
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen tunneling with an atypically small KIE measured in the mediated decomposition of the Co(CH3COOH)+ complex†
Simon U.
Okafor
 ,
Gabriele
Pinto
,
Michael
Brdecka
,
William
Smith
,
Tucker W. R.
Lewis
,
Michael
Gutierrez
and
Darrin J.
Bellert
*
,
Gabriele
Pinto
,
Michael
Brdecka
,
William
Smith
,
Tucker W. R.
Lewis
,
Michael
Gutierrez
and
Darrin J.
Bellert
*
Baylor University, 1311 S 5th St, Waco, TX 76706, USA. E-mail: Darrin_Bellert@baylor.edu
First published on 22nd October 2024
Abstract
Quantum mechanical tunneling (QMT) is a well-documented phenomenon in the C–H bond activation mechanism and is commonly identified by large KIE values. Herein we present surprising findings in the kinetic study of hydrogen tunneling in the Co+ mediated decomposition of acetic acid and its perdeuterated isotopologue, conducted with the energy resolved single photon initiated dissociative rearrangement reaction (SPIDRR) technique. Following laser activation, the reaction proceeds along parallel product channels Co(CH4O)+ + CO and Co(C2H2O)+ + H2O. An energetic threshold is observed in the energy dependence of the unimolecular microcanonical rate constants, k(E). This is interpreted as the reacting population surmounting a rate-limiting Eyring barrier in the reaction's potential energy surface. Measurements of the heavier isotopologue's reaction kinetics supports this interpretation. Kinetic signatures measured at energies below the Eyring barrier are attributed to H/D QMT. The below-the-barrier tunneling kinetics presents an unusually linear energy dependence and a staggeringly small tunneling KIE of ∼1.4 over a wide energy range. We explain this surprising observation in terms of a narrow tunneling barrier, wherein the electronic structure of the Co+ metal plays a pivotal role in enhanced reactivity by promoting efficient tunneling. These results suggest that hydrogen tunneling could play important functions in transition metal chemistry, such as that found in enzymatic mechanisms, even if small KIE values are measured.
1 Introduction
Quantum mechanical tunneling (QMT) is no longer considered an exotic dynamic feature but rather integral to the complete understanding of chemical reactivity. In fact, QMT has recently been dubbed the third reactivity paradigm placing it on par with the concepts of thermodynamic and kinetic control.1,2 QMT maintains a wide sphere of influence and measurements implicate the controlling role of hydrogen tunneling within enzymatic reactions,3–8 although its specific significance to enzyme catalysis relative to other reactions is under debate.9–11 QMT is likely the only kinetic pathway for both interstellar chemistry and reactions that occur under cryogenic conditions.12–16 QMT has been implicated in organometallic chemistry in both biomimetic systems8,17 as well as transition metal complexes.18–20 Moreover, modern studies have demonstrated QMT in atoms heavier than hydrogen, which is surprising given that tunneling probability is inversely dependent on the tunneling particle's mass.15,16,21–25In many cases, measurement of a large kinetic isotope effect (KIE) is the discriminating factor used to identify hydrogen QMT.20,26–28 The KIE is the ratio of rate constants (kH/kD) determined when the transferring hydrogen has been replaced with its heavier deuterium isotope. Substantial QMT contributions to the rate constants are implicated when the KIE magnitude is too large to be caused by the differences in zero-point vibrational energies between the two isotopologues. However, in the other extreme, KIE magnitudes that are small would appear to result from secondary effects and QMT, if present, may not be realized. A situation similar to this latter scenario was recently reported where hydrogen tunneling in an isomerization reaction exhibited a small primary KIE when measured in solid N2.12 Over-barrier kinetics are simply too improbable at such extreme low temperatures and hydrogen QMT must be the reactive process. In this case, the authors attributed the coupling of matrix N2 molecules to the tunneling H/D atoms as the cause of the unexpected low KIE.
The study presented here measures a primary hydrogen tunneling KIE that is also unexpectedly small but cannot be attributed to solvent effects. Rather, we suggest that the low KIE is caused by efficient hydrogen QMT. In this study, a transition metal mediates chemistry to adiabatically reduce reaction barriers and, we believe, to also promote the hydrogen tunneling process through barrier width reduction. The reduction in barrier width greatly improves tunneling efficiency and increases QMT probability29 at system energies that extend far below the transition barrier. We believe that efficient QMT may play important roles in metal atom catalysis but may go unrealized if the measured KIE is less than the expected 7–10 for room temperature reactions.
Focusing on the metal's role in QMT requires specialized instrumentation capable of significant energy and temporal resolution. This necessitates that studies are conducted in a collision-less gas-phase environment with tight control over the reaction's start-time and measurement of the reaction's temporal development. Additionally, there are requirements on the molecular system chosen for study – namely it must be relatively small with tunneling atoms that can be individually distinguished. Kinetic measurements made on such systems in this environment are sufficiently resolved to confidently infer reaction dynamics from the measured kinetics. Examining hydrogen tunneling in small metal containing molecular systems where the environment and available energy are strictly controlled provides the greatest opportunity to reveal fundamental insights of broad relevance.
The system presented here is the kinetic study of the Co+ cation mediated decomposition of CH3COOH and its perdeuterated isomer, CD3COOD. As the system's energy content increases, an abrupt change in the unimolecular rate constant's energy dependence is measured. The energy at this threshold is interpreted as a limit to an Eyring barrier's energy. Hydrogen QMT, however, extends chemical reactivity below the Eyring barrier. Measurements in this energetic QMT regime reveal an unexpected energy dependence and surprisingly low KIEs magnitudes for their typical association with hydrogen tunneling.
The study is the first part of an anticipated two-part series. This first manuscript focuses on our experimental kinetic observations and their interpretation with respect to Eyring barrier energies and QMT. The second paper will extend our experimental observations by computation of the reaction PES in tandem with the kinetic modeling of the important mechanistic steps in this chemical reaction.
2 Procedure
The SPIDRR technique has been described previously.30 Briefly, transition metal cations are generated through laser ablation of a pure metal target located within a high vacuum chamber. These metal ions are entrained in a pulse of helium gas that contains the vapor pressure of CH3COOH. Collisions between the cations and organic form electrostatic clusters where the Co+ cation is bound to the dipole moment of the CH3COOH molecule. The nozzle geometry, gas pulse conditions, and vacuum chamber pressure are such to ensure supersonic cooling of the newly formed clusters. These clusters enter collision-less space just a few millimeters past the expansion nozzle. This binary Co+–CH3COOH cluster is the reactant complex (RC) that will undergo unimolecular decay into the two products, Co(CH4O)+ and Co(C2H2O)+, observed in this study.The species within the pulsed molecular beam travel at the terminal velocity of helium through 79 cm of vacuum to enter into the parallel plates of a pulsed orthogonal accelerator (OA). These are quickly charged to 1750 V to extract cations into 1.86 m of field-free flight within a time-of-flight mass spectrometer (TOFMS). A hemispherical kinetic energy filter, or sector, is located at the end of the TOF which is voltage tuned to transmit the full kinetic energy of the ion beam (transmits all species formed in the source) or a portion of the kinetic energy (transmits products formed from a chemical reaction). The products arrival time to the detector (located just past the sector's exit aperture) is the same as the precursor from which each product was formed. Thus, products formed from an induced chemical reaction of various species within the molecular beam are distinguished. An example mass spectrum showing the species within the beam and the products from the Co(CH3COOH)+ cluster is provided in the ESI† as Fig. S1.
Pulsed, tunable laser radiation from a Nd3+:YAG pumped dye laser (together considered the activation laser (AL)) is directed through the OA, counterpropagating to the molecular beam's direction. The RC absorbs a quantum of photon energy to populate a metastable excited state. The Co+ cation is the chromophore and the electronic transitions excited are atomic-like 3d8 (3P) ← 3d8 (3F) or 3d74s ← 3d8 (3F).31 These are parity forbidden atomic transitions but gain oscillator strength in the RC. The excited electronic state is metastable since emission to lower states is forbidden, and the energy of the excited electronic state is below the adiabatic bond energy of the ground triplet electronic state. Thus, relaxation of the excited state occurs through internal conversion of the photon's energy populating the high vibrational levels of the ground triplet RC surface. Internal vibrational redistribution (IVR) of this energy statistically populates the many vibrational states of the ground potential well at the energy of the absorbed photon. This process (electronic excitation → internal conversion → IVR → statistical distribution of vibrational states) likely occurs within the first few nanoseconds of the reaction. This creates an energy-resolved density of vibrational states that can statistically explore the PES finding pathways to arrive at products. The ensuing nuclear rearrangement reactions occur on the microsecond time scale.
Photoexcitation of the RC occurs along the molecular beam axis. Photon absorption coincident with the extraction pulse to the OA is the start-time of the reaction and is defined as zero τ. Unimolecular decay of the RC into products begins as the RC is accelerated through the parallel plates of the OA. However, only those RCs that exit the OA (requires ∼3 μs) and enter the field-free region of the TOF will produce products with the same arrival time to the detector as the RC. One of the products’ integrated intensity, accumulated over the free flight of the TOF, is transmitted through the sector where it is detected and recorded at zero τ. The AL timing is then reduced from zero τ, firing earlier than the pulse to charge the OA. The RC still forms the same quantity of products resulting from the photon induced chemical reaction, but now a reduced quantity of those is formed in the field-free region of the TOF and ultimately detected. In such fashion, the photo-exited population's survival probability decreases as |τ| increases. The plot of integrated product intensities vs. −τ is analyzed to reveal rate limiting rate constants that are attributed to the decay of the precursor into the product transmitted through the sector to the detector.
Making measurements in such fashion guarantees that products detected result from single photon absorption by the RC. Two photon absorption, which likely occurs, results in a RC excited to levels higher than the adiabatic bond energy of the cluster. Thus, a two-photon excited RC simply dissociates into the Co+ cation + neutral CH3COOH. Energizing the RC through single photon absorption is verified by measuring a linear dependence between the product's intensity vs. the AL laser pulse fluence as shown in Fig. S2 (ESI†).
The energy resolution of the SPIDRR technique results from supersonic cooling such that the absorbed photon's energy is sufficiently greater than the residual thermal population unquenched by adiabatic expansion. This is readily determined in smaller Co+–ligand systems through vibronic spectroscopic techniques.32–37 However, the large density of vibrational eigenstates coupled with the relatively small rotational constants in larger systems makes resolved spectroscopy impossible. Thus, the residual thermal population of the ground state can only be assessed through indirect methods. The translational temperature of the ions within the molecular beam is determined through measurement of the precursor velocity distribution within the beam. These measurements routinely yield Mach numbers > 60 indicating substantial translational cooling.38 The rotational temperature is assumed low since the rotational eigenstate energy difference is small suggesting that cooling collisions with the helium carrier gas will efficiently remove residual energy. Electronic cooling in transition metal species is facilitated by strong spin–orbit coupling and there has never been a long-lived electronic metastable species detected through spectroscopic studies of Co+–ligand complexes. The degree of vibrational cooling within the RC is admittedly more challenging to justify. However, vibrational cooling is assessed here by changing the expansion conditions and re-measuring reaction rate constants. It has been shown that doping a small quantity of a polyatomic gas into a supersonic expansion more efficiently removes vibrational energy within the beam.39 Presumably, this is due to collisions with the doped gas where vibrational cooling is enhanced through vibrational energy transfer to the polyatomic. Thus, the supersonic cooling of vibrational degrees of freedom is assessed by doping 1% CH4 into the helium expansion and re-measuring reaction rates. If the measured rate constants are the same in both expansion environments, then it can be assumed that collisions with helium atoms in the neat helium expansion sufficiently cools residual vibrational energy in the encounter complex. Fig. S3 (ESI†) shows this comparison following absorption of an AL photon = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 950 cm−1 where the rate constants measured in both expansion environments agree. It is noteworthy that the reaction rate constant is very dependent on system energy at 13950 cm−1, as discussed below. Thus, the contribution of unquenched residual energy to the system's total energy will have its most significant impact when the system absorbs a quantum of photon energy = 13950 cm−1 or greater.
950 cm−1 where the rate constants measured in both expansion environments agree. It is noteworthy that the reaction rate constant is very dependent on system energy at 13950 cm−1, as discussed below. Thus, the contribution of unquenched residual energy to the system's total energy will have its most significant impact when the system absorbs a quantum of photon energy = 13950 cm−1 or greater.
3 Results
The unimolecular rearrangement reaction kinetics of two precursor isotopologues, Co(CH3COOH)+ and Co(CD3COOD)+, are measured at resolved internal energies. Each absorbs a single quantum of photon energy to initiate the Co+ mediated decomposition reaction yielding two products. The reaction mechanism thus bifurcates along two pathways which terminate in forming these species. The charged products detected have masses consistent with the following chemical reactions:
This assignment of charged product masses is supported by measuring the expected change in the transmission sector voltages associated with transmission of the analogous deuterium labeled products: Co(CD4O)+ and Co(C2D2O)+.
The integrated intensity of each charged product, acquired at an AL photon energy, is plotted vs. τ to reveal the temporal dependence of product formation. Measurements acquired at the indicated AL photon energies are shown in the left panels of Fig. 1. Each panel shows the product formation's intensity vs. time dependence following the Co(CH3COOH)+ reactant complex absorption of a single quantum of photon energy. The intensity of each product ion exponentially decreases as |τ| increases from zero μs.
 | ||
| Fig. 1 SPIDRR results in the Co+ mediated decomposition of CH3COOH (left side) and CD3COOD (right side) measured at various AL photon energies. | ||
This is consistent with the rate-limiting unimolecular decay of a long-lived intermediate in the Co+ mediated decomposition mechanism of CH3COOH. The temporal dependence of each product ion's formation is independently fit to a single exponential function in τ, which is plotted as the solid curve through the measured product intensities. Preexponential factors and rate constants k(E) are extracted from each fit. The relative preexponential factors determine the kinetic competition between the formation of each product measured at an AL energy. Thus, the product's intensities are normalized to the sum of the two preexponential constants and this reveals the fractioning into each exit channel. The formation of Co(CH4O)+ is preferred relative to Co(C2H2O)+ by a 55/45 ratio. This exit channel fraction (ECF) remains relatively constant at all measured AL energies. The rate constants extracted from each fit are indicated in the left panels of Fig. 1.
The right panels of Fig. 1 show the temporal dependence of Co(C2D2O)+ and Co(CD4O)+ product formation from the Co+ mediated decomposition of the heavier D4 isotopologue. These result from the precursor absorption of a single quantum of the indicated photon energies. As in the left panel, each measured temporal dependence was fit to a single exponential function in τ and seen as the solid curves through the observed intensities. It is acknowledged that the set of traces at lower AL photon energies are biexponentially dependent on τ. This may be due to two-state reactivity (TSR) which has been observed in a previous Co+ study by our laboratory.40,41 This will be addressed in a subsequent publication of this system. For this current study, however, we focus on the long-time component of the biexponential dependence and secure this rate constant by fitting a single exponential function to the product's intensities at values of |τ| greater than 20 μs, where the kinetic contributions from TSR are minimal. As in the lighter isomer, the ratio of one preexponential factor relative to the sum results in the same 55/45 ECF favoring the Co(CD4O)+ product. The ECF is independent of the isotopologue identity at the energies studied here. Moreover, Fig. 1 indicates that the rate constant for formation of the Co(C2H2O)+ product are slightly larger than the Co(CH4O)+ product measured at each AL photon energy and is consistent across both isotopologues.
In general, the measured rate constants for the formation of each product decreases with decreasing photon energy. However, there exists striking differences with the rate of decrease relative to the system energy content. This is easily observed in the three lower panels on the H4 side of Fig. 1 where the rate constants roughly double as the system energy is increased over this 900 cm−1 energy range. However, when the system's internal energy is further increased by 50 cm−1, from 13900 cm−1 to 13950 cm−1, the rate constants increase by factors of 1.9 and 2.6, over this narrow energy range. As an additional 150 cm−1 of energy is added to the system (top left panels in Fig. 1), the rate constants increase by an additional ∼1.5×. Thus, the chemical reaction kinetics of the Co(CH3COOH)+ system change significantly once a 13950 cm−1 energy threshold is surpassed. We interpret this rapid change in reaction rates as a transition in the controlling reaction dynamics that occurs at this threshold. We believe that 13950 cm−1 is an upper energy limit to an Eyring barrier, and upon transition of this energy, there is a change from QMT to over-the-barrier dynamics where RRKM theory governs reaction rates at these higher system energies.
An analogous picture unfolds through observation of the D4 side of Fig. 1. The lower four panels span a 700 cm−1 energy range where the rate constants gradually increase with increasing AL energy. However, the top two panels show that the rate constants nearly double between AL energies 14100 cm−1 and 14350 cm−1. Again, the abrupt change in the rate constant's energy dependence is interpreted as an upper limit to the Eyring barrier energy and the consequent transition from quantum to statistical control of the reaction dynamics.
These abrupt increases in rate constant values are more easily seen in Fig. 2 which plots each rate constant vs. the AL photon energy absorbed by the precursor. Thresholds are identified as the energy where the rate constants effectively double from their previous measurement. These threshold energies occur at 13950 cm−1 in the H4 isotopologue and 14350 cm−1 in the heavier D4 system. Both product channels in each isomer have the same threshold energy indicating that the changing dynamics within that isotopologue's chemistry affect each reaction pathway in the same fashion. The transition from quantum to statistical dynamic reaction control is the most likely phenomenon consistent with the measured changes in the reaction rate constant's energy dependence.
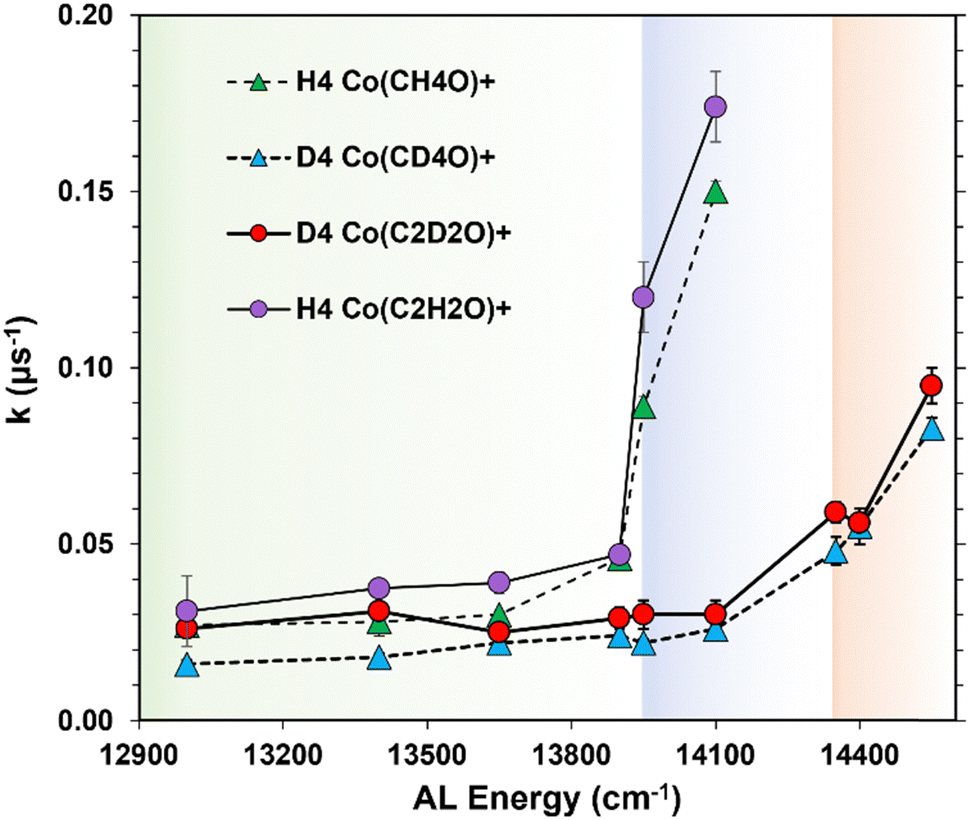 | ||
| Fig. 2 Energy dependence of the unimolecular rate constants. The shading interfaces indicate changing reaction dynamics. Hydrogen (deuterium) tunneling exists below 13950 cm−1 (14350 cm−1), whereas statistical control occurs at energies higher than these thresholds. | ||
This change in reaction dynamics manifests as a rapid change in reaction rate constants which occurs as an Eyring's barrier energy is breached. Without QMT, the reaction rate constants would behave in classical RRKM fashion and erupt from the horizontal axis in Fig. 2 once the Eyring threshold energy is surpassed. This is due to the large density of states in the reactant well that distributes into the growing sum of states at the transition state to ultimately form products. Both the sum and density of states increase rapidly with energy with a consequent increase in the rate of product formation. These statistical effects yield rate constants that eclipse rate constants caused by QMT. Thus, the Eyring barrier energy is identified as a threshold in a k(E) vs. E plot indicated by the energy where the measured rate constants energy dependence markedly changes (Fig. 2).
