
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into Ni3TeO6 calcination via in situ synchrotron X-ray diffraction†
Shubo
Wang
 *a,
Javier
Fernández-Catalá
ab,
Qifeng
Shu
c,
Marko
Huttula
a,
Wei
Cao
a and
Harishchandra
Singh
*a
*a,
Javier
Fernández-Catalá
ab,
Qifeng
Shu
c,
Marko
Huttula
a,
Wei
Cao
a and
Harishchandra
Singh
*a
aNano and Molecular Systems Research Unit, University of Oulu, Oulu, FIN-90014, Finland. E-mail: shubo.wang@oulu.fi; harishchandra.singh@oulu.fi
bMaterials Institute and Inorganic Chemistry Department, University of Alicante, Ap. 99, E-03080 Alicante, Spain
cProcess Metallurgy Research Unit, University of Oulu, FI-90014 Oulu, Finland
First published on 1st November 2024
Abstract
The versatility of metal tellurate chemistry enables the creation of unique structures with tailored properties, opening avenues for advancements in a wide range of applications. However, precise nanoengineering of Ni3TeO6, a ceramic Ni tellurate with a broad variety of properties, like electrical, magnetic, photocatalytic and multiferroic properties, demands a deep understanding of the synthesis process, which is strongly influenced by experimental parameters. This study delves into the formation mechanism of Ni3TeO6 nanoparticles during calcination of hydrothermally produced precursors, using in situ synchrotron X-ray diffraction, complemented by post-mortem TEM and XPS, and thermal analysis. The results reveal a reaction sequence involving dehydration and dehydroxylation of stoichiometric Ni/Te oxyhydroxide coordinated by Te. This oxyhydroxide can be schematically represented by a formula of (3Ni/Te)(OOH)4·H2O. Subsequently, preferential nucleation of Ni3TeO6 occurs. Further calcination after full crystallization of Ni3TeO6 leads to the formation of a different Ni tellurate (NiTeO4) phase as an impurity. These findings clarify the reactions occurring during calcination of Ni/Te mixed precursors, which have frequently been inferred from empirical and post-mortem reports but not confirmed via comprehensive and in situ guided explorations.
1. Introduction
There have been increasing interests in developing multifunctional nanomaterials due to the ability to tailor their properties through the precise control of composition, crystal structure, crystallinity, particle shape and size.1–3 To date, transition metal (M = Ni, Co, Mn, Cu) tellurates (MTOs) have emerged as highly attractive multifunctional nanomaterials. This is due to their diverse properties and broad applications, including electronics, energy, optics and catalysis.4–7 However, it has been reported frequently that a subtle variation in synthesis conditions could dramatically influence the phase purity, particle size, and crystalline structure of the resulting MTOs.4–18 This sensitivity and complexity in synthesis are likely due to the complex MTO chemistry. For example, six stable compounds of the Ni–Te–O system have already been reported so far, i.e. NiTeO3,14 NiTeO4,15 NiTe2O5,16 NiTe6O13,17 Ni2Te3O86,18 and Ni3TeO6.7–9,19 Among these, Ni3TeO6 (denoted as NTO) is the longest known one, dating back to 1967.19 NTO can be synthesized using various methods, including solid state reactions,7,8 sol–gel processes9 and hydrothermal synthesis.6 These methods typically require mixing reagents with a Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Te ratio of 3:1 by solid state and wet chemistry (sol–gel or hydrothermal) methods, followed by a calcination step.5–7,9–12
:Te ratio of 3:1 by solid state and wet chemistry (sol–gel or hydrothermal) methods, followed by a calcination step.5–7,9–12
Our previous studies demonstrated the possibility to tailor the morphology and functionality (antiferromagnetic, photoconductivity, and photocatalytic properties) of NTO using an efficient, cost-effective and versatile hydrothermal synthesis followed by calcination.20–22 This approach yielded NTO in the form of 2D nanosheets and nanoparticles (NP) by controlling the type and concentration of additives like CO(NH2)2 and NaOH during hydrothermal reactions. The overall reaction mechanism can be briefly described as follows:
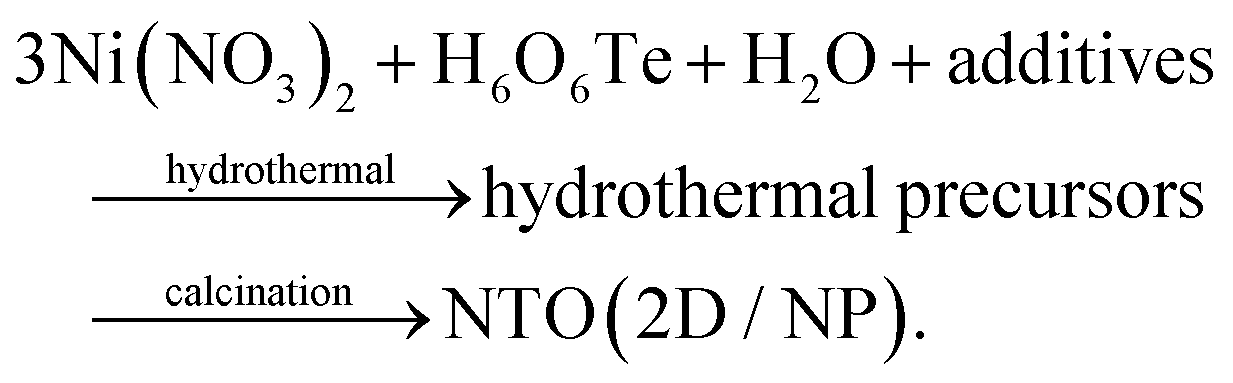
However, the details of intermediate formations remain unclear. For example, it is still unknown whether Ni3TeO6 nuclei form directly during hydrothermal synthesis or during the calcination stage, and what intermediates form between the reactant and the final product. Understanding these intermediate steps is critical for optimizing the synthesis process and achieving NTO products with enhanced functionalities.
To address this knowledge gap, we employed in situ synchrotron X-ray diffraction (SXRD) during calcination to track the phase transitions starting from the hydrothermally prepared NTO precursor. Additionally, post-mortem characterization techniques, such as transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS), and thermal analysis were also used to corroborate the findings from SXRD. These combined data provide valuable insights into the synthesis and tailoring of nanostructured Ni3TeO6, potentially applicable to other Ni tellurates as well.
2. Materials and methods
The precursor prepared by hydrothermal synthesis (designated as NTO-hydro) was synthesized using reagents, such as Ni(NO3)2·6H2O, H6O6Te and NaOH, as described previously in ref. 22. Calcination of NTO-hydro was conducted using an in situ SXRD system at 70 keV (λ = 0.1779 Å) photons at the Brockhouse high energy wiggler beamline, Canadian light sources (CLS), Canada.23 The sample was heated from room temperature at a rate of 10 °C min−1 to a calcination temperature of 600 °C and held for 2 h (Fig. 1a). The calcination parameters were chosen based on previous successful syntheses of Ni3TeO6 nanomaterials.20–22 2D diffraction data (Fig. 1b, c and Movie S1, ESI†) were collected using a transmission geometry,24,25 with an area detector positioned 1144 mm downstream of the sample. The calcinated powder was designated as NTO-600.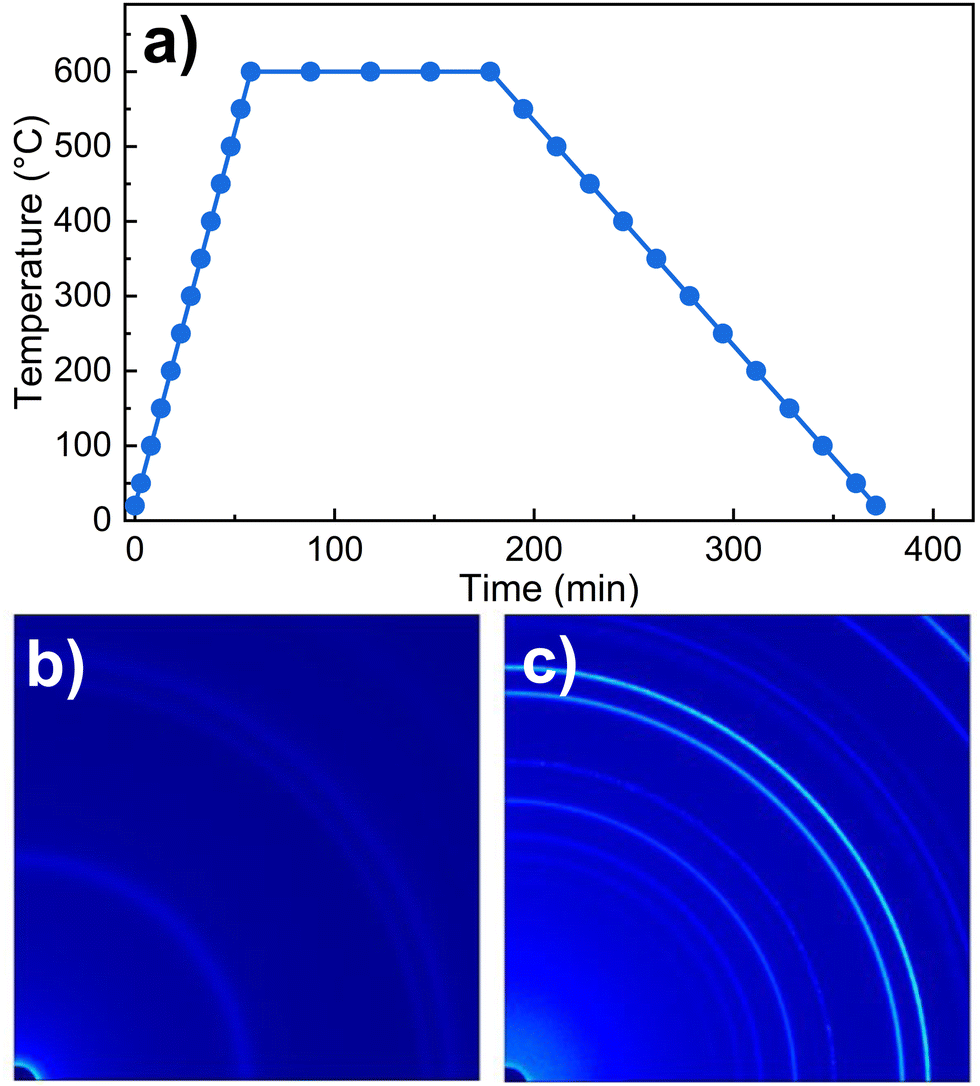 | ||
| Fig. 1 (a) A schematic temperature regime for in situ SXRD measurements, and the representative quarter of the collected 2D SXRD pattern for (b) NTO-hydro and (c) after calcination at 600 °C for 2 h, i.e. NTO-600. The circles in (a) highlight the temperatures where the SXRD patterns were collected. | ||
To complement the in situ SXRD analysis, the NTO-hydro precursor was additionally calcined in a muffle furnace at 450 °C for 5 min using the same heating rate as that used in the in situ experiment. The resulting powder was designated as NTO-450. Thermal analysis, using a STA449 F3 thermal analyzer (Netzsch-Gerätebau Gmbh) with Ar purge gas, was employed to determine the transition temperatures during calcination. The heating profiles were the same as that used in the SXRD measurements. TEM images for the NTO samples were acquired using a JEOL JEM-2200FS FETEM/STEM. XPS analyses were performed using a Thermo Fisher Scientific ESCALAB 250Xi XPS system with Avantage software for data acquisition and analysis. UV-vis spectra were collected using a Shimadzu UV-2600 spectrophotometer.
3. Results
Fig. 2a displays the color-coded SXRD profiles collected within a 2θ range of 1.8–7.5°. The SXRD pattern of NTO-hydro exhibits relatively diffuse Debye–Scherrer rings (Fig. 1b), resulting in broad peaks in the corresponding 1D profiles. This indicates a short-range order or a relatively poor crystallinity in the material. Indexing of this pattern reveals that the primary constituent is nickel oxyhydroxide, Ni2O2(OH)4 (PDF 00-013-0229). No clear presence of crystalline Ni3TeO6 nuclei is observed due to overlapping Bragg peaks (Fig. 2b). However, the asymmetric peak shapes and the presence of shoulder peaks suggest the possible existence of minor phases. | ||
| Fig. 2 (a) SXRD profiles collected during the in situ heating and cooling cycle, indexing results of the diffractogram of (b) NTO-hydro, prior to calcination, and (c) NTO-600, after calcination. The 2θ angles in the x-axis in (a) were converted to d (interplanar spacing values) in (b) and (c). In (c), the solid lines depict the simulated powder XRD patterns of all the reported Ni–Te–O compounds mentioned above and possible decomposition products NiO (PDF 00-044-1159) and TeO2 (PDF 00-042-1365). | ||
During the ramping up stage, the broad peaks become less detectable. Concurrently, new peaks emerge at around 3.98° and 6.83°. By referring the indexing results in Fig. 2c, the peak at around 3.98° can be assigned to the (110) reflection of Ni3TeO6 with the R3 space group. This suggests the preferential nucleation of Ni3TeO6 crystals21 from the hydrothermal precursor. The latter peak at 6.83° is more asymmetric and splits into two sharp peaks at higher temperatures, corresponding to the (12![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ) and (300) reflections of Ni3TeO6. Other diffraction peaks of Ni3TeO6 appear with increasing temperature and time.
) and (300) reflections of Ni3TeO6. Other diffraction peaks of Ni3TeO6 appear with increasing temperature and time.
After 60 min of isothermal calcination at 600 °C, well-crystalline Ni3TeO6 (a = b = 5.124 Å, c = 13.844 Å, c/a = 2.702, α = β = 90°, γ = 120°) is successfully obtained. The (110) reflection at 3.98° remains the strongest peak, contrasting with the database reference (PDF 04-009-2820, Fig. 2c) where the strongest peak is the (104) reflection at 3.72°. This comparative result indicates that the synthesized Ni3TeO6 shows (110) as the preferred orientation in this study. Meantime, a weak peak (marked by *) becomes detectable, indicating the presence of an impurity phase. The intensity of this peak increases with longer calcination duration up to 120 min. No significant changes are observed during the cooling stage. The impurity phase, exhibiting peaks at 2.01, 2.91 and 3.12° (corresponding to d values of 0.509, 0.344 and 0.327 nm), has been identified as a different nickel tellurate NiTeO4 with a monoclinic P121/c1 space group (a = 6.114 Å, b = 4.667 Å, c = 5.575 Å, α = γ = 90°, and β = 123.44°) as shown in Fig. 2c, with better visibility in Fig. S1a (ESI†). Quantitative analysis using the Rietveld refinement (Fig. S1b, ESI†) shows that the NiTeO4 phase content increases with increasing calcination durations and finally reaches ∼10.4 wt% (Table S1, ESI†). It again confirms a good fit to R3 Ni3TeO6 with lattice constants of a = b = 5.099 Å and c = 13.754 Å (c/a = 2.697) and P121/c1 NiTeO4 with lattice constants of a = 6.088 Å, b = 4.638 Å and c = 5.545 Å at room temperature, corresponding to thermal expansion coefficients of 5.29 × 10−5 °C−1 along the a and b axes and 1.51 × 10−4 °C−1 along the c axis for Ni3TeO6 and 4.82 × 10−5, 5.01 × 10−5, and 5.24 × 10−5 °C−1 along the a, b, and c axes for NiTeO4 during the cooling stage, respectively (Table S1, ESI†).
The TEM image reveals the characteristic morphology of NTO-hydro, i.e. flake-like shapes composed of numerous agglomerated nanoparticles and nanosheets (Fig. 3a). This NTO-hydro exhibits a characteristic greenish color and weak visible light absorption (Fig. 4), which is consistent typically with Ni (oxy)hydroxides.26 The corresponding selected area electron diffraction (SAED) in Fig. 3b displays relatively diffuse rings centered at d-spacings of 0.458, 0.257 and 0.149 nm, along with a few discrete diffraction spots scattered along the rings. These features are in good agreement with (001), (110) and (020) interplanar spacings of Ni2O2(OH)4 (Fig. 2b). No Te containing phase can be definitively resolved by TEM. This might be attributed to (i) the poor crystallinity of the sample and (ii) the use of a relatively low beam density to minimize beam damage to hydroxides.27 However, the uniform distribution of Ni, Te and O elements observed in the STEM elemental mapping (Fig. S2, ESI†) indicates the presence of Te within the sample. Based on this evidence and previous report on Te incorporation with edge metal atoms in transition metal hydroxides,28 it is reasonable to conclude that Te might incorporate into Ni2O2(OH)4, forming Te–OH and/or Te–O bonds.
 | ||
| Fig. 3 Bright field TEM (upper panel) and HRTEM (lower panel) images for (a) and (b) NTO-hydro, (c) and (d) NTO-450, and (e) and (f) NTO-600 samples. Insets show the SAED patterns (contrast was enhanced for clarity) from the circled areas. | ||
 | ||
| Fig. 4 (a) UV-vis absorbance spectra and (b) digital images of NTO-hydro, NTO-450 and NTO-600. The inset of (a) depicts the Tauc plot for NTO-600. | ||
In contrast to NTO-hydro, the morphology of NTO-450 (obtained after calcination at 450 °C with 5 min holding) barely changes (Fig. 3c). However, its color is seen to change significantly to dark olive or grey, exhibiting the strongest visible light absorption (Fig. 4). The SAED pattern (inset of Fig. 3c) reveals diffraction rings at d-spacings of 0.255, 0.217 and 0.147 nm, corresponding to the (110), (021) and (300) of Ni3TeO6. This represents the disappearance of Te incorporated Ni2O2(OH)4 and formation of Ni3TeO6. The HRTEM image in Fig. 3d reveals a much longer-range order compared to NTO-hydro, indicating the improved crystallinity of the material. It further suggests that the crystalline Ni3TeO6 nanoparticles are likely only a few nanometers in size. The color of this NTO-450 thus can be ascribed to the formation of this ultrafine nanocrystalline oxide structure.29,30
After calcination, NTO-600 exhibits heterogeneous nanoparticle morphology, as shown in Fig. 3e. It results in discrete diffraction spots in the corresponding SAED pattern. The HRTEM image (Fig. 3f) confirms the high crystallinity and presence of the Ni3TeO6 crystalline phase. The strong visible light absorption of NTO-600, starting at around 520 nm (Fig. 4a), is attributed to its band gap (Eg ≈ 2.43 eV) characteristic of Ni3TeO6. This relatively small Eg is induced by the excitation of photoelectrons from O 2p to Ni 3d orbitals, consistent with previous reports.9,22 The second band at 1.67 eV, corresponding to the broad absorption beyond 520 nm, aligns with electron transition from the occupied Ni 3d to empty Ni 3d orbitals.31
SXRD collects the bulk information as the high energy X-rays penetrate the entire sample volume. In contrast, XPS is surface-sensitive. The sampling depth of XPS is limited to approximately 3λ (λ is the inelastic mean free path for photoelectrons),32 estimated to be within 2.8–3.5 nm of the outmost surfaces for the studied materials with the applied Al Kα X-ray source. Considering the sample morphologies in Fig. 3, the XPS signal likely contains the relatively bulk information from NTO-hydro and NTO-450, while the signal from NTO-600 reflects the surface characteristics of the crystalline nanoparticles. Therefore, directly comparing the compositional information obtained from peak deconvolution of XPS spectra would be misleading. Consequently, only the fitted peak characteristics are listed in Table S2 (ESI†).
The XPS survey spectra of all three samples show all the expected peaks without any additional or impurity elements (Fig. S3, ESI†). High-resolution spectra in Fig. 5 reveal that the asymmetric peaks for Te and O narrow down significantly after calcination, while peaks for Ni exhibit no significant changes. This observation prompts us to focus on the Te 3d (Fig. 5b) and O 1s (Fig. 5c) spectra first. To achieve optimum fitting while avoiding overfitting, a minimum number of components were also employed for these spectra. Notably, the Te 3d and O 1s of NTO-hydro required one additional doublet/singlet peak, compared to the calcinated NTO-450 and NTO-600 samples.
 | ||
| Fig. 5 The peak deconvolution for high-resolution XPS spectra of (a), (d) and (g) Ni 2p, (b), (e) and (h) Te 3d and (c) (f) and (i) O 1s core levels for (a)–(c) NTO-hydro, (d)–(f) NTO-450 and (g)–(i) NTO-600, respectively. | ||
The Te 3d core level spectra exhibit two 3d5/2 and 3d3/2 spin orbit doublets with a characteristic peak area ratio of 3:2, despite their broadness. For NTO-hydro, the broadest Te 3d level is optimally fitted with three components at 577.1, 578.8 and 581.7 eV for the 3d5/2 level, labelled as Te1, Te2 and Te3, respectively. In contrast, NTO-450 and NTO-600 require only two components, Te1 and Te2, at 576.6 eV and 578.6 eV (NTO-450) and 576.1 eV and 577.3 eV (NTO-600) for the 3d5/2 level, respectively. These binding energies can be compared to reference compounds, i.e. TeO2 (Te4+) at 576.1 eV, Te(OH)6 (Te6+) at 576.6 eV, and TeO3 (Te6+) at 577.3 eV.33 The difference in binding energy, nearly 1.0 eV, between the fitted Te components in NTO-hydro and NTO-600 (Table S2, ESI†) suggests that components with the same name likely represent different Te species in these samples. For the crystalline NTO-600, Te1 and Te2 suggest the presence of a dominant Te6+ and a minor Te4+ species, consistent with our previous study.22 In NTO-hydro and NTO-450, Te1 corresponds to a Te6+ species, while Te2 and Te3 suggest a more complex bonding environment for Te involving oxygen and hydroxyl groups. Qualitatively, the broader peaks and higher binding energies in NTO-hydro and NTO-450 indicate a higher oxidation state of Te (≥6+) with few electrons per atom. Calcination promotes electron enrichment around Te, leading to a lower oxidation state (between 6+ and 4 +) and a shift to lower binding energies.
The changes of the O 1s spectra align with those of the Te 3d spectra (Fig. 5c). The O 1s in NTO-hydro must be fitted with three, O1, O2 and O3, components centered at 531.0, 532.7, and 535.7 eV, respectively. The first two components correspond to oxide lattice oxygen, hydroxyl groups and/or oxygen vacancies,34 which remain after calcination. The third component can be attributed to a mixture of adsorbed molecular water and free or vapor phase water,35 both of which disappear along with the aforementioned Te3 component during calcination. Upon calcination, the O1 and O2 peaks become narrower and shift towards lower binding energies. Notably, the binding energy difference for O1 (representing oxide lattice oxygen) between NTO-hydro and NTO-600 is only 0.2 eV (Table S2, ESI†), indicating a relatively consistent chemical environment due to the presence of short-range or long-range ordered oxides. A more significant change is seen in O2, which represents the hydroxyl groups and/or oxygen vacancies. This suggests that the enhanced crystallinity and atomic order in the material are associated with the dehydroxylation reaction.
The Ni 2p spectra demonstrate complex line shapes with satellite features at higher binding energy just beside the main peaks (Fig. 5a). Poorer fitting can be found at the binding energy region corresponding to the Ni 2p1/2 level compared to Ni 2p3/2, due to the overlap with Te 3p1/2.36 However, the line shapes of this core level are relatively consistent across the three samples. Smaller binding energy variations among the four fitted components are seen compared to Te 3d and O 1s levels, although intensity variations exist (Fig. 5 and Table S2, ESI†). These four nickel components (Ni1–4) can be assigned to Ni2+ species in Ni–O (Ni1) and Ni–OH (Ni2) bonding configurations and their respective satellites (Ni3 and 4),37–40 based on their binding energies, peaks, FWHM and the O 1s deconvolution. This agrees well with Ni in a 2+ oxidation state. However, the shift towards lower binding energy upon calcination suggests an interaction between Ni and Te, particularly with the Te3 and O3 components identified earlier. This interaction is likely responsible for the presence of Ni with a higher oxidation state (4+) within Ni2O2(OH)4 in NTO-hydro.
To understand the reactions occurring during the calcination step, we have also measured the heat flow and mass changes, as plotted in Fig. 6. The results are consistent with the SXRD data (Fig. 2), particularly the characteristic peak of the oxyhydroxide phase located around 2.2° (Fig. 2b), and its peak characteristics are summarized in Table S3 (ESI†). This reveals that calcination of NTO-hydro to NTO-600 is a complex, multistage process. The first stage (A) involves a weight loss of 4.0 wt% below 250 °C, likely attributed to the evaporation of absorbed and intercalated water (the O3 component in XPS spectra) coordinated mainly with Te (the Te3 component) in NTO-hydro. This dehydration process results in the broad, shallow endothermic grooves in the heat flow curve. Correspondingly, the oxyhydroxide diffraction exhibits a slight left shift in the peak center due to lattice expansion. There are only minor changes in the integrated peak area, but the peak intensity decreases while the peak width increases (Table S3, ESI†). Stage B follows, characterized by an endothermic process that causes a slight dip in the heat flow at 280–360 °C, coinciding with a weight loss of 7.7 wt%. The peak located initially at around 2.18° becomes significantly weaker and narrower, with a pronounced left-shift peak center. In the temperature range of 360 °C to 600 °C (stage C), the weight loss curve becomes smoother (1.7 wt%), while the heat flow curve displays a continuous exothermic characteristic. Meantime, the characteristic oxyhydroxide peak becomes progressively weaker and narrower, while its peak centre shifts left first and then right (Table S3, ESI†). These changes indicate the nucleation and growth of Ni3TeO6 (Fig. 1), which results in a sharp defined peak at 2.204°, corresponding to the (003) reflection of Ni3TeO6 after 60 min isothermal calcination at 600 °C (Fig. 2a). Further isothermal calcination up to 2 h leads to a further 1.4 wt% mass loss, which could possibly suggest the decomposition of Ni3TeO6.8,13
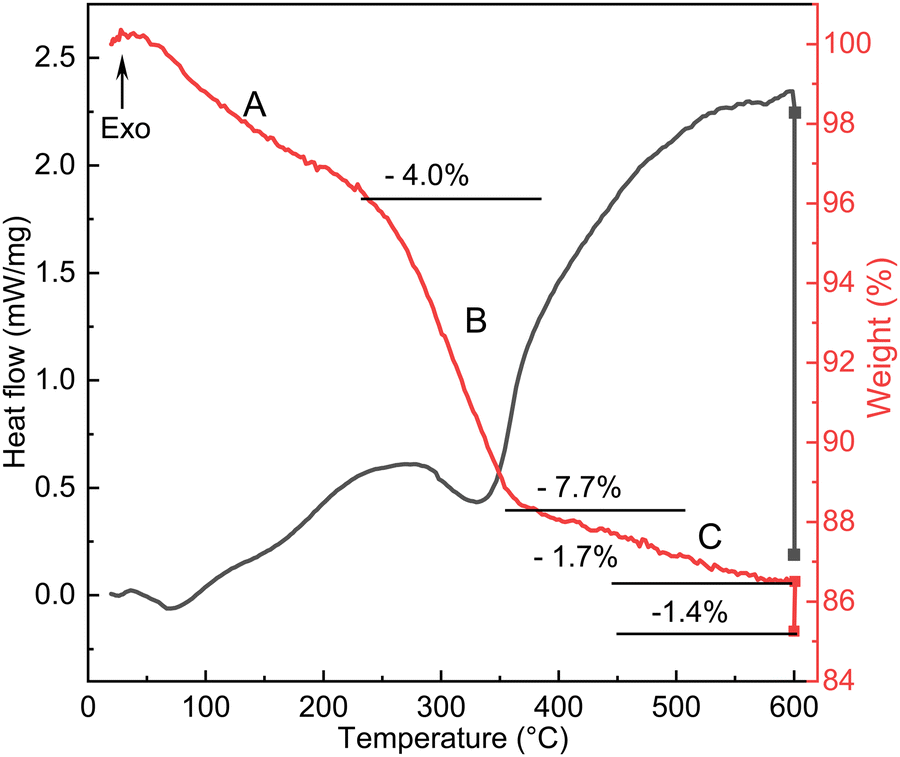 | ||
| Fig. 6 Heat flow and mass change curves during the calcination of NTO-hydro. | ||
4. Mechanism of nano-Ni3TeO6 calcination
While the SXRD profiles in Fig. 2 show no phase transition before 400 °C, some reactions likely occur according to the thermal analysis. It has been reported that the nickel hydroxide decomposition to oxide occurs at around 320 °C.41,42 Therefore, stage B can be attributed to the loss of hydroxyls through| Ni2O2(OH)4 → Ni2O4 + 2H2O, | (1) |
| Te(OH)6 → H2TeO4 + 2H2O → TeO3 + 3H2O | (2) |
Assuming a stoichiometric conversion of Te incorporated Ni oxyhydroxides to 3NiO/TeO3 for the subsequent formation Ni3TeO6, eqn (1) can be modified as follows:
| 3(Ni2O2(OH)4) + 2 incorporated Te → 2(3NiO/TeO3) + 6H2O. | (3) |
Based on these observations, we propose the following reaction sequence for NTO-hydro during calcination:
| (3Ni/Te)(OOH)4·H2O (orthorhombic) → (3Ni/Te)(OOH)4 + H2O | (4) |
| (3Ni/Te)(OOH)4 (orthorhombic) → (3Ni/Te)O6+ 2H2O | (5) |
In the temperature range of 360 °C to 600 °C (stage C), preferred nucleation of Ni3TeO6 occurs:
| (3Ni/Te)O6 (orthorhombic)→ Ni3TeO6 | (6) |
5. Conclusions
In conclusion, this study investigated the formation of Ni3TeO6 nanoparticles from a Ni/Te mixture precursor prepared by a hydrothermal method. In situ SXRD, complemented by TEM, XPS and thermal analysis, provided insights into the calcination process. The results reveal that the hydrothermally obtained powder precursor exhibits a short-range order and a nickel oxyhydroxide structure with incorporation of Te, schematically represented as (3Ni/Te)(OOH)4·H2O. Calcination of this complex oxyhydroxide is a multistage process, starting with dehydration followed by dehydroxylation and nucleation of Ni3TeO6. A Ni:Te stoichiometric ratio of 3:1 remains consistent with the initial aqueous solution, and preferential nucleation from the (110) plane of Ni3TeO6 is observed. However, an impurity tellurate phase appears after complete crystallization of Ni3TeO6 during the in situ SXRD experiment. This is likely due to the deviations from the ideal calcination environment. We believe that this study sheds light on the understanding of nanostructured Ni3TeO6 from its precursors during calcination. This knowledge can be applied to optimize the process and manipulate the size and morphology of Ni3TeO6 and other metal tellurates.
Data availability
Microscopic data supporting the finding are available within the article, and the raw SXRD profiles at different temperatures, and also the raw XPS, UV-Vis and thermal analysis spectra, are available within its ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the financial support from the University of Oulu, Research Council of Finland Profi #352788 H2FUTURE project, Strategic Research Council within the Research Council of Finland decision #358422 JustH2Transit, and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programs (grant agreement no. 101002219). TEM and XPS analyses were carried out with the support of the Centre for Material Analysis, University of Oulu, Finland. The authors thank Mr Al Rahemtulla and Doc. Graham King for the kind help during the beamtime, and Dr Christopher Campbell for proofreading the manuscript. SW thanks the Research Council of Finland research fellowship #363210. JFC thanks the MARSALAS21-09 postdoctoral researcher grant funded by MCIN/AEI/10.13039/501100011033 and the European Union Next Generation EU/PRTR.References
- H. Wang, X. Liang, J. Wang, S. Jiao and D. Xue, Multifunctional inorganic nanomaterials for energy applications, Nanoscale, 2020, 12, 14–42 RSC.
- A. V. Kabashin, A. Singh, M. T. Swihart, I. N. Zavestovskaya and P. N. Prasad, Laser-Processed Nanosilicon: A Multifunctional Nanomaterial for Energy and Healthcare, ACS Nano, 2019, 13, 9841–9867 CrossRef CAS PubMed.
- I. S. Hwang, V. Manikandan, R. P. Patil, M. A. Mahadik, W.-S. Chae, H.-S. Chung, S. H. Choi and J. S. Jang, Hydrogen-Treated TiO2 Nanorods Decorated with Bimetallic Pd–Co Nanoparticles for Photocatalytic Degradation of Organic Pollutants and Bacterial Inactivation, ACS Appl. Nano Mater., 2023, 6, 1562–1572 CrossRef CAS.
- H. Singh, A. K. Sinha, S. M. Gupta, M. N. Singh and H. Ghosh, Insight into the Growth Reaction Mechanism of Ceramic Co3TeO6: Synchrotron Structural and Thermal Analysis, J. Am. Ceram. Soc., 2016, 99, 3443–3448 CrossRef CAS.
- A. K. Patel, M. R. Panda, E. Rani, H. Singh, S. S. Samatham, A. Nagendra, S. N. Jha, D. Bhattacharyya, K. G. Suresh and S. Mitra, Unique Structure-Induced Magnetic and Electrochemical Activity in Nanostructured Transition Metal Tellurates Co1−xNixTeO4 (x = 0, 0.5, and 1), ACS Appl. Energy Mater., 2020, 3, 9436–9448 CrossRef CAS.
- A. Yamuna, N. Karikalan and T. Y. Lee, Effect of the Ni3TeO6 phase in a Ni2Te3O8/expanded graphite composite on the electrochemical monitoring of metribuzin residue in soil and water samples, J. Hazard. Mater., 2022, 435, 128988 CrossRef CAS PubMed.
- M. Numan, M. Salman Khan and S. Majumdar, Vacancy induced mixed valence state in nickel tellurate Ni3TeO6, Mater. Today Proc., 2022, 57, 151–156 CrossRef CAS.
- M. Ali, R. Mishra, A. S. Kerkar, S. R. Bharadwaj and D. Das, Gibbs energy of formation of solid Ni3TeO6 from transpiration studies, J. Nucl. Mater., 2002, 301, 183–186 CrossRef CAS.
- L. Xu, C. Qin, H. Xie, Y. Huang, L. Qin and H. J. Seo, Ilmenite-type semiconductor Ni3TeO6: preparation, optical property and photo-degradation ability, Mater. Lett., 2016, 184, 1–4 CrossRef CAS.
- R. Mathieu, S. A. Ivanov, P. Nordblad and M. Weil, Enhancement of antiferromagnetic interaction and transition temperature in M3TeO6 systems (M = Mn, Co, Ni, Cu), Eur. Phys. J. B, 2013, 86, 361 CrossRef.
- S. A. Ivanov, P. Nordblad, R. Mathieu, R. Tellgren, C. Ritter, N. V. Golubko, E. D. Politova and M. Weil, New type of incommensurate magnetic ordering in Mn3TeO6, Mater. Res. Bull., 2011, 46, 1870–1877 CrossRef CAS.
- M. Z. Iqbal, E. Carleschi, B. P. Doyle and R. J. Kriek, Photocharged Water Splitting Employing a Nickel(II) Tellurium Oxide (Photo)anode in Alkaline Medium, ACS Appl. Energy Mater., 2019, 2, 8125–8137 CrossRef CAS.
- R. Dawar, R. Babu, K. Ananthasivan and S. Anthonysamy, Thermodynamic characterization of Ni3TeO6, Ni2Te3O8 and NiTe2O5, J. Nucl. Mater., 2017, 493, 219–224 CrossRef CAS.
- K. Kohn, K. Inoue, O. Horie and S.-I. Akimoto, Crystal chemistry of MSeO3 and MTeO3 (M = Mg, Mn, Co, Ni, Cu, and Zn), J. Solid State Chem., 1976, 18, 27–37 CrossRef CAS.
- J. Isasi, New MM′O4 oxides derived from the rutile type: synthesis, structure and study of magnetic and electronic properties, J. Alloys Compd., 2001, 322, 89–96 CrossRef CAS.
- K. Krishnan, G. A. Rama Rao, K. D. Singh Mudher and V. Venugopal, Vaporization behaviour and Gibbs energy of formation of Ni2Te3O8, NiTe2O5 and Ni3TeO6, J. Alloys Compd., 1999, 288, 96–101 CrossRef CAS.
- J. T. S. Irvine, M. G. Johnston and W. T. A. Harrison, Lone-pair containment in closed cavities. The MTe6O13 (M = Mn, Ni, Co) family of ternary oxides, Dalton Trans., 2003, 2641–2645 RSC.
- A. Tiwari, D. C. Kakarla, B. Poojitha, P. Sahoo, H. L. Liu, A. Dixit, C. W. Wang, T. W. Yen, M. J. Hsieh, J. Y. Lin, J. Krishnamurthy, Y. C. Lai, H. Chou, T. W. Kuo, A. Pal and H. D. Yang, Spin–phonon-charge coupling in the two-dimensional honeycomb lattice compound Ni2Te3O8, Phys. Rev. B, 2023, 108, 075113 CrossRef CAS.
- R. E. Newnham and E. P. Meagher, Crystal structure of Ni3TeO6, Mater. Res. Bull., 1967, 2, 549–554 CrossRef CAS.
- R. Botella, J. Fernández-Catalá and W. Cao, Experimental Ni3TeO6 synthesis condition exploration accelerated by active learning, Mater. Lett., 2023, 352, 135070 CrossRef CAS.
- J. Fernández-Catalá, A. A. Kistanov, Y. Bai, H. Singh and W. Cao, Theoretical prediction and shape-controlled synthesis of two-dimensional semiconductive Ni3TeO6, npj 2D Mater. Appl., 2023, 7, 48 CrossRef PubMed.
- J. Fernández-Catalá, H. Singh, S. Wang, H. Huhtinen, P. Paturi, Y. Bai and W. Cao, Hydrothermal Synthesis of Ni3TeO6 and Cu3TeO6 Nanostructures for Magnetic and Photoconductivity Applications, ACS Appl. Nano Mater., 2023, 6, 4887–4897 CrossRef PubMed.
- A. Gomez, G. Dina and S. Kycia, The high-energy X-ray diffraction and scattering beamline at the Canadian Light Source, Rev. Sci. Instrum., 2018, 89, 063301 CrossRef CAS PubMed.
- S. Wang, A. A. Kistanov, G. King, S. Ghosh, H. Singh, S. Pallaspuro, A. Rahemtulla, M. Somani, J. Kömi, W. Cao and M. Huttula, In-situ quantification and density functional theory elucidation of phase transformation in carbon steel during quenching and partitioning, Acta Mater., 2021, 221, 117361 CrossRef CAS.
- S. Wang, E. Rani, F. Gyakwaa, H. Singh, G. King, Q. Shu, W. Cao, M. Huttula and T. Fabritius, Unveiling Non-isothermal Crystallization of CaO–Al2O3–B2O3–Na2O–Li2O–SiO2 Glass via In Situ X-ray Scattering and Raman Spectroscopy, Inorg. Chem., 2022, 61, 7017–7025 CrossRef CAS PubMed.
- Y. Qi, H. Qi, J. Li and C. Lu, Synthesis, microstructures and UV-vis absorption properties of β-Ni(OH)2 nanoplates and NiO nanostructures, J. Cryst. Growth, 2008, 310, 4221–4225 CrossRef CAS.
- R. F. Egerton, P. Li and M. Malac, Radiation damage in the TEM and SEM, Micron, 2004, 35, 399–409 CrossRef CAS PubMed.
- H. Han, K. M. Kim, J. H. Ryu, H. J. Lee, J. Woo, G. Ali, K. Y. Chung, T. Kim, S. Kang, S. Choi, J. Kwon, Y.-C. Chung, S. Mhin and T. Song, Boosting oxygen evolution reaction of transition metal layered double hydroxide by metalloid incorporation, Nano Energy, 2020, 75, 104945 CrossRef CAS.
- M. Horie, K. Nishio, K. Fujita, H. Kato, A. Nakamura, S. Kinugasa, S. Endoh, A. Miyauchi, K. Yamamoto, H. Murayama, E. Niki, H. Iwahashi, Y. Yoshida and J. Nakanishi, Ultrafine NiO Particles Induce Cytotoxicity in Vitro by Cellular Uptake and Subsequent Ni(II) Release, Chem. Res. Toxicol., 2009, 22, 1415–1426 Search PubMed.
- J. Liu, Y. Li, J. Ke, S. Wang, L. Wang and H. Xiao, Black NiO–TiO2 nanorods for solar photocatalysis: recognition of electronic structure and reaction mechanism, Appl. Catal., B, 2018, 224, 705–714 CrossRef CAS.
- D. Wang, J. Tang, Z. Zou and J. Ye, Photophysical and Photocatalytic Properties of a New Series of Visible-Light-Driven Photocatalysts M3V2O8 (M = Mg, Ni, Zn), Chem. Mater., 2005, 17, 5177–5182 CrossRef CAS.
- M. Långberg, F. Zhang, E. Grånäs, C. Örnek, J. Cheng, M. Liu, C. Wiemann, A. Gloskovskii, T. F. Keller, C. Schlueter, S. Kulkarni, H. Noei, D. Lindell, U. Kivisäkk, E. Lundgren, A. Stierle and J. Pan, Lateral variation of the native passive film on super duplex stainless steel resolved by synchrotron hard X-ray photoelectron emission microscopy, Corros. Sci., 2020, 174, 108841 CrossRef.
- M. K. Bahl, R. L. Watson and K. J. Irgolic, X-ray photoemission studies of tellurium and some of its compounds, J. Chem. Phys., 2008, 66, 5526–5535 CrossRef.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Systematic XPS studies of metal oxides, hydroxides and peroxides, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- G. Ketteler, S. Yamamoto, H. Bluhm, K. Andersson, D. E. Starr, D. F. Ogletree, H. Ogasawara, A. Nilsson and M. Salmeron, The Nature of Water Nucleation Sites on TiO2(110) Surfaces Revealed by Ambient Pressure X-ray Photoelectron Spectroscopy, J. Phys. Chem. C, 2007, 111, 8278–8282 CrossRef CAS.
- M. Salim, G. Khattak, N. Tabet and L. Wenger, X-Ray photoelectron spectroscopy (XPS) studies of copper–sodium tellurite glasses, J. Electron Spectrosc. Relat. Phenom., 2003, 128, 75–83 CrossRef CAS.
- H. W. Nesbitt, D. Legrand and G. M. Bancroft, Interpretation of Ni 2p XPS spectra of Ni conductors and Ni insulators, Phys. Chem. Miner., 2000, 27, 357–366 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, New interpretations of XPS spectra of nickel metal and oxides, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- K. Sakamoto, F. Hayashi, K. Sato, M. Hirano and N. Ohtsu, XPS spectral analysis for a multiple oxide comprising NiO, TiO2, and NiTiO3, Appl. Surf. Sci., 2020, 526, 146729 CrossRef CAS.
- X. Yu, J. Zhao, L.-R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, Hydrogen Evolution Reaction in Alkaline Media: Alpha- or Beta-Nickel Hydroxide on the Surface of Platinum?, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- Z.-H. Liang, Y.-J. Zhu and X.-L. Hu, β-Nickel Hydroxide Nanosheets and Their Thermal Decomposition to Nickel Oxide Nanosheets, J. Phys. Chem. B, 2004, 108, 3488–3491 CrossRef CAS.
- A. Tang, X. Li, Z. Zhou, J. Ouyang and H. Yang, Mechanochemical synthesis of Ni(OH)2 and the decomposition to NiO nanoparticles: thermodynamic and optical spectra, J. Alloys Compd., 2014, 600, 204–209 CrossRef CAS.
- C. Pico, A. Jerez, M. L. Veiga and E. Gutierrez-Rios, Contribution to the formulation of a structural model by the determination of kinetic data, J. Therm. Anal., 1979, 15, 191–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03765k |
| This journal is © the Owner Societies 2024 |