
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of dynamic nuclear polarisation using “off-the-shelf” Gd(III)-based polarising agents†
Daniel J.
Cheney
 a,
Paolo
Cerreia Vioglio
b,
Adam
Brookfield
c and
Frédéric
Blanc
*ade
a,
Paolo
Cerreia Vioglio
b,
Adam
Brookfield
c and
Frédéric
Blanc
*ade
aDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: frederic.blanc@liverpool.ac.uk
bDNP MAS NMR Facility, Sir Peter Mansfield Imaging Centre, University of Nottingham, Nottingham NG7 2RD, UK
cDepartment of Chemistry and Photon Science Institute, University of Manchester, Manchester M13 9PL, UK
dLeverhulme Research Centre for Functional Materials Design, Materials Innovation Factory, University of Liverpool, Liverpool, L69 7ZD, UK
eStephenson Institute for Renewable Energy, University of Liverpool, Liverpool L69 7ZF, UK
First published on 11th September 2024
Abstract
Complexes of paramagnetic metal ions, in particular Gd3+, have been demonstrated as efficient polarising agents for magic-angle spinning (MAS) dynamic nuclear polarisation (DNP). We recently demonstrated that commercially available and inexpensive Gd(NO3)3 is suitable for use as an “off-the-shelf” MAS DNP polarising agent, providing promising sensitivity enhancements to 1H, 13C, and 15N NMR signals. Here we expand upon this approach by investigating the impact of the Gd(NO3)3 concentration and by exploring a larger range of readily available Gd3+ sources. We found that a Gd(NO3)3 concentration of 20 mM in the case of 1H and 13C, and 40 mM in the case of 15N, offers optimum signal enhancements and is rationalised as a trade-off between DNP enhancements, polarisation build-up times, and electron paramagnetic resonance (EPR) spin–spin relaxation times. We determined that a range of different gadolinium compounds (GdCl3, Gd2(SO4)3, GdBr3, and Gd(OAc)3) are also suitable for use as polarising agents and yield 1H, 13C, and 15N signal enhancements of variable values. Gd(OAc)3 yields lower signal enhancements, which is proposed to be the result of greater local asymmetry at the Gd3+ centre leading to EPR line broadening, and the methyl group in the acetate ion acting as a relaxation sink and limiting the nuclear polarisation available.
Introduction
Dynamic nuclear polarisation (DNP) has been widely demonstrated as a highly efficient method for overcoming the typically limited sensitivity of solid-state nuclear magnetic resonance (NMR) spectroscopy under magic-angle spinning (MAS).1–3 By employing microwave irradiation to transfer polarisation from unpaired electrons to nuclear spins, improvements to the signal-to-noise ratio of orders of magnitude can be achieved in a variety of NMR applications, including inorganic materials4 and biomolecules.5 DNP is particularly important for low natural abundance nuclei such as 17O,6 nuclei with low gyromagnetic ratios such as 89Y,7 and surface sites.8,9Some of the most significant advances in DNP have been attributed to the rational design of polarising agents (PAs), in particular nitroxide biradicals for which polarisation transfer is driven by the highly efficient cross effect (CE) mechanism.10 One of the first major developments in PA design was the introduction of the water-soluble bis-nitroxide TOTAPOL,11 with subsequent optimisation focussing on increasing the rigidity of the linker, resulting in PAs such as AMUPol,12 TEKPol,13 AsymPol,14 and TEKPol2.15 More recently, HydrOPol and NaphPol have been shown to be the most efficient PAs to date, providing 1H enhancements of 330 and 249, respectively, at 9.4 T (400 MHz) and 100 K.16–18
Paramagnetic metal ion complexes have also been extensively investigated for use as PAs in DNP driven by the solid effect (SE) mechanism.19 Gadolinium(III) is the most promising of these, due to its weak spin–orbit coupling (SOC), negligible hyperfine coupling to its NMR active isotopes (155Gd and 157Gd), and high stability with respect to oxidation and reduction.20 [Gd(dota)(H2O)]−, which gives a 1H enhancement of −16 at 9.4 T, was used as a basis for an investigation into a wide range of Gd(III) chelates.21–24 [Gd(tpatcn)] was shown to be the strongest performer, giving a 1H enhancement of −36 at the same field. In addition, bis-gadolinium complexes have been considered as potential PAs for CE DNP.25 Paramagnetic metal ions have also been applied to the labelling of specific sites in proteins and nucleic acids to achieve site-selective enhancements22,26,27 Endogenous DNP has been carried out for materials doped with metal ions, with notable examples being inorganic glasses28,29 as well as 6Li, 7Li, and 17O DNP in battery anode and phosphor materials doped with manganese(II) and iron(III).30–33
A key limitation of DNP is the relative inaccessibility of PAs, with only a limited range of the most widely-used being commercially available. The synthesis of PAs is seldom trivial, with most requiring multi-step syntheses with low yields. For example, AMUPol is prepared using a four-step synthesis with a 24% yield,12 and [Gd(tpatcn)] requires a six-step synthesis with a 6% yield.23 Furthermore, radicals are known to exhibit poor stability in strongly reducing or acidic environments.34 Clearly there is a need for the development of PAs that are more widely accessible.
Recently, our group introduced the concept of “off-the-shelf” polarising agents: paramagnetic compounds that are readily commercially available, easily affordable, and can be used “as is” without any further synthesis.35 We initially selected gadolinium(III) nitrate ([Gd(NO3)3]) and achieved enhancements of −16 and −11 for 13C and −57 and −23 for 15N in glycine at 9.4 T and 14.1 T (600 MHz), respectively. Although these enhancements were smaller than those attainable using Gd(III) complexes with chelating ligands, it is hoped that the convenient nature of “off-the-shelf” PAs will contribute to DNP becoming more accessible.
In this work, we further explore other Gd(III) sources as well as the impact of the Gd(III) concentration, investigating the effect on the DNP efficiency, with the enhancement, build-up dynamics, and paramagnetic bleaching being considered. This will be accompanied by supporting electron paramagnetic resonance (EPR) data, as well as an investigation into the Gd(III) coordination sphere using high-resolution mass spectrometry (HRMS).
Materials and methods
Sample preparation
Five Gd(III) compounds from common suppliers were investigated as potential PAs: Gd(NO3)3·6H2O (Sigma-Aldrich, 99.99%), GdCl3·6H2O (Alfa-Aesar, 99.9%), Gd2(SO4)3·8H2O (Alfa-Aesar, 99.99%), GdBr3·xH2O (Alfa-Aesar, 99.99%), and Gd(OAc)3·xH2O (Alfa-Aesar, 99.9%). The number of waters of hydration, x, was determined using thermogravimetric analysis (TGA) to be 13 and 4 for GdBr3 and Gd(OAc)3, respectively.For the DNP experiments, solutions of 1.5 M 2-13C,15N-glycine (Sigma-Aldrich, 99% 13C labelled on the α-carbon, 98% 15N), doped with various concentrations of Gd(NO3)3, 10 mM Gd2(SO4)3, and 20 mM of all other Gd(III) compounds (so as to maintain a constant Gd3+ concentration), were prepared in the glass-forming mixture glycerol-d8/D2O/H2O (6/3/1 v/v/v) (“DNP juice”). The volume of glycerol was determined indirectly by mass, whereas water and water/glycerol mixtures were pipetted using miropipettes with an uncertainty of 1%. The samples were sonicated at 70 °C for 30 minutes to ensure complete dissolution of both glycine and the Gd(III) compound (although Gd2(SO4)3 was difficult to dissolve, indicating that the solubility limit may be exceeded at this concentration). 20 μL amounts of solution were placed into 3.2 mm sapphire rotors, closed with Vespel drive caps and sealed using polytetrafluoroethylene (PTFE) tape around the inside of the caps. The rotors were weighed before and after sample loading in order to determine the sample mass.
Solutions for EPR and HRMS analysis were prepared in a similar manner, albeit both with and without 1.5 M unlabelled glycine (Alfa Aesar, 99%) and 3/2 v/v glycerol/H2O as the solvent. EPR tubes with outer diameters of 5 mm were filled with each solution to a 1 cm height to ensure that the active region of the resonator was entirely filled. A small amount of formic acid was added to the HRMS samples to ensure complete dissolution.
DNP MAS NMR
DNP experiments were performed at ∼105 K on a commercial Bruker Biospin DNP system36 at a static magnetic field B0 = 14.1 T on a 600 MHz AVANCE III spectrometer with a gyrotron microwave (μW) source operating at a frequency of ω0S/2π = 395 GHz. Experiments were performed at a MAS rate ωr/2π of 10 kHz on either a 3.2 mm triple resonance HXY low-temperature MAS probe tuned to X = 13C and Y = 15N or a 3.2 mm double resonance HX low-temperature MAS probe tuned to X = 13C or 15N. 1H, 13C, and 15N NMR spectra were measured using Hahn echo pulse sequences, synchronised to one rotor period, with radiofrequency (rf) pulse amplitudes of ωH/2π = 66 kHz, ωC/2π = 60 kHz, and ωN/2π = 38 kHz on the triple resonance probe and ωH/2π = 88 kHz, ωC/2π = 74 kHz, and ωN/2π = 50 kHz on the double resonance probe. 1H → 13C cross polarisation (CP) spectra were recorded using Hartman–Hahn matched rf pulse amplitudes of ωH/2π = 68 kHz and ωC/2π = 60 kHz on the triple resonance probe and ωH/2π = 88 kHz and ωC/2π = 74 kHz on the double resonance probe, with a 70% → 100% linear amplitude ramp on the 1H pulse, and a CP contact duration of 2 ms. SPINAL-64 decoupling was applied during 13C and 15N NMR signal acquisition with a 1H rf-pulse amplitude of 63 kHz on the triple resonance probe, and 88 kHz on the double resonance probe.37 The stator and waveguide design is identical for both probes38 and there is no significant change in the effective microwave power at the sample and DNP enhancement as exemplified in the data given in Fig. S1 (ESI†). A train of 100 pre-saturation rf pulses, separated by 1 ms, was applied on all relevant spectrometer rf-channels before data collection.DNP build-up time constants (TB,ON) were measured by varying the length of the irradiation period following pre-saturation (using a standard pulse programme for saturation recovery), and fitting the resulting data with a stretched exponential function of the type  , where α is a stretching constant, and the final TB,ON values were calculated as
, where α is a stretching constant, and the final TB,ON values were calculated as  , with
, with  being the gamma function. Uncertainties in TB,ON values were taken to be the 95% confidence intervals calculated using MATLAB's curve fitting tool. The DNP enhancements, ε, defined as the ratio of the integrated NMR signal intensities with and without μW irradiation, were measured at the field position for optimum negative enhancement with an electron Larmor frequency of 395 GHz (previously determined to be 14.159 T for 1H, 14.175 T for 13C, and 14.179 T for 15N, with some minor adjustments of less than 2 mT being made where necessary to optimise the enhancement) with a build-up delay of 5 × TB,ON, and are reported as the optimum values as a function of the μW power curve. Uncertainties in the DNP enhancements were calculated as
being the gamma function. Uncertainties in TB,ON values were taken to be the 95% confidence intervals calculated using MATLAB's curve fitting tool. The DNP enhancements, ε, defined as the ratio of the integrated NMR signal intensities with and without μW irradiation, were measured at the field position for optimum negative enhancement with an electron Larmor frequency of 395 GHz (previously determined to be 14.159 T for 1H, 14.175 T for 13C, and 14.179 T for 15N, with some minor adjustments of less than 2 mT being made where necessary to optimise the enhancement) with a build-up delay of 5 × TB,ON, and are reported as the optimum values as a function of the μW power curve. Uncertainties in the DNP enhancements were calculated as  , where SNR is the signal-to-noise ratio.18 The contribution factor (θ), which accounts for paramagnetic bleaching, is defined as the ratio of the integrated intensities of the 13C CP MAS spectra with and without a Gd(III) compound, scaled by the sample masses in each case. Due to potential differences in T1ρ, the CP contact time was optimised for maximum signal on each sample. The recycle delay was set to five times the 1H T1 value. The 1H and 13C spectra were externally referenced to adamantane at 1.8 ppm39,40 and 29.45 ppm,41 respectively, while the 15N spectra were internally referenced to glycine at 33.4 ppm.42
, where SNR is the signal-to-noise ratio.18 The contribution factor (θ), which accounts for paramagnetic bleaching, is defined as the ratio of the integrated intensities of the 13C CP MAS spectra with and without a Gd(III) compound, scaled by the sample masses in each case. Due to potential differences in T1ρ, the CP contact time was optimised for maximum signal on each sample. The recycle delay was set to five times the 1H T1 value. The 1H and 13C spectra were externally referenced to adamantane at 1.8 ppm39,40 and 29.45 ppm,41 respectively, while the 15N spectra were internally referenced to glycine at 33.4 ppm.42
EPR
EPR data were recorded using Bruker E580 Elexsys pulsed spectrometers, equipped with a Bruker 4118X-MD5 Flexline resonator for X-band (9.5 GHz) measurements, and a Bruker QT-II resonator for Q-band (34 GHz) measurements. Experiments were carried out at 100 K (80 K for 40 and 60 mM Gd(NO3)3, and 30 K for 100 mM Gd(NO3)3), with cryogenic temperatures achieved with closed cycle cryofree cryostats from Bruker Biospin and Cryogenic Ltd. The presence of helium in the cryostats excluded oxygen from the samples. Echo-detected field-swept EPR spectra were recorded using a standard Hahn echo sequence of π/2–τ–π–τ where the π pulse was 32 ns, and τ was 180 ns and 300 ns at X-band and Q-band, respectively. Electron spin–spin relaxation time constants (T2e) were measured by increasing τ in 4 ns increments. In some cases, a longer π pulse of 60 ns was used in order to suppress ESEEM (Echo Spin Echo Envelope Modulation) from interacting nuclei modulating the magnetisation decay curve. The data were fit with an exponential function of the type . Electron spin–lattice relaxation time constants (T1e) were obtained using a three-pulse inversion recovery echo sequence π–T–π/2–τ–π–τ, with a four-step phase cycle in order to minimise effects from unwanted echoes, and with T increasing in 2 ns increments. The data were fit with a stretched exponential function of the type
. Electron spin–lattice relaxation time constants (T1e) were obtained using a three-pulse inversion recovery echo sequence π–T–π/2–τ–π–τ, with a four-step phase cycle in order to minimise effects from unwanted echoes, and with T increasing in 2 ns increments. The data were fit with a stretched exponential function of the type  , where β is a stretching constant, and the final T1e values were calculated as
, where β is a stretching constant, and the final T1e values were calculated as  , with
, with  being the gamma function.
being the gamma function.
The axial zero-field splitting (ZFS) components, D, of each Gd(III) compound were determined using echo-detected field swept spectra measured at 10 K with a Gd(III) concentration of 25 μM. Under these conditions, T2e was measured to be longer than 3 μs for both the central and satellite transitions, which is significantly longer than the echo delay. Therefore, the satellite signals are not expected to have decayed to a greater degree than the central transition, allowing for adequate fitting of both spectral components.43–45 The procedure for extracting D using spectral fitting has been described previously.46,47 The distribution of D values was assumed to be Gaussian in nature, with a width of σD, while the ratio of E (the transverse ZFS constant) to D was assumed to be randomly distributed between 0 and 1/3 (with all E/D values being equally probable). For a range of D and σD values (between 300 and 1950 MHz, and 50 and 600 MHz, respectively, and separated by 50 MHz in both cases), 1000 values of D and E were generated according to the above constraints, and EPR spectra at both X-band and Q-band were simulated using EasySpin.48 The average spectrum in each case was averaged, and compared to experimental data at X-band and Q-band in order to calculate the root mean square deviation (RMSD) using:
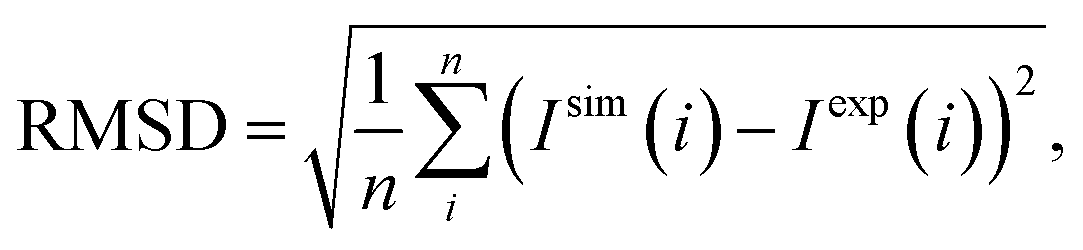 | (1) |
HRMS
HRMS data were recorded on an Agilent 6540 quadrupole-time-of-flight mass spectrometer using electrospray ionisation in the positive mode. A list of notable peaks is given in Table S1 (ESI†). Peaks arising from Gd(III) species are easily identifiable due to gadolinium's characteristic isotopic pattern (approximately 0.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.21:0.16:0.25:0.22 for 155Gd:156Gd:157Gd:158Gd:160Gd).
:0.21:0.16:0.25:0.22 for 155Gd:156Gd:157Gd:158Gd:160Gd).
Results and discussion
Impact of the Gd(NO3)3 concentration
The DNP enhancement of 2-13C,15N-glycine was previously determined to be driven by the solid effect, as shown by the Zeeman field profiles.35 Here, the measurements were carried out at the previously-determined optimum fields for negative enhancement, due to the positive enhancements being inaccessible within the range of the sweep coil. As an example, the DNP-enhanced MAS NMR spectra using 10 mM Gd(NO3)3 for 1H (indirectly detected via1H → 13C CP), 13C, and 15N (both directly detected via Hahn echoes) show enhancements (ε, taken as the ratio of the signal intensity under microwave irradiation to that without irradiation in the same sample) of −2.7 ± 0.1, −14 ± 0.2, and −23 ± 3, respectively (Fig. 1). The 1H DNP enhancement (Fig. 2(a) and Table S2, ESI†) initially increases as the Gd(NO3)3 concentration increases, up to −4.5 ± 0.1 at 20 mM, since more polarisation sources become available, but with higher concentrations, the enhancement decreases. A similar concentration dependence is seen for direct 13C polarisation (Fig. 2(b)) where the optimum concentration of 20 mM provides an enhancement of −23 ± 3. For 15N, on the other hand, 40 mM gives the highest enhancement of −37 ± 4 (Fig. 2(c)). It was experimentally observed that in no case did the optimum field for the negative enhancement change significantly, strongly suggesting that there is no contribution from the CE (which might be expected at higher concentration if the EPR spectrum is broadened and the electron–electron coupling becomes sufficiently strong).25 | ||
| Fig. 1 (a) CP-detected 13C, (b) direct Hahn echo detected 13C, and (c) direct Hahn echo detected 15N NMR spectra of 2-13C,15N-glycine in 6/3/1 v/v/v glycerol-d8/D2O/H2O, doped with 10 mM Gd(NO3)3, with (red) and without (black) microwave irradiation. All spectra were measured at ∼105 K and 14.1 T with a MAS frequency of 10 kHz. Signals marked with an asterisk originate from the glycerol solvent. | ||
 | ||
| Fig. 2 (a)–(c) DNP enhancements, (d)–(f), DNP build-up times, and (g), (h), (i) overall sensitivity enhancements for (a), (d), (g) 1H, (b), (e), (h) 13C, and (c), (f), (i) 15N of 1.5 M 2-13C,15N-glycine in 6/3/1 v/v/v glycerol-d8/D2O/H2O at ∼105 K and 14.1 T, with various concentrations of Gd(NO3)3. | ||
It is well-known that the DNP enhancement is, by itself, not an accurate metric for the overall DNP efficiency, due to other factors which influence the sensitivity.49–52 The overall improvement to the NMR sensitivity εθ (i.e. the increase in signal-to-noise ratio per square root unit time) was determined using:53
 | (2) |
 | ||
| Fig. 3 Contribution (paramagnetic bleaching) factors for 2-13C,15N-glycine as a function of (a) the Gd(NO3)3 concentration and (b) the Gd(III) source. | ||
For all three nuclei considered, it is seen that TB,ON becomes shorter as the Gd(NO3)3 concentration is increased (Fig. 2(d)–(f)), which is expected to be mainly due to paramagnetic relaxation enhancement. With build-up dynamics and paramagnetic bleaching taken into account, the optimum sensitivity enhancements are −7.2 ± 0.3 for 1H with 20 mM Gd(NO3)3, −24 ± 3 for 13C with 20 mM Gd(NO3)3, and −61 ± 12 for 15N with 40 mM Gd(NO3)3 (Fig. 2(g)–(i)).
The decrease in enhancement at higher Gd(III) concentrations can be rationalised by considering the EPR data. Gd(III) has an electronic configuration of 4f7 with each of the seven f-orbitals being singly occupied, leading to a total electron spin of S = 7/2. Due to the half-filled f-subshell, the spin–orbit coupling (SOC) is negligible, resulting in a g-factor close to the free electron value of 2.0023.54 There are eight Zeeman levels and seven single-quantum transitions. The central transition (ms = −1/2 → +1/2) is only broadened by a second-order zero-field splitting (ZFS) effect, which results in a narrow central EPR line.55 The satellite transitions on the other hand are broadened by a direct first-order ZFS effect, which leads to an additional broad satellite EPR signal.
The EPR spectra of various concentrations of Gd(NO3)3 at X-band (9.5 GHz) and Q-band (34 GHz) are shown in Fig. 4(a) and (b), respectively. The expected narrow central transition and broad satellite transitions are easily identifiable at Q-band, while at X-band, they are not easily distinguished. The g-factor was determined to be 1.990, close to the previously-reported value for GdCl3 in aqueous solution.47 No hyperfine splittings are resolvable due to the weak coupling to gadolinium's NMR-active isotopes. It has been shown using perturbation theory that the full-width at half-maximum (FWHM, Δh) of the central transition varies with the magnetic field, B0, as:
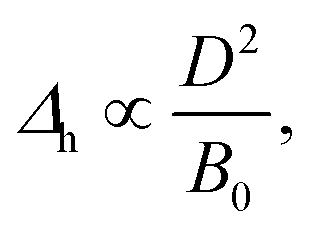 | (3) |
 | ||
| Fig. 4 Echo-detected field-swept EPR spectra of various concentrations of Gd(NO3)3 in 3/2 v/v glycerol/H2O, measured at (a) X-band and (b) Q-band at the temperatures indicated for each concentration. | ||
The electron spin–lattice (T1e) and spin–spin (T2e) relaxation times are important parameters for assessing a PA's suitability, particularly for SE DNP, since it is easier to saturate the forbidden zero- and double-quantum transitions for slowly relaxing electron spins. Both time constants were found to decrease as the Gd(NO3)3 concentration was increased (Fig. 5(a), (b) and Table S3, ESI†), which is expected due to stronger dipolar couplings between Gd(III) centres. For this reason, the data for Gd(III) concentrations of 40 mM or higher were collected at lower temperatures (80 K for 40 and 60 mM, 30 K for 100 mM), due to significant relaxation during echo delays at higher temperatures. This is also the cause of the lower signal-to-noise ratio for 60 mM Gd(NO3)3 at X-band, and the slightly distorted lineshape for 100 mM Gd(NO3)3 at Q-band. Previously-reported Gd(III) complexes were found to have Q-band T1e values ranging between 500 and 860 ns, and T2e values ranging between 300 and 470 ns, respectively.24 These were measured at 10 K and with a concentration of 25 μM, so no direct comparisons to our 100 K values can be made. However, we have also measured T1e and T2e of 25 μM Gd(NO3)3 at 10 K to be 219 μs and 3.7 μs, respectively. Therefore, the relaxation behaviour of Gd(NO3)3 is suitable for DNP applications. The broadened lineshape and faster relaxation are likely to be the main contributing factors to the reduced DNP enhancement when using higher Gd(NO3)3 concentrations.
 | ||
| Fig. 5 Dependence of (a), (c) T1e and (b), (d) T2e on (a), (b) the concentration of Gd(NO3)3 and (c), (d) the Gd(III) source, measured at X-band and Q-band at 100 K (80 K for 40 and 60 mM Gd(NO3)3, 30 K for 100 mM Gd(NO3)3), with and without 1.5 M glycine. | ||
Impact of the Gd(III) source
The DNP enhancements for 1H, 13C, and 15N were measured for a range of Gd(III) compounds: Gd(NO3)3, GdCl3, Gd2(SO4)3, GdBr3, and Gd(OAc)3 (Fig. 6(a)–(c) and Table S4, ESI†). This range was selected based on the commercial accessibility of these compounds, and provides a mixture of monodentate, bidentate, hard and soft ligands. It should be noted that GdCl3 has previously been investigated for use as a PA, albeit at 5 T, with a concentration of 10 mM, and with 3/2 v/v 13C3-glycerol/H2O as the solvent.22 For 1H and 15N, Gd(NO3)3, GdCl3, Gd2(SO4)3, and Gd(OAc)3 appear to give similar enhancements, comparable to the calculated uncertainties. However, for 13C, there appears to be some variation between compounds (which is larger than the uncertainties in the enhancements), with Gd(NO3)3 and GdCl3 giving the largest enhancements, and Gd2(SO4)3 and GdBr3 giving smaller enhancements. It is also noted that for all three nuclei, Gd(OAc)3 gives significantly smaller enhancements than all other Gd(III) compounds. | ||
| Fig. 6 (a)–(c) DNP enhancements, (d)–(f), DNP build-up times, and (g)–(i) overall sensitivity enhancements for (a), (d), (g) 1H, (b), (e), (h) 13C, and (c), (f), (i) 15N of 1.5 M 2-13C,15N-glycine in 6/3/1 v/v/v glycerol-d8/D2O/H2O at ∼105 K and 14.1 T, with various Gd(III) compounds. | ||
The polarisation build-up time constants (Fig. 6(d)–(f)) for Gd(NO3)3, GdCl3, and Gd2(SO4)3 are very similar, but those for GdBr3 and Gd(OAc)3 are noticeably faster. It was also noted that the 13C and 15N build-up times for Gd2(SO4)3 were slightly longer than all other Gd(III) compounds. This may indicate a lower-than-expected Gd(III) concentration in solution, owing to its lower solubility in DNP juice, which may limit its suitability as a DNP polarising agent. It was observed that, as expected, the contribution factor does not depend on the Gd(III) source (Fig. 3(b)). The overall sensitivity enhancements (Fig. 6(g)–(i)) for Gd(NO3)3, GdCl3, and GdBr3 were very similar (with the exception of the 13C enhancement for GdBr3, which is noticeably lower, the cause of which is not apparent). The sensitivity increase for Gd2(SO4)3 is slightly smaller for 1H and 15N, and significantly smaller for 13C, which is possibly due to its lower solubility with respect to the other Gd(III) compounds. Finally, in all cases, the overall sensitivity increase for Gd(OAc)3 is significantly smaller, in spite of the faster DNP build-up times.
One of the most important parameters affecting the DNP efficiency is the zero-field splitting constant and since the SE DNP enhancement is inversely proportional to the EPR linewidth,46 the DNP efficiency has an inverse quadratic dependence on D in the absence of additional line broadening mechanisms.23,24 Using established models for fitting the EPR spectra measured at 10 K and a Gd(III) concentration of 25 μM (echo-detected field-swept spectra shown in Fig. 7(a) and (b),46,47 a value of D = 1079 ± 53 MHz is yielded for Gd(NO3)3 (Fig. 8, contour plot shown in Fig. S4, ESI†). This is larger than the previously-reported value of 810 MHz, in part due to the difference in the measurement conditions (which were 100 K and 20 mM previously), and in part due to σD originally being assumed to be fixed at D/3.35 Similar values are seen for most of the other Gd(III) compounds (Table 1 and Fig. S4, ESI†), which leads to virtually identical EPR linewidths. T1e and T2e vary slightly with different Gd(III) sources (Fig. 5(c) and (d) and Table S3, ESI†), although there is no apparent trend, indicating that the counter-ion is not a significant source of electron spin relaxation.
 | ||
| Fig. 7 Echo-detected field-swept EPR spectra of 25 μM of various Gd(III) compounds in 3/2 v/v glycerol/H2O without (a) and (c) and with (b) and (d) 1.5 M glycine, at (a) and (b) X-band and (c) and (d) Q-band at 10 K. | ||
| Gd(III) compound | D/MHz | σ D/MHz | ||
|---|---|---|---|---|
| Without glycine | With glycine | Without glycine | With glycine | |
| Gd(NO3)3 | 1079 ± 53 | 1342 ± 60 | 311 ± 27 | 371 ± 34 |
| GdCl3 | 1038 ± 70 | 1295 ± 46 | 288 ± 39 | 350 ± 30 |
| Gd2(SO4)3 | 1044 ± 36 | 1389 ± 57 | 299 ± 20 | 373 ± 33 |
| GdBr3 | 1062 ± 63 | 1367 ± 35 | 300 ± 38 | 369 ± 23 |
| Gd(OAc)3 | 1050 ± 66 | 1535 ± 68 | 295 ± 39 | 433 ± 36 |
 | ||
| Fig. 8 Best fits to the (a) X-band and (b) Q-band echo detected field swept EPR spectra of 25 μM Gd(NO3)3 in 3/2 v/v glycerol/H2O at 10 K. | ||
It is well-established that the strength of the ZFS is dependent upon the symmetry about the gadolinium centre.23,24,58 While this is potentially accessible from liquid-state NMR spectra, this is significantly challenged by the paramagnetic nature of Gd(III). Moreover, the Gd(III) species of interest are formed in frozen glasses, precluding the use of powder X-ray diffraction for structural refinements. These challenges have previously been recognised in the case of coordination and organometallic chemistry, with mass spectrometry (MS) being suggested as a suitable alternative.59–61 Therefore, we turned to high-resolution MS (HRMS) with electrospray ionisation to gain some insight into possible Gd(III) species formed. Mass spectra were recorded for 20 mM of each Gd(III) compound (10 mM for Gd2(SO4)3) dissolved in 3/2 v/v glycerol/H2O. The relevant HRMS peaks and their assignments are given in Table S1 (ESI†). Here we make the assumption that the detected Gd(III) species exist in solution, and are not formed following ionisation of the solution and subsequent evaporation of the solvent from the charged droplets.
All salts showed a collection of peaks corresponding to [Gd(glycerol)2]3+ fragments, which is expected given that glycerol can act as either a bidentate or tridentate ligand for lanthanide ions, including Gd3+.62 It was previously noted35 that the nitrate anion also binds directly to the metal ion, as evidenced by the detection of [Gd(glycerol)2(NO3)3]2+ (Fig. 9(a)). This is reasonable given that nitrate is a hard Lewis base and can act as a bidentate ligand, so binds strongly to Gd3+ (a hard Lewis acid). Similarly, sulfate, also a hard ligand that can bind in the bidentate mode, is found to bind directly to the metal centre. The corresponding peaks for GdCl3 are much weaker, owing to chloride being a softer, more polarisable anion which, being monoatomic, can only bind in the monodentate mode. Surprisingly, despite the bromide ion being an even softer Lewis base than chloride, strong peaks corresponding to [Gd(glycerol)2Br]3+, are observed. Furthermore, no signals corresponding to [Gd(glycerol)2(OAc)]2+ are seen, despite the fact that acetate is a hard Lewis base. Therefore, the interactions between hard and soft Lewis acids and bases are unlikely to be the cause of the effects seen here. However, evidence of the formation of bis-gadolinium species is seen for Gd(OAc)3, many of which contain acetate ions. It is not possible to distinguish whether glycerol or acetate acts as the bridging ligand, although the absence of similar peaks for the other Gd(III) compounds makes the latter case more plausible, and there is literature precedence for the existence of such complexes.63 The emergence of these bis-gadolinium species may be the reason no evidence of acetate binding to a single Gd(III) centre are seen. The similarities in the ligand spheres for most of the Gd(III) compounds is consistent with their virtually identical ZFS constants.
 | ||
| Fig. 9 Structures of Gd(III) coordination spheres in 3/2 v/v glycerol/H2O, both (a) without and (b) with 1.5 M glycerol, determined using HRMS. | ||
In order to determine the Gd(III) structures formed in the DNP samples at hand, HRMS measurements were also carried out in the presence of 1.5 M glycine. Interestingly, all five compounds now show the same five collections of peaks, corresponding to the coordination of glycerol and glycine to the metal centre: [Gd(glycerol)2]3+, [Gd(glycine)(glycerol)]3+, [Gd(glycine)(glycerol)2]3+, [Gd(glycine)3]3+, and [Gd(glycine)2(glycerol)]3+ (Fig. 9(b)). It is possible that the former two of these do not nominally exist in solution and are the result of fragmentation during ionisation. Peaks corresponding to the counter-ion binding to the inner sphere do not appear for any of the compounds. This seems to imply that in the DNP sample of interest, the ligand sphere does not differ between Gd(III) compounds. The EPR spectra all appear to be broader than those measured in the absence of glycine (Fig. 7(c) and (d)). Despite the ZFS constants of most of the compounds once again being similar (albeit larger than those measured without glycine), the value for Gd(OAc)3 is notably larger than the others (Table 1 and Fig. S5, ESI†). It was previously observed that for Gd(tpatcn), water molecules could form hydrogen bonds to the carboxylate moieties of the chelating ligand, resulting in a second coordination sphere.64,65 It is possible that a similar phenomenon could occur here, with acetate anions forming hydrogen bonds to glycerol and glycine protons (with the latter being more likely due to the absence of this line broadening without glycine), as shown in Fig. 10. Since this has been shown to alter Gd–O bond lengths, this may provide an explanation for an apparent change in local symmetry (and hence a stronger ZFS interaction). It is also feasible that the relative concentrations of each Gd(III) species in solution may vary between compounds. It is not, however, possible to determine this from the HRMS peak intensities given that it is not a quantitative technique (due to differences in solvent evaporation rates and ionisation efficiencies of various species). At 100 K, the addition of glycine, in some cases, also results in a slight increase in T1e (Fig. 5(c) and Table S3, ESI†).
 | ||
| Fig. 10 Proposed structure of the Gd(III) coordination sphere for Gd(OAc)3 in 3/2 v/v glycerol/H2O with added glycine, showing hydrogen bonding between the acetate anion and glycine. | ||
A stronger ZFS interaction would certainly justify a significantly lower DNP enhancement with Gd(OAc)3. It is also possible that the methyl group in the acetate ion can act as a relaxation sink, limiting the nuclear polarisation that may be obtained, a fact that is well-known in the context of PA development.13,66–69 In addition, the presence of quadrupolar nuclei in the counter-ions (14N (nuclear spin I = 1), 35/37Cl (I = 3/2) and 79/81Br (I = 3/2)) could act as a relaxation sink (an effect which could tentatively be active for GdBr3, where a lower 13C enhancement was observed), but could also facilitate the spin-diffusion of enhanced polarisation. Recent studies have shown that the addition of simple salts such as sodium chloride or lithium chloride in large concentration can significantly increase DNP enhancements in aqueous solvents by promoting glass formation.70,71 However, it was suggested that it is the cation, rather than the anion, that causes these effects. Furthermore, such effects are unlikely to be significant for samples in DNP juice, where glass formation is already efficient.
Conclusions
This work has expanded upon our previous proof-of-concept demonstration of the use of simple gadolinium compounds as DNP polarising agents, establishing that 20 mM is the optimum concentration of Gd(NO3)3 for enhancing 1H and 13C, and 40 mM for enhancing 15N. We have shown using mass spectrometry that while the ligand sphere structures for different gadolinium compounds vary slightly in water/glycerol solution, the addition of glycine to the samples results in the predominant Gd(III) species becoming identical in all cases. This is supported by the EPR data, which shows that the electron spin relaxation times and zero-field splitting parameters are not affected by the choice of counter-ion, except for Gd(OAc)3 where a stronger ZFS interaction results in a broader EPR lineshape. In most cases, this in turn translates into similar DNP enhancements for Gd(NO3)3, GdCl3, and GdBr3, providing a broader choice when selecting a polarising agent, while in general, Gd2(SO4)3 and Gd(OAc)3 gave lower sensitivity improvements. The lower solubility of the former could be the cause of this effect, potentially limiting its suitability as a DNP polarising agent. The low enhancement for Gd(OAc)3 may be due to a less symmetrical local Gd(III) environment, or the methyl group in the acetate ion acting as a relaxation sink. Our enhancements are smaller than those obtained using previously-reported complexes, with values that are in line with those predicted based on their ZFS parameters.24 Nevertheless, it is promising that appreciable sensitivity enhancements can be achieved using a variety of gadolinium compounds, which is expected to be of significant benefit as a cost-saving factor in instances where modest enhancements, rather than the hundreds-fold enhancements provided by biradicals, are sufficient.Exploring other paramagnetic ions is one way in which the field of “off-the-shelf” polarising agents could progress. Mn2+ appears to be an obvious choice, having been introduced alongside [Gd(dota)(H2O)]−,21 and later used to enhance 6Li and 7Li in battery anode materials.31 However, a significant disadvantage of Mn2+ is the hyperfine coupling to the spin-5/2 55Mn, splitting the DNP enhancement profile into six lines each for the positive and negative lobes. This results in a six-fold reduction in the maximum enhancement that can be achieved (although this is partially offset by the larger proportion of electron spins that contribute to the central EPR transition in S = 5/2 versus S = 7/2 systems, and hence to the solid effect).21 Given the already modest enhancements with simple Gd3+ compounds, it is likely that enhancements provided by simple Mn2+ salts would be too small to be useful. This problem is not applicable to metals without abundant NMR-active isotopes, such as Fe3+ (S = 5/2)32,33 or Cr3+ (S = 3/2),72 which have also been used in DNP, although the latter requires high local symmetry in order to avoid a large g-anisotropy. Other factors such as the EPR relaxation behaviour and redox stability would also need to be taken into account, but these metal ions may be a worthy avenue for further investigation.
Finally, it should be noted that while these proof-of-concept studies demonstrate that simple Gd(III) compounds can be used as PAs without complex ligands, it is yet to be established how this approach could be applied to more complex substrates. Particularly the observation that the Gd3+ ion is observed to bind directly to glycine may mean that it may be unsuitable for biomolecules, to which Gd3+ and other metal ions are known to bind (as has already been exploited in DNP),27 potentially resulting in conformational changes. This approach is therefore more suited to the detection of small molecules in solution However, it is worth considering that in this work, the glycine concentration (1.5 M) was significantly higher than the Gd(III) concentration (at most 100 mM). Therefore, the majority of glycine molecules are not bound to the metal centre. This may not remain to be the case if DNP using simple Gd(III) compounds is applied to the detection of low-concentration metabolites.
Author contributions
Daniel J. Cheney: investigation, formal analysis, writing – original draft; Paolo Cerreia Vioglio: technical support (DNP); Adam Brookfield: technical support (EPR); and Frédéric Blanc: conceptualization, funding acquisition, supervision, writing – review & editing.Data availability
NMR data are available from the University of Liverpool Data Catalogue portal at https://datacat.liverpool.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leverhulme Trust for funding a Research Project Grant under RPG-2020-066. MAS DNP experiments were performed at the Nottingham DNP MAS NMR Facility, which is funded by the University of Nottingham and EPSRC [grant numbers EP/L022524/1 and EP/R042853/1]. EPR spectra were collected at the EPSRC National Service for EPR Spectroscopy at the University of Manchester [grant number NS/A000055/1], and we thank Dr Alice Bowen and Dr Muralidharan Shanmugan for additional support during these experiments. We thank Richard Roberts and Stephen Moss (University of Liverpool) for collecting the HRMS data, as well as Glynn Connolly (University of Liverpool) for confirming the level of hydration in the Gd(III) compounds via thermogravimetric analysis. Finally, we would like to acknowledge fruitful discussions with Dr Benjamin Duff (University of Liverpool), Dr Stuart Elliott (Imperial College London) and Dr Daniel Lee (University of Manchester).References
- A. S. Lilly Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102–103, 120–195 CrossRef PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef.
