
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IR spectrum of SiH3OH2+SiH4: cationic OH⋯HSi dihydrogen bond versus charge-inverted SiH⋯Si hydrogen bond†
Martin Andreas Robert
George
 and
Otto
Dopfer
*
and
Otto
Dopfer
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Berlin 10623, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 12th August 2024
Abstract
The low electronegativity of Si gives rise to a variety of nonconventional intermolecular interactions in clusters of silanes and their derivatives, which have not been well characterized yet. Herein, we characterize the structures of various isomers of bare and Ar-tagged SiH3OH2+SiH4 dimers composed of protonated silanol and silane by infrared photodissociation (IRPD) of mass-selected ions and dispersion-corrected density functional calculations (B3LYP-D3/aug-cc-pVTZ). The analysis of the IRPD spectra recorded in the OH stretch range reveals the competition between two types of nonconventional hydrogen bonds (H-bonds). The first one represents a OH⋯HSi ionic dihydrogen bond (DHB), in which SiH4 interacts with the H2O moiety of SiH3OH2+. The second one represents a charge-inverted SiH⋯Si ionic H-bond (CIHB), in which the SiH4 ligand interacts with the SiH3 moiety of SiH3OH2+. The latter may also be considered as a weak three-centre two-electron (3c–2e) bond. Although both types of H-bonds are computed to have comparable interaction strengths for SiH3OH2+SiH4 (D0 ≈ 35–40 kJ mol−1), DHB isomers dominate the population in the supersonic plasma expansion, while the abundance of CIHB isomers is roughly one order of magnitude lower, probably as a result of entropic factors.
1. Introduction
Silanes (SixHy), silanols (SixHyOz), and their derivatives are important molecules in inorganic chemistry, polymer and materials science, astrochemistry, plasma chemistry, and theoretical chemistry.1–9 Laboratory spectra of silanes and their ions are essential to analyze and control the complex chemistry of silane plasmas used in semiconductor industry.3,10–13 In addition, based on the detection of SiH4 in the interstellar environment,14 laboratory spectra of neutral and cationic SixHy(+) and SixHyOz(+) molecules are needed for comparison with astronomical spectra.Although Si is a group IV element like C, the chemical bonds in CxHy and SixHy (and their ions) are quite different.15 Part of these differences results from the lower electronegativity of Si (ENSi = 1.90) compared to those of H and C (ENH = 2.20, ENC = 2.55). In general, Si–Si and Si–H bonds are longer than corresponding C–H and C–C bonds and exhibit more often nonclassical Si–H–Si bridges.16–20 Such Si–H–Si bridges are three-center two-electron (3c–2e) bonds,21,22 in which two electrons in a bonding orbital form two stable bonds in a more or less linear Si–H–Si bridge. These bridges can also be considered as strong charge-inverted hydrogen bonds (CIHBs) with polarity Siδ+–Hδ−–Siδ+,23–25 because ENH is higher than ENSi. We have previously characterized such ionic CIHBs in a variety of SixHy+ cations in the gas phase using infrared photodissociation spectroscopy (IRPD).26–29 In addition, we recently presented the first spectroscopic and structural characterization of the highly elusive protonated silanol molecule (SiH3OH2+) based on the IRPD spectrum of its Ar-tagged cluster.30 While monosilanol (SiH3OH) is rather unstable with respect to intermolecular condensation reactions and has hardly been characterized structurally and spectroscopically,2 with the notable exception of a single IR band (νSiO = 859 cm−1),31 we could analyze the chemical bonding in SiH3OH2+ and assign it to a dative bond of H2O to the SiH3+ cation.
Herein, we report IRPD spectra of SiH3OH2+SiH4 and its Ar-tagged SiH3OH2+SiH4–Ar cluster to study the competition between two interesting and nonconventional types of intermolecular bonds between SiH3OH2+ and SiH4. The first one is the formation of a CIHB bond between SiH4 and the SiH3+ moiety of SiH3OH2+, which is typical for polysilane ions such as Si2H7+ or longer SiH3+(SiH4)n hydride wires,26,29 but is affected in SiH3OH2+SiH4 by the dative bond of H2O to SiH3+. The second binding motif is the formation of a cationic dihydrogen bond (DHB) of the type Siδ+Hδ−⋯Hδ+Oδ− between the H2O moiety of SiH3OH2+ and the SiH4 ligand, which represents a subclass of H-bond interactions.32–46 The DHB is a H-bond interaction between two oppositely charged H atoms, which can only occur when one of the two H atoms is bonded to a more electropositive atom (e.g., Si, B, transition metal) while the other H atom is bonded to a more electronegative atom (e.g., O in our case). DHBs in systems with transition metals and B in their crystalline forms have been studied extensively.32,34,35,47–56 In the gas phase, DHBs of the type BH⋯HO and BH⋯HN were investigated by IR spectroscopy and quantum chemical calculations.42,57–63 The first spectroscopic evidence for a DHB of the type SiH⋯HO was reported by Ishikawa and co-workers in the phenol-diethylmethylsilane (PhDEMS) dimer.64 Subsequently, SiH⋯HO bonds were found in phenol-triethylsilane (PhTES) and phenol-ethyldimethylsilane (PhEDMS), as well as in the related cationic Ph+DEMS and Ph+TES dimers.65,66 The SiH⋯HO DHB is considered to be an intermediate motif in chemical reactions such as H2O + SiH4 → SiH3OH + H2.67,68 While neutral DHBs with Ph are weak and comparable to dispersion interactions, cationic DHBs are much stronger due to the much higher acidity of Ph+ arising from its excess positive charge.66 To gain a better understanding of the nature of the SiH⋯HO ionic DHBs, it is necessary to collect further spectroscopic information. To this end, we study in this work a significantly smaller system containing an ionic DHB, SiH3OH2+SiH4, to reveal the intrinsic nature of the SiH⋯HO DHB, which is the dominant intermolecular interaction in this cluster, without any interference from aromatic or aliphatic hydrocarbon structures.
2. Experimental and computational techniques
IRPD spectra of bare and Ar-tagged SiH3OH2+SiH4 ions in the OH stretch range (2700–3800 cm−1) are obtained in a tandem quadrupole mass spectrometer coupled to an electron ionization (EI) source described elsewhere.69–72 SiH3OH2+SiH4 and SiH3OH2+SiH4–Ar clusters are generated in a pulsed supersonic plasma expansion of a SiH4/He/Ar gas mixture (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:200, 5 bar stagnation pressure) seeded with H2O vapor. The gas mixture is ionized by EI (and/or chemical ionization), resulting in the formation of a variety of hydrated silicon hydride cluster cations and their Ar complexes. The rather stable silyl cation (SiH3+) is the major primary EI product of SiH4. In a next step, protonated silanole is produced via barrierless addition of H2O to SiH3+ forming a rather stable chemical Si–O bond in SiH3OH2+.30 In subsequent three-body aggregation reactions occurring in the high-pressure region of the expansion, weakly-bound clusters of SiH3OH2+ with SiH4 and Ar are generated and cooled down to lower temperatures. A typical mass spectrum of the EI source reveals strong Arn+ cluster signals, accompanied by weaker peaks arising from OH1–2+, SixHy+, SixHyO+, and their Ar clusters (Fig. S1, ESI†). After extraction through a skimmer, bare or Ar-tagged SiH3OH2+SiH4 ions (m/z 81 or 121) are selected by the first quadrupole mass filter and irradiated in an adjacent octupole with tunable IR laser radiation generated by an optical parametric IR oscillator pumped by a nanosecond Q-switched Nd:YAG laser. The IR radiation is characterized by a pulse energy of ∼1–5 mJ in the employed spectral range, a repetition rate of 10 Hz, and a bandwidth of 1 cm−1. Calibration of the IR laser frequency (νIR) is accomplished by a wavemeter. Resonant vibrational excitation of SiH3OH2+SiH4(–Ar) induces the rupture of the weakest intermolecular bond (i.e., loss of Ar or SiH4). The resulting SiH3OH2+(SiH4) fragment ions are selected by the second quadrupole mass filter and monitored as a function of νIR to derive the IRPD spectra of SiH3OH2+SiH4(–Ar). To separate the fragment ions produced by metastable decay from those generated by laser-induced dissociation, the ion source is triggered at twice the laser frequency, and signals from alternating triggers are subtracted. The widths of the observed transitions result from unresolved rotational substructure and overlapping sequence hot bands of intramolecular fundamentals with low-frequency intermolecular modes and possibly lifetime broadening. Because the population of rotational levels of the parent ions generated in this supersonic plasma expansion cannot be described by a single rotational temperature due to the lack of thermal equilibrium (levels with larger rotational quantum numbers are populated according to a higher temperature), we cannot readily simulate a rotational band profile. In addition, as the time-of-flight in the octupole is of the order of one millisecond, we do not observe a kinetic shift and all photoexcited ions with a final energy larger than the dissociation energy contribute to the measured IRPD yield. The IRPD yield is normalized for laser intensity variations measured with a pyroelectric detector. Because the mass spectrum of the ion source is rather complex, collision-induced dissociation (CID) experiments are employed to confirm the composition of the investigated SiH3OH2+SiH4(–Ar) parent ions. To this end, the octupole is filled with 10−5 mbar of N2, allowing for collisions with mass-selected ions at a kinetic energy of 10 eV in the laboratory frame. Clearly, the CID spectrum of mass-selected SiH3OH2+SiH4–Ar (m/z 121) demonstrates the almost exclusive loss of Ar followed by loss of SiH4 resulting in SiH3OH2+ (m/z 49), confirming its composition (Fig. S2, ESI†). A very minor channel follows the other sequence (primary loss of SiH4 followed by loss of Ar).
:20:200, 5 bar stagnation pressure) seeded with H2O vapor. The gas mixture is ionized by EI (and/or chemical ionization), resulting in the formation of a variety of hydrated silicon hydride cluster cations and their Ar complexes. The rather stable silyl cation (SiH3+) is the major primary EI product of SiH4. In a next step, protonated silanole is produced via barrierless addition of H2O to SiH3+ forming a rather stable chemical Si–O bond in SiH3OH2+.30 In subsequent three-body aggregation reactions occurring in the high-pressure region of the expansion, weakly-bound clusters of SiH3OH2+ with SiH4 and Ar are generated and cooled down to lower temperatures. A typical mass spectrum of the EI source reveals strong Arn+ cluster signals, accompanied by weaker peaks arising from OH1–2+, SixHy+, SixHyO+, and their Ar clusters (Fig. S1, ESI†). After extraction through a skimmer, bare or Ar-tagged SiH3OH2+SiH4 ions (m/z 81 or 121) are selected by the first quadrupole mass filter and irradiated in an adjacent octupole with tunable IR laser radiation generated by an optical parametric IR oscillator pumped by a nanosecond Q-switched Nd:YAG laser. The IR radiation is characterized by a pulse energy of ∼1–5 mJ in the employed spectral range, a repetition rate of 10 Hz, and a bandwidth of 1 cm−1. Calibration of the IR laser frequency (νIR) is accomplished by a wavemeter. Resonant vibrational excitation of SiH3OH2+SiH4(–Ar) induces the rupture of the weakest intermolecular bond (i.e., loss of Ar or SiH4). The resulting SiH3OH2+(SiH4) fragment ions are selected by the second quadrupole mass filter and monitored as a function of νIR to derive the IRPD spectra of SiH3OH2+SiH4(–Ar). To separate the fragment ions produced by metastable decay from those generated by laser-induced dissociation, the ion source is triggered at twice the laser frequency, and signals from alternating triggers are subtracted. The widths of the observed transitions result from unresolved rotational substructure and overlapping sequence hot bands of intramolecular fundamentals with low-frequency intermolecular modes and possibly lifetime broadening. Because the population of rotational levels of the parent ions generated in this supersonic plasma expansion cannot be described by a single rotational temperature due to the lack of thermal equilibrium (levels with larger rotational quantum numbers are populated according to a higher temperature), we cannot readily simulate a rotational band profile. In addition, as the time-of-flight in the octupole is of the order of one millisecond, we do not observe a kinetic shift and all photoexcited ions with a final energy larger than the dissociation energy contribute to the measured IRPD yield. The IRPD yield is normalized for laser intensity variations measured with a pyroelectric detector. Because the mass spectrum of the ion source is rather complex, collision-induced dissociation (CID) experiments are employed to confirm the composition of the investigated SiH3OH2+SiH4(–Ar) parent ions. To this end, the octupole is filled with 10−5 mbar of N2, allowing for collisions with mass-selected ions at a kinetic energy of 10 eV in the laboratory frame. Clearly, the CID spectrum of mass-selected SiH3OH2+SiH4–Ar (m/z 121) demonstrates the almost exclusive loss of Ar followed by loss of SiH4 resulting in SiH3OH2+ (m/z 49), confirming its composition (Fig. S2, ESI†). A very minor channel follows the other sequence (primary loss of SiH4 followed by loss of Ar).
Quantum chemical calculations are performed at the dispersion-corrected B3LYP-D3/aug-cc-pVTZ level of theory for SiH4, SiH3OH2+, and various isomers of SiH3OH2+SiH4(–Ar) to determine their energetic, structural, vibrational, and electronic properties.73 This computational level reliably reproduces the properties of SiH3OH2+ and its SiH3OH2+–Arn≤5 complexes and the 3c–2e bonds in SixH4x−1+ hydride wires.29,30 Relative energies and equilibrium binding energies (Ee, De) are corrected for harmonic zero-point vibrational energies to derive E0 and D0 values. Gibbs free energies (G0) are evaluated at 298.15 K. Harmonic frequencies are scaled by factors of 0.9631 (0.9805) for frequencies above (below) 2000 cm−1 to optimize the agreement between calculated and measured frequencies of H2O.30 Natural bond orbital (NBO) analysis is employed to evaluate the charge distribution and charge transfer, as well as the second-order perturbation energies (E(2)) of donor–acceptor orbital interactions involved in the H-bonds. Calculated vibrational frequencies are compared with experimental values in Tables S1–S3 (ESI†), and calculated energies are listed in Tables S4–S9 (ESI†).
3. Results and discussion
3.1 Overview of IRPD spectra
IRPD spectra of SiH3OH2+SiH4 and SiH3OH2+SiH4–Ar recorded in the OH stretch range are compared in Fig. 1 to that of SiH3OH2+–Ar reported previously.30 The positions, widths, and suggested vibrational and isomer assignments are listed in Table 1. For a detailed discussion of the properties of SiH3OH2+ and SiH3OH2+–Ar, we refer to our previous work.30 Because of the strong bonds in bare SiH3OH2+, no IRPD spectrum can be obtained for this ion under the employed single-photon absorption conditions. Its symmetric and antisymmetric OH stretch fundamentals (νOHs/a) computed as 3550 and 3626 cm−1 are indicated by grey dashed lines in Fig. 1. The splitting of 76 cm−1 between νOHs and νOHa results from the coupling of the two equivalent local OH stretch oscillators. The SiH3OH2+–Ar spectrum shows two OH stretch bands C1 and E at 3400 and 3600 cm−1, which can readily be attributed to the Ar-bonded and free OH stretch modes (νOHb(Ar) and νOHf) of the global minimum structure, in which Ar forms an OH⋯Ar H-bond to one of the two OH groups of SiH3OH2+. The assignment of these two bands is not only supported by their frequencies and shifts from those of bare SiH3OH2+ but also by their band profiles. While the free OH stretch band has a symmetric profile, the Ar-bonded OH stretch band has a sharp P-branch head and a long blueshaded tail, which are typical for excitation of proton donor stretch modes.70,74,75 Excitation of a proton-donor stretch fundamental causes the H-bond to become stronger, leading to smaller rotational constants in the vibrational excited state, giving rise to a P-branch head. In addition, the stronger H-bond in the intramolecular excited state causes the intermolecular stretch and bend frequencies to be larger than in the ground vibrational state due to the larger radial force constant and larger angular anisotropy in the intermolecular potential. Hence, sequence hot bands of the proton-donor stretch fundamental with intermolecular modes appear to the blue of the fundamental transition. These effects to not operate for excitation of the free OH stretch mode and, as a result, such bands exhibit a symmetric band shape. Complexation with Ar removes the coupling between the two OH stretch oscillators. As a result, the νOHf band of SiH3OH2+–Ar occurs roughly at the average frequency of νOHs and νOHa of bare SiH3OH2+ predicted as 3588 cm−1, while the νOHb(Ar) band of SiH3OH2+–Ar is redshifted by almost 200 cm−1 from this value due to the formation of the OH⋯Ar H-bond. The SiH3OH2+SiH4 spectrum is dominated by strongly redshifted νOHb(SiH4) bands A1 and A2 (by around 750 cm−1) near 2850 cm−1 indicative of the formation of a much stronger OH⋯SiH4 H-bond. This observation is consistent with the larger polarizability of SiH4 compared to Ar (computed as α = 32.20 vs. 11.15 a03), because induction and dispersion interactions provide the major contribution to the intermolecular attraction. The intense A1 band peaks at 2830 cm−1 with a width of 40 cm−1, while the weak shoulder A2 has its maximum at 2872 cm−1. In addition, three bands D–F appear in the free OH stretch range above 3500 cm−1, suggesting the presence of at least two isomers. The strongest band E at 3602 cm−1 has almost the same frequency as band E in the SiH3OH2+ spectrum, indicating an assignment to the free OH stretch (νOHf) of a cluster with a strong OH⋯SiH4 H-bond. On the other hand, bands D and F at 3578 and 3695 cm−1 occur not far from the free OH stretch bands νOHs and νOHa of bare SiH3OH2+ predicted at 3550 and 3626 cm−1 suggesting the presence of an isomer, in which the SiH4 ligand is not attached to the OH2 side of SiH3OH2+ but to the SiH3 side. Ar-tagging of SiH3OH2+SiH4 causes modest blue shifts of the A1/A2 bands (to 2869/2910 cm−1) and produces an intense band C1 at 3456 cm−1 (with a width of 15 cm−1) characteristic for a complex with one OH⋯SiH4 and one OH⋯Ar H-bond. Such small blueshifts in proton donor stretch vibrations (like here for A1/A2) are characteristic for interior ion solvation, which is accompanied by small noncooperative effects on the H-bond strengths due to increased charge delocalization into a larger number of neutral ligands. The presence of the weaker bands C2 and E in the OH stretch range at 3495 and 3629 cm−1 is again indicative of a less stable isomer in which one OH group of the SiH3OH2+ core ion is not engaged in a H-bond (νOHf) while the other one forms an OH⋯Ar H-bond (νOHb(Ar)). In such an isomer, the SiH4 ligand does not bind to one of the OH groups of SiH3OH2+ but to its SiH3 moiety. The IRPD spectra of both bare and Ar-tagged SiH3OH2+SiH4 reveal a weak band B at ∼3190 and ∼3230 cm−1, respectively, which is not present in the SiH3OH2+–Ar spectrum. They may arise either from the βOH overtone of the H2O moiety or a combination band of the SiH4-bound OH stretch fundamentals with an intermolecular mode. The latter scenario may be supported by the blueshift in band B upon Ar-tagging (∼20 cm−1), which parallels the blue shifts of bands A1/A2 (∼40 cm−1). | ||
| Fig. 1 IRPD spectra of SiH3OH2+SiH4 and SiH3OH2+SiH4–Ar in the 2700–3800 cm−1 range recorded in the SiH4 and Ar loss channels, respectively, are compared to the IRPD spectrum of SiH3OH2+–Ar.30 The position, widths, and assignments of the transitions observed are listed in Table 1 and Tables S1–S3 (ESI†). | ||
| Isomera | Mode | SiH3OH2+–Arc | SiH3OH2+SiH4 | SiH3OH2+SiH4–Ar | |
|---|---|---|---|---|---|
| a Intermolecular binding motifs of isomers I–III (DHB) and IV (CIHB) of SiH3OH2+SiH4 and SiH3OH2+SiH4–Ar are assigned to the observed transitions (and do not apply to SiH3OH2+–Ar). b Tentative assignment. c Ref. 30. | |||||
| A1 | DHB | ν OH b(SiH4) | — | 2830 (40) | 2869 (15) |
| A2 | DHB | ν OH b(SiH4) | — | 2872 (40) | 2910 (40) |
| B | DHB/CIHB | 2βOHb | — | ∼3190 | ∼3230 |
| C1 | DHB | ν OH b(Ar) | 3400 (21) | — | 3456 (15) |
| C2 | CIHB | ν OH b(Ar) | — | — | 3495 (20) |
| D | CIHB | ν OH s | — | 3578 (20) | — |
| E | DHB/CIHB | ν OH f | 3600 (30) | 3602 (30) | 3629 (15) |
| F | CIHB |
ν
OH
ab |
— | 3695 (10) | — |
In summary, the initial analysis of the IRPD spectra of bare and Ar-tagged SiH3OH2+SiH4 reveals two routes of cluster growth. Along the predominant path, SiH4 and Ar form intermolecular H-bonds to the two available acidic OH groups of SiH3OH2+, while a minor route involves attachment of SiH4 to the SiH3 moiety, whereas Ar then forms a OH⋯Ar H-bond. To derive more details of this preliminary analysis based on the IRPD spectra alone, we resort to quantum chemical calculations.
3.2 Computational analysis and assignments
The tetrahedral SiH4 molecule has a computed Si–H bond length and vibrational frequencies (1.484 Å and ν1–4 = 2150, 966, 2151, 906 cm−1), in good agreement with available experimental data (1.480 Å and 2187, 975, 2191, 914 cm−1).76,77 Significantly, because of the negative partial charges of its H atoms (qH = −0.161 e), they are attracted by the positive charge centres of SiH3OH2+ (OH and Si) to form ionic SiH⋯HO and SiH⋯Si H-bonds.
 | ||
| Fig. 2 Calculated equilibrium structures (in Å and degrees) of SiH4, SiH3OH2+, SiH3OH2+–Ar(H), SiH3OH2+–Ar(Si), and SiH3OH2+SiH4(I–IV) in their ground electronic state (B3LYP-D3/aug-cc-pVTZ). | ||
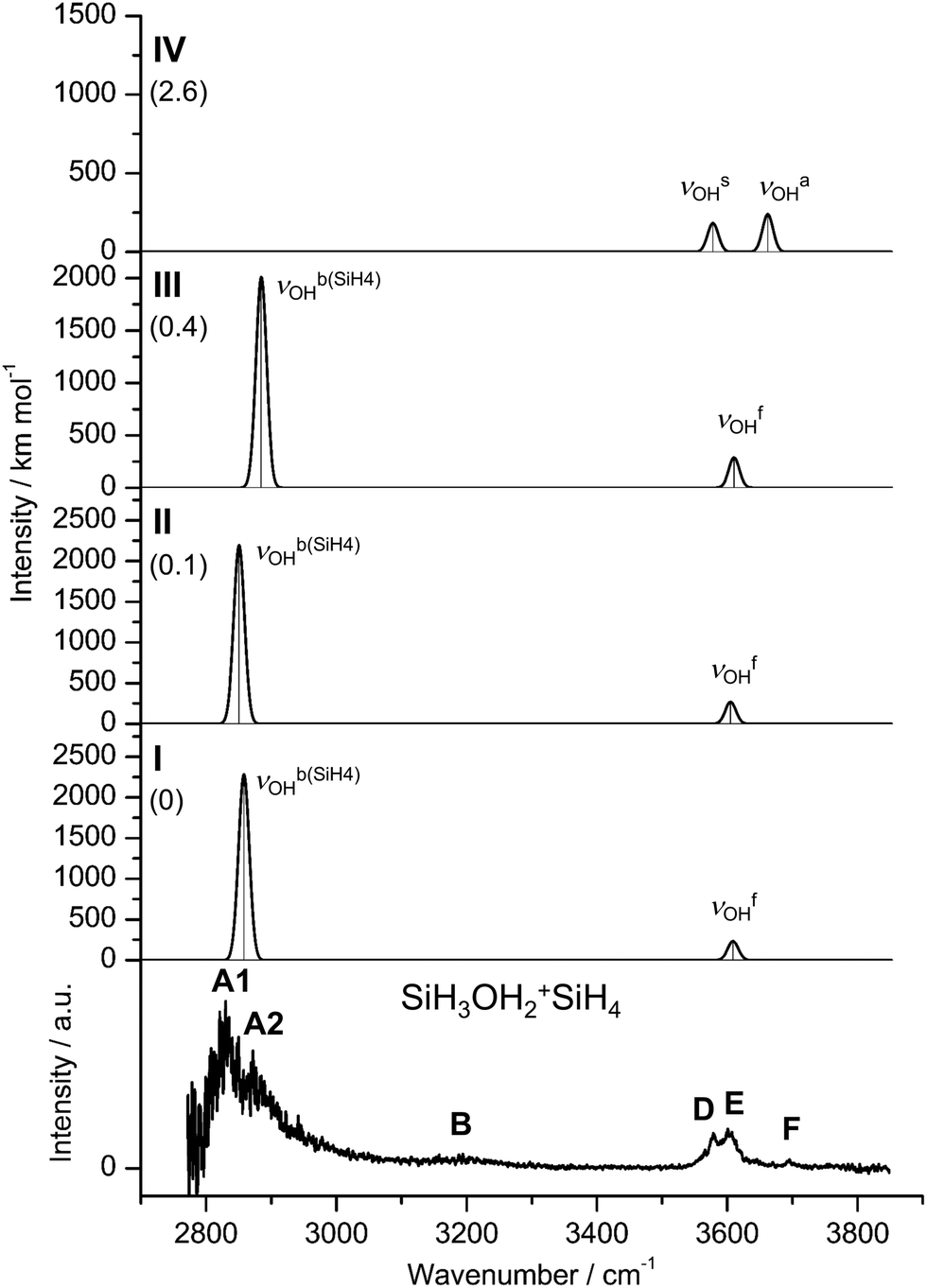 | ||
| Fig. 3 IRPD spectrum of SiH3OH2+SiH4 compared to linear IR absorption spectra of isomers I–IV calculated at the B3LYP-D3/aug-cc-pVTZ level. The positions of the transition observed in the IRPD spectrum of SiH3OH2+SiH4 and their vibrational assignment are listed in Table S2 (ESI†). Differences in relative energy (E0) are given in kJ mol−1 (in parentheses). | ||
In the three most stable isomers (I–III), SiH4 binds to one of the two equivalent OH groups of SiH3OH2+via a rather strong OH⋯HSi ionic DHB with very similar binding energies (D0 = 38.9, 38.7, 38.5 kJ mol−1, ΔD0 ≤ 0.4 kJ mol−1). These conformers differ mainly in the orientation of the SiH4 ligand. SiH4 binds with one of its negative H atoms (qH = −282, 286, −267 me) to a positive H atom (qH = 549, 548, 548 me) of the OH group at intermolecular bond distances of R = 1.397, 1.389, and 1.418 Å in almost linear OH⋯H ionic DHBs (θ = 173.3°, 175.8°, 175.0°). The SiH⋯H bond angles deviate significantly more from linearity (θ = 131.3°, 134.2°, 125.1°), indicating a rather small angular anisotropy of the potential for the orientation of the SiH3 group when optimizing dispersion and induction forces. The DHB involves substantial charge transfer from SiH3OH2+ to SiH4 (Δq = 81, 81, 80 me), consistent with the large E(2) energies describing the strong interaction between the bonding σSiH orbital and the antibonding σOH* orbital (99.4, 101.0, 96.1 kJ mol−1). Upon formation of the strong OH⋯HSi DHBs, both proton donor bonds are strongly elongated (ΔrSiH = 38, 38, 36 mÅ, ΔrOH = 38, 39, 36 mÅ). As a result, the νOHb modes are massively redshifted from νOHs = 3550 cm−1 in bare SiH3OH2+ down to νOHb(SiH4) = 2858, 2851, and 2885 cm−1 for I–III, respectively. These large redshifts of around 700 cm−1 are accompanied by a drastic increase in IR intensity by a factor of 10. In contrast, the free O–H bonds of all three isomers contract slightly (ΔrOH = −2 mÅ) causing corresponding minor blueshifts of the order of 20 cm−1 from the averaged free OH stretch frequency of SiH3OH2+ (3588 cm−1) to νOHf = 3608, 3605, and 3611 cm−1. These occur to the red of νOHa of SiH3OH2+ (3626 cm−1), because SiH4 complexation of one OH group removes the strong coupling between the two free and equivalent OH local modes in the monomer (ΔνOH = 76 cm−1). Furthermore, the DHB of SiH3OH2+ to SiH4 shortens the Si–O bond from 1.851 to 1.816 (I/III) and 1.815 Å (II), while the O–H–O bond angle increases slightly from 110.8° to 111.2°(I/III) and 111.0° (II).
In contrast to the DHB isomers I–III, SiH4 is attached in IV to the Si atom of SiH3OH2+via an ionic Si–H–Si (SiH···Si) CIHB (R = 1.898 Å, D0 = 36.2 kJ mol−1). This CIHB is only slightly less stable than the DHB by ΔE0 = 2.6 kJ mol−1 at the B3LYP-D3 level. Moreover, it is nonlinear (θ = 159.8°), as is typical for CIHBs,24 and may be considered as a rather asymmetric 3c–2e Si–H–Si bond (R = 1.535 and 1.898 Å). The Si–H proton donor bond is elongated from 1.484 to 1.535 Å (ΔrSiH = 51 mÅ). The NBO analysis reveals a larger charge transfer from SiH3OH2+ to SiH4 (159 me) and also a larger E(2) energy (167.9 kJ mol−1) from the bonding σSiH orbital to the lone pair orbital of Si (LPSi*) when compared to the DHB. In contrast to I–III, complexation with SiH4 in IV leads to a strong elongation of the Si–O bond by 98 mÅ and a minor contraction of the O–H bonds by 3 mÅ. As a result, both νOHa/s modes are blueshifted by 37 cm−1 to νOHs/a = 3587/3662 cm−1, while the coupling between both OH stretch oscillators remains similar (75 vs. 76 cm−1).
Comparison of the IRPD spectrum of SiH3OH2+SiH4 with the linear IR spectra computed for the four isomers I–IV in Fig. 3 immediately confirms the presence of isomers with a DHB, as the IRPD spectrum is dominated by the intense and strongly redshifted νOHb(SiH4) bands A1 and A2 caused by SiH4 binding to the OH2 group. The strongest transition A1 at 2830 cm−1 is assigned to the νOHb(SiH4) modes of I and II with deviations of 28 and 21 cm−1, while peak A2 at 2872 cm−1 may tentatively be attributed to the slightly less redshifted νOHb(SiH4) mode of III at 2885 cm−1. Alternatively, this weak satellite band may also arise from sequence hot bands of νOHb(SiH4) with intermolecular modes, which are typical for the excitation of proton donor stretch modes.70,74,75 The corresponding νOHf modes of I–III can be assigned to band E at 3602 cm−1 with minor deviations of 6, 3, and 9 cm−1. However, band F at 3695 cm−1 cannot be rationalized by any isomer with a DHB due to its high frequency. Instead, along with band D at 3578 cm−1, these transitions can arise from the coupled free OH stretch modes νOHs/a of IV predicted at 3587 and 3662 cm−1, respectively. From the integrated intensities of the bands A1/A2 and D (accounting for the shoulder of E) and the computed IR intensities, a population ratio of 10:1 can roughly be estimated for isomers with a DHB (I–III) and isomers with a CIHB (IV). The intensity ratios of bands D and E are consistent with this rough population ratio. While the predominant production of isomers I–III over IV is consistent with their slightly larger binding energies computed at the B3LYP-D3 level (D0 = 39 vs. 36 kJ mol−1), their rather large fractional abundance appears at first glance surprising in view of the similar computed stabilities. However, when considering the free entropy values, the ΔE0 difference between I and IV of 2.6 kJ mol−1 increases to ΔG0 = 4.1 kJ mol−1. Finally, their appear to be several more low-energy (local) DHB minima than CIHB minima, which may further enhance the population of the former type of isomers for statistical reasons. In addition, the DHB minima are doubly degenerate because of the two equivalent OH groups of SiH3OH2+, which may again favor the presence of DHB over CIHB isomers. To test whether the energy difference between both types of isomers changes with the computational level, single-point energy calculations are conducted at the CCSD(T)/aug-cc-pVTZ level, yielding actually a slightly larger binding energy (not corrected for zero-point energy) for IV than for I (De = 40.4 vs. 36.5 kJ mol−1). Similarly, the binding energies obtained at the GBS-QB3 level are also slightly in favor of IV (D0 = 34.4 vs. 33.0 kJ mol−1), while again the free energy values slightly favor I over IV (by 1.9 kJ mol−1). Hence, the considered computational levels predict rather similar binding energies for the DHB and CIHB isomers but entropy factors apparently favor the formation of DHB isomers, in agreement with the experimental observation. The weak transition B at 3194 cm−1 is probably an overtone or combination band and may be assigned for example to the first overtone of the βOH bending mode predicted at 3212, 3222, 3216, and 3228 cm−1 of I–IV neglecting anharmonicity effects. Finally, the B3LYP-D3 binding energies of 35–40 kJ mol−1 (∼2900–3350 cm−1) are of the same order as the photon energies of the transitions observed in the investigated spectra range (∼2800–3700 cm−1), indicating that single-photon dissociation of the SiH4 ligand is feasible for all isomers, even for those with no or only little rovibrational internal excitation.
 | ||
| Fig. 4 IRPD spectrum of SiH3OH2+SiH4–Ar compared to linear IR absorption spectra of I-Ar(I), IV-Ar(I) and I-Ar(II) together with their equilibrium structures (in Å) calculated at the B3LYP-D3/aug-cc-pVTZ level. The positions of the transition observed in the IRPD spectrum of SiH3OH2+SiH4–Ar and their vibrational assignment are listed in Table S3 (ESI†). Differences in relative energy (E0) are given in kJ mol−1. | ||
In the most stable I-Ar(I) isomer, Ar binds to the remaining free OH group of SiH3OH2+SiH4(I) via an OH⋯Ar H-bond (R = 2.228 Å, D0 = 14.0 kJ mol−1). Due to noncooperative three-body effects of interior ion solvation arising from enhanced charge delocalization, this H-bond is somewhat weaker than that in H-bonded SiH3OH2+–Ar with only one ligand (R = 2.176 Å, D0 = 16.1 kJ mol−1), with correspondingly smaller impact on the intramolecular properties of SiH3OH2+. The H-bond in I-Ar(I) slightly elongates the O–H proton donor bond by 7 mÅ and contracts the adjacent O–H bond by 5 mÅ, resulting in a νOHb(Ar) redshift of 160 cm−1 to 3448 cm−1 and a νOHb(SiH4) blueshift of 76 cm−1 to 2934 cm−1. As a consequence of the stronger O–H bond interacting with SiH4, Ar attachment leads to a destabilization of the OH⋯HSi DHB, which elongates by 16 mÅ. In the corresponding II/III-Ar(I) isomers (E0tot = 0.3 and 0.9 kJ mol−1), Ar is also H-bonded to the free OH group at similar distances (R = 2.223/2.226 Å) with similar binding energies (D0 = 13.8/13.5 kJ mol−1) and comparable shifts of the νOHb(Ar) (3442 cm−1) and νOHb(SiH4) modes (2925, 2958 cm−1) (Fig. S5 and S6, ESI†).
In the less stable I-Ar(II) isomer (E0 = 5.6 kJ mol−1), Ar is bound perpendicularly to the OH proton of I (θOHAr = 96.5°, R = 3.206 Å, D0 = 8.3 kJ mol−1) engaged in the DHB. As a result, this O–H bond contracts slightly (by 1 mÅ) leading to a small blueshift (by 12 cm−1) of the corresponding νOHb(SiH4) mode to 2870 cm−1 when compared to I. On the other hand, the DHB in I-Ar(II) is stronger than in I-Ar(II) (R = 1.401 vs. 1.413 cm−1), causing a larger redshift in νOHb(SiH4) (2870 vs. 2934 cm−1). In the corresponding II-Ar(II) isomer (E0tot = 5.9 kJ mol−1), Ar has a similar binding motif, resulting in a comparable blueshift of νOHb(SiH4) (2869 cm−1) so that it cannot be distinguished (Fig. S5, ESI†).
In the most stable IV-Ar(I) isomer featuring a Si–H–Si bond (E0tot = 5.3 kJ mol−1), Ar is slightly less H-bonded to one of the free OH groups (R = 2.287 Å, D0 = 11.3 kJ mol−1) than in I-Ar(I). This OH⋯Ar H-bond elongates the O–H proton donor bond by 6 mÅ and contracts the adjacent free O–H bond by 1 mÅ. As a result, νOHb(Ar) of IV-Ar(I) is redshifted by 105 cm−1 to 3482 cm−1 while νOHf is less redshifted by 29 cm−1 to 3633 cm−1. In IV-Ar(II) (E0tot = 5.6 kJ mol−1), Ar is bound to the other free OH group with a binding energy of D0 = 11.0 kJ mol−1, resulting in very similar shifts to νOHb(Ar) = 3482 cm−1 and νOHf = 3634 cm−1 as for IV-Ar(I).
In the less stable (I–III)-Ar(III) isomers (E0tot = 6.3, 6.4, 6.5 kJ mol−1), Ar is Si-bonded to the SiH3 group (D0 = 7.7, 7.8, 7.9 kJ mol−1), which affects the O–H bond of the DHB of I–III leading to small blueshifts of νOHb(SiH4) by 43, 45, and 45 cm−1 to 2901, 2896, and 2930 cm−1, respectively (Fig. S4–S6, ESI†). In IV-Ar(III) (E0tot = 10.7 kJ mol−1), Ar is bound almost perpendicularly to the H atom of the Si–H–Si bridge (R = 3.396 Å, D0 = 5.9 kJ mol−1, Fig. S7, ESI†). This affects the Si–H–Si bridge by shortening the Si–H and SiH⋯Si bonds by 2 and 3 mÅ but has almost no effect on the O–H bonds and their vibrational modes. The same is true for IV-Ar(IV) (E0tot = 12.6 kJ mol−1), in which Ar binds to the Si atom of the SiH4 ligand (R = 3.563 Å, D0 = 4.0 kJ mol−1). In (I–III)-Ar(IV) with E0tot = 10.1, 9.9, and 9.9 kJ mol−1, Ar is also bound to the SiH4 ligand (D0 = 3.9, 3.9, 3.6 kJ mol−1) leading to slight elongations (1–2 mÅ) of the O–H bonds involved in the DHB and small redshifts of the corresponding νOHb modes by 31, 29, and 28 cm−1 to 2827, 2822, and 2857 cm−1 for I-Ar(IV), II-Ar(IV) and III-Ar(IV), respectively (Fig. S4–S6, ESI†).
As mentioned above, the IR spectra predicted for (I–III)-Ar are almost identical. Hence, for simplicity only the IR spectra calculated for I-Ar(I,II) and IV-Ar(I) are compared in Fig. 4 to the IRPD spectrum of SiH3OH2+SiH4–Ar. However, the vibrational assignments for I-Ar(I,II) apply equally well to II-Ar(I,II) and III-Ar(I,II). The SiH3OH2+SiH4–Ar spectrum also shows a significant population of isomers with a DHB, as the strong transitions A1 and A2 can only be assigned to νOHb(SiH4) modes of (I–III)-Ar isomers. The blueshift of band A2 to 2910 cm−1 upon Ar-tagging can be explained by Ar binding to the free OH or SiH3 groups, which leads to a contraction of the O–H bonds involved in the DHB. Therefore, A2 is attributed to νOHb(SiH4) modes of the energetically favored (I–III)-Ar(I) isomers with deviations of 24, 15, and 48 cm−1. However, A2 could also be assigned to νOHb(SiH4) of (I–III)-Ar(III) (E0tot = 0.0, 0.3, 0.9 kJ mol−1), with deviations of 9, 14, and 20 cm−1. Moreover, the predicted redshifts of νOHb(Ar) of (I–III)-Ar(I) caused by Ar-tagging also agree well with transition C1 at 3456 cm−1, with only minor deviations of 8, 14, and 14 cm−1. The less blueshifted band A1 at 2869 cm−1 can only be explained by νOHb(SiH4) modes of (I–III)-Ar isomers, in which Ar has a minor effect on the O–H bond involved in the DHB. To this end, band A1 may be assigned to νOHb(SiH4) of (I/II)-Ar(II) (E0tot = 5.6 and 5.9 kJ mol−1) at 2870/2869 cm−1. However, band A1 could also be attributed to νOHb(SiH4) of (I–III)-Ar(IV) (E0tot = 10.1, 10.2, 10.8 kJ mol−1) although with larger deviations of 42, 47, and 12 cm−1, respectively. The associated νOHf modes of (I/II)-Ar(II) also agree well with band E at 3629 cm−1, with deviations of 14 and 18 cm−1. On the other hand, band E may also be assigned to νOHf of (I–III)-Ar(III/IV) with deviations of less than 20 cm−1. Transition C2 at 3495 cm−1 cannot be explained by isomers with a DHB (I–III) and thus can only be attributed to νOHb(Ar) modes of the energetically favored isomers IV-Ar(I,II) with Si–H–Si H-bonds (E0tot = 5.3 and 5.6 kJ mol−1) with minor deviations of 13 cm−1. The associated νOHf modes of IV-Ar(I,II) can also be attributed to band E with minor deviations of 4 and 5 cm−1. The IV-Ar(III,IV) isomers can be excluded, because their νOHs (3579/3580 cm−1) and νOHa modes (3663/3664 cm−1) are not observed. In summary, the measured IRPD spectrum of SiH3OH2+SiH4–Ar can be fully accounted for by the three lowest-energy isomers I-Ar(I,II) and IV-Ar(I) representing the DHB and the CIHB, although we cannot exclude the population of similar but less stable Ar isomers. From the experimental integrated peak areas and the calculated IR intensities of the bands A1/A2 and C2, the same population ratio as for bare SiH3OH2+SiH4 of 10:1 can roughly be estimated for (I–III)-Ar (DHB) and IV-Ar (CIHB), clearly favoring the DHB over the CIHB. The Ar binding energies of the isomers I–IV are in the range D0 = 11.3–14.0 kJ mol−1, while the SiH4 binding energies are in the range D0 = 36.2–38.9 kJ mol−1, yielding a total binding energy of the order of 50 kJ mol−1. This value is somewhat higher than the employed IR photon energy (<45 kJ mol−1) and thus can explain that IRPD of SiH3OH2+SiH4–Ar causes exclusively the loss of Ar (and not Ar plus SiH4). This consistency provides evidence that the computed interaction energies are in the correct range.
4. Further discussion
The analysis of the IRPD spectra of SiH3OH2+SiH4 clearly shows the preferential formation of the ionic OH⋯HSi DHB, while the population of isomers with a SiH⋯Si CIHB is substantially lower, in line with the respective computed SiH3OH2+⋯SiH4 interaction energies when accounting for entropy effects. The DHBs presented here correspond to the common definition of DHBs of the type Xδ−Hδ+⋯Hδ−Yδ+, where X is more electronegative than H whereas Y is more electropositive (ENX > ENH > ENY). In SiH4, the H atoms are bonded to the electropositive Si atom (Y), resulting in negatively charged H atoms that can combine with the positively charged H atoms bound to the electronegative O atom (X) of SiH3OH2+ to form a H-bond. The DHB can be associated with two MOs (HOMO−8/9) (Fig. S8, ESI†).In the ionic OH⋯HSi DHB, the calculated H⋯H distance (RHH = 1.4 Å) is much shorter than in neutral intermolecular DHBs (e.g., in amine-boranes and ReH5(PPh3)3indole) (RHH = 1.7–2.2 Å)46,53,56 but in a similar range of other ionic DHBs such as in Ph+DEMS and Ph+TEMS (RHH = 1.496–1.522 Å).66 Similar to other DHBs (θXHH = 150–170°, and θYHH = 95–115°/130),37,40,56 the O–H⋯H angle in the OH⋯HSi DHB is almost linear (θOHH = 173–176°) and the H⋯H–Si angle is bent (θSiHH = 125–134°). Due to the ionic character of the observed SiH⋯HO DHBs, the calculated binding energy (D0 = 38.5–38.9 kJ mol−1) is higher than those of neutral DHBs (16–25 kJ mol−1),33,53 but lower than of those of other ionic DHBs as, for example, observed in Ph+DEMS (D0 = 47.5–48.9 kJ mol−1) or Ph+TEMS (D0 = 49.1–50.9 kJ mol−1).66 The E(2) energies for the interaction between the bonding σSiH orbital and the antibonding σOH* orbital of SiH3OH2+SiH4 (E(2) = 96–101 kJ mol−1) also indicate slightly weaker DHBs compared to the DHBs of Ph+DEMS and Ph+TEMS (E(2) = 119/120 kJ mol−1).66 This view is also consistent with the experimentally observed redshifts of νOHb modes of SiH3OH2+SiH4 (Δν = 528/570 cm−1) compared to the more redshifted νOHb modes of Ph+DEMS and Ph+TEMS (Δν = 674 cm−1),66 which provide a direct experimental measure of the bond strength of the H-bonds. The DHBs of SiH3OH2+SiH4(I–III) appear to be slightly stronger than the CIHB in SiH3OH2+SiH4(IV) (D0 = 39 vs. 36 kJ mol−1 at B3LYP-D3), which is detected as a minor population. The H2O attached to the SiH3+ group significantly reduces the binding energy of the Si–H–Si H-bond preferred in silane ions, as can be seen by comparison with unperturbed Si2H7+ (D0 = 150 kJ mol−1) or Si3H11+ (D0 = 41 kJ mol−1) ions featuring one and two Si–H–Si H-bonds, respectively.26,29 Finally, the weak Si–H–Si H-bond of SiH3OH2+SiH4(IV) is strongly asymmetric and more linear compared to the much stronger symmetric bond in Si2H7+(RSiH = 1.535/1.898 vs. 1.625 Å, ϕSiHSi = 160° vs.144°).26,29
5. Conclusions
The analysis of IRPD spectra of mass-selected SiH3OH2+SiH4 and SiH3OH2+SiH4–Ar clusters in the OH stretch range (2700–3800 cm−1) using DFT calculations provides the first spectroscopic information about protonated silanol–silane complexes. The redshifted O–H stretch bands are clearly assigned to the energetically preferred structures (I–III) in which SiH4 is bonded to SiH3OH2+via a dihydrogen bond (DHB) of the type Siδ+Hδ−⋯Hδ+Oδ−. Significantly, this is the first spectroscopic and structural characterization of an ionic SiH⋯HO DHB in a small gas-phase cluster without any interference from aromatic or aliphatic hydrocarbon structures. In addition to the structures with a DHB, a small population of the order 10% of isomer IV with a charge-inverted hydrogen bond (CIHB) of the type Siδ+–Hδ−–Siδ+ is observed. Due to the OH2 group bonded to SiH3+, the CIHB is weaker than Si–H–Si H-bonds of related SixHy+ cations.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG, DO 729/9). The authors thank X. N. Truong for initial support in recording the IRPD spectra.References
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS.
- V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847–5910 CrossRef CAS.
- H. Chatham and A. Gallagher, J. Appl. Phys., 1985, 58, 159–169 CrossRef CAS.
- T. P. Martin and H. Schaber, J. Chem. Phys., 1985, 83, 855–858 CrossRef CAS.
- R. Singh, J. Phys.: Condens. Matter, 2008, 20, 045226 CrossRef.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2003, 90, 055502 CrossRef PubMed.
- P. D. Lickiss, Adv. Inorg. Chem., 1995, 42, 147–262 CrossRef CAS.
- J. Fischer, J. Baumgartner and C. Marschner, Science, 2005, 310, 825 CrossRef CAS PubMed.
- J. Nawrocki, J. Chromatogr. A, 1997, 779, 29–71 CrossRef CAS.
- M. J. Kushner, J. Appl. Phys., 1993, 74, 6538–6553 CrossRef CAS.
- M. J. Kushner, J. Appl. Phys., 1988, 63, 2532–2551 CrossRef CAS.
- M. L. Mandich, W. D. Reents and M. F. Jarrold, J. Chem. Phys., 1988, 88, 1703–1718 CrossRef CAS.
- G. Turban, Y. Catherine and B. Grolleau, Plasma Chem. Plasma Process., 1982, 2, 61–80 CrossRef CAS.
- D. M. Goldhaber and A. L. Betz, Astrophys. J., 1984, 279, L55–L58 CrossRef CAS.
- S. Patai and Z. Rappoport, Chem. Org. Silicon Compd., Wiley, Chichester, 1989 Search PubMed.
- K. Raghavachari, J. Chem. Phys., 1988, 88, 1688–1702 CrossRef CAS.
- K. Raghavachari, J. Chem. Phys., 1990, 92, 452–465 CrossRef CAS.
- L. A. Curtiss, H. Brand, J. B. Nicholas and L. E. Iton, Chem. Phys. Lett., 1991, 184, 215–220 CrossRef CAS.
- B. Ruscic and J. Berkowitz, J. Chem. Phys., 1991, 95, 2416–2432 CrossRef CAS.
- G. Maier, H. P. Reisenauer and J. Glatthaar, Chem. - Eur. J., 2002, 8, 4383–4391 CrossRef CAS PubMed.
- R. L. DeKock and W. B. Bosma, J. Chem. Educ., 1988, 65, 194–197 CrossRef CAS.
- J. E. McMurry and T. Lectka, Acc. Chem. Res., 1992, 25, 47–53 CrossRef CAS.
- M. Jablonski, Chem. Phys. Lett., 2009, 477, 374–376 CrossRef CAS.
- M. Jabłoński, Struct. Chem., 2020, 31, 61–80 CrossRef.
- S. Civiš, M. Lamanec, V. Špirko, J. Kubišta, M. Špet’ko and P. Hobza, J. Am. Chem. Soc., 2023, 145, 8550–8559 CrossRef PubMed.
- M. Savoca, J. Langer and O. Dopfer, Angew. Chem., Int. Ed., 2013, 52, 1568–1571 CrossRef CAS PubMed.
- M. A. R. George and O. Dopfer, Int. J. Mass Spectrom., 2019, 435, 51–60 CrossRef CAS.
- M. A. R. George, M. Savoca and O. Dopfer, Chem. – Eur. J., 2013, 19, 15315–15328 CrossRef CAS PubMed.
- M. A. R. George and O. Dopfer, Phys. Chem. Chem. Phys., 2024, 26, 6574–6581 RSC.
- M. A. R. George, N. X. Truong, M. Savoca and O. Dopfer, Angew. Chem., Int. Ed., 2018, 57, 2919–2923 CrossRef CAS PubMed.
- R. Withnall and L. Andrews, J. Phys. Chem., 1985, 89, 3261–3268 CrossRef CAS.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, J. Phys. Chem. A, 2004, 108, 5823–5830 CrossRef CAS.
- S. J. Grabowski, J. Phys. Org. Chem., 2013, 26, 452–459 CrossRef CAS.
- R. H. Crabtree, Science, 1998, 282, 2000–2001 CrossRef CAS.
- R. H. Crabtree, P. E. M. Siegbahn, O. Eisenstein, A. L. Rheingold and T. F. Koetzle, Acc. Chem. Res., 1996, 29, 348–354 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2016, 116, 8750–8769 CrossRef CAS PubMed.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS PubMed.
- X. Chen, J.-C. Zhao and S. G. Shore, Acc. Chem. Res., 2013, 46, 2666–2675 CrossRef CAS PubMed.
- B. G. de Oliveira, Phys. Chem. Chem. Phys., 2013, 15, 37–79 RSC.
- T. Kar and S. Scheiner, J. Chem. Phys., 2003, 119, 1473–1482 CrossRef CAS.
- A. Filippi, A. Troiani and M. Speranza, J. Phys. Chem. A, 1997, 101, 9344–9350 CrossRef CAS.
- G. N. Patwari, J. Phys. Chem. A, 2005, 109, 2035–2038 CrossRef CAS PubMed.
- G.-J. Zhao and K.-L. Han, J. Chem. Phys., 2007, 127, 024306 CrossRef PubMed.
- N. Mohan and C. H. Suresh, J. Phys. Chem. A, 2014, 118, 1697–1705 CrossRef CAS PubMed.
- V. Sumerin, F. Schulz, M. Nieger, M. Atsumi, C. Wang, M. Leskelä, P. Pyykkö, T. Repo and B. Rieger, J. Organomet. Chem., 2009, 694, 2654–2660 CrossRef CAS.
- J. Wessel, J. C. Lee Jr, E. Peris, G. P. A. Yap, J. B. Fortin, J. S. Ricci, G. Sini, A. Albinati, T. F. Koetzle, O. Eisenstein, A. L. Rheingold and R. H. Crabtree, Angew. Chem., Int. Ed. Engl., 1995, 34, 2507–2509 CrossRef CAS.
- C. A. Morrison and M. M. Siddick, Angew. Chem., Int. Ed., 2004, 43, 4780–4782 CrossRef CAS PubMed.
- Y. Meng, Z. Zhou, C. Duan, B. Wang and Q. Zhong, J. Mol. Struct.: THEOCHEM, 2005, 713, 135–144 CrossRef CAS.
- Y. Feng, S.-W. Zhao, L. Liu, J.-T. Wang, X.-S. Li and Q.-X. Guo, J. Phys. Org. Chem., 2004, 17, 1099–1106 CrossRef CAS.
- S. Marincean and J. E. Jackson, J. Phys. Chem. A, 2004, 108, 5521–5526 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1999, 103, 8861–8869 CrossRef CAS.
- E. Peris, J. C. Lee Jr and J. R. Rambo, J. Am. Chem. Soc., 1995, 117, 3485–3491 CrossRef CAS.
- T. Richardson, S. de Gala, R. H. Crabtree and P. E. M. Siegbahn, J. Am. Chem. Soc., 1995, 117, 12875–12876 CrossRef CAS.
- A. J. Lough, S. Park, R. Ramachandran and R. H. Morris, J. Am. Chem. Soc., 1994, 116, 8356–8357 CrossRef CAS.
- J. C. Lee Jr, E. Peris, A. L. Rheingold and R. H. Crabtree, J. Am. Chem. Soc., 1994, 116, 11014 CrossRef.
- W. T. Klooster, T. F. Koetzle, P. E. M. Siegbahn, T. B. Richardson and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 6337–6343 CrossRef CAS.
- G. N. Patwari, T. Ebata and N. Mikami, J. Chem. Phys., 2000, 113, 9885–9888 CrossRef.
- G. Naresh Patwari, T. Ebata and N. Mikami, J. Chem. Phys., 2001, 114, 8877–8879 CrossRef CAS.
- G. N. Patwari, T. Ebata and N. Mikami, J. Phys. Chem. A, 2001, 105, 8642–8645 CrossRef CAS.
- G. N. Patwari, T. Ebata and N. Mikami, Chem. Phys., 2002, 283, 193–207 CrossRef CAS.
- G. Naresh Patwari, T. Ebata and N. Mikami, J. Chem. Phys., 2002, 116, 6056–6063 CrossRef CAS.
- G. N. Patwari, A. Fujii and N. Mikami, J. Chem. Phys., 2006, 124, 241103 CrossRef PubMed.
- P. C. Singh and G. N. Patwari, J. Phys. Chem. A, 2007, 111, 3178–3183 CrossRef CAS PubMed.
- H. Ishikawa, A. Saito, M. Sugiyama and N. Mikami, J. Chem. Phys., 2005, 123, 224309 CrossRef PubMed.
- M. Uchida, T. Shimizu, R. Shibutani, Y. Matsumoto and H. Ishikawa, J. Chem. Phys., 2020, 153, 104305 CrossRef CAS PubMed.
- H. Ishikawa, T. Kawasaki and R. Inomata, J. Phys. Chem. A, 2015, 119, 601–609 CrossRef CAS PubMed.
- S.-W. Hu, Y. Wang, X.-Y. Wang, T.-W. Chu and X.-Q. Liu, J. Phys. Chem. A, 2004, 108, 1448–1459 CrossRef CAS.
- Y. Kawashima, R. D. Suenram and E. Hirota, J. Chem. Phys., 2016, 145, 114307 CrossRef.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- M. Fujii and O. Dopfer, Int. Rev. Phys. Chem., 2012, 31, 131–173 Search PubMed.
- O. Dopfer and M. Fujii, Chem. Rev., 2016, 116, 5432–5463 CrossRef CAS PubMed.
- M. J. Frisch, et al., GAUSSIAN16, revision C.02, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- N. Solcà and O. Dopfer, Chem. Phys. Lett., 2000, 325, 354–359 CrossRef.
- R. V. Olkhov and O. Dopfer, Chem. Phys. Lett., 1999, 314, 215–222 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standards and Technology, Gaithersburg, MD, 2001, https://webbook.nist.gov/, 20899 Search PubMed.
- D. R. J. Boyd, J. Chem. Phys., 2004, 23, 922–926 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02428a |
| This journal is © the Owner Societies 2024 |