
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of heavy atoms on the deactivation of electronic excited states of dye molecules near the surface of metal nanoparticles†
N.
Ibrayev
 *a,
E.
Seliverstova
a,
R.
Valiev
b,
A.
Aymagambetova
a and
D.
Sundholm
b
*a,
E.
Seliverstova
a,
R.
Valiev
b,
A.
Aymagambetova
a and
D.
Sundholm
b
aInstitute of Molecular Nanophotonics, Karaganda Buketov University, Karaganda 100024, Kazakhstan. E-mail: niazibrayev54@gmail.com
bDepartment of Chemistry, Faculty of Science, University of Helsinki, Helsinki FI-00014, Finland. E-mail: valievrashid@gmail.com
First published on 7th October 2024
Abstract
The influence of a heavy atom and the plasmon field on the efficiency of populating the lowest triplet state (T1) and on the phosphorescence intensity has been studied for fluorescein, 2Br-fluorescein, eosin and erythrosine, which have an increasing number of substituted heavy atoms. We show that the heavy atoms affect not only the rate constant of intersystem crossing (kISC) but also the rate constant of internal conversion (kIC). The calculations show that the C–H bonds in the meso position are the primary acceptors of the excitation energy of the lowest excited electronic singlet state (S1). Substitution of the meso hydrogen atoms with I or Br leads to a smaller kIC rate constant of 1 × 108 s−1 for fluorescein to 8 × 106 s−1 for eosin. Substitution with heavy atoms also leads to a larger ISC rate constant (kISC) between the T2 and S1 states because the spin–orbit coupling matrix element 〈S1|HSO|T2〉 increases by two orders of magnitude from 0.36 cm−1 for fluorescein to 35.0 cm−1 for erythrosine. The phosphorescence rate constant increases by three orders of magnitude from 4.8 × 10 s−1 for fluorescein to 3.3 × 104 s−1 for erythrosine, which is supported by experimental data. The plasmon effect increases the intensity of the xanthene dye emissions. The intensity and the quantum yield of fluorescence increase in the series fluorescein < 2Br-fluorescein < eosin < erythrosine. The intensity of the delayed fluorescence and phosphorescence grows in the same way. The enhancement factor of the phosphorescence intensity increases from 1.8 to 5.6 in the series from fluorescein to erythrosine. The differences in the plasmon effect originate from intensity borrowing to the radiative triplet–singlet transition (T1 → S0) from the singlet–singlet transitions (Sn → S0), which is more efficient when molecules have heavy atoms in the meso position.
1. Introduction
Triplet states of organic dyes play a decisive role in many photophysical and photochemical processes. The population of the triplet states depends on the size of the spin–orbit coupling (SOC), which can be enhanced by substituting heavy atoms to the molecule.1 Thus, introducing a heavy atom into a luminophore molecule increases the rate constant of intersystem crossing (kISC) that is responsible for populating triplet states from the singlet manifold. The intersystem crossing (ISC) rate constant between singlet and triplet states is proportional to the square of the SOC matrix element.2 The presence of a heavy atom leads to an increased phosphorescence intensity.Due to the sensitivity of the phosphorescence to temperature, molecular aggregation, and quenching by molecular oxygen, it is also utilized in other contexts. Due to the long phosphorescence lifetime, it is also used in biomedical applications and bioimaging.3,4 Phosphorescence plays a crucial role in studies of the electronic structure of organic molecules.2,5 Intense phosphorescence emission is necessary when constructing materials for chemiluminescent and luminescent sensors, OLED and PHOLED systems, up-conversion devices, and anti-counterfeiting and data-encryption materials.6–9
A modern method to enhance phosphorescence is to utilize the plasmon effect of metal nanoparticles (NPs). The impact of the plasmon effect of metal NPs on the phosphorescence of organic dyes was considered in ref. 10–16. We recently developed a quantum chemical method for calculating the phosphorescence quantum yield of molecules in the vicinity of plasmonic NPs.17 The calculations showed that the maximum quantum yield of phosphorescence is obtained when the molecule is 4–6 nm from the NP surface.17
Heavy atoms and the plasmon effect of metal NPs enhance the phosphorescence efficiency, which is observed as a decrease in the quantum yield of fluorescence, accompanied by an increase in the phosphorescence intensity. We investigate here the two effects separately and together for xanthene molecules by systematically increasing the number of heavy atoms. The studied molecules are fluorescein, 2Br-fluorescein, 4Br-fluorescein (eosin Y, 2′,4′,5′,7′-tetrabromofluorescein), and 4I-fluorescein (2′,4′,5′,7′-tetraiodofluorescein, erythrosine B), which are often used for labeling and probing biomolecules in biomedical research.18–21 Fluorescein and its derivatives have large absorption cross-sections, high fluorescence quantum yield, and the ability to attach to biomolecules, especially to amide groups.22 Since their lifetime and quantum yield of phosphorescence are sensitive to the surrounding medium, they can be used as detectors or media sensors in microenvironmental studies.23,24 The phosphorescence of erythrosine and the rose bengal dye is often used for determining the oxygen content in samples.25 The lowest excited triplet state of the xanthene dyes is used in photosensor reactions for generating reactive singlet oxygen species.26–28
Internal conversion (IC) between the S1 and S0 states competes with the ISC from S1 to Tn and affecting thereby the population of the lowest excited states. The IC rate constant (kIC) can be made smaller by replacing C–H bonds with C–D or C–X, where X is a heavier atom.29,30 The competition between the IC and ISC processes of the studied molecules has not been previously examined. In this work, we have also performed quantum chemical calculations of kIC for the xanthene dyes and studied how it changes when substituting fluorescein with heavy atoms.
2. Experimental and computational details
2.1. Materials and methods
The molecular structures of the studied xanthene dyes are shown in Fig. 1. They were purchased from Sigma Aldrich. Fluorescein was of BioReagent HPLC grade, while the other xanthene dyes had a purity of at least 95%. | ||
| Fig. 1 The molecular structures of the studied xanthene dyes. | ||
Silver island films (SIFs) were synthesized using silver nitrate (AgNO3), sodium hydroxide (NaOH), ammonium hydroxide (NH4OH), D-glucose, and polyvinyl alcohol (PVA) for the polymer films. All reagents were of analytical grade purity from Sigma Aldrich. Ultrapure water obtained from the Smart S15 UVF system (Drawell) was used in the sample preparations.
Films of the dyes in PVA were spin-coated at 3000 rpm either on the SIFs or on clean glass substrates, which were used as reference samples. The dye concentration in the 5 wt% polymer was 5 × 10−4 mol L−1. Microweighing was used to control the thickness of the films on the different substrates. The mass of the dye films on the glass surfaces and on the SIFs was almost identical.
The silver island films were prepared as described in ref. 31 and 32. The synthesized SIFs consisted of particles with an average diameter of 95 ± 31 nm,17,31 which was determined by scanning electron microscopy using a Mira 3LMU (Tescan). The absorption spectrum of the SIFs exhibits a broad band in the visible range of the spectrum.
2.2. Spectroscopical methods
The absorption and fluorescence spectra were measured using a Cary-300 and an Eclipse spectrometer (Agilent), respectively. Fluorescence decay kinetics were recorded in time-correlated single photon counting (TCSPC) mode using a TCSPC system (Becker&Hickl) with excitations by a diode laser at λgen = 488 nm and τ = 120 ps (Becker&Hickl). The lifetimes were estimated using the SPC Image software (Becker&Hickl). The quality of the fit was assessed by the χ2 test.Delayed fluorescence (DF) and phosphorescence spectra were measured after the Xe lamp flash (300 μs) using the Eclipse spectrometer (Agilent). The decay kinetics of the DF and the phosphorescence of the dye films were recorded using an FLS1000 spectrometer (Edinburgh Instr.). The samples were photoexcited at λgen = 532 nm using an Nd:YAG laser (SolarLS). The quantum yield of fluorescence (φfl) of the investigated samples was estimated by the absolute method31,33 using an integrating sphere AvaSphere 30-REFL coupled to an AvaSpec-ULS2048 spectrometer (Avantes), with the Nd:YAG laser and the OPO system (SolarLS) as a monochromatic light source.
2.3. Computational methods
Fluorescein, 2Br-fluorescein, eosin, and erythrosin in solution can exist in various charged forms across a wide pH range.18,34–36 However, as demonstrated in ref. 18, 36 and 37, the most intense transition between the singlet ground state (S0) and the first excited singlet state (S1) occurs for molecules in the dianionic form. The dianionic form of xanthene dyes was therefore chosen in the present study. Optimization of the molecular geometry in the S1 and T1 states was performed using the density functional theory (DFT) and time-dependent DFT (TD DFT) methods with the B3LYP functional,38 6-31++G(d,p) basis set and polarizable continuum model (PCM)39 with ethanol as solvent. The calculations were carried out with Gaussian 16.40 The B3LYP functional was chosen since it reproduced experimental data very well,37,41 and the 6-31++G(d,p) basis set with diffuse functions is appropriate for the present electronic structure calculations.The rate constants for IC (kIC), ISC (kISC), and for radiative (kr) electronic transitions were calculated using a computational approach developed by Valiev et al. as described in ref. 17,42–45. Estimating rate constants requires calculations of nonadiabatic coupling matrix elements (NACME) and spin–orbit coupling matrix elements (SOCME), which were calculated using Gaussian 16 and the MOLSOC code.46 The phosphorescence rate constants (kphos) were computed using the Dalton code.2,47
3. Results and discussion
3.1. The heavy atom effect on the deactivation of excited states of the xanthene dyes
The xanthene dyes absorb in the range of 400–600 nm, while they fluoresce in the range of 500–650 nm. Experimental studies (Fig. 2(a)) show that a bathochromic shift is observed in the absorption and the fluorescence spectra of the xanthene dyes when increasing the number and mass of the substituted heavy atoms (Table 1). Their fluorescence lifetime (τfl) also decreases when substituting heavy atoms to the meso positions (Table 1). The experimental fluorescence lifetime of fluorescein is 4.5 ± 0.15 ns, whereas for erythrosine, it is 0.6 ± 0.15 ns. | ||
| Fig. 2 (a) Normalized absorption (solid lines) and fluorescence (dashed lines) spectra of fluorescein and erythrosine in PVA. (b) The delayed fluorescence and phosphorescence spectra of fluorescein, 2Br-fluorescein and erythrosine in PVA. | ||
The measured quantum yields of fluorescence in ethanol solutions correlate well with data published in ref. 18 and 48. The quantum yield of luminescence decreases with increasing number and mass of halogen atoms in the xanthene dyes. The observed decrease in the fluorescence lifetime and the quantum yield can be attributed to the heavy atom effect.1 For Br- or I-substituted fluorescein dyes, the decay from the S1 state is faster than for unsubstituted fluorescein. The S1 state decays both to the ground state S0 and to the lower-lying triplet states due to the large SOC leading to faster ISC transitions and a higher concentration of molecules in the triplet state, which results in a stronger delayed fluorescence (DF) and phosphorescence (Fig. 2(b)).
