
Nuclear localization signal-tagged systems: relevant nuclear import principles in the context of current therapeutic design
Ritabrita
Goswami†
 a,
Aarohi
Gupta†
a,
Olga
Bednova
b,
Gaël
Coulombe
c,
Dipika
Patel
c,
Vincent M.
Rotello
*a and
Jeffrey V.
Leyton
*de
a,
Aarohi
Gupta†
a,
Olga
Bednova
b,
Gaël
Coulombe
c,
Dipika
Patel
c,
Vincent M.
Rotello
*a and
Jeffrey V.
Leyton
*de
aDepartment of Chemistry, University of Massachusetts, Massachusetts, USA. E-mail: rotello@chem.umass.edu
bDépartement de médecine nucléaire et radiobiologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Québec, Canada
cService des stages et du développement professionnel, Université de Sherbrooke, Sherbrooke, Québec, Canada
dÉcole des sciences pharmaceutiques, Université d’Ottawa, Ottawa, Ontario, Canada. E-mail: vleyton@uottawa.ca
eDepartment of Cellular and Molecular Medicine, University of Ottawa, Ottawa, Ontario, Canada
First published on 30th November 2023
Abstract
Nuclear targeting of therapeutics provides a strategy for enhancing efficacy of molecules active in the nucleus and minimizing off-target effects. ‘Active’ nuclear-directed transport and efficient translocations across nuclear pore complexes provide the most effective means of maximizing nuclear localization. Nuclear-targeting systems based on nuclear localization signal (NLS) motifs have progressed significantly since the beginning of the current millennium. Here, we offer a roadmap for understanding the basic mechanisms of nuclear import in the context of actionable therapeutic design for developing NLS-therapeutics with improved treatment efficacy.
 Ritabrita Goswami | Ritabrita Goswami received her BS in Chemistry from Lady Brabourne College, University of Calcutta, in 2015 and her MS in Chemistry from the Indian Institute of Technology Dhanbad in 2017, where she was introduced to nanocarrier-based systems for delivery and sensing applications. She is currently a PhD candidate under the supervision of Professor Vincent M. Rotello at the Department of Chemistry, University of Massachusetts Amherst. Her current research is focused on developing polymeric nanomaterials for intracellular delivery of therapeutics. |
 Aarohi Gupta | Aarohi Gupta received her BS in chemistry from Hindu College, University of Delhi, in 2015 and her MS in chemistry from Malaviya National Institute of Technology in 2017 where she was introduced to nanocarrier based drug delivery systems. She is currently a PhD candidate in the Rotello laboratory at the Department of Chemistry, UMass Amherst. Her current research is focused on developing targeted and responsive bioorthogonal nanocatalysts for localized production of bioactive agents. |
 Olga Bednova and Dipika Patel | Olga Bednova is a PhD candidate at the Faculty of Medicine and Health Sciences at the University of Sherbrooke. She received a bachelor's degree in biotechnology and master's degree in molecular and cellular biology from Moscow State University of Fine Chemical Technologies in Moscow, Russia. Her current domains of interest are drug discovery, antibody engineered fragments and antibody drug conjugates with enhanced ability to target the cell nucleus. Dipika Patel recently earned her bachelor's degree in Biochemistry from the University of Sherbrooke where she was instrumental in designing novel nuclear localization signals through the university's “co-op” program. She is currently completing a certificate in applied computer science with a focus in bioinformatics. |
 Gaël Coulombe | Gaël Coulombe is an undergraduate biochemistry student at the University of Sherbrooke. Due to the university's “co-op” program, he has acquired valuable practical experience in the field of molecular modelling and structural biology to understand importin-NLS interactions. |
 Vincent M. Rotello | Vincent Rotello is the Charles A. Goessmann Professor of Chemistry and a University Distinguished Professor at the University of Massachusetts at Amherst. He is a Fellow of the American Association for the Advancement of Science (AAAS) and of the Royal Society of Chemistry (U.K.). His research program focuses on using synthetic organic chemistry to engineer the interface between the synthetic and biological worlds, with over 650 peer-reviewed papers published to date. He is actively involved in bionanotechnology, and his research includes programs in delivery, imaging, diagnostics and nanotoxicology. |
 Jeffrey V. Leyton | Jeffrey Leyton is an associate professor at the School of Pharmaceutical Sciences at the University of Ottawa. He strives to unlock mysteries of cellular biology and create future medicines. His research program is dedicated to the study and discovery of strategies to target the cell nucleus for improving therapeutic efficacy. His primary focus lies in designing innovative nuclear localization signals and elucidating novel insights in nuclear import in parallel with applying these findings for developing novel nuclear-targeted therapeutics to effectively combat disease. His program also actively engages in research on antibody–drug conjugates, antibody engineering, imaging, and endosome escape. |
1. Introduction
The nucleus is the control center of the eukaryotic cell as it contains most of the genetic material necessary for host survival. Consequently, for many in therapeutic research, the nucleus is also the major susceptibility center of the cell. For example, approximately a quarter of all approved oncology drugs are active inside the nucleus.1 The major drug target is the DNA double helix, however targets inside the nucleus extend to DNA-interacting proteins such as topoisomerase I, RNA polymerase II, and nuclear kinases.2–4Importins are part of the karyopherin family of evolutionary-linked and yet highly diversified proteins based on α-solenoid structural domains. Importins play a crucial role in transporting cargo proteins from the cytoplasm into the nucleus. They collectively recognize and bind cargo containing a series of topogenic amino acid stretches traditionally named nuclear localization signals (NLS). The NLS recognition process by importins cues the transport of cargo proteins from the cytoplasm to the nucleus. Dr Gunter Blobel and colleagues introduced the concept of subcompartment-specific signals, which are encoded with the protein's primary amino acid sequence and govern its transport to specific subcompartments, including the nucleus.5 This mechanism of nuclear transport mediated by importins offers potential avenues for advancing therapeutic strategies targeting the nucleus.
The exploration of nanoparticle (NP)-based systems for drug delivery in cancer and other diseases has been extensive, due to their high drug-loading capacities, low off-target toxicities, and controlled release properties.6–9 Additionally, there has been a growing interest in directing NPs to the nucleus, as it holds great potential for targeted and efficient delivery of drug payloads, further enhancing the prospects of NP-based strategies.7,9
Achieving nuclear localization opens new possibilities for enhancing the efficacy of various therapeutic platforms. For example, free radical-generating modalities can maximize their therapeutic effect if nuclear localization were achieved. The cytotoxic effect of reactive oxygen free radicals generated by photodynamic therapies can be enhanced by localizing these agents close to desired targets (DNA, RNA, proteins) in the nucleus, given their limited lifespan and extremely short diffusion distance.10 Additional examples whereby efficient nuclear localization can potentially increase therapeutic efficacy include radionuclides emitting therapeutic (but short-range) Auger electrons, DNA vaccines including designs for COVID-19, and emerging nanotechnologies.11–13
Here, we describe some of the basic principles of nuclear import, and how these processes align with therapeutic design. The goal is to link key principles governing the complexities of nuclear import to therapeutic design aspects including NLS sequence variations, importin structural differences, and potentially exploitable nuclear import pathways in diseased cells. We will discuss significant advances in the NLS tagging of therapeutic platforms, highlighting innovative nuclear localization strategies and promising preclinical therapeutic impacts. By integrating these concepts, we hope to provide a useful roadmap to the research community and facilitate the clinical translation of NLS-therapeutics.
2. NLS-therapeutics face a barrier control
The nucleus requires a protective barrier from the cytoplasm to ensure the integrity and segregation of genetic material, regulate gene expression, and safeguard vital nuclear processes. Nuclear pore complexes (NPC) serve as channels that span the nuclear envelope, thereby providing a portal for proteins to access the nucleus. Mammalian cells typically have 2000–5000 NPCs per nucleus,14 indicating abundant access points for NLS-tagged therapeutics. However, NPCs are hydrophobic and narrow with passive diffusion limits of ∼30–50 kDa (∼5 nm diameter).15 NPCs comprise ≥30 unique proteins collectively termed nucleoporins (Nups).16 The Nups also provide important regulatory processes, contributing to multiple proposed ‘barrier control’ mechanisms.Although it is highly plausible that NPC barrier control impacts NLS-therapeutic nuclear import efficiency, we have not come across direct evaluations of these models within the context of NLS-therapeutics. Therefore, we consider it premature to delve extensively into the detailed description of these models at this stage. Furthermore, the barrier models and the mechanisms proposed for the traversal of NLS-cargo/importin complexes through NPCs also vary. There are recent reviews that provide insight into the structure and function of NPCs.17,18
3. NLS-therapeutics require energy to localize into the nucleus
The RanGTPase system is the driving force for directional nucleocytoplasmic transport. Human Ras-related nuclear protein (Ran) GTPase is a ∼24 kDa protein that hydrolyzes GTP (guanosine triphosphate) to GDP (guanosine diphosphate) and is essential for nuclear import.19,20 RanGTPase exists in two different states, where the nuclear pool of Ran is predominantly bound to GTP, and the Ran cytoplasmic pool is mainly bound to GDP.21,22 The two major proteins governing the two RanGTPase states are RanGAP1 (RanGTPase-activating protein) and RanGEF (RanGDP-GTP exchange factor), also known as RCCI (Regulator of chromosome condensation 1). RanGAP1 catalyzes the hydrolysis of GTP bound to Ran.23,24 The asymmetric distribution of these two regulators causes Ran to be mainly bound to GTP in the nucleus, but when Ran exits (there are also karyopherins that function to export cargoes out of the nucleus [‘exportins’]; reviews can be found here25,26) the nucleus it is rapidly hydrolyzed to the GDP-bound state in the cytoplasm. This process results in what is commonly referred to as the RanGTP/GDP gradient. Specifically, the GTP/GDP state of Ran determines the directionality that causes conformational changes in the nuclear transport complexes, enabling loading and unloading of NLS-cargoes (Fig. 1).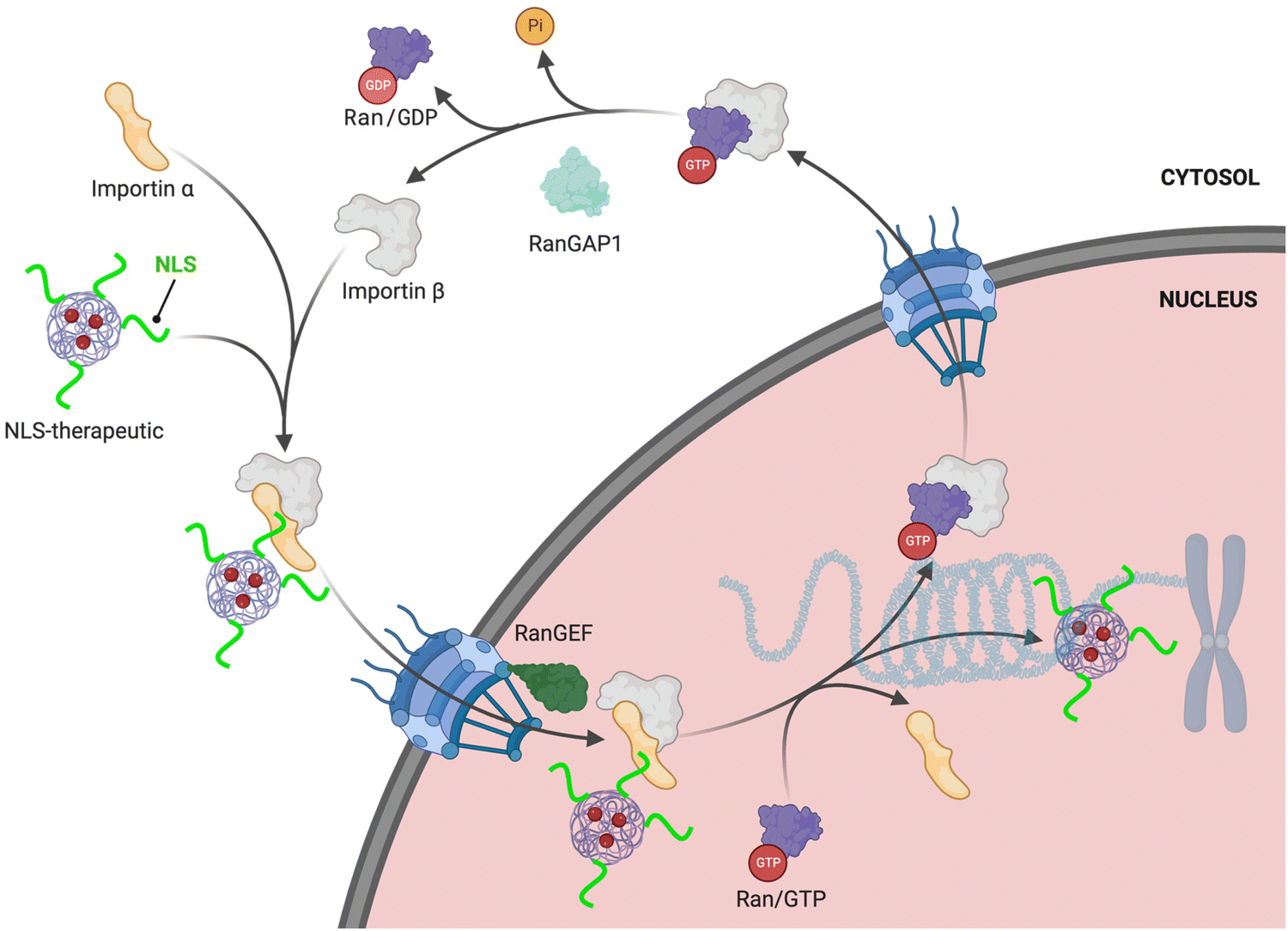 | ||
| Fig. 1 NLS-therapeutic nuclear import. Proposed scheme of NLS-therapeutic exploitation of the two cellular RanGTP/GDP states. | ||
4. The classical cooperative interplay between importin-α and importin-β for NLS-cargo binding
The most well described mechanism in nuclear import is the classical interplay between importin-α and importin-β. The human genome encodes seven importin-α isoforms, and their evolutionary links are well established. importin-α is a ∼59 kDa protein that consists of two key functional domains. All isoforms of importin-α share a fundamentally conserved architecture that consists of a small N-terminal, autoinhibitory, importin-β-binding (IBB) domain and the NLS-binding C-terminal region, composed of armadillo (ARM) repeats.27 ARMs are α-helical repeats that stack against each other and give importin-α an overall contorted cylindrical shape akin to a kidney bean that is ∼100 Å long with a diameter of 30–35 Å, with a ∼30° right hand twist.28 The structural twist provides importin-α with a shallow groove along its long axis. At each end of the groove are what are known as the major and minor NLS-binding pockets (Fig. 2). The binding pockets provide large surface areas that enable multivalent interactions to the NLS sequences (further described in subsequent section).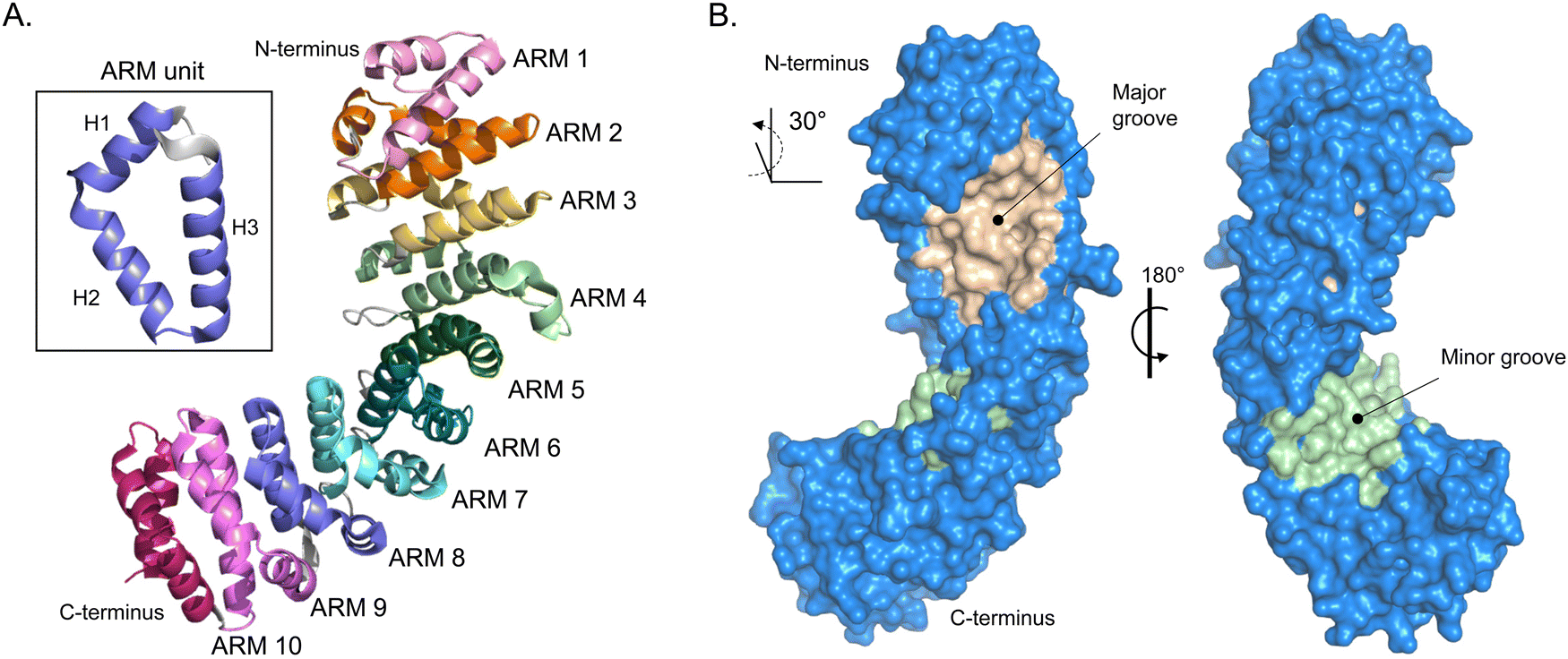 | ||
| Fig. 2 Importin-α structure. (A) Alpha-fold-generated ribbon model of importin-α (AF-P52293-F1 [accessed in Uniprot]) highlighting the alpha helical-rich 10 ARM repeats. Inset box, shows one ARM unit that is composed of helices 1–3 (H1–H3). (B) Crystal structure rendering of importin-α (PDB: 5WUN) with importin β-binding (IBB) domain deleted. Importin-α has a natural lengthwise 30° right-handed twist that brings to light the major (wheat) and minor (light green) NLS-binding grooves. | ||
The cytoplasm is the location of recognition and interaction of NLS-tagged cargoes. Cytosolic proteins bearing an NLS are first recognized and bound by importin-α. importin-α cannot localize to the nucleus on its own; hence, the IBB domain is a critical molecular liaison between the importin-α/NLS-cargo complex and importin-β. The IBB domain of importin-α occupies a large part of the NLS binding groove when not bound to NLS-cargoes, effectively acting as an internal NLS.28 This autoinhibition reduces the affinity of importin-α for NLS when importin-β is unavailable, preventing wasteful import. The IBB domain contains several basic amino acids that mimic the side chains found in classical NLSs, and mutation of critical residues affects the autoinhibitory capacity and NLS binding affinity.29,30 The trimeric import complex (importin-β/importin-α/NLS-cargo) is likely assembled cooperatively in the cytoplasm when both NLS-cargo and importin-β are simultaneously present to compete off the IBB domain and signal that the complex is ready for transport. (Fig. 3).
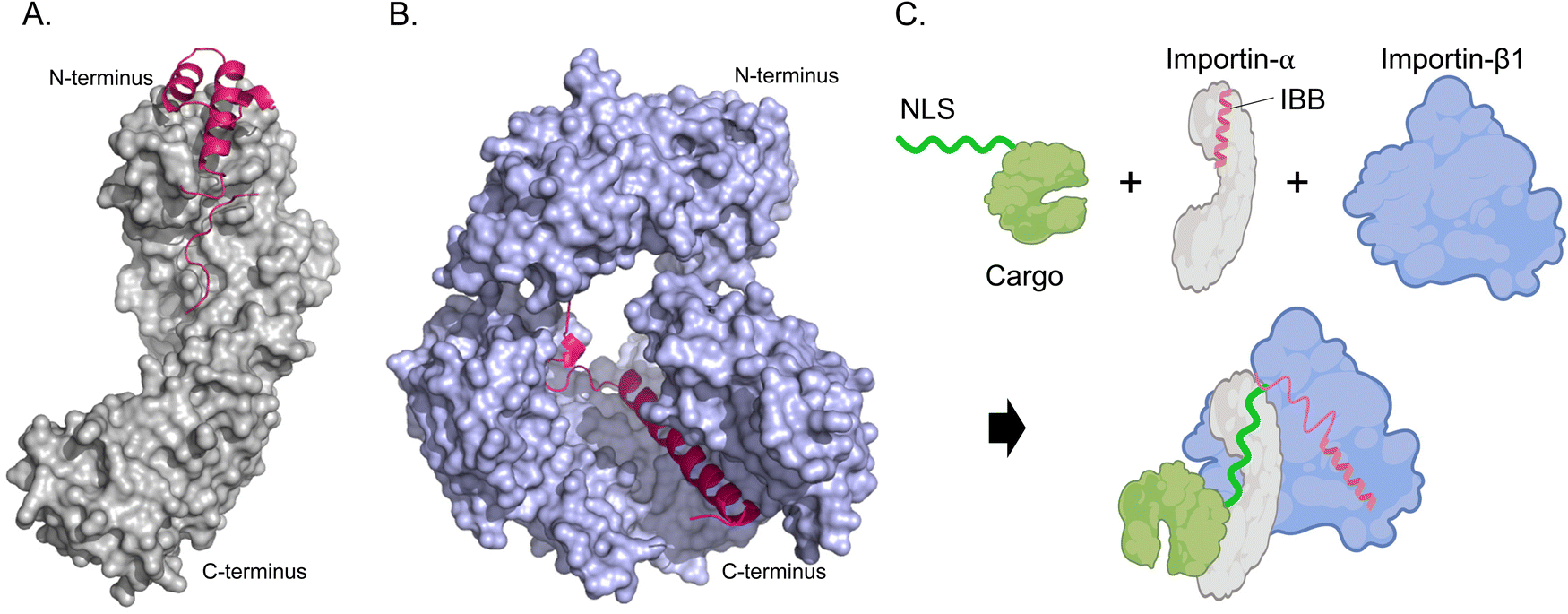 | ||
| Fig. 3 Mechanism of NLS-cargo, importin-α, and importin-β1 complex formation. (A) Crystal structure rendering of importin-α (PDB: 1IAL [ref. 28]) depicting the N-terminal IBB domain (hot pink) and C-terminal NLS-binding portion (grey). (B) Importin-β1 binding the IBB domain (hot pink) of importin-α (PDB: 1QGK [ref. 27]). Amino acids 275–335 and 840–875 are removed to better visualize the interaction of Importin-β1 and IBB. (C) Schematic depicting how a trimeric import complex of Importin-α/β1/NLS occurs when all three factors are present. | ||
This autoinhibitory role of the IBB domain in importin-α is important to consider during the design and development of NLS-tagged therapeutic agents. When designing NLS-therapeutics, it is crucial to ensure that the therapeutic cargo is efficiently loaded onto the importin-α to promote its nuclear import. Failure to do so would result in reduced efficacy of the therapeutic. Designers should also be aware that IBB-mediated autoinhibition of NLS-cargo binding is not a general feature for all NLSs, as certain cargoes, like STAT1, do not compete intramolecularly with the IBB domain of importin-α isoform 5.31
5. NLS-therapeutic-relevant NLS sequences and interaction modes for importin binding
(a) Relevant binding modes for importin-α
We delve now into the main characteristics of classical NLS binding to importin-α and the intricate interactions required for successful nuclear import. While non-classical systems exist and are significant in nuclear import, our focus lies on the classical NLS interactions due to their prominent usage in the current NLS-therapeutic design strategies. Notably, a recent review by the Chook group extensively covers the numerous and diverse consensus NLS sequences identified for the importin-β family members,32 shedding light on the complexities of non-classical NLS interaction modes. By highlighting a few of these complexities, we aim to underscore the compelling need for further research in this area in an NLS-therapeutic context.In 1984, the laboratory of Alan E. Smith reported that a minimal amino acid sequence of 126-PKKKRKV-132 from the simian virus 40 (SV40) large T-antigen, genetically fused to the protein β-galactosidase and needle-microinjected into mammalian cells was efficiently transported into the nucleus.33 As a result, the first NLS was identified.
The two main characteristics of classical NLSs are that the amino acid sequence is extended and contains a cluster of basic residues (lysine and arginine).34 The major NLS-binding pocket is an ideal surface for binding extended NLSs, being more like a groove than traditional pockets used for drugging with small molecules. The extended conformations of NLSs allow them to easily direct themselves along the lengthwise open groove caused by the 30° twist of importin-α (Fig. 2). The stacking of the ARM repeats that form the major (ARMs 2–4) and minor (ARMs 6–8) NLS-binding pockets have a concave surface providing hydrogen and ionic bonds and van der Waals interactions with precisely positioned NLS residues.
Within the major NLS-binding groove of importin-α isoforms, specific amino acid side chains are conserved and mapped to positions (P)1 through P6, forming a flat ladder of binding sites for the sequential extended and abundantly present cationic residues of classical NLSs (Fig. 4). Fig. 4 also shows the top 10 interactions for SV40 NLS within the major binding groove. Among the 10 most crucial interactions between the SV40 NLS and the major binding groove, asparagine residues play a vital role. The strongest interactions involve negatively charged aspartic acid residues forming salt bridges and hydrogen bonds with lysine residues K128 and K129 at the P2 and P3 positions, respectively. The presence of lysine at the P2 position is particularly crucial, as even a cationic arginine residue at this position significantly reduces NLS binding and nuclear import. Intriguingly, the arrangement of the ARMs creates a ‘capping’ effect at the groove edges, with C-terminus (P2) and N-terminus (P3) serving as ‘lateral caps’ for the NLS at the major site. K128 forms further strong hydrogen bonds with nearby glycine and threonine residues establishing itself as the most important binding position for the NLS, while the contacts made by K129 are not as numerous. Therefore, a basic residue, specifically a lysine, is crucial, and even an arginine residue at P2 severely reduces NLS binding and nuclear localization.33,35–37
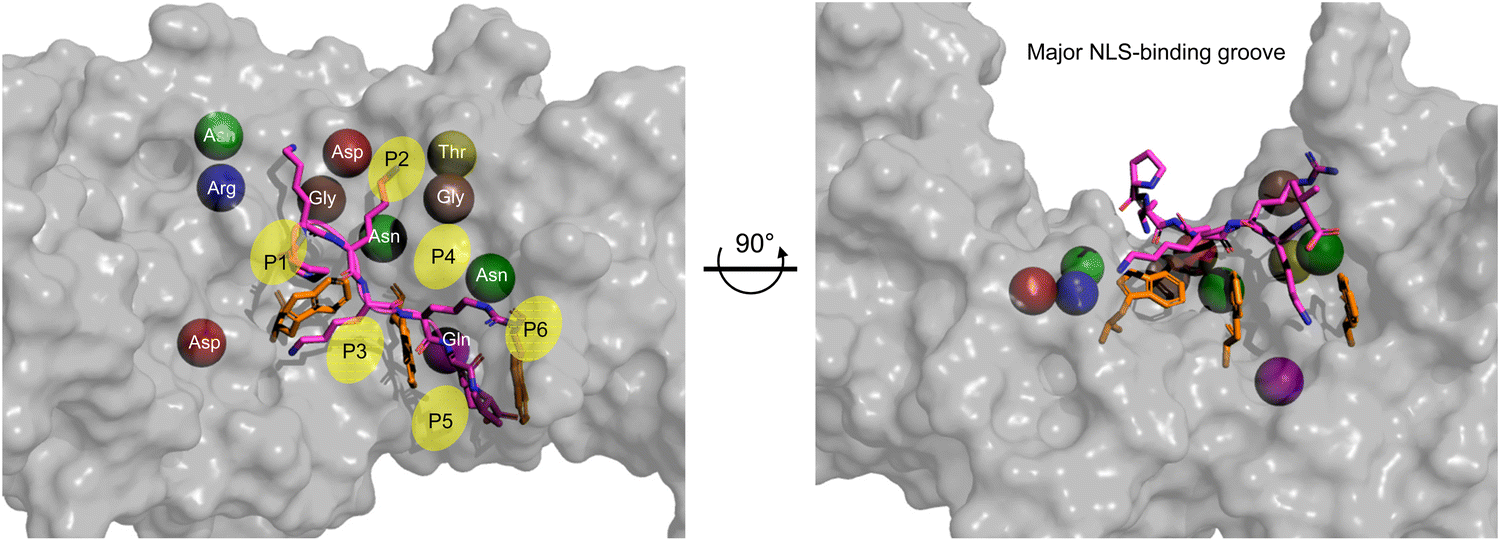 | ||
| Fig. 4 Monopartite NLS and top amino acid binding interactions in major groove. Zoom of the major NLS-binding groove for importin-α. The SV40 NLS shown in stick (magenta) and the positions P1–P6 indicated. The top 10 amino acids are shown as spheres and to better show their relative positions within the major groove. Ranking of most important amino acids for SV40 NLS binding based on SV40 in complex with mouse Imp-α1 (PDB: 1EJL [ref. 34]): Asp192 (red) > Asp270 (red) > Gln181 (purple) > Asn239 (green) > Gly191 (brown) > Gly150 (brown) > Asn188 (green) > Thr155 (yellow) > Asn146 (green) > Arg238 (blue). The three tryptophan residues that contribute van der Waals forces and cation-pi bonds and influence the topography of the NLS-binding groove are shown in orange sticks. The Molecular Operating Environment (MOE) software was used to determine contact energies between NLS residues and importin-α side chains. | ||
After the top 10 residues crucial for NLS binding, there is a substantial ∼3-fold drop-off in binding energy contributions from other residues. Tryptophan residues located on the H3 helices of ARMs 2, 3, and 4, respectively, collectively play an important role for two reasons. Firstly, they face inward toward the NLS in the major groove, creating a topography that requires NLSs to be elongated, with sequential basic amino acids fitting between what can be visualized as ‘camel humps’ forming shallow niches like saddles for the side chains to straddle (Fig. 4). Secondly, while individually providing modest van der Waals and cation–pi binding forces (there are also hydrogen bonds made that vary in strength), the cumulation of tryptophan residues contributes approximately 10–20% to the total binding affinity providing a minor but still appreciable contribution to overall binding.
In summary, classical monopartite NLS binding at the major site of importin-α follows two general rules. Firstly, NLSs need to be elongated to fit well within the topography of the major groove. Secondly, two basic residues, specifically lysine, are required at the P2 and P3 positions. While other NLS residues at positions P1, and P4–P6 also contribute to binding interactions, their variation among different classical NLS sequences is one explanation for the diversity of NLS-cargoes. Although compensations can occur with most of the canonical 20 amino acids found in humans, the consensus is that cationic sequences align well throughout the binding groove and hence NLSs have basic residue clusters. Although short classical NLSs also bind to the minor NLS-binding grooves with subtle differences to the P positions, crystal structure studies show better peptide ordering in the major site and, hence, it is accepted that classical NLSs prefer the major binding groove.34 In 2009, the Yanagawa group discovered a subset of classical NLSs that are specific for the minor pocket.38 However, these NLSs are much less prevalent in nature and information about their roles in nuclear import is limited.39
Bipartite NLSs display considerable variety in amino acid sequences. In contrast to the short classical NLSs, bipartite NLSs are long and contain N- and C-terminal basic amino acids that are both required to bind the N- and C-terminal NLS-binding pockets of importin-α for proper nuclear transport. Based on our analysis of 39 reported bipartite NLSs (most identified from the Rost group database of NLSs40), they stretch approximately 23 ± 7 amino acids in length and range from 15 to as long as 42 residues in length. The sequence of the region between the basic amino acid segments, which bind the major and minor NLS-binding grooves, exhibit significant variation and form interactions with diverse importin-α sidechains. In addition, bulky protein domains adjacent to the bipartite NLS can bind to ARMs outside the binding grooves and influence the specificities of importin-α isoforms,41,42 this specificity is a topic we believe relevant to current and future NLS-therapeutic design, as many strategies use bipartite NLS tags, and certain isoforms are overexpressed in various diseases (Section 6). Monopartite and bipartite NLSs are found in ∼20–30% and ∼12–30%, respectively, of nuclear localizing proteins.15 One of the earliest and, hence, often referred to example of a bipartite NLS is KRPAATKKAGQAKKKK from nucleoplasmin,43 and used in an NLS-therapeutic design we will describe later. The interaction modes follow suit as classical monopartite NLSs in the major and minor NLS-binding pockets. However, the concurrent utilization of two binding sites indeed confers a higher binding affinity compared to monopartite NLS and, hence, improved nuclear import efficiency.44 Therefore, it is tempting to speculate that a bipartite NLS-therapeutic design could potentially enhance nuclear localization and improve therapeutic efficacy. The aspect of monopartite versus bipartite in an NLS-therapeutic context will be touched on in subsequent sections.
(b) Relevant importin-β member NLS binding modes
Although most NLS-therapeutic designs thus far have utilized classical monopartite or bipartite NLS tags, it is important to consider the importin-β family to highlight the increased complexities associated with NLS-importin interactions. This understanding opens doors to novel strategies for future NLS-therapeutic designs.The importin-β members are composed of ‘HEAT’ repeats, a tandem repeat structural motif composed of two α-helical segments (termed A and B) linked by a short loop.27,45 The acronym HEAT comes from four proteins that were originally found to contain this repeated motif. The various functions of HEAT domain-containing proteins have been comprehensively reviewed.46 In a seminal analysis by Mosammaparast and Pemberton, it was found that the human importin-β family encompasses 20 members that function as nuclear importers and exporters, or have dual import and export functions.47 When compared to importin-α, the 10 specific importin β members KPNB1, TNPO1, TNPO2, TNPO3, IPO4, IPO5, IPO7, IPO8, IPO9, IPO11 [gene ID] have considerable increases in molecular weight (∼111 kDa vs. 59 kDa). Furthermore, there is an approximate 10 kDa size variation among these importin-β proteins, accompanied by an average of 19.9 ± 1.6 HEAT repeats. The A and B helices are located on the exterior and interior of the protein, respectively, and give the importin an overall structure called a helicoid (imagine a slinky toy). An important distinction between the importin-β and importin-α families is that the HEAT repeats provide more flexibility than the ARM repeats. This increased flexibility is most likely due to multiple functional requirements by the β proteins, such as the binding of multiple partners required for nuclear localization.
For this Review we will focus on the NLS interaction modes of specific importin-β members, TNPO1 (Transportin-1) and IPO7 (importin-7). These members are of particular interest due to their involvement in NLS-therapeutic applications, their well-known interaction with specific NLS sequences, and their overexpression in cancer cells. By exploring the interaction modes of these importin-β members, we can better understand potential key roles for NLS-therapeutics.
Transportin-1 is a nuclear transport receptor that facilitates NLS-cargo nuclear localization in a RanGTPase-dependent manner but independent of importin-α. Transportin-1 is involved in the nuclear import of various proteins, nuclear ribonucleoproteins (hnRNPs) A1 and A2, ribosomal proteins (RPL23A, RPS7, RPL5), and specific histones.48–51 These proteins include a 38-amino acid sequence located near the C-terminus termed M9, which acts as an import signal.52 Furthermore, transportin-1 mediates the nuclear import of SRP19, ADAR/ADAR1 isoform 1 and 5, and HIV-1 Rev.50,53 Therefore, transportin-1 is responsible for the nuclear import of diverse cargoes.
Through analysis of crystal structures of transportin-1 in complex with specific NLSs such as the M9 sequence, the Chook group elucidated essential features governing NLS recognition by this nuclear transport receptor.54 Similar to the classical NLS specific for importin-α, transportin-1-specific NLSs adopt an elongated conformation spanning ∼11 nm in a linear epitope that winds along the vertical axis of transportin-1 (Fig. 5). The bound structure indicates that the NLS occupies a stretch of ≥30 residues within the NLS-binding pocket of transportin-1. Although the NLS contains abundant glycine and serine residues and exhibits a cationic nature, it is predicted to be structurally disordered when not bound to transportin-1.
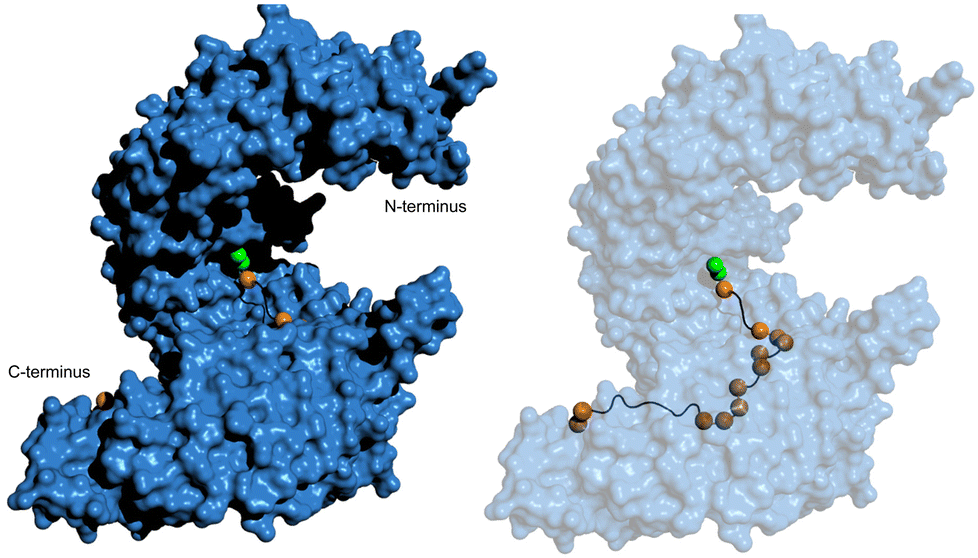 | ||
| Fig. 5 hPY-NLS binding mode with transportin-1. (left) Pymol (PDB: 2H4M [ref. 54]) depiction of the hPY-NLS interaction with transportin-1. (right) is transportin-1 set at 60% transparency to visualize hPY-NLS conformation within the interior binding groove. PY and hydrophobic residues depicted as green and orange spheres, respectively. | ||
A second requirement for an NLS to be recognized by transportin-1 is derived from the observation that the transportin-1/NLS-binding interface exhibits a highly negative charge. As with NLSs specific for importin-α, NLSs specific for transportin-1 with acidic characteristics are likely to face electrostatic repulsion and may not bind effectively. Analysis of all known transportin-1 NLSs performed by Lee et al., reveal an overall cationic character spanning at least 30 residues in six out of the seven NLSs evaluated.54
Thirdly, the analysis of transportin-1-specific NLSs revealed two conserved regions within the sequences. A C-terminal consensus sequence (R/K/H-X(2–5)-PY) is known as ‘PY-NLSs’. The PY-NLS class can be further subdivided if the central region is rich in hydrophobic (hPY-NLS) or basic (bPY-NLS) residues. hPY-NLSs prefer interactions with hydrophobic side chains at specific positions of transportin-1. Fig. 5 shows the hPY-NLS binding mode with transportin-1 as this NLS is relevant in current NLS-therapeutic design described later. In contrast, the bPY-NLSs contain a central motif enriched in basic residues, likely interacting with the highly acidic interface on transportin-1.
The second relevant example in the context of NLS-therapeutics is the importin-β family member, importin-7. Importin-7 forms a heterodimeric transport complex with importin-β that is essential for importing a linker-histone (H1.0), a highly cationic charged protein, into the nucleus through the NPC.55 This dimerization of autonomous transport receptors is crucial for the efficient transport of these proteins. The Halic group recently published the structure of the importin-7/importin-β complex with histone H1.0, shedding light on how the two β-members work together to chaperone the positively charged and disordered linear stretch of H1.0.56 Because histones function by packing the DNA to form nucleosomes,57 they are composed of regions that contain multiple stretches rich with basic amino acids that resemble classical NLSs.58 Significant electrostatic interactions between the linker of H1.0 and importin-7 stabilize the complex between these proteins. Structurally, the C-terminal tail of importin-7 binds with importin-β.
The Halic group revealed that importin-7 contains an abundance of acidic residues lining the inner concave surface and a unique C-terminal ∼90 amino acid chain with an acidic residue-rich loop. Due to the flexibility of importin-7, achieving an accurate structure was challenging, but the binding mode with H1.0 was still determined. The cryo-EM maps revealed a curved solenoid structure typical of the importin-β family of proteins for both importins, with importin-7 positioned in an anti-parallel manner with respect to importin-β. The H1.0 globular domains interact with importin-β HEAT repeats 3–16. Interestingly, the C-terminal tail of H1.0, having a net charge of +40, shaped the complex, interacting with various regions of importin-7. The H1.0 tail exhibited multiple conformations, explaining the less-defined density when bound by importin-7. However, it was clear that the interactions between the H1.0 tail and importin-7 stabilized the complex, and highlighting importin-7 as an interesting receptor and possibly a chaperone for H1.0 nuclear import.
Fig. 6 displays the computational modelling we performed of the importin-β, importin-7, and H1.0 complex to highlight key structural features and connect with an NLS-therapeutic design aspect described in Section 8. As the C-terminal tail was missing from the cryo-EM structures, an extended version was generated by adding residues 98–116 and followed by docking with the concave region of importin-7. This generated version reveals that interactions, namely with K108, K110, K114, and K115, comprise 87% of the total binding energy with importin-7. E735 of importin-7 makes extensive interactions with K108, K110, and K114. Although the full-length acidic-rich loop is not modelled, most likely electrostatic interactions will further contribute to the binding between importin-7 and H1.0. Taken together, this further supports the hypothesis that the importin-7 NLS interaction mode is driven by charge and not sequence as suggested by the Halic group.56
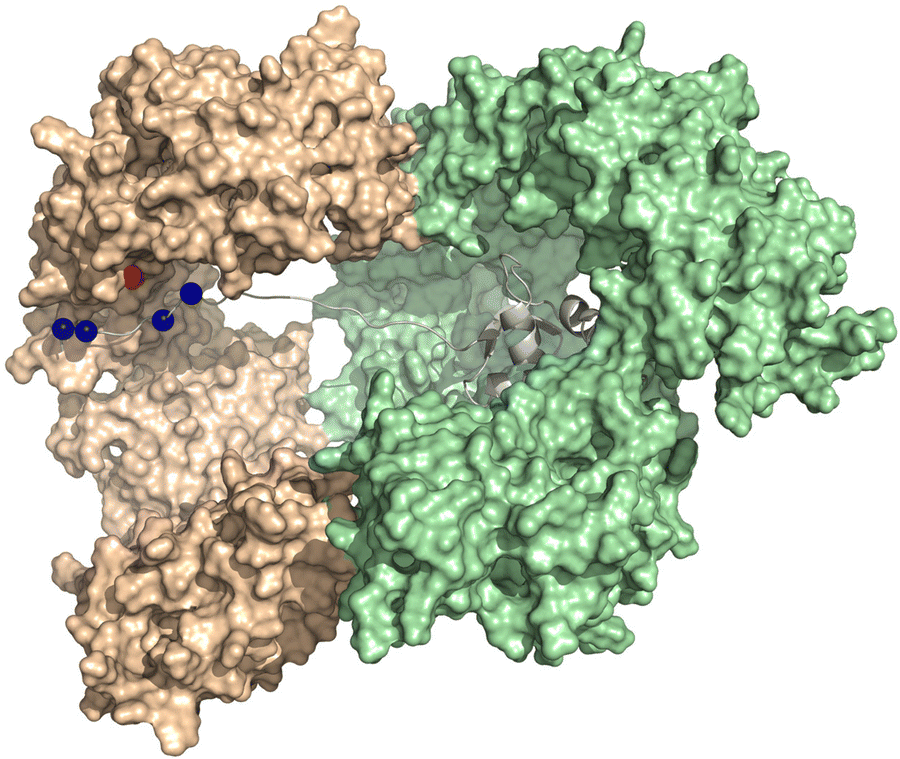 | ||
| Fig. 6 NLS binding mode with importin7. Depiction of the model generated in MOE-based on the solved the Cryo-EM structure of the importin-β (light green), importin-7 (wheat), and histone H1.0 (grey) complex (PDB: 6N88 [ref. 56]). Lysine residues K108, 110, 114, and 115 (blue spheres) of H1.0 and E735 (red sphere) of importin-7 are shown. The C-terminal acidic-rich loop is not modeled. | ||
6. The role of importins in disease
The overexpression of importins in various diseased cells Table 1, highlights the significance of considering importin-specific functions in NLS-therapeutic design. In cancer, where increased gene expression is often essential for cellular proliferation and progression, it is plausible that aberrant transcription necessitates enhanced nuclear localization of transcription factors. The differential expression and activity of importins in diseased cells further emphasizes the need to target overexpressed importins with potential heightened nuclear transport activity for delivering therapeutic payloads effectively into the nucleus. Despite the potential importance of importin-specific targeting, we cannot find a study to date that has demonstrated the capability of an NLS-tagged therapeutic to selectively exploit an overexpressed importin and boost nuclear localization. On the other hand, it may also be equally important to avoid importins that are downregulated or have different roles, as they may not contribute to the desired therapeutic outcomes. Therefore, exploring the roles of different importins and their dysregulation in NLS-therapeutic development offers valuable insights for future design strategies.| Importin (gene) | Disease | Notes |
|---|---|---|
| Importin-α's | ||
| Importin-α1 (KPNA2) | Invasive ductal breast carcinoma | Overexpression associated with shorter overall and recurrence-free survival.59 |
| Bladder cancer | Overexpression associated with increased proliferation, migration, invasion, aberrant cell cycle arrest, few apoptotic cells, and shorter patient survival.60 | |
| Alzheimer disease | Importin-α1 colocalized in Hirano bodies and resulted in the inability of NLS-cargoes to localize in the nucleus.61 | |
| Ovarian cancer | Overexpression associated with cell proliferation and tumorigenicity and poor prognosis.62,63 | |
| Prostate cancer | Overexpression associated with disease recurrence after radical prostatectomy.64 | |
| Oesophageal squamous cell carcinoma | Overexpression associated with poor tumour cell differentiation, proliferation, and invasiveness.65 | |
| Hepatocellular carcinoma | Overexpression identified as an independent predictor of early recurrence after tumor resection.66 | |
| Non-small cell lung cancer | Overexpressed and observed in the nucleus and linked to increased tumor cell migration and survival.67 | |
| Astrocytic glioma | Recurrent tumors expressed significantly increased levels of importin-α1 relative to the primary tumors. Decreased importin-α1 expression associated with longer overall and progression-free survival.68 | |
| Anaplastic oligoastrocytoma | Overexpression associated with poor prognosis. Reduced expression correlated with longer overall and progression-free survival.69 | |
| Importin-α3 (KPNA4) | Schizophrenia | Decreased expression associated with a single nucleotide polymorphism that is a marker for increased susceptibility to schizophrenia.70 |
| Impaired angiogenesis | Decreased expression associated with reduced nuclear import efficiency of hypoxia-inducible factor 1α and resulted in aberrant vascular endothelial growth factor expression. Decreased levels in ageing myocardial cells and associated with slower recovery after heart disease treatment.71 | |
| Breast cancer | Overexpressed in highly aggressive breast cancer subtypes and associated with poor prognosis.72 | |
| Prostate cancer | Overexpression mediated cancer metastasis through NF-κB transport.73 | |
| Head and neck of squamous cell carcinoma | Overexpression resulted in unrestrained nuclear localization signal-dependent transport activity and induced epidermal differentiation and promoted malignant phenotypes.74 | |
| Importin-α4 (KPNA3) | IgA nephropathy | Decreased levels inhibited glomerular endothelial cell activation, proliferation, and monocyte adhesion.75 |
| Importin-α5 (KPNA1) | Lesioned peripheral nerve | Observed as a critical adaptor to microtubules and directly participated in axonal injury signaling.76 |
| Importin-α6 (KPNA5) | Breast cancer | Residue mutations in ARM2 were frequently found in breast tumour samples and was associated with increased tumorigenesis and/or progression.77 The ARM2 mutations were thought to aberrantly influence NLS selectivity and nuclear import activity. |
| Importin-α7 (KPNA6) | Chronic myeloid leukemia | Overexpression observed.78 |
| Importin-α8 (KPNA7) | Infantile spasms and cerebellar malformation | A frequent mutation in an area near the NLS-binding site was associated with improper neuronal development.79 |
| Pancreatic cancer | Overexpression associated with increased tumour cell proliferation.80 | |
| Importin-β's | ||
| Importin-β1 (KPNB1) | Cervical cancer | Overexpression promoted increased cell proliferation and survival.81 |
| Gastric cancer | Increased levels promoted tumour cell proliferation and associated with poor prognosis.82 | |
| Hepatocellular carcinoma | Overexpression associated with poor survival and linked to tumour pathogenesis.83 | |
| B-cell lymphoma | Overexpression accelerated cell proliferation and drug resistance and was also associated with shorter survival.84 | |
| Multiple myeloma | Overexpression mediated myeloma cell proliferation and blocked apoptosis.85 | |
| Prostate cancer | Overexpression associated with tumour cell proliferation.86 | |
| Transportin-1 (TNPO-1) | Cervical cancer | TNPO-1-mediated nuclear import of proteins contributed to tumour immune evasion.87,88 |
| Amyotropic lateral sclerosis and frontotemporal dementia | Overexpressed and implicated in disease pathogenesis through abnormal nuclear transport.89 | |
| Importin-7 (IPO7) | Cervical cancer | Overexpression correlated with a poor prognosis and increased tumour cell proliferation.90 |
| Prostate carcinoma | Overexpression reported.91 | |
| Importin-8 (IPO8) | Acute myeloid leukemia | Importin-8 had increased nuclear transport activity of eIF4E, which activated the expression of genes involved in disease progression92 |
| Importin-11 (IPO11) | Non-muscle-invasive bladder cancer | Overexpression associated with poor prognosis.93 |
7. Current NLS-therapeutic design insights
The development of NLS-therapeutics, particularly in the context of NPs, has been driven by the understanding that efficient nuclear localization plays a pivotal role in maximizing the efficacy of delivered cytotoxic or gene-modifying agents that act within the nucleus.6,94 This rationale also extends to the emerging field of biological NLS-therapeutics, an area holding great promise.95 However, there is currently no optimal system in place to determine the ‘efficiency’ of nuclear localization and its direct therapeutic efficacy. In this section, we aim to link the insights discussed in Sections 5 and 6 regarding NLS sequences and interaction modes for importin binding and their relevance in disease, respectively. Specifically, we will highlight the most significant advancements in NLS-tagging for various therapeutic platforms, with a particular focus on structural elements such as NLS tag densities and orientations, conjugation mode, and diseased cellular environments.(a) Role of NP size and NLS conjugation density
The Chan group conducted a study on the nuclear localization efficiency of NLS-tagged NPs and found that properties such as particle size and NLS density significantly influence nuclear localization efficiency.96 In their work, they conjugated CdSeS/ZnS quantum dots with the NLS sequence DRQIKIWFQNRRMKKWKKK at various surface densities. While the publication mentioned that this NLS sequence was chosen based on its established ability to deliver small drugs encapsulated inside NPs into the cell nucleus,97 the specific origin of this NLS sequence was not disclosed. Our analysis of the sequence in the human proteome revealed potential homology to homeobox proteins, such as Homeobox B3 (Uniprot: F8VXG0), which are transcription factors involved in gene regulation and require nuclear import to bind target genetic DNA.98 We were unable to find further information relating to importin-α-specific nuclear import. An analysis algorithm of the sequence itself shows that the NLS is the sequence RRMKKWKKK.99 Interestingly, the study established that NPs of 4.8 and 8.0 nm could not localize inside the nucleus of cervical cancer HeLa cells through passive diffusion but were able to localize when tagged to the NLS at a specific density. Maximum nuclear localization, where half of the total intracellular CdSeS/ZnS NLS-NPs were transported into the nucleus, occurred at a surface NLS density of 20%. Any increases beyond 20% only resulted in modest enhancements in nuclear transport. The process of nuclear import was observed to occur within 2 h post-treatment, with a plateau reached at approximately 30 min.Similarly, the Lamprecht group studied chitosan NPs of different sizes: small (S-NP ≈ 25 nm) and large (L-NP ≈ 150 nm) tagged with different densities of the SV40 NLS CPKKKKKRKV (S-NPs: 0.25, 0.5, and 2.0 NLS per nm2 and L-NPs: 0.6, 0.9, and 2.0 NLS per nm2) for delivering loaded albumin-rhodamine to measure nuclear localization in murine L929 fibroblasts (Fig. 7).100 For the L-NPs, nuclear localization was enhanced only at the intermediate density of 0.9 NLS per nm2. Interestingly, there was an inverse relationship between NLS-tagging of S-NPs and nuclear localization efficiency. Taken together, the main outcome of this study was that tagging NPs at high NLS densities does not necessarily result in enhanced nuclear localization.
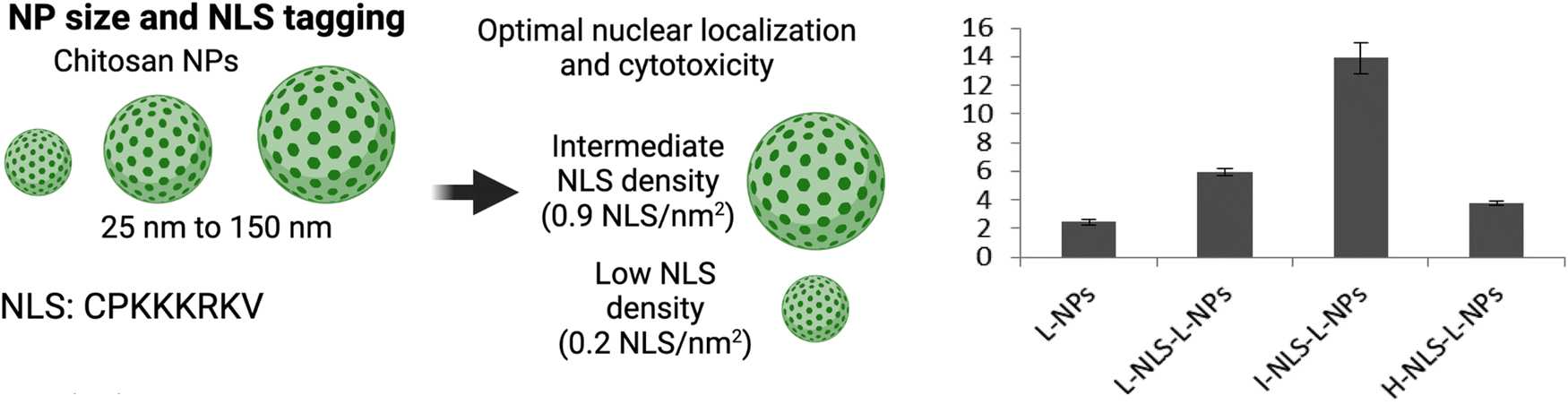 | ||
| Fig. 7 Effect of NP size and NLS conjugation density Left panel, representation of chitosan-based NPs of different size and the optimal NLS density for efficient nuclear localization. Right panel, nuclear localization efficiency (y-axis) of large NPs modified with low (L), intermediate (I), and high (H) NLS densities. Reproduced from ref. 100 with permission from ACS Publications, copyright 2015. | ||
These studies shed light on the influence of particle size and NLS density on the nuclear localization efficiency of NLS-tagged NPs, emphasizing the importance of carefully considering particle size and NLS density in NLS-therapeutic design to achieve efficient nuclear localization.
(b) Role of different NLS types and method of incorporation
The precise conditions to successfully utilize the properties of NLS peptides are often still unclear and vary with the type of NLS and the method of incorporation. The Rotello group studied the nuclear localization efficiencies of five different NLS types fused to eGFP delivered using gold NPs (Fig. 8).101 The NLS sequences chosen were (a) monopartite (SV40 [net-charge = +5], c-Myc [net-charge = +2],102 and TUS [Tus protein; KLKIKRPVK; net-charge = +5]103) and (b) bipartite (EGL-13 [MSRRRKANPTKLSENAKKLAKKEVEN; net-charge = +6]104 and NLP [nucleoplasmin (previously described); net-charge = +9]43), Interestingly, nuclear import efficiency differences were observed on quantitative comparison of the nuclear accumulation in HeLa cells (Fig. 8).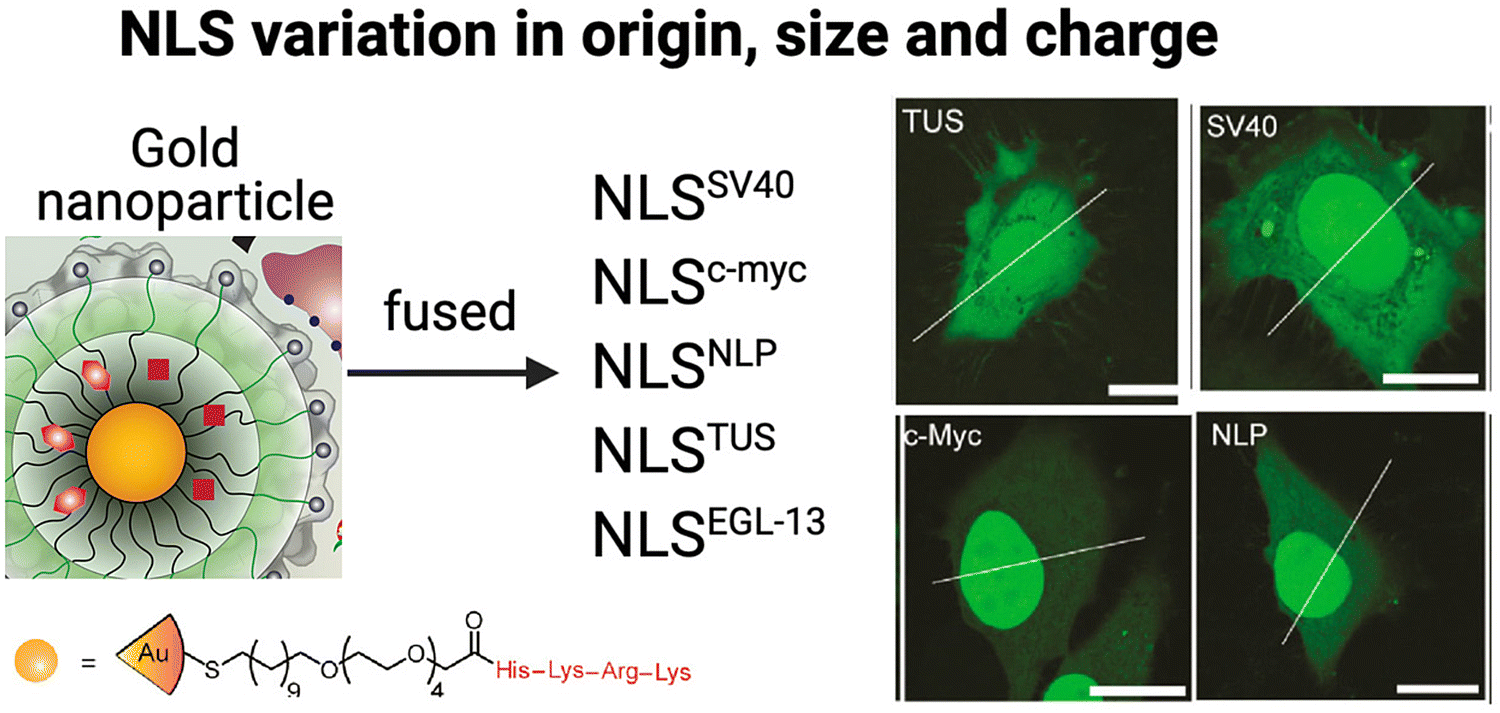 | ||
| Fig. 8 Effect of different NLS types Schematic representation of gold-based NPs tagged with five different NLS types and selected confocal microscopy images revealing fluorescence intensity differences associated with nuclear localization efficiencies compared to that in the cytoplasm in HeLa cells. Bars: 20 μm. Reproduced from ref. 101 with permission from ACS Publications, copyright 2015. | ||
This study demonstrated significant variations in nuclear import efficiencies among the different NLS sequences. Specifically, the NLSc-Myc-eGFP-NP exhibited a substantial 160% increase in nuclear fluorescence intensity compared to the surrounding cytoplasm. On the other hand, the most commonly used SV40 NLS tag, NLSSV40-eGFP-NP, showed a more modest 45% increase. NLSNLP-eGFP-NP and NLSEGL-13-eGFP-NP showed 85% and 70% increased nuclear intensity, respectively, while the NLSTUS-eGFP-NP showed a relatively modest 30% increase inside the nucleus. These findings highlight the significant impact of NLS origin, sequence length, and net-charge on nuclear localization efficiency of tagged therapeutic NPs. Hence, considering these factors is crucial for designing NLS-therapeutics to maximize nuclear localization and potentially, therapeutic efficacy.
The Read group studied four NLS types that interacted with importin-α and two importin-β members, aiming to compare the differences between classical and non-classical nuclear transport processes for improving NLS-therapeutic nuclear localization.105 They evaluated four NLS types: SV40-based classical NLSs [PKKKRKVEDPYC; net-charge = +3] and an ‘extended’ SV40 [SSDDEATADSQHSTPPKKKKRKVEDPYC; net-charge = 0], as well as non-classical NLSs, including the arginine-repeat NLS derived from the human T-cell leukaemia virus type 1 (HTLV) Rex protein106 [MPKTRRRPRRSQRKRPPTPWAHFPGFGQSLC; net-charge = +9], and the glycine-rich NLS from M9 [GNQSSNFGPMKGGNFGGRSSGPYGGGGQYFAKPRNQGGYC; net-charge = +2]. The arginine-repeat NLS type is known for its specific interaction with importin-β,107 while the M9 sequence is the hPY-NLS specific for transportin-1, as previously mentioned (Fig. 5).54,108 The goal of the investigation was to determine the efficacy of NLS-tagging, including the conjugation strategy and the choice of nuclear import route, in improving gene expression for NLS-therapeutics in the context of gene therapy. Two different methods of tagging the NLS types were used. The first method used a bifunctional peptide nucleic acid (PNA) linked to the NLS (PNA-NLS) and specifically conjugated to DNA via a thioether bond between a maleimide-activated PNA reactive to a C-terminal cysteine on the NLSs. The second method involved electrostatic complexation of the NLS to the net-negative charged DNA. For the latter, we surmise that this approach was taken because all the NLS types contained at least two cationic residues. Prior to conjugation, the NLS types were evaluated for binding their known importin receptors. Unexpectedly, the HTLV NLS bound strongly and at similar levels to importin-α1, importin-β, and transportin-1, which indicated that the HTLV NLS contained some degree of non-specificity. For the cysteine-specific conjugation, an average of 1.6 NLS peptides were attached to each DNA molecule out of the total DNA population, and this conjugation occurred for 80% of the DNA population. As expected, the SV40- and the SV40(extension)-PNA-NLS/DNA conjugates showed good binding for importin-α1. Like the earlier binding results, the HTLV-PNA-NLS/DNA conjugate also did not specifically bind importin-β. The M9-PNA-NLS/DNA conjugate bound well to transportin-1 but also to importin-α, albeit with 5-fold lower affinity. Nuclear localization efficiency was measured indirectly by transfecting HeLa cells and assessing the ability of the conjugates to express Luciferase after 24 h incubation. The experiments determined that the SV40(extension)-PNA-NLS/DNA conjugate improved Luciferase gene expression 7-fold compared to DNA alone, while there were no significant improvements observed with the other three NLS types. For the electrostatic complexation strategy, the SV40-, SV40(extension)-, and M9-NLS/DNA conjugates showed a modest 15–40% improvement in gene expression. Interestingly, the HTLV-NLS/DNA complex increased gene expression by approximately 200%.
The study by the read group aimed to compare the efficacy of different NLS types, specifically investigating the superiority of classical versus non-classical routes and employed two different methods for forming the NLS-conjugates. However, it is noteworthy that several of the attempted NLS-tagging approaches did not yield significant improvements in gene expression. Interestingly, the HTLV NLS, which was not specific for its importin-β receptors as a peptide alone, exhibited superior performance when electrostatically complexed with DNA. This observation suggests that the HTLV-NLS/DNA conjugate may have exploited both classical and non-classical nuclear import routes. This study provides important NLS-therapeutic design considerations while using different NLS types and conjugation strategies, as not all combinations may result in enhanced nuclear localization and gene expression.
It should be noted that although natural binding affinities of these various NLSs in a natural setting vary greatly, they were not explicitly involved in the design of the aforementioned NLS-therapeutics.
(c) Role of NLS orientation
Notable studies have investigated how the orientation of the NLS tag can affect the nuclear localization of NLS-therapeutics.109,110 DNA was coupled to the SV40 NLS at the C-terminal valine or at the N-terminal proline ends. Notably, the Emrick group explored how NLS orientation influences gene therapy efficacy. Specifically, the NLSs were directly synthesized on a polymer that possesses cationic properties to form nanoscale ‘polyplexes’ with DNA through electrostatic interactions.110 In one approach, the NLS was conjugated to the polymer starting from the N-terminal proline. In the other approach, the NLS was conjugated to the polymer starting at the C-terminal, resulting in a reverse orientation SV40 NLS (rNLS) (Fig. 9).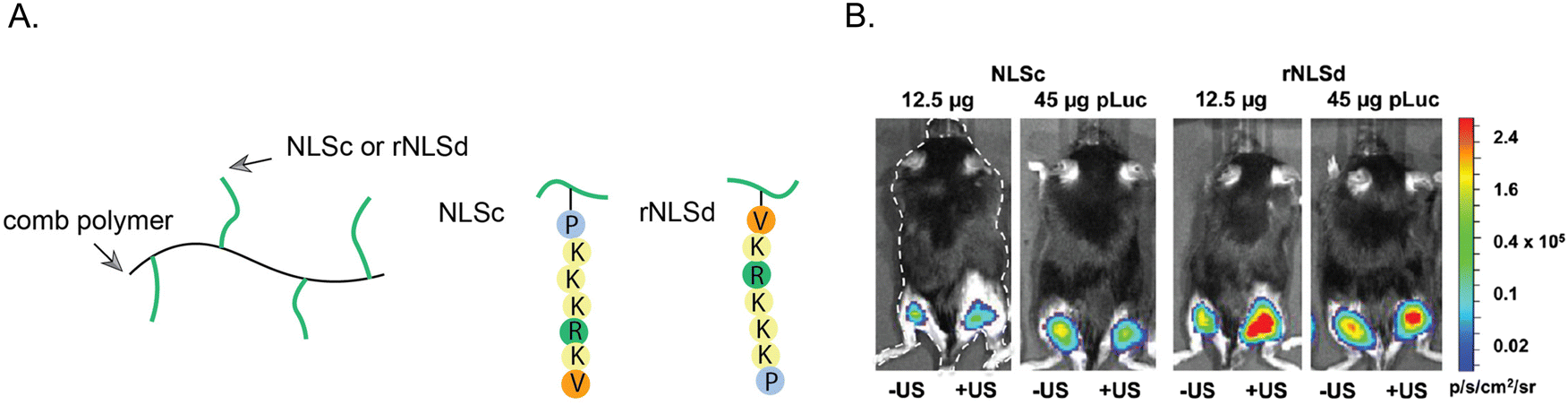 | ||
| Fig. 9 Effect of NLS orientation. (A) Representation of poly(cyclooctene-graft-oligopeptide) comb polymers tagged with SV40 NLS (NLSc) in the standard or reverse (rNLSd) orientations. (B) Luciferase expression from right hind legs upon treatment with NLSc- or rNLSd-tagged NPs and after ultrasound stimulus to facilitate deeper tissue penetration. Reproduce from ref. 110 with permission from ACS Publications, copyright 2014. | ||
In ovarian cancer SKOV3 cells, the rNLS/DNA conjugate showed a 3-fold increase in plasmid copy number per cell nucleus compared to the conventional NLS/DNA conjugate. Furthermore, cytotoxicity assays revealed that even at high concentrations, the rNLS/DNA conjugate maintained ∼100% cell viability, highlighting its potential for in vivo gene therapy applications. In an in vivo experiment involving intramuscular injection, the rNLS/DNA conjugate outperformed the standard NLS/DNA conjugate, resulting in an abundant 200–500% enhancement in luciferase expression. This work underscores the importance of exploring the reverse orientation of NLS sequences and offers valuable insights into the impact of NLS sequence orientation on NLS-therapeutic gene therapy applications in vivo.
(d) Role of intracellular environment on the effect of NLSs
Differences in the intracellular environment have a strong effect on NLS-therapeutic efficacy. The Lamprecht group has demonstrated that the nuclear delivery efficiency of NPs tagged with NLSs varies between cancer and normal cells.111 In this study, they showed that L-NPs had significantly improved nuclear localization when tagged with low-density SV40 NLS in L929 fibroblasts, primary human fibroblasts, and A549 lung cancer cells. When tagged with intermediate density NLS, the L-NPs had enhanced nuclear localization in human embryonic kidney 293 cells. However, any NLS tagging density significantly reduced nuclear localization of the L-NPs in glioma 261 cells. As expected, the S-NPs had no improvement with NLS tagging as these particles are small enough to passively diffuse into the nucleus.The authors propose that in glioma cell, overexpression of importin-α may lead to a scenario where multiple importin-α proteins compete to bind the same NP, potentially hindering nuclear import. However, it is important to note the study by Gousias et al. that focused on primary tissues rather than cell lines (noted in Table 1).68 In general, increased expression of importin-α plays a vital role in recognizing and binding NLS-containing cargo proteins, facilitating their transportation into the nucleus. Higher levels of importin-α expression would theoretically result in more available importin-α molecules to bind to NLS-therapeutics. This study further highlights the complexity of nuclear import dynamics, especially in the context of NLS-therapeutics. There remains, however, a need for further research to better understand the effects of importin-α expression (including different isoforms) on nuclear import efficiency and its implications for NLS-therapeutic design.
In another interesting and notable study, the Chen group evaluated NLS-therapeutic nuclear localization in the context of Ran expression levels.112 A PAMAM dendrimer-based platform with DNA payloads were tagged with increasing densities of NLS (1, 10, and 100 NLSs per molecule). The cervical cancer HeLa-derived cell line (KB) was used to generate RanGAP1 overexpressing cells (RanGAP1+). Importantly, the RanGAP1+ cells demonstrated increased GTPase activity. The 10-NLS-tagged PAMAM/DNA conjugate performed the best for localizing to the nucleus, supporting the previously described studies on the importance of an optimized NLS density (Section 5A). The PAMAM/DNA/10NLS also was not toxic to cells indicating its potential use for gene therapy applications. Importantly, the NLS-tagged conjugate that localized to the cell nucleus was almost four times better in RanGAP1+ cells compared to non-modified KB cells.
To evaluate the targeting ability and gene expression efficiency of the PAMAM/DNA(pDsRed-M-N1)/10 NLS conjugate in vivo, immunodeficient nude mice were subcutaneously implanted with RanGAP1+ cells to induce tumor formation. Once the tumors developed, mice received intravenous injections of the PAMAM/DNA/10NLS conjugate. After 48 h, the emission of red fluorescence was measured using a small animal imaging system to indirectly evaluate gene expression within the RanGAP1+ tumours. Notably, mice treated with the PAMAM/DNA/10NLS conjugate showed a significant increase in tumour fluorescence compared to mice injected with PAMAM/DNA alone, as well as in mice with tumours expressing normal levels of RanGAP1. These findings demonstrate that NP-based platforms can be administered intravenously when tagged with NLS and efficiently localize to the nucleus of tumour cells in vivo. Furthermore, the results suggest that the activity of the nuclear transport machinery can be augmented to enhance the targeting and gene expression capabilities of these conjugates.
(e) NLS in combination with additional therapeutic delivery technologies – the need for endosomal escape
Even with meticulous design, a perfectly engineered NLS-therapeutic may not effectively localize to the nucleus. The crucial step is engagement with the nuclear import machinery, which necessitates access to the cytosol. For most biopharmaceuticals, including NLS-therapeutics, endocytosis serves as the pathway for cell internalization.9 The process occurs through specific binding to target receptors on the cell surface or through nonspecific membrane penetration. However, biopharmaceuticals can become entrapped inside endosomes, followed by transfer to lysosomes where degradation takes place, rendering the therapeutics inactivate (as depicted in Fig. 10).113–115 To overcome this challenge, various nanocarriers such as inorganic NPs, liposomes and polymers, and cell-penetrating peptides (CPPs) have been developed over several years to facilitate membrane traversal,116 achieving selectivity, which is a requirement for cancer, is still a major problem. One promising approach is the combination of NLS with membrane traversal technologies, which has shown potential in enhancing nuclear targeting. In the following section, we will explore the latest advancements in the field of NLS combined with membrane traversal strategies.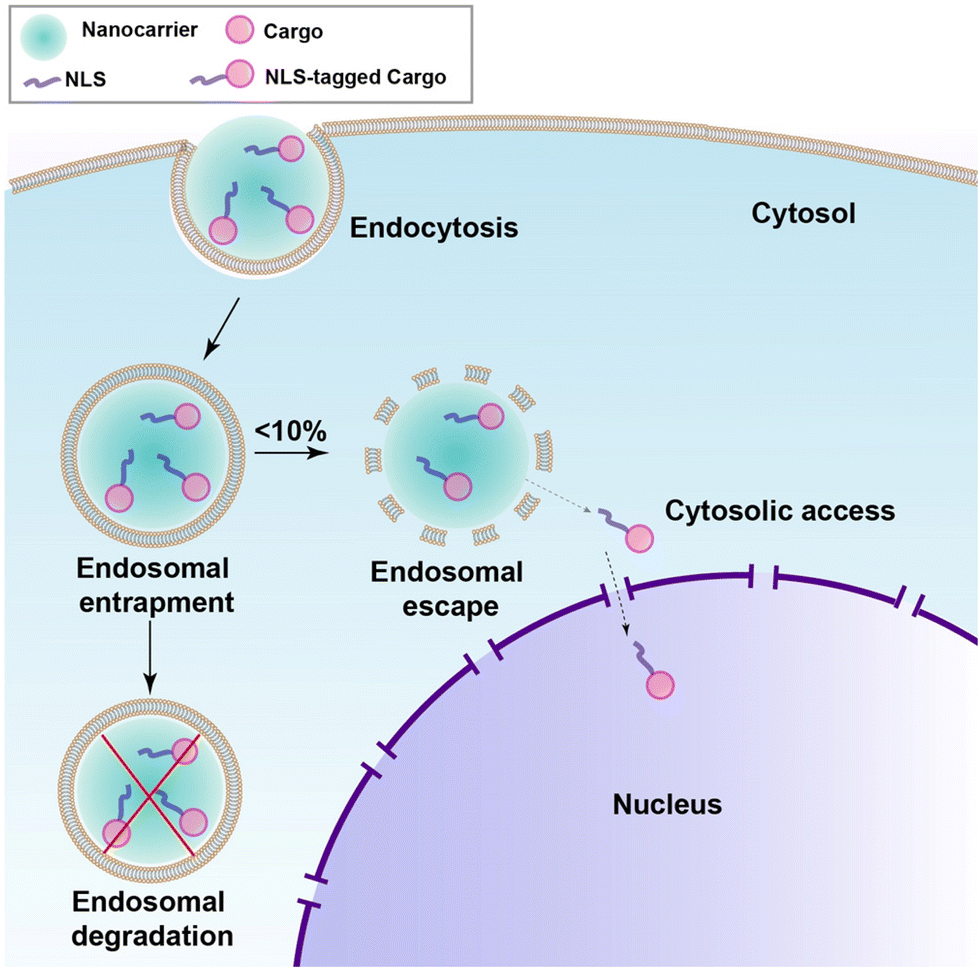 | ||
| Fig. 10 Schematic showing endosomal uptake of most nanocarriers leading to poor release in the cytosol and nucleus. | ||
The human immunodeficiency virus-1 transactivator of transcription (TAT) peptide (GRKKKRRQRRRPQ) combined with the SV40 NLS (TAT-NLS) was shown to enhance transfection efficiency and gene expression level for TAT-NLS-tagged agents.117 Specifically, negatively charged nanodiamonds (NDs) were tagged with TAT-NLSs. In HeLa and breast cancer MCF-7 cells, the NDs efficiently internalized and localized in the nucleus compared to non-TAT-NLS-tagged NDs. Furthermore, the TAT-NLS-NDs were loaded with antisense oligonucleotide ANA4625, a 20-mer with a 2′-O-methyoxy-ethoxy (MOE)-modified phosphorothioate backbone responsible for bcl-2 and bcl-xL gene knockdown and, thereby, inducing apoptosis. The BCL-2 protein inhibits apoptosis and promotes cell survival, while the BCL-XL protein also functions as an anti-apoptotic protein.118 The effects of these oligonucleotides on cell viability after delivery through a coupled endosome escape-active nuclear import were assessed in small cell and non-small cell lung cancer cell lines. These cell lines expressed different basal levels of bcl-2 and bcl-xL, and the assessment of therapeutic efficacy included evaluating apoptosis and the change in expression level upon treatment to the TAT-NLS-ND conjugates. Notably, the conjugate was able to abundantly reduce the amount of expressed protein products, and this activity induced apoptotic cell death up to 100-fold compared to non-TAT-NLS-tagged NDs. This study deserves attention as a potential NLS-therapeutic in lung cancer.
The Liu group developed a multifunctional endosome escape-nuclear import and pH-sensitive gene delivery system for regenerating the vasculature in dental pulp.119 They designed C-R9-G-NLSW, which consists of key distinct features: (1) cysteine (C) for coupling to a polymeric nanocarrier, (2) R9 CPP for cell membrane traversal to enhance cellular uptake and access to the cytoplasm, (3) glycine (G) residue to serve as spacer, (4) the SV40 NLS for active nuclear import, and (5) tryptophan (W) for fluorescence quantification and visualization of nuclear localization efficiency. The strategy worked successfully in vivo and will be discussed further in Section 8 below.
The Rebollo group developed a novel and interesting approach for applying the NLS-therapeutic concept.120 The Hippo signaling pathway plays a significant role in controlling cell proliferation and apoptosis. The pathway involves the inactivation of YAP (Yes Associated Protein), a co-transcriptional activator, through phosphorylation by a core group of kinases. When the Hippo pathway is inactive, YAP can localize to the nucleus and activate genes related to cell proliferation and survival.121 Dysregulation of the Hippo pathway, such as constitutive YAP nuclear localization is associated with tumor development.121 This study investigated the potential of targeting the interaction between YAP and TEAD, a DNA binding protein, as a strategy for cancer therapy.
A tri-functional peptide complex was developed that had membrane traversal, nuclear import, and YAP-TEAD inhibition properties. For YAP-TEAD inhibition, a series of peptides covering the relevant region where YAP binds to TEAD were synthesized and tested. The specific sequence for the best inhibiting peptide was KTANVPQTVPMRLRKLPD. The CPP (Mut3DPT-Sh18) was the first part of the long chimeric peptide. It was followed by the TEAD inhibitory sequence. The last part was a synthetic bipartite NLS. Strong cellular internalization and subsequent nuclear localization were observed in breast cancer MDA-MB231 cells (Fig. 11A).
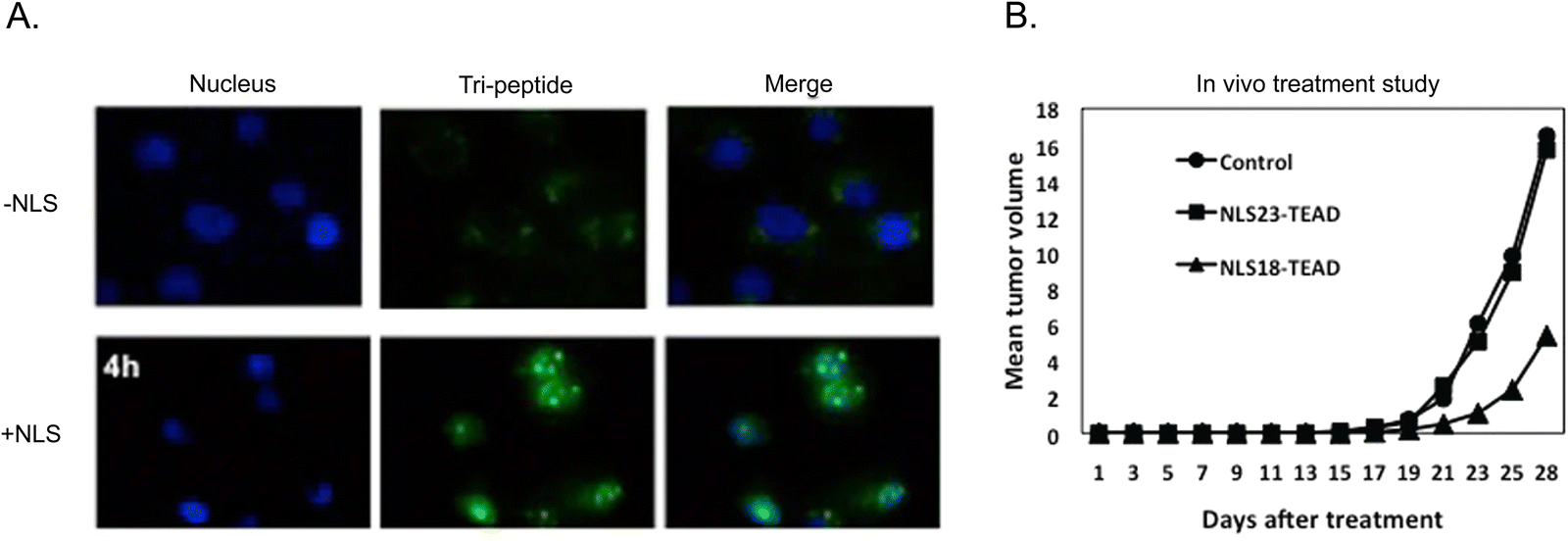 | ||
| Fig. 11 Multi-functional peptide-based NLS. (A) Merged channels of time-dependent nuclear localization of FITC-labelled TEAD inhibitor peptide with and without tagged NLS in breast cancer cells. (B) Anti-tumoral effect of two version of tri-peptide based on amino acid length differences for the CPP peptide component on breast cancer xenograft models. Reproduced from ref. 120 with permission from Springer Nature, copyright 2019. | ||
The apoptotic effect of the tri-functional peptide was evaluated. The peptide containing only membrane traversal and TEAD inhibition elements showed a weak apoptotic effect. In addition, CPP and NLS did not induce apoptosis. However, when the TEAD interfering sequence was combined with the CPP and NLS, the apoptotic effect significantly increased, reaching up to 63%. Therapeutic studies in mice bearing TN60-UNLP breast cancer xenografts showed that a dose of 5 mg kg−1 led to a significant reduction in tumour size (∼67%) compared to the control group treated with saline solution (Fig. 11B). It should be noted that treatment injections started only 5 days after the tumour cells were implanted. Notably, the peptide showed good stability and measurement of weight of the animals and the organs at the end of the treatment period demonstrated good tolerance to the administered peptide over four weeks.
In summary, the tri-functional peptide CPP-TEAD-inhibitor-NLS demonstrated promising potential as an anticancer NLS-therapeutic candidate. It possessed favorable characteristics, including solubility, efficient cell penetration, lack of toxicity, sufficient nuclear localization, and apoptotic and anti-tumoral effects in breast cancer xenograft models without inducing preclinical adverse events in treated mice. This NLS-therapeutic merits future studies such as pharmacokinetics, dosing, and overall clinical translation planning.
8. Applications of NLS-therapeutics in disease treatment
Chemotherapeutics, radionuclides, and biologics are frequently employed as payloads in NLS-therapeutics, representing prominent options for disease treatment.(a) Chemotherapeutic payloads
Doxorubicin (DOX) is used to treat a wide variety of cancers, such as breast and lung cancers, lymphoma, and acute lymphocytic leukemia.122 Dox intercalates into DNA and disrupts topoisomerase II-mediated DNA repair.123 In addition, Dox generates free radicals that damage cellular membranes, proteins, and DNA.123 Previous in vitro studies have demonstrated that only 26% of DOX enters the nucleus,124 an important issue since DOX dosage is limited due to cardiotoxicity.125 Multiple delivery platforms have been generated to improve the delivery of DOX to target diseased cells.126 Doxil is a liposome loaded with DOX and FDA-approved for the treatment of patients with ovarian cancer where the disease has progressed or recurred after traditional chemotherapy.127 However, only 0.4% of the DOX administered in the liposomal formulation enters the nucleus due to poor intracellular release and sequestration in lysosomes. As only 17–27% of patients respond to Doxil, more effective systems are needed to increase the efficiency of cellular and nuclear delivery of DOX and, hence, therapeutic efficacy.Dual-decorated cholesterol-modified glycol chitosan micelles provide targeted delivery of DOX into the tumor cell nucleus.128 The chitosan-based micelles were conjugated with folic acid to target overexpressed folate receptors (FR) on tumor (FR+) KB and (FR−) A549 cells and, tagged with the SV40 NLS and loaded with DOX and termed DOX/NFCHGC. Although there was no membrane traversal technology specifically integrated into the design, the authors report that the DOX/NFCHGC could penetrate through endosome/lysosome membranes, albeit with a delay, as cellular entry did not occur until 4 h post incubation and nuclear localization was not accomplished until 6 h.
Encouraging findings emerged from these cytotoxic delivery studies, underscoring the potential of the DOX-micelles as a targeted delivery platform. Notably, both the folate-modified and non-modified DOX-micelles demonstrated enhanced cytotoxicity compared to free DOX. This increased efficacy suggests that this chitosan-based micelle delivery platform effectively localized DOX inside the nucleus of tumor cells, resulting in a superior cytotoxic effect compared to the non-specific diffusion free DOX. Non-DOX-loaded micelles were not cytotoxic indicating the micelles themselves were not inducing cytotoxic effects. The folate-ligand modified, and DOX-loaded micelles showed good specificity as they significantly enhanced cytotoxicity in FR+ KB cells compared to FR− A549 cells.
The assessment of anti-tumour activity was performed in mice implanted with FR+ KB cells and treatment studies initiated with tumours were approximately 100 mm3. DOX/NFCHGC and several controls were intravenously injected at 3 mg kg−1. The results showed that treatment with DOX/NFCHGC micelles resulted in the most significant suppression of tumor growth compared to other groups. These findings indicate that folate-modified chitosan-based micelles improved the delivery of DOX into the tumour through active nuclear targeting. Interestingly, the authors note that because the tumour volume of mice treated with DOX/CHGC, which lacked the folate receptor targeting ligand, was smaller than that of the free DOX group, it is most likely that the enhanced permeability and retention (EPR) effect contributed to the anti-tumour activity. Importantly, the use of DOX-loaded chitosan-based micelles resulted in gradual increases in mouse body weight, indicating reduced side effects compared to free DOX that caused weight loss. This work demonstrates that cell surface receptor targeting, in combination, with NLS-mediated nuclear import incorporated into chitosan-based micelles, greatly improved the anti-tumor efficacy of DOX.
Antibody–drug conjugates (ADCs) utilize monoclonal antibody (mAb) selectivity to deliver a conjugated cytotoxin to receptors preferentially overexpressed on tumour cells, while sparing healthy cells, and increasing therapeutic efficacy relative to traditional chemotherapy.129 There are currently 13 ADCs approved for use in patients with refractory and/or relapsed metastatic cancers. Trastuzumab emtansine (T-DM1) is an approved ADC targeting the human epidermal growth factor receptor 2 (HER2). Although patients may initially respond to T-DM1 therapy, many patients experience disease progression soon after treatment completion.130,131 Incomplete trafficking of T-DM1 to the lysosome and inherent or acquired rapid recycling back to the cell surface result in low intracellular accumulation of T-DM1in tumor cells and, hence, poor tumour killing efficiency.
The Leyton group used an alternative strategy to redirect the routing of T-DM1 towards the nucleus, aiming to address three key objectives: (1) overcoming endosome entrapment, (2) investigating the potential of the nucleus as a reservoir for increased drug accumulation, and (3) determine if these activities are associated with increased cytotoxic potency. T-DM1 was tagged with the Accum technology, which is widely utilized to enhance intracellular drug delivery (Fig. 12A).132 Accum comprises a cholic acid component that activates sphingomyelinase, triggering the cleavage of sphingomyelin and generating ceramide within endosomes and lysosomes. This process disrupts the membranes of these compartments.133 By coupling the cholic acid to the SV40 NLS, tagged drugs can be specifically directed to the nucleus, as previously shown for conjugated antibody-conjugates.134 In the case of Accum-tagged T-DM1, it retained specific HER2 binding, underwent internalization, successfully escaped from endosomes, and effectively localized to the nucleus.135 Notably, Accum-T-DM1 exhibited an abundant 18-fold increase in cytotoxicity against SKBR3 breast cancer cells compared to T-DM1 alone.
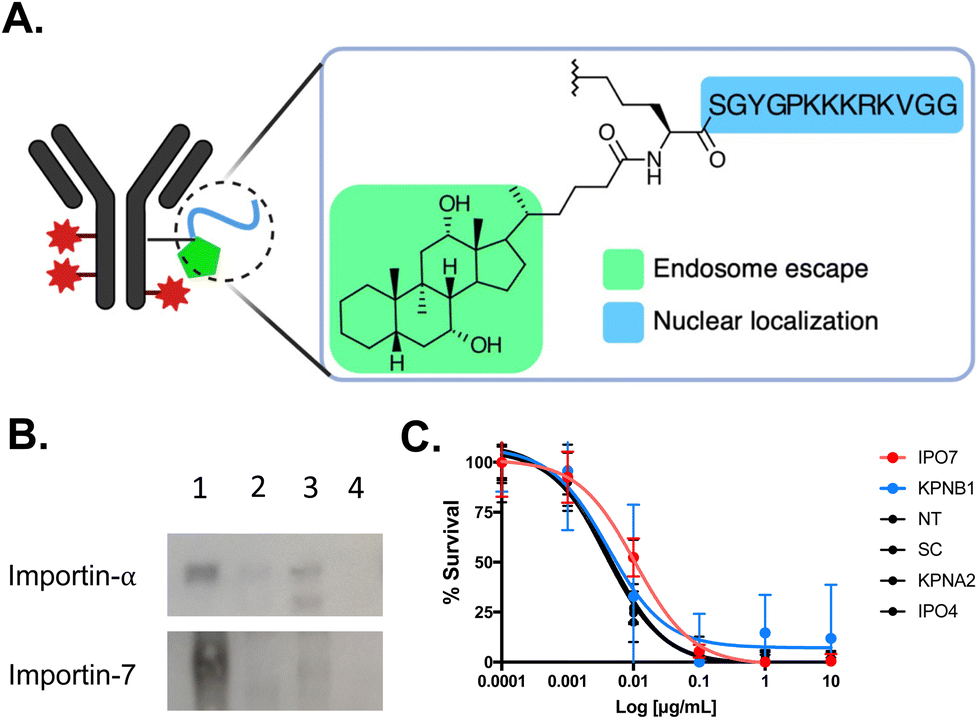 | ||
| Fig. 12 NLS-tagged ADC. (A) Schematic representation of T-DM1 modified with Accum (Acc). (B) Affinity purification followed by western blot demonstrating that Acc (lane 1) interacts preferentially with importin-7 and not importin-α. As anticipated, T-DM1 and unmodified trastuzumab (lanes 2 and 3, respectively) do not interact with importin-α. Lane 4 is beads only. (C) Cytotoxicity assays demonstrating that knockdown of the classical importin-α and importin-β1 did not affect Acc-T-DM1 cell killing potency. However, there was a 3-fold reduction in cytotoxicity in cells knockdown of importin-7 (IPO7). There was also no affect in cytotoxicity in cells knockdown of importin-4, which is another non-classical nuclear transport importin protein. Control assays were performed in non-transfected (NT) cells and in cells transfected with scrambled (SC) siRNA. Reproduced from ref. 135 with permission from Elsevier, copyright 2020. | ||
Within the spectrum of different importins in the human cell, the SV40 NLS sequence was preferentially recognized by the non-classical importin-7 in the context of Accum-T-DM1 (Fig. 12B). This reliance on importin-7 can be attributed to its electrostatic binding mechanism for NLS recognition, which is less dependent on specific amino acid sequences (Fig. 6).56 Additionally, the preference of importin-7 for SV40 NLS sequence in the context of Accum-T-DM1 (Fig. 12A) is likely due to the multiple NLS tags, each carrying a +5 cationic charge, which most likely contribute to electrostatic interactions driving the binding of importin-7 to Accum-T-DM1. Consequently, the nuclear localization and enhanced cytotoxicity of the Accum-tagged T-DM1 were primarily dependent on importin-7 (Fig. 12C). This study highlights that NLS-tagging can redirect ADCs to the nucleus and enhance their cytotoxic potency, but it also emphasizes the potential impact of NLS-tagging on the nuclear transport pathway of NLS-therapeutics.
(b) Radionuclide payloads
Nuclear targeting with theragnostic radionuclides allows for the application of high linear-energy transfer and short-range-emitting radionuclides such as α-particles and Auger electrons to transmit ionization densities that are lethal to cancer cells.136 NLS strategies provides decreased toxicity relative to traditional radiation techniques and aims to maximize DNA damage. The Reilly group developed an approach to exploit the Auger electron and gamma-ray emissions from the radionuclide indium-111 (111In) to image and kill cancer cells in vivo.137 They tagged SV40 NLS peptides to 111In-labeled trastuzumab and determined the therapeutic efficacy in a human breast cancer model. 111In-trastuzumab-NLS exhibited a 6-fold increase in killing SKBR3 cells compared to 111In-trastuzumab alone. The in vivo assessments demonstrated enhanced radioactivity accumulation within isolated nuclei of tumour cells from xenografts in mice that were intravenously injected with 111In-trastuzumab-NLS, as compared to unmodified 111In-trastuzumab. This increased nuclear localization in vivo, along with the superior killing efficacy observed in vitro, indicates the potential of 111In-trastuzumab-NLS to effectively target radiotherapeutic agents to HER2+ breast cancers.An 111In-labeled mAb specifically targeting CD123+ leukaemia stem cells (LSCs) was tagged with SV40 NLS peptides and evaluated for its ability to detect and eradicate primary acute myeloid leukemia (AML) engrafted into the bone marrow of mice.138–142 AML is a hematologic malignancy associated with poor outcomes and a high risk for relapse, mainly attributed to the survival of LSCs, responsible for disease reoccurrence following conventional chemotherapy.143 AML specimens obtained from patient bone marrow aspirations were intravenously injected into immunodeficient mice, resulting in bone marrow engraftment and high levels of circulating AML cells (Fig. 13A and B). Tracer doses of the anti-CD123 mAb (CSL360) or isotype control radiolabeled with 111In and tagged with NLS were administered intravenously (Fig. 13C). Small animal single photon emission computed tomography imaging conducted at 72 h post-injection revealed intense areas of radioactivity in the pelvis, spine, and the femoral heads (Fig. 13D),140,141 which are classic sites of human AML engraftment in the mouse.144 Notably, 111In-CSL360-NLS demonstrated potent therapeutic effects as early as 72 h post-injection, reducing the number of AML engrafted cells in the bone marrow by approximately 2-fold compared to 111In-IgG-NLS.139 Although there was a trend for improved survival in mice treated with 111In-CSL360-NLS, it was not substantial. This is most likely due to the exceptionally high leukemic burden in mice in the study as the mice were not expected to have a long lifespan due to the severity of the AML burden. The ability of 111In-CSL360-NLS to reduce the leukemic burden 2-fold by 72 h post-injection is notable. These studies demonstrate the use of an NLS-therapeutic in a treatment approach, effectively targeting and eradicating primary AML LSCs engrafted in mice.
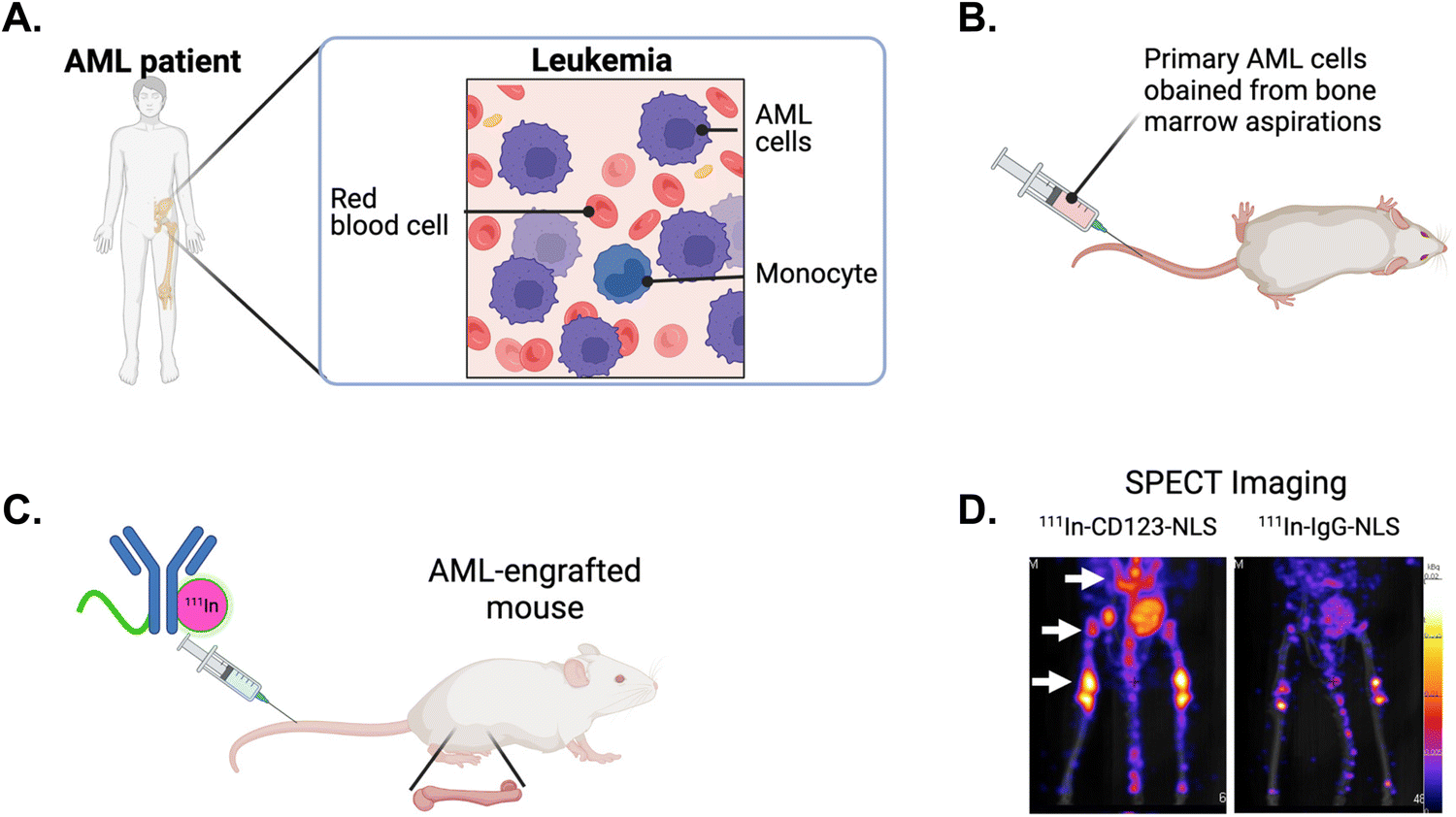 | ||
| Fig. 13 NLS-tagged radiolabeled antibody directed against leukemia stem cells (LSCs). (A) Schematic of acute myeloid leukemia (AML) in the bone marrow of patients and, (B) engrafted in mice. (C) NLS-tagged 111In-labeled mAb specific for LSC marker CD123 is intravenously injected into AML-engrafted mice. (D) Single photon emission computed tomography (SPECT) imaging showing the imaging of CD123+ LSCs engrafted in the bone marrow (arrows) in mice injected with 111In-specific CD123 (CSL360)-NLS or 111In-nonspecific IgG-NLS. Reproduced from ref. 140 with permission from Elsevier, copyright 2014. | ||
Building on the prior results above using the Accum technology in the context of ADCs, the Leyton group explored the potential of this strategy for improving the tumor targeting of radionuclide-mAb conjugates. An Accum-tagged mAb known as A14 to target interleukin-5 receptor-α (IL5Rα) in muscle invasive bladder cancer was radiolabeled with the positron-emitting radioisotope copper-64 (64Cu).145 The 64Cu-A14-Accum conjugate exhibited considerably increased nuclear localization compared to 64Cu-A14 and 64Cu-A14-NLS, the latter lacking endosome escape activity. Biodistribution analysis via positron emission tomography imaging revealed a significant ∼3-fold increase in tumor targeting for both I high and low IL5Rα-expressing tumours in mice intravenously injected with 64Cu-A14-Accum compared to 64Cu-A14 alone. Notably, the 64Cu-A14-NLS conjugate had poor tumor uptake and was linked to the inability of the conjugate to efficiently localize to the nucleus. These findings further emphasize the potential of the Accum technology in enhancing the targeting of tumor cell nucleus, this time in a theragnostic context.
(c) Biological payloads
As described earlier in this review, gene therapy has emerged as a promising domain with vast potential for NLS-therapeutic designs employing NP delivery platforms. Here, we describe further gene therapy applications using biological delivery platforms.The Chen group tagged the SV40 NLS onto human serum albumin engineered to be cationic (CHSA) with a pI = 8.9.146 The targeting rationale was to localize sufficient therapeutic in tumours via the EPR effect. Using electrostatic interactions to form conjugates, a CHSA/NLS/DNA agent was formed where the DNA sequence encoded tumour suppressor protein p53. The electrostatic interactions enabled the formation of conjugate spheres, ranging from 50–100 nm in diameter (Fig. 14A). The cationic nature of CHSA also helped interact and encapsulate the naturally anionic DNA and protect it from nuclease-mediated degradation. In vitro, the CHSA/NLS/DNA conjugate showed high cellular uptake and strong nuclear localization based on reporter gene expression assays. When intravenously injected in mice, the cationic CHSA agents had increased circulation in the bloodstream and were shown to be a crucial element in achieving sufficient tumor accumulation. As a result, CHSA/NLS/DNA exhibited up to 60% tumor growth inhibition in mice bearing sarcoma tumours (Fig. 14B). In contrast, mice injected with free oligonucleotides showed no tumour growth inhibition. Furthermore, there were no observable toxicities or inflammatory responses in mice. This study demonstrated a successful approach to protect plasmid DNA from degradation by nucleases in the bloodstream and improve circulation time and anti-tumour efficacy.
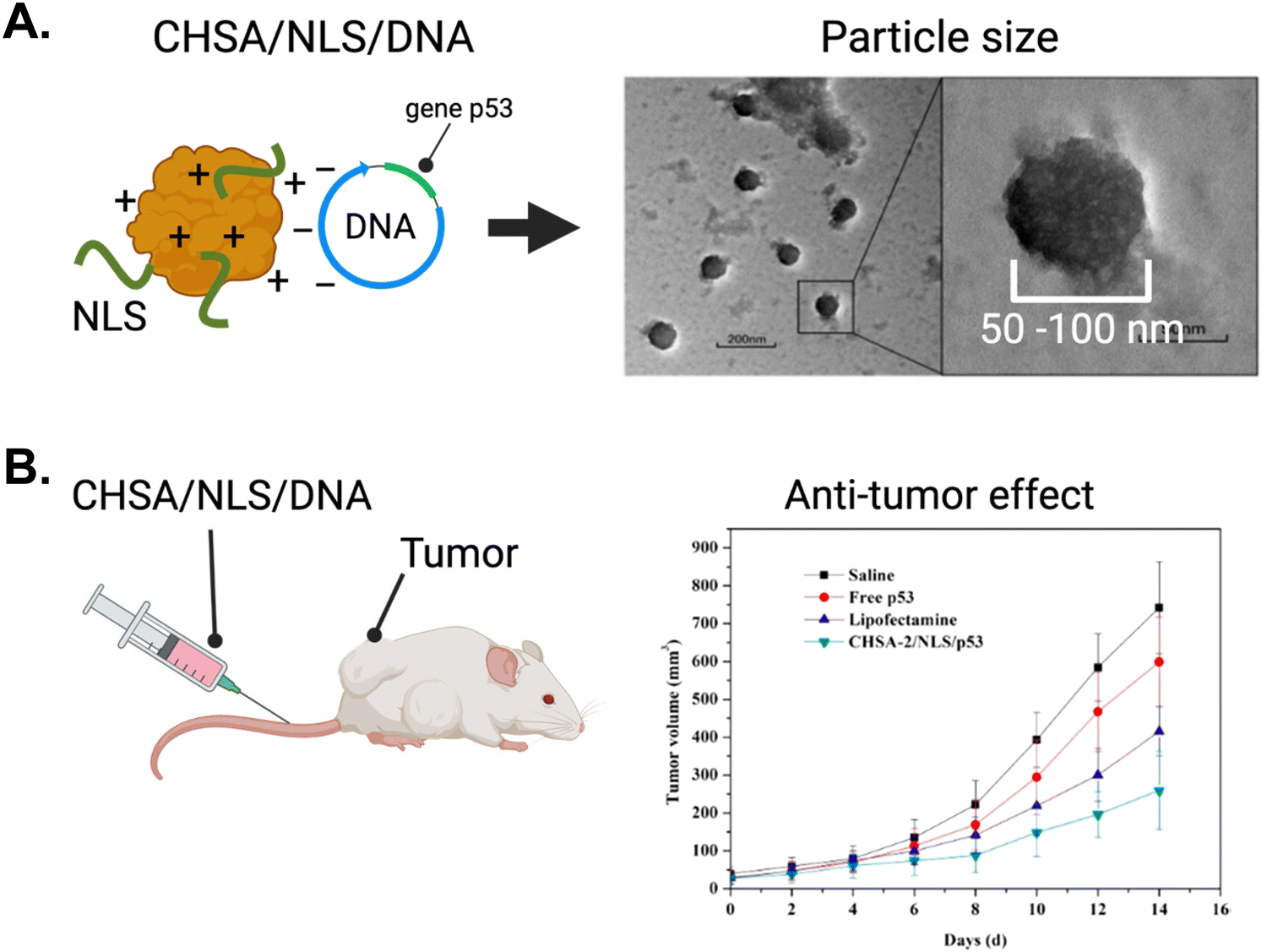 | ||
| Fig. 14 The application of gene therapy in oncology using NLS-system. (A) Left panel, schematic depicting the system for complexing cationic human serum albumin (CHSA) with NLS and DNA. Right panel, electron microscope picture depicting CHSA/NLS/DNA complex. (B) In vivo antitumor activity in mice receiving intravenous injection of CHSA/NLS/DNA vs controls. Reproduced from ref. 146 with permission from MDPI, copyright 2019. | ||
Gene therapy was investigated for dentistry, specifically endodontics, in a study focused on tissues involving the tooth root. A major challenge in regenerative endodontics is limited revascularization during pulp regeneration in the full-length tooth root canal.147 Linking back with the work from the Liu group describing the development of an NLS-therapeutic platform consisting of a peptide harbouring sequences that provides endosome escape and nuclear import activities C-R9-G-NLSW, (Section 7E), a poly(L-lysine)-based NP was developed and tagged with these peptides.119 The NP was loaded with DNA plasmids containing the gene to encode for the protein vascular endothelial growth factor (VEGF). Evaluating human dental pulp stem cells, the NP was able to increase nuclear localization by 36% and increased mRNA expression of VEGF approximately 4-fold compared to the non-peptide-modified NP/DNA agent. Furthermore, NP-treated dental pulp stem cells implanted into an in vivo human tooth root model resulted in an abundant increase in VEGF expression and, more importantly, a 69% increase in tissue regeneration of the tooth root canal. Therefore, innovative NLS-therapeutic approaches hold promise for addressing challenges in regenerative endodontics.
CRISPR/Cas9 technology has revolutionized gene editing by enabling precise modifications in the genome, including gene insertion, deletion, and regulation.148 Nonetheless, the clinical application of CRISPR/Cas9 faces a significant hurdle: effective and targeted in vivo delivery to specific tissues.149–151 As is the theme of this review, the effective targeting of agents active in the nucleus is important to enhance their therapeutic effects. Hence, similar considerations apply to the development of nuclear-targeting systems for the CRISPR/Cas9 technology.151
The Pcsk9 gene, which encodes the proprotein convertase subtilisin kexin 9 (PCSK9), is a key regulator of low-density lipoprotein (LDL) levels152 LDL proteins bind to and transport cholesterol throughout the body. Normally, LDL receptors on the surface of hepatocytes facilitate the uptake and internalization of LDL proteins and play an important role in clearing cholesterol from the bloodstream. However, when PCSK9 is released into the bloodstream by the liver, it binds to the LDL receptor, disrupting its normal internalization and recycling process. PCSK9 binding to the LDL receptor induces its lysosomal degradation and results in decreased LDL-mediated cholesterol clearance and increased levels of circulating cholesterol. Elevated cholesterol levels have been long associated with cardiovascular disease. Therefore, inhibiting PCSK9 activity could be a potential approach to lowering cholesterol levels and improving treatments for cardiovascular disease.
The Jiang group developed a method to effectively deliver CRISPR/Cas9 using a triple-layer targeting system.153 NLS-tagged Cas9 and Pcsk9-targeted single guide RNA (sgPcsk9) were complexed with gold NPs modified with TAT peptides and further encapsulated in a galactose-modified lipid layer to target these agents to the liver (Fig. 15A). Galactose targets the asialoglycoprotein receptor on hepatocytes.154 The NLS-therapeutic was termed Gal-PEG-lipid-coated gold Cas9/sgPcsk9 (Gal-LGCP) and it increased Cas9 and sgPcsk9 transport into the nuclei of hepatocytes. In comparison, the conjugate without TAT only localized to the periphery of the nucleus. Pcsk9 gene editing was significantly increased, while Gal-LGCP counterparts lacking TAT or NLS modification showed poor gene editing activities.
 | ||
| Fig. 15 Nuclear targeting of CRISPR/Cas9. (A) Schematic diagram of Gal-PEG-lipid-coated gold nanocluster that is complexed with Cas9-NLS fusion protein and single guide RNA targeting the Pcsk9 gene (termed Gal-LGCP). (B) Schematic of the intravenous administration of Gal-LGCP depositing in the target liver (depicted by yellow glow) and the resulting reduction in PCSK9 and LDL cholesterol serum levels. Reproduced from ref. 153 with permission from John Wiley and Sons, copyright 2019. | ||
In vivo, intravenously injected Gal-LGCP exhibited impressive circulation times, lasting sufficiently long to efficiently accumulate in the target liver. Small animal imaging showed fluorescence intensity from the liver was 8-fold higher for mice injected with Gal-LGCP compared to NPs not modified with galactose. Notably, sites of nonspecific sequestration, such as the spleen and lungs, showed considerably reduced amounts of Gal-LGCP compared to non-galactose modified NPs. Deep sequencing of extracted genetic material from hepatocytes taken from treated mice showed significantly increased Pcsk9 editing. In contrast, Pcsk9 editing was negligible in mice treated with NPs lacking an NLS. Blood taken and analyzed from mice treated with Gal-LGCP contained reduced levels of circulating PCSK9 and LDL-cholesterol compared to control groups treated with NPs lacking galactose, TAT, or NLS (Fig. 15B). These findings demonstrate the feasibility of delivering CRISPR/Cas9-based therapeutics to the liver using liver-specific targeting components in combination with active nuclear import.
9. Future areas of research
NLS-therapeutics have shown promise in a wide array of applications. There are, however, several areas that require further research and exploration to fully realize the potential of this approach. As an example, the current reliance on the SV40 NLS sequence has limitations, and future research provides an opportunity to develop more effective NLS-based approaches.Future areas of interest include identifying importin-α isoforms or importin-β members preferentially expressed in diseases of interest. As previously mentioned, different importin-α isotypes have distinct binding preferences for certain bipartite NLS-cargoes,41,42 and their expression patterns vary across cell types and disease conditions (Table 1). By characterizing the importin repertoire in specific disease contexts, researchers can identify potential importin-α isotypes or importin-β members that are highly relevant for therapeutic targeting. This knowledge will aid in the design of NLS sequences that are more selective for the desired importin and can potentially enhance nuclear import and therapeutic efficacy. In addition, other components of the nuclear transport machinery may have aberrant expressions and functions that, as we described, can boost NLS-therapeutic nuclear import.112
The exploration of NLS sequences can provide new insights and possibilities for enhancing NLS-therapeutics. As described above, bipartite NLS sequences enhance nuclear import efficiency and therapeutic efficacy.101,120 In addition, importin-β family member-specific NLS sequences have been explored in the context of NLS-therapeutics.105 These NLSs are drastically different in sequence, length, and chemical properties compared to classical importin-α-specific NLSs. Importin-βs that are capable of binding NLS-cargoes and facilitating their import independent of importin-α offer a potentially more kinetically favourable approach to nuclear import. However, specific importin-β NLS-therapeutics did not always improve nuclear import, primarily due to non-specificity issues.105 This inefficiency could be attributed to a limited knowledge of the role of particular importin-β family members in specific diseases. For example, importin-β, transportin-1, importin-7, and importin-11 have all been reported to be significantly overexpressed in cancers (Table 1), indicating the importance of understanding NLS in the context of geno/phenotype.
The potential for synergy between NLS tags presents another potential avenue of exploration. NLS-therapeutic nuclear import could be boosted through the tagging of a combination of different NLSs. For instance, the Rouse sarcoma virus Gag protein contains two independent NLSs. One NLS binds directly to importin-α and the other NLS binds to importin-11 and transportin-3 and utilizes a form of cooperative complex formation to boost nuclear import efficiency.155 Therefore, exploring the involvement of these import-β family members in specific diseases during NLS-therapeutic design presents significant opportunities for enhanced nuclear localization efficiency.
Underpinning all applications of NLS-based strategies is the need to understand fundamental aspects of NLS activity. Understanding the thermodynamics and kinetics of nuclear import is very important for NLS-therapeutic development. Timney et al., reported that importin-α concentration in normal cells was linearly related to import rates156 In artificially induced expression of importin-α, still higher importin-α concentrations was linear and eventually import rates saturated. Moreover, reducing the affinity for NLS-cargoes and importin-α significantly reduced nuclear import. This modulated affinity suggests that the nuclear import machinery is built for kinetically rapid nuclear localization and, hence, supports the strategy of targeting overexpressed importins in various diseases (refer to Table 1). However, the study also revealed that the intracellular space is highly complex, with significant competition of NLS-cargoes for importin-α. This competition may kinetically slow down nuclear import for an individual NLS-cargoes and NLS-therapeutics. Therefore, the design of NLS-therapeutics may incorporate bipartite NLSs that have increased binding affinity compared to monopartite NLSs. Alternatively, the integration of minor groove-specific NLSs into NLS-therapeutic design could be explored. A bioinformatic analysis indicated that the α-helical-containing minor groove-specific NLS is much less prominent than the classical major groove-binding NLS and, hence, is much less prevalent in nature.39 Consequently, there may be less competition in the intracellular environment for the minor groove, increasing the likelihood of efficient nuclear import for an NLS-therapeutic. Understanding of these thermodynamic and kinetic effects would substantially improve or ability to engineer NLS-therapeutics.
Understanding the cellular dynamics of NLS therapeutics is crucial. Determining how long these NLS agents remain within the nucleus can provide valuable information into their overall efficacy. While retention promoting factors (signals) for mRNAs are known,157 the exploration of retention mechanisms for NLS-therapeutics remains an intriguing opportunity for research. Studying the factors influencing natural nuclear retention of cellular molecules could provide researchers with the ability to design and optimize future NLS-therapeutics with enhanced therapeutic efficacy. Researchers have observed that diseased cells often exist in a ‘stressed’ state and the effects on nuclear localization may be advantageous for NLS-therapeutics. For instance, Miyamoto et al., demonstrated that conditions like UV irradiation, oxidative stress, and heat shock disrupt the Ran gradient in HeLa cells, leading to a halt in export functions.158 Consequently, there is an abundant increase and retention of importins within the nucleus. This natural setting of stress in diseased cells provides a favorable environment for the development of NLS-therapeutics.
Optimizing the construction and design of NLS-therapeutics is of utmost importance, as discussed throughout this review. While innovative NP systems have been explored, it is important to consider the NLS-tagging method itself, as factors such as NP size and NLS density and orientation can significantly impact nuclear import efficiency and therapeutic efficacy. Furthermore, parameters such as stability and safety must be carefully addressed. Although a few described NPs showed tolerability in mouse models,120,128 extensive testing will be necessary before any clinical translation can be attempted. In addition, it essential to combine NLS-therapeutic design with endosome/lysosome escape strategies, as cytosolic access is required for successful nuclear targeting and will enhance the overall efficacy and safety.
Lastly, it is crucial to delve into the complex and diverse landscape of nucleocytoplasmic transport pathways facilitated by the numerous importin-β family members. There are multiple parallel and simultaneous pathways, and it is being revealed that there are yet extensive NLS-cargoes to be discovered.159 Yet, the significance remains less well understood for both fundamental nuclear import and NLS-therapeutic design applications. High throughput methods can shed light on the redundancies and functional relationships between importin-β receptors, the biological process, the diseased cell state, and the nuclear import efficiency and therapeutic efficacy.135 Understanding the interplay between import receptors and their NLS-therapeutic cargo will be pivotal in developing improved nuclear targeting strategies and expanding our knowledge of nuclear import mechanisms, ultimately advancing therapeutic interventions in various disease contexts.
10. Concluding remarks
In summary, the field of NLS-therapeutics holds great potential for revolutionizing disease treatment through targeted and efficient delivery of therapeutic payloads to the nucleus of diseased cells. The comprehensive understanding of classical nuclear transport mechanisms, including the role of the diverse importin repertoire and wide range of accompanying NLS sequences, has paved the way for the development of innovative strategies and delivery platforms. Leading the charge are NP-based platforms, but biological agents such as antibodies as delivery vehicles are likewise advancing rapidly. As the field progresses, it is important to continue exploring the role of alternative bipartite NLS sequences based on diseases where specific importin-α isoforms are overexpressed, minor groove-specific monopartite NLSs, and importin-β family member-specific NLSs offer new possibilities for enhancing the construction and design of NLS-therapeutics. These different NLSs may prove better than the current SV40-based NLS, which dominates NLS-therapeutic design. This review has shed light on future directions for potential advancements in the field of NLS-therapeutics. Therefore, we believe that near-future research advancements will successfully address the current challenges and pave the way for a new era in precision medicine that will significantly impact patient outcomes.Author contributions
R. G. and A. G. performed literature reviews and wrote Section 7. O. B. performed literature review and wrote Sections 6 and 8. G. C. generated figures in Pymol. D. P. performed literature review for Section 6. V. R. and J. V. L. conceived of the topic, strategized on content and information organization, and edited the manuscript.Conflicts of interest
J. V. L. is listed on a patent for the technology described in ref. 132 and 134 and holds stock in the company Defence Therapeutics Inc. that commercially owns the rights to the technology.Acknowledgements
Relevant work from our laboratories described in this article was supported by the following agencies, and which is gratefully acknowledged: Canadian Institutes of Health Research (JVL) and National Institutes of Health (EB022641, DK121351, VMR).References
- https://www.cancer.gov/about-cancer/treatment/drugs, (accessed November 20, 2022).
- M. A. Bjornsti and S. H. Kaufmann, F1000Res, 2019, 8, 1–18 Search PubMed.
- M. Canavese, L. Santo and N. Raje, Cancer Biol. Ther., 2012, 13, 451–457 CrossRef CAS PubMed.
- R. D. Martin, T. E. Hebert and J. C. Tanny, Int. J. Mol. Sci., 2020, 21, 3354 CrossRef CAS PubMed.
- G. Blobel, ChemBioChem, 2000, 1, 86–102 CrossRef CAS PubMed.
- X. Fu, Y. Shi, T. Qi, S. Qiu, Y. Huang, X. Zhao, Q. Sun and G. Lin, Signal Transduction Targeted Ther., 2020, 5, 262 CrossRef PubMed.
- E. V. Munsell, N. L. Ross and M. O. Sullivan, Curr. Pharm. Des., 2016, 22, 1227–1244 CrossRef CAS PubMed.
- M. Sui, W. Liu and Y. Shen, J. Controlled Release, 2011, 155, 227–236 CrossRef CAS PubMed.
- X. Wang, Y. Qiu, M. Wang, C. Zhang, T. Zhang, H. Zhou, W. Zhao, W. Zhao, G. Xia and R. Shao, Int. J. Nanomed., 2020, 15, 9447–9467 CrossRef CAS PubMed.
- K. Han, W.-Y. Zhang, J. Zhang, Q. Lei, S.-B. Wang, J.-W. Liu, X.-Z. Zhang and H.-Y. Han, Adv. Funct. Mater., 2016, 26, 4351–4361 CrossRef CAS.
- B. M. Bavelaar, B. Q. Lee, M. R. Gill, N. Falzone and K. A. Vallis, Front. Pharmacol., 2018, 9, 996 CrossRef PubMed.
- M. D. Shin, S. Shukla, Y. H. Chung, V. Beiss, S. K. Chan, O. A. Ortega-Rivera, D. M. Wirth, A. Chen, M. Sack, J. K. Pokorski and N. F. Steinmetz, Nat. Nanotechnol., 2020, 15, 646–655 CrossRef CAS PubMed.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29 DOI:10.1002/adma.201700996.
- E. Grossman, O. Medalia and M. Zwerger, Annu. Rev. Biophys., 2012, 41, 557–584 CrossRef CAS PubMed.
- A. G. Floch, B. Palancade and V. Doye, Methods Cell Biol., 2014, 122, 1–40 CAS.
- S. K. Vasu and D. J. Forbes, Curr. Opin. Cell Biol., 2001, 13, 363–375 CrossRef CAS PubMed.
- G. Paci, J. Caria and E. A. Lemke, J. Cell Sci., 2021, 134, jcs247874 CrossRef CAS PubMed.
- S. Petrovic, G. W. Mobbs, C. J. Bley, S. Nie, A. Patke and A. Hoelz, Cold Spring Harbor Perspect. Biol., 2022, 14, a041264 CrossRef CAS PubMed.
- F. Melchior, B. Paschal, J. Evans and L. Gerace, J. Cell Biol., 1993, 123, 1649–1659 CrossRef CAS PubMed.
- M. S. Moore and G. Blobel, Nature, 1993, 365, 661–663 CrossRef CAS PubMed.
- D. Gorlich, N. Pante, U. Kutay, U. Aebi and F. R. Bischoff, EMBO J., 1996, 15, 5584–5594 CrossRef CAS PubMed.
- E. Izaurralde, U. Kutay, C. von Kobbe, I. W. Mattaj and D. Gorlich, EMBO J., 1997, 16, 6535–6547 CrossRef CAS PubMed.
- F. R. Bischoff, C. Klebe, J. Kretschmer, A. Wittinghofer and H. Ponstingl, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2587–2591 CrossRef CAS PubMed.
- C. Klebe, F. R. Bischoff, H. Ponstingl and A. Wittinghofer, Biochemistry, 1995, 34, 639–647 CrossRef CAS PubMed.
- J. G. Turner, J. Dawson and D. M. Sullivan, Biochem. Pharmacol., 2012, 83, 1021–1032 CrossRef CAS PubMed.
- T. Williams, L. H. Ngo and V. O. Wickramasinghe, Semin. Cell Dev. Biol., 2018, 75, 70–77 CrossRef CAS PubMed.
- G. Cingolani, C. Petosa, K. Weis and C. W. Muller, Nature, 1999, 399, 221–229 CrossRef CAS PubMed.
- B. Kobe, Nat. Struct. Biol., 1999, 6, 388–397 CrossRef CAS PubMed.
- M. T. Harreman, P. E. Cohen, M. R. Hodel, G. J. Truscott, A. H. Corbett and A. E. Hodel, J. Biol. Chem., 2003, 278, 21361–21369 CrossRef CAS PubMed.
- M. T. Harreman, M. R. Hodel, P. Fanara, A. E. Hodel and A. H. Corbett, J. Biol. Chem., 2003, 278, 5854–5863 CrossRef CAS PubMed.
- J. Nardozzi, N. Wenta, N. Yasuhara, U. Vinkemeier and G. Cingolani, J. Mol. Biol., 2010, 402, 83–100 CrossRef CAS PubMed.
- C. E. Wing, H. Y. J. Fung and Y. M. Chook, Nat. Rev. Mol. Cell Biol., 2022, 23, 307–328 CrossRef CAS PubMed.
- D. Kalderon, W. D. Richardson, A. F. Markham and A. E. Smith, Nature, 1984, 311, 33–38 CrossRef CAS PubMed.
- M. R. Fontes, T. Teh and B. Kobe, J. Mol. Biol., 2000, 297, 1183–1194 CrossRef CAS PubMed.
- W. H. Colledge, W. D. Richardson, M. D. Edge and A. E. Smith, Mol. Cell. Biol., 1986, 6, 4136–4139 CAS.
- D. Kalderon, B. L. Roberts, W. D. Richardson and A. E. Smith, Cell, 1984, 39, 499–509 CrossRef CAS PubMed.
- R. E. Lanford and J. S. Butel, Cell, 1984, 37, 801–813 CrossRef CAS PubMed.
- S. Kosugi, M. Hasebe, N. Matsumura, H. Takashima, E. Miyamoto-Sato, M. Tomita and H. Yanagawa, J. Biol. Chem., 2009, 284, 478–485 CrossRef CAS PubMed.
- C. W. Chang, R. M. Counago, S. J. Williams, M. Boden and B. Kobe, Traffic, 2013, 14, 1144–1154 CrossRef CAS PubMed.
- R. Nair, P. Carter and B. Rost, Nucleic Acids Res., 2003, 31, 397–399 CrossRef CAS PubMed.
- R. A. Pumroy, S. Ke, D. J. Hart, U. Zachariae and G. Cingolani, Structure, 2015, 23, 374–384 CrossRef CAS PubMed.
- R. S. Sankhala, R. K. Lokareddy, S. Begum, R. A. Pumroy, R. E. Gillilan and G. Cingolani, Nat. Commun., 2017, 8, 979 CrossRef PubMed.
- C. Dingwall, J. Robbins, S. M. Dilworth, B. Roberts and W. D. Richardson, J. Cell Biol., 1988, 107, 841–849 CrossRef CAS PubMed.
- T. S. Munasinghe, M. R. Edwards, S. Tsimbalyuk, O. A. Vogel, K. M. Smith, M. Stewart, J. K. Foster, L. A. Bosence, D. Aragao, J. A. Roby, C. F. Basler and J. K. Forwood, Nat. Commun., 2022, 13, 1604 CrossRef CAS PubMed.
- I. R. Vetter, A. Arndt, U. Kutay, D. Gorlich and A. Wittinghofer, Cell, 1999, 97, 635–646 CrossRef CAS PubMed.
- S. H. Yoshimura and T. Hirano, J. Cell Sci., 2016, 129, 3963–3970 CAS.
- N. Mosammaparast and L. F. Pemberton, Trends Cell Biol., 2004, 14, 547–556 CrossRef CAS PubMed.
- P. Barraud, S. Banerjee, W. I. Mohamed, M. F. Jantsch and F. H. Allain, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1852–1861 CrossRef CAS PubMed.
- N. E. Bernardes and Y. M. Chook, Biochem. Soc. Trans., 2020, 48, 2753–2767 CrossRef CAS PubMed.
- K. A. Dean, O. von Ahsen, D. Gorlich and H. M. Fried, J. Cell Sci., 2001, 114, 3479–3485 CrossRef CAS PubMed.
- J. Fritz, A. Strehblow, A. Taschner, S. Schopoff, P. Pasierbek and M. F. Jantsch, Mol. Cell. Biol., 2009, 29, 1487–1497 CrossRef CAS PubMed.
- E. Izaurralde, A. Jarmolowski, C. Beisel, I. W. Mattaj, G. Dreyfuss and U. Fischer, J. Cell Biol., 1997, 137, 27–35 CrossRef CAS PubMed.
- M. Arnold, A. Nath, J. Hauber and R. H. Kehlenbach, J. Biol. Chem., 2006, 281, 20883–20890 CrossRef CAS PubMed.
- B. J. Lee, A. E. Cansizoglu, K. E. Suel, T. H. Louis, Z. Zhang and Y. M. Chook, Cell, 2006, 126, 543–558 CrossRef CAS PubMed.
- S. Jakel, W. Albig, U. Kutay, F. R. Bischoff, K. Schwamborn, D. Doenecke and D. Gorlich, EMBO J., 1999, 18, 2411–2423 CrossRef CAS PubMed.
- N. Ivic, M. Potocnjak, V. Solis-Mezarino, F. Herzog, S. Bilokapic and M. Halic, Mol. Cell, 2019, 73, 1191–1203e1196 CrossRef CAS PubMed.
- M. Baake, M. Bauerle, D. Doenecke and W. Albig, Eur. J. Cell Biol., 2001, 80, 669–677 CrossRef CAS PubMed.
- M. Baake, D. Doenecke and W. Albig, J. Cell. Biochem., 2001, 81, 333–346 CrossRef CAS PubMed.
- E. Dahl, G. Kristiansen, K. Gottlob, I. Klaman, E. Ebner, B. Hinzmann, K. Hermann, C. Pilarsky, M. Durst, M. Klinkhammer-Schalke, H. Blaszyk, R. Knuechel, A. Hartmann, A. Rosenthal and P. J. Wild, Clin. Cancer Res., 2006, 12, 3950–3960 CrossRef CAS PubMed.
- C. Shi, L. Sun, S. Liu, E. Zhang and Y. Song, Med. Sci. Monit., 2020, 26, e921087 CAS.
- H. G. Lee, M. Ueda, Y. Miyamoto, Y. Yoneda, G. Perry, M. A. Smith and X. Zhu, Brain Res., 2006, 1124, 1–4 CrossRef CAS PubMed.
- L. Huang, H. Y. Wang, J. D. Li, J. H. Wang, Y. Zhou, R. Z. Luo, J. P. Yun, Y. Zhang, W. H. Jia and M. Zheng, Cell Death Dis., 2013, 4, e745 CrossRef CAS PubMed.
- M. Zheng, L. Tang, L. Huang, H. Ding, W. T. Liao, M. S. Zeng and H. Y. Wang, Obstet. Gynecol., 2010, 116, 884–891 CrossRef CAS PubMed.
- A. Mortezavi, T. Hermanns, H. H. Seifert, M. K. Baumgartner, M. Provenzano, T. Sulser, M. Burger, M. Montani, K. Ikenberg, F. Hofstadter, A. Hartmann, R. Jaggi, H. Moch, G. Kristiansen and P. J. Wild, Clin. Cancer Res., 2011, 17, 1111–1121 CrossRef CAS PubMed.
- M. Sakai, M. Sohda, T. Miyazaki, S. Suzuki, A. Sano, N. Tanaka, T. Inose, M. Nakajima, H. Kato and H. Kuwano, Anticancer Res., 2010, 30, 851–856 Search PubMed.
- K. Yoshitake, S. Tanaka, K. Mogushi, A. Aihara, A. Murakata, S. Matsumura, Y. Mitsunori, M. Yasen, D. Ban, N. Noguchi, T. Irie, A. Kudo, N. Nakamura, H. Tanaka and S. Arii, Ann. Surg. Oncol., 2011, 18, 2093–2103 CrossRef PubMed.
- C. I. Wang, C. L. Wang, C. W. Wang, C. D. Chen, C. C. Wu, Y. Liang, Y. H. Tsai, Y. S. Chang, J. S. Yu and C. J. Yu, Int. J. Cancer, 2011, 128, 2364–2372 CrossRef CAS PubMed.
- K. Gousias, A. J. Becker, M. Simon and P. Niehusmann, J. Neuro-Oncol., 2012, 109, 545–553 CrossRef CAS PubMed.
- K. Gousias, P. Niehusmann, G. Gielen, M. Simon and J. Bostrom, J. Clin. Neurosci., 2014, 21, 1719–1724 CrossRef CAS PubMed.
- P. Roussos, P. Katsel, K. L. Davis, S. G. Giakoumaki, T. Lencz, A. K. Malhotra, L. J. Siever, P. Bitsios and V. Haroutunian, Neuropsychopharmacology, 2013, 38, 533–539 CrossRef CAS PubMed.
- A. Ahluwalia, J. Narula, M. K. Jones, X. Deng and A. S. Tarnawski, J. Physiol. Pharmacol., 2010, 61, 133–139 CAS.
- G. Bertoli, C. Cava, C. Diceglie, C. Martelli, G. Rizzo, F. Piccotti, L. Ottobrini and I. Castiglioni, Breast Cancer Res. Treat., 2017, 161, 605–616 CrossRef CAS PubMed.
- J. Yang, C. Lu, J. Wei, Y. Guo, W. Liu, L. Luo, G. Fisch and X. Li, Oncogene, 2017, 36, 2868–2878 CrossRef CAS PubMed.
- M. Hazawa, K. Sakai, A. Kobayashi, H. Yoshino, Y. Iga, Y. Iwashima, K. S. Lim, D. Chih-Cheng Voon, Y. Y. Jiang, S. I. Horike, D. C. Lin and R. W. Wong, Oncogene, 2020, 39, 2212–2223 CrossRef CAS PubMed.
- H. Bao, H. Chen, X. Zhu, M. Zhang, G. Yao, Y. Yu, W. Qin, C. Zeng, K. Zen and Z. Liu, Kidney Int., 2014, 85, 624–635 CrossRef CAS PubMed.
- K. Ben-Yaakov, S. Y. Dagan, Y. Segal-Ruder, O. Shalem, D. Vuppalanchi, D. E. Willis, D. Yudin, I. Rishal, F. Rother, M. Bader, A. Blesch, Y. Pilpel, J. L. Twiss and M. Fainzilber, EMBO J., 2012, 31, 1350–1363 CrossRef CAS PubMed.
- X. Jiao, L. D. Wood, M. Lindman, S. Jones, P. Buckhaults, K. Polyak, S. Sukumar, H. Carter, D. Kim, R. Karchin and T. Sjoblom, Genes, Chromosomes Cancer, 2012, 51, 480–489 CrossRef CAS PubMed.
- C. Mascarenhas Cdo, A. Ferreira da Cunha, A. F. Brugnerotto, S. Gambero, M. H. de Almeida, M. F. Carazzolle, K. B. Pagnano, F. Traina, F. F. Costa and C. A. de Souza, Leuk. Lymphoma, 2014, 55, 1861–1869 CrossRef PubMed.
- A. R. Paciorkowski, J. Weisenberg, J. B. Kelley, A. Spencer, E. Tuttle, D. Ghoneim, L. L. Thio, S. L. Christian, W. B. Dobyns and B. M. Paschal, Eur. J. Hum. Genet., 2014, 22, 587–593 CAS.
- E. Laurila, E. Vuorinen, K. Savinainen, H. Rauhala and A. Kallioniemi, Exp. Cell Res., 2014, 322, 159–167 CrossRef CAS PubMed.
- P. J. van der Watt, C. P. Maske, D. T. Hendricks, M. I. Parker, L. Denny, D. Govender, M. J. Birrer and V. D. Leaner, Int. J. Cancer, 2009, 124, 1829–1840 CrossRef CAS PubMed.
- J. Zhu, Y. Wang, H. Huang, Q. Yang, J. Cai, Q. Wang, X. Gu, P. Xu, S. Zhang, M. Li, H. Ding and L. Yang, Tumour Biol., 2016, 37, 661–672 CrossRef CAS PubMed.
- L. Yang, B. Hu, Y. Zhang, S. Qiang, J. Cai, W. Huang, C. Gong, T. Zhang, S. Zhang, P. Xu, X. Wu and J. Liu, Med. Oncol., 2015, 32, 128 CrossRef PubMed.
- S. He, X. Miao, Y. Wu, X. Zhu, X. Miao, H. Yin, Y. He, C. Li, Y. Liu, X. Lu, Y. Chen, Y. Wang and X. Xu, J. Cancer Res. Clin. Oncol., 2016, 142, 561–572 CrossRef CAS PubMed.
- W. Yan, R. Li, J. He, J. Du and J. Hou, Cell. Signalling, 2015, 27, 851–859 CrossRef CAS PubMed.
- J. Yang, Y. Guo, C. Lu, R. Zhang, Y. Wang, L. Luo, Y. Zhang, C. H. Chu, K. J. Wang, S. Obbad, W. Yan and X. Li, Oncogene, 2019, 38, 4700–4714 CrossRef CAS PubMed.
- B. Yang, J. Chen, X. Li, X. Zhang, L. Hu, S. Jiang, Z. Zhang and Y. Teng, Cancer Lett., 2021, 515, 14–27 CrossRef CAS PubMed.
- B. Yang, J. Chen and Y. Teng, J. Immunol. Res., 2021, 2021, 9994004 Search PubMed.
- J. Brelstaff, T. Lashley, J. L. Holton, A. J. Lees, M. N. Rossor, R. Bandopadhyay and T. Revesz, Acta Neuropathol., 2011, 122, 591–600 CrossRef CAS PubMed.
- J. Chen, Y. Hu, Y. Teng and B. Yang, Front. Cell Dev. Biol., 2021, 9, 732786 CrossRef PubMed.
- J. Szczyrba, E. Nolte, M. Hart, C. Doll, S. Wach, H. Taubert, B. Keck, E. Kremmer, R. Stohr, A. Hartmann, W. Wieland, B. Wullich and F. A. Grasser, Int. J. Cancer, 2013, 132, 775–784 CrossRef CAS PubMed.
- L. Volpon, B. Culjkovic-Kraljacic, M. J. Osborne, A. Ramteke, Q. Sun, A. Niesman, Y. M. Chook and K. L. Borden, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5263–5268 CrossRef CAS PubMed.
- J. Zhao, W. Xu, M. He, Z. Zhang, S. Zeng, C. Ma, Y. Sun and C. Xu, Oncotarget, 2016, 7, 75648–75658 CrossRef PubMed.
- R. Mout, M. Ray, T. Tay, K. Sasaki, G. Yesilbag Tonga and V. M. Rotello, ACS Nano, 2017, 11, 6416–6421 CrossRef CAS PubMed.
- S. Aghevlian, A. J. Boyle and R. M. Reilly, Adv. Drug Delivery Rev., 2017, 109, 102–118 CrossRef CAS PubMed.
- P. S. Tang, S. Sathiamoorthy, L. C. Lustig, R. Ponzielli, I. Inamoto, L. Z. Penn, J. A. Shin and W. C. Chan, Small, 2014, 10, 4182–4192 CrossRef CAS PubMed.
- R. Misra and S. K. Sahoo, Eur. J. Pharm. Sci., 2010, 39, 152–163 CrossRef CAS PubMed.
- T. R. Burglin and M. Affolter, Chromosoma, 2016, 125, 497–521 CrossRef CAS PubMed.
- A. N. Nguyen Ba, A. Pogoutse, N. Provart and A. M. Moses, BMC Bioinf., 2009, 10, 202 CrossRef PubMed.
- S. N. Tammam, H. M. Azzazy, H. G. Breitinger and A. Lamprecht, Mol. Pharmaceutics, 2015, 12, 4277–4289 CrossRef CAS PubMed.
- M. Ray, R. Tang, Z. Jiang and V. M. Rotello, Bioconjugate Chem., 2015, 26, 1004–1007 CrossRef CAS PubMed.
- C. V. Dang and W. M. Lee, Mol. Cell. Biol., 1988, 8, 4048–4054 CAS.
- S. J. Kaczmarczyk, K. Sitaraman, T. Hill, J. L. Hartley and D. K. Chatterjee, PLoS One, 2010, 5, e8889 CrossRef PubMed.
- W. Hanna-Rose and M. Han, Development, 1999, 126, 169–179 CrossRef CAS PubMed.
- K. H. Bremner, L. W. Seymour, A. Logan and M. L. Read, Bioconjugate Chem., 2004, 15, 152–161 CrossRef CAS PubMed.
- H. Siomi, H. Shida, S. H. Nam, T. Nosaka, M. Maki and M. Hatanaka, Cell, 1988, 55, 197–209 CrossRef CAS PubMed.
- D. Palmeri and M. H. Malim, Mol. Cell. Biol., 1999, 19, 1218–1225 CrossRef CAS PubMed.
- V. W. Pollard, W. M. Michael, S. Nakielny, M. C. Siomi, F. Wang and G. Dreyfuss, Cell, 1996, 86, 985–994 CrossRef CAS PubMed.
- L. J. Branden, A. J. Mohamed and C. I. Smith, Nat. Biotechnol., 1999, 17, 784–787 CrossRef CAS PubMed.
- S. S. Parelkar, R. Letteri, D. Chan-Seng, O. Zolochevska, J. Ellis, M. Figueiredo and T. Emrick, Biomacromolecules, 2014, 15, 1328–1336 CrossRef CAS PubMed.
- S. N. Tammam, H. M. E. Azzazy and A. Lamprecht, J. Controlled Release, 2017, 253, 30–36 CrossRef CAS PubMed.
- K. Chen, L. Guo, J. Zhang, Q. Chen, K. Wang, C. Li, W. Li, M. Qiao, X. Zhao, H. Hu and D. Chen, Acta Biomater., 2017, 48, 215–226 CrossRef CAS PubMed.
- S. Du, S. S. Liew, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2018, 140, 15986–15996 CrossRef CAS PubMed.
- R. Goswami, T. Jeon, H. Nagaraj, S. Zhai and V. M. Rotello, Trends Pharmacol. Sci., 2020, 41, 743–754 CrossRef CAS PubMed.
- E. Xu, W. M. Saltzman and A. S. Piotrowski-Daspit, J. Controlled Release, 2021, 335, 465–480 CrossRef CAS PubMed.
- S. M. Farkhani, A. Valizadeh, H. Karami, S. Mohammadi, N. Sohrabi and F. Badrzadeh, Peptides, 2014, 57, 78–94 CrossRef CAS PubMed.
- H. M. Leung, M. S. Chan, L. S. Liu, S. W. Wong, T. W. Lo, C. H. Lau, C. Tin and P. K. Lo, ACS Sustainable Chem. Eng., 2018, 6, 9671–9681 CrossRef CAS.
- D. T. Chao and S. J. Korsmeyer, Annu. Rev. Immunol., 1998, 16, 395–419 CrossRef CAS PubMed.
- Q. Li, Z. Hu, Y. Liang, C. Xu, Y. Hong and X. Liu, Acta Biomater., 2021, 127, 252–265 CrossRef CAS PubMed.
- L. Dominguez-Berrocal, E. Cirri, X. Zhang, L. Andrini, G. H. Marin, S. Lebel-Binay and A. Rebollo, Sci. Rep., 2019, 9, 4771 CrossRef PubMed.
- K. Nakatani, T. Maehama, M. Nishio, H. Goto, W. Kato, H. Omori, Y. Miyachi, H. Togashi, Y. Shimono and A. Suzuki, J. Biochem., 2017, 161, 237–244 CAS.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS PubMed.
- S. Y. Kim, S. J. Kim, B. J. Kim, S. Y. Rah, S. M. Chung, M. J. Im and U. H. Kim, Exp. Mol. Med., 2006, 38, 535–545 CrossRef CAS PubMed.
- A. L. B. Seynhaeve, B. M. Dicheva, S. Hoving, G. A. Koning and T. L. M. Ten Hagen, J. Controlled Release, 2013, 172, 330–340 CrossRef CAS PubMed.
- S. M. Swain, F. S. Whaley and M. S. Ewer, Cancer, 2003, 97, 2869–2879 CrossRef CAS PubMed.
- A. E. Green and P. G. Rose, Int. J. Nanomed., 2006, 1, 229–239 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- J. Yu, X. Xie, X. Xu, L. Zhang, X. Zhou, H. Yu, P. Wu, T. Wang, X. Che and Z. Hu, J. Mater. Chem. B, 2014, 2, 2114–2126 RSC.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed.
- M. Barok, H. Joensuu and J. Isola, Breast Cancer Res., 2014, 16, 209 CrossRef PubMed.
- G. Li, J. Guo, B. Q. Shen, D. B. Yadav, M. X. Sliwkowski, L. M. Crocker, J. A. Lacap and G. D. L. Phillips, Mol. Cancer Ther., 2018, 17, 1441–1453 CrossRef CAS PubMed.
- A. E. El-Kadiry, S. Beaudoin, S. Plouffe and M. Rafei, Molecules, 2022, 27, 3807 CrossRef CAS PubMed.
- V. Shivanna, Y. Kim and K. O. Chang, Virology, 2015, 483, 218–228 CrossRef CAS PubMed.
- S. Beaudoin, A. Rondeau, O. Martel, M. A. Bonin, J. E. van Lier and J. V. Leyton, Mol. Pharmaceutics, 2016, 13, 1915–1926 CrossRef CAS PubMed.
- V. Lacasse, S. Beaudoin, S. Jean and J. V. Leyton, Mol. Ther.–Methods Clin. Dev., 2020, 19, 99–119 CrossRef CAS PubMed.
- A. Ku, V. J. Facca, Z. Cai and R. M. Reilly, EJNMMI Radiopharm. Chem., 2019, 4, 27 CrossRef PubMed.
- P. Chen, R. Cameron, J. Wang, K. A. Vallis and R. M. Reilly, J. Nucl. Med., 2003, 44, 1469–1478 CAS.
- C. Gao, J. V. Leyton, A. D. Schimmer, M. Minden and R. M. Reilly, Appl. Radiat. Isot., 2016, 110, 1–7 CrossRef CAS PubMed.
- J. V. Leyton, C. Gao, B. Williams, A. Keating, M. Minden and R. M. Reilly, Leuk. Res. Rep., 2015, 4, 55–59 Search PubMed.
- J. V. Leyton, M. Hu, C. Gao, P. V. Turner, J. E. Dick, M. Minden and R. M. Reilly, J. Nucl. Med., 2011, 52, 1465–1473 CrossRef CAS PubMed.
- J. V. Leyton, B. Williams, C. Gao, A. Keating, M. Minden and R. M. Reilly, Leuk. Res., 2014, 38, 1367–1373 CrossRef CAS PubMed.
- A. Zereshkian, J. V. Leyton, Z. Cai, D. Bergstrom, M. Weinfeld and R. M. Reilly, Nucl. Med. Biol., 2014, 41, 377–383 CrossRef CAS PubMed.
- N. Misaghian, G. Ligresti, L. S. Steelman, F. E. Bertrand, J. Basecke, M. Libra, F. Nicoletti, F. Stivala, M. Milella, A. Tafuri, M. Cervello, A. M. Martelli and J. A. McCubrey, Leukemia, 2009, 23, 25–42 CrossRef CAS PubMed.
- D. Bonnet and J. E. Dick, Nat. Med., 1997, 3, 730–737 CrossRef CAS PubMed.
- M. Paquette, S. Beaudoin, M. A. Tremblay, S. Jean, A. F. Lopez, R. Lecomte, B. Guerin, M. Bentourkia, R. Sabbagh and J. V. Leyton, Bioconjugate Chem., 2018, 29, 1352–1363 CrossRef CAS PubMed.
- G. Guan, B. Song, J. Zhang, K. Chen, H. Hu, M. Wang and D. Chen, Pharmaceutics, 2019, 11, 608 CrossRef CAS PubMed.
- M. A. Saghiri, A. Asatourian, C. M. Sorenson and N. Sheibani, J. Endod., 2015, 41, 797–803 CrossRef PubMed.
- M. Adli, J. Mol. Biol., 2019, 431, 1–2 CrossRef CAS PubMed.
- J. L. Gori, P. D. Hsu, M. L. Maeder, S. Shen, G. G. Welstead and D. Bumcrot, Hum. Gene Ther., 2015, 26, 443–451 CrossRef CAS PubMed.
- L. Jubair, S. Fallaha and N. A. J. McMillan, Mol. Ther., 2019, 27, 2091–2099 CrossRef CAS PubMed.
- C. A. Lino, J. C. Harper, J. P. Carney and J. A. Timlin, Drug Delivery, 2018, 25, 1234–1257 CrossRef CAS PubMed.
- N. G. Seidah, Z. Awan, M. Chretien and M. Mbikay, Circ. Res., 2014, 114, 1022–1036 CrossRef CAS PubMed.
- L. Zhang, L. Wang, Y. Xie, P. Wang, S. Deng, A. Qin, J. Zhang, X. Yu, W. Zheng and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12404–12408 CrossRef CAS PubMed.
- Y. Hu, M. T. Haynes, Y. Wang, F. Liu and L. Huang, ACS Nano, 2013, 7, 5376–5384 CrossRef CAS PubMed.
- B. L. Rice, M. S. Stake and L. J. Parent, J. Virol., 2020, 94, e00640 CrossRef CAS PubMed.
- B. L. Timney, J. Tetenbaum-Novatt, D. S. Agate, R. Williams, W. Zhang, B. T. Chait and M. P. Rout, J. Cell Biol., 2006, 175, 579–593 CrossRef CAS PubMed.
- M. Wegener and M. Muller-McNicoll, Semin. Cell Dev. Biol., 2018, 79, 131–142 CrossRef CAS PubMed.
- Y. Miyamoto, T. Saiwaki, J. Yamashita, Y. Yasuda, I. Kotera, S. Shibata, M. Shigeta, Y. Hiraoka, T. Haraguchi and Y. Yoneda, J. Cell Biol., 2004, 165, 617–623 CrossRef CAS PubMed.
- M. Kimura, Y. Morinaka, K. Imai, S. Kose, P. Horton and N. Imamoto, eLife, 2017, 6, e21184 CrossRef PubMed.
Footnote |
| † Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |