
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCold reactions of He+ with OCS and CO2: competitive kinetics and the effects of the molecular multipole moments
Fernanda B. V.
Martins†
 ,
Valentina
Zhelyazkova†
and
Frédéric
Merkt
*
,
Valentina
Zhelyazkova†
and
Frédéric
Merkt
*
ETH Zürich, Institute of Molecular Physical Science, CH-8093 Zürich, Switzerland. E-mail: frederic.merkt@phys.chem.ethz.ch
First published on 19th September 2024
Abstract
The reactions of He+ with OCS and CO2 have been studied at collision energies between ∼kB ⋅ 200 mK and ∼kB ⋅ 30 K by merging a beam of Rydberg He atoms with rotationally cold (∼3.5 K) seeded supersonic expansions containing either OCS or 13CO2 or a mixture of OCS (mole fraction 23.2%) and 13CO2 (76.8%). The observed product ions of the He+ + 13CO2 and He+ + OCS reactions are 13CO+, and CS+ and CO+, respectively. The He+ + OCS capture rate coefficient increases by ∼75% with decreasing collision energy over the investigated range, whereas that of He+ + 13CO2 decreases by ∼40%. The analysis of the experimental results using an adiabatic-channel capture model indicates that these opposite collision-energy dependences of the rate coefficients arise from the interaction between the charge of the ion and the electric multipole moments of OCS and 13CO2. From the relative product-ion yields observed when using the mixture of OCS and 13CO2, the He+ + OCS collisions are inferred to be ∼20% more reactive than those between He+ and 13CO2. The comparison of the calculated thermal rate coefficients with earlier experiments suggests that about half of the He+ + 13CO2 collisions are reactive.
1 Introduction
For over fifty years, cold ion–molecule reactions have been an object of interest in chemical physics and astrophysics.1–15 Their rate coefficients have been predicted to display a characteristic and sometimes strong collision-energy (Ecoll) dependence at near-zero Ecoll resulting from the interaction between the charge of the ion and the electric dipole and quadrupole moments of the neutral molecule.5–13 The effect of these charge–multipole-moment interactions makes the commonly used temperature- and Ecoll-independent Langevin rate coefficients kL16 unsuitable to describe many chemical processes occurring in the interstellar medium.6 Measuring and modeling the reaction rate coefficients of ion–molecule reactions at low temperatures and low collision energies offers insight into the fundamental aspects of cold chemistry.17–21Ion–molecule reactions have been studied from room temperature down to ∼10 K using ion guides and traps,22–24 supersonic flows,25,26 and, more recently, laser-cooled and sympathetically cooled ions in ion traps and Coulomb crystals.17,19,20,27–29 Even lower temperatures have been reached in ion–atom reactions in hybrid traps.30–32 Probing the sub-10 K region is experimentally challenging because stray electric fields and space-charge effects easily accelerate and heat up the ions. In the last decade, experimental investigations of cold ion–molecule reactions in a range of collision energies tunable from ∼kB ⋅ 60 K down to ∼kB ⋅ 200 mK have become possible through the development of a merged-beam technique in which the ion is substituted by the ion core of Rydberg atoms or molecules.33–36 The highly excited Rydberg electron ensures charge neutrality and shields the reactions taking place within its orbit from stray electric fields without otherwise influencing them.34,37–40
In recent studies of cold ion–molecule reactions, it was experimentally demonstrated that the partial-charge distribution in the neutral molecule dictates how the reaction rate coefficient varies with the collision energy near zero, confirming theoretical predictions made almost forty years ago.5–9 For polar molecules,36,41–43 the rotational-state-averaged rate coefficients k(Ecoll) increase as Ecoll approaches zero. For quadrupolar molecules, k(Ecoll) typically increases as Ecoll approaches zero for molecules with a positive zz component of the quadrupole-moment tensor (Qzz)35,44,45 and decreases if the molecular reactant has a negative Qzz.45,46 For molecules with neither a dipole moment nor a quadrupole moment, such as CH4, k(Ecoll) stays constant within the experimentally attainable Ecoll range and sensitivity.47
The work reported in this article was carried out as part of a series of systematic investigations of ion–molecule reactions at low collision energies and temperatures.34–36,40–43,45–48 It presents a comparative investigation of the He+ + OCS and the He+ + CO2 reactions in the Ecoll range from ∼kB ⋅ 30 K down to ∼kB ⋅ 200 mK. OCS and CO2 are both linear triatomic molecules with electronic ground state of 1Σ+ symmetry. Their relevant physical properties are presented in Table 1. OCS, unlike CO2, is not centrosymmetric and thus has a permanent electric dipole moment. CO2 is nonpolar but has a large (negative) quadrupole moment. We present experimental and theoretical studies of the effects of such different partial-charge distributions on the reaction rate coefficients of each reaction individually and also directly compare their rate coefficients by simultaneously measuring both reactions using an (OCS:13CO2) gas mixture of well-defined composition.
| Molecule | OCS | CO2 |
|---|---|---|
| a Value for 16O12C32S. b Value for 13C16O2. | ||
| Electronic ground state | 1Σ+ | 1Σ+g |
| Mass m (u) | 60.07a | 45.00b |
| Dipole moment |μel| (D) | 0.71549 | 0 |
| Quadrupole moment Qzz (D Å) | −0.29550 | −4.27851 |
| Polarizability volume α′ (Å3) | 5.09052 | 2.50753 |
| Polarizability anisotropy Δα′ (Å3) | 4.1654 | 2.1454 |
| Rotational constant B0 (cm−1) | 0.2028655![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
0.390237456b |
| Langevin rate kL (10−15 m3 s−1) | 2.7276a | 1.9360b |
Several investigations of the thermal reactions between He+ and carbonyl sulfide (OCS) and carbon dioxide (CO2) have been carried out since the 1960s. The He+ + OCS reaction was studied by monitoring the emission spectrum of OCS in helium flowing-afterglow experiments, and CO+57–59 and CS+57,58 were identified as the ionic products, but no branching ratios were reported. The He+ + CO2 reaction has been studied extensively at 300 K and above in drift-tube,60 ion-cyclotron resonance,61,62 selected-ion-flow-tube,63,64 flow-drift-tube,65 flowing-afterglow,66,67 and crossed-beam velocity-map-imaging68 experiments. The following reaction channels and branching ratios have been reported:
 | (1) |
Thermal rate constants k(T ≈ 300 K) between 0.85 (±20%) × 10−15 m3 s−1 (ref. 66) and 1.2 (±50%) × 10−15 m3 s−1 (ref. 67) have been reported. Rowe et al.69 conducted the lowest temperature experiment to date involving the He+ + CO2 reaction, and obtained k(70 K) = 1.6 (±30%) × 10−15 m3 s−1, which is only slightly lower than the Langevin rate constant kLCO2 of the He+ + CO2 reaction (see Table 1).
