
Vacuum-ultraviolet irradiation of pyridine:acetylene ices relevant to Titan astrochemistry†
Larissa
Lopes Cavalcante
 a,
Ellen C.
Czaplinski
b,
Helen E.
Maynard-Casely
c,
Morgan L.
Cable
b,
Naila
Chaouche-Mechidal
a,
Robert
Hodyss
b and
Courtney
Ennis
*ad
a,
Ellen C.
Czaplinski
b,
Helen E.
Maynard-Casely
c,
Morgan L.
Cable
b,
Naila
Chaouche-Mechidal
a,
Robert
Hodyss
b and
Courtney
Ennis
*ad
aDepartment of Chemistry, University of Otago, Dunedin 9054, New Zealand. E-mail: courtney.ennis@otago.ac.nz
bJet Propulsion Laboratory, California Institute of Technology, Pasadena, California 91109, USA
cAustralian Centre for Neutron Scattering, ANSTO, Kirrawee, New South Wales 2232, Australia
dMacDiarmid Institute for Advanced Materials and Nanotechnology, Wellington 6140, New Zealand
First published on 9th October 2024
Abstract
Nitrogen-containing polycyclic aromatic hydrocarbons (NPAHs) are important molecules for astrochemistry and prebiotic chemistry, as their occurrence spans from interstellar molecular clouds to planetary systems. Their formation has been previously explored in gas phase experiments, but the role of solid-state chemical reactions in their formation under cryogenic conditions remains elusive. Here, we explore the formation of NPAHs through vacuum ultraviolet (VUV) irradiation of pyridine:acetylene ices in amorphous and co-crystalline phases, with the aim to simulate conditions relevant to the interstellar medium and Titan's atmosphere. Our results show that the synthesis of ethynylpyridines from VUV-irradiated pyridine:acetylene amorphous ices is achievable at 18 K. In the co-crystal phase, photolysis at 110 K leads to the formation of NPAHs such as quinolizinium+ and precursors, reflecting a dynamical system under our conditions. In contrast, irradiation at 90 K under stable conditions did not produce volatile photoproducts. These results suggest that such chemical processes can occur in Titan's atmosphere and potentially in its stratosphere, where the co-condensation of these molecules can form composite ices. Concurrently, the formation of stable co-crystals can influence the depletion rates of pyridine, which suggests that these structures can be preserved and potentially delivered to Titan's surface. Our findings provide insights into the molecular diversity and chemical evolution of organic matter on Titan, crucial for future space exploration missions, such as the Dragonfly mission, which may uncover higher-order organics derived from pyridine precursors on Titan's surface.
1. Introduction
From molecular clouds to protostellar and planetary systems, nitrogen-containing polycyclic aromatic hydrocarbons (NPAHs) are relevant to astrochemistry and the origin of life.1–6 In the interstellar medium (ISM), NPAHs are considered candidates for the 6.2 μm unidentified infrared band detected in emission spectra.1,7 Within our solar system, interest in NPAHs has risen in the advent of exploration of Saturn's largest moon, Titan, with its atmosphere abundant in nitrogen and hydrocarbon species. To model Titan's atmospheric chemistry, complex reaction networks have been derived to interconnect an extensive molecular inventory of organic compounds.8–14 These models suggest that the formation of higher-order nitrogenous species occurs through ion–molecule and neutral pathways in the upper atmosphere.12,15–18 Here, products with molecular mass ranging from 170 to 310 Da have been detected during the Cassini–Huygens mission and attributed to (N)PAH compounds.19,20It is commonly observed that the highest mixing ratio for atmospheric species on Titan occurs at the altitude of their formation, which typically coincides with a peak in energy transferred from UV and particle radiation sources.11,21 However, molecular abundances are observed to decrease at lower stratosphere altitudes (60–100 km; temperature ca. 80–150 K21) due to the sequential freeze-out of polar and non-polar molecules. This fractional condensation process is thought to lead to aerosols possessing layers of composite molecular ice.
As possible domains within these aerosols, molecular co-crystals22–33 are structures composed of two (or more) species with a fixed stoichiometry that are stabilized by intermolecular interactions. If arranged in a favourable geometry between the constituent molecules, co-crystals have been hypothesised as environments conducive to efficient solid-state formation of complex organic molecules, and act as vessels for their preservation and transport to Titan's surface.34,35 Such chemistry may be feasible on Titan, as high-energy particles from Saturn's magnetosphere and galactic cosmic rays (GCRs) can penetrate Titan's atmosphere to drive chemical processing near the surface, where the temperature is around 90 K.21 It follows that co-crystals can act as cryominerals to study the irradiation-driven formation of NPAHs in simulating Titan chemistry. Having been previously subjects of laboratory studies, the benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (3:1),29 acetonitrile:acetylene (1:2),25 acetylene:ammonia (1:1),33 and pyridine:acetylene (1:1) co-crystals31 are all systems that comprise Titan species where established methods can now explore solid-state chemistry.
:acetonitrile (3:1),29 acetonitrile:acetylene (1:2),25 acetylene:ammonia (1:1),33 and pyridine:acetylene (1:1) co-crystals31 are all systems that comprise Titan species where established methods can now explore solid-state chemistry.
Precedence for such chemistry is provided by ion–molecule reactions between pyridine+ (C5H5N˙+) and acetylene (C2H2) in the gas-phase, where experimental studies have reported the room temperature formation of NPAH precursors 2-ethynylpyridine (C7H5N) and quinolizinium+ (C9H8N).36 Further, the synthesis of (iso)quinoline and quinolizinium+ was identified at higher temperatures3,6 applicable to circumstellar regions. However, at the low temperature at which co-crystals would exist on Titan, reactions require (near) barrierless kinetics to proceed. At Titan atmospheric conditions, Räp et al. (2022) confirmed that an exothermic ion–molecule reaction between pyridine+ and acetylene can occur at temperatures as cold as 150 K in the gas phase, leading to the formation of 2-ethynyl-H-pyridine, and quinolizinium+via a 2-vinyl-β-distonic-H-pyridine intermediate.5 Nevertheless, no photochemical experiments involving pyridine and acetylene at colder temperatures and in the solid state have been conducted to date. Without exposure to energetic particles, acetylene has been demonstrated to undergo solid-state cyclisation and cycloaddition reactions when adsorbed on simulated dust grain surfaces, resulting in the 100–180 K formation of benzene.37 Such studies confirm pathways for the formation of complex molecular cycles, such as (N)PAHs, are available for cooler environments such as Titan. Therefore, if co-crystals are indeed efficient in promoting this chemistry in cold ISM regions, as well as the icy moons of the outer solar system, it would affirm co-crystal deposits as significant targets for future spacecraft exploration, a terrain that may harbour materials central to Earth's prebiotic chemistry.38–41
The primary aim of this study is to explore the formation of NPAHs and their precursors through vacuum UV irradiation of mixed pyridine:acetylene (C5H5N:C2H2) ices in both amorphous and co-crystalline phases. By combining temperature-programmed desorption (TPD) mass spectrometry and infrared spectroscopy, we first investigate the photoproducts produced from the irradiation of a mixed C5H5N:C2H2 ice in an amorphous phase at 18 K. We then compare the identified products to those generated from the photolysis of the C5H5N:C2H2 co-crystal at 90 and 110 K. Here, we report the formation of ethynylpyridines as a product of the irradiation of the mixed amorphous ice at 18 K. These were detected by their mass signatures in TPD traces during ice sublimation in combination with the identification of nascent IR bands assigned by comparison to quantum-chemical vibrational frequency calculations. We further discuss the observation that specific NPAHs (and precursors) were formed during irradiation of the co-crystal at 110 K, while no volatile photoproducts were observed during irradiation at 90 K.
2. Experimental
2.1. Experimental setup and sample preparation
VUV photochemistry experiments were performed involving the C5H5N:C2H2 system (18, 90 and 110 K), and as control experiments: layered ice between C5H5N and C2H2, and pure C5H5N ices (18, 110 and 130 K). The thin films of molecular ices were produced in a stainless-steel high vacuum chamber evacuated to a base pressure of 10−9 torr using turbomolecular pumps backed by dry diaphragm pumps. A KBr optical window attached to the second stage of a closed-cycle helium cryocooler (Sumitomo RDK-101D) was cooled to 18 K to act as an infrared transparent substrate, as previously described.23 The surface temperature was monitored by a silicon diode positioned directly above the substrate linked to a temperature control unit (Lakeshore 330) that reports to an accuracy of ±0.5 K. A 50 Ω heater positioned beneath the substrate allowed temperature control to 300 K. Deposition of the molecular thin films were performed by introducing desired quantities of pyridine (C5H5N; Sigma: HPLC grade) and acetylene (C2H2; BOC: instrument grade) through a gas manifold connected to a leak valve before the chamber. Inline and independent mass flow controllers (MKC Type 1179A) allowed finely tuned gas deposition over 15–30 minutes. Gas mixtures were deposited at 18 K either simultaneously or sequentially resulting in either mixed or layered thin films, respectively, of ca. 1–5 μm thickness. The co-crystal was formed by annealing the deposited mixed ice at 18 K, due to the high volatility of acetylene at temperatures close to the co-crystal formation. To achieve the desired 1:1 molecular ratio for the deposited ices and to determine film thickness, IR absorption band areas for the precursors were measured for calculation of molecular column densities.42 Here, literature band strengths (A′) for amorphous C2H2 (ν3: 23.9 × 10−18 cm molecule−1 at 3240 cm−1)43 and amorphous C5H5N (ν14: 7.47 × 10−18 cm molecule−1 at 1438 cm−1)44 determined using a similar transmission IR geometry to the present setup were applied.
A vacuum-compatible rotary platform allowed the optical window to be rotated 90° for transmission IR experiments and UV photolysis experiments. For IR data acquisition, mid-IR spectra (500–4000 cm−1) were collected by a Thermo iG50 FTIR spectrometer equipped with an internal SiC thermal light source and a liquid-N2 cooled mercury–cadmium–telluride detector. The spectra were recorded in a sequential loop using OMNIC 9 software, with each spectrum averaged over 64 individual scans at 4 cm−1 resolution or 1 cm−1 for systems of high band density. For photolysis experiments, the ice films were irradiated by a H2/D2 lamp (Hamamatsu L11798) for a total of 48 hours, either at 18 K or after crystallization, with IR spectra taken after 6, 24, 30 and 48 hours. Broad-band emission ranges from a low energy 160 nm peak to a lower intensity but still significant contribution at the Lyman α (Ly-α: 121.6 nm) line.45 The flux of VUV photons irradiating the 10 mm KBr substrate was measured to be of the order of ∼1012 photons cm−2 s−1via an O2–O3 actinometry procedure (see the ESI† for details).
2.2. Temperature programmed desorption (TPD)
Following the photochemistry experiments, temperature programmed desorption (TPD) experiments were performed where the thin-film samples were warmed to 300 K at 1 K min−1 with the composition of the evolved gas-phase observed online using a quadrupole mass spectrometer (QMS; Hiden Analytical HAL/3F RC 301 PIC) applying 70 eV electron impact ionization. The mass-to-charge ratio (m/z) was recorded as: (i) full scans between m/z = 4–300 amu to identify active mass channels and (ii) in multiple ion detection (MID) mode to determine the temporal evolution of specific mass signals. By correlating the sublimation profiles and fragmentation patterns for target molecules with the depletion of associated IR vibrational bands, the desorbed molecular ions can be identified at higher sensitivity and selectivity using the TPD method. Based on previous studies on pyridine:acetylene binary systems in the gas phase,4 photochemical products: 2-ethynyl pyridine+ (C7H5N), 2-vinyl-β-H-pyridine+ (C7H7N), quinoline+ (C9H7N) and quinolizinium+ (C9H8N) were targeted via their primary molecular cation (parent or fragment signal) at m/z = 103, 105, 129 and 130, respectively. The main parent cation signals for C2H2+ (m/z = 26) and C5H5N+ (m/z = 79) were tracked, as well as the possible binary product from C5H5N, m/z = 156 (C10H8N2). Literature data for all known species, at 70 eV impact energy, was obtained from NIST Chemical Webbook (https://webbook.nist.gov/; accessed: september 4th, 2023).2.3. Computational methods
Geometry optimization and harmonic frequency calculations for the pure molecular crystals and co-crystal systems were performed by periodic-density functional theory (p-DFT) using the CRYSTAL17 (C17) package.46 The B3LYP hybrid functional was supplemented with triple-ζ quality 6-311G(d) basis sets, including D3(BJ) dispersion and dampening terms to account for long-range interactions that are imperative for molecular crystal systems.47,48 This code has been previously used to calculate vibrational frequencies for molecular crystals of astrophysical interest49,50 and to simulate vibrational spectra at sufficient accuracy to assign experimental data.23 Total energy convergence was achieved with a threshold RMS displacement and energy gradient of 8.2 × 10−3 and 3.3 × 10−2 eV, respectively, enabling accurate calculation of frequencies at the harmonic level. Starting geometries for C17 input files were transcribed from published crystallography data for C5H5N (Cambridge Structural Database (CSD) Refcode: PYRDNA01),51 C2H2 (Refcode: ACETYL09; deuterium substituted for hydrogen)52 and the C5H5N:C2H2 cocrystal (Refcode: WAFNIB)53 were used. The final p-DFT optimized geometries of all crystals showed minor deviations in bond lengths and angles when compared to the experimental structures. Convergence to potential energy surface (PES) minima was confirmed by the absence of imaginary frequencies. Finally, harmonic frequency data were converted to theoretical IR spectra for comparison to experimental vibrational bands and assignment of vibrational modes. The calculated IR spectra were scaled by a factor of 0.965 to account for the harmonic approximation applied.23 Structures for the proposed photolysis products were first optimized with the Gaussian 09 package54 by DFT at the B3LYP/6-311++G (2d, 2p) level of theory including GD3 dispersion correction.48,55 From the optimized structures, the harmonic IR spectra were calculated using the same level of theory with a scaling factor of 0.965 applied to calculated frequencies.3. Results
The main objective of this work is to investigate the formation of NPAHs and their precursors upon the VUV irradiation (160–121 nm) of mixed C5H5N:C2H2 ices in the amorphous and co-crystal phase. Here, we first confirm the formation of the co-crystal by gas deposition (Section 3.1) using FTIR spectroscopy with p-DFT calculation of diagnostic vibrational modes. Next, we describe the main photoproducts formed from the C5H5N:C2H2 ice photolysis after processing the amorphous ice mixture at 18 K (Section 3.2.1) and C5H5N:C2H2 co-crystal, at both 90 and 110 K (Section 3.2.2).3.1. Co-crystal formation by gas deposition.
In Fig. 1, the C5H5N:C2H2 co-crystal IR spectrum (at 110 K) was compared to the IR spectra of pure C2H2 (at 70 K) and pure C5H5N (at 110 K). To validate and assign the vibrational modes corresponding to each phase, we calculated the IR spectra of the orthorhombic C2H2 crystal (space group Acam),52 orthorhombic C5H5N crystal (space group Pna21)51 and monoclinic C5H5N:C2H2 co-crystal (space group P21/n)53 by p-DFT. The resulting vibrational bands are shown as bars under each corresponding experimental spectrum (Fig. 1(A)).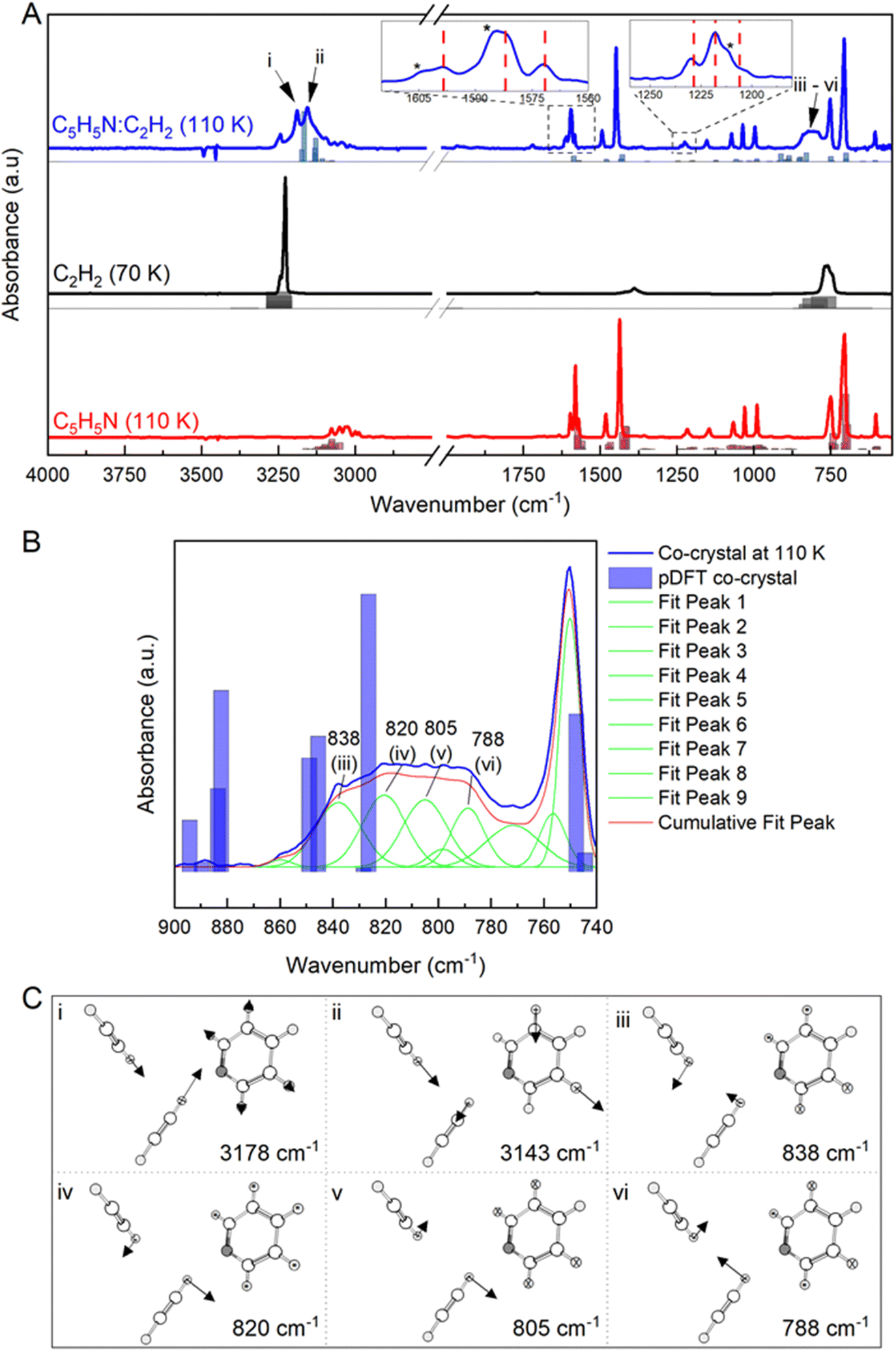 | ||
| Fig. 1 (A) Comparison between C5H5N:C2H2 co-crystal spectra at 110 K (blue) with pure C2H2 at 70 K (black) and pure C5H5N at 110 K (red). New bands attributed to the co-crystal include the CH stretching region at (i) 3178 and (ii) 3143 cm−1 and a broad feature centred at 810 cm−1 (iii–vi). The bands marked with (*) at 1603, 1584 and 1211 cm−1 indicate the vibrational modes of pyridine that undergo positional shift upon interaction with acetylene. The bars under the spectra correspond to the vibrational modes calculated by p-DFT for each phase based on the experimental crystal structures. (B) Deconvolution of the broad CCH bending feature in the co-crystal centred at 810 cm−1 resulting in four Gaussian component bands: 838 (iii), 820 (iv), 805 (v) and 788 (vi) cm−1. (C) Atom displacement plots for the co-crystal vibrational modes as confirmed by p-DFT calculated frequencies. Bands at (i) 3178 and (ii) 3143 cm−1 correspond to coupled C–H stretch of both C5H5N and C2H2 molecules within the co-crystal. The modes at at (iii) 838, (iv) 820, (v) 805 and (vi) 788 cm−1 correspond to CH bending modes. | ||
The appearance of new vibrational bands has been previously attributed as a diagnostic feature for co-crystal formation, resulting from a change in the molecular environment compared to the environment of the pure phases (e.g.25,27,31,32,56). In the present case, new bands were observed in the co-crystal spectrum (blue) at (i) 3178, (ii) 3143, and (iii–vi) a broad band centred ca. 810 cm−1 compared to the spectrum of C2H2 (black) and C5H5N (red) ices. Additionally, both red shifts (bathochromic) and blue shifts (hypsochromic) up to 6 cm−1 were observed for vibrational modes of the pure components, e.g. the bands at 1603, 1584 and 1211 (*) cm−1 (Fig. 1(A) and Table S1, ESI†). The vibrational mode at 1603 cm−1 can be attributed to a combination band (ν9 + ν10) of C5H5N, where the component normal vibrational modes ν9 and ν10 are expected to blue shift due to new H-bonds formed between C5H5N and C2H2, as previously observed for analogous systems.57
A similar effect is seen for the vibrational mode at 1584 cm−1, corresponding to the ring deformation (ν4) in C5H5N. The band at 1211 cm−1 corresponds to the ν16 in-plane bending mode in C5H5N, which has been previously observed in the amorphous phase at 1218 cm−1,43,58 but here red shifts upon crystallisation. IR spectra recorded during the temperature ramp between 20 and 160 K for the mixture of C2H2 and C5H5N are provided in Fig. S1 (ESI†). We also compared the co-crystal spectra with the spectra collected from a layered ice architecture, confirming a unique spectrum is obtained for the purported co-crystal system (Fig. S2, ESI†).
When comparing experimental spectra with p-DFT simulated vibrational profiles derived from empirical crystal structures, we found overall good agreement for both the pure component ices, as well as for the predicted co-crystal bands. To further model the vibrational character of each active mode attributed to the co-crystal, the broad 810 cm−1 feature was deconvoluted using Gaussian profiles and four components centred at (iii) 838, (iv) 820, (v) 805 and (vi) 788 cm−1 were fit, with a converged R2 of 0.998 (Fig. 1(B)).
The assignments for new bands identified in the co-crystal spectrum and the corresponding p-DFT calculated eigenvectors are shown in Fig. 1(C). The two highest wavenumbers at (i) 3178 and (ii) 3143 cm−1 were found to derive from the C–H stretching modes of C5H5N and C2H2 within the co-crystal. Similarly, the modes at (iii) 838, (iv) 820, (v) 805 and (vi) 788 cm−1 all correspond to the C–H bending of these components arranged within the co-crystal.
3.2. Photochemical products from ice irradiation.
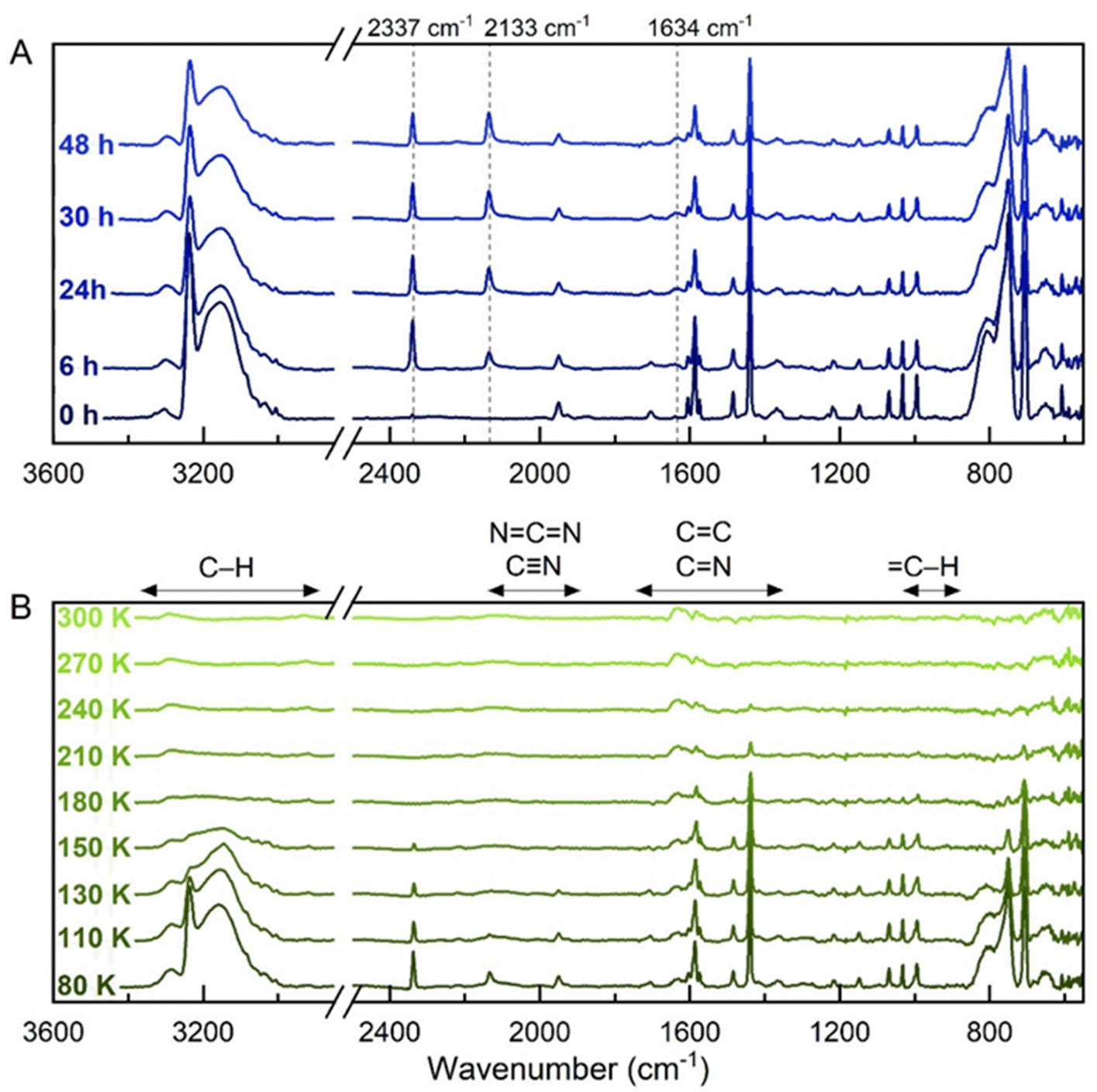 | ||
Fig. 2 Photochemical processing results after 48 hours VUV irradiation of the C5H5N and C2H2 mixed ice at 18 K. (A) IR spectra recorded after 0, 6, 24, 30 and 48 hours of irradiation. Dashed lines indicate build-up of CO2 (2337 cm−1) and CO (2133 cm−1) impurities, and of a band at 1634 cm−1. (B) IR spectra recorded during temperature ramp to 300 K. The main changes appear from 80 K onwards, with a progressive decrease in intensity for the main bands is observed to 300 K. Bands at 3290 and 2925 (C–H stretch), 1630 and 1588 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN stretches) cm−1 remain present at 300 K, indicating that refractory residues are formed. The vibrational mode regions for typical organic moieties are shown (B; top).59 C/CN stretches) cm−1 remain present at 300 K, indicating that refractory residues are formed. The vibrational mode regions for typical organic moieties are shown (B; top).59 | ||
During VUV photolysis at 18 K, a series of new bands at 2338, 2133 and 1634 cm−1 build up over the 48 h irradiation period. The first two bands are attributed respectively to atmospheric impurity carbon dioxide (CO2) and to carbon monoxide (CO); being the primary irradiation product of CO2. The deposition of ∼100 monolayers of CO2 is expected over the 48 h under our high-vacuum conditions, which attenuates the total UV flux reaching the target ice surfaces. Nevertheless, at early irradiation times interference from thin contamination layers will be negligible toward the bulk ice processing. As UV photons penetrate up to 800 monolayers of molecular ices,60 the presence of CO2 will only inflict minor hindrance to the later stages of the 48 h photolysis, when most processing would have already taken place. In the case of the band at 1634 cm−1, its intensity increase was accompanied by an overall decrease in the intensity of bands attributed to both component molecules (C2H2 and C5H5N), as well as the 3152 and 810 cm−1 bands from the mixed amorphous ice.
Next, the substrate was warmed to 300 K, where between 20 and 80 K, no changes were observed (Fig. S3, ESI†). From 80–180 K (Fig. 2(B)), the depletion of bands corresponding to the component molecules was observed, while above 200 K, there are identified bands at 1437, 989 and 710 cm−1 that slowly deplete until they vanish at 270 K. The bands at 3290, 2925, 1630 and 1588 cm−1 are attributed to C–H stretches and conjugated and aromatic CC or CN stretches that remain after temperature ramp up to 300 K, which are likely indicative of the formation of non-volatile residues.
The sublimation of molecules was also tracked by TPD mass spectrometry during the temperature ramp, with the detection of the targeted ion signals summarised in Fig. 3(A). The highest ion counts for the mass fragments among those tracked is m/z 103 (C7H5N+), with only trace levels recorded for m/z 105 (C7H7N+) and 130 (C9H8N+), and the absence of m/z 129 (C9H7N+) and 156 (C10H8N2+). The desorption of m/z 103 has an approximate onset of 155 K before reaching maximum counts at 225 K. The evaluation of a mass scan covering the 46–180 amu range during the TPD process confirmed that at peak temperature (225 K), the main ion signals correspond to m/z 52 attributed to C4H4+ and m/z 79 attributed to C5H5N+ of parent molecule C5H5N, and m/z 103 corresponding to the photoproduct C7H5N (Fig. 3(B)).
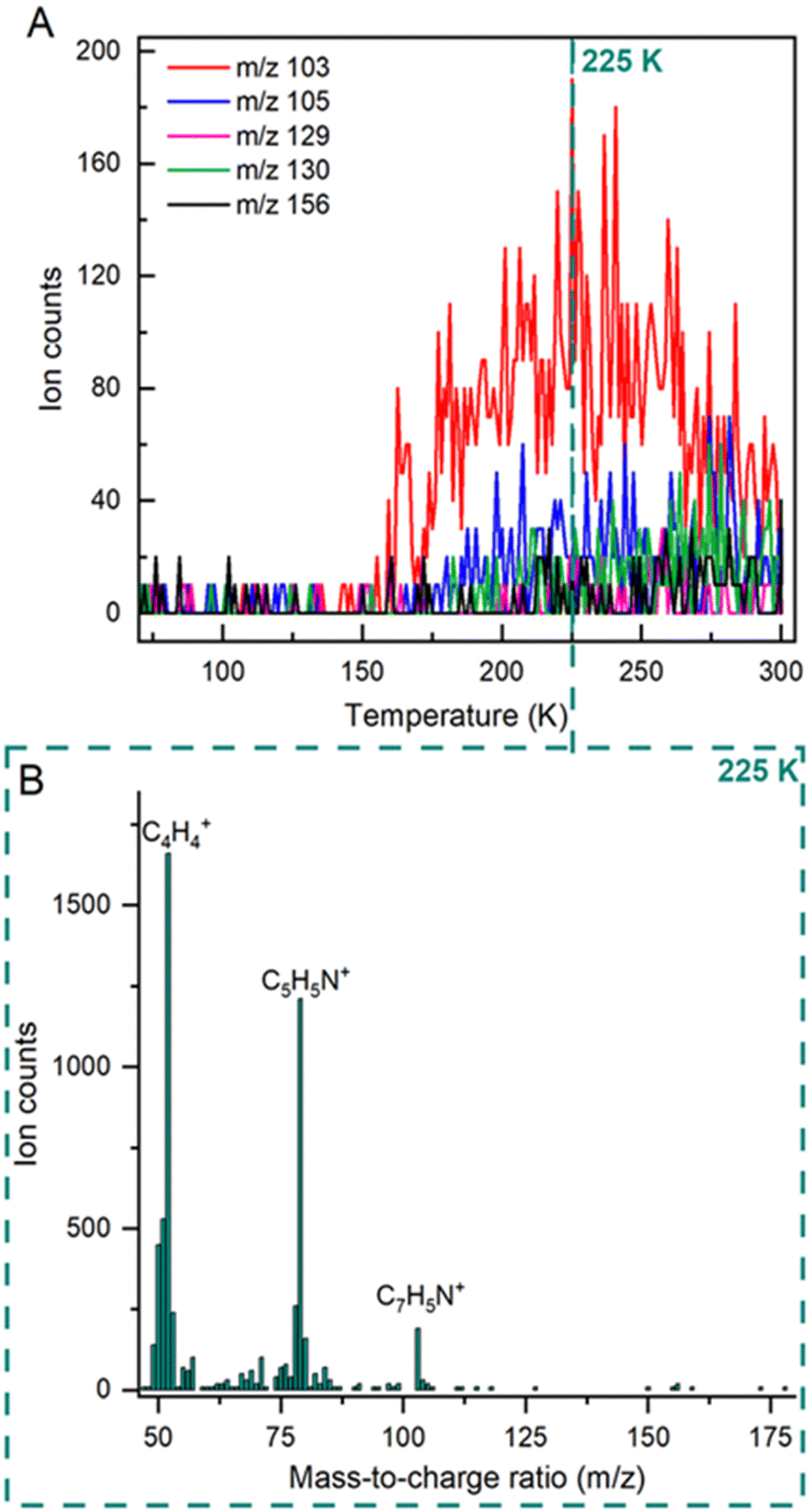 | ||
| Fig. 3 (A) Ion counts for target ions evaluated throughout by TPD between 70 and 300 K. The most abundant species, m/z 103, desorbs at onset 155 K, with only trace levels of m/z 105 and 130 detected and an absence of m/z 129 and 156 above the noise threshold. The peak ion count for the m/z 103 signal occurs at 225 K, as indicated by the reference line. (B) Mass spectrum between 46–180 amu recorded at 225 K, showing that the main ions present correspond to m/z 52 (C4H4+), m/z 79 (C5H5N+) and m/z 103 (C7H5N+). | ||
To determine the chemical identity of the m/z 103 C7H5N photoproduct, likely correlated to the IR profiles observed above 200 K, we calculated the IR difference spectrum from spectra taken at 210 and 240 K (Fig. 4) which encompasses the sublimation temperature range for this species. The difference spectrum shows the depletion of a broad feature centred at 3181 cm−1 as well as the depletion of bands at 3082, 3051, 3031, 1602, 1582, 1521, 1482, 1437, 1215, 1183, 1029, 988, 872, 747, 708, 670 and 605 cm−1.
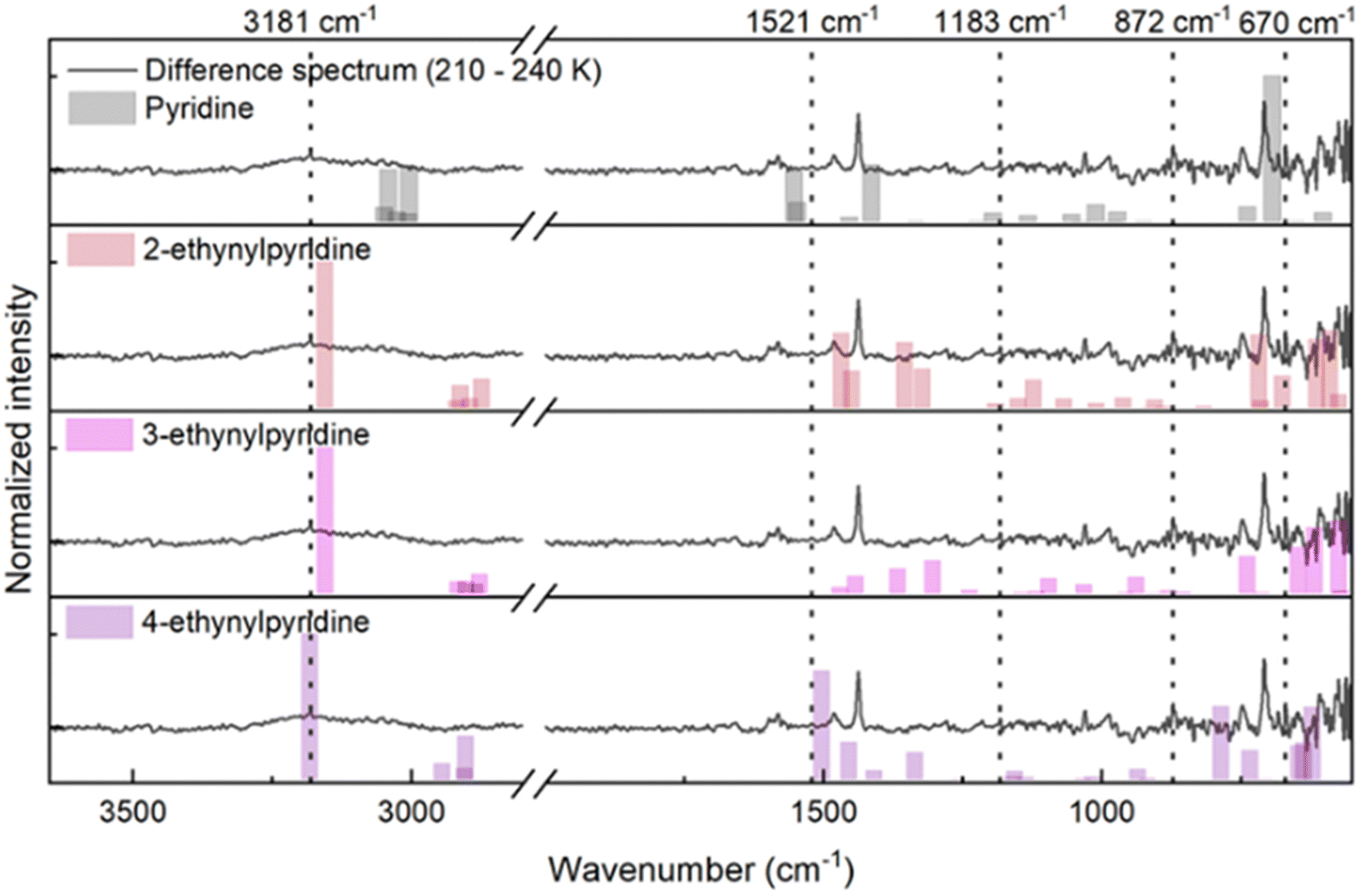 | ||
| Fig. 4 Difference spectrum (grey: 210–240 K) for the VUV photolysis of the C5H5N:C2H2 amorphous ice with calculated harmonic frequencies for pyridine, 2-ethynylpyridine, 3-ethynylpyridine and 4-ethynylpyridine (bars; top to bottom). Bands at 3082, 3051, 3031, 1602, 1582, 1482, 1437, 1215, 1029, 988, 747, 708, and 605 cm−1 are assigned to pyridine (top), whereas bands at 3181, 1521, 1183, 872 and 670 cm−1 (dashed lines) are assigned to isomers of ethynylpyridine. For the substituted pyridines, best agreement between calculated bands and those observed experimentally occurs for 2-ethynylpyridine (second) and 4-ethynylpyridine (bottom). | ||
Considering that the desorption of pyridine also occurs within this range (Fig. 3(B)), we neglect pyridine signatures from the difference spectrum by considering the bands reported for the crystalline phase (Table S1, ESI†) and calculated single molecule harmonic frequencies (Fig. 4 – top). The DFT calculated IR spectrum of pyridine shows good agreement with bands reported previously in condensed phase experiments,44 allowing us to assign the bands at 3082, 3051, 3031, 1602, 1582, 1482, 1437, 1215, 1029, 988, 747, 708, and 605 cm−1 to pyridine.
Finally, we compared the remaining bands at 3185, 1521, 1183, 872 and 670 cm−1 in the difference spectrum with the single molecule harmonic IR frequencies of 2-ethynylpyridine, 3-ethynylpyridine and 4-ethynylpyridine (Fig. 4) calculated by DFT. As C5H5N shows good agreement between the theoretical gas-phase monomer frequencies with the experimental band positions of the condensed molecular film, we assume that the substituted pyridines will also display only minor deviations. As a result, the closest fit between the remaining experimental bands and calculated frequencies occurs for 2-ethynylpyridine and 4-ethynylpyridine, suggesting that a mixture between these isomers could form during VUV photolysis of the binary ice leading to the m/z 103 ion signal in the TPD.
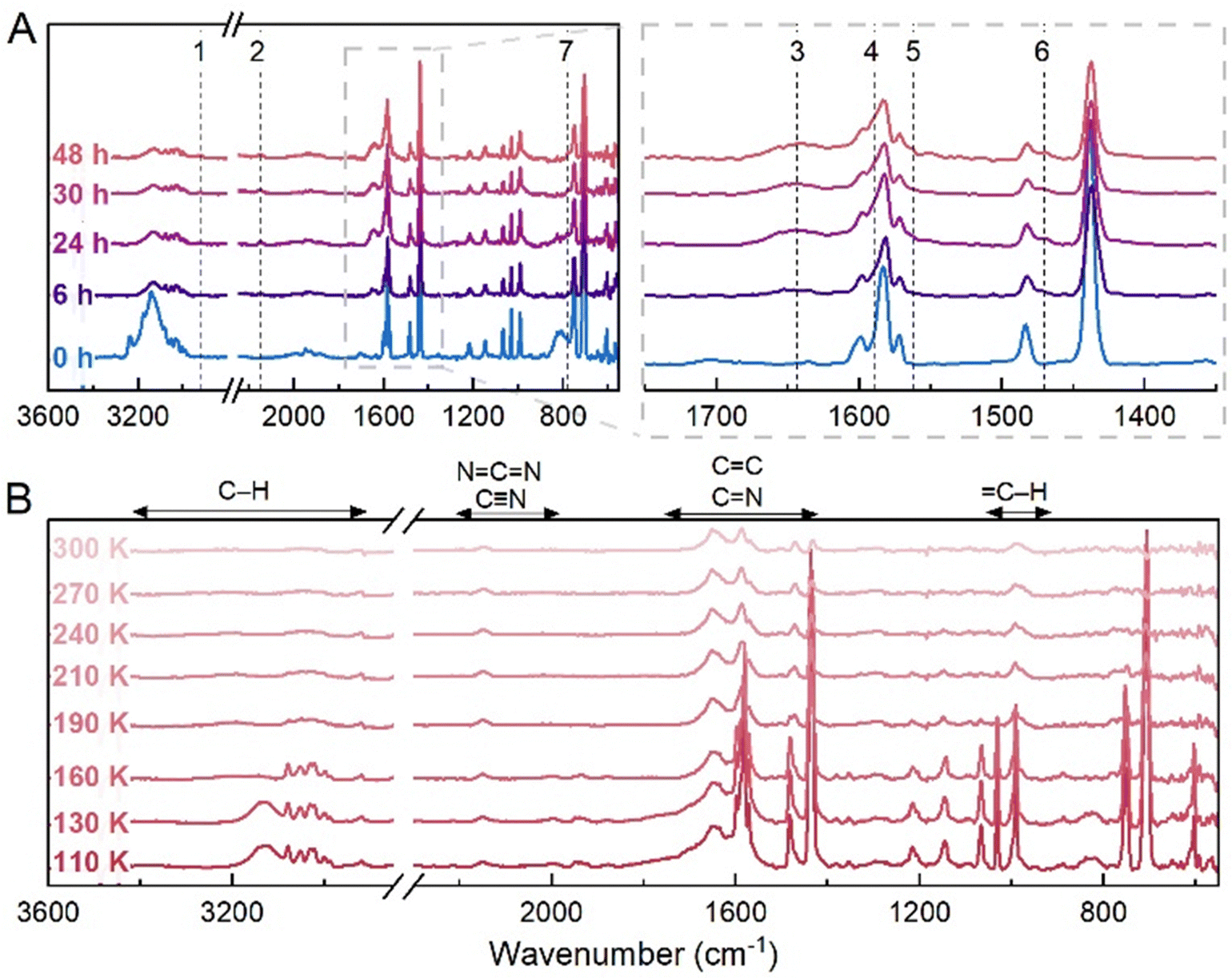 | ||
Fig. 5 Photochemical processing results after 48 hours VUV irradiation of the C5H5N:C2H2 co-crystal at 110 K. (A) IR spectra recorded after 0, 6, 24, 30 and 48 hours of irradiation. Dashed lines indicate the most prominent changes occurring in the spectra, with the appearance of bands at (1) 2921, (2) 2149, (3) 1643, (4) 1589, (5) 1561, (6) 1470 and (7) 776 cm−1. (B) IR spectra recorded during temperature ramp to 300 K. The main changes appear from 130 K onwards, where the co-crystal bands are observed to completely deplete. Above 160 K, bands corresponding to the vibrational modes of C5H5N also deplete, followed by a slow decrease in intensity for bands at 3079, 3052, 3028, 1437, 1148, 992, 775, 746 and 707 cm−1 to 300 K. Bands at 3003 (CH stretch), 2929 (CH stretch), 2143 (NCN or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch), 1651, 1587, 1568 (sh), 1504, 1467, 1430 (conjugated and aromatic CC or CN stretches), 989 (C–H deformation) cm−1 remain present at 300 K, indicating the formation of refractory residues. The vibrational mode regions for typical organic moieties are shown (B; top).59 N stretch), 1651, 1587, 1568 (sh), 1504, 1467, 1430 (conjugated and aromatic CC or CN stretches), 989 (C–H deformation) cm−1 remain present at 300 K, indicating the formation of refractory residues. The vibrational mode regions for typical organic moieties are shown (B; top).59 | ||
This alteration of bands suggests a depletion of the co-crystal and possible alteration of the crystalline environment. The depletion can be explained as a consequence of the present experimental conditions. Despite having been previously evaluated as a stable co-crystal system in the 90–150 K temperature range,31 we observed that the co-crystal is unstable at 110 K (Fig. S4, ESI†) under our conditions, with considerable depletion within 30 minutes of its formation. Nevertheless, the continued VUV irradiation of the increasingly amorphous solid resulted in the appearance of new bands at 2921, 2149, 1643, 1589, 1561, 1470 and 776 cm−1, which are distinct from those observed from the irradiation of the C5H5N and C2H2 mixed ice system at 18 K.
During temperature ramping (Fig. 5(B)), the decrease in intensity of bands corresponding to the component C5H5N and C2H2 molecules is primarily observed up to 190 K. This precedes the slow depletion of bands at 3079, 3052, 3028, 1437, 1148, 992, 775, 746 and 707 cm−1 between 190 and 240 K. Lastly, bands at 3003 (CH stretch), 2929 (CH stretch), 2143 (NCN or CN stretch), 1651, 1587, 1568 (sh), 1504, 1467, 1430 (conjugated and aromatic CC or CN stretches), 989 (C–H deformation) cm−1 persist at temperatures to 300 K, which indicates the formation of refractory residues.
Correlating with band changes observed in the IR spectra, the release of sublimed material was followed by obtaining ion counts for targeted molecular ions over the TPD experiment (Fig. 6(A)). Here, in contrast with the 18 K amorphous irradiation, the species with the overall highest ion counts is m/z 156, which displays a desorption onset at 200 K. Additionally, ions m/z 103, 105 and 130 were detected above the noise threshold between 150 and 200 K. Scanning the 90–180 amu mass range was performed at: 166 K, where the highest counts were recorded for m/z 130 (Fig. 6(B)); 225 K to allow comparison between sublimed species at this temperature with the earlier performed mixed ice profile (Fig. 6(C)); and at 266 K, where m/z 156 is the dominant species in the ion counts (Fig. 6(D)).
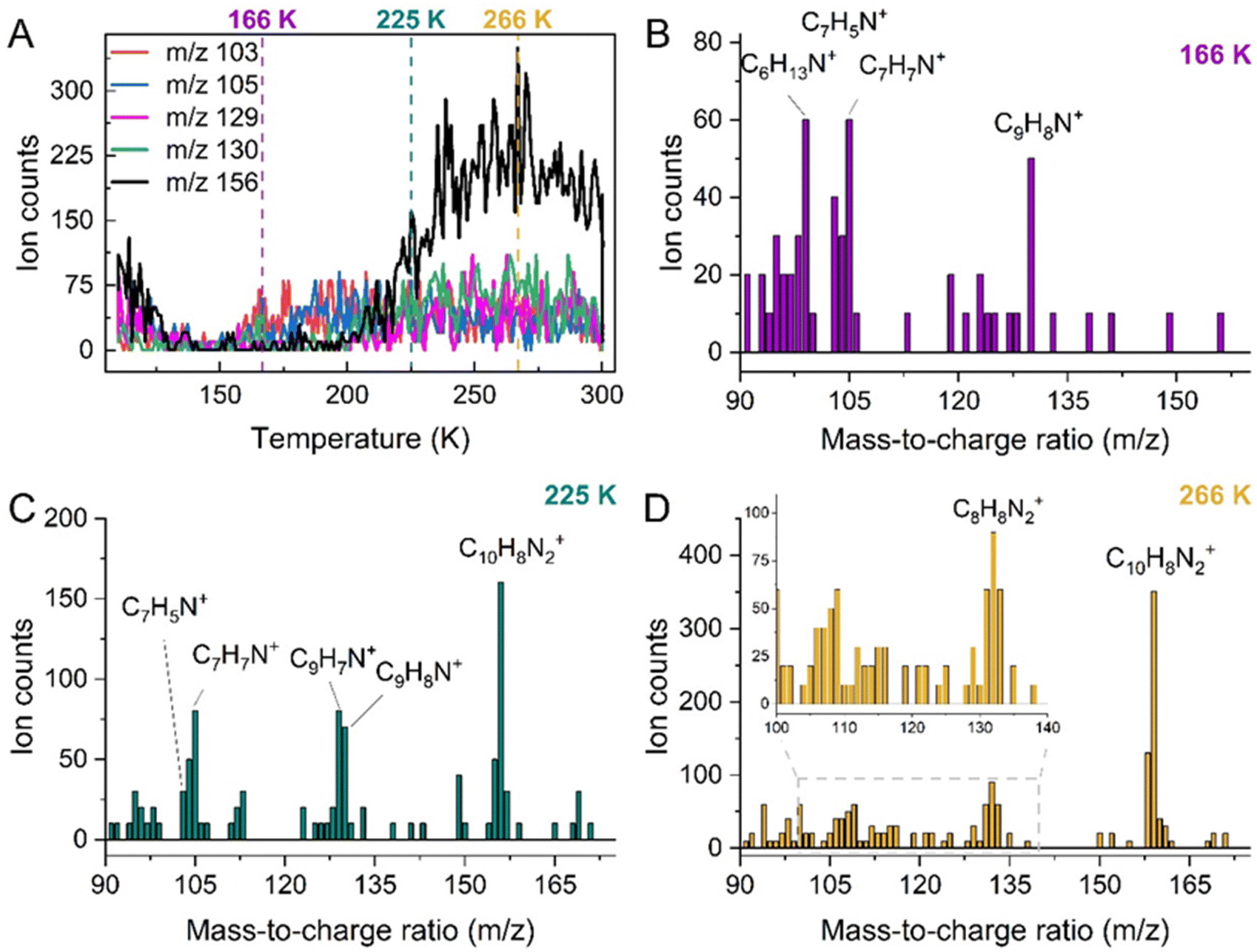 | ||
| Fig. 6 (A) Ion counts for target ions evaluated by TPD between 110 and 300 K. The most abundant species, m/z 156, desorbs at onset 200 K. Lower maximum counts were obtained for species m/z 103, 105 and 130, however, these were the most prominent ions between 150 and 200 K. No significant levels of m/z 129 and 156 were detected within this temperature range. Dashed lines indicate the temperatures at which the 90–175 amu mass scans were performed: 166, 225 and 266 K. (B) Mass spectrum at 166 K, indicating prominent ion counts at m/z 99 (C6H13N+); m/z 103 (C7H5N+); m/z 105 (C7H7N+); and m/z 130 (C9H8N+). (C) Mass spectrum at 225 K showing the presence of: m/z 103 (C7H5N+); m/z 105 (C7H7N+); m/z 129 (C9H7N+); m/z 130 (C9H8N+); and m/z 156 (C10H8N2+). (D) Mass spectrum at 266 K showing the presence of: m/z 132 (C8H8N2+); and m/z 156 (C10H8N2+). | ||
At 166 K, we detected the presence of species with m/z 103, 105 and 130, which correspond to photoproducts with empirical formulas C7H5N, C7H7N and C9H8N, respectively. Additionally, a signal with m/z 99 was detected and tentatively attributed to C6H13N+, although this species possesses a much higher H/C ratio than the other ions. At 225 K, the molecular ions m/z 103, 105, 129, 130 and 156 are all present, corresponding to C7H5N+, C7H7N+, C9H7N+, C9H8N+ and C10H8N2+. Lastly, the m/z 156 signal dominates the mass scan recorded at 266 K, corresponding to the C10H8N2+ molecular ion. Note the presence of m/z 132 was also detected above the noise threshold, which is attributed to the C8H8N2+ ion.
Due to the large number of different molecular ions identified by TPD over the temperature range of 150–200 K, it is difficult to correlate the identity of subliming photoproducts to the carriers of IR bands that undergo concomitant depletion. However, above 200 K, the most prominent molecular ion observed in the gas phase has a mass of 156 amu, which most likely corresponds to a parent molecule comprising two bonded pyridine rings with a molecular formula C10H8N2.
Compiling the IR difference spectrum between temperatures 250 and 275 K, two broad bands centred at 3220 and 3031 cm−1, as well as narrower bands at 2917, 2849, 1659, 1583, 1482, 1437, 1152, 992, 764, 705, 680, 653 and 617 cm−1, are associated with molecules desorbed over this temperature range (Fig. 7). Therefore, to confirm the identity of the m/z 156 photoproduct, we compare the IR bands of the sublimed species with single molecule harmonic frequencies for 2,2-bipyridine, 2,3-dipyridyl, 2,4-bipyridine, 3,3-bipyridine and 4,4-bipyridine (Fig. 7). After compiling the DFT calculated band positions for the bipyridines, the closest agreement between experiment and theory is for 2,3-dipyridyl, followed by 2,4-bipyridine and 3,3-bipyridine. However, the overlapping group modes for the structural isomers suggest that a distribution of bipyridine compounds is likely formed from the VUV exposure. Finally, the broad bands at 3031 and 1583 cm−1 could correspond to pyridine precursor trapped in a solid bipyridine matrix, while the few unassigned bands correspond to unknown species undergoing sublimation; the identity of these species was not further investigated.
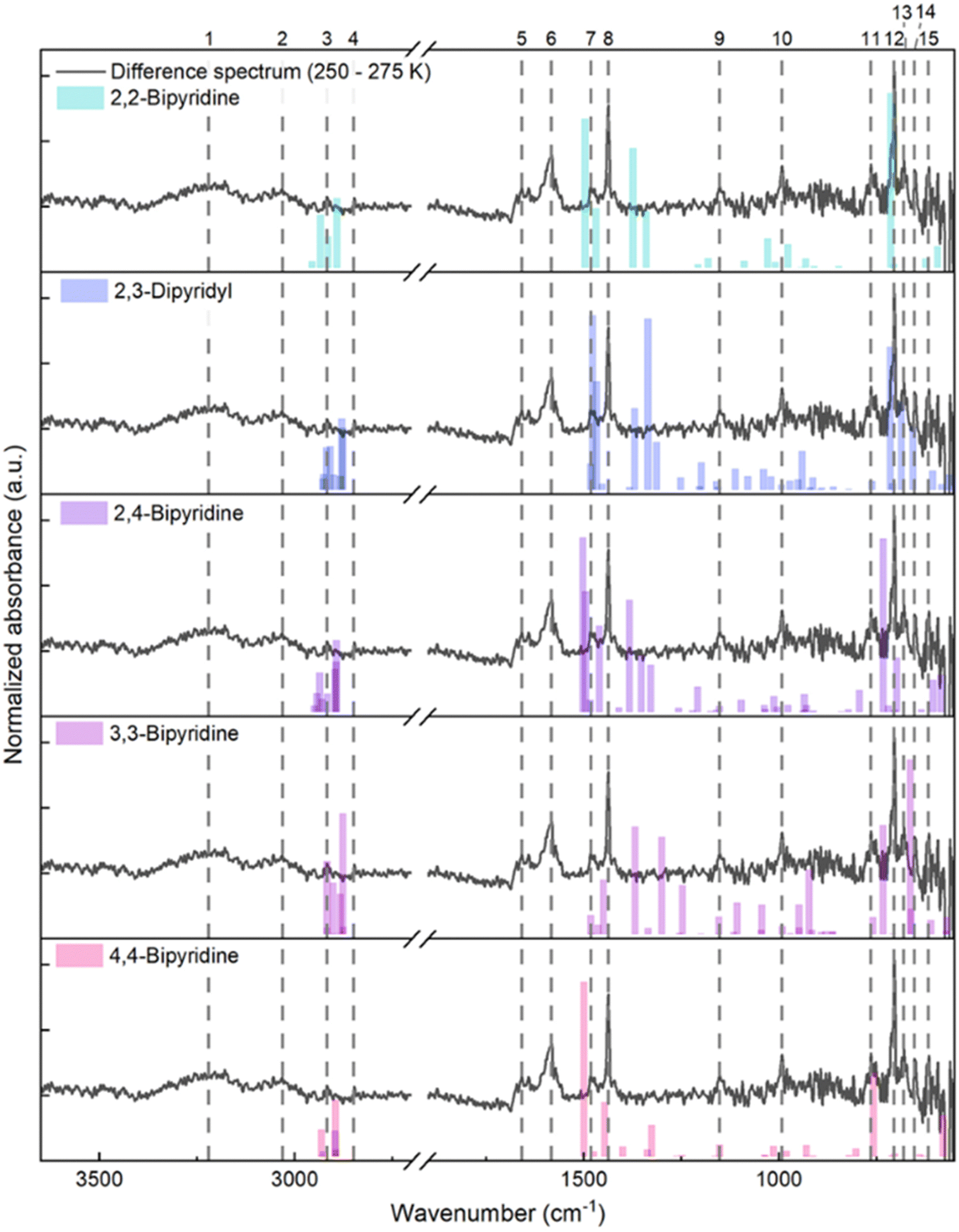 | ||
| Fig. 7 Difference spectrum (grey: 250–270 K) for the VUV photolysis of the C5H5N:C2H2 co-crystal ice at 110 K with the calculated harmonic frequencies for 2,2-bipyridine, 2,3-dipyridyl, 2,4-bipyridine, 3,3-bipyridine and 4,4-bipyridine (top to bottom). The difference spectrum shows two broad bands centred at (1) 3220 and (2) 3031 cm−1, and bands at (3) 2917, (4) 2849, (5) 1659, (6) 1583, (7) 1482, (8) 1437, (9) 1152, (10) 992, (11) 764, (12) 705, (13) 680, (14) 653 and (15) 617 cm−1, indicated by the dashed lines. Comparison to each bipyridine isomer reveals closest agreement with 2,3-dipyridyl, followed by 2,4-bipyridine and 3,3-bipyridine. Unassigned bands at 3220, 3031, 1659 and 1583 cm−1 correspond to unknown photo-product species that sublime in this temperature range. | ||
The IR spectra were collected up to 48 hours to identify changes over the exposure period (Fig. 8(A)), where after 6 h the co-crystal C–H stretch modes at 3177 and 3144 cm−1 were again observed to broaden forming an unresolved feature centred at 3132 cm−1. This indicates that the interaction between the hydrogen of C2H2 and the nitrogen of C5H5N within the co-crystal changed, impacting their associated CH stretch and bending vibrations. After 48 h, IR bands at 2921, 1644, 1470 and 977 cm−1 emerge, similar to the series of new bands observed during the irradiation of the co-crystal at 110 K.
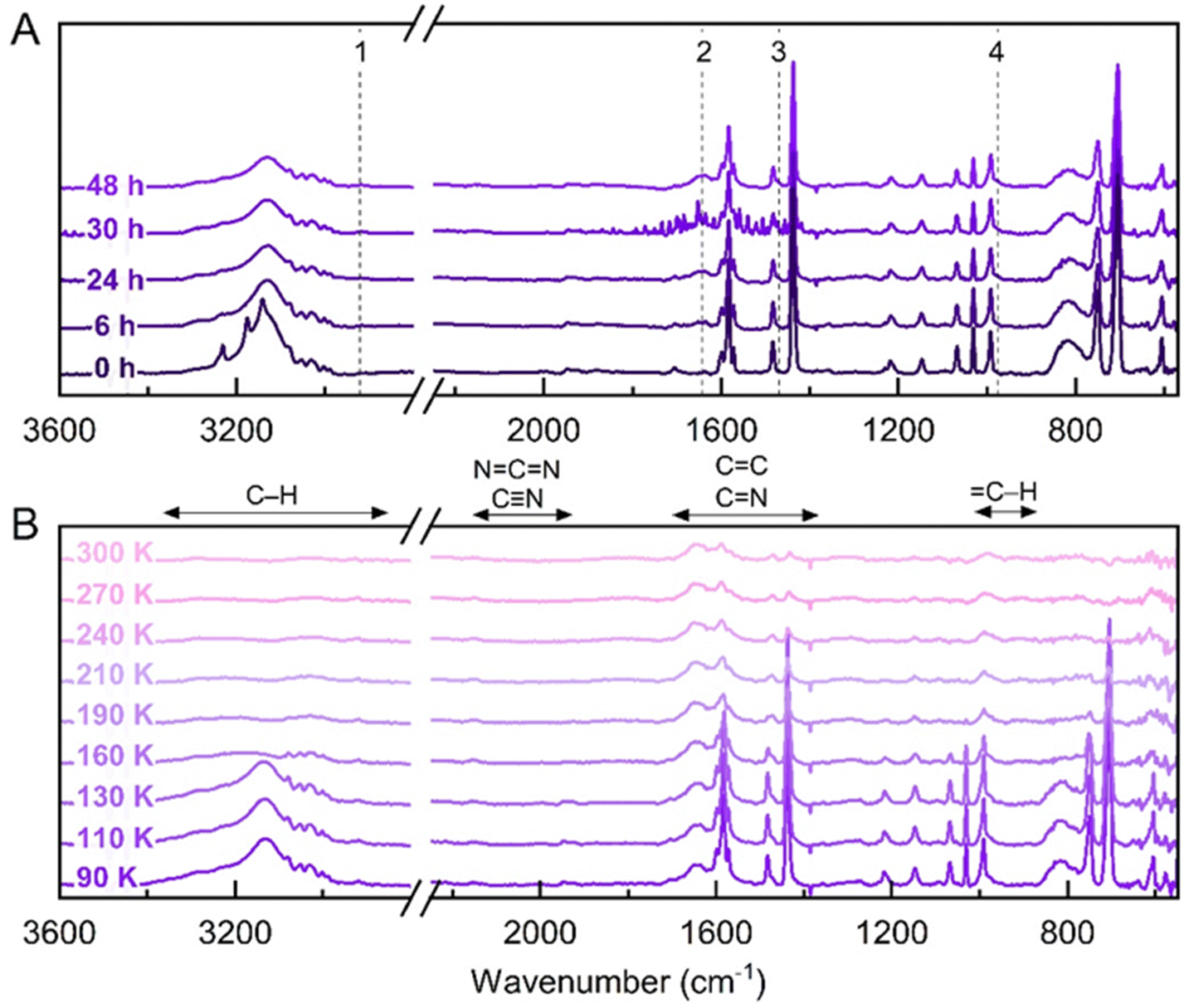 | ||
| Fig. 8 Photochemical processing results after 48 hours VUV irradiation of the C5H5N:C2H2 co-crystal at 90 K. (A) IR spectra recorded after 0, 6, 24, 30 and 48 hours of irradiation. Dashed lines indicate the most prominent changes occurring in the spectra, with the appearance of bands at (1) 2921, (2) 1644, (3) 1470, (4) 977 cm−1. The spectrum taken after 30 hours shows atmospheric water interference caused by variations in the dry N2 supply to the IR detector. (B) IR spectra recorded during temperature ramp to 300 K. The main changes appear from 130 K onwards, where the co-crystal bands are observed to completely deplete. After 160 K, bands corresponding to the vibrational modes within C5H5N also deplete, followed by a slight decrease in intensity for bands at 3201, 1295, 1214, 749, 706 and 610 cm−1 to 300 K. Bands at 3281, 3032, 2917 (CH stretch); 2155 (NCN or CN) 1650, 1583, 1471, 1437 (CC or CN stretch); 1149, 991 (C–H deformation) cm−1 remain present at 300 K, indicating refractory residues are formed. The vibrational mode regions for typical organic moieties are shown (B; top).59 | ||
During the temperature ramp to 300 K (Fig. 8(B)), a similar alteration of band profiles was observed for the co-crystal at 110 K. Here, fundamental bands associated with both component molecules decrease in intensity until 190 K. Remaining after C5H5N and C2H2 sublimation are bands at 3281, 3032 and 2917 cm−1 (CH stretch); 2155 cm−1 (NCN or CN stretch); 1650, 1583, 1471, 1437 cm−1 (conjugated or aromatic CC or CN stretch); and 1149 and 991 cm−1 (CH deformation), mostly corresponding to pure C5H5N vibrational modes. The remaining bands still visible at 300 K indicate the formation of non-volatile residues from this photolysis.
As for the aforementioned C5H5N:C2H2 systems, the release of volatile species was followed by their target ions evaluated throughout the TPD procedure. However, in striking contrast to the mixed amorphous ice and co-crystal irradiated at 110 K, there were no significant ion counts detected for any of the programmed photoproducts possessing m/z values above 82 amu (Fig. S5, ESI†).
4. Discussion
4.1. Effect of UV irradiation on the C5H5N:C2H2 ices
Arising over 48 h of VUV exposure, we report the appearance of new IR features from the various ice systems, as summarized in Table 1.| Ice | New bands within 48 h of irradiation (cm−1) | Non-volatile residues at 300 K (cm−1) |
|---|---|---|
| Mixed (18 K) | 1634 | 3290, 2925, 1630, 1588 |
| Co-crystal (90 K) | 2921, 1644, 1470, 977 | 3281, 3032, 2917, 2155, 1650, 1583, 1471, 1437, 1149, 991 |
| Co-crystal (110 K) | 2921, 2149, 1643, 1589, 1561, 1470 and 776 | 3003, 2929, 2143, 1651, 1587, 1568 (sh), 1504, 1467, 1430, 989 |
The broadband VUV lamp used in the experiments emits photons between 121.6–160 nm, which interact primarily with the C5H5N component, as evidenced by the VUV absorption cross-sections for the two main ice constituents.61–63 Additionally, previous photolysis studies showed that Lyman-α photons (121.6 nm) possess sufficient energy to ionize C5H5N,62 inferring that ion–molecule reactions could propagate within the irradiated ice environment. Consequently, over 48 hours of irradiation, photolytic reactions occurring within amorphous and crystalline phases can be first assessed by analysing the depletion of C5H5N over time. Thus, the photolytic destruction of C5H5N can be determined following the equation:42
| N(φ) = N0exp(−φσph) | (1) |
| N(φ) = A0 + A1φ + A2[exp(−φσph)] | (2) |
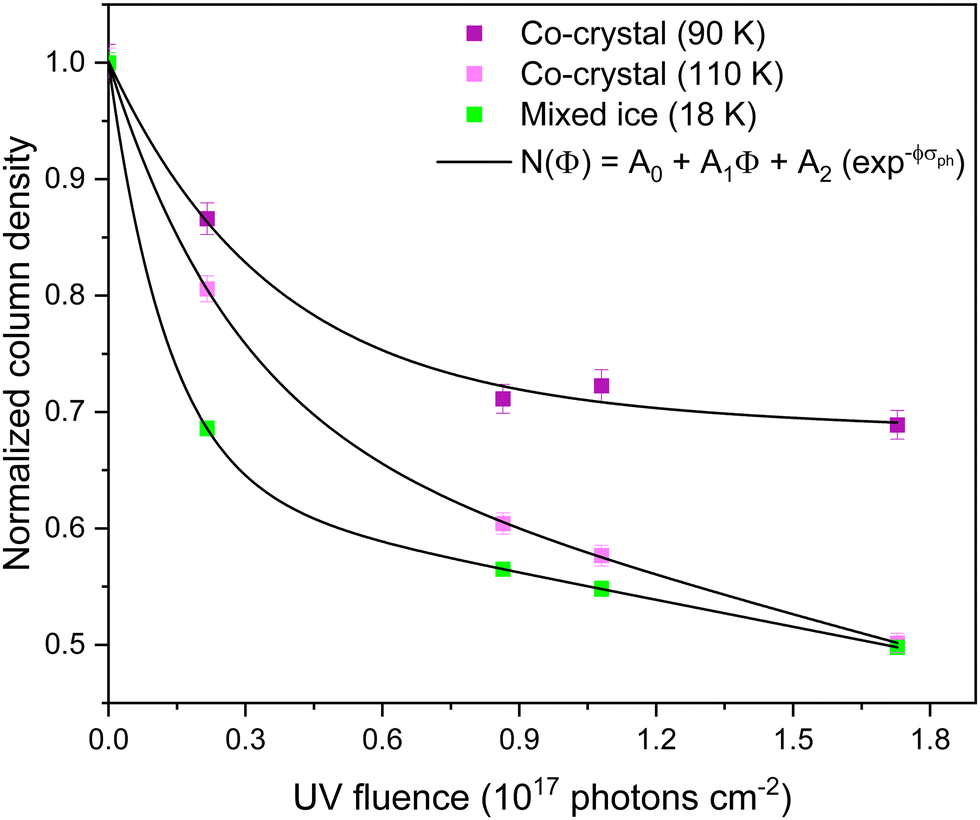 | ||
| Fig. 9 Normalized column density of pyridine calculated using the area of its ν14 vibrational band, as a function of UV fluence for the: C5H5N:C2H2 co-crystal irradiated at 90 K (purple; σph ∼ 2.84 (±1.27) × 10−16 cm2); C5H5N:C2H2 co-crystal irradiated at 110 K (pink; σph ∼ 3.59 (± 0.14) × 10−16 cm2) and mixed C5H5N and C2H2 ice irradiated at 18 K (green; σph ∼ 7.60 (±0.03) × 10−16 cm2). Pyridine is depleted in all systems through both photolysis and photo-desorption processes. | ||
From the C5H5N depletion data, the photolysis cross-sections are measured to be 7.60 (± 0.03), 3.59 (± 0.14), and 2.84 (± 1.27) × 10−16 cm2 for the C5H5N and C2H2 mixed ice irradiated at 18 K, the C5H5N:C2H2 co-crystal irradiated at 110 K, and at 90 K, respectively. The overall behaviour of the photolysis plots suggests a more rapid C5H5N depletion for the amorphous mixed ice and co-crystal at 110 K and slower depletion of pyridine for the 90 K system.
First considering the amorphous ice at 18 K, the formation of a new band at 1634 cm−1, combined with a faster depletion rate of C5H5N, indicates that ionised C5H5N rapidly reacts with neighbouring C2H2 molecules to form the observed ethynyl-pyridine products (m/z 103). The absence of photoproducts with higher mass is explained by the absence of excess of adjacent C2H2 molecules, preventing further reactions that would lead to the formation of NPAHs, and the low temperature environment reducing the mobility of reactive intermediates within the ice. This is consistent with what has been previously observed by Räp et al. (2022) for the reaction between C5H5N˙+ and C2H2 in the gas phase at 150 K, where it was seen that for lower concentrations of C2H2, the main product was 2-ethynyl-H-pyridine. However, upon a stepwise increase in acetylene abundance, the authors detected 2-vinyl-β-distonic-H-pyridine (m/z 105) followed by quinolizinium (m/z 130), validating the proposed sequential acetylene addition pathways toward NPAH formation.
In the case of the C5H5N:C2H2 co-crystal irradiated at 110 K, the availability of C2H2 for solid-state reactions changes due to its diffusion through the ice, caused by the instability of the co-crystal at this temperature. The co-crystal instability indirectly leads to a greater C5H5N depletion rate when compared with the ice at 90 K, reflecting a more dynamic environment that favours the formation of a broader range of photoproducts. These include a suite of ethynylpyridines, vinylpyridines and the quinolizinium species observed in the TPD measurements, as well as bipyridines. In the latter case, as the desorption of C2H2 continues, the formation of bipyridines (and potentially higher-order pyridine polymers) is favoured due to a C5H5N excess remaining in the ice. This is consistent with the bipyridine products observed in the irradiation of pure pyridine ice at the same 110 K temperature (Fig. S6, ESI†).
On the other hand, for the C5H5N:C2H2 co-crystal irradiated at 90 K, the lower photolysis cross-section indicates that recombination is a significant process occurring within the ice, further evidenced by the absence of higher mass photoproducts in the TPD results. This suggests that the diffusion of C5H5N˙+ and C2H2 molecules in the 90 K co-crystal is limited, as the position of reactive species is effectively locked within the highly ordered crystalline environment, where the distance between reactants in the crystal structure (between 2.46 and 2.53 Å)31 is sufficient to suppress two-body interactions. The slower decay of crystalline ices, compared to their amorphous counterparts, when exposed to ionizing radiation has been previously reported.65–70 This effect has been attributed to intermolecular interactions that stabilise the phase against radiation-induced decay, while also noticing that at higher temperatures the recombination process within the ices is favoured,65,66,70 which agrees with our results. Nonetheless, C5H5N is observed to deplete, even if at a lower rate compared to the 110 K co-crystal. Its consumption can be explained by photo-desorption processes at the surface, and by chemical processing as indicated by the appearance of new bands that remain present at 300 K, being directly attributed to the formation of refractory products.
The similarity of the IR profiles attributed to the non-volatile products formed in C5H5N:C2H2 co-crystals irradiated at both 90 and 110 K temperatures, and within the pure pyridine ices confirms that a similar set of reaction pathways are taking place (Fig. S7, ESI†), indicating that the polymerisation of pure pyridine in both co-crystals is the most likely source of such refractory residues. A similar residue IR spectrum was also obtained for the mixed amorphous phase irradiated at 18 K. However, this material appears absent of vinyl/vinylene groups (associated with C–H deformation modes) and shows overall lower band intensities. Further attempts to characterize the residue materials require ex situ analysis methods that will be performed in follow-up work.
4.2. Implications for Titan
The formation of ethynylpyridines, vinylpyridines and the NPAH quinolizinium within the investigated C5H5N:C2H2 cryominerals reassures that VUV-driven condensed phase reactions are accessible under both ISM and Titan atmosphere conditions. It has been previously suggested that the gas-phase ion–molecule reaction between C5H5N˙+ and C2H2 occurs in Titan's ionosphere where VUV photons are plentiful.71 Here, our results suggest that a similar chemistry could occur in the Titan stratosphere, where transport of higher-order organics and low temperature may promote the co-condensation of C5H5N and C2H2 to form composite ices, potentially in the form of a co-crystal.At these altitudes, as VUV irradiation is largely attenuated, Galactic Cosmic Rays (GCRs) particles play a more important role in depositing energy to ice materials and driving condensed-phase chemistry. At the same time, previous comparisons of the effect of VUV irradiation with electron impact ionization on ice chemistry showed that the formed products are overall similar, especially in the case of high-energy electron bombardment.64 As a result, we can assume that the solid-state reactions observed in our experiments are similarly representative of what would be observed within Titan's stratosphere.
Interestingly, previous gas-phase studies have reported the rapid destruction of C5H5N in the presence of excited N atoms under Titan-relevant conditions,72 affecting its overall abundance and impacting its ability to be delivered to Titan's surface. In the present work, we highlighted that the stability of a co-crystal at a given temperature has a major influence on solid-state chemistry, a property that can modify the product abundance ratios. Our results suggest that upon forming a stable co-crystal structure between C5H5N and C2H2 in the solid state, a slower depletion for C5H5N is observed. This may indicate that if effectively encased in a condensed material, the biologically important pyridine molecule can be preserved and delivered to Titan's surface for possible involvement in prebiotic chemistry.
It is then expected that most higher-order photochemical and radiolytic products generated within ice structures on Titan will remain intact during ice particle precipitation, since the energy of solar UV photons decreases with altitude. Hirai et al. (2023) have analysed the behaviour of similar organic residues after deposition on simulated methane lakes, indicating that aromatic compounds within organic aerosols either fractionate or coagulate into particulates upon wet-dry cycling, which likely contribute icy organic minerals located within the Titan landscape. Based on our laboratory simulations, we predict that abundances of higher-order organics derived from pyridine precursors, including NPAH compounds, likely have accumulated on Titan's surface over geological timescales, which may now be uncovered by future exploration including the upcoming Dragonfly mission.73
5. Conclusions
In the present study, we investigated the formation of NPAHs and pyridine-based NPAH precursors by VUV irradiation of the amorphous ice formed between C5H5N and C2H2 at 18 K, as well as the C5H5N:C2H2 co-crystal structure studied at 90 and 110 K. The co-crystal formation between C5H5N and C2H2 was confirmed via FTIR bands matched to p-DFT frequencies calculated from its known crystal structure.Our data shows that the irradiation of C5H5N:C2H2 ices, both in the amorphous phase and as a dynamic co-crystal under Titan stratospheric temperature, leads to the formation of: (i) ethynylpyridines, important precursors to NPAHs such as quinoline, (ii) vinylpyridines, and (iii) quinolizinium. When stabilized at 90 K, the co-crystal showed slower depletion of pyridine, which has direct implications for the delivery of this molecule to Titan's surface.
To our knowledge, this study is the first example of low-temperature photochemistry being performed on a co-crystal molecular ice system. As opposed to the inhomogeneous distribution of precursors in binary amorphous ices, or systems showing the segregation of molecular components into distinct phases upon warming, the formation of a co-crystal phase necessitates complete mixing of precursor material allowing transition to the co-crystal lattice. This unique crystalline arrangement leads to a narrow distribution of higher-order organic compounds, synthesized under VUV or particle irradiation. Additionally, non-volatile residues were also formed from all ice systems investigated, most likely due to pure pyridine polymerization. In a Titan context, these macromolecular compounds, harboured within precipitating aerosols, are transported to the surface to contribute to Titan's terrain of diverse cryominerals.
Studies toward the photochemistry of other pyridine-based co-crystals of potential relevance to Titan environment are to follow, which will advance our understanding of the formation and potential delivery of biologically important NPAHs to Titan's surface. Following these studies, we can provide targets for future reconnaissance of Titan's surface, where compounds central to the molecular origin of life may currently reside.
Author contributions
C. E. and L. L. C. conceived the experimental concept. L. L. C. conducted laboratory research and drafted the original manuscript. All authors contributed providing theoretical and experimental insight. H. E. M.-C. was the lead on crystallography content. All authors have reviewed and approved the final version of the manuscript. Corresponding author C. E. obtained funding for the research.Data availability
Further experimental data are available in the ESI.†Conflicts of interest
The authors confirm that there are no conflicts to declare.Acknowledgements
This research is supported by the Marsden Fund Council from Government New Zealand funding, managed by Royal Society Te Apārangi (Proposal: 21-UOO-123), and by an AINSE Ltd. Postgraduate Research Award (PGRA). We wish to additionally thank New Zealand eScience Infrastructure (NeSI) for high-performance computing resources (Project UOO03077). Part of this work was conducted at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration (80NM0018D0004). Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not constitute or imply its endorsement by the United States Government or the Jet Propulsion Laboratory, California Institute of Technology.References
- S. B. Charnley, Y.-J. Kuan, H.-C. Huang, O. Botta, H. M. Butner, N. Cox, D. Despois, P. Ehrenfreund, Z. Kisiel, Y.-Y. Lee, A. J. Markwick, Z. Peeters and S. D. Rodgers, Adv. Space Res., 2005, 36, 137–145 CrossRef CAS.
- P. Ehrenfreund, S. Rasmussen, J. Cleaves and L. Chen, Astrobiology, 2006, 6, 490–520 CrossRef CAS PubMed.
- D. S. N. Parker, R. I. Kaiser, O. Kostko, T. P. Troy, M. Ahmed, B.-J. Sun, S.-H. Chen and A. H. H. Chang, Phys. Chem. Chem. Phys., 2015, 17, 32000–32008 RSC.
- D. S. N. Parker and R. I. Kaiser, Chem. Soc. Rev., 2017, 46, 452–463 RSC.
- D. B. Rap, J. G. M. Schrauwen, A. N. Marimuthu, B. Redlich and S. Brünken, Nat. Astron., 2022, 6, 1059–1067 CrossRef.
- A.-R. Soliman, A. M. Hamid, I. Attah, P. Momoh and M. S. El-Shall, J. Am. Chem. Soc., 2013, 135, 155–166 CrossRef CAS PubMed.
- M. P. Bernstein, A. L. Mattioda, S. A. Sandford and D. M. Hudgins, Astrophys. J., 2005, 626, 909–918 CrossRef CAS.
- C. Sagan, W. R. Thompson and B. N. Khare, Acc. Chem. Res., 1992, 25, 286–292 CrossRef CAS PubMed.
- C. Sagan and W. Reid Thompson, Icarus, 1984, 59, 133–161 CrossRef CAS.
- E. de Vanssay, M. C. Gazeau, J. C. Guillemin and F. Raulin, Planet. Space Sci., 1995, 43, 25–31 CrossRef CAS PubMed.
- S. M. Hörst, J. Geophys. Res.: Planets, 2017, 122, 432–482 CrossRef.
- V. Vuitton, R. V. Yelle, S. J. Klippenstein, S. M. Hörst and P. Lavvas, Icarus, 2019, 324, 120–197 CrossRef CAS.
- V. Vuitton, O. Dutuit, M. A. Smith and N. Balucani, in Titan: Interior, Surface, Atmosphere, and Space Environment, ed. C. A. Griffith, E. Lellouch, I. Müller-Wodarg and T. E. Cravens, Cambridge University Press, Cambridge, 2014, pp. 224–284 Search PubMed.
- S. M. Mackenzie, S. P. D. Birch, S. Hörst, C. Sotin, E. Barth, J. M. Lora, M. G. Trainer, P. Corlies, M. J. Malaska, E. Sciamma-O’Brien, A. E. Thelen, E. Turtle, J. Radebaugh, J. Hanley, A. Solomonidou, C. Newman, L. Regoli, S. Rodriguez, B. Seignovert, A. G. Hayes, B. Journaux, J. Steckloff, D. Nna-Mvondo, T. Cornet, M. Y. Palmer, R. M. C. Lopes, S. Vinatier, R. Lorenz, C. Nixon, E. Czaplinski, J. W. Barnes, E. Sittler and A. Coates, Planet. Sci. J., 2021, 2, 112 CrossRef.
- V. A. Krasnopolsky, Icarus, 2014, 236, 83–91 CrossRef CAS.
- V. A. Krasnopolsky, Icarus, 2009, 201, 226–256 CrossRef CAS.
- E. Hébrard, M. Dobrijevic, J. C. Loison, A. Bergeat, K. M. Hickson and F. Caralp, Astron. Astrophys., 2013, 552, A132 CrossRef.
- J. C. Loison, M. Dobrijevic and K. M. Hickson, Icarus, 2019, 329, 55–71 CrossRef CAS.
- N. Balucani, A. Caracciolo, G. Vanuzzo, D. Skouteris, M. Rosi, L. Pacifici, P. Casavecchia, K. M. Hickson, J.-C. Loison and M. Dobrijevic, Faraday Discuss., 2023, 245, 327–351 RSC.
- R. P. Haythornthwaite, A. J. Coates, G. H. Jones, A. Wellbrock, J. H. Waite, V. Vuitton and P. Lavvas, Planet. Sci. J., 2021, 2, 26 CrossRef CAS.
- E. L. Barth, Planet. Space Sci., 2017, 137, 20–31 CrossRef CAS.
- H. E. Maynard-Casely, M. L. Cable, M. J. Malaska, T. H. Vu, M. Choukroun and R. Hodyss, Am. Mineral., 2018, 103, 343–349 CrossRef.
- T. A. Francis, H. E. Maynard-Casely, M. L. Cable, R. Hodyss and C. Ennis, J. Phys. Chem. A, 2023, 127, 2322–2335 CrossRef CAS PubMed.
- M. L. Cable, T. Runčevski, H. E. Maynard-Casely, T. H. Vu and R. Hodyss, Acc. Chem. Res., 2021, 54, 3050–3059 CrossRef CAS PubMed.
- M. L. Cable, T. H. Vu, M. J. Malaska, H. E. Maynard-Casely, M. Choukroun and R. Hodyss, ACS Earth Space Chem., 2020, 4, 1375–1385 CrossRef CAS.
- T. H. Vu, H. E. Maynard-Casely, M. L. Cable, R. Hodyss, M. Choukroun and M. J. Malaska, J. Appl. Crystallogr., 2020, 53, 1524–1530 CrossRef CAS.
- M. L. Cable, T. H. Vu, M. J. Malaska, H. E. Maynard-Casely, M. Choukroun and R. Hodyss, ACS Earth Space Chem., 2019, 3, 2808–2815 CrossRef CAS.
- H. E. Maynard-Casely, R. Hodyss, M. L. Cable, T. H. Vu and M. Rahm, IUCrJ, 2016, 3, 192–199 CrossRef CAS PubMed.
- C. A. McConville, Y. Tao, H. A. Evans, B. A. Trump, J. B. Lefton, W. Xu, A. A. Yakovenko, E. Kraka, C. M. Brown and T. Runčevski, Chem. Commun., 2020, 56, 13520–13523 RSC.
- C. Ennis, M. L. Cable, R. Hodyss and H. E. Maynard-Casely, ACS Earth Space Chem., 2020, 4, 1195–1200 CrossRef CAS.
- E. C. Czaplinski, T. H. Vu, M. L. Cable, M. Choukroun, M. J. Malaska and R. Hodyss, ACS Earth Space Chem., 2023, 7, 597–608 CrossRef CAS PubMed.
- E. Czaplinski, X. Yu, K. Dzurilla and V. Chevrier, Planet. Sci. J., 2020, 1, 76 CrossRef.
- A. C. Thakur and R. C. Remsing, arXiv, 2022, preprint, arxiv:2210.12188 DOI:10.48550/arxiv.2210.12188.
- I. Couturier-Tamburelli, N. Piétri and M. S. Gudipati, Astron. Astrophys., 2015, 578, A111 CrossRef.
- M. S. Gudipati, R. Jacovi, I. Couturier-Tamburelli, A. Lignell and M. Allen, Nat. Commun., 2013, 4, 1648 CrossRef PubMed.
- O. J. Shiels, P. D. Kelly, C. C. Bright, B. L. J. Poad, S. J. Blanksby, G. da Silva and A. J. Trevitt, J. Am. Soc. Mass Spectrom., 2021, 32, 537–547 CrossRef CAS PubMed.
- V. L. Frankland, A. D. James, J. D. C. Sánchez, T. P. Mangan, K. Willacy, A. R. Poppe and J. M. C. Plane, Icarus, 2016, 278, 88–99 CrossRef CAS.
- F. Raulin, C. Brassé, O. Poch and P. Coll, Chem. Soc. Rev., 2012, 41, 5380 RSC.
- C. He and M. A. Smith, Icarus, 2014, 238, 86–92 CrossRef CAS.
- C. He and M. A. Smith, Icarus, 2013, 226, 33–40 CrossRef CAS.
- A. Mann, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4566–4568 CrossRef CAS PubMed.
- K. I. Öberg, R. T. Garrod, E. F. Van Dishoeck and H. Linnartz, Astron. Astrophys., 2009, 504, 891–913 CrossRef.
- R. L. Hudson, R. F. Ferrante and M. H. Moore, Icarus, 2014, 228, 276–287 CrossRef CAS.
- R. L. Hudson and Y. Y. Yarnall, Icarus, 2022, 377, 114899 CrossRef CAS.
- R. Martín-Doménech, K. I. Öberg and M. Rajappan, Astrophys. J., 2020, 894, 98 CrossRef.
- F. Pascale, C. M. Zicovich-Wilson, F. López Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- C. Ennis, R. Auchettl, D. R. T. Appadoo and E. G. Robertson, Mon. Not. R. Astron. Soc., 2017, 471, 4265–4274 CrossRef CAS.
- C. Ennis, R. Auchettl, D. R. T. Appadoo and E. G. Robertson, Phys. Chem. Chem. Phys., 2018, 20, 23593–23605 RSC.
- D. Mootz and H. G. Wussow, J. Chem. Phys., 1981, 75, 1517–1522 CrossRef CAS.
- R. K. McMullan, Å. Kvick and P. Popelier, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 726–731 CrossRef.
- M. T. Kirchner, R. Boese, A. Gehrke and D. Bläser, CrystEngComm, 2004, 6, 360–366 RSC.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. L. Cable, T. H. Vu, H. E. Maynard-Casely, M. Choukroun and R. Hodyss, ACS Earth Space Chem., 2018, 2, 366–375 CrossRef CAS.
- H. Takahashi, K. Mamola and E. K. Plyler, J. Mol. Spectrosc., 1966, 21, 217–230 CrossRef CAS.
- L. Corrsin, B. J. Fax and R. C. Lord, J. Chem. Phys., 1953, 21, 1170–1176 CrossRef CAS.
- E. Hirai, Y. Sekine, N. Zhang, N. Noda, S. Tan, Y. Takahashi and H. Kagi, Geophys. Res. Lett., 2023, 50, e2023GL103015 CrossRef CAS.
- K. Öberg, Chem. Rev., 2016, 116, 9631–9663 CrossRef PubMed.
- S. Tixier, G. Cooper, R. Feng and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 2002, 123, 185–197 CrossRef CAS.
- G. Cooper, G. R. Burton and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 1995, 73, 139–148 CrossRef CAS.
- C. Q. Jiao, C. A. DeJoseph, R. Lee and A. Garscadden, Int. J. Mass Spectrom., 2006, 257, 34–40 CrossRef CAS.
- K. I. Öberg, E. F. Van Dishoeck and H. Linnartz, Astron. Astrophys., 2009, 496, 281–293 CrossRef.
- D. V. Mifsud, P. A. Hailey, P. Herczku, B. Sulik, Z. Juhász, S. T. S. Kovács, Z. Kaňuchová, S. Ioppolo, R. W. McCullough, B. Paripás and N. J. Mason, Phys. Chem. Chem. Phys., 2022, 24, 10974–10984 RSC.
- D. V. Mifsud, P. A. Hailey, P. Herczku, Z. Juhász, S. T. S. Kovács, B. Sulik, S. Ioppolo, Z. Kaňuchová, R. W. McCullough, B. Paripás and N. J. Mason, Eur. Phys. J. D, 2022, 76, 87 CrossRef CAS.
- W. Zheng, D. Jewitt and R. I. Kaiser, Chem. Phys. Lett., 2007, 435, 289–294 CrossRef CAS.
- W. Zheng, D. Jewitt and R. I. Kaiser, Astrophys. J., 2006, 639, 534–548 CrossRef CAS.
- W. Zheng, D. Jewitt and R. I. Kaiser, Astrophys. J., 2006, 648, 753–761 CrossRef CAS.
- D. V. Mifsud, P. Herczku, R. Rácz, K. K. Rahul, S. T. S. Kovács, Z. Juhász, B. Sulik, S. Biri, R. W. McCullough, Z. Kaňuchová, S. Ioppolo, P. A. Hailey and N. J. Mason, Front. Chem., 2022, 10, 1003163 CrossRef CAS PubMed.
- D. B. Rap, A. Simon, K. Steenbakkers, J. G. M. Schrauwen, B. Redlich and S. Brünken, Faraday Discuss., 2023, 245, 221–244 RSC.
- P. Recio, D. Marchione, A. Caracciolo, V. J. Murray, L. Mancini, M. Rosi, P. Casavecchia and N. Balucani, Chem. Phys. Lett., 2021, 779, 138852 CrossRef CAS.
- J. W. Barnes, E. P. Turtle, M. G. Trainer, R. D. Lorenz, S. M. MacKenzie, W. B. Brinckerhoff, M. L. Cable, C. M. Ernst, C. Freissinet, K. P. Hand, A. G. Hayes, S. M. Hörst, J. R. Johnson, E. Karkoschka, D. J. Lawrence, A. Le Gall, J. M. Lora, C. P. McKay, R. S. Miller, S. L. Murchie, C. D. Neish, C. E. Newman, J. Núñez, M. P. Panning, A. M. Parsons, P. N. Peplowski, L. C. Quick, J. Radebaugh, S. C. R. Rafkin, H. Shiraishi, J. M. Soderblom, K. S. Sotzen, A. M. Stickle, E. R. Stofan, C. Szopa, T. Tokano, T. Wagner, C. Wilson, R. A. Yingst, K. Zacny and S. C. Stähler, Planet. Sci. J., 2021, 2, 130 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: VUV calibration procedure, additional results of co-crystal formation, layered ice and pyridine ices controls; CRYSTAL17 output files for frequency calculation of C5H5N, C2H2 and C5H5N:C2H2 crystal structures. See DOI: https://doi.org/10.1039/d4cp03437f |
| This journal is © the Owner Societies 2024 |