
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The valence electron affinity of uracil determined by anion cluster photoelectron spectroscopy†
Connor J.
Clarke
 a,
E. Michi
Burrow
a and
Jan R. R.
Verlet
*ab
a,
E. Michi
Burrow
a and
Jan R. R.
Verlet
*ab
aDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: j.r.r.verlet@durham.ac.uk
bJ. Heyrovský Institute of Physical Chemistry, Czech Academy of Sciences, Dolejškova 3, 18223 Prague 8, Czech Republic
First published on 1st July 2024
Abstract
The unoccupied π* orbitals of the nucleobases are considered to play important roles in low-energy electron attachment to DNA, inducing damage. While the lowest anionic valence state is vertically unbound in all neutral nucleobases, it remains unclear even for the simplest nucleobase, uracil (U), whether its valence anion (U−) is adiabatically bound, which has important implications on the efficacy of damage processes. Using anion photoelectron spectroscopy, we demonstrate that the valence electron affinity (EAV) of U can be accurately measured within weakly solvating clusters, U−(Ar)n and U−(N2)n. Through extrapolation to the isolated U limit, we show that EAV = −2 ± 18 meV. We discuss these findings in the context of electron attachment to U and its reorganization energy, and more generally establish guidance for the determination of molecular electron affinities from the photoelectron spectroscopy of anion clusters.
Introduction
Low-energy electrons can induce strand breaks in DNA, even at electron energies below the ionization energy of individual components and below the dissociation energies of covalent bonds in the biopolymer.1–3 The initial step is believed to involve the electron attachment to valence states of components of DNA, forming temporary negative ions that can then lead to bond-rupture through a long-range dissociative electron attachment process.4–6 The nucleobases in particular have received much attention as the site for initial electron attachment via their low-lying valence states that are of π* character.6–10 In the gas phase, electron transmission spectroscopic experiments have mapped out the location of these π* resonances.11,12 However, such experiments are not sensitive to the subsequent dynamics that could stabilize the generated temporary negative ions. In particular, nuclear dynamics can compete with autodetachment to decrease the energy gap between the anion valence state and the neutral ground state, leading to a highly reactive radical anion that is long-lived, especially if the anion valence π* state is adiabatically bound. So, a natural question then arises: what is the adiabatic electron affinity of the lowest valence state, EAV, of a nucleobase? Even for the simplest nucleobase, uracil (U), this remains a debated question. Here, we seek to answer this question by applying anion photoelectron spectroscopy to a range of uracil clusters to determine EAV of U.The U nucleobase forms a stable anion in the gas phase,13,14 where the excess electron is very weakly bound in a diffuse orbital by the strong dipole moment of the molecule (μ ≈ 4.5 D). However, such a dipole-bound state (DBS)15 is not a valence state and, while non-valence states can partake in electron attachment at very low electron energies16–23 and population can pass from valence to non-valence state and vice versa,24,25 they are not thought to be relevant in the context of DNA damage because the environment will disrupt the orbital.26 From electron transmission spectroscopy, the lowest-energy shape resonance, 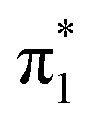 , is populated with incident electron energies around 220 meV.11 Upon geometric relaxation, electronic states may stabilize by hundreds of meV, and the
, is populated with incident electron energies around 220 meV.11 Upon geometric relaxation, electronic states may stabilize by hundreds of meV, and the  valence state was suspected to be adiabatically bound with respect to electron loss. However, anion photoelectron spectroscopic studies reported observation of only the DBS of U−.13 This was rationalized as the DBS being more stable than the
valence state was suspected to be adiabatically bound with respect to electron loss. However, anion photoelectron spectroscopic studies reported observation of only the DBS of U−.13 This was rationalized as the DBS being more stable than the  state, and an upper-bound for the EAV was found to be EAV < EAD ≈ +90 meV, where EAD is the electron binding energy of the uracil anion in its DBS. On the other hand, long-lived U− in its
state, and an upper-bound for the EAV was found to be EAV < EAD ≈ +90 meV, where EAD is the electron binding energy of the uracil anion in its DBS. On the other hand, long-lived U− in its  state was observed to form via Rydberg electron transfer to U(Ar)n clusters, following evaporation of the solvating argon atoms.27 This was presented as confirmation that the
state was observed to form via Rydberg electron transfer to U(Ar)n clusters, following evaporation of the solvating argon atoms.27 This was presented as confirmation that the 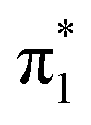 state is adiabatically bound with respect to electron loss, and they reported EAV = +62 ± 32 meV.27 Computationally, there have been a wide range of EAV values reported ranging from negative28–33 to positive.6,34–40 The seemingly most comprehensive treatment has been performed by Gu et al., who followed the W1BD composite method41 and found EAV = +24 ± 13 meV,42 suggesting a weakly adiabatically bound
state is adiabatically bound with respect to electron loss, and they reported EAV = +62 ± 32 meV.27 Computationally, there have been a wide range of EAV values reported ranging from negative28–33 to positive.6,34–40 The seemingly most comprehensive treatment has been performed by Gu et al., who followed the W1BD composite method41 and found EAV = +24 ± 13 meV,42 suggesting a weakly adiabatically bound  valence state.
valence state.
Solvation stabilizes valence-bound anions to a greater degree than dipole-bound anions.43 Consequently, the ground state of uracil–water cluster anions, U−(H2O)n, is observed to be the  valence state, as easily distinguished in photoelectron spectra by its greater spectral width compared to the signal arising from a DBS.44–46 By plotting the n-dependent EAV of these clusters, it therefore becomes possible to extrapolate towards n = 0 and obtain an estimate for the EAV of U. Two studies have performed this procedure, both yielding relatively large positive adiabatic electron affinities: EAV = +150 ± 120 meV44 and EAV = +159 meV46 (uncertainty not stated). However, the 0–0 transition for photodetachment from the
valence state, as easily distinguished in photoelectron spectra by its greater spectral width compared to the signal arising from a DBS.44–46 By plotting the n-dependent EAV of these clusters, it therefore becomes possible to extrapolate towards n = 0 and obtain an estimate for the EAV of U. Two studies have performed this procedure, both yielding relatively large positive adiabatic electron affinities: EAV = +150 ± 120 meV44 and EAV = +159 meV46 (uncertainty not stated). However, the 0–0 transition for photodetachment from the  valence state could not be discerned clearly for any of the clusters, and moreover, the validity of performing such an extrapolation is questionable. Water molecules interact very strongly with the uracil anion through a multitude of interactions (e.g. hydrogen bonding and dipolar) so that a simple linear extrapolation is unlikely to be valid even for small clusters. In principle, these concerns can be alleviated through the study of uracil–solvent cluster anions with more weakly interacting solvent molecules. To this end, we performed anion photoelectron spectroscopy on two different series of uracil–solvent cluster anions: U−(Ar)n and U−(N2)n, and compared our findings to results on U−(H2O)n. This approach, utilizing spectroscopic tags, also offers colder clusters and allowed us to unambiguously identify the 0–0 transition for photodetachment from the
valence state could not be discerned clearly for any of the clusters, and moreover, the validity of performing such an extrapolation is questionable. Water molecules interact very strongly with the uracil anion through a multitude of interactions (e.g. hydrogen bonding and dipolar) so that a simple linear extrapolation is unlikely to be valid even for small clusters. In principle, these concerns can be alleviated through the study of uracil–solvent cluster anions with more weakly interacting solvent molecules. To this end, we performed anion photoelectron spectroscopy on two different series of uracil–solvent cluster anions: U−(Ar)n and U−(N2)n, and compared our findings to results on U−(H2O)n. This approach, utilizing spectroscopic tags, also offers colder clusters and allowed us to unambiguously identify the 0–0 transition for photodetachment from the  valence state. Our determination for EAV is broadly in line with the ‘best’ computational predictions,6,42 and expands upon the results from Rydberg electron transfer experiments.27 It also provides a rigorous assessment and establishes protocols for the determination of small (be they positive or negative) adiabatic electron affinities using photoelectron spectroscopy of anion clusters.
valence state. Our determination for EAV is broadly in line with the ‘best’ computational predictions,6,42 and expands upon the results from Rydberg electron transfer experiments.27 It also provides a rigorous assessment and establishes protocols for the determination of small (be they positive or negative) adiabatic electron affinities using photoelectron spectroscopy of anion clusters.
Methods
Experimental
The experimental apparatus has been described in detail elsewhere.47 A solid sample of uracil (U) was placed inside a pulsed Even–Lavie valve48 and heated to approximately 220 °C. The valve was backed with argon or nitrogen at ∼10 bar pressure to produce the desired series of molecular clusters, U(Ar)n or U(N2)n. To induce formation of uracil–water clusters, U(H2O)n, a drop of water was added to the backing line. The molecular beam passed through a (thoriated tungsten) filament ring ionizer held at high current, attaching electrons and generating the corresponding progressions of cluster anions. A Wiley–McLaren time-of-flight mass spectrometer49 separated the cluster anions by their mass-to-charge ratio (m/z), and the targeted anion packet was intersected with a delayed laser pulse at the center of a velocity map imaging electron spectrometer. Nanosecond laser pulses were sourced from an Nd:YAG laser (Quantel, Q-smart 450), at the fundamental (1064 nm) or second harmonic (532 nm) wavelengths. Photoelectron spectra and angular distributions were reconstructed from the resulting photoelectron images using the polar onion peeling (POP) algorithm.50 The well-known energetics of atomic I− were used for calibration, and full details on our calibration process are included in the ESI.†Computational
Density functional theory (DFT) tends to produce a bound state of the uracil anion,42 making it an ideal choice for calculating the vibronic spectrum of valence U−. We opted for the long-range corrected CAM-B3LYP functional,51 with the diffuse aug-cc-pVDZ Dunning basis set.52 By omitting the extra-diffuse basis functions that are often added into calculations on U−, the optimized anion geometry settled into the buckled
state of the uracil anion,42 making it an ideal choice for calculating the vibronic spectrum of valence U−. We opted for the long-range corrected CAM-B3LYP functional,51 with the diffuse aug-cc-pVDZ Dunning basis set.52 By omitting the extra-diffuse basis functions that are often added into calculations on U−, the optimized anion geometry settled into the buckled  valence state, and a minimum was confirmed through vibrational frequency analysis. The planar equilibrium structure of neutral uracil was also calculated, allowing the vibronic spectrum associated with the S0 ←
valence state, and a minimum was confirmed through vibrational frequency analysis. The planar equilibrium structure of neutral uracil was also calculated, allowing the vibronic spectrum associated with the S0 ← 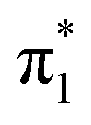 transition to be computed. Each transition was broadened by a Gaussian function with standard deviation σ = 20 meV, and summed to produce a representative photoelectron spectrum. The simulated spectrum was shifted to align with the measured 0–0 transition of U−(Ar)3. All calculations were performed with Gaussian 16.53
transition to be computed. Each transition was broadened by a Gaussian function with standard deviation σ = 20 meV, and summed to produce a representative photoelectron spectrum. The simulated spectrum was shifted to align with the measured 0–0 transition of U−(Ar)3. All calculations were performed with Gaussian 16.53
Results and discussion
Photoelectron spectra of uracil–argon cluster anions
Photoelectron spectra of a series of uracil–argon cluster anions, U−(Ar)n where n ≤ 25, were acquired using nanosecond laser pulses with photon energy hν = 1.165 eV. Fig. 1(a) shows a selection of the spectra corresponding to the smaller clusters, n ≤ 9. Photoelectron signal is plotted in terms of electron binding energy, defined as eBE = hν − eKE, where eKE is the measured electron kinetic energy. As found in earlier photoelectron spectroscopy experiments,13,44,45 the photoelectron spectrum of U− exhibits a single, sharp peak at low electron binding energy. This is the characteristic photoelectron signal arising from photodetachment of a DBS to reach the ground-state neutral, as the excess electron only weakly interacts with the neutral core. We additionally confirm that the electron emission is highly anisotropic (β2 = +2.0, see Fig. 1(b)) as expected from an s-like non-valence orbital.54,55 We determine that EAD = +75 ± 6 meV (details of our calibration process in the ESI†). | ||
Fig. 1 (a) Photoelectron spectra of uracil–argon cluster anions, U−(Ar)n, acquired using nanosecond laser pulses with hν = 1.165 eV. Blue arrows highlight the electron binding energy (eBE) associated with the 0–0 transition from the  valence state to the neutral ground state. (b) Corresponding photoelectron images from dipole-bound U− (left) and valence-bound U−(Ar)3 (right). The fixed direction of laser polarization vector is represented by ε. valence state to the neutral ground state. (b) Corresponding photoelectron images from dipole-bound U− (left) and valence-bound U−(Ar)3 (right). The fixed direction of laser polarization vector is represented by ε. | ||
The photoelectron spectra of U−(Ar)1 and U−(Ar)2 are similar to the spectrum of U−. The low-energy electrons (eBE > 0.5 eV) were produced from small contamination of uracil–water cluster anions of similar masses to U−(Ar)1–2, but given this signal does not interfere with the low binding energy peak associated with the dipole-bound state, this signal has no impact on the current discussion. It is clear that formation of the DBS of U− remains favorable in the presence of one or two argon atoms, and solvation appears to only increase EAD by a few meV per argon atom. Upon the addition of a third solvating argon atom, the photoelectron spectrum changes drastically. U−(Ar)3 exhibits a broad, structured photoelectron signal, with a far more isotropic photoelectron angular distribution (β2 ≈ +0.4, see Fig. 1(b)). This arises from photodetachment of the 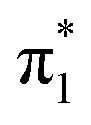 valence state and suggests that it has become the ground electronic state of the anion due to stabilization from the solvating argon atoms and agrees well with an earlier photoelectron spectroscopic measurement of the
valence state and suggests that it has become the ground electronic state of the anion due to stabilization from the solvating argon atoms and agrees well with an earlier photoelectron spectroscopic measurement of the 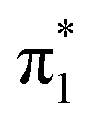 valence state of U−(Xe)1,45 which also shows a broad feature with underlying vibrational structure (although the structured peak was partially obscured by photoelectron signal arising from some accompanying DBS). The considerable spectral width of the
valence state of U−(Xe)1,45 which also shows a broad feature with underlying vibrational structure (although the structured peak was partially obscured by photoelectron signal arising from some accompanying DBS). The considerable spectral width of the  detachment feature arises from the substantially different equilibrium geometries between the anionic and neutral form of the uracil molecule: the neutral molecule is planar while the anion becomes non-planar.33
detachment feature arises from the substantially different equilibrium geometries between the anionic and neutral form of the uracil molecule: the neutral molecule is planar while the anion becomes non-planar.33
Despite the disparate geometries, it appears that a peak corresponding to the 0–0 transition is resolved in Fig. 1 (blue arrows), which represents a direct measure of EAV associated with the  state of U−(Ar)n. To reinforce assignment of the 0–0 transition peak, Fig. 2 recasts the photoelectron spectrum of U−(Ar)3 with an overlay of a computed (DFT/(U)CAM-B3LYP/aug-cc-pVDZ) vibronic photodetachment spectrum of the
state of U−(Ar)n. To reinforce assignment of the 0–0 transition peak, Fig. 2 recasts the photoelectron spectrum of U−(Ar)3 with an overlay of a computed (DFT/(U)CAM-B3LYP/aug-cc-pVDZ) vibronic photodetachment spectrum of the  valence state of U−. As the excess charge is expected to localize on the nucleobase, exclusion of the argon in the calculations was considered acceptable. Indeed, the vibrational structure displayed in photoelectron spectra of the larger U−(Ar)n>3 clusters are very similar, demonstrating the negligible effect of the argon atoms. The calculated vibronic spectrum shows good overall agreement, although the vibrational structure of the experimental spectrum was not perfectly captured. Nevertheless, the dominant vibrational mode can be clearly identified as v15, which corresponds to an out-of-plane motion (displacement vector shown in ESI†). Some disparity might be expected due to differences in anion–neutral geometries, which results in a broad Franck–Condon window made up of many combined excitations, and the neglect of anharmonicity. In the context of this study, it is more critical that we can conclude that the 0–0 transition is clearly distinguishable in the photoelectron spectra of U−(Ar)n, and therefore the associated EAV can be accurately determined for each cluster.
valence state of U−. As the excess charge is expected to localize on the nucleobase, exclusion of the argon in the calculations was considered acceptable. Indeed, the vibrational structure displayed in photoelectron spectra of the larger U−(Ar)n>3 clusters are very similar, demonstrating the negligible effect of the argon atoms. The calculated vibronic spectrum shows good overall agreement, although the vibrational structure of the experimental spectrum was not perfectly captured. Nevertheless, the dominant vibrational mode can be clearly identified as v15, which corresponds to an out-of-plane motion (displacement vector shown in ESI†). Some disparity might be expected due to differences in anion–neutral geometries, which results in a broad Franck–Condon window made up of many combined excitations, and the neglect of anharmonicity. In the context of this study, it is more critical that we can conclude that the 0–0 transition is clearly distinguishable in the photoelectron spectra of U−(Ar)n, and therefore the associated EAV can be accurately determined for each cluster.
 | ||
Fig. 2 Measured photoelectron spectrum of U−(Ar)3 (black), overlaid with the calculated (CAM-B3LYP/aug-cc-pVDZ) vibronic photodetachment spectrum (red) of the 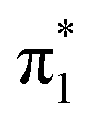 valence state of U−. valence state of U−. | ||
With increasing cluster size, the 0–0 transition shifts to higher electron-binding energy. This demonstrates that EAV increases with each incrementally added argon atom, directly reflecting the imposed anion-stabilizing effect. In particular, U− is stabilized through favorable interactions between the excess negative charge and the polarizability of the argon atoms. From the measured 0–0 transition energies, it is possible to extrapolate the measured EAV(n) to the isolated U− limit (i.e. n = 0), obtaining an estimate for EAV of U. However, in order to perform such an extrapolation, the behavior of EAV(n) should be considered in detail.
Electron-binding energy extrapolation of valence-bound anions in solvating clusters
Fig. 3 compiles the argon-induced anion-stabilizing effect, Estab(n) = EAV(X(Ar)n) − EAV(X), from a number of photoelectron spectroscopic studies on valence-bound X−(Ar)n clusters, where X = O,56 NO,57 Cl,58 Br,59 I,59 I2,60 and pyrazine.61 It is apparent that the more electron-dense anions are stabilized to a greater degree by the clustered argon atoms. The trend in Estab(n) (and therefore also EAV) for each anion cluster appears approximately linear at small n, but curves slightly at larger n, where the Estab(n) increases by a smaller amount per successive argon atom added. This curvature has been explained to be a result of various many-body interactions (mostly between the charged anion and the clustered argon atoms),58,59 ultimately adding a small destabilizing effect to the anion with respect to the neutral form. Through least-squares fitting, we found the power function Estab(n) ∼ n0.95 to reproduce the exhibited curvature very well for each series of anion clusters. The only exception to this is O−(Ar)n, which also interacts most strongly with the solvating Ar atoms. In the case of U, Estab(n) for U−(Ar)n is the most comparable to Br−(Ar)n, suggesting that the U−–Ar interaction strength is similar to Br−–Ar and the above power function is appropriate to use for extrapolation purposes. | ||
| Fig. 3 Stabilization energy Estab = EAV(X(Ar)n) − EAV(X), associated with the clustering of n argon atoms to different anions, X. Circles and triangles are used to distinguish between atomic and molecular anions, respectively. The dotted lines show fitted power functions of n associated with each series of anion clusters. For errors on specific data points, the author is directed to the relevant studies. Adapted from ref. 56–61, with the permission of AIP publishing. | ||
Determination of the electron affinity of the valence state of uracil
Fig. 4 presents the experimentally determined EAV associated with each measured U−(Ar)n cluster, demonstrating a slightly curved behavior at smaller cluster size. There is a clearly observable ‘kink’ in the trend around the U−(Ar)12 cluster, which is indicative of the (partial) closing of a solvation shell around U−.56,57 For now, we focus on the EAV values of the small clusters. The EAV values of U(Ar)3–5 were fit to the empirical function EAV(n) = kArn0.95 + EAV, where kAr was an optimized constant associated with the interaction strength between U− and Ar. Only clusters consisting of up to five argons were included in the fit, since a second less-defined kink may be present at n = 6. The fitting function, extrapolated to n = 0, is shown in Fig. 4. Extrapolation to n = 2 suggests that the EAV is below the electron binding energy of the DBS for this cluster, consistent with the absence of the valence state signal in the photoelectron spectrum of U−(Ar)2. From the full extrapolation, we find that EAV = −6 ± 24 meV, where the uncertainty in EAV was determined by the combined contributions of our experimental resolution and the extrapolation process.62 Unfortunately, our results on the U−(Ar)n clusters were unable to conclusively determine whether the
valence state signal in the photoelectron spectrum of U−(Ar)2. From the full extrapolation, we find that EAV = −6 ± 24 meV, where the uncertainty in EAV was determined by the combined contributions of our experimental resolution and the extrapolation process.62 Unfortunately, our results on the U−(Ar)n clusters were unable to conclusively determine whether the  state of U− is adiabatically bound or not. Nevertheless, we can conclude that EAV is very small. By directly observing the 0–0 transition associated with the
state of U− is adiabatically bound or not. Nevertheless, we can conclude that EAV is very small. By directly observing the 0–0 transition associated with the  valence state in weakly perturbed clusters, this extracted value is currently the most accurate experimentally-determined EAV, and is lower than, but within the error of the most reliable theoretical determination.42
valence state in weakly perturbed clusters, this extracted value is currently the most accurate experimentally-determined EAV, and is lower than, but within the error of the most reliable theoretical determination.42
 | ||
Fig. 4 Adiabatic electron affinity (EA) of uracil–argon clusters, U(Ar)n, extracted from photoelectron spectroscopy of the corresponding anions. Dots indicate EAV(n) associated with the  valence state of U−(Ar)n, whereas the crosses show the measured electron binding energies of the DBS anions, EAD. An extrapolation using the EAV(n) of U(Ar)3–5 is overlaid (black line). Inset is a magnified view for small n (red dashed area). Uncertainties in each data point were smaller than the size of the data point symbols, and were below ±10 meV for n = 0–5. valence state of U−(Ar)n, whereas the crosses show the measured electron binding energies of the DBS anions, EAD. An extrapolation using the EAV(n) of U(Ar)3–5 is overlaid (black line). Inset is a magnified view for small n (red dashed area). Uncertainties in each data point were smaller than the size of the data point symbols, and were below ±10 meV for n = 0–5. | ||
Comparison with other solvent clusters and the caveats of using strongly interacting solvents
Further estimates for EAV can be obtained by repeating the above procedure with different solvent molecules. Fig. 5 displays photoelectron spectra (hν = 1.165 eV) of U−(N2)n, with n ≤ 6. The vibrational structure in the detachment from the valence state is very similar to that observed from U−(Ar)n, allowing EAV(n) to be extracted from the distinct 0–0 transition feature in each spectrum. The nitrogen molecules interact more strongly with the uracil anion than do the argon atoms, as reflected in the greater degree of stabilization per added solvent molecule, as well as in the observation of the
valence state is very similar to that observed from U−(Ar)n, allowing EAV(n) to be extracted from the distinct 0–0 transition feature in each spectrum. The nitrogen molecules interact more strongly with the uracil anion than do the argon atoms, as reflected in the greater degree of stabilization per added solvent molecule, as well as in the observation of the  valence state for U−(N2)2. Despite the similar polarizabilities of N2 and Ar,63 the inherently anisotropic charge distribution of diatomic nitrogen allows for additional interactions with U−, e.g. through its significant quadrupole.64 The observed 0–0 transitions in the photoelectron spectra of U−(N2)n are also broader than in the case of U−(Ar)n, and so the uncertainty associated with finding the EAV was slightly larger. This spectral broadening again correlates with stronger interactions between the anion and N2, consistent with previous studies on NO− cluster anions.57 In particular, stronger interactions between U− and N2 (compared to U− and Ar) can act to both: encourage excitation of lower-frequency vibrational modes which cannot be spectrally resolved; and increase the degree of structural rearrangement in the transition from anion to neutral, leading to a broader Franck–Condon window. Additionally, some of the broadening may be attributed to the presence of multiple structural isomers for each anion cluster, as the N2 molecules preferentially bind to different sites of the uracil anion (with facile interchange), resulting in subtly different binding energies for the excess electron between isomers.
valence state for U−(N2)2. Despite the similar polarizabilities of N2 and Ar,63 the inherently anisotropic charge distribution of diatomic nitrogen allows for additional interactions with U−, e.g. through its significant quadrupole.64 The observed 0–0 transitions in the photoelectron spectra of U−(N2)n are also broader than in the case of U−(Ar)n, and so the uncertainty associated with finding the EAV was slightly larger. This spectral broadening again correlates with stronger interactions between the anion and N2, consistent with previous studies on NO− cluster anions.57 In particular, stronger interactions between U− and N2 (compared to U− and Ar) can act to both: encourage excitation of lower-frequency vibrational modes which cannot be spectrally resolved; and increase the degree of structural rearrangement in the transition from anion to neutral, leading to a broader Franck–Condon window. Additionally, some of the broadening may be attributed to the presence of multiple structural isomers for each anion cluster, as the N2 molecules preferentially bind to different sites of the uracil anion (with facile interchange), resulting in subtly different binding energies for the excess electron between isomers.
 | ||
Fig. 5 Photoelectron spectra of uracil–nitrogen cluster anions, U−(N2)n, acquired using nanosecond laser pulses with hν = 1.165 eV. Blue arrows highlight the electron binding energy (eBE) associated with the 0–0 transition from the  valence state to the neutral ground state. valence state to the neutral ground state. | ||
Fig. 6 presents the extracted EAV(n) of the U(N2)n clusters, alongside the earlier results on U(Ar)n. Over the range of small clusters measured, the curvature exhibited in the plot of EAV(n) was exacerbated with the N2 solvent. Once more, this follows from stronger intermolecular forces within the clusters. In fact, the strength of interaction between U− and N2 is comparable to the O−(Ar)n anion clusters shown in Fig. 3, which also expressed a pronounced curvature (compared to the more weakly interacting anion–argon clusters). However, there is an important difference between the U−(N2)n and O−(Ar)n: the latter series of clusters are comprised of atomic subunits, whereas both uracil and molecular nitrogen are polyatomic. Therefore, the many-body interactions that govern the exhibited curvature of the O−(Ar)n clusters are completely absent in O−(Ar)1, and so linear behavior is expected in the range 0 ≤ n ≤ 2 for these atomic clusters. In the case of U−(N2)n, the curvature results from a combination of many intermolecular interactions (e.g. between dipoles, quadrupoles, polarizabilities), which all remain present even in U−(N2)1. Consequently, there is no reason to suspect the exhibited curvature not to persist to the n = 0 limit. We found a relationship, EAV(n) = kN2n0.7 + EAV, to fit the data appropriately (fit included in Fig. 6), with the optimized coefficient kN2 > kAr. A value EAV = +1 ± 26 meV was extracted, consistent with the EAV derived from the U−(Ar)n series of clusters, and providing further evidence that the excess electron in the  valence state of U− is very weakly bound, if at all.
valence state of U− is very weakly bound, if at all.
 | ||
| Fig. 6 Adiabatic electron affinities (EA) for a selection of uracil–solvent clusters, U(Ar)n (black), U(N2)n (red), and U(H2O)n (blue), extracted from photoelectron spectroscopy of the corresponding anions. Extrapolations are overlaid as solid lines. Uncertainties in each data point were smaller than the size of the data point symbols for the U−(N2)n and U−(H2O)1 clusters. The uncertainties for U−(H2O)2–6 were larger (due to absence of the 0–0 transition), around 0.1 eV. | ||
Solvation-induced stabilization of U− has been the most extensively studied in uracil–water cluster anions,44,46,65 where even a single water molecule sufficiently stabilizes the  valence state to become the ground state of the uracil anion. A photoelectron spectrum of U−(H2O)1 is shown in Fig. 7 (hν = 1.165 eV), where the 0–0 transition peak arising from the
valence state to become the ground state of the uracil anion. A photoelectron spectrum of U−(H2O)1 is shown in Fig. 7 (hν = 1.165 eV), where the 0–0 transition peak arising from the  valence state was distinct but broad. Through its characteristic spectral signature, the formation of the solvent-stabilized DBS of U−(H2O)1 in our ion source was also observed; it will be the subject of a forthcoming communication and is briefly discussed further below. From the 0–0 transition associated with the
valence state was distinct but broad. Through its characteristic spectral signature, the formation of the solvent-stabilized DBS of U−(H2O)1 in our ion source was also observed; it will be the subject of a forthcoming communication and is briefly discussed further below. From the 0–0 transition associated with the  valence state, the solvating power of H2O is demonstrably much greater than either Ar or N2. Indeed, the anion-stabilizing effect of a single water molecule exceeded that of 25 argon atoms: EAV(U(H2O)1) > EAV(U(Ar)25). It is unsurprising that the interactions of U− with H2O are so vastly stronger, as clustering with water is additionally supported by long-range charge–dipole interactions as well as hydrogen bonding.
valence state, the solvating power of H2O is demonstrably much greater than either Ar or N2. Indeed, the anion-stabilizing effect of a single water molecule exceeded that of 25 argon atoms: EAV(U(H2O)1) > EAV(U(Ar)25). It is unsurprising that the interactions of U− with H2O are so vastly stronger, as clustering with water is additionally supported by long-range charge–dipole interactions as well as hydrogen bonding.
 | ||
Fig. 7 Photoelectron spectra of small uracil–water cluster anions, U−(H2O)n, acquired using nanosecond laser pulses with hν = 1.165 eV (n = 0, 1) and hν = 2.230 eV (n = 2, 3). Blue arrows highlight the electron binding energy (eBE) associated with the 0–0 transition from the  valence state to the neutral ground state. The contribution from the DBS of U−(H2O)1 is highlighted in red. valence state to the neutral ground state. The contribution from the DBS of U−(H2O)1 is highlighted in red. | ||
Larger clusters of U−(H2O)n≥2 did not produce distinguishable 0–0 transition features due to further spectral broadening, and therefore each EAV(n) associated with these clusters was estimated from the onset of the Gaussian-like photoelectron signal (taken as 10% of peak height). The resulting EAV(n) values are also displayed in Fig. 6, and exhibit an even more pronounced curvature with n. We do not present an extrapolation to the monomeric limit (n = 0) using the uracil–water cluster EAV(n) for several reasons: (i) there was no simple power function which suitably captured the experimental data; (ii) the 0–0 transition was not observable in all but one of the clusters, obscuring accurate determination of the EAV; and (iii) water molecules are far more selective towards the binding site of U− (or to other H2O molecules in the cluster), largely due to their tendency for hydrogen bonding. Nonetheless, linearly extrapolating the EAV(n) of the smallest U−(H2O)n clusters results in an estimated EAV ≈ +177 meV, similar to the electron affinities obtained in earlier studies that performed this procedure.44,46 Our results on the U−(Ar)n and U−(N2)n clusters highlight the problems associated with performing such a linear extrapolation: in particular, the EAV for some of the argon- and nitrogen-stabilized clusters are measurably lower than 177 meV. We conclude that, in general, linear behavior of the electron binding energy should not be assumed in clusters containing a polyatomic anion and a strongly solvating species such as water. In the case of U−, this leads to systematic errors up to hundreds of meV.
General discussion
The observed vibrational structure in the photoelectron spectra of U−(Ar)n and U−(N2)n also provides insight into the nuclear rearrangement undergone by uracil in the transition from its neutral to anionic valence state. The vertical detachment energy (VDE) of each anion cluster was extracted from the eBE at which there was most (fitted) photoelectron signal, and it was found to decrease with smaller n at the same rate as EAV(n). From the appropriate extrapolation, we find that VDE(U−) = +210 ± 30 meV. The extracted detachment energies of U− (EAD, EAV, and VDE) can be used to draw a schematic of the relevant diabatic potential energy surfaces along the ring-buckling coordinate that connects the neutral (S0) and anionic (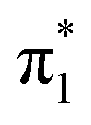 ) equilibrium geometries (Fig. 8). Encouragingly, electron transmission spectroscopy has measured the vertical attachment energy (VAE) of the
) equilibrium geometries (Fig. 8). Encouragingly, electron transmission spectroscopy has measured the vertical attachment energy (VAE) of the  valence state of U to be very close to our extracted VDE:VAE = 220 meV.11 Assuming the same harmonic diabatic surfaces for S0 and
valence state of U to be very close to our extracted VDE:VAE = 220 meV.11 Assuming the same harmonic diabatic surfaces for S0 and  valence states allows us to connect the buckling coordinate in a consistent picture as shown in Fig. 8. In the context of low-energy electron attachment into the
valence states allows us to connect the buckling coordinate in a consistent picture as shown in Fig. 8. In the context of low-energy electron attachment into the  resonance, U− forms in the planar geometry, but will rapidly stabilize via the buckling distortion. With only the slightest buckling away from planarity, the
resonance, U− forms in the planar geometry, but will rapidly stabilize via the buckling distortion. With only the slightest buckling away from planarity, the 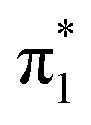 valence state energy approaches that of the neutral S0 state and then becomes vertically bound with respect to electron loss, greatly enhancing the lifetime of the generated anion. Regardless of whether the
valence state energy approaches that of the neutral S0 state and then becomes vertically bound with respect to electron loss, greatly enhancing the lifetime of the generated anion. Regardless of whether the  state is adiabatically bound in its minimum energy (fully-buckled) structure (i.e. EAV < 0 or EAV > 0), its vertical binding exceeds 200 meV and U− formed by electron attachment via the
state is adiabatically bound in its minimum energy (fully-buckled) structure (i.e. EAV < 0 or EAV > 0), its vertical binding exceeds 200 meV and U− formed by electron attachment via the 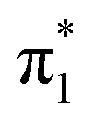 resonance is expected to persist on a timescale greatly surpassing the sub-picosecond lifetimes associated with shape resonance autodetachment.19,66,67
resonance is expected to persist on a timescale greatly surpassing the sub-picosecond lifetimes associated with shape resonance autodetachment.19,66,67
 | ||
Fig. 8 Schematic showing diabatic potential energy surfaces of neutral (S0) and anionic ( and DBS) uracil along the ring-buckling coordinate, Q, treated within a harmonic approximation. Energy gaps reflect the experimentally determined values, and the gray region represents the electron detachment continuum. Uncertainty in the measured value of EAV is shown with blue bars. and DBS) uracil along the ring-buckling coordinate, Q, treated within a harmonic approximation. Energy gaps reflect the experimentally determined values, and the gray region represents the electron detachment continuum. Uncertainty in the measured value of EAV is shown with blue bars. | ||
A competing decay pathway for the 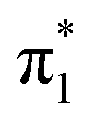 state is through internal conversion to the DBS.24 Within the harmonic approximation displayed in Fig. 8, an energetic barrier is expected to separate the two states, limiting the rate of internal conversion from the
state is through internal conversion to the DBS.24 Within the harmonic approximation displayed in Fig. 8, an energetic barrier is expected to separate the two states, limiting the rate of internal conversion from the  state to the DBS. Numerous computational studies have attempted to calculate the height of this barrier, with estimates ranging from tens to hundreds of meV.32,38,68 Within our simple linear response model in Fig. 8, it appears the barrier height is on the order of 10 s of meV. This barrier may have held important consequences for the earlier Rydberg electron transfer experiments that had suggested a positive value for the EAV.27 These experiments attached electrons to U(Ar)n clusters, and following evaporation of the clustered argon atoms, formed U− in the
state to the DBS. Numerous computational studies have attempted to calculate the height of this barrier, with estimates ranging from tens to hundreds of meV.32,38,68 Within our simple linear response model in Fig. 8, it appears the barrier height is on the order of 10 s of meV. This barrier may have held important consequences for the earlier Rydberg electron transfer experiments that had suggested a positive value for the EAV.27 These experiments attached electrons to U(Ar)n clusters, and following evaporation of the clustered argon atoms, formed U− in the  state. U− was kinetically trapped in the valence state for many microseconds (in order to be mass-separated), which is consistent with our combined observations of (i) a large vertical binding energy (VDE), and (ii) the suggested presence of a barrier between the valence- and dipole-bound states. We note that the lower threshold of their stated EAV (+30 meV) was derived from the intermolecular binding energy between uracil and argon, which was explained to be less than EAV such that even the smallest generated U−(Ar)n clusters could then have sufficient internal energy to evaporate off the clustered argon atom(s), but not enough to undergo autodetachment. Our extracted EAV for U− is also consistent with this picture, despite being less than 30 meV. Our extrapolation predicts that EAV ∼ 35 meV for U−(Ar)1 so that both electron and argon loss result in similarly stable products. It should also be noted that these loss channels will differ in their kinetics. In particular, electron loss from the valence state of any U−(Ar)n cluster is driven by a buckled-to-planar transformation of the uracil ring, which is expected to be inhibited by an energetic barrier. Therefore, we do not expect autodetachment to occur for up to many microseconds, giving sufficient time for competitive loss of the solvating argon.
state. U− was kinetically trapped in the valence state for many microseconds (in order to be mass-separated), which is consistent with our combined observations of (i) a large vertical binding energy (VDE), and (ii) the suggested presence of a barrier between the valence- and dipole-bound states. We note that the lower threshold of their stated EAV (+30 meV) was derived from the intermolecular binding energy between uracil and argon, which was explained to be less than EAV such that even the smallest generated U−(Ar)n clusters could then have sufficient internal energy to evaporate off the clustered argon atom(s), but not enough to undergo autodetachment. Our extracted EAV for U− is also consistent with this picture, despite being less than 30 meV. Our extrapolation predicts that EAV ∼ 35 meV for U−(Ar)1 so that both electron and argon loss result in similarly stable products. It should also be noted that these loss channels will differ in their kinetics. In particular, electron loss from the valence state of any U−(Ar)n cluster is driven by a buckled-to-planar transformation of the uracil ring, which is expected to be inhibited by an energetic barrier. Therefore, we do not expect autodetachment to occur for up to many microseconds, giving sufficient time for competitive loss of the solvating argon.
Nuclear rearrangement associated with electron attachment to the  state has been previously explored in U−(H2O)n clusters,65,69 where the reorganization energy λ was estimated from the difference between the adiabatic and vertical detachment energies, λ = VDE − EAV. Hydration of the uracil anion was found to significantly enhance the reorganization energy, since the preferred orientation of the water molecules is very sensitive to the overall charge state of the uracil. Extrapolation of λ(n) to the aqueous limit (n → ∞) culminated in an estimate of λ(aq) ≈ 1.2 eV for U(aq).65 This can be decomposed into two separate contributions: λ(aq) = λIS + λOS; where λIS represents the (inner-sphere) reorganization energy of the buckling uracil ring, and λOS represents the (outer-sphere) reorganization energy of the surrounding water molecules.69 The first term, λIS, is independent of the solvent and can therefore be accurately extracted from the EAV and VDE of U−, reported here (i.e.Fig. 8). We establish that λIS = 0.2 eV and conclude that the dominant contribution to the neutral-to-anion reorganization energy of U(aq) arises from restructuring of the intermolecular hydration sphere (λOS ≈ 1.0 eV), i.e. from the solvent response, rather than the solute. This differs to our previous prediction that the inner- and outer-sphere reorganization energies were roughly equal, which we arrived at by approximating λIS as the λ of U−(H2O)1.69 As clearly demonstrated here, even a single water molecule has a substantial effect on the reorganization energy of U−, and this approximation does not hold and underscores the dramatic limitations of cluster extrapolations using strongly-interacting solvent molecules.
state has been previously explored in U−(H2O)n clusters,65,69 where the reorganization energy λ was estimated from the difference between the adiabatic and vertical detachment energies, λ = VDE − EAV. Hydration of the uracil anion was found to significantly enhance the reorganization energy, since the preferred orientation of the water molecules is very sensitive to the overall charge state of the uracil. Extrapolation of λ(n) to the aqueous limit (n → ∞) culminated in an estimate of λ(aq) ≈ 1.2 eV for U(aq).65 This can be decomposed into two separate contributions: λ(aq) = λIS + λOS; where λIS represents the (inner-sphere) reorganization energy of the buckling uracil ring, and λOS represents the (outer-sphere) reorganization energy of the surrounding water molecules.69 The first term, λIS, is independent of the solvent and can therefore be accurately extracted from the EAV and VDE of U−, reported here (i.e.Fig. 8). We establish that λIS = 0.2 eV and conclude that the dominant contribution to the neutral-to-anion reorganization energy of U(aq) arises from restructuring of the intermolecular hydration sphere (λOS ≈ 1.0 eV), i.e. from the solvent response, rather than the solute. This differs to our previous prediction that the inner- and outer-sphere reorganization energies were roughly equal, which we arrived at by approximating λIS as the λ of U−(H2O)1.69 As clearly demonstrated here, even a single water molecule has a substantial effect on the reorganization energy of U−, and this approximation does not hold and underscores the dramatic limitations of cluster extrapolations using strongly-interacting solvent molecules.
Returning to our photoelectron spectrum of U−(H2O)1 shown in Fig. 7, signal arising from both the valence- and dipole-bound anions was present. This is in contrast to earlier photoelectron spectroscopic studies,44–46 which observed detachment from only the  valence state. Our observed ratio of DBS to valence state signal was sensitive to the ion source conditions, indicating that the DBS of U−(H2O)1 was generated in the source region of our experiment, and then persisted for the hundreds of microseconds that elapsed prior to photodetachment. But why was the DBS so long-lived in our experiment? From Fig. 7, the EAD of U(H2O)1 was measured to exceed 200 meV and, thus, the DBS is clearly stable against autodetachment. On the other hand, EAD < EAV for U(H2O)1, such that internal conversion from the DBS to the valence state (through buckling of the uracil ring and reorganization of the water molecule) is energetically favorable. Hence, we conclude that there is a significant energetic barrier separating the DBS from the
valence state. Our observed ratio of DBS to valence state signal was sensitive to the ion source conditions, indicating that the DBS of U−(H2O)1 was generated in the source region of our experiment, and then persisted for the hundreds of microseconds that elapsed prior to photodetachment. But why was the DBS so long-lived in our experiment? From Fig. 7, the EAD of U(H2O)1 was measured to exceed 200 meV and, thus, the DBS is clearly stable against autodetachment. On the other hand, EAD < EAV for U(H2O)1, such that internal conversion from the DBS to the valence state (through buckling of the uracil ring and reorganization of the water molecule) is energetically favorable. Hence, we conclude that there is a significant energetic barrier separating the DBS from the  state, allowing the DBS to be kinetically trapped for the duration of our experiment (note that the initial electron attachment in the source is to a planar neutral U). We also observed a very small amount of DBS signal from U−(N2)2 (which also produced valence anions), indicating that some kinetic trapping was possible in the more weakly solvated clusters too. Since the relative DBS population of U−(N2)2 appeared to be much smaller than that of U−(H2O)1, it is likely that the barrier separating the DBS from the valence state is larger when the solvent is water, as may be expected from the strong hydrogen-bonding interactions that can act to inhibit the buckling motion. There is also an interesting question regarding the position of the clustered water molecule in the DBS of U−(H2O)1; if the water molecule were to solvate the DBS orbital rather than the uracil molecule, one may expect a greater reorganization between the dipole- and valence-bound states, and hence a greater dividing barrier. This will be considered further in a future study.
state, allowing the DBS to be kinetically trapped for the duration of our experiment (note that the initial electron attachment in the source is to a planar neutral U). We also observed a very small amount of DBS signal from U−(N2)2 (which also produced valence anions), indicating that some kinetic trapping was possible in the more weakly solvated clusters too. Since the relative DBS population of U−(N2)2 appeared to be much smaller than that of U−(H2O)1, it is likely that the barrier separating the DBS from the valence state is larger when the solvent is water, as may be expected from the strong hydrogen-bonding interactions that can act to inhibit the buckling motion. There is also an interesting question regarding the position of the clustered water molecule in the DBS of U−(H2O)1; if the water molecule were to solvate the DBS orbital rather than the uracil molecule, one may expect a greater reorganization between the dipole- and valence-bound states, and hence a greater dividing barrier. This will be considered further in a future study.
As demonstrated for U−, extrapolating adiabatic electron affinities of anion–solvent clusters must be performed with due consideration of the strength of the anion–solvent interaction. Strong interactions lead to significantly non-linear behavior, and a linear extrapolation to n = 0 is inappropriate. If the interactions are too weak, then many solvent molecules may be required to render the anion a bound (or kinetically-trapped metastable) state, causing greater uncertainty in the extrapolated EA. For instance, U−(He)n and U−(Ne)n anion clusters can also provide an estimate for the EAV of U, but the weak solvating power of He and Ne means that the  valence state is unlikely to be observed until n ≥ 4, necessitating a more extreme extrapolation that may lead to poorer determination of EAV. Overall, the ideal solvent must strike a balance between strongly and weakly solvating the anion, permitting an approximately linear extrapolation from small clusters. In addition to argon and nitrogen, xenon also appears to be a suitable solvent probe for U−; as mentioned earlier, the valence state of U−(Xe)1 had been measured with photoelectron spectroscopy, although with some obfuscation from the co-generated DBS.45 From our clear characterizations of the 0–0 transition present in the photoelectron spectra of U−(Ar)n and U−(N2)n, we can infer that EAV(U(Xe)1) ≈ 120 meV.45 By then comparing the general anion solvating power of xenon with that of other solvent molecules,57 which is stronger than argon but weaker than water, it appears that an extrapolation from the electron affinities of U−(Xe)n is also likely to yield a value of EAV close to zero.
valence state is unlikely to be observed until n ≥ 4, necessitating a more extreme extrapolation that may lead to poorer determination of EAV. Overall, the ideal solvent must strike a balance between strongly and weakly solvating the anion, permitting an approximately linear extrapolation from small clusters. In addition to argon and nitrogen, xenon also appears to be a suitable solvent probe for U−; as mentioned earlier, the valence state of U−(Xe)1 had been measured with photoelectron spectroscopy, although with some obfuscation from the co-generated DBS.45 From our clear characterizations of the 0–0 transition present in the photoelectron spectra of U−(Ar)n and U−(N2)n, we can infer that EAV(U(Xe)1) ≈ 120 meV.45 By then comparing the general anion solvating power of xenon with that of other solvent molecules,57 which is stronger than argon but weaker than water, it appears that an extrapolation from the electron affinities of U−(Xe)n is also likely to yield a value of EAV close to zero.
Conclusions
The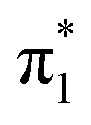 valence state of the uracil anion is significantly stabilized by clustered solvent molecules, even in the weakly-interacting case of U−(Ar)n. Earlier estimates for the EAV of isolated uracil utilized linear extrapolation techniques from the EAV(n) of uracil–water clusters, in which the solvent interacts strongly with the nucleobase anion. We demonstrate that such linear extrapolations are, in general, not accurate and should not be applied in the context of a polyatomic anion and a strongly-interacting solvent. Instead, weakly-interacting solvent molecules induce a nearly linear increase in the EAV(n), and can be used to perform a more suitable extrapolation. Our photoelectron spectroscopic measurements on U−(Ar)n and U−(N2)n clusters distinguished the 0–0 transition corresponding to photodetachment of the
valence state of the uracil anion is significantly stabilized by clustered solvent molecules, even in the weakly-interacting case of U−(Ar)n. Earlier estimates for the EAV of isolated uracil utilized linear extrapolation techniques from the EAV(n) of uracil–water clusters, in which the solvent interacts strongly with the nucleobase anion. We demonstrate that such linear extrapolations are, in general, not accurate and should not be applied in the context of a polyatomic anion and a strongly-interacting solvent. Instead, weakly-interacting solvent molecules induce a nearly linear increase in the EAV(n), and can be used to perform a more suitable extrapolation. Our photoelectron spectroscopic measurements on U−(Ar)n and U−(N2)n clusters distinguished the 0–0 transition corresponding to photodetachment of the  valence state of U−, allowing for an accurate determination of the associated EAV. With accountment for the subtle non-linearity in EAV(n) with increasing cluster size, our extrapolated value for the adiabatic electron affinity associated with the
valence state of U−, allowing for an accurate determination of the associated EAV. With accountment for the subtle non-linearity in EAV(n) with increasing cluster size, our extrapolated value for the adiabatic electron affinity associated with the  state of bare uracil, EAV = −2 ± 18 meV, using the combined values from U−(Ar)n and U−(N2)n. Within the uncertainty of our experiment, we are unable to conclude that the
state of bare uracil, EAV = −2 ± 18 meV, using the combined values from U−(Ar)n and U−(N2)n. Within the uncertainty of our experiment, we are unable to conclude that the  state is adiabatically stable, which has elsewise been suggested by Rydberg electron transfer experiments,27,70 and extrapolations from U−(H2O)n.44,46 Nonetheless, our results demonstrate the magnitude of EAV is very small and that any potential binding of the excess electron in the
state is adiabatically stable, which has elsewise been suggested by Rydberg electron transfer experiments,27,70 and extrapolations from U−(H2O)n.44,46 Nonetheless, our results demonstrate the magnitude of EAV is very small and that any potential binding of the excess electron in the  valence state of U− must be very weak. This finding appears to reveal why computational attempts to calculate (the sign of) EAV have shown such disagreement. Nevertheless, our reported value is close to the most rigorous computational efforts.42 We also offer insight into the barrier connecting the valence and non-valence states along the nuclear buckling coordinate. Our results are consistent with electron transmission spectroscopy, which, taken together with current insight, provides a direct measure of the intrinsic reorganization energy associated with U + e− → U− of λIS = 0.2 eV.
valence state of U− must be very weak. This finding appears to reveal why computational attempts to calculate (the sign of) EAV have shown such disagreement. Nevertheless, our reported value is close to the most rigorous computational efforts.42 We also offer insight into the barrier connecting the valence and non-valence states along the nuclear buckling coordinate. Our results are consistent with electron transmission spectroscopy, which, taken together with current insight, provides a direct measure of the intrinsic reorganization energy associated with U + e− → U− of λIS = 0.2 eV.
Data availability
The data which support the findings of this study will be available online at https://zenodo.org/10.5281/zenodo.12571786.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was funded by the EPSRC under grant EP/V007971/1 (EMB, JRRV), the OP JAK project no. CZ.02.01.01/00/22_008/0004649 (QUEENTEC) (JRRV), and the Durham University for a Scholarship (CJC). For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising.References
- B. Boudaïffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Science, 2000, 287, 1658–1660 CrossRef PubMed.
- F. Martin, P. D. Burrow, Z. Cai, P. Cloutier, D. Hunting and L. Sanche, Phys. Rev. Lett., 2004, 93, 068101 CrossRef PubMed.
- C. Liu, Y. Zheng and L. Sanche, Photochem. Photobiol., 2022, 98, 546–563 CrossRef CAS.
- J. Simons, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed.
- J. Gu, Y. Xie and H. F. Schaefer, J. Am. Chem. Soc., 2006, 128, 1250–1252 CrossRef CAS PubMed.
- J. Gu, J. Leszczynski and H. F. I. Schaefer, Chem. Rev., 2012, 112, 5603–5640 CrossRef CAS PubMed.
- M. A. Huels, I. Hahndorf, E. Illenberger and L. Sanche, J. Chem. Phys., 1998, 108, 1309–1312 CrossRef CAS.
- S. Denifl, S. Ptasińska, M. Probst, J. Hrušák, P. Scheier and T. D. Märk, J. Phys. Chem. A, 2004, 108, 6562–6569 CrossRef CAS.
- S. Denifl, S. Ptasińska, G. Hanel, B. Gstir, M. Probst, P. Scheier and T. D. Märk, J. Chem. Phys., 2004, 120, 6557–6565 CrossRef CAS.
- D. Svozil, P. Jungwirth and Z. Havlas, Collect. Czech. Chem. Commun., 2004, 69, 1395–1428 CrossRef CAS.
- K. Aflatooni, G. A. Gallup and P. D. Burrow, J. Phys. Chem. A, 1998, 102, 6205–6207 CrossRef CAS.
- A. M. Scheer, K. Aflatooni, G. A. Gallup and P. D. Burrow, Phys. Rev. Lett., 2004, 92, 068102 CrossRef CAS PubMed.
- J. H. Hendricks, S. A. Lyapustina, H. L. de Clercq, J. T. Snodgrass and K. H. Bowen, J. Chem. Phys., 1996, 104, 7788–7791 CrossRef CAS.
- C. Desfrançois, H. Abdoul-Carime and J. P. Schermann, J. Chem. Phys., 1996, 104, 7792–7794 CrossRef.
- C. Desfrançois, H. Abdoul-Carime and J.-P. Schermann, Int. J. Mod. Phys. B, 1996, 10, 1339–1395 CrossRef.
- T. Sommerfeld, J. Phys.: Conf. Ser., 2005, 4, 245 CrossRef CAS.
- M. A. Yandell, S. B. King and D. M. Neumark, J. Am. Chem. Soc., 2013, 135, 2128–2131 CrossRef CAS PubMed.
- J. P. Rogers, C. S. Anstöter and J. R. R. Verlet, Nat. Chem., 2018, 10, 341–346 CrossRef CAS PubMed.
- A. Kunin and D. M. Neumark, Phys. Chem. Chem. Phys., 2019, 21, 7239–7255 RSC.
- J. N. Bull, C. S. Anstöter and J. R. R. Verlet, Nat. Commun., 2019, 10, 5820 CrossRef CAS PubMed.
- G. Liu, S. M. Ciborowski, J. D. Graham, A. M. Buytendyk and K. H. Bowen, J. Chem. Phys., 2020, 153, 044307 CrossRef CAS.
- D. H. Kang, J. Kim, H. J. Eun and S. K. Kim, J. Am. Chem. Soc., 2022, 144, 16077–16085 CrossRef CAS PubMed.
- D. H. Kang, J. Kim, H. J. Eun and S. K. Kim, Acc. Chem. Res., 2022, 55, 3032–3042 CrossRef CAS PubMed.
- J. R. R. Verlet, C. S. Anstöter, J. N. Bull and J. P. Rogers, J. Phys. Chem. A, 2020, 124, 3507–3519 CrossRef CAS.
- C. J. Clarke and J. R. R. Verlet, Annu. Rev. Phys. Chem., 2024, 75, 89–110 CrossRef.
- M. Elena Castellani, C. S. Anstöter and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2019, 21, 24286–24290 RSC.
- C. Desfrançois, V. Periquet, Y. Bouteiller and J. P. Schermann, J. Phys. Chem. A, 1998, 102, 1274–1278 CrossRef.
- A. O. Colson, B. Besler, D. M. Close and M. D. Sevilla, J. Phys. Chem., 1992, 96, 661–668 CrossRef CAS.
- M. D. Sevilla, B. Besler and A.-O. Colson, J. Phys. Chem., 1995, 99, 1060–1063 CrossRef CAS.
- R. A. Bachorz, J. Rak and M. Gutowski, Phys. Chem. Chem. Phys., 2005, 7, 2116–2125 RSC.
- P. Dedíková, L. Demovič, M. Pitoňák, P. Neogrády and M. Urban, Chem. Phys. Lett., 2009, 481, 107–111 CrossRef.
- H. Motegi and T. Takayanagi, J. Mol. Struct., 2009, 907, 85–92 CrossRef CAS.
- C. S. Anstöter and S. Matsika, J. Phys. Chem. A, 2020, 124, 9237–9243 CrossRef PubMed.
- N. Russo, M. Toscano and A. Grand, J. Comput. Chem., 2000, 21, 1243–1250 CrossRef CAS.
- S. D. Wetmore, R. J. Boyd and L. A. Eriksson, Chem. Phys. Lett., 2000, 322, 129–135 CrossRef CAS.
- N. J. Saettel and O. Wiest, J. Am. Chem. Soc., 2001, 123, 2693–2694 CrossRef CAS PubMed.
- S. S. Wesolowski, M. L. Leininger, P. N. Pentchev and H. F. Schaefer, J. Am. Chem. Soc., 2001, 123, 4023–4028 CrossRef CAS PubMed.
- T. Sommerfeld, J. Phys. Chem. A, 2004, 108, 9150–9154 CrossRef CAS.
- R. A. Bachorz, W. Klopper and M. Gutowski, J. Chem. Phys., 2007, 126, 085101 CrossRef PubMed.
- D. Roca-Sanjuán, M. Merchán, L. Serrano-Andrés and M. Rubio, J. Chem. Phys., 2008, 129, 095104 CrossRef PubMed.
- J. M. L. Martin and G. de Oliveira, J. Chem. Phys., 1999, 111, 1843–1856 CrossRef CAS.
- J. Gu, Y. Xie and H. F. Schaefer, J. Chem. Theory Comput., 2014, 10, 609–612 CrossRef CAS PubMed.
- I. Anusiewicz, P. Skurski and J. Simons, J. Phys. Chem. A, 2020, 124, 2064–2076 CrossRef CAS PubMed.
- J. Schiedt, R. Weinkauf, D. M. Neumark and E. W. Schlag, Chem. Phys., 1998, 239, 511–524 CrossRef CAS.
- J. H. Hendricks, S. A. Lyapustina, H. L. de Clercq and K. H. Bowen, J. Chem. Phys., 1998, 108, 8–11 CrossRef CAS.
- S. Eustis, D. Wang, S. Lyapustina and K. H. Bowen, J. Chem. Phys., 2007, 127, 224309 CrossRef PubMed.
- J. P. Rogers, C. S. Anstöter, J. N. Bull, B. F. E. Curchod and J. R. R. Verlet, J. Phys. Chem. A, 2019, 123, 1602–1612 CrossRef CAS PubMed.
- U. Even, EPJ Techn. Instrum., 2015, 2, 17 CrossRef.
- W. C. Wiley and I. H. McLaren, Rev. Sci. Instrum., 1955, 26, 1150–1157 CrossRef CAS.
- G. M. Roberts, J. L. Nixon, J. Lecointre, E. Wrede and J. R. R. Verlet, Rev. Sci. Instrum., 2009, 80, 053104 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. N. Zare, Mol. Photochem., 1972, 4, 1–37 CAS.
- J. Simons, J. Phys. Chem. A, 2020, 124, 8778–8797 CrossRef CAS PubMed.
- S. T. Arnold, J. H. Hendricks and K. H. Bowen, J. Chem. Phys., 1995, 102, 39–47 CrossRef CAS.
- J. H. Hendricks, H. L. de Clercq, C. B. Freidhoff, S. T. Arnold, J. G. Eaton, C. Fancher, S. A. Lyapustina, J. T. Snodgrass and K. H. Bowen, J. Chem. Phys., 2002, 116, 7926–7938 CrossRef CAS.
- T. Lenzer, I. Yourshaw, M. R. Furlanetto, N. L. Pivonka and D. M. Neumark, J. Chem. Phys., 2001, 115, 3578–3589 CrossRef CAS.
- I. Yourshaw, Y. Zhao and D. M. Neumark, J. Chem. Phys., 1996, 105, 351–373 CrossRef CAS.
- K. R. Asmis, T. R. Taylor, C. Xu and D. M. Neumark, J. Chem. Phys., 1998, 109, 4389–4395 CrossRef CAS.
- J. K. Song, N. K. Lee and S. K. Kim, J. Chem. Phys., 2002, 117, 1589–1594 CrossRef CAS.
- Note: Applying a linear extrapolation (n = 3–5) instead obtains EAV = +2 meV, showing that the curvature has very little effect on the estimated energy.
- T. N. Olney, N. M. Cann, G. Cooper and C. E. Brion, Chem. Phys., 1997, 223, 59–98 CrossRef CAS.
- A. Halkier, S. Coriani and P. Jørgensen, Chem. Phys. Lett., 1998, 294, 292–296 CrossRef CAS.
- G. A. Cooper, C. J. Clarke and J. R. R. Verlet, J. Am. Chem. Soc., 2023, 145, 1319–1326 CrossRef CAS PubMed.
- D. A. Horke, Q. Li, L. Blancafort and J. R. R. Verlet, Nat. Chem., 2013, 5, 711–717 CrossRef CAS PubMed.
- G. J. Schulz, Rev. Mod. Phys., 1973, 45, 423–486 CrossRef CAS.
- Y. Yokoi, K. Kano, Y. Minoshima and T. Takayanagi, Comput. Theor. Chem., 2014, 1046, 99–106 CrossRef CAS.
- G. A. Cooper, C. J. Clarke and J. R. R. Verlet, J. Phys. B: At., Mol. Opt. Phys., 2023, 56, 185102 CrossRef.
- V. Periquet, A. Moreau, S. Carles, J. P. Schermann and C. Desfrançois, J. Electron Spectrosc. Relat. Phenom., 2000, 106, 141–151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02146k |
| This journal is © the Owner Societies 2024 |