This article is structured as follows: in Section 2 we describe the experimental setup and procedure. Section 3 presents the experimental results and Section 4 the theoretical analysis. The competitive kinetics study of the reaction between He+ and a mixture of OCS and 13CO2 is discussed in Section 5. The conclusions are presented in Section 6.
2 Experiment
We study ion–molecule reactions near zero collision energy using the merged-beam apparatus41 depicted in Fig. 1. A supersonic expansion of He atoms is generated using a cryogenic home-built valve delivering short gas pulses with a duration of ∼20 μs at a repetition rate of 25 Hz. An electric discharge populates the He* (1s)1(2s)1 3S1 metastable state near the valve orifice. The beam of He* atoms propagates with a velocity of ∼1000 m s−1 and is crossed at right angles by laser light (wavelength of ∼260 nm, pulse energy of ~400 μJ) in the presence of an electric field just before a curved Rydberg–Stark deflector. After photoexcitation of the He* atoms to a selected Rydberg–Stark state of principal quantum number n in the range between 25 and 50 (n = 35 was used to obtain the results presented in this article), the He(n) Rydberg atoms are trapped, deflected and accelerated to higher or lower velocities by applying the appropriate time-dependent potentials to the electrodes of the surface deflector.33,36,70 This deflector is used to merge the He(n) beam with a beam of ground-state (GS) neutral molecules (13CO2 or OCS) and set the forward velocity vHe(n) of the He(n) beam. The collision between the ground-state molecules and the He(n) atoms takes place in a reaction region embedded in a time-of-flight (TOF) mass spectrometer. | ||
| Fig. 1 Schematic representation of the merged-beam setup used to study the reactions between He+ and small molecules (here OCS and 13CO2) at low collision energies, consisting of the surface-electrode deflector and decelerator, the time-of-flight mass spectrometer, and a free-flight region used to measure the velocity distributions of the supersonic beams containing OCS and 13CO2 with two fast-ionization gauges (FG). | ||
The collision energy Ecoll = μHe(n)–GS(vHe(n) − vGS)2/2 is tuned by changing vHe(n) while keeping the velocity vGS of the ground-state beam constant (μHe(n)–GS is the reduced mass of the reactants). This beam is generated by a second home-built valve, which also delivers short (∼20 μs) gas pulses. The ∼0.6-m-long propagation distance of the short gas pulses ensures a large spatial dispersion, so that the small cloud of He(n) atoms overlaps with neutral reactants in a narrow velocity class. As a result, we achieve a collision-energy resolution of ∼kB ⋅ 200 mK near Ecoll = 0, which arises mainly from the velocity-distribution width of the deflected He(n)-atom beam.33,36
To study the He+ + OCS and He+ + CO2 reactions, we use seeded supersonic beams containing either OCS or 13CO2 or a (23.2:76.8) OCS:13CO2 mixture (mole ratio) prepared by weighing the gas samples. The simultaneous measurement of the two reactions in a single set of measurements using a mixture containing both OCS and 13CO2 serves three purposes: first, it enables us to verify that the measurements are not significantly affected by possible differences in the expansions of the different gas mixtures; second, the ∼3–5% seeding ratio ensures the same velocity for OCS and 13CO2; and third, it offers the prospect of directly comparing the rate coefficients of the two reaction systems. 13CO2 was chosen instead of 12CO2 to distinguish between the CO+ products from the two reaction systems, which made a competitive-kinetics investigation possible. Seeding the neutral molecules (∼3–5%) in a mixture71 of He and Ne allows one to efficiently cool the rotational degrees of freedom and avoid the formation of OCS clusters while imposing a similar velocity to the neutral reactants in all three different experiments. With this approach, we could also select a vGS value within the range of velocities vHe(n) that can be generated with the surface-deflector and thus vary vrel = vHe(n) − vGS through zero.
An electric-field pulse is used to extract the product ions of the reactions toward a microchannel-plate (MCP) detector along a time-of-flight tube and to detect them mass selectively as Ecoll is varied. Examples of TOF mass spectra are displayed in Fig. 2. The spectra plotted in black were recorded near Ecoll = 0 for the reactions between He(n = 35) atoms travelling at vHe(n) = 1050 m s−1 and OCS (a), 13CO2 (b), and a mixture of OCS and 13CO2 (c). The corresponding background spectra in red were recorded with the Rydberg-excitation laser turned off, i.e., in the absence of He(n) atoms. The ions which appear in these spectra correspond to products of the Penning-ionization reactions between He* and the background molecules in the chamber, i.e., H2O (O+, OH+ and H2O+), and either OCS (S+ and OCS+) or 13CO2 (13CO2+). The peaks which are only present in the spectra plotted in black, namely CO+ and CS+ in panels (a) and (c) and 13CO+ in panels (b) and (c), are from the products of the studied ion–molecule reactions. Two different ion products (CO+ and CS+) are observed in the He+ + OCS reaction system [panel (a)], corresponding to the two reaction channels He+ + OCS → He + S + CO+ and He+ + OCS → He + O + CS+. A single reaction channel, He+ + 13CO2 → He + O + 13CO+, is detected for the He+ + 13CO2 reaction system [see panel (b)]. The product ions resulting from the other reactive channels of He+ + 13CO2 shown in eqn (1) and from He+ + OC34S are not observed. However, the sensitivity of our experiment does not allow for the detection of ions that are formed in reaction channels with branching ratios below ∼10%.
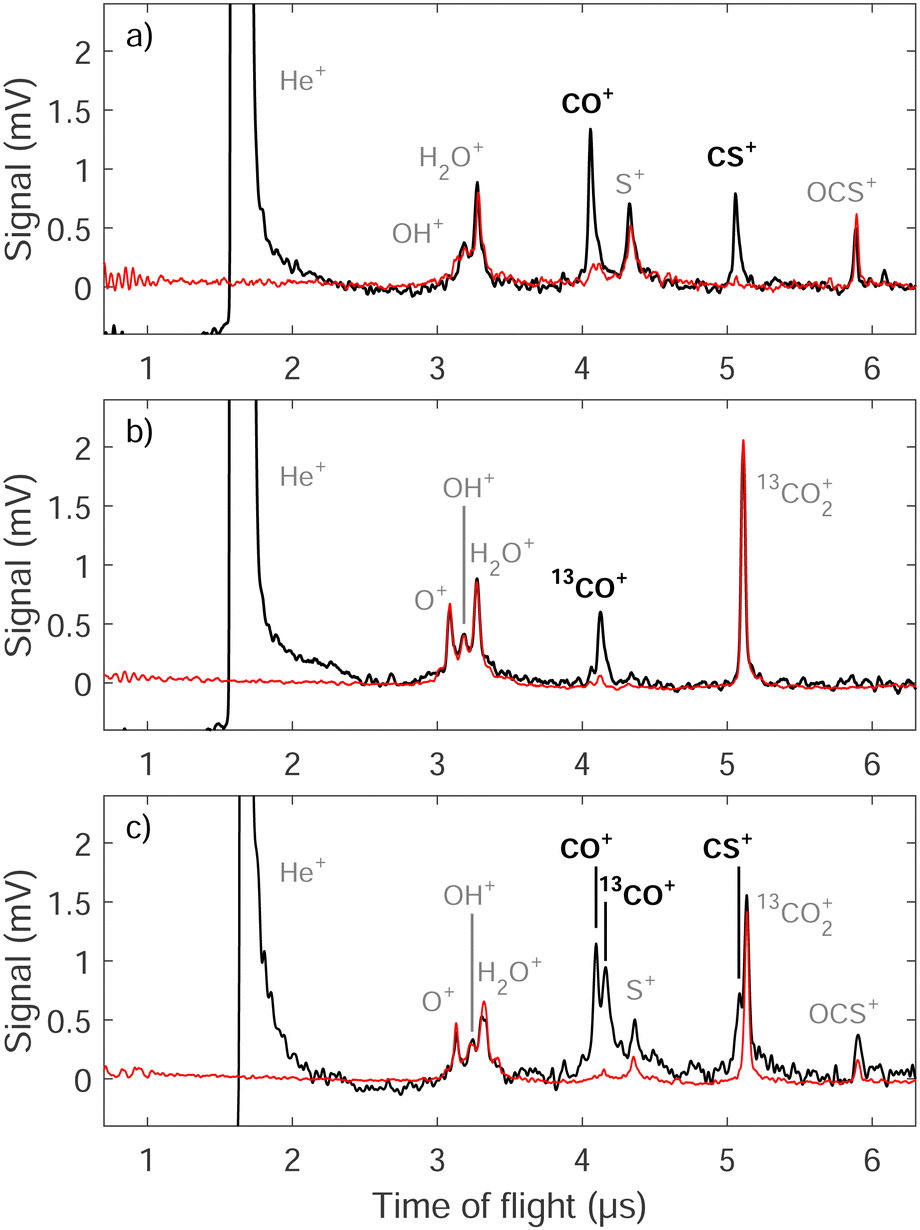 | ||
| Fig. 2 Time-of-flight mass spectra of the ion signals recorded after the reaction between He Rydberg atoms (n = 35) and (a) OCS, (b) 13CO2, and (c) a mixture of OCS and 13CO2. The spectra in red correspond to background measurements obtained with the Rydberg-excitation laser turned off. The reaction products are labeled in black bold font. See text for details. | ||
The velocity distribution of the ground-state beam was measured using two fast-ionization gauges (FG I and II in Fig. 1) as explained in detail in ref. 43. The effects of changing the ratio of He and Ne in the ground-state-beam expansion are illustrated in panel (a) of Fig. 3, which displays the velocity distributions obtained from measurements of the flight times using a fast-ionization gauge (FG I in Fig. 1). The distribution displayed in red corresponds to a beam containing ∼3% OCS seeded in a mixture of He:Ne (2:3), which resulted in a mean velocity of ∼960 m s−1. Seeding ∼3% OCS in a (1:1) mixture of He and Ne yielded a faster beam with a mean velocity of ∼1035 m s−1 (black trace). The vertical green lines correspond to the mean velocity of the OCS-molecule cloud overlapping with the He(n) atoms in the reaction region. These velocities were selected by applying appropriate time delays between the OCS and the He* gas-pulse triggers. The green shaded regions represent the estimated uncertainty of ±15 m s−1 in the velocity measurements using the fast-ionization gauges. To avoid turbulent parts of the beams and the formation of clusters, we typically targeted velocity classes which either corresponded to the mean velocity of the ground-state beam (960 m s−1), as in the distribution displayed in red, or to a faster velocity, as in the distribution displayed in black, for which vOCS ≈ 1070 m s−1. The experiments are carried out under conditions where the densities of the GS molecules by far exceed those of He(n) and the reaction probability of He(n) is less than 1%. Consequently, the product-ion signals follow pseudo-zero-order kinetics and directly correspond to the relative rate coefficients.
 | ||
| Fig. 3 (a) Velocity distributions, measured with the fast-ionization gauges, of supersonic beams consisting of ∼3% OCS seeded in He:Ne mixtures of 1:1 (black) and 2:3 (red) pressure ratios. (b) and (c) Corresponding total integrated product-ion signal (CO+ and CS+) of the He+ + OCS reaction as a function of (b) the He(n)-beam velocity and (c) the collision energy. The vertical green lines in panels (a) and (b) indicate the targeted OCS velocities in each beam (∼960 m s−1 and ∼1070 m s−1, respectively), which correspond to Ecoll = 0 in panel (c). The areas shaded in light green in panels (a) and (b) correspond to the experimental uncertainty of ±15 m s−1 in the targeted OCS beam velocities. The vertical error bars in panels (b) and (c) represent ±1σ and the horizontal bars in panel (c) the collision-energy resolution. | ||
3 Results
Panel (b) of Fig. 3 depicts the relative total product-ion yields of the He+ + OCS reaction as a function of vHe(n). The yield corresponding to the OCS beam with vOCS ≈ 960 m s−1 is depicted in red and that of the beam with vOCS ≈ 1070 m s−1 in black [see panel (a) of Fig. 3]. These product yields, corresponding to the sum of the integrated CO+ and CS+ signals, were obtained by recording TOF mass spectra such as those displayed in Fig. 2 as a function of the velocity of the He(n) beam. The integrated signals were normalized by the density of He(n) atoms in the reaction zone, as determined in a separate measurement using pulsed-field ionization.40,41 The data were further corrected for the different densities of the decelerated and accelerated He(n) beams, as explained in ref. 41. The green lines in panels (a) and (b) illustrate that changing the target velocity of the OCS beam from 960 m s−1 to 1070 m s−1 results in a shift of the He(n) mean velocity at which the reaction yield reaches a maximal value, which in both cases occurs when vOCS = vHe(n), i.e., at zero relative velocity. The same data are displayed in panel (c) as a function of the collision energy. The increase in product-ion yield as the relative velocity between the reactants approaches zero, observed in both data sets, is a typical signature of ion–molecule reactions involving polar molecules, as previously observed for CH3F,36 NH3,41,43 and NO.42Fig. 4 presents a comparison of the dependence on vrel of the product yields obtained in the measurements of the reactions of He+ with OCS [panel (a)], 13CO2 (b), and the OCS:13CO2 mixture (c). The integrated product-ion signal obtained in individual measurements is shown as as pale-color circles as a function of vHe(n) in the range between 800 m s−1 and 1200 m s−1. The corresponding averages of the product-ion signal are shown with dark-color circles. The vertical error bars correspond to one standard deviation.