4 Discussion
a The Eyring barrier energy and the tunneling KIE
The 13950 cm−1 threshold energy measured in this study is interpreted as an effective barrier height and an upper limit to the rate-limiting Eyring barrier's energy along the Co(CH3COOH)+ potential energy surface (PES). We define the Eyring barrier's energy as the difference between the v = 0 level of the reactant states and the v = 0 level of the activated complex structure located at the TS (often called the energy difference between the vibrationless levels of the reactant and TS). The effective barrier is an upper limit because our measured rate constants, at the Eyring barrier's energy, will contain contributions from both QMT and over-barrier statistics. The observed thresholds occur at energies where the contribution from QMT is negligible in comparison to RRKM statistics. This must be at an energy slightly above the v = 0 level of the activated complex which contains 20 vibrational modes, many of which are low frequency. This results in a sum of vibrational states at the TS that rapidly increases with energy.
The effective barrier energy in this system's heavier Co(CD3COOD)+ isotopologue is measured at 14350 cm−1. This value both supports our interpretation that the observed thresholds are related to Eyring barrier energies and assigns the type of QMT observed in this system. The threshold energy difference measured between the heavier and lighter isotopologues is 400 cm−1. This energy difference is consistent with the expected zero-point energy (ZPE) lowering associated with the heavier isotopologue. This energy difference will have a significant contribution from ZPE lowering in the reactant state and a reduced contribution from the ZPE lowering in the activated complex at the TS. The large 400 cm−1 energy difference observed between the threshold energies strongly suggests that the reaction coordinate of this rate-limiting portion of the PES follows a high-frequency vibrational mode which becomes imaginary at the TS. The high-frequency modes in the precursor are either C–H or O–H vibrations. If a 3000 cm−1 stretching frequency is assumed for these modes, then the expected ZPE lowering of the reactant state upon deuterium labeling will equal 900 cm−1 along this reaction coordinate. This value only accounts for the reactant state and thus represents a maximum energy difference between threshold values upon deuterium labeling. The measured energy difference, 400 cm−1, is less than the maximum but too large to be caused by a secondary KIE. This suggests that some ZPE lowering must be attributed to the activated complex structure upon deuterium substitution, and/or, that the assumed 3000 cm−1 stretching frequency is less than this value in the intermediate structure that precedes this rate-limiting transition state. More importantly, the large 400 cm−1 energy difference associates a transferring hydrogen to the rate limiting Eyring barrier on the Co(CH3COOH)+ PES. This indicates that rate constants measured at AL energies below 13950 cm−1 in the H4 isomer, or rate constants measured in the D4 isomer at energies below 14350 cm−1, are due to hydrogen and deuterium tunneling.
H/D QMT KIE values are acquired from the formation kinetics of each product at the AL energies studied. The third and fourth panels on each side of Fig. 1 shows the formation of the products measured at 13900 cm−1 and 13650 cm−1 resulting from H/D tunneling. This shows a side-by-side comparison of the changing tunneling kinetics associated with each isotopologue. The KIE measured at 13900 cm−1 is 1.9 ± 0.2 and 1.62 ± 0.2 for formation of Co(CH4O)+ and Co(C2H2O)+ products. These KIE values are remarkably small for their typical association with H/D QMT. The rate constant measurements made at 13650 cm−1 and their corresponding KIE values for the formation of each product are 1.4 ± 0.1 and 1.6 ± 0.1, which again are unusually small for hydrogen QMT.
Fig. 3 plots twelve KIE values vs. the AL energy at which each was measured. Each of the KIE values measured at energies below 13950 cm−1, the so-called tunneling energetic regime, are atypically small. Whereas, the KIE measured at 13950 cm−1, the effective energy of the rate-limiting hydrogen transfer barrier, is substantially larger with an average value of 4.4 ± 0.5. This large increase in KIE is due to the differences in energy dependence in the controlling dynamics between the two isotopologues. From Fig. 2, at a system energy equal to 13950 cm−1, the reaction dynamics in the lighter H4 isomer have just transitioned from quantum to primarily statistical control while the reaction kinetics of the heavier D4 isomer remains under quantum control. Consequently, KIE values rapidly increase as this interface between the controlling dynamic paradigms is crossed.
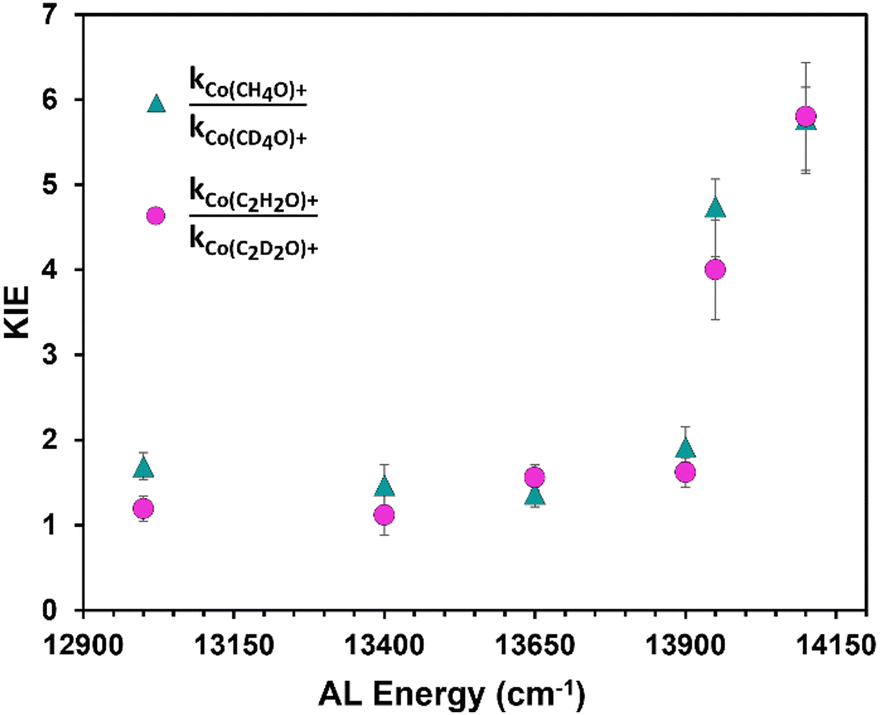 | ||
| Fig. 3 The KIE energy dependence. Measurements at energies below 13900 cm−1 result from pure H/D tunneling. | ||
b Barrier width reduction as the cause of these unusual observations
Fig. 2 indicates that the tunneling rate constants monotonically decrease with an apparent linear dependence on decreasing energy at absorbed photon energies below the Eyring threshold. We have previously observed this linear dependence measured between tunneling rate constants and system energy in our study of the Co+ mediated decomposition of CH3CHO.41,42 We have also measured a similar linear dependence in a Ni+ system that is under current study with preliminary results provided in Fig. S4 (ESI†). The linear dependence of the H/D tunneling rate constants on system energy appears prevalent in the chemistry of the metal mediated systems that we have studied, although such dependencies have not been reported by other groups, to our knowledge. We reinforce that this slight energy dependence of the microcanonical rate constants with system energy is not predicted by statistical rate theories. We therefore associate this slight energy dependence as a QMT effect within this ∼1000 cm−1 energy range below the Eyring barrier. The QMT KIEs displayed in Fig. 3 show a slight increase with decreasing energy over the 12900–13900 cm−1 range. This slight energy dependence appears unique in the literature but is reminiscent of the slight temperature dependence of certain enzymatic reactions where QMT was determined by a large, measured H/D KIE. However, the tunneling KIE measured here has an average value of only 1.4 over this 1000 cm−1 energy range. There have only been a few reports of such small H/D QMT KIE values in the literature, and they have been attributed to the tunneling H-atom coupled to a solvent molecule,12 or to systems where diffusion limited rates mask QMT.43
As these KIE-attenuating phenomena are not present in our high-vacuum gas-phase experiments, we instead attribute the small QMT KIE values measured in this study to a remarkable hydrogen tunneling efficiency that may be mediated by the Co+ cation electronic structure. Tunneling theories indicate that the width of the tunneling barrier is the governing factor influencing QMT probability, with reduced contributions from the tunneling particle's mass and the system's energy relative to the energy of the potential barrier. Thus, we presume that a potential barrier exists in this system with such a small width that it nearly negates the mass dependence of the tunneling particle.
Although elegant tunneling theories exist,44–47 here we employ a simple theoretical description of QMT to ascertain whether barrier width reduction could possibly predict properties consistent with the measured kinetic properties of this system. This is in keeping with the theme of this first publication in this series, whereas more complete theoretical descriptions are planned in a subsequent paper. The tunneling theory chosen utilizes free particle wavefunctions that penetrate a rectangular potential barrier and yield exact analytic solutions. This theory is the same as presented in undergraduate quantum courses to describe scanning tunneling electron microscopy (STEM) and details an electron's penetration through a barrier.
Within this rough theoretical description, hydrogen or deuterium atoms impinge upon a rectangular potential barrier with finite width and energy. Free particle wavefunctions are ascribed to the impinging particles. The probability of particle transmission through the barrier is then determined by solving the time-independent Schrödinger equation while taking proper account of the barrier interfaces such that the wavefunction, and its first derivative, are continuous through space. The square modulus of the resulting wavefunction then provides the probability of finding the particle on the opposite side of the barrier. The ratio of the squared modulus intensities for the transmitted particle relative to those particles that impinge upon the barrier yields an expression for the tunneling probability. This resulting function is dependent on the particle's mass (m) and energy (E), the barrier energy (U0), and the barrier's width (L).

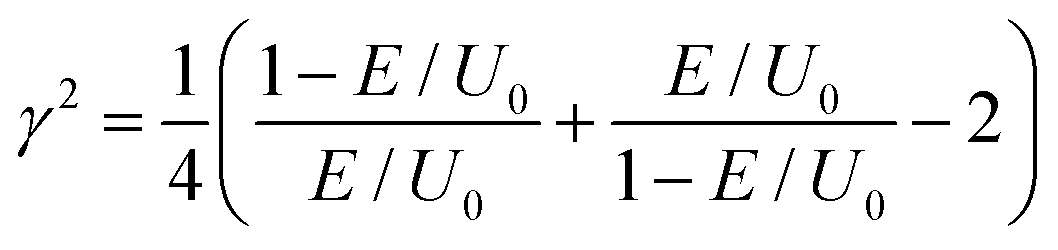 and
and 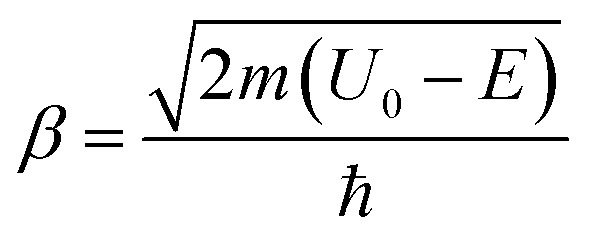 .
.
The measured barrier energies from this study, as well as the particle's mass and system energy, are input into this equation to determine the tunneling probability at various barrier widths. We assume that the tunneling probability scales with the first order tunneling rate constants measured in this study, and therefore, the corresponding KIEs will be well represented by the probability ratio of hydrogen to deuterium tunneling. The barrier width is then modified to minimize a squared residuals fit of the predicted tunneling probabilities ratio to the measured QMT KIEs. The predicted energy dependence of H-tunneling, D-tunneling, and the KIEs are plotted in Fig. 4, along with the measured QMT KIEs from the Co(CH4O)+ product channel in Fig. 3. The optimized barrier width that resulted from this analysis is 6 pm. With this narrow barrier, the tunneling probability T(L,E) for both the H and D atoms is significantly higher than one would expect, as shown by the dashed lines in Fig. 4. For the H atom, the barrier penetration probability exceeds 0.5 (green dashed line), while the D atom's tunneling probability approaches 0.35 at the Eyring barrier energy. Interestingly, the probabilities calculated at a 6 pm barrier width become quasi-independent of energy, and their ratio results in a very low KIE, consistent with our observations for the Co(CH3COOH)+ reactions described in this paper.
 | ||
| Fig. 4 The results from a simple tunneling model applied to our measurements. | ||
The purpose of this analysis is not to quantify the rate-limiting barrier width on the Co(CH3COOH)+ PES – this theory is too simple, and the assumptions unjustified for this expectation. Rather, this analysis suggests that it is possible that a reduction in barrier width can lead to the unusual kinetic behavior measured in this system. The analysis, moreover, indicates the need to apply more modern descriptions of tunneling in this system.
The minimal energy pathway (MEP) is the multidimensional curved pathway of lowest energy through the potential landscape. The shortest tunneling pathway, however, is a straight path on the concave side of the MEP from the reactants side of the barrier to the products. This short straight path does not necessarily contain the reaction saddle point and, consequently, will be of higher potential energy. Such theories as instanton theory,44,48–50 short curvature tunneling (SCT),45 and long curvature tunneling (LCT)46,47 have gone beyond the separable reaction coordinate hypothesis and instead use QMT treatments that optimize tunneling by considering various paths straying from the MEP. Central to these theories are high-quality PESs that are supported by measurement.
Drawing from the findings of pioneers in instanton theory, SCT and LCT, we speculate that perhaps it is the Co+ cation's electronic structure that reduces the potential energy barrier widths in the coordinates transverse to the MEP, allowing the reacting population greater exploration over the PES to ultimately find shorter pathways, thereby increasing tunneling efficiency. Providing computational support for our theory is a topic of our next publication, where calculations of the ground state PES and the structures associated with key intermediates and transition states may lead to a refinement of our hypothesis concerning the reduction of barrier width. This will complement the experimental findings reported herein and provide greater insight into this apparent innate ability of certain transition metals to promote hydrogen QMT.
5 Conclusions
Unexpected kinetic properties have been measured in the Co+ mediated decomposition of CH3COOH and its perdeuterated isomer. Reaction rates are observed to change rapidly at certain threshold energies. This is interpreted as a transition from quantum to statistical dynamic control over the chemical reaction which manifests as a rapid change in the reaction's first order rate constant's energy dependence. Compelling evidence for this interpretation are measurements of relatively small rate constants below the barriers, attributed to pure H/D tunneling, and the large shift in barrier energies between the isotopologues that is consistent with a primary KIE. A low tunneling KIE and an unexpected QMT rate constant energy dependence arise from such measurements. However, these can be partially explained by associating a very narrow barrier width to the rate-limiting hydrogen motion. We hypothesize that reduction of the barrier width must be due to the interaction with the metal's charge and electronic structure. Some corroboration of this postulate is provided by our current study of Ni+ mediated chemistries where a similar slight dependence of reaction rate constants on system energy is measured (Fig. S3, ESI†).These findings indicate that C–H bond activation mediated by the Co+ cation in its ground, triplet coupled, electronic configuration has significant tunneling contributions despite exhibiting a small KIE. Moreover, given the efficiency by which tunneling occurs in this system, QMT may extend to very low system energies. Our current studies of similar properties observed in Ni+ mediated chemistry suggest that such tunneling dynamics may be more general and a property attributable to certain oxidized 3d transition metals suggesting their ability as catalytic active sites in C–H functionalization chemistry.
Given the prevalence of oxidized, open-shell transition metals to function as the catalytic active sites in metalloenzymes, we speculate whether the reaction dynamics inferred here may extend to such systems. Kinetic studies of C–H functionalization by metalloenzymes have measured very large KIEs and this is regarded as the QMT hallmark. However, the contributions of hydrogen tunneling in metalloenzyme chemistry may go unrealized should a measurement yield a small KIE as this, on the contrary, may suggest highly efficient hydrogen QMT. This suggests that QMT may be more pervasive in metalloenzyme chemistry than what current literature indicates.
Rate constants measured below each effective barrier result from hydrogen/deuterium tunneling. Pure H/D tunneling rate constants, uncontaminated by contributions from over-barrier kinetics, are not commonly measured. Our technique, in addition to groups that make rate measurements in cryogenic fluids, are of the few that can achieve this goal. However, these low temperature measurements are not typically associated with catalysis and the rate constants acquired are rather small (on the order of events per second). The chemistry here is driven by the transition metal's ability to lower reaction energy requirements, which is the function of a catalytic active site. Moreover, this chemistry occurs at high energies (on the order of 25% of a C–H bond energy) and the tunneling rate constants indicate turnovers of tens of thousands per second. This research uniquely positions us to gain fundamental insight into QMT in general, and its role in catalysis, underscoring the significance of this research in advancing this field.
Data availability
Data for this article, in the form of the raw data files secured from our instrument, are available at Bellert, Darrin, 2024, “Co(AcOH)+”, https://doi.org/10.18738/T8/SLBSVC, Texas Data Repository, V1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgement is made to the donors of The American Chemical Society Petroleum Research Fund for support (or partial support) of this research.References
- P. R. Schreiner, Tunneling Control of Chemical Reactions: The Third Reactivity Paradigm, J. Am. Chem. Soc., 2017, 139(43), 15276–15283, DOI:10.1021/JACS.7B06035.
- P. R. Schreiner, Quantum Mechanical Tunneling Is Essential to Understanding Chemical Reactivity, Trends Chem., 2020, 2(11), 980–989, DOI:10.1016/J.TRECHM.2020.08.006.
- Z. X. Liang and J. P. Klinman, Structural Bases of Hydrogen Tunneling in Enzymes: Progress and Puzzles, Curr. Opin. Struct. Biol., 2004, 14(6), 648–655, DOI:10.1016/J.SBI.2004.10.008.
- Y. Cha, C. J. Murray and J. P. Klinman, Hydrogen Tunneling in Enzyme Reactions, Science, 1989, 243(4896), 1325–1330, DOI:10.1126/SCIENCE.2646716.
- Z. D. Nagel and J. P. Klinman, Tunneling and Dynamics in Enzymatic Hydride Transfer, Chem. Rev., 2006, 106(8), 3095–3118, DOI:10.1021/CR050301X.
- J. Basran, M. J. Sutcliffe and N. S. Scrutton, Enzymatic H-Transfer Requires Vibration-Driven Extreme Tunneling, Biochemistry, 1999, 38(10), 3218–3222, DOI:10.1021/BI982719D.
- C. R. Pudney, S. Hay, C. Levy, J. Pang, M. J. Sutcliffe, D. Leys and N. S. Scrutton, Evidence to Support the Hypothesis That Promoting Vibrations Enhance the Rate of an Enzyme Catalyzed H-Tunneling Reaction, J. Am. Chem. Soc., 2009, 131(47), 17072–17073, DOI:10.1021/JA908469M/SUPPL_FILE/JA908469M_SI_001.PDF.
- J. P. Layfield and S. Hammes-Schiffer, Hydrogen Tunneling in Enzymes and Biomimetic Models, Chem. Rev., 2013, 114(7), 3466–3494, DOI:10.1021/cr400400p.
- S. C. L. Kamerlin and A. Warshel, An Analysis of All the Relevant Facts and Arguments Indicates That Enzyme Catalysis Does Not Involve Large Contributions from Nuclear Tunneling, J. Phys. Org. Chem., 2010, 23(7), 677–684, DOI:10.1002/POC.1620.
- M. H. M. Olsson, W. W. Parson and A. Warshel, Dynamical Contributions to Enzyme Catalysis: Critical Tests of a Popular Hypothesis, Chem. Rev., 2006, 106(5), 1737–1756, DOI:10.1021/CR040427E.
- D. G. Truhlar, Tunneling in Enzymatic and Nonenzymatic Hydrogen Transfer Reactions, J. Phys. Org. Chem., 2010, 23(7), 660–676, DOI:10.1002/POC.1676.
- J. P. L. Roque, C. M. Nunes, P. R. Schreiner and R. Fausto, Hydrogen Tunneling Exhibiting Unexpectedly Small Primary Kinetic Isotope Effects, Chem. – Eur. J., 2024, 30(39), e202401323, DOI:10.1002/CHEM.202401323.
- P. R. Schreiner, H. P. Reisenauer, D. Ley, D. Gerbig, C. H. Wu and W. D. Allen, Methylhydroxycarbene: Tunneling Control of a Chemical Reaction, Science, 2011, 332(6035), 1300–1303, DOI:10.1126/SCIENCE.1203761/SUPPL_FILE/SCHREINER.SOM.PDF.
- H. Hidaka, M. Watanabe, A. Kouchi and N. Watanabe, Reaction routes in the CO-H2CO-dn-CH3OH-dm system clarified from H(d) exposure of solid formaldehyde at low temperatures, Astrophys. J., 2009, 702, 291–300, DOI:10.1088/0004-637X/702/1/291.
- M. P. Merini, T. Schleif and W. Sander, Heavy-Atom Tunneling in Bicyclo[4.1.0]Hepta-2,4,6-Trienes, Angew. Chem., 2023, 135(44), e202309717, DOI:10.1002/anie.202309717.
- M. Ertelt, D. A. Hrovat, W. T. Borden and W. Sander, Heavy-Atom Tunneling in the Ring Opening of a Strained Cyclopropene at Very Low Temperatures, Chem. – Eur. J., 2014, 20(16), 4713–4720, DOI:10.1002/chem.201303792.
- D. Mandal, D. Mallick and S. Shaik, Kinetic Isotope Effect Determination Probes the Spin of the Transition State, Its Stereochemistry, and Its Ligand Sphere in Hydrogen Abstraction Reactions of Oxoiron(IV) Complexes, Acc. Chem. Res., 2018, 51(1), 107–117, DOI:10.1021/acs.accounts.7b00442.
- J. M. Matxain and M. A. Huertos, Hydrogen Tunneling in Stoichiometric and Catalytic Reactions Involving Transition Metals, ChemCatChem, 2023, 15(24), e202300962, DOI:10.1002/cctc.202300962.
- M. A. Tepaske, A. Fitterer, H. Verplancke, D. Delony, M. C. Neben, B. de Bruin, M. C. Holthausen and S. Schneider, C−H Bond Activation by Iridium(III) and Iridium(IV) Oxo Complexes, Angew. Chem., Int. Ed., 2024, 63(7), e202316729, DOI:10.1002/ANIE.202316729.
- J. E. Schneider, M. K. Goetz and J. S. Anderson, Variable Temperature Kinetic Isotope Effects Demonstrate Extensive Tunnelling in the C–H Activation Reactivity of a Transition Metal-Oxo Complex, Chem. Commun., 2023, 59(55), 8584–8587, 10.1039/D3CC02130K.
- W. T. Borden, Advanced Review Reactions That Involve Tunneling by Carbon and the Role That Calculations Have Played in Their Study, WIREs Comput. Mol. Sci., 2016, 6, 20–46, DOI:10.1002/wcms.1235.
- C. Castro and W. L. Karney, Heavy-Atom Tunneling in Organic Reactions, Angew. Chem., 2020, 132(22), 8431–8442, DOI:10.1002/anie.201914943.
- S. Karmakar and A. Datta, Heavy-Atom Tunneling in Organic Transformations, J. Chem. Sci., 2020, 132(1), 1–22, DOI:10.1007/S12039-020-01809-X/FIGURES/6.
- A. Nandi and J. M. L. Martin, Heavy-Atom Tunneling in the Covalent/Dative Bond Complexation of Cyclo[18]Carbon-Piperidine, J. Phys. Chem. B, 2022, 126(8), 1799–1804, DOI:10.1021/ACS.JPCB.2C00218/ASSET/IMAGES/LARGE/JP2C00218_0003.JPEG.
- H. Inui, K. Sawada, S. Oishi, K. Ushida and R. J. McMahon, Aryl Nitrene Rearrangements: Spectroscopic Observation of a Benzazirine and Its Ring Expansion to a Ketenimine by Heavy-Atom Tunneling, J. Am. Chem. Soc., 2013, 135(28), 10246–10249, DOI:10.1021/JA404172S/SUPPL_FILE/JA404172S_SI_001.PDF.
- J. P. Klinman, A New Model for the Origin of Kinetic Hydrogen Isotope Effects, J. Phys. Org. Chem., 2010, 23(7), 606–612, DOI:10.1002/POC.1661.
- Z. D. Nagel and J. P. Klinman, Tunneling and Dynamics in Enzymatic Hydride Transfer, Chem. Rev., 2006, 106(8), 3095–3118, DOI:10.1021/CR050301X/ASSET/IMAGES/MEDIUM/CR050301XE00008.GIF.
- S. Hu, S. C. Sharma, A. D. Scouras, A. V. Soudackov, C. A. M. Carr, S. Hammes-Schiffer, T. Alber and J. P. Klinman, Extremely Elevated Room-Temperature Kinetic Isotope Effects Quantify the Critical Role of Barrier Width in Enzymatic C−H Activation, J. Am. Chem. Soc, 2014, 136, 42, DOI:10.1021/ja502726s.
- G. Qiu and P. R. Schreiner, The Intrinsic Barrier Width and Its Role in Chemical Reactivity, ACS Cent. Sci., 2023, 9(11), 2129–2137, DOI:10.1021/acscentsci.3c00926.
- M. G. Gutierrez, Z. Theis, T. W. R. Lewis and D. J. Bellert, A Molecular Beam Apparatus for Performing Single Photon Initiated Dissociative Rearrangement Reactions (SPIDRR) with Transition Metal Cation Bound Organic Clusters, Rev. Sci. Instrum., 2018, 89(7) DOI:10.1063/1.5024939.
- Atomic Spectra Database, NIST DOI:10.18434/T4W30F.
- D. Bellert, T. Buthelezi and P. J. Brucat, The Structure of Co + ˙OCO, Chem. Phys. Lett., 1998, 290(4), 316–322, DOI:10.1016/S0009-2614(98)00534-X.
- A. Kamariotis, T. Hayes, D. Bellert and P. J. Brucat, The Ground and Charge-Transfer Excited States of CoO+, Chem. Phys. Lett., 2000, 316(1–2), 60–66, DOI:10.1016/S0009-2614(99)01263-4.
- R. L. Asher, D. Bellert, T. Buthelezi and P. J. Brucat, The Ground State of CoAr, Chem. Phys. Lett., 1994, 227(3), 277–282, DOI:10.1016/0009-2614(94)00828-0.
- T. Buthelezi, D. Bellert, V. Lewis and P. J. Brucat, The Bond Length of CoKr, Chem. Phys. Lett., 1995, 242(6), 627–631, DOI:10.1016/0009-2614(95)00789-7.
- R. L. Asher, D. Bellert, T. Buthelezi and P. J. Brucat, Optical Excitation of Co+.cntdot.N2, J. Phys. Chem., 1995, 99(4), 1068–1072, DOI:10.1021/j100004a002.
- A. Kocak, G. Austein-Miller, W. L. Pearson, G. Altinay and R. B. Metz, Dissociation Energy and Electronic and Vibrational Spectroscopy of Co+(H2O) and Its Isotopomers, J. Phys. Chem. A, 2013, 117(6), 1254–1264, DOI:10.1021/JP305673T.
- V. A. Castleberry, S. J. Dee, O. J. Villarroel, I. E. Laboren, S. E. Frey and D. J. Bellert, The Low-Energy Unimolecular Reaction Rate Constants for the Gas Phase, Ni+-Mediated Dissociation of the C−C σ Bond in Acetone, J. Phys. Chem. A, 2009, 113(39), 10417–10424, DOI:10.1021/jp904561y.
- M. A. Ashraf, J. Kozubal and R. B. Metz, Bond Dissociation Energy and Electronic Spectroscopy of Cr + (NH3) and Its Isotopomers, J. Chem. Phys., 2018, 149(17), 174301, DOI:10.1063/1.5053691.
- T. W. R. Lewis, E. M. Mastin, Z. C. Theis, M. G. Gutierrez and D. J. Bellert, Measurement of Time Dependent Product Branching Ratios Indicates Two-State Reactivity in Metal Mediated Chemical Reactions, Phys. Chem. Chem. Phys., 2022, 24(4), 2300–2308, 10.1039/D1CP05473B.
- T. W. R. Lewis, E. M. Mastin, Z. C. Theis, S. U. Okafor, M. G. Gutierrez and D. J. Bellert, Two State Reactivity (TSR) and Hydrogen Tunneling Reaction Kinetics Measured in the Co+ Mediated Decomposition of CH3CHO, Phys. Chem. Chem. Phys., 2023, 25(35), 23477–23490, 10.1039/D2CP05042K.
- T. W. R. Lewis, E. M. Mastin, Z. C. Theis, M. G. Gutierrez and D. J. Bellert, Measurement of Time Dependent Product Branching Ratios Indicates Two-State Reactivity in Metal Mediated Chemical Reactions, Phys. Chem. Chem. Phys., 2022, 24(4), 2300–2308, 10.1039/D1CP05473B.
- T. Hama, H. Ueta, A. Kouchi and N. Watanabe, Quantum Tunneling Observed without Its Characteristic Large Kinetic Isotope Effects, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(24), 7438–7443 CrossRef CAS.
- K. Johannes, Theory and Simulation of Atom Tunneling in Chemical Reactions, WIREs Comput. Mol. Sci., 2014, 4, 158–168, DOI:10.1002/wcms.1165.
- D. H. Lu, T. N. Truong, V. S. Melissas, G. C. Lynch, Y. P. Liu, B. C. Garrett, R. Steckler, A. D. Isaacson, S. N. Rai, G. C. Hancock, J. G. Lauderdale, T. Joseph and D. G. Truhlar, POLYRATE 4: A New Version of a Computer Program for the Calculation of Chemical Reaction Rates for Polyatomics, Comput. Phys. Commun., 1992, 71(3), 235–262, DOI:10.1016/0010-4655(92)90012-N.
- B. C. Garrett, D. G. Truhlar, A. F. Wagner and T. H. Dunning, Variational Transition State Theory and Tunneling for a Heavy–Light–Heavy Reaction Using an Ab Initio Potential Energy Surface. 37Cl+H(D) 35Cl → H(D) 37Cl+35Cl, J. Chem. Phys., 1983, 78(7), 4400–4413, DOI:10.1063/1.445323.
- B. C. Garrett, N. Abusalbi, D. J. Kouri and D. G. Truhlar, Test of Variational Transition State Theory and the Least-action Approximation for Multidimensional Tunneling Probabilities against Accurate Quantal Rate Constants for a Collinear Reaction Involving Tunneling into an Excited State, J. Chem. Phys., 1985, 83(5), 2252–2258, DOI:10.1063/1.449318.
- J. O. Richardson, Ring-Polymer Instanton Theory, Int. Rev. Phys. Chem., 2018, 37(2), 171–216, DOI:10.1080/0144235X.2018.1472353.
- W. Fang, P. Winter and J. O. Richardson, Microcanonical Tunneling Rates from Density-of-States Instanton Theory, J. Chem. Theory Comput., 2021, 17(1), 40–55, DOI:10.1021/ACS.JCTC.0C01118/ASSET/IMAGES/MEDIUM/CT0C01118_M075.GIF.
- J. O. Richardson, Perspective: Ring-Polymer Instanton Theory, J. Chem. Phys., 2018, 148(20) DOI:10.1063/1.5028352/197026.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02722a |
| This journal is © the Owner Societies 2024 |