
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of solution stoichiometry on the thermodynamic stability of prenucleation FeS clusters†
Vincent F. D.
Peters
 a,
Janou A.
Koskamp
a,
Devis
Di Tommaso
b and
Mariette
Wolthers
*a
a,
Janou A.
Koskamp
a,
Devis
Di Tommaso
b and
Mariette
Wolthers
*a
aDepartment of Earth Sciences, Utrecht University, Princetonlaan 8A, 3584 CB Utrecht, The Netherlands. E-mail: m.wolthers@uu.nl
bSchool of Physical and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, UK
First published on 15th January 2025
Abstract
The significance of iron sulphide (FeS) formation extends to “origin of life” theories, industrial applications, and unwanted scale formation. However, the initial stages of FeS nucleation, particularly the impact of solution composition, remain unclear. Often, the iron and sulphide components' stoichiometry in solution differs from that in formed particles. This study uses ab initio methods to computationally examine aqueous FeS prenucleation clusters with excess Fe(II) or S(−II). The results suggest that clusters with additional S(−II) are more likely to form, implying faster nucleation of FeS particles in S(−II)-rich environments compared to Fe(II)-rich ones.
1 Introduction
Iron sulphides are a complex group of minerals which can go through a variety of (metastable) phases.1 FeS clusters found in nature are often linked to “origin of life” theories2,3 and their distinct chemical properties also make iron sulphides promising materials for industrial applications related to energy storage,4,5 CO2 conversion,6 and environmental remediation.7,8 However, their formation in natural waters can be problematic in piping in for instance geothermal energy systems due to scale formation.9,10 Hence, there is a need for knowledge and control on their formation in natural or engineered waters. The effect of solution stoichiometry on nucleation and the formation of prenucleation clusters in particular is poorly understood,11 even though in natural waters the concentration ratio between Fe(II) and S(−II) ions forming the materials might vary. For example, Fe(II) concentrations range from 75–200 μmol L−1 in sediment pore samples12 and 6–20 mmol L−1 in ground water rich with Fe(II).13 S(−II) concentration ranges from 0.05–100 μmol L−1 in layers of marine sediments depending on oxygen level14 and 17 mmol L−1 in high-temperature hydrothermal vent fluid.15 Hence the Fe(II)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S(−II) concentration ratio of (mixed) natural waters might range between 10−5 and 103.
:S(−II) concentration ratio of (mixed) natural waters might range between 10−5 and 103.
For several other minerals we have shown the importance of solution stoichiometry for nucleation and the formation of prenucleation clusters.16–18 In particular, the stability and lifetime of triple ion complexes from Ba2+, Ca2+, CO32− and SO42− determined by molecular dynamics simulations,18 was correlated to a stoichiometric effect on nucleation of minerals formed from these ions examined with, among others, dynamic light scattering. Therefore it is interesting to investigate how triple ion complexes with Fe(II) and S(−II) would form and if FeS nucleation would be similarly impacted by solution stoichiometry. Several reaction mechanisms for FeS formation have been proposed depending on pH.1 Our goal is to elucidate the mechanism of formation of FeS prenucleation clusters by determining the thermodynamic stability of intermediate states that might occur in excess of Fe(II) or S(−II) on path to nucleation. This should give an indication of how fast the first steps towards nucleation might occur in excess of either ion. Since here we focus our calculations on the prenucleation clusters of FeS, our work is mostly related to the formation of mackinawite (stoichiometric FeS), which is the first phase that forms in anoxic environments with Fe(II) and S(−II) ions and can transform to other phases like greigite or pyrite.1 As the most current FeS force field to our knowledge,19 is not developed enough for a bias-enhanced exploration of triple ion clusters with molecular dynamics calculation, we used ab initio methods. We reviewed several previously used ab initio methods for FeS complexes and calculated the reaction free energy of formation and thus stability of triple ion complexes with different stoichiometry with the goal to examine the effect of excess in Fe(II) or S(−II) on path to nucleation.
2 Methodology
2.1 Triple ion complexes
At stoichiometric conditions it is often assumed that first ion pairs are formed, which can then be treated as monomers following classical nucleation theory.20 However, here we assume that at non-stoichiometric conditions when either ion is in a large excess, it is more likely for nucleation to occur via single-ion addition. This leads initially to triple ion complexes. Note that we will also assume an anoxic environment and no redox reactions involving Fe(II) and S(−II).To obtain a more complete overview of a possible stoichiometry effect, we have looked into the reaction free energy to form triple ion complexes from three different cation–anion combinations. In one case we assume the divalent ions Fe2+ and S2−, which form an FeS ion pair and either Fe2S2+ or FeS22− triple ion complexes. This case is most similar to our previous research on triple ion complexes with Ba2+, Ca2+, SO42−, and CO32−18 as both the cation and anion are divalent. This ensures charge effects are similar in a surplus of either Fe(II) or S(−II). This ion pair however has limited direct experimental relevance since the presence of a S2− ion is questionable in most or all environments.21 With a pK1 of around 7 at 25 °C and 1 atm,22 H2S is expected to be the dominant species in acidic environments, while HS− would be dominant in alkaline environments. Within the scope of this work we only focused on alkaline environments. Hence as a second case, we investigated the pair of Fe2+ or HS−, which forms an FeSH+ ion pair and either Fe2SH3+ or Fe(SH)2 triple ion complexes. Due to the large difference in charge between these three triple ion complexes, it is expected that the neutral Fe(SH)2 would be more favourable to form. As a last case, we examined the FeOH+ and HS− ions, which form FeOHSH and subsequently either (FeOH)2SH+ or FeOH(SH)2− complexes. Fe(II) can be present as FeOH+ in alkaline environments.23 As FeOH+ can already be classified as an ion pair, technically we are now looking into the formation of triple and quintuple ion complexes instead. For simplicity in comparisons between the different cases we will consider FeOHSH as the ion pair and (FeOH)2SH+ or FeOH(SH)2− as triple ion complexes. Since now both triple ion complexes are monovalent, the charge is of equivalent magnitude in both cases. Note that in all cases we neglected any intermediate steps that might occur such as FeOHSH → FeSH+ + OH− or FeSH+ → FeS + H+, which would change one complex into another. We assume that by examing these distinct cases, we have also captured the behaviour involved for these cross reactions.
2.2 Computational details
We used several ab initio methods, which have been employed previously in related Fe(II) or FeS research.24–27 We have excluded multi-reference methods as these would be computationally unaffordable for larger clusters explicitly including the surrounding water. However, the reported methods have compared favourably to experimental or CCSD data on related complexes justifying the use of these single reference methods. Using the different levels of theory we computed the geometry and J-coupling for an Fe2S2 complex in the gas phase and the solvation free energy of H2O, HS−, OH−, and Fe2+ to determine which method is most appropriate. Additionally, we performed initial computations with all methods for the reaction free energy in one case (Fe2+ and S2−) to see if any stoichiometry effect would be influenced by the method. Lastly we performed more extensive computations with the most appropriate method on all three distinct cases.Using Gaussian1628 we have used mPW1B95 hybrid-meta-DFT method29 with a basis set of 6-31+g(d,p),30–32 which was applied previously to calculate the reaction free energy for a number of FeS complexes.24 MPW1B95 is a hybrid meta-GGA density functional that has proven to be quite effective for a variety of chemical systems, particularly in the areas of thermochemistry, kinetics, and non-covalent interactions.33 Therefore, we choose mPW1B95 for its ability to predict reaction energies. Furthermore, we have employed the MP234–38 method with a basis set of 6-31++g(d,p)30–32 which has recently been used to examine the configuration of H2O around Fe2+26 and the corresponding solvation energy.27 They have shown that MP2, a post-Hartree Fock method, is capable of predicting binding, clustering, and solvation energy of Fe2+ in line with experimental results, making it a suitable option for further reaction energy predictions. We will refer to the former method as mPW and the latter as MP2. While both methods use a different basis set for consistency with the original papers, it is unlikely this is of much influence as it only concerns the long-range interactions between hydrogen atoms and any observed differences in results is likely attributed to the difference in method. The total spin was fixed at high or low spin and the state with the lowest electronic energy was used. In cases with two Fe(II) we also computed the broken symmetry state, where the spin states on the two Fe(II) are antiparallel. We have accounted for solvent effects with a cluster–continuum model39 as detailed later on which combines explicit considerations of nearby H2O molecules with a continuum solvation model. For the continuum solvation model we used PCM with the integral equation formalism variant.40 As an alternative we have also used the SMD variation,41 which Gaussian16 recommends for computing the solvation energy. Geometry optimisations and frequency calculations were either performed in gas phase or continuum solvent as will be mentioned accordingly.
With VASP 6.442–44 we employed a DFT+U approach45 characterised by U = 5 with a PBE functional46 and the PAW method.47,48 This was previously used for calculations on the Fe2S2 complex25 and their results compared favourably to experimental data of the biological Fe2S2 complex.49 As a reference we also performed calculations without this Hubbard correction. We will refer to either U = 5 or PBE to denote the calculation with or without the inclusion of the U correction. We used a plane wave cutoff of 550 eV and PAW potentials considering 4s and 3d electrons of Fe, the 3s and 3p of S, the 2s and 2p of O, and the 1s of H as valence electrons. The calculations were performed at Γ-point and a Grimme correction (D2) was included to account for dispersion forces.50 To compensate for charge effects within the periodic boundary conditions implemented in VASP, we use a first order dipole correction51,52 and a box of 28 × 28 × 28 Å.53 The total spin was optimised during the calculation starting from a high spin configuration. In cases with two Fe(II) atoms we performed an additional computation starting from a broken symmetry state and continued with the state that had the lowest electronic energy. For a continuum solvation model we used VASPsol.54,55 Geometry optimisations and frequency calculations were only performed in gas phase, but single point calculations were performed in the solvent phase. VASPkit56 was used to extract the energy corrections from the frequency calculations, which uses the same equations as Gaussian16.
Magnetic coupling constants J were determined for Fe2S2 as described previously using:25
 | (1) |
2.3 Reaction free energy
To obtain the reaction free energy for reaction A + B → C we used: | (2) |
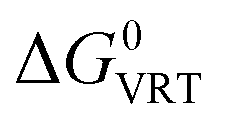 the difference in the vibrational, rotational, and translational contributions in the gas phase at 298 K under a standard-state pressure of 1 atm,
the difference in the vibrational, rotational, and translational contributions in the gas phase at 298 K under a standard-state pressure of 1 atm,  the difference in the solvation free energy
the difference in the solvation free energy  going from 1 mol L−1 gas to 1 mol L−1 in solution, and
going from 1 mol L−1 gas to 1 mol L−1 in solution, and 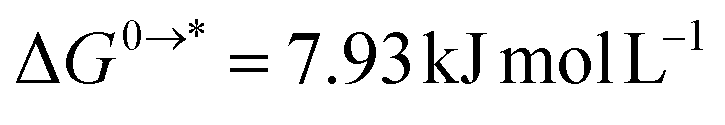 is associated to the standard-state conversion of 1 atm to 1 mol L−1. The solvation free energy
is associated to the standard-state conversion of 1 atm to 1 mol L−1. The solvation free energy 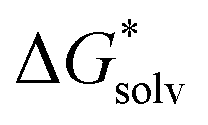 was obtained by a cluster–continuum model,39 which combines explicit inclusion of water molecules near the complexes with an implicit continuum solvent model. This relates the solvation energy of a solute X to the cluster formation X + nH2O → X(H2O)n, where n is the amount of explicit water molecules considered.
was obtained by a cluster–continuum model,39 which combines explicit inclusion of water molecules near the complexes with an implicit continuum solvent model. This relates the solvation energy of a solute X to the cluster formation X + nH2O → X(H2O)n, where n is the amount of explicit water molecules considered.  is then given by39:
is then given by39: | (3) |
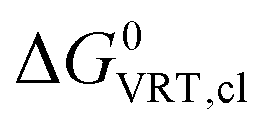 (X(H2O)n) are based on the clustering reaction and
(X(H2O)n) are based on the clustering reaction and  (X(H2O)n) is the solvation free energy of the entire cluster obtained from calculations with the continuum solvent model. The vaporisation free energy of water ΔGvap(H2O) is given by:39
(X(H2O)n) is the solvation free energy of the entire cluster obtained from calculations with the continuum solvent model. The vaporisation free energy of water ΔGvap(H2O) is given by:39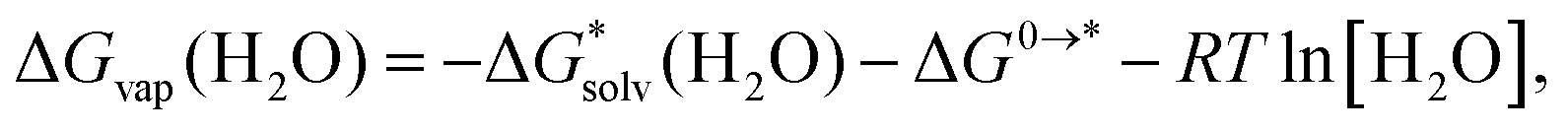 | (4) |
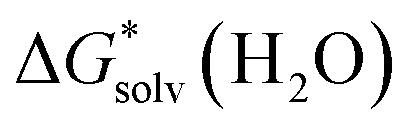 is the solvation free energy of H2O from calculations with the continuum solvent model, R is the ideal gas constant, T is the temperature 298 K, and [H2O] = 55.5 mol L−1.
is the solvation free energy of H2O from calculations with the continuum solvent model, R is the ideal gas constant, T is the temperature 298 K, and [H2O] = 55.5 mol L−1.
The amount of H2O molecules n is of importance for the exact value of  and
and 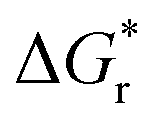 . According to Pliego and Riveros,39 by varying n and exploring all possible configurations a minimum in solvation free energy should be found. For the initial computations with all different methods, we instead used the same number of H2O and did not search for this optimum for each separate method. In Table 1 we show the amount of H2O used for each species. For OH−, HS−, and Fe2+ this number was based on the coordination number observed from coordination analysis of the radial distribution function in a preliminary short ab initio simulation using the PBE functional in VASP6.4. Starting configurations were based on a snapshot of this simulation. For FeS, Fe2S2+, and FeS22− this number was based on the coordination found in similar complexes.24 For S22− it was chosen to be the same as FeS22− as this would be consistent with the cationic complex where Fe2+ and Fe2S2+ also had the same coordination number.
. According to Pliego and Riveros,39 by varying n and exploring all possible configurations a minimum in solvation free energy should be found. For the initial computations with all different methods, we instead used the same number of H2O and did not search for this optimum for each separate method. In Table 1 we show the amount of H2O used for each species. For OH−, HS−, and Fe2+ this number was based on the coordination number observed from coordination analysis of the radial distribution function in a preliminary short ab initio simulation using the PBE functional in VASP6.4. Starting configurations were based on a snapshot of this simulation. For FeS, Fe2S2+, and FeS22− this number was based on the coordination found in similar complexes.24 For S22− it was chosen to be the same as FeS22− as this would be consistent with the cationic complex where Fe2+ and Fe2S2+ also had the same coordination number.
| n | n | ||
|---|---|---|---|
| Fe2+ | 6 | Fe2S2+ | 6 |
| S22− | 2 | FeS22− | 2 |
| HS− | 6 | FeS | 3 |
| OH− | 4 |
For the more extensive calculations using a single method for all three distinct reaction schemes, we did vary n and explored possible configurations. The amount n for most FeS complexes needed to be large to get close to an optimum, making the number of possible configurations large. To reduce the computational effort of exploring all configurations, we first performed several computations in a range of n and estimated possible configurations and from that used a more systematic approach. Starting from the minimum configuration of the initial assessment we added H2O where it increases the coordination around S, Fe, OH, or H2O in the first coordination shell and in such a way that it has the most hydrogen bonds. Then we determined which configuration had the lowest solvation free energy and added the next H2O to this configuration. If increasing a certain coordination was unfavourable for the addition of multiple H2O in a row, we did not add H2O in that specific coordination. This process continued until either  decreased minimally for multiple H2O or up to n ≈ 13–16, where the calculation became unmanageable. Note that we only checked the electronic energy of different spin states for small n and used the most stable spin state at small n for calculations at higher n. For the final configuration at high n we confirmed whether the other spin state was more stable or not.
decreased minimally for multiple H2O or up to n ≈ 13–16, where the calculation became unmanageable. Note that we only checked the electronic energy of different spin states for small n and used the most stable spin state at small n for calculations at higher n. For the final configuration at high n we confirmed whether the other spin state was more stable or not.
3 Results and discussion
3.1 Method comparison
| CCSD25 | PBE | U = 5 | mPW | MP2 | |
|---|---|---|---|---|---|
| d(Fe–Fe) | 2.58 | 2.21 | 2.56 | 2.20 | 2.56 |
| d(Fe–S) | 2.27 | 2.20 | 2.27 | 2.21 | 2.27 |
| θ(Fe–S–Fe–S) | 0.0 | 22.9 | 0.17 | 18.6 | 0.04 |
| J | — | +192 | −103 | −181 | −60 |
It seems that VASPsol poorly describes the solvation free energy for all ionic species. It is possible that VASPsol is less accurate for charged species and/or requires additional considerations accounting for charge. The SMD solvation model only performs well for the Fe2+-ion. This is somewhat surprising as it is the recommended solvation model by Gaussian16 for calculation of the solvation free energy. The discrepancy for OH− and HS− is likely exacerbated from the fact that the H2O solvation free energy is not well described and the cluster–continuum model relies on this value for the computation of all other solvation free energies. Overall MP2 with the PCM solvation model and geometry optimisation in the solvent phase performs the best. Unsurprisingly, this corresponds to the same solvation settings used to determine solvation energies of Fe2+ with MP2 using a slightly different cluster–continuum model.27 There they included up to 13 H2O molecules around the Fe2+-ion and found a value of −1889 kJ mol−1 after extrapolation, which is in line with the reference values. Hence it was expected that a more accurate value would be found in our calculations with MP2 and PCM if the number of H2O molecules are optimised. As MP2 also described the Fe2S2 complex adequately, this seemed the most appropriate method to do our more extensive calculations on.
To get an indication whether some of the results in Table 3 are not biased because the number of H2O molecules is not optimal, we varied this number in the case of HS− between 1 and 6 for MP2 with the two different solvation methods (SMD and PCM) and the two types of geometry optimisation (gas and solvent). For all solvation methods the optimal number of H2O was 2 and the solvation free energy changed (in the same order as the Table) to −270, −278, −289, and −300 kJ mol−1. While this is an improvement for all cases, it is still the best for MP2 with PCM and solvent-phase geometry. The fact that the energy also changes less than the other methods for different amounts of water, shows that it would be more robust. From now on, when we discuss MP2 the optimal solvation method (PCM and solvent-phase geometry) is implied.
| Method | Triple ion complex formation via: | ||
|---|---|---|---|
| Pair formation | Fe(II) addition | S(−II) addition | |
| PBE | −94 | −47 | −141 |
| U = 5 | −28 | −23 | −111 |
| mPW | −138 | −75 | −130 |
| MP2 | −137 | −48 | −127 |
For all methods there is a strong difference between Fe(II) or S(−II) addition with S(−II) addition being favoured. For PBE and U = 5, S(−II) addition is favoured over pair formation, while for mPW and MP2 it is in a similar range. These initial results suggest that the prenucleation cluster formation might be more rapid in the presence of an excess of S(−II) as opposed to an excess of Fe(II). One could argue that this result might be expected because it is known that S2− is an unstable ion in water21 and therefore it would be highly favourable to form any kind of new bond. Hence in the next section, the other reaction schemes are also examined.
3.2 Triple ion complexes
This section follows a more thorough analysis of the pair formation and triple ion complexes using only MP2 with PCM solvation and solvent-phase geometry. First the optimal amount of H2O n is examined by varying the configurations and coordination of water for all relevant complexes and ions in the reaction schemes. Then reaction free energies are presented and discussed. from the cluster–continuum model is plotted as a function of number n of H2O that is considered explicitly. The black datapoints represent the most stable configuration computed for that number n, while the grey datapoints represent less stable configurations. In some graphs less grey datapoints are included than others, because either there were fewer possible different configurations or, especially in the case of the complexes with two Fe(II), the computation could not converge in other geometries even when changing various settings. All complexes with Fe(II) are presented in high spin, though complexes in broken symmetry state differed less than 1 kJ mol−1 from a high spin state. The geometries of the most stable configurations are presented in Supplementary Information S3 (ESI†). We have also included a linear fit for the decrease in
from the cluster–continuum model is plotted as a function of number n of H2O that is considered explicitly. The black datapoints represent the most stable configuration computed for that number n, while the grey datapoints represent less stable configurations. In some graphs less grey datapoints are included than others, because either there were fewer possible different configurations or, especially in the case of the complexes with two Fe(II), the computation could not converge in other geometries even when changing various settings. All complexes with Fe(II) are presented in high spin, though complexes in broken symmetry state differed less than 1 kJ mol−1 from a high spin state. The geometries of the most stable configurations are presented in Supplementary Information S3 (ESI†). We have also included a linear fit for the decrease in  as in most cases the decrease was fairly linear (as indicated by R2). In some cases we only fitted until the dashed line as from that point increasing n did not lower
as in most cases the decrease was fairly linear (as indicated by R2). In some cases we only fitted until the dashed line as from that point increasing n did not lower  by as much. The dashed line was drawn at the minimum
by as much. The dashed line was drawn at the minimum  obtained and nmin indicates where the two lines cross and represents a minimal amount of H2O needed to describe the solvation free energy. For Fe2+, S2−, and (FeOH)2SH+ we did not include the data for n < 2 in the linear fit as they are significant outliers where the first H2O seemed to have a stronger effect. Additionally, for Fe2+, S2−, and FeOH+nmin is quite close to their computational limit of n ≈ 16 and one could argue that
obtained and nmin indicates where the two lines cross and represents a minimal amount of H2O needed to describe the solvation free energy. For Fe2+, S2−, and (FeOH)2SH+ we did not include the data for n < 2 in the linear fit as they are significant outliers where the first H2O seemed to have a stronger effect. Additionally, for Fe2+, S2−, and FeOH+nmin is quite close to their computational limit of n ≈ 16 and one could argue that 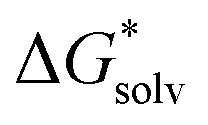 would still be lower for more n. However, additional H2O would either be added in equivalent positions as the configurations for n = 15 and 16 or in a further coordination shell (see Fig. S3–S5, ESI†) and therefore would likely only contribute to a small decrease in
would still be lower for more n. However, additional H2O would either be added in equivalent positions as the configurations for n = 15 and 16 or in a further coordination shell (see Fig. S3–S5, ESI†) and therefore would likely only contribute to a small decrease in  .
.
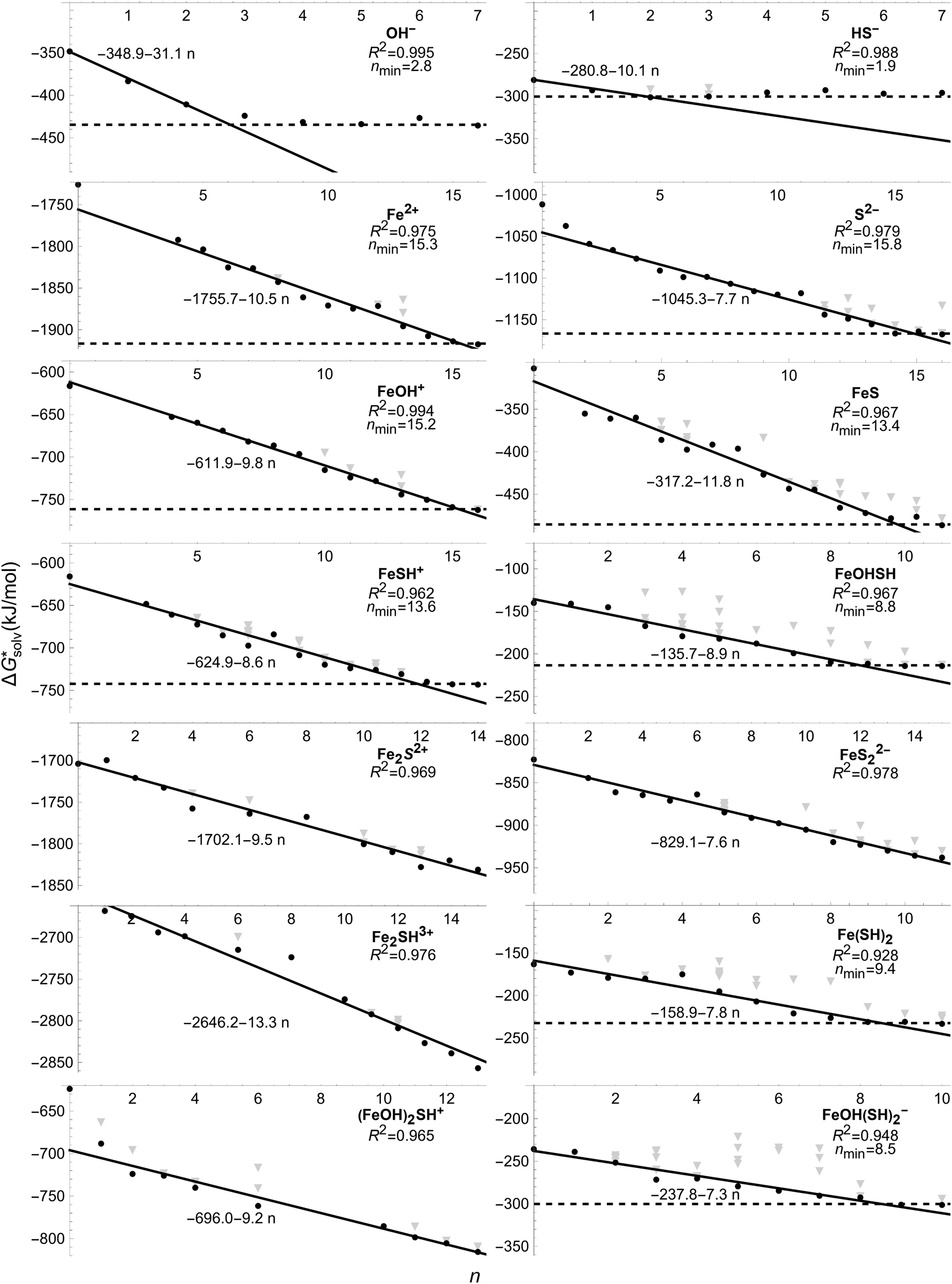 | ||
Fig. 1 The solvation free energy 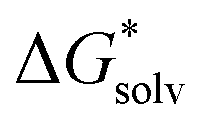 in kJ mol−1 is shown as a function of the number n of explicit H2O around the ion/complex. The black datapoints represent the most stable configuration computed for that number n, while the grey datapoints represent less stable configurations. The lines represent a fit for the most stable configurations. in kJ mol−1 is shown as a function of the number n of explicit H2O around the ion/complex. The black datapoints represent the most stable configuration computed for that number n, while the grey datapoints represent less stable configurations. The lines represent a fit for the most stable configurations. | ||
Now we look into the ions also shown in Table 3 and how these results correlate to related complexes. It is clear that OH− and HS− only require few H2O to adequately describe  and the graphs show that the values in Table 3 are representative for an optimal amount of H2O as well. Hence the results in the Figure are also in good agreement with reference values.59 As a consequence, most complexes with more than two of either OH− and HS− (Fe(SH)2, FeOHSH, and FeOH(SH)2−) require less n compared to the other complexes. The exception is (FeOH)2SH+, which needs more H2O as there are two Fe(II) present.
and the graphs show that the values in Table 3 are representative for an optimal amount of H2O as well. Hence the results in the Figure are also in good agreement with reference values.59 As a consequence, most complexes with more than two of either OH− and HS− (Fe(SH)2, FeOHSH, and FeOH(SH)2−) require less n compared to the other complexes. The exception is (FeOH)2SH+, which needs more H2O as there are two Fe(II) present.
For Fe2+ our result in  has lowered significantly compared to our result in Table 3, but the value is still in the expected reference range.57,58,60,61 We also compared this result with reported solvation free energies of Fe2+ using the MP2 method.27 Their
has lowered significantly compared to our result in Table 3, but the value is still in the expected reference range.57,58,60,61 We also compared this result with reported solvation free energies of Fe2+ using the MP2 method.27 Their  decreased more slowly at n = 12 with a value
decreased more slowly at n = 12 with a value 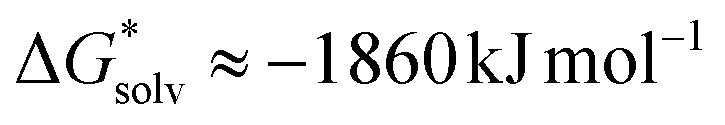 . This discrepancy is either because of the difference in the cluster–continuum model, an unknown difference in certain settings in Gaussian16, or a difference in input geometry which they did not disclose. When Fe2+ is paired with a single OH− or HS− very similar configurations are stable as just Fe2+ (see Fig. S3, S5 and S7, ESI†) and hence a similar nmin is needed to adequately describe
. This discrepancy is either because of the difference in the cluster–continuum model, an unknown difference in certain settings in Gaussian16, or a difference in input geometry which they did not disclose. When Fe2+ is paired with a single OH− or HS− very similar configurations are stable as just Fe2+ (see Fig. S3, S5 and S7, ESI†) and hence a similar nmin is needed to adequately describe  . The configuration of H2O around FeS (see Fig. S6, ESI†) is quite different as much more is coordinated around the S(−II)-ion, but a similar nmin was obtained. It is striking that for S2− a similarly large n was needed as for Fe2+ to adequately describe
. The configuration of H2O around FeS (see Fig. S6, ESI†) is quite different as much more is coordinated around the S(−II)-ion, but a similar nmin was obtained. It is striking that for S2− a similarly large n was needed as for Fe2+ to adequately describe  . This meant that for most complexes with two or more of either ions (Fe2S2+, FeS22−, and Fe2SH3+) a larger amount of n is needed than could be computed as multiple ions in the complex would require a significant amount of H2O around them.
. This meant that for most complexes with two or more of either ions (Fe2S2+, FeS22−, and Fe2SH3+) a larger amount of n is needed than could be computed as multiple ions in the complex would require a significant amount of H2O around them.
Extrapolation of 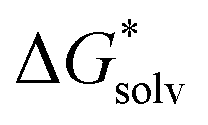 for the complexes where we did not compute with enough n is tricky as it requires an estimation of nmin. For Fe2SH3+ and (FeOH)2SH+ a roughly estimated value for nmin could be argued from the configurations of FeSH+ and FeOHSH respectively. For FeSH+ only two H2O are directly coordinated to the SH group and for FeOHSH no H2O are directly coordinated around the SH group (see Fig. S7 and S8, ESI†). As an estimate for nmin, we could expect a similar coordination around Fe(II) in Fe2SH3+ and around the FeOH groups in (FeOH)2SH+. We would not expect direct H2O coordination around the SH group at nmin as it is already bonded by two Fe(II) and this was not favourable in configurations we computed at lower n (see Fig. S7 and S8, ESI†). This would lead to nmin ≈ 2 × (13.3 − 2) ≈ 22.6 for Fe2SH3+ and nmin ≈ 2 × 8.8 ≈ 17.6 for (FeOH)2SH+. An extrapolation for Fe2S2+ and FeS22− based on the configuration of FeS alone is more challenging as the H2O configuration around FeS is much more divided between the Fe(II) and S(−II) groups (see Fig. S6, ESI†). However, since nmin of FeS and FeSH+ is similar, it is reasonable to expect that upon addition of Fe22+nmin is also similar, hence we use the same estimate nmin ≈ 22.6 for Fe2S2+. Furthermore, since Fe2+ and S2− have a similar nmin, we also estimate nmin for Fe2S2+ and FeS22− to be the same.
for the complexes where we did not compute with enough n is tricky as it requires an estimation of nmin. For Fe2SH3+ and (FeOH)2SH+ a roughly estimated value for nmin could be argued from the configurations of FeSH+ and FeOHSH respectively. For FeSH+ only two H2O are directly coordinated to the SH group and for FeOHSH no H2O are directly coordinated around the SH group (see Fig. S7 and S8, ESI†). As an estimate for nmin, we could expect a similar coordination around Fe(II) in Fe2SH3+ and around the FeOH groups in (FeOH)2SH+. We would not expect direct H2O coordination around the SH group at nmin as it is already bonded by two Fe(II) and this was not favourable in configurations we computed at lower n (see Fig. S7 and S8, ESI†). This would lead to nmin ≈ 2 × (13.3 − 2) ≈ 22.6 for Fe2SH3+ and nmin ≈ 2 × 8.8 ≈ 17.6 for (FeOH)2SH+. An extrapolation for Fe2S2+ and FeS22− based on the configuration of FeS alone is more challenging as the H2O configuration around FeS is much more divided between the Fe(II) and S(−II) groups (see Fig. S6, ESI†). However, since nmin of FeS and FeSH+ is similar, it is reasonable to expect that upon addition of Fe22+nmin is also similar, hence we use the same estimate nmin ≈ 22.6 for Fe2S2+. Furthermore, since Fe2+ and S2− have a similar nmin, we also estimate nmin for Fe2S2+ and FeS22− to be the same.
 obtained and extrapolated values if applicable. We have included the formation free energy of the ion pair itself as a reference. In Fig. 2 we have also schematically illustrated our (extrapolated) results and how they position on the path to nucleation. When compared to the MP2 values in Table 4, we notice that all reaction free energies for Fe2+ and S2− have higher absolute values in Table 5 than Table 4. Since all solvation energies
obtained and extrapolated values if applicable. We have included the formation free energy of the ion pair itself as a reference. In Fig. 2 we have also schematically illustrated our (extrapolated) results and how they position on the path to nucleation. When compared to the MP2 values in Table 4, we notice that all reaction free energies for Fe2+ and S2− have higher absolute values in Table 5 than Table 4. Since all solvation energies  have been lowered, it becomes less favourable to break bonds with H2O to form new bonds. However, we still observe that S(−II) addition is strongly favoured over Fe(II) addition. For the other cases in Table 5 S(−II) addition is similarly favoured over Fe(II) addition.
have been lowered, it becomes less favourable to break bonds with H2O to form new bonds. However, we still observe that S(−II) addition is strongly favoured over Fe(II) addition. For the other cases in Table 5 S(−II) addition is similarly favoured over Fe(II) addition.
| Ion pair | Triple ion complex formation via: | ||
|---|---|---|---|
| Pair formation | Fe(II) addition | S(−II) addition | |
| Fe2+ + S2− | −62 | 103 | 13 |
| Extrapolation | 16 | −51 | |
| Fe2+ + HS− | −35 | 92 | −29 |
| Extrapolation | 2 | ||
| FeOH+ + HS− | −13 | 42 | −30 |
| Extrapolation | −1 | ||
 | ||
| Fig. 2 Schematic representation of the formation of FeS complexes. | ||
For some of these reactions we can compare to literature values for (the logarithm of) stability constants logK, computed using the expression  . For FeSH+ formation, stability constants have been reported ranging from logK = 4.34 to 5.94 depending on experimental method and ionic strength.62–65 Using −35 kJ mol−1 we obtain logK = 6.05, which is quite close to experimental results. One report with logK = 5.07 also reported a stability constant for Fe2SH3+(logK = 10.07).64 We can compute this stability constant from our results by adding
. For FeSH+ formation, stability constants have been reported ranging from logK = 4.34 to 5.94 depending on experimental method and ionic strength.62–65 Using −35 kJ mol−1 we obtain logK = 6.05, which is quite close to experimental results. One report with logK = 5.07 also reported a stability constant for Fe2SH3+(logK = 10.07).64 We can compute this stability constant from our results by adding  of the pair formation with the respective (extrapolated) value for ion addition leading to logK = 5.79. This is quite a different value indicating that for Fe2SH3+ the estimated nmin might need to be higher. Increasing nmin by two H2O molecules (nmin = 24.6) would lead to logK = 10.46, which is much more similar to the literature value. Additionally, for Fe(SH)2 a stability constant of logK = 11.27 is obtained, which is of similar magnitude giving some validation to this value. For Fe(SH)2 other values were also reported (logK = 6.4566 or 8.967) which are substantially lower. However, in the same reports the stability constants for FeSH+ were considerably lower (logK < 366 or = 1.467) than most other reported values, which means these values are likely underestimated. Additionally, the existence of Fe(SH)2 in their experiments is controversial.64,66,67
of the pair formation with the respective (extrapolated) value for ion addition leading to logK = 5.79. This is quite a different value indicating that for Fe2SH3+ the estimated nmin might need to be higher. Increasing nmin by two H2O molecules (nmin = 24.6) would lead to logK = 10.46, which is much more similar to the literature value. Additionally, for Fe(SH)2 a stability constant of logK = 11.27 is obtained, which is of similar magnitude giving some validation to this value. For Fe(SH)2 other values were also reported (logK = 6.4566 or 8.967) which are substantially lower. However, in the same reports the stability constants for FeSH+ were considerably lower (logK < 366 or = 1.467) than most other reported values, which means these values are likely underestimated. Additionally, the existence of Fe(SH)2 in their experiments is controversial.64,66,67
For all cases, we find that S(−II) addition is favoured over Fe(II) addition, but this result is influenced by the estimate of nmin for the extrapolated values. Hence for the complexes with two Fe(II) we have calculated how much higher nmin needs to be for Fe(II) addition to become favoured over S(−II) addition. This means that  should be lowered by at least 66 kJ mol−1 for Fe2S2+. Similarly, for Fe2SH3+
should be lowered by at least 66 kJ mol−1 for Fe2S2+. Similarly, for Fe2SH3+ should be lowered by 31 kJ mol−1 and for (FeOH)2SH+ by 28 kJ mol−1. Using the linear fits for
should be lowered by 31 kJ mol−1 and for (FeOH)2SH+ by 28 kJ mol−1. Using the linear fits for  , we estimated that nmin should be increased respectively by 7.0, 2.3, and 3.1. For Fe2S2+ such a large increase would be unlikely and therefore S(−II) addition should be more favourable in this case even with the optimal nmin. For Fe2SH3+ and (FeOH)2SH+, the increases in nmin are smaller, so there is a reasonable uncertainty in our conclusion on whether S(−II) addition would be favoured over Fe(II) addition. It is worth reminding that for Fe2SH3+ our resulting stability constant logK = 11.27 for S(−II) addition is larger than the experimental result for Fe(II) addition (logK = 10.07), but we have overestimated other experimental values as well so this gives little confirmation for either conclusion.
, we estimated that nmin should be increased respectively by 7.0, 2.3, and 3.1. For Fe2S2+ such a large increase would be unlikely and therefore S(−II) addition should be more favourable in this case even with the optimal nmin. For Fe2SH3+ and (FeOH)2SH+, the increases in nmin are smaller, so there is a reasonable uncertainty in our conclusion on whether S(−II) addition would be favoured over Fe(II) addition. It is worth reminding that for Fe2SH3+ our resulting stability constant logK = 11.27 for S(−II) addition is larger than the experimental result for Fe(II) addition (logK = 10.07), but we have overestimated other experimental values as well so this gives little confirmation for either conclusion.
Overall though, S(−II) addition seems favoured in most cases and for most methods, which would imply that in an alkaline environment there would be faster nucleation in an S(−II) excess with respect to an Fe(II) excess. Thus in an S(−II) excess you would expect a large amount of small particles and in an Fe(II) excess you would expect to form a smaller amount of large particles. Additionally, a negatively charged particle seems more favourable to form than a positively charged particle. These results could be related to density functional theory calculations on mackinawite surfaces.68 Here an S-terminated {001} surface had the smallest surface energy and hence was the most stable surface, while an Fe-terminated {001} surface was the least stable.
4. Conclusions
We have computed the reaction free energy for the formation of complexes in excess of either Fe(II) or S(−II) using ab initio methods. Our results demonstrate that care should be taken when describing FeS complexes with ab initio methods in selecting the right ab initio method, solvation model, and explicit consideration of H2O. However, with the MP2 method this lead to solvation free energies for HS−, OH−, Fe2+ in line with experimental results and a reaction free energy for FeSH+ in reasonable agreement with experiments. It seemed complexes with an additional S(−II) are overall more favourable to form suggesting that in alkaline environments an S(−II) excess leads to faster nucleation compared to an Fe(II) excess. These results again showcase the importance of solution stoichiometry in nucleation and warrant further experimental investigation of this effect on FeS formation in particular.Author contributions
Vincent F. D. Peters: conceptualization, formal analysis, investigation, methodology, writing – original draft. Janou A. Koskamp: conceptualization, methodology. Devis di Tommaso: methodology, supervision, writing – review & editing. Mariette Wolthers: conceptualization, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the article and ESI.† Optimised structures are available at https://public.yoda.uu.nl/geo/UU01/HC7V0W.html.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank A. Muthuperiyanayagam and A. Živković for help with the ab initio calculations and A. Karami for discussions on FeS formation. This project has received funding to M. W., V. F. D. P., and J. A. K from the European Research Council (ERC) under the European Unions Horizon 2020 research and innovation programme (grant agreement no. 819588).Notes and references
- D. Rickard and G. W. Luther, Chem. Rev., 2007, 107, 514–562 CrossRef CAS PubMed.
- M. J. Russell and A. J. Hall, J. Geol. Soc., 1997, 154, 377–402 CrossRef CAS PubMed.
- C. Bonfio, L. Valer, S. Scintilla, S. Shah, D. J. Evans, L. Jin, J. W. Szostak, D. D. Sasselov, J. D. Sutherland and S. S. Mansy, Nat. Chem., 2017, 9, 1229–1234 CrossRef CAS PubMed.
- C. Modrzynski and P. Burger, Dalton Trans., 2019, 48, 1941–1946 RSC.
- J. Zhao, J. Syed, X. Wen, H. Lu and X. Meng, J. Alloys Compd., 2019, 777, 974–981 CrossRef CAS.
- A. Roldan, N. Hollingsworth, A. Roffey, H.-U. Islam, J. Goodall, C. Catlow, J. Darr, W. Bras, G. Sankar, K. Holt, G. Hogarth and N. De Leeuw, Chem. Commun., 2015, 51, 7501–7504 RSC.
- Y. Gong, J. Tang and D. Zhao, Water Res., 2016, 89, 309–320 CrossRef CAS PubMed.
- Y. Sun, J. Liu, X. Fan, Y. Li and W. Peng, Front. Environ. Sci., 2023, 11, 1212355 CrossRef.
- J. Jamero, S. Zarrouk and E. Mroczek, Geothermics, 2018, 72, 1–14 CrossRef.
- A. Hamza, I. Hussein, R. Jalab, M. Saad and M. Mahmoud, Energy Fuels, 2021, 35, 14401–14421 CrossRef CAS.
- Y. Liu, Z. Zhang, N. Bhandari, F. Yan, F. Zhang, G. Ruan, Z. Dai, H. Alsaiari, A. Lu, G. Deng, A. Kan and M. Tomson, SPE Int. Oilfield Chem. Symp. Proc., 2017, 2017-April, 361–376 Search PubMed.
- M. Ma, H. Wang, J. Xu, Y. Huang, D. Yuan, X. Zhang and Q. Song, ACS Omega, 2020, 5, 31551–31558 CrossRef CAS PubMed.
- C. Blodau, Acta Hydrochim. Hydrobiol., 2005, 33, 104–117 CrossRef.
- Y. Zheng, R. Anderson, A. Van Geen and J. Kuwabara, Geochim. Cosmochim. Acta, 2000, 64, 4165–4178 CrossRef CAS.
- K. Ding, W. Seyfried Jr., M. Tivey and A. Bradley, Earth Planet. Sci. Lett., 2001, 186, 417–425 CrossRef CAS.
- S. Y. M. H. Seepma, S. E. Ruiz-Hernandez, G. Nehrke, K. Soetaert, A. P. Philipse, B. W. M. Kuipers and M. Wolthers, Cryst. Growth Des., 2021, 21, 1576–1590 CrossRef CAS PubMed.
- V. F. D. Peters, A. Baken, S. Y. M. H. Seepma, J. A. Koskamp, A. Fernández-Martínez, A. E. S. van Driessche and M. Wolthers, Ind. Eng. Chem. Res., 2024, 63, 78–88 CrossRef CAS PubMed.
- J. A. Koskamp, S. Y. M. H. Seepma, V. F. D. Peters, D. Toroz, D. Di Tommaso and M. Wolthers, Chem. – Eur. J., 2024, 30, e202303860 CrossRef CAS PubMed.
- E. Moerman, D. Furman and D. J. Wales, J. Chem. Inf. Model., 2021, 61, 1204–1214 CrossRef CAS PubMed.
- D. Kashchiev and G. M. Van Rosmalen, Cryst. Res. Technol., 2003, 38, 555–574 CrossRef CAS.
- P. May, D. Batka, G. Hefter, E. Königsberger and D. Rowland, Chem. Commun., 2018, 54, 1980–1983 RSC.
- O. Suleimenov and T. Seward, Geochim. Cosmochim. Acta, 1997, 61, 5187–5198 CrossRef CAS.
- F. Furcas, B. Lothenbach, O. Isgor, S. Mundra, Z. Zhang and U. Angst, Cem. Concr. Res., 2022, 151, 106620 CrossRef CAS.
- S. Haider, D. Di Tommaso and N. H. De Leeuw, Phys. Chem. Chem. Phys., 2013, 15, 4310–4319 RSC.
- U. Terranova and N. H. De Leeuw, Phys. Chem. Chem. Phys., 2014, 16, 13426–13433 RSC.
- O. Boukar, J. J. Fifen, M. Nsangou, H. Ghalila and J. Conradie, New J. Chem., 2021, 45, 10693–10710 RSC.
- O. Boukar, J. J. Fifen, J. Conradie and M. M. Conradie, J. Mol. Model., 2024, 30, 52 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian–16 Revision C.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- Y. Zhao and D. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- T. Clark, J. Chandrasekhar, G. Spitznagel and P. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. Frisch, J. Pople and J. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- V. Rassolov, M. Ratner, J. Pople, P. Redfern and L. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- M. Head-Gordon, J. Pople and M. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- S. Sæbø and J. Almlöf, Chem. Phys. Lett., 1989, 154, 83–89 CrossRef.
- M. Frisch, M. Head-Gordon and J. Pople, Chem. Phys. Lett., 1990, 166, 281–289 CrossRef CAS.
- M. Frisch, M. Head-Gordon and J. Pople, Chem. Phys. Lett., 1990, 166, 275–280 CrossRef CAS.
- M. Head-Gordon and T. Head-Gordon, Chem. Phys. Lett., 1994, 220, 122–128 CrossRef CAS.
- J. R. Pliego and J. M. Riveros, J. Phys. Chem. A, 2001, 105, 7241–7247 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- S. Dudarev and G. Botton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6, 8245–8257 CrossRef CAS.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. Albers, S. Demeshko, K. Pröpper, S. Dechert, E. Bill and F. Meyer, J. Am. Chem. Soc., 2013, 135, 1704–1707 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067–16080 CrossRef CAS PubMed.
- G. Makov and M. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS PubMed.
- R. González Gómez, I. del Rosal, K. Philippot and R. Poteau, Theor. Chem. Acc., 2019, 138, 95 Search PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- Y. Marcus, Ions in Solution and their Solvation, Wiley Online Library, Hoboken, NJ, 1st edn, 2015, pp. 1–293 Search PubMed.
- R. Noyes, J. Am. Chem. Soc., 1962, 84, 513–522 CrossRef CAS.
- A. V. Marenich, C. P. Kelly, J. D. Thompson, G. D. Hawkins, C. C. Chambers, D. J. Giesen, P. Winget, C. J. Cramer and D. G. Truhlar, Minnesota Solvation Database (MNSOL) version 2012, 2020, Retrieved from the Data Repository for the University of Minnesota Search PubMed.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- W. R. Fawcett, J. Phys. Chem. B, 1999, 103, 11181–11185 CrossRef CAS.
- G. Luther and T. Ferdelman, Environ. Sci. Technol., 1993, 27, 1154–1163 CrossRef CAS.
- D. Wei and K. Osseo-Asare, J. Colloid Interface Sci., 1995, 174, 273–282 CrossRef CAS.
- G. W. Luther, D. Rickard, S. Theberge and A. Olroyd, Environ. Sci. Technol., 1996, 30, 671–679 CrossRef CAS.
- R. Al-Farawati and C. Van Den Berg, Mar. Chem., 1999, 63, 331–352 CrossRef CAS.
- W. Davison, N. Phillips and B. Tabner, Aquat. Sci., 1999, 61, 23–43 CrossRef CAS.
- D. Dyrssen, Mar. Chem., 1988, 24, 143–153 CrossRef CAS.
- A. J. Devey, R. Grau-Crespo and N. H. De Leeuw, J. Phys. Chem. C, 2008, 112, 10960–10967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03758h |
| This journal is © the Owner Societies 2025 |