The long-lived luminescence spectra exhibit two bands corresponding to DF and phosphorescence, which are bathochromically shifted to longer wavelengths in the series fluorescein < 2Br-fluorescein < eosin < erythrosine (Table 2). The DF of the investigated dyes has a thermally activated nature, which is well-known for the studied xanthene dyes.49,50 The DF is due to the thermally activated reverse ISC from T1 to the S1 state. The phosphorescence is stronger for erythrosine and eosin as compared to that of fluorescein and 2Br-fluorescein.
| Dye | λ max | I∼t−n | A 1 | τ ± 0.2 | A 2 |
|---|---|---|---|---|---|
| a Power-law exponent n was estimated when the decay curve of luminescence was approximated by a power law I∼t−n in the time range of t = 0–0.5 ms. b The lifetime τ was estimated when the decay curve of luminescence was approximated with mono-exponent law. | |||||
| Delayed fluorescence | |||||
| Fluorescein | 535 | 0.20 ± 0.01 | 16 | 3.80 | 84 |
| 2Br-fluorescein | 545 | 0.16 ± 0.01 | 12 | 3.70 | 88 |
| Eosin | 550 | 0.25 ± 0.01 | 6 | 3.20 | 94 |
| Erythrosine | 560 | — | — | 0.68 | — |
| Phosphorescence | |||||
| Fluorescein | 645 | 0.17 ± 0.01 | 12 | 3.30 | 88 |
| 2Br-fluorescein | 660 | — | — | 3.20 | — |
| Eosin | 680 | — | — | 3.00 | — |
| Erythrosine | 685 | — | — | 0.70 | — |
The decay kinetics of the DF of the fluorescein, 2Br-fluorescein and eosin films do not follow the mono-exponent decay law (Fig. 3). The DF lifetime (τ) decreases with increasing number and mass of halogen atoms. The shorter lifetimes of the DF and phosphorescence of erythrosine are due to an increasing deactivation channel from the T1 state to both the S0 and the S1 states because the molecules containing heavy atoms have a stronger SOC than fluorescein.
 | ||
| Fig. 3 The decay kinetics of the intensity of the delayed fluorescence of fluorescein and its derivatives in PVA. | ||
In condensed media, the decay of triplet states can occur via both monomolecular (1) and (2) and bimolecular (3) processes:
| T1 → S0 + hνphos phosphorescence; | (1) |
T1 ![[long arrow, wavy then straight]](https://www.rsc.org/images/entities/char_e0f6.gif) S1 → S0 + hνDF thermally activated DF; S1 → S0 + hνDF thermally activated DF; | (2) |
| T + T → [T⋯T] → S1 + S0 → 2S0 + hνADF triplet–triplet annihilation (TTA). | (3) |
The decay kinetics of the phosphorescence and the thermally activated DF is described by a mono-exponent decay. In homogeneous media, the TTA rate constant is time independent and the annihilation DF (ADF) decays exponentially with a lifetime given by τADF = τPhos/2, where τADF is the ADF lifetime and τPhos is the phosphorescence lifetime.51,52
In solid heterogeneous structures, the TTA process annihilates closely spaced triplet pairs. The luminescence intensity is then determined by the number of these pairs. Due to the different distance between the pairs, the TTA rate constant is time dependent implying that the intensity of the ADF decay becomes non-exponential. In the early stage of the decay, the intensity of the ADF can be approximated by a power law (I∼t−n).53,54
The DF intensity of fluorescein, 2Br-fluorescein, and eosin was approximated by a power law (I∼t−n) in the beginning of the emission (at t = 0–0.5 ms), while the shape of the later part of the DF decay is exponential. The power-law exponent (n) contribution (A1) of 16% is larger for fluorescein than for 2Br-fluorescein. For eosine, the A1 contribution is only 6% of the entire DF suggesting that the TTA process is more important in fluorescein films immediately after sample excitation than for eosin. This is also supported by the decay of the phosphorescence intensity of fluorescein that is well described by a power law in the initial part of the decay curve and by a mono-exponential decay law for longer times of the decay curve. The later part (t > 0.5 ms) of the DF intensity decay is due to reverse ISC from the T1 to the S1 state, since the lifetimes of the DF and the phosphorescence are similar. The triplet annihilation of the DF is more efficient in fluorescein than for other dyes because the SOC is weaker. This leads to slower transitions of the T1 states to the S0 and S1 states via phosphorescence or ISC. The almost absent TTA process in the eosin and erythrosine films can be explained by the heavy atom effect that causes a fast transition to the triplet state, which then rapidly undergoes the radiative T1 → S0 transition and reverse ISC T1 S1. This mechanism is supported by the short phosphorescence lifetimes of eosin and erythrosine (Table 2).
Quantum chemical calculations of the radiative and non-radiative rate constants of electronic transitions and of the energy levels were performed to understand the experimental data. The results of the calculations are summarized in Table 3. The calculated energies of the S1 state of the investigated molecules agree within 0.2 eV with the experimental data. The T1 state is the only excited state located below S1. The energy gap of 0.7–0.8 eV between the S1 and T1 states of the xanthene dyes is large.
| Fluorescein | 2Br-fluorescein | Eosin | Erythrosine | |
|---|---|---|---|---|
| S1 | 2.43 (2.34) | 2.42 (2.27) | 2.44 (2.25) | 2.39 (2.23) |
| T1 | 1.64 (1.92) | 1.67 (1.88) | 1.72 (1.82) | 1.67 (1.81) |
| T2 | 2.56 | 2.50 | 2.49 | 2.49 |
| 〈S1|HSO|T1〉 | 0.17 | 3.95 | 1.14 | 0.9 |
| 〈S1|HSO|T2〉 | 0.36 | 12.6 | 26.8 | 35.0 |
| k ISC(S1 → T1) | 2.5 × 103 | 3.0 × 106 | 3.5 × 105 | 2.0 × 105 |
| k IC(S1 → S0) | 1 × 108 | 1 × 108 | 8 × 106 | 6 × 107 |
| k r (S1 → S0) | 3.6 × 108 | 3.5 × 108 | 3.5 × 108 | 8 × 107 |
| k phos(T1 → S0) | 4.8 × 101 | 4.0 × 102 | 2.1 × 103 | 3.3 × 104 |
| τ calcphos | 21.00 | 2.50 | 0.47 | 0.03 |
The ISC rate constant between the S1 and T1 states (kISC(S1 → T1)) is small since it is proportional to the square of the small 〈S1|HSO|T1〉 matrix element.17,55 The ISC process cannot compete with IC and the radiative decay process of the S1 state. The 〈S1|HSO|T1〉 matrix element for fluorescein is also much smaller than for 2Br-fluorescein, eosin and erythrosine, whose kISC(S1 → T1) is significantly larger due to the presence of the heavy atoms (Fig. 4). The experimentally observed decrease in the quantum yield of fluorescence φfl in the series from fluorescein to erythrosine cannot be explained by considering only the transition channel between S1 and T1.
 | ||
| Fig. 4 The Jablonsky diagram for fluorescein and erythrosine. The rate constants are given in s−1. | ||
The calculations show that there is another triplet state (T2) slightly above S1. The energy gap ΔES1T2 between T2 and S1 is only 0.1 eV implying that the T2 state is populated through the thermally activated process, whose efficiency depends strongly on the Boltzmann factor.17 We did not consider the rate constant of the reversed ISC process from the T2 to the S1 state in the calculation of the fluorescence quantum yield φfl because the accuracy of the quantum chemical calculation of the excitation energies is ∼0.2 eV, which is not accurate enough to estimate a reliable Boltzmann factor. Calculations in ref. 37 showed that the excitation energy of the T2 state is very sensitive to environmental effects and in some cases, T2 may even be below the S1 state. The calculated 〈S1|HSO|T2〉 matrix element increases from 0.36 to 35.0 cm−1 in the series from fluorescein to erythrosine. The calculated phosphorescence rate constants (kphos(T1 → S0)) in Table 3 correlate well with the number and mass of the heavy atoms. The kphos(T1 → S0) rate constants increase in the series from fluorescein to erythrosine. The calculated phosphorescence lifetimes (τcalcphos) also correlate well with experimentally measured total lifetimes of phosphorescence τPhos, which are 3.3 ms for fluorescein and 0.7 ms for erythrosine. The total phosphorescence lifetime is not caused only by the phosphorescence radiative process but also through non-radiative channels.17
The kIC(S1 → S0) rate constants for eosin and erythrosine in Table 3 are significantly smaller than for fluorescein and 2Br-fluorescein. The kIC(S1 → S0) rate constants were obtained by using an approximate computational method that accounts only for contributions from the C–H bonds, which has been previously shown to be the dominating contribution to the IC rate constant.56 The main contribution to kIC(S1 → S0) originates from the C–H bonds in the meso-positions (Fig. 5).
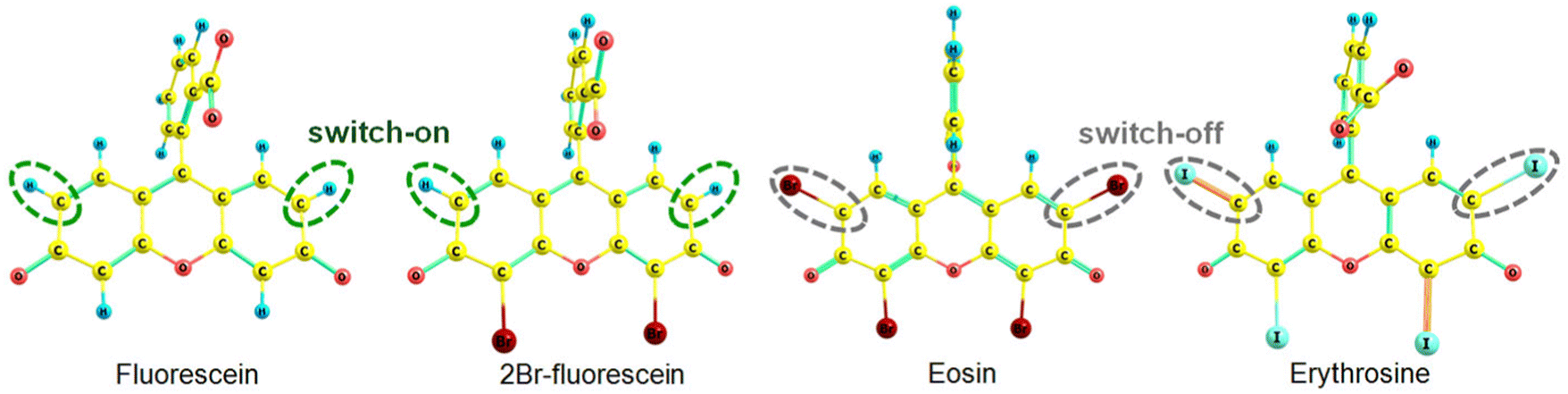 | ||
| Fig. 5 The most important accepting bonds in the internal conversion process are surrounded by the dashed lines. | ||
When hydrogen atoms in the meso positions are replaced by iodine or bromine atoms, the bonds to the meso carbon atoms do not contribute significantly to the IC rate constant, leading to a smaller kIC(S1 → S0) rate constant. The calculations show how the heavy atoms affect the IC rate constant of the studied xanthene dyes.
3.2. The plasmon effect
There were no noticeable changes in the position of the absorption and emission bands when the dye films were deposited on a SIF surface instead of on a glass surface. The measurements showed that the plasmon effect of the SIF significantly increases the absorbance of the studied dyes (Table 4), leading to an increase in the optical density (D) at the maximum of the absorption band.| Molecule | D SIF/Dglass | I SIFfl/Iglassfl | ϕ SIFfl/ϕglassfl | I SIFDF/IglassDF | ϕ SIFPhos/ϕglassPhos |
|---|---|---|---|---|---|
| Optical density, intensities, quantum yield | |||||
| Fluorescein | 1.1 | 1.8 | 1.83 | 1.7 | 1.8 |
| 2Br-fluorescein | 2.1 | 2.0 | 1.87 | 2.5 | 2.6 |
| Eosin | 2.5 | 4.1 | 3.73 | 4.1 | 4.0 |
| Erythrosine | 3.2 | 7.6 | 5.60 | 5.5 | 5.6 |
| τ SIFfl/τglassfl | τ SIFDF/τglassDF | τ SIFPhos/τglassPhos | |
|---|---|---|---|
| Lifetimes | |||
| Fluorescein | 0.96 | 0.59 | 0.92 |
| 2Br-fluorescein | 0.93 | 0.77 | 0.90 |
| Eosin | 0.86 | 0.98 | 0.97 |
| Erythrosine | 0.83 | 0.98 | 0.99 |
The larger D value is due to the presence of the silver particles and because the films have different thicknesses. To eliminate the influence of the thickness of the film, the absorption spectrum of the SIF with the dye films was measured and corrected for the absorption spectrum recorded for the same SIF before the dye film was deposited on it. The substrate with a pure dye film and with the SIF and the dye sample were weighted showing that their masses were almost identical. Thus, the observed change in the optical density is due to the plasmon effect of the SIF. The larger optical density leads to a significant increase in the fluorescence intensity.
The observed increase in the fluorescence intensity (Ifl) is not only due to the changes in D but also due to an increase in the quantum yield of fluorescence (φfl) and the decrease in the fluorescence lifetime (τfl). Thus, the stronger intensity is due to the plasmon enhancement of the radiative emission from the S1 state to the S0 state.31,57–59 The increase in Ifl and φfl grows in the series from fluorescein to erythrosine because the plasmon effect is larger for photo processes of lower efficiency.58,59
The enhancement of the DF and phosphorescence is larger for the xanthene dyes having heavy atoms. The amplification coefficients of the DF and the phosphorescence are between 1.7 and 5.6 for the studied dyes. The observed differences in the plasmon effect on the long-lived luminescence of the xanthene dyes are due to a larger intensity borrowing of the T1 → S0 transition from the S1 → S0 transition in the molecules with heavy atoms, which was also verified by calculating SOC values (Table 3).
4. Conclusion
The synergy of the effects of heavy atoms and plasmons on the luminescence properties of xanthene dyes was investigated. Experimental data showed that the presence of heavy atoms leads to a decrease in the lifetime of the excited singlet state and the fluorescence quantum yield. Substituting heavy atoms to the molecules results in a faster ISC transfer to the triplet state, as evidenced by the enhanced intensity and reduced lifetimes of the phosphorescence and the DF. Analysis of the decay of the phosphorescence and the DF shows that for unsubstituted fluorescein, the initial part of the decay is due to the TTA process. With increasing the number and mass of the heavy atoms, the contribution from TTA to the decay of the T1 state decreases because the phosphorescence and the DF are faster.The heavy atom effect was also studied using quantum chemical calculations. The influence on the rate constants of IC (kIC(S1 → S0)), phosphorescence (kphos(T1 → S0)), and ISC (kISC(S1 → T1)) was investigated. The calculations showed that substitution of a heavy atom in specific positions leads to a decrease in kIC(S1 → S0), from 1 × 108 s−1 for fluorescein to 6 × 107 s−1 for erythrosine and 8 × 106 s−1 for eosin. The slower IC process is due to fewer good acceptor modes of the electronic excitation energy for the non-radiative transition between the S1 and S0 states.
The rate constant of phosphorescence increases by an order of magnitude in the series from fluorescein to erythrosine because the number and mass of heavy atoms increase in that series. The 〈S1|HSO|T2〉 matrix element increases with the number and mass of the heavy atoms. The present study suggests that the lowest triplet state T1 is populated via two channels: through the ISC between the S1 and T1 states and through the IC between the T2 and T1 states. The calculated IC rate constant for the T2 T1 transition is about 1011 s−1.
The plasmon effect significantly enhances the absorbance, which is reflected in the increased optical density at the maximum of the absorption band. The increase in the intensity and the quantum yield of fluorescence grows in the series of fluorescein < 2Br-fluorescein < eosin < erythrosine. The enhancement of the DF and the phosphorescence is larger for the halogenated molecules. The phosphorescence of fluorescein is enhanced by a factor of 1.8, and for erythrosine, it is 5.6 times stronger. The differences in the plasmon effect can be attributed to the intensity borrowing of the intensity of the radiative triplet–singlet transition (T1 → S0) from singlet–singlet transitions (Sn → S0), which is larger for molecules with heavy atoms.
The obtained results show that the phosphorescence efficiency of organic dyes can be significantly enhanced by taking advantage of both the heavy-atom effect of substituents and the plasmonic effect of metal NPs. Strongly phosphorescent dyes are useful in biomedical research and in the development of sensor devices, OLED and PHOLED systems, anti-counterfeiting and data encryption materials.
Author contributions
N. I.: conceptualization, methodology, resources, project administration, writing – review and editing; E. S.: investigation, data curation, validation and visualization, writing – original draft, writing – review and editing; R. V.: methodology, conceptualization, calculations; modelling, software, writing – original draft, writing – review and editing; A. A.: investigation, formal analysis; D. S.: conceptualization, data validation, writing – review and editing. The authors have discussed the obtained results and contributed equally to the final version of the manuscript.Data availability
Quantum chemical software utilized: (1) The optimized molecular structures, excitation energies, and matrix elements of spin-orbital coupled interaction operators were computed using GAUSSIAN 16 [https://gaussian.com/gaussian16/] and MOLSOC [https://github.com/gaox-qd/pysoc]. The results of these calculations are compiled and discussed in detail in the main text of the article. (2) The intramolecular rate constants were determined using proprietary code developed by Valiev and colleagues: [https://pubs.rsc.org/en/content/articlelanding/2020/cp/d0cp03231j]. (3) The working expressions for intermolecular rate constants, along with the initial parameters, are provided and discussed within the main text. (4) All measured photophysical values are reported in the main text of the article, with detailed measurement procedures described.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan (Grant No. AP14870117). This work has been supported by the Academy of Finland through projects 340582 (RRV) and 340583 (DS). We thank CSC – IT Center for Science, Finland for computer time.References
- S. P. McGlynn, T. Azumi and M. Kinoshita, Molecular Spectroscopy of the Triplet State, Prentice-Hall, Inc., Englewood Cliffs, New Jersey, 1969 Search PubMed.
- G. Baryshnikov, B. F. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS.
- B. Chang, J. Chen, J. Bao, T. Sun and Z. Cheng, Chem. Rev., 2023, 123(24), 13966–14037 CrossRef CAS PubMed.
- R. Knoblauch, B. Bui, A. Raza and Chris D. Geddes, Phys. Chem. Chem. Phys., 2018, 20, 15518–15527 RSC.
- M. Stanitska, D. Volyniuk, B. Minaev, H. Agren and J. V. Grazulevicius, J. Mater. Chem. C, 2024, 12, 2662–2698 RSC.
- R. E. Bangle, H. Li and M. H. Mikkelsen, ACS Nano, 2023, 17, 24022–24032 CrossRef CAS.
- M. S. Weaver, M. A. Fusella, R. Saramak, R. Bushati, H. Mundoor, V. M. Menon, N. J. Thompson and J. J. Brown, J. Mater. Chem. C, 2022, 10, 4182–4186 RSC.
- Z. Zhou, X. Xie, Z. Sun, X. Wang, Z. An and W. Huang, J. Mater. Chem. C, 2023, 11, 3143–3161 RSC.
- M. Ji and X. Ma, Ind. Chem. Mater., 2023, 1, 582–594 RSC.
- T. M. Chmereva, M. G. Kucherenko, F. Yu Mushin and A. P. Rusinov, J. Appl. Spectrosc., 2024, 91, 1–9 CrossRef CAS.
- H. Mishra, B. L. Mali, J. Karolin, A. I. Dragan and C. D. Geddes, Phys. Chem. Chem. Phys., 2013, 15, 19538–19544 RSC.
- Y. Zhang, K. Aslan, M. J. R. Previte, S. N. Malyn and C. D. Geddes, J. Chem. Phys. B, 2006, 110, 25108–25114 CrossRef CAS.
- M. Meng, F.-L. Zhang, J. Yi, L.-H. Lin, C.-L. Zhang, N. Bodappa, C.-Y. Li, S.-J. Zhang, R. F. Aroca, Z.-Q. Tian and J.-F. Li, Anal. Chem., 2018, 90, 10837–10842 CrossRef CAS PubMed.
- S. Bidault, A. Devilez, V. Maillard, L. Lermusiaux, J.-M. Guigner, N. Bonod and J. Wenger, ACS Nano, 2016, 10, 4806–4815 CrossRef CAS PubMed.
- H. Wang, J. Jung, K. Chung, J. W. Lim, Y. You, J. Kim and D. H. Kim, J. Appl. Phys., 2017, 122, 153103 CrossRef.
- R. Lakshmanan, N. C. Shivaprakash and S. Sindhu, J. Lumin., 2018, 196, 136–145 CrossRef CAS.
- N. K. Ibrayev, R. R. Valiev, E. V. Seliverstova, E. P. Menshova, R. T. Nasibullin and D. Sundholm, Phys. Chem. Chem. Phys., 2024, 26, 14624–14636 RSC.
- E. Slyusareva, A. Sizykh, A. Tyagi and A. Penzkofer, J. Photochem. Photobiol., A, 2009, 208, 131–140 CrossRef CAS.
- Y. Kushida, T. Nagano and K. Hanaoka, Analyst, 2015, 140, 685–695 RSC.
- S. M. Derayea and D. M. Nagy, Rev. Anal. Chem., 2018, 37, 20170020 CAS.
- M. Kabeláč, F. Zimandl, T. Fessl, Z. Chval and F. Lankaš, Phys. Chem. Chem. Phys., 2010, 12, 9677–9684 RSC.
- K. N. Patterson, M. A. Romero-Reyes and J. M. Heemstra, ACS Omega, 2022, 7, 33046–33053 CrossRef PubMed.
- V. S. Maryakhina, Laser Phys., 2016, 26, 105603 CrossRef.
- V.-N. Nguyen, A. Kumar, M. H. Lee and J. Yoon, Coord. Chem. Rev., 2020, 425, 213545 CrossRef.
- S. K. Lam, M. A. Chan and D. Lo, Sens. Actuators, B, 2001, 73, 135–141 CrossRef.
- Y. Zhang, K. Aslan, M. J. Previte and C. D. Geddes, J. Fluoresc., 2007, 17, 345–349 CrossRef PubMed.
- E. Gandin, Y. Lion and A. Van de Vorst, J. Photochem. Photobiol., 1983, 37, 271–278 CrossRef.
- N. Macia, R. Bresoli-Obach, S. Nonell and B. Heyne, J. Am. Chem. Soc., 2019, 141, 684–692 CrossRef PubMed.
- V. L. Ermolaev, Opt. Spectrosc., 2016, 121, 567–584 CrossRef.
- R. R. Valiev, R. T. Nasibullin, V. N. Cherepanov, G. V. Baryshnikov, D. Sundholm, H. Ågren, B. F. Minaev and T. Kurtén, Phys. Chem. Chem. Phys., 2020, 22, 22314–22323 RSC.
- N. K. Ibrayev, E. V. Seliverstova, R. R. Valiev, A. E. Kanapina, A. A. Ishchenko, A. V. Kulinich, T. Kurten and D. Sundholm, Phys. Chem. Chem. Phys., 2023, 25, 22851–22861 RSC.
- K. Aslan, Z. Leonenko, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2005, 15, 643–654 CrossRef PubMed.
- C. Würth, M. G. González, R. Niessner, U. Panne, C. Haisch and U. R. Genger, Talanta, 2012, 90, 30–37 CrossRef.
- D. C. Neckers and O. M. Valdes-Aguilera, Adv. Photochem., 1993, 18, 315–394 CrossRef.
- J. Paczkowski, J. J. Lamberts, B. Paczowska and D. C. Neckers, J. Free Radicals Biol. Med., 1985, 1, 341–351 CrossRef PubMed.
- A. V. Rogova, F. N. Tomilin, M. A. Gerasimova and E. A. Slyusareva, Russ. Phys. J., 2020, 63, 1417–1423 CrossRef.
- B. Demirbay, G. Baryshnikov, M. Haraldsson, J. Piguet, H. Ågren and J. Widengren, Methods Appl. Fluoresc., 2023, 11, 045011 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. A. Gerasimova, F. N. Tomilin, E. Yu Malyar, S. A. Varganov, D. G. Fedorov, S. G. Ovchinnikov and E. A. Slyusareva, Dyes Pigm., 2020, 173, 107851 CrossRef.
- R. R. Valiev, R. T. Nasibullin, V. N. Cherepanov, A. Kurtsevich, D. Sundholm and T. Kurten, Phys. Chem. Chem. Phys., 2021, 23, 6344–6348 RSC.
- R. R. Valiev, V. N. Cherepanov, R. T. Nasibullin, D. Sundholm and T. Kurten, Phys. Chem. Chem. Phys., 2019, 21, 18495–18500 RSC.
- R. R. Valiev, V. N. Cherepanov, V. Y. Artyukhov and D. Sundholm, Phys. Chem. Chem. Phys., 2012, 14, 11508–115017 RSC.
- R. R. Valiev, V. N. Cherepanov, G. V. Baryshnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 6121–6133 RSC.
- S. G. Chiodo and M. Leopoldini, Comput. Phys. Commun., 2014, 185, 676–683 CrossRef.
- H. Ågren, B. F. Minaev and S. Knuts, J. Phys. Chem., 1994, 98, 3943–3949 CrossRef.
- G. R. Fleming, A. W. E. Knight, J. M. Morris, R. J. S. Morrison and G. W. Robinson, J. Am. Chem. Soc., 1977, 99, 4306–4311 CrossRef.
- D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- C. A. Parker, Photoluminescence of Solutions, Elsevier Publishing Co., Amsterdam, 1968 Search PubMed.
- M. Pope and Ch. E. Swenberg, Electronic Processes in Organic Crystals and Polymers, Oxford University Press, Oxford, 1999 Search PubMed.
- N. A. Efremov, S. G. Kulikov, R. I. Personov and Yu. V. Romanovskii, Chem. Phys. Lett., 1988, 128, 9–21 CAS.
- N. A. Efremov, S. G. Kulikov, R. I. Personov and Yu. V. Romanovskii, Phys. Solid State, 1989, 3, 103–111 Search PubMed.
- H. Yersin, R. Czerwieniec, U. Monkowius, R. Ramazanov, R. Valiev, M. Z. Shafikov, W. M. Kwok and C. Ma, Coord. Chem. Rev., 2023, 478, 214975 CrossRef.
- R. R. Valiev, B. S. Merzlikin, R. T. Nasibullin, A. Kurtzevitch, V. N. Cherepanov, R. R. Ramazanov, D. Sundholm and T. Kurtén, Phys. Chem. Chem. Phys., 2023, 25, 6406–6415 RSC.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef.
- E. Seliverstova, N. K. Ibrayev, G. Omarova, A. Ishchenko and M. Kucherenko, J. Lumin., 2021, 235, 118000 CrossRef.
- C. D. Geddes and J. R. Lakowicz, J. Fluoresc., 2002, 12, 121–129 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02621g |
| This journal is © the Owner Societies 2024 |