
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the photoswitch based on 5-deazaFMN and LOV2 from Avena sativa: a combined absorption and NMR spectroscopy study†
Sabrina
Panter‡
 a,
Jakob
Wörner‡
a,
Jing
Chen
a,
Boris
Illarionov
b,
Adelbert
Bacher
c,
Markus
Fischer
b and
Stefan
Weber
*a
a,
Jakob
Wörner‡
a,
Jing
Chen
a,
Boris
Illarionov
b,
Adelbert
Bacher
c,
Markus
Fischer
b and
Stefan
Weber
*a
aInstitut für Physikalische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: stefan.weber@pc.uni-freiburg.de
bInstitut für Lebensmittelchemie, Universität Hamburg, Grindelallee 117, 20146 Hamburg, Germany
cTUM School of Natural Sciences, Technische Universität München, Lichtenbergstr. 4, 85747 Garching, Germany
First published on 6th November 2024
Abstract
The LOV2 domain from Avena sativa (As) is a blue light receptor that undergoes adduct formation with the native flavin mononucleotide (FMN) cofactor [Salomon et al., Biochemistry, 2000, 39, 9401]. We report the photochemical changes of AsLOV2 through cofactor exchange with the FMN analogue 5-deazaFMN. Absorption spectroscopy shows that upon illumination a thermodynamically stable adduct is formed. We were able to confirm the structure of the adduct by introducing 13C-labelled 5-deazaFMN isotopologues in solution NMR experiments. Dark-adapted state recovery can be photo-induced, providing a photoswitch that is easy to manipulate. The robust photocycle is repeatable without significant loss. Based on the data presented we propose the system as an alternative to wild-type AsLOV2 for applications in optogenetics.
1 Introduction
Chromophores of blue-light sensitive proteins comprise p-coumaric acid utilised in xanthopsins such as the photoactive yellow protein,1 retinal found in channelrhodopsins2 and in sensory rhodopsins,3 as well as various flavin derivatives that are non-covalently bound to BLUF (“blue light using FAD”) domains, cryptochromes and LOV (“light, oxygen, voltage”) domain-based proteins.4–9 The latter proteins are ubiquitous in all kinds of organisms.3,10,11 Following their initial description in phototropin (phot) from Arabidopsis12 they were identified in a number of plant-based (Zeitlupe-family, LOV/LOV protein), bacterial (YtvA, LOV HK, SL2, LovK), fungal (VVD, WC-1) and algal (aureochrome) proteins, see10,13–15 for reviews. Phots are a notable exception among this group of proteins as they contain two LOV domains. LOV domains bind flavin mononucleotide (FMN) as the redox-active cofactor in a globular α/β fold, along with C- and N-terminal helices.11 These proteins are responsible for a variety of physiological functions using blue light as a trigger signal, including phototropism, leaf expansion, gene expression, stomatal opening and chloroplast accumulation.16 In most cases, the ultimate unfolding of the C-terminal helix (Jα helix) after light excitation is responsible for activating the respective effector domain.11The unfolding of the Jα helix is a well-defined structural alteration in LOV domains, such as phot1 LOV2 from Avena sativa (As), which therefore finds frequent application as a photoswitch in optogenetics.9,17 Peptides or proteins fused to the Jα helix of the LOV domain can be controlled by photo-activation.18 This technique provides regulation of biological processes such as cell signalling, gene expression, genome editing, protein stabilisation or localisation.19,20
The reversible photocycle of the wild-type LOV protein involves the adduct formation between the FMN cofactor and a conserved cysteine residue.21,22 The mechanism of this cycle, ultimately leading to structural changes in the terminal helices as mentioned above,23,24 is still not entirely understood. The dark-adapted state protein LOV445 has its absorbance maximum in the blue light regime at 445 nm. Upon light excitation, the fully oxidised FMN cofactor is transferred to an excited singlet state. Subsequent intersystem crossing (ISC) leads to a triplet state with an absorbance maximum at approximately 660 nm.5,11,25 Unlike suggested in earlier works,25,26 recent studies indicate that the adduct formation proceeds without prior protonation of N5 of the FMN by the neighbouring cysteine.27–29 The adduct LOV390 forms within a few seconds25,30via a C4a–S bond, as shown by NMR spectroscopy31 and X-ray structure analysis.22 It is hypothesised that the adduct formation comprises a transient FMNH˙ radical although unambiguous evidence is still lacking.27,32–34 Dark-adapted state recovery is thermodynamically driven and usually completed within seconds to hours depending on the organism.25,35–38 Evidence suggests that the protonation of FMN at N5 leads to a “flip” of the neighbouring glutamine.22,39–43 The induced polarity change is assumed to be transmitted to the N- or C-terminal helix by a change in the hydrogen bond network as shown by molecular dynamics (MD) simulations.40,44,45 In AsLOV2, the light excitation ultimately leads to an unbinding and unfolding of the Jα helix24 which activates the serine/threonine kinase effector domain of phot1.11
Reaction mechanisms of flavoproteins have been extensively studied by replacing the native flavin cofactors, e.g. with a 5-deazaflavin analogue.46–49 5-DeazaFMN is structurally similar to FMN, see Fig. 1, with N replaced by C–H at position 5. Thus, substituting the FMN cofactor with 5-deazaFMN maintains a binding situation comparable to that observed in the wild-type protein. However, this modification significantly affects the photochemical and electrochemical properties of the protein. The transformation of a dehalogenase into a nitrogenase by cofactor exchange serves as an astonishing example.50 Multiple cofactor replacement studies indicate that one-electron transfer reactions are often suppressed in flavoproteins with a 5-deazaflavin cofactor.51,52 For this reason, the 5-deazaflavin radical was believed to be too unstable for detection.46,53–55 Only recently, the indirect detection of the 5-deazaFMN radical was achieved through works conducted in our laboratory56 expanding the reactivity of 5-deazaflavins to single-electron transfers.
