
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the role of carboxylic acid surfactants in the growth inhibition effect during area-selective atomic layer deposition: the case of ZnO growth on Cu and Cu2O†
L. E.
López-González
 *,
R.
Ponce-Pérez
,
H.
Tiznado
* and
J.
Guerrero-Sánchez
*
*,
R.
Ponce-Pérez
,
H.
Tiznado
* and
J.
Guerrero-Sánchez
*
Centro de Nanociencias y Nanotecnología, Universidad Nacional Autónoma de México, Ensenada, BC 22860, Mexico. E-mail: luis.lopez@ens.cnyn.unam.mx; tiznado@ens.cnyn.unam.mx; guerrero@ens.cnyn.unam.mx
First published on 30th August 2024
Abstract
Herein, we report a detailed adsorption process of acetic acid (AA) as a model for the head group of carboxylic acid self-assembled monolayers on Cu and Cu2O (111) surfaces and the effect of diethyl zinc (DEZ) on its adsorption geometry on Cu2O (111) using quantum chemical calculations. The most stable adsorption configurations were obtained considering electrostatic potential compatibility from the molecule and surface. Overall, the adsorption behavior revealed bidentate binding as the most stable configuration. Weak van der Waals interactions are key in AA adsorption on Cu (111), while in Cu2O (111), coordination and hydrogen bonds dominated the interaction. AA adsorption geometry on Cu2O revealed that DEZ has no significative impact on the carbonyl-chemisorbed AA and bidentate adsorption modes. These results highlight the significance of the different adsorption modes for achieving area-selective deposition using atomic layer deposition and soft removal SAM molecules.
1. Introduction
A recently explored fabrication approach is area selective deposition (ASD), whose main advantage is that it allows growth only at specific regions in a self-aligned manner.1 The main process consists of depositing the material of interest on the surface where growth is desired (growth surface), avoiding deposition in the neighboring areas where growth is not required (non-growth surface).2 The main considerations for achieving area-selective deposition are related to differences in the physical or chemical properties, such as surface terminations and the composition of the material of interest.2 Typically, the control of surface functional groups is the approach employed to modify the deposition chemistry,3 either employing a substrate that inherently inhibits growth4 or changing surface terminations5 by anchoring small molecule inhibitors (SMIs)6 or using self-assembled monolayers (SAMs).7The atomic layer deposition (ALD) technique consists of cyclic and alternating exposures of the substrate to a precursor and a co-reactant that undergo self-limiting surface reactions. It allows atomic-level thickness control, conformality, and uniformity.8 Modifying surface functional groups by the approaches described above before or during the ALD process gives rise to area-selective atomic layer deposition (AS-ALD).9
SAMs are the most studied platform to passivate a surface for achieving AS-ALD, which consists of organic molecules that spontaneously and preferentially form a packed and ordered layer on specific surfaces.10 The molecules used to form SAMs have an amphiphilic character and consist of a head group that binds to the surface of interest, an alkyl chain that favors order and stability, and a tail group that defines wetting surface properties.8,11
The choice of a non-growth surface depends on its chemical compatibility with the inhibiting molecule.12 Cu, CuO, and Cu2O surfaces are frequently employed in testing SAMs for AS-ALD7,13 because of their low surface acidity14 when acid inhibitor molecules are intended to be employed. On the other hand, copper is typically employed as an interconnect material in Si-based electronic devices, and it is commonly used as a non-growth surface in the companionship of native SiO2 as a growth surface. Additionally, ZnO is a semiconductor material employed widely in electronic device fabrication, such as transistors, diodes, and sensors, and diethyl zinc (DEZ) is one of the most employed ZnO ALD precursors.
Even though AS-ALD has great potential, it faces challenges that must be overcome, such as high defectivity and low throughput.5 In the specific case of SAMs, selectivity is gradually lost with increasing ALD cycles. Conditions that may lead to selectivity loss are the thermal degradation of SAMs during ALD, the physisorption of ALD precursors onto the SAM, and structural changes in the SAM due to the interactions between highly reactive ALD precursor molecules and the SAMs during the ALD process.10
Carboxylic acid SAMs and SMIs have been recently studied as promising candidates for AS-ALD inhibitor molecules11,15 since they can strongly attach to several metal oxide surfaces16,17 but do not bind to native SiO2. For example, stearic acid (SA) can inhibit ZnO ALD growth and is easily removable.11 Despite these benefits, its ZnO ALD blocking ability gradually decreases. Speculations on the causes of this loss have pointed out the possibility of diffusion of the DEZ precursor through the SAM.11 However, it is yet to be elucidated.
In this context, the study of the adsorption of carboxylic acid surfactants on surfaces of interest such as Cu and Cu2O and the effect that co-adsorbed DEZ molecules have on the surface–carboxylic acid system is of profound interest to further understand the factors impacting selectivity loss in SAM- and SMI-mediated AS-ALD.
In this work, we present a detailed atomic scale analysis of the adsorption of the stearic acid molecule modeled as acetic acid (AA) as a model of the head group of the SA surfactant on Cu and Cu2O (111) surfaces and the effect of the DEZ ALD precursor molecule on the interactions of AA with the Cu2O surface using density functional theory (DFT) calculations. The results showed highly stable adsorption energy values when the surfactant molecule was adsorbed in bidentate coordination on Cu (111) and Cu2O (111). The non-covalent interaction (NCI) index analysis revealed that coordination and hydrogen bonds (HBs) were the most stabilizing interactions in chemisorbed states on both surfaces. The DEZ co-adsorption study revealed that when the AA was adsorbed in a carbonyl chemisorbed and bidentate manner, the interaction with the diethylzinc molecule was not strong enough to remove the inhibitor molecule from the surface, providing valuable insight into the behavior of carboxylic acid inhibitors in the presence of DEZ precursors for area-selective atomic layer deposition approaches.
2. Methods
The calculations were made in the periodic density functional theory framework employed in the PWsf code of the Quantum ESPRESSO package.18 The General Gradient Approximation (GGA) was used to model the exchange–correlation energies with the Perdew, Burke, Ernzerhof (PBE) gradient corrected functional.19 The dispersion-corrected van der Waals (vdW) interactions have been considered using the Grimme DFT-D3 framework.20,21 Electron–ion interactions were treated using Vanderbilt ultrasoft pseudopotentials.22 The choice of PBE using the Grimme DFT-D3 framework was made under the acknowledgment that it may overestimate the electronic properties in some metal oxides. However, we considered previously reported fair lattice parameter errors23 and adsorption energies for molecules on metal oxide surfaces.24,25 The complete optimization of kinetic energy cutoff, charge density cutoff, k-point mesh, and lattice parameters of bulk structures was carried out for the Cu and Cu2O bulk structures using PBE without dispersion corrections (DFT-D3) (Fig. S1 and S2, ESI†). The electronic states are expanded in plane waves with an optimized energy cutoff of 45 Ry and a charge density cutoff of 360 Ry for Cu, and 35 Ry and 280 Ry for Cu2O. The gamma-centered optimized k-point mesh is 9 × 9 × 9 and 6 × 6 × 6 for Cu and Cu2O, respectively. The optimized lattice parameter for Cu and Cu2O is a = 3.62 Å and a = 4.29 Å, respectively; these values are consistent with the ones reported experimentally for Cu (3.61 Å26) and Cu2O (4.26 Å27). In the geometry optimization, all force components should be smaller than 0.026 eV Å−1, and total energy differences must be less than 0.0026 eV. Surface models were constructed employing the supercell method and considering the optimized lattice parameters. Each supercell consists of a 4-atomic layer slab and a vacuum gap of 20 Å to avoid interactions between adjacent surfaces.28 The Brillouin zone integration has been done using a Methfessel–Paxton smearing of 0.02 Ry and a Monkhorst–Pack k-point grid29 of 3 × 3 × 1 and 2 × 2 × 1 for Cu and Cu2O, respectively. Adsorption was treated considering a 3 × 3 supercell for pristine copper and a 2 × 2 supercell for cuprous oxide (Fig. 1(b) and (c)). The Cu of the Cu–Si–Cu system in the experimental AS-ALD environment is polycrystalline, and the surface, when exposed to air, is natively oxidized. The experimental initial composition of Cu native oxide is Cu2O.30 On the other hand, in some studies, the pristine Cu surface has been employed for ASD approaches.31 Hence, we also considered the pristine Cu surface for this study. The most observed surfaces of Cu and Cu2O are (111), (110), and (001). The most stable and most studied surface of both materials is (111).32,33 Therefore, the Cu (111) and Cu2O (111) surfaces were used in this work. | ||
| Fig. 1 The optimized structures treated in this work. (a) Stearic acid and acetic acid, bulk and surface (111) models of (b) Cu and (c) Cu2O. | ||
The SA and DEZ molecules were optimized in a cube of 25 Å in length with a k-point at the gamma point. Considering the analysis of the electrostatic potential isosurface, we determined that the alkyl chain in the molecule will not contribute as an active adsorption site; then, we used the AA molecule as a model system, optimized similarly to SA and DEZ (Fig. 1(a)).
Vibrational frequencies for the AA and DEZ molecules in the adsorption models were calculated using the Vienna Ab initio Simulation Package (VASP).34 Tables of the molecule's bonds and angles before and after adsorption and vibrational frequencies are included in the ESI† in Tables S1 and S2, respectively.
AA adsorption on Cu and Cu2O surfaces can be physisorption, non-dissociative chemisorption, and dissociative chemisorption. The formation of bidentate adsorption is a dissociative process that requires a dehydrogenation reaction. The carboxyl hydrogen atom is taken out of the acetic acid molecule, binding to the surface during the adsorption process, as discussed by Nuzzo.35 Bidentate adsorption models were constructed by removing the hydrogen atom from the AA molecule and placing it onto the surface as the initial structures.
Based on the above description, molecule–surface adsorption was analyzed using the following two equations:
For non-dissociative adsorption models (physisorption and carbonyl chemisorbed AA):
| Eads = EAA–slab − (Eslab + EAA), |
For dissociative adsorption models (bidentate chemisorption):
| Eads = Eacetate+H+slab − (Eslab + EAA), |
In the case of chemisorption, a bond character was studied using Bader charge analysis and charge density differences.37 To understand the long-range interactions that emerge upon the molecule–surface interaction, we have used the NCI index, as implemented in the CRITIC238 code (extending the NCI method39 to periodic solid-state electron densities). Such analysis provides insight into hydrogen bonding, repulsion, and weak vdW molecule–surface interactions.
In the NCI index analysis, two scalar fields are calculated to map the local bonding properties: the electron density (ρ) and the reduced density gradient (RDG), defined as.38

RDG is a dimensionless quantity that describes the deviation from the homogeneous electron distribution.40 The RDG vs. ρ multiplied by the sign of the second Hessian eigenvalue (sign(λ2)) plot evidences the non-covalent interactions present in our systems in the region of low ρ and low RDG. These regions correspond to the decaying density tails (far from the nuclei). Covalent bonds can be visualized through electron density properties (i.e., the bonds’ critical point) using the atoms-in-molecules theory.37 These correspond to saddle points at RDG = 0, with a high ρ in the RDG vs. ρ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sign(λ2) graph. Non-covalent interactions induce a notable RDG drop to nearly zero (spikes at low ρ).41 The sign of the density Laplacian (∇2ρ), contributed by sign(λ2), is used to distinguish between different NCIs. Hydrogen bonds produce density accumulation (λ2 < 0), vdW interactions characterize a negligible density overlap (λ2 ≈ 0), and steric repulsions produce density depletion (λ2 > 0).41
sign(λ2) graph. Non-covalent interactions induce a notable RDG drop to nearly zero (spikes at low ρ).41 The sign of the density Laplacian (∇2ρ), contributed by sign(λ2), is used to distinguish between different NCIs. Hydrogen bonds produce density accumulation (λ2 < 0), vdW interactions characterize a negligible density overlap (λ2 ≈ 0), and steric repulsions produce density depletion (λ2 > 0).41
To understand the impact of the ALD precursor on ZnO deposition, we considered the most stable adsorption configurations of acetic acid on the Cu2O (111) surface. Also, we assessed the interaction of the DEZ molecule. Once we have the most stable configurations, we calculate the NCI, Bader charges analysis, and charge density differences plots.37,39
3. Results and discussion
3.1 Adsorption of acetic acid: energy and geometries
Initially, aiming to determine the most likely binding sites on the studied surfaces, the charge density maps colored by the electrostatic potential were obtained (see Fig. S3, ESI†). The pristine copper (111) surface presents an equally positive electrostatic potential in the surface Cu atoms, while the Cu2O (111) surface exhibited negative potential regions around the oxygen atoms, while copper showed a positive character.All surface copper atoms are equivalent in the Cu (111) surface model, while in Cu2O (111) (Fig. 1(b)), we can distinguish two types of Cu atoms: coordinatively saturated (CSA) and coordinatively unsaturated (CUS) (Fig. 1(c)).42
As expected, the carboxyl group has a tremendously negative electrostatic potential. On the other hand, the DEZ molecule shows a highly positive potential in the Zn atom, while the ethyl moieties exhibit a negative potential to a certain extent. All the adsorption models were constructed based on these molecule and surface considerations. To ensure that calculated geometries corresponded to a minimum on the potential energy surface, calculations of vibrational frequencies of the adsorbed molecules were carried out. The results, presented in Table S2 (ESI†), revealed no imaginary frequencies in the adsorption models, indicating that the calculated models were stable. Furthermore, vibrational mode wavenumber values were similar to the values experimentally observed, specifically for the C–O antisymmetric and symmetric stretches in the Cu2O adsorption models, experimentally observed at 1585 cm−1 and 1471 cm−1, respectively.11 Additionally, C–H stretches (frequency range: 2850–3120 cm−1) slightly shifted to higher wavenumber values. This was an expected behavior since acetic acid coverage in our models is not high enough to allow lateral interaction between the alkyl groups, which is typical in highly ordered self-assembled monolayers of amphipathic molecules.
 | ||
| Fig. 2 Most stable configurations for the adsorption of acetic acid on the Cu (111) surface. (a) Bidentate binding, (b) carbonyl chemisorbed configuration, (c) “laying” physisorption, and (d) “standing” physisorption. | ||
Bidentate binding of the molecule is likely the most stable adsorption mode for alkanoic acids in the formation of the self-assembled monolayer. The binding possibilities were described by Nuzzo35 in his early study of carboxylic acid SAMs, pointing out that the proton of the acid is added to the surface, propitiating carboxylate-like binding.
Moreover, carbonyl chemisorbed binding is also expected to occur as a stable adsorption mode. The model presented in Fig. 2(b) shows the carbonyl chemisorbed AA. The carbonyl group was coordinated to the Cu atom; however, the adsorption energy values were an order of magnitude less negative compared to those in Fig. 2(a), approaching physisorption values. This result might be attributed to electrostatic repulsions between hydrogens from the methyl group and copper atoms.12 On the other hand, most of the assessed configurations of AA showed physisorption likely induced by electrostatic repulsions, as described above. Fig. 2(c) presents a physisorption state where the AA molecule was oriented with the carboxyl and methyl groups parallel to the Cu surface like it is “lying down”. The methyl group relaxed slightly farther from the surface than the carboxylic group, implying an electrostatic repulsion effect. The adsorption energy value was −0.23 eV, which is similar to values reported by Zang and coworkers for the adsorption of acetic acid on the Cu (111) surface.43Fig. 2(d) shows the physisorption state where carboxyl and methyl groups are oriented perpendicular to Cu (111), with the carboxyl group facing the surface in a “standing” way. Even though hydroxyl and carbonyl groups were facing copper atoms almost on top, no coordination bond was observed. This was attributed to the directionality of the lone pair non-bonding electrons in oxygen atoms and electrostatic repulsion from the C atom of the carboxyl group near the Cu surface atoms.
 | ||
| Fig. 3 Most stable configurations for the adsorption of acetic acid on the Cu2O (111) surface: (a) carbonyl chemisorbed with CUS with an HB, (b) carbonyl chemisorbed with CUS, and (c) carbonyl chemisorbed with CSA with an HB, (d) and (e) bidentate binding with CUS and CSA atoms, respectively. | ||
The adsorption geometry of AA in a bidentate mode is presented in Fig. 3(d) and (e), where each oxygen atom of AA forms bonds with CUS and CSA atoms. The adsorption energy values are ≈−2.33 eV and −2.45 eV for Fig. 3(d) and (e), respectively. The more negative adsorption energy values calculated for the carbonyl chemisorbed AA with respect to bidentate adsorption models were attributed to the reaction energy barrier necessary to break the O–H bond.
An interesting aspect further explored was the initial geometry in a monodentate mode (dissociative chemisorption with one COO oxygen binding to a CUS Cu atom and H adsorbed on the surface), which, upon relaxation, spontaneously became the bidentate adsorption mode (Fig. S4, ESI†), indicating that bidentate adsorption mode is preferred after the dehydrogenation step at the studied coverage.
The carbonyl chemisorbed configuration is stable and has been observed experimentally for highly packed carboxylic acid SAMs, even though it does not involve the strongest Cu–O bond.45 At high coverages, the molecule gets constrained by steric effects, thus favoring lateral interactions. Such adsorption analysis is beyond the scope of this work because our main goal is to understand the loss of selectivity in area-selective atomic layer deposition that is not precisely designed at high coverages.
On the other hand, the transition metal character of copper apparently has an important contribution to Cu–O binding, as observed in the adsorption configuration presented in Fig. 3(c), where chemisorption is accomplished between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and the saturated copper atom. Moreover, the stabilization provided by the formation of HB interactions in the case of the Cu2O surface provides a geometric effect like the one observed for the bidentate adsorption, where rotational degrees of freedom of the molecule are reduced in comparison to the carbonyl chemisorbed configuration.
O group and the saturated copper atom. Moreover, the stabilization provided by the formation of HB interactions in the case of the Cu2O surface provides a geometric effect like the one observed for the bidentate adsorption, where rotational degrees of freedom of the molecule are reduced in comparison to the carbonyl chemisorbed configuration.
The nature of the formed bonds was further investigated using the NCI index and Bader charge analysis, which is presented in the next section.
3.2 Charge transfer and non-covalent interactions
The role of non-covalent interactions in the adsorption geometry and adsorption energy was studied through NCI index analysis. This is a powerful and helpful tool to study surface–molecule interactions. In general, the adsorption of AA on both studied surfaces revealed the presence of van der Waals interactions (attraction and repulsion components), coordination bonds, and hydrogen bonds.In Fig. 4, the NCI index results for the most stable adsorption configurations of AA on the Cu (111) surface are plotted as RDG against the density values multiplied by the sign of the second eigenvalue of the hessian matrix. The RDG minima on each plot indicate the presence of an interaction. The position on the density value axis denotes the strength or type of interaction, while the sign of λ2 indicates if it is an attraction (−) or a repulsion (+). In the inset squares, NCIs are depicted as RDG isosurfaces colored by the density value. van der Waals attractions are shown in green, and strong attractions are shown in blue, while weak repulsions are shown in yellow. Fig. 4(a) shows an RDG minimum at a density value of −0.08, attributed to the strong attractions of the Cu–O bonds, which are observed as intense blue donuts in the isosurface images, and at −0.04 corresponding to the Cu–H interactions.
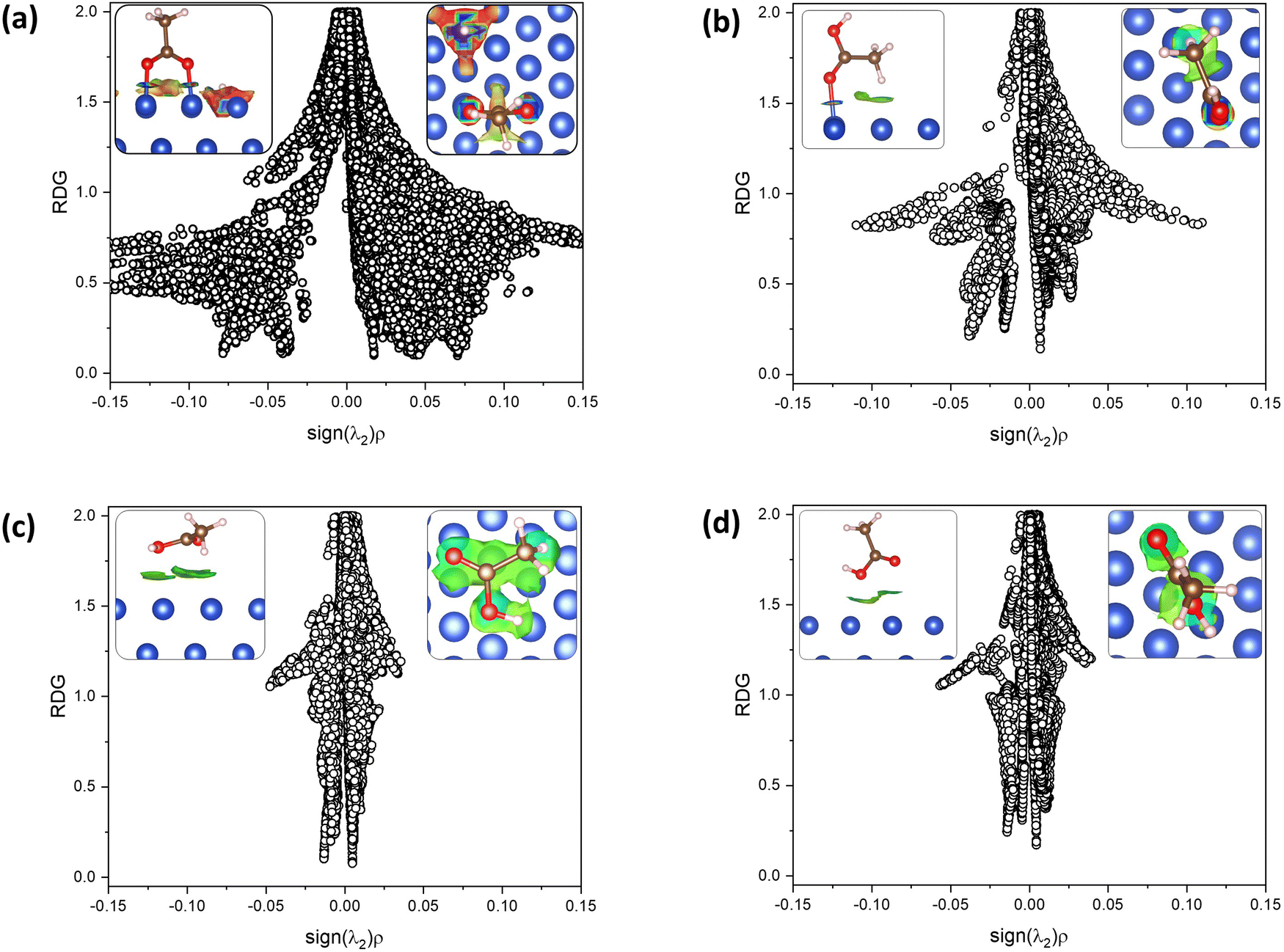 | ||
| Fig. 4 Non-covalent interaction index (NCI) analysis, represented as the plots of reduced density gradient (RDG) vs. the sign(λ2)ρ for the most stable adsorption models of AA in Cu (111): (a) bidentate binding, (b) carbonyl chemisorbed configuration, (c) “laying” physisorption, and (d) “standing” physisorption. RDG minima point out an interaction. The negative density values show attraction. The inset images represent the NCIs RDG isosurfaces colored by density values (isovalue 0.5) (side and top view for left and right, respectively). Each color depicts interactions: blue, green, yellow, and red define strong attractions, weak vdW attractions, vdW repulsions, and strong repulsions, respectively. | ||
Additionally, a weak minimum appeared at −0.025, corresponding to small van der Waals (vdW) attraction. In contrast, a clear minimum is observed at +0.025, showing a vdW repulsion component observed as the yellow isosurface region observed below the carbon atom in between two coppers; this is due to the partially positive character of the C atom due to the high electronegativity of the O atoms in the COO group. In Fig. 4(b), two negative minimums are observed in the plot, one near −0.05, corresponding to the strong attraction of the Cu–O bond, shown as the strong blue isosurface and the other at −0.025, corresponding to the vdW attractions, depicted as a light green-blue isosurface under the methyl group. The plot for the physisorption model in Fig. 4(c) shows a pronounced minimum at low negative density values (≈0) characteristic of vdW attractions observed as the big greenish to light blue isosurface appearing slightly more attractive in the oxygen regions. Finally, in Fig. 4(d), the second physisorption configuration is presented, where this time, the carboxylic group is facing the surface. Two minima in the zone of vdW attractions are observed as green and light blue isosurfaces, showing a more attractive zone in the OH group of the COOH and depicting the attraction to the two nearest Cu atoms.
Additionally, the van der Waals component in the Cu2O adsorption model was shown to be, in most cases, repulsive with a slight tendency to attraction. The major adsorption component was attributed to the coordination bonds and HB. In Fig. 5, the NCI index results are presented as RDG vs. sign(λ2)ρ plots with insets showing RDG isosurfaces colored by density values.
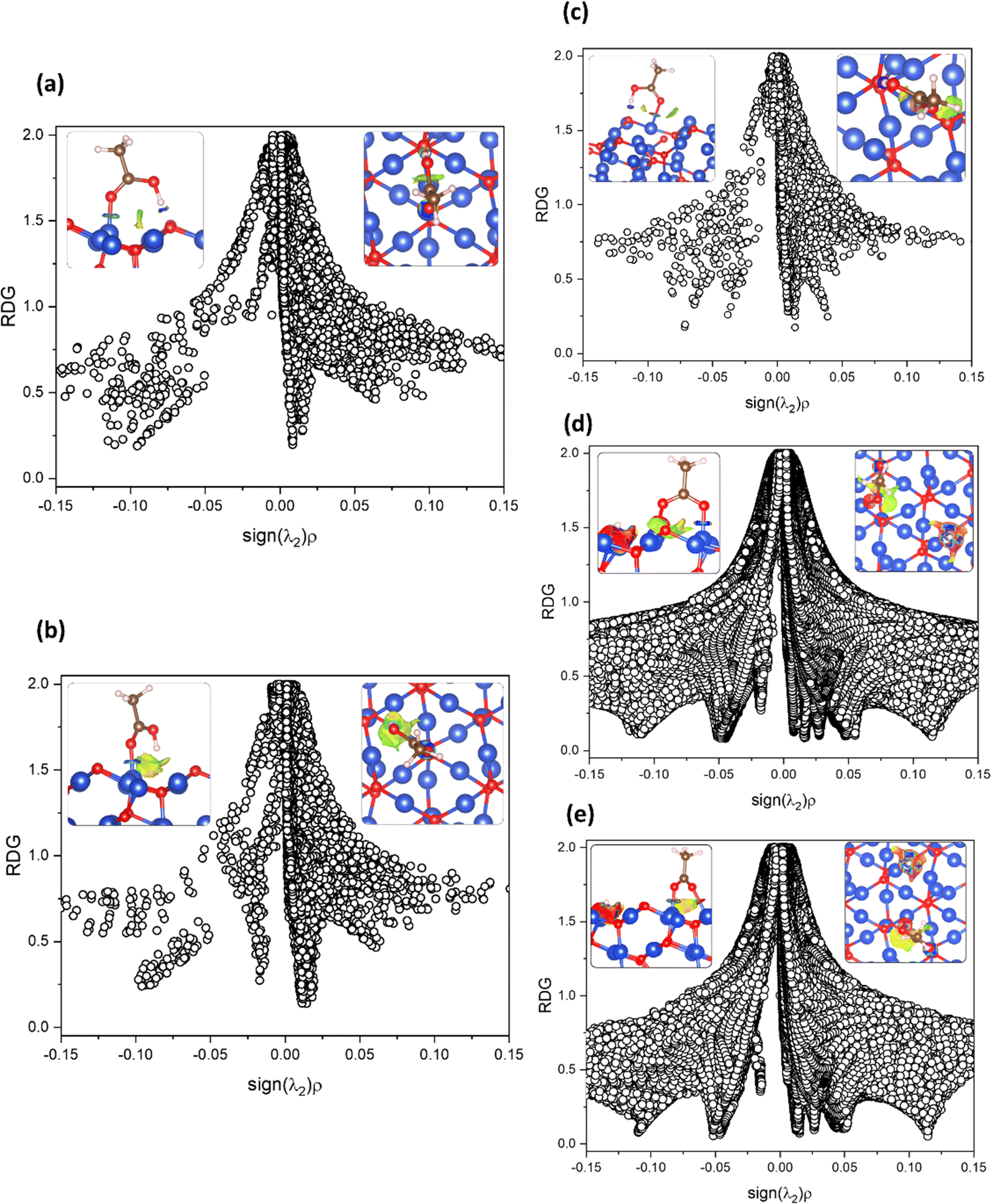 | ||
| Fig. 5 Non-covalent interaction index (NCI) analysis, represented as the plots of reduced density gradient (RDG) vs. the sign(λ2)ρ for most stable adsorption models of AA on Cu2O (111): (a) carbonyl chemisorbed with CUS with an HB, (b) carbonyl chemisorbed with CUS, and (c) carbonyl chemisorbed with CSA with an HB, (d) and (e) bidentate binding with CUS and CSA atoms, respectively. RDG minima point out an interaction. The negative density values show attraction. The inset images represent the NCIs RDG isosurfaces colored by density values (isovalue 0.5) (side and top view for left and right, respectively). Each color depicts interactions: blue, green, yellow, and red define strong attractions, weak vdW attractions, vdW repulsions and strong repulsions, respectively. | ||
The NCI index analysis showed the presence of attractions corresponding to the CUS Cu–O bond, indicated by a minimum near −0.10 for the carbonyl chemisorbed models and displayed as a strong blue donut isosurface (Fig. 5(a)–(c)). In Fig. 5(a), a strong attraction overlapped minimum at sign(λ2)ρ ≈ −0.10, corresponding to the HB observed as the small, strong blue isosurface between hydrogen and oxygen. Such observed strength for the HB could be attributed to the greatly favorable orientation of the OH group toward the surface oxygen encountering the almost ideal HB directionality. In Fig. 5(b), in addition to the strong Cu–O bond, there is a minimum at the attractive van der Waals zone displayed as a green to light-blue area in between the hydroxyl and the second layer of oxygen from the surface. Finally, Fig. 5(c) shows a difference in the strength of attraction corresponding to the CSA Cu–O bond, showing a less negative density value than the CUS Cu–O bonds from the Fig. 5(a) and (b) models. This difference is attributed to the higher electrostatic component of the CUS Cu–O bond. Lastly, a relatively strong attraction minimum was attributed to the HB formed (intense blue isosurface circle) between the molecule OH and O atom from the surface.
The models presented in Fig. 5(d) and (e) correspond to the adsorption in the bidentate form. Particularly for Fig. 5(d), there is a minimum at very negative density (strong attraction) values attributed to the CUS Cu–O bond, which is observed as the blue donut in the isosurface maps. On the other hand, a middle strength repulsion minimum is observed (sign(λ2)ρ ≈ +0.05) as a red region near the CSA Cu–O bond. This repulsion is attributed to the constrained orientation of the oxygen atom with respect to the other carboxyl oxygen atom, which formed a dihedral angle of 43.7°. Strong attractions are shown between Cu and H, accompanied by some repulsion components. Also, a green isosurface under the carboxyl C atom was observed, accompanied by a minimum in the plot near −0.03, corresponding to van der Waals attractions. Small vdW repulsion near 0 positive minimum and a yellow isosurface near the CSA Cu-bond oxygen and the opposite CSA were also observed.
In Fig. 5(e), we can see a difference between the two strong attraction minima observed, one of them being stronger, corresponding to the CUS Cu–O bond (sign(λ2)ρ ≈ −0.10), while the other corresponds to the CSA Cu-bond (sign(λ2)ρ ≈ −0.06). The differences in strength were attributed to the constraint wielded over the C–O–O–CSA Cu torsion angle caused by the orientation of the CSA atom. Even though it did not yield repulsion, as shown in Fig. 5(d), it is weaker than the CUS–Cu bond. This strength difference was attributed to the stronger electrostatic component presented by the dangling bond of the CUS atom in comparison to the CSA, as observed in the electrostatic potential map of the Cu2O surface (Fig. S3, ESI†). Cu–H strong attractions were also observed. Additionally, vdW repulsions dominated under the carboxyl C atom. There is a bigger repulsive component in Fig. 5(d) compared to Fig. 5(e), and the adsorption energy value is more stable for Fig. 5(e), showing that steric effects play an important role in adsorption energy.
To have a clearer vision of molecule–surface interactions, the nature of the formed bonds was later investigated in the models where chemisorption was present in Cu and Cu2O (111) surfaces by employing Bader charge analysis (Table 1). Partial charge transfer to oxygen was observed in the models corresponding to the copper surface. In the bidentate form (adsorption energy = −2.08 eV), the copper atoms showed a net charge of +0.23e in each of both Cu atoms due to the delocalized nature of the carboxyl group, while oxygens presented a charge of −1.76e. A small difference can be noted in the carbonyl chemisorbed model on the Cu (111) surface (adsorption energy = −0.39 eV), where the charge transfer to Cu is less than in the bidentate form, implying a stronger ionic character in the bidentate coordination bond than in the carbonyl chemisorbed form.
| Surface | Adsorption energy | Bond | Atom | Net charge (e) |
|---|---|---|---|---|
| Cu (111) | ||||
| (a) −2.08 eV | Cu–O–C | Cu | +0.23 | |
| O | −1.76 | |||
| (b) −0.39 eV | Cu–OC |
Cu | +0.15 | |
| O | −1.87 | |||
| Cu2O (111) | ||||
| (a) −3.44 eV | CuCUS–OC |
Cu | +0.61 | |
| O | −1.76 | |||
| (b) −3.04 eV | CuCUS–OC |
Cu | +0.60 | |
| O | −1.84 | |||
| (c) −2.81 eV | CuCSA–OC |
Cu | +0.69 | |
| O | −1.89 | |||
| (d) −2.33 eV | CuCUS–O | Cu | +0.62 | |
| O | −1.69 | |||
| CuCSA–O | Cu | +0.75 | ||
| O | −1.86 | |||
| (e) −2.45 eV | CuCUS–O | Cu | +0.62 | |
| O | −1.72 | |||
| CuCSA–O | Cu | +0.78 | ||
| O | −1.81 | |||
For the Cu2O (111) surface models with adsorption in the bidentate form, we observed a higher charge transfer from the Cu atoms to oxygen than in Cu (111). However, there was a significant charge difference between CSA and CUS, resulting in more positive changes for CSA Cu atoms in both bidentate adsorption models than for CUS–Cu bound O atoms, implying that CSA Cu atoms are better Lewis bases than CUS–Cu atoms. This agrees with the more positive electrostatic potential observed for CUS Cu than for CSA Cu (Fig. S3, ESI†). This behavior was also observed in the adsorption configurations of the carbonyl chemisorbed form, showing slightly more positive charge value in the CSA–Cu atoms that formed coordination bonds with the carbonyl group than the CUS–Cu forming the same type of bonds.
3.3 Effect of diethyl zinc on the adsorption geometry of acetic acid in Cu2O
The ability of the methyl-terminated carboxylic acid molecules to block the atomic layer deposition of ZnO was investigated through the interaction of the commonly employed ZnO ALD precursor molecule DEZ with the most stable adoption models of AA in Cu2O (111) as a model of native oxide. The systematic search for feasible co-adsorption structures was made based on the electrostatic potential maps colored by charge density (Fig. S5, ESI†), considering that the electrostatic potential of oxygen atoms (AA and Cu2O surface) is strongly negative and that of the Zn atom in DEZ showing a highly positive electrostatic potential. The DEZ molecule was positioned between the surface and the AA head group, with the Zn atom at an adequate distance to interact either with AA or the surface, in all assessed models. The geometry results are presented in Fig. 6. Fig. 6(a) and (b) exhibit the most stable configurations of the AA inhibitor in the carbonyl chemisorbed and bidentate adsorption configurations, respectively, interacting with the DEZ precursor in the side and top view (from left to right). In the first one, the DEZ molecule did not directly affect the surface-AA adsorption. Instead, the tendency of DEZ to interact with surface oxygen atoms was favored by the low coverage of AA. | ||
| Fig. 6 Effect of DEZ molecule adsorption in the geometry of the (a) carbonyl chemisorbed and (b) bidentate configurations of AA on Cu2O (111) (color code: C brown, H white, O red, Cu blue, and Zn grey). | ||
Fig. 6(b) shows the effect of the DEZ precursor when the AA molecule is adsorbed in a bidentate manner, revealing almost no effect in the adsorption geometry, indicating that the inherent reactivity of the DEZ molecule was not enough to break the bidentate binding mode of carboxylic acid SAMs revealing that even though this configuration did not have the most negative adsorption energy, it exhibited outstanding adsorption behavior to prevent ZnO ALD. This is of great impact, not just in AS-ALD methods that may employ carboxylic acid SAMs as growth-blocking agents, but also for methods that employ this molecule head group as a small molecule inhibitor.
There is a high probability that the dependence of the adsorption geometry on the inhibition effect may be observed, especially at low coverages. In practice, SAM order, i.e., adsorption geometry, can be controlled with higher adsorption times, favoring the most stable adsorption configurations,45 hence reaching the adsorption states where the DEZ precursor has no impact.
NCI index analysis was performed to identify how the molecules interact with each other and between surfaces. The results of the RDG isosurfaces colored by density values are presented in Fig. 7. The initial observation in Fig. 7(a) shows the continued presence of the CUS Cu–O coordination bond and HB, observed as blue donuts, confirming that the DEZ molecule could not remove AA from the surface. However, a small vdW attraction region is observed between DEZ and AA but not strong enough to remove it. Additionally, a blue donut isosurface was observed between Zn and O atoms, indicating strong attraction. Also, vdW attractions and repulsions are observed in the ethyl groups of the DEZ molecule.
 | ||
| Fig. 7 NCI index analysis is represented as the RDG isosurfaces colored by density values for the effect of DEZ in (a) carbonyl chemisorbed and (b) bidentate AA adsorption configurations on the Cu2O (111) surface. Each color depicts interactions: blue, green, yellow, and red define strong attractions, weak vdW attractions, vdW repulsions, and strong repulsions, respectively. | ||
Finally, Fig. 7(b) shows how the DEZ molecule did not impact the adsorption of the inhibition molecule adsorbed through bidentate binding. However, the presence of a small strong-blue isosurface between Zn and O atoms from AA was observed, indicating that even though the DEZ precursor was not strong enough to remove the AA molecule in this adsorption mode, it can bind to the carboxylic acid head group, which may lead to the nucleation of ZnO and a further loss of selectivity. In this context, precursor diffusion is an aspect that plays an important role in selectivity loss. To have a major picture of the interactions between DEZ and AA and the impact of ZnO ALD inhibition on Cu2O, charge density difference (Δρ) plots were obtained, as shown in Fig. 8.
 | ||
| Fig. 8 Charge density difference isosurfaces (isovalue = 0.003) plots showing the effect of DEZ on (a) carbonyl chemisorbed and (b) bidentate AA adsorption configurations on the Cu2O (111) surface. Yellow indicates charge accumulation, and blue depicts charge depletion. | ||
The charge distribution observed in Fig. 8(a) corroborates that in this model, the DEZ molecule tended to interact more preferably with the uncovered surface region than with the AA molecule. This is observed as a significant charge density accumulation area (yellow lobe) in the oxygen and CUS Cu atoms under the Zn and methyl group. On the other hand, the interaction bonds of the AA molecule remained are observed as charge accumulation in the O atoms (yellow lobes) and charge depletion (blue lobes) in the H atoms and CUS Cu.
Lastly, the interaction between the DEZ molecule and AA in the bidentate adsorption mode was also corroborated as shown in Fig. 8(b). This is observed as a depletion lobe in the Zn atom and a big accumulation zone in the O form carboxyl group. Also, we supported our previous affirmation that no change in the Cu–O bonds was observed after DEZ adsorption. Both results imply that even though DEZ tends to interact with the carboxyl group, the interaction is not strong enough to unbind the carboxylic acid inhibitor molecule from the surface, hence not affecting the ALD blocking effect. However, it is indeed a liability that may cause the presence of Zn in the Cu2O-monolayer interface. On the other hand, the diffusion of DEZ and posterior interaction with the carboxylic head group in highly packed monolayers may yield the formation of a non-anchoring group because water molecules would not permeate due to high hydrophobicity, as speculated in the previous work from our group.11 However, it is still unclear, and further studies must be done. In this matter, the results here provided that precursor–inhibitor interaction was not a cause of monolayer destruction and hence did not affect the ALD growth inhibition effect of the carboxylic acid molecule in the most stable adsorption modes (bidentate and carbonyl chemisorbed with an HB), which are present in highly packed monolayers (adsorption times equal to 48 h7). Yet, molecule adsorption was affected in some configurations, such as the model presented in Fig. 8(b), which may occur at low coverages or low adsorption times. This supports previous works on AS-ALD with SAMs where higher monolayer packing produced better-blocking results.8
4. Conclusions
In the present work, we extensively studied the adsorption of acetic acid as a model head group of carboxylic acid type surfactants on the Cu and Cu2O (111) surfaces and the interactions of this system with the diethylzinc molecule to further understand the blocking ability of such inhibitor molecules and their limitations with this ALD precursor.Highly stable adsorption energy values were found when the surfactant molecule was adsorbed in a bidentate manner on Cu (111) and Cu2O (111). The molecule resulted in highly stable carbonyl chemisorbed configuration on the Cu2O (111) surface, while on Cu (111), the carboxylic group tended to adsorb mainly through physisorption.
NCI index analysis revealed that coordination bonds and HB showed the most stabilizing interactions in chemisorbed states on both surfaces. The coordination bonds were stronger for CUS than for CSA Cu, which was also reflected in a small difference in adsorption energy. Bader charge analysis revealed mild charge transfer from Cu to AA in all the chemisorption configurations. However, it was more prominent in Cu2O.
The co-adsorption of diethylzinc with the most stable configurations of AA in Cu2O revealed two major conditions. When the molecule was adsorbed in a bidentate configuration, the interaction with the diethylzinc molecule was not strong enough to remove the inhibitor molecule from the surface. This provides valuable insight into the behavior of carboxylic acid inhibitors with the DEZ precursor, meaning that at high coverages, the DEZ precursor will not influence the inhibition ability. However, the presented results show that if diffusion through the SAM occurs, DEZ might interact with the headgroup, and the presence of Zn can be measured as observed in previous reports.
This work points out the significance of the different adsorption modes (that typically depend on SAM coverage or adsorption time) for achieving area-selective deposition using the ALD technique with soft removal SAM molecules.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DGAPA-UNAM projects IG101124 and IA100624 for the partial financial support. Calculations were performed at the DGCTIC-UNAM Supercomputing Center under projects LANCAD-UNAM-DGTIC-368 and LANCAD-UNAM-DGTIC-422. JGS acknowledges the LNS-BUAP project 202201042N and the THUBAT KAAL IPICYT supercomputing center project TKII-JGSA001 for their computational resources. LELG thanks DGAPA-UNAM for the postdoctoral position. H. T. acknowledges the financial support from DGAPA-UNAM grants no. IN108821, IN119023; FORDECYT grant no. 21077; CONAHCyT grant no. A1-S-21323, and FONCICyT grant no. 246648. We thank E. Murillo and Aldo Rodriguez-Guerrero for their technical support and useful discussions.References
- M. Pasquali, S. De Gendt and S. Armini, Understanding the impact of Cu surface pre-treatment on Octadecanethiol-derived self-assembled monolayer as a mask for area-selective deposition, Appl. Surf. Sci., 2021, 540, 148307 CrossRef CAS.
- I. Tezsevin, J. F. W. Maas, M. J. M. Merkx, R. Lengers, W. M. M. Kessels, T. E. Sandoval and A. J. M. Mackus, Computational Investigation of Precursor Blocking during Area-Selective Atomic Layer Deposition Using Aniline as a Small-Molecule Inhibitor, Langmuir, 2023, 39, 4265–4273 CrossRef CAS PubMed.
- D. Bobb-Semple, L. Zeng, I. Cordova, D. S. Bergsman, D. Nordlund and S. F. Bent, Substrate-dependent study of chain orientation and order in alkylphosphonic acid self-assembled monolayers for ALD blocking, Langmuir, 2020, 36, 12849–12857 CrossRef CAS PubMed.
- S. E. Atanasov, B. Kalanyan and G. N. Parsons, Inherent substrate-dependent growth initiation and selective-area atomic layer deposition of TiO2 using “water-free” metal–halide/metal alkoxide reactants, J. Vac. Sci. Technol., A, 2016, 34(1), 01A148 CrossRef.
- B. Karasulu, F. Roozeboom and A. Mameli, High-Throughput Area-Selective Spatial Atomic Layer Deposition of SiO2 with Interleaved Small Molecule Inhibitors and Integrated Back-Etch Correction for Low Defectivity, Adv. Mater., 2023, 35(25), 2301204 CrossRef CAS PubMed.
- J. Yarbrough, A. B. Shearer and S. F. Bent, Next generation nanopatterning using small molecule inhibitors for area-selective atomic layer deposition, J. Vac. Sci. Technol., A, 2021, 39, 021002 CrossRef CAS.
- F. S. M. Hashemi, C. Prasittichai and S. F. Bent, A New Resist for Area Selective Atomic and Molecular Layer Deposition on Metal–Dielectric Patterns, J. Phys. Chem. C, 2014, 118, 10957–10962 CrossRef CAS.
- D. Bobb-Semple, K. L. Nardi, N. Draeger, D. M. Hausmann and S. F. Bent, Area-Selective Atomic Layer Deposition Assisted by Self-Assembled Monolayers: A Comparison of Cu, Co, W, and Ru, Chem. Mater., 2019, 31, 1635–1645 CrossRef CAS.
- F. S. Minaye Hashemi, B. R. Birchansky and S. F. Bent, Selective Deposition of Dielectrics: Limits and Advantages of Alkanethiol Blocking Agents on Metal–Dielectric Patterns, ACS Appl. Mater. Interfaces, 2016, 8, 33264–33272 CrossRef CAS PubMed.
- T. L. Liu, L. Zeng, K. L. Nardi, D. M. Hausmann and S. F. Bent, Characterizing Self-Assembled Monolayer Breakdown in Area-Selective Atomic Layer Deposition, Langmuir, 2021, 37, 11637–11645 CrossRef CAS PubMed.
- L. E. López-González, J. Guerrero-Sánchez and H. Tiznado, Soft removal of stearic acid self-assembled monolayer for area-selective atomic layer deposition, Surf. Interfaces, 2023, 41, 103298 CrossRef.
- L. E. López-González, R. Ponce-Pérez, N. Takeuchi, H. Tiznado and J. Guerrero-Sánchez, Adsorption of sorbitan ester surfactant on copper and Cuprous oxide surfaces: A density functional theory study, Appl. Surf. Sci., 2022, 589, 153061 CrossRef.
- F. S. Minaye Hashemi, C. Prasittichai and S. F. Bent, Self-Correcting Process for High Quality Patterning by Atomic Layer Deposition, ACS Nano, 2015, 9, 8710–8717 CrossRef CAS PubMed.
- R. T. Sanderson, Chemical Bonds and Bonds Energy Physical Chemistry A Series of Monographs, Academic Press Inc, New York, 2nd edn, 1976, vol. 21 Search PubMed.
- S. Satyarthy, M. Hasan Ul Iqbal, F. Abida, R. Nahar, A. J. Hauser, M. M.-C. Cheng and A. Ghosh, Stearic Acid as an Atomic Layer Deposition Inhibitor: Spectroscopic Insights from AFM-IR, Nanomaterials, 2023, 13(19), 17739 CrossRef PubMed.
- W. R. Thompson and J. E. Pemberton, Characterization of Octadecylsilane and Stearic Acid Layers on AI2O3 Surfaces by Raman Spectroscopy, Langmuir, 1995, 11(5), 1720–1725 CrossRef CAS.
- H. R. Talesh Bahrami, A. Azizi and H. Saffari, Influence of different parameters of preparing self-assembled monolayers on copper surfaces in the dropwise condensation heat transfer: an experimental study, J. Braz. Soc. Mech. Sci. Eng., 2019, 41(3), 159 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, A. Smogunov and P. Umari, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502, DOI:10.1088/0953-8984/21/39/395502.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- D. Vanderbilt, Rapid Commun., 1990, 41, 7892–7895 CrossRef PubMed.
- Y. Hinuma, H. Hayashi, Y. Kumagai, I. Tanaka and F. Oba, Comparison of approximations in density functional theory calculations: Energetics and structure of binary oxides, Phys. Rev. B, 2017, 96(9), 094102 CrossRef.
- D. Gustinčič and A. Kokalj, A DFT study of adsorption of imidazole, triazole, and tetrazole on oxidized copper surfaces: Cu2O(111) and Cu2O(111)-w/o-CuCUS, Phys. Chem. Chem. Phys., 2015, 17, 28602–28615 RSC.
- L. I. Bendavid and E. A. Carter, CO2 adsorption on Cu2O(111): A DFT+U and DFT-D study, J. Phys. Chem. C, 2013, 117, 26048–26059 CrossRef CAS.
- H. M. Otte, Lattice parameter determinations with an X-ray spectrogoniometer by the Debye-Scherrer method and the effect of specimen condition, J. Appl. Phys., 1961, 32, 1536–1546 CrossRef CAS.
- S. S. Hafner and S. Nagel, The electric field gradient at the position of copper in Cu2O and electronic charge density analysis by means of K-factors, Phys. Chem. Miner., 1983, 9, 19–22 CrossRef CAS.
- J. Guerrero-Sánchez, N. Takeuchi and F. Zaera, Density Functional Theory Study of the Surface Adsorption and Dissociation of Copper(I) Acetamidinates on Cu(110) Surfaces, J. Phys. Chem. C, 2019, 123, 4341–4348 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. J. Díaz León, D. M. Fryauf, R. D. Cormia and N. P. Kobayashi, Low-Dimensional Materials and Devices 2016, SPIE, 2016, vol. 9924, p. 99240O Search PubMed.
- T. Suh, Y. Yang, P. Zhao, K. U. Lao, H. Y. Ko, J. Wong, R. A. Distasio and J. R. Engstrom, Competitive Adsorption as a Route to Area-Selective Deposition, ACS Appl. Mater. Interfaces, 2020, 12, 9989–9999 CrossRef CAS PubMed.
- B. Z. Sun, W. K. Chen and Y. J. Xu, Reaction mechanism of CO oxidation on Cu2O(111): A density functional study, J. Chem. Phys., 2010, 133, 154502 CrossRef PubMed.
- C. Li, F. Wang, S. F. Li, Q. Sun and Y. Jia, Stability and electronic properties of the O-terminated Cu2O(111) surfaces: First-principles investigation, Phys. Lett. A, 2010, 374, 2994–2998 CrossRef CAS.
- J. Hafner, Ab initio simulations of materials using VASP: Density-functional theory and beyond, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- D. L. Allara and R. G. Nuzzo, Spontaneously organized molecular assemblies. 1. Formation, dynamics, and physical properties of n-alkanoic acids adsorbed from solution on an oxidized aluminum surface, Langmuir, 1985, 1, 45–52 CrossRef CAS.
- R. Barzaga, M. P. Hernández, F. Aguilar-Galindo and S. Dĺaz-Tendero, Revealing the Interplay between Covalent and Non-Covalent Interactions Driving the Adsorption of Monosubstituted Thiourea Derivatives on the Au(111) Surface, J. Phys. Chem. C, 2020, 124, 9924–9939 CrossRef CAS.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- A. Otero-De-La-Roza, E. R. Johnson and V. Luaña, Critic2: A program for real-space analysis of quantum chemical interactions in solids, Comput. Phys. Commun., 2014, 185, 1007–1018 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A Program for Plotting Noncovalent Interaction Regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- A. Kokalj and S. Peljhan, Density functional theory study of adsorption of benzotriazole on Cu2O surfaces, J. Phys. Chem. C, 2015, 119, 11625–11635 CrossRef CAS.
- M. Zhang, R. Yao, H. Jiang, G. Li and Y. Chen, Insights into the mechanism of acetic acid hydrogenation to ethanol on Cu(111) surface, Appl. Surf. Sci., 2017, 412, 342–349 CrossRef CAS.
- M. Poberžnik, F. Chiter, I. Milošev, P. Marcus, D. Costa and A. Kokalj, DFT study of n-alkyl carboxylic acids on oxidized aluminum surfaces: From standalone molecules to self-assembled-monolayers, Appl. Surf. Sci., 2020, 525, 146156 CrossRef.
- M. S. Lim, K. Feng, X. Chen, N. Wu, A. Raman, J. Nightingale, E. S. Gawalt, D. Korakakis, L. A. Hornak and A. T. Timperman, Adsorption and desorption of stearic acid self-assembled monolayers on aluminum oxide, Langmuir, 2007, 23, 2444–2452 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03117b |
| This journal is © the Owner Societies 2024 |