
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energy transfer in near-infrared photoluminescent PbS/CdS quantum dot-based three-dimensional networks and films†
Denis
Pluta
 ac,
Rebecca T.
Graf
acd,
Dirk
Dorfs
bcde and
Nadja C.
Bigall
*bcde
ac,
Rebecca T.
Graf
acd,
Dirk
Dorfs
bcde and
Nadja C.
Bigall
*bcde
aInstitute of Physical Chemistry and Electrochemistry, Leibniz University Hannover, Callinstraße 3A, Hannover 30167, Germany
bInstitute of Physical Chemistry, University Hamburg, Grindelallee 117, Hamburg 20146, Germany. E-mail: nadja-carola.bigall@uni-hamburg.de
cLaboratory of Nano and Quantum Engineering, Leibniz University Hannover, Schneiderberg 39, Hannover 30167, Germany
dCluster of Excellence PhoenixD, Leibniz University Hannover, Hannover 30167, Germany
eCluster of Excellence CUI: Advanced Imaging of Matter, University Hamburg, Hamburg 20146, Germany
First published on 21st September 2024
Abstract
Nanocrystal (NC) assemblies, such as films or NC-based three-dimensional networks, provide insight into the nanoscopic properties of their building blocks, and are macroscopic materials. In NC assemblies in general, interactions between the building blocks have to be considered, since a decrease of the distance between the NCs allows phenomena such as energy transfer to occur. Novel quantum dot-based aerogels with optical properties in the near-infrared (NIR) region are not characterized in terms of these important properties in the literature, while the structural complexity of these networks raises questions about the interactions within. Since knowledge about the physical phenomena is vital for applications, we here investigate the photoluminescence (PL) of PbS/CdS QD-based assemblies, namely drop-cast films and aerogels with steady-state measurements at cryogenic temperatures and time-resolved measurements at room temperature. We find multiple emissive in-gap states (IGS), and a correlation between the number of nearest QD neighbors, the distance between the QDs in the assemblies and the non-radiative recombination rate by linking the observations to different energy transfer phenomena.
Introduction
The assembly of nanocrystals into macroscopic materials and the investigations of their properties are key steps to access their nanoscopic properties in applications. Next to assembly techniques where NCs are grouped into close packed films or incorporated in different materials, the formation of self-supporting three-dimensional networks, so-called aerogels, solely based on the NCs themselves, represent a very promising method. It introduces more variables for material optimization and additional properties like porosity, which are not possible to achieve with other assemblies.1,2To date, lots of scientific efforts have been put into the investigation of quantum dot-based aerogels, while the main focus was on the visible and ultraviolet range of the electromagnetic spectrum. On the other hand, aerogels synthesized from NIR active NCs remain vastly unexplored.1,3 Especially the investigation of their optical properties is interesting, since they can give compelling insight into the underlying electronic properties and energy transfer mechanisms. Such phenomena have previously been reported in the literature for other assembly types based on PbS and PbS/CdS NCs, which are the NC building blocks used in the present work.4–9
The insights gained by performing in depth optical characterization allow understanding of the present energy transfer phenomena and electronic structure of the materials studied, enabling future tailoring of these materials for potential applications in solar cells, photocatalysis, electrochemistry, sensing and more.10–16
Results and discussion
Synthesis of particles and assemblies
The synthesis of the PbS/CdS QDs and aerogels has been described in detail in a previous publication and in the Experimental section in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 We first synthesized PbS QDs via hot-injection based on the method from Hines and Scholes.18,19 The introduction of the CdS shell was then carried out by exchanging the lead ions in the outer layers of the PbS QDs with cadmium ions based on a method from Zhao et al.20 The resulting PbS/CdS QDs showed PL centered at 1142 nm (1.086 eV) with a PLQY of 29.8%. The as-synthesized QDs are stabilized with oleic acid and are stable in non-polar solvents. Since the gelation of the QD dispersions takes place in aqueous solution, a ligand exchange has to be performed, using 3-mercaptopropionic acid (MPA) and 11-mercaptoundecanoic acid (MUA), which was modified from the literature.21 The resulting QDs were dispersed in 0.01 M aqueous KOH. The formation of the three-dimensional network structures was initiated by the addition of an aqueous solution of YCl3, leading to the controlled destabilization of the QDs in solution.22 This leads to the formation of the networks, either by coalescence and the formation of crystal contacts between the building blocks or via linked surface ligands. The solvent of the hydrogels was then exchanged from water via anhydrous acetone to liquid CO2. After supercritical drying, the final aerogel materials were received.
17 We first synthesized PbS QDs via hot-injection based on the method from Hines and Scholes.18,19 The introduction of the CdS shell was then carried out by exchanging the lead ions in the outer layers of the PbS QDs with cadmium ions based on a method from Zhao et al.20 The resulting PbS/CdS QDs showed PL centered at 1142 nm (1.086 eV) with a PLQY of 29.8%. The as-synthesized QDs are stabilized with oleic acid and are stable in non-polar solvents. Since the gelation of the QD dispersions takes place in aqueous solution, a ligand exchange has to be performed, using 3-mercaptopropionic acid (MPA) and 11-mercaptoundecanoic acid (MUA), which was modified from the literature.21 The resulting QDs were dispersed in 0.01 M aqueous KOH. The formation of the three-dimensional network structures was initiated by the addition of an aqueous solution of YCl3, leading to the controlled destabilization of the QDs in solution.22 This leads to the formation of the networks, either by coalescence and the formation of crystal contacts between the building blocks or via linked surface ligands. The solvent of the hydrogels was then exchanged from water via anhydrous acetone to liquid CO2. After supercritical drying, the final aerogel materials were received.
In addition to the aerogels, closed-packed films of drop-cast QDs were also synthesized for comparison of their optical properties with those of the investigated gel type materials. They were prepared using the same PbS/CdS QDs, stabilized with MUA and MPA, as described above for the aerogel synthesis. As a substrate for the QD films, functionalized ITO slides were used, on which a scotch tape mold with a diameter of 6 mm was placed. In this mold a specific amount of the aqueous PbS/CdS QDs was drop-cast and dried under ambient conditions. All prepared assemblies of PbS/CdS QDs, including aerogels and films, were stored in a nitrogen filled glovebox.