 | ||
| Fig. 4 Integrated product-ion signals of the reactions between He+ and OCS [(a) and (d)], 13CO2 [(b) and (e)], and a mixture of OCS and 13CO2 [(c) and (f)] as a function of the He(n)-beam velocity [(a)–(c)] and the collision energy [(d)–(f)] for fixed velocities of the ground-state molecular beams (vOCS ≈ 960 m s−1, v13CO2 ≈ 1000 m s−1, vmix ≈ 990 m s−1). The data points plotted in pale and dark colors represent individual measurements and their weighted averages, respectively. The vertical error bars represent ±1σ and the horizontal bars in panels (d)–(f) the Ecoll resolution. The pale red and blue lines in panels (d)–(f) correspond to the calculated state-averaged capture rate coefficients. The maxima (minima) in product formation for the He+ + OCS (He+ + 13CO2) reaction occur when vHe(n) = vGS (i.e., for Ecoll = 0), as indicated by the green vertical lines in panels (a)–(c). | ||
Panels (d)–(f) present the same data as a function of Ecoll. In these panels, the vertical error bars correspond to one standard deviation and the horizontal bars to the experimental collision-energy resolution ΔEcoll [see eqn (1) in ref. 36]. The data were binned according to Ecoll, with bin sizes proportional to ΔEcoll. The colored lines represent the rotational-state-averaged reaction rate coefficient calculated using the capture-rate-coefficient model presented in Section 4. In our experiment, relative (as opposed to absolute) rate coefficients are measured, and therefore, for the comparison, the experimental data are scaled by a global factor to yield the best agreement with the calculated reaction rate coefficients.
Panel (a) of Fig. 4 corresponds to the results obtained for the He+ + OCS reaction for an OCS beam with vOCS = 960 m s−1 and already displayed in panels (b) and (c) of Fig. 3 but showing, in addition, the yields of the CO+ and CS+ products as pink and orange circles, respectively. Both product yields reveal the same dependence on the relative velocity between the reactants within the experimental uncertainties, which indicates that the branching ratio of the two product channels is independent of the collision energy. The observed CO+ branching ratio is (70 ± 8.5)%. We observe an increase of about 75% in the total He+ + OCS product yield as vHe(n) is varied from 800 m s−1 (1200 m s−1) up (down) to 960 m s−1, which is the mean velocity of the reacting OCS molecules in this experiment. The red circles in panel (d) show that the sum of the CO+ and the CS+ product yields increases by 75% as the collision energy is reduced from ∼kB ⋅ 30 K down to zero.
The product yield of the He+ + 13CO2 reaction [panel (b)] exhibits the opposite behavior, i.e., a decrease in the 13CO+ product formation (blue circles) by 40% as vHe(n) is scanned toward v13CO2, i.e., toward Ecoll = 0 [see panel (e)]. The trends of increasing (decreasing) rate coefficients for the He+ + OCS (He+ + 13CO2) reactions as vrel approaches zero are also observed in the experiments carried out with the OCS:13CO2 mixture, as shown in panels (c) and (f). This observation demonstrates that these trends are not influenced by the different gas-expansion conditions in these measurements, as expected, and that the main systematic sources of error are accounted for by the procedure used to normalize the product signal using the measured relative densities of the He(n) and the 13CO2/OCS reactants.
4 Analysis of the capture rate coefficients
To analyze the experimental results presented in Section 3, we use a capture model originally developed by Clary and coworkers5,6,9 and Troe and coworkers,7,8,10–13 as detailed in ref. 41,45,47. The rate-coefficient calculations rely on the use of potentials that include the interaction between the charge of the ion and the electric multipole moments of the neutral molecules. Descriptions of this model along with the relevant equations can be found, e.g., in ref. 41,42. Here, we only summarize the main aspects.We determine adiabatic long-range electrostatic interaction potentials between the ion (here He+) and the neutral molecule (here OCS or 13CO2) for each individual molecular rotational level i that is significantly populated at the rotational temperature Trot of the supersonic expansion. These potentials are given by
| Vi(L)(R) = VL(R) + ΔEi(R), | (2) |
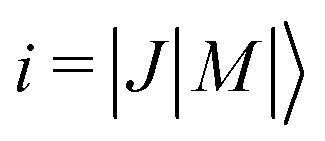 of the neutral molecule induced by the field of the ion. Without considering ΔEi, the Langevin potential VL(R) gives rise to a rate constant kL which is independent of the temperature and the collision energy, whereas the ΔEi terms introduce a collision-energy dependence of the reaction rate coefficients. The Stark shifts ΔEi of the rotational levels of OCS and 13CO2 result from the charge–dipole (∝μelR−2) and charge–quadrupole (∝QzzR−3) interactions and are calculated by diagonalizing the Hamiltonian matrix [see eqn (9)–(11) in ref. 41] in the appropriate rigid-rotor basis using the known values of the molecular dipole and quadrupole moments, as listed in Table 1. The ΔEi terms initially increase in magnitude at low fields as the distance between the ion and the molecule decreases and the molecule is exposed to the increasing (inhomogeneous) electric field of the ion.
of the neutral molecule induced by the field of the ion. Without considering ΔEi, the Langevin potential VL(R) gives rise to a rate constant kL which is independent of the temperature and the collision energy, whereas the ΔEi terms introduce a collision-energy dependence of the reaction rate coefficients. The Stark shifts ΔEi of the rotational levels of OCS and 13CO2 result from the charge–dipole (∝μelR−2) and charge–quadrupole (∝QzzR−3) interactions and are calculated by diagonalizing the Hamiltonian matrix [see eqn (9)–(11) in ref. 41] in the appropriate rigid-rotor basis using the known values of the molecular dipole and quadrupole moments, as listed in Table 1. The ΔEi terms initially increase in magnitude at low fields as the distance between the ion and the molecule decreases and the molecule is exposed to the increasing (inhomogeneous) electric field of the ion.
Panels (a) and (b) of Fig. 5 display the converged calculated Stark shifts of OCS and 13CO2, respectively, in the field of the approaching He+ ion. The results are depicted as a function of the electric field [e/(4πε0R2)] (bottom axis) and of the ion–molecule distance R (top axis) for all rotational levels with J ≤ 4, which are the ones significantly populated at Trot = 3.5 K. The rotational state density of OCS is higher than that of 13CO2 because B0OCS < B013CO2 and because, unlike in OCS [panel (a)], only even-J states are populated in 13CO2 [panel (b)] as a result of nuclear-spin statistics. Moreover, OCS has a permanent electric dipole moment. Consequently, its rotational levels are subject to larger Stark shifts than those of 13CO2. For example, the |00〉 state of OCS is shifted by −4 cm−1 at a field of ∼0.4 MV cm−1, whereas a field of ∼2.3 MV cm−1 is needed to produce the same shift in 13CO2.
 | ||
Fig. 5 (a) and (b) Stark shifts of the rotational levels  of OCS (a) and 13CO2 (b) with J ≤ 4 in the field of the He+ ion. (c) and (d) Rotational-state-specific rate coefficients calculated for the reactions of He+ with OCS (c) and 13CO2 (d) for J ≤ 2. State-averaged rate coefficients for the He+ + OCS (e) and the He+ + 13CO2 (f) reactions corresponding to the sum of the state-specific rate coefficients weighted by the probabilities of occupation of the rotational states for rotational temperatures Trot between 1.5 and 11.5 K and considering the collision-energy resolution. The bold green lines in panels (e) and (f) correspond to Trot = 3.5 K and the corresponding occupation probability of the rotational levels is given in the insets of panels (c) and (d). of OCS (a) and 13CO2 (b) with J ≤ 4 in the field of the He+ ion. (c) and (d) Rotational-state-specific rate coefficients calculated for the reactions of He+ with OCS (c) and 13CO2 (d) for J ≤ 2. State-averaged rate coefficients for the He+ + OCS (e) and the He+ + 13CO2 (f) reactions corresponding to the sum of the state-specific rate coefficients weighted by the probabilities of occupation of the rotational states for rotational temperatures Trot between 1.5 and 11.5 K and considering the collision-energy resolution. The bold green lines in panels (e) and (f) correspond to Trot = 3.5 K and the corresponding occupation probability of the rotational levels is given in the insets of panels (c) and (d). | ||
Negative (positive) Stark shifts lead to attractive (repulsive) potential terms which increase (decrease) the values of the rate coefficients. The state-specific reaction rate coefficients ki resulting from the modified He+ + OCS and He+ + 13CO2 interaction potentials are depicted in panels (c) and (d) of Fig. 5. They are displayed on an absolute scale (left axis) and normalized to their corresponding Langevin rates kL (right axis) as a function of the collision energy. The rate coefficients of OCS are typically several times larger than those of 13CO2, both on the absolute and on the kL-normalized scale. The rate coefficients of the |00〉 and |11〉 rotational states in OCS [panel (c)], for example, exhibit a strong Ecoll dependence and increase by factors of approximately 5 and 3, respectively, as the collision energy decreases from ~kB ⋅ 20 K down to zero. This behavior is the consequence of the strong high-field-seeking character of these rotational states, as seen in panel (a).
The Ecoll dependence of the state-specific rate coefficients of the He+ + 13CO2 reaction is less pronounced because the charge-quadrupole interaction is weaker than the charge-dipole interaction and the zero-field spacings of the rotational levels are larger than in OCS [see panel (b)]. For both molecules, states exhibiting a low-field-seeking behavior such as the |22〉 state of 13CO2 and the |20〉 state of OCS have vanishing rate coefficients at low collision energies [see panels (c) and (d) of Fig. 5].
The insets in panels (c) and (d) show the probabilities of occupation of the relevant 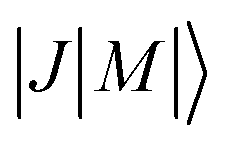 states of OCS and 13CO2 at the rotational temperature of the supersonic beam, which we estimate to be 3.5 K on the basis of the comparison with the calculated rotational-state-averaged rate coefficients and of similar experiments carried out with molecules such as N2, NO, and CO for which the rotational temperature could be determined spectroscopically. These insets illustrate that more J levels are populated and contribute to the averaged He+ + OCS rate coefficient compared to the He+ + 13CO2 reaction. To obtain the Ecoll-dependent state-averaged reaction rate coefficients of the He+ + OCS [He+ + 13CO2] reaction shown in panel (e) [(f)], we weighted the state-specific rate coefficients by the corresponding probabilities of occupation considering rotational temperatures between 1.5 K and 11.5 K. In both reactions, the calculations indicate that lowering the rotational temperature leads to an increase of the state-averaged rate coefficients, unlike in the He+ + NH3 (ND3)41 and D2+ + NH3 (ND3)43 reactions, and to a stronger Ecoll dependence of the reaction rate coefficient. This effect is explained by the fact that, for both OCS and 13CO2, the most strongly high-field-seeking states are the lower rotational levels, which have a higher probability of occupation at the lowest rotational temperatures.
states of OCS and 13CO2 at the rotational temperature of the supersonic beam, which we estimate to be 3.5 K on the basis of the comparison with the calculated rotational-state-averaged rate coefficients and of similar experiments carried out with molecules such as N2, NO, and CO for which the rotational temperature could be determined spectroscopically. These insets illustrate that more J levels are populated and contribute to the averaged He+ + OCS rate coefficient compared to the He+ + 13CO2 reaction. To obtain the Ecoll-dependent state-averaged reaction rate coefficients of the He+ + OCS [He+ + 13CO2] reaction shown in panel (e) [(f)], we weighted the state-specific rate coefficients by the corresponding probabilities of occupation considering rotational temperatures between 1.5 K and 11.5 K. In both reactions, the calculations indicate that lowering the rotational temperature leads to an increase of the state-averaged rate coefficients, unlike in the He+ + NH3 (ND3)41 and D2+ + NH3 (ND3)43 reactions, and to a stronger Ecoll dependence of the reaction rate coefficient. This effect is explained by the fact that, for both OCS and 13CO2, the most strongly high-field-seeking states are the lower rotational levels, which have a higher probability of occupation at the lowest rotational temperatures.
The state-averaged rate coefficients of both reactions at the estimated rotational temperature of the neutral molecules (Trot = 3.5 K) are displayed with a thick green line in panels (e) and (f) of Fig. 5, and are compared with experimental data in panels (d)–(f) of Fig. 4. The results of the experiments and calculations are in agreement for the three sets of experimental data depicted in Fig. 4 and for both the He+ + OCS and the He+ + 13CO2 reactions. We attribute the very different dependences on Ecoll and magnitudes of the rate coefficients of the He+ + OCS and He+ + 13CO2 reactions to the fact that OCS—unlike 13CO2—is a polar molecule with closely spaced rotational levels that are much more easily mixed by the inhomogeneous electric field of the approaching He+ ion.
At high electric fields, or, equivalently, at small ion–molecule distances, the quadratic Stark effect orients the dipole in OCS, which contributes to enhance the reaction rates, particularly at low collision energies. In contrast, the Stark shifts of 13CO2, which has a negative quadrupole moment, reduce the state-averaged rate coefficient as Ecoll approaches zero.4513CO2 thus displays the same behavior as N245 and CO,46 which also have negative quadrupole moments. Based on the calculations [see panel (d) of Fig. 5], one might be able to observe an increase of the rate coefficient in the He+ + 13CO2 reactions at collision energies Ecoll/kB ≪ 1 K, below the Ecoll/kB > 200 mK accessible in the present experiments.
Fig. 6 compares the He+ + 13CO2 thermal capture rate coefficients calculated as described in ref. 41 (blue line and dots) with earlier experimental thermal rate coefficients corresponding to the sum of the rates of the different product channels.61,63,65–67,69 Most of the experimental rate constants were measured at 300 K and only one at 70 K.69 All experimental values are significantly smaller than the calculated capture rates. Agreement of the calculated rates with the experimental ones can be reached by scaling the former by a factor of ∼0.45, corresponding to the black curve. These results would suggest that only about half of the capture processes are reactive in this system, which is surprising for a barrier-free exothermic reaction. Theoretical calculations including the short-range interactions would be needed to fully account for the observed rate coefficients and branching ratios.
 | ||
| Fig. 6 Comparison of the calculated thermal rate coefficients of the He+ + 13CO2 reaction (blue line and dots) and the experimental values of k(T) obtained in ref. 61,63,65–67,69 for He+ + CO2. The black line and dots correspond to the calculated k(T) scaled by a factor of 0.45. The inset shows k(T) for temperatures between ∼0 and 10 K. | ||
5 Competitive kinetics
As explained in Section 2, the use of an OCS:13CO2 mixture with precise composition offers the possibility of measuring the dependence of their relative rate coefficients on the collision energy. We can relate the experimentally determined and the calculated rate-coefficient ratios using the equation | (3) |
The calculations presented in Section 4 focus on the capture rate coefficients, and do not distinguish between capture processes leading to reaction products or (in)elastic collisions. Moreover, our experiment only monitors reactive channels and does not yield absolute rate coefficients because we do not measure the absolute concentrations of the reactants, only the relative ones. Using a mixture of molecular reactants with well-known concentrations, however, allows us to determine the ratio of the corresponding rate coefficients and its dependence on Ecoll and, viaeqn (3), to extract the ratio of reactivities η(i)/η(ii) assuming the validity of the capture model (see also ref. 72). This comparison relies on the assumptions that the detection efficiency is the same for all products, and that the density ratio of the reactants within the supersonic expansion is the same as in the prepared gas sample, which is equivalent to neglecting possible different expansion velocities and forward-velocity slips resulting from the different masses of the two species.
In Fig. 7, we show the ratio of the total product-ion yields of the He+ + OCS and the He+ + 13CO2 reactions measured using the seeded (23.2:76.8) OCS:13CO2 mixture (mole ratio) as a function of Ecoll. The purple dots correspond to the experimentally measured sum of the CO+ and the CS+ product signals from the He+ + OCS reaction divided by the 13CO+ product signal from the He+ + 13CO2 reaction and scaled by x13CO2/xOCS. These product-ion signals are individually shown in panel (f) of Fig. 4.
 | ||
| Fig. 7 Rate-constant ratios of the He+ + OCS and the He+ + 13CO2 reactions obtained experimentally using a OCS:13CO2 gas mixture (purple dots) and predicted using adiabatic capture theory (orange line). The purple line is obtained by assuming that the collisions of He+ with OCS are 1.2 times as reactive as those with 13CO2. The vertical error bars represent ±1σ and the horizontal bars the Ecoll resolution. | ||
The ratio of the calculated total capture rate coefficients of the two reactions (orange line) is smaller by about 20% than the experimentally measured ratio, but otherwise displays the same dependence on Ecoll. This observation suggests that the He+ + OCS and the He+ + CO2 collisions do not have the same probabilities of yielding products upon capture. Scaling the calculated rate-coefficient ratio by ηOCS/η13CO2 = 1.2 (purple line) leads to almost perfect agreement between calculations and experiment, which indicates that He+ + OCS capture processes are ∼20% more reactive than He+ + CO2 capture processes. This result is obtained by considering Trot = 3.5 K for both OCS and CO2, which leads to the best agreement for the data presented in panel (f) of Fig. 4. If we allow Trot to vary by ±1 K for each reactant, ηOCS/η13CO2 can vary from ∼1.05 to ∼1.3 while yielding reasonable agreement between the experimental and calculated results.
6 Conclusions
In this article, we have studied the reactions between He+ and CO2 and OCS at low collision energies using the Rydberg-merged-beam approach.34,36 The experiments were carried out using He:Ne-seeded supersonic beams of pure OCS and 13CO2, as well as a (23.2:76.8) mixture of OCS and 13CO2, to assess the roles of the dipole and quadrupole moments in these systems and to examine the relative rate constants under conditions of competitive kinetics. The studies of He+ + OCS and He+ + 13CO2 using a single expansion containing both OCS and 13CO2 further allowed us to verify that the observed kinetics do not depend on the composition of the supersonic expansions.
Two reaction channels with an Ecoll-independent branching ratio were observed in the He+ + OCS reaction, leading to the formation of CO+ (∼70%) and CS+ (∼30%), but only one product ion (13CO+) was observed in the He+ + 13CO2 reaction. Whereas the rate coefficient of the He+ + 13CO2 → He + 13CO+ + O reaction decreases by ∼40% between ∼kB ⋅ 30 K and zero, the overall rate coefficient of the He+ + OCS reaction increases by ∼75% over the same Ecoll range. The reaction probabilities upon capture were estimated to be ∼20% larger in the He+ + OCS system. Comparing calculated thermal rate coefficients of the He+ + CO2 reaction system with thermal rates at 70 K and 300 K determined in earlier studies suggests that only about half of the collisions are reactive in this system. Theoretical calculations including the short-range interactions would be needed for the full interpretation of the observed branching ratios and reaction probabilities.
Data availability
All essential data are included in the article. Further data may be obtained from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Daniel Zindel for the preparation of the gas mixtures, Josef A. Agner and Hansjürg Schmutz for the technical assistance, and Dr. Raphaël Hahn and David Schlander for fruitful discussions. This work was supported by the Swiss National Science Foundation (Grant No. 200020B 200478).References
- E. Herbst and W. Klemperer, Astrophys. J., 1973, 185, 505–533 CrossRef CAS.
- E. Roueff, Phys. Scr., 1987, 36, 319–322 CrossRef CAS.
- D. Smith, Chem. Rev., 1992, 92, 1473–1485 CrossRef CAS.
- T. P. Snow and V. M. Bierbaum, Ann. Rev. Anal. Chem., 2008, 1, 229–259 CrossRef CAS PubMed.
- D. C. Clary, D. Smith and N. G. Adams, Chem. Phys. Lett., 1985, 119, 320–326 CrossRef CAS.
- D. C. Clary, Ann. Rev. Phys. Chem., 1990, 41, 61–90 CrossRef CAS.
- J. Troe, J. Chem. Phys., 1987, 87, 2773–2780 CrossRef CAS.
- J. Troe, J. Chem. Phys., 1996, 105, 6249–6262 CrossRef CAS.
- T. Stoecklin, D. C. Clary and A. Palma, J. Chem. Soc., Faraday Trans., 1992, 88, 901–908 RSC.
- E. I. Dashevskaya, I. Litvin, E. E. Nikitin and J. Troe, J. Chem. Phys., 2005, 122, 184311 CrossRef CAS PubMed.
- M. Auzinsh, E. I. Dashevskaya, I. Litvin, E. E. Nikitin and J. Troe, J. Chem. Phys., 2013, 139, 084311 CrossRef CAS PubMed.
- M. Auzinsh, E. I. Dashevskaya, I. Litvin, E. E. Nikitin and J. Troe, J. Chem. Phys., 2013, 139, 144315 CrossRef CAS PubMed.
- E. I. Dashevskaya, I. Litvin, E. E. Nikitin and J. Troe, J. Chem. Phys., 2016, 145, 244315 CrossRef CAS PubMed.
- V. G. Anicich and W. T. Huntress, Jr., Astrophys. J., Suppl. Ser., 1986, 62, 553–672 CrossRef CAS.
- V. Wakelam, I. W. M. Smith, E. Herbst, J. Troe, W. Geppert, H. Linnartz, K. Öberg, E. Roueff, M. Agúndez, P. Pernot, H. M. Cuppen, J. C. Loison and D. Talbi, Space Sci. Rev., 2010, 156, 13–72 CrossRef CAS.
- P. Langevin, Ann. Chim. Phys., 1905, T5, 245–288 Search PubMed.
- S. Willitsch, Chemistry with controlled ions, Advances in Chemical Physics, John Wiley & Sons, Inc., 2017, vol. 162, pp. 307–340 Search PubMed.
- Cold Chemistry: Molecular Scattering and Reactivity Near Absolute Zero, ed. O. Dulieu and A. Osterwalder, The Royal Society of Chemistry, 2017 Search PubMed.
- L. S. Petralia, A. Tsikritea, J. Loreau, T. P. Softley and B. R. Heazlewood, Nat. Commun., 2020, 11, 173 CrossRef CAS PubMed.
- J. Toscano, H. J. Lewandowski and B. R. Heazlewood, Phys. Chem. Chem. Phys., 2020, 22, 9180–9194 RSC.
- A. Tsikritea, J. A. Diprose, T. P. Softley and B. R. Heazlewood, J. Chem. Phys., 2022, 157, 060901 CrossRef CAS PubMed.
- D. Gerlich, Low Temperatures and Cold Molecules, Imperial College Press, London, 2008, ch. 3, pp. 121–174 Search PubMed.
- R. Wester, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 154001 CrossRef.
- C. R. Markus, O. Asvany, T. Salomon, P. C. Schmid, S. Brünken, F. Lipparini, J. Gauss and S. Schlemmer, Phys. Rev. Lett., 2020, 124, 233401 CrossRef CAS PubMed.
- J. B. Marquette, B. R. Rowe, G. Dupeyrat, G. Poissant and C. Rebrion, Chem. Phys. Lett., 1985, 122, 431–435 CrossRef CAS.
- B. R. Rowe, A. Canosa and V. Le Page, Int. J. Mass Spectrom. Ion Processes, 1995, 149–150, 573–596 CrossRef.
- S. Willitsch, M. T. Bell, A. D. Gingell and T. P. Softley, Phys. Chem. Chem. Phys., 2008, 10, 7200–7210 RSC.
- O. A. Krohn, K. J. Catani, S. P. Sundar, J. Greenberg, G. da Silva and H. J. Lewandowski, J. Phys. Chem. A, 2023, 127, 5120–5128 CrossRef CAS PubMed.
- O. A. Krohn and H. J. Lewandowski, J. Phys. Chem. A, 2024, 128, 1737–1752 CrossRef CAS PubMed.
- P. Puri, M. Mills, C. Schneider, I. Simbotin, J. A. Montgomery Jr., R. Côté, A. G. Suits and E. R. Hudson, Science, 2017, 357, 1370–1375 CrossRef CAS PubMed.
- P. Puri, M. Mills, I. Simbotin, J. A. Montgomery Jr., R. Côté, C. Schneider, A. G. Suits and E. R. Hudson, Nat. Chem., 2019, 11, 615–621 CrossRef CAS PubMed.
- A. Voute, A. Dörfler, L. Wiesenfeld, O. Dulieu, F. Gatti, D. Peláez and S. Willitsch, Phys. Rev. Res., 2023, 5, L032021 CrossRef CAS.
- P. Allmendinger, J. Deiglmayr, J. A. Agner, H. Schmutz and F. Merkt, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 90, 043403 CrossRef.
- P. Allmendinger, J. Deiglmayr, O. Schullian, K. Höveler, J. A. Agner, H. Schmutz and F. Merkt, Chem. Phys. Chem., 2016, 17, 3596–3608 CrossRef CAS PubMed.
- P. Allmendinger, J. Deiglmayr, K. Höveler, O. Schullian and F. Merkt, J. Chem. Phys., 2016, 145, 244316 CrossRef PubMed.
- V. Zhelyazkova, F. B. V. Martins, J. A. Agner, H. Schmutz and F. Merkt, Phys. Rev. Lett., 2020, 125, 263401 CrossRef CAS PubMed.
- S. T. Pratt, J. L. Dehmer, P. M. Dehmer and W. A. Chupka, J. Chem. Phys., 1994, 101, 882–890 CrossRef CAS.
- E. Wrede, L. Schnieder, K. Seekamp-Schnieder, B. Niederjohann and K. H. Welge, Phys. Chem. Chem. Phys., 2005, 7, 1577–1582 RSC.
- M. Matsuzawa, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 82, 054701 CrossRef.
- F. B. V. Martins, V. Zhelyazkova, Ch. Seiler and F. Merkt, New J. Phys., 2021, 23, 095011 CrossRef CAS.
- V. Zhelyazkova, F. B. V. Martins, J. A. Agner, H. Schmutz and F. Merkt, Phys. Chem. Chem. Phys., 2021, 23, 21606–21622 RSC.
- V. Zhelyazkova, F. B. V. Martins, S. Schilling and F. Merkt, J. Phys. Chem. A, 2023, 127, 1458–1468 CrossRef CAS PubMed.
- R. Hahn, D. Schlander, V. Zhelyazkova and F. Merkt, Phys. Rev. X, 2024, 14, 011034 CAS.
- K. Höveler, J. Deiglmayr and F. Merkt, Mol. Phys., 2021, 119, e1954708 CrossRef.
- V. Zhelyazkova, F. B. V. Martins, M. Žeško and F. Merkt, Phys. Chem. Chem. Phys., 2022, 24, 2843–2858 RSC.
- F. B. V. Martins, V. Zhelyazkova and F. Merkt, New J. Phys., 2022, 24, 113003 CrossRef CAS.
- V. Zhelyazkova, F. B. V. Martins and F. Merkt, Phys. Chem. Chem. Phys., 2022, 24, 16360–16373 RSC.
- K. Höveler, J. Deiglmayr, J. A. Agner, H. Schmutz and F. Merkt, Phys. Chem. Chem. Phys., 2021, 23, 2676–2685 RSC.
- F. J. Lovas, J. S. Coursey, S. A. Kotochigova, J. Chang, K. Olsen and R. A. Dragoset, NIST Standard Reference Database 117, 2003.
- N. Chetty and V. W. Couling, Mol. Phys., 2011, 109, 655–666 CrossRef CAS.
- C. Graham, D. A. Imrie and R. E. Raab, Mol. Phys., 1998, 93, 49–56 CrossRef CAS.
- M. Gussoni, R. Rui and G. Zerbi, J. Mol. Struct., 1998, 447, 163–215 CrossRef CAS.
- T. N. Olney, N. M. Cann, G. Cooper and C. E. Brion, Chem. Phys., 1997, 223, 59–98 CrossRef CAS.
- M. P. Bogaard, A. D. Buckingham, R. K. Pierens and A. H. White, J. Chem. Soc., Faraday Trans., 1978, 74, 3008–3015 RSC.
- G. Herzberg, Molecular Spectra and Molecular Structure, Volume III, Electronic Spectra and Electronic Structure of Polyatomic Molecules, Van Nostrand Reinhold Company, New York, 2nd edn, 1966 Search PubMed.
- D. Bailly and C. Rossetti, J. Mol. Spectrosc., 1984, 105, 229–245 CrossRef CAS.
- M. Tsuji, M. Matsuo and Y. Nishimura, Int. J. Mass Spectrom. Ion Phys., 1980, 34, 273–286 CrossRef CAS.
- H. Sekiya, M. Tsuji and Y. Nishimura, Chem. Phys. Lett., 1983, 100, 494–498 CrossRef CAS.
- H. Sekiya, M. Tsuji and Y. Nishimura, Bull. Chem. Soc. Jpn., 1984, 57, 3329–3330 CrossRef CAS.
- H. Chatham, D. Hils, R. Robertson and A. C. Gallagher, J. Chem. Phys., 1983, 79, 1301–1311 CrossRef CAS.
- V. G. Anicich, J. B. Laudenslager, W. T. Huntress, Jr. and J. H. Futrell, J. Chem. Phys., 1977, 67, 4340–4350 CrossRef CAS.
- J. B. Laudenslager, W. T. Huntress, Jr. and M. T. Bowers, J. Chem. Phys., 1974, 61, 4600–4617 CrossRef CAS.
- N. G. Adams and D. Smith, J. Phys. B: At. Mol. Phys., 1976, 9, 1439–1451 CrossRef CAS.
- N. G. Adams and D. Smith, Int. J. Mass Spectrom. Ion Phys., 1976, 21, 349–359 CrossRef CAS.
- W. Lindinger, D. L. Albritton and F. C. Fehsenfeld, J. Chem. Phys., 1975, 62, 4957–4958 CrossRef CAS.
- R. C. Bolden, R. S. Hemsworth, M. J. Shaw and N. D. Twiddy, J. Phys. B: At., Mol. Opt. Phys., 1970, 3, 45–60 CrossRef CAS.
- F. C. Fehsenfeld, A. L. Schmeltekopf, P. D. Goldan, H. I. Schiff and E. E. Ferguson, J. Chem. Phys., 1966, 44, 4087–4094 CrossRef CAS.
- Q. Guo, Y. Zhi, J. Hu and S. X. Tian, J. Phys. Chem. A, 2024, 128, 3821–3829 CrossRef CAS PubMed.
- B. R. Rowe, J. B. Marquette and C. Rebrion, J. Chem. Soc., Faraday Trans., 1989, 85, 1631 RSC.
- V. Zhelyazkova, M. Žeško, H. Schmutz, J. A. Agner and F. Merkt, Mol. Phys., 2019, 117, 2980–2989 CrossRef CAS.
- C.-H. Chang and D. J. Nesbitt, J. Chem. Phys., 2015, 142, 244313 CrossRef PubMed.
- F. B. V. Martins, V. Zhelyazkova, A. Osterwalder and F. Merkt, Chimia, 2023, 77, 221–224 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |