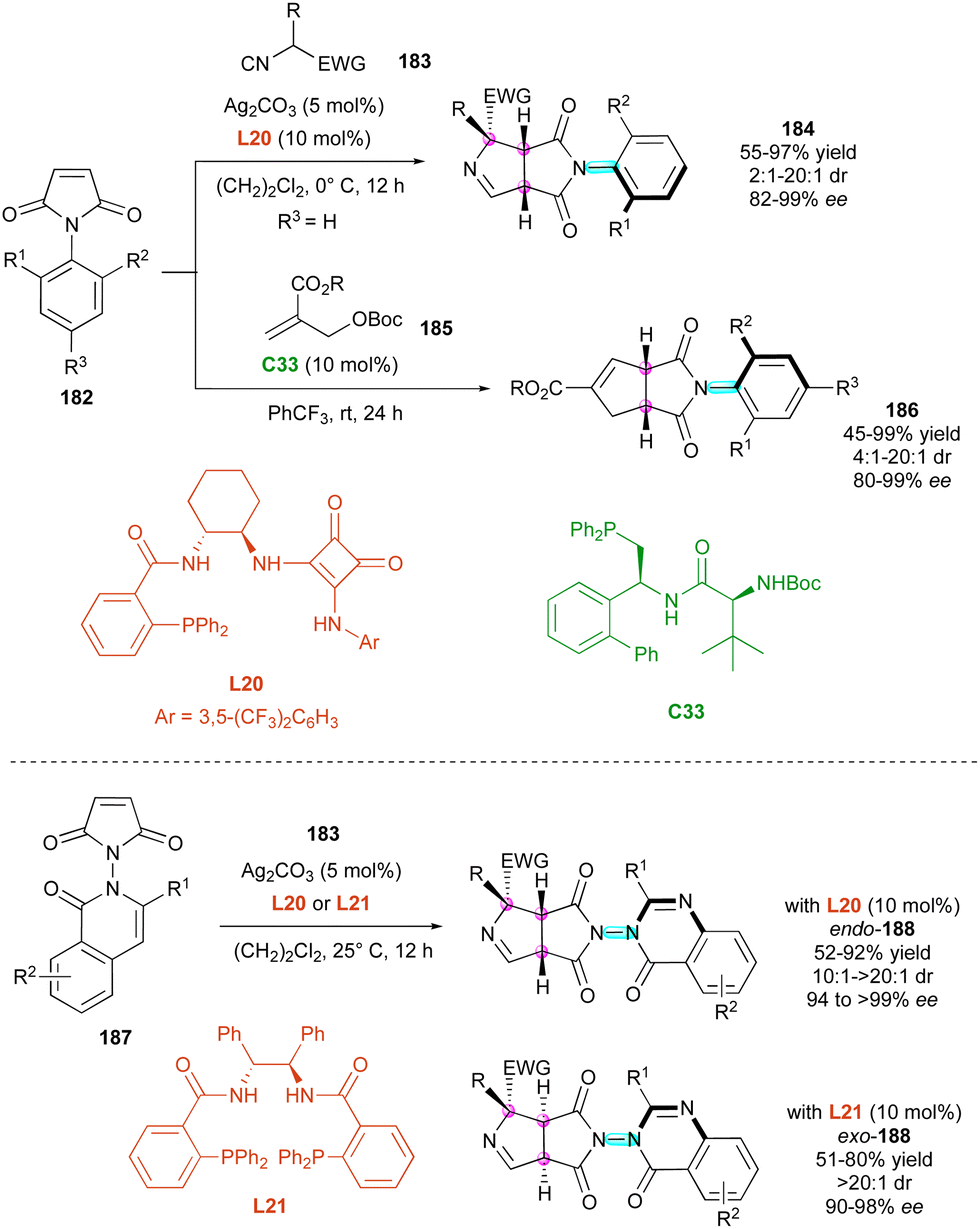 | ||
| Scheme 57 Phosphine- and silver-catalysed (3+2) atroposelective annulations. | ||
Concerning the dienophilic reactivity of maleimides, to the best of our knowledge, two pioneer examples by Corey and Yamamoto, in 1994 and 2001, respectively, were reported on the Diels–Alder enantioselective desymmetrisation of N-aryl maleimides but the authors did not discuss the possible control of the axial chirality.99 Since then, besides Bencivenni's formal (4+2) annulation in 2015 (vide supra),83 this approach remained without development until the recent contribution by our group (Scheme 58).100 We illustrated an unprecedented chiral NHC-catalysed pseudo-three component reaction between enal 189 and two equivalents of a prochiral N-aryl maleimide 182 under oxidative conditions, resulting in the formation of bis-succinimide adducts 190 with high stereocontrol of up to four consecutive stereogenic centres and two remote C–N stereogenic axes. The best conditions involved an inorganic base to generate the reactive NHC precursor of Breslow intermediate 191, which upon in situ oxidation by 3,3′-5,5′-tetra-tert-butyl-4,4′-diphenoquinone (TTBDQ) provided the key chiral azolium dienolate 192, triggering the formal (4+2) cycloaddition, followed by a completely diastereoselective conjugate addition to a second equivalent of N-aryl maleimide 182. Products 190 featuring multiple stereogenic elements, including two challenging C–N axes in the pentatomic series, were obtained as only one diastereomer out of 64 possible stereoisomers and with excellent enantioselectivity.
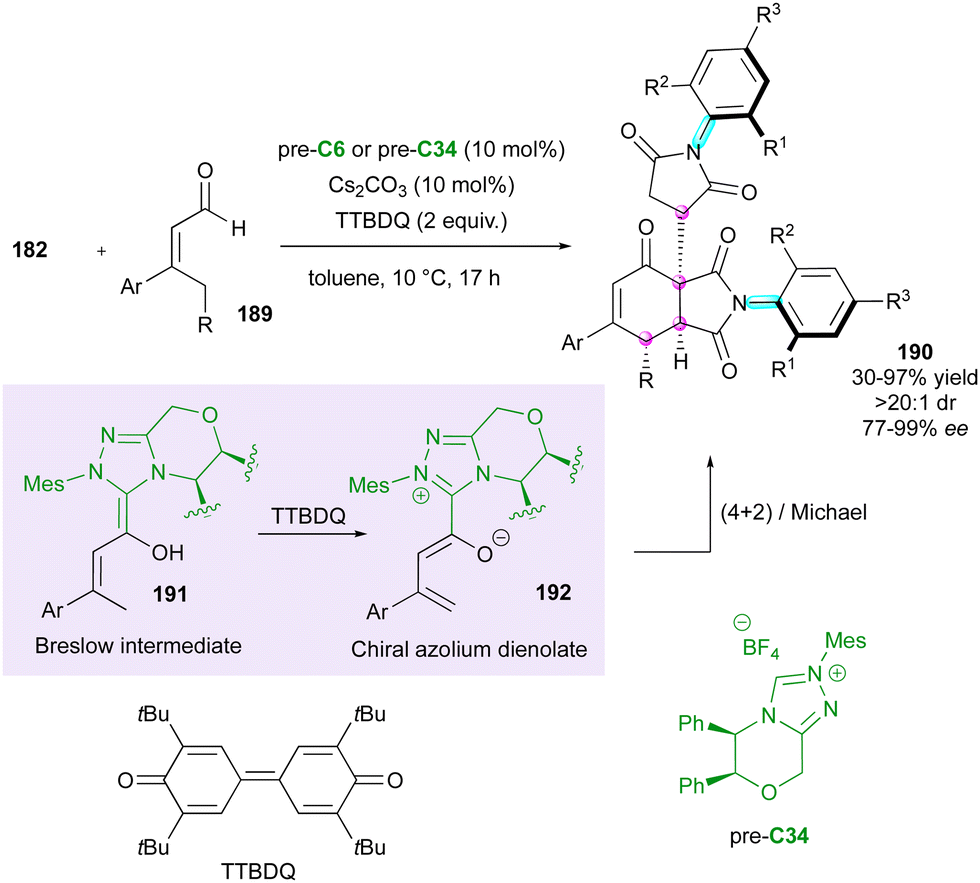 | ||
| Scheme 58 NHC-Catalysed formal (4+2) oxidative desymmetrisation of maleimides. | ||
Non maleimide/succinimide substrates
Closely related to maleimides, 4-aryl-1,2,4-triazole-3,5-dione (ATAD) derivatives can also be interesting candidates for atroposelective desymmetrisations, as shown by Tan's group in 2016.101 Capitalising on this discovery, the same group102 proposed an elegant consecutive three-component synthesis of complex axially chiral spiroxindole-urazole derivatives 197 bearing three stereogenic centres including a spirocyclic one (Scheme 59). This unique transformation involves, as the first step, the chiral bisthiourea C35-catalysed domino Diels–Alder cycloaddition between 3-vinylindoles 193 and methyleneindolinones 194 to afford spirooxindole intermediates 196, followed by a diastereoselective ene reaction with ATADs 195. Notwithstanding the long reaction times and the usually moderate chemical yields, good diastereoselectivities and excellent enantiopurities were obtained when an ortho-tert-butyl group is present on the ATAD.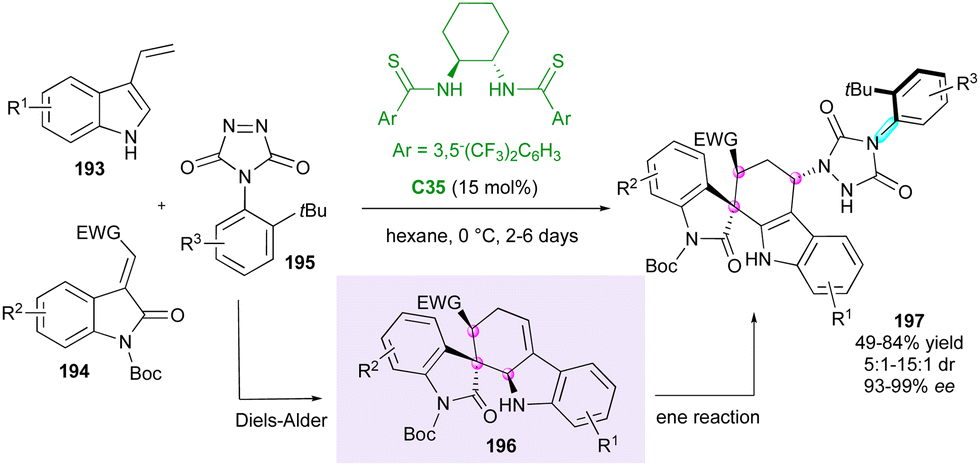 | ||
| Scheme 59 Three-component desymmetrisation of ATAD derivatives. | ||
Very recently, inspired by the peculiar reactivity of ATADs, Pan and collaborators103 proposed the original CPA-catalysed enantioselective dearomatisation of 1-substituted-2-naphthols 198 for the atroposelective desymmetrisation of 4-(2-t-butylphenyl)-1,2,4-triazole-3,5-dione derivatives 195 to give functionalised chiral urazole embedded naphthalenones 199 having both axial and central stereogenic elements in good to high yields with high diastereo- and enantioselectivities (Scheme 60). Both the free OH of the naphthol and the ortho-tBu substituent of the ATAD are crucial to ensure good efficiency but other phenolic partners are also tolerated, leading to the corresponding 1-naphthalenone 200, indole derivative 201, and sesamol 202.
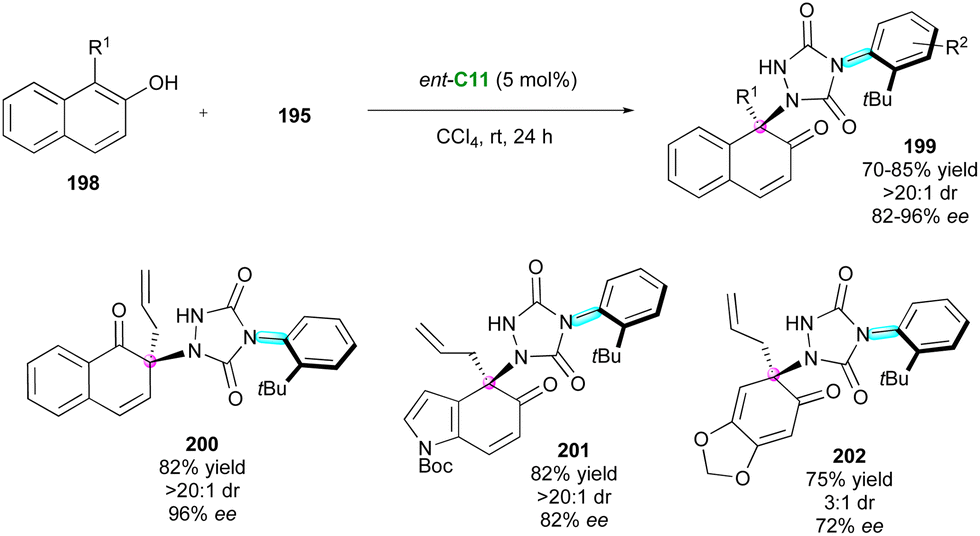 | ||
| Scheme 60 CPA-Catalysed dearomatisative desymmetrisation of urazoles. | ||
Concerning the atroposelective construction of axially and centrally chiral C–C heterobiaryls, in 2022, Xiao's group104 developed the first catalytic Minisci reaction of symmetric 5-arylpyrimidines 203 with α-amino acid-derived redox-active esters 204 (Scheme 61). This unprecedented metal-free radical desymmetrisation was possible using commercially available carbazolyl isophthalonitrile photoredox catalyst 4CzlPN C36 in conjunction with CPA organocatalyst C37 under blue LEDs irradiation. The resulting axially chiral heterobiaryl and centrally chiral α-branched amines 205 were generally obtained with excellent regio-, diastereo-, and enantioselectivity. The key to the success of this reaction is the photocatalyst-controlled formation of radical intermediate 207 and the efficient transfer of stereochemical information from CPA-pyrimidine salt 206. Interestingly, the reaction could be scaled up to 4.0 mmol with comparable results, and under sunlight irradiation, the cross-coupled amino pyrimidines were also smoothly obtained without deleterious effects on the efficiency and stereoselectivity.
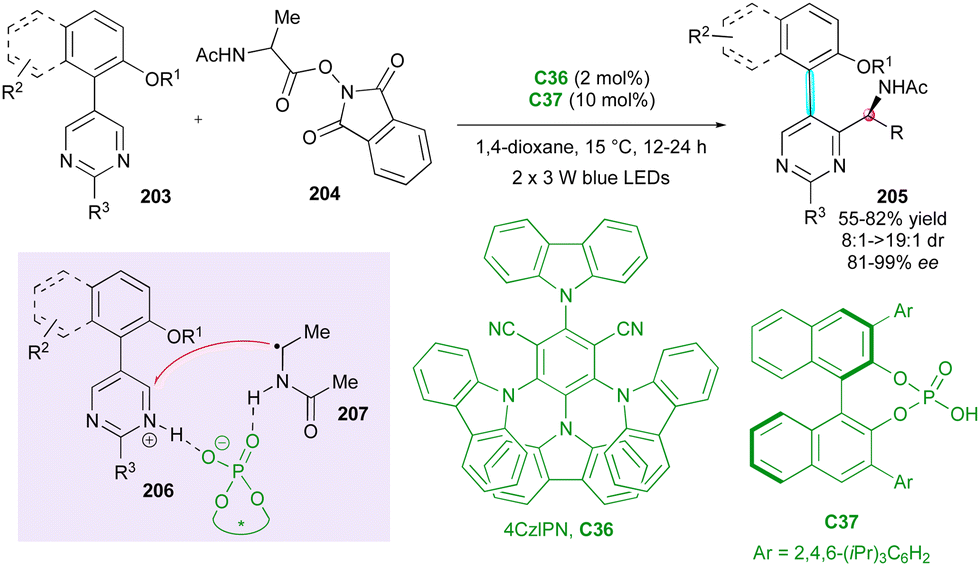 | ||
| Scheme 61 Photoinduced atroposelective radical Minisci reaction. | ||
Another interesting strategy for simultaneously controlling both the central and C–C axial chirality involves metal-catalysed enantioselective desymmetrising additions of various nucleophiles to axially prochiral dialdehydes. This approach emerged recently and concurrently by three different groups. Firstly, Liao's and Qian's groups105 proposed the intriguing silver-catalysed diastereo- and enantioselective (3+2) cycloaddition of activated isocyanides 183 to prochiral biaryl aldehydes 208 (Scheme 62). Employment of diphenyl phosphine-derived cinchonine chiral ligand L22 gave the best results in terms of yields and stereoselectivities, producing a wide range of atropisomeric biaryl aldehydes 209 containing a stereogenic C–C axis and two adjacent stereogenic centres in moderate to excellent diastereoselectivities and uniformly excellent enantioselectivities.
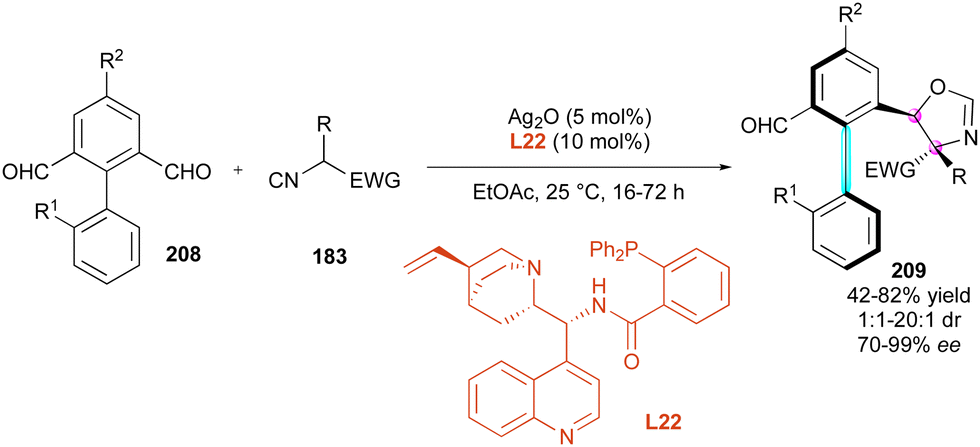 | ||
| Scheme 62 Ag-Catalysed (3+2) cycloaddition with activated isocyanides. | ||
Correspondingly, two closely related photoinduced cobalt-catalysed enantioselective reductive couplings were reported independently by the Cheng, Xiao106 and Zheng, Zhang groups,107 respectively (Scheme 63, top). The first metallaphotoredox approach combines visible light-induced cobalt-catalysed enantioselective reductive couplings with either aryl iodides or alkynes. The commercially available carbazolyl isophthalonitrile photocatalyst 4CzlPN C36 in the presence of (R,R)-BDPP chiral diphosphine ligand L23 and Hantzsch ester (HE) as the reducing agent gave the best results under blue LED irradiation. A broad substrate scope was achieved, leading to a large panel of atropisomeric biphenyl hydroxy aldehydes 211 or 212 in good yields with excellent stereocontrol of axial and central chirality. The origin of the stereoselectivity was corroborated based on DFT calculations for the stereo-determining transition state during Grignard addition of in situ-generated Co(II) intermediates 210 to biaryl dialdehyde 208. Simultaneously, Zheng, Zhang and collaborators proposed the same methodology under similar conditions with comparable results for the desymmetrising reductive arylation of biaryl dialdehydes 208, leading to benzylic alcohols 211 (Scheme 63, bottom).
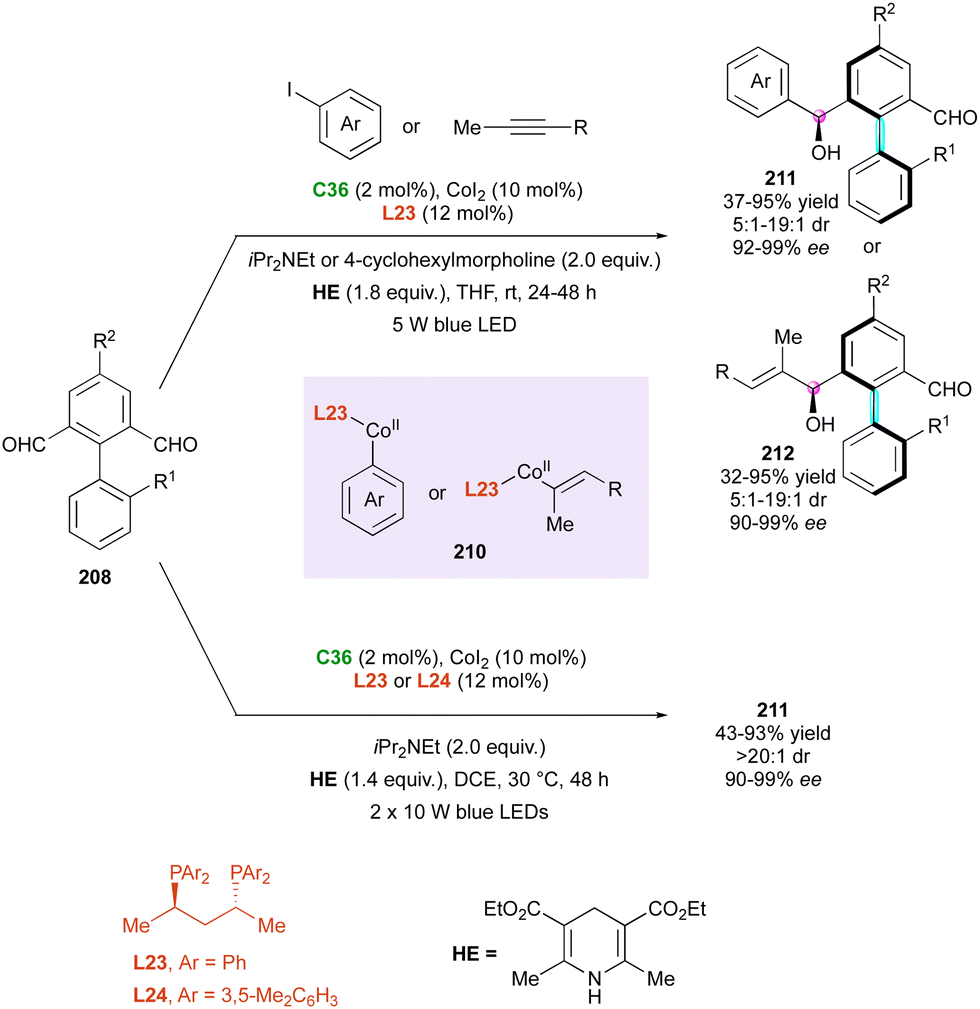 | ||
| Scheme 63 Photoinduced Co-catalysed enantioselective reductive couplings. | ||
More recently, this elegant cobalt-catalysed photo-reductive coupling was extended to the expedient synthesis of axially and centrally chiral more challenging diaryl ethers by the Li and Yu group (Scheme 64).108 Under the same experimental conditions with an (S,S)-BDPP chiral diphosphine ligand (ent-L23), pro-axially chiral biaryl ethers 213 were efficiently coupled with internal alkynes to give the expected atropisomeric hydroxy biaryl ethers 214 in good yields and usually with good to excellent diastereo- and enantioselectivities.
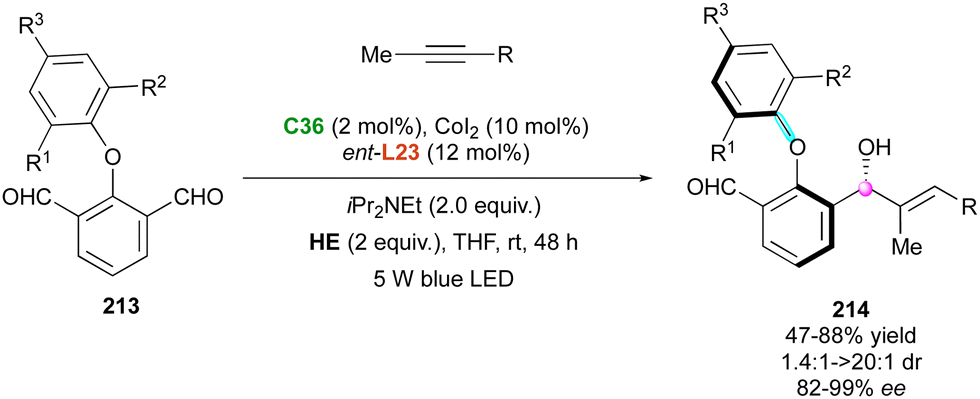 | ||
| Scheme 64 Photoinduced Co-catalysed enantioselective reductive coupling with biaryl ethers. | ||
In their work on the use of NHC organocatalysts for the desymmetrisation of pro-axially chiral dialdehydes 215,109 Zeng and coworkers reported the synthesis axially chiral diarylethers, four of which displayed additional central chirality due to the use of prochiral nucleophilic di- or triols 216 (Scheme 65). The diastereoselectivity in 217 was found to be excellent, and from a mechanistic point of view, the reaction with pre-catalyst ent-C6 could generate atropisomerically enriched Breslow acylazolium intermediates 218 under oxidative conditions using bisquinone (DQ) followed by a nucleophilic substitution with various prochiral alcohols.