Cryogenic photoluminescence measurements
In order to gain in depth knowledge about the electronic properties and structure and the influence the different assemblies have onto them, we measured the PL of the QD films at low temperatures, down to 4.2 K. Investigations of the low-temperature optical properties of PbS/CdS QDs and their assemblies are not very common in the literature, but since only the PbS core is optically active, we also reference previous studies of PbS core-only QDs here. It was previously shown that PbS QDs exhibit multiple emissive states.5,8,23–25 For PbS QDs Litvin et al. reported two emissive states, the 1s1s and an additional in-gap state (IGS). Depending on the size of the QDs studied, the population of these states changed, leading to the observation of PL from both states only for QDs between 4.4 and 6 nm in size. Bigger and smaller QDs only showed PL from either the 1s1s or the IGS, respectively. In addition, they observed a splitting of the 1s1s band only for QDs with a size of 6 nm and smaller.8 Kim et al. observed three PL components for PbS QDs and attributed them to the recombination from shallow traps near the 1s1s state and from two IGS, that are related to defects.24In Fig. 1 the PL measurements at 295 K, 150 K and 4.2 K are shown. All PL spectra were fit with Gaussian functions to determine the influence of the individual states on the overall PL and show their temperature dependencies. All fitting parameters can be found in Tables S1–S11 in the ESI.†
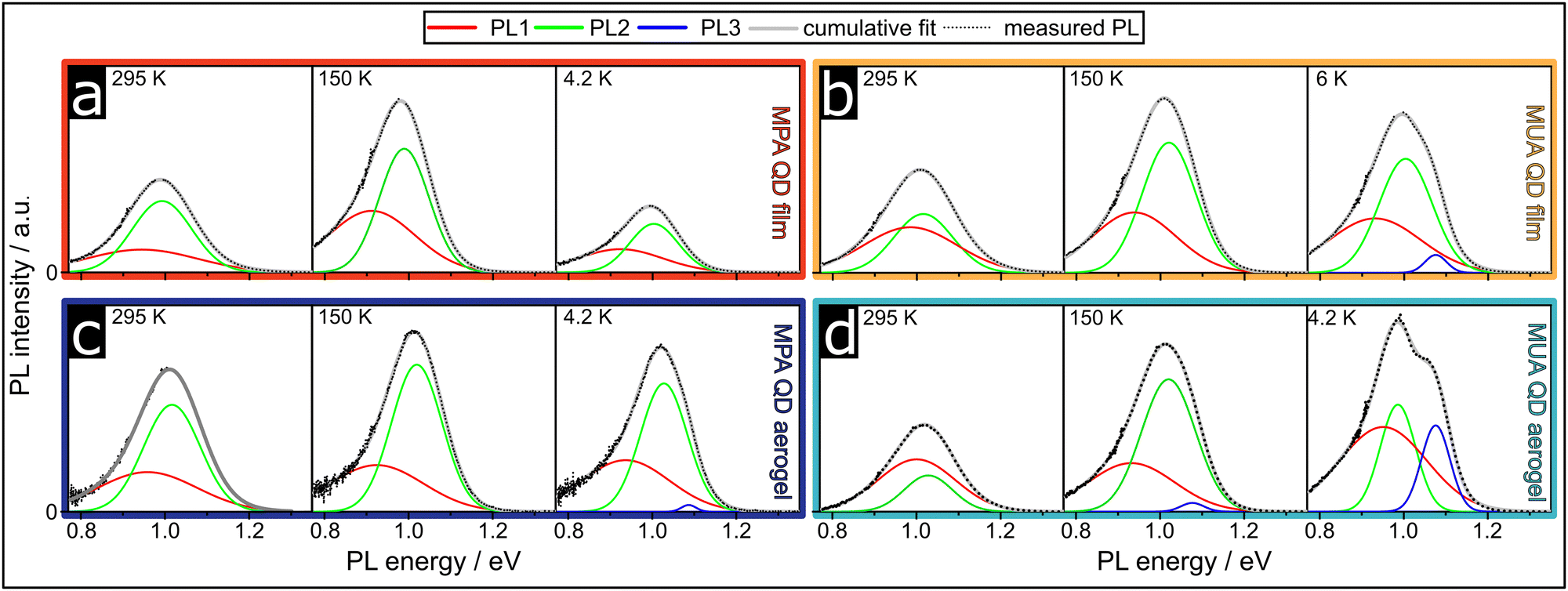 | ||
| Fig. 1 At lower temperatures the evolution of a third PL component for the MUA QD film, MPA QD film and MUA QD aerogel can be observed. Emission spectra measured at three temperatures for the (a) MPA QD film, (b) MUA QD film, (c) MPA QD aerogel and (d) MUA QD aerogel. The raw data were Jacobian and line shape corrected and fitted with two or three Gaussian contributions, indicated as PL1, PL2 and PL3. | ||
At room temperature, all four samples show two emissive states (PL1 and PL2) and their FWHM and peak center energy decrease with decreasing temperature, which was also observed in the literature for PbS QD assemblies of a similar size.8,23
The observation of two emissive states does not change for the MPA QD film with decreasing temperature and even the relative PL peak areas of PL1 and PL2 do not change significantly from 37% to 46% for PL1 from 295 K to 4.2 K. The relative PL peak areas for all samples and measured temperatures can be found in Fig. S1 in the ESI.†
In contrast, the MUA QD film and both the MUA and MPA QD aerogels show the evolution of a third PL component (PL3) at lower temperatures. While the relative PL peak area of PL 3 for the MPA QD aerogel and MUA QD film is rather small with 1% and 3%, respectively, it reaches almost 20% for the MUA QD aerogel at 4.2 K. The variation in the relative PL peak area of PL3 with decreasing temperatures is rather low, but the temperature it is first observed at is different for the three samples. While it is only observable at 40 K and lower for the MPA QD aerogel, it can already be observed at 90 K for the MUA QD film and even 150 K for the MUA QD aerogel (see Fig. S1 in the ESI†). The fact that PL3 is only observable at quite low temperatures could also explain why the observation of three PL components for PbS and PbS/CdS QDs in the literature is rather rare. Even though there are a number of cryogenic PL investigations published, a lot of them only show PL measurements down to 77 K, while in our measurements PL3 is not necessarily measurable yet.8,23
In Fig. 2 our proposed Jabłoński diagram for our studied PbS/CdS QD assemblies is shown, including the recombination pathways of the three PL components. Similar electronic structures have been proposed by Litvin et al. and Kim et al.8,24 They and other authors connect PL1 to the radiative recombination from IGS1 to the ground state. IGS1 is attributed to a defect state or a defect-induced electron or hole surface-trap.8,24 PL2 is connected to the recombination from IGS2 to the ground state, where IGS2 is either also attributed to a defect state or to the splitting of the 1s1s state by size-dependent intervalley splitting or electron–hole exchange interactions.8,26,27 Our interpretation of our findings is that the electronic structure as originally proposed by Litvin et al. and Kim et al. can be confirmed, and we also found the PL3 component, that was not observed by Litvin et al. That being said, we did not observe the PL3 component in all the samples, indicating that the recombination from the 1s1s state to the ground state, as proposed by Kim et al., depends on the complexity of the influencing factors, which have not been unraveled yet. Our findings suggest that surface effects and effects based on the coupling of QDs within the assemblies are relevant factors. The exact parameters leading to the radiative recombination from the third state cannot be revealed with the present study, nevertheless possible effects can be discussed. The intensity of the third state is highest for the MUA QD aerogel, while small contributions were also observed for the MPA QD aerogel and the MUA QD film. Since the third state can be observed in both aerogels, the influence of the gelation or drying process on the QD morphology is evident. While the gelation leads to the fusion of QDs into complex and branched structures, the supercritical drying is not expected to result in significant changes to the morphology. The formation of subpopulations is also possible, which differ in the extent the gelation process influences their morphology and even the distance to other QD neighbors. Nevertheless, gelation cannot be the only relevant factor, since the third component can also be observed within the MUA QD film.
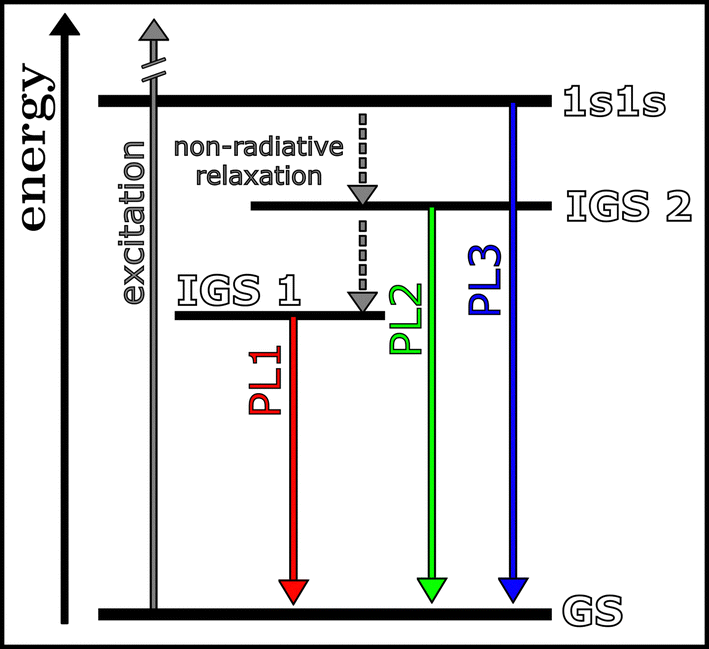 | ||
| Fig. 2 Jabłoński diagram indicating the origin of the three PL components, which were observed at low temperatures. The recombination takes place from three states (1s1s, in gap state (IGS) 1 and in gap state (IGS) 2) to the ground state (GS). | ||
Another discernible parameter is the ligand coverage, ligand removal, and associated surface defect states. Since ligands are removed during gelation to facilitate the destabilization and attachment, surface atoms can be present with unsaturated dangling bonds, which can act as defect sites.28 This hypothesis is supported by the claim from Kim et al., attributing the two in-gap states to defects.24
In addition, the internal conversion from the 1s1s state to IGS1 and IGS2 is favored in general, leading to the depopulation of the 1s1s state, and therefore less radiative recombination from the 1s1s state. One of the parameters relevant for the internal conversion rate is the difference in energy ΔE between the two states, in this case the 1s1s state and IGS2, as stated by the energy gap law.29,30 We can estimate the energy difference between 1s1s and IGS2 by calculating the difference between the PL energy position of the PL2 and PL3 peak. The calculated average difference (calculated from the Gaussian fits of the measurements taken between 4.2 and 40 K) for the MPA QD aerogel, the MUA QD film and the MUA QD aerogel are 59.6 meV, 67.8 meV and 88.7 meV, respectively. Sung et al. previously investigated the trapping rate in the InP/ZnSe/ZnS QDs and showed the expected exponential dependency of the trapping rate and the energy difference between the band edge and trap state. They showed that an increase in ΔE from 87 meV to 111 meV decreases the trapping rate by roughly 40%.31 Since we found a similar difference in ΔE, the values from the literature are applicable here and indicate the influence that the seemingly small changes in ΔE can have.
This explanation is assuming that the individual QDs are independent from each other, which is certainly not the case for the assemblies studied here, due to the close proximity of the QDs. It has often been described in the literature that homo FRET or NRET is observed in PbS and PbS/CdS nanocrystal-based assemblies. The transfer of charge carriers from smaller QDs to bigger QDs introduces an additional mechanism that influences the population of the relative emissive states and demands a closer look at the recombination and transfer rates.
In Fig. S2 in the ESI† the temperature-dependent integrated PL intensity, normalized to the value at 295 K is shown. The trend observed for the MPA QD film is qualitatively similar to the observations made by Lü et al.4 At first the intensity increases with decreasing temperature and then decreases again. This was explained by the different temperature-dependencies of the non-radiative recombination and energy transfer rates, leading to a maximum of total PL intensity at intermediate temperature, where the non-radiative recombination rate is already decreased but energy transfer is still possible. This model is not completely in agreement with the proposed model in this study, because the authors propose that only non-radiative recombination from IGS1 to the ground state or energy transfer to bigger QDs in the population, acting as acceptors, is possible. We also observe radiative recombination from IGS1, but in general the temperature dependency of the relevant recombination rates can also explain our findings. The relative contributions of the three PL components show that the ratio between PL1 and PL2 for the MPA QD film and aerogel does not change drastically when changing the temperature. Since the ratio between the radiative and non-radiative recombination rates directly influence the PL intensities, or more precisely the PLQY, both show different temperature dependencies or at least the non-radiative recombination rates show a more complex temperature dependency. Since the non-radiative recombination rate consists of multiple processes, this is very likely and a deeper investigation of the kinetics of the charge carrier recombination is necessary to obtain more insight into the underlying recombination processes.
The observations made for the MUA QD aerogel are different. Instead of a decrease in the PL intensity at intermediate temperatures, it plateaus between 150 and 70 K and then increases again below 70 K with a sharp increase below 20 K. The PL of the MUA QD film shows a similar temperature dependency, even though a small decrease instead of a plateau can be observed. The MPA QD aerogel shows both a decrease at intermediate temperatures and an increase at low temperatures.
It is likely that the initial increase in the integrated PL intensity, observed for all samples, is attributed to the reduction in the non-radiative recombination rate, as proposed before.4 The decrease in the PL intensity observed for the MPA QD aerogel and especially the MPA QD film at low temperatures could be linked to the energy transfer rate and its temperature dependency, since the assembly structure, and therefore the energy transfer mechanisms, are expected to be different.
Fluorescence lifetime measurements
The investigation of PL lifetimes can lead to a deeper understanding of the recombination processes of the studied material and give insight into the kinetics of the different recombination pathways. In particular, the kinetics of the energy transfer processes within the QD population are significant, which has been shown in the literature by measuring the PL lifetimes at multiple emission bands, corresponding to different sizes of QD within the population.4–7,32 This allows investigation of the size-dependent recombination kinetics of the QDs within the assembly. We measured the PL lifetime at 5 positions of the emission bands of the MUA and MPA QD films and aerogels and their PLQY. The results are shown in Fig. 3a together with the corresponding PL spectra in Fig. 3c.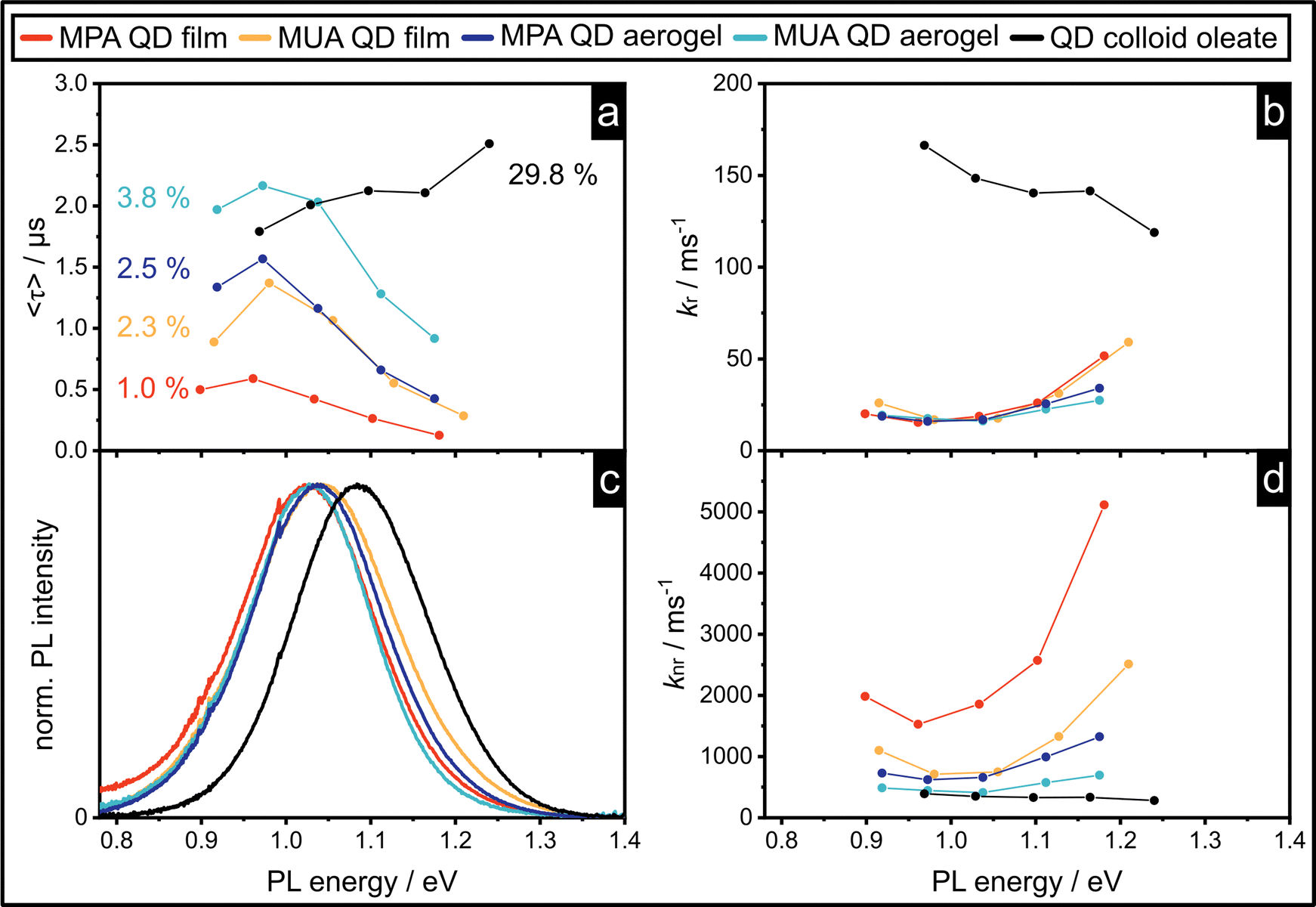 | ||
| Fig. 3 The radiative recombination rates of all interacting samples are similar, while the non-radiative recombination rates show significant differences manifesting in the measured PLQYs and fluorescence lifetimes. (a) and (c) Emission spectra and amplitude weighted average lifetimes of the colloids, QD films and aerogels measured at five positions of the emission peak of the respective sample. The measured PLQY of the sample is indicated. (b) and (d) Radiative recombination rates, kr, and non-radiative recombination rates, knr, calculated with equation (3) and (4), respectively. | ||
For the colloidal, oleate stabilized PbS/CdS QD sample the amplitude-weighted average PL lifetime decreases, as the PL energy decreases and the particles increase in size, which has been described before.25 In contrast, the PL lifetime of the assemblies increases with increasing size of the QDs, which can be explained by energy transfer within the population. The smaller QDs act as donors and the energy is transferred to the bigger QDs. This additional fast, non-radiative process decreases the lifetime of the smaller donor QDs. Since the probability of this process decreases the bigger the QDs are within the population, the energy transfer rate kET decreases with increasing size. This leads to the observed decreasing PL lifetime with increasing QD size. This size dependency is underlined by the fact that the smaller QDs show bi-exponential PL decay, while the bigger QDs show mono-exponential behavior4,7 (see Table S12 in the ESI†). While the smaller QDs show two lifetime components, which can be defined as τ1 = (kr + kET)−1 and τ2 = (kr + knr)−1, the bigger QDs only show one component with τ2 = (kr + knr)−1, which is in agreement with models in the literature.4
Even though the qualitative trend of the size dependency and the number of lifetime components is similar for all PbS/CdS QD assemblies, there is a significant difference between the absolute lifetimes measured for each sample. Only mentioning the extreme cases, they are in the range of 100–500 ns for the MPA QD film and 1–2 μs for the MUA QD aerogel. Additionally, the PLQY increases in the same order as the PL lifetimes, from the MPA QD film to MUA QD film to MPA QD aerogel to MUA QD aerogel.
In Fig. 3b and d and in Table 1 the radiative and non-radiative recombination rates kr and knr are shown, which have been calculated from the PL lifetime τ and the PLQY Φ by using eqn (1)–(4).
 | (1) |
 | (2) |
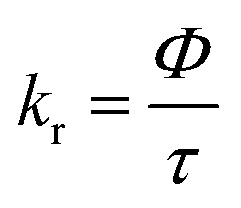 | (3) |
| knr = τ−1 − kr | (4) |
| Sample | Average kr/ms−1 | Average knr/ms−1 |
|---|---|---|
| QD colloid oleate | 143 | 337 |
| MUA QD aerogel | 21 | 522 |
| MPA QD aerogel | 22 | 866 |
| MUA QD film | 30 | 1280 |
| MPA QD film | 26 | 2611 |
The distinction between radiative (kr) and non-radiative (knr) recombination rates shows that kr is very similar for all assemblies (Table 1), which is plausible since the radiative recombination process remains the same in all four samples, because all assemblies consist of the same PbS/CdS QDs stabilized with thiol ligands. At the same time, a significant difference between the calculated value of kr for the PbS/CdS QD oleate colloid in comparison to the assemblies is observed. This is most likely caused by the inhomogeneous quenching of the colloidal QDs after undergoing ligand exchange and transfer to the aqueous phase. knr on the other hand, shows a significant difference for the four assembled samples (Table 1). It increases from the MUA QD aerogel to MPA QD aerogel to MUA QD film to MPA QD film, which leads to the decrease in the average amplitude weighted lifetime (〈τ〉) in the same order. As mentioned before, energy transfer from smaller to larger QDs within the QD population significantly influences the size-dependency of the PL lifetimes. Since the assemblies have different microstructures, the possible energy transfer mechanisms are expected to be different, and we propose these different mechanisms and structures to be the cause of the differences in knr. The calculation of kr and knr is expected to include an error, based on the significant deviations of the optical properties of the QDs within the population. While the PL lifetime is size dependent, the PLQY can only be measured for the whole population. In reality, the PLQY will most likely also show size-dependency. For this reason, the average values of kr and knr have been calculated and shown in Table 1 in order to increase the relevance of the comparison between the samples. This allows a qualitative assessment of the data and the observed phenomena within the assemblies.
Thinking about the influence of the assembly structure on energy transfer phenomena, there are two parameters that have the biggest influence: the distance between the donor and acceptor and the probability of an acceptor being close to a donor.
The distance is rather straight forward, especially regarding the MUA and MPA QD film, since the ligand length will be the relevant parameter here. MUA and MPA are chemically very similar, since they both have a thiol and a carboxylic acid group at each end, but the carbon chain length is significantly different, with a C3-chain for MPA and a C11-chain for MUA. Regarding the Tanford formula, these chain lengths translate to a fully stretched ligand length (omitting the functional groups) of roughly 1.5 nm and 0.5 nm for MUA and MPA respectively.33 In close-packed films the QDs are only separated by their ligand shell, leading to the expectation that the QDs in the MPA QD film are very close to each other (∼1 nm), while in the MUA QD film, this distance is expected to be greater (∼3 nm). In principle, the same is true for the aerogels, but here the network formation is also very important. We previously discussed the structural differences when using MUA and MPA and the results indicate that the ligand removal and formation of crystal–crystal contacts is significantly more efficient with MPA ligands. One of the reasons will be the length of the ligands again, since the QDs are simply closer to each other once the Y3+-ions are initiating the network formation, which is one of the critical steps for the gelation.22 The resulting network structure leads to direct crystal–crystal contacts between QDs in the MPA QD aerogel, while some MUA ligands are still separating the individual QDs in the MUA QD aerogel.17 This is shown by the non-crystalline residue between the QDs in the TEM image and is illustrated schematically in Fig. 4. With these observations and assumptions, we can order the average estimated distances between the QDs within the assemblies from short to long as follows: MPA QD aerogel, MPA QD film, MUA QD aerogel, and MUA QD film.
 | ||
| Fig. 4 The inter-particle distance and the number of nearest neighbours influence the energy transfer within the QD assemblies. (a)–(d) Schematic representation of the assembly structure and the indicated proposed energy transfer mechanisms including FRET, Dexter energy transfer and delocalization of charge carriers. TEM images of the (e) MUA QD aerogel and (f) MPA QD aerogel. | ||
The probability for an acceptor and donor being close to each other within the assembly can be evaluated statistically. A QD in a close-packed film will have a total of 12 (for a close-packed structure) or 8 (for a body-centered cubic structure) nearest neighbors, while the formation of a close-packed structure is more likely and has been described before.34 In contrast, the microstructure of the aerogels shows that there are often only few QDs next to each other. This is also very much expected, since the high porosity of the gels will lead to an overall less dense structure by definition.
Combining the distance and the number of nearest neighbors, we can explain the measured absolute lifetimes of the assemblies given in Fig. 3a. Depending on the distance, different energy transfer mechanisms are expected and depending on the number of nearest neighbors in the assemblies, the likelihood of that mechanism being triggered post excitation is influenced. This is illustrated in Fig. 4 for all four assemblies.
For the two MUA samples the distance between the QDs is expected to be in the range of 2–3 nm, and therefore FRET is expected to be the dominating energy transfer mechanism, which has been described numerous times in the literature for PbS and PbS/CdS closed-packed QD films.5,7,9,32 FRET therefore presents an additional non-radiative pathway for the QDs in the assembly, and the PL lifetimes are shortened by the addition of the energy transfer rate (kET) to the non-radiative recombination rates. In the MUA QD film, the QDs are expected to be packed tightly and the number of nearest neighbors per QD and therefore possible transfer partners is high, leading to an overall decrease in the PL lifetimes to roughly 0.5–1.5 μs. In the MUA QD aerogel on the other hand, FRET is also expected due to the distance, but since there are fewer nearest neighbors per QD, there are also fewer transfer partners per QD and therefore kET decreases. This would explain the longer PL lifetimes between 1–2 μs measured for the MUA QD aerogel.
Qualitatively, the same can be said for the MPA assemblies regarding the number of nearest neighbors. Here the number of possible transfer partners is also higher in the QD film, compared to the aerogel, decreasing kET for the MPA QD aerogel, compared to the MPA QD film. Additionally, we expect the domination of different energy transfer mechanisms for the MPA QD film and aerogel, because of their smaller inter-particle distances and contacts. For the MPA QD film, Fang et al. previously studied MPA treated films of PbS/CdS QDs and proposed charge tunneling as a possible alternative to FRET with the shorter inter-particle distances expected here.9 Energy transfer processes, such as Dexter energy transfer, are significantly faster than FRET and therefore kET would be increased significantly.9,35 In the MPA QD aerogel, the particles form a continuous network and the PbS cores are only separated by the thin CdS shell, enabling completely different processes to occur in theory. It was also previously shown that thin-shell PbS/CdS QDs exhibit quasi-type II band alignment.9 For CdSe/CdS nanorod-based gel networks the type II band alignment leads to an increased PL lifetime, because of the delocalization of the electron wave function over several nanorods and less electron–hole overlap and therefore a decrease in the radiative recombination rate.36,37 In a similar quasi-type II system with PbS instead of CdSe, the delocalization might be less pronounced, since the exciton binding energy of bulk PbS is significantly higher than the bulk value for CdSe (3968 and 15 meV).38–40 Nevertheless, the electronic barrier would be smaller and the rates of energy transfer processes based on tunneling of charge carriers would be increased. In comparison to the films or the MUA QD aerogel we do not measure a decrease in kr for the MPA QD aerogel, which shows a continuous network of fused QDs. This means that delocalization of the electron over multiple QDs in the network does not occur.
Similar to our observations, the distance dependency and changes based on the formation of inter-particle crystal contacts have also been observed by Cui et al. They studied coupled CdSe/CdS QD molecules and found that resonance energy transfer is active for both ligand-linked and fused dimers. Additionally, they attributed the observed shortening of the PL lifetimes to the strong distance dependency of the tunneling of an electron.41
The influence of the shell thickness on the energy transfer efficiency is another relevant parameter, which has previously been investigated by Fang et al.9 They observed a difference in the sensitivity of the PbS/CdS QD films regarding the inter-particle distance, which was controlled via the length of the surface ligands and measured in the lifetime of the samples. While thin shell PbS/CdS QD films showed a decrease from 389 ns (oleic acid ligands) to 346 ps (MPA ligands), the thick shell PbS/CdS QD films showed a reduction from 0.46 μs (oleic acid ligands) to 0.25 μs (1,4-benzenedithiol ligands). Therefore, the lifetime of the thin shell samples decreases by a factor of 1000, while the lifetime of the thicker shell samples only decreases by a factor of 2. This observation was explained by the delocalization of the electronic wave function into the CdS shell only when the shell is thin, while the exciton is localized in the type I structure when the shell thickness is increased. Additionally, the core–core distances increase with increasing shell thickness. Since the energy transfer efficiency is directly dependent on the donor–acceptor distance, even a small increase of a few nm can change the energy transfer efficiency and therefore the exciton lifetime significantly.9
In conclusion, the distance between the QDs and the number of nearest neighbors per QD within the assemblies drastically influence the type and activity of energy transfer mechanisms, leading to the differences observed in the PL lifetime and in extension the non-radiative recombination rates knr.
Assemblies of mixed size QDs
To further support our findings, we prepared assemblies with different ratios of QDs with two different sizes. The small QDs are the same size as the ones used for the synthesis of the assemblies discussed above with an average diameter of 3.7 nm and a PbS core size of 3 nm, while the bigger QDs used here were synthesized from 4.7 nm PbS QDs and have a final PbS core size of 4 nm after the cation exchange reaction, forming the CdS shell. The size distribution of the QDs in the assembly is increased by mixing the two synthesized batches in practice, allowing us to observe the energy transfer from the smaller to the larger QDs more effectively. The steady state optical properties of the as-synthesized oleate-stabilized colloids are shown in Fig. 5a.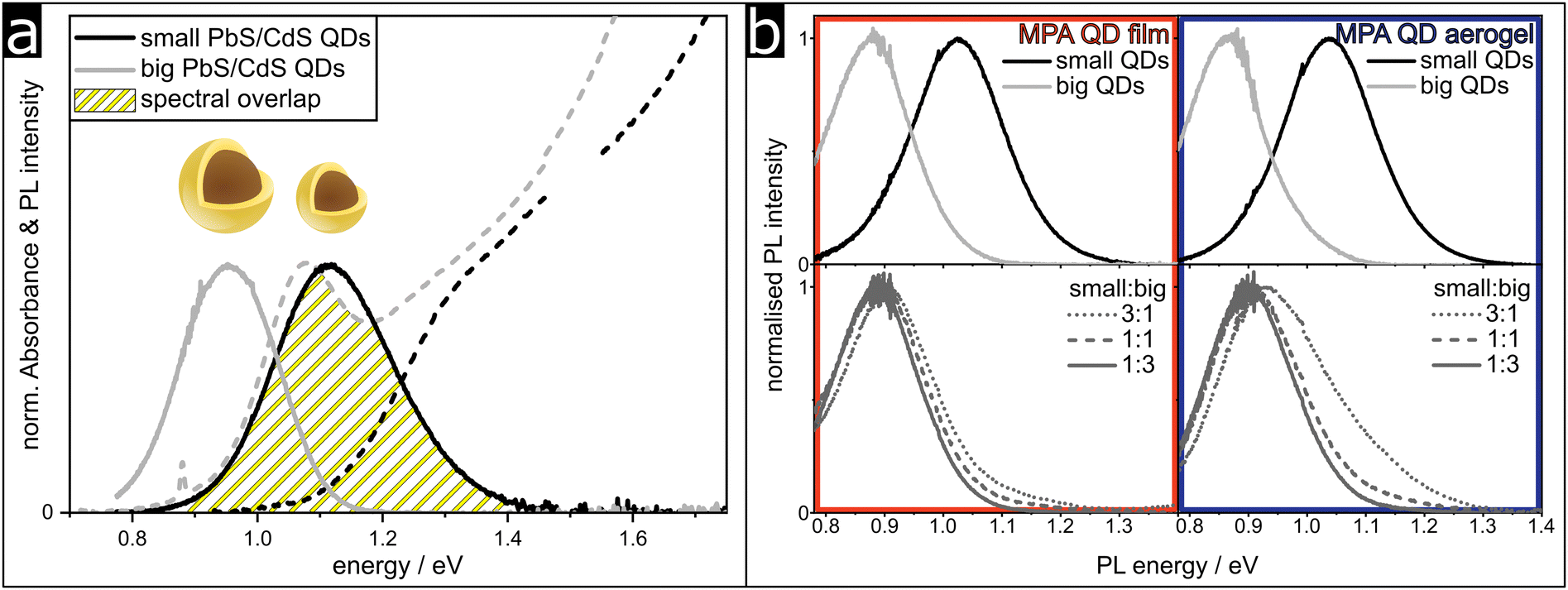 | ||
| Fig. 5 The preparation of assemblies from a mix of small and big PbS/CdS QDs leads to the disproportional decrease in the small QDs’ PL. (a) PL and absorbance spectra of small (3.7 nm) and big (4.7 nm) PbS/CdS QDs. The spectral overlap of the small QDs’ PL and the big QDs’ absorbance is highlighted. (b) PL spectra of MPA QD films and MPA QD aerogels, prepared with both small and big QDs in the ratios 3:1, 1:1 and 1:3. | ||
The yellow hatched area highlights the spectral overlap between the PL of the small QDs and the absorbance of the big QDs. The spectral overlap is a crucial factor for efficient energy transfer and the spectra show that the combination of these different sized QDs leads to a much improved overlap compared to the small QDs alone. Therefore, a more efficient energy transfer from the small QDs to the big QDs is expected, leading to the quenching of the small QDs’ PL. In Fig. 5b the PL spectra of the mixed MPA samples are shown. Independent of the mixing ratio, the PL of the small QDs is suppressed compared to the PL of the big QDs, as expected. With an increasing amount of bigger QDs the PL of the small QDs further decreases, because firstly there are simply less small QDs present. Secondly, the efficiency of the energy transfer also increases with the decreasing small to big QD ratio, because the probability of an acceptor being near a donor is increased. The quenching of the small QDs’ PL is more significant for the MPA QD film compared to the MPA QD aerogel, which fits well with the explanation of the number of nearest neighbors having an impact on the probability of energy transfer.
Conclusion
We have performed in-depth PL characterization of PbS/CdS QD-based three-dimensional networks and compared them to drop-cast QD films by measuring the steady-state PL at cryogenic temperatures and performed time-resolved measurements at room temperature. There are a total of three emissive states contributing to the PL of most assemblies, while most of the time in the literature only two emissive states are described. In addition, we found a correlation between the non-radiative recombination rates, the number of nearest neighbors and the distance between the building blocks. We explain these findings with the variation in the energy transfer rates, because the structures specify the active energy transfer mechanisms, such as Dexter energy transfer and Förster resonance energy transfer (FRET).Author contributions
Sample preparation was performed by D. P. The TEM images were recorded by R. T. G. The optical characterization was carried out by D. P. The project was initiated and supervised by N. C. B. The original draft was written by D. P. and reviewed and edited by all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. C. B. and D. D. would like to thank the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for funding under Germany's excellence strategy within the cluster of excellence PhoenixD (EXC 2122, project ID 390833453) and the cluster of excellence CUI: Advanced Imaging of Matter (EXC 2056, project ID 390715994). D. P. and R. T. G. are thankful for the support from the Hannover School for Nanotechnology (HSN). The authors thank the Laboratory of Nano and Quantum Engineering (LNQE) for providing the TEM facility. We acknowledge financial support from the Open Access Publication Fund of Universität Hamburg.References
- C. Ziegler, A. Wolf, W. Liu, A. K. Herrmann, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2017, 56, 13200 CrossRef CAS.
- P. Rusch, D. Zámbó and N. C. Bigall, Acc. Chem. Res., 2020, 53, 2414 CrossRef CAS.
- H. Lu, G. M. Carroll, N. R. Neale and M. C. Beard, ACS Nano, 2019, 13, 939 CAS.
- W. Lü, I. Kamiya, M. Ichida and H. Ando, Appl. Phys. Lett., 2009, 95, 1 Search PubMed.
- X. Huang, Z. Peng, Q. Guo, X. Song, J. Qiu and G. Dong, J. Am. Ceram. Soc., 2019, 102, 3391 CrossRef CAS.
- A. P. Litvin, E. V. Ushakova, P. S. Parfenov, A. V. Fedorov and A. V. Baranov, J. Phys. Chem. C, 2014, 118, 6531 CrossRef CAS.
- A. P. Litvin, A. A. Babaev, P. S. Parfenov, E. V. Ushakova, M. A. Baranov, O. V. Andreeva, K. Berwick, A. V. Fedorov and A. V. Baranov, J. Phys. Chem. C, 2017, 121, 8645 CrossRef CAS.
- A. P. Litvin, P. S. Parfenov, E. V. Ushakova, A. L. Simões Gamboa, A. V. Fedorov and A. V. Baranov, J. Phys. Chem. C, 2014, 118, 20721 CrossRef CAS.
- H.-H. Fang, D. M. Balazs, L. Protesescu, M. V. Kovalenko and M. A. Loi, J. Phys. Chem. C, 2015, 119, 17480 CrossRef CAS.
- J. S. Steckel, S. Coe-Sullivan, V. Bulović and M. G. Bawendi, Adv. Mater., 2003, 15, 1862 CrossRef CAS.
- Q. Ma and X. Su, Analyst, 2010, 135, 1867 RSC.
- A. J. Nozik, M. C. Beard, J. M. Luther, M. Law, R. J. Ellingson and J. C. Johnson, Chem. Rev., 2010, 110, 6873 CrossRef CAS PubMed.
- L. Sun, J. J. Choi, D. Stachnik, A. C. Bartnik, B. R. Hyun, G. G. Malliaras, T. Hanrath and F. W. Wise, Nat. Nanotechnol., 2012, 7, 369 CrossRef CAS PubMed.
- Y. Yan, R. W. Crisp, J. Gu, B. D. Chernomordik, G. F. Pach, A. R. Marshall, J. A. Turner and M. C. Beard, Nat. Energy, 2017, 2, 1 Search PubMed.
- E. H. Sargent, Adv. Mater., 2005, 17, 515 CrossRef CAS.
- S. Keuleyan, E. Lhuillier, V. Brajuskovic and P. Guyot-Sionnest, Nat. Photonics, 2011, 5, 489 CrossRef CAS.
- D. Pluta, H. Kuper, R. T. Graf, C. Wesemann, P. Rusch, J. A. Becker and N. C. Bigall, Nanoscale Adv., 2023, 5005 RSC.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844 CrossRef CAS.
- J. Zhang, R. W. Crisp, J. Gao, D. M. Kroupa, M. C. Beard and J. M. Luther, J. Phys. Chem. Lett., 2015, 6, 1830 CrossRef CAS PubMed.
- H. Zhao, M. Chaker, N. Wu and D. Ma, J. Mater. Chem., 2011, 21, 8898 RSC.
- T. Kodanek, H. M. Banbela, S. Naskar, P. Adel, N. C. Bigall and D. Dorfs, Nanoscale, 2015, 7, 19300 RSC.
- D. Zámbó, A. Schlosser, P. Rusch, F. Lübkemann, J. Koch, H. Pfnür and N. C. Bigall, Small, 2020, 16, 1906934 CrossRef PubMed.
- J. R. Caram, S. N. Bertram, H. Utzat, W. R. Hess, J. A. Carr, T. S. Bischof, A. P. Beyler, M. W. B. Wilson and M. G. Bawendi, Nano Lett., 2016, 16, 6070 CrossRef CAS PubMed.
- D. Kim, T. Kuwabara and M. Nakayama, J. Lumin., 2006, 119–120, 214 CrossRef CAS.
- E. V. Ushakova, A. P. Litvin, P. S. Parfenov, A. V. Fedorov, M. Artemyev, A. V. Prudnikau, I. D. Rukhlenko and A. V. Baranov, ACS Nano, 2012, 6, 8913 CrossRef CAS PubMed.
- A. L. Efros, M. Rosen, M. Kuno, M. Nirmal, D. Norris and M. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 4843 CrossRef CAS.
- J. M. An, A. Franceschetti and A. Zunger, Nano Lett., 2007, 7, 2129 CrossRef CAS.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 364 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 285 CrossRef.
- S. J. Jang, J. Chem. Phys., 2021, 155, DOI:10.1063/5.0068868.
- Y. M. Sung, T. G. Kim, D. J. Yun, M. Lim, D. S. Ko, C. Jung, N. Won, S. Park, W. S. Jeon, H. S. Lee, J. H. Kim, S. Jun, S. Sul and S. Hwang, Small, 2021, 17, 2102792 CrossRef CAS.
- Z. Lingley, S. Lu and A. Madhukar, J. Appl. Phys., 2014, 115, 084302 CrossRef.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020 CrossRef CAS.
- E. Marino, T. E. Kodger, G. H. Wegdam and P. Schall, Adv. Mater., 2018, 30, 1 Search PubMed.
- J. J. Choi, J. Luria, B. R. Hyun, A. C. Bartnik, L. Sun, Y. F. Lim, J. A. Marohn, F. W. Wise and T. Hanrath, Nano Lett., 2010, 10, 1805 CrossRef CAS PubMed.
- S. Sánchez-Paradinas, D. Dorfs, S. Friebe, A. Freytag, A. Wolf and N. C. Bigall, Adv. Mater., 2015, 27, 6152 CrossRef PubMed.
- P. Rusch, B. Schremmer, C. Strelow, A. Mews, D. Dorfs and N. C. Bigall, J. Phys. Chem. Lett., 2019, 10, 7804 CrossRef CAS PubMed.
- D. A. Baghdasaryan, V. A. Harutyunyan, D. B. Hayrapetyan, E. M. Kazaryan, S. Baskoutas and H. A. Sarkisyan, Nanomaterials, 2022, 12, 3690 CrossRef CAS PubMed.
- U. Elfurawi, Optical and Electronic Properties of PbS Colloidal Nanocrystals, PhD thesis, University of Nottingham, 2012 Search PubMed.
- P. Geiregat, C. Rodá, I. Tanghe, S. Singh, A. Di Giacomo, D. Lebrun, G. Grimaldi, J. Maes, D. Van Thourhout, I. Moreels, A. J. Houtepen and Z. Hens, Light: Sci. Appl., 2021, 10, 112 CrossRef CAS PubMed.
- J. Cui, Y. E. Panfil, S. Koley, D. Shamalia, N. Waiskopf, S. Remennik, I. Popov, M. Oded and U. Banin, Nat. Commun., 2019, 10, 5401 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02427c |
| This journal is © the Owner Societies 2024 |