
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantiopure molecules form apparently racemic monolayers of chiral cyclic pentamers†
Benjamin R.
Heiner
 a,
Kaitlyn M.
Handy
a,
Angela M.
Devlin
b,
Jewel L.
Soucek
a,
Alexander M.
Pittsford
a,
David A.
Turner
c,
Jacob P.
Petersen
d,
Allen G.
Oliver
a,
Steven A.
Corcelli
a and
S. Alex
Kandel
*a
a,
Kaitlyn M.
Handy
a,
Angela M.
Devlin
b,
Jewel L.
Soucek
a,
Alexander M.
Pittsford
a,
David A.
Turner
c,
Jacob P.
Petersen
d,
Allen G.
Oliver
a,
Steven A.
Corcelli
a and
S. Alex
Kandel
*a
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: skandel@nd.edu
bDepartment of Chemistry and Biochemistry, Creighton University, Omaha, NE 68179, USA
cIntel Corporation, Hillsboro, OR 97124, USA
dEvident Scientific, Webster, TX 77598, USA
First published on 18th September 2024
Abstract
Ultra-high vacuum scanning tunneling microscopy (UHV-STM) was used to investigate two related molecules pulse-deposited onto Au(111) surfaces: indoline-2-carboxylic acid and proline (pyrrolidine-2-carboxylic acid). Indoline-2-carboxylic acid and proline form both dimers and C5-symmetric “pinwheel” pentamers. Enantiomerically pure S-(−)-indoline-2-carboxylic acid and S-proline were used, and the pentamer structures observed for both were chiral. However, the presence of apparently equal numbers of ‘right-‘ and ‘left-handed‘ pinwheels is contrary to the general understanding that the chirality of the molecule dictates supramolecular chirality. A variety of computational methods were used to elucidate pentamer geometry for S-proline. Straightforward geometry optimization proved difficult, as the size of the cluster and the number of possible intermolecular interactions produced an interaction potential with multiple local minima. Instead, the Amber force field was used to exhaustively search all of phase space for chemically reasonable pentamer structures, producing a limited number of candidate structures that were then optimized as gas-phase clusters using density functional theory (DFT). The binding energies of the two lowest-energy pentamers on the Au(111) surface were then calculated by plane-wave DFT using the VASP software, and STM images predicted. These calculations indicate that the right- and left-handed pentamers are instead two different polymorphs.
1 Introduction
Studies of molecular chirality at surfaces benefit a wide range of fields, including enantioselective heterogeneous catalysts,1–3 2D chiral crystal engineering,4–6 chirality in 2D nanomaterials,7,8 and determination of the fundamental role of chirality in crystallization.9,10 Understanding enantioselective crystallization has significant practical applications, for instance, the development of single-enantiomer active pharmaceutical compounds.11,12 Chiral molecular self-assemblies on metal surfaces have been studied via a variety of surface science techniques,13 particularly scanning tunneling microscopy (STM),1,13–26 as the submolecular resolution affords insight into the structural details of two-dimensional crystal structures.Any arrangement of enantiopure molecules necessarily remains chiral; this includes monolayer or other structures formed when molecules adsorb onto a surface.6,17,27–29 Indeed, many examples exist where enantiopure adsorbates transfer their molecular chirality to the surface, which then exhibits global chirality.16,30–35 So far, we have only read of two exceptions to this phenomenon. In the first, Walba et al. observed apparently heterochiral domains forming when depositing the enantiopure samples of the chiral liquid crystalline 4-[(S)-2-ethoxypropoxy]phenyl 4-(decyloxy)benzoate; they determined these were instead “quasi-enantiomorphous” domains resulting from different monolayer structures.36 In the second, the S-enantiomer of a chiral diacetylene isophthalic acid forms an enantiomorphous lamella structure that is not correlated to the surface. Zhang et al. explained that the self-assembled monolayers did not follow the molecular chirality due to the stereocenter being forced away from interacting with the surface, which was believed to drive the organization.37 In both systems, chirality appears to be lost in STM images, ultimately because the chiral center does not propagate to form structures that are apparent to the microscope.
Much of the work in our group revolves around studying molecular “families,” or studying many derivatives of the same molecular backbone to probe the effect different functional groups have on the intermolecular interactions that drive self-assembly.38 One such molecule, the achiral indole-2-carboxylic acid, adsorbed to the surface of Au(111) in chiral cyclic pentamers after pulse deposition.39,40 However, under vapor deposition conditions it forms catemer chains, thus showing that pulse deposition can lead to interesting metastable assemblies on surfaces.41 In this study, we look at another molecule in the same family, S-(−)-indoline-2-carboxylic acid (S-ICA), the chiral analog of indole-2-carboxylic acid. We found that the chirality of S-ICA does not clearly transfer to the monolayer, but instead forms a monolayer that appears to be a racemic mixture of chiral pinwheel-like pentamers. We continue the study by investigating a similar family of molecules (related by removing the fuzed benzene ring) that include S-(−)-pyrrolidine-2-carboxylic acid (S-proline). Structures of these molecules are shown in Fig. 1. The S-proline molecule also did not pass chirality to the monolayer, instead showing the same mixture of left- and right-handed pinwheel pentamers.
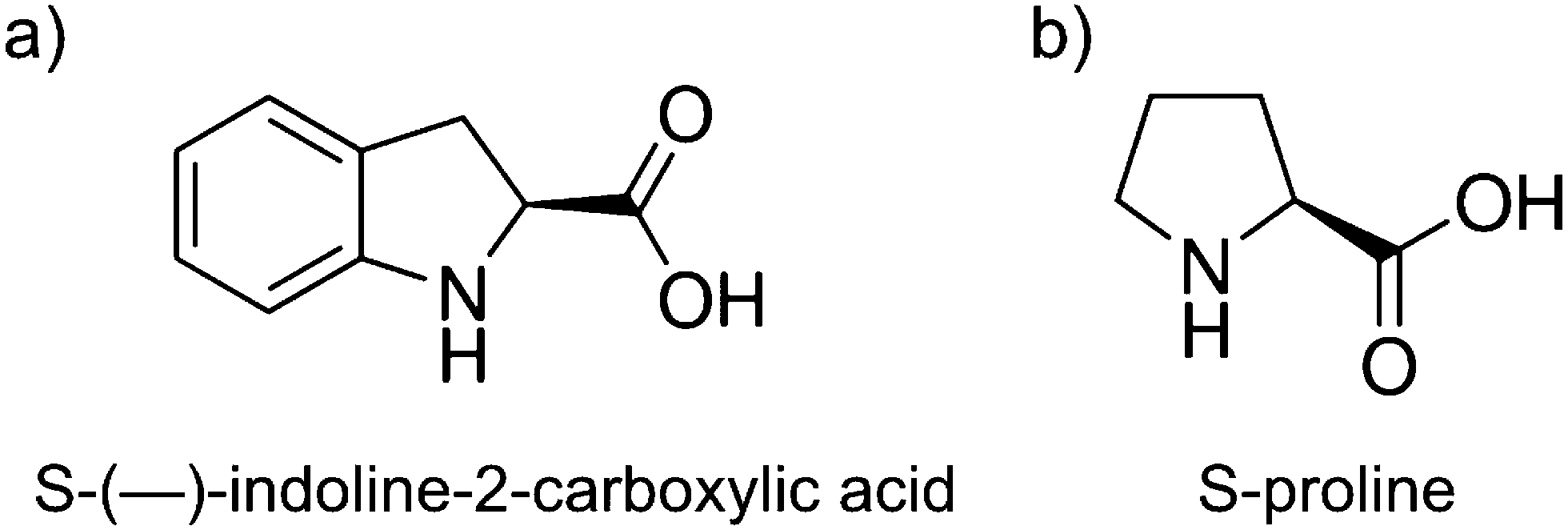 | ||
| Fig. 1 Molecular structures of the molecules used in this study: (a) S-indoline-2-carboxylic acid (S-ICA) and (b) S-proline. | ||
2 Methods
2.1 Experimental
Au(111)-on-mica thin films were cleaned in high vacuum with two cycles of Ar+ sputtering (0.55 kV for 15 min) and subsequent annealing at 350–400 °C, which we found to yield flat, clean surfaces of Au(111) with the characteristic “herringbone” reconstruction. Cleaned samples were allowed to cool before being transferred to a load-lock chamber for preparation of the monolayer. All molecules used in this study were purchased from Sigma Aldrich and used without further purification: S-(−)-indoline-2-carboxylic acid (S-ICA) ≥99%, L-proline (S-proline) ReagentPlus® ≥99%. Solutions with a concentration of 10 mM were prepared in methanol (unless otherwise noted). Droplets of solutions of interest were delivered via a pulsed-solenoid valve (Parker Instruments, Series 9, Iota One Driver, 0.5 mm diameter nozzle) onto the cleaned Au(111) substrate kept at room temperature in a load-lock chamber. Before the deposition, the load-lock chamber was at pressures <5 × 10−6 Torr. During the deposition, the pressure rose significantly and then returned to the baseline within a minute. The sample was then transferred to an Omicron LT-STM, kept at a base pressure of 5 × 10−10 Torr, and was cooled to 77 K. All images were acquired with a Pt/Ir tip in constant current mode with a tunneling current of 10 pA and a tip-sample bias of +1.0 V unless otherwise noted.2.2 Computational
Because the clusters we are studying are quite large, DFT became computationally expensive. To narrow the size of the problem, we used a less accurate but much more affordable force-field method to achieve reasonable starting points for the more expensive electronic-structure calculations. This dramatically lowered our computational cost while still allowing us to probe tens of thousands of pentamer configurations.The simulations of proline pentamers used the Amber22 force field in the Gaussian 16 software package.42,43 A single zwitterionic proline monomer was first geometrically optimized, then used as the building block to generate pentamers. As a C5-symmetric pentamer can be defined by three angles and a distance, a script was used to produce ∼18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 starting configurations for proline pentamers that covered all of geometric phase space. An initial single-point energy calculation allowed us to rank each cluster by starting energy. Geometric optimization of the lowest 3000 starting configurations in the Amber force field resulted in 60 unique configurations. These configurations were then geometrically optimized using DFT at the B3LYP/6-31g(d) level of theory.
000 starting configurations for proline pentamers that covered all of geometric phase space. An initial single-point energy calculation allowed us to rank each cluster by starting energy. Geometric optimization of the lowest 3000 starting configurations in the Amber force field resulted in 60 unique configurations. These configurations were then geometrically optimized using DFT at the B3LYP/6-31g(d) level of theory.
All pentamer-on-gold calculations were performed utilizing VASP.44–47 A slab of Au(111) was constructed with the dimensions of 6 × 5 × 3 gold atoms with 10 Å of vacuum above and below the slab. The lattice constant was found to be 4.2 Å. All calculations were conducted with a PBE functional with an energy cutoff (encut) of 500 eV. The k points used were [7, 7, 1] with an energy difference of 1 × 10−5 eV.
Pentamer configurations were placed on the surface of the gold and single-point calculations were performed for both nitrogen-up and nitrogen-down orientations of the pentamer. The pentamer was raised off the surface at varying heights. For the nitrogen-up orientation, the height is defined as the height of the top of the surface to the lowest oxygen in the pentamer. As for the nitrogen-down orientation, the height is defined as the height of the top of the surface to the lowest carbon atom. Single-point calculations were conducted for the gold slab, each pentamer, and the pentamer on the gold slab. All binding energies were calculated by:
| Ebindingenergy = Eslab&pentamer − Eslab − Epentamer | (1) |
3 Results and discussion
3.1 Indoline-2-carboxylic acid monolayers
Fig. 2 shows an STM image of the S-ICA monolayer after pulse deposition. The majority species cluster on the surface was the tightly packed pinwheel-like structures seen in Fig. 2. These pentamers are similar in structure to those we observed previously in the monolayer of indole-2-carboxylic acid and isatin.39,48 These pentameric clusters are quite stable, remaining on the surface after thermal annealing to 30–40 °C, but desorbing when annealed to 65–70 °C. S-ICA pentamers also form via vapor deposition, so they are not preparation-method dependent as previously observed for indole-2-carboxylic acid pentamers.39,41 | ||
| Fig. 2 STM image (139 × 139 Å) of S-ICA pulse deposited in methanol on the Au(111) surface, with red (R-) and blue (S-) circles indicating the handedness of each pentamer. The pentamers appear to be approximately hexagonally packed, with the unit cell rhombus measuring 17.2 ± 1.4 and 60 ± 6.0°. | ||
Fig. 3(A) and (B) shows pulsed deposited S-ICA onto Au(111) using acetonitrile as the solvent. Fig. 3(A) shows that the pentamer structure is still present on the surface of the gold, so the pentamer structure is observed using at least two different solvents. However, when acetonitrile is used as the pulse-deposition solvent, a new phase is observed: parallel dimer chains, highlighted in Fig. 3(A) and (B). Dimer chains were not observed for S-ICA using methanol as the solvent for pulse deposition, but are a common structural motif for carboxylic acids.49 X-ray crystallography data for S-ICA confirms a zwitterionic dimer-based crystal structure (see ESI†). The parallel chains observed in Fig. 3(A) and (B) are similar to those in the crystal structure of S-ICA.
 | ||
| Fig. 3 (A) and (B) Monolayer of S-ICA pulse deposited in acetonitrile consists of both phases of pentamers and dimer chains; representative pentamers are colored in light blue and dimer chains are colored in green and blue (200 × 200 Å and 148 × 148 Å, respectively). | ||
In our previous studies of clusters and monolayers produced through pulse deposition, the choice of solvent has been shown to affect the range of structures formed, though the mechanism by which this occurs remains uncertain.48,50–52 Simple chemical intuition (e.g., zwitterions favored in polar solvents) does not have strong predictive power.53–58
Analysis of STM images was done in an automated fashion with minimal user input. First, the centroid of each pentameric cluster was determined, and the molecular features in the STM image fit to a function of the form sin5θ + ϕ. The chirality can then be measured by the dependence of ϕ on radius (distance from the centroid)—that is, for left-handed pentamers ϕ increases with radius, while ϕ decreases for right-handed pentamers. The calculated chirality is shown in Fig. 2, with pentamers circled in red and blue for R- and S-pentamers, respectively. Once the orientation and chirality of every pentamer in the image have been determined in this fashion, clusters of like chirality can be overlapped (and reoriented as necessary), and the numerical average of the STM image calculated. These composites are what is shown in Fig. 4(A) and (B). Fig. 4(C) is a tight zoom on the S pentamer with a line drawn from the centroid of the pentamer cluster through the top molecule. As all mirror planes must contain the C5 axis, the lack of mirror symmetry in Fig. 4(C) makes the chirality of the cluster evident. We automated a count, Fig. 2, of 226 pentamer clusters over 5 scanned areas and determined the chirality split to be 118:108 (R:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) S). This is indistinguishable from a binomial distribution that assumes equal probabilities, which gives a most likely result of 113 with a standard deviation of 7.5 (see ESI†).
S). This is indistinguishable from a binomial distribution that assumes equal probabilities, which gives a most likely result of 113 with a standard deviation of 7.5 (see ESI†).
 | ||
| Fig. 4 (A) Composite of left-handed pentamers shown in the image in Fig. 2 (25 × 25 Å). (B) Composite of right-handed pentamers shown in the image in Fig. 2. The pentamer is distinct from that in (A). (C) One lobe of the composite left-handed pentamer with a line drawn from the centroid of the pentamer cluster through the centroid of a single lobe. As there is a clear imbalance of electron density on either side of the line, it does not constitute a mirror plane, defining the structure as chiral. | ||
The mixing of the R- and S-pentamers of the S-ICA monolayer shown in Fig. 2 is peculiar. In addition to the equal total numbers of R- and S-pentamers, there is no obvious segregation of homochiral pentamers into separate domains. This is analyzed quantitatively, showing only a slight deviation from a random mixture for nearest-neighbor and next-nearest-neighbor pentamers (see ESI,† Fig. S1). This implies that there are no significant short-range interactions between left- and right-hand pinwheels, so that one geometry does not seed the growth of a domain; or, alternately, there must be a very large number of “seeds” such that many domains grow and merge. As we do observe the characteristic “herringbone” reconstruction of the gold surface underneath the monolayer, the kinks that accompany this reconstruction could be one source of such nucleation sites. Diffusion of pentamers may also be limited because of their size, the high surface coverage, or the peculiar condition that result during the pulse-deposition process.59 In contrast, most of the extant literature reports that when mirror-image enantiomorphs are present on the surface, they phase separately into two homochiral domains (conglomerates).16,60–62 These conglomerates typically consist of homochiral molecular adsorbates, in which both enantiomers self-assemble in different domains.62
A racemic mixture of pentameric structures is surprising given that the deposited molecule is purely the S-enantiomer. Typically, for a racemic mixture of 2D crystals to form, a racemic mixture of the starting molecule must be adsorbed.61S-ICA should not racemize during or after deposition as the energetic barrier to switch to R-ICA is far higher than kT, especially at our operating temperatures.63,64 Supramolecular chirality usually results from the chirality of the starting molecule,65 except in limited cases.36,37 Because S-ICA is a small molecule, the stereocenter would be forced to interact with the surface, so we would expect the chirality of the molecule to be passed to the surface, forming an enantiomorphous monolayer.16,17
3.2 S-Proline monolayers
S-Proline and S-ICA have very similar chemical structures, Fig. 1(a) and (b), S-proline differing only in the absence of the benzene ring fused to the five-membered heterocycle. Because the amine and carboxylic acid groups remain in the same positions, we expected the chemical interactions to be quite similar to those of S-ICA. Indeed, when we scan S-proline after pulse deposition, Fig. 5, we find both the pentamer and dimer phases present in the image. Additionally, we observe that the proline pentamers are chiral and form an apparently racemic mixture, just as the S-ICA pentamers do. | ||
| Fig. 5 STM images of S-proline pulse deposited on Au(111). (A) Pentamers and dimer chains are observed (450 × 450 Å). (B) Representative pentamers are colored in light blue, and right-handed and left-handed pentamers are present in the monolayer. The pentamers appear to be approximately hexagonally packed, with the unit cell rhombus measuring 18.4 ± 1.6 Å and 60 ± 8.8° (169 × 169 Å). | ||
The adsorption of amino acids on metal surfaces has been well studied. The copper surface is more reactive and commonly results in the deprotonation of carboxylic acids, resulting in adsorption in the anionic form.66–74S-Proline adsorbes on Pd(111) in the zwitterionic form,75 but zwitterionization does not typically occur on the less reactive gold surface76,77 (except at elevated temperatures).78 However, monolayers of proline have been observed on the Au(111) surface79 that consisted of a motif reminiscent of the crystal structure of proline, in which zwitterionic molecules are bound by NH⋯O hydrogen bonds to form a two-dimensional network.80 Given this evidence, we believe it likely that at our experimental conditions—in which molecules are deposited on the gold surface at room temperature upon adsorption—proline adsorbs on the gold surface in the zwitterionic form.
A significant difference between the current study and the aforementioned studies of amino acids on metal surfaces is the incorporation of the solution to deliver the molecule to the surface. Previous work from our group has resulted in the formation of kinetically-locked structures that have been attributed to the non-equilibrium solvent evaporation conditions present in the pulse deposition process.39,51,81 It is possible that pentamer formation can occur independent of the non-equilibrium adsorption kinetics of solution deposition.48,82 Nonetheless, for the case of S-proline pentamers, non-equilibrium adsorption kinetics are likely the cause of pentamer formation in our experiments because Seljamae-Green et al. do not observe pentamers of S-proline on a vapor-deposited monolayer.79
In addition to there being no measurable preference for right-handed or left-handed pinwheels, both for the S-ICA and the S-proline, there is also no evidence for separation of these pinwheels into homochiral domains. In Fig. S1 (ESI†), the likelihood of neighboring left- and right-handed pentamers is very close to what would be expected from a random distribution. This implies that there are no significant short-range interactions between left- and right-hand pinwheels, so that one geometry does not seed the growth of a domain; or, alternately, there must be a very large number of “seeds” such that many domains grow and merge.
Fig. 6(A) and (B) shows the composite images for the S- and R-pentamers of proline. In exactly the same way as the S-ICA pentamers, the line in Fig. 6(C) does not define a mirror plane, causing the pentamer structure to be chiral.
 | ||
| Fig. 6 Composite of (A) left- and (B) right-handed pentamers shown in the image in Fig. 5(A) (25 × 25 Å). (C) One lobe of the composite left-handed pentamer with a line drawn from the centroid of the pentamer cluster through the centroid of a single lobe. As there is a clear imbalance of electron density on either side of the line, it does not constitute a mirror plane, defining the structure as chiral. | ||
3.3 Molecular modeling
We used proline in our computational approach to simplify calculations and lower computational cost. S-ICA and proline are identical except for the aforementioned fused benzene ring, so the strongly interacting functional groups (carboxylic acid, amine) are shared. We expect that, if undertaken, the S-ICA calculations would yield similar results.In the solid state, proline is in the zwitterionic form.80 The zwitterion is also the majority species adsorbed on the surface of Au(111), though XPS of a monolayer does reveal small amounts of co-existing neutral and anionic species.79 In our observations, proline pentamers are C5 symmetric, which is not consistent with a mixture of different charge states. Based on the XPS data, we modeled all prolines as zwitterionic.
Gas phase DFT optimizations for systems of this size are feasible. However, we found that a geometry optimization procedure produced different solutions depending on the starting geometry used, so we could not be confident that the global minimum-energy structure or all local minima had been found. An exhaustive search of phase space was not possible, given the time required for each individual electronic structure calculation. Instead, we used Amber force-field calculations to find the type of structural configurations of proline that made physical and chemical sense, and thus to narrow down the number of starting configurations to test with DFT.
To identify all potential configurations, and thus probe phase space for these pentamers, we exhaustively sampled starting geometries of C5 symmetric pentamer clusters. We forced all clusters to be C5 symmetric to match our observation of C5 symmetric pentamers in the STM images. For C5-symmetric clusters, only three angles and one distance are needed to fully specify the internal cluster geometry, so a grid covering all values of the angles and a physically reasonable range of distances was generated, translating to ∼18000 potential pentamer geometries. We performed single-point energy calculations on each geometry using the Amber22 force field.42 The 3000 lowest energy configurations were then geometrically optimized using the same force field. Our study of the optimized clusters found that all of the original 18000 structures had either broken symmetry or converged to one of 60 C5-symmetric clusters. Each of these clusters corresponds to a local minimum in the potential energy, as defined using Amber. The six lowest-energy clusters are shown in Fig. 7.
 | ||
| Fig. 7 Six lowest-energy stable or metastable C5-symmetric S-proline pentamer structures predicted using the AMBER force field. | ||
It is important to note that a classical force field will not produce quantitatively accurate results, especially given the importance of hydrogen bonding in these molecular clusters. The results are also necessary qualitative in nature as they are performed on gas-phase clusters; that is, in the absence of a surface. Au(111) is, however, both flat and non-reactive, and in several previous studies, we have found that gas-phase calculations provide good predictions for observed self-assembled structures.38 Additional evidence for a molecule-based model of self-assembly is that molecules adsorb incommensurately, and with clusters in random orientations. This is emphatically not the case for other crystal faces of gold or other metals. For Au(111) specifically, the self-assembly of molecules can often be substantively understood based on molecule–molecule interactions while ignoring the molecule–surface interactions. This can be true even if the molecule–surface interactions are large in magnitude (as we would expect given the polarizability of gold and the large molecular dipole of proline); all that is required is that these interactions do not have significant lateral corrugation, whether directly or through substrate-mediated interactions.83–85
3.4 DFT
Unlike 18000 starting configurations, 60 configurations are not computationally prohibitive for DFT calculations. Using the results of the Amber calculations as starting points, we optimized the geometry of the clusters using the B3LYP/6-31g(d) level of theory. All 60 configurations optimized to one of three results, which are shown in Fig. 8(A)–(C). We believe that this approach is the most practical way to identify multiple local energy minima, though it can not guarantee it will find all of them. The geometry of the cluster in Fig. 8(C) is significantly different from the other two and is much less stable, so we consider only Fig. 8(A) (configuration A) and Fig. 8(B) (configuration B) to explain the pentamers observed experimentally.
 | ||
| Fig. 8 The three pentamer configurations that all DFT optimizations conformed to. In all three cases, nitrogen is on the far side of the cluster. Configurations (A) and (B) are apparently opposite chiralities, but are not mirror images, and so are diastereomers. NH⋯O hydrogen-bond contact distances are 1.63 Å in configuration A and 1.57 Å in configuration B. (C) is ignored as it is qualitatively unlike the other two, and because of its significantly less favorable binding energy. Binding energies were calculated by subtracting five times the total monomer energy from each pentamer energy. (D) Configurations A and B from an axial view with the nitrogen-up and -down sides labeled. | ||
While configurations A and B appear qualitatively similar, they are not mirror images. This can be seen by comparing the top view, that is with respect to the C5 axis of symmetry, of both clusters in Fig. 8(A) and (B). Then, the NH2-to-CO2 axis of each proline is oriented differently: clockwise for configuration A and counterclockwise for B. The clusters should be described as diastereomeric rather than enantiomeric as they have opposite handedness but are not mirror images.
The experimental images of proline and ICA show equal numbers of opposite-handedness pentamers. One hypothesis is that this results from a mixture of A and B diastereomers, with the two configurations appearing as near-mirror images due to the specifics of STM contrast. Alternately, either or both could be flipped over, adsorbed with the oxygen atoms (instead of nitrogen) in proximity to the surface, which, of course, also flips the perceived handedness of the cluster. To address these questions, we turned to DFT calculations of the surface-adsorbed clusters using VASP software.
3.5 VASP
A proline pentamer adsorbed on a gold slab is large enough that using DFT to perform a full geometry optimization is prohibitively expensive. Instead, calculations with VASP were aimed at answering specific questions important to understanding the experimental results: (1) what is the difference in binding energy when a pentamer is flipped over, nitrogen-up vs. nitrogen-down; (2) would STM be able to distinguish between clusters in nitrogen-up and nitrogen-down orientations; and (3) are configurations A and B predicted to appear as mirror images when observed using STM?Using the DFT-optimized pentamer clusters, a series of single-point energy calculations was performed while varying the cluster–surface distance to determine the energy minimum of absorption. The potential curves for both orientations are shown in Fig. 9 for configurations A and B. We initially expected to find a strong preference for the nitrogen-down orientation because we hypothesized a nitrogen–surface interaction, but we instead see that the minimum energies in Fig. 9 are similar for both configurations A and B in both the nitrogen-up and -down orientations.
 | ||
| Fig. 9 Calculations for both accepted clusters from Fig. 8. Single-point energy calculations were taken of both clusters from varying distances and at nitrogen-up and nitrogen-down orientations. | ||
The formation of any pentamers at all is likely due to kinetic factors at play in the pulse deposition process, as they have not been reported in early observations of this system.71,86 On the sole basis of calculated energies, then, all four possibilities (both configurations and both orientations) could be present in a mixture on the surface. To discriminate between them, we simulated constant-current STM images using the output of the VASP calculations, and these images are shown in Fig. 10. From these, we conclude that despite not being true enantiomers, configurations A and B are predicted to appear as mirror images in STM measurements, in both the nitrogen-down (Fig. 10(A) and (B)) and nitrogen-up (Fig. 10(C) and (D)) orientations. However, for both configurations, there is a large contrast between the two orientations. Of the two, the simulated images of the nitrogen-up orientations of configuration A and B, as seen in Fig. 10(C) and (D), are a better match to the experimental data. Fig. 11 shows the same simulated images as Fig. 10 with models of the molecules overlaid on the images. The main contrast seems to originate from the carboxyl oxygens, particularly when those atoms are in proximity to the surface (in the nitrogen-up orientation).
 | ||
| Fig. 10 Simulated STM images using VASP for both accepted cluster configurations from Fig. 8 in both nitrogen-down and nitrogen-up orientations. (A) Configuration A/nitrogen-down, (B) configuration B/nitrogen-down, (C) configuration A/nitrogen-up, (D) configuration B/nitrogen-up. | ||
 | ||
| Fig. 11 Simulated STM images from Fig. 10 with overlaid molecular models. (A) Configuration A/nitrogen-down, (B) configuration B/nitrogen-down, (C) configuration A/nitrogen-up, (D) configuration B/nitrogen-up. | ||
In comparing the experimental STM images (Fig. 6) and calculated images (Fig. 10) for proline, we note that agreement is by no means quantitative: the experimentally observed molecular features are larger and less well resolved than what is predicted from the calculations. This could be the effect of limited experimental resolution, but another likely confounding factor is the simplifications made in the VASP calculation, most importantly that the geometry of the surface-adsorbed cluster was not fully optimized. However, we propose that the qualitative findings of the calculations are what is most significant: that nitrogen-up and nitrogen-down oriented clusters would be readily distinguished in STM images, while different diastereomeric configurations can appear very similar in structure, but with apparently opposite chirality.
4 Conclusion
The adsorption of both S-indoline-2-carboxylic acid and S-proline on the Au(111) surface creates pinwheel-shaped cyclic pentamers. Even though both molecules were enantiomerically pure, upon adsorption both left- and right-handed pinwheels form in an apparently racemic mixture. Electronic structure calculations confirm two C5-symmetric diastereomers with comparable binding energies and predict that STM images of the two look nearly identical, but are mirror-imaged. Calculated STM images also rule out that there is a mixture of adsorption orientations. We conclude that the observed loss of chirality results from a near-equal mixture of diastereomeric clusters, where the apparent chirality of a pentamer on the surface is not derived from the molecular stereocenter.Considering the relatively small NH⋯O hydrogen bond distances for both proposed pentamer configurations, we conclude that molecule–molecule interactions, specifically hydrogen bonding, drive pentamer formation for these molecules instead of being stabilized by molecule–surface interactions.
These results show that even for small organic molecules, the intermolecular interaction potential can be complex, leading to multiple local energy minima and ultimately structural polymorphism during self-assembly. This study presents one computational approach to investigate the complexities of these interactions. In addition, it highlights the importance of polymorphism in the propagation of chiral structure in self-assembly. A better fundamental understanding of chiral self-assembly may inform efforts to more effectively realize directed enantioselective crystallization, which has important implications in the development of single-enantiomer pharmaceuticals and enantioselective heterogeneous catalysts.
Data availability
CCDC 2356146 contains the supplementary crystallographic data for this paper.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work has been provided by the National Science Foundation NSF grants no. CHE-1807313 and CHE-2108186. The authors would like to thank Professor William Schneider for helpful discussions.Notes and references
- F. Zaera, J. Phys. Chem. C, 2008, 112, 16196–16203 CrossRef CAS.
- C. Baddeley, T. E. Jones, A. G. Trant and K. E. Wilson, Top. Catal., 2011, 25, 17–28 CrossRef.
- G. Kyriakou, S. K. Beaumont and R. M. Lambert, Langmuir, 2011, 27, 9687–9695 CrossRef CAS PubMed.
- J. Huan, X. Zhang and Q. Zeng, Phys. Chem. Chem. Phys., 2019, 21, 11537–11553 RSC.
- Y. Zheng, W. Luo, L. Yu, S. Chen, K. Mao, Q. Fang, Y. Yang, C. Wang, H. Zhu and B. Tu, J. Phys. Chem. B, 2022, 126, 723–733 CrossRef CAS PubMed.
- A. Bera, S. Henkel, J. Mieres-Perez, Y. Andargie Tsegaw, E. Sanchez-Garcia, W. Sander and K. Morgenstern, Angew. Chem., Int. Ed., 2022, 61, e202212245 CrossRef CAS PubMed.
- F. Purcell-Milton, R. McKenna, L. J. Brennan, C. P. Cullen, L. Guillemeney, N. V. Tepliakov, A. S. Baimuratov, I. D. Rukhlenko, T. S. Perova, G. S. Duesberg, A. V. Baranov, A. V. Fedorov and Y. K. Gun'Ko, ACS Nano, 2018, 12, 954–964 CrossRef CAS PubMed.
- L. Ji, Y. Zhao, M. Tao, H. Wang, D. Niu, G. Ouyang, A. Xia and M. Liu, ACS Nano, 2020, 14, 2373–2384 CrossRef CAS PubMed.
- I. Kuzmenko, H. Rapaport, K. Kjaer, J. Als-Nielsen, I. Weissbuch, M. Lahav and L. Leiserowitz, Chem. Rev., 2001, 101, 1659–1696 CrossRef CAS PubMed.
- B. Sung, A. de la Cotte and E. Grelet, Nat. Commun., 2018, 9, 1405 CrossRef PubMed.
- H. Caner, E. Groner, L. Levy and I. Agranat, Drug Discovery Today, 2004, 9, 105–110 CrossRef CAS PubMed.
- A. Calcaterra and I. D'Acquarica, J. Pharm. Biomed. Anal., 2018, 147, 323–340 CrossRef CAS PubMed.
- F. Zaera, Chem. Soc. Rev., 2017, 46, 7374–7398 RSC.
- N. Jiang, Y. Wang, Q. Liu, Y. Zhang, Z. Deng, K.-H. Ernst and H.-J. Gao, Phys. Chem. Chem. Phys., 2010, 12, 1318–1322 RSC.
- R. Raval, Chem. Soc. Rev., 2009, 38, 707–721 RSC.
- J. A. A. W. Elemans, I. De Cat, H. Xu and S. De Feyter, Chem. Soc. Rev., 2009, 38, 722–736 RSC.
- K.-H. Ernst, Phys. Status Solidi B, 2012, 249, 2057–2088 CrossRef CAS.
- A. Bauer, M. Maier, W. M. Schosser, J. Diegel, F. Paschke, Y. Dedkov, F. Pauly, R. F. Winter and M. Fonin, Adv. Mater., 2020, 32, 1907390 CrossRef CAS PubMed.
- J. C. Martín Romano, P. Casado Aguilar, A. L. Vázquez de Parga, M. Garnica, O. Rodríguez de la Fuente, J. M. Rojo and M. A. Niño, Phys. Chem. Chem. Phys., 2022, 24, 8022–8031 RSC.
- L. Xie, Y. Ding, D. Li, C. Zhang, Y. Wu, L. Sun, M. Liu, X. Qiu and W. Xu, J. Am. Chem. Soc., 2022, 144, 5023–5028 CrossRef CAS PubMed.
- H. Liu, H. Li, Y. He, P. Cheng, Y.-Q. Zhang, B. Feng, H. Li, K. Wu and L. Chen, Nat. Commun., 2023, 14, 2100 CrossRef CAS PubMed.
- M.-X. Shi, J. Xu, K. Sun, M.-L. Tao, J.-Y. Yang, D.-X. Yang, Z.-L. Wang, Z. Li, J.-Z. Wang, Q.-K. Xue and S. Meng, Nano Res., 2022, 15, 5316–5321 CrossRef CAS.
- I. Baltaci, M. G. H. Schulte and C. Westphal, J. Phys. Chem. C, 2020, 124, 23648–23656 CrossRef CAS.
- L. M. Rodríguez, P. Gómez, M. Más-Montoya, J. Abad, A. Tárraga, J. I. Cerdá, J. Méndez and D. Curiel, Angew. Chem., Int. Ed., 2020, 60, 1782–1788 CrossRef PubMed.
- Y. Cao, J. Mieres-Perez, J. F. Rowen, E. Sanchez-Garcia, W. Sander and K. Morgenstern, Nat. Commun., 2023, 14, 4500 CrossRef CAS PubMed.
- Z. Tessari, K. Tahara and S. De Feyter, J. Phys. Chem. C, 2023, 127, 9248–9256 CrossRef CAS.
- J. Lipton-Duffin, M. Abyazisani and J. MacLeod, Chem. Commun., 2018, 54, 8316–8319 RSC.
- K. Tahara, Y. Kubo, S. Hashimoto, T. Ishikawa, H. Kaneko, A. Brown, B. E. Hirsch, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2020, 142, 7699–7708 CrossRef CAS PubMed.
- Q. Han, Z. Li, K. Sun, M.-L. Tao, M.-X. Shi, D.-X. Yang, J.-X. Xia, J.-J. Wan and J.-Z. Wang, Phys. Chem. Chem. Phys., 2022, 24, 10292–10296 RSC.
- D. G. Yablon, J. Guo, D. Knapp, H. Fang and G. W. Flynn, J. Phys. Chem. B, 2001, 105, 4313–4316 CrossRef CAS.
- S. De Feyter, A. Gesquière, P. C. M. Grim, F. C. De Schryver, S. Valiyaveettil, C. Meiners, M. Sieffert and K. Müllen, Langmuir, 1999, 15, 2817–2822 CrossRef CAS.
- A. Miura, P. Jonkheijm, S. De Feyter, A. Schenning, E. Meijer and F. De Schryver, Small, 2004, 1, 131–137 CrossRef PubMed.
- M. M. Knudsen, N. Kalashnyk, F. Masini, J. R. Cramer, E. Lægsgaard, F. Besenbacher, T. R. Linderoth and K. V. Gothelf, J. Am. Chem. Soc., 2011, 133, 4896–4905 CrossRef CAS PubMed.
- M. L. Liriano, A. M. Larson, C. Gattinoni, J. Carrasco, A. E. Baber, E. A. Lewis, C. J. Murphy, T. J. Lawton, M. D. Marcinkowski, A. J. Therrien, A. Michaelides and E. C. H. Sykes, J. Chem. Phys., 2018, 149, 034703 CrossRef PubMed.
- M. Maeda, K. Sato, S. De Feyter and K. Tahara, Nanoscale, 2023, 15, 19569–19576 RSC.
- D. M. Walba, F. Stevens, N. A. Clark and D. C. Parks, Acc. Chem. Res., 1996, 29, 591–597 CrossRef CAS.
- J. Zhang, A. Gesquière, M. Sieffert, M. Klapper, K. Müllen, F. C. De Schryver and S. De Feyter, Nano Lett., 2005, 5, 1395–1398 CrossRef CAS PubMed.
- B. R. Heiner, A. M. Pittsford and S. A. Kandel, Chem. Commun., 2023, 59, 170–178 RSC.
- N. A. Wasio, R. C. Quardokus, R. D. Brown, R. P. Forrest, C. S. Lent, S. A. Corcelli, J. A. Christie, K. W. Henderson and S. A. Kandel, J. Phys. Chem. C, 2015, 119, 21011–21017 CrossRef CAS.
- M. Wolf, V. Hayes, C. R. Gerber, P. G. Quardokus, J. J. Ortiz-Garcia, C. Plummer and R. C. Quardokus, J. Vac. Sci. Technol., A, 2020, 38, 022413 CrossRef CAS.
- F. De Marchi, D. Cui, J. Lipton-Duffin, C. Santato, J. M. MacLeod and F. Rosei, J. Chem. Phys., 2015, 142, 101923 CrossRef PubMed.
- D. Case, H. Aktulga, K. Belfon, I. Ben-Shalom, J. Berryman, S. Brozell, D. Cerutti, T. I. Cheatham, G. Cisneros, V. Cruzeiro, T. Darden, R. Duke, G. Giambasu, M. Gilson, H. Gohlke, A. Goetz, R. Harris, S. Izadi, S. Izmailov, K. Kasavajhala, M. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. Simmerling, N. Skrynnikov, J. Smith, J. Swails, R. Walker, J. Wang, J. Wang, H. Wei, R. Wolf, X. Wu, Y. Xiong, Y. Xue, D. York, S. Zhao and P. Kollman, Amber, 2022 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- A. M. Silski, R. D. Brown, J. P. Petersen, J. M. Coman, D. A. Turner, Z. M. Smith, S. A. Corcelli, J. C. Poutsma and S. A. Kandel, J. Phys. Chem. C, 2017, 121, 21520–21526 CrossRef CAS.
- T. Beyer and S. L. Price, J. Phys. Chem. B, 2000, 104, 2647–2655 CrossRef CAS.
- J. P. Petersen, R. D. Brown, A. M. Silski, S. A. Corcelli and S. A. Kandel, J. Phys. Chem. C, 2019, 123, 13610–13614 CrossRef CAS.
- A. M. Silski-Devlin, J. P. Petersen, J. Liu, G. A. Turner, J. C. Poutsma and S. A. Kandel, J. Phys. Chem. C, 2020, 124, 5213–5219 CrossRef CAS.
- A. M. Silski, J. P. Petersen, R. D. Brown, S. A. Corcelli and S. A. Kandel, J. Phys. Chem. C, 2018, 122, 25467–25474 CrossRef CAS.
- R. Gatti, J. M. MacLeod, J. A. Lipton-Duffin, A. G. Moiseev, D. F. Perepichka and F. Rosei, J. Phys. Chem. C, 2014, 118, 25505–25516 CrossRef CAS.
- W. Mamdouh, H. Uji-I, J. S. Ladislaw, A. E. Dulcey, V. Percec, F. C. De Schryver and S. De Feyter, J. Am. Chem. Soc., 2005, 128, 317–325 CrossRef PubMed.
- T. Sirtl, W. Song, G. Eder, S. Neogi, M. Schmittel, W. M. Heckl and M. Lackinger, ACS Nano, 2013, 7, 6711–6718 CrossRef CAS PubMed.
- S. Lei, et al. , Angew. Chem., Int. Ed., 2008, 47, 2964–2968 CrossRef CAS PubMed.
- M. Lackinger, S. Griessl, W. M. Heckl, M. Hietschold and G. W. Flynn, Langmuir, 2005, 21, 4984–4988 CrossRef CAS PubMed.
- L. Kampschulte, M. Lackinger, A.-K. Maier, R. S. K. Kishore, S. Griessl, M. Schmittel and W. M. Heckl, J. Phys. Chem. B, 2006, 110, 10829–10836 CrossRef CAS PubMed.
- R. C. Quardokus, N. A. Wasio, R. D. Brown, J. A. Christie, K. W. Henderson, R. P. Forrest, C. S. Lent, S. A. Corcelli and S. Alex Kandel, J. Chem. Phys., 2015, 142, 101927 CrossRef PubMed.
- T. Balandina, M. W. van der Meijden, O. Ivasenko, D. Cornil, J. Cornil, R. Lazzaroni, R. M. Kellogg and S. De Feyter, Chem. Commun., 2013, 49, 2207–2209 RSC.
- S. Dutta and A. J. Gellman, Chem. Soc. Rev., 2017, 46, 7787–7839 RSC.
- Y. Fang, B. D. Lindner, I. Destoop, T. Tsuji, Z. Zhang, R. Z. Khaliullin, D. F. Perepichka, K. Tahara, S. D. Feyter and Y. Tobe, J. Am. Chem. Soc., 2020, 142, 8662–8671 CrossRef CAS PubMed.
- R. H. Janke, G. Haufe, E. U. Würthwein and J. H. Borkent, J. Am. Chem. Soc., 1996, 118, 6031–6035 CrossRef CAS.
- J. Weckesser, A. De Vita, J. V. Barth, C. Cai and K. Kern, Phys. Rev. Lett., 2001, 87, 961011 CrossRef PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- T. Eralp, A. Shavorskiy and G. Held, Surf. Sci., 2011, 605, 468–472 CrossRef CAS.
- T. Eralp, A. Shavorskiy, Z. V. Zheleva, G. Held, N. Kalashnyk, Y. Ning and T. R. Linderoth, Langmuir, 2010, 26, 18841–18851 CrossRef CAS PubMed.
- S. Barlow, K. Kitching, S. Haq and N. Richardson, Surf. Sci., 1998, 401, 322–335 CrossRef CAS.
- S. M. Barlow, S. Louafi, D. Le Roux, J. Williams, C. Muryn, S. Haq and R. Raval, Langmuir, 2004, 20, 7171–7176 CrossRef CAS PubMed.
- V. Humblot, C. Méthivier and C.-M. Pradier, Langmuir, 2006, 22, 3089–3096 CrossRef CAS PubMed.
- M. Forster, M. S. Dyer, M. Persson and R. Raval, J. Am. Chem. Soc., 2009, 131, 10173–10181 CrossRef CAS PubMed.
- S. Stepanow, T. Strunskus, M. Lingenfelder, A. Dmitriev, H. Spillmann, N. Lin, J. V. Barth, C. Wöll and K. Kern, J. Phys. Chem. B, 2004, 108, 19392–19397 CrossRef CAS.
- T. Schmitt, L. Hammer and M. A. Schneider, J. Phys. Chem. C, 2016, 120, 1043–1048 CrossRef CAS.
- M. Abyazisani, J. Bradford, N. Motta, J. Lipton-Duffin and J. MacLeod, Langmuir, 2019, 35, 7112–7120 CrossRef CAS PubMed.
- F. Gao, Y. Wang, L. Burkholder and W. Tysoe, Surf. Sci., 2007, 601, 3579–3588 CrossRef CAS.
- S. Clair, S. Pons, A. P. Seitsonen, H. Brune, K. Kern and J. V. Barth, J. Phys. Chem. B, 2004, 108, 14585–14590 CrossRef CAS.
- N. Zhu, T. Osada and T. Komeda, Surf. Sci., 2007, 601, 1789–1794 CrossRef CAS.
- D. Schwarz, R. van Gastel, H. J. Zandvliet and B. Poelsema, J. Phys. Chem. C, 2013, 117, 1020–1029 CrossRef CAS.
- R. T. Seljamäe-Green, G. J. Simpson, F. Grillo, J. Greenwood, S. M. Francis, R. Schaub, P. Lacovig and C. J. Baddeley, Langmuir, 2014, 30, 3495–3501 CrossRef PubMed.
- J. J. Koenig, J.-M. Neudörfl, A. Hansen and M. Breugst, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2018, 74, 1067–1070 CrossRef CAS PubMed.
- R. D. Brown, J. M. Coman, J. A. Christie, R. P. Forrest, C. S. Lent, S. A. Corcelli, K. W. Henderson and S. A. Kandel, J. Phys. Chem. C, 2017, 121, 6191–6198 CrossRef CAS.
- S. Karan, Y. Wang, R. Robles, N. Lorente and R. Berndt, J. Am. Chem. Soc., 2013, 135, 14004–14007 CrossRef CAS PubMed.
- E. Hansen, A. R. Rosales, B. Tutkowski, P.-O. Norrby and O. Wiest, Acc. Chem. Res., 2016, 49, 996–1005 CrossRef CAS PubMed.
- J. Wyrick, D.-H. Kim, D. Sun, Z. Cheng, W. Lu, Y. Zhu, K. Berland, Y. S. Kim, E. Rotenberg, M. Luo, P. Hyldgaard, T. L. Einstein and L. Bartels, Nano Lett., 2011, 11, 2944–2948 CrossRef CAS PubMed.
- E. C. H. Sykes, P. Han, S. A. Kandel, K. F. Kelly, G. S. McCarty and P. S. Weiss, Acc. Chem. Res., 2003, 36, 945–953 CrossRef CAS PubMed.
- R. T. Seljamäe-Green, G. J. Simpson, F. Grillo, J. Greenwood, S. M. Francis, R. Schaub, J. E. Gano, H. A. Früchtl, P. Lacovig and C. J. Baddeley, Langmuir, 2015, 31, 262–271 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2356146. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp02094d |
| This journal is © the Owner Societies 2024 |