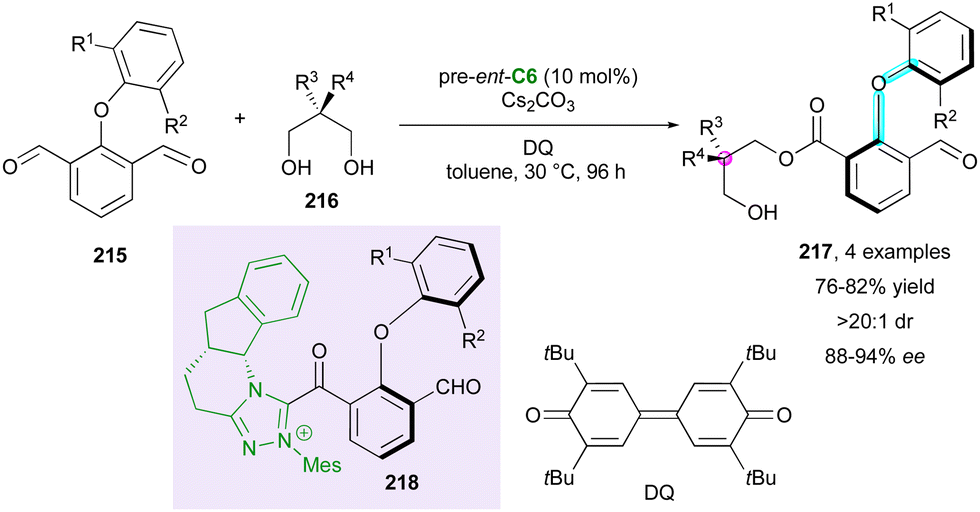 | ||
| Scheme 65 NHC-Catalysed atroposelective synthesis of diaryl ethers. | ||
During their study on the CPA-catalysed atroposelective Friedländer reaction of 2-aminoaryl ketone 219, Cheng and collaborators110 revealed its potential for the desymmetrisation of a small series of 4-substituted cyclohexanones (Scheme 66). The overall cyclodehydration process was triggered by catalyst C15, leading to atropisomeric 9-aryltetrahydroacridines 220 and evolved through centrally chiral tertiary alcohol 221, from which a central-to-axial chirality conversion occurred with relatively modest efficiency in terms of diastereo- and enantioselectivity.
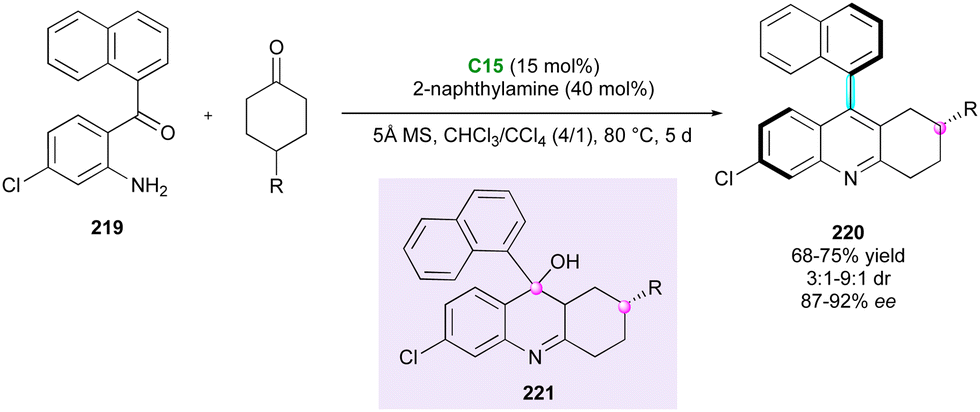 | ||
| Scheme 66 CPA-Catalysed atroposelective Friedländer reaction. | ||
While investigating the NHC-catalysed enantioselective acylation of propargylic triols 222 (R1 = H) (Scheme 67), Wong and Zhao's team unexpectedly discovered the formation of bridged biarylbenzofurans 223 with an eight-membered lactone when reacted with enals in the presence of azolium pre-catalyst pre-C4 under basic conditions.111 This unique class of bridged atropisomers, featuring a benzofuran ring and a single stereogenic centre, has a broad scope, but with triol containing an internal alkyne (222, R1 = Me), the corresponding axially chiral products with two stereogenic centres were obtained in relatively low yield (32%) and dr (1.5:1 to 3:1).
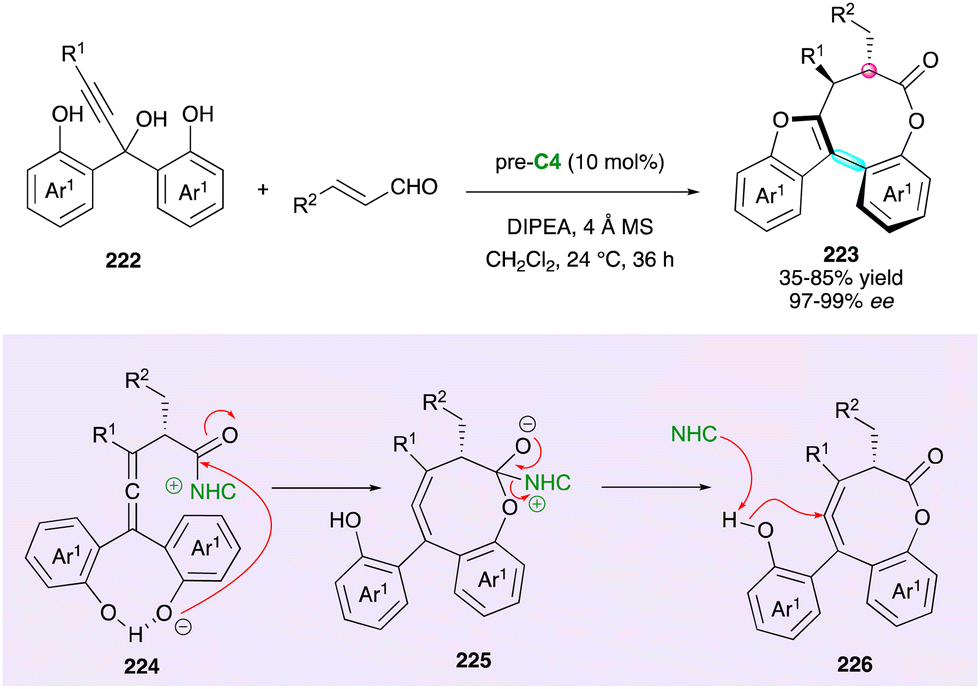 | ||
| Scheme 67 Atroposelective synthesis of benzofurans fused with an eight-membered lactone. | ||
The mechanistic investigations by DFT calculations suggested the direct propargylic substitution of the in situ-generated azolium enolate, resulting in the key centrally chiral allene acylazolium intermediate 224, which undergoes a sequential bi-directional heterocyclisation through 225 and 226 to the final products 223 with both axial and central stereogenic elements.
Although much less widespread, the simultaneous control of axial chirality and chirality centrered on a heteroelement has recently found some interesting solutions. The first example in 2018 by the Cramer group112 described the enantioselective desymmetrisation of phosphine oxides 227 by C–H arylation with o-quinone diazides catalysed by iridium(III) complex C38 bearing an axially chiral cyclopentadienyl ligand and phthaloyl tert-leucine C39 as a co-catalyst (Scheme 68). This method notably enables the selective formation of biaryl C–C atropisomers 228 with a stereogenic phosphorus atom, achieving excellent yields and generally good diastereo- and enantioselectivities. Enantiospecific reductions produce chiral phosphorus(III) compounds, featuring structures and biaryl backbones that are valuable as monodentate ligands in enantioselective catalysis.
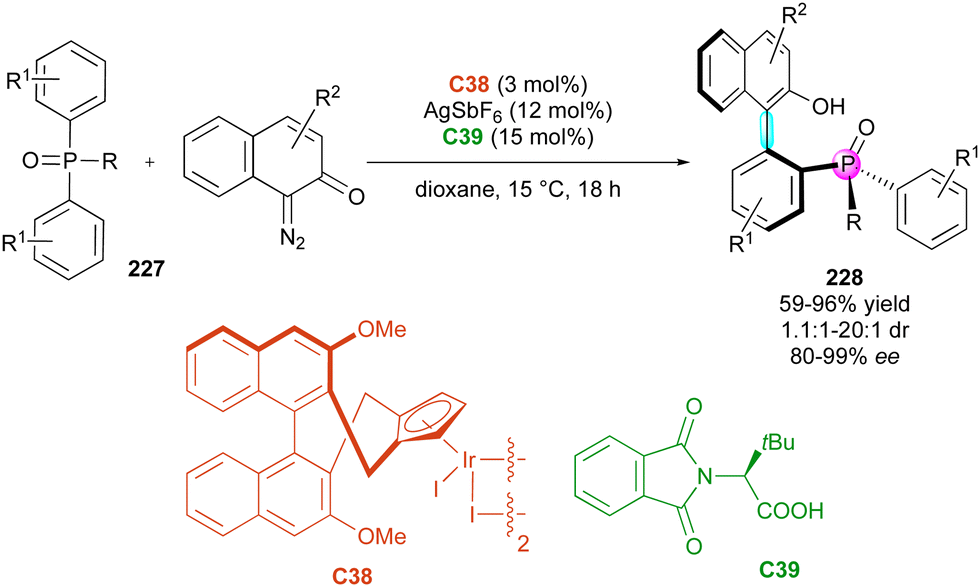 | ||
| Scheme 68 Enantioselective C–H arylation of phosphine oxides. | ||
More recently, the Yu and Li group113 proposed the modular assembly of diverse P-stereogenic and axially chiral thiophosphine derivatives 230 by the palladium-catalysed enantioselective desymmetrisation of cyclic phosphonium salts 229 in the presence of chiral phosphoramidite ligand L25 (Scheme 69). The overall process involved the cleavage of the C–P bond, followed by concomitant C–H bond functionalisation with diverse aryl boronic acids and final treatment with S8. This unprecedented domino sequence was postulated to evolve via key chiral phosphapalladacycle 231 and the resulting atropisomeric binaphthyl phosphine sulfides 230, which are usually obtained with high diastereo- and atroposelectivities, can be easily reduced to the corresponding chiral phosphines and used as efficient ligands in the phosphine-catalysed enantioselective (3+2) cycloaddition. The same authors reported an extension of this approach using benzoxazoles/benzothiazoles instead of the boronic acid partners.114 This palladium-catalysed enantioselective cleavage of C–P bond/intermolecular C–H bond functionalisation allowed related P-stereogenic and axially chiral phosphinooxazoles ligands to be obtained after a reduction step.
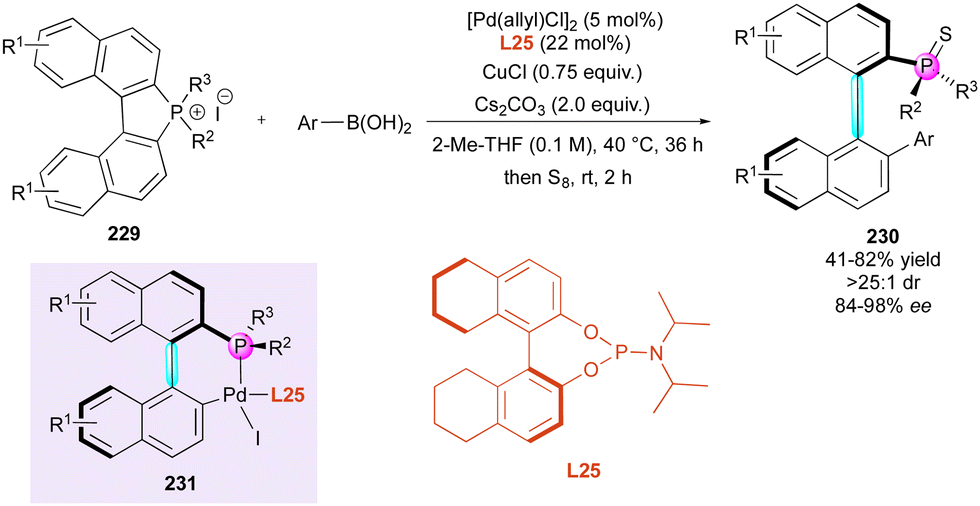 | ||
| Scheme 69 Pd-Catalysed assembly of P-stereogenic and axially chiral phosphinooxazoles. | ||
Another related Pd-catalysed desymmetrisation was recently reported by Huang and coworkers (Scheme 70)115via ring-opening of the cyclic biarylsulfonium salts 232 with phosphines, which led to the formation of atropisomeric phosphines 233 with a controlled stereogenic axis and phosphorous centre. Interestingly, a methyl was needed at the ortho position of the aryl substituent (Ar) of the phosphine, probably for better discrimination regarding the phenyl substituent. Indeed, when a less bulky group was introduced, such as a methoxy, the stereoselectivity dropped remarkably (50% ee and 6:1 dr vs. >87% ee and >10:1 dr for o-Me substituted aryls). Nevertheless, simple recrystallisation could be performed to improve the enantiopurity of products 233.
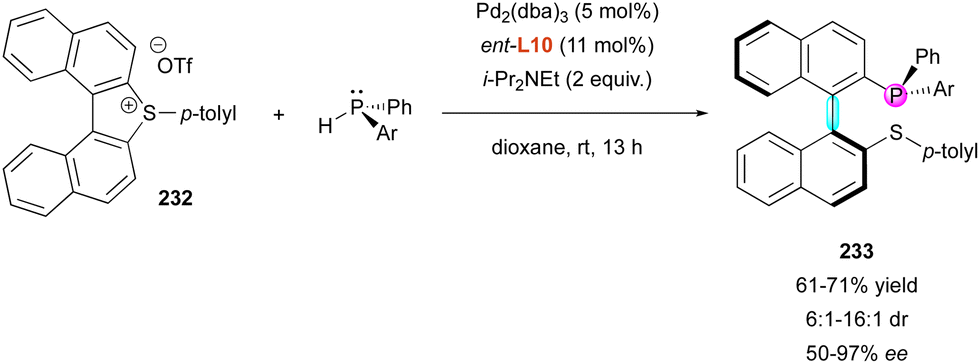 | ||
| Scheme 70 Pd-Catalysed enantioselective phosphination of cyclic biarylsulfonium salts. | ||
The use of silafluorenes 234 as a substrate by Gu's group allowed access to rare biaryl atropisomers 235, displaying one silicon-stereogenic centre, via rhodium-catalysed enantioselective ring-opening (Scheme 71).116 The use of methanol as an additive allowed excellent diastereoselectivities to be achieved, supposedly acting as a ligand to the rhodium catalyst.
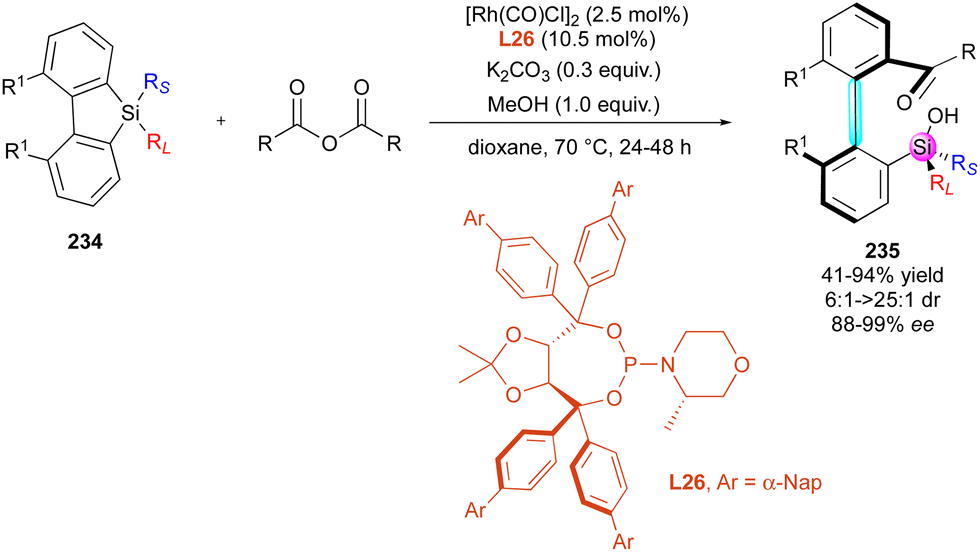 | ||
| Scheme 71 Rh-Catalysed enantioselective ring-opening of silafluorenes. | ||
Complementarily, the desymmetrisation of biaryl dihydrosilanes 236 displaying di-ortho-substituted methyl groups was realised by the He group117 based on intramolecular C–H silylation, generating a new type of axially chiral biaryl silicon-stereogenic silanes 237 in moderate yields with high enantioselectivities (Scheme 72). This unprecedented transformation is catalysed by a Rh(I) complex in the presence of Josiphos L27 as a chiral ligand and a norbornene derivative as a hydrogen acceptor (NBE-OMe), giving access to unusual atropisomeric six-membered ring bridged silicon-stereogenic dibenzooxasilines 237, in which the configuration of the stereogenic axis is controlled by the adjacent silicon-stereogenic centre with a large dihedral angle of 39°.
Fang and coworkers demonstrated that biaryl dienes 238 are also suitable substrates for desymmetrisation (Scheme 73).118 The Ni-catalysed hydrocyanation of 238 using L28 as a chiral diphosphine ligand was reported with good enantio- and diastereoselectivity (one example with 3:1 dr, most of them with >20:1 dr). The authors showed the wide scope of the reaction with intriguing R1 and R2 substituents, such as halogens, nitrile, vinyl and heterocycles. Interestingly, a couple of products 239 could be used as the ligand and organocatalyst for enantioselective transformations.
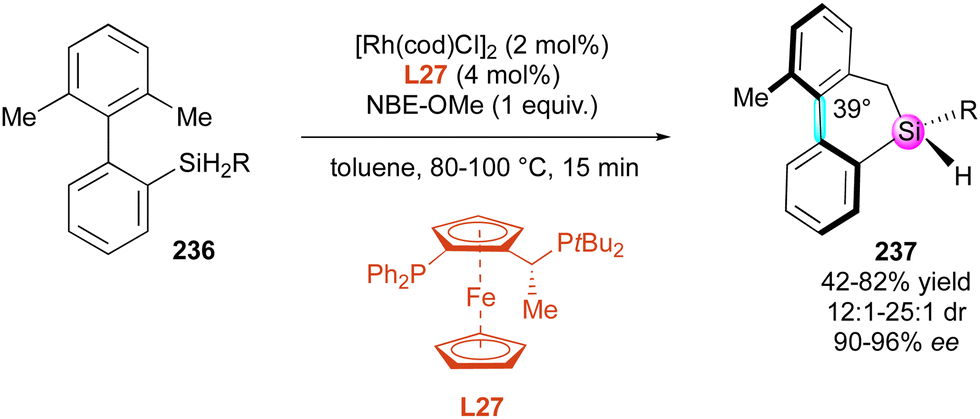 | ||
| Scheme 72 Rh-Catalysed intramolecular atroposelective C–H silylation. | ||
The combination of axial and planar chirality is a rarely encountered scenario, but inspired by the pioneer Uemura's diastereoselective desymmetrisation of planar chiral chromium complexes by enantiotopic ortho lithiation,119 Kamikawa, Takahashi and Ogasawara developed a smart strategy to synthesise axially chiral N-arylindole compounds 241 through enantioselective desymmetrising ring-closing metathesis (RCM) (Scheme 74).120 They began with functionalised prochiral planar (arene)chromium complexes bearing an indolyl substituent, which is fixed in the anti-configuration relative to the chromium atom. The reactions were conducted in benzene at 40 °C with 10 mol% of in situ-generated chiral molybdenum catalyst C40. The presence of an electron-rich phosphine coordinated to the chromium was essential for achieving high yields and enantioselectivities. Under these conditions, chiral chromium complexes were efficiently produced, and both axial and planar stereogenic elements were simultaneously generated and controlled during the transformation.
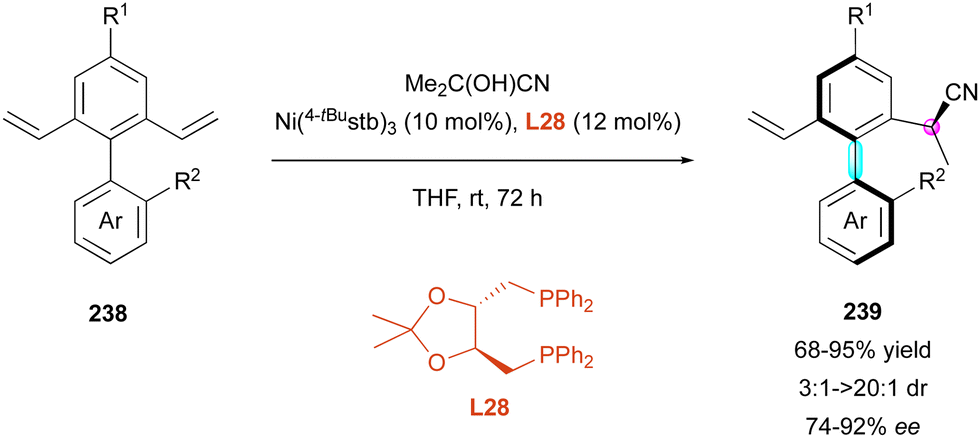 | ||
| Scheme 73 Desymetrisation via Ni-catalysed hydrocyanation of biaryl dienes. | ||
More recent complementary results in this field were reported by the Wu, Li and Zhang group in 2021, with the gold-catalysed enantioselective hydroarylation of ortho-alkynylaryl ferrocene derivatives 242, allowing the simultaneous control of axial and planar chirality (Scheme 75).121 Due to the linear coordination geometry of the Au(I) catalyst, control of the enantioselectivity is more challenging and the bulky chiral sulfoximine ligand is required for this reaction. The optimisation of the key 6-endo-dig cyclisation leading to the corresponding ferrocenyl atropisomers 243 resulted in the use of an enantiopure chiral Au(Sadphos)Cl precatalyst C41 and NaBArF as an additive in dichloromethane at 0 °C for 24 h. The diastereoselectivity was improved by using cyclohexylmethyl as a protecting group for naphthol (R1) but substitution at the ortho-position of the aryl alkyne (R3) was detrimental for the enantioselectivity. The Re face of the alkyne is shielded by the naphthyl of the ligand, leading to the addition of the ferrocenyl by the Si face to secure the observed planar chirality, while control of the axial chirality is the result of the impeded rotation of the naphthalene ring provided by the presence of the alkoxy substituent (–OR1). DFT calculations supported the proposed reaction mechanism, where the enantioselectivity is achieved through π-stacking between the naphthyl groups of the catalyst and the substrate.
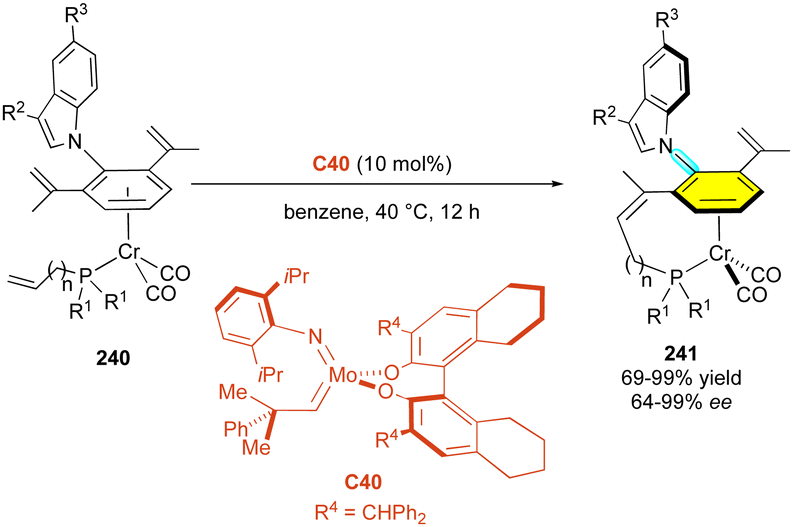 | ||
| Scheme 74 Enantioselective ring-closing metathesis. | ||
Following this first achievement in the desymmetrisation of ferrocene derivatives, the Li, Liu and Liang groups performed the synthesis of a library of ferrocenyl derivatives 246 exhibiting both axial and planar chirality using an enantioselective Catellani reaction (Scheme 76).122 The catalytic system involved Pd(OAc)2 (10 mol%) as the catalyst, tri(2-furyl)phosphine (TFP, 20 mol%) as the ligand and excess of chiral norbonene mediator L29 (3 equiv.) at reflux in toluene for 24 h to perform the reaction between 2-iodophenyl(methyl)carbamates 244 and 1-bromonaphthalenes 245. Axially and planarly chiral products 246 were obtained in high enantio- and diastereoselectivity with a great tolerance for electron-donating and electron-withdrawing groups on the iodo-aryl partner. An axial-to-planar diastereoinduction model was proposed governed by the steric repulsion between the ester moiety of the chiral ligand and the naphthyl, in agreement with the observed diastereoselectivity.
 | ||
| Scheme 75 Au-Catalysed hydroarylation reaction. | ||
Using a similar Catellani strategy, in 2023, the Zhou group described a domino sequence for the reaction between 2-iodophenethylferrocenes 247 (M = Fe) and ortho,ortho′-disubstituted 2-bromoaryls 248 (Scheme 77).123 In only 5 h in toluene at 120 °C, by using Pd(OPiv)2, MePhos and a catalytic amount of chiral enantiopure norbornene derivative L30 (10 mol%), they obtained a library of fused five- to seven-membered rings 249 combining axial and planar stereogenicity with excellent enantio- and diastereoselectivity and in good to excellent yields. This method was efficiently extended to ruthenocenes 247 (M = Ru), for which a dual cascade was observed and two sets of axial and planar stereogenic elements were obtained, always with high stereoselectivity. An axial-to-planar diastereoinduction model was also proposed for this reaction. Once the chiral axis is formed, the steric repulsion between the pivalic group and the ferrocene ring disfavored the formation of the (Ra, Sp) product.
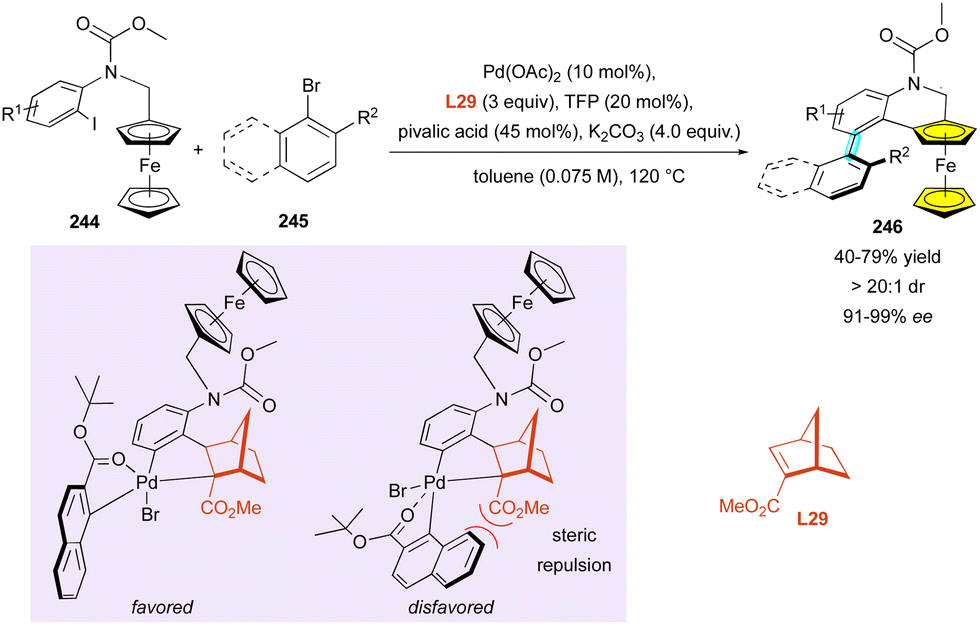 | ||
| Scheme 76 Pd-Catalysed enantioselective Catellani reaction. | ||
Finally, although the desymmetrisation of [2.2]paracyclophanes are rarely encountered in the literature,124 the Davies group newly reported their work on the reaction of donor/acceptor carbenes 251 with [2.2]paracyclophanes 250 (Scheme 78).125 The reaction proceeded by rhodium-catalysed enantioselective cyclopropanation, followed by ring opening, leading to the formation of original centrally and planarly chiral [2.2]paracyclophanes 252 bearing a seven-membered ring. A very low catalytic loading (0.5 mol%) of C42 allowed the construction of 18 examples in varying yields (7–78%) and very good enantioselectivity (78% to 98% ee) always with perfect diastereoselectivity. The reaction tolerated diverse (hetero)aromatic rings with both electron-donating and withdrawing substituents, decorated at the para, meta and ortho positions. Interestingly, the other enantiomer could be obtained using ent-C42 as the catalyst without any loss of efficiency.
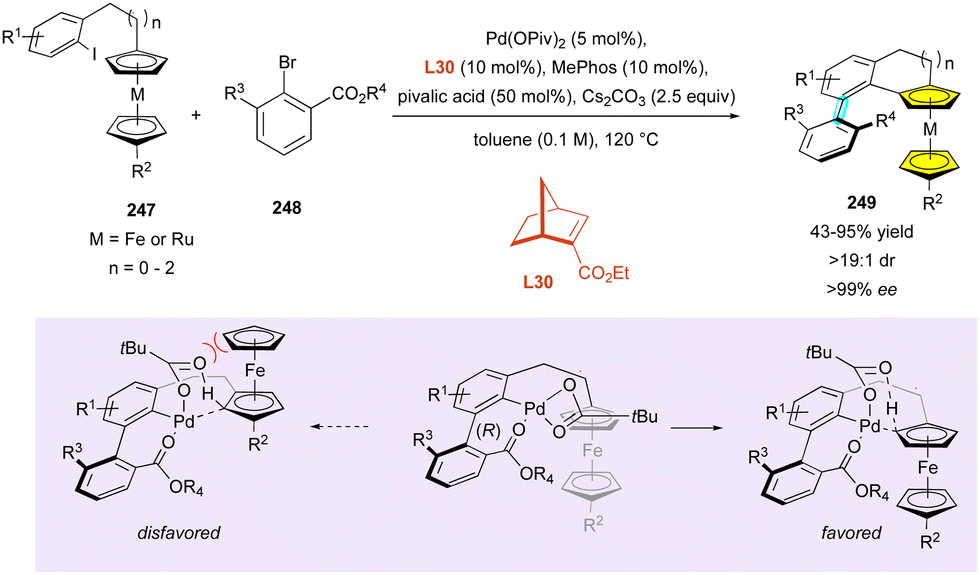 | ||
| Scheme 77 Pd-Catalysed enantioselective Catellani reaction. | ||
Simultaneous construction of both stereogenic elements
Organocatalysed transformations
:1 dr) and uniformly good to high enantioselectivity (76–98% ee).
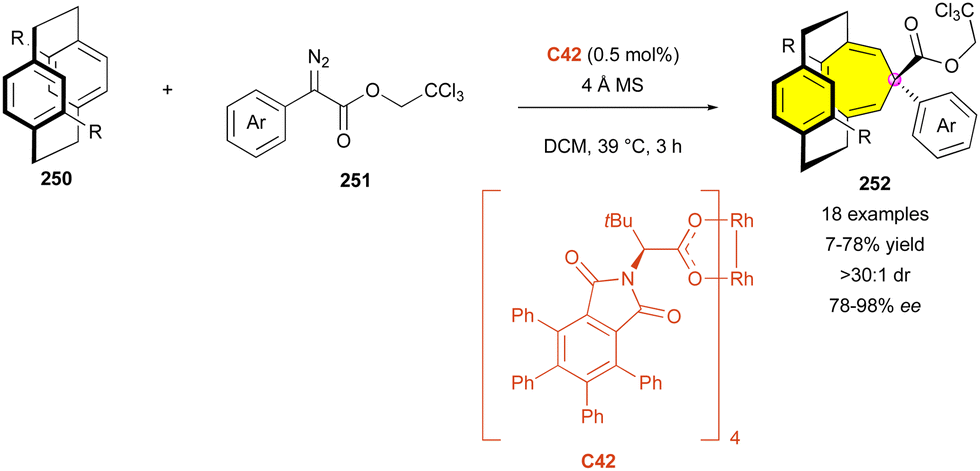 | ||
| Scheme 78 Rh(II)-Catalysed enantioselective ring expansion of [2.2]paracyclophanes. | ||
Closely following these pioneering results, the Seidel group described the efficient catalytic enantioselective synthesis of C–N atropisomeric isoindolinones 260 due to the condensation of 2-acylbenzaldehydes 258 and anilines 259 following a biomimetic pathway (Scheme 80).127 In the presence of only 1 mol% of CPA catalyst (S)-TRIP ent-C37, the reactions reached completion within 10 min and provided products with up to 98% enantiomeric excess. Three examples of anilines with an ortho t-butyl group formed atropisomeric products with decent diastereoselectivity, thereby enabling the simultaneous generation of axial and central stereoelements from achiral substrates by a simple dehydration sequence.
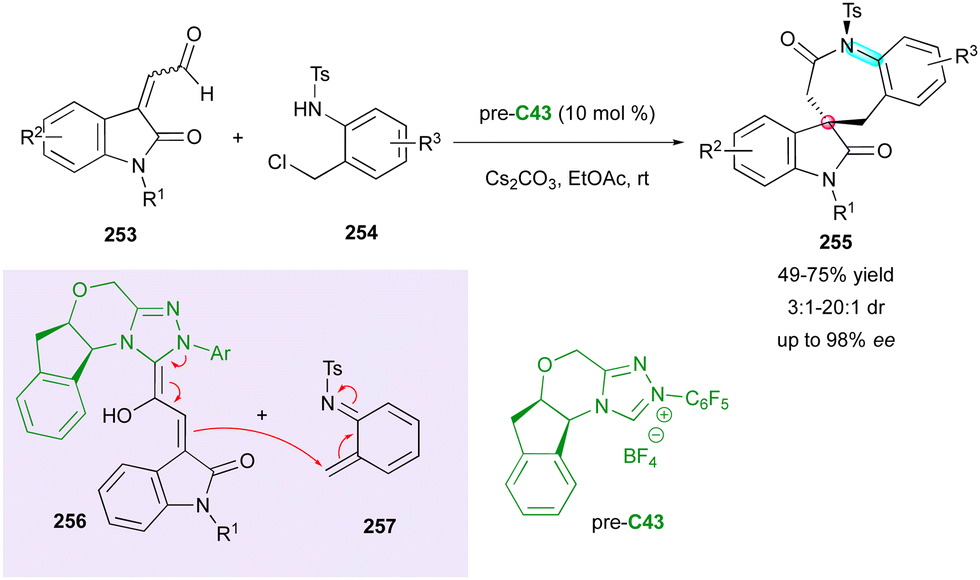 | ||
| Scheme 79 Enantioselective access to spirobenzazepinones bearing axial and spirocentral chirality. | ||
Similarly, in 2023, the Qi group reported a rare example of atroposelective access to dihydropyridinones scaffold 262 with a stereodefined C–N axis and bearing central chirality, starting from achiral 2-aminomaleate derivatives 261 and simple enals (Scheme 81).128 This transformation uses the reactivity of the NHC catalyst in the presence of base (DBU) and oxidant (TTBDQ) to afford the desired products with high enantiomeric excess (up to 99% ee) in moderate to high yield (11–90%) and diastereoselectivity (1.4:1–15:1 dr). However, a bulky tert-butyl substituent is a requirement for high control of the stereoselectivity, which limits its applicability.
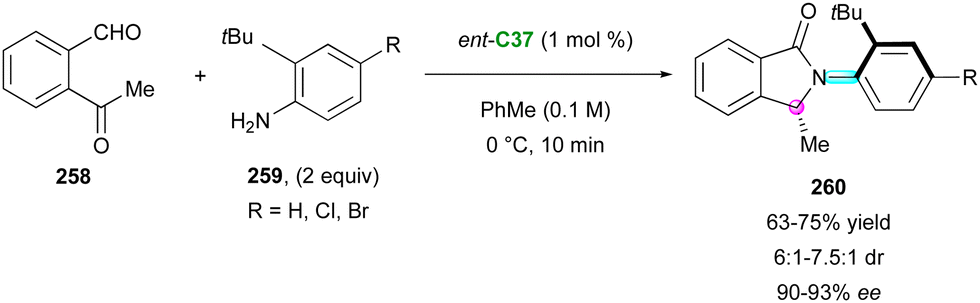 | ||
| Scheme 80 Enantioselective condensation between 2-acylbenzadehydes and o-tBu-anilines. | ||
Correspondingly, the Biju group harnessed the power NHC catalysis for the creation of a stereogenic N–N axis together with a stereogenic carbon centre (Scheme 82).129 Here, the presence of the benzyl group in precatalyst C44 was sufficient to dictate the approach of enamine 263 with excellent enantioselectivity and variable diastereoselectivity. Substantial rotational barriers were calculated (≈250 kJ mol−1), justifying the very high configurational stability of 264 and leading to a large range of products (51 examples) with moderate to very good yields.
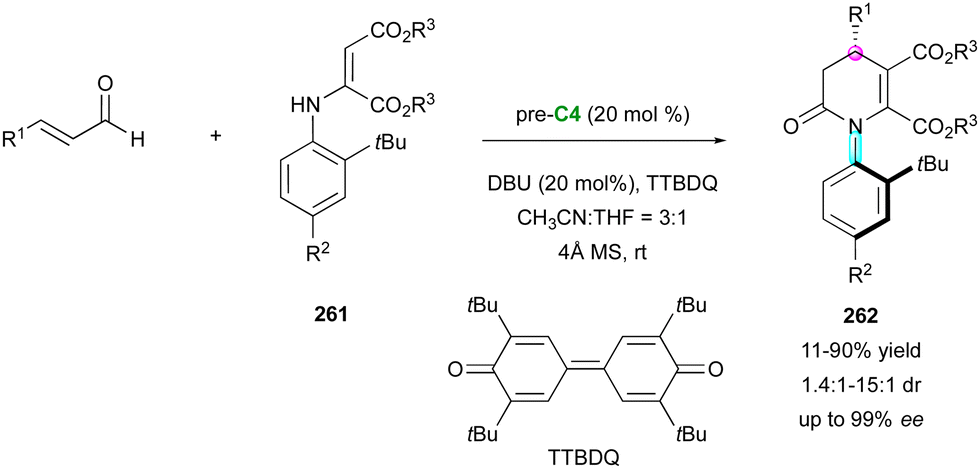 | ||
| Scheme 81 Enantioselective synthesis of dihydropyridinones bearing a stereogenic C–N axis. | ||
Very recently, a related strategy involving oxidative NHC-organocatalysis was disclosed by Wong and Lu for the atroposelective synthesis of rare N–N axially and centrally chiral indoles and pyrroles 266 (Scheme 83).130 This concept is based on the enantioselective construction of the 5,6-dihydropyrimidine ring using chiral α,β-unsaturated acylazoliums from enals as C3-synthons towards the aza-Michael addition of readily available isothioureas 265, leading to the key centrally chiral acylazolium intermediate 267, which evolved by intramolecular N-acylation to the final N–N atropisomers 274. The reaction is efficient in terms of yield and enantioselectivity even if only moderate to good diastereoselectivities were generally observed. The experimental evidence corroborated by DFT calculations clearly indicates an interesting negative nonlinear effect (NLE), suggesting that a second NHC molecule helps to activate the isothiourea function in the enantio-differentiating transition state. The desired products 266 showed reasonable atropostability with a diastereomerisation barrier of 155 kJ mol−1 (for R1 = Ph, R2 = H, R3 = Me).
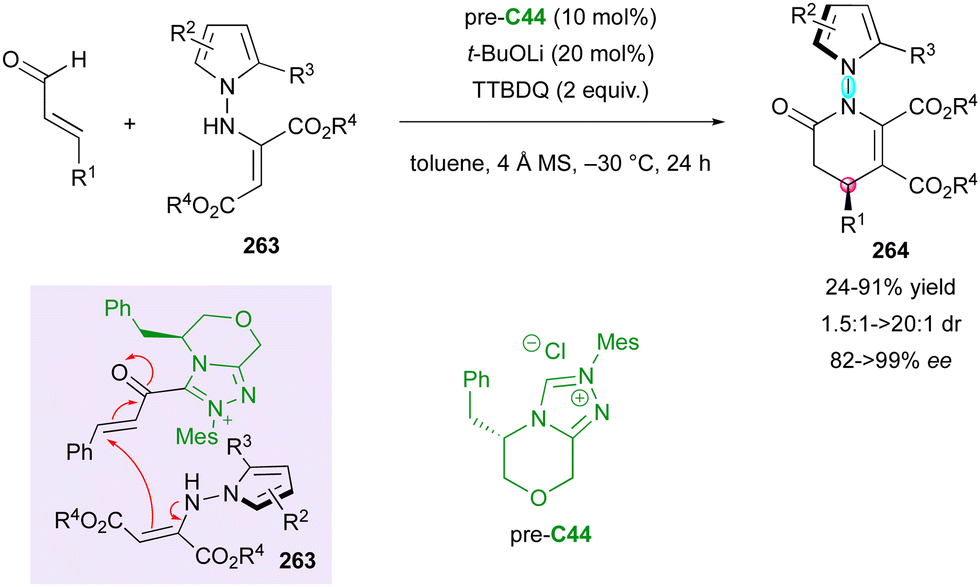 | ||
| Scheme 82 NHC-Catalysed diastereoselective (3+3) annulation. | ||
The Wong and Zhao group successfully applied their NHC-catalysed atropoenantioselective benzofurannulation (see Scheme 67) to the synthesis of related indole-derived bridged biaryls 269 (Scheme 84).108 Starting from racemic propargylic tosylamidophenols (±)-268, they observed highly chemoselective sequential intramolecular O-acylation and N-alkylation from key acyl azolium intermediate 270, producing eight-membered ring bridged lactones 269 featuring central and axial stereogenic elements.
 | ||
| Scheme 83 Organocatalytic atroposelective synthesis of N–N axially chiral indoles and pyrroles. | ||
The highly electrophilic reactivity of the allene function combined with its intrinsic axial chirality makes this intriguing structural unit a particularly efficient intermediate for the control of multi-chirality (see Schemes 21–23). Also, the in situ generation of chiral vinylidene-quinone methides (VQMs)131 from 2-alkynylnaphthols by organocatalysis has recently attracted interest for the simultaneous control of multiple stereogenic elements involving cycloadditions or nucleophilic additions with the transfer of chirality.132
In this context, in 2019, Liu, Yan and Li described the enantioselective one-pot construction of intriguing molecules 273 bearing a stereogenic carbon atom, an axially chiral styrene with a stereoisomeric double bond (Scheme 85).133 They employed 5H-oxazol-4-ones 271 as nucleophilic partners towards 2-alkynylnaphthols 272. This method enabled the rapid construction of a series of stereochemically complex products 273 with excellent E,Z selectivity, diastereoselectivity (>20:1 dr), and enantioselectivity (up to 96% ee). The reaction proceeds under mild reaction conditions, tolerates a range of functional groups, and is applicable to gram-scale synthesis. Two years later, the same strategy was successfully employed by Wang with pyrazolones 274 as nucleophiles, with in situ-generated chiral VQM intermediates 277 under hydrogen-bonding organocatalysis with C47 yielding fully enantiocontrolled products 276 within a shorter reaction time (5 to 6 h).134 Benzyl derivatives (R1) are preferable in terms of enantioselectivity, but modifications on other positions are very well tolerated. Gram-scale synthesis was performed smoothly without the loss of efficiency or stereoselectivity.
 | ||
| Scheme 84 Atroposelective synthesis of indole-derived bridged biaryls. | ||
Simultaneously, the Yan group reported an intramolecular nucleophilic addition on transient chiral VQMs as a unique example of the fully controlled enantioselective synthesis of three different stereogenic elements generated simultaneously from properly functionalised achiral substrates using organocatalysis (Scheme 86).135 They smartly designed 1-(2-(9H-carbazol-9-yl)phenyl)naphthalen-2-ols 278, which undergo concomitant electrophilic bromination, leading to chiral VQM intermediates 280, followed by annulation to yield a new class of chiral azepines 279 displaying two chiral axes (C–C and C–N) and a stereogenic nitrogen atom, which together resulted in an inherent helical shape of the fused heterocyclic moiety. Low temperature (−78 °C) and N-bromosuccinimide (NBS) quickly afforded azepines 279 in excellent yields and stereoselectivities. A gram-scale reaction could be achieved and the reaction tolerated both electron-withdrawing and donating groups without the loss of enantioselectivity.
 | ||
| Scheme 85 Enantioselective intermolecular nucleophilic addition on VQM intermediates. | ||
The Hong group was inspired by this intramolecular approach, who reported an alternative enantioselective domino strategy for the construction of spiroketal lactones 283 containing both a C–C stereogenic axis and a spirocyclic quaternary stereogenic centre (Scheme 87).136 The key chiral VQM intermediates 284 were generated via electrophilic halogenation of functionalised 2-alkynylnaphthols 281 using N-haloimides 282 in the presence of bifunctional thiourea catalyst C48, triggering an intramolecular domino annulation, which resulted in the construction of axial and central chirality by the transfer of axial chirality. This elegant spiroannulation tolerated a broad functional diversity, producing a large new family of atropisomeric and centrally chiral spiroketals 283 in good yields (up to 99%) with high enantio- and diastereoselectivities (up to 98%, >20:1 dr).
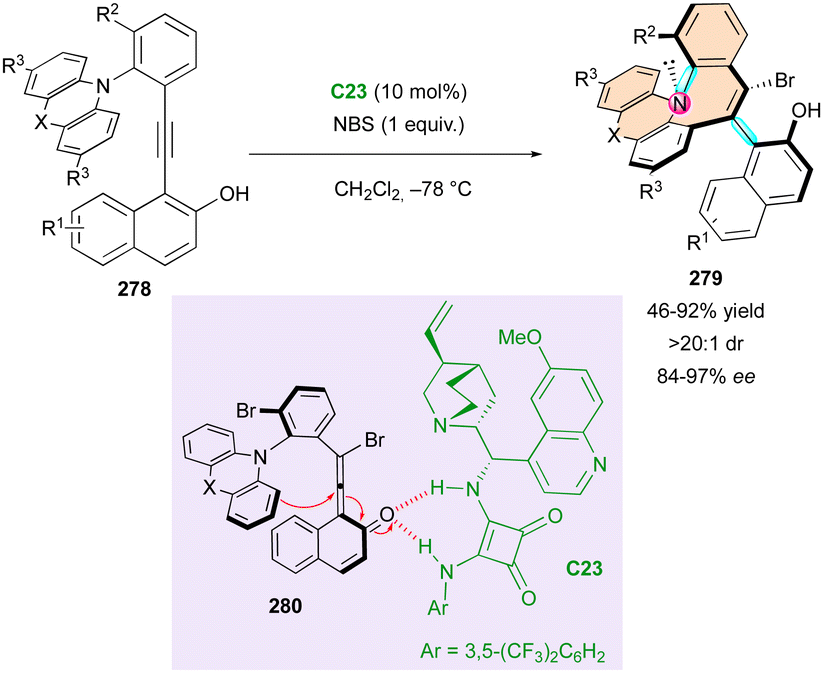 | ||
| Scheme 86 Enantioselective intramolecular nucleophilic addition on chiral VQM intermediates. | ||
In a related intramolecular trapping of a chiral VQM intermediate, the Zhou group performed the enantioselective synthesis of a new class of naphthyl-2H-chromene derivatives 287 bearing both central and axial chirality (Scheme 88).137 CPA catalyst C49 was used as a dual hydrogen-bonding activator, performing the enantioselective nucleophilic addition of 2-alkynylnaphthols 285 to o-quinone methides 286 (o-QMs) as C-electrophiles, which resulted in the generation of the corresponding chiral all-carbon tetrasubstituted VQM intermediates 288 instead of the hetero-Diels–Alder reaction with a triple bond. Based on DFT calculations, the reaction seems to evolve by an intramolecular dearomative Michael-type addition to spirocyclobutene intermediate 289, suffering 4π electrocyclic ring opening, which determines the resulting configuration of the stereogenic axis in 290 by central-to-axial chirality conversion. The last step is an oxa-6π-electrocyclisation, providing stable chiral naphthyl-2H-chromene derivatives 287via an axial-to-central chirality conversion. This efficient synthesis provided the product in excellent yield (72–99%) with high enantioselectivity (81–99%) and moderate to high diastereoselectivity (4:1 to >20:1 dr).
 | ||
| Scheme 87 Enantioselective synthesis of spiroketal lactones with both axial and central chirality. | ||
Recently, a new class of conformationally stable C(sp2)–C(sp3) atropisomers was discovered in some natural products,138 attracting interest from chemists for the enantioselective synthesis of these unusual molecular frameworks. In 2017, the Bencivenni group reported the first case of thermodynamic control of the C(sp2)–C(sp3) stereogenic axis (Scheme 89).139 They performed the enantioselective Friedel–Crafts-type alkylation of properly functionalised inden-1-ones 219 with 8-substituted 2-naphthols 292 using chiral quinidine derivative C30 as the catalyst. Interestingly, under these conditions, with bulky R2 groups, a single antiperiplanar conformational diastereomer ap-293 combining axial and central chirality was obtained with very high enantioselectivity (up to 98% ee). This clearly indicates that the barrier for the rotation of the C(sp2)–C(sp3) bond is high enough to prevent atropodiastereomerisation to sp-293. This experimental observation was corroborated by DFT calculations, indicating that the bulkiest are the groups on the 8-postion of naphthol 292 and on the 4-postion of inden-1-ones 291, making the atropodiastereomerisation barrier higher. A step further was possible when R2 = phenanthrenyl, albeit in the low yield of 15%, obtaining a 1:1 mixture of two diastereomers ap-(R,M)-294 and ap-(R,P)-294 featuring two stereogenic axes and one stereogenic centre in 94% and 65% ee, respectively.
 | ||
| Scheme 88 Enantioselective synthesis of axially and centrally chiral naphthyl-2H-chromenes. | ||
Some years later, the Jørgensen group published one of the first case of multi-stereogenic synthesis including a conformationally stable C(sp2)–C(sp3) bond and two consecutive stereogenic centres (Scheme 90).140 This new approach was developed with two different chiral pyrrolidine catalysts, C50 or C51, in combination with pivalic acid. This catalytic system provided the addition of 5H-benzo[a]pyrrolizine-3-carbaldehydes 295 to olefin-bearing electron-withdrawing group 296 as Michael acceptors, leading to the corresponding cyclised product 297 with moderate yield (up to 69%) and moderate to excellent enantio- and diastereoselectivity (up to 99% ee, from 1:1 to >20:1 dr). The plausible mechanism involves enamine intermediate 298 from aldehyde 295 with the pyrrolidine catalyst in acid condition followed by a Michael/Mannich-type cyclisation sequence with the olefin rendering the final chiral atropisomeric heterocycle 297 after regeneration of the catalyst by elimination from intermediate 299.
 | ||
| Scheme 89 Enantioselective synthesis of stable C(sp2)–C(sp3) atropisomers via Friedel–Crafts-type alkylation. | ||
 | ||
| Scheme 90 Enantioselective organocatalytic synthesis of conformationally stable C(sp2)–C(sp3) atropisomers. | ||
The synthesis of small molecules with a stable helical shape, particularly when incorporating five-membered rings, presents a significant challenge in organic chemistry. The inherent instability of these configurations complicates their production and necessitates the development of new methodologies. In the following very recent study, our group introduced a padlocking technique employing the enantioselective organocatalytic domino furannulation shown before (Scheme 91). This method allows the controlled synthesis of helically chiral tetracyclic structures 304, containing one or two five-membered rings, from carefully designed achiral fused-tricyclic precursors 303 (Scheme 92).143 Through this approach, we achieved simultaneous control of both the central and helical chirality, addressing a crucial need in this field. Moreover, applying this protocol to achiral naphthofuran 305 having a low barrier to rotation of only 55.7 kJ mol−1, we were pleased to observe the clean formation of products 306 displaying three different stereogenic elements, in good yield, excellent enantioselectivity and in a decent 5:1 diastereomeric ratio, relative to the configuration of the stereogenic C–C bond. Noteworthy, the generation of this still rare family of enantioenriched compounds featuring axial, central and helicoidal stereogenic elements was achieved through a one-step synthesis.
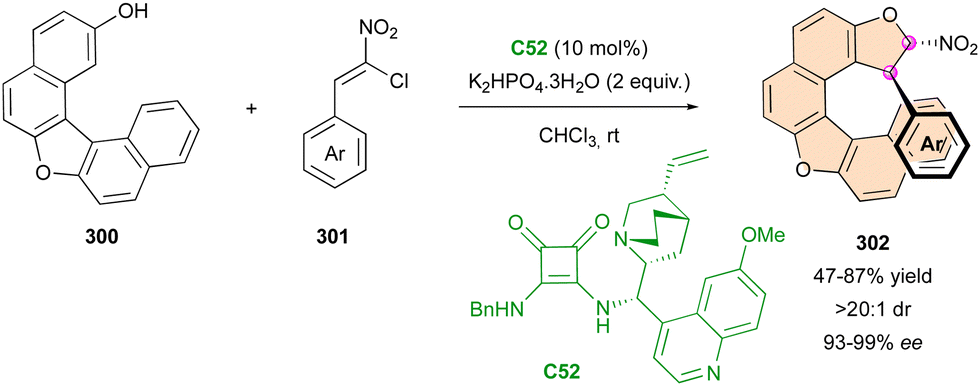 | ||
| Scheme 91 Helicoselective heteroannulation of naphthol derivatives. | ||
A conceptually related sequential oxidative helicoselective heteroannulation was reported in 2023 independently by the Li144 and Yang145 groups, who proposed two complementary elegant methods to access azahelicene structures based on a CPA-catalysed three-component Povarov reaction followed by oxidative aromatisation of the transient helicoidal tetrahydroquinolines featuring two stereogenic centres. Although the overall one-pot oxidative process is efficient and general for the preparation of a large series of aza[4]- and aza[5]helicenes, the corresponding multi-chiral helicenoid tetrahydroquinolines were isolated only once by Li (310) and twice by Yang (307 and 308) (Scheme 93).
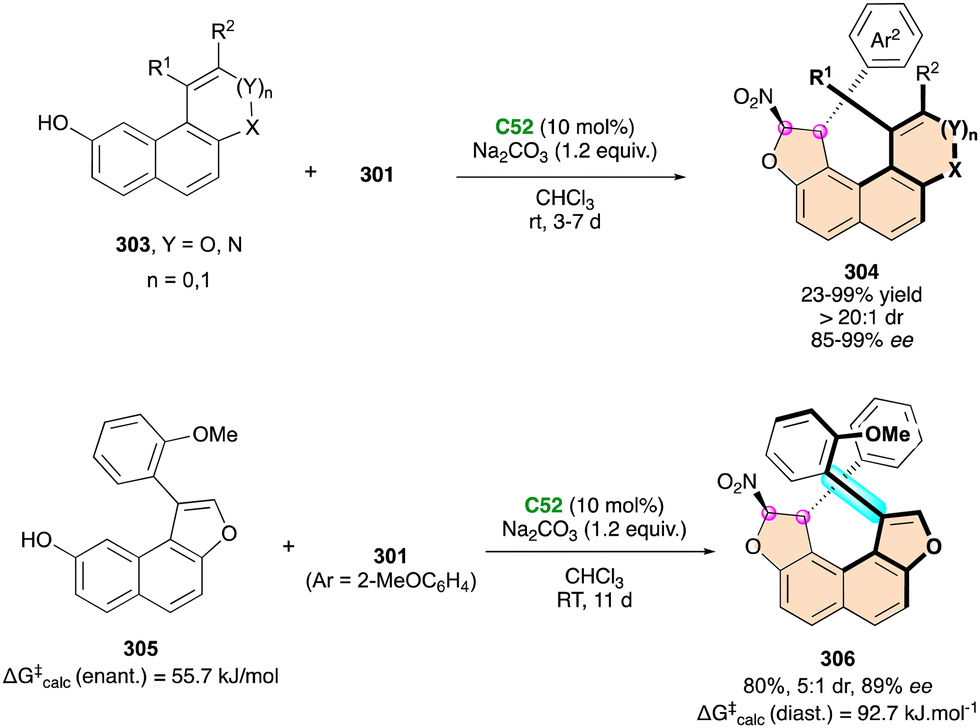 | ||
| Scheme 92 Enantioselective construction of configurationally locked helically-chiral tetracyclic scaffolds. | ||
In both contributions, the proposed selective functionalisation of benzo[c]phenanthrene-2-amine 309, in the presence of benzaldehyde and enamides or 3-vinyl-indole gave the expected centrally and helically chiral tetrahydroquinolines 307, 308 and 310, respectively, in good yield and high stereocontrol. Alternatively, in the study by Li, the relative 1,3-configuration of 310 was not determined, while Yang was able to separate the two diastereomers of 308 and 312 and showed that their respective DDQ oxidation afforded the same aza[5]helicene 311 (Scheme 94). This result is in agreement with the helical chirality being determined by the formation of the C-1 stereocentre of tetrahydroquinoline 308. Finally, the mechanistic studies proposed by Yang suggested that the (4+2) cycloaddition reaction could occur in a stepwise manner, fully catalysed by the CPA with perfect stereocontrol.
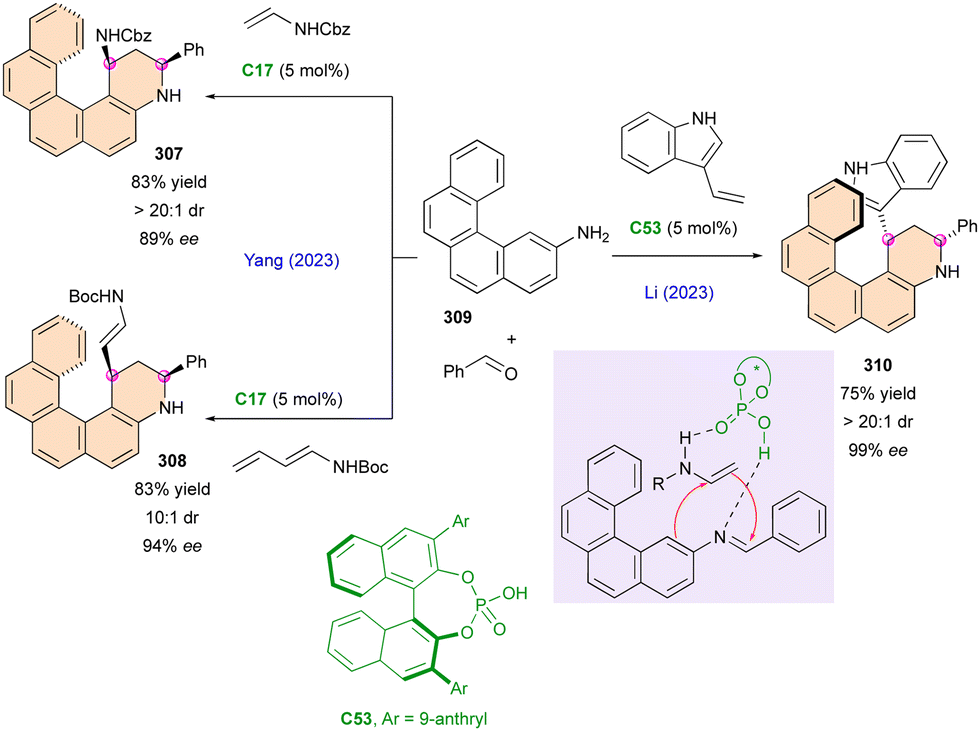 | ||
| Scheme 93 Helicoselective Povarov reaction. | ||
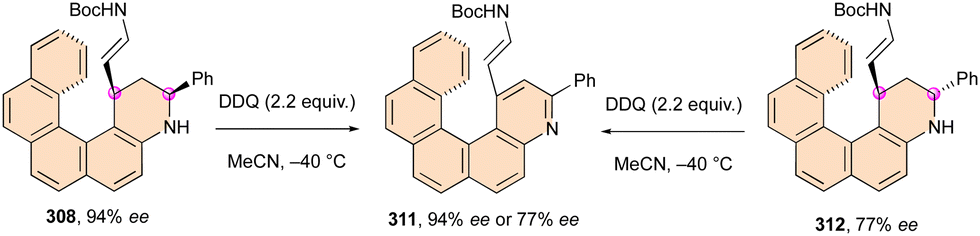 | ||
| Scheme 94 DDQ oxidation of diastereomeric helicoidal tetrahydroquinolines. | ||
C–H peri-functionalisation of 2-hydroxybenzo[c]phenanthrene derivatives 317 was reported by Liu and Wang for the efficient assembly of 1,12-disubstituted carbo[4]helicenes (Scheme 96).147 Hence, organocatalyst C54 promoted the simultaneous atropo- and helicoselective C–H amination of 317 with highly electrophilic diazocarboxamide partners 318, delivering the desired carbo[4]helicenes 319 in moderate yield but excellent diastereo- and enantioselectivities. DFT calculations revealed that a dual hydrogen-bonding activation between the thiourea N–H group and phenolate moiety for one part and between the alkylammonium ion and the diazocarboxamide for the other part accounted for the observed stereochemistry. In the major transition state 320, supplementary favorable C–H–π interactions between the pyrenyl substituent of the catalyst and phenanthrenol substrate 317 were postulated. Remarkably, the authors reported the use of prochiral diazocarboxamide 318 as a substrate for the control of an additional stereogenic C–N bond in final structure 321.
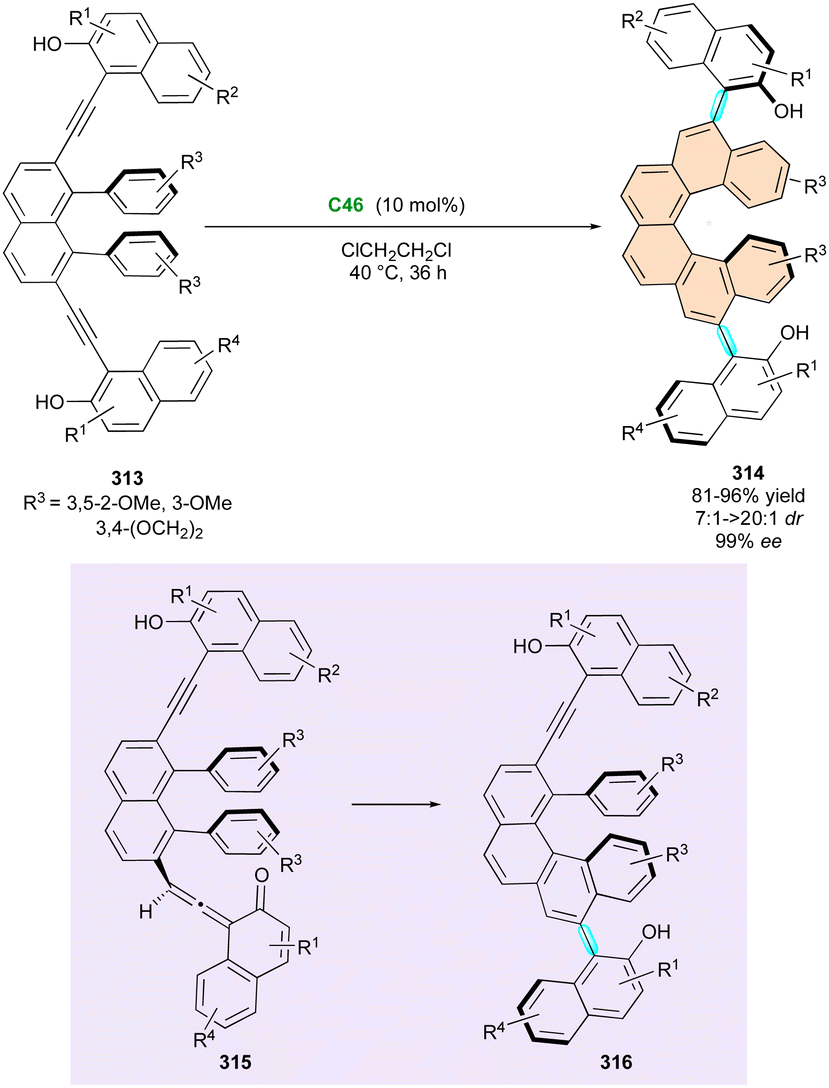 | ||
| Scheme 95 Enantioselective synthesis of axially chiral carbo[6]helicenes. | ||
:1) within one hour. Subsequently, the reaction conditions were further optimised to enhance diastereoselectivity by increasing the reaction temperature to 60 °C and adding di-tert-butyl acetylenedicarboxylate 323 dropwise. Central chirality was introduced at the ring-fused position of bicyclic rhodacyclopentene intermediate 325 through the oxidative coupling of the metal centre with an enyne. The configuration of the stereogenic axis was not controlled at this stage, but during the following reaction with the alkyne.
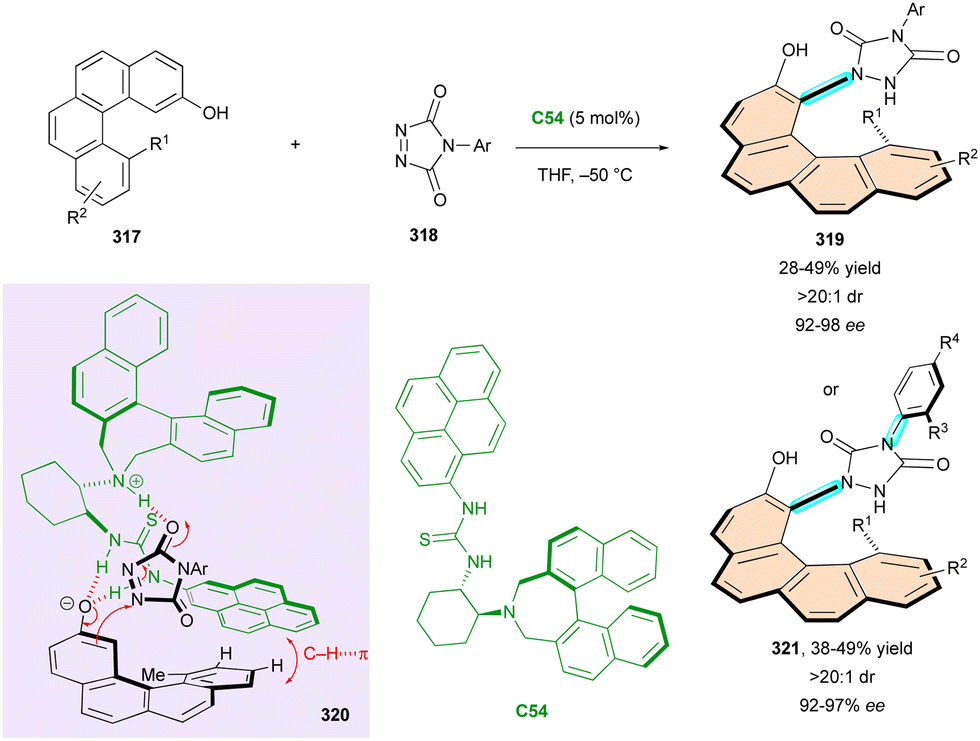 | ||
| Scheme 96 Atropo- and helicoselective C–H amination of benzophenanthrols. | ||
Functionalised alkynes are also prone to react with electron-deficient alkenes following a [2+2] cycloaddition/retroelectrocyclisation (CA-RE) sequence to synthetically useful 1,3-butadiene intermediates.149 This attractive strategy was elegantly exploited by Cao and Feng in 2023, starting with ortho-alkynylnaphthols 327 and 1-azadienes 326 to obtain axially and centrally chiral 1,2-dihydrobenzofuro[3,2-b]pyridines 328 (Scheme 98).150 The catalytic process was performed using a L31-gadolinium(III) complex as the Lewis acid, triggering the in situ formation of reactive vinylidene quinone methides (VQMs) 329 as precursors of [2+2] cycloadduct intermediate 330. In most cases, centrally and axially chiral dihydrobenzofuro-pyridines 328 were obtained with decent to high stereocontrol together with good yields after 1 to 7 days of reaction at room temperature. Lower stereoselectivities were observed when changing the N-protecting group to a methyl sulfonyl and the enantiocontrol was negatively affected with ortho-substituted aryl (Ar1 and Ar2). The proposed mechanism emphasises the formation of axially chiral intermediate 331, which after tautomerisation gives 331, evolving by a stereoselective 1,6-conjugate addition with an overall axial-to-central conversion of chirality and leading to centrally and axially chiral dihydrobenzofuro-pyridines 328 with decent to high stereocontrol.
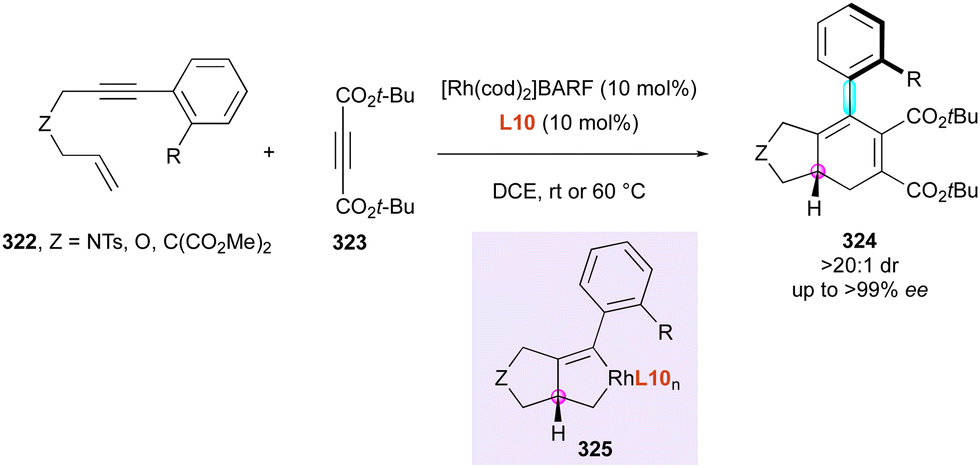 | ||
| Scheme 97 Rh-Catalysed [2+2+2] atroposelective cycloaddition. | ||
1-Alkynyl tosylindoles 334 were identified by Li and collaborators as powerful partners for the development of an efficient enantioselective Rh(III)-catalysed (3+2) annulation, initiated by a C–H activation of an arylnitrone 333 acting as an electrophilic directing group (Scheme 99, top).151 The enantiopure bis-methoxy Cramer-type (R)-CpxRh(III) catalyst C55 proved to be optimal to give the expected pentatomic C–N atropisomeric cycloadducts 335 bearing a distal C-centred point-chirality with excellent diastereo- and enantioselectivity. Extrapolation to ortho-tosyloxy-naphthyl-substituted alkynes 336 proceeded with the same global efficiency when Ar2 is a bulky aromatic substituent to form the corresponding axially and centrally chiral indene derivatives 337 (Scheme 99, bottom). The experimental observations combined with DFT calculations allowed the identification of two enantio-determining steps under the catalyst control and highlighted the crucial role of the tosyl substituent in 336, given that changing the 2-OTs group to 2-OMs led essentially to no reaction. Firstly, the alkyne insertion in 338 determines the atroposelectivity and the folding of the tosyl group allows a stabilising π–π stacking, which reduces steric repulsion with the nitrone group. Secondly, the diastereoselectivity of the reaction is dictated by 339via the migratory insertion of Rh-alkenyl to nitrone driven by the oxygen-tethering effect.
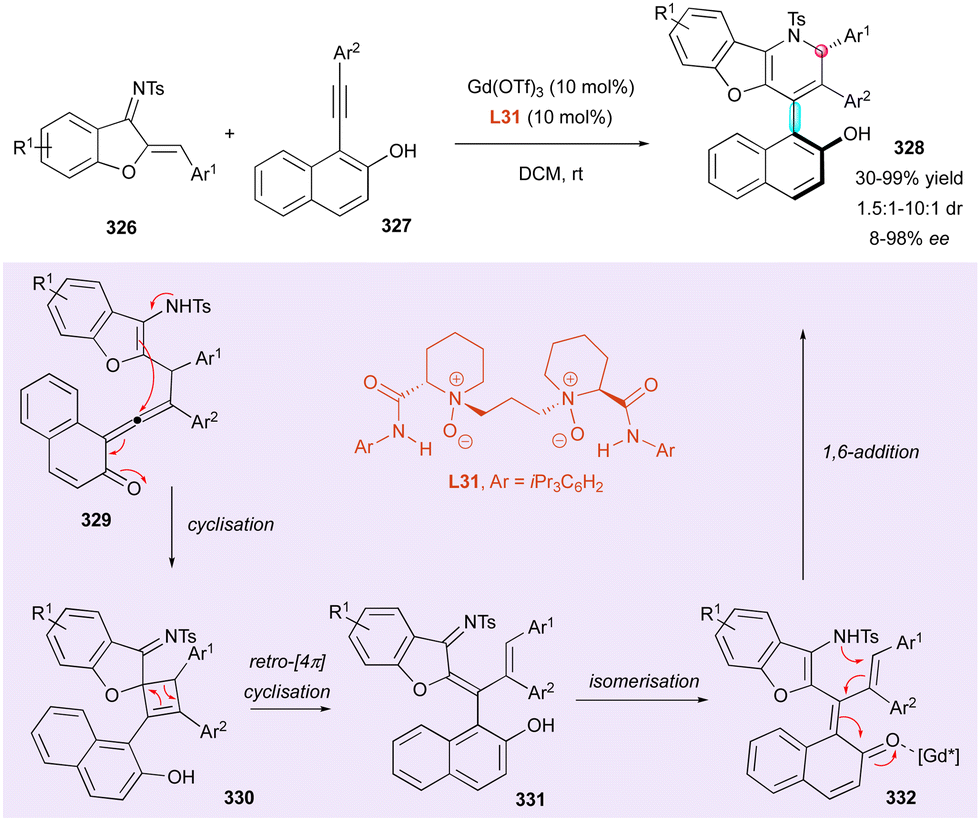 | ||
| Scheme 98 Gd-Catalysed [2+2] cycloaddition/retroelectrocyclisation cascade. | ||
Enantioselective synthesis of axially chiral allenes bearing central chirality
Metal-catalysed approaches
In complement with previous strategies for the control of multiple stereogenic elements in the central, axial, planar, and helicoidal series, axially chiral allenes with point chirality also constitute targets of recent interest.In 2012, Davies and colleagues exploited the well-established transition-metal-catalysed functionalisation of propargylic alcohols152 for the enantioselective synthesis of a few centrally and axially chiral products (R,aS)-343 from racemic propargylic tertiary alcohols (±)-341 with moderate to good diastereoselectivity and high enantiomeric excess (Scheme 100).153 Mechanistically, the reaction proceeds through a rhodium-catalysed [2,3]-sigmatropic rearrangement in the presence of N-sulfonyl (S)-proline ligand L32, with migration of the tertiary alcohol revealing both stereogenic elements. This method has not been broadly exemplified and the diastereoselectivity seems to be dependent on the substrate, but it represents one rare example of the enantioselective synthesis of centrally and axially chiral allenes.
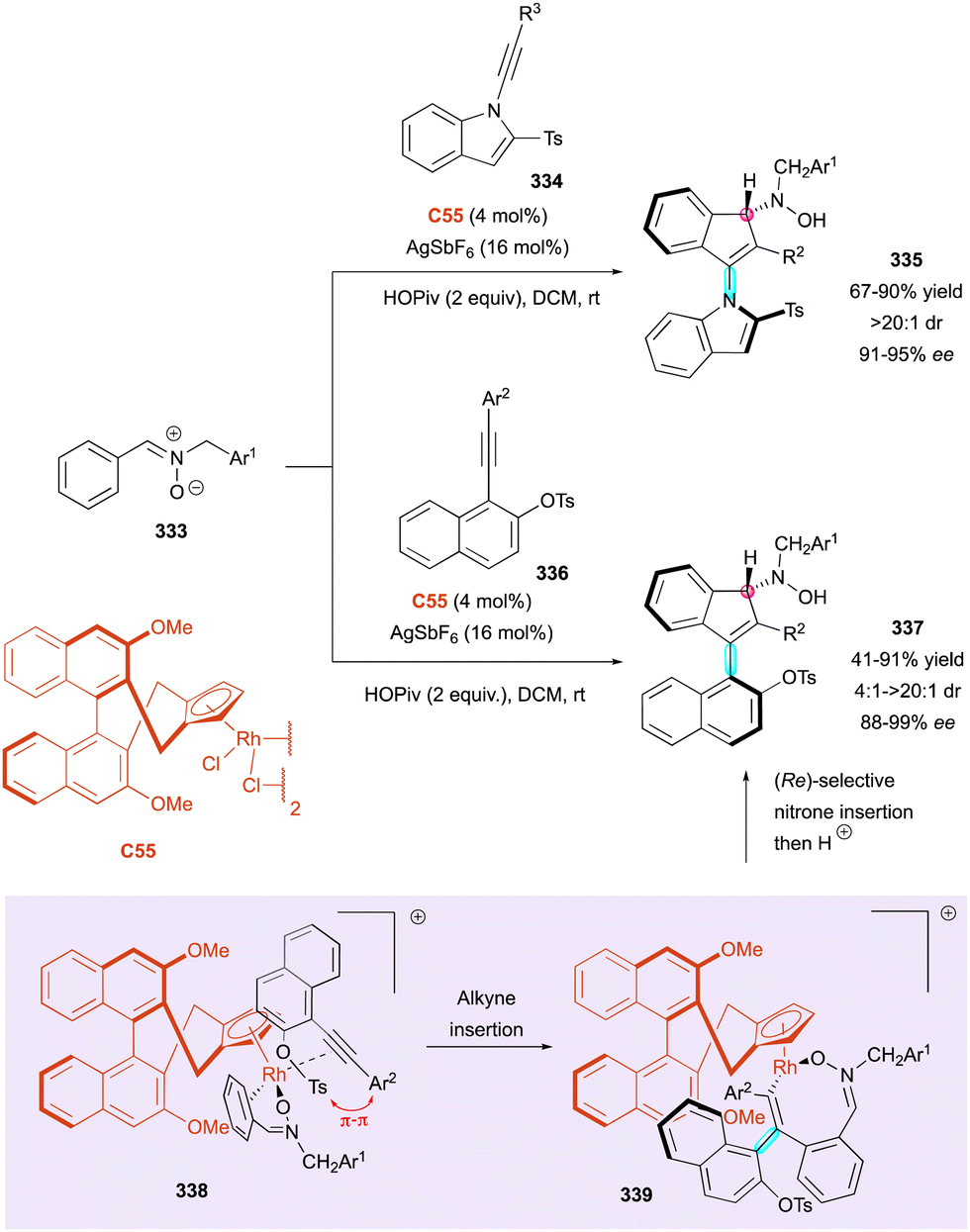 | ||
| Scheme 99 Rh-Catalysed (3+2) cycloaddition to C–N and C–C axially and centrally chiral indenes. | ||
Similarly, four years later, Liu, Feng, and coworkers designed racemic propargyl vinyl ethers (±)-344 capable of undergoing sigmatropic Claisen rearrangement for the synthesis of centrally and axially chiral allenes (R,aR)-345via a KR process, also producing (S)-344 in up to 99% ee (Scheme 101).154 The nickel-catalysed reaction in the presence of chiral N,N′-dioxide ligand L33 allowed the isolation of several examples with excellent stereoselectivity (9:1 to 50:1 dr and 92% to 99% ee). Interestingly, the size of the carbocycle in the substrate plays a crucial role in the reactivity of the transformation. Indeed, for (±)-344a (n = 1, R1 = Ph, R2 = Me), the reaction was completed after one hour at −20 °C, whereas with (±)-344b (n = 3, R1 = Ph, R2 = Me), almost a day at 35 °C was necessary for complete transformation. Additionally, the diastereoselectivity seemed to be impacted by the presence of bulky R1 and R2 substituents.
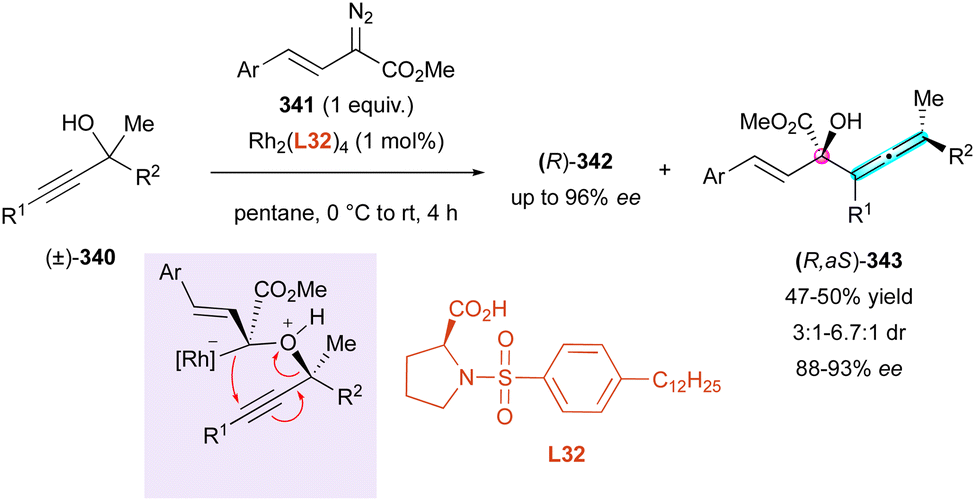 | ||
| Scheme 100 Enantioselective Rh-catalysed ylide formation/[2,3]-sigmatropic rearrangement. | ||
In 2021, using a copper-catalysed cross-coupling/alkynylogous aldol sequence, the Sun group displayed access to chiral cyclic α-allenols 348 containing both central and axial stereogenic elements (Scheme 102).155 A copper(II) salt combined with BOX ligand L34 was used to perform this enantioselective transformation starting from 2-prop-2yn-1-ylbenzaldehydes 346 (R2 = H) and α-phenyl diazoacetates 347 to give five-membered ring products 348 in good to excellent yields, enantiomeric excess and diastereoselectivity. Modifications of the diazo partner were well tolerated, but the utilisation of ketones (R2 = Me) and the formation six-membered rings (n = 2) were far less efficient or even not feasible for four-membered analogues (n = 0). The copper–carbene migratory insertion to the alkyne was enantio-determining for the control of both the axial and central chirality.
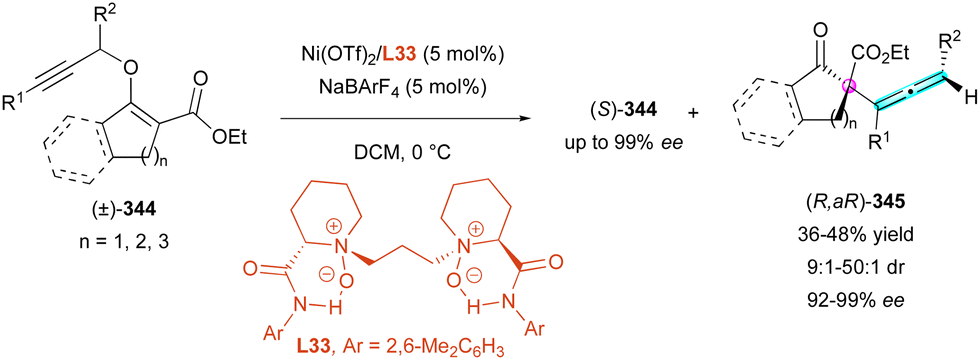 | ||
| Scheme 101 Enantioselective Claisen rearrangement for the synthesis of chiral allenes. | ||
Capitalising on the stereoselective formation of chiral allenes by the hydrofunctionalisation of conjugated enynes, Lin, He, and collaborators showed that this transformation can be combined with a DKR process when racemic fluorinated esters (±)-350 are used as pronucleophiles (Scheme 103).156 This method relied on an enantioselective dual Cu/Pd synergistic catalysis with two complementary chiral ligands L35 and L36 for palladium and copper, involving the reversible in situ formation of nucleophilic copper-enolate intermediate 353 and electrophilic π-allylpalladium complex 354, respectively. Both stereogenic elements could be controlled efficiently, leading to the formation of a large number of tertiary fluoride-tethered allenes 352 with very high stereoselectivity (8:1 to >20:1 dr and 90% to >99% ee). Interestingly, the nature of the Ar groups of biaryl diphosphine ligand L35 was crucial given that a simple phenyl gave almost no diastereoselectivity.
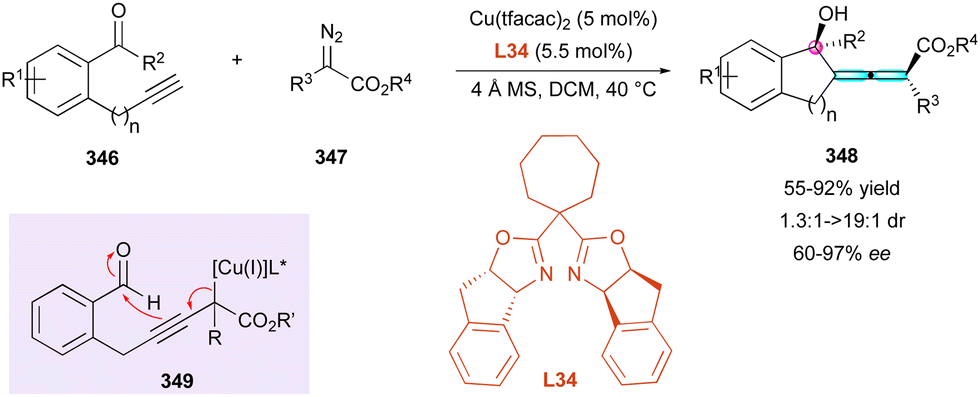 | ||
| Scheme 102 Cu-Catalysed cross-coupling/alkynylogous aldol sequence. | ||
One year later, Wang's group reported the elegant three-component dual photoredox and chromium-catalysed radical 1,4-functionalisation of 1,3-enynes for the preparation of chiral α-allenols 356 bearing a C-centred stereogenic element (Scheme 104).157 The conditions were optimised using benzaldehyde (R1 = Ph), a slight excess of both TIPS-1,3-enyne 351 (R2 = TIPS) and isopropyl dihydropyridine (DHP) 355 (R3 = iPr) as a radical precursor. Chromium dichloride and chiral oxazoline ligand L37 with 4-CzlPN C35 as the photocatalyst under blue LED irradiation for 12 h at room temperature afforded α-allenols 356 in excellent yield, enantio- and diastereoselectivity. This method tolerated both aryl and aliphatic aldehydes but changing the 1,3-enyne partner for a less hindered silyl or aryl derivative often resulted in the loss of diastereoselectivity. Finally, other radical sources such as redox active esters and trifluoroborate salts were also successfully investigated but the transfer was limited to secondary and tertiary radicals. The authors suggested that both stereogenic elements are formed simultaneously during the addition of propargyl chromium(III) intermediate 357 to the aldehyde.
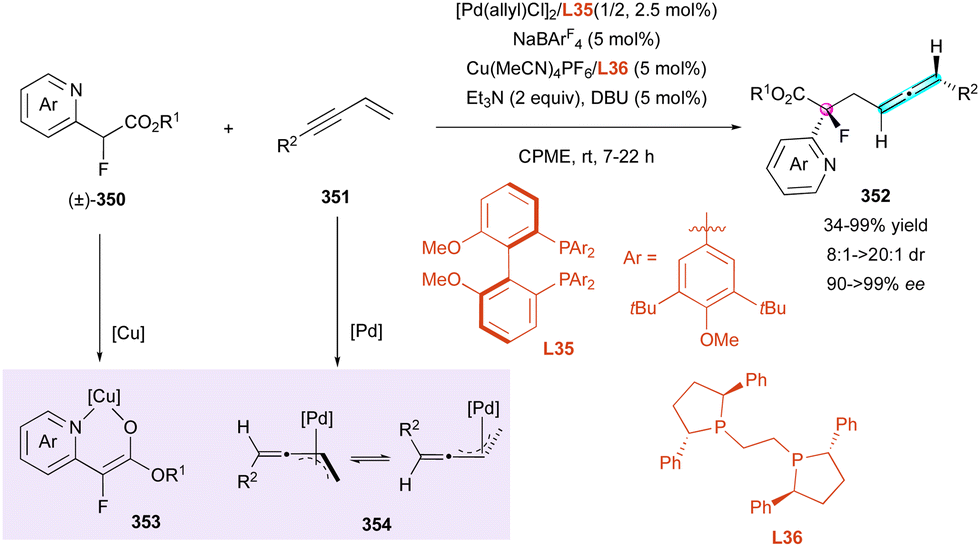 | ||
| Scheme 103 Dual Cu/Pd-catalysed stereodivergent synthesis of fluoride-containing allenes. | ||
Fu and co-workers reported the nickel-catalysed enantioconvergent and diastereoselective cross-coupling between trifluoro alkyl bromides 359 and TMS-substituted 1,3-enynes 360 for the simultaneous installation of central and axial stereogenic elements in allenes 361 (Scheme 105).158 In addition to not being highly sensitive to moisture or air, this protocol allows the coupling between a variety of CF3-substituted alkyl bromides, including possibly sensitive moieties such as primary alkyl chloride/bromide, secondary carbamate and epoxide. The mechanism is supposed to proceed via the formation of chiral nickel complex 362159 with controlled central chirality, which is converted to axial chirality upon the formation of complex 363. Then, diastereoselective cross-coupling with CF3-alkyl radical affords 364 as precursors of 361 after a final reductive elimination step (Scheme 106).
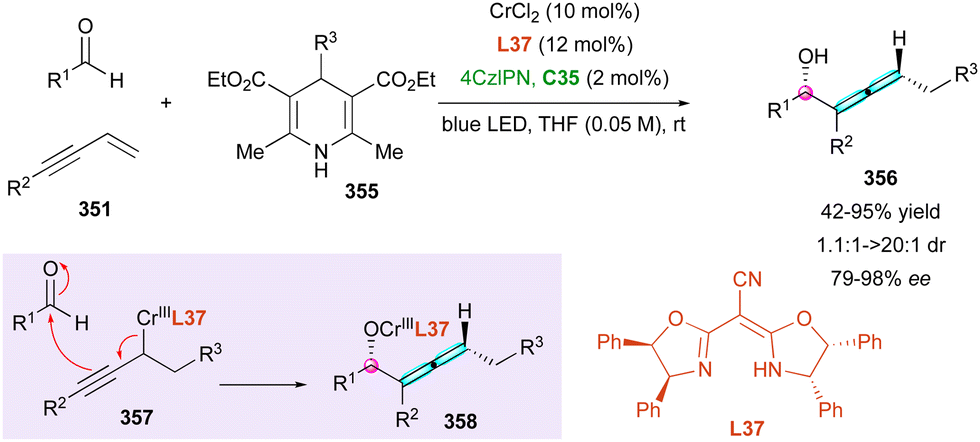 | ||
| Scheme 104 Photoinduced and Cr dual-catalysed cascade reaction. | ||
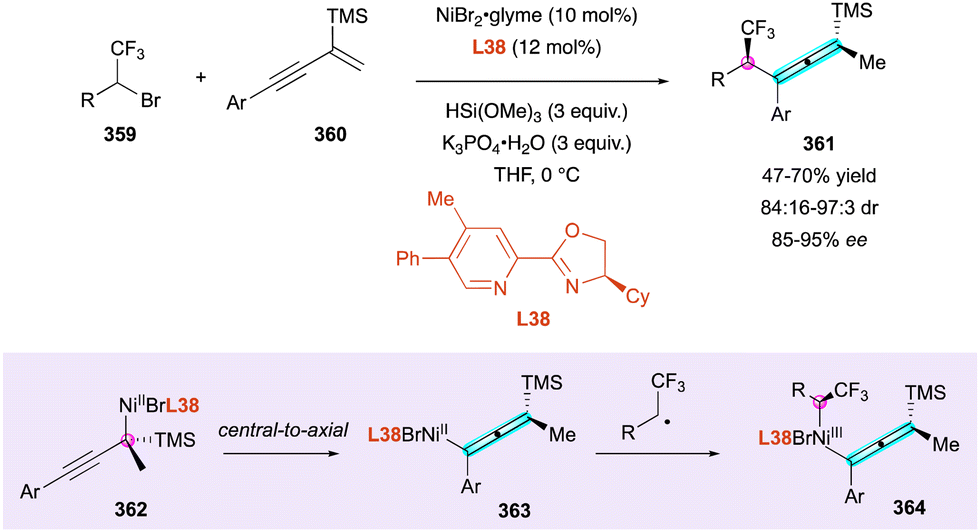 | ||
| Scheme 105 Ni-Catalysed enantioconvergent allenylation of alkyl electrophiles. | ||
In complement to the rearrangement of propargylic derivatives, in 2016, the Feng group engaged racemic allenes 365 in an enantioselective gold-catalysed alleno-aldol reaction with isatins 366, creating a stereocentre, while controlling the configuration of the allene moiety in 367.160 The interaction between allenoates and the gold complex allowed epimerisation via the formation of key reactive gold-alkenyl enolate intermediate 368 as the precursor of the final allene. Thus, the authors presented a number of alleno-oxindoles 367 obtained with mostly excellent yields (up to 99%) although sterically challenging R2 substituents led to lower yields (33% yield for R1 = Et, R2 = i-pentyl, R3 = Me, R4 = H) and an aryl R2 substituent could not be introduced. Variable diastereoselectivity from 3:1 to 99:1 was observed together with decent enantioselectivity not exceeding 90% ee. Lower selectivity was observed in the case of sterically hindered isatins with R4 = 4-Cl or 4-Br.
Finally, Ma, Zhang, and colleagues simultaneously published their work on the DKR of allenic acetates (±)-369 through the synergistic Pd/Cu-catalysed enantioselective allenylation of imines 370 (Scheme 107).161 Here, the chiral environment of both Pd and Cu complexes combining SegPhos [(S)- or (R)-L10] and Phosferrox (L39) ligands, respectively, allowed efficient stereocontrol of the reaction via racemisation of the allene. The reaction could be performed with cyclic and acyclic imines (not shown) with a surprising inversion of the stereocentre configuration. Interestingly, both configurations of allenes 371/372 could be obtained depending on the enantiomer of SegPhos used, always with good diastereoselectivity (3:1 to >20:1 dr) and enantioselectivity (87% to >99% ee), although (R)-L10 seemed to achieve better selectivity. Very recently, the authors extended this method with the same nucleophiles 370 to (3E,5Z)-5-bromo-1,3,5-trienes 373 electrophilic partners, allowing the stereodivergent formation of similar products 374 with an additional C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double-bond between both stereogenic elements.162
C double-bond between both stereogenic elements.162
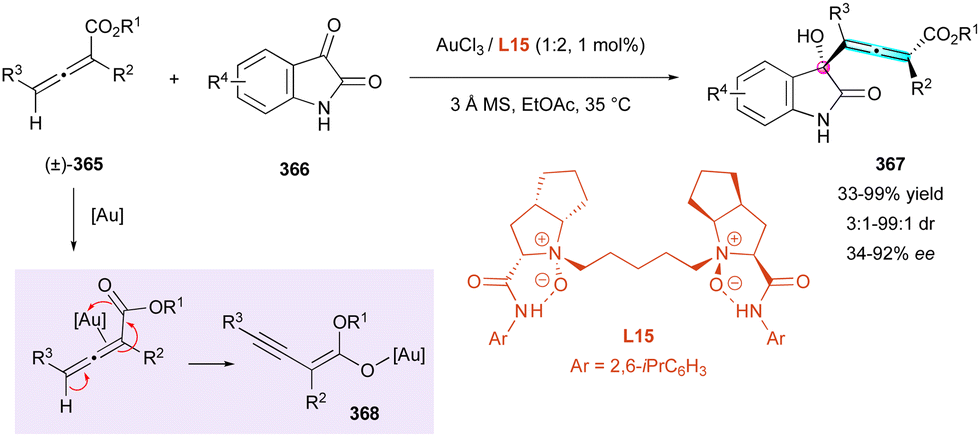 | ||
| Scheme 106 Au-Catalysed enantioselective alleno-aldol reaction of allenoates with isatins. | ||
Organocatalysed approaches
The organocatalysed enantioselective synthesis of chiral allenes featuring a stereocentre has been also the focus of interest in the last two decades.163 One of the first examples of the organocatalytic approach was reported by the Tang group in 2010.164 They proposed a synthetic way to perform bromolactonisation of conjugated (Z)-enyne-carboxylic acids 375 using an electrophilic brominating agent such as N-bromosuccinimide (NBS), promoted by chiral urea catalyst C56 (Scheme 108). This reaction exclusively gave chiral allenic lactones 376 in generally high yield (44–88%) and stereoselectivity (>10:1 dr, up to 99% ee), but was only compatible with (Z)-enyne isomers 375, where the other double bond geometry giving rise to a racemic mixture. A very broad range of substrates could be used, containing electron-withdrawing or electron-donating group, with different linkers (n = 0, 1), leading either to mono-6-membered ring or fused bicyclic seven-membered ring lactones, respectively.
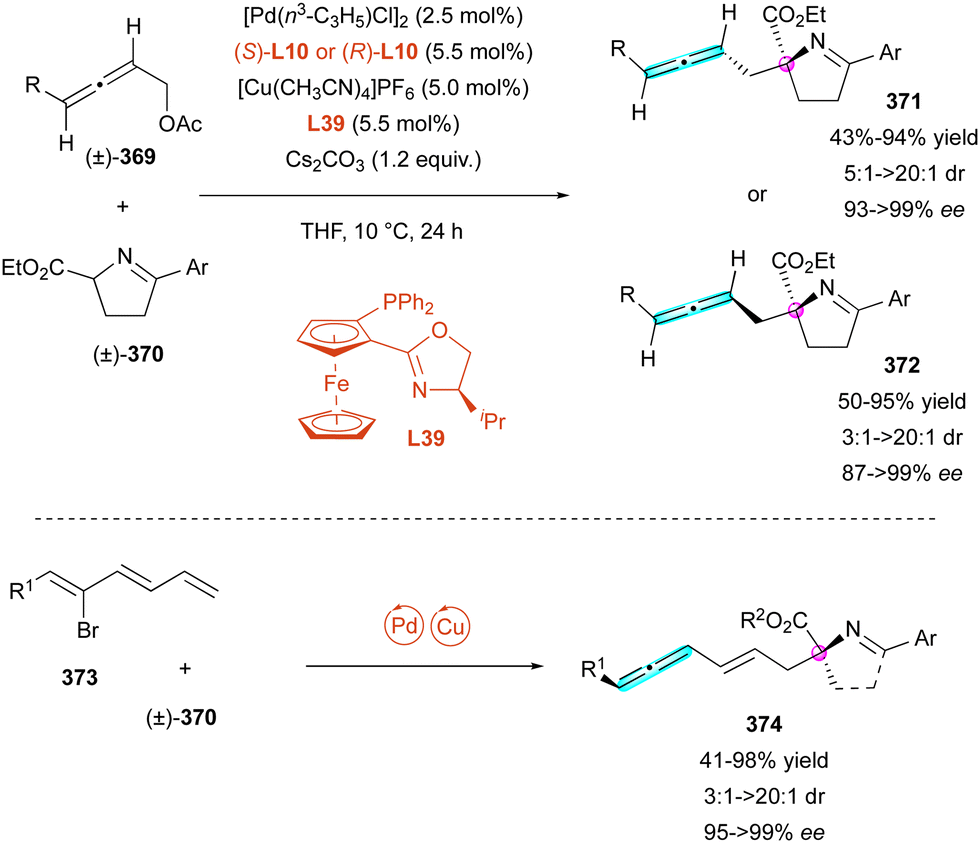 | ||
| Scheme 107 Pd-Catalysed stereoselective construction of chiral allenes. | ||
Following their interest in the organocatalytic synthesis of chiral allenoates by formal SN2′ nucleophilic substitutions of propargylic derivatives,165 the Sun group became interested more recently in the stereocontrolled synthesis of tetrasubstituted allenes 379 endowed with a quaternary stereocentre (Scheme 109).166 Their strategy is based on the ability of racemic propargylic alcohols 377 to generate highly reactive propargylic cation intermediate 380, which is prone to participate in an efficient intermolecular C–C bond formation with 1,3-dicarbonyl pronucleophiles 378 in the presence of CPA catalyst C57 or C58. The presence of a 4-heteroatom (X = O, S) in propargylic alcohol 377 is mandatory to assist the generation and stabilisation of the key propargylic para-quinone methide (p-QM) cation 380 associated with its chiral counter anion, which tolerates a broad variety of electron-rich, electron-neutral, and electron-deficient aryl moieties (Ar1, Ar2), leading to tetrasubstituted allenes 379 with good yield (62–96%) and good enantio- and diastereoselectivity (up to 97% ee, >6:1 dr). In 2019, the same group extended the reaction to generate aza-p-quinone methides as key intermediates (XNH) with comparable efficiency.167
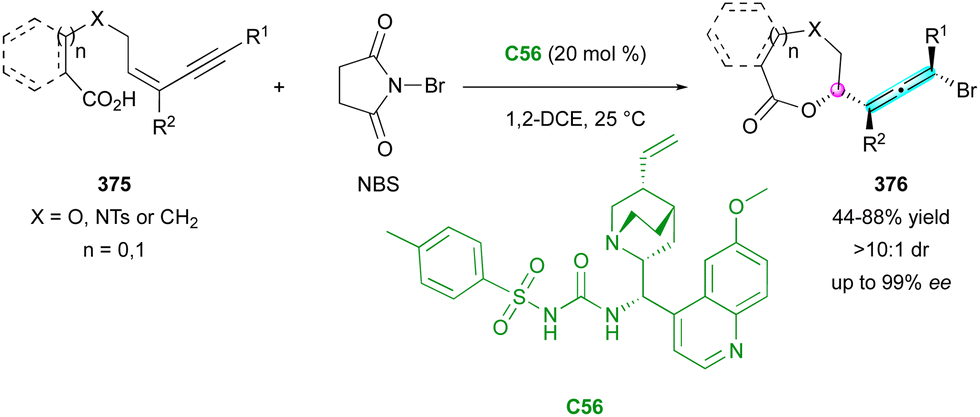 | ||
| Scheme 108 Enantioselective bromolactonisation of conjugated (Z)-enynes. | ||
A similar work was simultaneously achieved by the Li group, reporting the synthesis of vicinal axially chiral tetrasubstituted allenes 384–385 bearing a heteroatom-funtionalised quaternary carbon stereocentre.168 This was accomplished through the organocatalytic stereocontrolled addition of thiazolones 382 and azalactones 383 as nucleophiles to propargylic alcohols 381 (XO) catalysed by various CPA C59 and C53 (Scheme 110). The developed optimisation led to the use of non-standard conditions, such as PhCF3 as the solvent for thiazolone nucleophiles 382, but provided excellent yields (65–97%) for a large extended scope with high enantio- and diastereoselectivity (up to >99% ee, >10:1 dr). In their following article, the authors extended this method to para-amino-substituted propargylic alcohols 381 (XNH) with a comparable efficiency only when an adamentane carbonyl is used as the N-protecting group,169 leading to analogous centrally and axially chiral allenes 386.
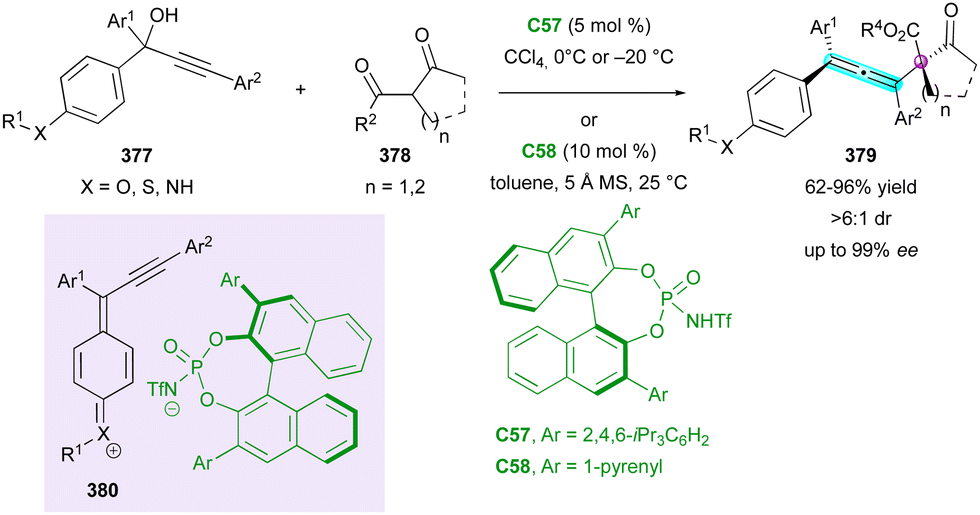 | ||
| Scheme 109 Organocatalytic synthesis of chiral tetrasubstituted allenes from racemic propargylic alcohols and 1,3-dicarbonyls. | ||
In 2020, alternative contributions were proposed independently by the Wang170 and Lin groups,171 who developed the CPA-catalysed enantioselective synthesis of α-tetrasubstituted α-amino allenoates 389 and 391 bearing a quaternary stereogenic centre (Scheme 111). Electrophilic β,γ-alkynyl-α-imino esters 387 in combination with C-nucleophilic heterocycles such as 2,3-disubstituted indoles 390 and 1-substituted 2-naphthols 388, in the presence of molecular sieves as a crucial dehydrating agent, allowed high efficiency to be achieved both in terms of yield and stereoselectivity. It was shown that the chiral backbone and the steric environment of the CPA-catalyst were the most influent parameters to ensure good stereocontrol because of the planar approach of both partners, directed by the hydrogen bond network created by CPA.
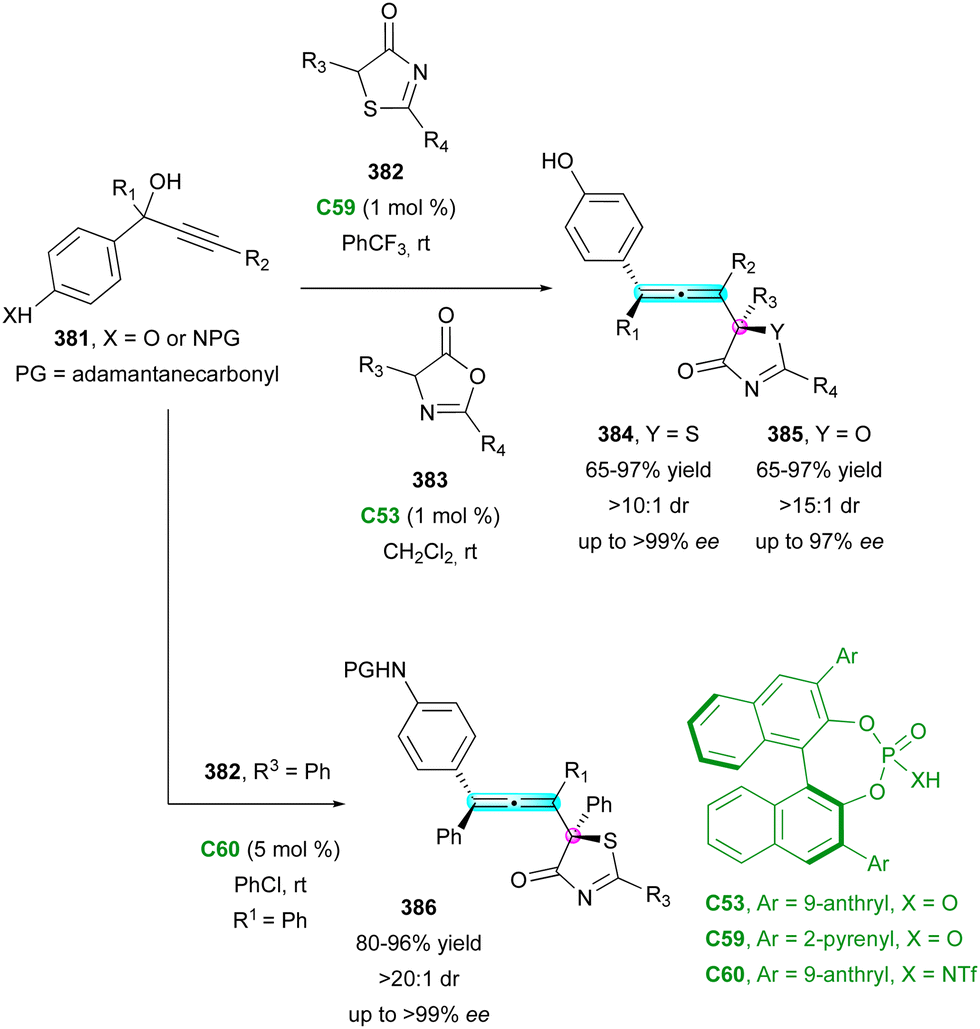 | ||
| Scheme 110 Organocatalytic synthesis of chiral tetrasubstituted allenes featuring a heteroatom-functionalised quaternary carbon stereocentre. | ||
One year later, the Li group extended the reactivity of these electrophilic β,γ-alkynyl-α-imino esters 393 (Scheme 112).172 They studied the stereoselective transformation of isoxazol-5(4H)-one derivatives 392 as pro-nucleophiles for regio- and stereoselective addition, leading to the corresponding α-amino allenoates 394 with both axial and central chirality in good yields (66–91%) under similar conditions with CPA-catalyst (S)-TRIP ent-C37. However, in this case, no dehydrating agent was needed to preserve a high diastereo- and enantioselectivity (up to 94% ee, >20:1 dr) and the reaction was limited to the N-Boc function as the protecting group, and 3-aryl substituted isoxazolones.
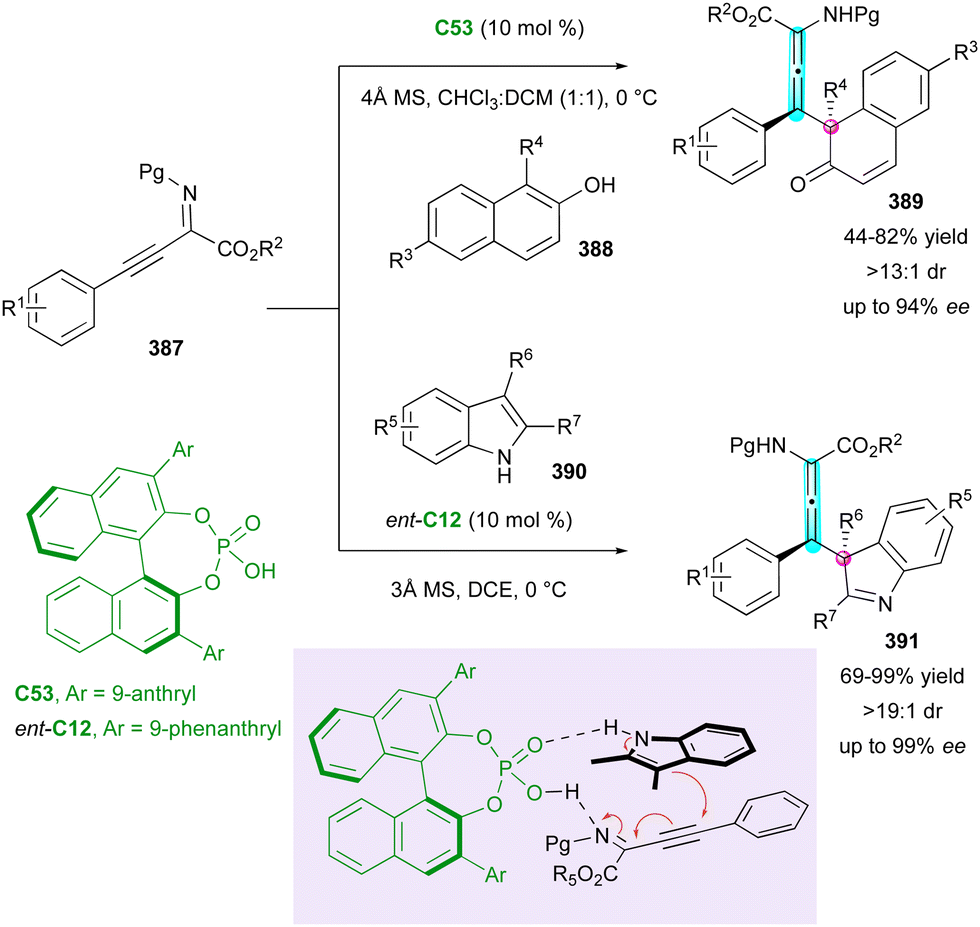 | ||
| Scheme 111 Organocatalytic enantioselective dearomatisation reaction for the synthesis of axial chiral allenoates. | ||
Related oxazolones 395 were used by Peng and Yang as nucleophilic partners with activated 1,3-enynes 396 under CPA catalysis for enantioselective and diastereodivergent access to axially chiral allenes 397a and b bearing two stereogenic carbon atoms (Scheme 113).173 Both catalysts C61 and C62 could catalyse the first Michael addition with oxazolones 395 activated as their Münchnone-type intermediate 398, affording the same major stereoisomer for Michael adduct 399, regardless of the structure of the organocatalyst. Then, the proton transfer generates the allene function, simultaneously creating the axial chirality and with diastereodivergency whose origin presumably comes from the difference in the rigidity between the Binol (more flexible) and the Spinol (more rigid) backbones of the catalyst. This was supported by DFT calculations.
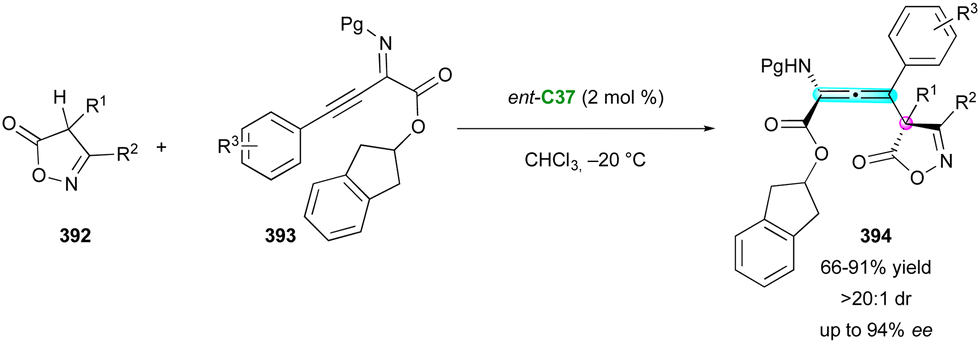 | ||
| Scheme 112 Organocatalytic enantioselective γ-additions of isoxazole-5(4H)-ones to access to axially and centrally chiral tetrasubstituted amino allenoates. | ||
In 2013, Maruoka's group made a step forward, demonstrating the feasibility of synthesising stereodefined tetrasubstituted allenes 402 featuring a stereogenic centre by the diastereo- and enantioselective alleno-Mannich-type reaction of racemic allenyl dicarboxylates (±)-400 with tosylimines 401 under phase transfer catalysis (PTC).174 Chiral cumulenolate key intermediate 403 is generated through the utilisation of chiral ammonium bromide C63, triggering nucleophilic addition to sulfonyl imine 396 (Scheme 114). The only limitation is the absence of reactivity for p-NO2-phenyl imines and the poor to moderate diastereocontrol in some cases, but the constant elevated enantiomeric excess is impressive for the first example reported in the literature.
Miller and co-workers disclosed a similar reaction employing pyridylalanine-based peptide catalyst C64 with racemic allenyl amido esters or thioesters (±)-404 (X = O, S) and N-acylimines 405. In this case, the deep mechanistic study clearly demonstrated the intervention of an addition mechanism through chiral pyridinium dienolate intermediate 407 rather than the formation of a cumulenolate (Scheme 115).175 Within this system, the presence of the anilide substituent in 404 as a hydrogen donor significantly improved the diastereoselectivity by participating in the H-bonding network compared to an ester function. The catalyst also controls the enantioselectivity of the reaction by creating hydrogen bonding interactions with the imine protecting group of 405. Both the diastereo- and enantioselectivity were improved compared to the previous Maruoka's method (up to 42:1 dr and up to 98% ee).
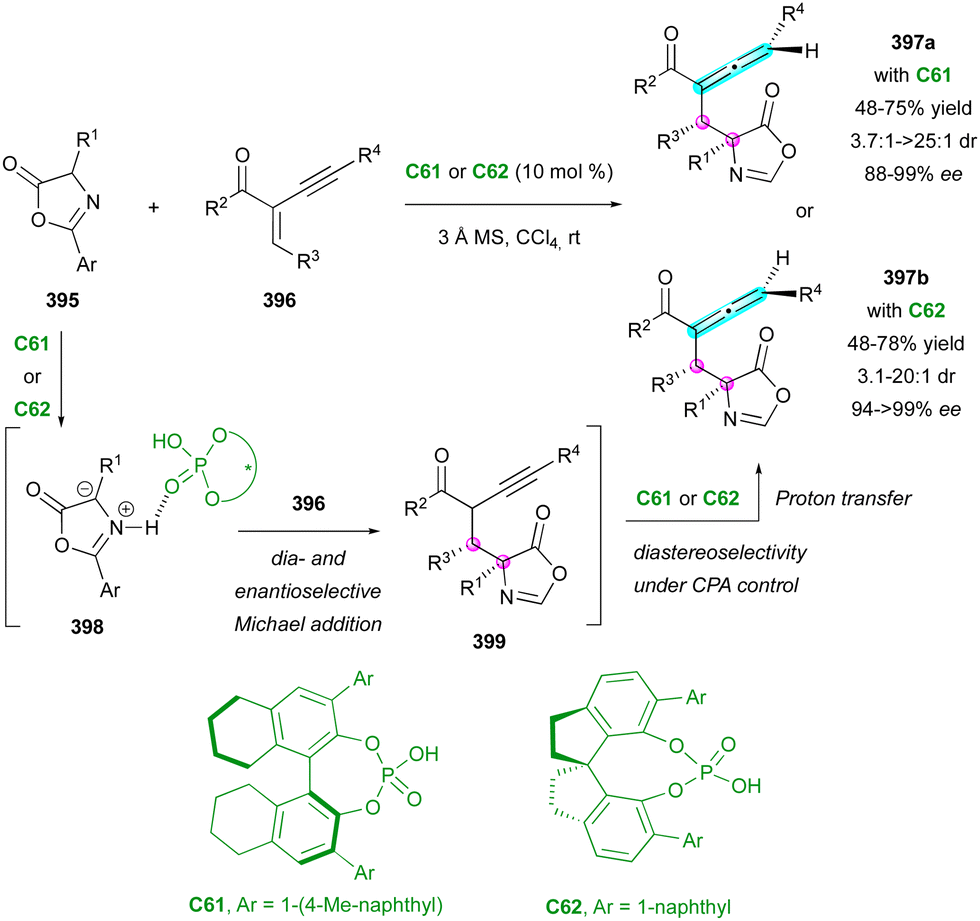 | ||
| Scheme 113 CPA-Catalysed stereodivergent synthesis of trisubstituted allenes. | ||
Guo and collaborators proposed a related approach via the enantioselective allylic alkylation of trisubstituted racemic allenoates (±)-408 performed by a tertiary amine catalyst to simultaneously control the axial and central chirality (Scheme 116).176 The use of β-isocinchonine C65 as the catalyst resulted in aza-Michael addition to Morita–Baylis–Hillman carbonates (MBH) 409, leading to electrophilic intermediates 411 with the simultaneous generation of tert-butoxide, which deprotonated the trisubstituted allenoates to transient allenic anions 412. This triggered a diastereofacial nucleophilic addition by the Re face, leading to tetrasubstituted allenes 410 and release of the catalyst. For most examples, the diastereoselectivity was excellent (dr > 20:1), but the introduction of a nitro substituent in the ortho position of the aryl group of the MBH carbonate resulted in a diminished diastereomeric ratio (dr = 4:1).
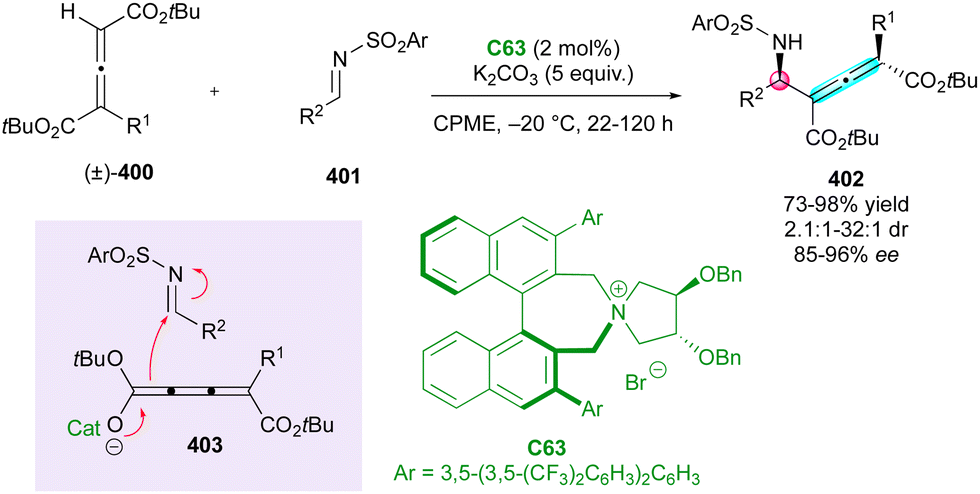 | ||
| Scheme 114 Enantioselective PTC for the synthesis of tetrasubstituted allenes. | ||
The pioneering contribution by the List group for the development of enantioselective synthesis177 was reported in 2016 with the proposal of a new alkynylogous Mukaiyama aldol reaction, allowing enantioselective control of the two different stereogenic elements of α-hydroxy allenoates 414 (Scheme 117).178 Silyl alkynyl ketene acetals 413 were selected as pro-nucleophiles towards simple aromatic aldehydes, using their previously developed silylated disulfonimide (DSI)-catalyst C66 as a chiral Lewis acid. After a challenging optimisation, they could perform the regio- (>20:1), enantio- (up to 96% ee) and diastereoselective (3.7:1 to 27:1 dr) transformation, proceeding with moderate to good yield (33–96%) (Schemes 116 and 117).
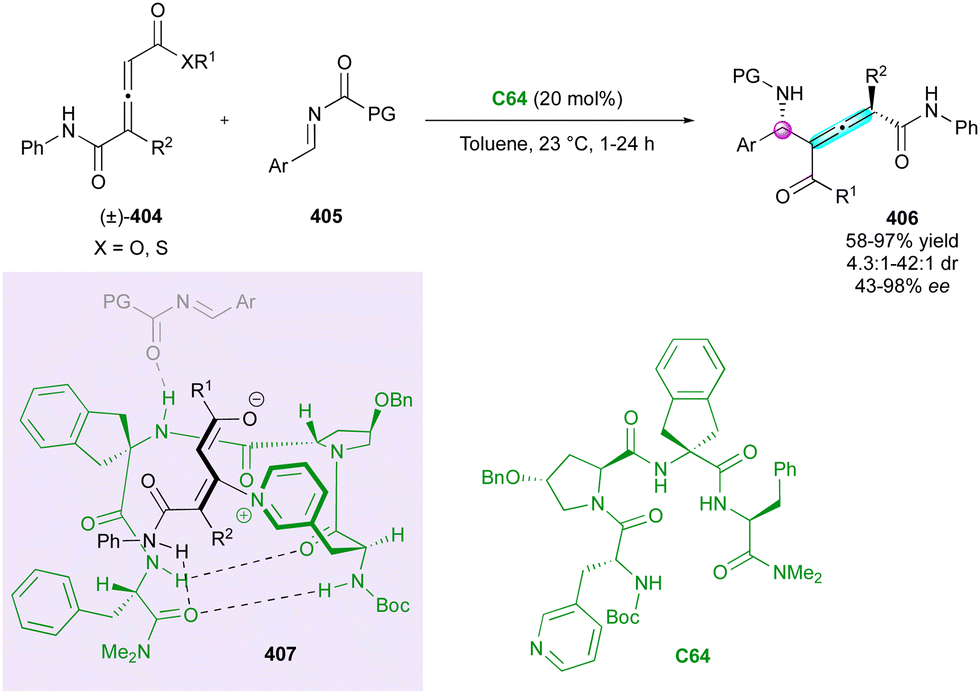 | ||
| Scheme 115 Pyridylalanine-based peptide-catalysed enantioselective addition of allenoates to N-acylimines. | ||
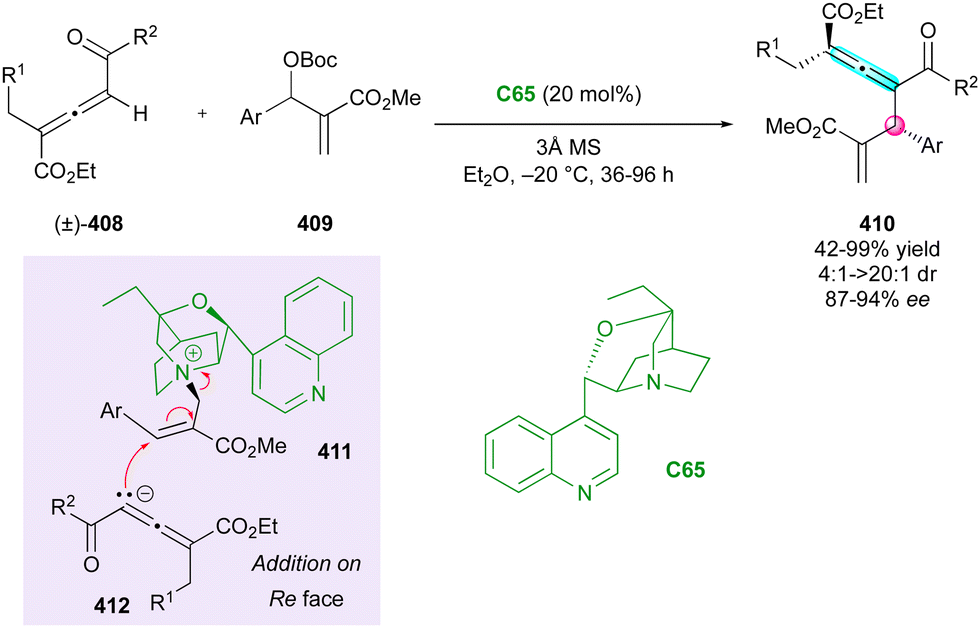 | ||
| Scheme 116 Organocatalytic enantioselective C(sp2)–H allylic alkylation. | ||
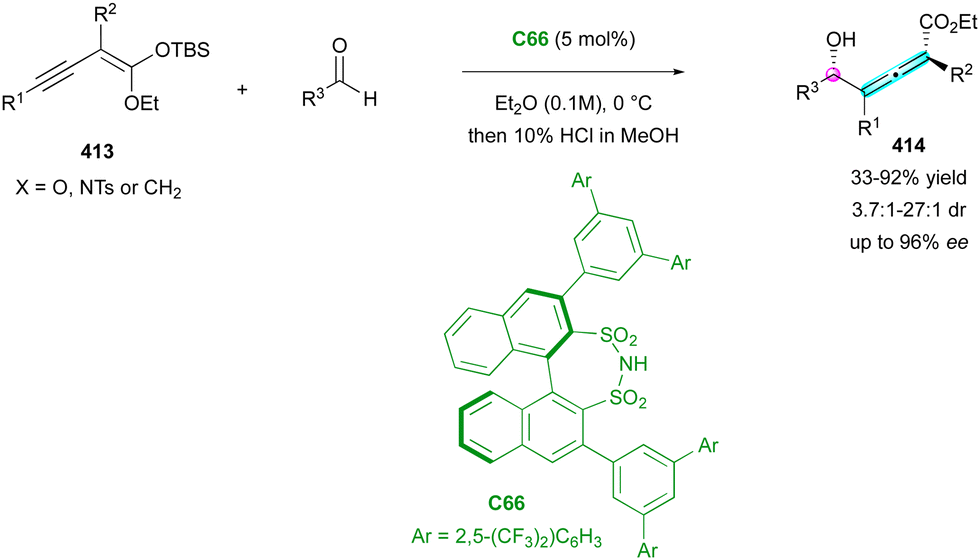 | ||
| Scheme 117 Catalytic enantioselective alkynylogous Mukaiyama aldol reaction. | ||
Conclusions
In conclusion, the enantioselective synthesis of molecules bearing different multiple stereogenic elements represents a significant recent advancement in the field of organic chemistry. The reviewed studies emphasise the importance of developing efficient and selective methodologies for constructing complex chiral molecules in enantioenriched form, given that they already play a crucial role in catalyst/ligand design, drug discovery, natural product synthesis, materials science, and various other applications. However, although this field has undoubtedly made remarkable progress in last years, there are still challenges to be addressed, such as scalability, substrate scope, and broader applicability, in particular for molecules displaying helical and/or planar chirality in combination with other stereogenic elements. Also, the enantioselective synthesis of molecules bearing more than two different stereogenic elements is in its infancy and still represents a formidable challenge. The reviewed literature not only sheds light on the current state of the art but also paves the way for future research directions in the pursuit of more eco-compatible, selective, and broadly applicable methods for producing complex chiral molecular architectures.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Agence Nationale pour la Recherche (ANR-21-CE07-0036), Aix-Marseille Université, the Centre National de la Recherche Scientifique (CNRS), and Centrale Marseille is gratefully acknowledged.Notes and references
- For a review, see: G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed.
- (a) G. Bringmann, M. Rübenacker, P. Vogt, H. Busse, L. Aké Assi, K. Peters and H. G. von Schnering, Phytochem., 1991, 30, 1691 CrossRef CAS; (b) S. Fayez, T. Bruhn, D. Feineis, L. A. Assi, P. P. Kushwah, S. Kumar and G. Bringmann, RSC Adv., 2022, 12, 28916 RSC.
- A. Richieu, P. A. Peixoto, L. Pouységu, D. Deffieux and S. Quideau, Angew. Chem., Int. Ed., 2017, 56, 13833 CrossRef CAS PubMed.
- (a) M. Isaka, M. Tanticharoen, P. Kongsaeree and Y. Thebtaranonth, J. Org. Chem., 2001, 66, 4803 CrossRef CAS PubMed; (b) I. L. Jones, F. K. Moore and C. L. L. Chai, Org. Lett., 2009, 11, 5526 CrossRef CAS PubMed.
- S. Obermaier, W. Thiele, L. Fürtges and M. Müller, Angew. Chem., Int. Ed., 2019, 58, 9125 CrossRef CAS PubMed.
- (a) J. Kohno, Y. Koguchi, M. Nishio, K. Nakao, M. Kuroda, R. Shimizu, T. Ohnuki and S. Komatsubara, J. Org. Chem., 2000, 65, 990 CrossRef CAS PubMed; (b) A. Coste, A. Bayle, J. Marrot and G. Evano, Org. Lett., 2014, 16, 1306 CrossRef CAS PubMed.
- L. D. Fader, E. Malenfant, M. Parisien, R. Carson, F. Bilodeau, S. Landry, M. Pesant, C. Brochu, S. Morin, C. Chabot, T. Halmos, Y. Bousquet, M. D. Bailey, S. H. Kawai, R. Coulombe, S. LaPlante, A. Jakalian, P. K. Bhardwaj, D. Wernic, P. Schroeder, M. Amad, P. Edwards, M. Garneau, J. Duan, M. Cordingley, R. Bethell, S. W. Mason, M. Bös, P. Bonneau, M.-A. Poupart, A.-M. Faucher, B. Simoneau, C. Fenwick, C. Yoakim and Y. Tsantrizos, ACS Med. Chem. Lett., 2014, 5, 422 CrossRef CAS PubMed.
- Y. Ikeura, T. Doi, A. Fujishima and H. Natsugari, Chem. Commun., 1998, 2141 RSC.
- B. A. Lanman, A. T. Parsons and S. G. Zech, Acc. Chem. Res., 2022, 55, 2892 CrossRef CAS PubMed.
- (a) M. Omote, Y. Nishimura, K. Sato, A. Ando and I. Kumadaki, Tetrahedron Lett., 2005, 46, 319 CrossRef CAS; (b) L.-S. Zheng, K.-Z. Jiang, Y. Deng, X.-F. Bai, G. Gao, F.-L. Gu and L.-W. Xu, Eur. J. Org. Chem., 2013, 748 CrossRef CAS; (c) R. Oost, J. Rong, A. J. Minnaard and S. R. Harutyunyan, Catal. Sci. Technol., 2014, 4, 1997 RSC; (d) S. Kitagaki, K. Sugisaka and C. Mukai, Org. Biomol. Chem., 2015, 13, 4833 RSC; (e) X.-F. Bai, T. Song, Z. Xu, C.-G. Xia, W.-S. Huang and L.-W. Xu, Angew. Chem., Int. Ed., 2015, 54, 5255 CrossRef CAS PubMed; (f) Y. Zhang, S.-Z. Nie, J.-J. Ye, J.-P. Wang, M.-M. Zhou, C.-Q. Zhao and Q. Li, J. Org. Chem., 2019, 84, 8423 CrossRef CAS PubMed; (g) L. Ling, Z. Song, H. Shan, C. Wang, S. Li, Y. Wang, J. Hu, Q. Chen, H. Zhang and Y. Yang, Chem. Commun., 2023, 59, 2739 RSC.
- Y. Hao, Z.-H. Li, Z.-G. Ma, R.-X. Liu, R.-T. Ge, Q.-Z. Li, T.-M. Ding and S.-Y. Zhang, Chem. Sci., 2023, 14, 9496 RSC.
- (a) M. Hasegawa, Y. Nojima, Y. Nagata, K. Usui, K.-I. Sugiura and Y. Mazaki, Eur. J. Org. Chem., 2023, e202300656 CrossRef CAS; (b) S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. van Leeuwen, E. Otten and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 17278 CrossRef CAS PubMed.
- (a) T. M. T. Tuyet, T. Harada, K. Hashimoto, M. Hatsuda and A. Oku, J. Org. Chem., 2000, 65, 1335 CrossRef CAS PubMed; (b) R. S. Ward and D. D. Hughes, Tetrahedron, 2001, 57, 4015 CrossRef CAS; (c) M. Penhoat, V. Levacher and G. Dupas, J. Org. Chem., 2003, 68, 9517 CrossRef CAS PubMed; (d) I. G. Stará, Z. Alexandrová, F. Teplý, P. Sehnal, I. Starý, D. Šaman, M. Buděšínský and J. Cvačka, Org. Lett., 2005, 7, 2547 CrossRef PubMed; (e) W. Zhang, H. Xu, H. Xu and W. Tang, J. Am. Chem. Soc., 2009, 131, 3832 CrossRef CAS PubMed; (f) S. Postikova, M. Sabbah, D. Wightman, I. T. Nguyen, M. Sanselme, T. Besson, J.-F. Brière, S. Oudeyer and V. Levacher, J. Org. Chem., 2013, 78, 8191 CrossRef CAS PubMed; (g) R. Raghunathan, E. Kumarasamy, A. Iyer, A. Ugrinova and J. Sivaguru, Chem. Commun., 2013, 49, 8713 RSC; (h) P. Aillard, P. Retailleau, A. Voituriez and A. Marinetti, Chem. Commun., 2014, 50, 2199 RSC; (i) Z. Zuo, J. Liu, J. Nan, L. Fan, W. Sun, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 15385 CrossRef CAS PubMed; (j) M. Šámal, S. Chercheja, J. Rybáček, J. V. Chocholoušová, J. Vacek, L. Bednárová, D. Šaman, I. G. Stará and I. Starý, J. Am. Chem. Soc., 2015, 137, 8469 CrossRef PubMed; (k) D. Waghray, G. Bagdziunas, J. Jacobs, L. Van Meervelt, J. V. Grazulevicius and W. Dehaen, Chem. – Eur. J., 2015, 21, 18791 CrossRef CAS PubMed; (l) X. Xue and Z. Gu, Org. Lett., 2019, 21, 3942 CrossRef CAS PubMed; (m) J. Nejedlý, M. Šámal, J. Rybáček, I. G. Sánchez, V. Houska, T. Warzecha, J. Vacek, L. Sieger, M. Buděšínský, L. Bednárová, P. Fiedler, I. Císařová, I. Starý and I. G. Stará, J. Org. Chem., 2020, 85, 248 CrossRef PubMed.
- (a) B. H. Lipshutz, F. Kayser and Z.-P. Liu, Angew. Chem., Int. Ed. Engl., 1994, 33, 1842 CrossRef; (b) G. Brigmann, P. A. Keller and K. Rölfing, Synlett, 1994, 423 CrossRef; (c) O. Kitagawa, H. Izawa, K. Sato, A. Dobashi, T. Taguchi and M. Shiro, J. Org. Chem., 1998, 63, 2634 CrossRef CAS PubMed; (d) A. Fürstner and M. Méndez, Angew. Chem., Int. Ed., 2003, 42, 5355 CrossRef PubMed; (e) P.-E. Broutin and F. Colobert, Org. Lett., 2003, 5, 3281 CrossRef CAS PubMed; (f) A. I. Meyers, T. D. Nelson, H. Moorlag, D. J. Rawson and A. Meier, Tetrahedron, 2004, 60, 4459 CrossRef CAS; (g) C. G. Newton, E. Braconi, J. Kuziola, M. D. Wodrich and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 11040 CrossRef CAS PubMed; (h) D. Meidlinger, L. Marx, C. Bordeianu, S. Choppin and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 9160 CrossRef CAS PubMed; (i) A. Urbano, A. M. del Hoyo, A. Martínez-Carrión and M. C. Carreño, Org. Lett., 2019, 21, 4623 CrossRef CAS PubMed; (j) S. Jin, J.-Y. Wang, Y. Tang, H. Rouh, S. Zhang, T. Xu, Y. Wang, Q. Yuan, D. Chen, D. Unruh and G. Li, Front. Chem., 2022, 10, 860398 CrossRef CAS PubMed; (k) A. Mondal, N. O. Thiel, R. Dorel and B. L. Feringa, Nat. Catal., 2022, 5, 10 CrossRef CAS.
- (a) B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978 CrossRef CAS PubMed; (b) G. Gao, F.-L. Gu, J.-X. Jiang, K. Jiang, C.-Q. Sheng, G.-Q. Lai and L.-W. Xu, Chem. – Eur. J., 2011, 17, 2698 CrossRef CAS PubMed.
- (a) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871 CrossRef CAS PubMed; (b) L.-J. Li, J.-J. Chen, C.-F. Feng, H.-Y. Li, X. Wang, H. Xu and H.-X. Dai, Org. Lett., 2020, 22, 9169 CrossRef CAS PubMed.
- (a) A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, Tetrahedron, 1991, 47, 1677 CrossRef CAS; (b) G. Bringmann, M. Breuning, S. Tasler, H. Endress, C. L. J. Ewers, L. Göbel, K. Peters and E.-M. Peters, Chem. – Eur. J., 1999, 5, 3029 CrossRef CAS; (c) G. Bringmann, M. Breuning, S. Tasler, H. Endress, C. L. J. Ewers, L. Göbel, K. Peters and E.-M. Peters, Chem. – Eur. J., 1999, 5, 3029 CrossRef CAS; (d) K. Kamikawa, M. Furusyo, T. Uno, Y. Sato, A. Konoo, G. Bringmann and M. Uemura, Org. Lett., 2001, 3, 3667 CrossRef CAS PubMed; (e) V. Chan, J. G. Kim, C. Jimeno, P. J. Carroll and P. J. Walsh, Org. Lett., 2004, 6, 2051 CrossRef CAS PubMed; (f) A. Yubuta, A. Tsurusaki and K. Kamikawa, Chem. Commun., 2021, 57, 6600 RSC; (g) D. Enders, S. Nolla and J. W. Bats, Synlett, 2005, 2679 CrossRef CAS; (h) R. Senda, Y. Watanabe, S. Miwa, A. Sato and O. Kitagawa, J. Org. Chem., 2023, 88, 9579 CrossRef CAS PubMed; (i) B. Yang, X. Tan, Y. Ge, Y. Lia and C. He, Org. Chem. Front., 2023, 10, 4862 RSC.
- H. Yang, W.-L. Xu, X.-Y. Zeng, J. Chen, L. Yu and L. Zhou, Org. Lett., 2021, 23, 9315 CrossRef CAS PubMed; J. Dong, A. Ostertag and C. Sparr, Angew. Chem., Int. Ed., 2022, 61, e202212627 CrossRef PubMed; Q. Ni, Z. Zhu, Y. Fan, X. Chen and X. Song, Org. Lett., 2021, 23, 9548 CrossRef PubMed; H. Homma, S. Harada, T. Ito, A. Kanda and T. Nemoto, Org. Lett., 2020, 22, 8132 CrossRef PubMed; Y. Kwon, A. J. Chinn, B. Kim and S. J. Miller, Angew. Chem., Int. Ed., 2018, 57, 6251 CrossRef PubMed.
- X.-F. Bai, Y.-M. Cui, J. Cao and L.-W. Xu, Acc. Chem. Res., 2022, 55, 2545 CrossRef CAS PubMed.
- In the following excellent review, Zhang and Shi summarised only examples of atropisomers bearing one other stereogenic element (central, planar, helical chirality). H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2023, 63, e202311053 CrossRef PubMed.
- During the evaluation process of this manuscript, several methodologies have been released in the literature: (a) P. Wu, W.-T. Zhang, J.-X. Yang, X.-Y. Yu, S.-F. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202410581 Search PubMed; (b) Y. Wu, Z. Wang, Y. Shan, Y. Ma, T. Li, C. Yuan, H. Guo and B. Mao, Chem. Sci., 2024, 15, 9703 RSC; (c) X. Hu, Y. Zhao, T. He, C. Niu, F. Liu, W. Jia, Y. Mu, X. Li and Z.-Q. Rong, Chem. Sci., 2024, 15, 13541 RSC; (d) J. Hou, W. Hao, Y. Chen, Z. Wang and W. Yao, J. Org. Chem., 2024, 89, 9068 CrossRef CAS PubMed; (e) T.-T. Wang, J. Cao and X. Li, Org. Lett., 2024, 26, 6179 CrossRef CAS PubMed; (f) T. von Münchow, Y.-R. Liu, R. Parmar, S. E. Peters, S. Trienes and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202405423 CrossRef PubMed; (g) X. Wang, S.-J. Wang, X. Xin, H. An, Z. Tu, H. Yang, M. W. Wong and S. Lu, Chem. Sci., 2024, 15, 13240 RSC; (h) X. Wang, S.-J. Wang, X. Xin, H. An, Z. Tu, H. Yang, M. W. Wong and S. Lu, Chem. Sci., 2024, 15, 13240 RSC; (i) Q. Liu, K. Teng, Y. Zhang, Y. Lv, Y. Robin Chi and Z. Jin, Angew. Chem., Int. Ed., 2024, 63, e202406386 Search PubMed.
- D. Leow, S. Lin, S. K. Chittimalla, X. Fu and C.-H. Tan, Angew. Chem., Int. Ed., 2008, 47, 5641 CrossRef CAS PubMed.
- P. Dorizon, C. Martin, J.-C. Daran, J.-C. Fiaud and H. B. Kagan, Tetrahedron: Asymmetry, 2001, 12, 2625 CrossRef CAS.
- K. Akagawa, N. Nishi, I. Yoshikawa and K. Kudo, Eur. J. Org. Chem., 2015, 5055 CrossRef CAS.
- Y. Zhao, H. Wang, B. Wua and Y.-G. Zhou, Org. Chem. Front., 2019, 6, 3956 RSC.
- Y. Zhao, X.-Q. Wang, Y.-J. Yu and Y.-G. Zhou, J. Org. Chem., 2021, 86, 1262 CrossRef CAS PubMed.
- A.-N. Alba, P. Gómez-Sal, R. Rios and A. Moyano, Tetrahedron: Asymmetry, 2009, 20, 1314 CrossRef CAS.
- M. Akiyama, K. Akagawa, H. Seinob and K. Kudo, Chem. Commun., 2014, 50, 7893 RSC.
- G. Yang, Y. He, T. Wang, Z. Li and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316739 CrossRef CAS PubMed.
- J. Bie, M. Lang and J. Wang, Org. Lett., 2018, 20, 5866 CrossRef CAS PubMed.
- (a) S. Barik, R. C. Das, K. Balanna and A. T. Biju, Org. Lett., 2022, 24, 5456 CrossRef CAS PubMed; (b) For an isolated example of a closely related C–N axially chiral fused-dihydropyridinone, see: Y. Chu, M. Wu, F. Hu, P. Zhou, Z. Cao and X.-P. Hui, Org. Lett., 2022, 24, 3884 CrossRef CAS PubMed.
- R. Mi, Z. Ding, S. Yu, R. H. Crabtree and X. Li, J. Am. Chem. Soc., 2023, 145, 8150 CrossRef CAS PubMed.
- L. Cui, Y. Wang, Z. Fan, Z. Li and Z. Zhou, Adv. Synth. Catal., 2019, 361, 3575 CrossRef CAS.
- C. Ma, F.-T. Sheng, H.-Q. Wang, S. Deng, Y.-C. Zhang, Y. Jiao, W. Tan and F. Shi, J. Am. Chem. Soc., 2020, 142, 15686 CrossRef CAS PubMed.
- Q. Shi, F. Fang and D.-J. Cheng, Adv. Synth. Catal., 2024, 366, 1 CrossRef.
- J. Clayden and L. W. Lai, Angew. Chem., Int. Ed., 1999, 38, 2556 CrossRef CAS PubMed.
- V. Chan, J. G. Kim, C. Jimeno, P. J. Carroll and P. J. Walsh, Org. Lett., 2004, 6, 2051 CrossRef CAS PubMed.
- Z. Gao, J. Qian, H. Yang, X.-C. Hang, J. Zhang and G. Jiang, Chem. Commun., 2020, 56, 7265 RSC.
- S.-L. Li, Q. Wu, C. Yang, X. Li and J.-P. Cheng, Org. Lett., 2019, 21, 5495 CrossRef CAS PubMed.
- X. Li, J.-P. Cheng and G. Zheng, Org. Lett., 2021, 23, 3997 CrossRef PubMed.
- F. Jiang, K.-W. Chen, P. Wu, Y.-C. Zhang, Y. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 15104 CrossRef CAS PubMed.
- S. Yang, J.-B. Huang, D.-H. Wang, N.-Y. Wang, Y.-Y. Chen, X.-Y. Ke, H. Chen, S.-F. Ni, Y.-C. Zhang and F. Shi, Precis. Chem., 2024, 2, 208 CrossRef CAS.
- X. Yuan, X. Wu, F. Peng, H. Yang, C. Zhu and H. Fu, Chem. Commun., 2020, 56, 12648 RSC.
- C. Ma, F. Jiang, F.-T. Sheng, Y. Jiao, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014 CrossRef CAS PubMed.
- F.-T. Sheng, Z.-M. Li, Y.-Z. Zhang, L.-X. Sun, Y.-C. Zhang, W. Tan and F. Shi, Chin. J. Chem., 2020, 38, 583 CrossRef CAS.
- J.-Y. Wang, C.-H. Gao, C. Ma, X.-Y. Wu, S.-F. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2023, e202316454 Search PubMed.
- (a) V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140 CrossRef CAS PubMed; (b) S. Shaaban, H. Li, F. Otte, C. Strohmann, A. P. Antonchick and H. Waldmann, Org. Lett., 2020, 22, 9199 CrossRef CAS PubMed; (c) For a recent review, see: W. Tan, X.-Y. Wu and F. Shi, ChemCatChem, 2024, 16, e202401022 CrossRef.
- P. Wu, L. Yu, C.-H. Gao, Q. Cheng, S. Deng, Y. Jiao, W. Tan and F. Shi, Fundam. Res., 2023, 10, 237 CrossRef PubMed.
- Y. Xia, M. Liu, C. Qian, P. Li, M. Dong and W. Li, Org. Chem. Front., 2023, 10, 30 RSC.
- H.-Q. Wang, S.-F. Wu, J.-R. Yang, Y.-C. Zhang and F. Shi, J. Org. Chem., 2023, 88(12), 7684 CrossRef CAS PubMed.
- A. Kim, A. Kim, S. Park, S. Kim, H. Jo, K. M. Ok, S. K. Lee, J. Song and Y. Kwon, Angew. Chem., Int. Ed., 2021, 60, 12279 CrossRef CAS PubMed.
- A. Kim, C. Lee, J. Song, S. K. Lee and Y. Kwon, Nat. Commun., 2023, 14, 5502 CrossRef CAS PubMed.
- C.-X. Hu, L. Chen, D. Hu, X. Song, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2020, 22, 8973 CrossRef CAS PubMed.
- (a) G. T. Wong, D. Manfra, F. M. Poulet, Q. Zhang, H. Josien, T. Bara, L. Engstrom, M. Pinzon-Ortiz, J. S. Fine, H. J. Lee, L. Zhang, G. A. Higgins and E. M. Parker, J. Biol. Chem., 2004, 279, 12876 CrossRef CAS PubMed; (b) I. H. Hall, A. R. K. Murthy and S. D. Wyrick, J. Pharm. Sci., 1986, 75, 622 CrossRef CAS PubMed; (c) H. J. Lee and K. Maruoka, Chem. Rec., 2022, 22, e202200004 CrossRef CAS PubMed.
- Y. Liu, Y.-L. S. Tse, F. Y. Kwong and Y.-Y. Yeung, ACS Catal., 2017, 7, 4435 CrossRef CAS.
- J. Liu, X. Yang, Z. Zuo, J. Nan, Y. Wang and X. Luan, Org. Lett., 2018, 20, 244 CrossRef CAS PubMed.
- X. Wang, Y. Luo, J. Zhao and S. Luo, Org. Biomol. Chem., 2023, 21, 6697 RSC.
- J.-Y. Du, T. Balan, T. D. W. Claridge and M. D. Smith, J. Am. Chem. Soc., 2022, 144, 14790 CrossRef CAS PubMed.
- N. Kotwal, Tamanna, A. Changotra and P. Chauhan, Org. Lett., 2023, 25, 7523 CrossRef CAS PubMed.
- Y. Wang, Y. Huang, X. Bao, X. Wei, S. Wei, J. Qu and B. Wang, Chem. Sci., 2024, 15, 8880 RSC.
- J.-H. Wu, S. Fang, X. Zheng, J. He, Y. Ma, Z. Su and T. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309515 CrossRef CAS PubMed.
- J. A. Carmona, V. Hornillos, P. Ramírez-López, A. Ros, J. Iglesias-Sigüenza, E. Gómez-Bengoa, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2018, 140, 11067 CrossRef CAS PubMed.
- V. Hornillos, J. A. Carmona, A. Ros, J. Iglesias-Sigüenza, J. López-Serrano, R. Fernández and J. M. Lassaletta, Angew. Chem., Int. Ed., 2018, 57, 3777 CrossRef CAS PubMed.
- J. A. Carmona, P. Rodriguez-Salamanca, R. Fernandez, J. M. Lassaletta and V. Hornillos, Angew. Chem., Int. Ed., 2023, 62, e202306981 CrossRef CAS PubMed.
- C. Rodríguez-Franco, A. Ros, P. Merino, R. Fernández, J. M. Lassaletta and V. Hornillos, ACS Catal., 2023, 13, 12134 CrossRef PubMed.
- A. Romero-Arenas, V. Hornillos, J. Iglesias-Sigüenza, R. Fernández, J. López-Serrano, A. Ros and J. M. Lassaletta, J. Am. Chem. Soc., 2020, 142, 2628 CrossRef CAS PubMed.
- M. Xiong, Z. Yan, S.-C. Chen, J. Tang, F. Yang and D. Xing, ACS Catal., 2024, 14, 7243 CrossRef CAS.
- Y. Li, Y.-C. Liou, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202212595 CrossRef CAS PubMed.
- Z.-J. Zhang, N. Jacob, S. Bhatia, P. Boos, X. Chen, J. C. DeMuth, A. M. Messinis, B. Bongsuiru Jei, J. C. A. Oliveira, A. Radović, M. L. Neidig, J. Wencel-Delord and L. Ackermann, Nat. Commun., 2024, 15, 3503 CrossRef CAS PubMed.
- Z.-J. Zhang, M. M. Simon, S. Yu, S.-W. Li, X. Chen, S. Cattani, X. Hong and L. Ackermann, J. Am. Chem. Soc., 2024, 146, 9172 CrossRef CAS PubMed.
- T. Bhattacharya, S. Ghosh, S. Dutta, S. Guin, A. Ghosh, H. Ge, R. B. Sunoj and D. Maiti, Angew. Chem., Int. Ed., 2024, 63, e202310112 CrossRef CAS PubMed.
- S. H. Jang, H. W. Kim, W. Jeong, D. Moon and Y. H. Rhee, Org. Lett., 2018, 20, 1248 CrossRef CAS PubMed.
- Z. Gao, C.-X. Yan, J. Qian, H. Yang, P. Zhou, J. Zhang and G. Jiang, ACS Catal., 2021, 11, 6931 CrossRef CAS.
- Y. Zhang, Y.-Q. Liu, L. Hu, X. Zhang and Q. Yin, Org. Lett., 2020, 22, 6479 CrossRef CAS PubMed.
- H. Hu, Y. Peng, T. Yu, S. Cheng, S. Luo and Q. Zhu, Org. Lett., 2021, 23, 3636 CrossRef CAS PubMed.
- D. Ji, J. Jing, Y. Wang, Z. Qi, F. Wang, X. Zhang, Y. Wang and X. Li, Chem, 2022, 8, 3346 CAS.
- Q. Ren, M. Lang, H. Liu, X. Li, D. Wu, J. Wu, M. Yang, J. Wei, Z. Ren and L. Wang, Org. Lett., 2023, 25, 7745 CrossRef CAS PubMed.
- E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110 CrossRef PubMed.
- C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Tetrahedron, 2022, 106–107, 132629 CrossRef.
- J. Bennett, P. L. Pickering and N. S. Simpkins, Chem. Commun., 2004, 1392 RSC.
- D. P. Curran, H. Qi, S. J. Geib and N. C. DeMello, J. Am. Chem. Soc., 1994, 116, 3131 CrossRef CAS.
- (a) W.-L. Duan, Y. Imazaki, R. Shintani and T. Hayashi, Tetrahedron, 2007, 63, 8529 CrossRef CAS; (b) for the utilisation of a N,N-bidentate ligand, see: J. Lai, J. Yang, C. Yang, R. Csuk, B. Song and S. Li, Org. Chem. Front., 2022, 9, 183 RSC.
- For a personal account, see: N. Di Iorio, S. Crotti and G. Bencivenni, Chem. Rec., 2019, 19, 2095 CrossRef CAS PubMed.
- N. Di Iorio, P. Righi, A. Mazzanti, M. Mancinelli, A. Ciogli and G. Bencivenni, J. Am. Chem. Soc., 2014, 136, 10250 CrossRef CAS PubMed.
- (a) N. Di Iorio, F. Champavert, A. Erice, P. Righi, A. Mazzanti and G. Bencivenni, Tetrahedron, 2016, 72, 5191 CrossRef CAS; (b) N. Di Iorio, L. Soprani, S. Crotti, E. Marotta, A. Mazzanti, P. Righi and G. Bencivenni, Synthesis, 2017, 1519 CAS.
- F. Eudier, P. Righi, A. Mazzanti, A. Ciogli and G. Bencivenni, Org. Lett., 2015, 17, 1728 CrossRef CAS PubMed.
- Desymmetrization of N-pyrazolyl maleimides via organo enantioselective Michael addition of pyrazolones was also reported, albeit with no diastereoselectivity: J. Geng, X. Wei, B. He, Y. Hao, J. Qu and B. Wang, Molecules, 2023, 28, 4279 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, L. Lin, Q. Yao, X. Liua and X. Feng, Chem. Commun., 2015, 51, 10554 RSC.
- J. Wang, H. Chen, L. Kong, F. Wang, Y. Lan and X. Li, ACS Catal., 2021, 11, 9151 CrossRef CAS.
- For recent reviews, see: (a) Y. He, J. Chen, X. Jiang and S. Zhu, Chin. J. Chem., 2022, 40, 651 CrossRef CAS; (b) X. Yin, S. Li, K. Guo, L. Song and X. Wang, Eur. J. Org. Chem., 2023, e202300783 CrossRef CAS; (c) X.-Y. Sun, B.-Y. Yao, B. Xuan, L.-J. Xiao and Q.-L. Zhou, Chem. Catal., 2022, 2, 3140 CrossRef CAS.
- X.-W. Gu, Y.-L. Sun, J.-L. Xie, X.-B. Wang, Z. Xu, G.-W. Yin, L. Li, K.-F. Yang and L.-W. Xu, Nat. Commun., 2020, 11, 2904 CrossRef CAS PubMed.
- F. Sun, T. Wang, G.-J. Cheng and X. Fang, ACS Catal., 2021, 11, 7578 CrossRef CAS.
- S. Barik, S. Shee, S. Das, R. G. Gonnade, G. Jindal, S. Mukherjee and A. T. Biju, Angew. Chem., Int. Ed., 2021, 60, 12264 CrossRef CAS PubMed.
- H.-C. Liu, H.-Y. Tao, H. Cong and C.-J. Wang, J. Org. Chem., 2016, 81, 3752 CrossRef CAS PubMed.
- Y. Wu, B. Xu, B. Liu, Z.-M. Zhang and Y. Liu, Org. Biomol. Chem., 2019, 17, 1395 RSC.
- S. Zhang, Z.-H. Luo, W.-T. Wang, L. Qian and J.-Y. Liao, Org. Lett., 2022, 24, 4645 CrossRef CAS PubMed.
- H. Wang, Y. Wei, Y. Li, S. Long, L.-J. Sun, S. Li and Y.-W. Lin, Org. Lett., 2022, 24(36), 6494 CrossRef CAS PubMed.
- W.-T. Wang, S. Zhang, W. Lin, Z.-H. Luo, D. Hu, F. Huang, R. Bai, Y. Lan, L. Qian and J.-Y. Liao, Org. Chem. Front., 2024, 11, 3308 RSC.
- (a) E. J. Corey, S. Sarshar and D.-H. Lee, J. Am. Chem. Soc., 1994, 116, 12089 CrossRef CAS; (b) K. Uemae, S. Masuda and Y. Yamamoto, J. Chem. Soc., Perkin Trans. 1, 2001, 1002 RSC.
- M. Barday, J. Rodrigues, P. Bouillac, J. Rodriguez, M. Amatore and T. Constantieux, Adv. Synth. Catal., 2023, 365, 148 CrossRef CAS.
- J. W. Zhang, J.-H. Xu, D.-J. Cheng, C. Shi, X.-Y. Liu and B. Tan, Nat. Commun., 2016, 7, 10677 CrossRef PubMed.
- L.-L. Zhang, J.-W. Zhang, S.-H. Xiang, Z. Guo and B. Tan, Org. Lett., 2018, 19, 6022 CrossRef PubMed.
- C. Parida, S. K. Dave, K. Das and S. C. Pan, Adv. Synth. Catal., 2023, 365, 1185 CrossRef CAS.
- D. Liang, J.-R. Chen, L.-P. Tan, Z.-W. He and W.-J. Xiao, J. Am. Chem. Soc., 2022, 144, 6040 CrossRef CAS PubMed.
- F. Huang, L.-F. Tao, J. Liu, L. Qian and J.-Y. Liao, Chem. Commun., 2023, 59, 4487 RSC.
- H. Jiang, X.-K. He, X. Jiang, W. Zhao, L.-Q. Lu, Y. Cheng and W.-J. Xiao, J. Am. Chem. Soc., 2023, 145, 6944 CrossRef CAS PubMed.
- T. Liang, Y. Wu, J. Sun, M. Li, H. Zhao, J. Zhang, G. Zheng and Q. Zhang, Chin. J. Chem., 2023, 41, 3253 CrossRef CAS.
- Y. Wang, R. Mi, S. Yu and X. Li, ACS Catal., 2024, 14, 4638 CrossRef CAS.
- Y. Liu, L. Yuan, L. Dai, Q. Zhu, G. Zhong and X. Zeng, J. Org. Chem., 2024, 11, 7630 CrossRef PubMed.
- Y.-D. Shao, D.-D. Han, W.-Y. Maa and D.-J. Cheng, Org. Chem. Front., 2020, 7, 2255 RSC.
- S. Lu, J.-Y. Ong, H. Yang, S. B. Poh, X. Liew, C. S. D. Seow, M. W. Wong and Y. Zhao, J. Am. Chem. Soc., 2019, 141, 17062 CrossRef CAS PubMed.
- Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS PubMed.
- L. Pang, Q. Sun, Z. Huang, G. Li, J. Liu, J. Guo, C. Yao, J. Yu and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202211710 CrossRef CAS PubMed.
- L. Pang, C. Wang, C. Ma, J. Liu, M. Shi, C. Yao, J. Yu and Q. Li, Org. Lett., 2023, 25, 7705 CrossRef CAS PubMed.
- J. Li, Y. Yan, X. Chen, Z. Huang and Y. Huang, Chem. Sci., 2024, 15, 6943 RSC.
- X. Bi, J. Feng, X. Xue and Z. Gu, Org. Lett., 2021, 23, 3201 CrossRef CAS PubMed.
- Y. Guo, M.-M. Liu, X. Zhu, L. Zhu and C. He, Angew. Chem., Int. Ed., 2021, 60, 13887 CrossRef CAS PubMed.
- C. Li, S.-Z. Cai, J. Ye and X. Fang, Org. Lett., 2024, 18, 3867 CrossRef PubMed.
- H. Koide, T. Hata and M. Uemura, J. Org. Chem., 2002, 67, 1929 CrossRef CAS PubMed.
- K. Kamikawa, S. Arae, W.-Y. Wu, C. Nakamura, T. Takahashi and M. Ogasawara, Chem. – Eur. J., 2015, 21, 4954 CrossRef CAS PubMed.
- P.-C. Zhang, Y.-L. Li, J. He, H.-H. Wu, Z. Li and J. Zhang, Nat. Commun., 2021, 12, 4609 CrossRef CAS PubMed.
- Y. An, X.-Y. Zhang, Y.-N. Ding, Y. Li, X.-Y. Liu and X.-Y. Liu, Org. Lett., 2022, 24, 7294 CrossRef CAS PubMed.
- J. Ye, L. Li, Y. You, C. Jiao, Z. Cui, Y. Zhang, S. Jia, H. Cong, S. Liu, H.-G. Cheng and Q. Zhou, JACS Au, 2023, 3, 384 CrossRef CAS PubMed.
- (a) V. Dočekal, F. Koucký, I. Císařová and J. Veselý, Nat. Commun., 2024, 15, 3090 CrossRef PubMed; (b) M.-L. Delcourt, S. Felder, E. Benedetti and L. Micouin, ACS Catal., 2018, 8, 6612–6616 CrossRef CAS.
- D. Ly, J. Basca and H. M. L. Davies, ACS Catal., 2024, 14, 6423–6431 CrossRef CAS PubMed.
- L. Wang, S. Li, M. Blümel, A. R. Philipps, A. Wang, R. Puttreddy, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 11110 CrossRef CAS PubMed.
- C. Min, Y. Lin and D. Seidel, Angew. Chem., Int. Ed., 2017, 56, 15353 CrossRef CAS PubMed.
- Y. Li, X.-Y. Duan, C. Yang, Y. Wei, J. Li, X. Ren and J. Qi, J. Org. Chem., 2023, 88, 11299 CrossRef CAS PubMed.
- S. S. Ranganathappa, B. S. Dehury, G. K. Singh, S. Shee and A. T. Biju, ACS Catal., 2024, 14, 6965 CrossRef CAS.
- S.-J. Wang, X. Wang, X. Xin, S. Zhang, H. Yang, M. W. Wong and S. Lu, Nat. Commun., 2024, 15, 518 CrossRef CAS PubMed.
- (a) J. Rodriguez and D. Bonne, Chem. Commun., 2019, 55, 11168 RSC; (b) W. Qin, Y. Liu and H. Yan, Acc. Chem. Res., 2022, 55, 2780 CrossRef CAS PubMed.
- for a pioneer contribution, see: M. Furusawa, K. Arita, T. Imahori, K. Igawa, K. Tomooka and R. Irie, Tetrahedron Lett., 2013, 54, 7107 CrossRef CAS.
- A. Huang, L. Zhang, D. Li, Y. Liu, H. Yan and W. Li, Org. Lett., 2019, 21, 95 CrossRef CAS PubMed.
- W. Zhang, S. Wei, W. Wang, J. Qu and B. Wang, Chem. Commun., 2021, 57, 6550 RSC.
- S. Huang, H. Wen, Y. Tian, P. Wang, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 21486 CrossRef CAS PubMed.
- S. Xu, A. Huang, Y. Yang, Y. Wang, M. Zhang, Z. Sun, M. Zhao, Y. Wei, G. Li and L. Hong, Org. Lett., 2022, 24, 2978 CrossRef CAS PubMed.
- B.-B. Gou, Y. Tang, Y.-H. Lin, L. Yu, Q.-S. Jian, H.-R. Sun, J. Chen and L. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202208174 CrossRef CAS PubMed.
- (a) S.-G. Li, Y.-T. Wang, Q. Zhang, K.-B. Wang, J.-J. Xue, D.-H. Li, Y.-K. Jing, B. Lin and H.-M. Hua, Org. Lett., 2020, 22, 7522 CrossRef CAS PubMed; (b) Q.-F. He, Z.-L. Wu, X.-J. Huang, T.-Q. Xia, G. Tang, W. Tang, L. Shi, W.-C. Ye and Y. Wang, J. Org. Chem., 2021, 86, 5870 CrossRef CAS PubMed.
- (a) N. Di Iorio, G. Filippini, A. Mazzanti, P. Righi and G. Bencivenni, Org. Lett., 2017, 19, 6692 CrossRef CAS PubMed; (b) For a pioneer observation of a dynamic equilibration in C(sp2)–C(sp3) stereogenic dihydropyridines, see: O. Quinonero, M. Jean, N. Vanthuyne, C. Roussel, D. Bonne, C. Constantieux, C. Bressy, X. Bugaut and J. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 1401 CrossRef CAS PubMed.
- G. Bertuzzi, V. Corti, J. A. Izzo, S. Ričko, N. I. Jessen and K. A. Jørgensen, J. Am. Chem. Soc., 2022, 144, 1056 CrossRef CAS PubMed.
- P. Liu, X. Bao, J.-V. Naubron, S. Chentouf, S. Humbel, N. Vanthuyne, M. Jean, L. Giordano, J. Rodriguez and D. Bonne, J. Am. Chem. Soc., 2020, 142, 16199 CrossRef CAS PubMed.
- V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140 CrossRef CAS PubMed.
- A. Gaucherand, E. Yen-Pon, D. García-López, J.-V. Naubron, S. Chentouf, M. Giorgi, S. Humbel, M. Jean, J. Rodriguez and D. Bonne, Chem. Sci., 2024, 15, 7300 RSC.
- C. Li, Y.-B. Shao, X. Gao, Z. Ren, C. Guo, M. Li and X. Li, Nat. Commun., 2023, 14, 3380 CrossRef CAS PubMed.
- W. Liu, T. Qin, W. Xie, J. Zhou, Z. Ye and X. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303430 CrossRef CAS PubMed.
- S. Jia, S. Li, Y. Liu, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 18496 CrossRef CAS PubMed.
- X. Liu, B. Zhu, X. Zhang, H. Zhu, J. Zhang, A. Chu, F. Wang and R. Wang, Nat. Commun., 2024, 15, 732 CrossRef CAS PubMed.
- T. Shibata, M. Otomo, Y.-K. Tahara and K. Endo, Org. Biomol. Chem., 2008, 6, 4296 RSC.
- For reviews on CA-RE of alkynes with alkenes: (a) S.-I. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994 RSC; (b) T. Michinobu and F. Diederich, Angew. Chem., Int. Ed., 2018, 57, 3552 CrossRef CAS PubMed.
- W. Xiao, F. Li, X. Liu, W. Cao and X. Feng, Org. Lett., 2023, 25, 8005 CrossRef CAS PubMed.
- F. Wang, J. Jing, Y. Zhao, X. Zhu, X.-P. Zhang, L. Zhao, P. Hu, W.-Q. Deng and X. Li, Angew. Chem., Int. Ed., 2021, 60, 16628 CrossRef CAS PubMed.
- For a recent review, see: S. Du, A.-X. Zhou, R. Yang, X.-R. Song and Q. Xiao, Org. Chem. Front., 2021, 8, 6760 RSC.
- Z. Li, V. Boyarskikh, J. H. Hansen, J. Autschbach, D. G. Musaev and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 15497 CrossRef CAS PubMed.
- Y. Liu, X. Liu, H. Hu, J. Guo, Y. Xia, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 4054 CrossRef CAS PubMed.
- G. Xu, Z. Wang, Y. Shao and J. Sun, Org. Lett., 2021, 23, 5175 CrossRef CAS PubMed.
- S.-Q. Yang, Y.-F. Wang, W.-C. Zhao, G.-Q. Lin and Z.-T. He, J. Am. Chem. Soc., 2021, 143, 7285 CrossRef CAS PubMed.
- F.-H. Zhang, X. Guo, X. Zeng and Z. Wang, Nat. Commun., 2022, 13, 5036 CrossRef CAS PubMed.
- A. Hossain, R. L. Anderson, C. S. Zhang, P.-J. Chen and G. C. Fu, J. Am. Chem. Soc., 2024, 146, 7173 CrossRef CAS PubMed.
- Z. Wang, H. Yin and G. C. Fu, Nature, 2018, 563, 379 CrossRef CAS PubMed.
- G. Wang, X. Liu, Y. Chen, J. Yang, J. Li, L. Lin and X. Feng, ACS Catal., 2016, 6, 2482 CrossRef CAS.
- J. Zhang, X. Huo, J. Xiao, L. Zhao, S. Ma and W. Zhang, J. Am. Chem. Soc., 2021, 143, 12622 CrossRef CAS PubMed.
- J. Zhang, Y. Luo, E. Zheng, X. Huo, S. Ma and W. Zhang, J. Am. Chem. Soc., 2024, 146, 9241 CrossRef CAS PubMed.
- For a recent review, see: T. T. Nguyen, Org. Biomol. Chem., 2023, 21, 252 RSC.
- W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed.
- H. Qian, X. Yu, J. Zhang and J. Sun, J. Am. Chem. Soc., 2013, 135, 18020 CrossRef CAS PubMed.
- D. Qian, L. Wu, Z. Lin and J. Sun, Nat. Commun., 2017, 8, 567 CrossRef PubMed.
- M. Chen, D. Qian and J. Sun, Org. Lett., 2019, 21, 8127 CrossRef CAS PubMed.
- P. Zhang, Q. Huang, Y. Cheng, R. Li, P. Li and W. Li, Org. Lett., 2019, 21, 503 CrossRef CAS PubMed.
- L. Zhang, Y. Han, A. Huang, P. Zhang, P. Li and W. Li, Org. Lett., 2019, 21, 7415 CrossRef CAS PubMed.
- J. Yang, Z. Wang, Z. He, G. Li, L. Hong, W. Sun and R. Wang, Angew. Chem., Int. Ed., 2020, 59, 642 CrossRef CAS PubMed.
- A. G. Woldegiorgis, Z. Han and X. Lin, Org. Lett., 2021, 23, 6606 CrossRef CAS PubMed.
- F. Li, S. Liang, Y. Luan, X. Chen, H. Zhao, A. Huang, P. Li and W. Li, Org. Chem. Front., 2021, 8, 1243 RSC.
- J. Wang, S. Zheng, S. Rajkumar, J. Xie, N. Yu, Q. Peng and X. Yang, Nat. Commun., 2020, 11, 5527 CrossRef CAS PubMed.
- T. Hashimoto, K. Sakata, F. Tamakuni, M. J. Dutton and K. Maruoka, Nat. Chem., 2013, 5, 240 CrossRef CAS PubMed.
- C. T. Mbofana and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 3285 CrossRef CAS PubMed.
- Y. Hu, W. Shi, B. Zheng, J. Liao, W. Wang, Y. Wu and H. Guo, Angew. Chem., Int. Ed., 2020, 59, 19820 CrossRef CAS PubMed.
- J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC.
- A. Tap, A. Blond, V. N. Wakchaure and B. List, Angew. Chem., Int. Ed., 2016, 55, 8962 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |