
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl ether-free polymer electrolytes for electrochemical and energy devices†
Eun Joo
Park
 a,
Patric
Jannasch
b,
Kenji
Miyatake
cd,
Chulsung
Bae
e,
Kevin
Noonan
f,
Cy
Fujimoto
g,
Steven
Holdcroft
h,
John R.
Varcoe
i,
Dirk
Henkensmeier
jkl,
Michael D.
Guiver
*m and
Yu Seung
Kim
*a
a,
Patric
Jannasch
b,
Kenji
Miyatake
cd,
Chulsung
Bae
e,
Kevin
Noonan
f,
Cy
Fujimoto
g,
Steven
Holdcroft
h,
John R.
Varcoe
i,
Dirk
Henkensmeier
jkl,
Michael D.
Guiver
*m and
Yu Seung
Kim
*a
aLos Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: yskim@lanl.gov
bLund University, SE-221 00 Lund, Sweden
cUniversity of Yamanashi, Kofu 400-8510, Japan
dWaseda University, Tokyo 169-8555, Japan
eRensselaer Polytechnic Institute, Troy, NY 12180, USA
fCarnegie Mellon University, Pittsburgh, PA 15213, USA
gSandia National Laboratories, Albuquerque, NM 87123, USA
hSimon Fraser University, Burnaby, BC V5A 1S6, Canada
iUniversity of Surrey, Guildford GU2 7XH, UK
jKorea Institute of Science and Technology (KIST), Seoul 02792, South Korea
kKIST School, University of Science and Technology (UST), Seoul 02792, South Korea
lKU-KIST School, Korea University, Seoul 02841, South Korea
mState Key Laboratory of Engines, Tianjin University, Tianjin 300072, China. E-mail: michael.guiver@outlook.com
First published on 26th April 2024
Abstract
Anion exchange polymers (AEPs) play a crucial role in green hydrogen production through anion exchange membrane water electrolysis. The chemical stability of AEPs is paramount for stable system operation in electrolysers and other electrochemical devices. Given the instability of aryl ether-containing AEPs under high pH conditions, recent research has focused on quaternized aryl ether-free variants. The primary goal of this review is to provide a greater depth of knowledge on the synthesis of aryl ether-free AEPs targeted for electrochemical devices. Synthetic pathways that yield polyaromatic AEPs include acid-catalysed polyhydroxyalkylation, metal-promoted coupling reactions, ionene synthesis via nucleophilic substitution, alkylation of polybenzimidazole, and Diels–Alder polymerization. Polyolefinic AEPs are prepared through addition polymerization, ring-opening metathesis, radiation grafting reactions, and anionic polymerization. Discussions cover structure–property–performance relationships of AEPs in fuel cells, redox flow batteries, and water and CO2 electrolysers, along with the current status of scale-up synthesis and commercialization.
 Eun Joo Park | Eun Joo Park is a Research Scientist at Los Alamos National Laboratory. She obtained her BS in chemistry from University of Toronto in 2010, MA in Science Education from Teachers College, Columbia University in 2012. She received her PhD in chemistry in 2016 from Rensselaer Polytechnic Institute, where she focused on synthesis of functional polymers via post-polymerization modification under supervision of Prof. Chulsung Bae. Her research interests are design, synthesis and characterization of ionic polymers used in electrochemical devices. |
 Patric Jannasch | Patric Jannasch received his PhD degree in Chemical Engineering from Lund University, Sweden, in 1996. He was appointed full professor of Polymer Technology at the same university in 2010, and was a guest professor at the University of Tartu (Estonia) during 2017–2022. His current research focuses on molecular design, synthesis, and function of polymer electrolytes and ion-exchange membranes for fuel cells, water electrolysers, and flow batteries, as well as on new bio-based monomers, polymers and plastics from renewable resources. He presently serves as editor of Solid State Ionics. |
 Kenji Miyatake | Prof. Kenji Miyatake received his PhD degree in polymer chemistry from Waseda University, Japan in 1996. He was a postdoc (JSPS overseas research fellow) at McGill University, Canada, from 1999 to 2001. In 2001, he was appointed an associate professor in Clean Energy Research Center at the University of Yamanashi, where he currently serves as a professor. He also holds a professor position at Waseda University. He is a Fellow of the Royal Society of Chemistry. He serves as an associate editor of Fuel Cells (Wiley). His research interest involves new functional polymers for energy device applications. |
 Steven Holdcroft | Steven Holdcroft is a Professor and Canada Research Chair, and former President of the Canadian Society for Chemistry. He researches materials for electrochemical energy conversion & storage. He is the author of 300 peer-reviewed articles and inventor of 20+ patents. He has received the Macromolecular Science and Engineering Division Award of the Chemical Institute of Canada (CIC), and the CSC RioTinto Alcan Award for Electrochemistry. In 2021, he was elected to the fellowship of the Royal Society of Canada and in 2024 awarded the CIC Montreal Medal for his contributions to the Canadian chemical community. |
 John R. Varcoe | John Varcoe is a Professor of Materials Chemistry at the University of Surrey and Director of Research for the School of Chemistry and Chemical Engineering. He is also a Fellow of the Royal Society of Chemistry. He was awarded his BSc and PhD chemistry degrees at the University of Exeter. For the past 25 years, his research has focused on ion-exchange membranes, with specialisms in radiation grafted materials, anion-exchange membranes, and Raman microscopy. His group's membranes and ionomers have been tested in many types of electrochemical devices including fuel cells, hydrogen and carbon dioxide electrolysers, and electrodialysis systems. |
 Dirk Henkensmeier | Dirk Henkensmeier received a PhD in chemistry at Hamburg University and worked at LG Chem, Sartorius and Paul Scherrer Institute, before joining Korea Institute of Science and Technology (KIST) in 2009, where he is a principal researcher. Concurrently he is a full professor at University of Science and Technology and teaches at Korea University. His research focus is on polymers and membranes for energy conversion and storage, especially for fuel cells, water electrolysis and flow batteries. He is a co-organizer of the EMEA workshop series and a section board member of MDPI's Membranes. |
1. Introduction
Anion exchange polymers (AEPs) are anion-conductive polymers with cationic functionality appended to the chain such as quaternary ammonium or metal cation complexes. The concentration of cations in an AEP is expressed by its ion exchange capacity (IEC), measured as milliequivalents per gram of the polymer (mequiv. g−1 of dry AEP). Generally, an increase in IEC results in higher anion conductivity and water uptake of an AEP, but a decrease in its mechanical strength in film form.For over 60 years, AEPs have been widely used for electrodialysis (ED) and reverse electrodialysis (RED), in the form of anion exchange resin, an anion exchange membrane (AEM), and as one component of a bipolar membrane (BPM) (Fig. 1a and b).1,2 In ED and RED applications, AEPs selectively separate monovalent and multivalent ions from wastewater. Since 2006, AEPs have been also used for membrane capacitive deionization (MCDI) for water purification or valuable metal ion recovery.3,4 In MCDI, a cation exchange membrane (CEM) is attached to the cathode, and an AEM is attached to the anode (Fig. 1c). The role of ion exchange membranes is to prevent co-ions from being flushed out of electrodes during the ion removal process. Thus, MCDI improves device efficiency compared to conventional CDI.
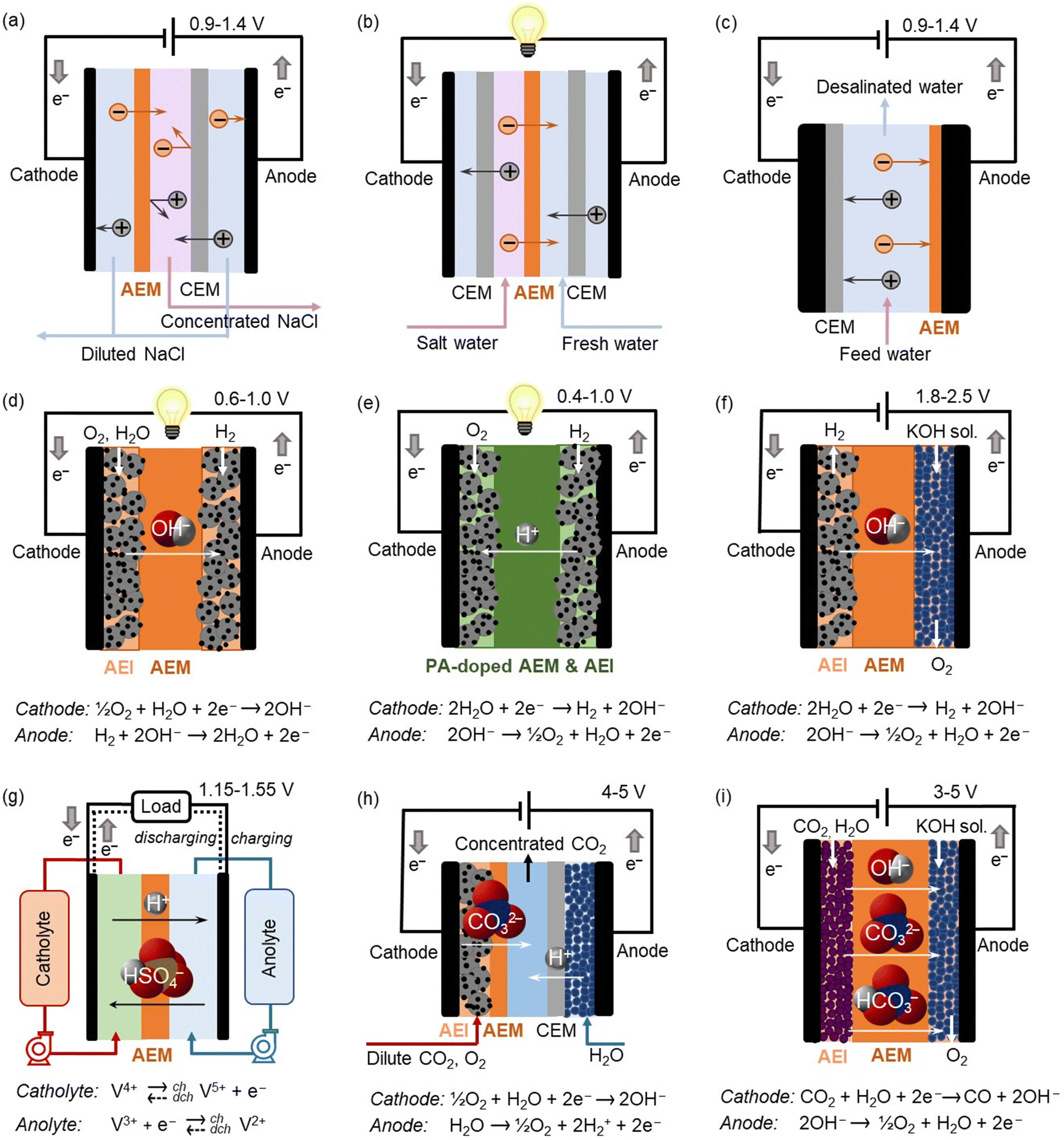 | ||
| Fig. 1 Schematic diagrams, cell reactions and operating cell voltages of electrochemical devices using AEPs: (a) ED, (b) RED, (c) MCDI, (d) AEMFC, (e) ion-pair HT-PEMFC, (f) AEMWE, (g) vanadium RFB, (h) electrochemical CO2 capture, and (i) CO2 electrolyser. | ||
In 2004, the U.S. Department of Energy (DOE) increased federal funding for hydrogen and fuel cell research, development, and demonstration through the hydrogen fuel initiative. This increased funding allowed the hydrogen program to expand and address a broad range of barriers facing the widespread use of hydrogen and fuel cell technologies in transportation. One of the focused R&D areas was developing AEM fuel cells (AEMFCs) that have the potential to operate with platinum group metal-free (PGM-free) catalysts.5 In AEMFC applications, AEPs are used both as a hydroxide-conducting medium in the form of a membrane and as an anion exchange ionomer (AEI) at the electrodes (Fig. 1d).
As interest in fuel cell vehicles shifted from light-duty to heavy-duty vehicles, such as class 8 long-haul trucks and other heavy-duty transportation applications, including trains, ships, and aviation in the late 2010s, heat rejection in fuel cell vehicles based on low-temperature proton-exchange membrane fuel cells (LT-PEMFCs) became a challenging issue. For LT-PEMFCs, operating fuel cells at a higher cell voltage at rated power with a fuel cell/battery hybrid strategy can mitigate the negative impact of the heat dissipation issue. Alternatively, polybenzimidazole (PBI) or AEPs can be used as a proton exchange membrane (PEM) after phosphoric acid (PA) doping.6,7 Since PA-doped polymers can conduct protons without water, the operating temperature of fuel cells can exceed 100 °C (Fig. 1e), effectively resolving the heat dissipation issue. Compared to PBI-based fuel cells, AEP-based ion-pair fuel cells have shown higher stability under dynamic operating conditions due to stronger ion-pair interactions between cationic functional groups of AEPs and PA.8,9
In 2021, the U.S. DOE announced the energy earthshots initiative, aiming to accelerate breakthroughs in more abundant, affordable, and reliable clean energy solutions within this decade. The first energy earthshot, “Hydrogen Shot,” seeks to reduce the cost of clean hydrogen by 80% to $1 per 1 kilogram in one decade (“1 1 1”). To achieve this goal, AEM water electrolysers (AEMWEs) have been drawing substantial interest. AEMWEs use PGM-free catalysts and less expensive metal interconnects while enabling differential pressure operation and operating at higher current densities than conventional liquid alkaline electrolysers.10 AEPs play critical roles in the performance and durability of AEMWEs. Unlike in AEMFCs, a low-concentration of liquid electrolyte is often added in AEMWEs to reduce cell resistance, prevent ionomer adsorption, and increase local pH, thereby improving hydrogen and oxygen evolution reactions (HER and OER, respectively, Fig. 1f).11
The second energy earthshot, “Long Duration Storage Shot,” aims to reduce the cost of energy storage systems by 90% within a decade. Electrical energy storage is recognized as an underpinning technology for maintaining power network stability and reliability from energy generation and load balance maintenance. Redox flow batteries (RFBs) store energy in two soluble redox couples contained in external liquid electrolyte tanks. During the charging phase, one electrolyte is oxidized at the anode, while another is reduced at the cathode, converting electrical energy into chemical energy of the electrolyte. AEMs can be used as separators in RFBs, offering high coulombic efficiency (CE) by providing superior barrier properties against cations. During the charging and discharging of the RFBs, both anions and protons are transported through the AEM via diffusion and the Grotthuss mechanism (Fig. 1g).12
The third energy earthshot, “Carbon Negative Shot,” aims to develop energy-efficient carbon capture from dilute sources, transferring it to durable storage or converting it into products. Using AEPs for both direct air capture (DAC) of CO2 and its electrochemical reduction is highly promising. In the CO2 capture device, CO2 dissolved in water as (bi)carbonate is regenerated using electrochemical cells with several configurations (Fig. 1h).13,14 CO2 electrolysers that convert CO2 into carbon monoxide and other value-added chemical products can utilize a membrane electrode assembly (MEA) containing AEPs for highly efficient electrochemical CO2 reduction.15 AEPs are typically used for AEMs, AEIs in the catalyst layers, and as a component of BPMs. In AEP-based CO2 electrolysers, the cationic functional groups of AEPs facilitate anion transport from the cathode to the anode, enabling the CO2 reduction reaction (CO2RR) to occur in a basic environment. This environment suppresses the competing HER by decreasing the concentration of protons at the catalyst surface (Fig. 1i).
AEPs are critical materials not only for the emerging technologies mentioned above but also for other applications, including reverse osmosis (RO), sensors, photo-electrolysis, and metal ion batteries. Fig. 2 shows the number of publications about AEPs over the last 25 years. The total number of publications on AEPs has increased fivefold from 2000 to 2023, with over 1500 AEP papers published each year since 2021. The largest portion of AEP-related papers in 2023 is in the field of fuel cells (584 papers), followed by water electrolysis (343 papers). Compared to the number of publications five years ago, the relative proportions in ED, RED, MCDI, fuel cell, and RFB have remained similar. However, the proportions in water electrolysis and CO2 capture/electrolysis have increased approximately fourfold, reflecting the current trend of research. Another notable research trend from Fig. 2 is that modern AEP research has become more device-specific rather than focusing on general material development. In 2000, only 23% of papers were device-specific, but by 2023, 88% of the papers on AEPs were related to specific device applications. This indicates that the design of AEPs is evolving to meet the specific requirements of the electrochemical devices.
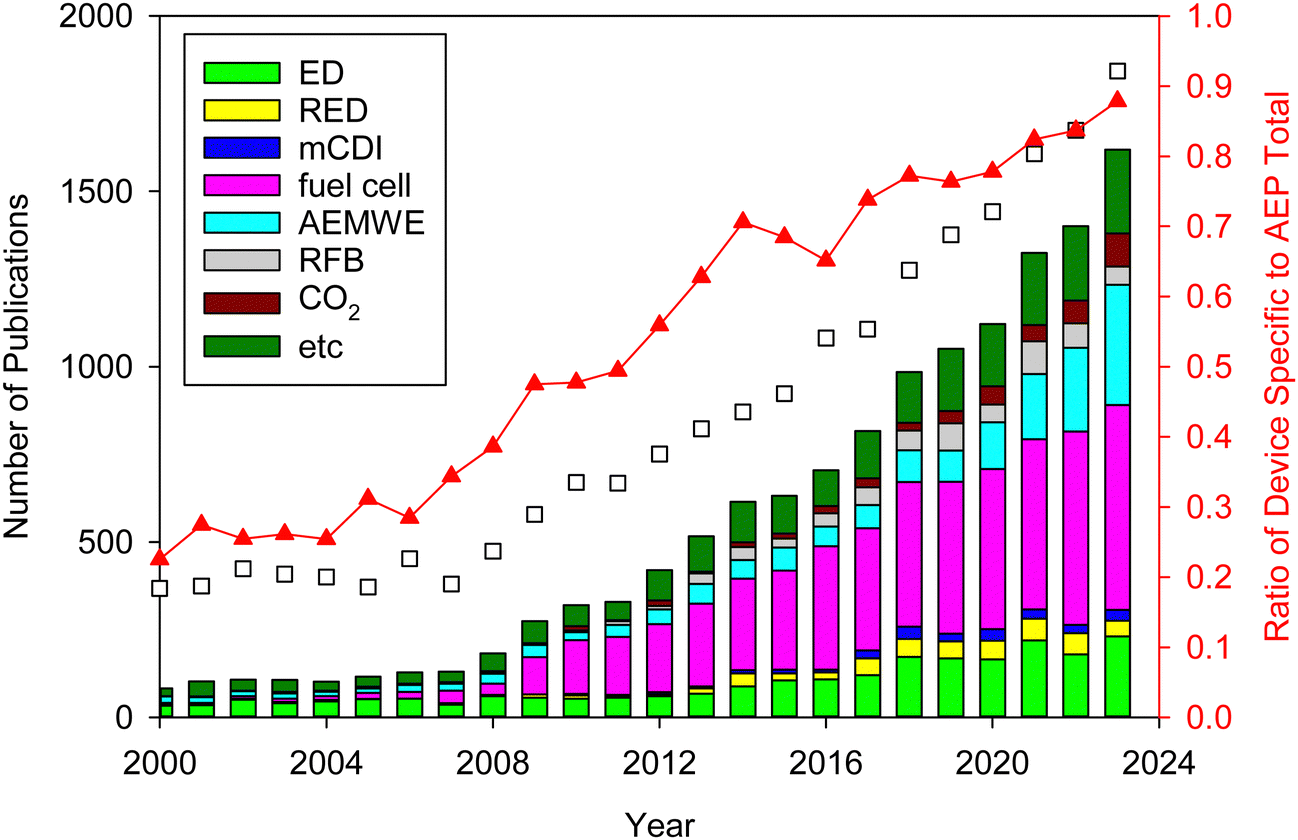 | ||
| Fig. 2 Number of publications on AEPs for various applications over the last 25 years. The related articles were identified using the keywords “anion exchange membrane” and corresponding applications from Web of Science™, Thomson Reuters (coloured bar). The total number of publications on AEPs (black square) represents those found with the keyword “anion exchange membrane”. The “etc.” category includes supercapacitor, metal ion batteries, gas separation, actuators, sensors, photo-electrolysis, microbial/enzymatic fuel cells, metal ion recovery, and RO. | ||
2. Chemical stability of AEPs
The electrochemical applications using AEP materials require essential properties tailored to specific applications, such as ion conductivity, permselectivity, chemical stability, thermal stability, mechanical properties, and gas permeability (high gas permeability for ionomers, low for membranes). The most critical property for electrochemical applications is the chemical stability of AEPs under the operating conditions of the devices. Fig. 3 shows the typical pH and potential ranges of various electrochemical devices utilizing AEPs. While AEMs for gas separation applications do not face a chemical stability issue, the AEPs for RO, ED, RED, and MCDI applications require chemical stability under medium to high pH conditions (pH 4–13), especially in the presence of multivalent metal cations. AEPs for AEMFC, AEMWE, and CO2 capture/electrolyser applications need chemical stability under higher pH conditions (pH > 13). Conversely, AEPs for HT-PEMFC applications require chemical stability under low pH conditions (pH 1–3) and elevated temperatures (100–200 °C). AEPs for aqueous RFB applications need chemical stability under lower pH conditions (pH < 1), particularly in the presence of multivalent metal cations with high oxidation states and potentials. In addition to alkaline stability, AEPs for AEMWE and CO2 electrolyser applications require high electrochemical oxidative stability at >1.8 V. It is important to note that although Fig. 3 depicts typical pH and potential ranges of electrochemical devices, AEPs may encounter even harsher environments under specific cell configurations and operations. For example, a few recent studies reported the operating voltage of BPM-based MCDIs, EDs, or AEMWEs exceeding >2 V.16–18 Furthermore, the cleaning of ED cells with acid and base solutions may result in membrane degradation due to the extreme pH conditions.19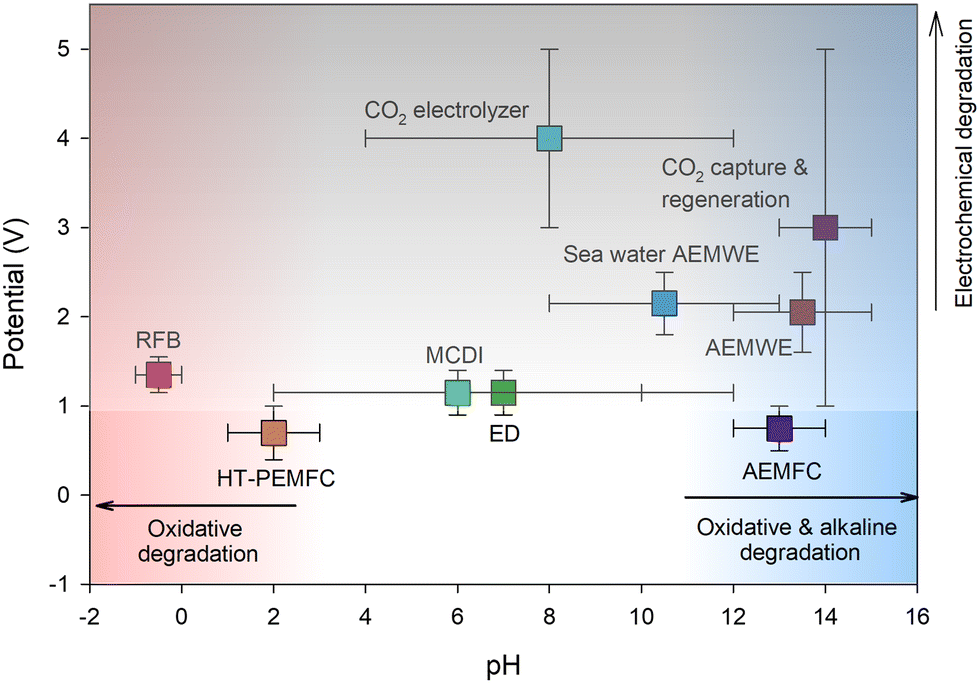 | ||
| Fig. 3 Typical pH and potential ranges of various electrochemical devices using AEPs. | ||
2.1. AEP synthesis before 2012
Before 2012, AEPs were primarily prepared for ED separation processes using one of two synthetic routes. The first route involves chemical modification of perfluoroacid precursors through multiple steps, forming anion-conducting counterparts of Nafion™. In 1986, Matsui et al. prepared quaternized perfluorinated AEMs by modifying the sulfonic acid functional groups of Nafion™.20 The multiple steps include (i) conversion of a carboxylic ester to an amide, (ii) reduction of the carbonyl group to methylene, and (iii) quaternization using methyl iodide (Scheme 1a). Other perfluorinated AEPs were also prepared from the carboxylic ester precursor (Scheme 1b)15 and sulfonamide precursor (Scheme 1c)21,22 of Nafion™, incorporating various ammonium structures, including guanidinium and hexylammonium. Perfluorinated AEMs can also be prepared in a single step via amination of the sulfonyl fluoride precursor under anhydrous conditions (Scheme 1d and e).23,24 However, attempting to prepare AEMs via amination of sulfonyl fluoride under aqueous conditions is unsuccessful, as it leads to hydrolysis of sulfonyl fluoride into sulfonic acid paired with an ammonium counterion.25,26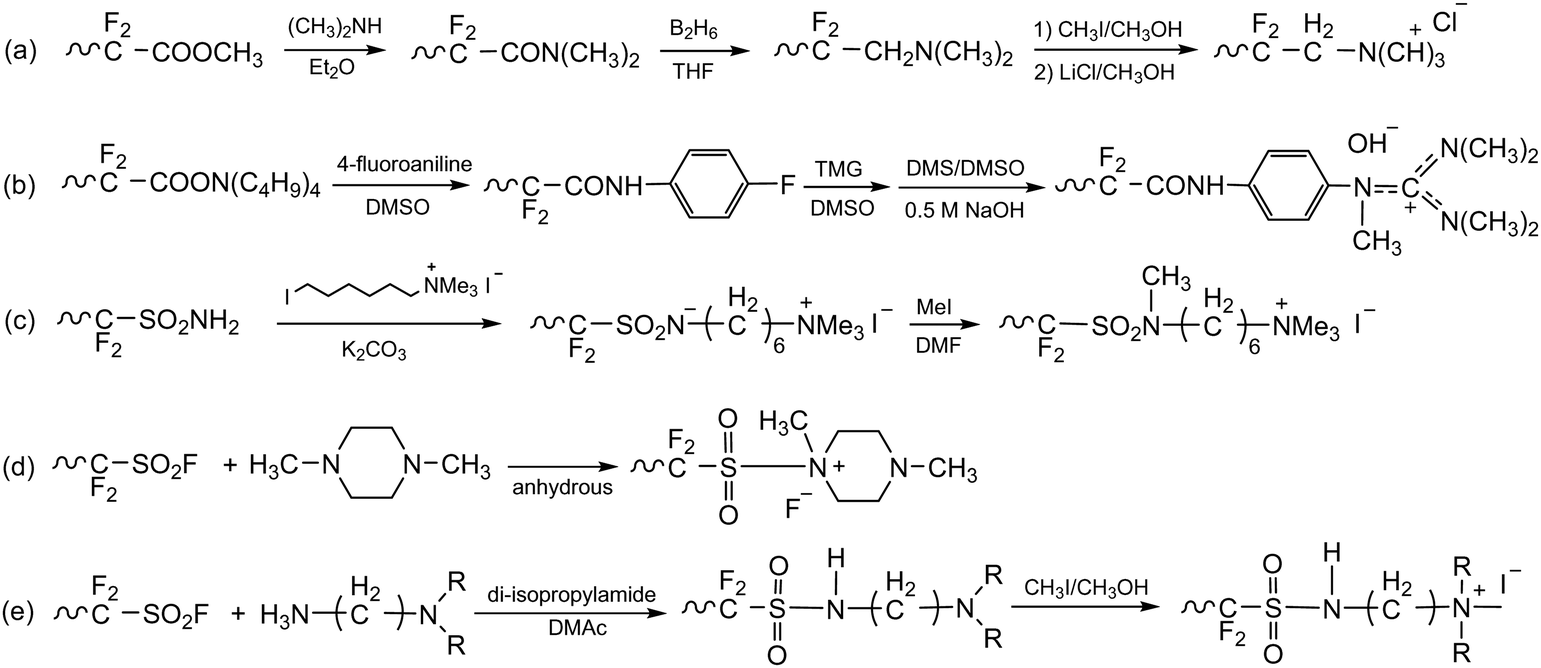 | ||
| Scheme 1 AEPs from chemical modification of perfluorinated polymer precursors: (a) quaternized perfluorinated AEM,20 (b) guanidinium perfluorinated AEP,21 (c) sulfonamide-linked alkyl ammonium perfluorinated AEM from sulfonamide precursor,22 (d) methylpiperazinium perfluorinated AEM,23 and (e) sulfonamide-linked alkyl ammonium perfluorinated AEM from sulfonylfluoride precursor.24 | ||
The quaternized perfluorinated AEPs exhibited excellent chemical stability under chlorine and acidic conditions, making them suitable for ED and RED applications. For example, quaternized perfluorinated AEMs with difluoroethane linkages remained chemically stable in chlorine-saturated water at 60 °C for 1000 hours without structural change.20 These membranes also demonstrated stability in 6 N hydrochloric and nitric acids at 60 °C. However, the mechanical properties of these quaternized AEMs deteriorated under basic conditions. Additionally, the difluoroethylene linkage in the AEMs is susceptible to hydroxide attack.27,28 Although amide and sulfonamide linkages in other perfluorinated AEMs showed improved chemical stability under basic conditions, slow hydrolysis may occur under AEMFC operating conditions,29 limiting their use in alkaline devices over extended time.
The second route involves post-polymerization modification of a hydrocarbon polymer with a desired cationic group. Quaternized polystyrenes are typically prepared by chloromethylation of polystyrene or copolymerization of styrene and vinylbenzyl chloride, followed by amination to form benzylammonium cation (Scheme 2a). A commercial polystyrene AEP (Selemion®, AGC) is prepared using this method. In 1985, Zschochke and Qullmalz synthesized a quaternized poly(aryl ether sulfone) via chloromethylation and subsequent quaternization of benzyl chloride (Scheme 2b).30 Due to the high toxicity and carcinogenic nature of the common chloromethylation agent, chloromethyl methyl ether, and its tendency to undergo crosslinking via methylene bridges,31 bromination of methylbenzene-containing poly(aryl ether sulfone)s or poly(p-phenylene oxide) (PPO) has been adopted for obtaining quaternized polymers (Scheme 2c).32–34 However, it was shown that the free-radical bromination pathway leads to radical-induced side reactions that produce polymers of lower molecular weight.35,36 Despite this, due to the commercial availability and versatile applicability of ether-containing aromatic polymers, functionalization using chloromethylation and bromination was the most popular synthetic pathways over the last decade, before the emergence of diverse aryl ether-free poly(arylene) AEPs.37–45 Another method includes lithiation chemistry to prepare quaternized poly(aryl ether sulfone) (Scheme 2d).46 The bis(dimethylamine)-containing side chain was functionalized onto the lithiated polymer, followed by methylation to introduce quaternary ammonium groups and produce AEMs.
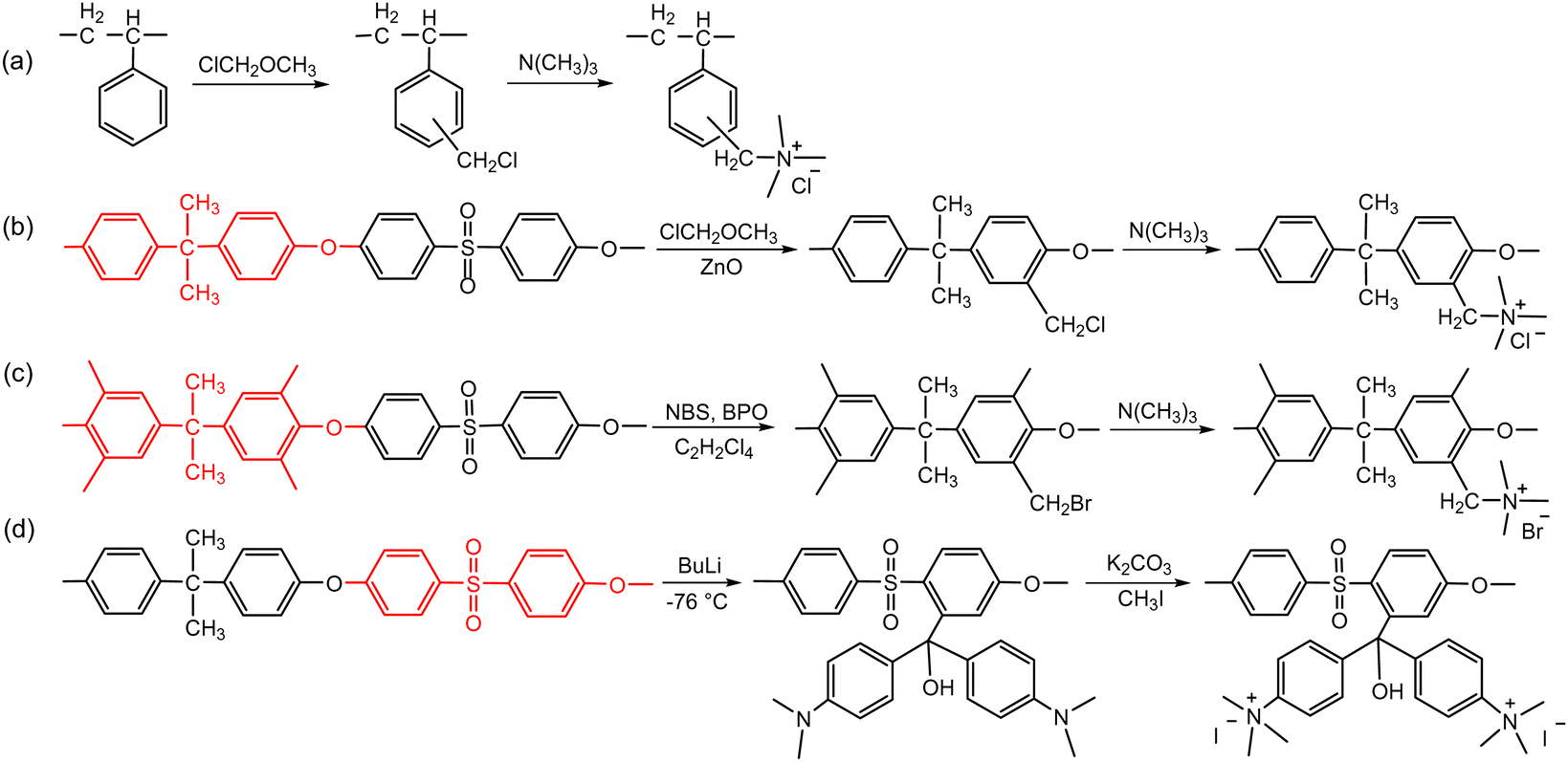 | ||
| Scheme 2 AEPs from polymer functionalization: (a) quaternized polystyrene, (b) quaternized poly(aryl ether sulfone) via chloromethylation,30 (c) quaternized poly(aryl ether sulfone) via bromination of benzyl groups,32 and (d) quaternized poly(aryl ether sulfone) via lithiation.46 | ||
The AEPs prepared from post-polymerization modification showing reasonably high stability under low to medium pH conditions are commercially available as water purification membranes. However, chemical stability tests of these polymers under alkaline conditions often reveal significantly reduced IEC values or mechanical failure of the membranes, indicating the need for improved chemical structure.
2.2. Alkaline stability of cationic groups
The generally lower alkaline stability of AEPs, compared to non-functionalized polymers, suggests that the chemical degradation of the cation headgroups predominates over that of the polymer backbone. Therefore, it has been assumed that the alkaline stability of AEPs is determined by the stability of the positively charged groups.47,48A hydroxide ion, acting as a strong nucleophilic base, can cause degradation through several different chemical mechanisms and pathways. The chemical degradation mechanism of quaternary ammonium groups under high pH conditions was well established before 2012. The attack of a hydroxide ion at the β-H-atom of the alkyl ammonium leads to a Hofmann elimination reaction, resulting in the formation of a tertiary amine from the neighbouring carbon, along with the formation of an alkene, a tertiary amine, and water (Scheme 3a). Another common degradation mechanism involves a nucleophilic substitution reaction at the α-C-atom of the quaternary ammonium. This reaction either yields methanol and a tertiary amine-functionalized polymer from methyl substitution (Scheme 3b) or a tertiary amine and an alcohol-functionalized polymer from the SN2 reaction (Scheme 3c). The degradation mechanism via the ylide pathway begins with a hydroxide ion abstracting a proton from a methyl group of ammoniums to produce a water molecule and a ylide intermediate. This intermediate then undergoes rearrangement to produce either an amine or an alcohol as a by-product (Scheme 3d).49
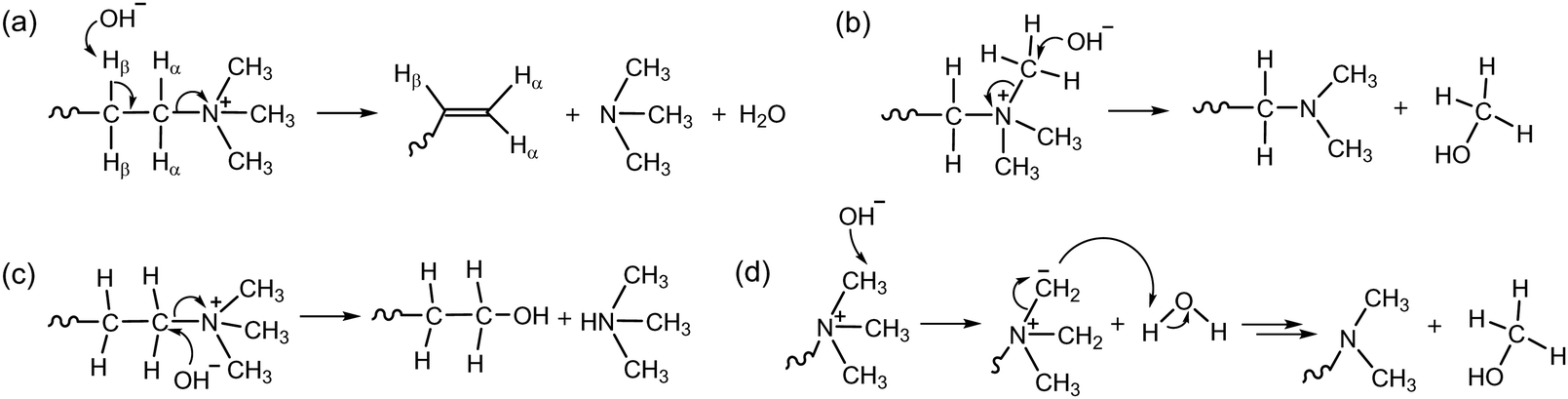 | ||
| Scheme 3 Degradation mechanisms of quaternary ammonium initiated by a hydroxide ion: (a) E2 (Hofmann) elimination, (b) nucleophilic methyl substitution, (c) SN2 reaction, and (d) ylide intermediate formation. | ||
The chemical degradation of various heterocyclic ammonium groups has also been reported. Methyl imidazolium undergoes ring-opening degradation triggered by the nucleophilic attack of a hydroxide ion on the imidazolium ring at the C2 position (the carbon adjacent to both nitrogen atoms), leading to the degradation of the cyclic cations (Scheme 4a).50,51 Methyl pyridinium degrades through a nucleophilic ring-opening reaction, which results in the replacement of the hydrogen by a hydroxide ion, followed by ring closure and dehydrogenation of the hydroxy group (Scheme 4b).52,53 The degradation of aliphatic-heterocyclic ammonium, such as dimethyl pyrrolidinium or piperidinium, can occur through three pathways: exocyclic demethylation (Scheme 4c), ring-opening elimination (Scheme 4d), and ring-opening substitution (Scheme 4e).54–56 The ring-opening reactions are predominant, but the degradation rate is influenced by ring size and the geometry of the transition state.
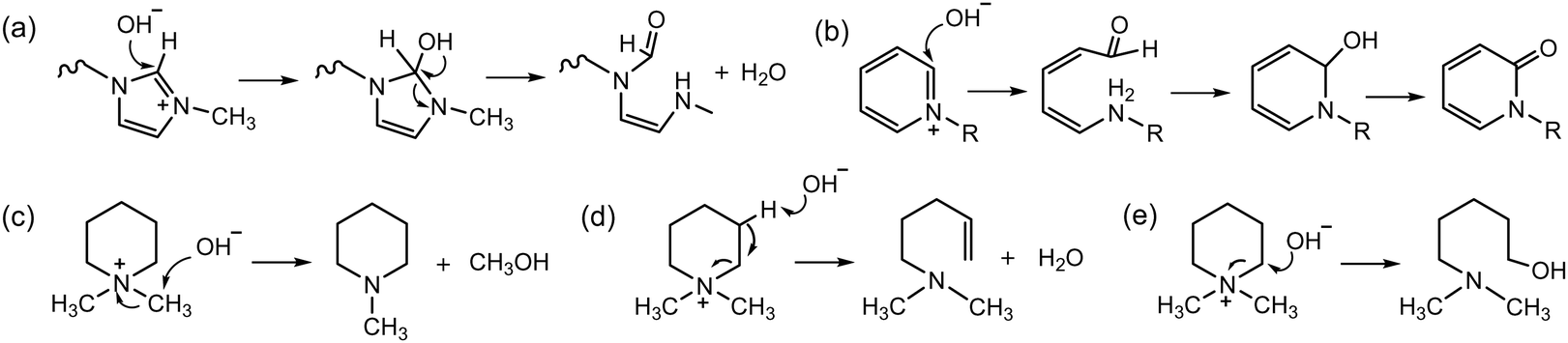 | ||
| Scheme 4 Degradation mechanisms of heterocyclic cations: (a) ring opening of imidazolium, (b) nucleophilic ring opening of pyridinium, (c) nucleophilic (methyl) substitution of dimethyl piperidinium (DMP), (d) ring-opening E2 elimination of DMP, and (e) ring-opening nucleophilic substitution of DMP. | ||
Research on the chemical stability of cationic functional groups has been further expanded to include quaternary ammonium with different substituents and more stable cationic structures (Fig. 4).57–60 The study of benzyltrimethyl ammonium (BTMA) has been extensively conducted because it is the simplest, synthetically accessible quaternary ammonium cation and thus the most commonly used in the early stages of AEP development.61–64 Phenyltrimethyl ammonium is much less stable than BTMA; for example, while 90% of BTMA solids remained in 5 M NaOH at 80 °C after 29 days, only 30% of phenyltrimethyl ammonium remained under the same conditions.65 Benzyl-tethered ammonium cations, such as benzyl pyridinium, benzyl pyrrolidinium, benzyltrimethyl guanidinium, benzylmethyl diazabicyclo[2.2.2] octane (DABCO), and benzyl 1,2-dimethylimidazolium, are less stable and prone to substitution. Alkyltrimethyl ammonium, phenyl-substituted imidazolium, and some heterocyclic cationic groups with high activation energy toward hydroxide attack, including piperidinium, quinuclidinium, and 6-azoniaspiro[5.5]undecane, have been reported to be more stable than BTMA under high pH conditions.48,58,66–71 Extensive discussions of headgroup stability have been reviewed recently.72
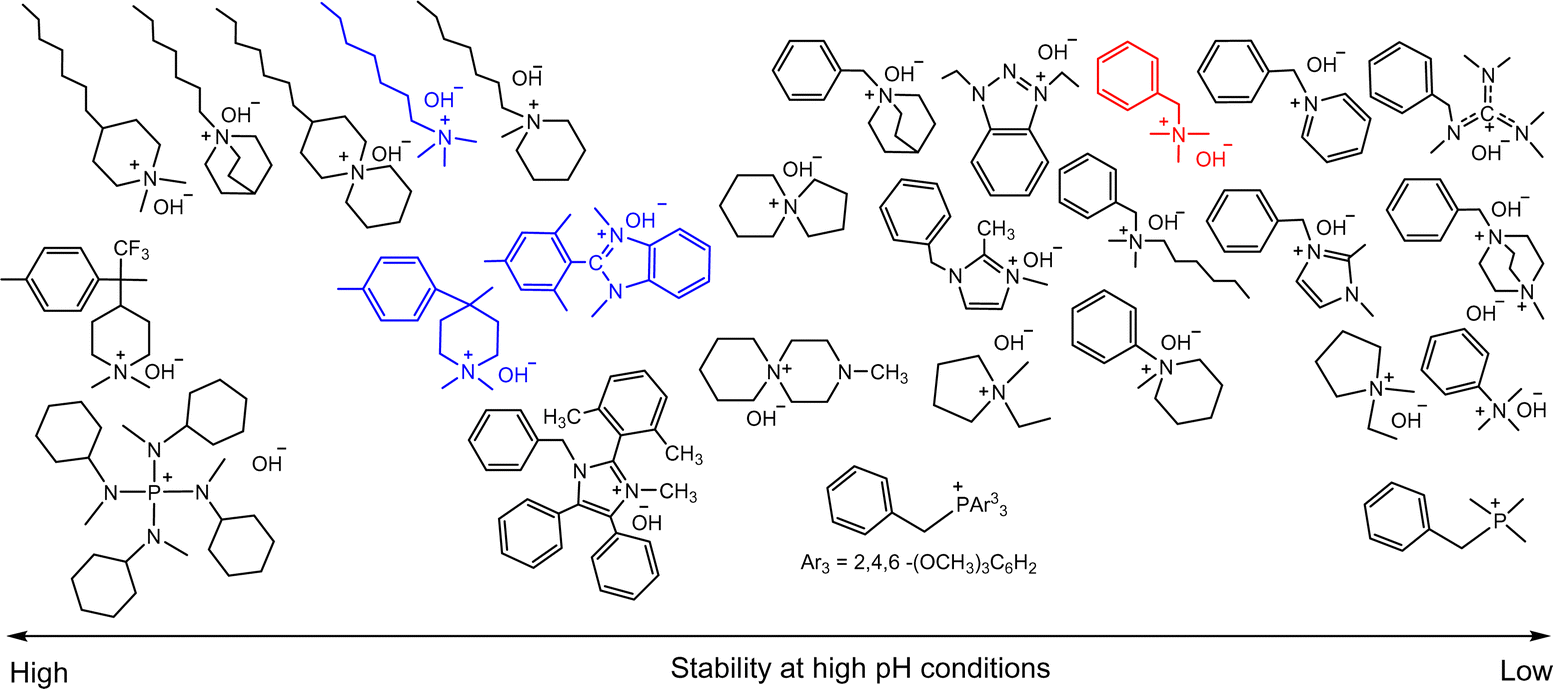 | ||
| Fig. 4 Chemical stability comparison of quaternary ammoniums under high pH conditions. Red: BTMA (control), Blue: popular cationic groups in modern AEPs. | ||
2.3. Alkaline stability of backbones
Polyvinylidene fluoride (PVDF)-based proton exchange materials are among the most promising alternatives to perfluorinated sulfonic acid polymers (PFSAs) due to their high hydrophobicity, inertness to electrocatalytic activity, and high chemical and mechanical stability.73,74 However, under high pH conditions, the PVDF backbone composed of difluoroethane, rapidly degrades through dehydrofluorination (1,2-HF elimination) and subsequent oxidation reactions (Scheme 5a).75,76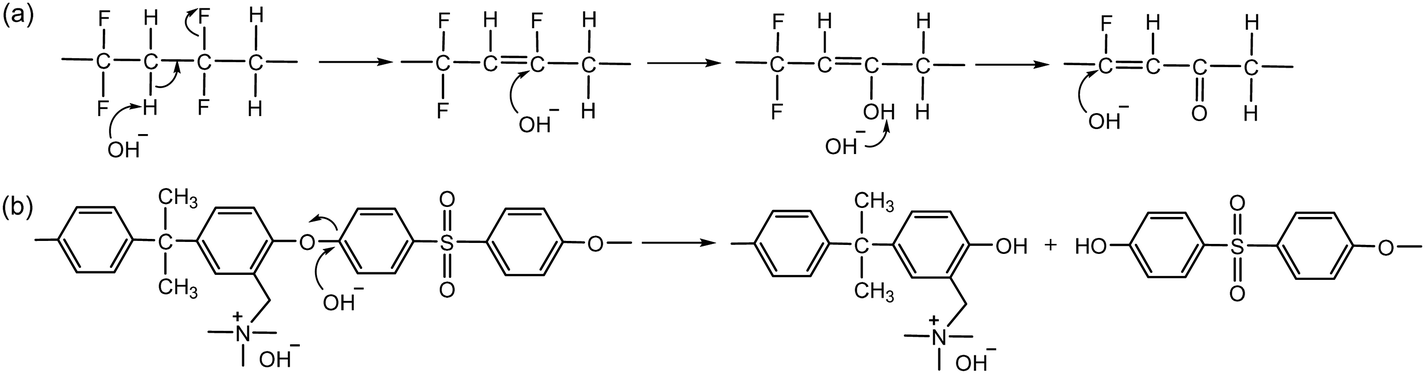 | ||
| Scheme 5 Degradation mechanisms of polymer backbone under high pH conditions: (a) dehydrofluorination and subsequent oxidation reactions of PVDF and (b) aryl ether cleavage of benzyl ammonium functionalized poly(aryl ether sulfone). | ||
Polystyrene and poly(aryl ether sulfone)s were among the first choices during the early AEP development period, as their sulfonated derivatives are chemically stable under high pH conditions (40% NaOH at 70–80 °C for 300 hours).77 However, several studies showed notably better alkaline stability of quaternized polystyrenes compared to quaternized poly(aryl ether sulfone) under high pH conditions.78–80 Only a few studies reported more than 100 hours of AEMFC durability using a quaternized poly(aryl ether sulfone) AEM, whereas several studies reported more than 200 hours of AEMFC durability using quaternized polystyrene or other polyolefinic AEMs during the early AEP development period.81–84 In 2012 and 2013, Fujimoto et al.85 and Ramani et al.86 independently reported that the backbone of quaternized poly(aryl ether sulfone)s cleaves at the aryl ether linkage under high pH conditions (Scheme 5b).
Follow-up studies investigated the effect of substituent groups on the barrier energy of the aryl ether cleavage reaction. The energy barrier for trimethylammonium-functionalized phenyl ether (85.8 kJ mol−1) was found to be lower than that for nucleophilic benzyl substitution (90.8 kJ mol−1) (Fig. 5a).87 The energy barrier was substantially lower in the presence of electron-withdrawing sulfone groups (32.6 kJ mol−1), but higher with an electron-donating methyl group (77.0 kJ mol−1) (Fig. 5b).88 These calculations are in good agreement with experimental data from MEA testing, where an MEA fabricated with a quaternized AEM containing two methyl groups on an adjacent aryl ring of ether showed much higher durability than an MEA using a quaternized poly(aryl ether sulfone) AEM.89 This indicates that the polymer backbone of electron-rich quaternized PPOs has better alkaline stability than that of electron-deficient aryl sulfone-containing polymers under high pH conditions.
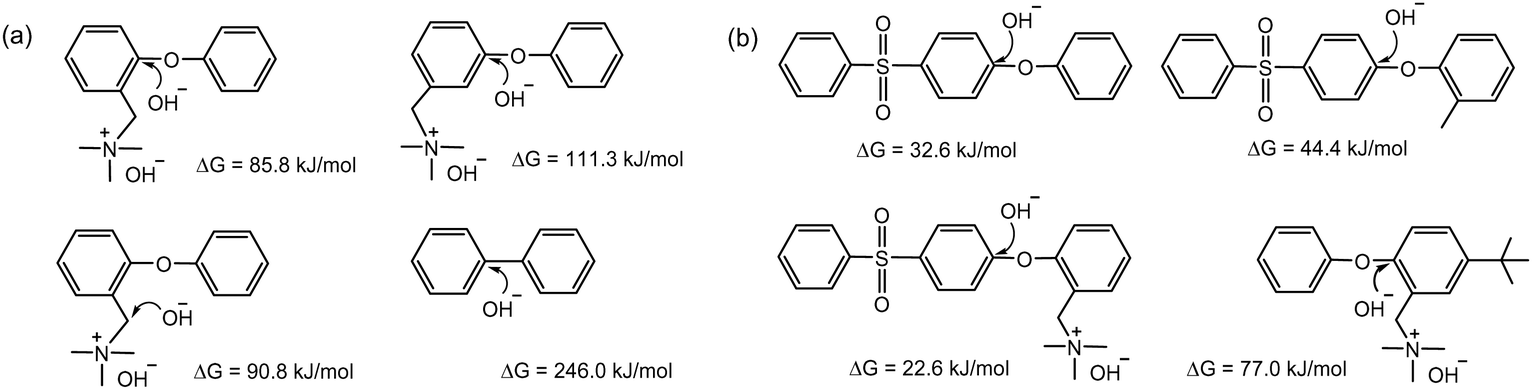 | ||
| Fig. 5 The energy barriers of aromatic fragments and hydroxide ions: (a) comparison between aryl backbone and quaternary ammonium. Geometry optimization by ωB97XD functional with a 6-311++G(2d,2p) basis set. (b) Effect of sulfone group substituent. Geometry optimization by M06-2X functional with a 6-31+G(d) basis set. | ||
Although several model compound studies on dimethyl phenylene oxide indicated no trace of the aryl ether cleavage reaction occurring, backbone degradation of quaternized PPOs was observed under high pH conditions.90,91 Ramani et al.35,92 and Becerra-Arciniegas et al.93 observed that quaternized PPO showed different backbone degradation depending on their synthetic process; the quaternized PPO prepared by bromination (Br-PPO-TMA) showed more substantial backbone degradation than the quaternized PPO prepared by chloroalkylation (Cl-PPO-TMA). This disparity was explained by the structural differences between Br-PPO-TMA and Cl-PPO-TMA that may influence the different rates of backbone degradation: (i) Cl-PPO-TMA has an extra methyl group in its backbone, increasing electron density, and (ii) the presence of unreacted bromine directly substituted on the benzene ring in Br-PPO-TMA accelerates the backbone degradation (Scheme 6).
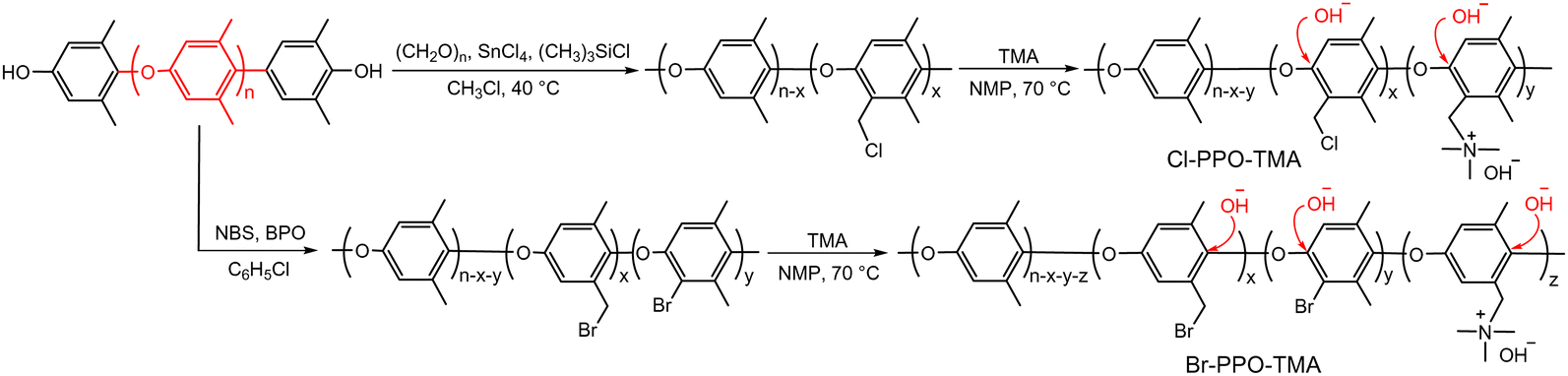 | ||
| Scheme 6 Synthetic pathways for quaternized PPOs via chloromethylation and bromination. Denoted are the reaction sites of aryl ether cleavage. | ||
Without cationic or electron-withdrawing groups in close proximity, the polymer backbone remains chemically stable in alkaline conditions. Several unfunctionalized aryl ether-free polymers, including both polyaromatic and vinyl polymers, did not show any sign of degradation at a high base concentration (10 equiv. of NaOCH3 in THF/methanol).88 The result suggests that aryl ether-free polymers are chemically stable to serve as a backbone for AEP materials.
2.4. Chemical stability at low pH
The chemical stability of AEPs under low pH conditions is known to be higher than that under high pH conditions, as there is no strong nucleophile in the acidic system. Several papers reported reasonably high chemical stability of quaternized polystyrene94 and poly(aryl ether sulfone).95–103 AEMs under vanadium RFB operating conditions need to be highly acidic to solvate multivalent vanadium ions with high oxidation states. The chemical degradation of AEPs detected in RFBs appears to be associated more with oxidative degradation than with acid-assisted degradation. Notable aryl ether cleavage reactions were observed for quaternized poly(aryl ether sulfone)s or poly(aryl ether ketone)s under the oxidative conditions of VO2+ in concentrated H2SO4 solution (Scheme 7a).104–106 The rate of backbone degradation of quaternized poly(aryl ether sulfone)s by VO2+ is similar to that of sulfonated poly(aryl ether sulfone)s,106 which contrasts starkly with the rate of backbone degradation of quaternized poly(aryl ether sulfone)s by hydroxide ions being much greater than that of sulfonated poly(aryl ether sulfone)s. The similar oxidative degradation rates between sulfonated and quaternized polyaromatics suggest that oxidative polymer degradation occurs in the aryl ether sulfone fragment rather than in the ionic groups. The oxidative degradation of polyaromatics primarily occurs at the electron-rich aryl groups because strong oxidizers such as hydroxyl and vanadium oxide radicals are highly electrophilic (Scheme 7a and b).107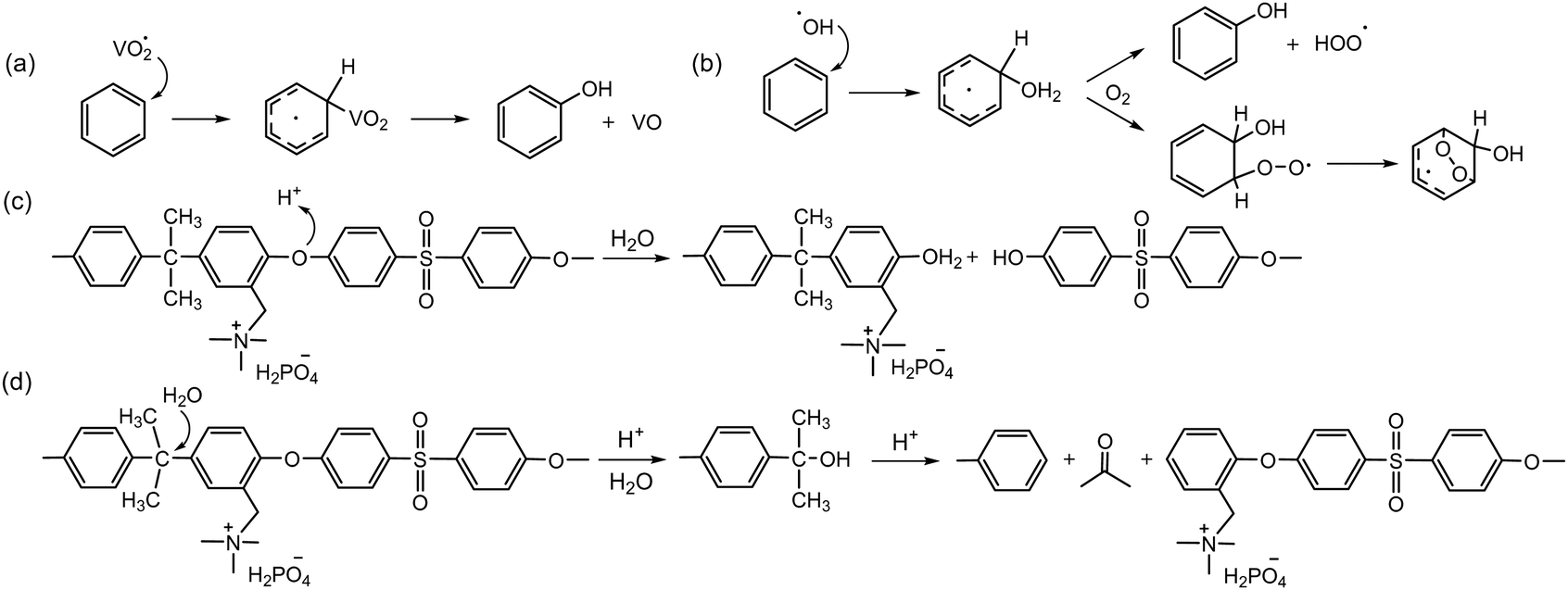 | ||
| Scheme 7 Degradation mechanisms of polymer backbone under low pH conditions: (a) and (b) suggested oxidative degradation of poly(aryl ether sulfone) by VO2+ in H2SO4, (c) aryl ether cleavage of quaternized poly(aryl ether sulfone) in 85% PA at 160 °C, and (d) C–C bond scission via retro-Friedel–Crafts alkylation in 85% PA at 160 °C. | ||
In HT-PEMFCs, the polymer backbone degradation of quaternized poly(aryl ether sulfone)s may occur in a protonic environment at a high temperature, around 160 °C. To date, only a limited number of studies have reported on chemical degradation under HT-PEMFC operating conditions. A different degradation mechanism of PA-doped poly(aryl ether sulfone) has been proposed: the aryl ether cleavage reaction via protonation of the ether oxygen followed by nucleophilic attack of water (Scheme 7c), as well as the scission of C–C bond at the isopropylidene carbon via a retro-Friedel–Crafts alkylation process (Scheme 7d).108
2.5. Electrochemical oxidative stability
Radical generation during the operation of electrochemical devices in aqueous conditions has been commonly observed, especially active oxygen species, e.g., hydroxyl radical (HO˙), reportedly formed with hydrogen peroxide by permeating O2 reacting with Pt/H2 to form H2O2 under alkaline conditions and at relatively low electrode potentials, ca. <1 V.109 Hydroxyl radicals can be further produced by hydroxide ion transfer. Wierzbicki et al. observed both HO˙ and HOO˙ on the cathode and H˙ on the anode under AEMFC operating conditions.110 The oxidative degradation of quaternized polystyrene and other polyaromatics by reactive oxygen radicals is well documented. Superoxide anion radicals can be formed from carbanions followed by subsequent reduction (Scheme 8a).111,112 Reactive oxygen radicals can abstract hydrogen from vulnerable positions (Scheme 8b) or add to aromatic compounds, forming hydroxycyclohexadienyl radicals (Scheme 8c).113 These oxidative radical species attack the vulnerable ternary carbon connected to the benzyltrimethyl ammonium hydroxide, resulting in chain scission (Scheme 8d).114,115 The rate of chain scission increases with exposure to higher oxygen concentrations. Similar oxidation reactions can occur with the dimethyl phenyl group in quaternized PPO (Scheme 8e) and with the diphenyl isopropylidene unit, which has a higher electron concentration in quaternized poly(aryl ether sulfone)s (Scheme 8f). Holdcroft et al. showed that backbone chain scission of sulfonated polyphenylene and formation of benzoic acid can occur through hydroxyl radical-induced oxidative degradation (Scheme 8g),116 with the degradation mainly occurring in non-sulfonated phenyl rings rather than in electron-withdrawing sulfonic acid-containing phenyl rings. Unlike highly nucleophilic hydroxide ions, radicals generated in an electrochemical environment, such as HO˙ and H˙, are electrophilic and tend to attack electron-rich sites of the substrate.117 Consequently, AEPs with high electron density, such as those containing methoxy or piperidino groups, have lower resistance to radical-induced oxidation.113 On the other hand, AEPs with electron-withdrawing groups possess a high electrophilic character and remain stable against oxygen radical attack.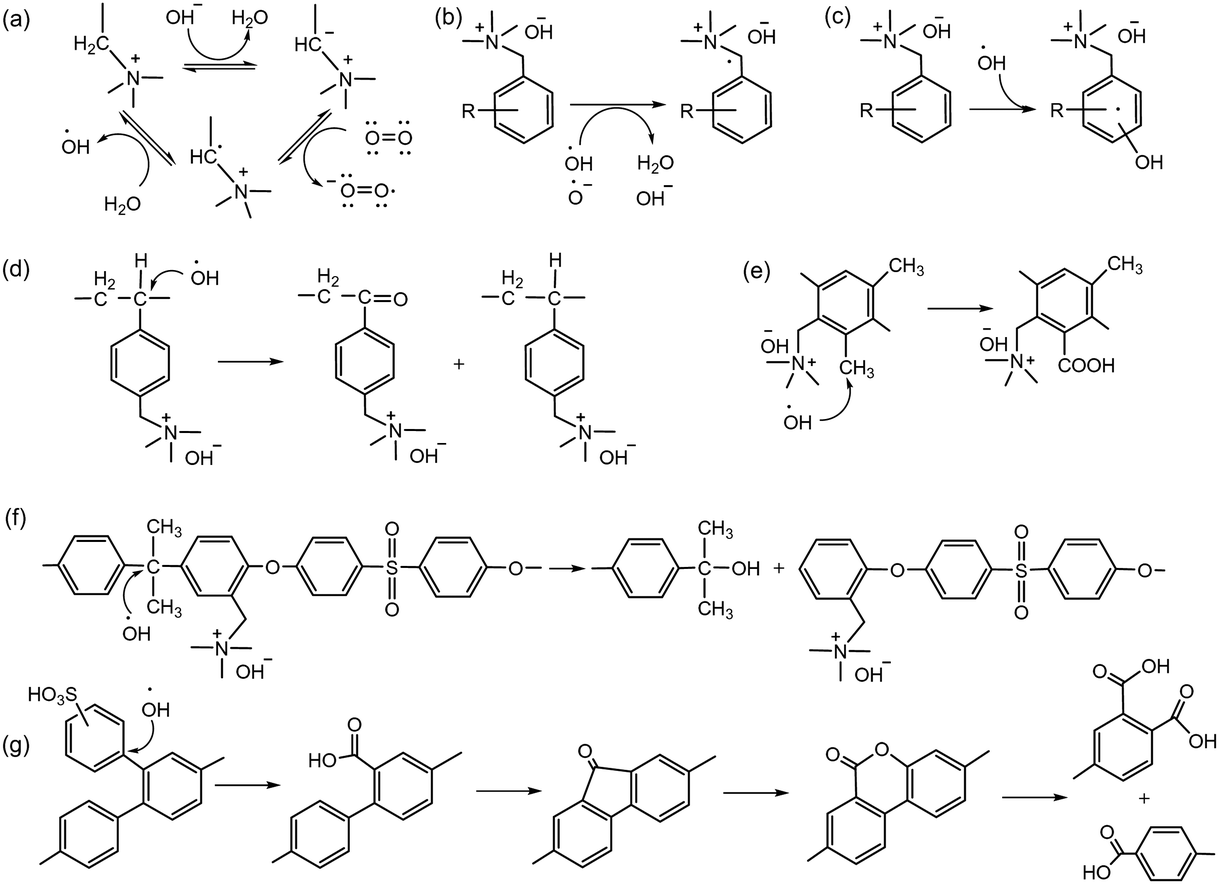 | ||
| Scheme 8 Degradation mechanisms by oxidative radicals: (a) reactive oxygen generation under high pH conditions, (b) electrochemical degradation of benzyltrimethyl ammonium by hydrogen abstraction, and (c) electrochemical degradation of benzyltrimethyl ammonium by radical addition. Different mechanism of radical-induced degradation of (d) the backbone of quaternized polystyrene, (e) quaternized PPO, (f) diphenyl isopropylidene unit of quaternized poly(aryl ether sulfone), and (g) sulfonated polyphenylene. | ||
Another critical oxidation process of AEPs is the catalytic oxidation of phenyl groups. The catalytic oxidation of benzene by transition metals and carbon-based catalysts under oxidative conditions is well documented.118–121 The electrochemical oxidation of phenyl groups to form phenol has been observed in quaternized poly(biphenyl) AEPs in AEMFC and AEMWE after prolonged operation at high cell voltages (0.9 V for AEMFC and 2.1 V for AEMWE) (Fig. 6a).122,123 Electrochemical oxidation of methylphenyl by noble metal catalysts has also been reported124–126 and may occur under the operating conditions of the device with applied voltages.127 The electrochemical oxidation of methylphenyl groups to form benzoate was observed with quaternized PPO AEPs in AEMWE, which substantially increased the degradation rate at the anode of the AEMWE (Fig. 6b).93,128 With electrochemical oxidation potentials, other AEP fragments can also degrade under device operating conditions (Fig. 7). For example, the electrochemical oxidation of cyclic alkanes and cyclic olefins, such as norbornene, occurs at approximately 1.0 V, resulting in the production of cycloaliphatic ketones and aliphatic (di)carboxylic acids.129 The electrochemical oxidation of heterocycles occurs at around 1.5 V or higher, depending on the chemical environments.130–132
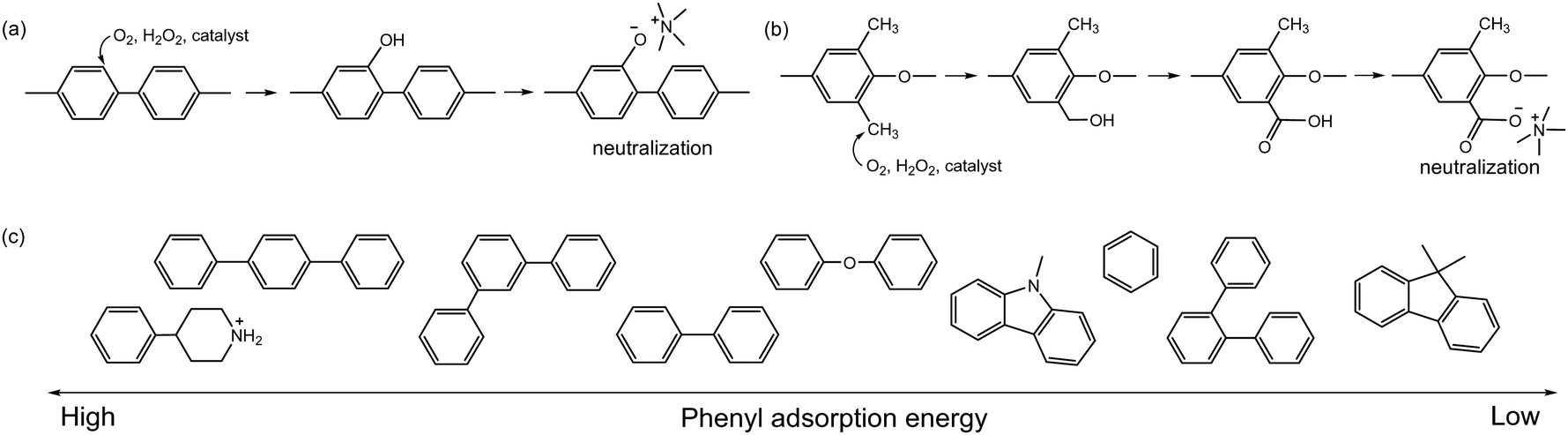 | ||
| Fig. 6 Electrochemical oxidation: (a) phenyl oxidation to form phenol, (b) methylphenyl oxidation to form carboxylic acid, (c) phenyl adsorption energy of polyaromatic fragments on Pt(111) surface. The adsorption energy was calculated by density functional theory (DFT) using optPBE-vdW. | ||
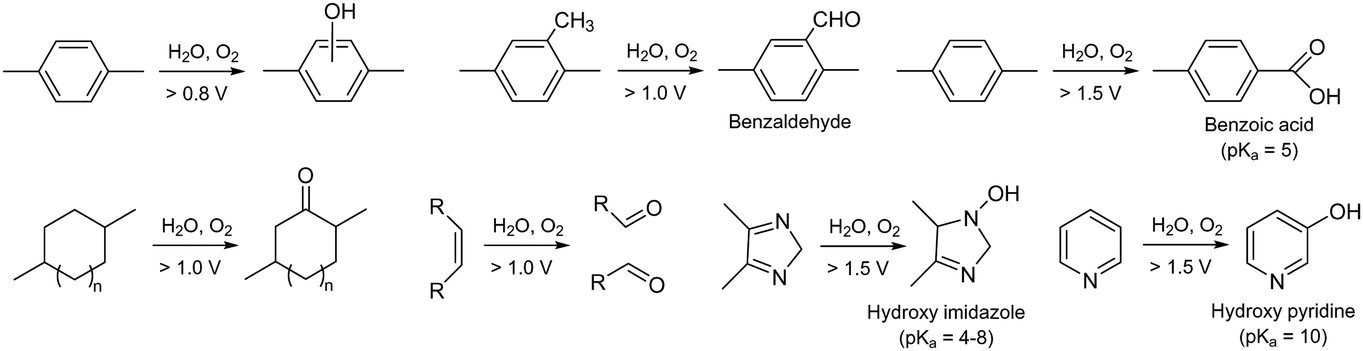 | ||
| Fig. 7 Electrochemical oxidation process and oxidation potentials of various ionomer fragments. | ||
The detection of electrochemical oxidation products is challenging since the oxidation occurs at the catalyst and ionomer interface. However, the impact of electrochemical oxidation on device performance can be significant because the oxidative products can lower the local pH near the catalyst's active site. The device operating potential and the adsorption energy of the phenyls and other AEP fragments onto the catalyst surface are the two most important factors for catalytic oxidation. Consequently, AEPs in the electrodes that are exposed to high potentials, such as those in the anode of AEMWEs and CO2 electrolysers, are much more vulnerable to the catalytic oxidation of AEPs. Matanovic et al. investigated the adsorption energy of the phenyl groups derived from AEP fragments (Fig. 6c) and found that more durable device performance was achieved with ionomers having lower phenyl adsorption energy.133,134 The adsorption behaviours of ionomers are also significantly influenced by the type of catalysts and their surface and electronic structures. Matanovic et al. noted that Pt alloyed catalysts exhibit less aromatic adsorption characteristics, attributed to electron transfer between phenyl and metal surfaces induced by alloying.135 The same research group observed that the adsorption energy of benzyltrimethyl groups on the surface of the La0.85Sr0.15CoO3 perovskite catalyst was substantially lower than on Pt or IrO2 catalysts.122
2.6. Demand for aryl ether-free AEPs
The literature survey in the previous sections suggests that the degradation pathways of AEPs are dependent upon the operating conditions of electrochemical devices. The two most critical factors are the pH environment and the electrode potential required for electrochemical reactions. In general, aryl ether-free polymers exhibit considerably higher oxidative stability than aryl ether-containing polymers.136,137 For AEMFC, AEMWE, and electrochemical CO2 conversion/capture devices, the alkaline stability of AEPs is most critical, and most aryl ether-containing AEPs lack chemical stability for durable operation in these applications. High electrochemical oxidative stability is required for AEMWE and CO2 electrolyser/capture devices. Given that the nature of degradation by hydroxide ions (alkaline, nucleophilic) and hydroxyl radicals (oxidative, electrophilic) are different, AEPs with balanced chemical stability specific to an electrochemical device can enable long-term operation. AEIs in the catalyst layers require higher electrochemical oxidative stability compared to AEMs.138 However, some studies indicate that supplying a liquid alkaline solution to the electrode can mitigate the electrochemical oxidation of ionomers by reducing ionomer fragment adsorption.122,139,140 Therefore, if an electrochemical device can incorporate a supplemental liquid alkaline solution, the requirements for electrochemical stability may be less stringent.The chemical stability studies on AEPs indicate that aryl ether-free AEPs are excellent candidates for various electrochemical devices. Alternative material candidates may include ion-solvating polymers with basic functional groups, such as benzimidazole. Reasonably high performance and durability of ion-solvating membranes have been demonstrated in RFB,141–144 HT-PEMFC,145–147 and electrolyser148–151 applications. However, these polymers can only be used in electrochemical devices supplying liquid electrolytes, which have additional stability issues associated with highly concentrated liquid electrolytes. Therefore, aryl ether-free AEPs remain a strong contender for use in modern electrochemical devices. In the following sections, we will discuss various synthetic pathways that have been implemented to prepare cation-functionalized aryl ether-free AEPs, demonstrating outstanding material properties, chemical stability, and successful performance in electrochemical devices.
3. Chemistry of polyaromatics
Polyaromatics are a class of polymers whose main chains are composed of phenylene units. They are best known for their excellent thermal and mechanical properties, making them suitable for various applications. The most promising design of polyaromatics for high chemical stability and longevity in alkaline conditions involves backbones that are devoid of labile aryl ether linkages, predominantly consisting of C–C bonds.152 In this section, we will focus on the notable chemistry of polymerization used to prepare quaternized aryl ether-free polyaromatics. This includes the reaction mechanism, the representative AEPs from each method, and their general properties for electrochemical devices.3.1. Acid-catalysed polyhydroxyalkylation
For the synthesis of AEPs, it is desirable to have a polymerization method that leads to high molecular weight growth, allows for various monomer choices, and is tolerant of any functional groups present. Acid-catalysed polyhydroxyalkylation offers many advantages in these factors with respect to other reactions and has been widely adopted for the synthesis of aryl ether-free polyaromatic-based AEPs in recent years.When strong Brønsted or Lewis acids interact with electrophiles and undergo superelectrophilic activation, they can form highly reactive electron-deficient compounds known as superelectrophiles.153 The early demonstrations of superelectrophiles for Friedel–Crafts alkylation used selective carbonyl compounds, including benzaldehyde, and electron-rich aromatics.154 The enhancement of the reactivity of carbonyl electrophiles heavily depends on the acidity of the system; while 100% sulfuric acid (pKa = −12) is not sufficiently strong to activate most carbonyl compounds, superacids such as trifluoromethanesulfonic acid (TFSA, pKa = −14) or even stronger acids are required. During the reaction, a carbonyl compound is protonated by high acidity proton donating species, forming extremely electron deficient carboxonium/hydroxycarbenium intermediates that react with nucleophilic aromatics via hydroxyalkylation (Scheme 9).155
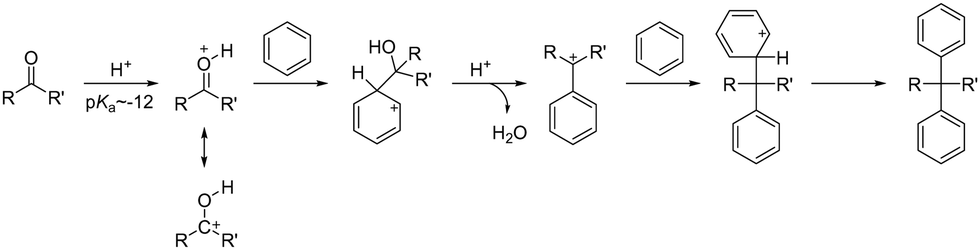 | ||
| Scheme 9 General reaction mechanism of acid-catalysed Friedel–Crafts hydroxyalkylation. | ||
The reactivity of electrophiles can be further increased by the presence of electron-withdrawing substituents, such as trifluoromethyl or nitro groups, in close proximity to the carbonyl group. An electronegatively substituted carbonyl compound was used to form a linear, high molecular weight, yet still soluble polymer using an aromatic hydrocarbon in a superacid environment via polyhydroxyalkylation.156 Several carbonyl-containing compounds and aromatic compounds were screened for their reactivity in a superacid medium to form high molecular weight polymers (number-averaged molecular weight, Mn, >50 kg mol−1).155,157–159 According to the proposed mechanism, based on theoretical and experimental studies,155,159 the substituents of carbonyl compounds (R and R′ in Scheme 9) influence the rate-determining step of polymerization, which can be either the aromatic electrophilic substitution or the subsequent dehydration reaction. Strong electron-withdrawing groups on both substituents can increase the electrophilicity of the carbonyl carbon, allowing for a fast reaction with an aromatic compound, but they may also increase the activation energy required for the formation of carbocation from dehydration, thus slowing down the rate of polymerization. Therefore, a number of ketone monomers containing a trifluoromethyl group along with an electron-donating methyl or phenyl group (e.g., trifluoroacetone and trifluoroacetophenone) have been used for the formation of ultrahigh molecular weight polymers (Mn > 106 g mol−1).158
In the case of aromatic monomers, H–Ar–H, the nucleophilicity of the aromatic compound is another factor that determines the polymerization rate.160 Highly nucleophilic aromatic compounds lead to the continuous growth of polymer chains via this step-growth polymerization by reacting with carbonyl electrophiles. Symmetrical and electron-rich phenyl compounds, such as biphenyl, p-terphenyl, and diphenyl ether, are typically used to enable the formation of high molecular weight polymers. Depending on the reactivity of the monomers, the stoichiometric ratio between carbonyl and aromatic monomers can be adjusted to increase the polymerization rate, but a large excess of one monomer over the other may result in low molecular weight polymers or cross-linking.160 Anhydrous dichloromethane is mainly used as a polymerization solvent due to its inertness towards strong acids and its solvating power for most of the monomers utilized.
The first report using the acid-catalysed polyhydroxyalkylation chemistry to produce AEPs was in 2015 by Bae et al. They employed biphenyl and a ketone monomer containing a bromoalkyl precursor for subsequent quaternization using trimethylamine (BPN1-m, Scheme 10).161 Alkylhalide is tolerant in a superacid environment; although the reactivity of a bromoalkyl-containing ketone monomer is lower than that of trifluoroacetone, the polymerization proceeds efficaciously, growing to high molecular weight polymers at room temperature in 12 hours (Mn > 70 kg mol−1 and weight-averaged molecular weight, Mw, >100 kg mol−1). Copolymers bearing a repeat unit of biphenyl and non-functional trifluoroacetone have been prepared to adjust the IEC of the polymers (1.5–2.6 mequiv. g−1). Despite being composed of rigid aromatic rings, the polyaromatic-based AEPs from the superacid-catalyzed polyhydroxyalkylation, such as BPN1-m, are readily soluble in common organic solvents due to the insertion of a kinked Csp3 in every repeating unit. The quaternized polymers have high solubility in polar aprotic organic solvents for easy processability, and high tensile strength (up to 35 MPa at 50 °C, 50% relative humidity (RH)). The hydroxide conductivity at 80 °C of BPN1-100 (IEC = 2.6 mequiv. g−1) was 122 mS cm−1, with a water uptake of 145%.161 The AEPs did not show any change in IEC values or hydroxide conductivity after immersion in 1 M NaOH at 80 °C for 30 days, demonstrating high chemical stability in an alkaline environment and opening up the possibility of using this chemistry for the synthesis of alkaline stable AEPs.
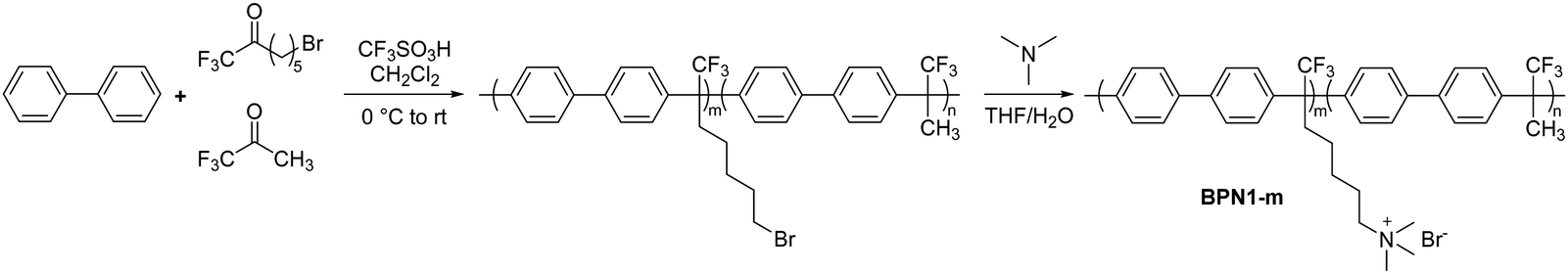 | ||
| Scheme 10 Representative reaction schematic of acid-catalysed polyhydroxyalkylation to synthesize biphenyl-based AEP BPN1-m. | ||
Since then, various combinations of monomer choices for both aromatic and carbonyl monomers have been utilized to form AEPs in recent years, as summarized in Fig. 8.66,71,162–175 Most polymerization reactions have been done in one-pot mode to obtain precursor polymers, either containing bromoalkyl or tertiary amine groups, followed by quaternization using a tertiary amine or alkylation using an alkyl bromide to form their corresponding AEPs. The high tolerance of the acid catalyst allows the inclusion of cation- and secondary amine-containing monomers, which can be used as AEPs once polymerized71,169,172,174 or simply quaternized via a post-grafting reaction using alkyl ammoniums.176 For the superacid catalysts, TFSA alone or in combination with trifluoroacetic acid (TFA) has been used for polymerization, where TFA can serve as a solvent medium, covering a wide range of acidity to increase the reactivity of monomers.177 The ratio of the ketone monomer varies depending on the reaction conditions and electrophilic reactivity, but it is nearly stoichiometric with respect to aromatic monomers (approximately 1.0–1.2 equivalents of the carbonyl monomer for superelectrophilic activation; up to 1.5 equivalents for less reactive monomers173). To promote molecular weight growth of polymers for membrane applications, non-functional high-reactivity monomers (e.g., p-terphenyl, 2,2,2-trifluoroacetophenone) have been incorporated as repeat units of copolymers. Poly(terphenyl piperidinium) with 85% ionic repeat units exhibits a much higher molecular weight (intrinsic viscosity 4.71 dl g−1, Mw = 70.5 kg mol−1)166 compared to the terphenyl piperidinium homopolymer (0.39 dl g−1) (Fig. 9a).66
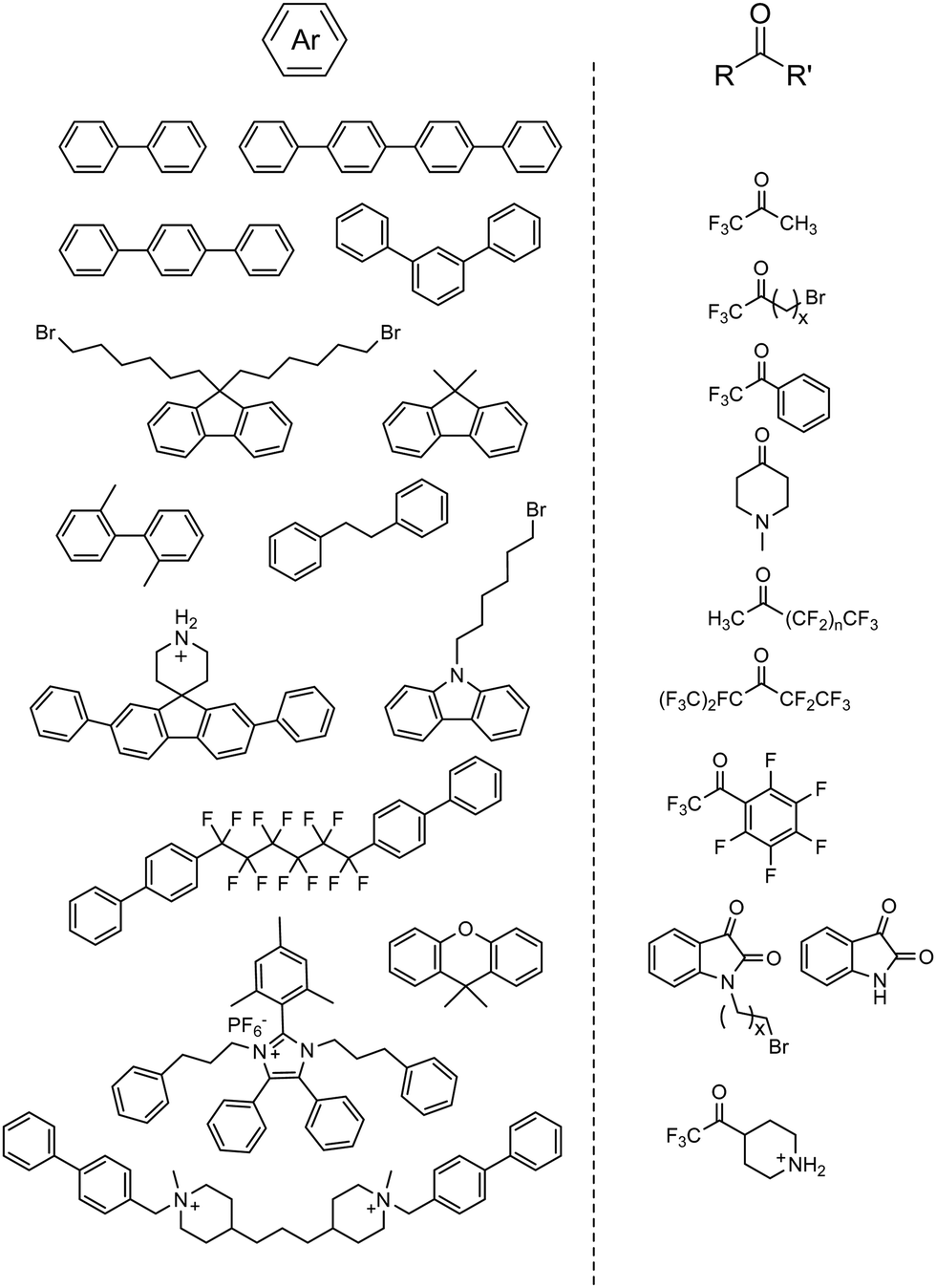 | ||
| Fig. 8 Representative aromatic hydrocarbon and carbonyl-containing monomers used for acid-catalysed polymerization. | ||
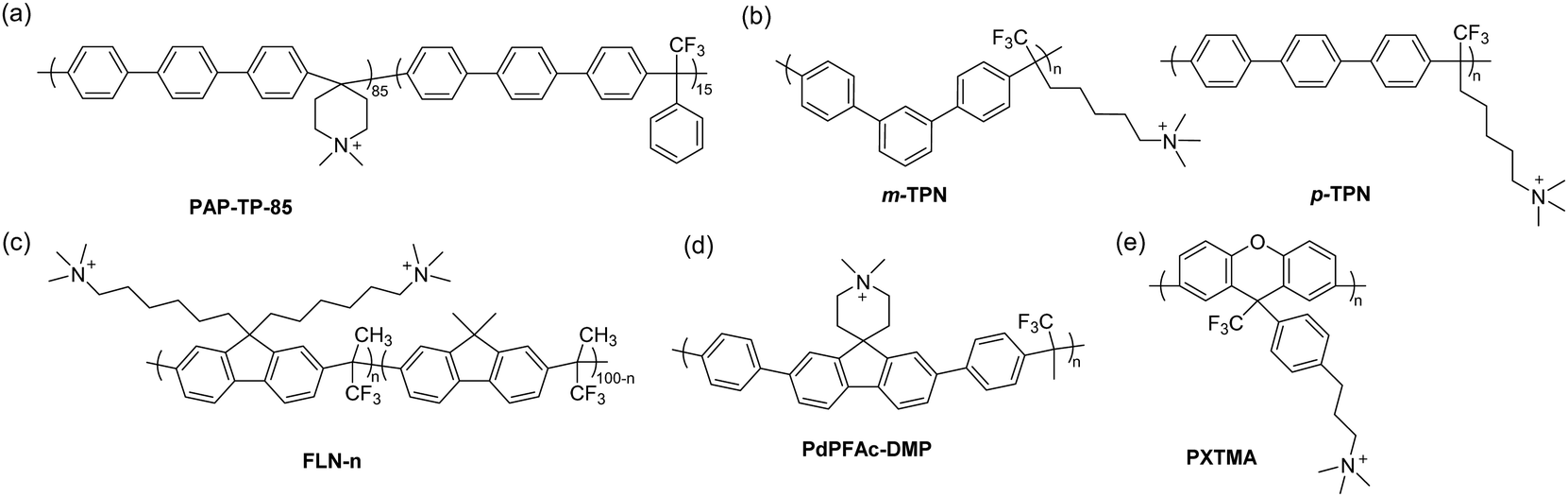 | ||
| Fig. 9 Selected examples of polyaromatic-based AEPs synthesized by acid-catalysed polyhydroxyalkylation: (a) poly(terphenyl piperidinium) copolymer, (b) poly(terphenyl alkylene)s, (c) bis-quaternary ammonium functionalized fluorene-based AEPs, (d) N-alicyclic cation-containing polyfluorene, and (e) polyxanthene-based AEPs. | ||
The effect of the polymer backbone arrangement and morphology on membrane properties was investigated using two different terphenyl monomers, p-terphenyl and m-terphenyl, to synthesize their respective AEMs (Fig. 9b).163m-TPN, consisting of meta-terphenyl in the backbone, exhibited higher molecular weight, better solubility, water uptake, hydroxide conductivity, and fuel cell performance in AEMFC than para-terphenyl containing p-TPN. This improvement is ascribed to the favourable morphology and polymer chain packing for ion transport, as confirmed by wide-angle X-ray scattering, likely from the kinked structure of m-terphenyl.163 The alkaline stability of m-TPN (IEC = 2.2 mequiv. g−1) was found to be higher than that of BPN1-100. It showed no signs of degradation in the 1H NMR spectrum and titrated IEC in 1 M NaOH at 95 °C for 60 days, while a slight decrease was observed in BPN1-100 under the same condition, likely due to the higher IEC (2.6 mequiv. g−1).178
9,9-Bis(6-bromohexyl)fluorene has been a popular choice for the aromatic component in AEPs because it allows the inclusion of two quaternary ammonium groups per repeat unit, increasing the resulting IEC of the polymer up to 3.5 mequiv. g−1 when polymerized with trifluoroacetone (Fig. 9c).164 The ionic repeat unit was optimized to be 55% by incorporating non-functionalized 9,9-dimethylfluorene as a co-monomer, adjusting the IEC to 2.5 mequiv. g−1, which showed a high hydroxide conductivity of 127 mS cm−1 at 80 °C. Fluorene-based AEPs also have the advantage of being effective electrode AEIs, as their fused ring structure has minimal adsorption energy to the surface of electrocatalysts compared to other phenyl-containing monomers, such as biphenyl.134 This offers benefits of high oxidative stability of AEIs, especially for electrochemical devices operated at high voltage conditions.122,123 The wide range of IECs of fluorene-based ionomers can aid in controlling water management in electrodes or operate at low RH conditions by using an asymmetric electrode approach.179 Recently, fluoroalkyl-containing fluorene ionomers were reported to effectively control electrode hydrophobicity for appropriate water management in AEMFCs.173 The downsides of employing fluorene in the AEP structure would be that the monomer synthesis to include the bis(bromohexyl) group requires tedious purification steps,164 and AEMs using only fluorene as an aromatic monomer tend to result in brittle membranes, likely due to the bulky structure of fluorene interfering with chain entanglement.
Because of the high regioselectivity of the polymerization, using aromatic monomers containing unsubstituted terminal phenyl groups is highly effective. An ortho-methyl substituted biphenyl, designed to increase the fractional free volume of the polymer chains for efficient hydrogen diffusion on the anode electrode, did not form sufficiently high molecular weight polymers, unlike its unsubstituted biphenyl counterpart.165 Several studies reported the polymerization of aromatic monomers having electron-deficient groups, such as unsubstituted phenyl rings at both terminal positions. With this approach, even aromatic monomers that are challenging to polymerize, i.e., those containing electron-deficient substituents or ionic moieties that affect electron density, can be used in acid-catalysed polymerization. Miyatake and co-workers reported perfluorohexylene-containing AEMs by synthesizing a fluorinated monomer bearing terminal biphenyl groups.175 The AEM exhibited a well-defined phase-separated morphology due to the incorporation of the hydrophobic perfluorohexylene group in the backbone and showed high hydroxide conductivity (115 mS cm−1 at 80 °C, IEC = 2.0 mequiv. g−1). The membrane properties remained unchanged after immersion in 8 M KOH at 80 °C for 1000 hours, confirming that the phenylene-based backbone with a perfluoroalkyl moiety is highly alkali stable. Jannasch et al. prepared an N-alicyclic cation-containing fluorene monomer with terminal phenyl groups, which directly produced AEMs incorporating a piperidinium cation (Fig. 9d).174 PdPFAc-DMP with an IEC of 1.9 mequiv. g−1 showed a hydroxide conductivity of 84 mS cm−1 at 80 °C with a water uptake of 57%. Indications of cation degradation were observed in the 1H NMR spectra after immersion in 1 M NaOH at 80 °C for 30 days, occurring in the piperidinium ring attached to the fluorene backbone. Zhang and co-workers demonstrated the direct polymerization of an imidazolium-containing monomer fully substituted with phenyl groups, sequentially polymerized with additional biphenyl and trifluoroacetone to form ionene segmented block copolymers (more about ionenes in Sections 3.3 and 3.4).172 The AEM exhibited a hydroxide conductivity of 57 mS cm−1 at 80 °C with an IEC of 1.2 mequiv. g−1. Similarly, Wang et al. synthesized a bis-piperidinium monomer with terminal biphenyl groups, polymerizing it with a superacid to form poly(bis-alkylimidazolium) ionenes (IEC = 2.3 mequiv. g−1, σ80°C = 45 mS cm−1).180 The approach of using terminal phenyl groups expands the library of aromatic monomers available for polymerization, but the downside of this approach might be that aromatic monomers need to be prepared via metal-catalysed coupling reactions165,175 or via multistep synthesis.172,174
Superacid catalysts are also effective for the synthesis of another type of polyaromatic, polyxanthene, where its backbone is composed of a pyran ring fused with two phenyl rings.181 This tricyclic aromatic structure in the polymer backbone may exhibit intrinsic microporosity, favouring both ion and gas transport.182 4,4′-Biphenol and bromopropyl trifluoroacetophenone were polymerized via polyhydroxyalkylation followed by cyclodehydration to form xanthene, which was then converted into AEMs with a tertiary amine (Fig. 9e).182 Although the polymer contains C–O–C bonds as part of the main chain, no sign of backbone degradation was detected after immersion in 1 M NaOH at 80 °C for 720 hours, likely due to the absence of electron-withdrawing groups near the aryl ether bonds. The TMA-containing AEM, PXTMA, showed the highest IEC of 2.3 mequiv. g−1 and hydroxide conductivity of 129 mS cm−1 at 80 °C among the reported polyxanthene AEMs. Another polyxanthene-based AEP, synthesized using 9,9-dimethylxanthene and isatin with TFSA, was subsequently functionalized using bromobutyl trimethylammonium bromide.183 The hydroxide conductivity at 80 °C of this AEM with an IEC of 2.1 mequiv. g−1 was found to be 205 mS cm−1, ascribed to the twisted, intrinsically microporous arrangement of the xanthene backbone for efficient ion transport, as confirmed by microstructure characterization using atomic force microscopy (AFM) and small-angle X-ray scattering (SAXS).
To enhance the mechanical properties of polymers for membrane applications, various branching and crosslinking strategies have been employed for acid-catalysed AEPs. A branched AEM was reported using 2.5% of 1,3,5-triphenylbenzene as a branching node with p-terphenyl and N-methyl piperidine for polymerization. The resulting cast thin films (20 μm) exhibited improved membrane properties compared to previously reported terphenyl piperidinium-based AEMs.184 Other strategies include using the thiol–ene click reaction with a hexane-tethered piperidine polymer and a dithiol compound185 and in situ crosslinking of 9-vinyl-carbazole during polymerization with N-methyl piperidine.186
Acid-catalysed polyhydroxyalkylation can be done in a one-pot, metal-free condition at low to room temperature to produce highly conductive and alkaline-stable AEPs. The acid catalyst is tolerant to various monomers with functionality including haloalkyl, amine, and even quaternary ammonium, but the strong acidity and hygroscopic nature of TFSA may restrict the choice of reactors, especially for larger-scale reactions. It is also important to note that successful superelectrophilic activation for high molecular growth of polymers can primarily be achieved by trimethylfluoro-containing carbonyl compounds, with only a few exceptions, which limits the choice for synthesizing non-fluorinated AEPs. Nevertheless, this polymerization method has led to the development of several commercially available products, such as PiperION by Versogen and TM1 by Orion Polymer, demonstrating its practical utility for various applications.
3.2. Metal-promoted coupling reaction
Metal-promoted C–C coupling reactions have been used to prepare phenylene-based polymers carrying quaternary ammonium precursors, such as halides or tertiary amino groups. Classical Ullmann-type condensation reactions, which have been used to prepare polyaromatics utilizing copper catalysts,187 require high reaction temperatures. However, the yield and selectivity are often insufficient for polymer synthesis, even with the most reactive aryl iodide. Among various metal promoters/catalysts for coupling reactions, Ni(0) is the most promising in terms of the reactivity and selectivity for intermolecular C–C coupling reactions or reductive dehalogenative polycondensation, and thus, has been utilized for the formation of high-molecular-weight polyaromatics. Typically, Ni(0) is used as complexes with organic ligands such as cyclooctadiene (cod) and triphenylphosphine (PPh3) because of their stability in inert conditions, easy handling, high reactivity, and good solubility in organic solvents, although they are expensive and require high purity. Ni(cod)2 is one of the most extensively used polymerization promoters as it can be used with aromatic chlorides, which are usually less reactive than bromides and iodides but are more versatile and widely available. In addition to functionalized polyphenylene-based ionomers, quaternized polyaromatics containing heterocyclic groups (e.g., poly(arylimidazoliums)) have also been prepared via Ni(0)-mediated coupling reaction (refer to Scheme 16 in Section 3.4).A general synthetic scheme of polyaromatics used in AEPs, utilizing Ni(0)-promoted coupling reactions, is shown in Scheme 11a.188–203 Typically, pre-aminated, tertiary amino group-containing aromatic monomers and hydrophobic aromatic monomers are copolymerized. The polymerization reaction usually proceeds rapidly and quantitatively within 3–8 hours at 80 °C in polar aprotic solvents such as DMSO and DMAc. During the reaction, Ni(cod)2 undergoes ligand exchange with 2,2′-bipyridine (bpy), which then oxidatively reacts with aryl chlorides to form adducts (1) (Scheme 11b).204,205 The complexes form bisaryl complexes (2) and dichloro complexes (3) through a disproportionation reaction. The former yields the corresponding biaryl as the dimer (4), while the latter provides NiCl2. The successive reaction from (1) to (5) provides precursor polymers (6), which are then quaternized to produce the final AEP products. As the polymerization reaction is based on oxidative addition, and Ni(0) is recovered as Ni(II) or NiCl2, a stoichiometric amount of Ni(cod)2 is required as a promoter, as it does not provide high molecular weight polymers with catalytic amounts. In fact, an excess of Ni(cod)2, often 1.5 to 2 times the equimolar amount, is typically utilized for efficient polymerization due to its susceptibility to water and oxygen in the environment.
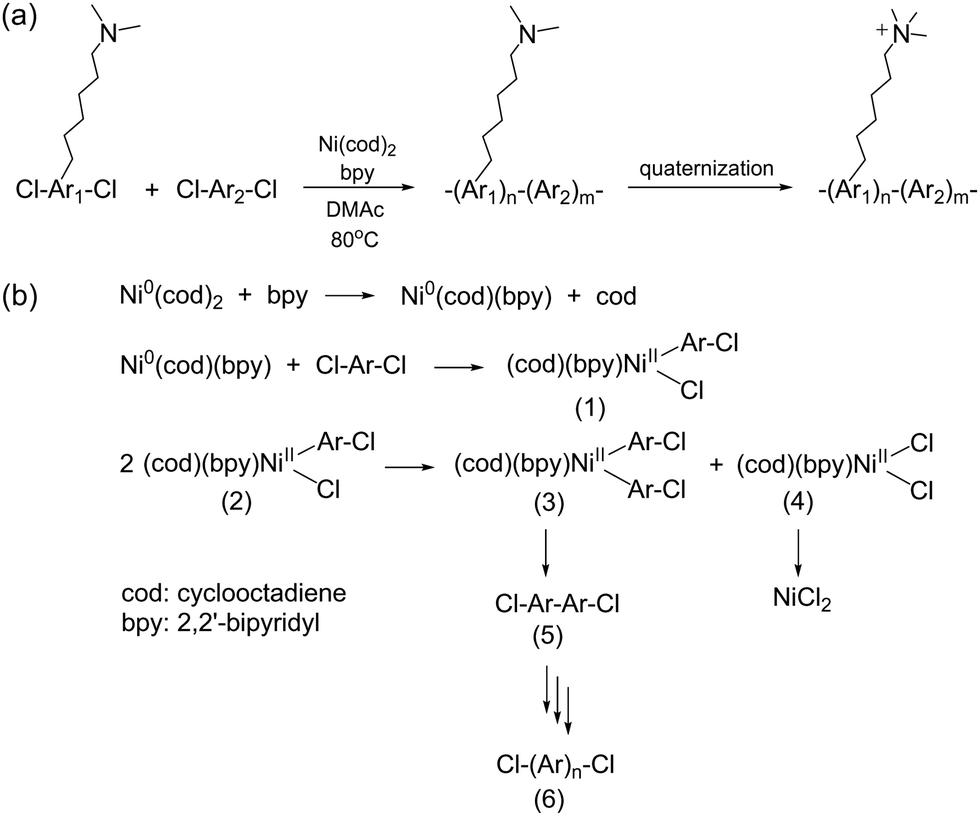 | ||
| Scheme 11 (a) Synthesis of polyaromatic-based AEPs via a Ni(0)-promoted coupling reaction and (b) polymerization mechanism of polyaromatics promoted by Ni(cod)2. | ||
The IEC of the resulting AEPs can be tailored for different applications by adjusting the molar ratio of repeating units. These units are composed of hydrophilic components containing pendent quaternary ammonium groups and hydrophobic aromatic units. AEPs are commonly prepared from tertiary aminated monomers (e.g., containing dimethylaminoalkyl or alicyclic aminoalkyl groups) as precursors for the quaternized ammonium groups. These groups do not interfere with the Ni(0)-promoted aryl halide insertion reactions and remain intact during the polycondensation reactions. The tertiary amino groups are readily quaternized by a typical Menshutkin reaction with alkyl halides. For the methylating reagent, although methyl iodide is most commonly used, dimethyl sulfate is sometimes preferable despite its carcinogenic properties, since trace amounts of iodide ions could remain in the AEPs even after subsequent ion exchange reactions, which may deteriorate some of the favourable properties of the resulting AEMs.
Another approach for producing polyaromatic-based quaternized AEPs involves chloromethylate precursor copolymers that contain electron-rich phenylene rings, followed by a reaction with tertiary amines to introduce quaternary ammonium groups. This method is versatile and has been widely practiced with many polyaromatics, as aromatic groups (without electron-withdrawing groups) are reactive in electrophilic Friedel–Crafts reactions using chloromethyl methyl ether. However, as mentioned in Section 2, the toxic nature and technical problems of using chloromethyl methyl ether make this approach less attractive compared to using tertiary aminated monomers. Alternatively, in situ generation of chloromethyl groups using paraformaldehyde, trimethylchlorosilane and Lewis acid catalyst, such as tin chloride, has been also used for AEP synthesis.206
The molecular weight of the polyaromatic-based precursor copolymers, as measured by gel permeation chromatography (GPC), typically falls within the range of several tens of thousands for Mn and several hundreds of thousands for Mw. These copolymers often have a somewhat higher molecular weight distribution, or polydispersity index (PDI), which is likely due to the relatively rapid polymerization reaction promoted by Ni(0). Typical PDIs are around 4 but can be higher than 10, depending on the reactivity of the monomer and the polymerization conditions. The coupling reaction can produce polymers with sufficiently high molecular weights to afford thin flexible membranes by solution casting. Since the Ni(0)-promoted aryl C–C coupling reaction is very fast and many aryl chloride monomers are highly reactive, the resulting copolymers are randomized rather than sequenced, even when there are considerable differences in the reactivity of the comonomers. Terpolymers composed of two different hydrophobic components, introduced to include additional functionality, have also been investigated.201,203 Furthermore, some semi-block copolymer AEPs have employed pre-synthesized telechelic (chlorine-terminated) hydrophobic oligomers, which were then copolymerized with hydrophilic monomers.207 In most cases, traces of metal catalysts can be easily removed by appropriate workup with mineral acids (e.g., hydrochloric or nitric acid) after the polymerization reaction to the level that does not affect the properties of the resulting membranes and the performance/durability of the devices.
A remaining challenge of using the Ni(0)-promoted coupling reaction for the synthesis of polyaromatic-based AEPs is enabling the polymerization reaction with catalytic, or less than stoichiometric, amounts of costly Ni(cod)2. One report reveals that the amount of Ni(cod)2 could be reduced to half of the conventional amount by using excess bpy ligands.205 Another approach involves using ZnX2, where X = Cl or Br, as a catalyst in the presence of reducing agents (e.g., Zn powder) and ligands (such as bpy or PPh3). In this method, the in situ reduction of Ni(II) to Ni(0) and ligand exchange reactions produce reactive Ni(0) species (Scheme 12). Iodide-containing additives, like NaI or Et4NI, also facilitate the reduction of Ni(II).205 The polymerization is effective for non-ionic monomers, yielding corresponding high molecular weight polyaromatics, but it is less successful with functionalized monomers, such as those that are brominated or tertiary aminated. Employing appropriate protecting groups may prevent side reactions and increase the reactivity of these functional monomers, although the accompanying protecting and deprotecting reactions are cumbersome.
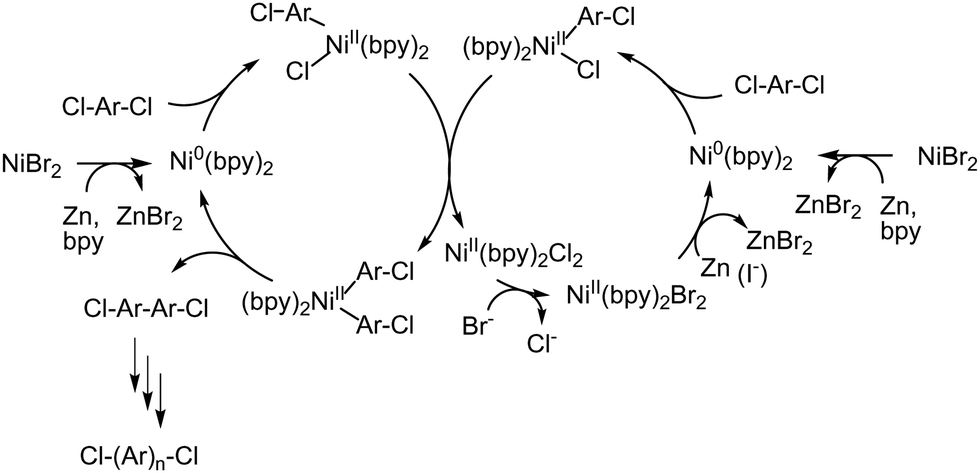 | ||
| Scheme 12 Possible polymerization mechanism for the synthesis of polyphenylenes from dichloroaromatics catalysed by Ni(II) in the presence of Zn as a reducing agent. | ||
A few other metal-promoted polymerization reactions have been developed for AEPs. The most successful and promising one is based on Suzuki coupling using Pd(0) catalyst.162,208,209 The polymerization reaction requires dibrominated and diboronic acid (or in many cases, as pinacol boronate, Bpin) aromatic compounds as reactive monomers. It proceeds quantitatively with a catalytic amount (ca. 2 mol% of the total monomers) of Pd(PPh3)4 under basic conditions. A typical polycondensation reaction shown in Scheme 13 is carried out in toluene at 100 °C for 1.5 to 5 days to provide the pendent brominated precursors (Mw = ca. 30 to 80 kg mol−1, PDI = ca. 2–3). These precursors are then quaternized with tertiary amines, such as trimethylamine and methylpiperidine to obtain desired AEPs with targeted IECs.
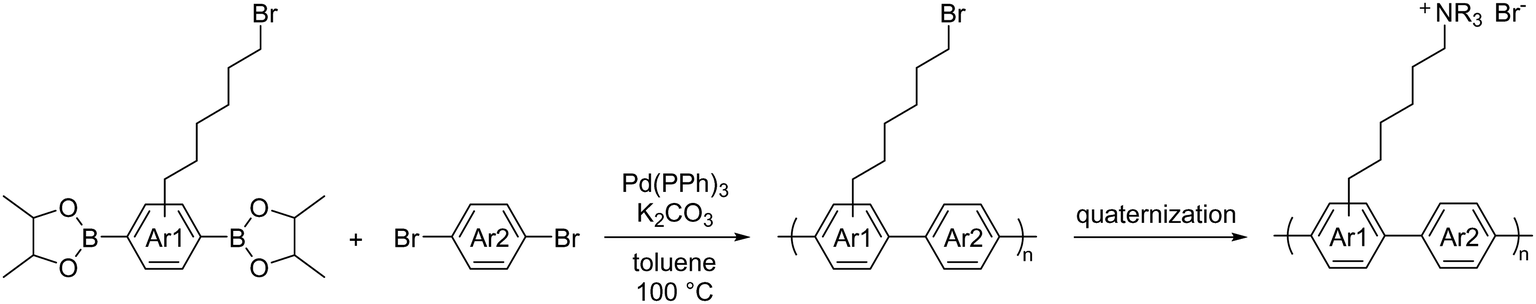 | ||
| Scheme 13 Synthesis of polyaromatic-based AEPs via Suzuki coupling reaction using Pd(0) catalyst. | ||
Fig. 10 lists some recent polyaromatic-based AEPs that have been synthesized via metal-promoted polycondensation reactions, most commonly using Ni(cod)2. Fluorenyl groups are often employed as hydrophobic components and/or scaffolds for pendent ammonium groups since the halogenated fluorenyl monomers are readily available and highly reactive in both Ni(0)- and Pd(0)-promoted polymerization reactions. The hydrophobic groups are often partially fluorinated to augment the differences in hydrophobicity between the units. This approach helps to develop a phase-separated morphology, creating efficient ion transport pathways.188,197
 | ||
| Fig. 10 Selected examples of polyaromatic-based AEPs synthesized via metal promoted polycondensation (PFBFF+ of which structure is not shown is a copolymer of PFB+ and PFF+). | ||
Most polyaromatic-based AEMs exhibit high anion conductivity in water within the temperature range of 20–80 °C. Fig. 11 plots water uptake and hydroxide ion conductivity (both measured at 30 °C in water) as a function of the IEC obtained by titration for recent polyaromatic-based AEMs. These membrane samples feature various main chain and side chain structures, ionic groups, and copolymer/terpolymer compositions. The water uptake demonstrates a roughly linear relationship with the IEC, where two distinct trend lines are observed (Fig. 11a). The trend line with higher slope corresponds to AEMs containing perfluoroalkyl groups (i.e., QPAF-1,188 QPAF-4,192 and BAF-QAF197) as hydrophobic component, while the lower slope line is of those containing alkyl and fluorenyl hydrophobic groups (i.e., PFB+,162 PFF+,162 PFBFF+,162 PFPE,208 and PFPB209). An interesting and counterintuitive finding is that AEPs with more hydrophobic perfluorinated main chains tend to show higher water uptake. The hydroxide ion conductivity of the membranes does not show a strong correlation with the IEC (Fig. 11b). In fact, the highest conductivity (89 mS cm−1) is exhibited by the QPAF-1 membrane, which has a relatively low IEC of 1.33 mequiv. g−1.193 The conductivity appears to be more closely related to the water uptake (Fig. 11c), where two different trend curves are obtained, similar to the water uptake/IEC relationship. Both curves exhibit similar conductivity plateaus when the water uptake exceeds approximately 100%, likely due to a dilution effect.
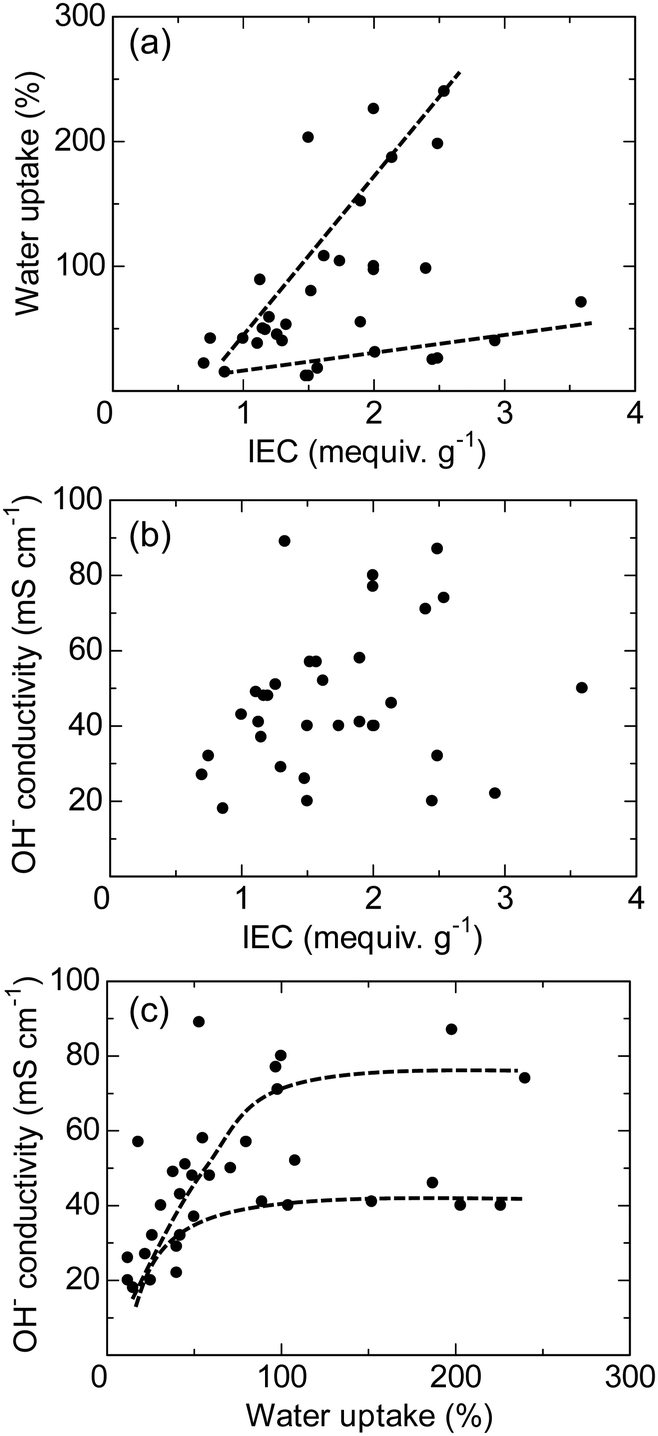 | ||
| Fig. 11 (a) Water uptake, (b) hydroxide conductivity as a function of the IEC, and (c) hydroxide ion conductivity as a function of water uptake for Ni-catalysed polyaromatic-based AEMs (the data were measured at 30 °C).162,188,192,193,197,208,209 | ||
Polyaromatic-based AEMs exhibit Arrhenius-type temperature dependence for hydroxide ion conductivity in water, at least up to 80 °C, reaching conductivity values higher than 160 mS cm−1. The activation energy, estimated from the slopes of the lines, is approximately 10–12 kJ mol−1. Since the membranes absorb large amounts of water, and the absorbed water molecules are predominantly located in the vicinity of the ammonium groups or within the hydrophilic channels, it is reasonable to assume that the hydroxide ions migrate via the Grotthuss mechanism, including the H-bonding between water molecules/hydroxide ions.
The alkaline stability of AEPs synthesized by metal-promoted polycondensation reactions has been evaluated in hot (60–90 °C) and concentrated (1–10 M) alkaline solutions (NaOH or KOH aqueous solution). A negligible decrease in conductivity was observed over one month or even longer under most conditions. However, in more concentrated solutions (e.g., >8 M KOH), the conductivity gradually decreases by up to 20%201 depending on the ammonium structure. Post-structural analyses, including 1H NMR spectra and mechanical properties, suggest that the aryl ether-free polyphenylene backbones prepared by metal-promoted coupling reactions remain stable in alkaline solutions even when the ammonium groups degrade to some extent. Despite the disadvantages, such as the requirement for dichloro-containing monomers, a stoichiometric amount of the Ni catalyst, or the use of an expensive Pd catalyst, metal-promoted polycondensation has been employed as a tool to demonstrate various structures of AEPs with high backbone chemical stability.
3.3. Ionenes by nucleophilic substitution
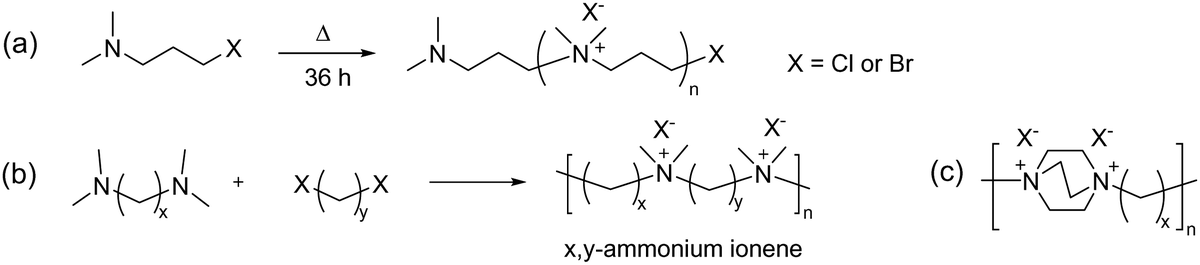 | ||
| Fig. 12 Early examples of ionenes: (a) the first ionene reported in 1933, (b) ionenes using different aliphatic chain lengths of dihalide and diamine monomers, and (c) ionene structure derived from DABCO. | ||
Following the pioneering work of Marvel and Rembaum, a large number of different aliphatic ionenes have been synthesized and studied, although most of these ionenes have not been utilized for membrane applications.213–217 Due to their high chain flexibility and high ionic content, aliphatic ionenes are typically water-soluble and act as polyelectrolytes in solution.213 Hence, in order to be employed as AEMs these ionenes need to be efficiently immobilized by, for example, blending218–223 or through covalent coupling with hydrophobic polymers to form segmented block224 or graft225 copolymers. Besides ammonium ionenes, phosphonium-based ionenes have been prepared through nucleophilic substitution polymerizations of tertiary diphosphines and dibromoalkanes.226 However, these polymerizations are complicated because of the sensitivity of the diphosphites to air and water.214 To date, no membrane properties of this type of ionenes have been reported.
Early work by Müllen et al. demonstrated the synthesis of a spiro-ionene employing benzobis(tetrahydropyrrole), a tricyclic secondary diamine, and 1,2,4,5-tetrabromomethylbenzene in a repetitive Menshutkin-type cyclo-quaternization reaction.228 Because this reaction involves a secondary amine, it is necessary to employ a non-nucleophilic base to deprotonate the secondary amine to form the nucleophile. From the solubility properties, it was inferred that the ionomer had a rod-like shape and exhibited polyelectrolyte effects. Aiming to prepare AEM materials with high alkali stability, Jannasch and co-workers developed a cyclo-quaternization polymerization involving 1,2,4,5-tetrabromomethylbenzene and two different secondary bis-piperidines using N,N-diisopropylethylamine (DIPEA) as the base to produce spiro-ionene 1 and 2, respectively (Fig. 13a).218 Despite the potential side reactions, the cyclization reaction was very efficient, yielding high molecular weight polymer products (Mn = up to 80 kg mol−1) with IECs of 4.0–4.6 mequiv. g−1 in quantitative yields after merely 1–2 hours. The spiro-ionenes were film-forming and thermally stable up to 300 °C, with no degradation detected by 1H NMR analysis after storage in 1 M KOD/D2O at 80 °C. However, signs of degradation through ring-opening elimination and ring-opening substitution (Scheme 4d and e, respectively) emerged when the temperature was raised to 120 °C, with spiro-ionene 2 (10% total ionic loss after 336 hours) being more stable than spiro-ionene 1. The authors hypothesized that the flexible trimethylene bridge of the former ionene greatly facilitates ring relaxation compared to the latter.218 Water-stable AEMs were obtained by blending spiro-ionene 2 with PBI to form ammonium-benzimidazolate complexes for ionic crosslinking under basic conditions (Fig. 13b). A transparent AEM blend containing 70 wt% spiro-ionene 2 gave a hydroxide conductivity of 120 mS cm−1 at 90 °C.218
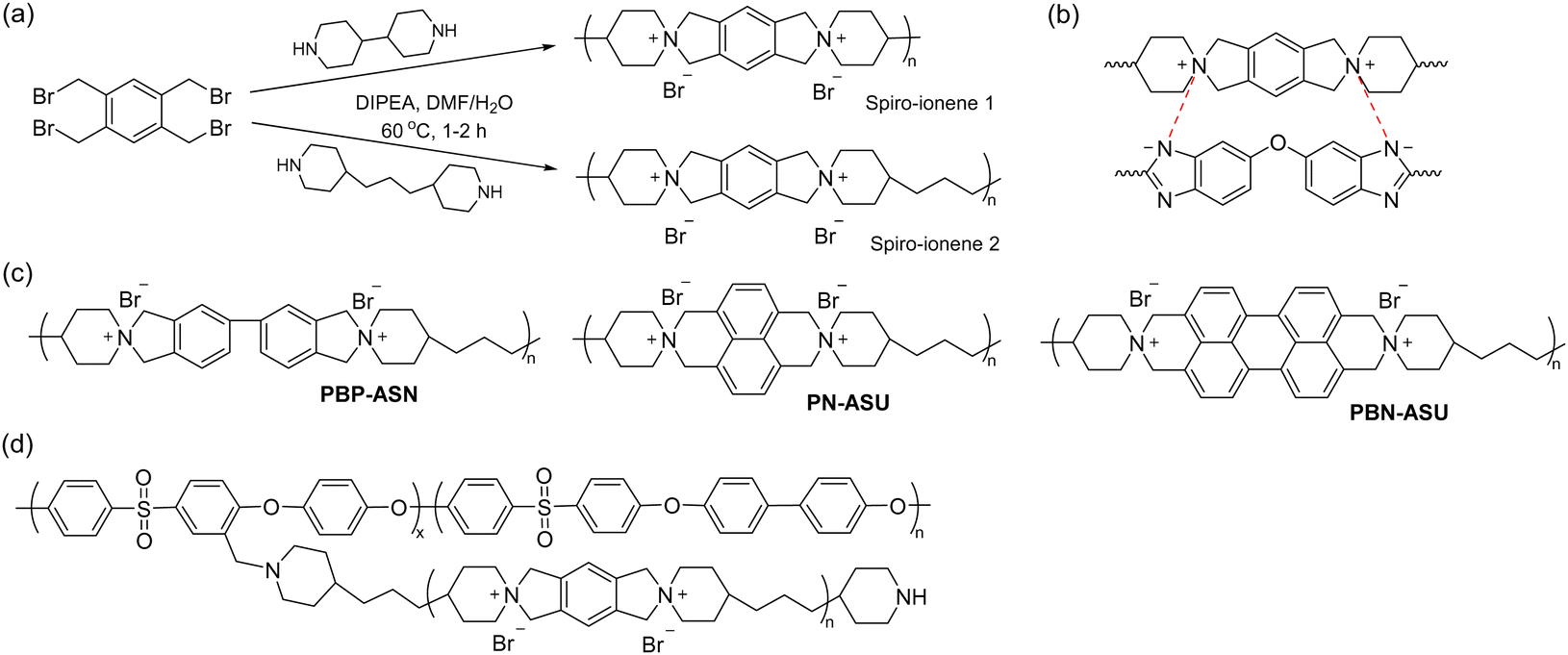 | ||
| Fig. 13 Chemical structures of spirocyclic ionenes used for AEP: (a) spiro-ionenes from bis-piperidines, (b) ionic crosslinking of spiro-ionenes with PBI to form water-stable AEMs, (c) ionenes based on biphenyl, naphthalene and bi-naphthalene, respectively, and (d) the grafting approach of oligomeric ionene to polysulfone for AEM formation. | ||
Li and co-workers prepared a family of ionenes based on biphenyl (PBP-ASN), naphthalene, and bi-naphthalene, respectively (Fig. 13c), and benchmarked the properties against spiro-ionene 2.219 These spiro-ionenes combine 5-/6- and 6-/6-membered rings, respectively, in their spirocyclic arrangement, which are expected to influence the stability of the materials. Model compound testing and DFT calculations indicated that the 5-/6-membered rings, originating from the tetrabenzylbrominated phenyl and biphenyl monomers, were more stable than the corresponding 6-/6-membered rings of the naphthalene-based monomers, probably due to the strongly electron-withdrawing effect of the naphthalene rings.219 Additionally, the naphthalene-based monomers produced ionenes with lower molar masses than those derived from phenyl and biphenyl monomers. Ionically crosslinked blends based on spiro-ionene 2 and PBP-ASN, each containing 20% of a naphthalene-based PBI, were prepared and reached a hydroxide conductivity just above 50 mS cm−1 at 80 °C. After 1000 hours in 5 M NaOH at 80 °C, the blend containing spiro-ionene 2 retained more than 60% of its original conductivity, whereas the blend with PBP-ASN only maintained 40% of its original value, suggesting that spiro-ionene 2 is the more stable ionene.
Jana et al. prepared ionically crosslinked AEM blends of spiro-ionene 2 and a pyridine-containing PBI.220 An AEM (IEC = 2.39 mequiv. g−1) containing 70% of the ionene reached a hydroxide conductivity of 129 mS cm−1 at 90 °C and retained 80% of the conductivity after 500 hours in 2 M KOH at 60 °C. Fu and co-workers employed molecular dynamics (MD) and DFT calculations to study the effect of the degree of deprotonation and backbone flexibility of the PBI component in blends of spiro-ionene 2 and PBI as AEMs. The results showed that electrostatic interactions between the ammonium cations and the deprotonated imidazole play a central role in restricting the water uptake of the AEMs. The electrostatic interactions were found to be enhanced by protonation but weakened by the chain flexibility of the PBI.229 In subsequent work, the same group used MD simulations to study the mechanism of hydroxide conductivity in blend AEMs composed of spiro-ionene 2 and a naphthalene-based PBI.230
Besides blending, water-soluble spiro-ionenes can also be immobilized by grafting onto hydrophobic polymers. Following this strategy, Liu et al. tethered oligomeric spiro-ionene 2 to PES (Fig. 13d).225 First, an oligomeric spiro-ionene 2 terminated with secondary piperidine rings was prepared.218 Next, a benzylbrominated PES was prepared by first conducting a K2CO3 catalysed polycondensation of bis(4-fluorophenyl)sulfone, 4,4′-dihydroxybiphenyl, and methylhydroquinone, followed by a radical-mediated benzylbromination using benzoylperoxide and N-bromosuccinimide. The grafting reaction was subsequently performed in a DMSO solution at 90 °C using DIPEA as the catalyst. AEMs prepared from the graft copolymers retained 84% of the hydroxide conductivity and 86% of their IEC after immersion in 2 M NaOH at 80 °C for 864 hours. NMR analysis revealed that nucleophilic substitution was the dominant degradation mechanism. AFM and TEM analysis showed a distinct microphase-separated morphology. The AEM with IEC = 1.75 mequiv. g−1 exhibited a hydroxide conductivity of 96 mS cm−1 at 80 °C, a water uptake of 33%, and a dimensional swelling ratio of 8%.225
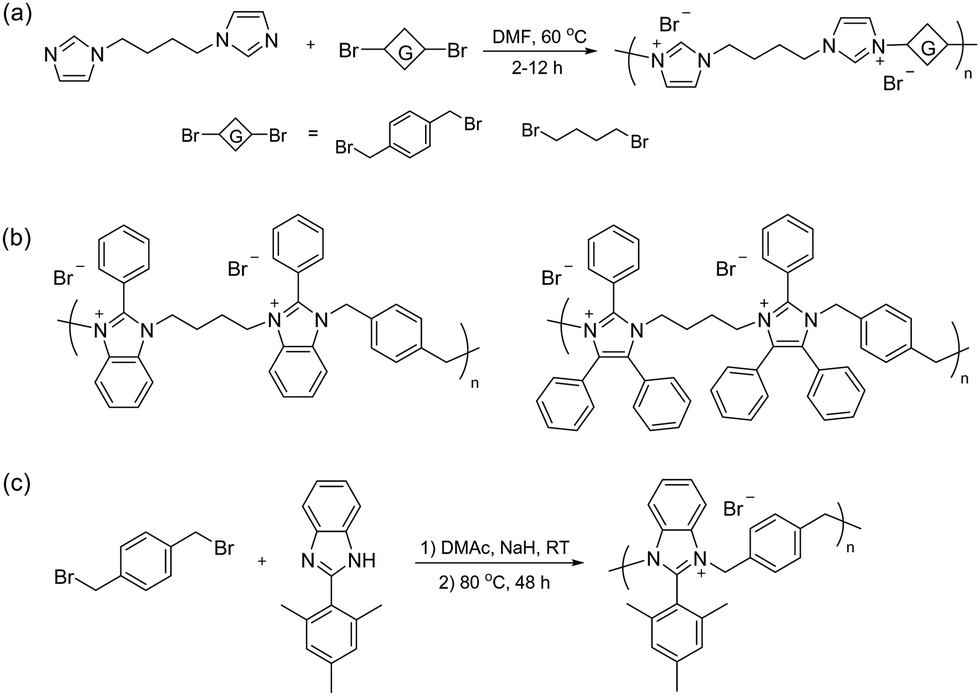 | ||
| Fig. 14 Chemical structures of imidazolium and benzimidazolium-based ionenes: (a) nucleophilic substitution polymerization to prepare imidazolium ionenes, (b) sterically-protected phenyl-substituted benzimidazolium and imidazolium ionenes, and (c) sterically-protected benzimidazolium ionene. | ||
Holdcroft et al.,233 and later Coates and co-workers,234 have shown that imidazolium and benzimidazolium cations can be efficiently sterically protected from hydroxide attack by appropriate substitution, especially by incorporating bulky groups at the C2 position. Using butyl-bridged bis-(2-phenylbenzimidazole) and bis-(2,4,5-triphenylimidazole), Yang et al. carried out nucleophilic substitution polymerizations with 1,4-dibromomethylbenzene to prepare phenyl-substituted benzimidazolium and imidazolium ionenes, respectively (Fig. 14b).221 These ionenes were then blended with PBI to produce water-stable and mechanically strong AEMs with IECs in the range of 1.0–1.5 mequiv. g−1, water uptake of 39–57%, and area dimensional swelling of 14–17% at 80 °C. A triphenylimidazolium-based AEM with an IEC of 1.5 mequiv. g−1 achieved a hydroxide conductivity of 52 mS cm−1 at 80 °C. Alkali-stability testing in 1 M KOH at 80 °C showed that the imidazolium-containing AEMs retained conductivity better than the benzimidazolium-based AEMs. However, all the AEMs suffered from a significant conductivity loss during the first 200 hours. 1H NMR analysis revealed that the benzimidazolium ionene had degraded (as will be discussed later in Section 3.4.1), while the corresponding imidazolium ionene was much more stable.
Henkensmeier and co-workers synthesized a sterically-protected benzimidazolium ionene by polymerizing 2-(2,4,6-trimethylphenyl)benzimidazole and 1,4-dibromomethylbenzene by first deprotonating the benzimidazole using NaH, and then adding the dibromo monomer (Fig. 14c).223 The former monomer was obtained by reacting 2,4,6-trimethylbenzoic acid and 1,2-diaminobenzene in polyphosphoric acid at 200 °C.233 However, the resulting ionene was not film-forming, likely due to its moderate molecular weight (Mw = 40 kg mol−1) and the highly rigid backbone structure. Self-supporting blend AEMs were instead obtained after co-casting with PBI, which reduced the IEC from 2.67 mequiv. g−1 for the neat ionene, to 1.33–1.78 mequiv. g−1 for the blend AEMs. The water uptake and conductivity of the blend AEMs ranged from 32–49% and 0.35–0.64 mS cm−1 at 60 °C, respectively, in the Cl− form. The AEMs were subsequently evaluated in vanadium RFB, and hence the hydroxide conductivity and alkali stability of these materials were not evaluated.
Ionenes synthesized by nucleophilic substitution have emerged as an important class of AEPs. Depending on the structure of quaternary ammonium, an ionene can exhibit enhanced alkaline stability, as shown for the spiro-ionene structures. However, the location of the anion exchange groups in the backbone and the high-water uptake make these materials less ideal for membrane applications as free-standing films. Consequently, various blending strategies, inducing ionic crosslinking, have been developed to improve membrane stability and evaluate ionene-based AEPs in electrochemical devices.
3.4. Polybenzimidazoliums and polyimidazoliums
 | ||
| Scheme 14 (a) Deprotonation and alkylation of m-PBI, (b) alkylation to PDMBI, and (c) the degradation route of dimethylbenzimidazolium hydroxide. | ||
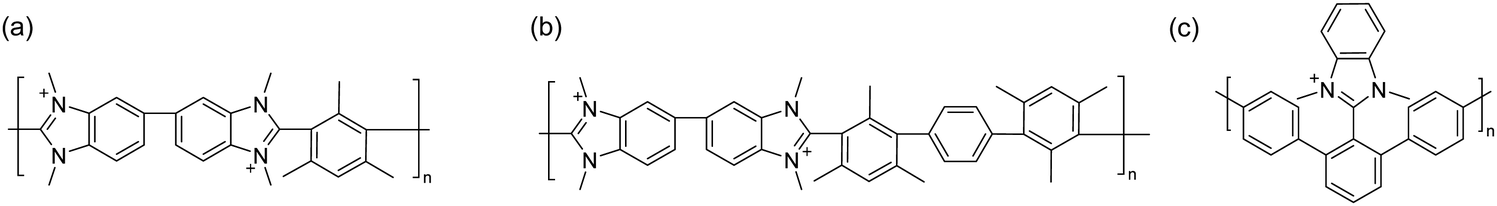 | ||
| Fig. 15 Structures of C2-phenyl-substituted benzimidazolium polymers: (a) mesitylene-polydimethylbenzimidazolium (Mes-PDMBI); (b) hexamethyl-p-terphenylene-polydimethylbenzimidazolium (HMT-PDMBI); and (c) polyphenylene benzimidazolium (PPMB). | ||
Given that the methyl groups attached to the C2 position of mesitylene (Fig. 15b) are paramount to stabilizing PBI, it was speculated that bulkier groups could provide even greater protection against hydroxide attack. Consequently, the methyl groups were replaced with phenyl groups to instil even greater steric hindrance around the C2-positon (Fig. 15c).247,250 These m-terphenyl-protected benzimidazolium AEMs, poly(4,4′′-[2′-(1,3-dimethyl-1H-benzimidazolium-2-yl)-m-terphenylene]) (PPMB), are reported to exhibit an exceptional half-lifetime stability of more than 3000 hours in 3 M NaOH at 80 °C. This enhanced stability is believed to be due to the suppression of ring-opening by hydroxide ion attack at the C2 position.
 | ||
| Scheme 15 Synthesis of hexamethylterphenylene polyimidazolium (HMT-PMPI). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours) than their methyl derivatives, but to the detriment of reduced IEC and dilution of the charge carriers. A reduction in charge carrier concentration can be compensated by introducing bis-imidazolium cations into the polymer, which can achieve theoretical IECs as high as 3.0 mequiv. g−1 (i.e., when fully methylated).253 Such sterically-protected poly(arylimidazolium) AEPs were prepared by Yamamoto-coupling polymerization (the Ni-mediated coupling reaction discussed in Section 3.2) of dichloro-imidazole monomers as shown in Scheme 16a. This synthetic route is substantially different from the previously reported poly(arylimidazolium) AEPs, which were based on bis-diketone/dialdehyde/ammonium polycondensation. The biaryl-coupling polymerization route typically results in low molecular weight polymers because of the limited solubility of highly rigid aromatic backbones. However, the adopted synthetic strategy allows for the preparation of an intermediary polymer in its semi-alkylated form, which exhibited much higher solubility and resulted in high molecular weight poly(arylimidazole)s (Mw = 140 kg mol−1). N-Alkylation of the polymers was achieved by reacting them with various alkylating reagents to produce different poly(arylimidazolium) AEPs. These sterically-protected poly(bis-arylimidazolium) polymers demonstrated high stability in caustic solutions; for example, a butylated derivative is reported to exhibit a half-life exceeding 8000 hours in 10 M KOH at 80 °C.253
000 hours) than their methyl derivatives, but to the detriment of reduced IEC and dilution of the charge carriers. A reduction in charge carrier concentration can be compensated by introducing bis-imidazolium cations into the polymer, which can achieve theoretical IECs as high as 3.0 mequiv. g−1 (i.e., when fully methylated).253 Such sterically-protected poly(arylimidazolium) AEPs were prepared by Yamamoto-coupling polymerization (the Ni-mediated coupling reaction discussed in Section 3.2) of dichloro-imidazole monomers as shown in Scheme 16a. This synthetic route is substantially different from the previously reported poly(arylimidazolium) AEPs, which were based on bis-diketone/dialdehyde/ammonium polycondensation. The biaryl-coupling polymerization route typically results in low molecular weight polymers because of the limited solubility of highly rigid aromatic backbones. However, the adopted synthetic strategy allows for the preparation of an intermediary polymer in its semi-alkylated form, which exhibited much higher solubility and resulted in high molecular weight poly(arylimidazole)s (Mw = 140 kg mol−1). N-Alkylation of the polymers was achieved by reacting them with various alkylating reagents to produce different poly(arylimidazolium) AEPs. These sterically-protected poly(bis-arylimidazolium) polymers demonstrated high stability in caustic solutions; for example, a butylated derivative is reported to exhibit a half-life exceeding 8000 hours in 10 M KOH at 80 °C.253
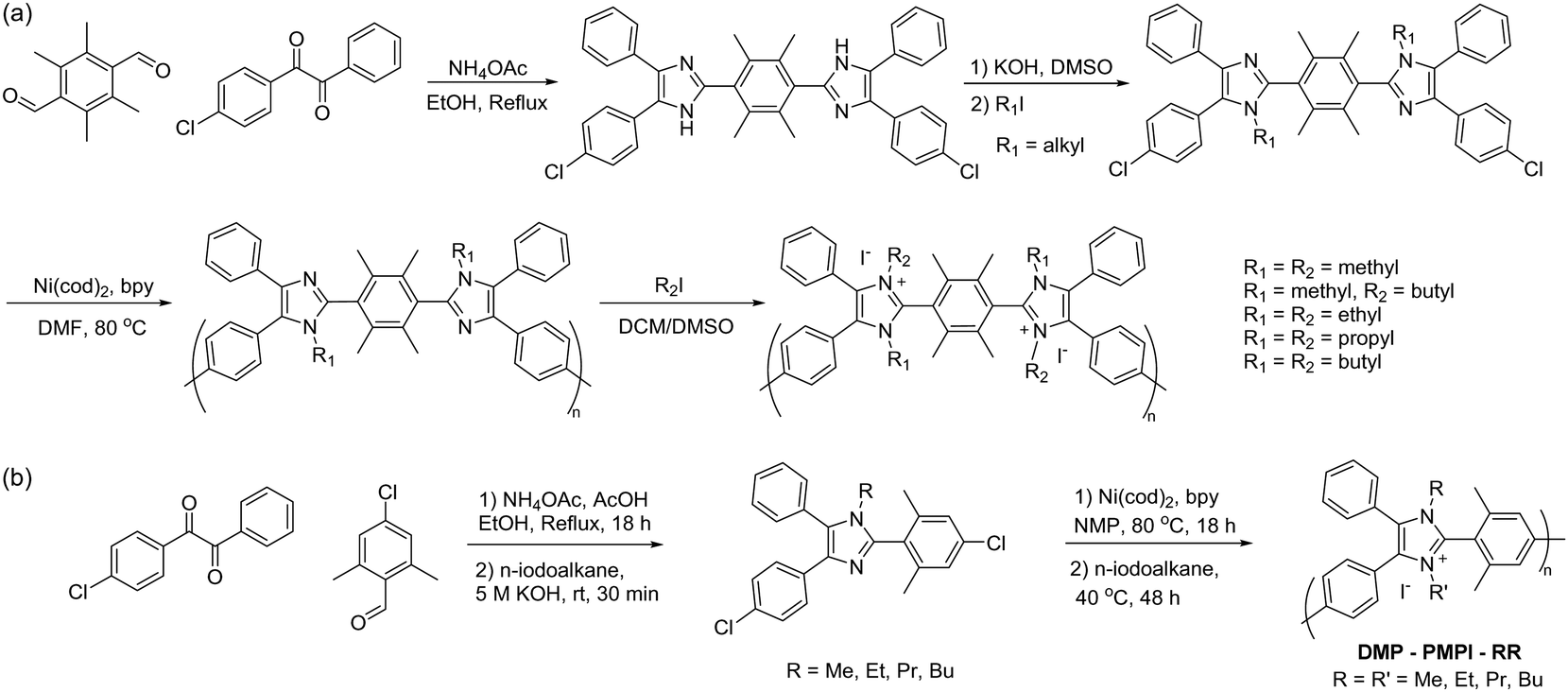 | ||
| Scheme 16 (a) Synthesis of C2-protected poly(bisarylimidazolium) AEPs (PAIM). (b) Synthesis of aryl imidazole monomers and the poly(arylimidazolium) (DMP-PMPI-RR) having N,N′-dialkyl functionality. | ||
Having established a viable route for designing robust poly(imidazolium) AEPs through C2-protection of the imidazolium, it became paramount to be able to tune these polymers for specific properties. The water content, or more specifically, the number of water molecules per ion, affects every physicochemical property of an AEM.215,254 To achieve higher ionic conductivity, a high ionic content of AEMs is considered necessary. However, increasing the IEC enhances the hydrophilicity and water content, which in turn induces significant changes in AEM properties due to osmotic pressure, such as deleterious volumetric and morphological transformations. Furthermore, reduced solvation of hydroxide ions under low hydration conditions decreases the shielding of their negative charge, promoting mechanisms of nucleophilic attack. Consequently, water content is a critical parameter in considering the electrochemical applications of AEMs,255,256 and a deeper understanding of the structure–property relationships is required to fully comprehend the influence of the molecular structure of polyimidazolium ionenes on water content and the effect of water. Due to the rapidly rising interest in this class of polymers, attention to the complex interplay between ionic content and conductivity/hydration relationships is still in the earlier stages of investigation.
A pioneering case study, published in 2020, presented a cross-correlation of ionic conductivity and water uptake in poly(arylimidazolium) ionenes. These polymers were prepared by Yamamoto coupling of penta-substituted imidazolium monomers having steric protection at the imidazolium C2-position (Scheme 16b).257 The monomers having one N-alkyl function were synthesized in several steps. N-Alkylation occurs at either of the two nitrogen sites, resulting in the monomer being obtained as a mixture of the two structural isomers. Consequently, polymerizations yield polymers with mixed repeat unit configurations forming either of three possible configurations. The ionic conductivities of these poly(imidazolium) membranes vary significantly with IEC, and this trend holds regardless of repeat unit structure and coupling configuration. Polymerization and quaternization are reported to be near-quantitative in yield, but the conversion of the second N-alkylation decreases with increasing alkyl chain length, consistent with the general trend of decreasing reactivity with increasing alkyl chain length of n-haloalkanes in quaternization reactions. This leads to a decreasing degree of quaternization and decreasing ionic content with increasing alkyl chain length. AEMs in their hydroxide counterion forms convert to their mixed carbonate form upon exposure to air containing CO2. Therefore, the cross-correlation of water uptake and ionic conductivity was presented using the chloride form, which remained stable in the air. The observed trends of σ(Cl) ∼ IEC for these polymers are in agreement with those of comparable poly(arylimidazolium)s. AEMs with higher IEC exhibit a lower range of ion conductivity (σ) and hydration number (λ) within certain limits of temperature and humidity. Decreasing the IEC correlates with an increase in the effective activation energy of ion transport at a constant λ, leading to the conclusion that AEMs with lower IEC are more dependent on environmental conditions, such as RH and temperature.
![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n) of AB-PAI polymers, as n is often crucial for the effective processing of polymers. Advantageously, it was found that steric encumberment of the imidazole C2 introduced in the AB-type monomer did not inhibit polycondensation. Partially quaternized, primary chain AB-PAI polyelectrolytes, such as ionenes, have zwitterionic imidazolate-imidazolium character, owing to the conjugate base of imidazole (pKa ≈ 7). Therefore, AB-PAI polymers and their derivatives could be useful in complex and heterogeneous ion transport applications.
n) of AB-PAI polymers, as n is often crucial for the effective processing of polymers. Advantageously, it was found that steric encumberment of the imidazole C2 introduced in the AB-type monomer did not inhibit polycondensation. Partially quaternized, primary chain AB-PAI polyelectrolytes, such as ionenes, have zwitterionic imidazolate-imidazolium character, owing to the conjugate base of imidazole (pKa ≈ 7). Therefore, AB-PAI polymers and their derivatives could be useful in complex and heterogeneous ion transport applications.
 | ||
| Scheme 17 First instance of AB polymerization to synthesize poly(aryleneimidazolium) (AB-PAI), demonstrating molecular weight control and end group functionalization. | ||
3.5. Diels–Alder polymerization
Diels–Alder polymerization was first employed to develop ion-conducting polymers for fuel cell applications by Sandia National Laboratories in 2001. Polyphenylene composed of para- or meta-linkages of phenylene units can be synthesized by [4+2] Diels–Alder cycloaddition of a bis-diene (such as bistetracyclone, BTC) and a bis-dienophile (1,4-diethynylbenzene, DEB), reported by Frietag,267 Ried,268 and Stille et al.269–272 The mechanism of this reaction is shown in Scheme 18; depending on the diene and dienophile used, either para- or meta-linkages are generated. For instance, a para-linkage is formed when R2 of the diene is on the opposite side of R3 of the approaching dienophile. The [4+2] cycloaddition forms a carbonyl-bridged intermediate that subsequently extrudes carbon monoxide, driven by the formation of a more energetically stable aromatic centre. The resultant polymer contains pendent phenyl rings and a mixture of backbone para/meta-linkages that inhibit molecular weight limiting π–π interactions,273 leading to good solubility in organic solvents270 and favourable mechanical, film-forming properties.274,275 Proton conducting membranes have been developed by sulfonating the Diels–Alder poly(phenylene)(DAPP) backbone, controlling the ratio of chlorosulfonic acid to the polymer repeat units.276 | ||
| Scheme 18 Two possible regioisomers of the [4+2] Diels–Alder polycondensation of bis(tetracyclone) and p-diethynylbenzene. | ||
In the early 2000s, no alkaline-stable AEMs had been reported; the primary degradation mode was believed to be hydroxide nucleophilic attack of the cationic ammonium group, either by Hofmann elimination or direct displacement, as discussed in Section 2.3.277–279 Hibbs et al. developed the first example of an AEM with the DAPP backbone by polymerizing a methylated bis(tetracyclone) (tetramethylbis(cyclopentadienone), TMBTC) with DEB (Scheme 19).280 The pendent methyl groups were brominated via radical bromination, and then the bromo functional groups were nucleophilically replaced by trimethylammonium via the Menshutkin reaction using trimethylamine, forming benzyltrimethyl ammonium-substituted DAPP (TMA-MDAPP). This conversion was carried out heterogeneously by immersing cast films of bromomethylated DAPP in concentrated aqueous trimethylamine at room temperature for 48 hours. However, the values of theoretical IEC, calculated by 1H NMR of bromomethyl groups, and the experimental IEC via titration were disproportionate, with the titrated values being 25–45% lower than theoretical ones. The lower-than-expected titrated IEC was attributed to incomplete nucleophilic substitution due to the slow heterogeneous reaction between the bromomethyl groups and trimethylamine, with the remaining bromomethyl groups converting to hydroxymethyl groups over time and exposure to humid environments. The highest IEC reported for TMA-MDAPP was 1.57 mequiv. g−1 (films with a higher targeted IEC dissolved in the trimethylamine bath), exhibiting a water uptake of 120% and hydroxide conductivity of 50 mS cm−1 at room temperature, comparable to the highest values reported for AEMs at that time. Hibbs compared the durability of PES-based AEM and TMA-MDAPP in 4 M NaOH at 60 °C for 30 days, reporting that the IEC and mechanical properties of TMA-MDAPP did not change over the course, whereas the PES AEM became extremely brittle.
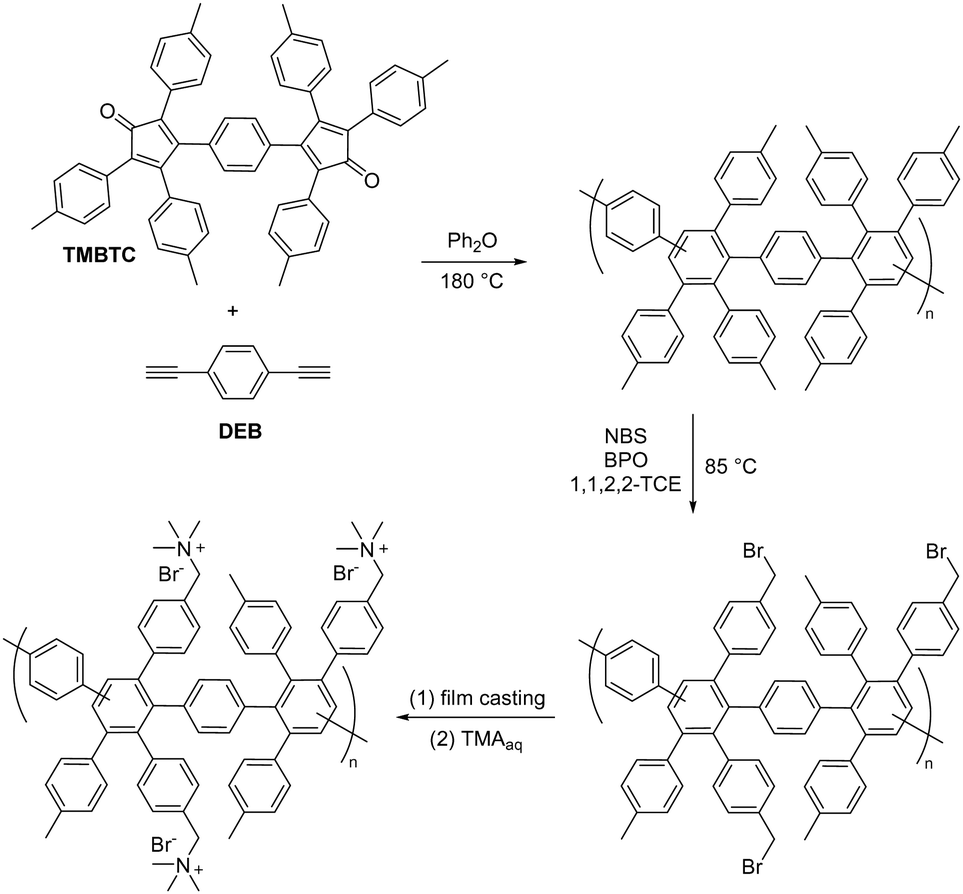 | ||
| Scheme 19 Synthesis scheme of DAPP AEM functionalized with BTMA groups. | ||
The BTMA group is unstable above 60 °C under alkaline conditions, leading to efforts to improve the stability of organic cations by examining resonance stabilized guanidinium281 and imidazolium282 or sterically bulky phosphonium cation.283 However, a novel approach was demonstrated in the mid-1990s by Tomoi, where increasing the separation between the aromatic backbone and ammonium group by methylene spacers (more than 3 carbons) led to improved alkaline stability.284 Building on this, Hibbs sought to combine the alkaline-stable DAPP backbone with this long-chain spacer approach to further optimize alkaline stability.285 The first step began with the Friedel–Crafts acylation of DAPP with bromohexanoyl chloride, to form DAPP with attached 6-bromo-1-phenylhexan-1-one. Since the attached ketone could react with hydroxide either by enolate formation (due to the acidic α-proton of ketones) or by direct nucleophilic attack of the ketone to form a geminal diol, it was reduced to a methylene group. The resultant bromohexyl functionalized DAPP was cast as films and immersed in aqueous trimethylamine to form trimethylammonium hexyl DAPP (TMAH-DAPP, Scheme 20). As observed with TMA-MDAPP, the measured IEC of TMAH-DAPP was slightly lower than theoretical values, likely due to the incomplete heterogeneous conversion of the hexylbromo groups. The stability of TMAH-DAPP was monitored by immersing the films in 4 M KOH at 90 °C for two weeks. Periodically, the film was removed from the bath, and chloride ions were exchanged to measure chloride conductivity and then re-immersed in the alkali solution. After one week, the chloride conductivity of TMAH-DAPP reduced by 5%, but did not change during the second week, suggesting that the change in conductivity was due to the reorganization of the hydrophilic/hydrophobic domains, rather than to cation group degradation. This conclusion was further supported since the IEC of TMAH-DAPP remained unchanged throughout the experiment.286
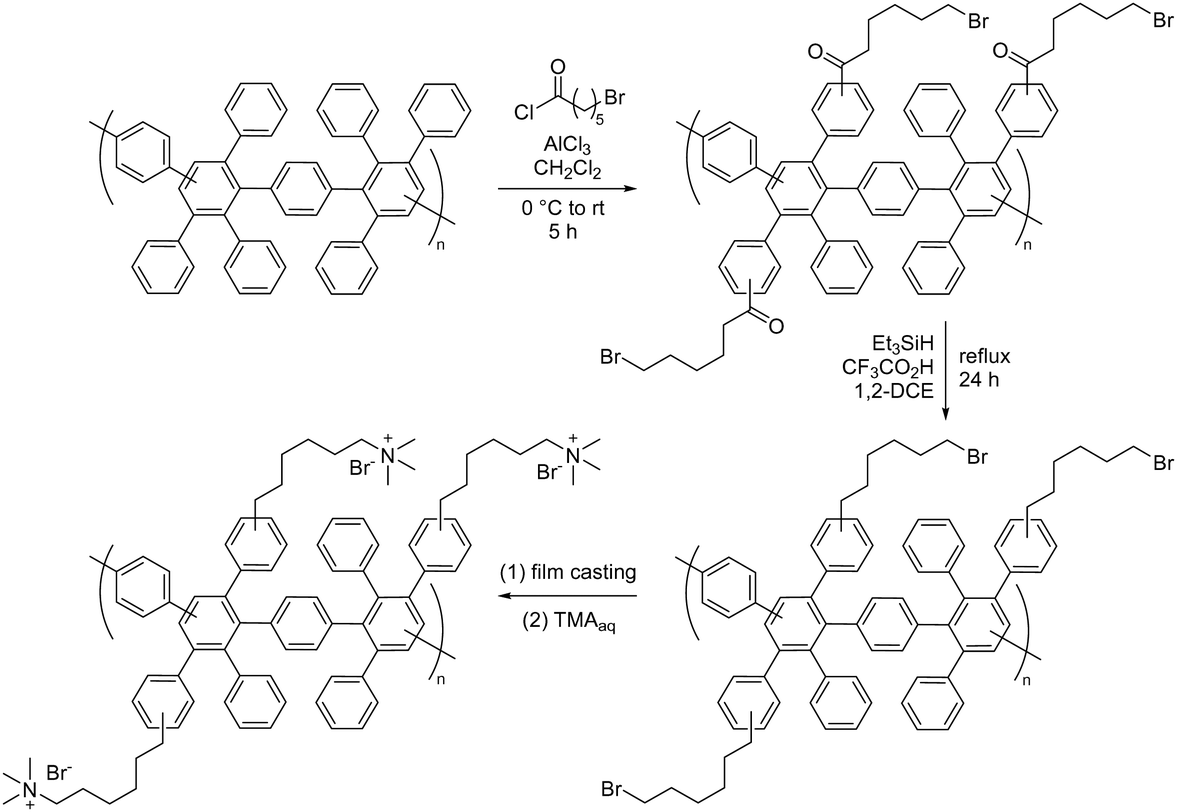 | ||
| Scheme 20 Synthesis of TMAH-DAPP AEMs. | ||
The functionalization of DAPP has focused on inducing the formation of hydrophilic ionic channels through the aggregation of neighbouring ionic functional groups attached to the pendent aryl rings. A novel approach in ionomeric DAPP materials involves controlling the position of the ionic groups on the DAPP backbone. This is achieved by attaching a strong electron-withdrawing moiety, such as pentafluorobenzoyl, to the backbone to inhibit electrophilic aromatic substitution reaction (i.e., sulfonation) on the pendent aryl rings.137 Incorporating fluorine into the backbone serves as an internal reference, helping to determine the degree of functionalization with pentafluorobenzoyl groups using 19F NMR spectroscopy. The polymer was functionalized to attach up to six pentafluorobenzoyl groups, followed by sulfonation on the backbone to induce ionic aggregation.
Compared to other quaternized polyaromatic polymers, quaternized DAPPs exhibit higher gas permeability is desirable for ionomeric binders, but it is less advantageous for AEM applications, as increased gas crossover can lead to higher overpotential in the device. Additionally, the bulky structure of these polymers hinders chain entanglement, resulting in poor mechanical properties at a given molecular weight. However, quaternized DAPPs also offer distinctive advantages, including all-phenylene and non-ionic backbones, good processibility, chemical robustness, and a wide range of IECs.
4. Chemistry of vinyl polymers
Vinyl polymers have a long history as commodity polymers, displaying excellent chemical stability stemming from their fully saturated backbone. This section explores various synthetic strategies for preparing polyolefin-based AEPs.4.1. Addition polymerization
Addition polymers, broadly speaking, encompass polymers obtained through a chain-growth mechanism, forming a structure where the repeat unit contains the same atoms as the monomer. The range of vinyl monomers that can be polymerized via vinyl-addition polymerization is exclusively limited. Many cyclic olefins necessitate highly reactive catalysts, often accompanied by co-catalysts and activators for polymerization. Moreover, these catalysts often have poor functional group tolerance.287 Only a select number of cyclic olefins, such as norbornene and cyclopentene, have been identified as monomers capable of forming functional polymers. In the context of AEP synthesis, addition polymerization has primarily been reported for the synthesis of quaternized polynorbornene.Norbornene, a reactive bicyclic olefin with high ring strain, has the capability to polymerize through three independent pathways, resulting in different norbornene-based polymers: ring-opening metathesis polymerization (ROMP), isomerization polymerization, and addition polymerization.288–290 ROMP, the most well-known method for polymerizing norbornene, will be discussed in detail in Section 4.2. Isomerization polymerization of norbornene can be catalysed by radical, cationic, and anionic initiators,291 but it has been scantily studied due to the low molecular weight and poorly defined structures of the resulting polymers. Vinyl-addition or addition polymerization of norbornene (Scheme 21), first reported in the 1960s,292 produces polymer backbones with a bicyclic structure by only opening the double bond of the vinyl group, which is distinct from polyolefins generated from ROMP. Although the polymerization reactivity is lower than that of ROMP,293 addition polymerization can grow norbornene into high molecular weight polymers, featuring rigid and fully saturated backbones. Catalysed by late transition metal catalysts including Ti, Zr, Co, Cr, Ni, and Pd,294 the polymers composed of polyolefin backbones are known for their chemical inertness, good mechanical strength, and high thermal stability, including an extremely high Tg (norbornene homopolymer is >300 °C).
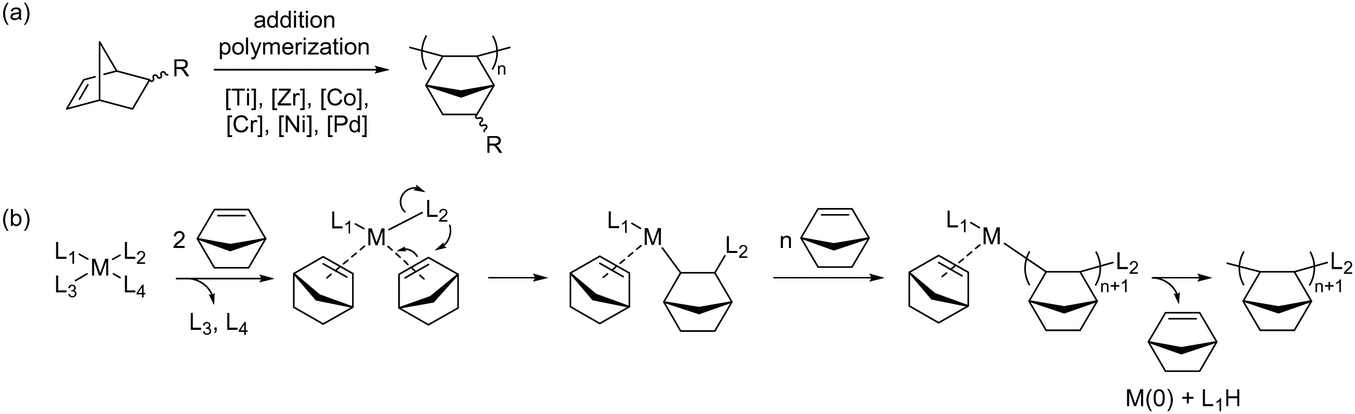 | ||
| Scheme 21 (a) Vinyl-addition polymerization of functional norbornene using transition metal catalysts and (b) suggested mechanism of norbornene polymerization using a metal catalyst. | ||
The known disadvantages of polynorbornenes are their high melting points, poor solubility in organic solvents, and poor adhesion. To increase thermal processability, norbornene is often copolymerized with other alkene monomers, e.g., ethene or propene. Its adhesion properties can be enhanced by polar group substitution, such as with triethoxylsilane,295 making it suitable for use in discs, films, fibres, binders, packaging, and optical applications.296
Scheme 21b illustrates the suggested mechanism for the addition polymerization of norbornene.297,298 The norbornene monomers initially coordinate with the metal catalyst, replacing weakly coordinating labile ligands (L3, L4), forming a complex. Subsequently, norbornene insertion occurs for chain initiation. Chain propagation then proceeds to increase the molecular weight of the polymer, and the polymerization terminates via the reductive elimination of the catalyst. Catalysts based on nickel and palladium exhibit the highest reactivity towards addition polymerization and the highest tolerance to substituted norbornene monomers, making them the most employed catalysts.289,290
The post-polymerization modification of substituted alkyl halides with tertiary amines via the Menshutkin reaction is currently the most applied method to introduce anion exchange groups to the polynorbornene prepared by addition polymerization. Alkyl halide (either Cl or Br)-substituted norbornenes can be synthesized by Diels–Alder cycloaddition reactions of cyclopentadiene and terminal 1-haloalkenes as a mixture of endo (major) and exo (minor) isomers in a ratio of endo:exo = 85:15 (Scheme 22).299 A variety of catalysts (e.g., bis(β-ketonaphthylamino) Ni/B(C6F5)3,300 (η3-allyl)Pd(PPh3)Cl/Li[FABA],301 Pd(t-Bu3P)MeCl,302 and trans-[Ni(C6F5)2(SbPh3)2]303) have been successfully used to polymerize alkyl halide-substituted norbornene, producing copolymers in combination with other norbornene monomers, which were converted into anion exchange groups. Very recently, a direct coordination–insertion polymerization of cationic norbornene monomers has been reported, using the Pd(t-Bu3P)MeCl catalyst.304
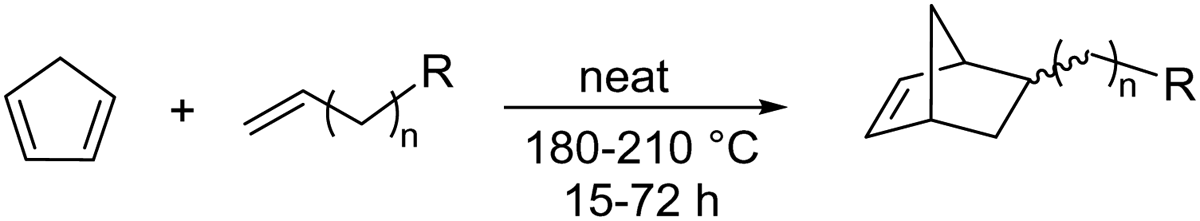 | ||
| Scheme 22 Substituted norbornene monomer synthesis. | ||
Addition polymerization was initially employed in the synthesis of AEPs in 2015 when He and co-workers reported quaternary ammonium-containing addition-type norbornene copolymers for direct methanol fuel cells (Fig. 16a).300 For norbornene monomers, alkyl- and bromoalkyl-substituted 5-norbornene-2-methanol was used for copolymerization with a Ni catalyst. The polymers underwent functionalization via chloromethylation using chloromethyl methyl ether and were quaternized using tertiary amines of varying chain lengths. The resulting AEM demonstrated an IEC of 1.8 mequiv. g−1 and low dimensional swelling of 0.9–3.3%, but the anion conductivity at 80 °C was only 4.1 mS cm−1.
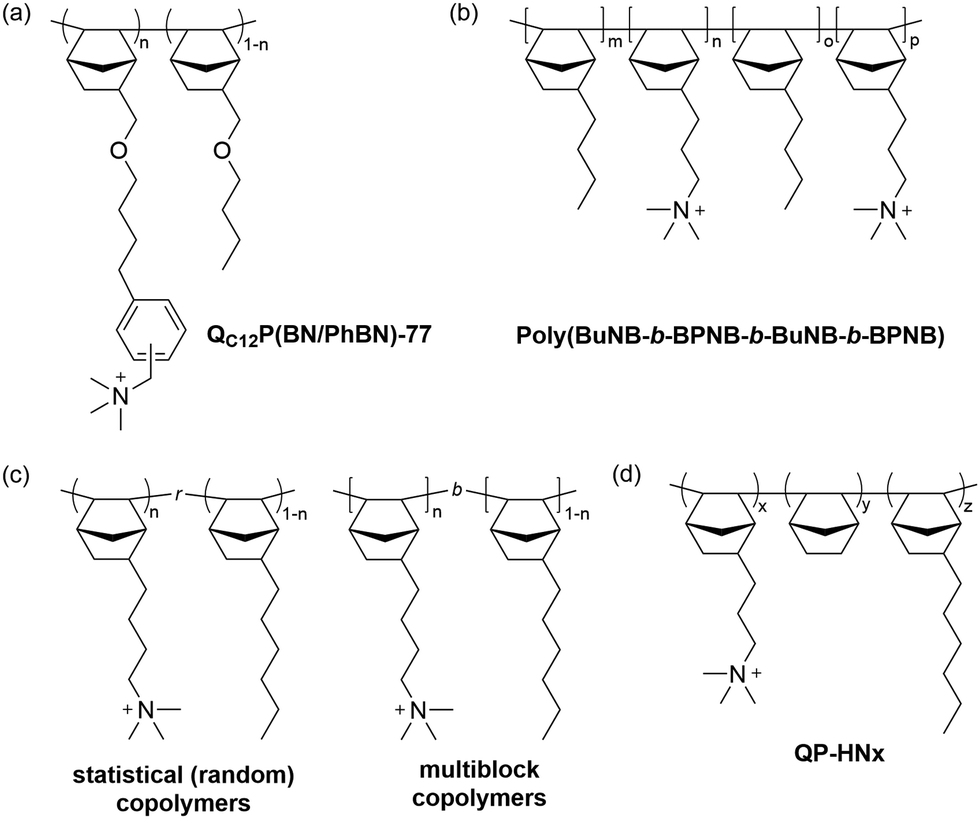 | ||
| Fig. 16 Selected examples of polynorbornene AEPs using addition polymerization: (a) quaternary ammonium-functionalized polynorbornene reported in 2015, (b) tetrablock copolymers of polynorbornene, (c) statistical copolymers synthesized by one-pot addition polymerization and multiblock copolymers made via sequential addition, and (d) solvent-processable random copolymers of polynorbornene. | ||
Kohl et al. synthesized tetrablock copolymers of polynorbornene through the sequential addition of butylnorbornene and bromopropylnorbornene utilizing a palladium catalyst with an activator (Fig. 16b).301 The bromopropyl-containing tetrablock copolymer was cast as a film and quaternized by immersion in an aqueous trimethylamine solution. The Mn of the polymers was from 38 to 115 kg mol−1, as determined by GPC. The molecular weight growth of each block was also monitored to confirm the sequential growth of block copolymers. The AEMs showed phase-separated morphology confirmed by SAXS and TEM, attributed to the hydrophobic–hydrophilic block structure. The AEMs with the IEC value of 1.9 mequiv. g−1 and the highest molecular weight (Mn = 115 kg mol−1) showed the highest hydroxide conductivity of 122.7 mS cm−1 at 80 °C, higher than that of the AEM with higher IEC (2.6 mequiv. g−1, σ80°C = 80 mS cm−1, Mn = 45 kg mol−1), likely due to the higher molecular weight causing lower water uptake and better chain entanglement for higher hydroxide mobility.301 The polynorbornene AEMs exhibited high alkaline stability, showing minimal sign of a change of ionic conductivity (less than 1%) observed over 1400 hours after immersion in 1 M NaOH at 80 °C, demonstrating the high potential of this polyolefinic AEMs for alkaline electrochemical devices.
As demonstrated, based on how monomers are introduced to the polymerization reaction, either one-pot or sequentially, addition-polymerized polynorbornene can form either random (statistical) or multiblock copolymers (Fig. 16c). In addition to the tetrablock copolymers, in a follow-up study Kohl et al. prepared AEMs based on homopolymer and random copolymers of polynorbornene with similar IECs (3.5–4.5 mequiv. g−1) and compared their hydroxide conductivity, water uptake, dimensional swelling, and membrane morphology from polymer nanostructures. While only the block copolymer exhibited well-defined microphase-segregated morphology, its hydroxide conductivity (201 mS cm−1 at 80 °C) was comparable to that of the random copolymer (194 mS cm−1 at 80 °C), as well as other membrane properties (WU 119% vs. 114%, dimensional swelling 32% vs. 31% at room temperature, respectively). Usually, higher IEC membranes have increased concentration of unbound water, enabling more favourable proton conduction while lower IEC membranes only contain primarily bound water. However, this study indicates that AEMs with very high IECs and an excess volume of unbound water lead to flooding of the ionic channels, and the phase-separated morphology does not play a significant role in improving membrane properties. All AEMs showed less than 1.4% loss of ion conductivity after immersion in 1 M NaOH at 80 °C for 1000 hours.
Noonan and co-workers conducted a similar study, systematically comparing a statistical copolymer and a series of multiblock copolymers of addition-polymerized polynorbornene to investigate the impact of polymer composition on hydroxide transport in AEM.302 Two monomers, hexyl-substituted norbornene, and bromobutyl-substituted norbornene were copolymerized via addition polymerization using a palladium catalyst to form two different polynorbornene architectures. Four block copolymers were prepared, from diblock to pentablock, with the targeted molecular weight (Mn = 80–130 kg mol−1) and the IEC value (1.7 mequiv. g−1). All block copolymers showed higher hydroxide conductivity and water uptake than those of random copolymers owing to their phase-separated microstructure and more interconnected networks for facile ion transport. The tetrablock copolymer showed the highest conductivity of 105 mS cm−1 at 80 °C and a water uptake of 85%, while the random copolymer showed 60 mS cm−1 at 80 °C with a water uptake of 30%. The block copolymer and microstructure might have shown a more accentuated impact on conductivity and water uptake in this study302 because the IEC values of the AEMs were lower than those of the other study305 (1.7 vs. 3.5 mequiv. g−1). The location of the ionic block also affects the water uptake; it was found to be more effective in controlling water uptake if the ionic block is located in the middle block of multiblock copolymers.
Saito and co-workers investigated a series of random copolymers of norbornene for AEMs (Fig. 16d).303 The composition of different chain lengths of bromoalkyl and alkyl-containing norbornene monomers, along with unsubstituted norbornene used in polymerization was tailored to control the AEM properties. The molecular weight of the synthesized terpolymers was remarkably high (Mn > 160 kg mol−1) owing to the highly reactive Ni-based catalyst306 to ensure high mechanical stability of the membranes. The AEM was made with trimethylammonium propyl group and unsubstituted norbornene with an IEC of 2.0 mequiv. g−1, showed a hydroxide conductivity of 109 mS cm−1 and a water uptake of 71% at 80 °C. The membranes showed no significant difference in their hydroxide conductivity and mechanical properties (tensile strength, elongation at break) after immersion in 1 M NaOH at 80 °C for 1000 hours.
Functionalized polynorbornenes often exhibit poor solubility in organic solvents due to the saturated olefinic backbone structure; therefore, quaternization of polynorbornene is mostly done under heterogeneous conditions by immersing the precast film in an aqueous amine solution to prepare AEMs. The resulting quaternized polymers are typically insoluble in any organic solvents, limiting their use as AEIs for electrodes to only particulate dispersions.307 In a follow-up study, Saito et al. further tuned the ratio of the terpolymer with a hexyl-substituted monomer and prepared quaternized polynorbornene, which was solvent-processable in an alcohol-based dispersing agent. The polynorbornene ionomer used for AEMFC and AEMWE demonstrated outstanding performance compared to those cells using state-of-the-art phenyl-containing AEIs, which can be ascribed to the phenyl-free backbone structure of polynorbornene.
Various crosslinking strategies have been employed for the addition of polynorbornene to avoid excessive water uptake and membrane swelling and increase the mechanical properties of polyolefin-based AEPs. Crosslinking of AEMs with high IEC (>3.5 mequiv. g−1) is necessary because excessive membrane water uptake limits their physical stability. The tetrablock copolymer (Fig. 16b) reacted with N,N,N′,N′-tetramethyl-1,6-hexanediamine to form both quaternary ammonium functional groups and crosslinks (Fig. 17a).308 Different mole ratios of the crosslinking agent (4–50 mol%) were investigated, and the AEM with an IEC of 3.5 mequiv. g−1 and a low degree of crosslinking of 5% showed good control of water uptake with the highest hydroxide conductivity of 198 mS cm−1 at 80 °C, which is higher than non-crosslinked block copolymers. By incorporating polytetrafluoroethylene (PTFE) reinforcement, the membrane demonstrated one of the highest performance in AEMFCs (3.4 W cm−2 at 80 °C, 500 hours continuous operation).309
 | ||
| Fig. 17 Selected examples of crosslinked addition polynorbornene AEPs: (a) diamine-crosslinked tetrablock copolymers of polynorbornene, (b) triamine-crosslinked polynorbornene ionomers with an epoxy group for enhanced adhesion, and (c) dithiol-crosslinked polynorbornene. | ||
Crosslinking also helped to improve the adhesion properties of quaternized polynorbornene when used as catalyst AEIs. Polynorbornene was crosslinked by a triamine crosslinker to react with an epoxy binder to induce adhesion between the ionomer and catalyst particles (Fig. 17b).310 The study reported that the approach of crosslinking quaternary ammonium AEIs helped to improve the durability of AEMWE operation by improving catalyst adhesion on the electrode. You et al. demonstrated the synthesis of crosslinked AEMs through copolymerization of vinyl-substituted norbornene, which reacts with dithiol reagents by UV-initiated thiol–ene click reaction (Fig. 17c).311 After quaternization, a crosslinked AEM was obtained, and the strategy helped to solve the trade-off between high IEC/hydroxide conductivity and membrane dimensional stability. Other crosslinking strategies include using bis(quaternary ammonium siloxane) linkage,312 bis(siloxane imidazole) linkage,313 and bisimidazole cationic crosslinker.314
Norbornene-based polyolefins are promising materials for AEPs, and their synthesis has been explored intensively in recent years. With appropriate catalysts, selected norbornene monomers with haloalkyl groups can be readily polymerized in either random or block fashion. The synthesized cyclic olefinic polymers have a fully saturated backbone, which is ideal for achieving high alkaline stability and very low adsorption energy on catalyst surface in electrochemical device applications,179 although its radical oxidative stability is still questionable.140 In addition, end groups present in these and many other polyolefin systems have not been examined in detail. These groups will likely impact the alkaline and oxidative stability of the system, especially for low molecular weight AEPs, and should be identified where possible. Some challenges and needs that have been addressed290 to make further progress in addition polymerization chemistry for polynorbornene, which can also be applied for AEP applications include: (a) development of more tolerant and air-stable catalysts with enhanced activity, (b) simplified synthesis of more reactive exo-norbornene monomers than endo-isomers, and (c) improvement of both catalysts and functionalized monomers for better-controlled microstructures, compositions, and molecular weight of resulting polymers.
4.2. Ring-opening metathesis polymerization
Post-polymerization modification of pendent alkyl halides or tertiary amines via the Menshutkin reaction or methylation, respectively, is by far the most common strategy to obtain quaternary ammonium groups to prepare AEPs. However, monomers can also be prefunctionalized with cationic groups and polymerized directly. Copolymerization of a cationic monomer with a neutral monomer/crosslinker serves as a straightforward approach to balance conductivity, mechanical strength, and water uptake. Direct polymerization of cationic monomers is particularly valuable if the cationic moiety is difficult to synthesize, or when synthetic methods to append the cation to the polymer backbone may not be highly efficient.215 One benefit of this approach is that a cationic monomer can be carefully prepared and purified for incorporation into an AEP. The polymerization chemistry and cationic functional group must be compatible, where enchainment of the monomer can be accomplished in the presence of the functionality. Olefin metathesis has emerged as a valuable tool for this process.215Olefin metathesis refers to the redistribution or exchange of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and is a critically important reaction in the construction of polymers, macrocycles, and a wide range of complex organic substrates.315 The widely accepted mechanism by which olefin metathesis occurs was described by Chauvin and Hérrison in 1971 (Scheme 23a).316 This involves a [2+2] cycloaddition where a metal–carbene reacts with an alkene to form a metallocyclobutane intermediate, followed by cycloelimination to form either the original alkene or a new olefin. Metathesis of strained cyclic olefins leads to polymeric materials as shown in Scheme 23b, driven by the release of ring-strain, which is termed ROMP.317 Alternatively, dienes can be combined with the loss of a small molecule (typically ethylene) for the preparation of polyolefins (acyclic diene metathesis polymerization, ADMET).318 The kinetic profile for these reactions is different, as ROMP proceeds via a chain-growth mechanism, and ADMET proceeds via a step-growth process. To date, AEMs have been primarily synthesized using ROMP.
C bonds and is a critically important reaction in the construction of polymers, macrocycles, and a wide range of complex organic substrates.315 The widely accepted mechanism by which olefin metathesis occurs was described by Chauvin and Hérrison in 1971 (Scheme 23a).316 This involves a [2+2] cycloaddition where a metal–carbene reacts with an alkene to form a metallocyclobutane intermediate, followed by cycloelimination to form either the original alkene or a new olefin. Metathesis of strained cyclic olefins leads to polymeric materials as shown in Scheme 23b, driven by the release of ring-strain, which is termed ROMP.317 Alternatively, dienes can be combined with the loss of a small molecule (typically ethylene) for the preparation of polyolefins (acyclic diene metathesis polymerization, ADMET).318 The kinetic profile for these reactions is different, as ROMP proceeds via a chain-growth mechanism, and ADMET proceeds via a step-growth process. To date, AEMs have been primarily synthesized using ROMP.
 | ||
| Scheme 23 (a) General olefin metathesis mechanism and (b) polymerization chemistries possible using olefin metathesis. | ||
Heteroatom-free polyolefin backbones can be obtained using olefin metathesis, and a wide range of catalysts can be used to effect ROMP or ADMET, though molybdenum and ruthenium catalysts are the most popular.315 Ruthenium catalysts are most common in AEP synthesis,215 as they are less oxophilic than molybdenum catalysts, signifying that polymerizations can be carried out in the presence of a wide range of functional groups such as alcohols, esters, amides, and ketones, to name a few.315 Commercially available Ru-complexes known as Grubbs catalysts (1st, 2nd, and 3rd generation) are typically used in the preparation of AEMs (abbreviated as Ru cat in Scheme 24).319–321 Living polymerization of highly strained cycloolefins is possible with fast-initiating catalysts,321,322 and a few examples of block copolymer AEMs have been reported.323,324
 | ||
| Scheme 24 Synthetic approaches to ROMP-based AEMs: (a) general scheme for polymerization of norbornene monomers, (b) general scheme for polymerization of cyclooctene monomers, and (c) crosslinking approach with difunctional monomers. | ||
The typical monomers used in AEP synthesis are norbornenes, cyclooctenes, and dicyclopentadiene (Scheme 24). Some of the first polymers to be synthesized via olefin metathesis were ring-opened polynorbornenes and polyalkenamers.325,326 Ring-opened polynorbornenes were commercialized in the 1970s under the trade name Norsorex, and polyoctenamers, prepared from ROMP of cis-cyclooctene, are commercial products sold under the trade name Vestenamer.315 Polydicyclopentadiene, derived from polymerization of endo-dicyclopentadiene, is a thermoset polymer, which has also found use in a range of applications.315 The resultant polyolefins all have alkenes present in the repeat unit of the polymer backbone, and the relative concentrations of cis and trans content in the polymer backbone can be influenced by the choice of polymerization catalyst. Generally, Ru catalysts lead to mixtures of cis/trans content in the polymer backbone. Stereospecific polymerizations of norbornene, endo-dicyclopentadiene, and tetracyclododecene are possible,327–332 but Ru catalysts are typically non-stereospecific.333,334 The alkene units present in the polymer backbone are susceptible to oxidation, so hydrogenation and crosslinking can be used to prepare stable saturated hydrocarbons. Hydrogenation of ring-opened polynorbornenes has been accomplished using tosyl hydrazide (>95%) at 120 °C,333 and with 5 wt% reduced Pd on CaCO3 at 100 °C with 400–600 psi of H2. Hydrogenation of polyalkenamers can also be accomplished with tosyl hydrazide at high temperatures (often with tri-n-propylamine),322,335 or with iridium catalysts and high pressures of H2.336 Hydrogenation of ring-opened polynorbornenes and polyoctenamers can also result in semicrystalline polymers,322,333,334,337 which is an important consideration in the design of AEMs.
Functional norbornene monomers are prepared from Diels–Alder cycloaddition reactions and functional cyclooctene monomers are typically prepared from 1,5-cyclooctadiene. Multistep synthetic pathways are often needed to obtain cationic monomers, which can be used directly in polymerization. Copolymerization of the cationic monomer with a neutral comonomer is common, as shown in Scheme 24a and b, affording linear polymers that can be soluble in organic solvents. Sometimes, the combination of hydrophilic headgroups and hydrophobic polyolefins can make solution processing difficult, particularly after hydrogenation. Crosslinking is also commonly employed with ROMP as this can help control swelling and water uptake in these materials. This can be accomplished by copolymerization of a cationic monomer with dicyclopentadiene (Scheme 24a) or by synthesizing a difunctional cationic monomer, as shown in Scheme 24c.
Historically, the first reports of ROMP-based AEMs appeared in 2009–2010.336,338,339 Coates developed a straightforward approach to a norbornene monomer bearing a trimethylammonium group.338 The β-position was blocked with a methyl group to prevent Hofmann degradation in the polymer.338 The resultant monomer was copolymerized with dicyclopentadiene to yield a crosslinked polymer film, xPNB-NMe3 (Fig. 18a). Upon conversion to the hydroxide form, this material did not swell appreciably in MeOH, and had a σ22°C = 18 mS cm−1, which, at the time, was one of the highest hydroxide conductivities for an AEM.338 Since then, the Coates group has primarily reported on cyclooctene-based ROMP polymers (Fig. 18a),336,339–341 where the resultant cationic polyoctenamers were converted into polyethylene-based AEMs using an Ir-catalysed hydrogenation. The ratio of cationic monomer was optimized with cyclooctene comonomer, and the final optimized PE-NMe3 and recently synthesized PE-pip have higher water uptake and significantly higher hydroxide conductivities than the original crosslinked xPNB-NMe3 (PE-NMe3σ22°C = 48 mS cm−1 and WU = 132%, PE-pip σ22°C = 41 mS cm−1 and WU = 87%).336,340
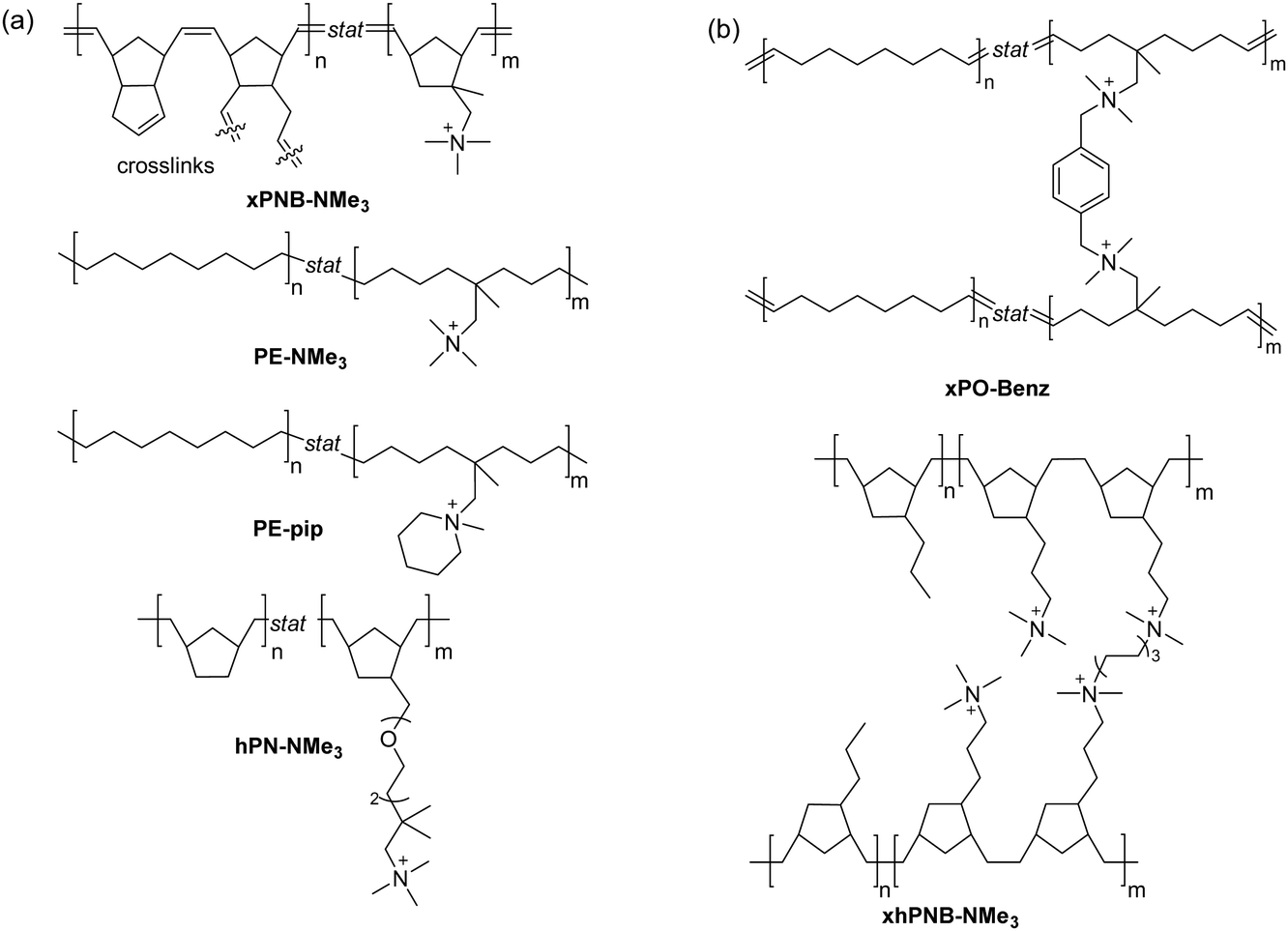 | ||
| Fig. 18 Selected examples of ammonium-based AEMs prepared using ROMP: (a) linear polymers and polymers crosslinked using dicyclopentadiene and (b) crosslinked films via ammonium linkages. | ||
Beyer and co-workers reported a hydrogenated polynorbornene derivative where ether linkages were used to tether the ammonium cation to the polymer backbone (hPN-NMe3 in Fig. 18a).342 The goal was to partially mimic some of the structural features of Nafion™ (semicrystalline backbone, ether linkages to the side chain) to see if the performance characteristics could be replicated. Exceptional conductivities for these materials were noted (optimized structure σ80°C = 177 mS cm−1 with WU80°C = 82%), but the mechanical properties of the film were a concern for long-term fuel cell operation.342
Multiple reports on crosslinked AEMs derived from both norbornenes and cyclooctenes have appeared.323,339,343,344 Coates reported on a crosslinked polyalkenamer comprised of dicyclooctene tethered with two benzyl ammonium groups (Fig. 18b).339 Copolymerization with cyclooctene (1.5 equiv.) afforded the highly conductive xPO-Benz, which exhibited a σ22°C = 68.7 mS cm−1, outperforming PE-NMe3.336,339 The high conductivity was attributed to the high IEC, which was made possible by crosslinking. Kohl et al. also recently reported on a crosslinked polynorbornene material xhPNB-NMe3, which had high ionic conductivity (Fig. 18b).323 In that work, block copolymers of 5-bromopropylnorbornene and 5-propylnorbornene were synthesized and hydrogenated using tosyl hydrazide. Then, N,N,N′,N′-tetramethyl-1,6-hexanediamine was used to crosslink the material via a reaction with two bromoalkyl groups, and the remaining pendents were replaced with trimethylammonium groups.323 The crosslink density was key in these materials, and the authors noted that with 20 mol% crosslinker, the highest hydroxide conductivity of 99 mS cm−1 at 25 °C and 195 mS cm−1 at 80 °C (WU = 115%) was obtained.323 No detectable degradation in conductivity was noted in 1 M NaOH at 80 °C over 792 hours. Finally, although the conductivity for this optimized material was high, the AEMFC open-cell voltage was still somewhat low (0.70 V), which was most likely due to the high gas crossover from the high water uptake.323 The peak power density (PPD) was 126 mW cm−2 for the optimized membrane, demonstrating how conductivity and mechanical properties must be optimized for specific applications.323 Beyond these reports, pyrrolidinium-based crosslinked ROMP membranes have also been described.343,344
Coates and co-workers reported on an imidazolium-fused cyclooctene monomer in 2018, which was combined with the Grubbs second-generation metathesis catalyst to produce cyclic oligomers (cPO-imid in Fig. 19).345 The formation of macrocycles was attributed to the moderate ring strain of the fused imidazolium monomer (−7.5 kcal mol−1), which slows propagation and thus renders secondary metathesis competitive, and to the planar imidazolium creating “U-turns” in the propagating chain.345 To synthesize an AEM, a crosslinked imidazolium polymer was prepared using a bifunctional imidazolium crosslinker. Upon hydrogenation, this imidazolium-based AEM had good alkaline stability and hydroxide conductivity (σ22°C = 37 mS cm−1, WU = 94%).345 In a follow-up study, imidazolium-functionalized trans-cyclooctene monomers were synthesized.346 These have increased ring-strain relative to the previous derivatives, and both statistical and block copolymer PE-imidazolium could be synthesized (Fig. 19).346 Much higher conductivities were noted for the statistical copolymers, partially attributed to the more disordered block copolymer microphase segregation, which may impede ion mobility.346 A crosslinked variant of the statistical copolymer was highly conductive (σ22°C = 49 mS cm−1, WU = 115%).346 Interestingly, the authors noted that the crosslinked films showed no degradation after 30 days in 1 M KOH at 80 °C, while the non-crosslinked films became brittle after 7 days under identical conditions.346 Several other reports on imidazolium-based ROMP AEMs have appeared.347,348
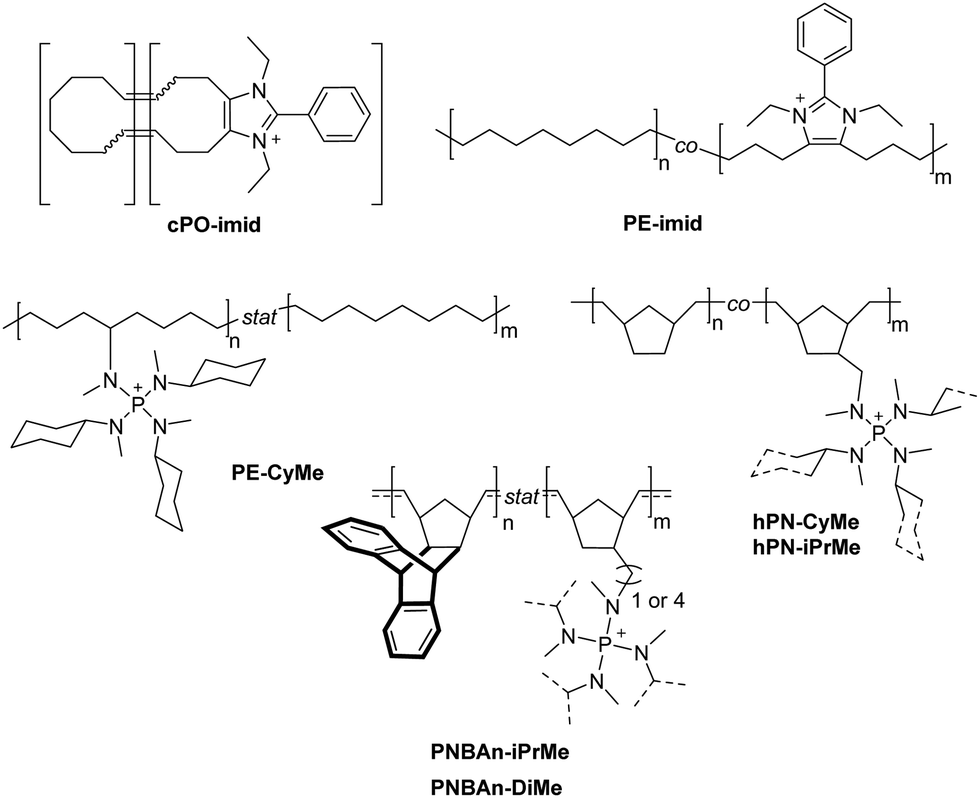 | ||
| Fig. 19 Selected examples of imidazolium- and phosphonium-based AEMs prepared using ROMP. | ||
The exceptional stability of tetraaminophosphonium has led to its exploration as cationic moieties in AEMs. The bulky P(N(Cy)Me)4[Cl] was reported to be highly stable as a phase-transfer catalyst under alkaline conditions (t1/2 = 67 hours at 100 °C in C6H5Cl/50 wt% NaOH).349 This cation was appended to a cyclooctene monomer and polymerized. After hydrogenation, the PE-CyMe copolymer was obtained (Fig. 19).64 The hydroxide conductivity for PE-CyMe was σ22°C = 22 mS cm−1 with moderate WU (52%).64 PE-CyMe was exceptionally stable to alkaline media, with hydroxide conductivity retained even after 20 weeks in 15 M KOH at 22 °C.64 The fairly complicated synthesis of PE-CyMe led to some more recent improvements in monomer synthesis. Noonan and co-workers reported a simple synthesis of tetraminophosphonium norbornenes in 2020.324 Both block and statistical copolymers with norbornene as a comonomer were synthesized (hPN-CyMe and hPN-iPrMe in Fig. 19), but the diblock copolymers were brittle.324 Statistical copolymers were much more effective AEMs, and hPN-CyMe (Fig. 19) had a hydroxide conductivity of 19 mS cm−1 at 25 °C with a water uptake of 82%.324 The hPN-iPrMe had higher hydroxide conductivity (27 mS cm−1) at 25 °C and slightly lower water uptake (∼75%).324 This was partially attributed to the increased IEC because of the more compact cation, but also to the increased crystallinity of the polymer as hPN-CyMe was completely amorphous, but hPN-iPrMe had some crystalline domains as evidenced by AFM studies and differential scanning calorimetry measurements. Going beyond 20 mol% of the cationic monomer was challenging for these materials without excessive swelling.64,324
Noonan and co-workers followed up on the hPN copolymers and demonstrated that copolymerization of a rigid norbornene anthracene cycloadduct with tetraaminophosphonium norbornenes was an effective approach to decrease segmental mobility in polynorbornene-based AEMs.350 The PNBAn-iPrMe swelled less (WU = 49%) at 25 °C and had higher hydroxide conductivity (30 mS cm−1) than the counterpart without anthracene (hPN-iPrMe).350 The authors also noted that the unsaturated PNBAn derivatives (with double bonds) had higher conductivities than the saturated ones, providing further support that decreasing segmental mobility is beneficial in these phosphonium copolymers.350 Stability studies in 10 M KOH also revealed the need for the bulky isopropyl groups around the P center for improved hydroxide stability.350 Though mechanical embrittlement of all the PNBAn copolymers was noted over time, the study demonstrated how modification of the ring-opened polynorbornene backbone can be used to enhance AEM performance.350
Various cobaltocenium AEMs have been synthesized via ROMP.351–354 These metallocene-based cations consist of a Co(III) metal centre sandwiched between two cyclopentadienyl ligands. These have been investigated as an alternative to the commonly used ammonium headgroups in AEMs. Two reports have appeared that highlight how to tailor the cyclopentadienyl ligands with substituents to impart good alkaline stability to cobaltoceniums.353,355 Yan and Coughlin demonstrated that the permethylated cyclopentadienyl ligand (C5Me5 or Cp*) can be used to make an ultra-stable Cp*2Co+ cation355 when compared to the C5H5 (Cp) parent derivative Cp2Co+. They conducted a stability test at 140 °C in 1 M NaOD/D2O and demonstrated that only 8.5% of Cp*2Co+ had degraded after 1000 hours, whereas the parent Cp2Co+ degraded completely in 1 week. Under the same conditions, the well-known benzyltrimethyl ammonium had degraded by 18% in 24 hours.
Tang and co-workers also systematically explored the impact of alkylation on cyclopentadienyl ligands for cobaltocenium alkaline stability.353 They found, as expected based on Yan and Coughlin's work, that increasing methylation offers marked improvements in alkaline stability.349,353 They also noted that either 1 or 2 t-butyl groups on the cyclopentadienyl ring can lead to alkaline-stable cations. The authors carried out stability tests in 5 M KOH/CD3OH, which is similar to reports from Coates and co-workers who have developed a protocol to examine cation headgroup stability and compare different families of cations.59,356 In Tang's work, the studies were also carried out under air rather than N2, which also impacts cobaltocenium stability.353 The authors found that the octamethyl cobaltocenium only degraded by ∼11% and ∼19% after 553 and 1025 hours, in 5 M KOH/CD3OH at 80 °C. They also discovered that the tetra-tert-butyl cobaltocenium (1,3-orientation of substituents on the Cp ring) only degraded ∼8% after 1025 hours. Under the same conditions, the benzyltrimethyl ammonium was almost completely degraded in just over 700 hours.
In early work, Tang and co-workers synthesized PE-Cp2Co using ROMP, and that material had hydroxide conductivities ranging from ∼20–90 mS cm−1 from 22–90 °C and water uptake ranging from 35–160% between 20 and 80 °C (Fig. 20).351 These AEMs could be crosslinked to mitigate dimensional swelling in water.352 As noted above, the parent Cp2Co+ has limited stability in alkaline media, and so the authors followed up on that work with ROMP polymers bearing more stable octamethyl cobaltocenium as a headgroup.353 The synthesized PE polymer exhibited good hydroxide conductivity (33–87 mS cm−1 from 22–80 °C). This polymer retained 91% of its conductivity after immersion in 3 M KOH at 60 °C for 30 days and was evaluated in an AEMFC. A PPD of 350 mW cm−2 was noted with an OCV of almost 1.0 V. Another ROMP-based AEM, PN-Cp2Co (Fig. 20), had a high chloride conductivity of 50 mS cm−1 at 90 °C and only 13.2% water uptake (95% RH) for an IEC of 1.5 mequiv. g−1.354 The cobaltocenium monomer was prepared in a unique manner, from lithium halogen exchange on an exo-4-bromophenyl substituted norbornene. Although the hydroxide conductivity of the polymer was not reported, this study highlighted the potential of polynorbornene polymers for metal-based AEMs.
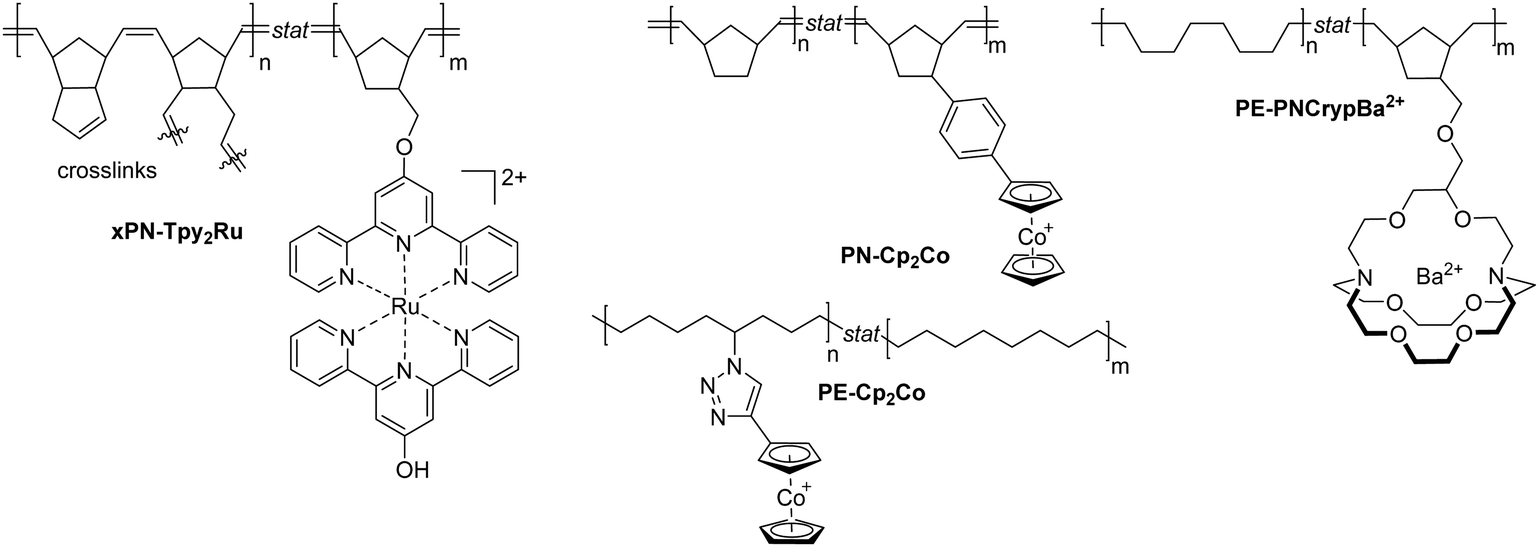 | ||
| Fig. 20 Selected examples of metal-based AEMs prepared using ROMP. | ||
The first example of a transition metal cation implemented in AEMs was a bis(terpyridine)Ru(II) functionalized polynorbornene made in 2012 by Hickner and Tew.357 These cations were directly polymerized via ROMP with dicyclopentadiene as a crosslinking comonomer (xPN-Tpy2Ru in Fig. 20). The optimized AEM had a 1:5 ratio of cationic monomer to hydrophobic/crosslinking monomer, resulting in relatively high water uptake (126%) and hydroxide conductivity of 28.6 mS cm−1 at 30 °C (IEC of 1.4 mequiv. g−1).357 The membrane was stable in 1 M NaOH at room temperature for 6 months.357 A follow-up study by Hickner and Tew on the copolymerization of this monomer included 1,5-cyclooctadiene and dicyclopentadiene as comonomers in polymerization.358
A 2017 study by Kwasny and Tew explored how altering the site of crosslinking in the polymer backbone impacted water uptake.359 Membranes were prepared from heteroleptic and homoleptic Ru complexes, where polymerizable norbornene units were either on one or both terpyridine ligands.359 Crosslinker to non-crosslinker and ionic to non-ionic monomer were held constant, and a substantially lower water uptake of 30–35% was obtained by utilizing a homoleptic ruthenium cation in comparison to the heteroleptic derivative (water uptake of ∼230%).359 The homoleptic complex also resulted in a decrease in Cl− conductivity.359 The authors also examined Ni- and Co-terpyridine complexes for AEMs as part of that work.359 Tew et al. also synthesized bis(terpyridine)Ni(II) AEMs through RAFT polymerization and crosslinking via thiol–ene click reaction to create phase-separated morphology.360 To our knowledge, the alkaline stability of these different terpyridine-based metal complexes has not been compared to ammonium headgroups.
You and co-workers recently reported on a cryptand-based AEM prepared using ROMP (Fig. 20).361 The authors noted that the cryptand is key to having a high binding constant for metal cations in water and developed a synthetic method to append this structure to a norbornene monomer and copolymerize it with cyclooctene to afford PE-PNCryp.361 Then, the choice of metal salt for binding was examined, and the only one that resulted in minimal leaching was Ba2+. This was attributed to the high binding constant of Ba2+ with the cryptand compared to Na+ and K+.362 The optimized PE-PNCrypBa2+ had a hydroxide conductivity of 23 mS cm−1 at 40 °C. A series of PECryp Ba2+ copolymers prepared by a different method was even more conductive than the PE-PNCrypBa2+ copolymer. The corresponding physical studies suggested that the ionic clustering network in the PECryp above the Tg is more robust. The PECrypBa2+ copolymers retained 70% of their chloride conductivity after immersion in 15 M KOH at 40 °C and 60 °C for 1600 hours. These are unprecedentedly harsh alkaline conditions and highlight the potential of this framework for future exploration. The authors noted that this membrane performed well in AEMWE with 0.1 M KOH, similar to the commercially available PiperION.
ROMP is a useful tool to make polyalkenamers, which can subsequently be hydrogenated to form heteroatom-free polymer backbones. A wide range of functionalities is tolerated in ROMP, and strained cycloalkenes bearing cationic groups can be directly polymerized, which is advantageous for the creation of new materials. The synthesis of AEPs decorated with main group and metal-based cations is a particularly important outcome of using ROMP chemistry. The stability of metal cations in AEPs under device operating conditions is a critical subject of research for the practical use of these polymers, as this will dictate their potential success in devices.
A direct benefit of the ROMP approach is the double bond present along the polymer backbone, which can be used to bring about crosslinking and facilitate the preparation of polymers with high IECs. However, the Ru catalysts used for ROMP are somewhat expensive, and a two-step process is needed to achieve a fully saturated hydrocarbon backbone (e.g., polymerization and hydrogenation), which adds an additional step to the overall process.
4.3. Radiation grafting method
Radiation-grafting is a useful method for the chemical modification of pre-formed, inert polymer substrates such as films,363 powders,364 and fibres,365 to form useful functional materials. Radiation-grafted (RG) polymers are being investigated for use in a wide variety of applications across many fields (e.g., clean energy, environmental remediation, healthcare) including electrolyte membranes for low- and high-temperature polymer electrolyte fuel cells and membrane-based water electrolysers,366–370 CO2 electrolysis to high-value chemicals;371 CO2 adsorbents;365 ion exchange membranes and separators for ED/RED,372–374 RFBs,367,375,376 actuators,377 Li-ion batteries,378 and supercapacitors;367 biofouling-resistant membranes for microfiltration;379 materials for the recovery, extraction, and separation of inorganics including heavy metals ions;380,381 chromatography materials for protein purification;382 biomaterials for tissue engineering and engineered skin;383 and UV absorbers.384Scheme 25 summarizes the key stages behind the most commonly encountered pre-irradiation grafting (PIG) method:367,369,370 (1) irradiation of inert polymer substrates to “activate” them (functionalize with radicals or peroxide groups, both of which can initiate copolymerisation); (2) monomer grafting onto the substrates (after an N2 purge to remove all traces of O2); and (3) an optional post-graft functionalization process.
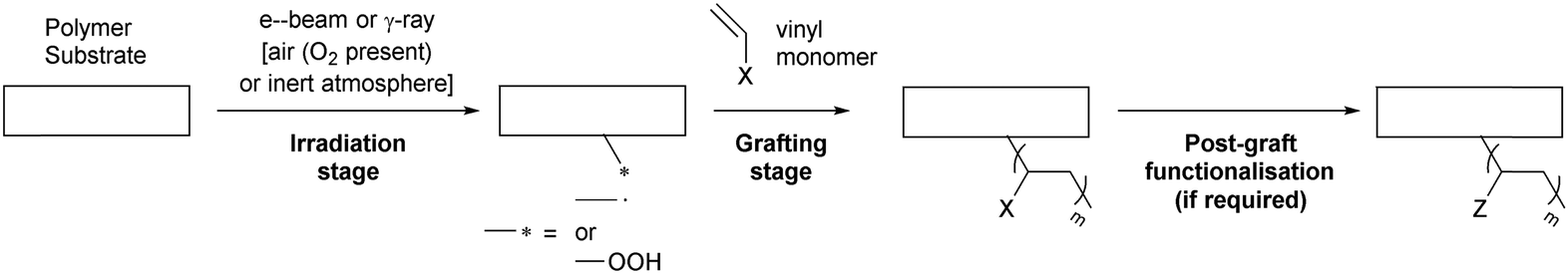 | ||
| Scheme 25 The key stages behind the pre-irradiation PIG. It is known as the peroxidation-PIG method if the polymer substrate is irradiated in air (which can result in an ether links between the substrate and grafted chains [not shown]). If the polymer substrate is submerged in the grafting mixture containing the monomer before irradiation treatment, this is the MIG. | ||
The radiation is commonly a high dose-rate electron-(e−)-beam (β radiation), a low dose-rate γ-ray, or an ultraviolet (UV) source. Radiation grafting of polymer substrates using lower energy UV electromagnetic radiation typically leads to a bias towards surface grafting, unless the films are thin enough to allow for reasonable levels of grafting into the bulk.374 The use of high energy and high dose-rate radiation (e.g., e−-beams) leads to bulk grafting where there is a high concentration of short-length (low molecular weight) grafted chains, while the use of high energy lower dose-rate radiation (e.g., γ-rays) also leads to bulk grafting, but where there is a lower concentration of longer-length grafted chains;114,385 the latter scenario is less desirable if chemical degradation is a concern (e.g., oxidative cleavage of entire grafted chains, vide infra) as there is a greater loss of functionality per cleaved chain.
With large polymer substrates, low dose-rate point sources such as γ-rays are also subject to inverse square law limitations (radiation intensity is inversely proportional to the square of the distance from the source), therefore requiring the periodic suspension of the irradiation process so that they can be moved around to ensure equal absorbed doses on all parts of the sample. With high energy (1–10 MeV) commercial e-beam facilities, more commonly known for sterilizing plastics and medical equipment,386 as well as for fabricating heat-shrink polymers,387 the e-beam source is often wider than the samples being irradiated.
Homogeneous irradiation is more easily controlled by passing the substrates at a controlled speed through the e-beam, with higher doses being enabled by multiple passes; this also helps with minimising sample heating (due to the high energy radiation), where the radicals/peroxide groups would be decomposed as they form. It has recently been found that irradiation of polymer films whilst being kept cool (<−10 °C) led to a higher degree of grafting (DoG (%) = % mass of grafted chains normalized to the mass of substrate used) and membranes with better properties;388,389 the radicals formed during radiation are preserved more during the lower temperature irradiation.
With fluorinated (e.g., PTFE, FEP) or partially fluorinated (e.g., ethylene-tetrafluoroethylene ETFE) substrates, low total absorbed radiation doses need to be used (typically <50 kGy) as high energy radiation can cleave the backbone C–C bonds, leading to mechanically weak functionalized membranes. Non-fluorinated substrates (e.g., polyethylene) can tolerate higher total absorbed doses (>100 kGy), but such high total doses can lead to radiation crosslinking (this can be desirable and undesirable depending on the required final membrane properties).388,390
If the irradiation step is conducted with the substrate exposed to air (rather than vacuum or inert atmospheres), then it is known as the peroxidation-PIG method.367,369,370 This typically requires a higher total radiation dose for a target DoG, compared to when irradiation of polymer substrates was conducted in inert atmospheres.389,391 Therefore, this can place a limit on practical grafting levels when functionalising polymer substrates containing C–F bonds (see discussion vide supra on upper limits of radiation doses that can be used).392 Peroxidation-PIG also leads to the formation of peroxide groups on the chains of irradiated polymer substrates, which can lead to the formation of ether (C–O–C) links between the polymer chains on the substrate and the grafted polymer chains (not shown in Scheme 25 to aid clarity).370 A major practical advantage of using the peroxidation-PIG method is that an onsite radiation source is not needed. The offsite irradiated and peroxidated polymer substrates can be transported back to a laboratory in dry ice and stored in low-temperature freezers (<−40 °C) for 6–12 months (storage time achievable depends on the polymer substrate used).393 Such peroxidation-PIG grafting processes often use commercial e-beam facilities, as large volumes of substrate can be treated in a single beaming session.
If inert atmosphere PIG is used, then fewer main chain bond scissions occur (cf. peroxidation-PIG), especially as lower radiation doses can be used, while more radiation crosslinking can also occur (cf. same radiation dose peroxidation-PIG);389 grafting times can also be quicker. This can lead to RG-AEMs (Scheme 26) with enhanced mechanochemical stabilities. When the polymer substrates are grafted immediately after irradiation, the radicals on the polymer chains of the substrate can directly co-graft without the formation of ether links. However, if the inert atmosphere-irradiated polymer substrates are cold stored in the air, peroxide groups will eventually form (as with the peroxidation-PIG).389 An alternative to PIG is the mutual-(simultaneous)-grafting method (MIG), where the polymer substrate is submerged in the grafting mixture to combine the irradiation and grafting stages into a single step.367,369,370 However, this requires a radiation facility on site and typically uses a low dose-rate γ-ray source (with radiation security issues to attend to); commercial e-beam facilities are often reluctant to irradiate containers containing toxic and flammable monomers. A major downside to MIG is that there can be a high level of homopolymerisation occurring; this is where the monomer polymerizes without the chains being covalently grafted onto the substrate. This can lead to a large amount of effort to remove the grafted substrates from the homopolymer (it has been known for the substrate to be encased in a block of homopolymer), as well as leading to materials with lower degrees of grafting. Despite this, MIG can be useful for small-scale trials of new RG-material chemistries, and it tends to lead to reduced levels of radiation crosslinking.391
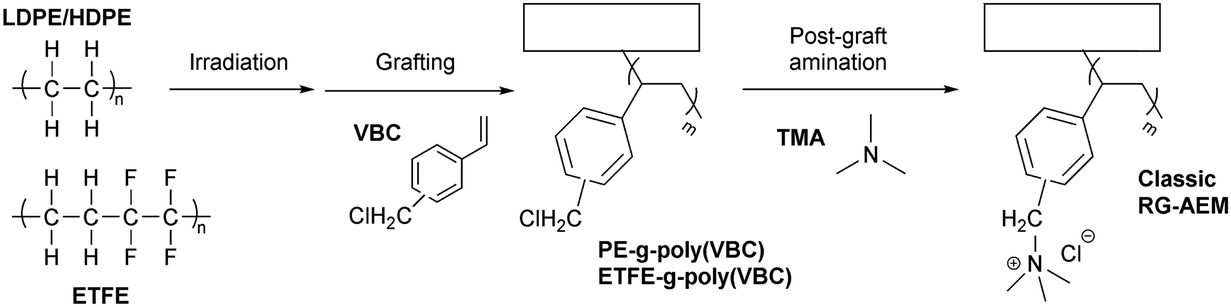 | ||
| Scheme 26 The PIG of vinylbenzyl chloride (VBC) onto ETFE and polyethylene (PE, either low- or high-density) film substrates with subsequent post-grafting amination with trimethylamine to form a classical benzyltrimethylammonium radiation-grafted anion-exchange membrane (TMA-RG-AEM). | ||
With the radiation-grafting of dense substrates, such as ETFE films, the grafting front mechanism can operate.385,394 This is where the monomer initially co-polymerizes onto the surface of the film (after low grafting durations), after which the monomer can penetrate further and further into the core of the film (after longer grafting durations), until homogeneous grafting levels are achieved throughout the bulk of the newly formed membrane. If such a mechanism is operating, then this can place an upper limit to the thickness of the films (or diameters of powders/fibres) that can be homogeneously grafted through the bulk; a good rule of thumb range is 100–200 μm maximum film thicknesses, depending on the substrate and grafting conditions. With less dense films, such as low-density polyethylene (LDPE), where the monomer can diffuse more quickly into the core of the film, then the grafting does not always follow such a front mechanism.392 Note that for RG-AEMs, PVDF is not a suitable substrate as it is unstable in alkali conditions (see Section 2.3).395 There is also some interest in making RG-AEMs from non-fluorinated non-polyethylene substrates such as cellulose acetate, and Nylon-6,6 nanofibrous sheets.396,397
The crystallinity of the substrate can also influence the final properties of RG-PEMs and RG-AEMs, as their final nano-morphologies can be complex.398–402 The radicals formed on irradiation tend to be more stable in the crystalline domains, but they can migrate to the interphase and amorphous domains,388,389 where they will react with any oxygen species, and grafting will predominantly propagate into the amorphous domains.403 For example, Sproll et al. reported that ETFE with a smaller number of larger crystallites produced an RG-PEM with better conductivity performance in a fuel cell;404 this is partly due to better connected amorphous domains.
The most common monomer used to fabricate RG-AEMs is vinylbenzyl chloride (Scheme 26).114,371–373,388,389,391,392,394,395,405–410 On amination/quaternization, cationic groups are introduced, resulting in an anion-conducting material, and the most commonly encountered amine is trimethylamine (TMA), which yields benzyltrimethylammonium headgroups. This class of RG-AEM has ease of handling, high hydroxide conductivities, and rapid water diffusion dynamics.411–413 Disadvantages include high levels of swelling in water without additional crosslinking,373 especially laterally (area swelling as opposed to thickness swelling), which will be a particular problem for their use in devices such as AEMFC that can experience humidity cycles. Without mitigations, this will also mean that pre-swelling of the RG-AEM may be needed if they are to be used in large-area AEMWE cells.414
A diversity of quaternization agents for VBC-based RG-substrates has been investigated including aliphatic heterocyclic types like N-methylpiperidine (MPIP) and quinuclidine (QUN), 2-butyl-1,1,3,3-tetramethyl-butylguanidine (TMBG), as well as aromatic types such as 1,2-dimethylimidazole (DMI) and pyridine (PYR) (Scheme 27).115,415–417 Alternative quaternization agents include 1,2,4,5-tetramethylimidazole (TMI, the cation found in Sustainion® AEMs), N-methylpyrrolidine (MPY), N-methylmorpholine (MMPH), 1,4-dimethylpiperazine (DMPZ), and DABCO, the latter two have the potential to form crosslinks as they contain two tertiary amine groups.115,415,418,419 RG-AEMs made with isoindolinium headgroups have also been fabricated,420 as have those with more complex imidazolium and pyridinium groups.407 As well as affecting the conductivity and alkali stability of the RG-AEMs (vide infra), the choice of cations can affect their water uptakes: e.g., the water uptakes of TMA types are generally less than MPIP analogues.372,415 Crosslinking can be used to reduce the excessive levels of swelling on hydration, but this can have a negative impact on other properties such as conductivity.373,421
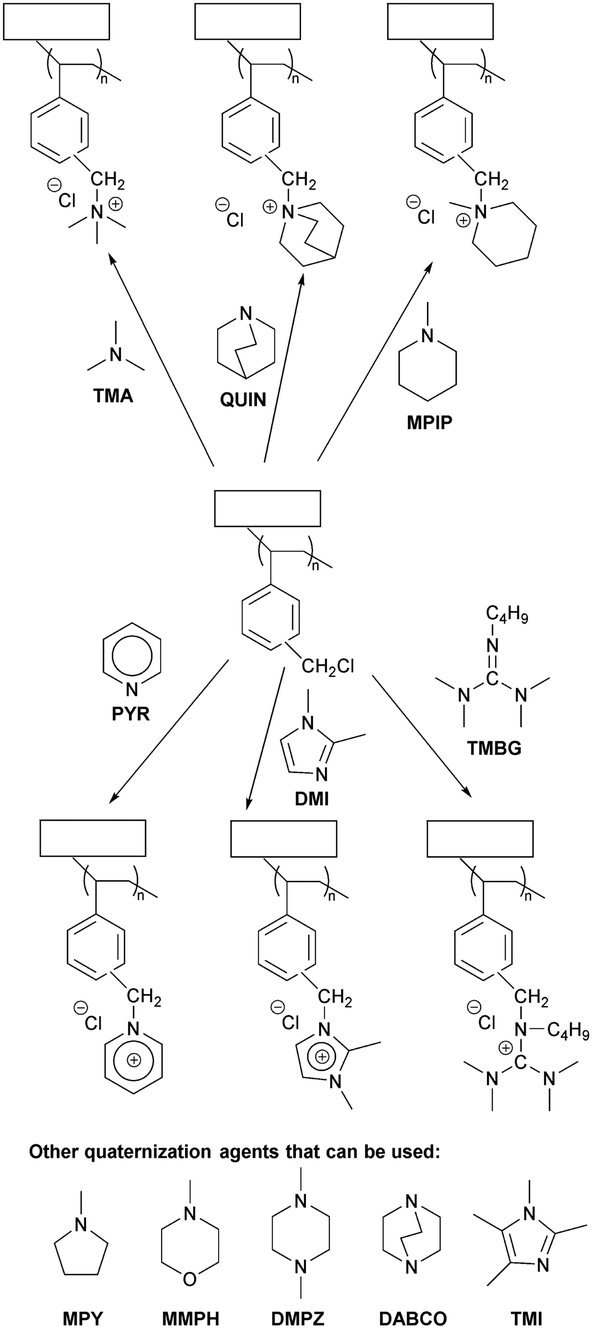 | ||
| Scheme 27 Different N-containing reagents that can be used for post-graft quaternization of VBC-based RG-substrates. | ||
Despite the possibility of hydroxide-derived nucleophilic substitution and elimination degradation pathways (SN2, E2, and Hofmann eliminations), VBC-grafted TMA- and MPIP-RG-AEMs are generally stable in hydroxide forms at 60 °C (at medium to high RH) and at higher temperatures when well hydrated.422,423 An LDPE-TMA-RG-AEM has even been shown to maintain a hydroxide conductivity of ca. 300 mS cm−1 over several hours at 110 °C when hydrated under pressurized conditions.424 An ETFE-TMA-RG-AEM made using 3-VBC (meta isomer, synthesised in-house due to commercial unavailability) appeared to be more alkali stable compared to a benchmark made with commercially available 4-VBC (para isomer), but the 3-VBC monomer did not graft very well, leading to a low IEC RG-AEM.425 Similarly, the grafting of a monomer containing a butyl-spacer between the vinylbenzene group and the chloride leaving group led to a more alkali stable ETFE-MPY-RG-AEM (cf. a VBC-grafted MPY-benchmark), but again this had a low IEC due to poor monomer grafting characteristics.426
Benzyl-DMI and -PYR cations have poorer stabilities in alkali and will not be suitable for application in AEMWEs.427,428 A benzyltriethylammonium (TEA) RG-AEM was also less stable in alkali than the TMA benchmark, especially at lower hydration levels, due to elimination reactions on the ethyl groups.429 Obviously, the RG-AEM contains benzene-rings and thus has the potential to be oxidized when in contact with OER anodes of AEMWEs (as mentioned in Section 2.5, leading to the formation of weakly acidic phenolic groups resulting in chemical instabilities and poor in situ performances).430
With RG-PEMs made from styrene, the reactive C–H bonds in the grafted hydrocarbon chains that are in the alpha position to the benzene rings are highly susceptible to radical attack, which leads to the cleavage of entire grafted chains; this is one of the reasons RG-PEMs made via co-grafting of α-methylstyrene (AMS) and methylacrylonitrile (MAN) are a preferred formulation (MAN is needed to boost the grafting of AMS and also to lower gas crossover reactions, while AMS contains α-C–CH3 groups and not labile α-C–H groups).370,431 Due to the historic low stability of AEMs to alkali, with degradations over 10 s to 100 s of hours, radical/oxidative degradation (ROD) pathways have been generally neglected. However, with the recent development of alkali-stable AEMs, a debate has raged on the significance of RODs.110 A recent study has also proven that significant concentrations of radicals can be observed in AEMFCs.432 Espiritu et al. have reported that grafted poly(vinylbenzyltrimethylammonium hydroxide) cations degrade more (loss of IEC) in O2 purged D2O than they do in N2 purged D2O;115 degradations included the loss of complete benzyltrimethylammonium groups (not just the loss of trimethylamine). A LDPE-TMA-RG-AEM also showed a higher degradation of conductivity in O2 gas compared to N2 gas.433
Espiritu et al. conducted a detailed degradation study on LDPE- and HDPE-based TMA-RG-AEMs (MIG with low dose-rate γ-ray) and an ETFE-TMA-RG-AEM (peroxidation-PIG with an e−-beam), where the hydroxide forms were submerged in (CO2-free) air-saturated deionised water.114 This study, which also involved 17O solid-state NMR, confirms that loss of entire grafted chains can occur via ROD, where higher dose-rates lead to a lower rate of IEC loss (due to there being more grafted chains of shorter length). These ROD reactions were the main contributor to IEC loss at lower pHs, over E2 and SN2 attack on the headgroups. An LDPE-DABCO-RG-AEM was also evaluated, which was observed to have slightly less stability.
Other classes of RG-AEM have been reported that do not involve the radiation-grafting of VBC monomer. Several papers from the Takasaki Advanced Radiation Research Institute have documented work on RG-AEMs made using vinylimidazole monomers (vinyl group attached to one of the N-atoms in the imidazole ring), followed by a methyl iodide quaternization reaction to yield the imidazolium headgroups.399–401,434 As 1-vinylimidazole and 1-methyl-2-vinylimidazole graft poorly on their own, a co-monomer boosting strategy was employed; this involves adding a co-monomer that easily grafts, such as styrene or MAN, to the grafting mixture to enhance the grafting levels of the poorer grafting primary monomer.370,434,435 There are concerns with the alkali stability of such pendent imidazolium groups, as they can undergo OH−-derived ring-opening reactions, especially if there is a C–H group in the C2 position.427
Further alternative RG-AEM chemistries involve imidazolium head groups where they are not linked to the grafted chains via an imidazolium N atom.435 An anilinium-type RG-AEM has also been fabricated using N,N-dimethylaminostyrene, with subsequent methylation with methyl iodide, to yield trimethylammonium groups that are directly bound to benzene rings on the grafted chains (and not connected to the benzene rings via methylene CH2 groups);436 however, the resulting RG-AEMs had very poor alkali stability. Finally, RG-AEMs made using the grafting of vinylpyridines, with subsequent methylation, have also been produced.377 Given that such unprotected pyridinium groups have low stability in alkali, these were being prepared for use in ionic polymer–metal composite actuators.
Despite only a small number of evaluations of RG-AEMs in AEMWEs (see Section 6.2), they have been studied much more for use in AEMFCs with precious-metal-free electrodes.437–441 They show a lot of promise as the complex nano-morphologies of RG-AEMs provide a good range of mechanical properties and gas permeability, along with high conductivities and facile water transport behaviours, leading to high performances in devices.393 The RG method also allows for the introduction of multiple different functional group chemistries in controlled ratios,373,431,434,442 which yields the ability to fabricate highly functional, tailored polymer materials. However, RG-AEMs can suffer from excess swelling, especially at high IECs, and due to the underlying radical polymerization mechanism, not all monomers are amenable for grafting (most reports have focused on monomers with styrene groups or carbonyl-containing groups such as with acrylates and arylamides, where the latter may not be useful for application in high pH environments). The RG method also requires access to high-energy radiation facilities and may only be amenable to batch processes on scale-up, rather than use with continuous processes such as roll-to-roll. For improvements and tailoring of RG-AEMs for specific use in AEMWEs, the following adaptations should be researched: (1) covalent and ionic crosslinking to reduce swelling in water to yield more dimensional stability;373,443,444 (2) incorporation of co-monomers to improve stability towards ROD including introducing antioxidants442,445 or MAN (the latter to reduce gas crossover);446 (3) incorporation of co-monomers such as styrene to modify nanomorphologies to alter conduction and water transport properties as well as to tailor mechanical properties;447 (4) incorporation of aliphatic sidechains (non-aromatic) on the grafted component.
4.4. Anionic polymerization
Styrene-diene block copolymers are an important class of thermoplastic elastomers. Using two industrially important chemicals as raw materials, the sequential living anionic polymerization of styrene and 1,3-butadiene (or other dienes such as isoprene) is known to construct a diverse range of diblock and triblock copolymers.448–451 The covalent bond connection between incompatible polystyrene and polydiene generates a wide array of microdomain morphology, as shown in Scheme 28. Polystyrene chains function as a hard block, providing mechanical strength and physically crosslinked domains, while polydiene chains serve as a soft matrix, providing elasticity to the material at ambient temperature. Among block copolymers, poly(styrene-b-butadiene-b-styrene) and its hydrogenated form poly(styrene-b-(ethylene-co-butylene)-b-styrene) (SEBS) (SBS of Scheme 28 and SEBS of Scheme 29, respectively) are commercially available and have been extensively studied for polymer microstructure-mechanical property relationship investigation for decades.449,450 | ||
| Scheme 28 Synthesis of styrene-diene block copolymers and microdomain morphology based on the composition of each polymer; spherical (S), cylindrical (C), gyroid (G), and lamella (L), and volume fraction of A polymer (fA) in AB diblock copolymers. Reproduced from ref. 448, with the permission of the American Institute of Physics, copyright 1999. | ||
 | ||
| Scheme 29 Synthesis of SEBS AEMs by (a) chloromethylation and (b) transition metal-catalysed C–H borylation–Suzuki coupling approaches. | ||
In SEBS, the midblock poly(ethylene-co-butylene) is composed of inert saturated C–C and C–H bonds. Thus, a selective functionalization of aromatic rings of polystyrene chain with ionic group could create ionic block copolymers using the commercial polymer as a starting material platform. For example, sulfonation of the polystyrene block of SEBS transforms the commercial elastomeric material into an elastic proton exchange membrane in one step.452,453 In general, the chemical functionalization of SEBS to prepare AEM involves the chloromethylation route shown in Scheme 29a. The reaction of SEBS with chloromethyl methyl ether in the presence of a Lewis acid catalyst incorporates a –CH2Cl group into the benzylic position of the polystyrene end block. Subsequent substitution of the benzyl chloride in the polymer with trimethylamine gives a BTMA-functionalized SEBS (SEBS-BTMA). As stated in Section 2.1, although commonly practiced in laboratories, chloromethylation in polymer substrates frequently results in low efficiency and side reactions, including extensive cross-linking.454–456 To overcome the limitations of chloromethylation, Bae and co-workers reported a transition metal-catalysed C–H borylation route for the synthesis of quaternary ammonium-functionalized SEBS AEMs (Scheme 29b).457 Once the Bpin group is incorporated into the aromatic ring of polystyrene by replacing a C–H bond with a C–B bond, the Bpin group is replaced by functionalized aromatics through a Pd-catalysed Suzuki-coupling reaction. Although this synthetic approach allows the incorporation of a wide array of quaternary ammoniums at the terminal benzylic position, the requirement for expensive transition metal catalysts (Ir and Pd) would limit the scalability of the process.
Among the commonly practiced organic reactions for polymers, the most frequently used catalysis is an acid-catalysed reaction, employing either Brønsted or Lewis acid. Therefore, for the chemical modification of polymers to be practical and scalable, acid-catalysed functionalization would be highly desirable. Furthermore, among quaternary ammonium structures in AEMs, the insertion of a long alkyl chain spacer between the polymer chain and quaternary ammonium structure is known to enhance alkaline stability and the mobility of ionic groups compared to BTMA functionality.57,58,458 Accordingly, the incorporation of a long spacer-tethered quaternary ammonium group to SEBS has been studied by utilizing Friedel–Crafts reactions. The Friedel–Crafts acylation of SEBS yields a ketone-functionalized polymer (SEBS-acylC5-Br of Scheme 30b), which can be subsequently reduced to –CH2– by reaction with triethylsilane. After being cast into a film, immersion of SEBS-acylC5-Br into a solution of trimethylamine forms SEBS-acylC5-TMA as an AEM.459 Although each reaction in this method is well-established in organic chemistry, the Friedel–Crafts acylation with an acid chloride that has terminal alkyl bromide moiety needs caution because both acid chloride and alkyl bromide can undergo Friedel–Crafts acylation and alkylation, respectively, in the presence of strong Lewis acid catalyst. If both reactions occur on polystyrene chains, crosslinking of polymer chains would occur, causing reduced solubility and gelation. The Friedel–Crafts alkylation with a brominated tertiary alcohol substrate eliminates that possible side reaction because Brønsted acid would react only with tertiary alcohol functionality, forming a tertiary carbocation, without affecting the terminal –CH2Br group (Scheme 30a).460 Compared to the Friedel–Crafts acylation method, the alkylation approach also has the advantage of a shorter reaction scheme by eliminating the reduction step of the acyl group in SEBS.
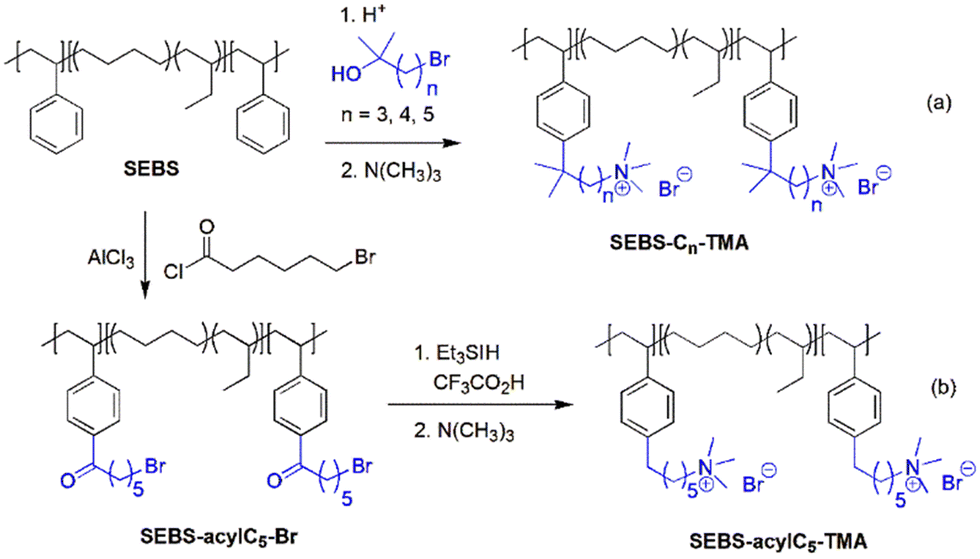 | ||
| Scheme 30 Synthesis of SEBS AEMs by (a) Friedel–Crafts alkylation and (b) Friedel–Crafts acylation-reduction approaches. | ||
Other methods of synthesizing styrene-diene block copolymer AEMs through anionic polymerization include radical-induced bromination of poly(4-methylstyrene) using NBS and AIBN (Scheme 31a). Once diblock copolymer PB-b-P4MS was prepared by living anionic polymerization, followed by hydrogenation using p-toluenesulfonyl hydrazide. Subsequent radical bromination at the benzylic position generates a –CH2Br moiety at the side chain of the polystyrene block. Amination of the block copolymer film in a solution of trimethylamine converts the benzyl bromide to benzyltrimethylammonium bromide in the polymer (PE-b-PS(BTMA) of Scheme 31a).461 Kraton Performance Polymers Inc. (Houston, TX) specializes in the synthesis of poly(4-tert-butylstyrene)-b-poly(2-methylbutylene)-b-poly(4-methylstyrene)-b-poly(2-methylbutylene)-b-poly(4-tert-butylstyrene) (tbS-MB-4mS-MB-tbS of Scheme 31b). The midblock sulfonated version of the symmetric pentablock copolymer is commercially available with the trade name Nexar® and has attracted significant attention as a functional material for water transport and separation applications. The tertiary butyl group at the end block of the polystyrene chain increases Tg and enhances mechanical strength, while the low Tg poly(2-methylbutylene) block provides flexibility. The midblock poly(4-methylstyrene) was functionalized with benzyltrimethylammonium bromide using a similar protocol of radical-induced bromination and investigated as an AEM (Scheme 31b).462
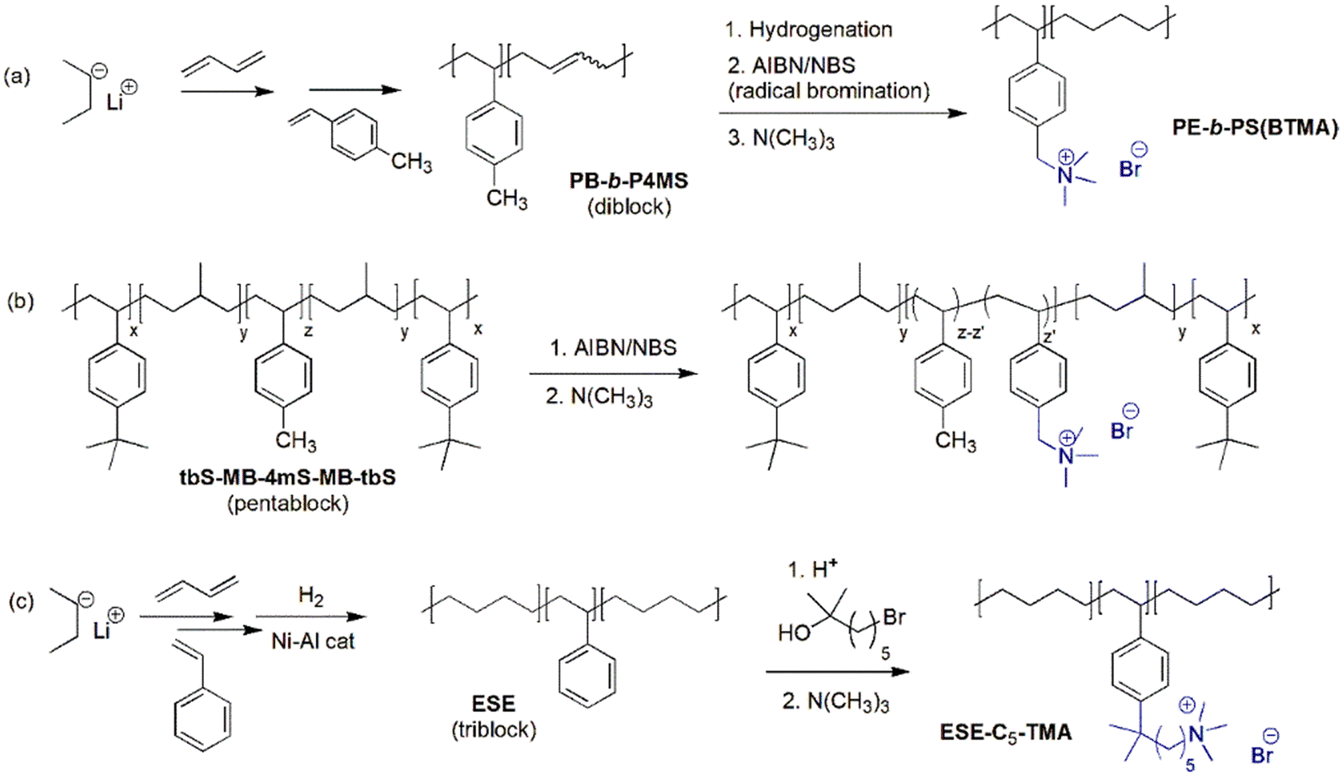 | ||
| Scheme 31 Additional synthetic methods of styrene-diene block copolymer AEMs. | ||
Most styrene-diene triblock copolymer AEMs have polystyrene chains as an end block because rigid polystyrene functions as a hard block component in ABA triblock copolymer systems. An interesting approach to using semicrystalline polyethylene as a hard block component has been reported.463 In this system, a sequence of anionic polymerization of butadiene and styrene in cyclohexane solvent generates a triblock copolymer poly(butadiene)-b-polystyrene-b-poly(butadiene). Although minor formation of poly(1,2-butadiene) is unavoidable, the polybutadiene end block consists primarily 1,4-configuration unit (over 90% based on 1H NMR spectroscopic analysis). This high percentage of 1,4-configuration in polybutadiene allows the conversion of the soft polymer chain at the end of the triblock copolymer to a rigid polyethylene block upon hydrogenation (ESE of Scheme 31c). The bromoalkyl functionalization using Friedel–Crafts alkylation chemistry460 and subsequent amination results in triblock copolymer AEMs where midblock polystyrene is functionalized with a quaternary ammonium group. (ESE-C5-TMA). This midblock functionalized AEM is equivalent to complemental SEBS AEM where the end block polystyrene is functionalized with an ionic group (SEBS-Cn-TMA of Scheme 30a).
Living anionic polymerization affords polymers with well-defined block composition and narrow molecular weight distribution but requires strict reaction conditions of an inert atmosphere and purification of reagents and solvents. Due to the strong nucleophilicity of anionic catalyst (e.g., sec-BuLi), the polymerization is not tolerant to common precursor groups typically used for AEPs, such as halides and amine, limiting the choice of polymers for AEP synthesis. Thus, the synthesis of AEP block copolymers from anionic polymerization first requires the installation of a precursor functional group by post-functionalization of the aromatic rings of the styrene block, such as chloromethylation or Friedel–Crafts reactions, which needs to be subsequently converted to alkaline stable quaternary ammonium groups.
5. Commercialization and milestones
5.1. Scale-up synthesis and markets
000 m2 were in service, and a service life of longer than 3 years indicates a replacement business of up to 215000 m2 year−1.465 Based on a price of 850 US$ per m2 for the 300 μm-thick Nafion 954 membranes used in this application, this represents a potential business of 180 million US$ per year.
For AEM, the situation is similar. While AEMs are an essential component for AEMWE, their main markets are currently in the field of water treatment, such as seawater desalination, tap water softening, or ED, which are already established markets. Although the requirements for conductivity, thermal and alkaline stability are quite different, it would be attractive for membrane producers to use the same membrane platform to serve the well-established water treatment/purification markets and the just-starting AEMWE market. In this light, it is not surprising that Fumatech, which strongly supports the energy community by providing a variety of ion exchange membranes, is part of BWT Holding (Best Water Technology), a company with over 5000 employees.
The European Union aims to install 40 GW of new electrolysis capacities by 2030. Using a very rough estimation, if the average electrolysis performance is 2 A cm−2 at 2 V (i.e., 4 W cm−2), the required active membrane area would be 1000000 m2, translating to a production volume of 125000 m2 year−1 (2023–2030). A cell design published by Loh et al. for a 100 cm2 cell indicates that the total area required for the cell is up to 30–76% larger than the active area.466 As an estimate, 200000 m2 year−1 may be needed to satisfy the European membrane demand for water electrolysis – if all installed systems would be AEMWE.
| Company | Country | Trade name | Comment | Key properties |
|---|---|---|---|---|
| Dioxide materials | USA | Sustainion® | Imidazolium-functionalized polystyrene, various grades available471 | Sustainion® X37-50 |
| Non-reinforced | ||||
| Thickness: 50 μm | ||||
| IEC: 1.1;472σ25°C: 72 (1 M KOH)472 | ||||
| Ionomr | Canada | AEMION/AEMION + ® | Poly(bis-arylimidazolium)-based, various grades available253,473 | AF3-HWK9-75-X |
| Reinforced (woven Polyether ether ketone) | ||||
| Thickness: 75 μm | ||||
| IEC: 1.9–2.7;474σ: >39474 | ||||
| Orion polymer | USA | TM1 | TMA-based polyphenylene membrane, various grades available163 | TM1 |
| Non-reinforced | ||||
| IEC: 2.1; σ80°C: 130 (water)163 | ||||
| TailorMem | Czechia | Hollex | Non-reinforced PSEBS-DABCO | Hollex ADL 911 NR |
| Non-reinforced | ||||
| IEC: 0.76–0.94;475,476σ30°C: >30,476 75 (water)475 | ||||
| Versogen | USA | PiperION | Piperidinium-functionalized polymer, various grades available166 | PI-20 |
| Non-reinforced | ||||
| Thickness: 20 μm | ||||
| IEC: 2.35 (PAP membrane which appears to be PI-20)477 | ||||
| σ: 71 (30 °C),469 58 (25 °C, PAP membrane which appears to be PI-20)477 | ||||
| PI-15 | ||||
| Reinforced with ePTFE | ||||
| Thickness: 15 μm | ||||
| σ 30°C: 77469 |
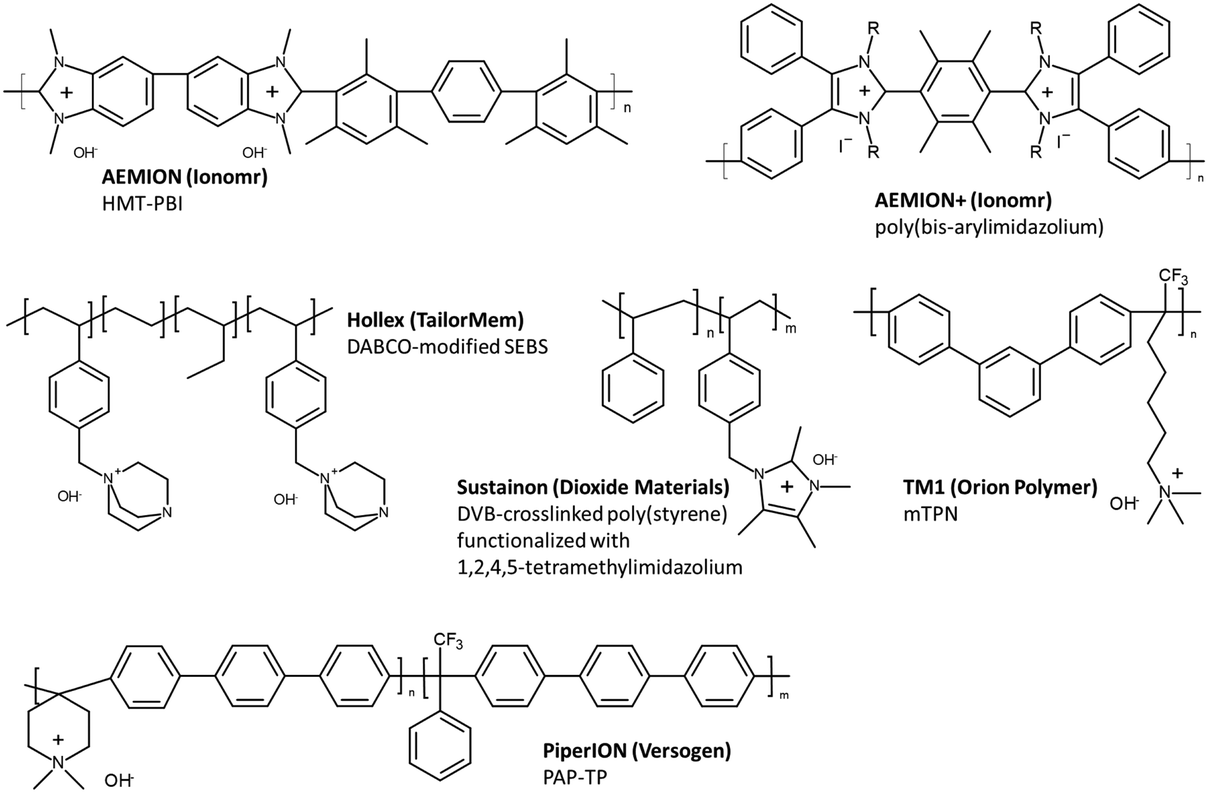 | ||
| Fig. 21 Chemical structures of commercial aryl ether-free AEMs. | ||
:1:2 in a Friedel–Crafts polymerization, followed by functionalization with vinylbenzylchloride as a crosslinking point and permethylation. Moving to the kg-scale (Fig. 22a) required several changes.478 While the monomer concentration remained unchanged during polymerization, the polymer concentration was increased to 50% for the functionalization step, possibly to reduce the amount of the solvent and to gain more robust polymer fibres in the precipitation step. Another concern is the dissipation of heat in large reactors. While small flasks can be efficiently stirred with magnetic stirrers and heat transfer to the wall of the flask is efficient, cooling large reactors can be challenging. Especially when the viscosity of the polymerization mixture increases, the temperature distribution becomes uneven. The choice of optimal stirrer designs and additional cooling circuits becomes necessary. The endpoint of polymerisation is often defined by reaching a certain viscosity. Qualitatively, Song et al. stopped the lab reaction after 9 hours when the magnetic stir bar stopped stirring. In the kg scale, the stirring paddle stopped moving after 24 hours.
 | ||
| Fig. 22 (a) Upscale production of a polymer made by polymerizing m-terphenyl, para-terphenyl and 1-methylpiperidine-4-one in a ratio of 1:1:2, followed by functionalization with vinylbenzylchloride as crosslinking point and permethylation; (b)–(d) pilot-scale membrane production of the membrane. Reproduced with permission.478 | ||
For radical polymerisations, especially with polymerizing undiluted monomers, auto-acceleration by the Trommsdorff–Norrish Effect, or simply the gel effect, poses a safety threat and increases the polymer molecular weight.479 When the viscosity increases, which may also happen locally, the mobility of the reactive chain ends decreases, and termination reactions slow down. At the same time, because the mobility of small monomers is still high, chains continue to grow, resulting in an overall accelerated reaction rate and a temperature increase. If this heat cannot be dissipated, the reactor may experience a thermal runaway situation.
Based on information from IRENA, Chatenet et al. reported that the active areas of AEMWE and AWE are in the range of <300 cm2 and 1–3 m2, respectively, and will increase to 1000 cm2 and 3 m2, respectively.480 Traditionally, many electrolysers are produced in a circular cell design, in contrast to fuel cells, which usually have square cell designs. While circular designs appear to have advantages in cell sealing and pressure distribution, square designs will lead to less material losses when cutting membrane sheets from a roll. If electrolyser companies request circular membranes of 1000 cm2 area, it will be necessary to provide sheets of >36 cm width and length, which may necessitate investments into casting equipment for some membrane companies. On the other hand, the standard roll widths for Nafion 212 membranes are 61 cm and the half roll width of 30.5 cm is made by splitting a roll. Song et al. showed the production of a 107 cm broad AEM (Fig. 22d),478 and ETFE films are available in 150 cm roll width.481
In roll-to-roll casting, membranes are typically prepared by pouring a polymer solution onto a moving support film, for example, a PET film. The thickness of the wet solution is then controlled by passing a blade, and the solvents are evaporated by passing through a drying unit. Finally, some online inspections may control the thickness or absence of pinholes. At the end of the production line, the membrane is rolled up, and sometimes an additional cover sheet is added to fully protect the membrane.482
Blade- or knife-coating (Fig. 23a) may lead to slight wave-like variations in the thickness. An alternative to blade or knife coating is the application of the polymer solution to the support by slot-die coating (Fig. 23b).483 In this method, the polymer solution is pumped into a long reservoir that spans the entire membrane width and has a slit opening on the bottom, through which the polymer solution falls onto the support film. Control parameters include the polymer flow rate, the gap width, and the distance between the slit and the support. A potential issue with this method is that the polymer solution is fed into the slot-die in the middle of the tool, which can result in different flow velocities and thus shear rates close to the solution inlet and at the ends of the tool.484 This can result in a different degree of orientation for the polymer chains in the middle and at the edges of the roll, especially when membranes are melt-processed. For example, Brack and Scherer reported different shrinking behaviour, when membrane stripes sampled at different width positions of an ETFE roll (which presumably was prepared by slot-die-coating of an ETFE melt) were heated.481
 | ||
| Fig. 23 Continuous roll-to-roll membrane production using (a) knife-coating and (b) slot-die coating. | ||
A special case is RG membranes, which do not require casting solutions. While all steps ((i) irradiation, (ii) grafting by immersion in a solution containing the monomers, (iii) final functionalization by reaction with an amine) can be easily done by moving the membrane from one roll to another through a radiation source or reaction bath, the kinetic control of the grafting step could be a potential challenge for mass production and will necessitate online control, e.g. online IR measurements to ensure the degree of grafting. In addition, any change in monomer concentration and solution viscosity, and possible formation of homopolymers, also need to be monitored during the process.
Industry prefers to cast membranes continuously for hours or even days before changing to another membrane type. One reason is that the first several meters of membrane leaving the casting machine are often not within the specifications. It takes time for the heat profile in the drying unit to become uniform, and the gap width of the casting knife may need adjustment. At the end of the casting process, some polymer may be lost in the pumps and tubes, resulting in significant losses for small batches. Therefore, to produce continuously, large solution volumes need to be prepared and used for hours or days, and the shelf-life of the solutions needs consideration. One potential problem is agglomeration and sedimentation, not only of additives like inorganic nanoparticles,485 but also of the polymer itself. For example, while PBI is fully soluble in DMAc, obtained solutions may gel suddenly. To suppress this gelation, which occurs due to an increasing number of chain-chain interactions, LiCl is added to the solutions to shield the hydrogen bond acceptor groups.486
The inclusion of porous supports increases the complexity of the casting process. To some extent, the application of porous supports is easier on a large scale, because the porous membrane will be under constant tension between the two roles, thus resisting wrinkling and allowing for easy pressing into the casted wet film. However, this requires high precision of the porous material. If one side of the porous film is slightly thicker than the other, the thicker side will be stretched when the film is wound up on a roll. When such support is embedded into the straight, freshly cast AEM, the curvature of the porous film will result in wrinkles.
One example is methyl iodide, which is commonly used for the quaternization of tertiary amine groups like polymer-bound dimethylamine, DABCO, or MPIP. Methyl iodide has a very high vapour pressure (∼23 times higher than that of water at 20 °C) and is toxic when inhaled, and is suspected of causing cancer.
Another example is polar aprotic solvents, which are commonly used to cast ion-conducting polymers. DMAc, DMF, and NMP have been identified by the European Chemicals Agency as Substances of Very High Concern.487 One factor to be considered is whether a substance is classified as carcinogenic, mutagenic, or toxic for reproduction, category 1A or 1B. One of the hazard statements for NMP is H360: May Damage Fertility or the Unborn Child. In addition to H360, the hazard statements for DMF and DMAc also include H351: Suspected of Causing Cancer. In December 2022, US EPA published a new risk determination for NMP and announced plans to propose a rule to regulate NMP.488
Ideally, casting solutions should be changed into water- or alcohol-based systems. Water-based systems could reduce capital expenditures, because water can be evaporated in open casting systems without the need to condense the evaporated solvents from the exhaust or take precautions like using an inert atmosphere to avoid explosive solvent vapour mixtures with air. However, the challenge is more complex than simply preparing solutions and obtaining a membrane from them, as there can be a strong correlation between membrane properties and casting solvents for ion exchange membranes. It has been shown for Nafion™ that the choice of solvents has a strong effect on the membrane's mechanical strength, which depends on the degree of crystallization and entanglement.489,490
5.2. Property measurement
The properties of AEPs, especially anion conductivity and water uptake, are heavily impacted by counter anions. Even other traits, such as dimensional stability, mechanical properties, and gas permeability, can all be influenced by counter ions because they change the polarity of the polymers. Understanding the balance between hydroxide, carbonate, and bicarbonate ions is critically important, especially for devices like electrochemical CO2 capture and electrolysers. When it comes to devices designed to separate anionic species, membrane properties with different counter ions are typically reported. Furthermore, even for electrochemical devices relying on hydroxide conduction, polymer properties with other counter ions are often reported because quaternized polymers with a hydroxide counter ion are less processable and more sensitive to carbonation.Because the properties of AEPs are highly influenced by carbonate equilibrium,491 standardized protocols become critical. Back in the early days before 2015, most of the property measurements for AEP materials were adopted by modifying PEM measurement protocols.492 However, AEP-specific property measurement techniques have been developed to compare the material properties with different chemistry. In this section, we briefly review measurements of five key properties.
000 kHz is applied to perturb the system to obtain the impedance data. The most common cell geometries for AEM conductivity are two-probe in-plane window cells493 and four-probe in-plane or through-plane geometries.494,495 In two-probe cell geometry, the current-generating electrodes serve as the voltage-measuring probes, while the four-probe in-plane geometry is connected separately. These two- and four-probe cells provide accurate halogenated and carbonated anionic conductivity,496–498 but accurate measurement of hydroxide conductivity of the AEMs in the trace of carbonate and bicarbonate ions is challenging due to the fast reaction rate of hydroxide ions with atmospheric CO2, replacing hydroxide ions with less mobile carbonate and bicarbonate ions.499 The hydroxide conductivity was measured in deionized water purging and blanketing with an inert gas such as nitrogen or argon,163 but even a short exposure to ambient air may cause carbonation. Ziv and Dekel proposed a method that measures hydroxide conductivity more accurately with an ex situ test set-up that forces the release of (bi)carbonate ions by applying an external electric current through the membrane.500 While the method was initially developed to analyse the conductivity of AEMs for AEMFC with humidified gas streams to transport the released (bi)carbonates away, membranes for AEMWE can be conveniently measured in water without the need for expensive equipment.469
The hydroxide conductivity of AEMs can also be measured with an MEA single cell. In this method, the ohmic resistance of MEAs is normally obtained by current interrupt501 or high-frequency resistance methods.502,503 The hydroxide conductivity is obtained by subtracting the non-membrane resistance from the measured ohmic resistance.504 Some high-performance AEIs are too brittle to cast into free-standing films.165,173 The anionic conductivity of the AEIs can be estimated by the solution (or dispersion) conductivity505 or sheet resistance of the electrode.506 The conductivity of AEIs from the solution and electrode does not reflect the true anionic conductivity; the solution conductivity depends on the dielectric constant of the solvent, and sheet resistance is a function of the tortuosity of the ionomer. Nevertheless, these methods provide useful conductivity-related information for comparison with the standard material that has the known conductivity value. The anion conductivity of some AEI materials that do not form free-standing films can still be measured by preparing a thin film coating on a substrate.507–509
The gas permeability is strongly affected by the hydration level of membranes. Increasing the membrane hydration level increases permeability; therefore, AEMs used in water electrolysers need to be fully hydrated for the measurement. Oxygen crossover is of less concern for hydrocarbon-based AEMs because of its low permeability compared to hydrogen.514,515
The polymer structure analysis using NMR spectroscopy is also an effective method to calculate IEC. The peak integration of a 1H NMR spectrum is typically used for IEC calculation.522 The sample of interest needs to be fully dissolved in a deuterated solvent (e.g., DMSO-d6, MeOD) for the solution NMR analysis. The method cannot be used for AEPs with limited solubility, such as crosslinked/reinforced polymers, block copolymers, and grafted copolymers. AEPs prepared by heterogeneous quaternization (cast as a film and then quaternized by immersion in aqueous amine solution) also show limited solubility; the degree of functionalization of precursor groups in these polymers is often analysed using NMR before quaternization using a less polar solvent, e.g., CDCl3, and compared with the titrated IEC value after quaternization to confirm quantitative conversion into cation.285 Solid-state NMR techniques have not been used much for measuring IEC due to their insufficient resolution for integration.523
The evaluation of ionomer adsorption on catalyst surfaces can be conducted through either direct or indirect methods. Direct measurements offer precise information about the adsorbed species but are often challenging due to the intricate behaviour of these species and the difficulty in separating them from bulk ionomers. On the other hand, indirect measurements, while sensitive, provide information specific to the measuring conditions during electrochemical responses.
Cationic species, commonly adsorbed at the anode of fuel cells and electrolysers, can be directly measured using techniques like infrared reflection absorption spectroscopy139 and neutron reflectometry.532 Alternatively, cyclic voltammetry can be employed as an indirect method to gauge the reduction in electrochemical surface area (ECSA) caused by adsorbed cationic group species. However, it is important to note that the ECSA reduction might not precisely correlate quantitatively with the adsorption area of cationic groups, as the adsorption sites for hydrogen and cationic groups may not be identical. Additionally, the ECSA is typically measured at relatively low potentials, and the adsorption of cationic groups at high potentials, such as the anode of electrolyser, may exhibit different behaviours.
For ionomer fragment adsorption, the electrochemical oxidized products of ionomers are examined by 1H NMR.122,123,533 This involves MEA testing at high potentials, followed by dissolving the ionomer in the NMR solvent to observe the structural changes induced by electrochemical oxidation. While this method offers detailed insights into the ionomer's structural alterations, it may pose challenges when dealing with insoluble ionomers. In such cases, X-ray photoelectron spectroscopy can be employed to detect oxidation products.127,140,534 Electrochemical analysis, a sensitive technique for measuring electro-oxidative current density, involves studying cyclic voltammograms of the electrodes. These voltammograms can reveal the delayed potential of catalyst oxide formation and the electrochemical oxidative current density at high potentials,135 typically exceeding 1.2 V, which differs from normal cyclic voltammograms used in catalyst studies.
5.3. Milestones
The property milestones of AEMs were first set by the US DOE the Hydrogen and Fuel Cell Technologies Office (HFTO) for AEMFCs in 2013,535 focusing on the alkaline stability of AEMs. The target performance is retaining ≥99% of the original IEC after 1000 hours in an alkaline solution at a temperature higher than 80 °C. In 2016, the US DOE ARPA-E Integration and Optimization of Novel Ion-Conducting Solids (IONICS) program set the AEM milestones in the 11 categories (Table 2). In 2019, the US DOE HFTO Alkaline Membrane Fuel Cell Workshop suggested a few modifications to the IONICS technical target based on the participants’ opinions.536| Number | Metric | ARPA-E (2016) | HFTO (2019) |
|---|---|---|---|
| 1 | Membrane chemical stability (at ≥80 °C immersed in a pH ≥ 14 solution) | ≥1000 hours with ≤2% loss in IEC, ionic ASR, spectroscopic measures of membrane state, and mechanical properties | ≥1000 hours with ≤5% loss in IEC and conductivity: should include spectroscopic characterization. |
| 2 | Component area over which property values are achieved to within ≥90% uniformity | ≥100 cm2 | Delete this target or decrease priority |
| 3 | Ionic ASR (hydroxide form, 80 °C, liquid equilibrated) | ≤0.04 Ω cm2 | Add a target at 40 °C: 0.04 Ω cm2 |
| 4 | Ionic ASR (80 °C, ≤50% RH, under air exposure, i.e., in presence of 400 ppm CO2) | ≤0.08 Ω cm2 | Change to a measurement at 80% RH under CO2-free air exposure. |
| 5 | Mechanical durability during humidity cycling | ≥20000 RH cycles |
Delete this target. |
| 6 | Electronic ASR | ≥1000 Ω cm2 | No change proposed |
| 7 | Humidity stability factor | >5 | No change proposed |
| 8 | Swelling in liquid water at 25 °C | <50% | To be measured as linear swell in X–Y plane in water, membrane in OH− form. |
| 9 | Pressure differential (bar) | ≥1 | Delete for fuel cell and flow battery applications; electrolyser industry input on burst test or other relevant test and metrics needed. |
| 10 | H2 crossover and O2 crossover | ≤25 nmol cm−2 s−1 | Change to <5 mA cm−2 for H2 crossover, eliminate O2 crossover target. |
| 11 | Cost for membrane that can be practically integrated in a device | ≤20 $ m−2 | No change proposed |
| 12 | H2O transport | n/a | New target proposed for water transport: >4 mmol cm−2 s−1. |
| 13 | Ionic permeability | n/a | New target proposed for flow batteries to limit permeability of other ions: <7 × 10−8 cm2 s−1. |
In 2016, the participants in the Alkaline Membrane Fuel Cell Workshop sponsored by US DOE HFTO suggested the AEM property milestone should be connected with PGM loading.537 Based on the suggestion, US DOE HFTO published the milestones for AEMFC538 and AEMWE (Table 3).539 The 2026 target performance of AEMWE is ∼60% compared to that of the PEMWE.
| Year | Milestone |
|---|---|
| AEMFC | |
| 2021 | Initial performance: 100 mW cm−2 at 0.8 V with ≤0.2 mgPGM cm−2, H2/air, T ≥ 80 °C, P ≤ 250 kPa |
| 2022 | Initial performance: 0.65 V at 1000 mA cm−2 on H2/O2; durability: ≤10% voltage degradation over 1000 hours; T ≥ 80 °C, P ≤ 150 kPa; total PGM loading: ≤0.2 mg cm−2 |
| 2024 | Membrane: H2 crossover ≤15 mA cm−2 during 1000 hours OCV hold at 70% RH and ≥80 °C, H2/N2 |
| 2025 | Initial performance: 1000 mW cm−2 at 0.65 V; H2/air (CO2-free) with total PGM loading ≤0.125 mg cm−2, H2/air, T ≥ 80 °C, P ≤ 250 kPa |
| 2030 | Initial performance: ≥600 mW cm−2 under H2/air (maximum pressure of 1.5 atm) in PGM-free MEA |
| Ultimate | Initial performance: ≥1000 mW cm−2 at rated power; PGM-free; H2/air; T ≥ 80 °C, P ≤ 250 kPa |
| AEMWE | |
| 2026 | ≥2.0 A cm−2 at 1.8 V with a degradation rate of <4 mV kh−1 (tested at least 25 cm2) in both water and supporting electrolyte feed (∼0.5 M) |
| PEMWE | |
| 2026 | ≥3.0 A cm−2 at 1.8 V with a degradation rate of 2.3 mV kh−1 over at least 1000 hours (tested at least 25 cm2) |
6. Device performance
6.1. Anion exchange membrane fuel cells
AEMFC performance is typically measured by polarization curves. Because AEMFC is not yet a mature technology, most AEMFC performance has been measured under H2/O2 conditions to decouple CO2-related complications and cell degradation. Most papers reported the PPD as a metric of the performance and voltage degradation rate at a constant current as a metric of cell durability. The properties of materials constituting the MEAs and their operating conditions affect the fuel cell performance as a result,5,540,541 and thus it is difficult to assess AEM and AEI independently from the PPD measured with different MEAs under different operating conditions. However, since AEMs and AEIs play a critical role in fuel cell performance and durability, it is useful to survey these components that show high performance in AEMFCs. In this section, we analyse the data that report more than 0.5 W cm−2 PPD to provide information regarding the implementation of aryl ether-free AEMs and AEIs for high-performance AEMFCs.Before 2015, the PPDs of most AEMWEs were less than 0.3 W cm−2. The first high-performance AEMFC was reported by Zhuang and co-workers using poly(aryl ether sulfone) membrane and ionomers, which achieved a PPD of 1 W cm−2 at 80 °C.542 Since then, about 10 articles reported more than 0.5 W cm−2 PPD using aryl ether-containing polyaromatic membranes (Table S1, ESI†). The first aryl ether-free polyaromatic AEM that showed more than 0.5 W cm−2 in AEMFCs is a perfluoroalkylene with a pendent ammonium group, reported by Miyatake and co-workers in 2017.192 Subsequently, several research groups reported MEAs that exhibited more than 0.5 W cm−2 with aryl ether-free polyaromatic AEM and polyfluorene (FLN) AEI combinations (Table S2, ESI†). A lightly-branched AEM with enhanced stability exhibited AEMFC performance of approximately 2 W cm2 PPD operating at 100 °C, and 195 hours of durability with 140 mV h−1 voltage decay rate, and showed balanced water management.543 Scott and co-workers reported a PPD of 0.61 W cm−2 using radiation-grafted aryl ether-free polyolefinic AEM in 2015.433 Approximately 15 articles report more than 0.5 W cm−2 PPD using various aryl ether-free polyolefinic AEMs as of August 2023 (Table S3, ESI†).
At least ten articles reported more than 0.5 W cm−2 PPD for PGM-free catalysed AEMFCs under H2/CO2-free air conditions (Table S4, ESI†). The AEMFCs that showed high performance mostly used either radiation-grafted or acid-catalysed phenyl piperidinium, probably because of the commercial availability of the latter. However, one should note that those AEMs have high IEC (2.1–2.9 mequiv. g−1), low thickness (15–25 μm) and relatively high operating temperature (∼80 °C).
Fig. 24a shows the number of publications of high-performance AEMWEs as a function of types of AEMs. The number of publications reporting aryl ether-containing polyaromatic AEMs in high-performance AEMFCs ranges from 1 to 4 each year. The number of publications for aryl ether-free polyaromatic (aryl ether-free PAr) AEMs substantially increased in recent years, suggesting that tailoring aryl ether-free PAr structure using different chemistries has now become well-established. Also, note that 80% of the AEMs in the aryl ether-free PAr publications used the acid-catalysed polyhydroxyalkylation route.
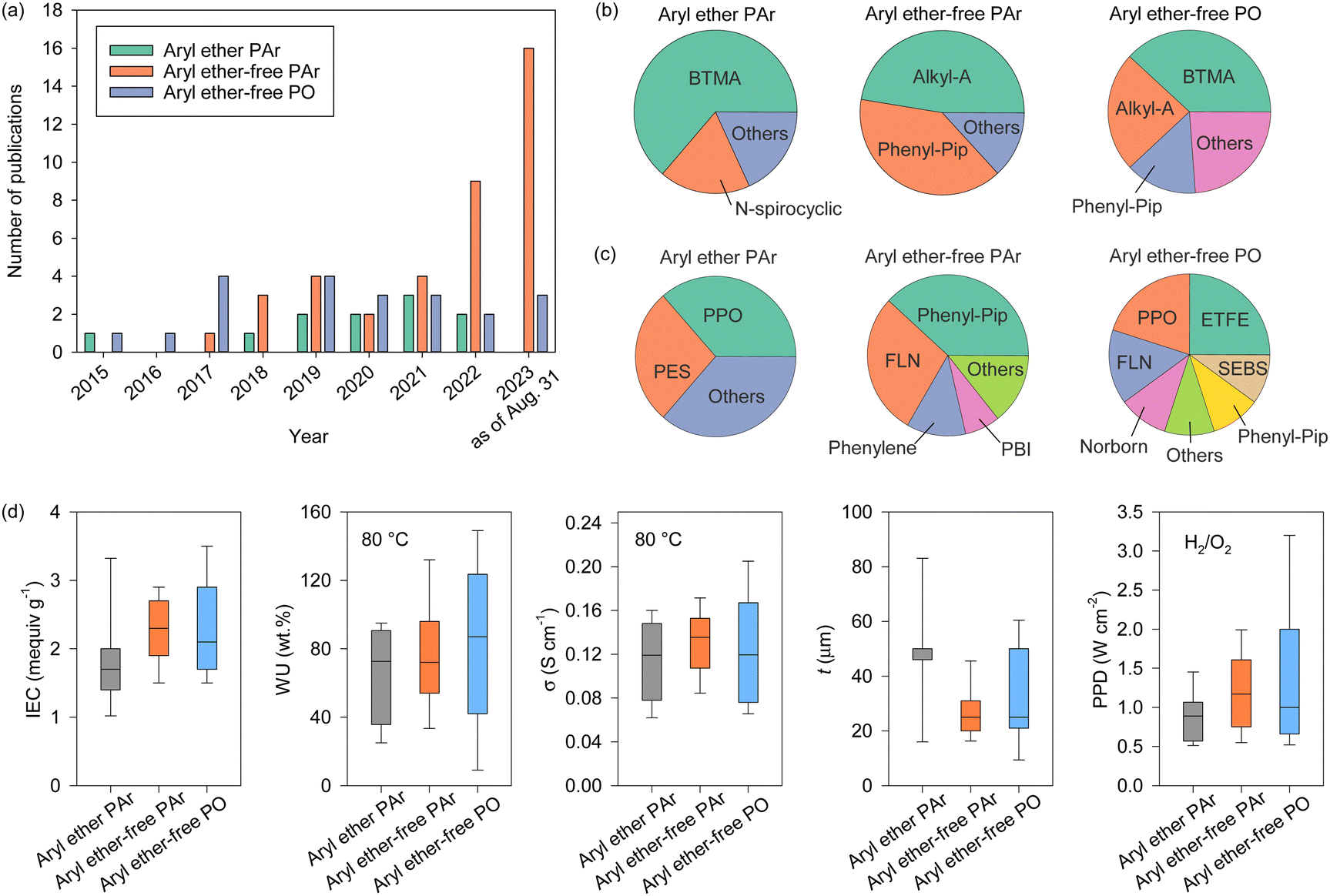 | ||
| Fig. 24 Summary of AEMFC performance using aryl ether-free polymers. (a) The number of publications that reported the PPD of more than 0.5 W cm−2. (b) The cationic functional group of the AEMs used for high-performance AEMFCs, (c) the AEI in MEAs used for high-performance AEMFCs, (d) the average IEC, WU, σ, t, and PPD of the AEMs used for high-performance AEMFCs. The WU and σ are data at 80 °C. | ||
Fig. 24b shows the cationic functional groups of the AEMs in the high-performance AEMFCs. For aryl ether-PAs, most studies used BTMA, likely because quaternization with trimethylamine in the chloromethylated or methylbrominated phenyl is well-established. For aryl ether-free PArs, alkaline-stable cationic functional groups such as alkyl ammonium (alkyl-A)168,230,544–554 or phenyl-piperidinium (phenyl-PiP)184,478,551,555–561 are prevalent as cationic functional groups. Other alkaline-stable cationic functional groups such as benzimidazolium562 and alkyl piperidinium563–565 have been frequently used as well. For aryl ether-free polyolefins (aryl ether-free POs), BTMA, alkyl-A, and phenyl-Pip are the most widespread functional groups. BTMA is commonly used for radiation-grafted polymers and other polyolefinic AEMs. Overall, the most prevalent cationic functional group is alkyl-A (appearing in 23 publications), followed by phenyl-PiP (appearing in 17 publications) and BTMA (appearing in 15 publications).
Fig. 24c illustrates the type of AEIs in the catalyst layers used for high-performance AEMFCs.566 For most of the aryl ether PAr studies, the same materials used for the AEM are employed as the ionomeric binders and quaternized PPO and poly(aryl ether sulfone) being the two most usual choices of aryl ether-containing AEI. The ionomeric binder that exhibited the highest performance in AEMFCs when paired with an aryl ether PAr-based AEM was a radiation-grafted ETFE powder.567 For aryl ether-free PArs, polyaromatics with the phenyl-Pip functional group were widely utilized. It is worth noting that phenyl-PiP is the most prevalent cationic group (appearing in 19 publications) compared to alkyl piperidinium, despite its lower alkaline stability (see Fig. 3). This is probably due to the higher fuel cell performance of phenyl-PiP, as competitive adsorption of piperidinium reduces phenyl adsorption on electrocatalysts.530 FLN ranks as the second most popular ionomer (appearing in 14 publications), possibly because its weaker interaction with electrocatalysts enhances AEMFC performance. Kim and co-workers demonstrated that the AEMFC performance using FLN ionomers is significantly higher than that using poly(phenylene) ionomers due to less phenyl adsorbing characteristics of fluorene.164 The superiority of the polyfluorene backbone to poly(phenylene) ionomers was also discussed by Lee and colleagues, who prepared poly(fluorenyl aryl piperidinium) AEMs by combining phenyl-PiP and FLN, resulting in high fuel cell performance.170 Similar FLN ionomers showed the highest H2/O2 AEMFC performance for polyaromatic-based MEAs (2.6 W cm−2), although the cell was evaluated under very high flow rate and back pressures.556 For aryl ether-free PArs, various AEIs, including quaternized PPO, ETFE, FLN, polynorbornene, and phenyl-Pip were utilized. The wider range of ionomeric binders used for aryl ether-free PO-based MEAs is likely due to the low solubility of quaternized polyolefins. Note that ETFE and most norbornene-based AEIs reported are used as particulate dispersion. Using a particulate ionomer, Kohl and co-workers demonstrated the highest performance (PPD = 3.5 W cm−2).307
Fig. 24d illustrates the properties of AEMs used in high-performance AEMFCs. The average IEC of the aryl ether PAr AEMs was 1.7 mequiv. g−1, notably lower than those of the aryl ether-free PArs and POs (2.3 and 2.0 mequiv. g−1, respectively). The higher IEC for the aryl ether-free systems reflects the trend of using more conductive AEMs to enhance AEMFC performance.568 Due to their higher IEC, these aryl ether-free systems also exhibited higher water uptake (WU) and hydroxide conductivity (σ). The average thickness of the aryl ether PAr AEMs was 48 μm, thicker than the aryl ether-free PAr AEMs (25 μm) and aryl ether-free PO AEMs (24 μm). Owing to their higher conductivity and lower thickness, the aryl ether-free system demonstrated higher PPD compared to the aryl ether PArs-based AEMFCs. The average PPDs of the aryl ether-free PAr and aryl ether-free PO-based AEMFCs were comparable, at 1.24 and 1.31 W cm−2, respectively. The relatively high variation of PPD for the aryl ether-free PO-based AEMFCs can be attributed to the substantially high performance of MEAs using particulate AEIs.
In assessing the durability of AEMFCs, we note the following key points: (i) MEAs using more alkaline-stable cationic groups, such as alkyl-A, demonstrated greater durability than those using less alkaline-stable cationic groups like BTMA;435,569 (ii) MEAs using AEMs with lower IEC and WU are more durable than those with higher IEC and WU;567,570 (iii) MEAs using aryl ether-free AEMs exhibited greater durability compared to those with aryl ether-containing AEMs;85,571,572 (iv) While high current density operation poses more challenges in water management, comparable AEMFC durability can still be achieved with appropriate humidity control.547,573
Table S5 (ESI†) presents the durability of AEMFC using aryl ether PAr AEMs from 2012 to 2023. Only three cases with a PPD greater than 0.5 W cm−2 demonstrated durability exceeding 100 hour.567,569 The most durable MEA used an N-spirocyclic-functionalized PES AEM, showing only 7% voltage loss after 550 hours of operation at 61 °C.567 The second most durable MEA used an imidazolium-functionalized AEM, exhibiting 70% voltage loss after 960 hours at 60 °C.569 Both cells used low IEC (∼1.4 mequiv. g−1) to maintain cell durability. In contrast to the lower durability of aryl ether PAr-based MEAs, those based on aryl ether-free AEMs showed high durability (Table S6, ESI†). The longest durability reported in AEMFC used an alkyl-A-functionalized polynorbornene AEM (10 μm-thick) with polynorbornene AEI, operating for up to 3600 hours at 75 °C.574 A ferrocenium-functionalized polyethylene AEM also demonstrated remarkable durability, with only a 4% voltage loss after 500 hours of operation at 120 °C.575
In analysing the durability of AEMFCs, we focused on MEAs with a PPD greater than 0.5 W cm−2, as detailed in Tables S5 and S6 (ESI†) (Fig. 25). We identified five MEAs with aryl ether PAr AEMs, eleven MEAs with aryl ether-free PAr AEMs, and six MEAs with aryl ether-free PO AEMs. The average IEC of the aryl ether PAr AEMs was substantially lower (1.5 mequiv. g−1) compared to the aryl ether-free AEMs (2.5 mequiv. g−1). Interestingly, the aryl ether PAr AEMs used in durability evaluations were thinner than the AEMs used for those performance evaluations. For instance, the average thickness of aryl ether PArs for durability tests was 20 μm, compared to 24 μm for performance tests. This enhanced durability with thinner AEMs is likely attributed to improved water management facilitated by higher water back-diffusion.567,576 Operating at slightly higher temperatures and thinner AEMs, the PPD of aryl ether-free PO AEM-based MEAs was higher (2.0 W cm−2) than that of others (1.0 W cm−2 for aryl ether PAr MEA and 1.5 W cm−2 for aryl ether-free PAr MEAs). MEAs using aryl ether-free PAr and PO AEMs were tested for longer operation times. The average duration for aryl ether PAr, aryl ether-free PAr, and aryl ether-free PO MEAs was 100, 400, and 610 hours, respectively, suggesting superior durability with aryl ether-free AEMs.
 | ||
| Fig. 25 Summary of AEMFC durability using aryl ether-free AEPs. (a) Analysis in terms of AEMs. AEMs were selected from Tables S5 and S6 (ESI†) with the criterion of PPD higher than 0.5 W cm−2. (b) Analysis in terms of the cationic functional groups and the form of AEIs. AEIs were selected from Tables S5 and S6 (ESI†) with the criteria of PPD higher than 0.5 W cm−2 and duration is 120 hours. | ||
In our analysis of AEIs, we selected the MEAs that achieved a PPD of greater than 0.5 W cm−2 and operated for a duration of ≥120 hours. We observed that the voltage degradation rate strongly depends on the type of cationic group and the method of AEI preparation (whether dissolved/dispersed in solution or ground to powder and used as particulate). Interestingly, only four types of AEIs demonstrated both high performance and durability, BTMA, phenyl-Pip, alkyl-A, and particulate ionomers, as compared in Fig. 25b. The cationic functional groups in the particulate ionomers were either BTMA or alkyl-A. Particulate ionomers exhibit distinctive characteristics, including high gas transport properties, and therefore can be categorized separately from ionomers prepared as dispersions. The AEMs for the BTMA AEI cells were slightly thinner (20 μm) compared to those for the phenyl-PIP (25 μm) and alkyl-A (30 μm) AEI cells. Despite having thinner AEMs, the BTMA AEI cells were lower (1.1 W cm−2) than the cells using phenyl-Pip (1.6 W cm−2) and alkyl-A (1.5 W cm−2) AEIs at similar operating temperatures. The MEA using particulate ionomers utilized thinner AEMs and showed higher PPDs. The most notable finding from this analysis is the voltage degradation rate; the BTMA cells exhibited more than two times higher voltage degradation rate (1.1 mV h−1) than those of phenyl-Pip (0.47 mV h−1) and alkyl-A (0.26 mV h−1) cells. The particulate AEI cells showed the lowest voltage degradation rate (019 mV h−1), suggesting that water and gas transport in the electrodes significantly influence AEMFC durability.
6.2. Water electrolysers
AEMs used for AEMFCs can be adapted for use in AEMWEs. We surveyed the AEMs used in high-performance AEMWEs over the last three years, focusing on those with high performance (Tables S7 and S8, ESI†). It is interesting to note that the AEMWE performance has been reported under both water and liquid electrolyte-feed conditions. This is because the performance of AEMWE greatly improves with liquid electrolyte-feed conditions. The main cause of this is the subject of ongoing work, but it is still under debate. The possible reasons include increasing catalyst-electrolyte contact area,577,578 lowering cell resistance,11 enhanced basicity,579,580 and mitigating ionomer adsorption140 by liquid electrolytes.Fig. 26a displays the types of AEMs used in high-performance AEMWEs. Compared to the number of publications on AEMs for AEMFCs, there have been fewer numbers on AEMs for AEMWEs, reflecting the more recent focused interest in AEMWEs. Aryl ether-free PAr AEMs outnumbered aryl ether PAr and aryl ether-free PO AEMs, although the difference in publication numbers was not significant. alkyl-A and phenyl-PiP are the two most commonly used cationic functional groups in AEMs for AEMWE applications, similar to their use in AEMFCs. However, imidazolium and DABCO are also widely used. The relatively high number of articles on imidazolium and DABCO can be attributed to the frequent use of commercially available AEMs such as X37-50 (Sustainion®) and Fumion (AGC Inc.) in high-performance AEMWEs.
 | ||
| Fig. 26 Summary of AEMWE performance using different types of AEMs and AEIs. (a) The number of publications of high AEMWE performance over the last three years (the current density of pure water and 1 M KOH-fed PGM-catalysed AEMWEs at 1.8 V: >0.4 A cm−2). (b) The properties of AEMs used in high performance AEMWEs. (c) The number of publications of AEIs as a function of the type of AEI binders in MEAs used for the pure water-fed AEMWEs, and the number of publications of AEIs as a function of the type of AEIs in MEAs used for the 0.1 M KOH-fed AEMWEs. | ||
The property requirements of AEMs used for AEMWEs slightly differ from those in AEMFCs, as shown in Fig. 26b. For AEMWEs, the average IEC of aryl ether-free PO AEMs is lower (1.3 mequiv. g−1) compared to their use in AEMFCs (2.5 mequiv. g−1). This discrepancy is because commercial AEMs with relatively low IECs have shown high performance in the presence of a liquid electrolyte. The AEM thickness for AEMWEs is about 20 μm thicker than that for AEMFCs, suggesting that more resistant AEMs are needed for AEMWEs. This requirement is due to the higher mechanical demands under AEMWE operating conditions, the need to prevent gas crossover, and a less strict requirement for low membrane resistance when liquid electrolyte is present. The operating temperatures of AEMWEs reported in research papers are similar to those of AEMFCs (50–90 °C). However, it is important to note that the operating temperature of AEMWEs in a commercial unit tends to be at the lower end of this range for longer-term operations.
Fig. 26c compares the AEI used in AEMWEs under pure water and 1 M KOH feed conditions. Under both conditions, aryl ether-free PAr AEIs were more frequently used than other types of AEIs. Interestingly, aryl ether PAr AEIs were often used for 1 M KOH-feed conditions, which could be due to several reasons. First, adverse phenyl adsorption on the catalyst may be mitigated under the liquid alkaline solution feed conditions.140 Secondly, the degradation of AEI is less of a concern with liquid electrolyte feed. Third, commercially available aryl ether PAr AEIs are more accessible for catalyst developers. Unlike AEMFCs, no significant correlation between the reported AEMWE performance and the types of AEIs used.
Before 2015, several papers reported the performance and durability of AEMWEs under relatively mild conditions. The performance reported during this period was poor, yet those studies provided insights into performance-limiting factors. In 2012, Wang and co-workers reported on the 1 M KOH-fed AEMWE performance of a commercial A201 AEM (Tokuyama Corporation), which operated for over 500 hours at 50 °C and a constant density of 0.2 A cm−2.581 This work suggested that AEMWE operations exceeding 500 hours are possible with alkaline unstable aryl ether-containing ionomer (aminated Radel®) under KOH-fed conditions. A UV-grafted LDPE-DABCO-RG-AEM was tested in a 1 wt% K2CO3-fed AEMWE, yielding less than 0.05 A cm−2 at 1.75 V.582 However, the AEMWE cell demonstrated durability for 500 hours at 45 °C and outlet pressure of 20 bar. Since 2013, the team led by Scott and Mamlouk at Newcastle University also tested AEMWEs containing RG-AEMs.583–586 These studies generally involved RG-AEMs alongside non-RG-ionomers (such as SEBS or PPO-based ionomers). The best performance reported in 2021 was 0.13 A cm−2 at 1.75 V with an LDPE-TMA-RG-AEM (IEC = 2.3 mequiv. g−1) and a SEBS-based ionomer (IEC = 1.9 mequiv. g−1); other conditions included a Pt/C cathode, NiCo2O4-based cathode, 0.1 M supporting electrolyte, and testing at 40 °C. Around 0.8 A cm−2 was achieved by 2.0 V. In 2014, Kim et al. reported more than 2000 hours of long-term performance of pure water-fed AEMWEs at 50 °C and 0.2 A cm−2 using aryl ether-free DAPP AEM.87 Although the performance of the AEMWE was modest (0.2 A cm−2 at 2.2 V), this work was the first to demonstrate long-term performance with 100 psi differential pressure. In the same year, Comotti et al. reported the outstanding PGM-free catalysed AEMWE durability (1000 hours, voltage degradation rate: ∼0.15 mV h−1) under 1 wt% K2CO3 feed conditions.87 This paper highlighted the potential for long-term operation using PGM-free Ni/(CeO2-La2O3)/C HER and CuCoOx anode catalysts. After 2015, more publications reported aryl ether-free AEPs to enhance the durability of AEMWEs.515
We compiled the data from recent papers on AEMWE MEAs that showed high durability (≥100 hours with a low voltage degradation rate), presented in Table S9 (ESI†) and Fig. 27. The performance and durability of AEMWEs are strongly influenced by liquid electrolytes and PGM-free catalysts (Fig. 27a). A 1 M KOH feed solution significantly enhanced cell performance with an average current density at 1.8 V for the 1 M KOH-fed AEMWEs was 1.2 A cm−2, considerably higher than that for the pure water-fed AEMWEs (0.75 A cm−2). PGM-free catalysed AEMWEs achieved a comparable current density (0.72 A cm−2) under 1 M KOH-fed conditions. However, durability test durations showed more variation between pure water and 1 M KOH-fed AEMWEs. The average duration for pure-water-fed AEMWEs was only 180 hours, whereas 1 M KOH-fed AEMWEs exhibited a much longer average duration of approximately 1000 hours. Li and co-workers conducted comparative durability tests of AEMFC and AEMWE using the same AEM and AEI for each cell, demonstrating that AEMWE under 1 M KOH feed conditions has significantly better durability than that of AEMFC.587 Holdcroft and co-workers investigated the pure water-fed AEMWEs using polybenzimidazolium-based AEM and AEIs. They found that a crosslinked derivative of the hexamethyl-p-terphenyl (HMT-PMBI) membrane, with four times lower volumetric swelling, did not enhance voltage stability with pure water feed, suggesting that excessive swelling of HMT-PMBI membrane is not the primary cause of performance loss. However, the hydrophobic and lower water uptake properties of the benzylated version of the non-crosslinked polybenzimidazolium AEI reduced dimensional swelling, leading to a fourfold increase in the lifetime of the AEMWE system operating with pure water. This indicates that the development of AEIs and catalyst layer is crucial for maximizing AEMWE lifetimes.184 This result suggests that the dimensional stability of the state-of-the-art aryl ether-free AEMs is not the primary factor limiting the durability of AEMWEs. Several AEMWEs with 1 wt% K2CO3 or 0.1 M KOH feeding conditions showed stable operation for over 300 hours, while AEMWEs with 1 M KOH feed demonstrated more than 1000 hours of stable operation. These results indicate that a major limiting factor for AEMWEs with low-concentration KOH feed is not necessarily the alkaline stability of the AEM, but rather issues related to the AEI. Several potential reasons for this include the accumulation of carbonated species in electrodes from water-feed AEMWEs, making it challenging to remove all the carbonated species from pure water and thereby increasing cell resistance of pure water-fed AEMFCs.430 Another possibility is phenyl adsorption and subsequent electrochemical oxidation of AEIs.122 Liquid electrolytes aid in the desorption of phenyl groups from the electrocatalysts.140 A third possibility is catalyst reconstruction with the pure water-fed AEMWEs due to the relatively low local pH environment.588 Although supplying highly concentrated KOH-supporting electrolytes (>1 M KOH) raises concerns about AEM and AEI degradation, lifetimes exceeding 1000 hours have been demonstrated. Nonetheless, more alkaline-stabile AEM and AEI need to be developed and tested. In this context, alkaline-stable non-quaternized ion-solvating membranes have been proposed as an alternative approach.150,589,590 Current durability tests generally lack post-mortem analysis of AEM/AEI components, which is necessary to understand the degradation mechanisms of AEPs under the AEMWE's operating conditions.
 | ||
| Fig. 27 Summary of AEMWE durability using aryl ether-free polymers: (a) the AEMWE performance as a function of liquid electrolytes, (b) the performance and durability of the pure water-fed AEMWEs,430,588,591–593,595–597 and (c) the performance and durability of the liquid electrolyte-fed AEMWEs.310,471,473,478,587,594,597–606 | ||
Since 2020, there have been notable improvements in pure water-fed AEMWEs. In 2020, Kim and co-workers reported high performance of AEMWEs using a high IEC polystyrene ionomer.430 The high IEC (3.3 mequiv. g−1) increases the local pH in the electrodes, thereby improving cell performance. The Ni–Fe anode catalysed AEMWE reached a current density of 2.7 A cm−2 at 1.8 V. However, the durability of the cell was compromised due to excessive water uptake by the AEI, leading to the loss of catalyst particles from the electrodes. In 2021, Boettcher et al. reported a durability of 180 hours using a commercially available aryl ether-free PiperION ionomer.591 Despite a high voltage degradation rate of 0.67 mV h−1, the cell operated at a reasonably high current density of 0.5 A cm−2. The same year, a similar durability (170 hours with a voltage loss rate of 0.7 mV h−1) at the constant current density of 0.5 A cm−2 was reported with a commercial ionomer (Sustainion®).592 In 2022, Mustain and co-workers achieved stable performance in a polynorbornene-based cell at 1 A cm−2 for 500 hours (voltage degradation rate: 93.5 μV h−1). They hypothesized that performance loss in polynorbornene-based AEM and AEI for pure water-fed AEMWEs might be due to the incomplete removal of salt or other impurities when exposed to KOH solution.593 Zhuang et al. reported a stable AEMWE performance at a relatively low current density (0.2 A cm−2) at low cell voltage (0.55 V) using a phosphate buffer solution. They suggested that the electrocatalysts might be reconstructed at neutral pH, requiring periodical replenishing of the cell with KOH and buffer solution to maintain stable performance at a relatively high operating temperature of 80 °C. The AEMWE showed high performance (2 A cm−2 at 1.8 V) with pure water-fed conditions.588 More recently in 2023, Kwon and co-workers reported stable, pure water-fed, PGM-free catalysed AEMWEs using a commercial AEM (Sustainion®). The high performance (current density at 1.8 V = 0.6 A cm−2) and low current density loss with PGM-free catalysts are encouraging. The performance and durability of pure water-fed AEMWEs are summarized in Fig. 27b. Notably, all the high-performing pure water-fed AEMWEs used aryl ether-free AEMs and AEIs, indicating that aryl ether-containing polymer electrolytes may not be practical for pure water-fed AEMWEs due to their apparent instability.
The durability of AEMWEs under 1 wt% K2CO3 or 0.1 M KOH feed conditions have been reported as much superior, mainly because the electrodes are unaffected by CO2 contamination (Fig. 27c). In 2021, Ayers et al. reported 400 hours of stable operation with near-zero voltage degradation in an AEMWE using a commercial AEM (Durion®, Xergy) at 0.75 A cm−2 under 1 wt% K2CO3 feed conditions.594 This work utilized a 30 μm-thick polyphenylene-based composite membrane in a 28 cm2 cell and demonstrated durability under differential pressures (60–100 psig). The following year, Kohl and co-workers demonstrated 600 hours of durability under 0.1 M NaOH feed condition. They used a 30 μm-thick polynorbornene composite AEM and Ni–Fe anode catalysts for the MEA and demonstrated improved performance after the durability test at the current density of 1 A cm−2. In the same year, Choi and co-workers reported 3000 hours of durability for a PGM-free anode-catalysed AEMWE operating at 0.5 A cm−2 under 0.1 M KOH conditions, with a voltage degradation rate of only 11.3 μV h−1. These reports attest that 1 wt% K2CO3 or dilute alkali metal solution feed is sufficient for the long-duration operation of AEMWEs.
Using higher concentration KOH further improves AEMWE performance (Fig. 27c). The first demonstration of high durability in 1 M KOH-fed PGM-free catalysed AEMWEs was reported by Masel and co-workers in 2018. They successfully operated AEMWEs using a commercially available AEM (Sustainion®) at a high current density (1 A cm−2) and 60 °C for 2000 hours, with a voltage degradation rate of only 5 μV h−1.471 In 2019, Holdcroft et al. reported stable performance for 150 hours in 1 M KOH-fed AEMWEs. The Ni alloy-based, PGM-free electrodes showed high performance and durability at a high current density (1 A cm−2).600 In 2021, Meroueh et al. used the same membrane and extended the duration to 12000 hours with a voltage degradation rate of 1 μV h−1.602 Arico and co-workers reported high durability of 2000 hours using another commercially available membrane (FAA-3-50, Fumasep®) during start-stop cycles. Despite some recoverable performance loss during the test, a very small unrecoverable loss was observed at the end of the durability test.603 In 2023, Wang and co-workers reported high-performance PGM-free AEMWEs using a commercial AEM (Sustainion®). The current density of the cell reached 1.56 A cm−2 at 80 °C with stable performance at 0.5 A cm−2 for 100 hours.607 In 2023, two notable results were published using non-commercial AEMs. Xu and co-workers reported high-performance AEMWEs (5.3 A cm−2 at 1.8 V) with stable performance for 3000 hours. They used phenyl-piperidinium AEM having relatively high IEC (2.5 mequiv. g−1) with crosslinking to reduce the water uptake of the AEM.478 Zhang and co-workers used a poly(biphenyl alkylene) membrane to demonstrate 3500 hours of stability at 1 A cm−2 where the AEMWE cell showed a very low voltage degradation rate (6.5 μV h−1).604 Also in 2023, Holdcroft et al. reported approximately 5000 hours of operation of an MEA using a polyimidazolium-based commercial AEM (Aemion+®), Nafion™ binder, and PGM-based catalysts at a current density of 0.6 A cm−2 and with a low H2 crossover less than 0.4%.473 The voltage degradation rate over the long-term test was 13 μV h−1. This study suggests that future research should focus on developing active and stable materials for catalysts, catalyst layers, and the integration of catalyst layers into MEAs, particularly as the anode components are highly susceptible to oxidative conditions. Also, more recent results by Peng et al.605 and Lee et al.606 demonstrated a relatively low degradation rate (5–50 μV h−1) of 1 M KOH-fed AEMWEs employing aryl ether-free AEMs at 1.5 and 2 A cm−2, achieving a practical level of hydrogen generation.
Although the MEAs using currently available AEMs and AEIs have shown promising performance and durability, there are several technical challenges related to polymer electrolytes that must be addressed to enable the practical adoption of this technology for hydrogen production. Firstly, the demonstration of the long-term performance of liquid electrolyte-fed AEMWEs at temperatures above 60 °C is rare. While some ex situ tests claim that certain aryl ether-free AEMs and AEIs are stable for over 1000 hours at 80 °C or higher, proving this durability at elevated operating temperatures in practical settings is a task that may be achievable in the coming years. Secondly, stable long-term performance (exceeding 1000 hours) of pure water-fed AEMWEs needs to be demonstrated. This challenge is more daunting, as the performance degradation mechanism in pure water-fed AEMWEs is not well understood. Nonetheless, demonstrating high durability in pure water-fed systems is critical for commercialization. Thirdly, the capability of AEMWEs to operate under differential pressure conditions should be established. While current demonstrations focus on performance and durability, the ability to operate effectively under varying pressure conditions, ranging from 5 to 100 bar, is essential for practical application. This requires careful consideration of mechanical properties and AEM thickness. Lastly, achieving higher current densities, ideally up to 2 A cm−2, is desirable to increase the hydrogen production rate. Operating at such high current densities may lead to accelerated component degradation and performance decline due to extensive gas bubble formation. In particular, the electrochemical oxidation of ionomers and the dissolution of PGM-free catalysts at high anode potentials are major concerns.
6.3. Redox flow batteries
AEMs are being explored in a variety of flow batteries, such as zinc-bromine,608 zinc/cerium,609 various types of aqueous organic,478,610–614 and non-aqueous organic RFB,615,616 but the all-vanadium RFB is the most extensively investigated type. Although Hwang and Ohya explored a vanadium RFB using an AEM as early as 1997,617 it was not until a decade later other researchers began to follow suit. Still, fewer than 20 articles per year are reporting new AEMs for use in RFBs (Fig. 28a). It is noteworthy that AEMs have been shown to be more efficient than PEMs at blocking vanadium cations. Among the commercial membranes in the vanadium RFB field, Fumatech's Fumasep FAP-450, an AEM, is recognized as one of the standard options, alongside Nafion™.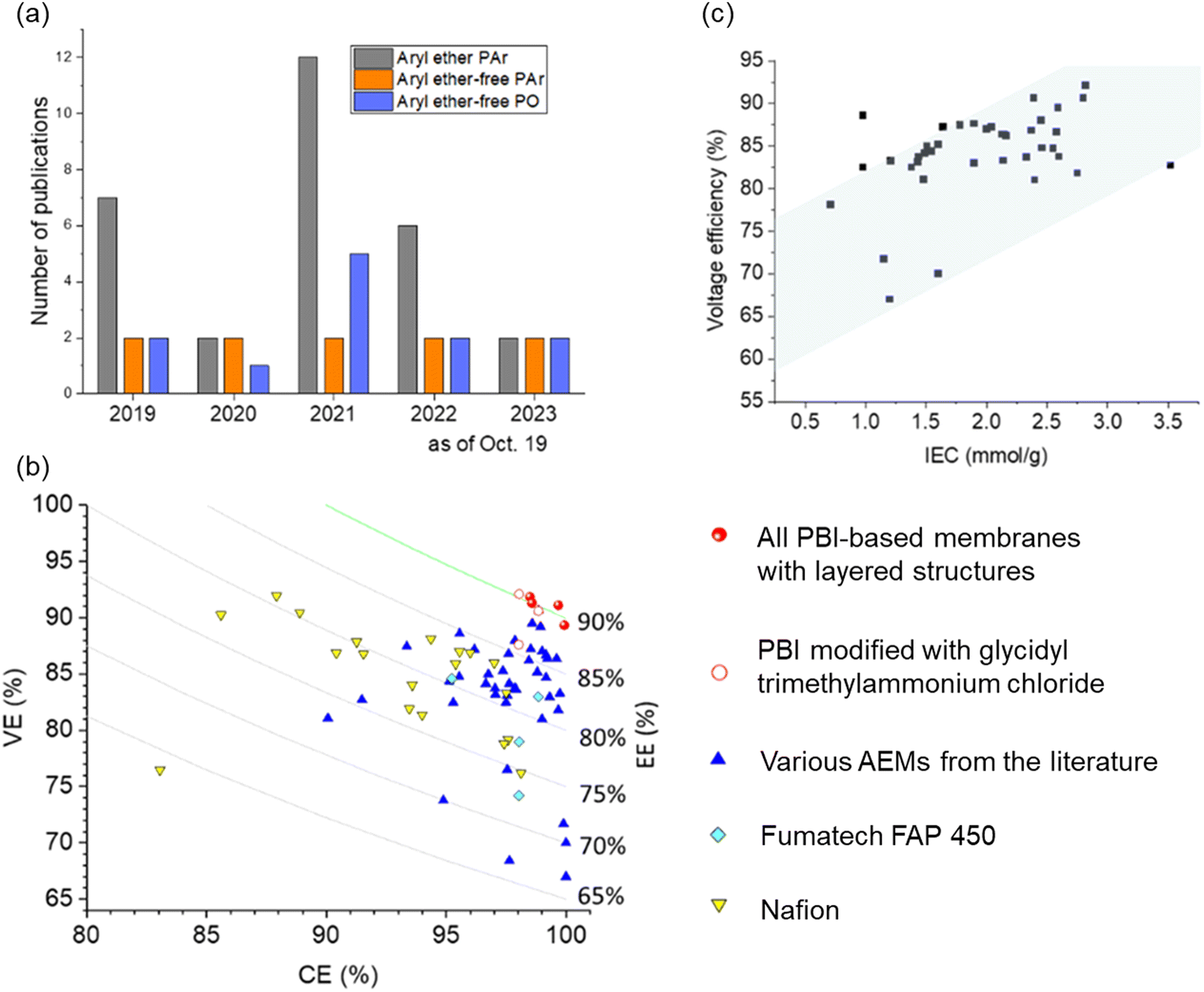 | ||
| Fig. 28 (a) Number of publications on new AEMs for RFB over the last 5 years; (b) vanadium RFB performances of cells operating at 80 mA cm−2. 50 cells using AEMs95,102,621–638 including commercial AEM Fumasep FAP-450624,628,630,636 are compared with 7 cells using Nafion 115,622,630–632,634,636,637 3 cells using Nafion 117,95,621,638 10 cells using Nafion 212102,623–629,633,635 and 4 cells using layered PBI membranes.639,640 CE, VE, and EE are coulombic, voltage, and energy efficiency, respectively; (c) voltage efficiency vs. IEC. Please see Table S10 in ESI,† for detailed data. | ||
A primary motivation for using AEMs in vanadium RFBs is to enhance the CE, which is significantly influenced by the membrane's permeability to vanadium ions. Nafion membranes, commonly used in vanadium RFBs, are cation exchange membranes that readily transport vanadium ions, resulting in lower CE (Fig. 28b). The positively charged cation headgroups in AEMs effectively hinder the transport of vanadium cations. This is supported by general trends; the average CE of cells with Nafion™ membrane is 93.1 ± 4.3% compared to 97.5 ± 2.2% of cells with AEMs. However, vanadium electrolyte solutions typically contain 2–3 M sulphuric acid and about 1.5 M vanadium sulphate. The high ionic strength of these electrolytes reduces the effectiveness of Donnan exclusion.618,619 Membranes with an open morphology with large hydrophilic domains, as in Nafion™ or other highly swollen membranes, tend to facilitate the transfer of co-ions. In this context, the typically lower degree of phase separation in AEMs is beneficial. Additionally, reduced access of highly oxidative VO2+ ions to the polymer chains in AEMs enhances their chemical resistance and, consequently, the lifetime of the polymers in vanadium RFBs.620
Although most AEMs exhibit lower conductivity compared to a commercial CEM like Nafion™, it suggests that AEMs have higher resistance, which could potentially lead to a lower VE. However, optimized AEMs are better at blocking the crossover of vanadium ions than Nafion™, which allows for the use of thinner membranes to retain VE. Consequently, because EE is the product of CE and VE, the average EE of the cells, as compared in Fig. 28, is 81.0 ± 5.6% for those using AEMs and 78.8 ± 4.6% for cells using Nafion™.
In vanadium RFBs, the capacity loss due to the crossover of vanadium ions can be recovered by mixing the discharged electrolytes, making EE a crucial parameter. Among the over 70 membranes shown in Fig. 28a, the highest EE was achieved with PBI-based membranes. Du et al. developed a PBI-based AEM by reacting a PBI with glycidyl trimethylammonium chloride.627 An even slightly higher EE was obtained by cells featuring a thin (1–2 μm) PBI layer to block vanadium crossover, supported by a porous or highly swollen gel-type PBI layer.639,640 The superior performance of PBI-based cells can be attributed to the acid-doped PBI carrying charges directly on the polymer backbone, which inhibits phase separation. The narrow spacing between the PBI chains limits the transport of vanadium ions through size exclusion, in addition to electrostatic repulsion. The inclusion of quaternary ammonium side chains, as demonstrated by Du et al., results in a more open membrane morphology, leading to an increase in VE and a decrease in CE.
Comparing published membrane conductivities can be misleading. For instance, the AEM with the highest EE in Fig. 28 has a conductivity of only 9.7 mS cm−1,627 yet achieved a VE of 92%. In contrast, a poly(ether ether ketone) (PEEK)-based AEM developed by Yang et al. had a higher conductivity of 65 mS cm−1, but a lower VE of 86%, and an EE of 85%.635 Since membrane thickness is optimized to balance VE and CE, the vanadium RFB community often focuses more on area-specific resistance (ASR, Ω cm2) as a key parameter. Other complicating factors include variation in sulphuric acid electrolyte concentration across studies and a significant increase in ASR when transitioning membranes from sulphuric acid solution to vanadium-containing electrolyte. In some cases, membrane A may exhibit a lower ASR in sulphuric acid than membrane B, but the reverse order is observed in vanadium electrolytes.641 Comparing VE values achieved with different membranes could be a potential solution, but VE is also heavily influenced by cell resistance and electrode activity. This is evident when comparing performances obtained with the Fumasep FAP-450 membrane, where VE ranges from 74.2% to 84.6%. A plot of VE values against IEC yields scattered results, suggesting a potential positive correlation between VE and IEC. Considering the variation in VE values for Fumasep FAP-450, the scattering is within a reasonable range.
Similar to the alkaline degradation of AEM backbones, it has been suggested that aromatic ether bonds are also susceptible to reaction with VO2+ ions, leading to chain scission and membrane failure due to embrittlement. Therefore, AEMs based on newer chemistries that avoid aromatic ether bonds, such as polyphenylene-based AEMs,478,628,631,637,642 and polybenzimidazolium-based AEMs, show promise.643,644
6.4. High-temperature PEMFCs
AEMs when paired with PA, play a crucial role in ion-pair HT-PEMFCs. To facilitate proton conduction under high temperature and anhydrous conditions, AEMs are doped with PA through the conventional imbibing process, resulting in the formation of PA-doped ion-pair membranes. There are distinct differences between ion-pair systems and conventional HT-PEMFCs based on PA-doped PBI. In PA-doped PBI systems, a proton of PA is transferred to benzimidazole, producing protonated benzimidazole, known as benzimidazolium. Conversely, ion-pair systems involve the abstraction of a proton from PA through interaction with a hydroxide anion, forming a quaternary ammonium-biphosphate anion interaction. The interaction, with a strength of approximately 110 kcal mol−1, significantly exceeds that between benzimidazole and PA, which is around 15 kcal mol−1.7 Due to this robust ion-pair interaction, ion-pair systems can achieve a greater doping level of acid at a given number of base sites. Initially developed methylated benzimidazole membranes were mechanically unstable and could only be utilized effectively through blending with a PBI membrane.645 The first ion-pair system without blending was demonstrated with BTMA-functionalized DAPP AEM in 2016.7 Other approaches to form ion-pair structure include using quaternary ammonium-modified polymers with intrinsic microporosity (PIM)646,647 and incorporating ionic liquid moieties.648–655 The ion-pair approaches prove beneficial for the stable operation of HT-PEMFCs, as they considerably reduce the risk of leaching out PA.7The number of publications for ion-pair HT-PEMFCs has increased since 2016 (Fig. 29a). Table S11 (ESI†) summarizes the performance and durability of MEAs in ion-pair HT-PEMFCs. The most prevalent polymer backbones for ion-pair membranes are aryl ether-free PAr and PBI, each constituting 45% of the total, while aryl ether PAr AEMs make up the remaining 10% (Fig. 29b). No polyolefinic AEMs have been reported yet, likely due to their lower thermal stability. Various cationic headgroups including alkyl-A, BTMA, imidazolium, piperidinium, and pyrrolidinium were used. Unlike in AEMWE and AEMFC applications, the alkaline stability of the cations is not a primary requirement as HT-PEMFCs operate under low pH conditions. Fig. 29c shows the PPDs of ion-pair HT-PEMFCs as a function of operating temperatures. Most PPDs were reported at 120, 160, and 200 °C under H2/O2 and H2/air conditions. The range of PPDs at a given operating temperature is relatively broad (0.5 to 1 W cm−2), as performance depends on several factors, including electrodes and other operating conditions such as reactant flow rate and back-pressure.
 | ||
| Fig. 29 Summary of performance and durability of ion-pair HT-PEMFC MEAs: (a) the number of recent publications on ion-pair HT-PEMFC. (b) The AEMs and AEIs used in ion-pair HT-PEMFCs. (c) PPD of ion-pair HT-PEMFC MEAs under H2/O2 and H2/air conditions. (d) The performance and durability comparison of the reported ion-pair HT-PEMFCs at 160 °C.7,9,108,186,649,656–661 | ||
Fig. 29d summarizes the performance and durability of ion-pair HT-PEMFCs. In 2016, Kim and co-workers introduced the term ‘ion-pair HT-PEMFCs’. They utilized a BTMA functionalized DAPP membrane and achieved the PPD of 0.75 W cm−2 at 160 °C under H2/O2 conditions.7 They demonstrated that ion-pair HT-PEMFCs can operate stably under dynamic operating temperature conditions with a water vapour pressure (PH2O) of 9.7 kPa and showcased 500 hours long-term durability at 120 °C and 0.4 V, with minimal current degradation rate (0.33 mA cm2 h−1). In 2020, Li et al. reported a slightly improved performance of ion-pair HT-PEMFCs over PBI-HT-PEMFCs.108 However, rapid degradation occurred within 30 hours at 160 °C and 0.2 A cm−2, indicating the instability of aryl ether-containing polysulfone in HT-PEMFC applications. In the same year, Wang and co-workers introduced a polybenzimidazolium membrane for ion-pair MEAs.656 The fuel cell displayed moderate (PPD160°C = 0.64 W cm−2 under H2/O2 conditions) and showed no degradation during 390 hours of operation at 160 °C. In 2021, a study using a similar polybenzimidazolium membrane demonstrated high PA retention of the membrane at 80 °C and 40% RH, with negligible degradation over 200 hours under anhydrous conditions at 160 °C.649 Kim and colleagues reported improved ion-pair MEA performance (PPD160°C = 1.2 W cm−2 under H2/O2 conditions) in the same year, achieved with a PA-functionalized ionomer instead of a PTFE binder.658 The performance reached 240 °C where PPD reached 1.75 W cm−2, and the cell operated stably at a constant current density of 0.6 A cm−2 over 500 hours at 160 °C with a voltage degradation rate of 0.35 mV h−1. The same group also reported the highest cell performance of the HTMA-DAPP-biphosphate ion-pair PEM and protonated PA-functionalized ionomer (PPD160°C = 1.7 and 0.78 W cm−2 under H2/O2 and H2/air conditions, respectively) with stable operation at 160 °C and 0.6 A cm−2 for 2500 hours.9 In 2020, Arges et al. demonstrated stable ion-pair MEA performance in the presence of 25% CO at 220 °C.657 This study was significant for illustrating practical high-temperature operation, which aids in increasing the operating temperatures of electrochemical hydrogen pumps that have high CO concentrations.662 In 2023, Sun and co-workers demonstrated stable fuel cell performance using phenyl-piperidinium-based AEM at 120 °C for 1000 hours. They obtained high fuel cell performance at 120 °C (PPD120°C = 1.5 W cm−2, 80 kPa), suggesting that phenyl-piperidinium AEM are promising for highly performing ion-pair HT-PEMFCs.186
6.5. CO2 and CO electrolysers
AEPs are extensively used in CO2 and CO electrolysers. The primary challenge in CO2 electrolysis lies in achieving efficiency and selectivity of the CO2 reduction reaction (CO2RR). Consequently, the bulk of research has centred on the CO2RR activity of various electrocatalysts under different system conditions, leading to the identification of some highly active electrocatalysts for CO2 electrolysers in flow cells or MEA designs. Prior to 2017, the literature on CO2 electrolysers using AEMs was sparse (Fig. 30a). However, post-2019, the literature on CO2 and CO electrolyser performance has increased, with at least ten articles per year, and the trend is upward. A significant advantage of AEM-based CO2 electrolysers is their high CO2RR activity under near-neutral conditions. By contrast, in CEM-based CO2 electrolysers, CO2 quickly transforms into (bi)carbonate, leading to carbonate salt build-up in the cathode flow field and within gas diffusion electrodes. This hampers CO2 access to the catalyst, leading to increased hydrogen faradaic efficiencies.663 To counteract this issue in AEM-based CO2 electrolysers, the approach using BPMs was demonstrated; BPM-based CO2 electrolysers can reconvert any formed (bi)carbonates back to CO2 by supporting protons to the cathode chamber. The reversed-bias BPM technique has been employed in various studies to enhance CO2 utilization in CO2 electrolysis systems.664–667 About 62% of the articles on CO2 and CO electrolysers involve AEMs, while 13% and 25% utilize BPMs and CEMs, respectively (Fig. 30b). It is noteworthy that most AEMs used for CO2 electrolysers are commercially available, indicating that CO2 electrolyser research is more on the CO2RR and device performance than on specific AEMs tailored for these devices (Table S12, ESI†). | ||
| Fig. 30 Summary of performance and durability of CO2 and CO electrolysers. (a) The number of publications of CO2 and CO electrolysers over the last seven years, (b) membrane types that was used for the CO2 and CO electrolysers, (c) AEMs used for CO2 and CO electrolysers, (d) supporting electrolyte that was used for the AEM-based CO2 and CO electrolysers, (e) the current density of CO2 and CO electrolysers, and (f) durability of CO2 and CO electrolysers. | ||
Several studies have examined the role of AEMs and AEIs in CO2 electrolysers’ performance. Masel and co-workers observed that CO2 electrolysers utilizing four acidic membranes-Nafion™, CMI-7001, SPEEK, and PA-doped PBI exhibited relatively low CO production rates compared to hydrogen evolution. Among AEM-based CO2 electrolysers, those with imidazolium functionalized polymers demonstrated the highest CO selectivity.668 Schmidt et al. used various polymeric membranes and ionomer combinations, concluding that balancing CO2RR and HER demands an alkaline environment at the cathode to achieve high CO2RR selectivity. However, fully alkaline cells showed increased CO2 migration from the cathode to the anode. They suggested that a BPM system with an acidic membrane and an AEI-bonded cathode catalyst could address this issue effectively.669 Rabinowitz and Kanan noted that the formation of carbonates not only impacts the energy balance by consuming hydroxide ions but also reduces the efficiency of oxygen evolution by lowering pH at the anode.670 To address this issue, forward bias BPMs can be employed to consume carbonate anions at the BPM interface. However, it is important to note that using BPMs introduces an additional overpotential for water dissociation. Janáky and co-workers underscored the importance of thin membranes in enhancing CO-forming capability by comparing the performance of cells with a thin membrane (PiperION, 15 μm thick) versus thicker ones (e.g., Sustainion®, 50–70 μm thick). The PiperION membrane, with its low resistance (0.36 Ω cm2), facilitated a high CO formation current density of 630 mA cm−2 in their zero-gap electrolyser.671 Jiao and co-workers explored the impact of AEMs’ alcohol permeation rate on the carboxylate production rates in CO electrolysers. They reported that membranes with higher diffusion rates for the products increased the molar production ratio towards carboxylates and the target product concentration.672 Ju et al. investigated the influence of different ionomers on the cathode performance of CO2 electrolysers, concluding that a balance of anionic conductivity and hydrophobicity is crucial for high CO faradaic efficiency.673 Broekmann and co-workers compared the ionomer performance between Nafion™ (cationic) and Femion (anionic), emphasizing that electrolyte management – particularly preventing K2CO3/KHCO3 precipitate formation – is more critical than ionomer hydrophobicity.674 Seger et al. investigated the effect of cationic groups on CO selectivity.371 They found that AEMs, while not directly affecting catalytic activity, create a local environment around the cathode catalyst layers through water management. The benzyl-N-methylpiperidinium-head group of RG-AEMs yielded the highest CO selectivity (>80%) compared to TMA or benzyl-N-methylpyrrolidinium head groups, thanks to improved water and ionic transport. The same research groups also demonstrated that minor variations in synthetic conditions of RG-AEMs can result in spectroscopically identical AEMs with markedly different hydration properties and CO2RR performance.413
In AEM-based CO2 and CO electrolysers, supporting electrolytes are commonly used alongside ionomers to suppress HER and enhance the CO2RR at the cathode and OER at the anode. The most frequently used supporting electrolytes include KOH and KHCO3 (Fig. 30c), with CsHCO3 also being a popular choice due to its effectiveness in mitigating salt deposition caused by salt crossover to the cathode.675,676 The supporting electrolytes significantly influenced the faradaic selectivity of CO2RR products. For instance, Hori et al. demonstrated that the formation of CH4via CO2RR electrocatalysts is favoured in environments with high concentrations of bicarbonate, a phenomenon attributed to variations in the buffer capacity of different electrolytes.677 In contrast, C2 products are more prevalent in dilute KHCO3 solutions. While achieving high faradaic CO2RR efficiency alongside low OER overpotentials remains a challenge, several studies have reported CO2 electrolyser systems that operate without supporting electrolytes.678–681 The primary rationale behind these ionomer-only electrolyte systems is to avoid the inevitable formation of salts associated with supporting electrolytes, which can diminish the stability and efficiency of the overall system.682 The choice of electrocatalysts plays a critical role in determining the CO2RR products. For example, Au and Ag catalysts exhibit high faradaic efficiency for CO production, accounting for nearly 50% of the AEM-based CO2 electrolysers' output (Fig. 30d). In contrast, catalysts based on Pb, Sn, and Pd favour the production of formic acid, while Cu-based catalysts are known for generating CH4, C2H4, and various alcohols.683
The reported performance of AEM-based CO2 and CO electrolysers typically involves lower current density compared to water electrolysers (Fig. 30e). The most frequently observed current density is 200–299 mA cm−2, accounting for 35% of reports, closely followed by 100–199 mA cm−2, which represents 29% of cases. Notably, only 10% of the reports indicate current density exceeding 500 mA cm−2. In 2018, Jiao and co-workers demonstrated a Cu-catalysed and Fumatech AEM-incorporated CO electrolyser, achieving C2+ faradaic efficiency of 91% with C2+ partial current density over 630 mA cm−2 by optimizing the triple-phase boundary at the electrode–electrolyte interface. In 2019, Strasser et al. showcased a Ni–N–C catalysed CO2 electrolyser capable of operating up to 700 mA cm−2.684 However, it was observed that the maximum faradaic efficiency (90%) of the Selemion AEM incorporated electrolyser was achieved in the 100 and 200 mA cm−2 range. Beyond this, an increase in current density led to a gradual decrease in CO efficiency. Later, The same research group reported CO2 electrolysers capable of operating at up to 700 mA cm−2, achieving a remarkable C2+ energy efficiency of 100% using a coupled tandem electrolyser approach.685 Additionally, piperidinium-based AEM-based CO2 electrolysers have demonstrated the capability to operate efficiently at high current density, such as 500 mA cm−2.671,686
A significant challenge in CO2 electrolysers is their limited durability. Notably, nearly half of the relevant studies did not report on the longevity of the electrolysers. Merely 7% of the publications document a lifespan exceeding 200 hours (Fig. 30f). The primary reason for the low stability is the salt accumulation and consequent clogging of flow channels. Additionally, the high operating cell voltage of CO2 electrolysers, around 3.0 V, can potentially lead to the electrochemical oxidation of AEMs. In 2017, Masel and co-workers reported exceptional durability in CO2 electrolysers, ranging from 550 to 4380 hours.668,678 These highly durable cells were operated under deionized water or 0.01 M KHCO3, conditions that are near neutral. The environment allowed the methylimidazolium-based polymers to operate for extended periods without degradation from hydroxide attack. Another noteworthy instance of durability in a CO2 electrolyser was achieved using a toluene-functionalized Cu catalyst. Zheng et al. observed that while a pristine Cu catalyst failed to maintain a stable voltage beyond 50 hours of electrolysis, a toluene-modified Cu catalyst exhibited remarkable stability for over 400 hours. This stability was coupled with an ethylene faradaic efficiency of 50% by supplying 0.1 M KHCO3.687
6.6. Other applications
DAC or CO2 separation from coal-fired power plants, is pivotal in decarbonizing the economy. Current DAC methods primarily utilize amine solution-based or solid adsorbents. However, materials based on quaternary ammonium offer distinct advantages over these amine-based techniques. Employing a moisture-swing process driven by water evaporation energy, quaternary ammonium materials can efficiently capture atmospheric CO2. This process involves sorption in low-humidity conditions and desorption when humidity increases, operating without external heat input. The DAC efficiency with these materials can reach 100% in dry conditions, exceeding amine-based counterparts.688,689 Yan et al. demonstrated an electrochemically driven CO2 separator utilizing a hydrogen-powered cell with an anion-conducting membrane.690 In their configuration, the cathode facilitates the ORR, generating hydroxide ions that remove CO2 from air by forming carbonates. Meanwhile, at the anode, the HOR generates protons, establishing a low pH environment. The formed carbonates at the cathode migrate to the anode via the membrane, where they convert into bicarbonates and eventually CO2 due to the pH gradient. The shorted membrane was prepared by blending a phenyl piperidinium AEP with electronically conductive carbon additives, enabling electron transport through the membrane and facilitating a compact and high-performance module. Through optimization, the shorted membrane cell demonstrated >99% CO2 removal from 2000 sccm air over a continuous operation period of 450 hours. Simari and co-workers utilized a quaternary ammonium-functionalized polyepichlorohydrin membrane for CO2 sorption, demonstrating superior capture efficiency under simulated flue gas conditions and effective regeneration with minimal energy consumption in mild N2 environments.691 Singh and co-workers developed an integrated system, combining electrochemical CO2 capture from flue gas and its reduction to value-added products and fuels, using an AEM.692 Their approach involved capturing CO2 in an organic liquid (KOH-saturated ethylene glycol, and choline hydroxide) and transporting HCO3− across an AEM. A successful and continuous integration of migration-assisted moisture gradient CO2 capture and electrochemical CO2 reduction was demonstrated. In a recent study, Freeman et al. investigated DAC using the Fumasep FAA-3 membrane, and a reactive transport model suggested that carbon support rates in AEMs are primarily limited by moisture-swing reaction kinetics.693 They concluded that polymer design optimization could enhance the moisture-swing effect, or coupling moisture gradient with electrochemical force could lead to more energy-efficient and rapid CO2 separation.Aryl ether-free AEMs are increasingly utilized in diffusion dialysis for acid recovery. These AEMs reject most cations through electrostatic repulsion and offer significant advantages over aryl ether-containing polymers694,695 and polyolefinic AEMs.696,697 Such benefits include lower swelling, higher selectivity, and higher chemical stability.185,698–700 The superior membrane properties of aryl ether-free polyaromatics allow for higher IEC, ranging from 2.0–2.8 mequiv. g−1, compared to those of other types of AEMs (0.8–2.0 mequiv. g−1), resulting in better performance in acid recovery applications.
In research focusing on the selective removal of heavy metal ions701–704 or minerals,705–711 AEPs are employed in processes like MCDI or adsorption. In the MCDI process, the membrane traps co-ions into intraparticle pores, enhancing the accumulation of counterions in macropores. The application of an inverted voltage upon discharging, facilitated by ion-exchange membranes, prevents the re-adsorption of desorbed ions on the counter electrode, thereby increasing electrode regeneration efficiency. While commercial polyolefinic AEPs are commonly used in this context, less focus has been placed on AEP materials due to the strong dependence of cell performance on cell configuration and adsorbing materials.712,713 Nonetheless, the importance of AEP properties such as IEC has been increasingly recognized as key parameters for the device performance.714
7. Perspective and outlook
This review presents a comprehensive overview of the chemical stability of AEPs and the various synthetic approaches to polyaromatic and vinyl-derived AEPs, charting the evolution of these chemistries to improve performance, chemical stability, and durability. The merits and weaknesses of each method to prepare aryl ether-free polymer electrolytes are summarized in Table 4. Additionally, this review explores the scale-up of AEPs and AEMs for prospective applications in the market. The final section discusses the performance of these devices in mainstream applications, including fuel cells, electrolysers, and flow batteries.| AEP synthesis method | Merits | Weaknesses |
|---|---|---|
| Chemistry of polyaromatics | ||
| Acid-catalysed polyhydroxyalkylation | One-pot, low to room temperature, metal-free polymerization | Limited choice of reactors for a large-scale reaction due to the high acidity of the catalyst |
| High tolerance of the acid catalyst towards functional monomers | Limited choice of monomers for high molecular weight growth | |
| Metal-promoted coupling reaction | Various choices of monomers for polymerization | Requirement of a stoichiometric amount of the Ni catalyst or the use of an expensive Pd catalyst |
| Ionenes including polybenzimidazoliums and polyimidazoliums | Incorporation of highly alkaline stable groups in the backbone | High water uptake and even water solubility |
| Difficulty in forming free-standing film and requirement of post-modification strategies | ||
| Diels–Alder polymerization | All phenylene backbone with high processability | Require high molecular weight to achieve good mechanical properties |
| High gas permeability due to the bulky backbone structure | ||
| Chemistry of vinyl polymers | ||
| Addition polymerization | Various polymer structures including random and block copolymers | Limited choice of monomers |
| Fully saturated backbones at the polymerization stage | Questionable radical stability | |
| Ring opening metathesis polymerization | High functional group tolerance including cations | The requirement of an expensive Ru catalyst |
| The requirement of a two-step process to achieve a fully saturated backbone | ||
| Radiation grafting method | AEM with complex nano-morphology | Limited choice of monomers for radical grafting |
| Synthesis of high-performing particulate AEI | The requirement of access to high-energy radiation facilities | |
| Difficult to control IEC precisely | ||
| Anionic polymerization | Well-defined block composition and narrow molecular weight distribution | Requirement of strict inert atmosphere and purity of monomers and solvents |
| Limited choice of monomers for polymerization | ||
| Requirement of post-functionalization | ||
The last fifteen years have witnessed a resurgent interest in AEPs for a variety of applications beyond the core AEMFC application, which still accounts for the largest share of research papers. Over the last five years, research on AEMWE and CO2 electrolysis has proportionately increased compared to other applications like ED, RED, and RFB, in part due to new opportunities and directed funding in these areas. The intended applications for AEPs span a broad range of environmental conditions under which they must operate stably. The structures of AEPs, in general, need to be specifically tailored to meet the requirements in which they operate. Fig. 3 summarizes the environmental regimes under which AEP applications operate.
In most applications, AEPs are in the form of AEMs, whose function is to maintain separation between reactants while allowing trans-membrane selective ion transport. Consequently, mechanical properties must be robust enough to maintain this separation boundary over a long period under specific environmental operating conditions. AEMs operating in entirely liquid environments include RO, RFB, ED, and MCDI. Since AEMs carry various amounts of fixed ionic charge, they are prone to liquid uptake, enhancing ion conductivity but becoming detrimental in excess. This often correlates with dimensional swelling, which weakens the mechanical strength and leads to AEM rupture, wrinkling, and defects at contact points. However, liquid uptake and dimensional swelling effects can be decoupled to some extent by various strategies, such as non-swelling mechanical supports, dipolar chain interactions, hydrogen bonding, and branched or crosslinked polymer chain architecture.
In addition to the AEM applications, aryl-ether free AEPs have potential uses in creating high-performance and durable BPMs suitable for a range of electrochemical processes. These processes include nitrate reduction, CO2 reduction, water electrolysers, and ionic separations like MCDI and ED.
AEP processability is crucial since the majority of AEMs are fabricated through solution casting followed by solvent evaporation. Crosslinking is necessarily done after the casting step, either in the presence of the casting solvent or in the dry state. AEPs containing a high proportion of rigid structural units, polar groups, or hydrogen bonding sites may require higher-boiling solvents for viable casting solutions, posing challenges for complete solvent removal during membrane formation. In the case of branched chain architecture, introducing free volume and increased chain entanglement is limited to low degrees of branching before gel formation occurs.
AEMs operating in more complex mixed-phase gas–liquid environments include AEMFC, AEMWE, and CO2 electrolysers, where electrocatalytic reactions occur on the membrane surface. This leads to potentially more reactive species, such as free radicals and nascent hydrogen and oxygen, imposing a more demanding chemical degradative stress on the AEPs. AEIs are used in these applications where catalysts are employed, serving multiple functions critical to the performance and durability of AEMFCs and other electrochemical devices. Firstly, they act as polymeric binders surrounding catalyst particles, requiring material compatibility with the AEM to avoid the delamination issue encountered in earlier work on PEMFC counterparts. Fortunately, much research and some commercially available AEIs have better compatibility with AEMs, being non-fluorinated. Secondly, unlike AEMs where low gas permeability is desired to prevent hydrogen crossover, AEIs should have high gas permeability. This is crucial, especially in AEMFCs during the hydrogen oxidation reaction at the cathode, where hydrogen permeation is limited by cationic group adsorption. Thirdly, AEIs function as a boundary phase, where gas, water, electron, and ion transport occur simultaneously. IEC values between AEM and AEI can be substantially different, and AEIs are typically dispersed in low-boiling solvents and mixed with catalysts to form inks. Some interesting characteristics, such as high gas permeability and low voltage degradation rates, have been observed for AEI particulates suspended in a solvent, providing new research opportunities for exploring high free-volume particulates as AEIs.
The chemical stability of AEPs under various application operating conditions is a critical requirement to ensure durability for commercial viability. Fig. 3 illustrates the typical pH and potential ranges of various electrochemical devices utilizing AEPs. AEM applications that do not employ catalysts, such as RO, ED, RED, and MCDI, operate in environments composed of multivalent metal cations, requiring chemical stability typically within a pH range of 4–13. On the other hand, various fuel cell and electrolyser AEM applications employing catalysts, namely AEMFC, AEMWE, and CO2 capture/electrolyser, have more stringent operating environments with pH levels exceeding 13, demanding high AEP chemical stability. Furthermore, high electrochemical stability at >2 V is required for AEMWE and CO2 electrolysers. Further research regarding the electrochemical stability of the fragments of AEPs under various electrode potentials and operating conditions needs to be explored.129 In addition to strongly basic environments, AEMs are also used in strongly acidic environments. HT-PEMFC applications operate in a pH range of 1–3 at elevated temperatures of 100–200 °C, while RFB applications operate at a pH below 1.
Many of the earlier AEPs were based on the chloromethylation–quaternization structural modification of readily available hydrocarbon polymers, such as polystyrene, polysulfone, or PPO, due to their accessibility through a relatively simple process. While these AEPs may be appropriate for use in applications in less aggressive environments (e.g., ED), they are less suited to more stringent fuel cell and electrolyser application conditions where high pH and electrocatalytic oxygen radicals occur. Aryl ether-containing AEPs are known to undergo degradation in high-pH environments.
Beyond the polymer backbone, the stability of the cation headgroup must be considered, along with its relationship and proximity to the backbone, and its accessibility to hydroxide ion nucleophilic attack, as degradation relations do not necessarily occur in isolation between backbone and headgroup. In terms of oxidative stability, AEPs with high electron density are more susceptible to radical-induced oxidation, while those with electron-withdrawing groups have high electrophilic character and remain more stable toward oxygen radical attack.
In the last decade, aryl ether-free AEPs have become well-established, representing major progress in the wider field. Notable classes of these AEPs are based on hindered poly(benzimidazolium), polyphenylene, polynorbornene, and terphenyl piperidinium, which have considerably higher oxidative stability than aryl ether-containing polymers and are thus better suited for fuel cell and electrolyser applications. Higher oxidative stability is particularly required for electrolysers due to the higher electrode potentials in the devices, as degradation pathways are dependent upon operating conditions.
As noted earlier, the nature of degradation by hydroxide ion (alkaline, nucleophilic) and hydroxyl radical (oxidative, electrophilic) is different, highlighting the need to design AEPs with balanced chemical stability for each specific electrochemical device and operating condition. Contemporary research is primarily focused on these four classes of AEMs for fuel cell and electrolyser applications, with the outlook anticipating further tuning of specific properties tailored to individual application operating environments. Properties such as ion conductivity and mechanical strength are influenced by IEC, water uptake and dimensional swelling, polymer chain architecture, crosslinking, chain interactions, type of cation headgroup, and chemical composition of the AEMs.
The kinetics of water uptake and dimensional swelling of AEMs may vary depending on the temperature and time for measurement. It is important to note that polymer chain structures, their strength of intra-and intermolecular interactions, how dry the membranes are to begin with, and the solvents used to cast the membrane may also play crucial roles in water uptake and dimensional swelling.
The IEC of AEMs for AEMWEs may be somewhat lower than for AEMFCs when liquid electrolyte is supplied, while the AEM may be thicker to reduce gas crossover and withstand desirable differential pressure operations. There are likely more opportunities for improving alkaline stability by exploring new cation headgroups, such as metallocenes, or decreasing the susceptibility of cation headgroups to nucleophilic hydroxide attack, as seen in the hindered access approach used in poly(benzimidazolium) AEMs. Increasing oxidative stability can also be addressed by specific chemical structures of AEPs that avoid susceptible units and the possibility of using anti-oxidative additives. Other AEM work highlights opportunities for aligned through-membrane conduction channels,575 although more scalable processes are essential for commercialization.
The main body of AEP (AEM and AEI) structural design for the past decade has been applied to AEMFC. However, similar structural design principles are usually applicable to AEMWE and CO2 electrolysers, but with some structural and membrane fine-tuning according to the specific environment of the application. In particular, AEPs for AEMFC, AEMWE, and CO2 capture/electrolyser applications all need chemical stability under higher pH conditions (pH > 13), which are highlighted in this review. As mentioned above, thicker AEMs are preferable for AEMWE to reduce gas crossover and mechanical stress from differential pressure. Since these AEMs are immersed in liquid, dimensional swelling must be controlled, either by AEM mechanical support, or mitigated by crosslinking or branching, and by a lower IEC. In many cases, commercially available AEMs are employed. Also, since the AEMs are thicker and require more material, there is more price sensitivity and a requirement for cheaper materials. In the case of CO2 electrolysers, most investigations have focused on the CO2RR and device performance, using commercially available AEMs, rather than on specific AEMs tailored for these purposes (Table S12, ESI†).
The exploration of AEIs within catalyst layers has been comparatively limited compared to AEMs, yet their significance in enhancing device performance is increasingly recognized. Beyond the essential attributes required of AEMs – such as ion conductivity, mechanical resilience, dimensional integrity, and alkaline endurance – three specific criteria stand out for AEIs. Firstly, AEIs can regulate pH conducive to electrochemical reactions. This adjustment is facilitated by factors like cationic and non-cationic functional groups, IEC, and the ratio of ionomer to catalyst within the catalyst layers. Generally, a higher pH is favoured for AEMWEs as opposed to AEMFCs. However, a significantly lower pH proves advantageous for CO2RR in the CO2 electrolysers.715–720 Secondly, AEIs should exhibit minimal interactions with catalysts. Interactions between ionomers and catalysts not only influence catalytic efficiency but also impact device durability. Notably, the adsorption of cationic AEI groups at the anode of electrochemical devices is prominent, leading to electrochemical oxidative degradation at high electrode potentials. Opportunities exist for designing AEIs with lower adsorption energy on the surface of catalysts, exemplified by fluorene-based AEIs having fused ring structures.134 This class of AEIs boasts high oxidative stability, which is particularly beneficial for electrolyser operating at high electrode potentials. Thirdly, AEIs with high gas permeability are desirable for a swift reactant or product transport. Conventional methods involve partial fluorination to enhance gas solubility.173,556,721 Modern advancements include the development of ionomers with particulate structures406,437,722,723 and increased free volume.165,509,724
The industrial implementation of AEMs and AEIs for fuel cells and electrolysers requires AEPs with the desired performance and functional characteristics, along with high stability, to ensure device durability. The principles of successful commercial adoption dictate that processes for making AEMs and AEIs should be cost-effective, simple, scalable, and sustainable.
Glossary of acronyms
| ADMET | Acyclic diene metathesis polymerization |
| AEM | Anion exchange membrane |
| AEMFC | Anion exchange membrane fuel cell |
| AEMWE | Anion exchange membrane water electrolyser |
| AEI | Anion exchange ionomer |
| AEP | Anion exchange polymers |
| AFM | Atomic force microscopy |
| AMS | α-Methylstyrene (AMS) |
| ASR | Area specific resistance |
| Bpin | Pinacol boronate |
| BPM | Bipolar membrane |
| BTC | Bistetracyclone |
| BTMA | Benzyltrimethyl ammonium |
| CE | Coulombic efficiency |
| CEM | Cation exchange membrane |
| DABCO | Diazabicyclo[2.2.2] octane |
| DAC | Direct air capture |
| DAPP | Diels–Alder poly(phenylene) |
| DEB | Diethynylbenzene |
| DFT | Density functional theory |
| DIPEA | N,N-Diisopropylethylamine |
| DMAc | Dimethylacetamide |
| DMF | Dimethylformamide |
| DMI | 1,2-Dimethylimidazole |
| DMSO | Dimethyl sulfoxide |
| DMP | Dimethyl piperidinium |
| DMPZ | 1,4-Dimethylpiperazine |
| DOE | Department of Energy |
| ECSA | Electrochemical surface area |
| ED | Electrodialysis |
| EE | Energy efficiency |
| ETFE | Ethylene tetrafluoroethylene |
| FTIR | Fourier transform infrared |
| GPC | Gel permeation chromatography |
| HER | Hydrogen evolution reaction |
| HFTO | The hydrogen and fuel cell technologies office |
| HMT | Hexamethyl-p-terphenylene |
| HT-PEMFC | High temperature-proton exchange membrane fuel cell |
| IEC | Ion exchange capacity |
| LDPE | Low-density polyethylene |
| LT-PEMFC | Low temperature-proton exchange membrane fuel cell |
| MAN | Methylacrylonitrile |
| MCDI | Membrane capacitive deionization |
| MD | Molecular dynamics |
| MEA | Membrane electrode assembly |
| MIG | Mutual-(simultaneous)-grafting method |
| MMPH | N-Methylmorpholine |
| MPIP | N-Methylpiperidine |
| NMP | N-Methyl-2-pyrrolidone |
| MPY | N-Methylpyrrolidine |
| NMR | Nuclear magnetic resonance |
| OER | Oxygen evolution reaction |
| PBI | Polybenzimidazole |
| PDI | Polydispersity index |
| PE | Polyethylene |
| PEEK | Poly(ether ether ketone) |
| PEM | Proton exchange membrane |
| PES | Poly(ether sulfone) |
| PFSA | Perfluorosulfonic acid |
| PGM | Platinum group metal |
| PIG | Pre-irradiation grafting |
| PIP | Piperidinium |
| PPD | Peak power density |
| PPO | Poly(p-phenylene oxide) |
| PTFE | Polytetrafluoroethylene |
| PVDF | Polyvinylidene fluoride |
| PYR | Pyridine |
| QUN | Quinuclidine |
| RED | Reverse electrodialysis |
| RFB | Redox flow battery |
| RG | Radiation-grafted |
| RH | Relative humidity |
| RO | Reverse osmosis |
| ROD | Radical/oxidative degradation |
| ROMP | Ring opening metathesis polymerization |
| SAXS | Small angle X-ray scattering |
| SBS | Poly(styrene-b-butadiene-b-styrene) |
| SEBS | Poly(styrene-b-(ethylene-co-butylene)-b-styrene) |
| TFA | Trifluoroacetic acid |
| TFSA | Trifluoromethanesulfonic acid |
| TMBG | 2-Butyl-1,1,3,3-tetramethyl-butylguanidine |
| TMBTC | Tetramethylbis(cyclopentadienone) |
| TMI | 1,2,4,5-Tetramethylimidazole |
| UV | Ultraviolet |
| VBC | Vinylbenzyl chloride |
| VE | Voltage efficiency |
Conflicts of interest
CB is a founder of Orion Polymer. SH is a scientific advisor to Ionomr Innovations Inc.Acknowledgements
This work is partly funded from the U.S. Department of Energy (DOE) Office of Energy Efficiency and Renewable Energy Hydrogen Fuel Cell Technologies Office through the Hydrogen from Next-Generation Electrolysers of Water (H2NEW) Consortium. EJP thanks the support by the Laboratory Directed Research and Development program of Los Alamos National Laboratory under project number 20230496ECR. PJ was supported by the Swedish Foundation for Strategic Research (project ARC19-0026). KM thanks the financial support by MEXT (KAKENHI 23H02058 and Data Creation and Utilization Type Material Research and Development Project JPMXP1122712807) and JST (GteX JPMJGX23H2). KN was supported by the Center for Alkaline-based Energy Solutions (CABES), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, and Basic Energy Sciences under Award DE-SC0019445. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. JRV acknowledges EPSRC grant EP/T009233/1 for funding his time. DH was supported by KIST internal projects (2E32591, 2E33281, 2E33284). MDG acknowledges funding from the National Natural Science Foundation of China (21875161), through the National Industry-Education Platform for Energy Storage, and the State Key Laboratory of Engines (SKLE), Tianjin University, Tianjin 300072, China. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or U.S. Government.References
- S. Al-Amshawee, M. Y. B. Yunus, A. A. M. Azoddein, D. G. Hassell, I. H. Dakhil and H. Abu Hasan, Chem. Eng. J., 2020, 380, 122231 CrossRef CAS.
- J. G. Hong, B. P. Zhang, S. Glabman, N. Uzal, X. M. Dou, H. G. Zhang, X. Z. Wei and Y. S. Chen, J. Membr. Sci., 2015, 486, 71–88 CrossRef CAS.
- J.-B. Lee, K.-K. Park, H.-M. Eum and C.-W. Lee, Desalination, 2006, 196, 125–134 CrossRef CAS.
- B. Shrimant, T. Kulkarni, M. Hasan, C. Arnold, N. Khan, A. N. Mondal and C. G. Arges, ACS Appl. Mater. Interfaces, 2024, 16, 11206–11216 CrossRef CAS PubMed.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. S. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- N. Seselj, D. Aili, S. Celenk, L. N. Cleemann, H. A. Hjuler, J. O. Jensen, K. Azizi and Q. F. Li, Chem. Soc. Rev., 2023, 52, 4046–4070 RSC.
- K. S. Lee, J. S. Spendelow, Y. K. Choe, C. Fujimoto and Y. S. Kim, Nat. Energy, 2016, 1, 16120 CrossRef CAS.
- K. H. Lim, I. Matanovic, S. Maurya, Y. Kim, E. S. De Castro, J. H. Jang, H. Park and Y. S. Kim, ACS Energy Lett., 2023, 8, 529–536 CrossRef CAS.
- K. H. Lim, A. S. Lee, V. Atanasov, J. Kerres, E. J. Park, S. Adhikari, S. Maurya, L. D. Manriquez, J. Jung, C. Fujimoto, I. Matanovic, J. Jankovic, Z. D. Hu, H. F. Jia and Y. S. Kim, Nat. Energy, 2022, 7, 248–259 CrossRef CAS.
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Ann. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS PubMed.
- J. Liu, Z. Kang, D. Li, M. Pak, S. M. Alia, C. Fujimoto, G. Bender, Y. S. Kim and A. Z. Weber, J. Electrochem. Soc., 2021, 168, 054522 CrossRef CAS.
- L. Zeng, T. S. Zhao, L. Wei, H. R. Jiang and M. C. Wu, Appl. Energy, 2019, 233, 622–643 CrossRef.
- Q. D. Shu, M. Haug, M. Tedesco, P. Kuntke and H. V. M. Hamelers, Environ. Sci. Technol., 2022, 56, 11559–11566 CrossRef CAS PubMed.
- P. Zhu, Z.-Y. Wu, A. Elgazzar, C. Dong, T.-U. Wi, F.-Y. Chen, Y. Xia, Y. Feng, M. Shakouri, J. Y. Kim, Z. Fang, T. A. Hatton and H. Wang, Nature, 2023, 618, 959 CrossRef CAS PubMed.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O'Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- S. Z. Oener, L. P. Twight, G. A. Lindquist and S. W. Boettcher, ACS Energy Lett., 2021, 6, 1–8 CrossRef CAS.
- L. Chen, Q. Xu, S. Z. Oener, K. Fabrizio and S. W. Boettcher, Nat. Commun., 2022, 13, 3846 CrossRef CAS PubMed.
- T. Kulkarni, A. M. I. Al Dhamen, D. Bhattacharya and C. G. Arges, ACS ES&T Eng., 2023, 3, 2171–2182 Search PubMed.
- K. S. Barros, M. C. Martí-Calatayud, V. Pérez-Herranz and D. C. R. Espinosa, Desalination, 2020, 492, 114628 CrossRef CAS.
- K. Matsui, E. Tobita, K. Sugimoto, K. Kondo, T. Seita and A. Akimoto, J. Appl. Polym. Sci., 1986, 32, 4137–4143 CrossRef CAS.
- D. S. Kim, C. H. Fujimoto, M. R. Hibbs, A. Labouriau, Y. K. Choe and Y. S. Kim, Macromolecules, 2013, 46, 7826–7833 CrossRef CAS.
- A. M. Park, Z. R. Owczarczyk, L. E. Garner, A. C. Yang-Neyerlin, H. Long, C. M. Antunes, M. R. Sturgeon, M. J. Lindell, S. J. Hamrock, M. A. Yandrasits and B. S. Pivovar, Polym. Electrolyte Fuel Cells 17 (PEFC 17), 2017, 80, 957–966 CAS.
- M. S. J. Jung, C. G. Arges and V. Ramani, J. Mater. Chem., 2011, 21, 6158–6160 RSC.
- M. A. Vandiver, J. L. Horan, Y. Yang, E. T. Tansey, S. Seifert, M. W. Liberatore and A. M. Herring, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1761–1769 CrossRef CAS.
- D. M. Hillman, S. H. Stephens, S. D. Poynton, S. Murphy, A. L. Ong and J. R. Varcoe, J. Mater. Chem. A, 2013, 1, 1018–1021 RSC.
- A. Bosnjakovic, M. Danilczuk, S. Schlick, P. N. Xiong, G. M. Haugen and S. J. Hamrock, J. Membr. Sci., 2014, 467, 136–141 CrossRef CAS.
- M. F. Rabuni, N. M. N. Sulaiman, M. K. Aroua and N. A. Hashim, Ind. Eng. Chem. Res., 2013, 52, 15874–15882 CrossRef CAS.
- N. A. Hashim, Y. T. Liu and K. Li, Chem. Eng. Sci., 2011, 66, 1565–1575 CrossRef.
- A. G. Divekar, M. R. Gerhardt, C. M. Antunes, L. Osmieri, A. C. Yang-Neyerlin, A. Z. Weber, B. S. Pivovar, G. Bender and A. M. Herring, Electrochim. Acta, 2022, 406, 139812 CrossRef CAS.
- P. Zschocke and D. Quellmalz, J. Membr. Sci., 1985, 22, 325–332 CrossRef CAS.
- E. Avram, M. A. Brebu, A. Warshawasky and C. Vasile, Polym. Degrad. Stab., 2000, 69, 175–181 CrossRef CAS.
- J. L. Yan and M. A. Hickner, Macromolecules, 2010, 43, 2349–2356 CrossRef CAS.
- Z. Zhao, J. H. Wang, S. H. Li and S. B. Zhang, J. Power Sources, 2011, 196, 4445–4450 CrossRef CAS.
- N. W. Li, T. Z. Yan, Z. Li, T. Thurn-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888–7892 RSC.
- C. G. Arges, L. H. Wang, M. S. Jung and V. Ramani, J. Electrochem. Soc., 2015, 162, F686–F693 CrossRef CAS.
- J. L. Yan, L. Zhu, B. L. Chaloux and M. A. Hickner, Polym. Chem., 2017, 8, 2442–2449 RSC.
- J.-S. Park, S.-H. Park, S.-D. Yim, Y.-G. Yoon, W.-Y. Lee and C.-S. Kim, J. Power Sources, 2008, 178, 620–626 CrossRef CAS.
- M. Tanaka, K. Fukasawa, E. Nishino, S. Yamaguchi, K. Yamada, H. Tanaka, B. Bae, K. Miyatake and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 10646–10654 CrossRef CAS PubMed.
- G. Wang, Y. Weng, D. Chu, R. Chen and D. Xie, J. Membr. Sci., 2009, 332, 63–68 CrossRef CAS.
- J. Wang, S. Li and S. Zhang, Macromolecules, 2010, 43, 3890–3896 CrossRef CAS.
- J. Zhou, M. Unlu, J. A. Vega and P. A. Kohl, J. Power Sources, 2009, 190, 285–292 CrossRef CAS.
- C. X. Lin, Y. Z. Zhuo, A. N. Lai, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Membr. Sci., 2016, 513, 206–216 CrossRef CAS.
- X. Lin, L. Wu, Y. Liu, A. L. Ong, S. D. Poynton, J. R. Varcoe and T. Xu, J. Power Sources, 2012, 217, 373–380 CrossRef CAS.
- T. W. Xu and W. H. Yang, J. Membr. Sci., 2001, 190, 159–166 CrossRef CAS.
- L. Zhu, J. Pan, C. M. Christensen, B. Lin and M. A. Hickner, Macromolecules, 2016, 49, 3300–3309 CrossRef CAS.
- N. Li, Q. Zhang, C. Y. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45, 2411–2419 CrossRef CAS.
- B. Bauer, H. Strathmann and F. Effenberger, Desalination, 1990, 79, 125–144 CrossRef CAS.
- J. Xue, J. Zhang, X. Liu, T. Huang, H. Jiang, Y. Yin, Y. Qin and M. D. Guiver, Electrochem. Energy Rev., 2022, 5, 348–400 CrossRef CAS.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CrossRef CAS.
- Y. Ye and Y. A. Elabd, Macromolecules, 2011, 44, 8494–8503 CrossRef CAS.
- B. Lin, H. Dong, Y. Li, Z. Si, F. Gu and F. Yan, Chem. Mater., 2013, 25, 1858–1867 CrossRef CAS.
- A. A. Zagorodni, D. L. Kotova and V. F. Selemenev, React. Funct. Polym., 2002, 53, 157–171 CrossRef CAS.
- J. Hnat, M. Paidar, J. Schauer, J. Zitka and K. Bouzek, J. Appl. Electrochem., 2011, 41, 1043–1052 CrossRef CAS.
- G. Cerichelli, G. Illuminati and C. Lillocci, J. Org. Chem., 1980, 45, 3952–3957 CrossRef CAS.
- G. Cospito, G. Illuminati, C. Lillocci and H. Petride, J. Org. Chem., 1981, 46, 2944–2947 CrossRef CAS.
- G. Illuminati and C. Lillocci, J. Org. Chem., 1977, 42, 2201–2203 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 RSC.
- W. You, K. M. Hugar, R. C. Selhorst, M. Treichel, C. R. Peltier, K. J. T. Noonan and G. W. Coates, J. Org. Chem., 2021, 86, 254–263 CrossRef CAS PubMed.
- K. Y. Zhang, W. S. Yu, X. L. Ge, L. Wu and T. W. Xu, J. Membr. Sci., 2023, 678, 121672 CrossRef CAS.
- O. I. Deavin, S. Murphy, A. L. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584–8597 RSC.
- S. A. Nunez and M. A. Hickner, ACS Macro Lett., 2013, 2, 49–52 CrossRef CAS PubMed.
- M. R. Sturgeon, C. S. Macomber, C. Engtrakul, H. Long and B. S. Pivovar, J. Electrochem. Soc., 2015, 162, F366–F372 CrossRef CAS.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS PubMed.
- B. R. Einsla, S. Chempath, L. R. Pratt, J. M. Boncella, J. A. Rau, C. S. Macomber and B. Pivovar, ECS Trans., 2007, 11, 1173–1180 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- H.-S. Dang and P. Jannasch, ACS Appl. Energy Mater., 2018, 1, 2222–2231 CrossRef CAS.
- A. Allushi, P. Thanh Huong, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 27164–27174 RSC.
- A. Allushi, T. H. Pham and P. Jannasch, J. Membr. Sci., 2021, 632, 119376 CrossRef CAS.
- D. Pan, P. Thanh Huong and P. Jannasch, ACS Appl. Energy Mater., 2021, 4, 11652–11665 CrossRef CAS.
- D. Pan, P. M. Bakvand, T. H. Pham and P. Jannasch, J. Mater. Chem. A, 2022, 10, 16478–16489 RSC.
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst, X. Lu, W. Xu, M. Tader, A. V. Soudackov, H. Zhang, M. Krumov, E. Murray, P. Xu, J. Hitt, L. Xu, H.-Y. Ko, B. G. Ernst, C. Bundschu, A. Luo, D. Markovich, M. Hu, C. He, H. Wang, J. Fang, R. A. DiStasio, Jr., L. F. Kourkoutis, A. Singer, K. J. T. Noonan, L. Xiao, L. Zhuang, B. S. Pivovar, P. Zelenay, E. Herrero, J. M. Feliu, J. Suntivich, E. P. Giannelis, S. Hammes-Schiffer, T. Arias, M. Mavrikakis, T. E. Mallouk, J. D. Brock, D. A. Muller, F. J. DiSalvo, G. W. Coates and H. D. Abruña, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed.
- B. Soresi, E. Quartarone, P. Mustarelli, A. Magistris and G. Chiodelli, Solid State Ionics, 2004, 166, 383–389 CrossRef CAS.
- H. Su, S. Pasupathi, B. Bladergroen, V. Linkov and B. G. Pollet, Int. J. Hydrogen Energy, 2013, 38, 11370–11378 CrossRef CAS.
- G. J. Ross, J. F. Watts, M. P. Hill and P. Morrissey, Polymer, 2000, 41, 1685–1696 CrossRef CAS.
- J. Sharma, C. Totee, V. Kulshrestha and B. Ameduri, Eur. Polym. J., 2023, 201, 112580 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- B. C. Lin, L. H. Qiu, J. M. Lu and F. Yan, Chem. Mater., 2010, 22, 6718–6725 CrossRef CAS.
- M. L. Guo, J. Fang, H. K. Xu, W. Li, X. H. Lu, C. H. Lan and K. Y. Li, J. Membr. Sci., 2010, 362, 97–104 CrossRef CAS.
- F. X. Zhang, H. M. Zhang and C. Qu, J. Mater. Chem., 2011, 21, 12744–12752 RSC.
- J. R. Varcoe, R. C. T. Slade and E. Lam How Yee, Chem. Commun., 2006, 1428–1429 RSC.
- K. Fukuta, presented in part at the 2011 AMFC Workshop, 2011.
- Y. S. Li and T. S. Zhao, Int. J. Hydrogen Energy, 2012, 37, 4413–4421 CrossRef CAS.
- Y. Zhao, H. M. Yu, D. L. Yang, J. Li, Z. G. Shao and B. L. Yi, J. Power Sources, 2013, 221, 247–251 CrossRef CAS.
- C. Fujimoto, D. S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423, 438–449 CrossRef.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- Y. K. Choe, C. Fujimoto, K. S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161, F615–F621 CrossRef CAS.
- A. Marinkas, I. Struzynska-Piron, Y. Lee, A. Lim, H. S. Park, J. H. Jang, H. J. Kim, J. Kim, A. Maljusch, O. Conradi and D. Henkensmeier, Polymer, 2018, 145, 242–251 CrossRef CAS.
- J. J. Han, W. F. Song, X. Q. Cheng, Q. Cheng, Y. Y. Zhang, C. F. Liu, X. R. Zhou, Z. D. Ren, M. X. Hu, T. S. Ning, L. Xiao and L. Zhuang, ChemSusChem, 2021, 14, 5021–5031 CrossRef CAS PubMed.
- C. G. Arges, L. Wang, J. Parrondo and V. Ramani, J. Electrochem. Soc., 2013, 160, F1258 CrossRef CAS.
- R. A. Becerra-Arciniegas, R. Narducci, G. Ercolani, S. Antonaroli, E. Sgreccia, L. Pasquini, P. Knauth and M. L. Di Vona, Polymer, 2019, 185, 121931 CrossRef CAS.
- T. Mohammadi and M. Skyllas-Kazacos, J. Appl. Electrochem., 1997, 27, 153–160 CrossRef CAS.
- Y. Ahn and D. Kim, J. Ind. Eng. Chem., 2019, 71, 361–368 CrossRef CAS.
- G. Shukla and V. K. Shahi, J. Membr. Sci., 2019, 575, 109–117 CrossRef CAS.
- Z. Wang, S. Zhang, Q. Liu, Y. Chen, Z. Weng and X. Jian, J. Membr. Sci., 2021, 633, 119416 CrossRef CAS.
- E. J. Park, S. Maurya, U. Martinez, Y. S. Kim and R. Mukundan, J. Membr. Sci., 2021, 617, 118565 CrossRef CAS.
- X. Hao, Z. Zhou, Y. Chen, L. Xiong and D. Chen, Mater. Chem. Phys., 2022, 277, 125490 CrossRef CAS.
- Z. Wang, S. Zhang, Q. Liu, L. Zhuo, Z. Liu, P. Xu, D. Wang, Z. Weng and X. Jian, J. Membr. Sci., 2022, 656, 120646 CrossRef CAS.
- B. Zhang, X. Zhang, Q. Liu, M. Zhao, Z. Yang, Y. Fu, E. Zhang, K. Wang, G. Wang, Z. Zhang and S. Zhang, J. Power Sources, 2022, 548, 232095 CrossRef CAS.
- Y. Chen, Y. Li, J. Xu, S. Chen and D. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 18923–18933 CrossRef CAS PubMed.
- J. Qian, S. Cai, J. Hu, C. Wang and G. Li, Ind. Eng. Chem. Res., 2023, 62, 2719–2728 CrossRef CAS.
- Y. Xing, L. Liu, C. Y. Wang and N. W. Li, J. Mater. Chem. A, 2018, 6, 22778–22789 RSC.
- X. Hao, N. Chen, Y. Chen and D. Chen, Polym. Degrad. Stab., 2022, 197, 109864 CrossRef CAS.
- D. Y. Chen and M. A. Hickner, Phys. Chem. Chem. Phys., 2013, 15, 11299–11305 RSC.
- S. Olivella, A. Sole and J. M. Bofill, J. Chem. Theory Comput., 2009, 5, 1607–1623 CrossRef CAS PubMed.
- H. Tang, K. Geng, J. Hao, X. Zhang, Z. Shao and N. Li, J. Power Sources, 2020, 475, 228521 CrossRef CAS.
- A. Laconti, H. Liu, C. Mittelsteadt and R. McDonald, ECS Trans., 2006, 1, 199 CrossRef CAS.
- S. Wierzbicki, J. C. Douglin, A. Kostuch, D. R. Dekel and K. Kruczala, J. Phys. Chem. Lett., 2020, 11, 7630–7636 CrossRef CAS PubMed.
- J. Parrondo, Z. Wang, M.-S. J. Jung and V. Ramani, Phys. Chem. Chem. Phys., 2016, 18, 19705–19712 RSC.
- Y. Z. Zhang, J. Parrondo, S. Sankarasubramanian and V. Ramani, ChemSusChem, 2017, 10, 3056–3062 CrossRef CAS PubMed.
- T. Nemeth, T. Nauser and L. Gubler, ChemSusChem, 2022, 15, e20221571 CrossRef PubMed.
- R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Mater. Chem. A, 2017, 5, 1248–1267 RSC.
- R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Power Sources, 2018, 375, 373–386 CrossRef CAS.
- T. Holmes, T. J. G. Skalski, M. Adamski and S. Holdcroft, Chem. Mater., 2019, 31, 1441–1449 CrossRef CAS.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef CAS PubMed.
- L. Balducci, D. Bianchi, R. Bortolo, R. D'Aloisio, M. Ricci, R. Tassinari and R. Ungarelli, Angew. Chem., Int. Ed., 2003, 42, 4937–4940 CrossRef CAS PubMed.
- J. H. Yang, G. Sun, Y. J. Gao, H. B. Zhao, P. Tang, J. Tan, A. H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793–798 RSC.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- D. H. Deng, X. Q. Chen, L. Yu, X. Wu, Q. F. Liu, Y. Liu, H. X. Yang, H. F. Tian, Y. F. Hu, P. P. Du, R. Si, J. H. Wang, X. J. Cui, H. B. Li, J. P. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. G. Zhou, L. T. Sun, J. Q. Li, X. L. Pan and X. H. Bao, Sci. Adv., 2015, 1, e150046 Search PubMed.
- D. G. Li, I. Matanovic, A. S. Lee, E. J. Park, C. Fujimoto, H. T. Chung and Y. S. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 9696–9701 CrossRef CAS PubMed.
- S. Maurya, A. S. Lee, D. G. Li, E. J. Park, D. P. Leonard, S. Noh, C. Bae and Y. S. Kim, J. Power Sources, 2019, 436, 226866 CrossRef CAS.
- C. Chen, F. Chen, L. Zhang, S. Pan, C. Bian, X. Zheng, X. Meng and F.-S. Xiao, Chem. Commun., 2015, 51, 5936–5938 RSC.
- R. Peng, S. Li, X. Sun, Q. Ren, L. Chen, M. Fu, J. Wu and D. Ye, Appl. Catal., B, 2018, 220, 462–470 CrossRef CAS.
- C.-S. Chen, T.-C. Chen, H.-C. Wu, P.-H. Huang and H.-M. Kao, J. Catal., 2021, 402, 275–288 CrossRef CAS.
- R. A. Krivina, G. A. Lindquist, M. C. Yang, A. K. Cook, C. H. Hendon, A. R. Motz, C. Capuano, K. E. Ayers, J. E. Hutchison and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2022, 14, 18261–18274 CrossRef CAS PubMed.
- J. Parrondo and V. Ramani, J. Electrochem. Soc., 2014, 161, F1015–F1020 CrossRef CAS.
- J. Nikl, K. Hofman, S. Mossazghi, I. C. Möller, D. Mondeshki, F. Weinelt, F.-E. Baumann and S. R. Waldvogel, Nat. Commun., 2023, 14, 4565 CrossRef CAS PubMed.
- I. G. Irtegova, V. F. Starichenko, I. A. Kirilyuk and I. A. Grigor′′ev, Russ. Chem. Bull., 2002, 51, 2065–2069 CrossRef CAS.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- L. Xue, W. Hu, C. Feng, N. Chen, H. Chen and Z. Hu, Desalination Water Treatment, 2020, 188, 212–222 CrossRef CAS.
- S. Maurya, C. H. Fujimoto, M. R. Hibbs, C. N. Villarrubia and Y. S. Kim, Chem. Mater., 2018, 30, 2188–2192 CrossRef CAS.
- I. Matanovic, S. Maurya, E. J. Park, J. Y. Jeon, C. Bae and Y. S. Kim, Chem. Mater., 2019, 31, 4195–4204 CrossRef CAS.
- I. Matanovic, H. T. Chung and Y. S. Kim, J. Phys. Chem. Lett., 2017, 8, 4918–4924 CrossRef CAS PubMed.
- Y. Y. Zhao, E. Tsuchida, Y. K. Choe, J. Wang, T. Ikeshoji and A. Ohira, J. Membr. Sci., 2015, 487, 229–239 CrossRef CAS.
- S. Maurya, E. Baca, K. K. Bejagam, H. Pratt, T. Anderson, R. Mukundan and C. Fujimoto, J. Power Sources, 2022, 520, 230805 CrossRef CAS.
- Y. S. Kim, Adv. Sci., 2023, 10, 2303914 CrossRef CAS PubMed.
- H. T. Chung, U. Martinez, I. Matanovic and Y. S. Kim, J. Phys. Chem. Lett., 2016, 7, 4464–4469 CrossRef CAS PubMed.
- G. A. Lindquist, J. C. Gaitor, W. L. Thompson, V. Brogden, K. J. T. Noonan and S. W. Boettcher, Energy Environ. Sci., 2023, 16, 4373–4387 RSC.
- T. Luo, O. David, Y. Gendel and M. Wessling, J. Power Sources, 2016, 312, 45–54 CrossRef CAS.
- R. Ye, D. Henkensmeier, S. J. Yoon, Z. Huang, D. K. Kim, Z. Chang, S. Kim and R. Chen, J. Electrochem. Energy Conversion Storage, 2018, 15, 010801 CrossRef.
- X. L. Zhou, T. S. Zhao, L. An, L. Wei and C. Zhang, Electrochim. Acta, 2015, 153, 492–498 CrossRef CAS.
- S. Maurya, S. D. Abad, E. J. Park, K. Ramaiyan, Y. S. Kim, B. L. Davis and R. Mukundan, J. Membr. Sci., 2023, 668, 121233 CrossRef CAS.
- S. H. Eberhardt, T. Lochner, F. N. Buechi and T. J. Schmidt, J. Electrochem. Soc., 2015, 162, F1367–F1372 CrossRef CAS.
- A. T. Pingitore, F. Huang, G. Qian and B. C. Benicewicz, ACS Appl. Energy Mater., 2019, 2, 1720–1726 CrossRef CAS.
- D. Aili, D. Henkensmeier, S. Martin, B. Singh, Y. Hu, J. O. Jensen, L. N. Cleemann and Q. Li, Electrochem. Energy Rev., 2020, 3, 793–845 CrossRef CAS.
- K. Likit-anurak, I. Karki, B. I. Howard, L. Murdock, N. Mukhin, J. Brannon, A. Hepstall, B. Young, S. Shimpalee, B. Benicewicz and B. Meekins, ACS Appl. Energy Mater., 2023, 6, 5429–5434 CrossRef CAS.
- B. Hu, Y. Huang, L. Liu, X. Hu, K. Geng, Q. Ju, M. Liu, J. Bi, S. Luo and N. Li, J. Membr. Sci., 2022, 643, 120042 CrossRef CAS.
- M. R. Kraglund, M. Carmo, G. Schiller, S. A. Ansar, D. Aili, E. Christensen and J. O. Jensen, Energy Environ. Sci., 2019, 12, 3313–3318 RSC.
- M. L. A. Trisno, A. Dayan, S. J. Lee, F. Egert, M. Gerle, M. R. Kraglund, J. O. Jensen, D. Aili, A. Roznowska, A. Michalak, H. S. Park, F. Razmjooei, S.-A. Ansar and D. Henkensmeier, Energy Environ. Sci., 2022, 15, 4362–4375 RSC.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- G. A. Olah, Angew. Chem., Int. Ed. Engl., 1993, 32, 767–788 CrossRef.
- G. A. Olah, G. Rasul, C. York and G. K. S. Prakash, J. Am. Chem. Soc., 1995, 117, 11211–11214 CrossRef CAS.
- O. Hernández-Cruz, M. G. Zolotukhin, S. Fomine, L. Alexandrova, C. Aguilar-Lugo, F. A. Ruiz-Treviño, G. Ramos-Ortíz, J. L. Maldonado and G. Cadenas-Pliego, Macromolecules, 2015, 48, 1026–1037 CrossRef.
- M. Zolotukhin, S. Fomine, R. Salcedo and L. Khalilov, Chem. Commun., 2004, 1030–1031 RSC.
- M. T. Guzmán-Gutiérrez, M. H. Rios-Dominguez, F. A. Ruiz-Treviño, M. G. Zolotukhin, J. Balmaseda, D. Fritsch and E. Prokhorov, J. Membr. Sci., 2011, 385–386, 277–284 CrossRef.
- A. R. Cruz, M. C. G. Hernandez, M. T. Guzmán-Gutiérrez, M. G. Zolotukhin, S. Fomine, S. L. Morales, H. Kricheldorf, E. S. Wilks, J. Cárdenas and M. Salmón, Macromolecules, 2012, 45, 6774–6780 CrossRef CAS.
- L. I. Olvera, M. T. Guzmán-Gutiérrez, M. G. Zolotukhin, S. Fomine, J. Cárdenas, F. A. Ruiz-Trevino, D. Villers, T. A. Ezquerra and E. Prokhorov, Macromolecules, 2013, 46, 7245–7256 CrossRef CAS.
- M. T. Guzmán-Gutiérrez, D. R. Nieto, S. Fomine, S. L. Morales, M. G. Zolotukhin, M. C. G. Hernandez, H. Kricheldorf and E. S. Wilks, Macromolecules, 2011, 44, 194–202 CrossRef.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS PubMed.
- W.-H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS PubMed.
- W.-H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS PubMed.
- S. Maurya, S. Noh, I. Matanovic, E. J. Park, C. Narvaez Villarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC.
- E. J. Park, S. Maurya, A. S. Lee, D. P. Leonard, D. Li, J. Y. Jeon, C. Bae and Y. S. Kim, J. Mater. Chem. A, 2019, 7, 25040–25046 RSC.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- R. Ren, S. Zhang, H. A. Miller, F. Vizza, J. R. Varcoe and Q. He, J. Membr. Sci., 2019, 591, 117320 CrossRef CAS.
- M. S. Cha, J. E. Park, S. Kim, S.-H. Han, S.-H. Shin, S. H. Yang, T.-H. Kim, D. M. Yu, S. So, Y. T. Hong, S. J. Yoon, S.-G. Oh, S. Y. Kang, O.-H. Kim, H. S. Park, B. Bae, Y.-E. Sung, Y.-H. Cho and J. Y. Lee, Energy Environ. Sci., 2020, 13, 3633–3645 RSC.
- T. H. Pham, A. Allushi, J. S. Olsson and P. Jannasch, Polym. Chem., 2020, 11, 6953–6963 RSC.
- N. Chen, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, C. Hu, J. Y. Bae, E. S. Sim, Y.-C. Chung, J.-H. Jang, S. J. Yoo, Y. Zhuang and Y. M. Lee, Nat. Commun., 2021, 12, 2367 CrossRef CAS PubMed.
- N. Chen, C. Hu, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, J. Y. Bae, J. H. Park and Y. M. Lee, Angew. Chem., Int. Ed., 2021, 60, 7710–7718 CrossRef CAS PubMed.
- B. Xue, W. Cui, S. Zhou, Q. Zhang, J. Zheng, S. Li and S. Zhang, Macromolecules, 2021, 54, 2202–2212 CrossRef CAS.
- S. Adhikari, D. P. Leonard, K. H. Lim, E. J. Park, C. Fujimoto, O. Morales-Collazo, J. F. Brennecke, Z. Hu, H. Jia and Y. S. Kim, ACS Appl. Energy Mater., 2022, 5, 2663–2668 CrossRef CAS.
- D. Pan, J. S. Olsson and P. Jannasch, ACS Appl. Energy Mater., 2022, 5, 981–991 CrossRef CAS.
- A. Mohamed Ahmed Mahmoud and K. Miyatake, ACS Appl. Polym. Mater., 2023, 5, 2243–2253 CrossRef CAS.
- S. Adhikari, I. Matanovic, D. Leonard, J. M. Klein, T. Agarwal and Y. S. Kim, ACS Macro Lett., 2023, 13, 28–33 CrossRef PubMed.
- M. G. Zolotukhin, S. Fomine, L. M. Lazo, R. Salcedo, L. E. Sansores, G. G. Cedillo, H. M. Colquhoun, J. M. Fernandez-G and A. F. Khalizov, Macromolecules, 2005, 38, 6005–6014 CrossRef CAS.
- E. J. Park, C. B. Capuano, K. E. Ayers and C. Bae, J. Power Sources, 2018, 375, 367–372 CrossRef CAS.
- D. P. Leonard, S. Maurya, E. J. Park, L. Delfin Manriquez, S. Noh, X. Wang, C. Bae, E. D. Baca, C. Fujimoto and Y. S. Kim, J. Mater. Chem. A, 2020, 8, 14135–14144 RSC.
- T. Wang, J. Dong, N. Yu, W. Tang, Y. Jin, Y. Xu and J. Yang, Eur. Polym. J., 2022, 173, 111271 CrossRef CAS.
- L. I. Olvera, M. G. Zolotukhin, O. Hernández-Cruz, S. Fomine, J. Cárdenas, R. L. Gaviño-Ramírez and F. A. Ruiz-Trevino, ACS Macro Lett., 2015, 4, 492–494 CrossRef CAS PubMed.
- D. Pan, S. Chen and P. Jannasch, ACS Macro Lett., 2023, 12, 20–25 CrossRef CAS PubMed.
- Q. Wang, L. Huang, Z. Wang, J. Zheng, Q. Zhang, G. Qin, S. Li and S. Zhang, Macromolecules, 2022, 55, 10713–10722 CrossRef CAS.
- X. Wu, N. Chen, H.-A. Klok, Y. M. Lee and X. Hu, Angew. Chem., Int. Ed., 2022, 61, e202114892 CrossRef CAS PubMed.
- B. Liu, T. Li, Q. Li, S. Zhu, Y. Duan, J. Li, H. Zhang and C. Zhao, J. Membr. Sci., 2022, 660, 120816 CrossRef CAS.
- W. Yuan, L. Zeng, T. Zhang, Y. Zhou, J. Wang, S. Jiang, L. Li, Q. Liao and Z. Wei, Adv. Funct. Mater., 2023, 33, 2307041 CrossRef CAS.
- M. Jayakannan and S. Ramakrishnan, Macromol. Rapid Commun., 2001, 22, 1463–1473 CrossRef CAS.
- H. Ono, J. Miyake, S. Shimada, M. Uchida and K. Miyatake, J. Mater. Chem. A, 2015, 3, 21779–21788 RSC.
- A. G. Wright, T. Weissbach and S. Holdcroft, Angew. Chem., Int. Ed., 2016, 55, 4818–4821 CrossRef CAS PubMed.
- A. M. A. Mahmoud, A. M. M. Elsaghier, K. Otsuji and K. Miyatake, Macromolecules, 2017, 50, 4256–4266 CrossRef CAS.
- M. Ozawa, T. Kimura, R. Akiyama, J. Miyake, J. Inukai and K. Miyatake, Bull. Chem. Soc. Jpn., 2017, 90, 1088–1094 CrossRef CAS.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- A. M. Ahmed Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- M. Ozawa, T. Kimura, K. Otsuji, R. Akiyama, J. Miyake, M. Uchida, J. Inukai and K. Miyatake, ACS Omega, 2018, 3, 16143–16149 CrossRef CAS PubMed.
- D. Koronka, A. M. A. Mahmoud and K. Miyatake, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1059–1069 CrossRef CAS.
- D. Koronka, A. Matsumoto, K. Otsuji and K. Miyatake, RSC Adv., 2019, 9, 37391–37402 RSC.
- T. Kimura, A. Matsumoto, J. Inukai and K. Miyatake, ACS Appl. Energy Mater., 2020, 3, 469–477 CrossRef CAS.
- D. Koronka and K. Miyatake, RSC Adv., 2021, 11, 1030–1038 RSC.
- A. M. Ahmed Mahmoud and K. Miyatake, J. Membr. Sci., 2022, 643, 120072 CrossRef CAS.
- Y. Shirase, A. Matsumoto, K. L. Lim, D. A. Tryk, K. Miyatake and J. Inukai, ACS Omega, 2022, 7, 13577–13587 CrossRef CAS PubMed.
- Y. Ozawa, Y. Shirase, K. Otsuji and K. Miyatake, Mole. Syst. Des. Eng., 2022, 7, 798–808 RSC.
- A. M. Ahmed Mahmoud and K. Miyatake, ACS Appl. Energy Mater., 2022, 5, 15211–15221 CrossRef CAS.
- Y. Ozawa and K. Miyatake, Bull. Chem. Soc. Jpn., 2023, 96, 16–23 CrossRef CAS.
- T. Yamamoto, S. Wakabayashi and K. Osakada, J. Organomet. Chem., 1992, 428, 223–237 CrossRef CAS.
- A. F. Nugraha, S. Kim, F. Wijaya, B. Bae and D. Shin, Polymers, 2020, 12, 1614 CrossRef CAS PubMed.
- A. Jasti, S. Prakash and V. K. Shahi, J. Membr. Sci., 2013, 428, 470–479 CrossRef CAS.
- I. Hosaka, M. Kusakabe and K. Miyatake, Chem. Lett., 2018, 47, 257–259 CrossRef CAS.
- F. Xu, Y. Chen, B. Lin, J. Li, K. Qiu and J. Ding, ACS Macro Lett., 2021, 10, 1180–1185 CrossRef CAS PubMed.
- H. Lim, I. Jeong, J. Choi, G. Shin, J. Kim, T.-H. Kim and T. Park, Appl. Surf. Sci., 2023, 610, 155601 CrossRef CAS.
- C. Gibbs, E. Littmann and C. Marvel, J. Am. Chem. Soc., 1933, 55, 753–757 CrossRef CAS.
- A. Rembaum, W. Baumgartner and A. Eisenberg, J. Polym. Sci., Part B: Polym. Lett., 1968, 6, 159–171 CrossRef CAS.
- J. C. Salamone and B. Snider, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 3495–3501 CrossRef CAS.
- S. R. Williams and T. E. Long, Prog. Polym. Sci., 2009, 34, 762–782 CrossRef CAS.
- J. E. Bara and K. E. O'Harra, Macromol. Chem. Phys., 2019, 220, 1900078 CrossRef.
- W. You, K. J. T. Noonan and G. W. Coates, Prog. Polym. Sci., 2020, 100, 101177 CrossRef CAS.
- K. E. O'Harra and J. E. Bara, Polym. Int., 2021, 70, 944–950 CrossRef.
- J. S. Lee, A. Hocken and M. D. Green, Mol. Syst. Des. Eng., 2021, 6, 334–354 RSC.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- X. Qiao, X. Wang, S. Liu, Y. Shen and N. Li, J. Membr. Sci., 2021, 630, 119325 CrossRef CAS.
- A. Das, B. Sana, R. Bhattacharyya, P. Chandra Ghosh and T. Jana, ACS Appl. Polym. Mater., 2022, 4, 1523–1534 CrossRef CAS.
- J. Dong, N. Yu, X. Che, R. Liu, D. Aili and J. Yang, Polym. Chem., 2020, 11, 6037–6046 RSC.
- N. Yu, J. Dong, T. Wang, Y. Jin, W. Tang and J. Yang, Polymer, 2022, 240, 124491 CrossRef CAS.
- I. Strużyńska-Piron, M. Jung, A. Maljusch, O. Conradi, S. Kim, J. H. Jang, H.-J. Kim, Y. Kwon, S. W. Nam and D. Henkensmeier, Eur. Polym. J., 2017, 96, 383–392 CrossRef.
- K. M. Lee, R. Wycisk, M. Litt and P. N. Pintauro, J. Membr. Sci., 2011, 383, 254–261 CrossRef CAS.
- F. H. Liu, Q. Yang, X. L. Gao, H. Y. Wu, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2020, 595, 117560 CrossRef CAS.
- S. T. Hemp, M. Zhang, M. Tamami and T. E. Long, Polym. Chem., 2013, 4, 3582–3590 RSC.
- N. Chen, Y. Jin, H. Liu, C. Hu, B. Wu, S. Xu, H. Li, J. Fan and Y. M. Lee, Angew. Chem., Int. Ed., 2021, 60, 19272–19280 CrossRef CAS PubMed.
- K. Müllen, J. Lex, R. C. Schulz and F. Walter, Polym. Bull., 1990, 24, 263–269 CrossRef.
- G. Zhang, R. Li, X. Wang, X. Chen, Y. Shen and Y. Fu, Sep. Purif. Technol., 2022, 291, 120950 CrossRef CAS.
- X. Chen, R. Li, G. Zhang, Y. Sun, Q. Hao, T. Guo, Y. Shen, Y. Fu and S. Dai, Ind. Eng. Chem. Res., 2023, 62, 8793–8803 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- T. K. Carlisle, J. E. Bara, A. L. Lafrate, D. L. Gin and R. D. Noble, J. Membr. Sci., 2010, 359, 37–43 CrossRef CAS.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753–10756 CrossRef CAS PubMed.
- K. M. Hugar, H. A. I. V. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- K. Brinker and I. Robinson, US2895948A, 1959.
- H. Vogel and C. S. Marvel, J. Polym. Sci., 1961, 50, 511–539 CrossRef CAS.
- T.-S. Chung, J. Macromol. Sci., Part C, 1997, 37, 277–301 Search PubMed.
- J. S. Wainright, J. T. Wang, D. Weng, R. F. Savinell and M. Litt, J. Electrochem. Soc., 1995, 142, L121 CrossRef CAS.
- D. J. Jones and J. Rozière, J. Membr. Sci., 2001, 185, 41–58 CrossRef CAS.
- Y. Iwakura, K. Uno and Y. Imai, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2605–2615 CrossRef.
- M. J. Sansone, US Pat., 4898917, 1990 Search PubMed.
- C. S. Marvel and H. A. Vogel, US Pat., 3174947, 1965 Search PubMed.
- M. Hu, E. M. Pearce and T. K. Kwei, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 553–561 CrossRef CAS.
- D. Henkensmeier, H.-J. Kim, H.-J. Lee, D. H. Lee, I.-H. Oh, S.-A. Hong, S.-W. Nam and T.-H. Lim, Macromol. Mater. Eng., 2011, 296, 899–908 CrossRef CAS.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, Polym. Chem., 2011, 2, 1641–1643 RSC.
- A. G. Wright and S. Holdcroft, ACS Macro Lett., 2014, 3, 444–447 CrossRef CAS PubMed.
- A. G. Wright, J. Fan, B. Britton, T. Weissbach, H.-F. Lee, E. A. Kitching, T. J. Peckham and S. Holdcroft, Energy Environ. Sci., 2016, 9, 2130–2142 RSC.
- X. Cao, D. Novitski and S. Holdcroft, ACS Mater. Lett., 2019, 1, 362–366 CrossRef CAS.
- E. M. Schibli, A. G. Wright, S. Holdcroft and B. J. Frisken, J. Phys. Chem. B, 2018, 122, 1730–1737 CrossRef CAS PubMed.
- H. Ma, H. Zhu and Z. Wang, J. Polym. Sci. Polm. Chem., 2019, 67, 1087–1096 CrossRef.
- H. Long and B. Pivovar, J. Phys. Chem. C, 2014, 118, 9880–9888 CrossRef CAS.
- J. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS PubMed.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- T. Luo, S. Abdu and M. Wessling, J. Membr. Sci., 2018, 555, 429–454 CrossRef CAS.
- T. J. Omasta, L. Wang, X. Peng, C. A. Lewis, J. R. Varcoe and W. E. Mustain, J. Power Sources, 2018, 375, 205–213 CrossRef CAS.
- C. E. Diesendruck and D. R. Dekel, Curr. Opin. Electrochem., 2018, 9, 173–178 CrossRef CAS.
- P. Overton, W. Li, X. Cao and S. Holdcroft, Macromolecules, 2020, 53, 10548–10560 CrossRef CAS.
- H. Vogel and C. S. Marvel, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 1531–1541 CrossRef.
- K. Y. Wang, M. Weber and T.-S. Chung, J. Mater. Chem. A, 2022, 10, 8687–8718 RSC.
- O. Olabisi and K. Adewale, Handbook of thermoplastics, CRC Press, 2016 Search PubMed.
- L. Xiao, H. Zhang, E. Scanlon, L. S. Ramanathan, E.-W. Choe, D. Rogers, T. Apple and B. C. Benicewicz, Chem. Mater., 2005, 17, 5328–5333 CrossRef CAS.
- E. Chauveau, C. Marestin and R. Mercier, Polymer, 2014, 55, 6435–6438 CrossRef CAS.
- E. Chauveau, C. Marestin, V. Martin and R. Mercier, Polymer, 2008, 49, 5209–5214 CrossRef CAS.
- S. Saxer, C. Marestin, R. Mercier and J. Dupuy, Polym. Chem., 2018, 9, 1927–1933 RSC.
- P. Overton, A. Konovalova, K. Fraser and S. Holdcroft, Macromolecules, 2023, 56, 2801–2808 CrossRef CAS.
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- W. Ried and D. Freitag, Naturwissenschaften, 1966, 53, 306 CrossRef CAS.
- W. Ried and K. H. Bönnighausen, Chem. Ber., 1960, 93, 1769–1773 CrossRef CAS.
- H. Mukamal, F. W. Harris and J. K. Stille, J. Polym. Sci., Part A-1: Polym. Chem., 1967, 5, 2721–2729 CrossRef CAS.
- G. K. Noren and J. K. Stille, J. Polym. Sci.: Macromol. Rev., 1971, 5, 385–430 CAS.
- J. K. Stille and G. K. Noren, Macromolecules, 1972, 5, 49–55 CrossRef CAS.
- H. F. VanKerckhoven, Y. K. Gilliams and J. K. Stille, Macromolecules, 1972, 5, 541–546 CrossRef CAS.
- J. M. Tour, Adv. Mater., 1994, 6, 190–198 CrossRef CAS.
- C. Fujimoto, E. Sorte, N. Bell, C. Poirier, E. J. Park, S. Maurya, K.-S. Lee and Y. S. Kim, Polymer, 2018, 158, 190–197 CrossRef CAS.
- T. J. G. Skalski, B. Britton, T. J. Peckham and S. Holdcroft, J. Am. Chem. Soc., 2015, 137, 12223–12226 CrossRef CAS PubMed.
- C. H. Fujimoto, M. A. Hickner, C. J. Cornelius and D. A. Loy, Macromolecules, 2005, 38, 5010–5016 CrossRef CAS.
- A. A. Zagorodni, D. L. Kotova and V. F. Selemenev, React. Funct. Polym., 2002, 53, 157–171 CrossRef CAS.
- B. Bauer, H. Strathmann and F. Effenberger, Desalination, 1990, 79, 125–144 CrossRef CAS.
- V. Neagu, I. Bunia and I. Plesca, Polym. Degrad. Stab., 2000, 70, 463–468 CrossRef.
- M. R. Hibbs, C. H. Fujimoto and C. J. Cornelius, Macromolecules, 2009, 42, 8316–8321 CrossRef CAS.
- D. S. Kim, A. Labouriau, M. D. Guiver and Y. S. Kim, Chem. Mater., 2011, 23, 3795–3797 CrossRef CAS.
- B. Qiu, B. Lin, L. Qiu and F. Yan, J. Mater. Chem., 2012, 22, 1040–1045 RSC.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499–6502 CrossRef CAS PubMed.
- M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161–1167 CrossRef CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- E. J. Park, S. Maurya, M. R. Hibbs, C. H. Fujimoto, K.-D. Kreuer and Y. S. Kim, Macromolecules, 2019, 52, 5419–5428 CrossRef CAS.
- A. R. Lavoie and R. M. Waymouth, Tetrahedron, 2004, 60, 7147–7155 CrossRef CAS.
- C. Janiak and P. G. Lassahn, J. Mol. Catal. A: Chem., 2001, 166, 193–209 CrossRef CAS.
- F. Blank and C. Janiak, Coord. Chem. Rev., 2009, 253, 827–861 CrossRef CAS.
- M. V. Bermeshev and P. P. Chapala, Prog. Polym. Sci., 2018, 84, 1–46 CrossRef CAS.
- M. V. Bermeshev, B. A. Bulgakov, A. M. Genaev, J. V. Kostina, G. N. Bondarenko and E. S. Finkelshtein, Macromolecules, 2014, 47, 5470–5483 CrossRef CAS.
- R. G. Schultz, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 541–546 CrossRef CAS.
- T. Hasan, K. Nishii, T. Shiono and T. Ikeda, Macromolecules, 2002, 35, 8933–8935 CrossRef CAS.
- T. Hasan, T. Ikeda and T. Shiono, Macromolecules, 2004, 37, 7432–7436 CrossRef CAS.
- F. Blank, J. K. Vieth, J. Ruiz, V. Rodríguez and C. Janiak, J. Organomet. Chem., 2011, 696, 473–487 CrossRef.
- W. S. R. Lago, C. Aymes-Chodur, A. P. Ahoussou and N. Yagoubi, J. Mater. Sci., 2017, 52, 6879–6904 CrossRef CAS.
- D. A. Barnes, G. M. Benedikt, B. L. Goodall, S. S. Huang, H. A. Kalamarides, S. Lenhard, L. H. McIntosh, K. T. Selvy, R. A. Shick and L. F. Rhodes, Macromolecules, 2003, 36, 2623–2632 CrossRef CAS.
- P. Huo, W. Liu, X. He, Z. Wei and Y. Chen, Polym. Chem., 2014, 5, 1210–1218 RSC.
- S. Martínez-Arranz, A. C. Albéniz and P. Espinet, Macromolecules, 2010, 43, 7482–7487 CrossRef.
- X. He, J. Liu, H. Zhu, Y. Zheng and D. Chen, RSC Adv., 2015, 5, 63215–63225 RSC.
- M. Mandal, G. Huang and P. A. Kohl, J. Membr. Sci., 2019, 570–571, 394–402 CrossRef CAS.
- R. Selhorst, J. Gaitor, M. Lee, D. Markovich, Y. Yu, M. Treichel, C. Olavarria Gallegos, T. Kowalewski, L. F. Kourkoutis, R. C. Hayward and K. J. T. Noonan, ACS Appl. Energy Mater., 2021, 4, 10273–10279 CrossRef CAS.
- M. Lehmann, D. Leonard, J. Zheng, L. He, X. Tang, X. C. Chen, K. H. Lim, S. Maurya, Y. S. Kim and T. Saito, ACS Appl. Energy Mater., 2023, 6, 1822–1833 CrossRef CAS.
- J. H. Hsu, C. R. Peltier, M. Treichel, J. C. Gaitor, Q. Li, R. Girbau, A. J. Macbeth, H. D. Abruña, K. J. T. Noonan, G. W. Coates and B. P. Fors, Angew. Chem., Int. Ed., 2023, 62, e202304778 CrossRef CAS PubMed.
- M. Mandal, G. Huang, N. U. Hassan, W. E. Mustain and P. A. Kohl, J. Mater. Chem. A, 2020, 8, 17568–17578 RSC.
- K. R. Gmernicki, E. Hong, C. R. Maroon, S. M. Mahurin, A. P. Sokolov, T. Saito and B. K. Long, ACS Macro Lett., 2016, 5, 879–883 CrossRef CAS PubMed.
- M. Mandal, G. Huang, N. Ul Hassan, X. Peng, T. L. Gu, A. H. Brooks-Starks, B. Bahar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 167, 054501 CrossRef.
- M. Mandal, G. Huang and P. A. Kohl, ACS Appl. Energy Mater., 2019, 2, 2447–2457 CrossRef CAS.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637 CrossRef CAS.
- M. Chen, M. Mandal, K. Groenhout, G. McCool, H. M. Tee, B. Zulevi and P. A. Kohl, J. Power Sources, 2022, 536, 231495 CrossRef CAS.
- T. Wang, Y. Wang and W. You, J. Membr. Sci., 2023, 685, 121916 CrossRef CAS.
- X. He, X. Jiang, Z. Wang, Y. Deng, Z. Han, Y. Yang and D. Chen, Polym. Eng. Sci., 2018, 58, 13–21 CrossRef CAS.
- Q. Li, X. He, L. Huang, Y. Lu, S. Zou, J. Ye, L. Huang, N. Yu, Z. Fu, X. Zang and D. Chen, J. Appl. Polym. Sci., 2023, 140, 1–17 CAS.
- X. He, J. Zou, Y. Wen, B. Wu, X. Zang, J. Deng, Z. Qin, G. Yang, J. Xu and D. Chen, Int. J. Hydrogen Energy, 2022, 47, 69–80 CrossRef CAS.
- L. Delaude and A. F. Noels, Kirk-Othmer Encyclopedia of Chemical Technology, 2005 Search PubMed.
- P. Jean-Louis Hérisson and Y. Chauvin, Die Makromolekulare Chemie, 1971, 141, 161–176 CrossRef.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- P. Atallah, K. B. Wagener and M. D. Schulz, Macromolecules, 2013, 46, 4735–4741 CrossRef CAS.
- P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039–2041 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903–2906 CrossRef CAS PubMed.
- T.-L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- R. Walker, R. M. Conrad and R. H. Grubbs, Macromolecules, 2009, 42, 599–605 CrossRef CAS PubMed.
- W. T. Chen, M. Mandal, G. Huang, X. M. Wu, G. H. He and P. A. Kohl, ACS Appl. Energy Mater., 2019, 2, 2458–2468 CrossRef CAS.
- M. Treichel, C. Tyler Womble, R. Selhorst, J. Gaitor, T. M. S. K. Pathiranage, T. Kowalewski and K. J. T. Noonan, Macromolecules, 2020, 53, 8509–8518 CrossRef CAS.
- W. L. Truett, D. R. Johnson, I. M. Robinson and B. A. Montague, J. Am. Chem. Soc., 1960, 82, 2337–2340 CrossRef CAS.
- G. Natta, G. Dall’Asta and G. Mazzanti, Angew. Chem., Int. Ed. Engl., 1964, 3, 723–729 CrossRef.
- S. Hayano, H. Kurakata, Y. Tsunogae, Y. Nakayama, Y. Sato and H. Yasuda, Macromolecules, 2003, 36, 7422–7431 CrossRef CAS.
- S. Hayano, T. Sugawara and Y. Tsunogae, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3153–3158 CrossRef.
- S. Hayano, Y. Takeyama, Y. Tsunogae and I. Igarashi, Macromolecules, 2006, 39, 4663–4670 CrossRef CAS.
- S. Hayano and Y. Tsunogae, Macromolecules, 2006, 39, 30–38 CrossRef CAS.
- B. Autenrieth, H. Jeong, W. P. Forrest, J. C. Axtell, A. Ota, T. Lehr, M. R. Buchmeiser and R. R. Schrock, Macromolecules, 2015, 48, 2480–2492 CrossRef CAS.
- B. Autenrieth and R. R. Schrock, Macromolecules, 2015, 48, 2493–2503 CrossRef CAS.
- B. Ai-Samak, V. Amir-Ebrahimi, A. G. Carvill, J. G. Hamilton and J. J. Rooney, Polym. Int., 1996, 41, 85–92 CrossRef.
- Y. Nakama, S. Hayano and K. Tashiro, Macromolecules, 2021, 54, 8122–8134 CrossRef CAS.
- Z. Wu and R. H. Grubbs, Macromolecules, 1994, 27, 6700–6703 CrossRef CAS.
- H. A. Kostalik, T. J. Clark, N. J. Robertson, P. F. Mutolo, J. M. Longo, H. D. Abruña and G. W. Coates, Macromolecules, 2010, 43, 7147–7150 CrossRef CAS.
- L.-B. W. Lee and R. A. Register, Macromolecules, 2005, 38, 1216–1222 CrossRef CAS.
- T. J. Clark, N. J. Robertson, H. A. Kostalik Iv, E. B. Lobkovsky, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 12888–12889 CrossRef CAS PubMed.
- N. J. Robertson, H. A. Kostalik, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400–3404 CrossRef CAS PubMed.
- C. R. Peltier, W. You, D. F. Volcanjk, Q. H. Li, A. J. Macbeth, H. D. Abruna and G. W. Coates, ACS Energy Lett., 2023, 8, 2365–2372 CrossRef CAS.
- W. You, J. M. Ganley, B. G. Ernst, C. R. Peltier, H. Y. Ko, R. A. DiStasio, R. R. Knowles and G. W. Coates, Chem. Sci., 2021, 12, 3898–3910 RSC.
- S. C. Price, X. Ren, A. M. Savage and F. L. Beyer, Polym. Chem., 2017, 8, 5708–5717 RSC.
- Y. Zhao, L. Feng, J. Gao, Y. Zhao, S. Wang, V. Ramani, Z. Zhang and X. Xie, Int. J. Hydrogen Energy, 2016, 41, 16264–16274 CrossRef CAS.
- C. Wang, B. Mo, Z. He, Q. Shao, D. Pan, E. Wujick, J. Guo, X. Xie, X. Xie and Z. Guo, J. Membr. Sci., 2018, 556, 118–125 CrossRef CAS.
- W. You, K. M. Hugar and G. W. Coates, Macromolecules, 2018, 51, 3212–3218 CrossRef CAS.
- W. You, E. Padgett, S. N. MacMillan, D. A. Muller and G. W. Coates, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9729–9734 CrossRef CAS PubMed.
- C. Cheng, X. He, S. Huang, F. Zhang, Y. Guo, Y. Wen, B. Wu and D. Chen, Int. J. Hydrogen Energy, 2020, 45, 19676–19690 CrossRef CAS.
- F. Zhang, X. He, C. Cheng, S. Huang, Y. Duan, C. Zhu, Y. Guo, K. Wang and D. Chen, Int. J. Hydrogen Energy, 2020, 45, 13090–13100 CrossRef CAS.
- R. Schwesinger, R. Link, P. Wenzl, S. Kossek and M. Keller, Chem. – Eur. J., 2006, 12, 429–437 CrossRef PubMed.
- J. C. Gaitor, M. Treichel, T. Kowalewski and K. J. T. Noonan, ACS Appl. Polym. Mater., 2022, 4, 8032–8042 CrossRef CAS.
- T. Zhu, S. Xu, A. Rahman, E. Dogdibegovic, P. Yang, P. Pageni, M. P. Kabir, X.-D. Zhou and C. Tang, Angew. Chem., Int. Ed., 2018, 57, 2388–2392 CrossRef CAS PubMed.
- T. Zhu and C. Tang, Polym. Chem., 2020, 11, 4542–4546 RSC.
- T. Zhu, Y. Sha, H. A. Firouzjaie, X. Peng, Y. Cha, D. M. M. M. Dissanayake, M. D. Smith, A. K. Vannucci, W. E. Mustain and C. Tang, J. Am. Chem. Soc., 2020, 142, 1083–1089 CrossRef CAS PubMed.
- H. Yuan, Y. Liu, T.-H. Tsai, X. Liu, S. B. Kim, R. Gupta, W. Zhang, S. P. Ertem, S. Seifert, A. M. Herring and E. B. Coughlin, Polyhedron, 2020, 181, 114462 CrossRef CAS.
- S. Gu, J. Wang, R. B. Kaspar, Q. Fang, B. Zhang, E. Bryan Coughlin and Y. Yan, Sci. Rep., 2015, 5, 11668 CrossRef CAS PubMed.
- K. M. Hugar, W. You and G. W. Coates, ACS Energy Lett., 2019, 4, 1681–1686 CrossRef CAS.
- Y. Zha, M. L. Disabb-Miller, Z. D. Johnson, M. A. Hickner and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 4493–4496 CrossRef CAS PubMed.
- M. L. Disabb-Miller, Y. Zha, A. J. DeCarlo, M. Pawar, G. N. Tew and M. A. Hickner, Macromolecules, 2013, 46, 9279–9287 CrossRef CAS.
- M. T. Kwasny and G. N. Tew, J. Mater. Chem. A, 2017, 5, 1400–1405 RSC.
- M. T. Kwasny, L. Zhu, M. A. Hickner and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 328–339 CrossRef CAS.
- H. Zhang, X. Wang, Y. Wang, Y. Zhang, W. Zhang and W. You, Angew. Chem., Int. Ed., 2023, 62, e202217742 CrossRef CAS PubMed.
- J.-M. Lehn and F. Montavon, Helv. Chim. Acta, 1978, 61, 67–82 CrossRef CAS.
- A. Bozzi and A. Chapiro, Int. J. Radiat. Appl. Instrumentation, Part C. Radiat. Phys. Chem., 1988, 32, 193–196 CrossRef CAS.
- S. D. Poynton, R. C. T. Slade, T. J. Omasta, W. E. Mustain, R. Escudero-Cid, P. Ocón and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 5124–5130 RSC.
- A. Abbasi, M. M. Nasef, S. Kheawhom, R. Faridi-Majidi, M. Takeshi, E. Abouzari-Lotf and T. Choong, Radiat. Phys. Chem., 2019, 156, 58–66 CrossRef CAS.
- N. Rajabalizadeh Mojarrad, S. Sadeghi, B. Yarar Kaplan, E. Güler and S. Alkan Gürsel, ACS Appl. Energy Mater., 2020, 3, 532–540 CrossRef CAS.
- M. M. Nasef, S. A. Gürsel, D. Karabelli and O. Güven, Prog. Polym. Sci., 2016, 63, 1–41 CrossRef CAS.
- T. Zhou, R. Shao, S. Chen, X. He, J. Qiao and J. Zhang, J. Power Sources, 2015, 293, 946–975 CrossRef CAS.
- M. M. Nasef, Chem. Rev., 2014, 114, 12278–12329 CrossRef CAS PubMed.
- L. Gubler, Adv. Energy Mater., 2014, 4, 1300827 CrossRef.
- C. A. Giron Rodriguez, B. Ó. Joensen, A. B. Moss, G. O. Larrazábal, D. K. Whelligan, B. Seger, J. R. Varcoe and T. R. Willson, ACS Sustainable Chem. Eng., 2023, 11, 1508–1517 CrossRef CAS PubMed.
- A. Chakraborty, I. Salam, M. Choolaei, J. Lee, C. Crean, D. K. Whelligan, R. Bance-Soualhi and J. R. Varcoe, Mater. Adv., 2023, 4, 2099–2105 RSC.
- R. Bance-Soualhi, M. Choolaei, S. A. Franklin, T. R. Willson, J. Lee, D. K. Whelligan, C. Crean and J. R. Varcoe, J. Mater. Chem. A, 2021, 9, 22025–22038 RSC.
- D. V. Golubenko, B. Van der Bruggen and A. B. Yaroslavtsev, J. Power Sources, 2021, 511, 230460 CrossRef CAS.
- M. Abdiani, E. Abouzari-Lotf, T. M. Ting, P. Moozarm Nia, S. S. Sha'rani, A. Shockravi and A. Ahmad, J. Power Sources, 2019, 424, 245–253 CrossRef CAS.
- O. Nibel, T. Rojek, T. J. Schmidt and L. Gubler, ChemSusChem, 2017, 10, 2767–2777 CrossRef CAS PubMed.
- J. H. Park, M. J. Han, D. S. Song and J. Y. Jho, ACS Appl. Mater. Interfaces, 2014, 6, 22847–22854 CrossRef CAS PubMed.
- Y. Ding, X. Shen, J. Zeng, X. Wang, L. Peng, P. Zhang and J. Zhao, Solid State Ionics, 2018, 323, 16–24 CrossRef CAS.
- L. Chen, Z. Hou, X. Lu, P. Chen, Z. Liu, L. Shen, X. Bian and Q. Qin, J. Appl. Polym. Sci., 2013, 128, 3949–3956 CrossRef CAS.
- J. Ao, H. Zhang, X. Xu, F. Yao, L. Ma, L. Zhang, B. Ye, Q. Li, L. Xu and H. Ma, RSC Adv., 2019, 9, 28588–28597 RSC.
- E. M. Kornacka, G. Przybytniak, L. Fuks, M. Walo and K. Łyczko, Radiat. Phys. Chem., 2014, 94, 115–118 CrossRef CAS.
- M. Grasselli and E. E. Smolko, Radiat. Phys. Chem., 2022, 194, 110055 CrossRef CAS.
- A. Elizalde-Cárdenas, M. González-Torres, G. Leyva-Gómez, H. Cortés, O. González-Mendoza, M. A. Pérez-Díaz, C. Pineda and R. M. Ribas-Aparicio, Mater. Lett., 2022, 324, 132783 CrossRef.
- L. Dong, X. Liu, Z. Xiong, D. Sheng, C. Lin, Y. Zhou and Y. Yang, Appl. Surf. Sci., 2018, 444, 497–504 CrossRef CAS.
- V. Sproll, T. J. Schmidt and L. Gubler, Polym. Int., 2016, 65, 174–180 CrossRef CAS.
- K. Czaja and M. SudoŁ, Radiat. Phys. Chem., 2011, 80, 514–521 CrossRef CAS.
- R. Bryant, Radiat. Phys. Chem., 2020, 174, 108895 CrossRef CAS.
- A. S. Barbosa, A. L. G. Biancolli, A. J. C. Lanfredi, O. Rodrigues, F. C. Fonseca and E. I. Santiago, J. Membr. Sci., 2022, 659, 120804 CrossRef CAS.
- A. L. G. Biancolli, S. Bsoul-Haj, J. C. Douglin, A. S. Barbosa, R. R. de Sousa, O. Rodrigues, A. J. C. Lanfredi, D. R. Dekel and E. I. Santiago, J. Membr. Sci., 2022, 641, 119879 CrossRef CAS.
- P. Posadas, J. L. Valentín, R. Benavente, E. Blázquez-Blázquez, A. Urtiaga, J. A. Álvarez and M. L. Cerrada, Radiat. Phys. Chem., 2023, 204, 110694 CrossRef CAS.
- A. L. G. Biancolli, A. S. Barbosa, Y. Kodama, R. R. de Sousa, A. J. C. Lanfredi, F. C. Fonseca, J. F. Q. Rey and E. I. Santiago, J. Power Sources, 2021, 512, 230484 CrossRef CAS.
- L. Wang, J. J. Brink, Y. Liu, A. M. Herring, J. Ponce-González, D. K. Whelligan and J. R. Varcoe, Energy Environ. Sci., 2017, 10, 2154–2167 RSC.
- L. Wang, X. Peng, W. E. Mustain and J. R. Varcoe, Energy Environ. Sci., 2019, 12, 1575–1579 RSC.
- W. H. Lee, C. Crean, J. R. Varcoe and R. Bance-Soualhi, RSC Adv., 2017, 7, 47726–47737 RSC.
- T. N. Danks, R. C. T. Slade and J. R. Varcoe, J. Mater. Chem., 2003, 13, 712–721 RSC.
- A. J. Samaniego, A. K. Arabelo, M. Sarker, F. Mojica, J. Madrid, P.-Y. A. Chuang, J. Ocon and R. Espiritu, J. Appl. Polym. Sci., 2021, 138, 49947 CrossRef CAS.
- E. Abouzari-lotf, H. Ghassemi, M. M. Nasef, A. Ahmad, M. Zakeri, T. M. Ting, A. Abbasi and S. Mehdipour-Ataei, J. Mater. Chem. A, 2017, 5, 15326–15341 RSC.
- Y. Zhao, K. Yoshimura, S. Sawada, T. Motegi, A. Hiroki, A. Radulescu and Y. Maekawa, Macromolecules, 2022, 55, 7100–7109 CrossRef CAS.
- Y. Zhao, K. Yoshimura, H. Takamatsu, A. Hiroki, Y. Kishiyama, H. Shishitani, S. Yamaguchi, H. Tanaka, S. Koizumi, A. Radulescu, M.-S. Appavou and Y. Maekawa, J. Electrochem. Soc., 2019, 166, F472 CrossRef CAS.
- K. Yoshimura, Y. Zhao, A. Hiroki, Y. Kishiyama, H. Shishitani, S. Yamaguchi, H. Tanaka, S. Koizumi, J. E. Houston, A. Radulescu, M.-S. Appavou, D. Richter and Y. Maekawa, Soft Matter, 2018, 14, 9118–9131 RSC.
- Y. Zhao, K. Yoshimura, H. Shishitani, S. Yamaguchi, H. Tanaka, S. Koizumi, N. Szekely, A. Radulescu, D. Richter and Y. Maekawa, Soft Matter, 2016, 12, 1567–1578 RSC.
- T. P. Pandey, A. M. Maes, H. N. Sarode, B. D. Peters, S. Lavina, K. Vezzù, Y. Yang, S. D. Poynton, J. R. Varcoe, S. Seifert, M. W. Liberatore, V. Di Noto and A. M. Herring, Phys. Chem. Chem. Phys., 2015, 17, 4367–4378 RSC.
- B.-S. Ko, K. Yoshimura, A. Hiroki and Y. Maekawa, J. Polym. Sci., 2021, 59, 108–116 CrossRef CAS.
- V. Sproll, G. Nagy, U. Gasser, J. P. Embs, M. Obiols-Rabasa, T. J. Schmidt, L. Gubler and S. Balog, Macromolecules, 2016, 49, 4253–4264 CrossRef CAS.
- A. L. Gonçalves Biancolli, D. Herranz, L. Wang, G. Stehlíková, R. Bance-Soualhi, J. Ponce-González, P. Ocón, E. A. Ticianelli, D. K. Whelligan, J. R. Varcoe and E. I. Santiago, J. Mater. Chem. A, 2018, 6, 24330–24341 RSC.
- L. Wang, E. Magliocca, E. L. Cunningham, W. E. Mustain, S. D. Poynton, R. Escudero-Cid, M. M. Nasef, J. Ponce-González, R. Bance-Souahli, R. C. T. Slade, D. K. Whelligan and J. R. Varcoe, Green Chem., 2017, 19, 831–843 RSC.
- B.-S. Ko, K. Yoshimura, S. Warapon, H. Shishitani, S. Yamaguchi, H. Tanaka and Y. Maekawa, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 503–510 CrossRef CAS.
- H. Koshikawa, K. Yoshimura, W. Sinnananchi, T. Yamaki, M. Asano, K. Yamamoto, S. Yamaguchi, H. Tanaka and Y. Maekawa, Macromol. Chem. Phys., 2013, 214, 1756–1762 CrossRef CAS.
- T. A. Sherazi, J. Yong Sohn, Y. Moo Lee and M. D. Guiver, J. Membr. Sci., 2013, 441, 148–157 CrossRef CAS.
- M. Mamlouk, J. A. Horsfall, C. Williams and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 11912–11920 CrossRef CAS.
- A. Zhegur-Khais, F. Kubannek, U. Krewer and D. R. Dekel, J. Membr. Sci., 2020, 612, 118461 CrossRef CAS.
- Y. Zheng, U. Ash, R. P. Pandey, A. G. Ozioko, J. Ponce-González, M. Handl, T. Weissbach, J. R. Varcoe, S. Holdcroft, M. W. Liberatore, R. Hiesgen and D. R. Dekel, Macromolecules, 2018, 51, 3264–3278 CrossRef CAS.
- T. R. Willson, C. A. Giron Rodriguez, Q. Xu, J. Frow, F. Foglia, K. Smith, R. Ravikumar, M. Vinothkannan, N. Mahmoudi, I. Salam, A. P. Periasamy, D. K. Whelligan, M. Mamlouk, H. Lin, B. Seger and J. R. Varcoe, J. Mater. Chem. A, 2023, 11, 20724–20740 RSC.
- M. Najibah, J. Kong, H. Khalid, J. Hnát, H. S. Park, K. Bouzek and D. Henkensmeier, J. Membr. Sci., 2023, 670, 121344 CrossRef CAS.
- J. Ponce-González, D. K. Whelligan, L. Wang, R. Bance-Soualhi, Y. Wang, Y. Peng, H. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724–3735 RSC.
- E. Abouzari-Lotf, M. V. Jacob, H. Ghassemi, M. Zakeri, M. M. Nasef, Y. Abdolahi, A. Abbasi and A. Ahmad, Sci. Rep., 2021, 11, 3764 CrossRef CAS PubMed.
- T. A. Sherazi, S. Zahoor, R. Raza, A. J. Shaikh, S. A. R. Naqvi, G. Abbas, Y. Khan and S. Li, Int. J. Hydrogen Energy, 2015, 40, 786–796 CrossRef CAS.
- B.-S. Ko, J.-Y. Sohn and J. Shin, Polymer, 2012, 53, 4652–4661 CrossRef CAS.
- J. Fang, Y. Yang, X. Lu, M. Ye, W. Li and Y. Zhang, Int. J. Hydrogen Energy, 2012, 37, 594–602 CrossRef CAS.
- K. Aggarwal, N. Gjineci, A. Kaushansky, S. Bsoul, J. C. Douglin, S. Li, I. Salam, S. Aharonovich, J. R. Varcoe, D. R. Dekel and C. E. Diesendruck, ACS Mater. Au, 2022, 2, 367–373 CrossRef CAS PubMed.
- L. Wang and M. A. Hickner, Polym. Chem., 2014, 5, 2928–2935 RSC.
- K. M. Meek, C. M. Reed, B. Pivovar, K.-D. Kreuer, J. R. Varcoe and R. Bance-Soualhi, RSC Adv., 2020, 10, 36467–36477 RSC.
- J. Müller, A. Zhegur, U. Krewer, J. R. Varcoe and D. R. Dekel, ACS Mater. Lett., 2020, 2, 168–173 CrossRef PubMed.
- J. C. Douglin, J. R. Varcoe and D. R. Dekel, J. Power Sources Adv., 2020, 5, 100023 CrossRef.
- J. Ponce-González, J. R. Varcoe and D. K. Whelligan, ACS Appl. Energy Mater., 2018, 1, 1883–1887 CrossRef.
- J. Ponce-González, I. Ouachan, J. R. Varcoe and D. K. Whelligan, J. Mater. Chem. A, 2018, 6, 823–827 RSC.
- O. M. M. Page, S. D. Poynton, S. Murphy, A. Lien Ong, D. M. Hillman, C. A. Hancock, M. G. Hale, D. C. Apperley and J. R. Varcoe, RSC Adv., 2013, 3, 579–587 RSC.
- X. Li, Y. Yu, Q. Liu and Y. Meng, Int. J. Hydrogen Energy, 2013, 38, 11067–11073 CrossRef CAS.
- S. Willdorf-Cohen, A. Zhegur-Khais, J. Ponce-González, S. Bsoul-Haj, J. R. Varcoe, C. E. Diesendruck and D. R. Dekel, ACS Appl. Energy Mater., 2023, 6, 1085–1092 CrossRef CAS PubMed.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- K. Jetsrisuparb, H. Ben youcef, A. Wokaun and L. Gubler, J. Membr. Sci., 2014, 450, 28–37 CrossRef CAS.
- S. Wierzbicki, J. C. Douglin, R. K. Singh, D. R. Dekel and K. Kruczała, ACS Catal., 2023, 13, 2744–2750 CrossRef CAS.
- R. Espiritu, M. Mamlouk and K. Scott, Int. J. Hydrogen Energy, 2016, 41, 1120–1133 CrossRef CAS.
- T. Hamada, K. Yoshimura, K. Takeuchi, S. Watanabe, Y. Zhao, A. Hiroki, T. Hagiwara, H. Shishitani, S. Yamaguchi, H. Tanaka, A. Radulescu, K. Ohwada and Y. Maekawa, Macromol. Chem. Phys., 2021, 222, 2100028 CrossRef CAS.
- A. M. A. Mahmoud, K. Yoshimura and Y. Maekawa, J. Membr. Sci., 2021, 620, 118844 CrossRef CAS.
- T. Hamada, K. Yoshimura, A. Hiroki and Y. Maekawa, J. Appl. Polym. Sci., 2018, 135, 46886 CrossRef.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- N. Zion, J. C. Douglin, D. A. Cullen, P. Zelenay, D. R. Dekel and L. Elbaz, Adv. Funct. Mater., 2021, 31, 2100963 CrossRef CAS.
- R. Ren, X. Wang, H. Chen, H. A. Miller, I. Salam, J. R. Varcoe, L. Wu, Y. Chen, H.-G. Liao, E. Liu, F. Bartoli, F. Vizza, Q. Jia and Q. He, Angew. Chem., Int. Ed., 2021, 60, 4049–4054 CrossRef CAS PubMed.
- J. C. Douglin, R. K. Singh, S. Haj-Bsoul, S. Li, J. Biemolt, N. Yan, J. R. Varcoe, G. Rothenberg and D. R. Dekel, Chem. Eng. J. Adv., 2021, 8, 100153 CrossRef CAS.
- X. Peng, T. J. Omasta, E. Magliocca, L. Wang, J. R. Varcoe and W. E. Mustain, Angew. Chem., Int. Ed., 2019, 58, 1046–1051 CrossRef CAS PubMed.
- T. de Wild, T. Nemeth, P. Becker, D. Günther, T. Nauser, T. J. Schmidt and L. Gubler, J. Power Sources, 2023, 560, 232525 CrossRef CAS.
- A. Shirole, P. M. Bakvand and P. Jannasch, ACS Appl. Energy Mater., 2023, 6, 7240–7249 CrossRef CAS.
- J. Zhou, P. Chen, Q. Weng, J. Fang, X. Chen and Z. An, Int. J. Hydrogen Energy, 2016, 41, 5765–5775 CrossRef CAS.
- Y. Buchmüller, Z. Zhang, A. Wokaun and L. Gubler, RSC Adv., 2014, 4, 51911–51915 RSC.
- D. Henkensmeier, H. Ben youcef, F. Wallasch and L. Gubler, J. Membr. Sci., 2013, 447, 228–235 CrossRef CAS.
- T. Hamada, Y. Zhao, K. Yoshimura, A. Radulescu, K. Ohwada and Y. Maekawa, ChemistrySelect, 2021, 6, 8879–8888 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- K. I. Winey, D. A. Gobran, Z. Xu, L. J. Fetters and E. L. Thomas, Macromolecules, 1994, 27, 2392–2397 CrossRef CAS.
- G. Kim and M. Libera, Macromolecules, 1998, 31, 2569–2577 CrossRef CAS.
- A. Hirao, R. Goseki and T. Ishizone, Macromolecules, 2014, 47, 1883–1905 CrossRef CAS.
- S. Elamathi, G. Nithyakalyani, D. Sangeetha and S. Ravichandran, Ionics, 2008, 14, 377–385 CrossRef CAS.
- J. Won, S. W. Choi, Y. S. Kang, H. Y. Ha, I.-H. Oh, H. S. Kim, K. T. Kim and W. H. Jo, J. Membr. Sci., 2003, 214, 245–257 CrossRef CAS.
- P. Dai, Z.-H. Mo, R.-W. Xu, S. Zhang and Y.-X. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20329–20341 CrossRef CAS PubMed.
- Q. H. Zeng, Q. L. Liu, I. Broadwell, A. M. Zhu, Y. Xiong and X. P. Tu, J. Membr. Sci., 2010, 349, 237–243 CrossRef CAS.
- L. Sun, J. Guo, J. Zhou, Q. Xu, D. Chu and R. Chen, J. Power Sources, 2012, 202, 70–77 CrossRef CAS.
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, M. R. Sturgeon, H. Long, B. S. Pivovar and C. Bae, J. Electrochem. Soc., 2017, 164, F1279–F1285 CrossRef CAS.
- C. Xiao Lin, X. Qin Wang, E. Ning Hu, Q. Yang, Q. Gen Zhang, A. Mei Zhu and Q. Lin Liu, J. Membr. Sci., 2017, 541, 358–366 CrossRef.
- J. Y. Jeon, S. Park, J. Han, S. Maurya, A. D. Mohanty, D. Tian, N. Saikia, M. A. Hickner, C. Y. Ryu, M. E. Tuckerman, S. J. Paddison, Y. S. Kim and C. Bae, Macromolecules, 2019, 52, 2139–2147 CrossRef CAS.
- Y. Li, Y. Liu, A. M. Savage, F. L. Beyer, S. Seifert, A. M. Herring and D. M. Knauss, Macromolecules, 2015, 48, 6523–6533 CrossRef CAS.
- S. P. Ertem, B. R. Caire, T.-H. Tsai, D. Zeng, M. A. Vandiver, A. Kusoglu, S. Seifert, R. C. Hayward, A. Z. Weber, A. M. Herring, E. B. Coughlin and M. W. Liberatore, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 612–622 CrossRef CAS.
- C. Trant, S. Hwang, C. Bae and S. Lee, Macromolecules, 2020, 53, 8548–8561 CrossRef CAS.
- H. Strathmann, AIChE J., 2001, 47, 1077–1087 CrossRef CAS.
- W. Grot, Fluorinated ionomers, William Andrew, 2011 Search PubMed.
- A. Loh, X. Li, S. Sluijter, P. Shirvanian, Q. Lai and Y. Liang, Hydrogen, 2023, 4, 257–271 CrossRef CAS.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Conversion Storage, 2020, 18, 024001 CrossRef.
- W. Jiang, A. Y. Faid, B. F. Gomes, I. Galkina, L. Xia, C. M. S. Lobo, M. Desmau, P. Borowski, H. Hartmann, A. Maljusch, A. Besmehn, C. Roth, S. Sunde, W. Lehnert and M. Shviro, Adv. Funct. Mater., 2022, 32, 2203520 CrossRef CAS.
- H. Khalid, M. Najibah, H. S. Park, C. Bae and D. Henkensmeier, Membranes, 2022, 12, 989 CrossRef CAS PubMed.
- J. Park, L. Wang, S. G. Advani and A. K. Prasad, J. Electrochem. Soc., 2012, 159, F864 CrossRef CAS.
- J. J. Kaczur, H. Yang, Z. Liu, S. A. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 263 CrossRef PubMed.
- Z. Liu, S. D. Sajjad, Y. Gao, H. Yang, J. J. Kaczur and R. I. Masel, Int. J. Hydrogen Energy, 2017, 42, 29661–29665 CrossRef CAS.
- M. Moreno-Gonzalez, P. Mardle, S. Zhu, B. Gholamkhass, S. Jones, N. Chen, B. Britton and S. Holdcroft, J. Power Sources Adv., 2023, 19, 100109 CrossRef CAS.
- Ionomr, 2023, https://ionomr.com/wp-content/uploads/2023/2005/FM-6063-B-Properties-of-2023rd-Gen-Aemion-Water-Electrolysis-Membranes-2023rd-Gen-2023-2001-2011.pdf.
- J. Hnát, M. Plevová, J. Žitka, M. Paidar and K. Bouzek, Electrochim. Acta, 2017, 248, 547–555 CrossRef.
- TailorMem, 2023, tailormem.com/#products.
- X. Luo, S. Rojas-Carbonell, Y. Yan and A. Kusoglu, J. Membr. Sci., 2020, 598, 117680 CrossRef CAS.
- W. J. Song, K. Peng, W. Xu, X. Liu, H. Q. Zhang, X. Liang, B. J. Ye, H. J. Zhang, Z. J. Yang, L. Wu, X. L. Ge and T. W. Xu, Nat. Commun., 2023, 14, 2732 CrossRef CAS PubMed.
- J. W. Gooch, Encyclopedic dictionary of polymers, Springer Science & Business Media, 2010 Search PubMed.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- H.-P. Brack and G. G. Scherer, Macromol. Symp., 1998, 126, 25–49 CrossRef CAS.
- D. E. Curtin, R. D. Lousenberg, T. J. Henry, P. C. Tangeman and M. E. Tisack, J. Power Sources, 2004, 131, 41–48 CrossRef CAS.
- T. Steenberg, H. A. Hjuler, C. Terkelsen, M. T. R. Sánchez, L. N. Cleemann and F. C. Krebs, Energy Environ. Sci., 2012, 5, 6076–6080 RSC.
- D. Igali, A. Perveen, D. Zhang and D. Wei, Processes, 2020, 8, 1524 CrossRef.
- N. N. Krishnan, D. Henkensmeier, J. H. Jang and H.-J. Kim, Macromol. Mater. Eng., 2014, 299, 1031–1041 CrossRef CAS.
- D. Aili, J. Yang, K. Jankova, D. Henkensmeier and Q. Li, J. Mater. Chem. A, 2020, 8, 12854–12886 RSC.
- É. Ujaczki, V. Stark-Rogel, M. Olbrich, M. Fuetsch and J. Backmann, Environ. Sci. Eur., 2022, 34, 101 CrossRef.
- Risk Evaluation for N-Methylpyrrolidone (NMP) (https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/risk-evaluation-n-methylpyrrolidone-nmp-0)).
- R. Moore Iii and C. Martin, Macromolecules, 1988, 21, 1334–1339 CrossRef.
- Y. S. Kim, C. F. Welch, R. P. Hjelm, N. H. Mack, A. Labouriau and E. B. Orler, Macromolecules, 2015, 48, 2161–2172 CrossRef CAS.
- T. D. Myles, K. N. Grew, A. A. Peracchio and W. K. S. Chiu, J. Power Sources, 2015, 296, 225–236 CrossRef CAS.
- Y. S. Kim and K.-S. Lee, Polym. Rev., 2015, 55, 330–370 CrossRef CAS.
- F. Wang, M. Hickner, Y. S. Kim, T. A. Zawodzinski and J. E. McGrath, J. Membr. Sci., 2002, 197, 231–242 CrossRef CAS.
- Z. Xie, C. Song, B. Andreaus, T. Navessin, Z. Shi, J. Zhang and S. Holdcroft, J. Electrochem. Soc., 2006, 153, E173 CrossRef CAS.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254 CrossRef CAS.
- A. Amel, N. Gavish, L. Zhu, D. R. Dekel, M. A. Hickner and Y. Ein-Eli, J. Membr. Sci., 2016, 514, 125–134 CrossRef CAS.
- M. Irfan, E. Bakangura, N. U. Afsar, M. M. Hossain, J. Ran and T. Xu, J. Power Sources, 2017, 355, 171–180 CrossRef CAS.
- K. Zhang, M. B. McDonald, I. E. A. Genina and P. T. Hammond, Chem. Mater., 2018, 30, 6420–6430 CrossRef CAS.
- A. M. Kiss, T. D. Myles, K. N. Grew, A. A. Peracchio, G. J. Nelson and W. K. S. Chiu, J. Electrochem. Soc., 2013, 160, F994 CrossRef CAS.
- N. Ziv and D. R. Dekel, Electrochem. Commun., 2018, 88, 109–113 CrossRef CAS.
- F. N. Büchi, A. Marek and G. G. Scherer, J. Electrochem. Soc., 1995, 142, 1895 CrossRef.
- T. E. Springer, T. A. Zawodzinski, M. S. Wilson and S. Gottesfeld, J. Electrochem. Soc., 1996, 143, 587 CrossRef CAS.
- K. R. Cooper and M. Smith, J. Power Sources, 2006, 160, 1088–1095 CrossRef CAS.
- B. S. Pivovar and Y. S. Kim, J. Electrochem. Soc., 2007, 154, B739 CrossRef CAS.
- N. Yaghini, L. Nordstierna and A. Martinelli, Phys. Chem. Chem. Phys., 2014, 16, 9266–9275 RSC.
- D. R. P. Morris, S. P. Liu, D. Villegas Gonzalez and J. T. Gostick, ACS Appl. Mater. Interfaces, 2014, 6, 18609–18618 CrossRef CAS PubMed.
- F. Wang, D. Wang and Y. Nagao, ChemSusChem, 2021, 14, 2694–2697 CrossRef CAS PubMed.
- U. N. Shrivastava, A. Zhegur-Khais, M. Bass, S. Willdorf-Cohen, V. Freger, D. R. Dekel and K. Karan, J. Phys. Chem. C, 2020, 124, 23469–23478 CrossRef CAS.
- X. Luo, D. I. Kushner, J. Li, E. J. Park, Y. S. Kim and A. Kusoglu, Adv. Funct. Mater., 2021, 31, 2008778 CrossRef CAS.
- E. J. Park, S. Komini Babu and Y. S. Kim, Front. Energy Res., 2022, 10, 945654 CrossRef.
- M. Bernt, J. Schröter, M. Möckl and H. A. Gasteiger, J. Electrochem. Soc., 2020, 167, 124502 CrossRef CAS.
- P. D. Beattie, V. I. Basura and S. Holdcroft, J. Electroanal. Chem., 1999, 468, 180–192 CrossRef CAS.
- S. S. Kocha, J. Deliang Yang and J. S. Yi, AIChE J., 2006, 52, 1916–1925 CrossRef CAS.
- T. Sakai, H. Takenaka, N. Wakabayashi, Y. Kawami and E. Torikai, J. Electrochem. Soc., 1985, 132, 1328 CrossRef CAS.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
- L. Wang, S. Rojas-Carbonell, K. Hu, B. P. Setzler, A. R. Motz, M. E. Ueckermann and Y. Yan, Front. Energy Res., 2022, 10, 887893 CrossRef.
- C. G. Arges, J. Parrondo, G. Johnson, A. Nadhan and V. Ramani, J. Mater. Chem., 2012, 22, 3733–3744 RSC.
- M. R. Hibbs, M. A. Hickner, T. M. Alam, S. K. McIntyre, C. H. Fujimoto and C. J. Cornelius, Chem. Mater., 2008, 20, 2566–2573 CrossRef CAS.
- F. Karas, J. Hnát, M. Paidar, J. Schauer and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 5054–5062 CrossRef CAS.
- T. Weissbach, A. G. Wright, T. J. Peckham, A. Sadeghi Alavijeh, V. Pan, E. Kjeang and S. Holdcroft, Chem. Mater., 2016, 28, 8060–8070 CrossRef CAS.
- Y. Mei, Z. Yao, L. Ji, P. H. Toy and C. Y. Tang, J. Membr. Sci., 2018, 549, 295–305 CrossRef CAS.
- A. D. Mohanty, Y.-B. Lee, L. Zhu, M. A. Hickner and C. Bae, Macromolecules, 2014, 47, 1973–1980 CrossRef CAS.
- X. Ge, Y. He, K. Zhang, X. Liang, C. Wei, M. A. Shehzad, W. Song, Z. Ge, G. Li, W. Yu, L. Wu and T. Xu, Research, 2021, 2021, 9762709 CrossRef CAS PubMed.
- C. G. Arges, V. Ramani, Z. Wang and R. J. Ouimet, Front. Energy Res., 2022, 10, 871851 CrossRef.
- K.-D. Kreuer and P. Jannasch, J. Power Sources, 2018, 375, 361–366 CrossRef CAS.
- S. Willdorf-Cohen, A. N. Mondal, D. R. Dekel and C. E. Diesendruck, J. Mater. Chem. A, 2018, 6, 22234–22239 RSC.
- H. T. Chung, Y.-K. Choe, U. Martinez, J. H. Dumont, A. Mohanty, C. Bae, I. Matanovic and Y. S. Kim, J. Electrochem. Soc., 2016, 163, F1503 CrossRef CAS.
- X. Chen, I. T. McCrum, K. A. Schwarz, M. J. Janik and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 15025–15029 CrossRef CAS PubMed.
- D. Li, H. T. Chung, S. Maurya, I. Matanovic and Y. S. Kim, Curr. Opin. Electrochem., 2018, 12, 189–195 CrossRef CAS.
- I. Matanovic and Y. S. Kim, Curr. Opin. Electrochem., 2023, 38, 101218 CrossRef CAS.
- K. Fraser, K. Dosaev, E. Savinova, S. Holdcroft and T. Asset, ChemCatChem, 2024, 16, e202301304 CrossRef CAS.
- J. H. Dumont, A. J. Spears, R. P. Hjelm, M. Hawley, S. Maurya, D. Li, G. Yuan, E. B. Watkins and Y. S. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 1825–1831 CrossRef CAS PubMed.
- A. Y. Faid, A. O. Barnett, F. Seland and S. Sunde, Int. J. Hydrogen Energy, 2022, 47, 23483–23497 CrossRef CAS.
- R. A. Krivina, G. A. Lindquist, S. R. Beaudoin, T. N. Stovall, W. L. Thompson, L. P. Twight, D. Marsh, J. Grzyb, K. Fabrizio, J. E. Hutchison and S. W. Boettcher, Adv. Mater., 2022, 34, 2203033 CrossRef CAS PubMed.
- B. Pivovar, Alkaline Membrane Fuel Cell Workshop, 2012 Search PubMed.
- B. Pivovar and Y. S. Kim, Anion Exchange Membrane Workshop, 2019 Search PubMed.
- B. Pivovar and Y. S. Kim, Alkaline Membrane Fuel Cell Workshop, 2016 Search PubMed.
- S. T. Thompson, D. Peterson, D. Ho and D. Papageorgopoulos, J. Electrochem. Soc., 2020, 167, 084514 CrossRef CAS.
- DOE, DE-FOA-0002922 Bipartisan Infrastructure Law: Clean Hydrogen Electrolysis, Manufacturing, and Recycling, 2023.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2015, 8, 177–181 RSC.
- J. Xue, J. C. Douglin, K. Yassin, T. Huang, H. Jiang, J. Zhang, Y. Yin, D. R. Dekel and M. D. Guiver, Joule, 2024, 8, 1–21 CrossRef.
- T. Wang, Y. Zhao, S. Wang, S. Cheng, S. Yang, H. Wei and Y. Ding, J. Power Sources, 2023, 557, 232590 CrossRef CAS.
- S. C. Xu, Y. Gu, T. Y. Ma, X. Su, J. Y. Chen and R. H. He, ACS Sustainable Chem. Eng., 2023, 11, 10402–10412 CrossRef CAS.
- R. X. Ma, Y. F. Kang, T. Wang, T. Jiang, H. Y. Yin, C. Liu, H. B. Wei and Y. S. Ding, J. Membr. Sci., 2023, 678, 121667 CrossRef CAS.
- D. P. Leonard, M. Lehmann, J. M. Klein, I. Matanovic, C. Fujimoto, T. Saito and Y. S. Kim, Adv. Energy Mater., 2023, 13, 2203488 CrossRef CAS.
- X. F. Wan, X. T. Wei, J. M. Ge, L. F. Lu, Z. Q. Liu and Y. Q. Zhu, J. Membr. Sci., 2023, 680, 121760 CrossRef CAS.
- J. Y. Chen, S. C. Xu, X. Su, W. Wei, Y. J. Li, R. L. Du and R. H. He, ACS Appl. Polym. Mater., 2023, 5, 6222–6231 CrossRef CAS.
- N. Xie, T. Wang, S. H. Du, Q. Weng, K. Zheng, T. Zhang, X. M. Ning, P. Chen, X. B. Chen and Z. W. An, J. Power Sources, 2023, 574, 233121 CrossRef CAS.
- J. M. Li, Q. Liu, L. Tian, W. L. Ma, F. H. Wang, Z. Q. Wang and H. Zhu, Int. J. Hydrogen Energy, 2022, 47, 32262–32272 CrossRef CAS.
- X. F. Li, K. Yang, Z. M. Wang, Y. H. Chen, Y. G. Li, J. Guo, J. F. Zheng, S. H. Li and S. B. Zhang, Macromolecules, 2022, 55, 10607–10617 CrossRef CAS.
- L. L. Ma, L. Li, M. H. Yuan, L. Bai, A. R. Zhang, X. M. Yan, G. H. He and F. X. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 5748–5757 CrossRef CAS.
- X. Z. Wang, W. T. Chen, T. T. Li, X. M. Yan, Y. Zhang, F. Zhang, X. M. Wu, B. Pang, J. N. Li and G. H. He, J. Mater. Chem. A, 2021, 9, 7522–7530 RSC.
- X. Y. Wu, N. J. Chen, C. Hu, H. A. Klok, Y. M. Lee and X. L. Hu, Adv. Mater., 2023, 35, 2210432 CrossRef CAS PubMed.
- C. Hu, J. H. Park, N. Y. Kang, X. Zhang, Y. J. Lee, S. W. Jeong and Y. M. Lee, J. Mater. Chem. A, 2023, 11, 2031–2041 RSC.
- Z. Yu, W. T. Gao, Y. J. Liu, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Colloid Interface Sci., 2023, 651, 404–414 CrossRef CAS PubMed.
- X. Li, B. Zhang, J. Guo, Y. Chen, L. Dai, J. Zheng, S. Li and S. Zhang, J. Mater. Chem. A, 2023, 11, 10738–10747 RSC.
- G. D. Xu, J. Pan, X. Y. Zou, Z. Y. Jin, J. L. Zhang, P. D. Fang, Q. H. Zhang, Z. Sun and F. Yan, Adv. Funct. Mater., 2023, 33, 2302364 CrossRef CAS.
- J. J. Wang, W. T. Gao, Y. S. L. Choo, Z. H. Cai, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Colloid Interface Sci., 2023, 629, 377–387 CrossRef CAS PubMed.
- W. T. Gao, X. L. Gao, W. W. Gou, J. J. Wang, Z. H. Cai, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2022, 655, 120578 CrossRef CAS.
- Q. L. Wei, X. Z. Cao, P. Veh, A. Konovalova, P. Mardle, P. Overton, S. Cassegrain, S. Vierrath, M. Breitwieser and S. Holdcroft, Sustainable Energy Fuels, 2022, 6, 3551–3564 RSC.
- M. L. Guo, T. Ban, Y. J. Wang, Y. N. Wang, Y. Y. Zhang, J. S. Zhang and X. L. Zhu, J. Membr. Sci., 2022, 647, 120299 CrossRef CAS.
- J. J. Chen, C. H. Shen and S. J. Gao, Desalination, 2023, 557, 116600 CrossRef CAS.
- J. J. Zhang, K. Y. Zhang, X. Liang, W. S. Yu, X. L. Ge, M. A. Shehzad, Z. J. Ge, Z. J. Yang, L. Wu and T. W. Xu, J. Mater. Chem. A, 2021, 9, 327–337 RSC.
- S. Favero, I. E. L. Stephens and M.-M. Titirci, Adv. Mater., 2023, 2308238 Search PubMed.
- A. C. Yang-Neyerlin, S. Medina, K. M. Meek, D. J. Strasser, C. He, D. M. Knauss, W. E. Mustain, S. Pylypenko and B. S. Pivovar, J. Electrochem. Soc., 2021, 168, 044525 CrossRef CAS.
- Y. S. Kim, ACS Appl. Polym. Mater., 2021, 3, 1250–1270 CrossRef CAS.
- X. J. Zhang, Y. J. Cao, M. Zhang, Y. G. Wang, H. Y. Tang and N. W. Li, J. Membr. Sci., 2020, 598, 117793 CrossRef CAS.
- X. M. Chu, Y. Shi, L. Liu, Y. D. Huang and N. W. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- L. Liu, X. M. Chu, J. Y. Liao, Y. D. Huang, Y. Li, Z. Y. Ge, M. A. Hickner and N. W. Li, Energy Environ. Sci., 2018, 11, 435–446 RSC.
- K. Yang, X. M. Chu, X. J. Zhang, X. F. Li, J. F. Zheng, S. H. Li, N. W. Li, T. A. Sherazi and S. B. Zhang, J. Membr. Sci., 2020, 603, 118025 CrossRef CAS.
- N. J. Chen, J. H. Park, C. Hu, H. H. Wang, H. M. Kim, N. Y. Kang and Y. M. Lee, J. Mater. Chem. A, 2022, 10, 3678–3687 RSC.
- N. Ul Hassan, M. J. Zachman, M. Mandal, H. A. Firouzjaie, P. A. Kohl, D. A. Cullen and W. E. Mustain, ACS Catal., 2022, 12, 8116–8126 CrossRef CAS.
- X. Liu, N. Xie, J. Xue, M. Li, C. Zheng, J. Zhang, Y. Qin, Y. Yin, D. R. Dekel and M. D. Guiver, Nat. Energy, 2022, 7, 329–339 CrossRef CAS.
- H. S. Shiau, I. V. Zenyuk and A. Z. Weber, J. Electrochem. Soc., 2017, 164, E3583–E3591 CrossRef CAS.
- H. Ito, N. Kawaguchi, S. Someya, T. Munakata, N. Miyazaki, M. Ishida and A. Nakano, Int. J. Hydrogen Energy, 2018, 43, 17030–17039 CrossRef CAS.
- A. Kiessling, J. C. Fornaciari, G. Anderson, X. Peng, A. Gerstmayr, M. R. Gerhardt, S. McKinney, A. Serov, Y. S. Kim, B. Zulevi, A. Z. Weber and N. Danilovic, J. Electrochem. Soc., 2021, 168, 084512 CrossRef CAS.
- B. Mayerhöfer, F. D. Speck, M. Hegelheimer, M. Bierling, D. Abbas, D. McLaughlin, S. Cherevko, S. Thiele and R. Peach, Int. J. Hydrogen Energy, 2022, 47, 4304–4314 CrossRef.
- I. Gatto, A. Caprì, C. Lo Vecchio, S. Zignani, A. Patti and V. Baglio, Int. J. Hydrogen Energy, 2023, 48, 11914–11921 CrossRef CAS.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed.
- M. Faraj, M. Boccia, H. Miller, F. Martini, S. Borsacchi, M. Geppi and A. Pucci, Int. J. Hydrogen Energy, 2012, 37, 14992–15002 CrossRef CAS.
- Z. Feng, P. O. Esteban, G. Gupta, D. A. Fulton and M. Mamlouk, Int. J. Hydrogen Energy, 2021, 46, 37137–37151 CrossRef CAS.
- G. Gupta, K. Selvakumar, N. Lakshminarasimhan, S. M. Senthil Kumar and M. Mamlouk, J. Power Sources, 2020, 461, 228131 CrossRef CAS.
- G. Gupta, K. Scott and M. Mamlouk, J. Power Sources, 2018, 375, 387–396 CrossRef CAS.
- X. Wu and K. Scott, Int. J. Hydrogen Energy, 2013, 38, 3123–3129 CrossRef CAS.
- M. Liu, X. Hu, B. Hu, L. Liu and N. W. Li, J. Membr. Sci., 2022, 642, 119966 CrossRef CAS.
- C. Lei, K. C. Yang, G. Z. Wang, G. W. Wang, J. T. Lu, L. Xiao and L. Zhuang, ACS Sustainable Chem. Eng., 2022, 10, 16725–16733 CrossRef CAS.
- M. Makrygianni, S. Aivali, Y. Xia, M. R. Kraglund, D. Aili and V. Deimede, J. Membr. Sci., 2023, 669, 121331 CrossRef CAS.
- X. Hu, B. Hu, C. Niu, J. Yao, M. Liu, H. Tao, Y. Huang, S. Kang, K. Geng and N. Li, Nat. Energy, 2024 DOI:10.1038/s41560-023-01447-w.
- G. A. Lindquist, S. Z. Oener, R. Krivina, A. R. Motz, A. Keane, C. Capuano, K. E. Ayers and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2021, 13, 51917–51924 CrossRef CAS PubMed.
- F. Razmjooei, T. Morawietz, E. Taghizadeh, E. Hadjixenophontos, L. Mues, M. Gerle, B. D. Wood, C. Harms, A. S. Gago, S. A. Ansar and K. A. Friedrich, Joule, 2021, 5, 1776–1799 CrossRef CAS.
- N. Ul Hassan, Y. W. Zheng, P. A. Kohl and W. E. Mustain, J. Electrochem. Soc., 2022, 169, 044526 CrossRef.
- A. R. Motz, D. G. Li, A. Keane, L. D. Manriquez, E. J. Park, S. Maurya, H. Chung, C. Fujimoto, J. Jeon, M. K. Pagels, C. Bae, K. E. Ayers and Y. S. Kim, J. Mater. Chem. A, 2021, 9, 22670–22683 RSC.
- L. Wan, J. Liu, Z. Xu, Q. Xu, M. B. Pang, P. C. Wang and B. G. Wang, Small, 2022, 18, 2200380 CrossRef CAS PubMed.
- P. Thangavel, H. Lee, T.-H. Kong, S. Kwon, A. Tayyebi, J.-H. Lee, S. M. Choi and Y. Kwon, Adv. Energy Mater., 2023, 13, 2203401 CrossRef CAS.
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378–1381 CrossRef CAS PubMed.
- Z. Xu, V. Wilke, J. J. Chmielarz, M. Tobias, V. Atanasov, A. S. Gago and K. A. Friedrich, J. Membr. Sci., 2023, 670, 121302 CrossRef CAS.
- M. J. Jang, S. H. Yang, M. G. Park, J. Jeong, M. S. Cha, S. H. Shin, K. H. Lee, Z. Y. Bai, Z. W. Chen, J. Y. Lee and S. M. Choi, ACS Energy Lett., 2022, 7, 2576–2583 CrossRef CAS.
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, ACS Appl. Energy Mater., 2019, 2, 7903–7912 CrossRef CAS.
- N. Chen, S. Y. Paek, J. Y. Lee, J. H. Park, S. Y. Lee and Y. M. Lee, Energy Environ. Sci., 2021, 14, 6338–6348 RSC.
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. R. Ni and L. Meroueh, Int. J. Hydrogen Energy, 2021, 46, 3379–3386 CrossRef CAS.
- S. C. Zignani, M. Lo Faro, A. Carbone, C. Italiano, S. Trocino, G. Monforte and A. S. Arico, Electrochim. Acta, 2022, 413, 140078 CrossRef.
- Z. Jiang, G. Yi, X. Yao, Y. Ma, X. Su, Q. Liu and Q. Zhang, Chem. Eng. J., 2023, 467, 143442 CrossRef CAS.
- A. W. Tricker, T. Y. Ertugrul, J. K. Lee, J. R. Shin, W. Choi, D. I. Kushner, G. Wang, J. Lang, I. V. Zenyuk, A. Z. Weber and X. Peng, Adv. Energy Mater., 2024, 14, 2303629 CrossRef CAS.
- C. Hu, N. Y. Kang, H. W. Kang, J. Y. Lee, X. Zhang, Y. J. Lee, S. W. Jung, J. H. Park, M.-G. Kim, S. J. Yoo, S. Y. Lee, C. H. Park and Y. M. Lee, Angew. Chem., Int. Ed., 2024, 63, e202316697 CrossRef CAS PubMed.
- Z. P. Wang, K. Chi, S. X. Yang, J. W. Xiao, F. Xiao, X. X. Zhao and S. Wang, Small, 2023, 19, 2301403 CrossRef CAS PubMed.
- M. Q. Li, H. Su, Q. G. Qiu, G. Zhao, Y. Sun and W. J. Song, J. Chem., 2014, 2014, 321629 Search PubMed.
- K. Amini and M. D. Pritzker, Electrochim. Acta, 2020, 356, 134785 CrossRef.
- B. Hu, C. DeBruler, Z. Rhodes and T. L. Liu, J. Am. Chem. Soc., 2017, 139, 1207–1214 CrossRef CAS PubMed.
- Y. Xiao, L. Hu, L. Gao, M. T. Di, X. J. Sun, J. Liu, X. M. Yan and G. H. He, Chem. Eng. J., 2022, 432, 134268 CrossRef CAS.
- M. T. Tsehaye, X. Yang, T. Janoschka, M. D. Hager, U. S. Schubert, F. Alloin and C. Iojoiu, Membranes, 2021, 11, 367 CrossRef CAS PubMed.
- T. Hagemann, J. Winsberg, M. Grube, I. Nischang, T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, J. Power Sources, 2018, 378, 546–554 CrossRef CAS.
- X. L. Lv, P. Sullivan, H. C. Fu, X. X. Hu, H. H. Liu, S. Jin, W. J. Li and D. W. Feng, ACS Energy Lett., 2022, 7, 2428–2434 CrossRef CAS.
- F. Alkhayri and C. A. Dyker, J. Electrochem. Soc., 2021, 168, 070501 CrossRef CAS.
- G. D. Charlton, S. M. Barbon, J. B. Gilroy and C. A. Dyker, J. Energy Chem., 2019, 34, 52–56 CrossRef.
- G. J. Hwang and H. Ohya, J. Membr. Sci., 1997, 132, 55–61 CrossRef CAS.
- D. Y. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, ACS Appl. Mater. Interfaces, 2013, 5, 7559–7566 CrossRef CAS PubMed.
- S. J. Seo, B. C. Kim, K. W. Sung, J. Shim, J. D. Jeon, K. H. Shin, S. H. Shin, S. H. Yun, J. Y. Lee and S. H. Moon, J. Membr. Sci., 2013, 428, 17–23 CrossRef CAS.
- Z. Mai, H. Zhang, H. Zhang, W. Xu, W. Wei, H. Na and X. Li, ChemSusChem, 2013, 6, 328–335 CrossRef CAS PubMed.
- M. Abdiani, E. Abouzari-Lotf, T. M. Ting, P. M. Nia, S. S. Sha'rani, A. Shockravi and A. Ahmad, J. Power Sources, 2019, 424, 245–253 CrossRef CAS.
- M. S. Cha, S. W. Jo, S. H. Han, S. H. Hong, S. So, T. H. Kim, S. G. Oh, Y. T. Hong and J. Y. Lee, J. Power Sources, 2019, 413, 158–166 CrossRef CAS.
- Y. Chen, Z. C. Liu, M. J. Lin, Q. L. Lin, B. H. Tong and D. Y. Chen, Science China-Chemistry, 2019, 62, 479–490 CrossRef CAS.
- H. Cho, H. M. Krieg and J. A. Kerres, Membranes, 2019, 9, 31 CrossRef PubMed.
- S. H. Roh, M. H. Lim, T. Sadhasivam and H. Y. Jung, Electrochim. Acta, 2019, 325, 134944 CrossRef CAS.
- H. Cho, V. Atanasov, H. M. Krieg and J. A. Kerres, Polymers, 2020, 12, 915 CrossRef CAS PubMed.
- Y. Du, L. Gao, L. Hu, M. T. Di, X. M. Yan, B. G. An and G. H. He, J. Membr. Sci., 2020, 603, 118011 CrossRef CAS.
- T. S. Wang, J. Y. Jeon, J. Han, J. H. Kim, C. Bae and S. Kim, J. Membr. Sci., 2020, 598, 117665 CrossRef CAS.
- B. G. Zhang, F. F. Wang, S. S. Guan, M. H. Zhao, E. L. Zhang, G. S. Wang, Z. G. Zhang, X. Q. Liu and S. H. Zhang, J. Power Sources, 2020, 477, 229011 CrossRef CAS.
- M. Charyton, C. Iojoiu, P. Fischer, G. Henrion, M. Etienne and M. L. Donten, Membranes, 2021, 11, 436 CrossRef CAS PubMed.
- W. Q. Tang, T. Mu, X. F. Che, J. H. Dong and J. S. Yang, ACS Sustainable Chem. Eng., 2021, 9, 14297–14306 CrossRef CAS.
- W. Q. Tang, Y. F. Yang, X. L. Liu, J. H. Dong, H. H. Li and J. S. Yang, Electrochim. Acta, 2021, 391, 138919 CrossRef CAS.
- Z. W. Tao, C. Y. Wang, S. J. Cai, J. F. Qian and J. Li, ACS Appl. Energy Mater., 2021, 4, 14488–14496 CrossRef CAS.
- Z. Q. Wang, S. H. Zhang, Q. Liu, Y. I. Chen, Z. H. Weng and X. G. Jian, J. Membr. Sci., 2021, 633, 119416 CrossRef CAS.
- S. Yang, X. F. Chu, B. Xue, T. Lv, B. C. Lin and Z. H. Zhang, ACS Appl. Energy Mater., 2021, 4, 6787–6796 CrossRef CAS.
- Y. Ahn and D. Kim, J. Ind. Eng. Chem., 2022, 110, 395–404 CrossRef CAS.
- X. F. Che, W. Q. Tang, J. H. Dong, D. Aili and J. S. Yang, Sci. China: Mater., 2022, 65, 683–694 CAS.
- A. K. Singh, P. Sharma, K. Singh and V. K. Shahi, J. Power Sources, 2022, 520, 230856 CrossRef CAS.
- X. H. Do, S. Abbas, M. M. Ikhsan, S. Y. Choi, H. Y. Ha, K. Azizi, H. A. Hjuler and D. Henkensmeier, Small, 2022, 18, e2206284 CrossRef PubMed.
- T. T. Bui, M. Shin, M. Rahimi, A. Bentien, Y. Kwon and D. Henkensmeier, Carbon Energy, 2024, e473 CrossRef.
- M. Mara Ikhsan, S. Abbas, X. H. Do, S.-Y. Choi, K. Azizi, H. A. Hjuler, J. H. Jang, H. Y. Ha and D. Henkensmeier, Chem. Eng. J., 2022, 435, 134902 CrossRef CAS.
- A. Khataee, D. Pan, J. S. Olsson, P. Jannasch and R. W. Lindstrom, J. Power Sources, 2021, 483, 229202 CrossRef CAS.
- B. Shanahan, T. Bohm, B. Britton, S. Holdcroft, R. Zengerle, S. Vierrath, S. Thiele and M. Breitwieser, Electrochem. Commun., 2019, 102, 37–40 CrossRef CAS.
- E. Lallo, A. Khataee and R. W. Lindstroem, Processes, 2022, 10, 270 CrossRef CAS.
- H. Cho, E. Hur, D. Henkensmeier, G. Jeong, E. Cho, H. J. Kim, J. H. Jang, K. Y. Lee, H. A. Hjuler, Q. Li, J. O. Jensen and L. N. Cleemann, Eur. Polym. J., 2014, 58, 135–143 CrossRef CAS.
- T. Huang, X. Qiu, J. Zhang, X. Li, Y. Pei, H. Jiang, R. Yue, Y. Yin, Z. Jiang, X. Zhang and M. D. Guiver, J. Power Sources, 2022, 527, 231143 CrossRef CAS.
- H. Tang, K. Geng, L. Wu, J. Liu, Z. Chen, W. You, F. Yan, M. D. Guiver and N. Li, Nat. Energy, 2022, 7, 153–162 CrossRef CAS.
- M. D. Guiver, M. Yahia, M. M. Dal-Cin, G. P. Robertson, S. Saeedi Garakani, N. Du and N. Tavajohi, Macromolecules, 2020, 53, 8951–8959 CrossRef CAS PubMed.
- F. Liu, S. Wang, D. Wang, G. Liu, Y. Cui, D. Liang, X. Wang, Z. Yong and Z. Wang, J. Power Sources, 2021, 494, 229732 CrossRef CAS.
- G. Skorikova, D. Rauber, D. Aili, S. Martin, Q. Li, D. Henkensmeier and R. Hempelmann, J. Membr. Sci., 2020, 608, 118188 CrossRef CAS.
- F. Liu, S. Wang, H. Chen, J. Li, X. Wang, T. Mao and Z. Wang, Renewable Energy, 2021, 163, 1692–1700 CrossRef CAS.
- Y. Xiao, X. Shen, R. Sun, S. Wang, J. Xiang, L. Zhang, P. Cheng, X. Du, Z. Yin and N. Tang, J. Power Sources, 2022, 543, 231802 CrossRef CAS.
- P. Wang, J. Lin, Y. Wu and L. Wang, J. Power Sources, 2023, 560, 232665 CrossRef CAS.
- Y. Jin, X. Che, Y. Xu, J. Dong, C. Pan, D. Aili, Q. Li and J. Yang, J. Electrochem. Soc., 2022, 169, 024504 CrossRef CAS.
- Y. Xiao, H. Chen, R. Sun, L. Zhang, J. Xiang, P. Cheng, H. Han, S. Wang and N. Tang, Polymers, 2023, 15, 3197 CrossRef CAS PubMed.
- C. Gao, M. Hu, L. Wang and L. Wang, Polymers, 2020, 12, 515 CrossRef CAS PubMed.
- G. Venugopalan, K. Chang, J. Nijoka, S. Livingston, G. M. Geise and C. G. Arges, ACS Appl. Energy Mater., 2020, 3, 573–585 CrossRef CAS.
- V. Atanasov, A. S. Lee, E. J. Park, S. Maurya, E. D. Baca, C. Fujimoto, M. Hibbs, I. Matanovic, J. Kerres and Y. S. Kim, Nat. Mater., 2021, 20, 370–377 CrossRef CAS PubMed.
- F. Arslan, K. Chuluunbandi, A. T. S. Freiberg, A. Kormanyos, F. Sit, S. Cherevko, J. Kerres, S. Thiele and T. Boehm, ACS Appl. Mater. Interfaces, 2021, 13, 56594–56606 CrossRef PubMed.
- J. Jiang, Z. Li, M. Xiao, S. Wang, K. Miyatake and Y. Meng, J. Membr. Sci., 2022, 660, 120878 CrossRef CAS.
- Q. Ju, H. Tang, G. Chao, T. Guo, K. Geng and N. Li, J. Mater. Chem. A, 2022, 10, 25295–25306 RSC.
- G. Venugopalan, D. Bhattacharya, E. Andrews, L. Briceno-Mena, J. Romagnoli, J. Flake and C. G. Arges, ACS Energy Lett., 2022, 7, 1322–1329 CrossRef CAS.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- D. A. Salvatore, D. M. Weekes, J. F. He, K. E. Dettelbach, Y. G. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- K. L. Yang, M. R. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
- K. Xie, R. K. Miao, A. Ozden, S. J. Liu, Z. Chen, C. T. Dinh, J. E. Huang, Q. C. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O'Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed.
- B. Eriksson, T. Asset, F. Spanu, F. Lecoeur, M. Dupont, F. A. Garcés-Pineda, J. R. Galán-Mascarós, S. Cavaliere, J. Rozière and F. Jaouen, J. Electrochem. Soc., 2022, 169, 034508 CrossRef CAS.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- A. Pătru, T. Binninger, B. Pribyl and T. J. Schmidt, J. Electrochem. Soc., 2019, 166, F34 CrossRef.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098–4105 RSC.
- S. Overa, B. S. Crandall, B. Shrimant, D. Tian, B. H. Ko, H. Shin, C. Bae and F. Jiao, Nat. Catal., 2022, 5, 738–745 CrossRef CAS.
- J. Wang, T. R. Willson, S. Brückner, D. K. Whelligan, C. Sun, L. Liang, X. Wang, P. Strasser, J. Varcoe and W. Ju, Electrochim. Acta, 2023, 461, 142613 CrossRef CAS.
- M. Liu, H. Hu, Y. Kong, I. Z. Montiel, V. Kolivoška, A. V. Rudnev, Y. Hou, R. Erni, S. Vesztergom and P. Broekmann, Appl. Catal., B, 2023, 335, 122885 CrossRef CAS.
- S. Garg, Q. Xu, A. B. Moss, M. Mirolo, W. Deng, I. Chorkendorff, J. Drnec and B. Seger, Energy Environ. Sci., 2023, 16, 1631–1643 RSC.
- A. G. Fink, E. W. Lees, Z. Zhang, S. Ren, R. S. Delima and C. P. Berlinguette, ChemElectroChem, 2021, 8, 2094–2100 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Utiliz., 2017, 20, 208–217 CrossRef CAS.
- W. Li, Z. Yin, Z. Gao, G. Wang, Z. Li, F. Wei, X. Wei, H. Peng, X. Hu, L. Xiao, J. Lu and L. Zhuang, Nat. Energy, 2022, 7, 835–843 CrossRef CAS.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- T. Möller, M. Filippi, S. Brückner, W. Ju and P. Strasser, Nat. Commun., 2023, 14, 5680 CrossRef PubMed.
- K. Ye, G. Zhang, X.-Y. Ma, C. Deng, X. Huang, C. Yuan, G. Meng, W.-B. Cai and K. Jiang, Energy Environ. Sci., 2022, 15, 749–759 RSC.
- Z. Liu, X. Lv, S. Kong, M. Liu, K. Liu, J. Zhang, B. Wu, Q. Zhang, Y. Tang, L. Qian, L. Zhang and G. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202309319 CrossRef CAS PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- Y. Kaneko and K. S. Lackner, Phys. Chem. Chem. Phys., 2022, 24, 14763–14771 RSC.
- L. Shi, Y. Zhao, S. Matz, S. Gottesfeld, B. P. Setzler and Y. Yan, Nat. Energy, 2022, 7, 238–247 CrossRef CAS.
- I. Nicotera, A. Policicchio, G. Conte, R. G. Agostino, M. H. U. Rehman, E. Lufrano and C. Simari, J. CO2 Utiliz., 2022, 63, 102135 CrossRef.
- A. Prajapati, R. Sartape, M. T. Galante, J. Xie, S. L. Leung, I. Bessa, M. H. S. Andrade, R. T. Somich, M. V. Rebouças, G. T. Hutras, N. Diniz and M. R. Singh, Energy Environ. Sci., 2022, 15, 5105–5117 RSC.
- J. L. Wade, H. Lopez Marques, W. Wang, J. Flory and B. Freeman, J. Membr. Sci., 2023, 685, 121954 CrossRef CAS.
- J. Liao, H. Ruan, X. Gao, Q. Chen and J. Shen, J. Membr. Sci., 2021, 621, 118999 CrossRef CAS.
- W. Xia, Y. Yang, X. Shang, X. Yang, S. Wang, F. Gong, L. Wang, X. Wang and X. Chen, Desalination, 2022, 529, 115646 CrossRef CAS.
- C. Cheng, Z. Yang, Y. He, A. N. Mondal, E. Bakangura and T. Xu, J. Membr. Sci., 2015, 493, 645–653 CrossRef CAS.
- M. Irfan, N. U. Afsar, E. Bakangura, A. N. Mondal, M. I. Khan, K. Emmanuel, Z. Yang, L. Wu and T. Xu, Sep. Purif. Technol., 2017, 178, 269–278 CrossRef CAS.
- X. Du, Z. Wang, H. Zhang, Y. Yuan, H. Wang and Z. Zhang, J. Membr. Sci., 2021, 619, 118805 CrossRef CAS.
- B. Liu, Y. Duan, T. Li, J. Li, H. Zhang and C. Zhao, Sep. Purif. Technol., 2022, 282, 120032 CrossRef CAS.
- L. Xu, H. Wang, L. Min, W. Xu and W. Zhang, Sep. Purif. Technol., 2023, 312, 123396 CrossRef CAS.
- Q. Dong, X. Guo, X. Huang, L. Liu, R. Tallon, B. Taylor and J. Chen, Chem. Eng. J., 2019, 361, 1535–1542 CrossRef CAS.
- C. Wang, T. Li, G. Yu and S. Deng, Chemosphere, 2021, 284, 131341 CrossRef CAS PubMed.
- C. Wang, L.-F. Ren, D. Ying, J. Jia and J. Shao, ACS ES&T Water, 2023, 3, 185–194 Search PubMed.
- A. Saravanan, P. R. Yaashikaa, P. Senthil Kumar, S. Karishma, P. Thamarai, V. C. Deivayanai, G. Rangasamy, R. Selvasembian and T. M. Aminabhavi, Sep. Purif. Technol., 2023, 317, 123897 CrossRef CAS.
- P. Nativ, O. Lahav and Y. Gendel, Desalination, 2018, 425, 123–129 CrossRef CAS.
- R. McNair, L. Cseri, G. Szekely and R. Dryfe, ACS Appl. Polym. Mater., 2020, 2, 2946–2956 CrossRef CAS PubMed.
- Q. Wu, D. Liang, S. Lu, J. Zhang, H. Wang, Y. Xiang and D. Aurbach, ACS Appl. Mater. Interfaces, 2021, 13, 46537–46548 CrossRef CAS PubMed.
- K. Singh, S. Sahin, J. G. Gamaethiralalage, R. L. Zornitta and L. C. P. M. de Smet, Chem. Eng. J., 2022, 432, 128329 CrossRef CAS.
- A. Siekierka and M. Bryjak, Membranes, 2022, 12, 103 CrossRef CAS PubMed.
- C. Carey, J. C. Díaz, D. Kitto, C. Espinoza, E. Ahn and J. Kamcev, J. Membr. Sci., 2023, 669, 121301 CrossRef CAS.
- Y. Zhang, X. Bu, X. Dong, Y. Wang and Z. Chen, Desalination, 2023, 556, 116571 CrossRef CAS.
- X. Zhao, H. Wei, H. Zhao, Y. Wang and N. Tang, J. Electroanal. Chem., 2020, 873, 114416 CrossRef CAS.
- S. Kumar, N. M. Aldaqqa, E. Alhseinat and D. Shetty, Angew. Chem., Int. Ed., 2023, 62, e202302180 CrossRef CAS PubMed.
- J. Xi, H. Ming, S. Liu, X. Shen, C. Geng, W. Gao, J. Meng, Y. Gao, Z. Zhao, J. Lv, Y. Guan and J. Liang, Environ. Technol., 2023, 44, 3585–3591 CrossRef CAS PubMed.
- J. Herranz, A. Pătru, E. Fabbri and T. J. Schmidt, Curr. Opin. Electrochem., 2020, 23, 89–95 CrossRef CAS.
- M. Mandal, ChemElectroChem, 2021, 8, 1448–1450 CrossRef CAS.
- B. Mayerhöfer, K. Ehelebe, F. D. Speck, M. Bierling, J. Bender, J. A. Kerres, K. J. J. Mayrhofer, S. Cherevko, R. Peach and S. Thiele, J. Mater. Chem. A, 2021, 9, 14285–14295 RSC.
- M. A. Blommaert, D. Aili, R. A. Tufa, Q. Li, W. A. Smith and D. A. Vermaas, ACS Energy Lett., 2021, 6, 2539–2548 CrossRef CAS PubMed.
- M. A. Blommaert, S. Subramanian, K. Yang, W. A. Smith and D. A. Vermaas, ACS Appl. Mater. Interfaces, 2022, 14, 557–563 CrossRef CAS PubMed.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- M. Hu, Q. Li, H. Peng, H. Ma, L. Xiao, G. Wang, J. Lu and L. Zhuang, J. Power Sources, 2020, 472, 228471 CrossRef CAS.
- T. J. Omasta, A. M. Park, J. M. LaManna, Y. Zhang, X. Peng, L. Wang, D. L. Jacobson, J. R. Varcoe, D. S. Hussey, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2018, 11, 551–558 RSC.
- G. Huang, M. Mandal, N. U. Hassan, K. Groenhout, A. Dobbs, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2020, 167, 164514 CrossRef CAS.
- B. Wang, J. Pan, X. Zou, J. Zhao, G. Xu, Z. Jin, Z. Sun and F. Yan, J. Mater. Chem. A, 2022, 10, 13355–13367 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cs00186e |
| This journal is © The Royal Society of Chemistry 2024 |