
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined experimental and computational study of the reactivity of the methanimine radical cation (H2CNH˙+) and its isomer aminomethylene (HCNH2˙+) with propene (CH3CHCH2)†
Vincent
Richardson
 *ab,
David
Sundelin
c,
Claire
Romanzin
de,
Roland
Thissen
de,
Christian
Alcaraz
de,
Miroslav
Polášek
f,
Jean-Claude
Guillemin
g,
Jan
Žabka
f,
Wolf D
Geppert
c and
Daniela
Ascenzi
*b
*ab,
David
Sundelin
c,
Claire
Romanzin
de,
Roland
Thissen
de,
Christian
Alcaraz
de,
Miroslav
Polášek
f,
Jean-Claude
Guillemin
g,
Jan
Žabka
f,
Wolf D
Geppert
c and
Daniela
Ascenzi
*b
aDepartment of Physics, The Oliver Lodge, University of Liverpool, Oxford St, Liverpool, L69 7ZE, UK. E-mail: vincent.richardson@liverpool.ac.uk
bDepartment of Physics, University of Trento, Via Sommarive 14, I-38123, Trento, Italy. E-mail: daniela.ascenzi@unitn.it
cDepartment of Physics, Stockholm University, Stockholm, Sweden
dInstitut de Chimie Physique, UMR8000, CNRS, Université Paris-Saclay, Orsay, France
eSynchrotron Soleil, L’Orme des Merisiers, Saint Aubin, France
fJ. Heyrovsky Institute of Physical Chemistry of the Czech Academy of Sciences, Prague, Czechia
gUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR – UMR6226, F-35000 Rennes, France
First published on 30th July 2024
Abstract
The gas phase reactivity of the radical cation isomers H2CNH˙+ (methanimine) and HCNH2˙+ (aminomethylene) with propene (CH3CHCH2) has been investigated by measuring absolute reactive cross sections and product branching ratios, under single collision conditions, as a function of collision energy (in the range ∼0.07–11.80 eV) using guided ion beam mass spectrometry coupled with VUV photoionization for selective isomer generation. Experimental results have been merged with theoretical calculations to elucidate reaction pathways and structures of products. The H2CNH˙+ isomer is over a factor two more reactive than HCNH2˙+. A major channel from both isomers is production of protonated methanimine CH2NH2+via hydrogen-atom transfer reaction but, while H2CNH˙+ additionally gives charge and proton transfer products, the HCNH2˙+ isomer leads instead to protonated vinylimine CH2CHCHNH2+, produced alongside CH3˙ radicals. The reactions have astrochemical implications in the build up of chemical complexity in both the interstellar medium and the hydrocarbon-rich atmospheres of planets and satellites.
1 Introduction
Propene (a.k.a. propylene, CH3CHCH2) is the second simplest alkene (after ethene, C2H4) and has been detected in many different astronomic environments. It was first discovered towards Taurus Molecular Cloud (TMC-1),1 where the observed column density of 4 × 1013 cm−2 was in the range of other well-known interstellar hydrocarbons. Following this, propene has been detected towards nine different other dark clouds including Lupus-1A, L1495B, L1521F, and Serpens South 1a,2 indicative of a ubiquitous presence in such objects. In addition to dark interstellar clouds, warm (75 K) propene has recently been observed towards the hot corino IRAS 16293-2422B,3 though attempts to observe a range of propene derivatives have so far proven unsuccessful.4Propene has also been observed in Titan's stratosphere by the composite infrared spectrometer (CIRS) onboard the Cassini spacecraft, its vertical abundance slowly increasing with altitude from 2.0 ± 0.8 ppbv at 125 km, to 4.6 ± 1.5 ppbv at 200 km.5 Further to this, the presence of propene has been predicted by photochemical models of Titan's atmosphere.6,7 Notably, propene also displays a different latitudinal trend than propane (C3H8) and propyne (CH3CCH). Whereas the latter two show abundance maxima at the winter pole, the one of propene is located at the equator.7
The abundance, distribution and ubiquitous presence of propene in many astronomic environments raises the question of its formation and destruction mechanisms. Earlier investigations have predicted the formation of propene by dissociative recombination of the C3H7+ cation, while theoretical calculations predicted that this latter species can be efficiently formed from the propargyl ion (CH2CCH+) by two consecutive radiative association with molecular hydrogen.8 However, later combined experimental and theoretical studies have concluded that these radiative association reactions do not produce protonated propene (C3H7+) efficiently under interstellar conditions due to substantial activation energy barriers,9 leading to the proposal of the following radical formation mechanism:10
| CH3˙ + CH˙ → CH3CH | (1) |
| CH3CH + CH2 → CH3CHCH2 | (2) |
Most importantly, propene can serve as a basis for the production of more complex species via ion-neutral elongation reactions followed by dissociative recombination of the resulting larger ions. It is well established that hydrocarbon cations as well as radicals (e.g. CH, C2H) can lead to chain elongation reactions with propene to give C4 and C5 hydrocarbons.12,13 Additionally, an investigation of the reactions of 2-, 3-, and 4-dehydroanilinium radical cations with propene both experimentally, using an ion trap, and theoretically, through ab initio calculations at the M06-2X/6-31G(2df,p) and G3XK levels, found that such reactions led to the formation of heavier ions under elimination of smaller radicals.14 In this way, reactions of nitrogen-containing radicals and radical ions which could lead to more complex ions and, subsequently, neutrals in Titan's atmosphere can ultimately lead to the tholins, which are thought to make up the orange-coloured haze that envelopes Titan. To assess the importance of these reactions, it is vital to have detailed and accurate data on the reaction rates and branching ratios of those processes, since these parameters serve as the key inputs for the large chemical reaction network models used to simulate the chemistry of interstellar clouds as well as planetary and satellite atmospheres.
Furthermore, as the chemical complexity of the species increases, so does the potential impact of isomer-specific reactivity, both in terms of reactants and products. A prime example of this is the [CNH3]˙+ radical cation isomers methylenimine (H2CNH˙+) and aminomethylene (HCNH2˙+), which have been shown to exhibit quite different reactivity with a range of smaller hydrocarbons species such as CH4,15 C2H216 and C2H4.17 Though the majority of the m/z 29 signal recorded by the Ion Neutral Mass Spectrometer (INMS) onboard the Cassini spacecraft was assigned to the C2H5+ ion, models predict a density of H2CNH˙+ and its isomers of 1.1 × 10−2 cm−3 in Titan's ionosphere.18 Importantly, as many protonated nitrile ions have been shown to be largely unreactive with unsaturated hydrocarbons,18 the reactivity of the [CNH3]˙+ radical cations are potentially key to the formation of larger nitrogen-containing hydrocarbons on Titan. Additionally, as neutral methanimine is ubiquitous in the ISM19–21 and possesses a low ionization energy of 9.97 eV,22 the reactivity of both isomers is also expected to be significant in other astrochemical environments.
This paper presents a combined computational and experimental reactivity study of both H2CNH˙+ and HCNH2˙+ with propene using dissociative single photon ionization of neutral precursors which have been shown to selectively generate the charged isomeric species.15–17 We report reactive cross sections (CSs) and branching ratios (BRs) for a number of pathways involving both common gas-phase processes such as proton, charge and H-atom transfer as well as a number of bond-forming processes leading to an increase in chemical complexity. All pathways are rationalized through comparison with relevant potential energy surfaces (PESs) obtained by ab initio calculations.
2 Experimental methodology
The data on the reactivity of the two [CNH3]˙+ isomers has been collected using the CERISES apparatus23,24 in combination with the DESIRS beamline25 of the SOLEIL synchrotron in Saint-Aubin (France). CERISES is a guided ion beam tandem mass spectrometer consisting of two octopoles located between two quadrupole mass filters. This allows for the mass-selection of reagent and product ions of ion-neutral reactions, with the neutral reagent being introduced into a scattering cell surrounding the final part of the first octopole.Neutral reagent pressures used were of the order of 1 × 10−7 bar, in order to ensure operation close to the single collision regime. In this way, we are able to reduce the contribution from secondary collisions and limit attenuation of the parent beam to under 10%. Absolute pressures were measured using a MKS 398H differential manometer.
HCNH2˙+ ions are generated through dissociative photoionisation of cyclopropylamine,15,26 while H2CNH˙+ ions are generated via direct ionisation of methanimine,16 using the tunable output of the DESIRS beamline. Data are collected as a function of the photon energy which, in turn, acts as a proxy for the internal energy of the reagent ions. The photon energies used were in the range of 9.5–14 eV, with a resolution of 20–40 meV defined by the monochromator slit setting. Photons of energies greater than 15.7 eV were removed by an Argon gas filter.27 Photon energies in the absolute scale were obtained using the absorption lines of argon around 11.823 and 14.304 eV,28,29 with systematic shifts of 10–20 meV above the tabulated values.
The collision energy available to the reactants depends on both the ionic charge (in this case +1) and the potential difference between the ion source and reaction cell. The retarding potential method30 has been used to determine the maximum of the first derivative of the parent ion yield, the corresponding voltage of which defines the zero of the kinetic energy in the laboratory frame, which can then be converted into centre-of-mass collision energies (ECM). By changing the potentials of the reaction cell and all subsequent elements, we are able to scan a collision energy range from ∼0.07 to ∼11.80 eV in the centre of mass frame. The FWHM of the collision energy is 0.06 eV in the ECM frame.
Data as a function of both photon and collision energies were collected in the “multi-scan” mode where the signals for all ionic species of interest are collected at a given point before moving to the next one. In this way, we are able to drastically reduce any potential effects from drifts in source or reaction cell pressure.
3 Theoretical methodology
The mechanisms for the reactions of H2CNH˙+ and HCNH2˙+ leading to the observed products were studied using GAUSSIAN 16, Revision D.01.31 Geometries for intermediate structures, i.e. minima and transition states (TSs), were optimized at the MP2/6-31G(d) and MP2/6-311++G(d,p) levels of theory. The identity of TSs and minima were checked by frequency calculations and zero-point energy corrections were applied to the obtained energies. Intrinsic reaction coordinate (IRC) calculations were performed at the MP2/6-31G(d) level to ensure that the TSs connect the correct minima. In instances where the IRC calculations proved unsuccessful due to program failure, the geometry of the molecule was slightly distorted in both directions along the normal coordinate of the negative frequency vibration (corresponding to the reaction coordinate that passes through the transition state) prior to optimization in order to ensure transition states are connected to the relevant minima.For barrierless dissociation processes, relaxed potential energy surface (PES) scans were performed along the bond dissociation axis. In some cases, this requires the freezing of one or two angles and/or dihedral angles in order to prevent the scan from leading either to the previous adduct or a separate minimum entirely. Single point energy calculations were carried out for all stationary points at the CCSD(T)/6-311++G(d,p) level with zero-point energy corrections taken from the optimized geometries calculated at the MP2/6-311++G(d,p) level. Reactants and product geometries and energies were also calculated at the same level (including zero-point energy corrections) and the reactants' energies subtracted from the optimizations of the TS and minima, resulting in the relative energies Erel. Cartesian coordinates, structures and electronic energies of reagents, products, minima and TSs are reported in the ESI.†
In calculations of van der Waals clusters one often encounters basis set superposition errors (BSSEs), which result in an artificial strengthening of the intermolecular interaction. To test for BSSEs, we performed calculations on all three levels of theory employed in this work using counterpoise corrections on the following clusters: V1, V3, V5, W1, W2 and W3. The structures of these different species are detailed in Section 5, but here we only note that they cover both entrance (reactant) and exit (product) clusters. The BSSE corrections are either considerably lower (in the case of V5 and W3) or in the range of the error bars of the relative translation energies of the reactants (for V1, V3, W2 and W1) and are also insufficiently large to affect the validity of the predictions based on the calculated PES. Further details are given in the ESI.†
4 Experimental results
The generation of HCNH2˙+ (m/z 29) via dissociative photoionization of cyclopropylamine (c-C3H5NH2) is described by the following equation:| c-C3H5NH2 + hv → HCNH2˙+ + C2H4 | (3) |
The characterization of the process has been described previously,15,26 with the AE of this channel previously measured as 10.2 ± 0.1 eV. The generation of the H2CNH˙+ isomer through direct photoionization of methanimine has also been described in detail previously,16 with an appearence energy (AE) of 10.01± 0.08 eV. In both cases the AE have been measured using the same set-up here presented, and the interested reader is directed to the relevant references for further details.15,16 Here, we will focus on the reactivity of both isomers with propene (CH3CHCH2), as well as on the potential impact of isobaric contaminants on the observed product channels.
4.1 Identification of product channels
The reaction of HCNH2˙+ with propene yields products at m/z 30, 43, 56 and 70, while the reaction of H2CNH˙+ shows an additional product at m/z 42. Assignments for the m/z 30, 42, 43, 56 and 70 products are given by reactions (4)–(11) respectively, while literature and computational reaction enthalpies for the pathways identified in Section 5 are given in Table 1. The charge transfer reaction for the HCNH2˙+ isomer is not considered in reaction (5) due to its significant endothermicity, as shown in Table 1.| HCNH2˙+/H2CNH˙+ + CH3CHCH2 → CH2NH2+ + [C3H5] m/z 30 | (4) |
| → CH3CHCH2˙++ [CNH3] m/z 42 | (5) |
| → [C2H4N]+ + C2H5˙ m/z 42 | (6) |
| → CH3CHCH3+ +H2CN˙ m/z 43 | (7) |
| → CH3CHCH3+ + HCNH˙ m/z 43 | (8) |
| → [C2H5N]˙+ + C2H4 m/z 43 | (9) |
| → [C3H6N]+ + CH3˙ m/z 56 | (10) |
| → [C4H8N]+ + H˙ m/z 70 | (11) |
| Products | m/z | Eqn | ΔH° with HCNH2˙+ (eV) | ΔH° with H2CNH˙+ (eV) | ||
|---|---|---|---|---|---|---|
| Lit.a (298 K) | Calc.b (0 K) | Lit.a (298 K) | Calc.b (0 K) | |||
| a Unless stated otherwise, all formation enthalpies have been taken from ATcT.32 b Present work. c [CNH3] should be read as HCNH2 for the reaction of HCNH2˙+ and as H2CNH for the reaction of H2CNH˙+. d The formation enthalpy for the CH2CNH2+ ion, having the structure of protonated ketenimine, has been taken from.33 e The values refer to production of the lowest energy trans-HCNH isomer. The cis-HCNH isomer is ∼0.20 eV higher in energy.32 f The formation enthalpy for the NH2CHCH2+ ion has been taken from.34 g No literature values are available for the ionic products. h The formation enthalpies for all [C3H6N]+ isomers have been calculated using the relative energies from35 in combination with the formation enthalpy for the CH3CH2CNH+ isomer given in.36 For CH2CHCHNH+ see also.37 | ||||||
| CH2NH2+ + CH2CHCH2˙ | 30 | (4a) | −1.37 ± 0.04 | −1.28 | −1.55 ± 0.04 | −1.48 |
| CH2NH2+ + CH3CCH2˙ | 30 | (4b) | −0.49 ± 0.04 | −0.67 ± 0.04 | −0.59 | |
| CH2NH2+ + CH3CHCH˙ | 30 | (4c) | −0.34 ± 0.04 | −0.23 | −0.52 ± 0.04 | −0.43 |
| CH3CHCH2˙+ + [CNH3]c | 42 | (5) | +1.53 ± 0.04 | +1.35 | −0.20 ± 0.03 | −0.20 |
| CH2CNH2+ + C2H5˙ | 42 | (6) | −0.41 ± 0.12d | −0.38 | ||
| CH3CHCH3+ + H2CN˙ | 43 | (7) | −0.26 ± 0.03 | −0.20 | ||
| CH3CHCH3+ + HCNH˙ | 43 | (8) | +0.27 ± 0.04e | +0.61 | +0.09 ± 0.04e | +0.41 |
| NH2CHCH2˙+ + C2H4 | 43 | (9a) | −1.80 ± 0.11f | −1.55 | ||
| CH2NHCH2˙+ + C2H4 | 43 | (9b) | −1.27 | |||
| CH2NHCHCH2+ + CH3˙ | 56 | (10a) | −1.18 ± 0.10h | −1.10 | ||
| c−CH2NHCHCH2+ + CH3˙ | 56 | (10b) | −1.14 ± 0.10h | −1.14 | ||
| CH2CHCHNH2+ + CH3˙ | 56 | (10c) | −1.56 ± 0.10h | −1.56 | ||
| c-NH2CHCHCH2+ + CH3˙ | 56 | (10d) | −0.53 ± 0.10h | −0.51 | ||
| c-CH(CH3)CH2NHCH+ + H˙ | 70 | (11a) | −0.81 | |||
| CH2CHCH2NHCH2+ + H˙ | 70 | (11b) | −0.74 | |||
| CH3CHCHNHCH2+ + H˙ | 70 | (11c) | −0.98 | |||
| CH2C(CH3)NHCH2+ + H˙ | 70 | (11d) | −0.90 | |||
| CH3CHCHCHNH2+ + H˙ | 70 | (11e) | −1.22 | |||
| CH2CHCH2CHNH2+ + H˙ | 70 | (11f) | −0.98 | |||
| c-NH2CH(CH3)CHCH+ + H˙ | 70 | (11g) | −0.32 | |||
To the best of our knowledge, the reaction of the H2CNH˙+ isomer with CH3CHCH2 has not been studied previously. The reaction of the HCNH2˙+ isomer with CH3CHCH2 has been studied using FT-ICR,38 where a major product (BR = 0.93) was observed at m/z 56 in addition to a minor m/z 70 product (BR = 0.07), but without any CSs or reaction rates being reported. In the same study, the majority of the m/z 56 flux was assigned to the CH2CHCHNH2+ isomer on the basis of reactions with differently deuterated propenes, with at least one unknown pathway via NH2CHCH(CH3)CH2˙+, a covalently-bound adduct of the reactants, being inferred to account for the small amount of scrambling observed. In this work, we report CSs for the various channels, and we identify two new products, one at m/z 30 (assigned to the hydrogen atom transfer (HAT) process, channel (4)) and another one at m/z 43 (assigned to a mixture of proton transfer and bond-forming adduct pathways, channels (7)–(9)).
A detailed analysis of potential isobaric impurities in the reagent ion beams is provided in the ESI.† Notably, the generation of H2CNH˙+ is free from isobaric contaminants. In contrast, C2H5+, 13CH2CH2˙+ and H13CNH+ ions (m/z = 29) can form during the generation of HCNH2˙+. Contamination by C2H5+ can be ignored by considering only data taken at Ephot < 12.5 eV, while the contamination from 13CH2CH2˙+ is negligible and does not affect any of the recorded mass channels. However, contamination from H13CNH+ results in a product at m/z 43, which overlaps with a recorded product channel. We have determined that, while H13CNH+ is present at all Ephot, the resultant contamination in the m/z 43 channel is <30% at Ephot above 11.0 eV, decreasing to below 25% at Ephot = 11.5 eV. This is the lowest photon energy at which trends as a function of collision energy have been measured. The error bars for this channel have been adjusted to reflect this greater uncertainty.
4.2 Experimental results: data as a function of the photon energy (Ephot)
For both isomers, data as a function of Ephot at low collision energies (ECM = 0.12 ± 0.06 eV for HCNH2˙+ and 0.09 ± 0.06 for H2CNH˙+) has been collected for the m/z 30, 43, 56 and 70 products. Additionally, m/z 42 product data has been collected for H2CNH˙+. Results for both isomers are shown in Fig. 1. For all product channels, the large error bars for the HCNH2˙+ isomer at low photon energies are the result of the small reactant ion flux.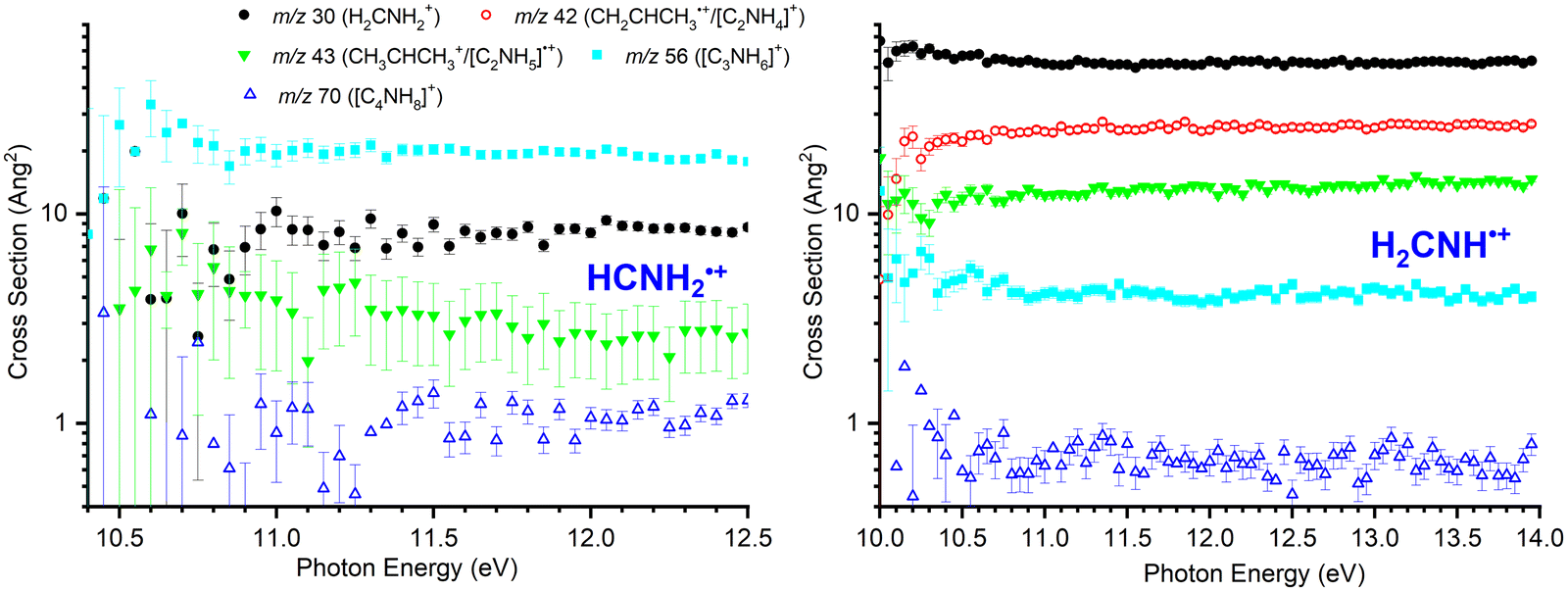 | ||
| Fig. 1 Left: Cross sections as a function of Ephot for the reaction of HCNH2˙+, generated through dissociative photoionization of cyclopropylamine, with CH3CHCH2 at ECM = 0.12 ± 0.06 eV. Right: Cross sections as a function of Ephot for the reaction of H2CNH˙+, generated through direct photoionization of methanimine, with CH3CHCH2 at ECM = 0.09 ± 0.06 eV. | ||
For the HCNH2˙+ isomer (Fig. 1, left panel), the major m/z 56 product ([C3H6N]+, reaction (10)) is approximately independent of Ephot, as are the medium intensity m/z 30 (CH2NH2+, reactions (4)) and 43 ([C3H7]+, reaction (8)/[C2H5N]˙+, reaction (9)) products. The m/z 70 product ([C4H8N]+, reaction (11)) is very minor throughout, but again shows no significant dependence on the photon energy. As none of the products show any marked trends as a function of Ephot, we infer that the reactivity of this isomer is largely independent of the internal energy of the reactant ion. However, the possibility that increasing the photon energy above that required for fragmentation does not lead to any increase in the internal energy of the HCNH2˙+ fragments cannot be dismissed.
For H2CNH˙+ (Fig. 1, right panel), the major ionic product at all photon energies is m/z 30 (CH2NH2+, reaction (4)) corresponding to the H-abstraction channel, which is approximately independent of Ephot. The next most intense channel is charge transfer detected at m/z 42 (CH3CHCH2+, reaction (5)), which shows a sharp increase in intensity above the appearance threshold of the parent ion before reaching a plateau above Ephot = 10.5 eV. Products with lower intensities are observed at m/z 43, corresponding either to the proton transfer ([C3H7]+, reaction (7)) or the formation of [C2H5N]˙+viareaction (9) and at m/z 56, corresponding to the formation of [C3H6N]+ (reaction (10)). In both cases, CSs are broadly independent of Ephot. Finally, a minor channel is detected at m/z 70 ([C4H8N]+, reaction (11)) that can be due to H-ejection from one or more covalently-bound adducts. This channel has a much smaller CS than any of the others and once again shows little-to-no dependence on the photon energy and by extension, due to the direct nature of the ionization for this isomer, the internal energy of the reactant ion.
4.3 Experimental results: data as a function of the collision energy (ECM)
In Fig. 2 absolute CSs data as a function of ECM are presented for HCNH2˙+ (left panel, measured at Ephot = 11.5 eV) and for H2CNH˙+ (right panel, measured at Ephot = 11.1 eV).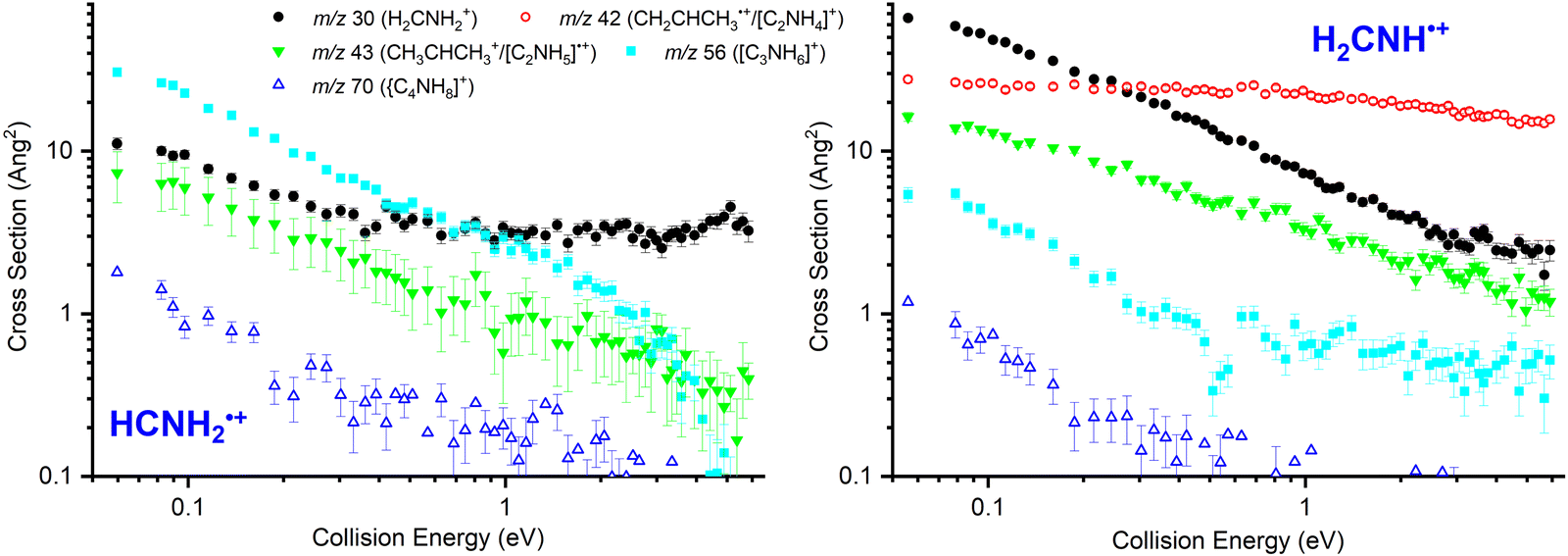 | ||
| Fig. 2 Left: Cross sections as a function of ECM for the reaction of HCNH2˙+, generated via dissociative photoionization of cyclopropylamine, with CH3CHCH2 at Ephot = 11.5 eV. Right: Cross sections as a function of ECM for the reaction of H2CNH˙+, generated by direct photoionization of methanimine, with CH3CHCH2 at Ephot = 11.1 eV. | ||
For the HCNH2˙+ isomer the CSs of products at m/z 56 ([C3H6N]+, reaction (10)) and at m/z 70 ([C4H8N]+, reaction (11)) decrease with increasing ECM, consistent with the expected barrierless formation via one or more covalently-bound adduct. The m/z 43 product ([C3H7]+, reaction (8)/[C2H5N]˙+, reaction (9)) also shows a decrease with increasing ECM, though this is more gradual than for the m/z 56 product, with a non-zero contribution at high collision energies indicative of a pathway proceeding either via a covalently-bound adduct or a van der Waals complex with an additional direct component. Given that the proton transfer from this isomer is ∼0.3 eV endothermic (see Table 1), the contribution at higher energies is consistent with an endothermic proton transfer, though no clear appearance threshold is observed. Finally, the m/z 30 product shows an initial decrease with increasing ECM followed by a plateau at higher collision energies (above ECM ∼ 0.5 eV), consistent with a combination of complex/adduct and direct mechanisms.
For the H2CNH˙+ isomer (Fig. 2, right panel), the m/z 30 product (CH2NH2+, reaction (4)) decreases with ECM, but with a non-zero contribution at high ECM corresponding to a direct pathway. The m/z 42 product (CH3CHCH2+, reaction (5)) shows no strong dependence on ECM, typical for a charge transfer process, while the m/z 43 product ([C3H7]+, reactions (7) and (8)/[C2H5N]˙+, reaction (9)) shows a similar trend to the m/z 30 product, albeit with a greater relative contribution at high ECM. The CSs for products at m/z 56 ([C3H6N]+, reaction (10)) and at m/z 70 ([C4H8N]+) decrease with increasing ECM, consistent with pathways proceeding via one or more covalently-bound adduct. However, we note that the CS of m/z 56 remains non-zero at higher collision energies, indicative of some contribution via one or more direct mechanism.
The observed trends and differing reactivity (both in terms of absolute CSs and relative BRs) of the two isomers are considered alongside proposed mechanisms in Section 5.
4.4 From CSs to rate constants
For applications where the rate constants are needed instead of CSs (e.g. in astrochemical modelling), Table 2 presents the BRs and the total rate constants (ktot(Eave)), determined from the measured CSs following a well-established method.17,39,40 For both isomers, the rates have been reported at both low (Eave = 0.12 ± 0.03 eV) and high (Eave = 3.01 ± 0.06 eV) representative average energies. The ktot(Eave) so obtained can be compared with values predicted by the Langevin (kL41) and Su-Chesnavich (kSC42–44) capture models. In both cases, as the ion is treated as a point charge, no differentiation is made between the two isomers.HCNH2˙+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
H2CNH˙+b |
||||
|---|---|---|---|---|---|
| Branching ratios (BRs) | |||||
| Product m/z | Eqn | E ave = 0.12 ± 0.03 eV | E ave = 3.01 eV | E ave = 0.12 ± 0.03 eV | E ave = 3.01 eV |
| a Generated via dissociative photoionization of cyclopropylamine at at Ephot = 11.5 eV, see Fig. 2, left. b Generated via direct photoionization of methanimine at at Ephot = 11.1 eV, see Fig. 2, right. | |||||
| 30 | (4) | 0.23 ± 0.02 | 0.65 ± 0.15 | 0.53 ± 0.01 | 0.13 ± 0.02 |
| 42 | (5) and (6) | — | — | 0.28 ± 0.01 | 0.77 ± 0.07 |
| 43 | (7)–(9) | 0.15 ± 0.03 | 0.18 ± 0.02 | 0.14 ± 0.01 | 0.08 ± 0.01 |
| 56 | (10) | 0.59 ± 0.04 | 0.15 ± 0.05 | 0.04 ± 0.00 | 0.02 ± 0.01 |
| 70 | (11) | 0.03 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 |
| Total rate constants (ktot) | ||||
|---|---|---|---|---|
| E ave = 0.12 ± 0.03 eV | E ave = 3.01 eV | E ave = 0.12 ± 0.03 eV | E ave = 3.01 eV | |
| k tot(Eave) | (4.0 ± 2.2)×10−10 | (2.5 ± 0.5)×10−10 | (9.9 ± 5.3)×10 | (1.3 ± 0.2)×10−9 |
The values obtained from these models, using an average polarizability of 5.99 Å345 and a dipole moment of 0.36 D45 for propene, are kL = 1.38 × 10−9 cm3 molecule−1 s−1 and kSC = 1.49 × 10−9 cm3 molecule−1 s−1, at 300 K. Notably, the kSC rate is approximately 4.0 and 1.5 times higher than the experimental values for the HCNH2˙+ and H2CNH˙+ isomers, respectively. However, it should be stressed that the capture model values represent upper limits to the actual rate coefficients, and a direct comparison with our experimental values should be made with caution as they refer to energy-dependent rate coefficients estimated at a collision energy of 0.12 ± 0.03 eV. With this caveat in mind, these results underscore the significance of experimental, isomer-specific data, as a reliance on capture theory often fails to replicate the significantly different reactivities among various isomeric ions.
5 Computational results
Theoretical calculations show that the HCNH2˙+ isomer is lower in energy than the H2CNH˙+ isomer by −18.82 kJ mol−1 (−0.20 eV), consistent with literature values46 giving a difference in the heat of formations of the two isomers, calculated at the UQCISD(T)/6-311++G(3df, 2p) and RCCSD(T)/cc-pVTZ levels of theory, equal to 16 kJ mol−1. All single point energies are calculated with the GAUSSIAN suite of programs at the CCSD(T)/6-311++G** level of theory, with ZPEs calculated at the MP2/6-311++G** level. Full details have been given in Section 3. Unless noted otherwise, relative enthalpies are given (in eV and in parenthesis after each structure) with respect to the sum of the separated reactants having the highest energy, i.e. H2CNH˙+ plus CH3CHCH2. For the sake of readability, the rotamers of a given isomer, formed by rotations around single bonds, are treated as a single species in the following description and in the PES figures included in the main manuscript. Rotamer-specific pathways and PESs are given in the ESI† (Fig. S5–S10).5.1 H2CNH˙+ PES
For the H2CNH˙+ isomer, two product channels can proceed via direct mechanisms. The lowest energy of these is the H-abstraction of a CH3 hydrogen to give CH2NH2+ (m/z 30) in combination with CH2CHCH2 (−1.48, black lines in Fig. 3), with the higher energy channel being charge transfer to give CH3CHCH2+ (m/z 42) plus H2CNH (−0.20, red line in Fig. 3). Proton transfer can proceed either directly or through a complex mediated mechanism via the van der Waals intermediates V1 (−0.77) and V2 (−0.77), separated by the transition state V1-2 (−0.78), see light green lines in Fig. 3. The lower energy of V1-2 with respect to the structures it joins is an artefact induced by the zero-point energy correction. Such behaviour is sometimes observed if the transition state is very close to the minima it connects, both geometrically and energetically. If one excludes ZPE, the energy of the transition state exceeds those of the adjacent minima by 0.08 and 0.06 eV, respectively.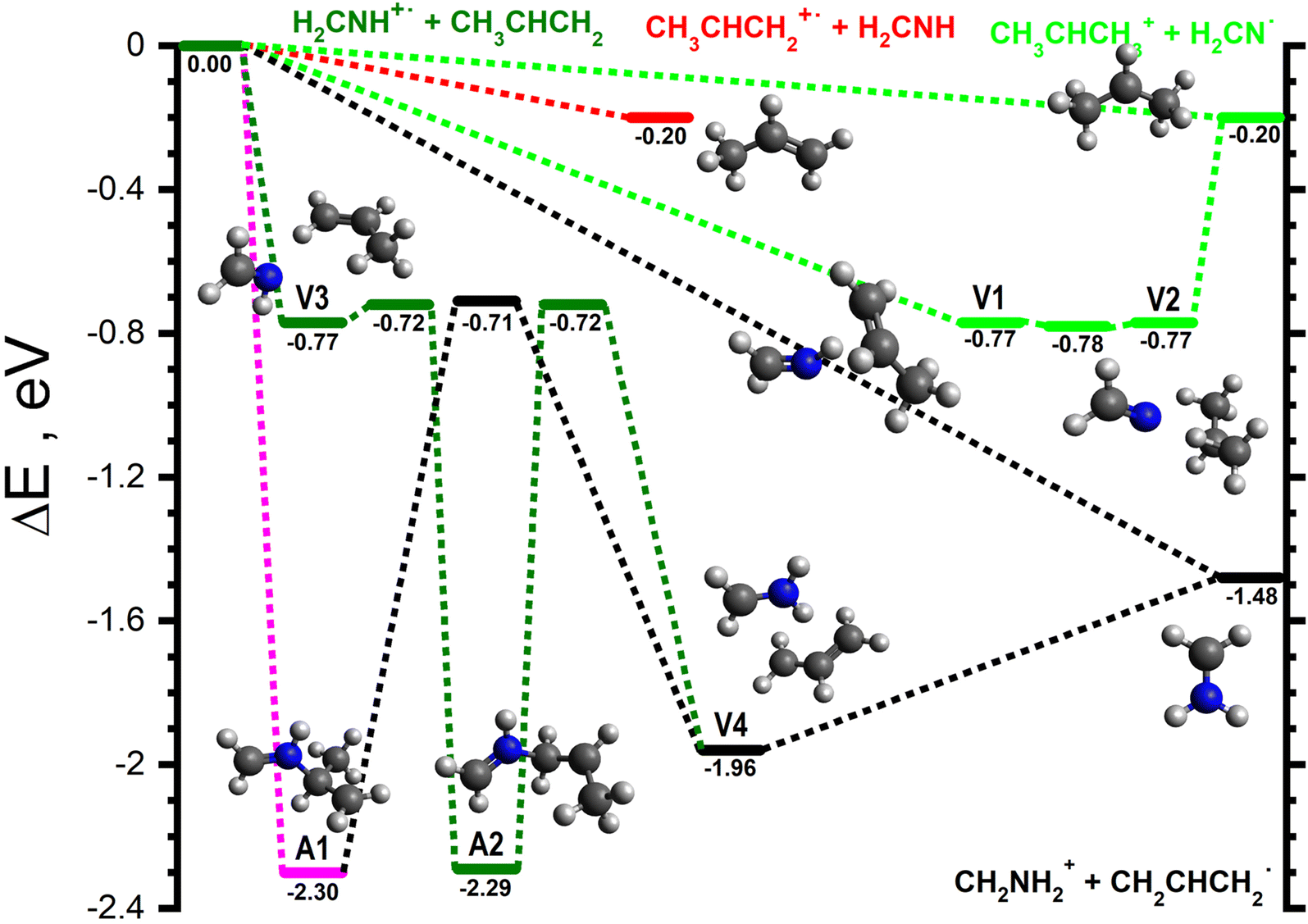 | ||
| Fig. 3 Schematic potential energy surface for the reaction of H2CNH˙+ with CH3CHCH2via a series of van der Waals adducts, as well as those leading to the initial covalently-bound adducts A1 and A2. The zero of the energy scale (in eV) is given by the sum of the energies of the separated H2CNH˙+ plus CH3CHCH2 reactants. Calculations are at the CCSD(T)/6-311++G(d,p) level of theory. Molecular structures are shown only for intermediates and products, with TS structures given in the (ESI†). | ||
Pathways involving the formation of one or more covalent bond proceed through different covalently bound adducts. The first one, CH2NHCH(CH3)CH2˙+, hereafter referred to as A1 (−2.30), can form directly from the reactants via a barrierless radical attack of the central carbon (pink line in Fig. 3). Similarly, the CH2NHCH2CHCH3˙+ adduct, hereafter referred to as A2 (−2.29), can form from V3 (−0.77), a van der Waals complex of the reactants, by radical attack of the terminal CH2 group viaV3-A2 (−0.72) (dark green in Fig. 3). The rotamer-specific pathways for A1 and A2 are shown in Fig. S5–S7 of the ESI.†
As well as the direct pathway to form CH2NH2+ in combination with CH2CHCH2˙ noted above, this pair of products can also form from both A1 and A2. In both cases this proceeds viaV4 (−1.96), a van der Waals complex of the products that can dissociate without a barrier and which can be formed from A1viaA1-V4 (−0.71, black lines in Fig. 3) and from A2viaA2-V4 (−0.72, dark green lines in Fig. 3).
The majority of the adduct pathways for the H2CNH˙+ isomer proceeds viaA2 and its rotamers. The lowest energy rearrangement (dark blue lines in Fig. 4) is the [1,4] cyclisation of A2 to give c-CH(CH3)CH2NHCH2˙+ (−2.03), hereafter referred to as A3, viaA2-3 (−1.20). A3 can then eject one of the H atoms of the CH2 group viaA3-V5 (−0.55) to give V5 (−0.81), a van der Waals complex of c-CH(CH3)CH2NHCH+ (m/z 70) and H˙ that can dissociate into the separated products (−0.81) without a barrier.
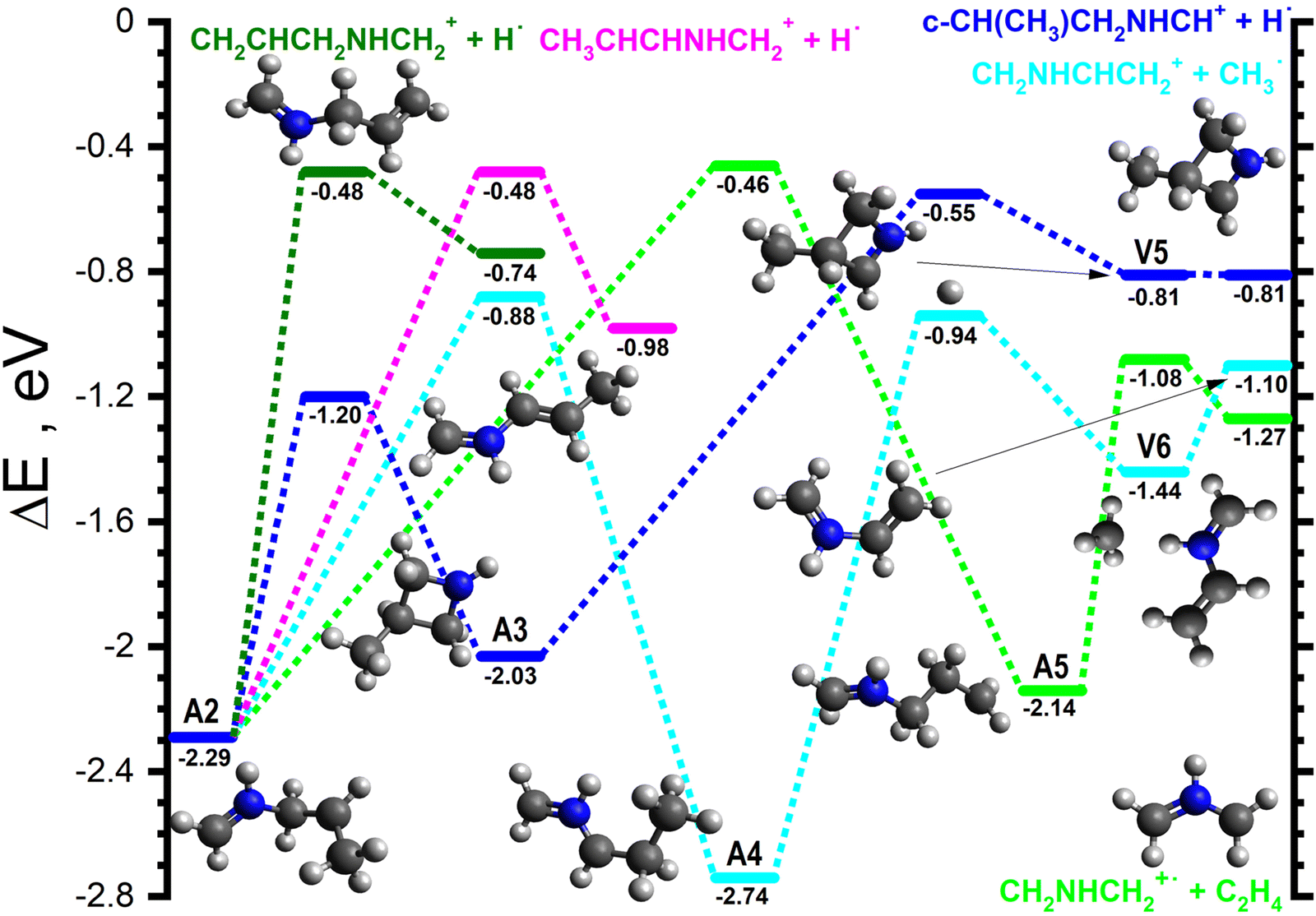 | ||
| Fig. 4 Schematic potential energy surface for the reaction of H2CNH˙+ with CH3CHCH2 from the initial covalently-bound adduct A2. The zero of the energy scale (in eV) is given by the sum of the energies of the separated H2CNH˙+ plus CH3CHCH2 reactants. Calculations are at the CCSD(T)/6-311++G(d,p) level of theory. Molecular structures are shown only for intermediates and products, with TS structures given in the (ESI†). | ||
The next lowest energy rearrangement (cyan lines in Fig. 4) is a [1,2] H-shift from A2 to give CH2NHCHCH2CH3˙+ (−2.74), hereafter referred to as A4viaA2-4 (−0.88). The latter can in turn fragment along the CH2–CH3 bond viaA4-V6 (−0.94) to give V6 (−1.44), a van der Waals complex of CH2NHCHCH2+ (m/z 56) and CH3˙ which can dissociate into the separated products (−1.10) without a barrier. The final rearrangement of note (light green lines in Fig. 4) is an alternative [1,2] H-shift from A2 to give CH2NHCH2CH2CH2˙+ (−2.14), hereafter referred to as A5viaA2-5 (−0.46). A5 can then fragment into CH2NHCH2˙+ (m/z 43) and C2H4 (−1.27) viaA5-P (−1.08).
Additionally, there are two direct fragmentation pathways from A2, the lowest energy of which (pink lines in Fig. 4) involves the ejection of one of the central CH2 hydrogens viaA2-P1 (−0.48) to give CH3CHCHNHCH2+ (m/z 70) plus H˙ (−0.98). The higher energy fragmentation (dark green lines in Fig. 4) involves the ejection of one of the CH3 hydrogens viaA2-P2 (−0.48) to give CH2CHCH2NHCH2+ (m/z 70) plus H˙ (−0.74).
Starting from A1 and its rotamers, the lowest energy pathway (cyan lines in Fig. 5) involves a cyclisation of A1 to give c-CH2NHCH(CH3)CH2˙+ (−2.13), hereafter referred to as A6, viaA1-6 (−1.16). A6 can then fragment along the CH–CH3 bond viaA6-V7 (−1.12) to give V7 (−1.26), a van der Waals complex of c-CH2NHCHCH2+ (m/z 56) and CH3˙, that can dissociate into the separated products (−1.14) without a barrier. The next lowest energy pathway (dark green lines in Fig. 5) proceeds via the fragmentation of the CH–CH3 bond of A1viaA1-V6a (−0.63) to give V6a (−1.20), a van der Waals complex of CH2NHCHCH2+ and CH3˙ (see Fig. 5), that can fragment into the separated products (−1.10) without a barrier. Another fragmentation of note (dark blue lines in Fig. 5) involves the ejection of the CH hydrogen of A1viaA1-P (−0.42) to give CH2C(CH3)NHCH2+ (m/z 70) plus H˙ (−0.90). Finally, A1 can interconvert into A2viaA1-2 (−1.18, pink lines in Fig. 5).
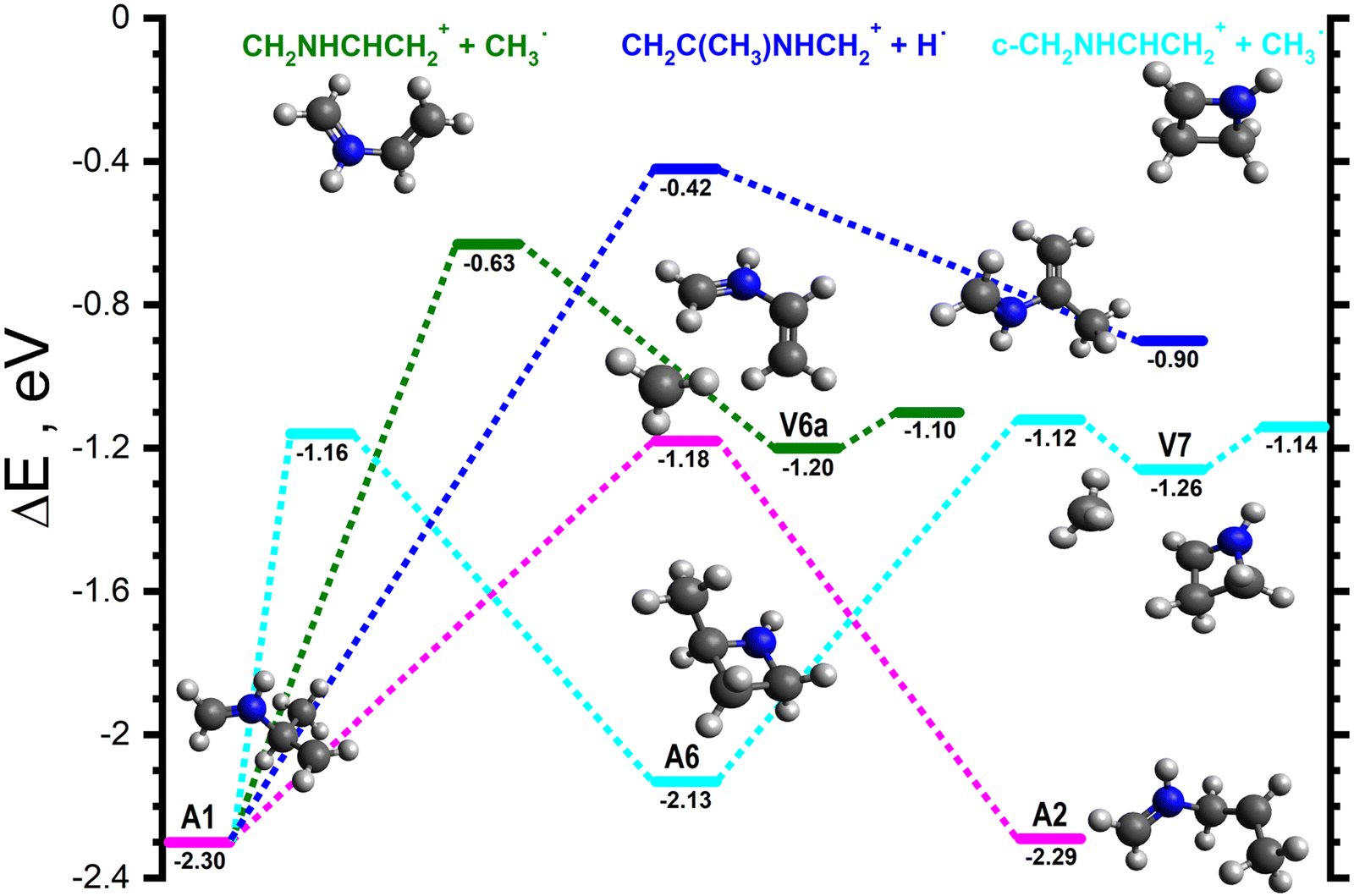 | ||
| Fig. 5 Schematic potential energy surface for the reaction of H2CNH˙+ with CH3CHCH2 from the initial covalently-bound adduct A1. The zero of the energy scale (in eV) is given by the sum of the energies of the separated H2CNH˙+ plus CH3CHCH2 reactants. Calculations are at the CCSD(T)/6-311++G(d,p) level of theory. Molecular structures are shown only for intermediates and products, with TS structures given in the (ESI†). | ||
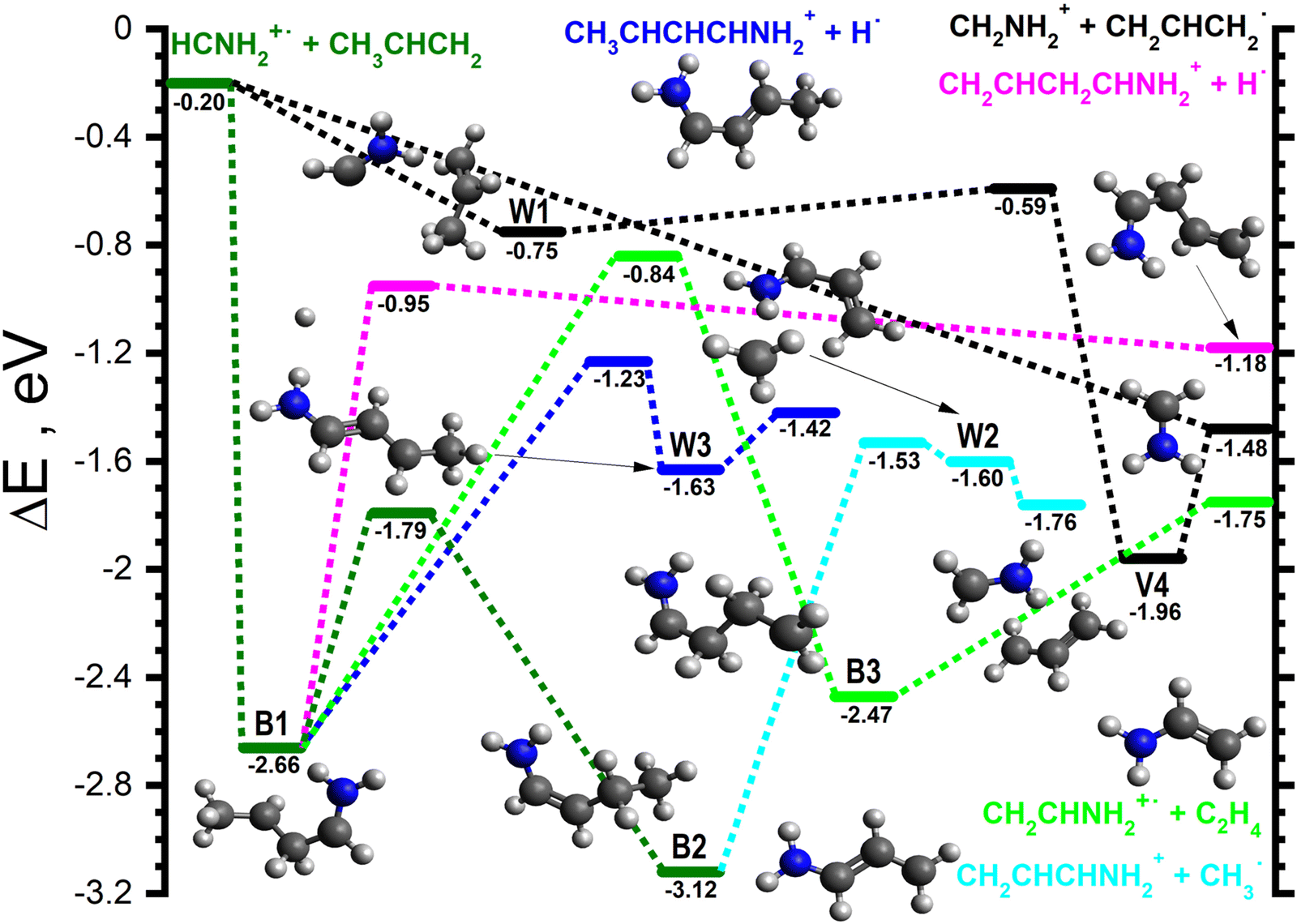 | ||
| Fig. 6 Schematic potential energy surface for the reaction of HCNH2˙+ with CH3CHCH2. To allow for a direct comparison with the PESs shown in Fig. 3–5, the zero of the energy scale (in eV) is given by the sum of the energies of the separated H2CNH˙+ plus CH3CHCH2 reactants. Calculations are at the CCSD(T)/6-311++G(d,p) level of theory. Molecular structures are shown only for intermediates and products, with TS structures given in the (ESI†). | ||
5.2 HCNH2˙+ PES
For the HCNH2˙+ isomer, the majority of the observed reaction pathways proceeds through one or more covalently-bound adduct, with the notable exception of HAT (black lines in Fig. 6). This channel proceeds following the formation of W1 (−0.75), a van der Waals complex of the reactants, which can then proceed to V4 (−1.96), a van der Waals complex of CH2NH2+ (m/z 30) and CH2CHCH2˙ that is common to the PESs of both isomers (see Fig. 3), viaW1-V4 (−0.59). V4 can then fragment into the separated products (−1.48) without a barrier. This pathway is also able to proceed directly at collision energies where complex formation is not feasible.The only covalently-bound adduct that can form from the HCNH2˙+ isomer without a barrier is CH3CHCH2CHNH2˙+ (−2.66, dark green lines in Fig. 6), hereafter referred to as B1. The lowest energy pathway from B1 involves an initial [1,2] H-shift to give CH3CH2CHCHNH2˙+ (−3.12), hereafter referred to as B2, viaB1-2 (−1.79). B2 can then cleave the CH2–CH3 bond viaB2-W2 (−1.53, cyan lines in Fig. 6) to give W2 (−1.60), a van der Waals complex of CH2CHCHNH2+ (m/z 56) and CH3˙, than can dissociate into the separated products (−1.76) without a barrier. The higher energy of W2 with respect to the products is due to the ZPE correction, without which W2 is approximately 0.05 eV below the products.
The next lowest energy pathway stems from a rotamer of B1via the ejection of one of the CH2 hydrogens (see the dark blue lines in Fig. 6) viaB1-W3 (−1.23) to give W3 (−1.63), a van der Waals complex of CH3CHCHCHNH2+ (m/z 70) and H˙ that can dissociate into the separated products (−1.42) without a barrier. There is an alternative pathway, slightly higher in energy (pink lines in Fig. 6) to form a m/z 70 product that involves the direct ejection of one of the CH3 hydrogens of B1viaB1-P (−0.95) to give CH2CHCH2CHNH2+ plus H˙ (−1.18).
As the proton transfer process is endothermic for this isomer, the only pathway leading to a m/z 43 product (light green lines in Fig. 6) involves a [1,2] H-shift from B1 to give CH2CH2CH2CHNH2˙+ (−2.47), hereafter referred to as B3, viaB1-3 (−0.84). B3 can fragment into CH2CHNH2˙+ (m/z 43) plus C2H4 (−1.75) without a barrier. In addition to the pathways presented here, we have identified pathways leading to higher energy product isomers (both ionic and neutral) which are described in the ESI,† since they are not expected to play a significant role in the reactivity of either isomers.
6 Discussion
As described for other systems,15–17 we observe different reactivity for the two [CNH3]˙+ isomers, indicative of a high level of isomer selectivity in their generation methods. Unlike in the Computational Results section, when mentioning barrier heights in the following, the energies are given (in eV) relative to the sum of the energies of CH2CHCH3 and the relevant isomer of [CNH3]˙+.6.1 Reactivity of the H2CNH˙+ isomer
The reactivity of this isomer is dominated by pathways proceeding either directly or via one or more van der Waals complex. At low collision energies, the major product (BR = 0.53) is CH2NH2+ (m/z 30), formed via HAT. This ion can be produced in combination with either CH2CHCH2˙ (see Fig. 3), CH3CCH2˙ or CH3CHCH˙ (both shown in Fig. S4, ESI†), corresponding to reactions (4a)–(4c) in Table 1. Although all of these pathways can proceed without a barrier, formation of CH2CHCH2˙ is expected to dominate due to its greater exothermicity (−1.48 eV compared to −0.59 eV for CH3CCH2˙ and −0.43 eV for CH3CHCH˙), as well as the possibility for this isomer to form directly from the reactants (as shown in Fig. 3). Additional pathways proceeding via multiple covalently-bound adducts can be disregarded due to the presence of comparatively higher energy barriers (see Fig. 3 and Fig. S5, S6, ESI†).The two other main products for this isomer are the m/z 42 channel (BR = 0.28) corresponding to the exothermic charge transfer reaction (5) and the m/z 43 (BR = 0.14) proton transfer channel (7). The former can proceed directly from the reactants, consistent with the collision energy profile observed in Fig. 2, while the latter can proceed either directly or via the van der Waals complexes V1 and V2 (see Fig. 3). Although there is an additional channel leading to the isobaric m/z 43 products CH2NHCH2˙+ and C2H4 (reaction (9b)), see light green lines in (Fig. 4), this pathway involves a comparatively high barrier (−0.46 eV) and multiple rearrangement steps, and so is expected to be minor.
For the minor channel at m/z 56 ([C3H6N]+, BR = 0.04) observed at low collision energies we have identified barrierless pathways leading to the formation of the isomeric ions CH2NHCHCH2+ (reaction (10a), cyan lines in Fig. 4) and c-CH2NHCHCH2+ (reaction (10b), cyan lines in Fig. 5). The lowest energy pathway corresponds to the cyclic isomer, but previous measurements on similar systems16 have shown that such process can be kinetically inhibited by even comparatively small amounts of internal energy. Due to the relatively small signal-to-noise ratio for this channel, we therefore refrain from drawing any conclusions about the yields of the two [C3H6N]+ isomers.
Finally, calculations have identified submerged pathways leading to the formation of various [C4H8N]+ isomers (detected at m/z 70, BR = 0.01) viareactions (11a)–(11d). Once again, given the small CS and the lack of clear energetic preferences based on the PES, no preferential pathway can be suggested.
6.2 Reactivity of the HCNH2˙+ isomer
For this isomer, reactivity is dominated by processes proceeding via one or more covalently-bound adduct. The major product at low collision energies is [C3H6N]+ (m/z 56) formed in combination with CH3˙ (BR = 0.59). The most plausible pathway corresponds to the formation of CH2CHCHNH2+, protonated 2-propen-1-imine, viareaction (10c) (see cyan lines in Fig. 6), but the reader is referred to the ESI† for the description of a further higher energy pathway.The second most significant product at low collision energies is CH2NH2+ (m/z 30), formed via HAT (BR = 0.23). Unlike for the H2CNH˙+ isomer, we have only been able to identify pathways leading to the formation of the CH2CHCH2˙ and CH3CHCH˙ isomers as neutral products, corresponding to reactions (4a) and (4c) respectively. As the formation of CH2CHCH2˙ is the more exothermic pathway and has the lower barrier height, we expect the majority of the reactive flux to correspond to the formation of this isomer. We also note that, as the sole pathway that can proceed directly for this isomer, this is by far the most significant channel at higher collision energies, where covalent adduct formation is not viable.