 | ||
| Fig. 1 Structure of flavins and 5-deazaflavins: FMN: X = N, R = ribityl-5′-PO3H2; 5-deazaFMN: X = CH, R = ribityl-5′-PO3H2; 5-deazariboflavin: X = CH, R = ribityl. | ||
The generation of a photoswitch by cofactor exchange using 5-deazaFMN in AsLOV2 was initially introduced by our group.57 As shown by UV/vis spectroscopy, the dark-adapted state recovery of the adduct is not thermodynamically driven as in the wild-type protein, resulting in a virtually unlimited stability of the adduct. Regeneration of the dark-adapted state can be induced photochemically. Although the mechanism of dark-adapted state recovery is not entirely understood, there is strong evidence that the abstraction of the proton from N5 is a rate-limiting step.25,36,58,59 The incorporation of 5-deazaFMN has a significant impact on the pKA of the proton at position 5. It is presumably too high for the abstraction by a neighbouring basic group as it is the case in the native protein. This is a possible explanation for why the dark-adapted state recovery is not thermodynamically driven in LOV2 reconstituted with 5-deazaFMN.48
This study was extended to further LOV domains: Aureochrome1a (Au1a) from Ochromonas danica (Od) and YtvA from Bacillus subtilis (Bs) were investigated by Kalvaitis et al.49 and Silva-Junior et al.,48 respectively. Combining optical spectroscopy and quantum mechanics/molecular mechanics (QM/MM) calculations, Silva-Junior et al.48 proposed a mechanism for the photocycle of BsYtvA reconstituted with 5-deazaFMN: The cofactor is photoexcited into the S1 state and proceeds to undergo ISC into the T1 state. A hydrogen is transferred from the close cysteinyl residue Cys62 to C5 of 5-deazaFMN, forming a biradical. Following ISC into the S0 ground state, the final adduct is formed via a C4a–S bond. Bond formation between C5 of 5-deazaFMN and the cysteinyl thiol group is ruled out based on a higher energy barrier. According to these calculations, the photo-induced dark-adapted state recovery is only accessible through a C4a adduct.48 The hypothesis of adduct formation via a C4a–S bond is supported by crystallographic data of the 5-deazaFMN-OdAu1a adduct.49 Comparison of electron density maps of protein crystals grown under dark and illuminated conditions clearly indicates the involvement of C4a of 5-deazaFMN in the adduct bond.
Motivated by these findings, we aim to expand the existing data on AsLOV2 reconstituted with 5-deazaFMN (5-deazaFMN-LOV2). By employing UV/vis spectroscopy, we explore the possibilities of a AsLOV2 photoswitch, supporting the aforementioned study of BsYtvA.48 As wild-type AsLOV2 is an established tool in optogenetics,9,60–63 the protein reconstituted with 5-deazaFMN could provide accessibility to more controllable and customised photoswitches. Moreover, we provide unambiguous evidence for a C4a–S bond formation in the protein based on a solution NMR study with selectively 13C-labelled cofactors in combination with time-dependent density functional theory (TD-DFT) calculations.
2 Experimental
2.1 Sample preparation
2.2 Spectroscopy
NMR experiments with proteins were conducted using an Avance III HD NMR spectrometer (Bruker, Ettlingen, Germany) operating at 14 T/600 MHz and a BBFO broadband probe head optimised for the detection of heteronuclei. All experiments were conducted at 278 K. 13C experiments were recorded using a 30° probe pulse and power-gated decoupling. Distortionless enhancement by polarisation transfer (DEPT) experiments were recorded using a standard pulse sequence with a 135° probe pulse and JC,H = 145 Hz.69 For all experiments a recycle delay of 2 s was chosen. The spectra were referenced using the methine and methylene signals of residual glycerol.70 The samples of AsLOV2 reconstituted with [4a-13C1]-5-deazaFMN (0.62 mM) and [5-13C1]-5-deazaFMN (0.58 mM) were prepared in H2O/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v) containing 50 mM K/Na phosphate, pH 7.0, 0.02% NaN3. For measurements of the adduct state, the samples were irradiated for 7 min with a LED (M405L3, Thorlabs GmbH, Bergkirchen, Germany) at 405 nm with 4.2 mW and transferred into a NMR tube.
:1, v:v) containing 50 mM K/Na phosphate, pH 7.0, 0.02% NaN3. For measurements of the adduct state, the samples were irradiated for 7 min with a LED (M405L3, Thorlabs GmbH, Bergkirchen, Germany) at 405 nm with 4.2 mW and transferred into a NMR tube.
2.3 Determination of extinction coefficient
The extinction coefficient of 5-deazaFMN-LOV2 was determined by unfolding six protein samples of known absorbance through addition of sodium dodecyl sulfate (SDS).71 The samples were cooled to 277 K. The protein sample (300 μL) was added to SDS (13%) dissolved in 50 mM K/Na phosphate, pH 7 (300 μL). After 90 min, the maximum absorbance of the samples containing the free cofactor was determined, see Fig. S1 (ESI†). The maximum absorbances were averaged and compared with the known extinction coefficient of 5-deazaFMN of ε396nm = 12.000 mM−1 cm−1.72 For verification, a sample of wild-type AsLOV2 was used. By comparison with the extinction coefficient of AsLOV2 (13.800 mM−1 cm−1 at 447 nm21), an extinction coefficient of 12.520 mM−1 cm−1 for FMN was found (data not shown) which is in accordance to the literature (ε445nm = 12.500 mM−1 cm−1).732.4 Target analysis of the dark-adapted-state-to-adduct-state conversion
The absorbance spectra recorded following intervals of steady-state illumination were analysed through target analysis. A self-written script utilising the Python-based package pyglotaran74 was employed for the analysis. Species-associated spectra (SAS) were calculated by assuming a mono-exponential decay for the reaction of the dark-adapted state to the adduct state. The spectrum at t = 0 is attributed to the pure dark-adapted state. As the adduct state has a non-zero extinction coefficient at 402 nm, a back reaction to the dark-adapted state at a slower rate is expected. But as an equilibrium is reached under continuous illumination, the reaction back to the dark-adapted state cannot be resolved and was excluded from the model.2.5 Determination of quantum yields
Quantum yields were determined using cuvettes of type 109.004F (Hellma, Müllheim, Germany) with a type SCS1.22 magnetic stirrer (Starna GmbH, Pfungstadt, Germany). For illumination, two LEDs were used: M430L5 (Thorlabs GmbH, Bergkirchen, Germany) operated at 34.0 mW for the illumination at 433 nm and M340L5 (Thorlabs GmbH, Bergkirchen, Germany) operated at 1.4 mW for the illumination at 341 nm as well as an optical fibre (Streppel Glasfaser-Optik, Wermelskirchen, Germany). Quantum yields of adduct formation and dark-adapted state recovery were determined according to a method described by Stadler et al.75 For the adduct formation, the illumination set-up was calibrated to determine the photon flux I0 = 8.17 × 10−5 mol L−1 s−1 at 433 nm and 277 K. For calibration, a solution of K3[Fe(C2O4)3]·3H2O in H2SO4 (0.05 M) with known quantum yield75 was used, see Fig. S2 (ESI†). The solution was bubbled with argon for 5 min prior to measurement. Three samples of 5-deazaFMN-LOV2 in degassed 50 mM K/Na phosphate, pH 7 buffer solution were illuminated. The absorbance changes at the illumination wavelength were fitted with a mono-exponential decay to obtain the decay rate constant kfit, see Fig. S3 (ESI†). The quantum yield Φ was calculated from the averaged decay rate constant, the initial protein concentration c0, the initial absorbance at the illumination wavelength and I0:
and I0: | (1) |
2.6 Computational methods
For theoretical calculations of absorbance spectra of possible adduct structures using TD-DFT, simplified models for adduct 1, 2a and 2b (see Tables S1–S3 and Fig. S6–S8 for optimised structures, ESI†) were used: 7,8,10-trimethyl-4a-(methylthio)-5,10-dihydropyrimido[4,5-b]quinoline-2,4(3H,4aH)-dione, 7,8,10-trimethyl-5-(methylthio)-5,10-dihydropyrimido[4,5-b]quinoline-2,4(1H,3H)-dione and 7,8,10-trimethyl-5-(methylthio)-5,10-dihydropyrimido[4,5-b]quinoline-2,4(3H,4aH)-dione, respectively. 5-Deazalumiflavin was employed as model for 5-deazaFMN. All geometries were optimised with ORCA 4.0.1.276,77 utilising a B3LYP functional78 and a TZVP basis set.79,80 The optimised geometries were used as input structures for TD-DFT calculations as well as calculations of chemical shifts, which were conducted in vacuo utilising a B3LYP functional as well as a def2-TZVP81 and def2/J basis set82 with a RIJCOSX approximation.83 Trimethylsilane (TMS) was employed as a chemical shift reference.3 Results & discussion
3.1 UV/vis characterisation of the photocycle of AsLOV2 reconstituted with 5-deazaFMN
The adduct formation of 5-deazaFMN-LOV2 was monitored by recording absorbance changes under steady-state illumination near its visible absorbance maximum, see Fig. 2. The absorbance maxima at 375 nm, 402 nm and 423 nm decrease with progressing irradiation time while the maximum at 339 nm increases and is gradually shifted to 334 nm. No significant changes in the band shape are visible, apart from a broadening of the fine structure around 334 nm. The gradual disappearance of the long-wavelength absorbance maximum can be attributed to the reduction of the 5-deazaflavin's aromatic system due to adduct formation. After 980 s, 5-deazaFMN-LOV2 has reached a steady state. The adduct state is thermodynamically stable and shows no significant absorbance changes during several days, see Fig. S9 (ESI†). The dark-adapted state recovery can be photo-induced by illuminating the photoadduct near its UV absorbance maximum, see Fig. 3. The spectral changes indicate that the dark-adapted state can be recovered up to approximately 70%. This incomplete adduct cleavage can be attributed to overlapping absorbance spectra of the dark-adapted state and the adduct state of 5-deazaFMN-LOV2. A quantitative comparison of the absorbance maxima is described in the following section. | ||
| Fig. 2 Steady-state illumination of 5-deazaFMN-LOV2 (spectrum of the dark-adapted state in blue). The absorbance changes from blue to orange. The illumination time is indicated for each spectrum. The emission profile of the LED used for illumination is indicated by the light blue area. | ||
 | ||
| Fig. 3 Dark-adapted state recovery by steady-state illumination of the AsLOV2-5-deazaFMN adduct. The absorbance changes from blue to orange. The illumination time is indicated for each spectrum. The emission profile of the LED used for illumination is indicated by the light blue area. | ||
To calculate the SAS of the adduct state, the illumination series shown in Fig. S10 (ESI†) was analysed by target analysis. In order to ensure linearity between concentration and absorbance, a maximal optical density lower than 1.0 was chosen for the initial sample. The spectrum before illumination was attributed to 5-deazaFMN-LOV2 in its pure dark-adapted state. The subsequent spectra of the illumination series are linear combinations of absorbance spectra of the dark-adapted and adduct state of the protein. Consequently, the SAS of the adduct state acquired by target analysis corresponds to its pure absorbance spectrum, see Fig. 4. The absorbance at 402 nm is significantly decreased while the absorbance at 334 nm is slightly increased compared to the dark-adapted state.
 | ||
| Fig. 4 Absorbance spectra of the dark-adapted (blue) and adduct state (orange) of 5-deazaFMN-LOV2 determined by target analysis as well as the experimentally acquired spectrum of the dark-adapted state (dashed line). | ||
The extinction coefficient ε of 5-deazaFMN-LOV2 in its dark-adapted state was determined experimentally by unfolding a protein sample and comparing the absorbance maxima of the protein with that of the free cofactor with known extinction coefficient.72 Comparison of the SAS in Fig. 4 allows the determination of extinction coefficients of both the dark-adapted and adduct states, see Table 1. The extinction coefficient at 334 nm of the adduct state is slightly higher than that of the dark-adapted state leading to simultaneous activation of adduct state and dark-adapted state formation by illumination at this wavelength. Consequently, it is not possible to achieve complete regeneration of the dark-adapted state following adduct formation.
| ε max/(mM cm)−1 (λ/nm) | ||
|---|---|---|
| Dark-adapted state | 14.4 ± 0.7 (339) | 15.3 ± 0.7 (402) |
| Adduct state | 17.3 ± 0.8 (334) | 0.40 ± 0.02 (402) |
To determine the applicability of the 5-deazaFMN-LOV2 system as a potential photoswitch, the repeatability of consecutive adduct state formations and dark-adapted state recoveries was investigated using UV/vis spectroscopy. Fig. 5 illustrates three illumination cycles during which the absorbance of 5-deazaFMN-LOV2 at 405 nm and 340 nm were recorded. Initially, the pure dark-adapted state is converted to the adduct state with approximately 90% conversion. The illumination of the adduct state leads to an equilibrium of both states, as explained above, with approximately 70% dark-adapted state regeneration. During the following illumination cycles, the sample oscillates between both states. No signs of sample degradation induced by illumination can be detected, thereby proving the robustness of the photocycle. This is in accordance with findings using OdAu1a, which showed a similar repeatability.49
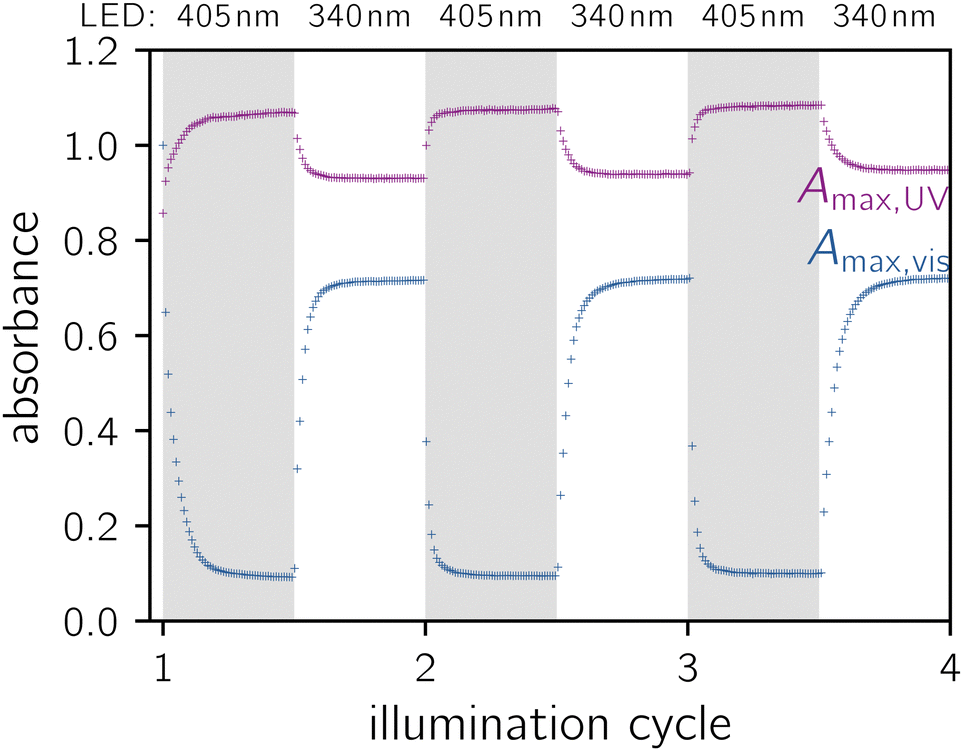 | ||
| Fig. 5 Absorbance of 5-deazaFMN-LOV2 at the visible light (402 nm) and the UV absorbance maximum. As the UV absorbance maximum shifts between 334 nm and 339 nm depending on the state of the protein, the maximal absorbance regardless of wavelength is depicted. Each illumination cycle comprises 100 absorbance measurements. Between each measurement, the sample was illuminated for 20 s by the LED indicated at the top of the graph. | ||
Furthermore, the quantum yield of the adduct formation of 5-deazaFMN-LOV2 was determined. By averaging the decay rate constant of three illumination series of 5-deazaFMN-LOV2, a quantum yield of 0.09 was determined, see eqn (1) and Fig. S3 (ESI†). For BsYtvA a similar value of 0.03–0.05 was found.48 This result is lower than the quantum yield of AsLOV2 harboring FMN (Φ = 0.44)21 which indicates that the native cofactor is more effective in inducing the adduct formation. For completeness, the dark-adapted state recovery was also analysed, although this method only provides a lower limit of the quantum yield due to the simultaneous activation of dark-adapted state recovery and adduct formation at 340 nm. By averaging the decay rate constants of two samples of the 5-deazaFMN-LOV2 adduct, a quantum yield of 0.35 was determined, see Fig. S5 (ESI†).
3.2 Characterisation of the adduct formation using NMR and TD-DFT
The exchange of N by C–H at position 5 in 5-deazaFMN allows for different bond formations between the cofactor and the cysteinyl residue upon adduct formation. Similar to the wild-type protein carrying FMN, C5 can be protonated and a bond between C4a and the thiol group of cysteine can be formed, see adduct 1 in Fig. 6. It is also conceivable that C5 forms a bond with the thiol group. In this instance, N1 and C4a are potential protonation sites, corresponding to adducts 2a and 2b, respectively. | ||
| Fig. 6 Proposed adduct formations of 5-deazaFMN and Cys450 in AsLOV2 with R1 = ribityl-5′-PO3H2 and R2 = Cys450. The bond formation between the cysteinyl thiol group and C4a (adduct 1) or C5 (adducts 2a and 2b) of 5-deazaFMN is considered. | ||
A first approach to solving the adduct structure is given by comparing the absorbance spectrum of the 5-deazaFMN-LOV2 adduct to predictions from TD-DFT for the proposed chemical structures shown in Fig. 6. In order to achieve this objective, 5-deazalumiflavin was employed as a simplified reference model for the 5-deazaFMN cofactor. TD-DFT calculations in vacuo yielded the absorbance spectra depicted in Fig. 7 that are compared to the experimental absorbance spectra of 5-deazaFMN-LOV2 before illumination and in its adduct state after illumination.
 | ||
| Fig. 7 Normalised absorbance spectra calculated using TD-DFT for models of adduct 1 (orange), adduct 2a (yellow) and adduct 2b (red) of 5-deazaFMN-LOV2 compared to a model of 5-deazalumiflavin (reference, blue). The experimental absorbance spectrum of 5-deazaFMN-LOV2 before and after 980 s of illumination at 403 nm is shown as a light blue and light orange area, respectively. | ||
A comparison of the experimental spectrum of the dark-adapted state with the calculated spectrum of 5-deazalumiflavin reveals a difference of absorbance maxima of 25 nm. We assume that this shift is mainly due to solvent effects that are not taken into account by the TD-DFT calculation in vacuo. The absorbance maxima of adducts 2a and 2b coincide at 293–294 nm, whereas the absorbance maximum of adduct 1 is shifted to 317 nm. These results clearly distinguish the different bond models, C4a–S and C5–S, from one another. The experimental absorbance maximum of 334 nm is in better agreement with the adduct model of a C4a–S bond. The shift between experimental and calculated spectrum amounts to 17 nm, which is comparable to the shift of the reference. This comparison suggests that adduct 1 corresponds to the structure of the 5-deazaFMN-LOV2 adduct.
To gather unambiguous experimental evidence, the 13C NMR shift changes of 5-deazaFMN-LOV2 after illumination were combined with the signal patterns of DEPT135 experiments. LOV2 was reconstituted with two 5-deazaFMN isotopologues 13C-labelled either at position 4a or position 5. Fig. 8 and 9 illustrate spectra of the dark-adapted state (orange), the adduct state (blue) and the adduct state using a DEPT135 pulse sequence (green) for [4a-13C1]-5-deazaFMN-LOV2 and [5-13C1]-5-deazaFMN-LOV2, respectively. Table 2 presents a summary of the experimental and calculated chemical shift values. In the spectra of the dark-adapted state, chemical shifts for C4a and C5 of 111.12 ppm and 143.61 ppm are observed. The signals were assigned according to the chemical shifts obtained from the respective cofactor in aqueous solution, 112.04 ppm and 142.93 ppm for C4a and C5, respectively, see Fig. S11 (ESI†). DFT calculations predict values of 121.8 ppm and 145.3 ppm for these positions in the simplified model of 5-deazalumiflavin, which corresponds reasonably well to the experimental data. Differences in chemical shifts, mainly for C4a, can be attributed to the DFT calculations being performed in vacuo.
 | ||
| Fig. 8
13C NMR spectra of LOV2 reconstituted with [4a-13C1]-5-deazaFMN: before illumination (orange, 71680 scans), after illumination (blue, 61440 scans) and after illumination using the DEPT135 sequence (green, 49152 scans). Signals of residual glycerol are denoted with asterisks. | ||
 | ||
| Fig. 9
13C NMR spectra of LOV2 reconstituted with [5-13C1]-5-deazaFMN: before illumination (orange, 71680 scans), after illumination (blue, 40960 scans) and after illumination using the DEPT135 sequence (green, 40960 scans). Signals of residual glycerol are denoted with asterisks. | ||
| Model/sample | δ(C4a)/ppm | δ(C5)/ppm |
|---|---|---|
| Dark-adapted state (exp.) | 111.12 | 143.61 |
| 5-DeazaFMN in solution (exp.) | 112.04 | 142.93 |
| Dark-adapted state (DFT) | 121.8 | 145.3 |
| Adduct state (exp.) | 44.82 | 33.70 |
| Adduct 1 (DFT) | 51.8 | 31.5 |
| Adduct 2a (DFT) | 93.1 | 48.5 |
| Adduct 2b (DFT) | 50.2 | 53.3 |
Following illumination, the signals of both C4a and C5 are observed to shift to 44.82 ppm and 33.70 ppm, respectively. The chemical shift of C4a is found to be consistent with the predictions for adduct 1 (51.8 ppm) and 2b (50.2 ppm) but in disagreement with the predicted value for adduct 2a (93.1 ppm). The chemical shift of C5 indicates the formation of adduct 1 (31.5 ppm) which shows better correspondence than adduct 2a (48.5 ppm) and adduct 2b (53.3 ppm). These results are confirmed by the signal phases of C4a and C5 obtained from DEPT135 experiments which differentiate methyl (CH3), methylene (CH2) and methine (CH) resonances. For CH and CH3, absorptive resonances are expected, while for CH2, the resonance is emissive. Quaternary carbons do not show any resonance in DEPT135 spectra.69,84 The DEPT135 spectra in Fig. 8 and 9 show an emissive signal for C5 and no signal for C4a. We can conclude that C4a is a quaternary carbon and C5 a methylene group in the adduct state. These conditions can only be met by adduct 1, where C4a is a quaternary carbon by forming the sulfide bond and C5 is additionally protonated. These findings prove unambiguously that the adduct state of 5-deazaFMN-LOV2 is formed via a C4a–S bond. To the best of our knowledge, this has not yet been demonstrated experimentally under physiological conditions.
This result is in accordance with findings for OdAu1a and BsYtvA reconstituted with 5-deazaFMN,48,49 which provides strong evidence that LOV domains from different organisms share similar behaviour towards the 5-deazaFMN cofactor. Extending cofactor replacement studies to more LOV domains are required to prove this hypothesis. This could result in the identification of a novel subgroup of photoswitches that are easier to manipulate between dark-adapted and adduct states than the respective wild-type LOV domains. It would be advantageous to achieve a higher conversion between the adduct state and reformation of the dark-adapted state. For this purpose, a minimal overlap of absorption between dark-adapted and adduct states in the UV and visible region is essential. It is possible that 5-deazaFMN cofactors that are shifted bathochromically could provide this feature to a certain degree.
Incorporating artificial cofactors by refolding of the protein may not be a successful approach for optogenetic experiments or other applications. An in vivo synthesis of the artificial cofactor bound to the LOV domain would be desirable. The expression of LOV1 from Chlamydomonas reinhardtii reconstituted with roseoflavin in E. coli was successfully demonstrated by Mathes et al.85 The E. coli strain was modified to express a riboflavin transporter to take up the riboflavin analogue from the medium. Additionally, a riboflavin biosynthetic gene was removed from the strain to completely prevent this pathway. This procedure could be modified by using commercially available 5-deazariboflavin to synthesise the 5-deazaFMN-LOV2 system in vivo. Furthermore, the elucidation of the biosynthetic pathway of the natural 5-deazaflavin cofactor F420 enabled the high-yield production of this cofactor in E. coli.86,87 Although not readily applicable, these advances may ultimately lead to the biosynthetic expression of LOV proteins reconstituted with natural 5-deazaflavin cofactors.
When used as an optogenetic tool, the effector domain is fused to the Jα helix in AsLOV2. For the presented system to be applicable, the unfolding of this helix must remain functional after cofactor exchange. The cofactor exchange in OdAu1a blocks the protonation of N5 of the cofactor which is thought to be an important step in the signalling pathway. Ultimately, the light-induced dimerisation is prohibited in 5-deazaFMN-OdAu1a.49 These results raise the question of whether a similar effect of cofactor exchange can be found in AsLOV2 which would reduce the applicability of the system. Further experiments are needed to clarify the consequences of reconstituting AsLOV2 with 5-deazaFMN.
4 Conclusions
This comprehensive study, which combines optical and NMR spectroscopy, investigates the adduct formation of AsLOV2 reconstituted with 5-deazaFMN providing mechanistic detail. The NMR study, combined with DFT calculations, provides clear evidence of photo-induced bond formation between C4a of 5-deazaFMN and the cysteinyl thiol group. To our knowledge, this is the first study to use DEPT pulse sequences to elucidate the structure of protein adducts. The absorption properties of the resulting 5-deazaFMN-LOV2 adduct are compared with those of its dark-adapted state. Furthermore, we demonstrate the robustness of the cycle between the dark-adapted state and the adduct state, which is an important prerequisite for the applicability of the system. We therefore propose this photoswitch system as a promising alternative to the wild-type AsLOV2.Author contributions
Conceptualisation: J. W., S. P., A. B., M. F. and S. W.; data curation: S. P. and J. W.; formal analysis: S. P., J. W. and J. C.; funding acquisition: M. F. and S. W.; investigation: S. P., J. W. and J. C.; methodology: S. P., J. W. and S. W.; project administration: M. F. and S. W.; resources: S. P., J. W., J. C., B. I., A. B., M. F., S. W.; software: S. P.; supervision: A. B., M. F. and S. W.; validation: S. P., J. W. and J. C.; visualisation: S. P.; writing – original draft: S. P., B. I. and S. W.; writing – review & editing: J. W., B. I., A. B. and M. F.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Lars Kuhn for providing technical support and for helpful discussions. SW thanks the SIBW/DFG for financing NMR instrumentation that is operated within the MagRes Center of the Albert-Ludwigs-Universität Freiburg (Germany). SW and MF acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) (project number 459493567: WE 2376/12-1 and FI824/13-1).Notes and references
- T. E. Meyer, Biochim. Biophys. Acta, Bioenerg., 1985, 806, 175–183 CrossRef CAS PubMed.
- E. B. Purcell and S. Crosson, Curr. Opin. Microbiol., 2008, 11, 168–178 CrossRef CAS PubMed.
- P. Hegemann, Annu. Rev. Plant Biol., 2008, 59, 167–189 CrossRef CAS PubMed.
- T. Kottke, A. Xie, D. S. Larsen and W. D. Hoff, Annu. Rev. Biophys., 2018, 47, 291–313 CrossRef CAS PubMed.
- A. Losi and W. Gärtner, Photochem. Photobiol., 2017, 93, 141–158 CrossRef CAS PubMed.
- S.-Y. Park and J. R. H. Tame, Biophys. Rev., 2017, 9, 169–176 CrossRef CAS PubMed.
- S. Masuda, Plant Cell Physiol., 2013, 54, 171–179 CrossRef CAS PubMed.
- I. Chaves, R. Pokorny, M. Byrdin, N. Hoang, T. Ritz, K. Brettel, L.-O. Essen, G. T. J. van der Horst, A. Batschauer and M. Ahmad, Annu. Rev. Plant Biol., 2011, 62, 335–364 CrossRef CAS PubMed.
- A. Losi, K. H. Gardner and A. Möglich, Chem. Rev., 2018, 118, 10659–10709 CrossRef CAS PubMed.
- J. Herrou and S. Crosson, Nat. Rev. Microbiol., 2011, 9, 713–723 CrossRef CAS PubMed.
- A. Flores-Ibarra, R. N. A. Maia, B. Olasz, J. R. Church, G. Gotthard, I. Schapiro, J. Heberle and P. Nogly, J. Mol. Biol., 2024, 436, 168356 CrossRef CAS PubMed.
- E. Huala, P. W. Oeller, E. Liscum, I. S. Han, E. Larsen and W. R. Briggs, Science, 1997, 278, 2120–2123 CrossRef CAS PubMed.
- N. Suetsugu and M. Wada, Plant Cell Physiol., 2013, 54, 8–23 CrossRef CAS PubMed.
- J. M. Christie, L. Blackwood, J. Petersen and S. Sullivan, Plant Cell Physiol., 2015, 56, 401–413 CrossRef CAS PubMed.
- J. M. Christie and M. D. Zurbriggen, New Phytol., 2021, 229, 3108–3115 CrossRef PubMed.
- A. Losi and W. Gärtner, Annu. Rev. Plant Biol., 2012, 63, 49–72 CrossRef CAS PubMed.
- M. D. Hoffmann, F. Bubeck, R. Eils and D. Niopek, Adv. Biosyst., 2018, 2, 1800098 CrossRef.
- A. C. McCue and B. Kuhlman, Curr. Opin. Struct. Biol., 2022, 74, 102377 CrossRef CAS PubMed.
- T. Iwata and S. Masuda, in Optogenetics. Light-Sensing Proteins and Their Applications in Neuroscience and Beyond, ed. H. Yawo, H. Kandori, A. Koizumi and R. Kageyama, Springer, Singapore, 2nd edn, 2021, ch. 11, pp. 189–206 Search PubMed.
- X. Li, C. Zhang, X. Xu, J. Miao, J. Yao, R. Liu, Y. Zhao, X. Chen and Y. Yang, Nucleic Acids Res., 2020, 48, e33 CrossRef CAS PubMed.
- M. Salomon, J. M. Christie, E. Knieb, U. Lempert and W. R. Briggs, Biochemistry, 2000, 39, 9401–9410 CrossRef CAS PubMed.
- S. Crosson and K. Moffat, Plant Cell, 2002, 14, 1067–1075 CrossRef CAS PubMed.
- J. P. Zayner, C. Antoniou and T. R. Sosnick, J. Mol. Biol., 2012, 419, 61–74 CrossRef CAS PubMed.
- S. M. Harper, L. C. Neil and K. H. Gardner, Science, 2003, 301, 1541–1544 CrossRef CAS PubMed.
- T. E. Swartz, S. B. Corchnoy, J. M. Christie, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, J. Biol. Chem., 2001, 276, 36493–36500 CrossRef CAS PubMed.
- J. T. M. Kennis, S. Crosson, M. Gauden, I. H. M. van Stokkum, K. Moffat and R. van Grondelle, Biochemistry, 2003, 42, 3385–3392 CrossRef CAS PubMed.
- C. W. M. Kay, E. Schleicher, A. Kuppig, H. Hofner, W. Rüdiger, M. Schleicher, M. Fischer, A. Bacher, S. Weber and G. Richter, J. Biol. Chem., 2003, 278, 10973–10982 CrossRef CAS PubMed.
- E. Schleicher, R. M. Kowalczyk, C. W. M. Kay, P. Hegemann, A. Bacher, M. Fischer, R. Bittl, G. Richter and S. Weber, J. Am. Chem. Soc., 2004, 126, 11067–11076 CrossRef CAS PubMed.
- C. Thöing, A. Pfeifer, S. Kakorin and T. Kottke, Phys. Chem. Chem. Phys., 2013, 15, 5916–5926 RSC.
- W. Holzer, A. Penzkofer, M. Fuhrmann and P. Hegemann, Photochem. Photobiol., 2002, 75, 479–487 CrossRef CAS PubMed.
- M. Salomon, W. Eisenreich, H. Dürr, E. Schleicher, E. Knieb, V. Massey, W. Rüdiger, F. Müller, A. Bacher and G. Richter, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12357–12361 CrossRef CAS PubMed.
- C. Neiß and P. Saalfrank, Photochem. Photobiol., 2003, 77, 101–109 CrossRef.
- C. Bauer, C.-R. Rabl, J. Heberle and T. Kottke, Photochem. Photobiol., 2011, 87, 548–553 CrossRef CAS PubMed.
- R. J. Kutta, K. Magerl, U. Kensy and B. Dick, Photochem. Photobiol. Sci., 2015, 14, 288–299 CrossRef CAS PubMed.
- M. Kasahara, T. E. Swartz, M. A. Olney, A. Onodera, N. Mochizuki, H. Fukuzawa, E. Asamizu, S. Tabata, H. Kanegae, M. Takano, J. M. Christie, A. Nagatani and W. R. Briggs, Plant Physiol., 2002, 129, 762–773 CrossRef CAS PubMed.
- B. D. Zoltowski, B. Vaccaro and B. R. Crane, Nat. Chem. Biol., 2009, 5, 827–834 CrossRef CAS PubMed.
- S. Raffelberg, M. Mansurova, W. Gärtner and A. Losi, J. Am. Chem. Soc., 2011, 133, 5346–5356 CrossRef CAS PubMed.
- H. Guo, T. Kottke, P. Hegemann and B. Dick, Biophys. J., 2005, 89, 402–412 CrossRef CAS PubMed.
- D. Nozaki, T. Iwata, T. Ishikawa, T. Todo, S. Tokutomi and H. Kandori, Biochemistry, 2004, 43, 8373–8379 CrossRef CAS PubMed.
- A. Möglich and K. Moffat, J. Mol. Biol., 2007, 373, 112–126 CrossRef PubMed.
- B. D. Zoltowski, C. Schwerdtfeger, J. Widom, J. J. Loros, A. M. Bilwes, J. C. Dunlap and B. R. Crane, Science, 2007, 316, 1054–1057 CrossRef CAS PubMed.
- A. T. Vaidya, C.-H. Chen, J. C. Dunlap, J. J. Loros and B. R. Crane, Sci. Signaling, 2011, 4, ra50 CrossRef CAS PubMed.
- S. Endres, J. Granzin, F. Circolone, A. Stadler, U. Krauss, T. Drepper, V. Svensson, E. Knieps-Grünhagen, A. Wirtz, A. Cousin, P. Tielen, D. Willbold, K.-E. Jaeger and R. Batra-Safferling, BMC Microbiol., 2015, 15, 30 CrossRef PubMed.
- E. Peter, B. Dick and S. A. Baeurle, Proteins: Struct., Funct., Bioinf., 2012, 80, 471–481 CrossRef CAS PubMed.
- A. Ganguly, W. Thiel and B. R. Crane, J. Am. Chem. Soc., 2017, 139, 2972–2980 CrossRef CAS PubMed.
- P. Hemmerich and V. Massey, FEBS Lett., 1977, 84, 5–21 CrossRef CAS PubMed.
- J. Fisher, R. Spencer and C. Walsh, Biochemistry, 1976, 15, 1054–1064 CrossRef CAS PubMed.
- M. R. Silva-Junior, M. Mansurova, W. Gärtner and W. Thiel, ChemBioChem, 2013, 14, 1648–1661 CrossRef CAS PubMed.
- M. E. Kalvaitis, L. A. Johnson, R. J. Mart, P. Rizkallah and R. K. Allemann, Biochemistry, 2019, 58, 2608–2616 CrossRef CAS PubMed.
- Q. Su, P. A. Boucher and S. E. Rokita, Angew. Chem., Int. Ed., 2017, 56, 10862–10866 CrossRef CAS PubMed.
- C. T. Walsh, Annu. Rev. Biochem., 2017, 86, 1–19 CrossRef CAS PubMed.
- M. V. Shah, J. Antoney, S. W. Kang, A. C. Warden, C. J. Hartley, H. Nazem-Bokaee, C. J. Jackson and C. Scott, Catalysts, 2019, 9, 868 CrossRef CAS.
- H. J. Duchstein, H. Fenner, P. Hemmerich and W. R. Knappe, Eur. J. Biochem., 1979, 95, 167–181 CrossRef CAS PubMed.
- M. Goldberg, I. Pecht, H. E. A. Kramer, R. Traber and P. Hemmerich, Biochim. Biophys. Acta, Gen. Subj., 1981, 673, 570–593 CrossRef CAS PubMed.
- P. F. Heelis, B. J. Parsons, G. O. Phillips and A. J. Swallow, Int. J. Radiat. Biol., 1989, 55, 557–562 CrossRef CAS PubMed.
- J. Wörner, S. Panter, B. Illarionov, A. Bacher, M. Fischer and S. Weber, Angew. Chem., Int. Ed., 2023, 62, e202309334 CrossRef PubMed.
- S. Hecht, G. Richter, A. Bacher, M. Joshi, W. Röhmisch, G. Greiner, R. Frank, S. Weber, W. Eisenreich and M. Fischer, in Flavins and Flavoproteins 2005, ed. T. Nishino, R. Miura, M. Tanokura and K. Fukui, ARchiTect Inc., Tokyo, Japan, 1st edn, 2005, pp. 569–574 Search PubMed.
- T. Kottke, B. Dick, R. Fedorov, I. Schlichting, R. Deutzmann and P. Hegemann, Biochemistry, 2003, 42, 9854–9862 CrossRef CAS PubMed.
- S. B. Corchnoy, T. E. Swartz, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, J. Biol. Chem., 2003, 278, 724–731 CrossRef CAS PubMed.
- Y. I. Wu, D. Frey, O. I. Lungu, A. Jaehrig, I. Schlichting, B. Kuhlman and K. M. Hahn, Nature, 2009, 461, 104–108 CrossRef CAS PubMed.
- M. Yazawa, A. M. Sadaghiani, B. Hsueh and R. E. Dolmetsch, Nat. Biotechnol., 2009, 27, 941–945 CrossRef CAS PubMed.
- A. Möglich, R. A. Ayers and K. Moffat, J. Mol. Biol., 2009, 385, 1433–1444 CrossRef PubMed.
- D. Strickland, X. Yao, G. Gawlak, M. K. Rosen, K. H. Gardner and T. R. Sosnick, Nat. Methods, 2010, 7, 623–626 CrossRef CAS PubMed.
- D. E. O'Brien, L. T. Weinstock and C. C. Cheng, J. Hetercycl. Chem., 1970, 7, 99–105 CrossRef.
- K. Sanada, H. Ube and M. Shionoya, J. Am. Chem. Soc., 2016, 138, 2945–2948 CrossRef CAS PubMed.
- S. S. Neti and C. D. Poulter, J. Org. Chem., 2016, 81, 5087–5092 CrossRef CAS PubMed.
- E. E. Carlson and L. L. Kiessling, J. Org. Chem., 2004, 69, 2614–2617 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- D. Doddrell, D. Pegg and M. Bendall, J. Magn. Reson., 1982, 48, 323–327 CAS.
- J. Lu, P. Wang, Q. Wang, Y. Wang and M. Jiang, Molecules, 2018, 23, 1177 CrossRef PubMed.
- M. C. McKean, J. D. Beckmann and F. E. Frerman, J. Biol. Chem., 1983, 258, 1866–1870 CrossRef CAS PubMed.
- R. Spencer, J. Fisher and C. Walsh, Biochemistry, 1976, 15, 1043–1053 CrossRef CAS PubMed.
- J. Kozioł, Methods Enzymol., 1971, 18, 253–285 Search PubMed.
- I. H. M. van Stokkum, J. Weißenborn, S. Weigand and J. J. Snellenburg, Photochem. Photobiol. Sci., 2023, 22, 2413–2431 CrossRef CAS PubMed.
- E. Stadler, A. Eibel, D. Fast, H. Freißmuth, C. Holly, M. Wiech, N. Moszner and G. Gescheidt, Photochem. Photobiol. Sci., 2018, 17, 660–669 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- M. R. Bendall, D. M. Doddrell and D. T. Pegg, J. Am. Chem. Soc., 1981, 103, 4603–4605 CrossRef CAS.
- T. Mathes, C. Vogl, J. Stolz and P. Hegemann, J. Mol. Biol., 2009, 385, 1511–1518 CrossRef CAS PubMed.
- G. Bashiri, J. Antoney, E. N. M. Jirgis, M. V. Shah, B. Ney, J. Copp, S. M. Stuteley, S. Sreebhavan, B. Palmer, M. Middleditch, N. Tokuriki, C. Greening, C. Scott, E. N. Baker and C. J. Jackson, Nat. Commun., 2019, 10, 1558 CrossRef PubMed.
- M. V. Shah, H. Nazem-Bokaee, J. Antoney, S. W. Kang, C. J. Jackson and C. Scott, Sci. Rep., 2021, 11, 21774 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02714k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |