
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling heparin's enhancement of amyloid aggregation in a model peptide system†
Suhas
Gotla
 ,
Anushka
Poddar
,
Ilana
Borison
and
Silvina
Matysiak
*
,
Anushka
Poddar
,
Ilana
Borison
and
Silvina
Matysiak
*
Fischell Department of Engineering, University of Maryland, College Park, Maryland, USA. E-mail: matysiak@umd.edu; Tel: +1 301 405 0313
First published on 7th August 2024
Abstract
A coarse-grained (CG) model for heparin, an anionic polysaccharide, was developed to investigate the mechanisms of heparin's enhancement of fibrillation in many amyloidogenic peptides. CG molecular dynamics simulations revealed that heparin, by forming contacts with the model amyloidogenic peptide, amyloid-β's K16LVFFAE22 fragment (Aβ16–22), promoted long-lived and highly beta-sheet-like domains in the peptide oligomers. Concomitantly, heparin-Aβ16–22 contacts suppressed the entropy of mixing of the oligomers’ beta-domains. Such oligomers could make better seeds for fibrillation, potentially contributing to heparin's fibril-enhancing behaviour. Additionally, reductions in heparin's flexibility led to delayed aggregation, and less ordered Aβ16–22 oligomers, thus offering insights into the contrasting inhibition of fibrillation by the relatively rigid polysaccharide, chitosan.
1 Introduction
Glycosaminoglycans (GAGs) are linear anionic polysaccharides found abundantly in physiological environments like the extracellular matrix.1,2 In addition to sharing these microenvironments with amyloid-forming peptides, GAGs are well known to co-aggregate with amyloid fibrils.3,4 Many GAGs, including the highly anionic model GAG heparin, are also known to enhance the fibrillation of peptides including the Alzheimer's disease-related amyloid-β (Aβ),5,6 several disease-related peptides,7–9 and peptide hormones.10–13 Beyond merely enhancing fibrillation, heparin has also been shown to induce specific fibril polymorphs and trigger condensate-to-fibril transitions.14,15 Yet, the mechanisms by which heparin and other GAGs modulate peptide aggregation remain elusive.An interesting contrast to the enhancement of amyloid aggregation by heparin is the inhibition of amyloid aggregation by another pyranosic linear polysaccharide, chitosan. Where heparin is flexible and polyanionic, chitosan is relatively rigid and displays pH-dependent polycationicity. These opposite effects with heparin and chitosan have been reported for several peptides including Aβ42 (net charge −3 under physiological conditions),16,17 tau298–317 (net charge +3 under physiological conditions),7 α-synuclein (net charge −9 under physiological conditions)8 and amylin (net charge +6 under physiological conditions).18 The diverse net charges displayed by these peptides, from −9 to +6, show that the opposite effects of heparin and chitosan on their aggregation occur independently of the peptides’ net charge. However, the mechanistic details of this phenomenon are not well characterized.
Atomistic molecular simulations, while adept at tackling such gaps in mechanistic understandings, have so far been limited to small numbers of peptides19–21 and short timescales.22 This is a pervasive limitation of classical atomistic forcefields, where rugged free energy landscapes lead to poor sampling and high computational costs. Much of these costs can be overcome with coarse-grained (CG) molecular dynamics simulations, whereby groups of atoms are coarsely represented by their average chemical characteristics as “beads”.
The forcefield for CG molecular dynamics developed in our lab, ProMPT, allows for the unbiased folding of secondary and super-secondary protein structures while retaining an explicit solvent architecture.23 In this work, we developed a ProMPT parameter set for the model GAG heparin (Fig. 1(a)), which was found to closely match the characteristic torsional angles of heparin in atomistic simulations, and experimentally measured radii of gyration.24 The development of this heparin model thus enabled us to study the effects of GAGs on amyloid aggregation using the ProMPT forcefield for CG molecular dynamics.
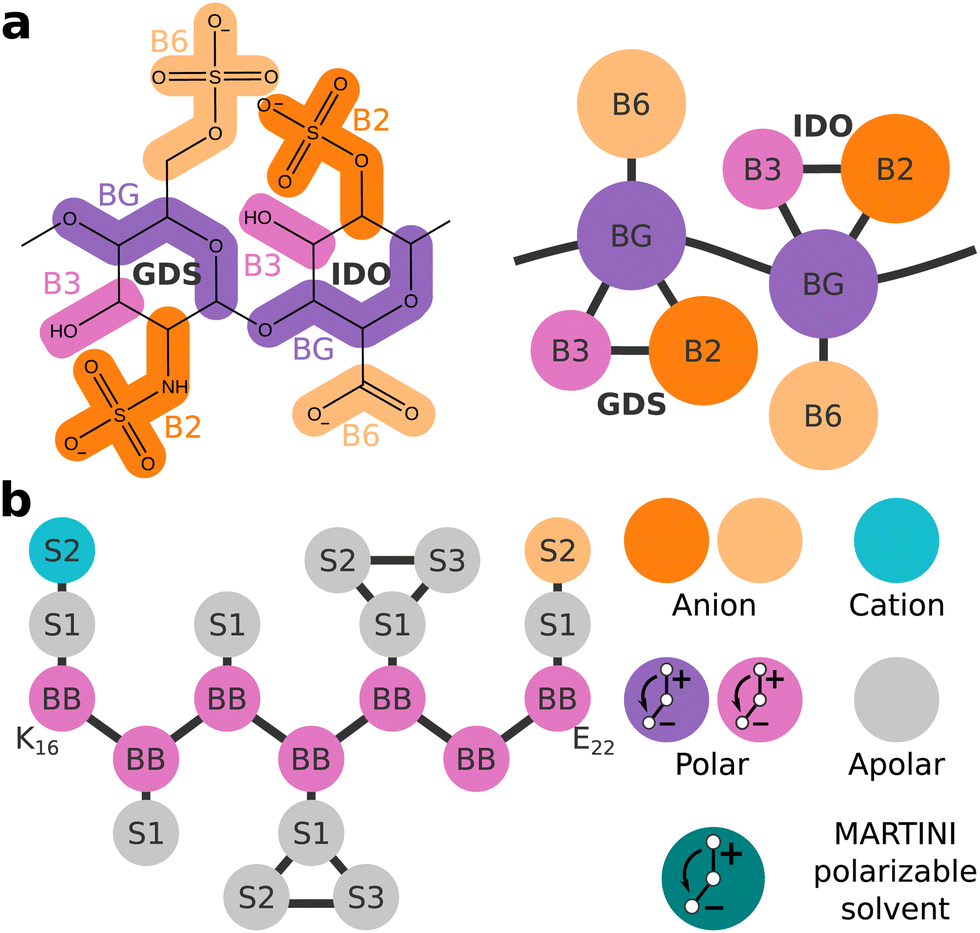 | ||
| Fig. 1 (a) Mapping of the CG heparin model. (b) Schematic representation of the CG Aβ16–22 fragment K16LVFFAE22 (left). A key for the bead types, and a schematic depiction of the CG MARTINI polarizable solvent model (right). The anionic and cationic dummy particles of any polar bead X would be named Xm and Xp, respectively. | ||
We chose the extensively studied amyloidogenic fragment of Aβ, K16LVFFAE22 (referred to henceforth as Aβ16–22),25 as the model peptide for this study. The Aβ16–22 fragment spans the heparin-binding basic patch of full-length Aβ at K16,5,26 thus making it suitable for our goal of studying the role of heparin in amyloid aggregation. Fig. 1(b) shows a schematic representation of Aβ16–22 as defined in ProMPT.
Through CG molecular dynamics simulations, we studied how the presence of heparin, heparin's degree of polymerization (dp), and rigidity influenced the aggregation of ordered Aβ16–22 oligomers. We also discussed our results on the effects of heparin's rigidity on Aβ16– 22's ordered aggregation in the context of previous studies, including one from our group,17 on chitosan's inhibition of Aβ16–22 aggregation.5,16
2 Methods
2.1 CG forcefield details
While heparin is a polydisperse molecule (with molecular weights of up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da depending on the source27), low molecular weight heparin (LMWH) is a formulation with a consistent average molecular weight of ∼5000 Da, i.e., heparin chains roughly dp18 in length.28 Therefore, most of our simulations employ dp18 heparin—a stand-in for laboratory/pharmaceutical grade LMWHs.
000 Da depending on the source27), low molecular weight heparin (LMWH) is a formulation with a consistent average molecular weight of ∼5000 Da, i.e., heparin chains roughly dp18 in length.28 Therefore, most of our simulations employ dp18 heparin—a stand-in for laboratory/pharmaceutical grade LMWHs.
Further details on the parametrization of the model's nonbonded and bonded interactions, validation of the model against experimental values of radii of gyration, and characteristic torsion data from atomistic simulation are presented in Section 1 of the ESI.†
2.2 CG simulation details
Starting with a fully extended conformation, a single Aβ16–22 peptide was placed in a water box and subjected to 10000 steps of steepest descent energy minimization and 10 ns of NPT simulation to generate a random initial structure. The coordinates of Aβ16–22 from the final frame, a collapsed conformation without helical or beta-sheet-like secondary structure, served as the initial configuration when building the simulation cell. Similarly, starting with the CG-mapped structure of the PDB record 3IRI,33 a single dp18 heparin was placed in a water box and subjected to 10000 steps of steepest descent energy minimization and 10 ns of NPT simulation to generate a random initial structure (the dp18 heparin structure was clipped to obtain coordinates for shorter heparin fragments). These coordinates of Aβ16–22 molecules and heparin molecules were randomly inserted, at least 1 nm apart, in a 9 × 9 × 9 nm cubic periodic box to initialize each system. Each system was solvated with roughly, 7600 particles of MARTINI polarizable solvent, and monovalent ions to balance net charges where necessary.
These initial system configurations were equilibrated with 10000 steps of steepest descent energy minimization, followed by 50000 steps of NPT simulation at 0.01 ps timesteps keeping the positions of solute molecules restrained with spring potentials. Finally, 3000 ns of NPT production MD was performed with timesteps of 0.01 ps. Four independent trials were performed, each 3 μs long and equilibrated with a unique velocity seed.
We used the leapfrog integrator in conjunction with the Nose–Hoover thermostat at 350 K with a time constant of 1 ps.34 Solvent, heparin, and Aβ16–22 molecules were coupled to separate temperature baths. Pressure was maintained at 1 bar with an isotropic Parrinello–Rahman barostat, 5 ps time constants, and compressibility of 3.5 × 10−5 bar−1.35 Long-range electrostatics were computed with the Particle Mesh Ewald scheme with a relative electrostatic permittivity of 2.5.36 Neighbour lists for short-range interaction calculations were updated every 10 steps. LINCS was used to constrain the dummy bonds within the MARTINI polarizable solvent particles, and the bonds within the aromatic rings of the protein molecules.37 All simulations were performed using the GROMACS 2019.4 simulation engine.38
2.3 Analysis
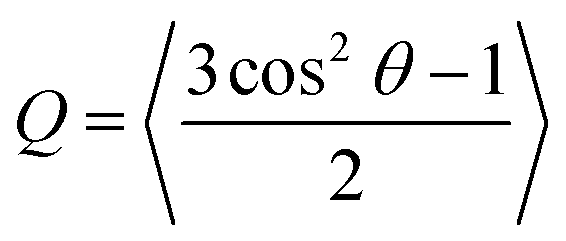 | (1) |
 | (2) |
A characteristic aggregation time, τagg was calculated from this time series by fitting a sigmoid curve:
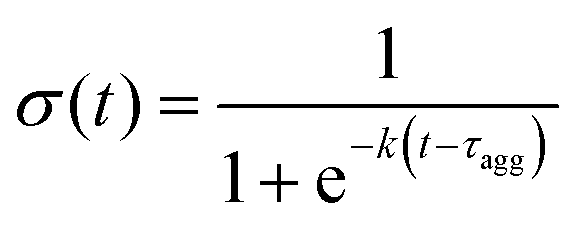 | (3) |
3 Results and discussion
First, we investigated how a single heparin chain, with a degree of polymerization of 18 (dp18), would influence Aβ16–22 aggregation at different concentrations—16, 24 and 32 mM, mapping to numbers of peptides (Npep), 8, 12 and 16, respectively in a 9 × 9 × 9 nm periodic solvent box.In each simulation, a single consolidated aggregate comprising all the available Aβ16–22 peptides was obtained. Representative structures of consolidated aggregates in water and with heparin at Npep = 16 are shown in Fig. 2(a) and (b), respectively. Aβ16–22 oligomers with characteristic hydrophobic cores were obtained in both cases (Fig. S7, ESI†). Conforming to its spheroid shape, heparin bound to the periphery of the Aβ16–22 oligomer, primarily with the cationic side chains of Aβ16–22 's K16 residue, and secondarily with peptide backbones though the B6 and B2 beads of heparin's GDS and IDO subunits, respectively (Fig. S8, ESI†). The co-existence of electrostatic interactions between heparin and K residues, and polar interactions of heparin with the backbones of diverse amino acids was also reported in a recent atomistic simulation study of heparin with the R3 fragment of the tau peptide.21 It is well-known from past experimental studies that heparin chains are structurally integral to mature amyloid fibrils.5,6,14 Our results suggest that heparin may also be an integral component of pre-fibrillar oligomers.
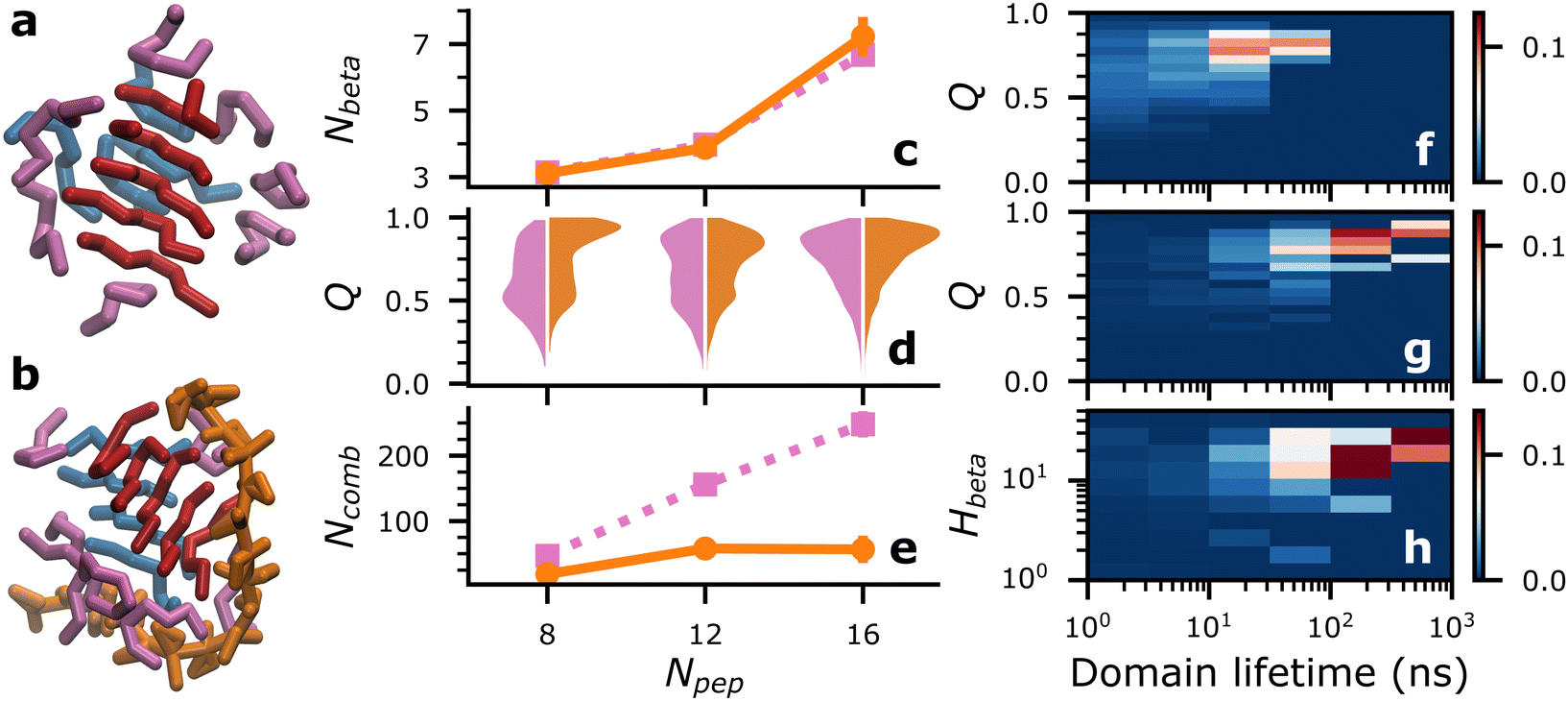 | ||
| Fig. 2 (a) and (b) Representative snapshots of Aβ16–22 aggregates in water (a) and with dp18 heparin (b) at Npep = 16. Beta-domains are coloured in red and blue, while all other peptides are coloured in pink, and heparin is coloured in orange. (c)–(e) Average number of beta-strands (Nbeta), violin plots of distributions of Q, and average number of beta-domain combinations, Ncomb, respectively, for Aβ16–22 in water (pink, ■ dotted lines) and with dp18 heparin (orange, •, solid lines) at varying Npep. (f)–(h) Probability distributions of beta-domains’ Q against their lifetimes for Aβ16–22 aggregation at Npep = 16 in water (f) and with dp18 heparin (g). Probability distribution of heparin contacts per peptide, Hbeta, and lifetimes of beta-domains for Aβ16–22 at Npep = 16 with dp18 heparin (h). | ||
While we cannot access the timescales and system sizes required to sample fibrillar structures, we can study the fibril-like qualities of early oligomeric species. A key feature of amyloid fibrils is an enhancement in beta-sheet secondary structures. In our simulations, Aβ16–22 oligomers contain transient patches of beta-sheet-like local order characterized by four or more contiguous backbone–backbone contacts between three or more peptides (such as the red and blue peptide domains in Fig. 2(a) and (b)). We call such patches “beta-domains”, and their constituent peptides “beta-strands”. While adept at detecting locally aligned peptides within oligomers, our beta-domain concept cannot discriminate between collapsed and extended structures. Consequently, the average number of beta-strands, Nbeta, in water and with heparin were nearly identical (Fig. 2(c)). Unless specified otherwise, data for Nbeta and all metrics in all figures were reported from the final 1 μs of four independent replica simulations, and averages were reported with error bars of ±2 standard errors.
Looking beyond raw beta-strand counts, we looked for more subtle structural differences in the orientation of beta-strands within the beta-domains with the order parameter, Q. A Q value of 1 corresponds to a set of peptides oriented in a perfectly parallel (or antiparallel) manner, while a value of 0 corresponds to randomly oriented peptides. In practice, highly ordered beta-sheets display Q values in the 0.75–0.95 range.40
Relative to Aβ16–22 in water, Fig. 2(d) shows a consistent increase in populations of beta domains with Q ≥ 0.75 in the presence of a single chain of dp18 heparin across all peptide concentrations studied. Therefore, we concluded that heparin enhanced fibril-like order in oligomers, a trend that agrees with the enhanced fibrillation reported in previous experimental studies.5,41
We could also track how the different peptide strands, identified by their peptide id in the simulations’ topologies, combined and recombined to form beta-domains. An example of two beta-domains recombining would be the domains identified by peptide id's {1,3,4,8}, and {2,5,9} recombining into two new domains {1,3,4}, and {2,5,9,8}. Importantly, by our definition, {1,3,4,8} and {8,1,4,3} are identical combinations, i.e., the order of peptide id's is irrelevant.
Fig. S10 (ESI†) illustrates, over a 300 ns period of self-assembly, the differences in the propensities for beta-domain recombination—rampant in Aβ16–22 aggregation in water, and significantly retarded in Aβ16–22 aggregation in with dp18 heparin. These differences were quantified by the numbers of beta-domain combinations, Ncomb, and their lifetimes. Across different Npep, Ncomb in water far exceeded that in the presence of heparin (Fig. 2(e)). Complementarily, the mean lifetimes of the beta-domains were higher in the presence of heparin than without (Fig. S11, ESI†). These data indicated that the entropy of mixing of beta-domains, Smix = kBln(Ncomb) where kB is the Boltzmann constant, was suppressed by heparin.
Differences in the beta-domains’ mixing propensities could be traced back to their level of order and their interactions with heparin. At Npep = 16, beta-domains with long lifetimes generally had higher Q (Fig. 2(f) and (g)), but heparin significantly shifted the ensemble of beta-domains towards Q ≥ 0.75, and lifetimes over 100 ns (see ESI† for data at other Npep values). Long-lived beta-domains also had, on average, more heparin contacts per beta-strand (Hbeta), indicating that beta-domain mixing was suppressed by the energetic costs of breaking peptide–heparin contacts (Fig. 2(h)). The following mechanism was summarized: peptide–heparin contacts prolonged beta-domain lifetimes, which then allowed the constituent beta-strands to arrange themselves into highly ordered configurations. By this mechanism, heparin could potentially spawn highly ordered oligomeric seeds for fibrillation, which could explain heparin's fibril-enhancing properties for Aβ and other peptides.5,6,9,11–13
Aβ16–22 at Npep = 16 was self-assembled in the presence of a series of short heparin chains (dp2, dp4, and dp8), to test if limiting heparin–peptide interactions would lead to commensurate effects on Q and beta-domain mixing, compared to dp18 heparin. Compared to baseline values for Aβ16–22 in water, Nbeta varied by less than ±1 (Fig. S12a, ESI†), but populations of beta-domains with Q ≥ 0.75 rose in proportion with heparin dp (Fig. 3(a)). Simultaneously, Hbeta and beta-domain lifetimes rose in proportion with heparin dp (orange marks in Fig. 3(c)), thus confirming our proposed mechanism that heparin-peptide interactions prolong beta-domain lifetimes, allowing them to sample highly ordered structures.
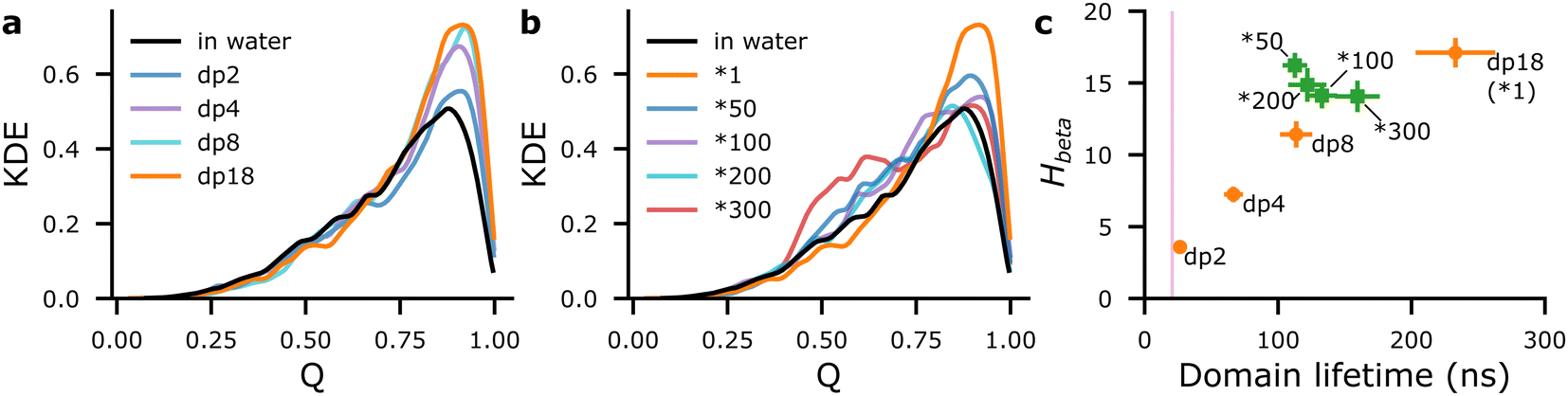 | ||
| Fig. 3 Distributions of Q for Aβ16–22 at Npep = 16 at varying heparin dp (a) and with rigid heparin analogues (b), represented by kernel density estimates (KDE). Scatter plots of mean Hbeta against mean lifetimes of beta-domains for Aβ16–22 at Npep = 16 at varying heparin lengths (orange • annotated by dp) and with rigid analogues of dp18 heparin (green ■ annotated by *X, where X is the rigidity factor). The pink region in (c) indicates mean domain lifetime ±2 SE for Npep = 16Aβ16–22 in water. | ||
N comb also decreased as heparin's dp, Hbeta, populations of domains with Q ≥ 0.75, and domain lifetimes increased, confirming the thermodynamic interpretation that heparin suppresses the Smix of the beta-domains (Fig. S13a, ESI†). Heparin dp-dependent increases in Q and lifetime potentially signal proportionate fibril-enhancement, which has been reported in previous experimental studies of amylin and PACAP27 peptide fibrillation with heparins of varying lengths.9,13
Next, we aimed to understand how heparin's rigidity influenced its effects on ordered Aβ16–22 aggregation. This analysis was motivated by the knowledge that chitosan, a charged polysaccharide of greater rigidity than heparin (persistence lengths of chitosan and heparin are 6 nm42 and 4.5 nm,43 respectively), is a strong inhibitor of Aβ fibrillation5,16—a contrast to heparin's fibril enhancing properties. In particular, we hypothesized that increasing heparin's rigidity would suppress its ability to spawn beta-domains with Q ≥ 0.75 in Aβ16–22 oligomers.
To test this hypothesis, we self-assembled Aβ16–22 at Npep = 16 with a series of rigid dp18 heparin analogues—heparin50, heparin100, heparin200 and heparin300—created by increasing the force constants of the glycosidic backbone angles by “rigidity factors” of 50, 100, 200, and 300, respectively. While these rigid heparins were increasingly biased towards extended conformations, their morphologies were not affected in strict proportion to their rigidity factors. Rather, the most prominent effect on the rigid heparins was the exclusion of collapsed conformations that were common in the reference heparin (henceforth referred to as heparin1), where heparin's radius of gyration, Rgyr, was between 1.6–1.8 nm (Fig. 4(a)).
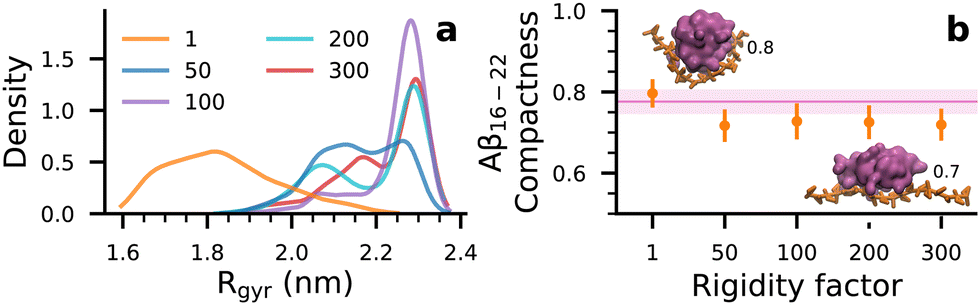 | ||
| Fig. 4 (a) Kernel density distributions of Rgyr of heparins at varying rigidity factors, over four independent 3 μs long trajectories. (b) Compactness of Aβ16–22 oligomers at Npep = 16 with different heparin rigidities (orange •). Pink regions denote the compactness of peptide aggregates in water. Insets show snapshots of oligomers (pink volumes) with compactness 0.8 and 0.7 with heparin1 and heparin200 (orange strands), respectively. | ||
Although differences in Nbeta were within ±1 beta-strand (Fig. S12b, ESI†), populations of Q ≥ 0.75 beta-domains with the rigid heparins were indeed lower than that with dp18 heparin1 (Fig. 3(b)). Moreover, the populations of beta-domains in the 0.4 ≤ Q ≤ 0.75 range were much higher in the presence of the rigid heparins, particularly heparin300, compared to Aβ16–22 in water. Thus, increasing heparin's rigidity not only suppresses its ability to spawn highly-ordered beta-domains (Q ≥ 0.75) but also increases the likelihood of poorly ordered domains (0.4 ≤ Q ≤ 0.75) in Aβ16–22 oligomers.
As for lifetimes, beta-domains formed with rigid heparins were intermediate between those formed in water and in the presence of heparin1 (green marks in Fig. 3(c)). Similarly, beta-domains’ Smix with the rigid heparins was lower than with heparin1, and higher than in water (Fig. S13b, ESI†). Altogether, rigid heparins spawned oligomers containing poorly ordered and short-lived beta-domains, which could serve as poor seeds that inhibit fibrillation at long timescales, as seen with chitosan.5,16 These results also conform to our previous work where chitosan was shown to suppress beta-strand counts and extended conformations—both of which count towards the order parameter Q—among Aβ16–22 peptides.17
Interestingly, beta-domains with rigid heparins had about as many heparin contacts, indicated by overlapping error bars in Fig. 3(c), and similar distributions of contacts across bead types (Fig. S8 and S9, ESI†) as those with heparin1. Yet, beta-domain lifetimes were lower, as if the rigid heparins were dp8 in length (Fig. 3(c)). To explain this discrepancy we looked at the shapes of the Aβ16–22 at different heparin rigidities and considered their implications.
In Fig. 4(b), shapes of Aβ16–22 oligomers are described by their compactness, i.e., the ratios of the smallest and largest moments of inertia where 1 corresponds to a spherical form and 0 corresponds to a rod-like form. Snapshots of oligomers in Fig. 4(b) serve as visual references for the shapes of oligomers at compactness values 0.8 and 0.7. When heparin chains were collapsible (heparin1 in Fig. 4(a)), peptide–heparin contacts were maximized (Fig. 3(b)) while slightly promoting the spherical character of the Aβ16–22 oligomers relative to Aβ16–22 in water. This enhancement in compactness implies a corresponding reduction in hydrophobic solvent accessible surface area. On the other hand, in trying to maximize contacts with extended and non-collapsible heparin chains (heparin50, heparin100, heparin200 and heparin300 in Fig. 4(a)), Aβ16–22 oligomers also adopted more extended rod-like shapes. The promotion of rod-like oligomers implied a corresponding increase in hydrophobic solvent accessible surface area. Thus, we reason that competition between heparin–peptide interactions and the hydrophobic effect would lead to frustration in the Aβ16–22 oligomer, which could be responsible for the lower beta-domain lifetimes and associated shifts in the ensemble from highly ordered (Q ≥ 0.75) to poorly ordered (0.40 ≤ Q ≤ 0.75) structures.
Finally, we characterised how heparin's rigidity affected the early kinetic pathways of Aβ16–22 self-assembly leading up to the consolidation of peptides into a single oligomer. The motivation for this analysis was the jump in the time taken for consolidation, τagg, from tens of nanoseconds in heparin1 to hundreds of nanoseconds among its rigid analogues (Table 1).
| Rigidity factor | τ agg (ns) |
|---|---|
| 1 | 50 ± 20 |
| 50 | 200 ± 100 |
| 100 | 400 ± 200 |
| 200 | 400 ± 200 |
| 300 | 600 ± 400 |
The consolidation of peptides into a single aggregate is achieved via a combination of two modes: the heparin-independent mode, where peptides consolidate in the bulk without heparin's involvement, and the heparin-dependent mode, where peptides consolidate on heparin's surface. To illustrate the role of rigidity in the heparin-dependent mode of consolidation, we focus on two simulations of Aβ16–22 aggregation at Npep = 16: one with heparin1 (Fig. 5(a), (c) and (e)), and another with heparin200 (Fig. 5(b), (d) and (f)). In both cases, unconsolidated peptide aggregates condensed at distant sites along the length of the heparin chain within the first 25 ns.
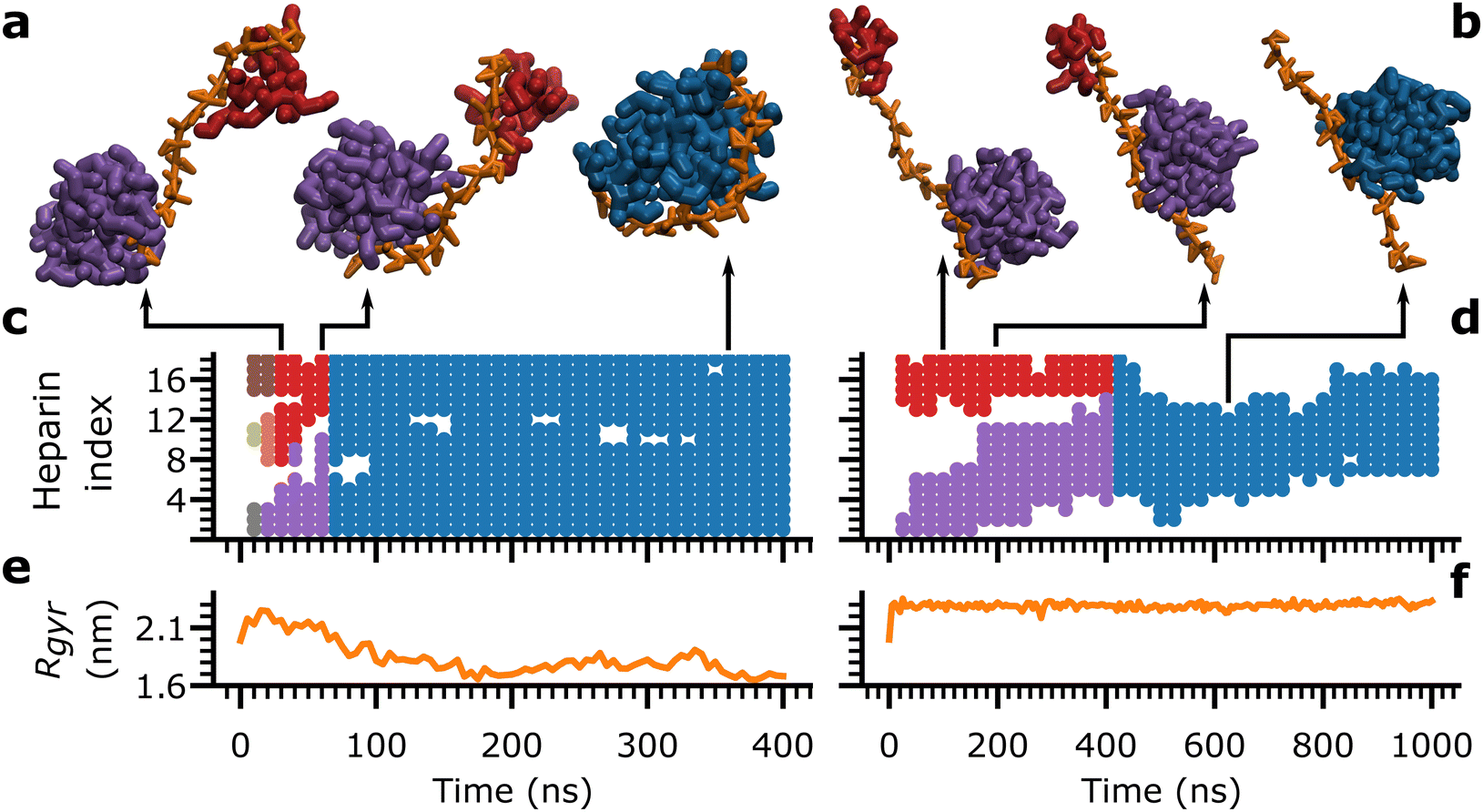 | ||
| Fig. 5 (a) and (b) Snapshots of Aβ16–22 aggregates (red, purple or blue) bound to heparin (orange chain) at representative time-points. (c) and (d) Heparin subunit indices bound by peptide aggregates, marked in their respective colours from their snapshots in (a) and (b), over time. (e), (f) Rgyr of heparin molecules over time, concurrent with (a) and (b). Data from a single trial of Aβ16–22 aggregation at Npep = 16 with dp18 heparin1 is shown on the left (a), (c) and (e). Data from a single trial of Aβ16–22 aggregation at Npep = 16 with dp18 heparin200 is shown on the right (b), (d) and (f). | ||
Each peptide aggregate, identified by its constituent peptide id's, was assigned a unique colour, as in the snapshots in Fig. 5(a) and (b) depicting key moments in the consolidation process. Concurrently, the indices of the heparin subunits in contact with each peptide aggregate were marked with the aggregate's assigned colour, as a time series (Fig. 5(c) and (d)). A third plot concurrently tracked the associated heparin's Rgyr as a function of time (Fig. 5(d) and (e)).
In heparin1, there were three aggregates at time = 10 ns (brown, olive, and grey), which merged into two (red and purple) and finally consolidated into a single aggregate (blue) around time = 70 ns (Fig. 5(a) and (c)). At the same time, heparin1 underwent a gradual collapse from time = 0 ns to time = 150 ns (Fig. 5(e)). Thus, the bending of heparin seemed to be the dominant force for peptide consolidation. In contrast, heparin200 maintained an extended conformation throughout (Fig. 5(f)) ruling out any contribution from its bending motions. Instead, a consolidated aggregate (blue) formed around the 400 ns mark exclusively by the larger purple aggregate crawling along the heparin200 chain towards the red aggregate (Fig. 5(b) and (d)). These aggregation events with heparin1 and heparin200 were captured in Movies S1 and S2 (ESI†), respectively.
In effect, the heparin-dependent consolidation is composed of two pathways: one where heparin bends to merge aggregates at distant sites along the heparin chain, and another where aggregates crawl along heparin chains to merge. Heparin index occupancy and Rgyr data for all trials with all heparins (ESI,† Fig. S16–S20) demonstrate that there is a mix of both pathways where heparin is flexible (heparin1), but the balance between the two pathways shifts towards peptide crawling at higher rigidity factors. Even in Fig. 5(c), there is evidence of the red and purple aggregates crawling along heparin1 between 30 and 60 ns, albeit somewhat obscured by heparin1 's dramatic collapse.
Thus, the delay in the consolidation of peptides into a single aggregate among rigid heparins (Table 1) was attributed to the inhibition of their bending motions and the associated reliance on aggregate-crawling as the pathway for consolidation. Looking back, we can associate the early kinetic pathways among rigid heparins with less ordered (Fig. 3) and more frustrated and rod-like oligomers (Fig. 4(b)). In other words, heparin's ability to enhance ordered Aβ16–22 aggregation is severely compromised without its flexibility.
4 Conclusions
In conclusion, we describe a potential mechanism underlying heparin's fibril-enhancing activities on Aβ,5,6 and other amyloidogenic peptides.12,13,44 We showed that contacts of Aβ16–22 peptides with heparin promoted long-lived and highly ordered beta-domains, i.e., locally ordered beta-sheet-like regions within oligomers. A thermodynamic interpretation was also outlined, whereby heparin suppressed the entropy of mixing, Smix of the beta-domains within the peptide oligomers. By promoting ordered oligomers, heparin could spawn better seeds for fibrillation relative to peptide aggregation in water.Additionally, results from our tests of Aβ16–22 aggregation with rigid heparin analogues may help us understand why chitosan is an inhibitor of Aβ aggregation.5,16 As with the rigid heparins, rigid chitosan chains would increase the peptides’ reliance on crawling, thus delaying their consolidation, ultimately resulting in frustrated and less ordered aggregates. Furthermore, chitosan tends to self-assemble into hydrogel networks. The complex topologies of hydrogel networks could exacerbate the delays in aggregation to such an extent that peptide aggregates could become quasi-sequestered, which we observed in our previous simulations of Aβ16–22 aggregation with chitosan.17
As mentioned in the introduction, the two opposite effects, of fibril enhancement by heparin and fibril inhibition by chitosan, have been demonstrated in several proteins including Aβ42,5,16 tau298–317,7 α-synuclein8 and amylin,18 whose net-charges range from −9 for α-synuclein to +6 for amylin. In light of these peptides’ diversity in sequence and net-charge, we posit that the mechanisms of polysaccharide rigidity-dependent peptide aggregation demonstrated here, with the net-charge 0 model peptide Aβ16–22, may be independent of the protein sequence and net-charge.
The methods and insights generated in this paper may also help us understand and potentially harness heparin's more recently discovered abilities to induce specific fibril polymorphs and modulate liquid–liquid phase separation of proteins.14,15
Author contributions
S. M. conceived and acquired funding for this research. S. G. performed investigations and drafted the manuscript. S. G. and S. M. co-edited the manuscript and performed data analyses. A. P. and I. B. performed investigations and data analyses towards heparin model development.Data availability
Outputs of molecular dynamics simulations generated in this article are available on Zenodo at https://doi.org/10.5281/zenodo.11479853. Input files for the simulations and a brief tutorial are available at https://github.com/suhasgotla/heparin_amyloid_self-assembly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by NSF award CHE-2202281 and supercomputing resources provided by the University of Maryland (https://hpcc.umd.edu).Notes and references
- R. U. Margolis, R. W. Ledeen, M. Sbaschnig-Agler, M. C. Byrne, R. L. Klein, B. H. Douglas II and R. K. Margolis, J. Neurochem., 1987, 49, 1839–1844 CrossRef CAS PubMed.
- W. L. Kiang, T. Krusius, J. Finne, R. U. Margolis and R. K. Margolis, J. Biol. Chem., 1982, 257, 1651–1659 CrossRef CAS PubMed.
- J. Daz-Nido, F. Wandosell and J. Avila, Peptides, 2002, 23, 1323–1332 CrossRef PubMed.
- H. Wang, P. Cao and D. P. Raleigh, J. Mol. Biol., 2013, 425, 492–505 CrossRef CAS PubMed.
- J. J. Valle-Delgado, M. Alfonso-Prieto, N. S. de Groot, S. Ventura, J. Samitier, C. Rovira and X. Fernàndez-Busquets, FASEB J., 2010, 24, 4250–4261 CrossRef CAS PubMed.
- B. Klajnert, M. Cortijo-Arellano, M. Bryszewska and J. Cladera, Biochem. Biophys. Res. Commun., 2006, 339, 577–582 CrossRef CAS PubMed.
- M. Islam, E. Argueta, E. P. Wojcikiewicz and D. Du, ACS Chem. Neurosci., 2022, 13, 3034–3043 CrossRef CAS PubMed.
- S. Mehra, D. Ghosh, R. Kumar, M. Mondal, L. G. Gadhe, S. Das, A. Anoop, N. N. Jha, R. S. Jacob, D. Chatterjee, S. Ray, N. Singh, A. Kumar and S. K. Maji, J. Biol. Chem., 2018, 293, 12975–12991 CrossRef CAS PubMed.
- S. Jha, S. M. Patil, J. Gibson, C. E. Nelson, N. N. Alder and A. T. Alexandrescu, J. Biol. Chem., 2011, 286, 22894–22904 CrossRef CAS PubMed.
- E. Bazar and R. Jelinek, ChemBioChem, 2010, 11, 1997–2002 CrossRef CAS PubMed.
- N. Quittot, M. Sebastiao and S. Bourgault, Biochem. Cell Biol., 2017, 95, 329–337 CrossRef CAS PubMed.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. R. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef CAS PubMed.
- M. Sebastiao, N. Quittot, I. Marcotte and S. Bourgault, Biochemistry, 2019, 58, 1214–1225 CrossRef CAS PubMed.
- Y. Tao, Y. Sun, S. Lv, W. Xia, K. Zhao, Q. Xu, Q. Zhao, L. He, W. Le, Y. Wang, C. Liu and D. Li, Nat. Commun., 2022, 13, 4226 CrossRef CAS PubMed.
- D. K. Garg and R. Bhat, Biophys. J., 2022, 121, 2568–2582 CrossRef CAS PubMed.
- H. Liu, B. Ojha, C. Morris, M. Jiang, E. P. Wojcikiewicz, P. P. N. Rao and D. Du, Biomacromolecules, 2015, 16, 2363–2373 CrossRef CAS PubMed.
- S. Gotla and S. Matysiak, Phys. Chem. Chem. Phys., 2023, 25, 10113–10120 RSC.
- Q.-Y. Meng, H. Wang, Z.-B. Cui, W.-G. Yu and X.-Z. Lu, Molecules, 2020, 25, 1314 CrossRef CAS PubMed.
- M. Schäffler, S. Samantray and B. Strodel, Int. J. Mol. Sci., 2023, 24, 11238 CrossRef PubMed.
- S. Samantray and B. Strodel, J. Phys. Chem. B, 2021, 125, 5511–5525 CrossRef CAS PubMed.
- X. Dong, R. Qi, Q. Qiao, X. Li, F. Li, J. Wan, Q. Zhang and G. Wei, Phys. Chem. Chem. Phys., 2021, 23, 20406–20418 RSC.
- B. Khurshid, A. U. Rehman, R. Luo, A. Khan, A. Wadood and J. Anwar, ACS Omega, 2022, 7, 15132–15144 CrossRef CAS PubMed.
- A. Sahoo, P.-Y. Lee and S. Matysiak, J. Chem. Theory Comput., 2022, 18, 5046–5055 CrossRef CAS PubMed.
- S. Khan, J. Gor, B. Mulloy and S. J. Perkins, J. Mol. Biol., 2010, 395, 504–521 CrossRef CAS PubMed.
- J. J. Balbach, Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed and R. Tycko, Biochemistry, 2000, 39, 13748–13759 CrossRef CAS PubMed.
- X. Zhou, Y. Wang, W. Zheng, G. Deng, F. Wang and L. Jin, Front. Mol. Biosci, 2022, 9, 824146 CrossRef CAS PubMed.
- A. V. Nogueira, D. L. Drehmer, M. Iacomini, G. L. Sassaki and T. R. Cipriani, Carbohydr. Polym., 2017, 157, 72–78 CrossRef CAS PubMed.
- D. H. Atha, B. Coxon, V. Reipa and A. K. Gaigalas, J. Pharm. Sci., 1995, 84, 360–364 CrossRef CAS PubMed.
- S. O. Yesylevskyy, L. V. Schäfer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, e1000810 CrossRef PubMed.
- D. H. de Jong, G. Singh, W. F. D. Bennett, C. Arnarez, T. A. Wassenaar, L. V. Schäfer, X. Periole, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2013, 9, 687–697 CrossRef CAS PubMed.
- S. J. Ganesan and S. Matysiak, J. Chem. Theory Comput., 2014, 10, 2569–2576 CrossRef CAS PubMed.
- S. J. Ganesan, H. Xu and S. Matysiak, Phys. Chem. Chem. Phys., 2016, 18, 17836–17850 RSC.
- S. Khan, K. W. Fung, E. Rodriguez, R. Patel, J. Gor, B. Mulloy and S. J. Perkins, J. Biol. Chem., 2013, 288, 27737–27751 CrossRef CAS PubMed.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- P. H. Nguyen, M. S. Li, G. Stock, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 111–116 CrossRef CAS PubMed.
- G. M. Castillo, W. Lukito, T. N. Wight and A. D. Snow, J. Neurochem., 1999, 72, 1681–1687 CrossRef CAS PubMed.
- G. Berth, H. Dautzenberg and M. G. Peter, Carbohydr. Polym., 1998, 36, 205–216 CrossRef CAS.
- G. Pavlov, S. Finet, K. Tatarenko, E. Korneeva and C. Ebel, Eur. Biophys. J., 2003, 32, 437–449 CrossRef CAS PubMed.
- N. N. Jha, A. Anoop, S. Ranganathan, G. M. Mohite, R. Padinhateeri and S. K. Maji, Biochemistry, 2013, 52, 8800–8810 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Heparin model development, supporting data on the properties of Aβ16–22 aggregates and movies depicting aggregation pathways. See DOI: https://doi.org/10.1039/d4cp02331e |
| This journal is © the Owner Societies 2024 |