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K.-N. Hu, C.-G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. A. van der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Chem. Phys., 2008, 128, 052211 CrossRef PubMed.
- A. G. M. Rankin, J. Trébosc, F. Pourpoint, J. P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2019, 101, 116–143 CrossRef PubMed.
- U. Akbey and H. Oschkinat, J. Magn. Reson., 2016, 269, 213–224 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS.
- N. J. Brownbill, D. Lee, G. De Paëpe and F. Blanc, J. Phys. Chem. Lett., 2019, 10, 3501–3508 CrossRef CAS.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2010, 15459–15461 CrossRef CAS.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- L. Gkoura and A. Equbal, J. Magn. Reson. Open, 2023, 16–17, 100125 CrossRef.
- C. Song, K.-N. Hu, C.-G. Joo, T. M. Swager and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 11385–11390 CrossRef CAS PubMed.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- F. Mentink-Vigier, I. Marin-Montesinos, A. P. Jagtap, T. Halbritter, J. van Tol, S. Hediger, D. Lee, S. Th. Sigurdsson and G. De Paëpe, J. Am. Chem. Soc., 2018, 140, 11013–11019 CrossRef CAS.
- D. J. Kubicki, G. Casano, M. Schwarzwälder, S. Abel, C. Sauvée, K. Ganesan, M. Yulikov, A. J. Rossini, G. Jeschke, C. Copéret, A. Lesage, P. Tordo, O. Ouari and L. Emsley, Chem. Sci., 2016, 7, 550–558 RSC.
- G. Stevanato, G. Casano, D. J. Kubicki, Y. Rao, L. E. Hofer, G. Menzildjian, H. Karoui, D. Siri, M. Cordova, M. Yulikov, G. Jeschke, M. Lelli, A. Lesage, O. Ouari and L. Emsley, J. Am. Chem. Soc., 2020, 142, 16587–16599 CrossRef CAS.
- A. Venkatesh, G. Casano, Y. Rao, F. De Biasi, F. A. Perras, D. J. Kubicki, D. Siri, S. Abel, H. Karoui, M. Yulikov, O. Ouari and L. Emsley, Angew. Chem., Int. Ed., 2023, 62, e202304844 CrossRef PubMed.
- A. Venkatesh, G. Casano, R. Wei, Y. Rao, H. Lingua, H. Karoui, M. Yulikov, O. Ouari and L. Emsley, Angew. Chem., Int. Ed., 2024, 63, e202317337 CrossRef CAS PubMed.
- D. Jardón-Álvarez and M. Leskes, Prog. Nucl. Magn. Reson. Spectrosc., 2023, 138–139, 70–104 CrossRef PubMed.
- A. Martorana, G. Bellapadrona, A. Feintuch, E. Di Gregorio, S. Aime and D. Goldfarb, J. Am. Chem. Soc., 2014, 136, 13458–13465 CrossRef CAS.
- B. Corzilius, A. A. Smith, A. B. Barnes, C. Luchinat, I. Bertini and R. G. Griffin, J. Am. Chem. Soc., 2011, 133, 5648–5651 CrossRef CAS.
- M. Kaushik, T. Bahrenberg, T. V. Can, M. A. Caporini, R. Silvers, J. Heiliger, A. A. Smith, H. Schwalbe, R. G. Griffin and B. Corzilius, Phys. Chem. Chem. Phys., 2016, 18, 27205–27218 RSC.
- G. Stevanato, D. J. Kubicki, G. Menzildjian, A.-S. Chauvin, K. Keller, M. Yulikov, G. Jeschke, M. Mazzanti and L. Emsley, J. Am. Chem. Soc., 2019, 141, 8746–8751 CrossRef CAS PubMed.
- Y. Rao, C. T. Palumbo, A. Venkatesh, M. Keener, G. Stevanato, A.-S. Chauvin, G. Menzildjian, S. Kuzin, M. Yulikov, G. Jeschke, A. Lesage, M. Mazzanti and L. Emsley, J. Phys. Chem. C, 2022, 126, 11310–11317 CrossRef.
- M. Kaushik, M. Qi, A. Godt and B. Corzilius, Angew. Chem., Int. Ed., 2017, 56, 4295–4299 CrossRef PubMed.
- P. Wenk, M. Kaushik, D. Richter, M. Vogel, B. Suess and B. Corzilius, J. Biomol. NMR, 2015, 63, 97–109 CrossRef PubMed.
- J. Heiliger, T. Matzel, E. C. Çetiner, H. Schwalbe, G. Kuenze and B. Corzilius, Phys. Chem. Chem. Phys., 2020, 22, 25455–25466 RSC.
- A. L. Paterson, F. A. Perras, M. F. Besser and M. Pruski, J. Phys. Chem. C, 2020, 124, 23126–23133 CrossRef.
- B. Thomas, D. Jardón-Álvarez, R. Carmieli, J. van Tol and M. Leskes, J. Phys. Chem. C, 2023, 127, 4759–4772 CrossRef PubMed.
- T. Chakrabarty, N. Goldin, A. Feintuch, L. Houben and M. Leskes, ChemPhysChem, 2018, 19, 2139–2142 CrossRef PubMed.
- T. Wolf, S. Kumar, H. Singh, T. Chakrabarty, F. Aussenac, A. I. Frenkel, D. T. Major and M. Leskes, J. Am. Chem. Soc., 2019, 141, 451–462 CrossRef PubMed.
- A. Harchol, G. Reuveni, V. Ri, B. Thomas, R. Carmieli, R. H. Herber, C. Kim and M. Leskes, J. Phys. Chem. C, 2020, 124, 7082–7090 CrossRef.
- D. Jardón-Álvarez, G. Reuveni, A. Harchol and M. Leskes, J. Phys. Chem. Lett., 2020, 11, 5439–5445 CrossRef.
- W. R. Couet, R. C. Brasch, G. Sosnovsky and T. N. Tozer, Magn. Reson. Imaging, 1985, 3, 83–88 CrossRef.
- S. J. Elliott, B. B. Duff, A. R. Taylor-Hughes, D. J. Cheney, J. P. Corley, S. Paul, A. Brookfield, S. Pawsey, D. Gajan, H. C. Aspinall, A. Lesage and F. Blanc, J. Phys. Chem. B, 2022, 126, 6281–6289 CrossRef PubMed.
- M. Rosay, L. Tometich, S. Pawsey, R. Bader, R. Schauwecker, M. Blank, P. M. Borchard, S. R. Cauffman, K. L. Felch, R. T. Weber, R. J. Temkin, R. G. Griffin and W. E. Maas, Phys. Chem. Chem. Phys., 2010, 12, 5850–5860 RSC.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef.
- A. Purea, C. Reiter, A. I. Dimitriadis, E. de Rijk, F. Aussenac, I. Sergeyev, M. Rosay and F. Engelke, J. Magn. Reson., 2019, 302, 43–49 CrossRef.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Magn. Reson. Chem., 2008, 46, 582–598 CrossRef PubMed.
- D. A. Hirsh, A. V. Wijesekara, S. L. Carnahan, I. Hung, J. W. Lubach, K. Nagapudi and A. J. Rossini, Mol. Pharmaceutics, 2019, 16, 3121–3132 CrossRef.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef.
- P. Bertani, J. Raya and B. Bechinger, Solid State Nucl. Magn. Reson., 2014, 61–62, 15–18 CrossRef.
- A. Raitsimring, A. Dalaloyan, A. Collauto, A. Feintuch, T. Meade and D. Goldfarb, J. Magn. Reson., 2014, 248, 71–80 CrossRef.
- L. Garbuio, K. Zimmermann, D. Häussinger and M. Yulikov, J. Magn. Reson., 2015, 259, 163–173 CrossRef PubMed.
- D. Ossadnik, S. Kuzin, M. Qi, M. Yulikov and A. Godt, Inorg. Chem., 2023, 62, 408–432 CrossRef PubMed.
- J. A. Clayton, K. Keller, M. Qi, J. Wegner, V. Koch, H. Hintz, A. Godt, S. Han, G. Jeschke, M. S. Sherwin and M. Yulikov, Phys. Chem. Chem. Phys., 2018, 20, 10470–10492 RSC.
- A. M. Raitsimring, A. V. Astashkin, O. G. Poluektov and P. Caravan, Appl. Magn. Reson., 2005, 28, 281–295 CrossRef.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef PubMed.
- H. Takahashi, D. Lee, L. Dubois, M. Bardet, S. Hediger and G. De Paepe, Angew. Chem., Int. Ed., 2012, 51, 11766–11769 CrossRef PubMed.
- H. Takahashi, C. Fernández-de-Alba, D. Lee, V. Maurel, S. Gambarelli, M. Bardet, S. Hediger, A.-L. Barra and G. De Paëpe, J. Magn. Reson., 2014, 239, 91–99 CrossRef PubMed.
- F. Mentink-Vigier, S. Paul, D. Lee, A. Feintuch, S. Hediger, S. Vega and G. De Paëpe, Phys. Chem. Chem. Phys., 2015, 17, 21824–21836 RSC.
- D. Lee, S. Hediger and G. De Paëpe, Solid State Nucl. Magn. Reson., 2015, 66–67, 6–20 CrossRef PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, D. Gajan, F. Rascón, M. Rosay, W. E. Maas, C. Copéret, A. Lesage and L. Emsley, Chem. Sci., 2012, 3, 108–115 RSC.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Oxford University Press, Oxford, 1970 Search PubMed.
- B. Corzilius, Phys. Chem. Chem. Phys., 2016, 18, 27190–27204 RSC.
- J. H. Ardenkjær-Larsen, I. Laursen, I. Leunbach, G. Ehnholm, L.-G. Wistrand, J. S. Petersson and K. Golman, J. Magn. Reson., 1998, 133, 1–12 CrossRef.
- O. Haze, B. Corzilius, A. A. Smith, R. G. Griffin and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 14287–14290 CrossRef.
- M. A. Hope, Y. Zhang, A. Venkatesh and L. Emsley, J. Magn. Reson., 2023, 353, 107509 CrossRef CAS PubMed.
- S. W. Hunsucker, R. C. Watson and B. M. Tissue, Rapid Commun. Mass Spectrom., 2001, 15, 1334–1340 CrossRef CAS PubMed.
- N. Geue, Anal. Chem., 2024, 96, 7332–7341 CrossRef CAS.
- J. S. McIndoe and K. L. Vikse, J. Mass Spectrom., 2019, 54, 466–479 CrossRef CAS.
- N. G. Naumov, M. S. Tarasenko, A. V. Virovets, Y. Kim, S.-J. Kim and V. E. Fedorov, Eur. J. Inorg. Chem., 2006, 298–303 CrossRef CAS.
- M. Ahrén, L. Selegård, F. Söderlind, M. Linares, J. Kauczor, P. Norman, P.-O. Käll and K. Uvdal, J. Nanopart. Res., 2012, 14, 1006 CrossRef.
- C. Gateau, M. Mazzanti, J. Pécaut, F. A. Dunand and L. Helm, Dalton Trans., 2003, 2428–2433 RSC.
- M. Botta, Eur. J. Inorg. Chem., 2000, 399–407 CrossRef CAS.
- H. Sato, S. E. Bottle, J. P. Blinco, A. S. Micallef, G. R. Eaton and S. S. Eaton, J. Magn. Reson., 2008, 191, 66–77 CrossRef CAS.
- V. Kathirvelu, C. Smith, C. Parks, M. A. Mannan, Y. Miura, K. Takeshita, S. S. Eaton and G. R. Eaton, Chem. Commun., 2009, 454–456 RSC.
- A. Zagdoun, G. Casano, O. Ouari, G. Lapadula, A. J. Rossini, M. Lelli, M. Baffert, D. Gajan, L. Veyre, W. E. Maas, M. Rosay, R. T. Weber, C. Thieuleux, C. Coperet, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2012, 134, 2284–2291 CrossRef CAS.
- H. Sato, V. Kathirvelu, A. Fielding, J. P. Blinco, A. S. Micallef, S. E. Bottle, S. S. Eaton and G. R. Eaton, Mol. Phys., 2010, 105, 2137–2151 CrossRef.
- M. Juramy, P. C. Vioglio, F. Ziarelli, S. Viel, P. Thureau and G. Mollica, Solid State Nucl. Magn. Reson., 2022, 122, 101836 CrossRef CAS.
- F. Ferrer, M. Juramy, R. Jabbour, S. Cousin, F. Ziarelli, G. Mollica, P. Thureau and S. Viel, J. Phys. Chem. Lett., 2023, 14, 9619–9623 CrossRef CAS PubMed.
- B. Corzilius, V. K. Michaelis, S. A. Penzel, E. Ravera, A. A. Smith, C. Luchinat and R. G. Griffin, J. Am. Chem. Soc., 2014, 136, 11716–11727 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparisons between double and triple-resonance probes, data tables, list of HRMS peaks, EPR spectral fits. See DOI: https://doi.org/10.1039/d4cp02924k |
| This journal is © the Owner Societies 2024 |