
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of H2 occupancy modes in small and large cages of H2–tetrahydrofuran hydrates on the hydrates’ stability and H2 storage capacity†
Ruyi
Zheng
 ab,
Sohaib
Mohammed
a,
Yang
Jia
a,
Rituparna
Hazra
a and
Greeshma
Gadikota
*a
ab,
Sohaib
Mohammed
a,
Yang
Jia
a,
Rituparna
Hazra
a and
Greeshma
Gadikota
*a
aSchool of Civil and Environmental Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: gg464@cornell.edu
bFrontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an, Shaanxi 710054, China
First published on 24th February 2025
Abstract
Hydrogen storage as hydrates is one of the most environmentally benign approaches to store hydrogen as it requires only water and traces of promoters. However, the scalability of storing hydrogen via hydrate formation is hindered by the limited understanding of the structure, dynamics and energetics of hydrogen and promoters in the hydrate cages. In this study, molecular dynamics simulation configurations with different occupancy modes of H2 and tetrahydrofuran (THF) in the hydrate cages are investigated under the following scenarios: (i) two H2 molecules occupying the small cages, (ii) occupancy of H2 molecules in the THF-free large cages, and (iii) co-occupancy of H2 and THF in one large cage. Exploring these scenarios reveals the impact of occupancy modes on the dynamic motion of guest and water molecules and on the hydrate structure stability. The results show that the occupancy of two H2 molecules in the small cages reduced the stability of the hydrate structure, triggered the inter-cage hopping of H2 molecules through pentagonal faces, and increased the probability of hydrogen bond formation between THF and cage H2O molecules. The thermodynamic stability of hydrate cages is increased when the THF-free large cages are occupied by H2 molecules and the tetrahedral feature of H2 distribution in the large cages is enhanced when the number of H2 in one cage increased from two to three. When the large cages are co-occupied by H2 and THF, the inter-cage migration of H2 originating from large cages demonstrated two different features, i.e., ballistic motion (MSD ∝ t2) due to the tunneling migration behavior in the initial stage and diffusive motion in the late stage. The ballistic migration of H2 molecules is more favorable for achieving a higher hydrogen storage capacity. The decay rate of THF orientation is reduced when the interaction between THF and H2O molecules is stronger. The mechanistic insights provided by this study are crucial to advancing hydrogen storage as hydrates for a sustainable energy future.
1. Introduction
Hydrogen (H2) is a promising energy carrier with a high energy density (∼142 MJ kg−1), making it an essential component in the integrated energy system which includes balancing the seasonal fluctuation of energy demand and supply. However, safe and efficient H2 storage is challenged due to its high flammability, low density, and small molecular size. Numerous approaches have been proposed for hydrogen storage, e.g., geological storage, high-pressure-tank storage, and hydrogen storage materials including hydrate-based storage.1–7 Hydrate-based H2 storage is a less explored but transformative approach owing to: (i) the ease of recovering the stored H2 with high purities, (ii) the formation of hydrates at mild pressure and temperature, (iii) environmentally benign features of hydrates compared to other H2 storage materials, and (iv) stable trapping of H2 molecules in the hydrate cages, which reduces the risk of H2 leakage.Clathrate hydrates are ice-like crystalline solids formed by guest molecules and cages of H2O molecules that are connected through hydrogen bonds and in which the guest molecules are trapped. Depending on the guest species and thermodynamic conditions, various structures of clathrate hydrates might be formed.8 In this context, the sII structure has been extensively detected in H2-containing hydrates.9,10 A unit cell of the sII hydrate is composed of 16 small cage and 8 large cages, as shown in Fig. 1. Each small cage is made of 12 pentagonal faces and each large cage consists of 12 pentagonal and 4 hexagonal faces. The radius of the small and large cages is 3.91 and 4.73 Å respectively, which results in an average lattice parameter of a sII unit cell around 17.31 Å. Both the large and small hydrate cages can accommodate multiple H2 molecules, which makes it conducive for storing H2 in the hydrate form. It has been reported that a H2 storage capacity of 5.3 wt% is achieved for the H2 hydrates formed in pure water.11
 | ||
| Fig. 1 Schematic representation of the approach and methodology used to probe the mechanisms underlying the influence of THF on H2 hydrate formation. | ||
The formation of pure H2 hydrates requires high pressure and low temperature conditions. For instance, a temperature of 140 K is required to form H2 hydrates at the ambient pressure, while 300 MPa is needed to form pure H2 hydrates at 280 K7,11 (see Fig. 1). Therefore, it is essential to enable H2 hydrate formation under moderate conditions to make this approach economically viable. Thermodynamic promoters have been considered to enhance the formation of H2 hydrates under mild conditions as they increase the stability of hydrate structures by occupying the large cages.12–14 As indicated in Fig. 1, the addition of THF to H2 hydrates could reduce the hydrate formation pressure from 300 MPa to 5 MPa at a temperature of around 280 K.9 The presence of thermodynamic promoters greatly improves the economic viability of the hydrate-based H2 storage approach, however, at the cost of reduced storage capacity. Some representative findings about the storage capacity of H2 in pure H2 hydrates and binary H2–THF hydrates can be found in Table S1 (ESI†). Generally, the storage capacity is higher when more H2 molecules can be stably accommodated in the small and large hydrate cages. However, the conclusions on the occupancy modes of H2 in the small and large cages (e.g., single or multiple H2 occupying one hydrate cage) demonstrate great discrepancy, especially for the binary H2–THF hydrates. Some researchers suggested that a high storage capacity of H2 is achievable in the H2–THF hydrates because small cages could accommodate two H2 molecules or because multiple H2 molecules might occupy the large cages in the presence or absence of promoters. For instance, Lee and co-workers inferred from the NMR results that the small cages in H2–THF hydrates are occupied by two H2 molecules and the H2 occupancy in the large cages could be increased by reducing the concentration of THF, which resulted in a tunable H2 storage capacity of up to 4.0 wt%.15 By using ice powders and solid THF in their experiments, Sugahara and co-workers16 observed a maximum H2 storage capacity of 3.4 wt%, with the small cages occupied by one H2 and multiple occupancy of H2 molecules in the large ones. Nishikawa and co-workers detected H2 occupancy in large cages of H2–THF hydrates using in situ Raman spectra without the quenching procedure.17 In contrast, it was concluded by other researchers that the small cages in H2–THF hydrates could only accommodate one H2 molecule.18–20 For instance, by using similar experimental procedures to Lee and co-workers,15 Strobel and co-workers concluded that the storage capacity of H2 in the H2–THF hydrates increased with pressure but asymptotically approached 1.0 wt%, with one H2 in the small cage and the large cages being empty.20
To reveal the mechanisms of different occupancy modes of H2 in the small and large cages, theoretical modeling and molecular dynamics (MD) simulations are conducted to understand the influence of promoters on the storage capacity of H2. Some research findings support the multiple occupancy of H2 in the small and large cages by analyzing the variation in energy or hydrate cage volume. For example, Patchkovskii and Tse concluded that the occupancy of two H2 molecules in the small cages and four H2 in the large ones favors the stability of the H2 hydrate structure due to the increased surface contact between H2 and cage H2O molecules.21 Sebastianelli and co-workers also suggested that the strong attractive interaction between H2 and cage H2O contributed to the negative ground-state energy when two H2 molecules occupied the small cage.22 Alavi and co-workers suggested that the configuration energy did not change significantly with the occupancy modes of H2 and THF based on classical MD simulation.23 By using ab initio molecular scale simulations, Tachikawa and co-workers concluded that the small and large cages could hold up to 2 and 5 H2 molecules, respectively.24 Koh and co-workers revealed that the small cages only needed to expand by around 3% in volume to provide thermodynamically stable room for two H2 molecules.25 On the other hand, some researchers argued against the multiple occupancy of H2 in the small and large cages.26 It was suggested that the H2 molecules are tightly confined in the repulsive potential field with two H2 molecules occupying the small cages.27 Liu and co-workers also indicated that the occupancy of two H2 molecules in the small cage is less favorable and the most stable occupancy mode is the single occupancy of H2 in the small cage and single H2 + THF in the large cage.28 Kang and co-workers concluded that the small cage is occupied by one H2 molecule, whereas the large cages free of THF could accommodate two to three H2 molecules.29
Despite considerable experimental observations and simulation results, several knowledge gaps associated with the storage capacity of H2 in H2–THF hydrates remain. The three key points with respect to the storage capacity of H2 in the hydrate forms that need to be resolved are: (i) feasibility of occupancy of two H2 molecules in one small hydrate cage; (ii) whether the large cages free of THF can be occupied by H2 when the concentration of THF is lower than the stoichiometric value, i.e., 5.56 mol%; (iii) the viability of co-occupancy of THF and H2 molecules in the same large cages. Also, the influence of hydrogen bonds between THF and cage H2O molecules on hydrate stability and dynamic motion of H2 and THF molecules in the small and large cages needs refinement. In this work, the classical MD simulation approach is applied to systematically analyze the abovementioned three factors by combining the dynamic motion of guest and water molecules with the interaction energy, as illustrated in Fig. 1, which is crucial for the understanding of mechanisms underlying the storage capacity of H2 in the hydrate form. The cage occupancy modes investigated in this work are based on those either suggested based on previous experimental observations or the ones that are reported viable in simulation work and under realistic conditions. Resolving the knowledge gaps articulated above will unlock the mechanisms underlying the role of promoters in enabling H2 storage as hydrates at moderate temperatures and pressures.
2. Methodology
Eight different configurations of binary H2–THF hydrates with various occupancy modes of small and large cages are investigated in this work using MD simulations, as listed in Table 1. The occupancy modes of H2 and THF in each configuration have been reported by either experiments or simulations. HS1–THF8 configuration denotes that each of the 16 small cages in a unit cell of sII hydrates is occupied by one H2 molecule, whereas all the 8 large cages are occupied by THF molecules.9,18–20 HS2–THF8 configuration is composed with 2 H2 molecules occupying each small cage and all large cages occupied by THF.15 By comparing these two configurations, insights into the impact of various occupancy modes in the small cages on the dynamic motion and structural change are obtained. Configuration HS2–THF7–HL4 represents the occupancy modes reported by Lee and co-workers,15 with 7 large cages in a unit cell occupied by THF and the other large one occupied by four H2 molecules. Each of the small cages is occupied by two H2 molecules. In HS1–THF7, each of the small cages accommodates one H2 molecule and seven of the large cages in a unit cell are occupied by THF, which denotes the observations by Strobel and co-workers that H2 molecules could not enter the large cages even when the concentration of THF was lower than the stoichiometric value of 5.56 mol%.20 As reported by Kang and co-workers, the large cages free of THF could be occupied by 2–3 H2 molecules, which are investigated in configurations HS1–THF7–HL2 and HS1–THF7–HL3, respectively.29 HS1–(THF + HL)8 denotes the occupancy mode suggested by Kaur and Ramachandran, i.e., each small cage accommodates one H2 molecule and each large cage is co-occupied by one THF and one H2 molecule.30 Liu and co-workers reported the same occupancy mode as HS1–(THF + HL)8, but at a lower temperature condition of 140 K.28 To investigate the impact of pressure and temperature conditions, configuration HS1–(THF + HL)8–140 K was designed, with a pressure and temperature of 0.1 MPa and 140 K, respectively. The pressure and temperature conditions in other configurations are 12 MPa and 270 K, respectively.| Configurationa | Cage occupancy | Ref. | Summary/novel findings | |
|---|---|---|---|---|
| Small cages (16) | Large cages (8) | |||
| a HS, HL, and THF stand for H2 molecules in the small cages, and large cages, and occupancy of THF molecules in the large cages, respectively. b P = 0.1 MPa, T = 140 K. | ||||
| HS1–THF8 | Each by one H2 | Each by one THF | 9 and 18–20 | No inter-cage hopping of H2 molecules; hydrate structure is stable but with low storage capacity |
| HS2–THF8 | Each by two H2 | Each by one THF | 15 | Active inter-cage hopping of H2; high probability of hydrogen bond formation between THF and H2O; reduced stability of hydrate structure; slower decay of THF orientation due to stronger THF–H2O interactions |
| HS2–THF7–HL4 | Each by two H2 | 7 by THF, 1 by four H2 | 15 | |
| HS1–THF7 | Each by one H2 | 7 by THF, 1 empty | 20 | Thermodynamic stability of hydrate is enhanced by occupying three H2 molecules in the THF-free large cages |
| HS1–THF7–HL2 | Each by one H2 | 7 by THF, 1 by two H2 | 29 | |
| HS1–THF7–HL3 | Each by one H2 | 7 by THF, 1 by three H2 | 29 | |
| HS1–(THF + HL)8 | Each by one H2 | All by one THF + one H2 | 30 | The inter-cage motion of H2 molecules evolved from ballistic to diffusive regime with co-occupancy of THF and H2 in large cages |
| HS1–(THF + HL)8–140 Kb | 28 | |||
The molecular dynamics simulation package of GROMACS 2020.631 is used. According to the sensitivity analysis on the size of the simulation box, we did not detect a significant change in the pairwise energy between water and H2 molecules in the large and small cages with the number of unit cells in the simulation box increasing from 2 × 2 × 2 to 5 × 5 × 5 (see Fig. S1, ESI†). Therefore, a simulation box containing 2 × 2 × 2 unit cells of sII hydrates is adopted in this work, with dimensions of 3.462 × 3.462 × 3.462 nm. The periodic boundary condition is employed in all three directions of the simulation box. The coordinates of O and H atoms of cage H2O molecules determined by Takeuchi and co-workers32 are applied in this work as the configuration of hydrate cages. The center of mass of H2 (or H2 clusters in the cases of multiple occupancy) and promoter molecules are placed in the center of the hydrate cages. The interaction between guest and water molecules are regulated by the force fields. In this work, the TIP4P/Ice potential function33 is applied for H2O molecules, which has been reported reliable in reproducing the phase boundary conditions of gas hydrates.34 The three-site model developed by Alavi et al. is employed for H2 molecules.35 As for THF molecules, the general AMBER force field (GAFF) is adopted, with the partial charges and atom positions optimized.12 The H2O, H2, and THF molecules are regarded as rigid and constrained using the LINCS algorithm in GROMACS. The parameters of the potential functions are summarized in Table S2 in the ESI.†
A typical simulation algorithm includes an energy minimization step performed with the initial simulation configuration, followed by a 100 ps NPT ensemble on the optimized configuration to reach the equilibrium state, which is further continued for 1.0 ns in the production stage. The Nose–Hoover thermostat and Berendsen barostat are used for the temperature and pressure coupling, respectively. A time step of 0.2 fs is applied in the equilibrium and production stages. The non-bonded interactions, e.g., van der Waals force and electrostatic forces are modeled using 12-6 Lennard-Jones (LJ) and coulombic models, respectively.36
3. Results and discussions
3.1. Simulation validation
To validate the developed MD simulation models in this work, the distribution and average distance of H2 molecules in the small and large cages of pure H2 hydrates are compared with those in the literature. In the pure H2 hydrates, the small cages are occupied by two H2 and the large ones by four H2 molecules, with a simulation pressure and temperature of 0.1 MPa and 100 K respectively, which is within the phase boundary conditions.35 The initial configuration of the pure H2 hydrates in the simulation box and that at the end of simulation time are illustrated in Fig. S2 in the ESI.†The two-dimensional (2D) density maps of H2 molecule distribution in the small and large cages of pure H2 hydrates are shown in Fig. 2. The number density is truncated at 20 nm−2 to highlight the distribution feature of H2. The distribution of two H2 molecules in each small cages separated into two regions away from the cage centers, while that for the four H2 molecules accumulated in four regions, demonstrating a tetrahedral structure in three dimensions (see Fig. S3 in the ESI†). With a closer observation of the 3D distribution profile of the four H2 molecules in Fig. S3 (ESI†), it is found that the vertices of the tetrahedral structure pointed to the four hexagonal faces of the large cages, which is consistent with Burnham and co-workers.37 Also, the average distance between the two H2 molecules in the small cages was 2.5–2.6 Å and that between the four H2 molecules in the large cage is 3.0–3.1 Å, consistent with previous studies.21,27,28,37 Furthermore, the lattice parameter obtained in this work (1.726 nm) is slightly higher than 1.699 nm obtained by Alavi and co-workers35 at the same cage occupancy mode and simulation pressure–temperature conditions. This higher lattice parameter is a result of a different force field of H2O, i.e., SPC/E applied by Alavi and co – workers. The H2 distribution and hydrate cage structure results indicate reliability of the developed simulation model.
 | ||
| Fig. 2 2D density maps of H2 molecules distribution in the (a) small and (b) large cages at T = 100 K, P = 0.1 MPa. The density maps are averaged over 1.0 ns of the simulation time. | ||
3.2. Occupancy of two H2 in small cages
The impact of different occupancy modes of H2 in the small cages on the dynamic motion and distribution of H2, THF and cage water molecules, e.g., probability of hydrogen bond formation between THF–oxygen (Ot) and water–hydrogen (Hw), pair-wise interaction energy between different molecules, and stability of hydrate structure is analyzed by comparing the simulation results of various configurations in Table 1. It is noted that the impact of H2 occupancy in small cages is not significantly influenced by the occupancy modes of H2 and THF in the large cages. For instance, the findings by comparing results of configurations HS1–THF8 and HS2–THF8 are similar to those by comparing results of configurations HS1–THF7–HL3 and HS2–THF7–HL4. Therefore, the simulation results of configurations HS1–THF8 and HS2–THF8 are presented as an example to analyze the impact of the occupancy mode of H2 in the small cages. | ||
| Fig. 3 MSD curves of H2 with small cages occupied by one H2 (HS1–THF8) and two H2 molecules (HS2–THF8). | ||
 | ||
| Fig. 4 2D density maps of H2 distribution in configurations: (a) HS1–THF8 and (b) HS2–THF8. | ||
With a closer look into the evolution of the H2 migration, it is found that the migration may occur to only one of the H2 molecules occupying a single cage, as shown in Fig. 5. Fig. 5(a1), (a2) and (b1), (b2) demonstrate the cumulative positions of the first and second H2 molecules at t = 0 and t = 1.0 ns, respectively. It can be observed that the first H2 molecule is retained in the original cage (marked by the green label) throughout the simulation time of 1.0 ns. As for the second H2 molecule, its inter-cage hopping extended to 4 cages in addition to the original one, with the trajectory described by the blue arrows. It is clearly shown that this H2 molecule first migrated to a neighboring small cage and the inter-cage hopping from the original small cage to the first cage was through a pentagonal face. Then it continued migrating to two other neighboring small cages through the pentagonal faces. The fourth cage where this H2 molecule appears is the center large cage which is stably occupied by a THF molecule throughout the simulation time. Two observations are noted in this process: (i) H2 migrates from cage to cage and the migration occurs mostly in the small cages; (ii) H2 hops into the large cage occupied by the THF molecule through a pentagonal face. For pure H2 sII hydrates, it is suggested that the energy barrier for the H2 hopping through a pentagonal face was much higher than a hexagonal one.24,38,40 However, it is also observed that the H2 migrated to a neighboring cage through a pentagonal face.41 The temporary breaking of the hydrogen bonds in the pentagonal faces may facilitate inter-cage hopping of H2 molecules and some of these hydrogen bonds restored after the migration.42 Therefore, the observation of H2 migration through pentagonal faces in configuration HS2–THF8 may be attributed to the distorted cage network by the newly formed hydrogen bond between the Ot–Hw in THF and cage water molecules.
 | ||
| Fig. 5 Cumulative positions of the first (a1) and (a2) and the second (b1) and (b2) H2 molecule in one small cage of configuration HS2–THF8. | ||
 | ||
| Fig. 6 RDF curves of (a) Ow–Ow pairs in H2O molecules and (b) Ot–Hw pairs in THF and H2O molecules respectively, with different occupancy modes of H2 in the small cages. | ||
The enlarged volume of hydrate cages by the increased number of H2 in the hydrate cages also results in slightly more active translational motion of THF. Interestingly, it is found that the decay rate of THF orientation decreases with the increased occupancy of H2 in the small cages (see Fig. S4 in the ESI†). Alavi and Ripmeester47 suggested that the hydrogen bond between THF and cage H2O molecules slows down the decay of THF orientation, which is consistent with the observation in this work by comparing the probability of hydrogen bond formation as demonstrated in Fig. 6(b). The probability of hydrogen bond formation between THF and cage H2O molecules is greatly elevated when the small cages are occupied by two H2 molecules, as indicated by the larger area below the RDF curves of O–H pairs around a distance of 0.21 nm. However, this relationship between THF–H2O hydrogen bonding and the decay of THF orientation is not true when comparing configurations HS1–(THF + HL)8 and HS1–(THF + HL)8–140 K, which will be further discussed in Section 3.4.
The long-range electrostatic and short-range LJ interactions between water (W), H2 in the small cages (HS), and THF molecules for in configurations HS1–THF8 and HS2–THF8 is also compared. It is noted that the pair-wise interaction energy (i.e., sum of long-range and short-range interaction energies) is averaged by dividing with the number of molecules for convenient comparison. From the energetics perspective, it is seen that the strongest interaction is between water molecules due to the hydrogen bonding, followed by the interaction between THF and cage water molecules owing to the large size of THF and temporary hydrogen bonds between THF and water molecules. The pair-wise interaction energy between water, H2, and THF with different occupancy modes in the small cages are compared in Fig. 7. It is seen that the pair-wise interaction energy of W–HS, HS–THF, and THF–THF are negative due to the overall attractive forces. The absolute value of W–HS and THF–THF interaction energy decreased with the occupancy ratio of H2 in the small cages due to the expansion of hydrate cages. The absolute value of HS–THF interaction energy is higher because the two H2 molecules in one small cage are distributed closer to cage edge as shown in Fig. 4(b), which shortened the distance between H2 and THF. It is noticeable that the interaction energy between the H2 molecules turned from negative to positive when the occupancy of H2 in the small cage increased from one to two. As discussed in Section 3.2, this repulsive force between H2 molecules facilitated the inter-cage hopping of H2 molecules.
 | ||
| Fig. 7 Pair-wise interaction energy between water (W), H2 in the small cages (HS), and THF in configurations HS1–THF8 and HS2–THF8. | ||
3.3. Occupancy of H2 in THF-free large cages
Configurations HS1–THF7, HS1–THF7–HL2, and HS1–THF7–HL3 are compared to investigate the impact of H2 occupancy in the large cage free of THF. It is found that the number of H2 molecules in the large cage has no noticeable influence on the probability of hydrogen bond formation between THF and cage water molecules. The RDF curves of Ow–Ow pairs in cage H2O molecules are demonstrated in Fig. 8(a). It is seen that the tetrahedral structure of the water cage is slightly enhanced with more H2 occupying the THF-free large cages, which is consistent with the slightly less active translational motion of H2O molecules (see Fig. S5 in the ESI†). Therefore, the occupancy of three H2 molecules in the THF-free large cages results in a slightly more stable hydrate structure. The occupancy modes of H2 in the THF-free large cages influence the distribution of H2 molecules, as shown in Fig. 8(b). It is seen that the H2 molecules became more orderly distributed in the large cages with the number of H2 molecules increased from two to three. When observing the 2D density maps of H2 distribution in these two configurations, it is found that the tetrahedral feature of H2 in the large cages becomes more pronounced when the number of H2 molecules is increased from two to three, which may suggest that the tetrahedral distribution of H2 in the large cages is favorable for the stability of the hydrate structure. | ||
| Fig. 8 RDF of (a) Ow–Ow pairs in cage H2O molecules and (b) H–Ow pairs of H2 and cage H2O with various occupancy modes of H2 in the THF-free large cages. | ||
It is interesting to note that even though the occupancy of three H2 molecules in the THF-free large cages contributes to the thermodynamic stability of the hydrate structure, the tunability of H2 storage in the THF-free large cages is not always captured in the experiments.20 This observation may be because of the high energy barrier to kinetically enclathrate H2 molecules into the large cages. It has been found that the occupancy of H2 in the THF-free large cages is feasible when ice powders16 are used to form the H2–THF hydrates or a gas exchange approach is applied to form H2 hydrates, e.g., enclathrating H2 into hydrate cages by exchanging with N2.48,49 The potential mechanisms for these methods of enhancing the storage capacity of H2 in the large cages may be due to the reduced energy barrier induced by the pre-existence of cavities in the ice powders and hydrate cages. Therefore, it is important to develop new promoters and techniques to overcome the kinetic barriers for H2 molecules enclathrating in the promoter-free large cages, which is essential to enhance the storage capacity of hydrate-based hydrogen storage technology.
3.4. Co-occupancy of H2 and THF in one large cage
It is seen in Fig. 5 that the probability of co-occupancy of H2 and THF in one large cage is low based on the short time the H2 molecule is retained in the center large cage, although co-occupancy of THF and small gas molecules, e.g., H2 and He in the same large cages has been suggested viable according to the ab initio or grand canonical Monte Carlo simulations.30,50 In this section, configurations HS1–(THF + HL)8 and HS1–(THF + HL)8–140 K are applied to explore the feasibility of co-occupancy of H2 and THF in one large cage by observing its impact on the dynamic motion of guest and cage water molecules and the hydrate cage stability.The 2D density maps of H2 molecules originally occupying the small and large cages are demonstrated in Fig. 9. By comparing with the typical 2D density maps of one or two H2 molecules in small cages in Fig. 4, the 2D density maps of H2 distribution in Fig. 9(a) suggested that some of the small cages are occupied by two H2 molecules during the simulation. Given that all the small cages are originally occupied by one H2 molecule, the extra one H2 molecule migrates from the large cages, which can be confirmed in the 2D density maps of H2 molecules originated from the large cages in Fig. 9(b). In Fig. 9(b), it is seen that the H2 molecules originated from the large cages not only showed up in the small cages throughout the simulation time, but also demonstrated a bar-like feature in the density map. With a closer observation of one H2 molecule initially co-occupying a large cage with THF, it turns out this bar-like distribution of the H2 molecule is a result of the tunneling behavior of H2 through the hexagonal faces of the large cages, as shown in Fig. 10. The example in Fig. 10 demonstrates that this H2 molecule goes back and forth through one hexagonal face in the first 0.7 ns and then moves to another hexagonal face and continues the tunneling behavior to the end of simulation.
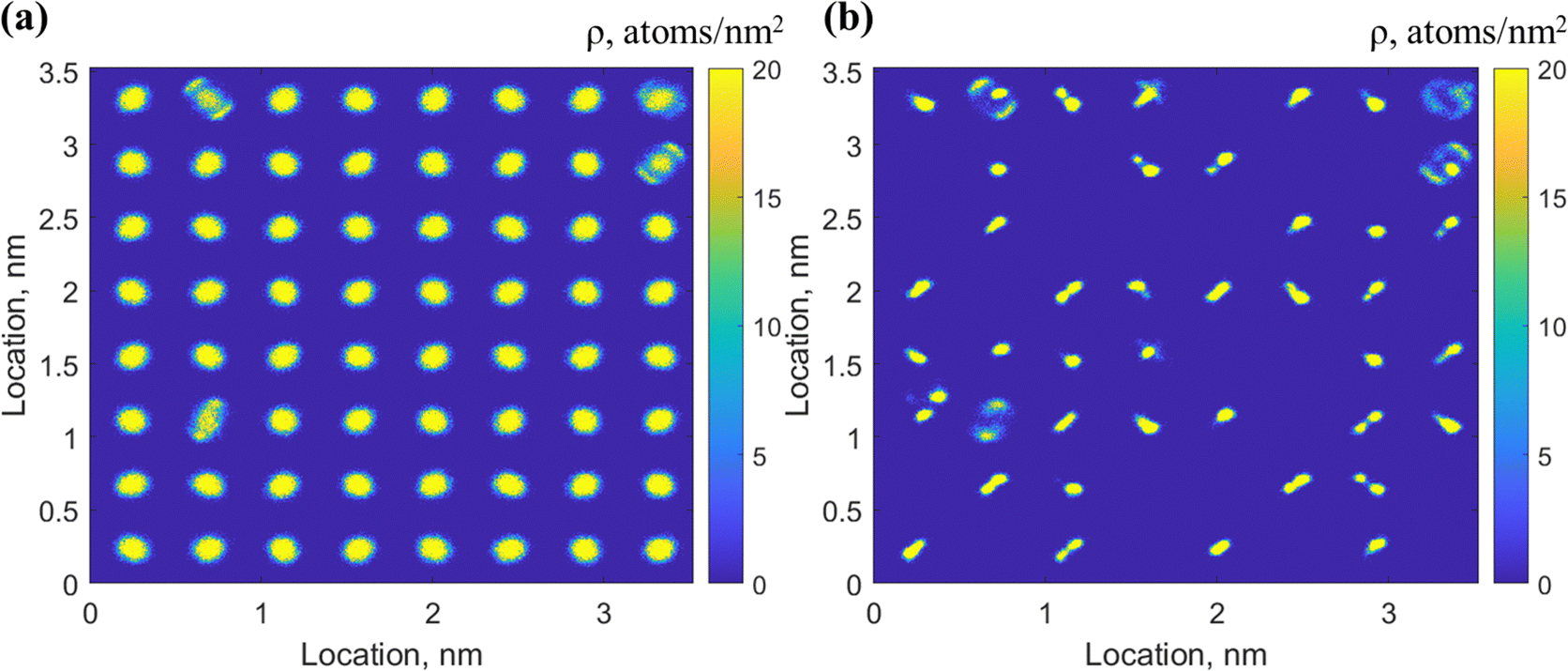 | ||
| Fig. 9 2D density maps of H2 distribution originally seated in the (a) small and (b) large cages in configuration HS1–(THF + HL)8. | ||
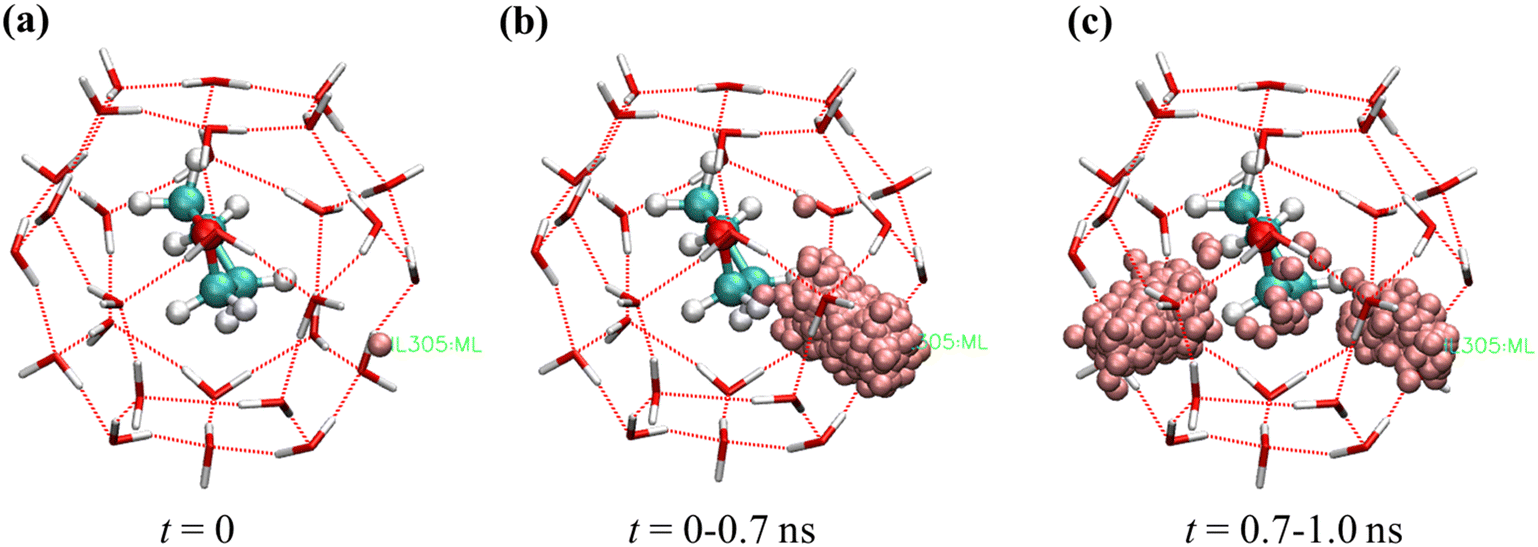 | ||
| Fig. 10 Evolution of the distribution of one H2 molecule originated from a large cage in configuration HS1–(THF + HL)8 (a) in the beginning of the simulation, (b) from 0–0.7 ns, (c) from 0.7–1.0 ns. | ||
The impact of pressure and temperature conditions is explored using configuration HS1–(THF + HL)8–140K, where the temperature and pressure are 140 K and 0.1 MPa, respectively. From the RDF curves of Ow–Ow pairs in cage H2O molecules in Fig. 11, it is seen that the hydrate cage structure is more stable at P = 0.1 MPa and T = 140 K compared with that at P = 12 MPa and T = 270 K. The peaks in the RDF curve not only became sharper, which indicates more orderly distribution of water molecules in the hydrate cages, they also shifted slightly to the left due to the shrinkage of hydrate cages at lower temperature, which is consistent with the smaller lattice parameter (by 1.04%) at the lower temperature conditions.
 | ||
| Fig. 11 RDF curves of Ow–Ow pairs in cage H2O molecules with co-occupancy of THF and H2 in large cages at different pressure–temperature conditions (12 MPa and 270 K for the black curve; 0.1 MPa and 140 K for the red curve). | ||
The impact of the lower temperature and lower pressure condition on the dynamic motion of H2 molecules initially co-occupying the large cages with THF can be found in Fig. 12. The MSD curves in Fig. 12(a) shows that the motion of H2 molecules is greatly reduced when the temperature is decreased to 140 K. However, different from the MSD curves of H2 in configuration HS2–THF8 (see Fig. 3, where the MSD increased linearly, indicating a diffusive motion), the MSD curves in Fig. 12 suggest a ballistic motion51,52 of H2 molecules (MSD ∝ t2), which is more pronounced for configuration HS1–(THF + HL)8–140 K. By observing the 2D density map of these H2 molecules in Fig. 12(b), it is seen that the migration of H2 molecules is featured with mostly tunneling behavior, which was also through the hexagonal faces as shown in Fig. S6 in the ESI.† Given the fact that the ballistic motion feature is more noticeable in configuration HS1–(THF + HL)8–140 K (see Fig. 12(a)) and that the tunneling behavior is more remarkable in the migration of these H2 molecules, it is indicated that the tunneling motion of H2 molecules originally co-occupying the large cages with THF leads to the ballistic behavior in the MSD curves.
 | ||
| Fig. 12 MSD curves of H2 molecules originally co-occupying the large cages with THF (a) and 2D density map of H2 distribution in configuration HS1–(THF + HL)8–140 K (b). | ||
To further demonstrate the relationship between different H2 inter-cage migration features (i.e., diffusion and tunneling) and features in the MSD curves (i.e., MSD ∝ t and MSD ∝ t2), the simulation time of configuration HS1–(THF + HL)8 is prolonged to 50 ns, with the MSD curves and the 2D density maps of H2 molecule distribution illustrated in Fig. 13. It is shown that the MSD ∝ t2 relationship is captured in the initial stage, ending up with a linear relationship. By observing the 2D density maps of H2 molecules at two different stages, i.e., in the first and last 2 ns, it is found that the probability of tunneling motion of H2 molecules is greatly reduced in the last 2 ns (see Fig. 13(b)), which corresponds to the MSD ∝ t feature in the MSD curve. Therefore, it is concluded that the MSD ∝ t2 feature in the MSD curve of H2 molecules results from the tunneling motion of H2 molecules through the hexagonal faces, whereas the inter-cage hopping of H2 molecules through diffusion leads to a linear feature in the MSD curves, which also applies to inter-cage hopping of H2 molecules initially occupying the small cages (e.g., configuration HS2–THF8). Also, the probability of hydrogen bond formation between THF and H2O molecules is low when the ballistic motion dominates as shown in Fig. 14(b), which means that the hydrate structure is less likely distorted in the ballistic regime. In contrast, when the hydrate structure is less stable, the ballistic motion of H2 molecules in the initial stage will be gradually replaced by the diffusive motion. These results may indicate that the stable co-occupancy of H2 and THF in one large cage is possible, accompanied by the ballistic motion of H2 molecules. In other words, the ballistic motion is favorable for a higher storage capacity of hydrogen in the hydrate form, due to the possible co-occupancy of H2 and THF in one hydrate cage. However, it requires a more stable hydrate structure, i.e., at higher pressure or lower temperature conditions.
 | ||
| Fig. 13 MSD curve of H2 molecules originally co-occupying the large cages with THF and 2D density maps of H2 distribution in the first 2 ns (a) and last 2 ns (b) with the simulation time of configuration HS1–(THF + HL)8 prolonged to 50 ns. | ||
 | ||
| Fig. 14 RACF curve of (a) THF and (b) RDF of O–H pairs in THF and H2O molecules. | ||
The impact of temperature and pressure on the decay rate of THF orientation is demonstrated in Fig. 14(a). It is seen that the decay rate is significantly reduced in configuration HS1–(THF + HL)8–140 K. As discussed in Section 3.2, it is suggested that the slower decay of THF orientation in H2–THF hydrates is due to the higher probability of hydrogen bond formation between THF and cage water molecules.42 However, it is observed in Fig. 14(b) that the probability of hydrogen bond formation in configuration HS1–(THF + HL)8–140 K is greatly reduced. Therefore, the relationship between stronger hydrogen bonds of THF–H2O and smaller decay rate of THF orientation is not valid. To explore the cause of slower decay of THF orientation, the pair-wise interaction energy between THF and cage water molecules in different configurations is compared. It is found that the decay rate of THF orientation is regulated by the interaction energy between THF and H2O molecules in general. For instance, the pair-wise interaction energy of THF–H2O is −54.95 kJ mol−1 and −57.52 kJ mol−1 respectively for configurations HS1–(THF + HL)8 and HS1–(THF + HL)8–140 K. The higher interaction force between THF and H2O arises from the short-range LJ interaction, which may be attributed to the shorter distance between THF and H2O with the shrunk cages. Another example is that the pair-wise interaction energy of THF–H2O for configurations HS1–THF8 and HS2–THF8 is −55.45 kJ mol−1 and −56.37 kJ mol−1, respectively. This stronger interaction between THF and H2O results in slower decay of the THF orientation (see Fig. S4, ESI†).
4. Conclusions
The influence of different occupancy modes of H2 in the small and large cages on the dynamic motion of gas and water molecules and hydrate structure stability to inform the storage capacity of H2 in H2–THF hydrate, is elucidated using classical molecular dynamics (MD) simulations. Eight configurations with occupancy modes determined or suggested by previous experiments and simulation research are used to investigate the impact of (i) occupancy of two H2 molecules in the small cages, (ii) occupancy of H2 molecules in the THF-free large cages, and (iii) co-occupancy of H2 and THF in one large cage. The following conclusions are drawn based on the configurations designed to address key knowledge gaps in hydrogen storage as hydrates:(1) Occupancy of two H2 molecules in small cages: the presence of two H2 molecules in the small cages induced strong repulsive forces between the two H2 molecules within a confined space, leading to active inter-cage hopping. The inter-cage migration occurred predominantly among small cages via pentagonal faces, with occasional migration into THF-occupied large cages. This phenomenon indicates that the distortion of the hydrate framework due to THF–water hydrogen bonding reduces the energy barrier for H2 migration through pentagonal faces. Structurally, the occupancy of two H2 molecules caused a 1.03% increase in lattice parameter, corresponding to a 3% expansion in cage volume. This expansion weakened the stability of the hydrate framework, as reflected by broader RDF peaks and disrupted hydrogen bonding among water molecules.
(2) Occupancy of H2 molecules in THF-free large cages: the occupancy of two or three H2 molecules in large cages free of THF resulted in enhanced tetrahedral arrangements of H2 molecules, particularly when three H2 molecules were present. The tetrahedral distribution is energetically favorable and contributes to slightly improved hydrate stability. The findings suggest that higher occupancy in THF-free large cages can stabilize the hydrate framework, albeit the experimental realization of such configurations may face kinetic challenges. Approaches such as using ice powders or gas exchange methods could overcome these barriers by reducing the energy required for enclathration.
(3) Co-occupancy of H2 and THF in large cages: the inter-cage migration of H2 molecules when co-occupying large cages with THF demonstrated two motion features, i.e., ballistic motion (MSD ∝ t2) due to the tunneling behavior in the initial stage and diffusive motion (MSD ∝ t) in the late stage. Prolonged simulations confirmed that the ballistic regime is associated with greater hydrate stability due to reduced hydrogen bond distortion. The probability of hydrogen bond formation between THF and H2O is low when the tunneling migration of H2 molecules is dominant in the inter-cage hopping, which may indicate that the tunneling migration behavior is enabled by a more stable hydrate structure, and is more favorable for achieving a higher storage capacity of H2 hydrates due to the co-occupancy mode. The decay rate of THF orientation is regulated by the interaction energy between THF and cage water molecules. A stronger interaction between THF and H2O molecules leads to slower decay of THF orientation.
By systematically analyzing the dynamic motion of H2 and THF molecules and their interactions with the hydrate framework, this study bridges longstanding knowledge gaps in understanding hydrogen storage capacity and hydrate stability. The insights into the effects of occupancy modes on molecular dynamics and structural integrity provide valuable guidance for optimizing hydrate-based hydrogen storage systems under moderate temperature and pressure conditions, paving the way for advancing sustainable hydrogen storage solutions that align with the goals of a sustainable energy future.
Author contributions
Ruyi Zheng: writing – original draft, concept, data curation, formal analysis, investigation, methodology, validation; Sohaib Mohammed: writing – review & editing, formal analysis, methodology, validation; Yang Jia: writing – editing, validation; Rituparna Hazra: writing – review & editing, methodology; Greeshma Gadikota: writing – concept, review & editing, supervision, conceptualization, resources, formal analysis.Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Conflicts of interest
G. G. is the co-founder of Carbon To Stone, a company focused on commercializing technologies for industrial carbon management.Acknowledgements
This work was supported as part of the Multi-Scale Fluid-Solid Interactions in Architected and Natural Materials (MUSE), an Energy Frontier Research Center funded by the U. S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0019285.References
- A. Gupta, G. V. Baron, P. Perreault, S. Lenaerts, R. G. Ciocarlan, P. Cool, P. G. Mileo, S. Rogge, V. Van Speybroeck, G. Watson and P. Van der Voort, Hydrogen clathrates: Next generation hydrogen storage materials, Energy Storage Mater., 2021, 41, 69–107 CrossRef.
- R. Zheng, T. C. Germann, L. Huang and M. Mehana, Driving mechanisms of quartz wettability alteration under in situ H2 geo-storage conditions: Role of organic ligands and surface morphology, Int. J. Hydrogen Energy, 2024, 59, 1388–1398 CrossRef CAS.
- R. Zheng, T. C. Germann, M. Gross and M. Mehana, Molecular insights into the impact of surface chemistry and pressure on quartz wettability: resolving discrepancies for hydrogen geo-storage, ACS Sustainable Chem. Eng., 2024, 12(14), 5555–5563 CrossRef CAS.
- N. Heinemann, J. Alcalde, J. M. Miocic, S. J. Hangx, J. Kallmeyer, C. Ostertag-Henning, A. Hassanpouryouzband, E. M. Thaysen, G. J. Strobel, C. Schmidt-Hattenberger and K. Edlmann, Enabling large-scale hydrogen storage in porous media–the scientific challenges, Energy Environ. Sci., 2021, 14(2), 853–864 RSC.
- R. Zheng, T. C. Germann, M. Gross and M. Mehana, Hydrogen diffusion in slit pores: Role of temperature, pressure, confinement, and roughness, Energy Fuels, 2024, 38(21), 21642–21650 CrossRef CAS.
- V. V. Struzhkin, B. Militzer, W. L. Mao, H. K. Mao and R. J. Hemley, Hydrogen storage in molecular clathrates, Chem. Rev., 2007, 107(10), 4133–4151 CrossRef CAS PubMed.
- Y. Zhang, G. Bhattacharjee, R. Kumar and P. Linga, Solidified hydrogen storage (Solid-HyStore) via clathrate hydrates, Chem. Eng. J., 2022, 431, 133702 Search PubMed.
- R. Zheng, Z. Wang, X. Li, Z. Fan and S. Negahban, Structural and dynamic analyses of CH4–C2H6–CO2 hydrates using thermodynamic modeling and molecular dynamic simulation, J. Chem. Thermodyn., 2022, 169, 106749 CrossRef CAS.
- L. J. Florusse, C. J. Peters, J. Schoonman, K. C. Hester, C. A. Koh, S. F. Dec, K. N. Marsh and E. D. Sloan, Stable low-pressure hydrogen clusters stored in a binary clathrate hydrate, Science, 2004, 306(5695), 469–471 CrossRef CAS PubMed.
- W. L. Mao, H. K. Mao, A. F. Goncharov, V. V. Struzhkin, Q. Guo, J. Hu, J. Shu, R. J. Hemley, M. Somayazulu and Y. Zhao, Hydrogen clusters in clathrate hydrate, Science, 2002, 297(5590), 2247–2249 CrossRef CAS PubMed.
- W. L. Mao and H. K. Mao, Hydrogen storage in molecular compounds, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(3), 708–710 CrossRef CAS PubMed.
- A. A. Atamas, H. M. Cuppen, M. V. Koudriachova and S. W. de Leeuw, Monte Carlo calculations of the free energy of binary sII hydrogen clathrate hydrates for identifying efficient promoter molecules, J. Phys. Chem. B, 2013, 117(4), 1155–1165 Search PubMed.
- B. Lal and O. Nashed, Chemical additives for gas hydrates, Springer Nature, 2019 Search PubMed.
- H. P. Veluswamy, R. Kumar and P. Linga, Hydrogen storage in clathrate hydrates: Current state of the art and future directions, Appl. Energy, 2014, 122, 112–132 CrossRef CAS.
- H. Lee, J. W. Lee, D. Y. Kim, J. Park, Y. T. Seo, H. Zeng, I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Tuning clathrate hydrates for hydrogen storage, Nature, 2005, 434(7034), 743–746 CrossRef CAS PubMed.
- T. Sugahara, J. C. Haag, P. S. Prasad, A. A. Warntjes, E. D. Sloan, A. K. Sum and C. A. Koh, Increasing hydrogen storage capacity using tetrahydrofuran, J. Am. Chem. Soc., 2009, 131(41), 14616–14617 CrossRef CAS PubMed.
- A. Nishikawa, T. Tanabe, K. Kitamura, Y. Matsumoto, K. Ohgaki and T. Sugahara, In situ Raman spectra of hydrogen in large cages of hydrogen+ tetrahydrofuran mixed hydrates, Chem. Eng. Sci., 2013, 101, 1–4 Search PubMed.
- S. Hashimoto, S. Murayama, T. Sugahara, H. Sato and K. Ohgaki, Thermodynamic and Raman spectroscopic studies on H2 + tetrahydrofuran + water and H2 + tetra-n-butyl ammonium bromide + water mixtures containing gas hydrates, Chem. Eng. Sci., 2006, 61(24), 7884–7888 CrossRef CAS.
- K. C. Hester, T. A. Strobel, E. D. Sloan, C. A. Koh, A. Huq and A. J. Schultz, Molecular hydrogen occupancy in binary THF − H2 clathrate hydrates by high resolution neutron diffraction, J. Phys. Chem. B, 2006, 110(29), 14024–14027 CrossRef CAS PubMed.
- T. A. Strobel, C. J. Taylor, K. C. Hester, S. F. Dec, C. A. Koh, K. T. Miller and E. D. Sloan, Molecular hydrogen storage in binary THF − H2 clathrate hydrates, J. Phys. Chem. B, 2006, 110(34), 17121–17125 CrossRef CAS PubMed.
- S. Patchkovskii and J. S. Tse, Thermodynamic stability of hydrogen clathrates, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(25), 14645–14650 CrossRef CAS PubMed.
- F. Sebastianelli, M. Xu, Y. S. Elmatad, J. W. Moskowitz and Z. Bačić, Hydrogen molecules in the small dodecahedral cage of a clathrate hydrate: quantum translation − rotation dynamics of the confined molecules, J. Phys. Chem. C, 2007, 111(6), 2497–2504 Search PubMed.
- S. Alavi, J. A. Ripmeester and D. D. Klug, Molecular-dynamics simulations of binary structure II hydrogen and tetrahydrofurane clathrates, J. Chem. Phys., 2006, 124, 014704 Search PubMed.
- H. Tachikawa, T. Iyama and H. Kawabata, Maximum capacity of the hydrogen storage in water clusters, Phys. Chem. Liq., 2009, 47(1), 103–109 Search PubMed.
- D. Y. Koh, H. Kang, J. Jeon, Y. H. Ahn, Y. Park, H. Kim and H. Lee, Tuning cage dimension in clathrate hydrates for hydrogen multiple occupancy, J. Phys. Chem. C, 2014, 118(6), 3324–3330 Search PubMed.
- Y. Krishnan, M. R. Ghaani, A. Desmedt and N. J. English, Hydrogen inter-cage hopping and cage occupancies inside hydrogen hydrate: Molecular-dynamics analysis, Appl. Sci., 2020, 11(1), 282 Search PubMed.
- J. Wang, H. Lu, J. A. Ripmeester and U. Becker, Molecular-dynamics and first-principles calculations of Raman spectra and molecular and electronic structure of hydrogen clusters in hydrogen clathrate hydrate, J. Phys. Chem. C, 2010, 114(49), 21042–21450 Search PubMed.
- J. Liu, J. Hou, J. Xu, H. Liu, G. Chen and J. Zhang, Ab initio study of the molecular hydrogen occupancy in pure H2 and binary H2 - THF clathrate hydrates, Int. J. Hydrogen Energy, 2017, 42(27), 17136–17143 Search PubMed.
- D. W. Kang, W. Lee, Y. H. Ahn and J. W. Lee, Exploring tuning phenomena of THF-H2 hydrates via molecular dynamics simulations, J. Mol. Liq., 2022, 349, 118490 CrossRef CAS.
- S. P. Kaur and C. N. Ramachandran, Hydrogen-tetrahydrofuran mixed hydrates: A computational study, Int. J. Hydrogen Energy, 2018, 43(42), 19559–19566 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1, 19–25 CrossRef.
- F. Takeuchi, M. Hiratsuka, R. Ohmura, S. Alavi, A. K. Sum and K. Yasuoka, Water proton configurations in structures I, II, and H clathrate hydrate unit cells, J. Chem. Phys., 2013, 138, 124504 CrossRef PubMed.
- J. L. Abascal, E. Sanz, R. García Fernández and C. Vega, A potential model for the study of ices and amorphous water: TIP4P/Ice, J. Chem. Phys., 2005, 122, 234511 CrossRef CAS PubMed.
- M. M. Conde and C. Vega, Determining the three-phase coexistence line in methane hydrates using computer simulations, J. Chem. Phys., 2010, 133, 064507 CrossRef CAS PubMed.
- S. Alavi, J. A. Ripmeester and D. D. Klug, Molecular-dynamics study of structure II hydrogen clathrates, J. Chem. Phys., 2005, 123, 024507 CrossRef PubMed.
- R. Zheng, X. Li and S. Negahban, Molecular-level insights into the structure stability of CH4-C2H6 hydrates, Chem. Eng. Sci., 2022, 247, 117039 CrossRef CAS.
- C. J. Burnham, Z. Futera and N. J. English, Quantum and classical inter-cage hopping of hydrogen molecules in clathrate hydrate: Temperature and cage-occupation effects, Phys. Chem. Chem. Phys., 2017, 19(1), 717–728 RSC.
- T. J. Frankcombe and G. J. Kroes, Molecular dynamics simulations of type-sII hydrogen clathrate hydrate close to equilibrium conditions, J. Phys. Chem. C, 2007, 111(35), 13044–13052 CrossRef CAS.
- H. Cao, N. J. English and J. M. MacElroy, Diffusive hydrogen inter-cage migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates, J. Chem. Phys., 2013, 138, 094507 Search PubMed.
- S. Alavi and J. Ripmeester, Hydrogen-gas migration through clathrate hydrate cages, Angew. Chem., Int. Ed., 2007, 46(32), 6102 CrossRef CAS PubMed.
- S. Y. Willow and S. S. Xantheas, Enhancement of hydrogen storage capacity in hydrate lattices, Chem. Phys. Lett., 2012, 525, 13–18 CrossRef.
- P. D. Gorman, N. J. English and J. M. MacElroy, Dynamical cage behaviour and hydrogen migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates, J. Chem. Phys., 2012, 136, 044506 CrossRef PubMed.
- S. Alavi, R. Susilo and J. A. Ripmeester, Linking microscopic guest properties to macroscopic observables in clathrate hydrates: Guest-host hydrogen bonding, J. Chem. Phys., 2009, 130, 174501 CrossRef PubMed.
- S. Alavi, K. Udachin and J. A. Ripmeester, Effect of guest–host hydrogen bonding on the structures and properties of clathrate hydrates, Chem. – Eur. J., 2010, 16(3), 1017–1025 CrossRef CAS PubMed.
- S. Alavi, K. Shin and J. A. Ripmeester, Molecular dynamics simulations of hydrogen bonding in clathrate hydrates with ammonia and methanol guest molecules, J. Chem. Eng. Data, 2015, 60(2), 389–397 CrossRef CAS.
- S. Alavi and J. A. Ripmeester, Molecular simulations of clathrate hydrates, Mol. Sci. Charact., 2022, 2, 283–367 Search PubMed.
- S. Alavi and J. A. Ripmeester, Effect of small cage guests on hydrogen bonding of tetrahydrofuran in binary structure II clathrate hydrates, J. Chem. Phys., 2012, 137, 054712 CrossRef PubMed.
- H. Lu, J. Wang, C. Liu, C. I. Ratcliffe, U. Becker, R. Kumar and J. Ripmeester, Multiple H2 occupancy of cages of clathrate hydrate under mild conditions, J. Am. Chem. Soc., 2012, 134(22), 9160–9162 Search PubMed.
- S. Park, D. Y. Koh, H. Kang, J. W. Lee and H. Lee, Effect of molecular nitrogen on multiple hydrogen occupancy in clathrate hydrates, J. Phys. Chem. C, 2014, 118(35), 20203–20208 Search PubMed.
- N. I. Papadimitriou, I. N. Tsimpanogiannis, A. K. Stubos, A. Martin, L. J. Rovetto and C. J. Peters, Unexpected behavior of helium as guest gas in sII binary hydrates, J. Phys. Chem. Lett., 2010, 1(6), 1014–1017 CrossRef CAS.
- S. Han, M. Y. Choi, P. Kumar and H. E. Stanley, Phase transitions in confined water nanofilms, Nat. Phys., 2010, 6(9), 685–689 Search PubMed.
- S. Mohammed, H. Asgar, C. J. Benmore and G. Gadikota, Structure of ice confined in silica nanopores, Phys. Chem. Chem. Phys., 2021, 23(22), 12706–12717 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04820b |
| This journal is © the Owner Societies 2025 |