Minor products are observed at m/z 43 (BR = 0.15) and m/z 70 (BR = 0.03). Due to the endothermicity of the proton transfer process, reaction (8), the former can be assigned to the formation of [C2H5N]˙+ and the latter to the formation of various [C4H8N]+ isomers. For the m/z 43 channel, we have identified only one barrierless pathway leading to the CH2NHCH2˙+ isomer (reaction (9b)). For the m/z 70 channel, although several isomers are possible (CH3CHCHCHNH2+via reaction (11e), CH2CHCH2CHNH2+viareaction (11f) and c-NH2CH(CH3)CHCH+viareaction (11g)) the most probable pathway is reaction (11e) that gives the CH3CHCHCHNH2+ isomer, as it is both the most exothermic (−1.22 eV) and has the smallest barrier height (−1.03 eV).
Considering the relative intensities of the different adduct pathways for this isomer, the dominance of the m/z 56 channel is easily explained by the lower barrier heights and greater overall exothermicity. What is more challenging to explain, however, is the limited contribution from the m/z 70 channel. We tentatively attribute this to the relatively high kinetic strain associated with trans radical H-ejection steps from linear species.
6.3 Similarities and differences in the reactivity of H2CNH˙+ and HCNH2˙+ with propene
The greater contribution from direct pathways (m/z 30, 42 and 43 products, cumulative BR = 0.95) in the case of the H2CNH˙+ isomer compared to HCNH2˙+ (m/z 30, BR = 0.23) is rationalised by the fact that, for the HCNH2˙+ isomer, both the charge and proton transfer channels are endothermic (by 1.35 and 0.61 eV respectively). The endothermicitiy of the charge transfer channel is attributed to the absence of a stable HCNH2 neutral species, while the endothermicity of the proton transfer channel can be explained by a combination of two factors. Firstly, the presence of the NH2 and CH moieties in the reactant ion precludes proton transfer to form the lower energy H2CN˙ isomer of [CH2N]˙. Consequently, proton transfer can only proceed to yield the higher energy HCNH˙ isomer. Secondly, the lower energy of HCNH2˙+ compared to H2CNH˙+ implies that this pathway is also higher in energy than the equivalent pathway from H2CNH˙+.The lower BR for the HAT channel from HCNH2˙+ can be explained by a combination of a smaller exothermicity, due to the lower energy for this isomer (by 0.20 eV) compared to H2CNH˙+, and the orientational effect, as detailed in a previous study using CH4 as neutral partner.15
There are, however, a pair of similarities between the reactivities of the two isomers. Firstly, we note that none of the channels recorded with either isomer exhibit a strong photon energy dependence. This indicates that both reactions proceed with limited dependence on the internal energy of the reactant ions. Secondly, given the lack of symmetry around the C–C double bond of CH3CHCH2, we note the preference for CH3-ejection over H-ejection from reaction intermediates for both isomers. As CH3-ejection is not kinetically favoured by either terminal or central attack, this is instead rationalised by the energetic preference for CH3-ejection, both in terms of reaction enthalpies (0.34 eV preference for formation of CH2CHCHNH2+ compared to CH3CHCHCHNH2+, see Table 1) and barrier heights (0.30 eV difference between maximum barriers B2-W2 and B1-W3, see Fig. 6).
Finally, we compare our results with those measured previously,38 where HCNH2˙+ was generated via electron-impact ionization. By employing D-labelled propenes, the authors concluded that the predominant process involves the loss of the CH3 group of propene. From this observation, they inferred that the primary reaction pathway proceeds from B1 to B2 before fragmenting to yield the CH2CHCHNH2+ isomer, in agreement with the conclusions drawn here on the basis of the calculated PES. Likewise, the same earlier study38 proposed a pathway involving central attack to rationalise the minor loss of CD2H˙ with CD2CHCH3 and CH3˙ with CH2CHCD3, consistent with the higher energy pathways originating from B7, as described in detail in the ESI.† The former observation could also be compatible with a small degree of H/D scrambling via the interconversion between B1 and B3.
7 Conclusions and astrochemical implications
Absolute cross sections and branching ratios for the reaction of HCNH2˙+ and H2CNH˙+ isomers with propene (CH2CHCH3) have been measured as a function of both collision and photon energies, with the latter acting as a proxy for the internal energy of the reagent ion. HCNH2˙+ ions have been generated via dissociative ionization of cyclopropylamine, while H2CNH˙+ ions have been generated by direct photoionization of methanimine (H2CNH). Experimental data have been merged with theoretical calculations to elucidate reaction pathways and structures of products.The H2CNH˙+ isomer is overall more reactive with propene than HCNH2˙+, with collision energy dependent total rate constants at 0.12 ± 0.03 eV equal to 9.9 ± 5.3 × 10−10 and 4.0 ± 2.2 × 10−10 cm3 molecule−1 s−1, respectively. For both isomers we observe a significant HAT channel at this collision energy, which gives protonated methanimine CH2NH2+ in combination with [C3H5]˙ radicals, with BR = 0.53 ± 0.01 for H2CNH˙+ and 0.23 ± 0.02 for HCNH2˙+. CH2NH2+ is a key ion in the synthesis of astrochemical methanimine, since it is predicted to recombine dissociatively with electrons to yield H2CNH plus H,47 and although not (yet) detected in space, its high-resolution rovibrational and rotational spectra have been recently measured in the laboratory.48
The HAT pathway from both isomers has also been observed with other hydrocarbons such as CH415 and C2H4,17 and we underline here its relevance for improving gas phase models to estimate the production of methanimine, a key prebiotic molecule and an important component in the build up of chemical complexity in both the ISM and the hydrocarbon-rich atmospheres of planets and satellites such as Titan.49,50 The fact that both isomers react significantly via HAT to produce CH2NH2+ allows for the recycling of methanimine in environments where the presence of energetic photons/particles leads to its destruction via ionization and isomerization. Collisions with hydrocarbons, yielding CH2NH2+ as the main product regardless of the starting isomer, enable the regeneration of methanimine via dissociative recombination with electrons.
Aside from the production of CH2NH2+, the chemistry of the two isomers is quite different. The other major channels for H2CNH˙+ are charge and proton transfer leading to CH3CHCH2˙+ plus H2CNH (BR = 0.28 ± 0.01) and CH3CHCH3+ plus H2CN˙ (BR = 0.14 ± 0.01), respectively. In both cases, the large cross sections at higher collision energies are consistent with direct pathways.
In contrast, in the case of the HCNH2˙+ isomer, the primary product observed at low collision energy has been identified as protonated vinylimine CH2CHCHNH2+, produced alongside CH3˙ radicals via complex-mediated mechanisms with a BR = 0.59 ± 0.04. Vinylimine (CH2CHCHNH) has been tentatively detected during a spectral survey of the molecular cloud G +0.693 in the central molecular zone of our galaxy51 (please note that in the paper the molecule has been incorrectly named as allylimine) and the reaction of HCNH2˙+ with CH3CHCH2, followed by dissociative recombination with electrons, may well contribute to its synthesis. This is noteworthy as it provides a mechanism for the elongation of the imine hydrocarbon chain, thereby leading to an increase in complexity while preserving chemical functionality. It should be noted that, due to the larger proton affinity of imines (∼868 kJ mol−1 for methanimine and ∼915 kJ mol−1 for vinylimine37) with respect to ammonia (∼853.6 kJ mol−1 ref. 52) the alternative gas phase mechanism for neutralization of complex organic molecules via proton transfer to NH3, recently proposed in53 and applied, for instance, in the modelling of hot cores,54 is not operative in the case of protonated imines.
For both isomers, there are also further minor pathways involving the formation of either C–C or C–N bonds. For the H2CNH˙+ isomer, this is in the form of the m/z 56 and 70 products (BRs ≤0.04 and ≤0.01) while, for the HCNH2˙+ isomer, the m/z 43 and 70 products (BRs = 0.15 ± 0.03 and ≤0.03) correspond to minor bond-forming process. As the measured cross sections show negative collision energy dependences, indicative of barrierless and exothermic processes, these processes are therefore feasible at the low temperatures relevant to both the ISM and the ionospheres of planets and their satellites.
Data availibility
Details on the geometries for the calculated structures described in the paper are given in the ESI,† with experimental data available at 10.5281/zenodo.12653872.Conflicts of interest
No conflict of interests to be declared.Acknowledgements
We are grateful to the DESIRS beamline team for their assistance during the synchrotron measurements and to the technical staff of SOLEIL for the smooth running of the facility under projects no. 20180118 and 20190249. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska Curie grant agreement no 811312 for the project “Astro-Chemical Origins” (ACO), from MUR PRIN 2020 project no. 2020AFB3FX “Astrochemistry beyond the second period elements”. D. A. also acknowledges financial support from the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender no. 1409 published on 14.9.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU – Project Title P20223H8CK “Degradation of space-technology polymers by thermospheric oxygen atoms and ions: an exploration of the reaction mechanisms at an atomistic level” – CUP E53D23015560001. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or European Commission. Neither the European Union nor the granting authority can be held responsible for them. V. R. acknowledges funding for a PhD fellowship from the Dept. Physics, University of Trento. WDG and DS acknowledge support from the Swedish research council (grant no. 2019 04332). J.-C. G. thanks the Programme National “Physique et Chimie du Milieu Interstellaire” (PCMI) of CNRS/INSU with INC/INP co-funded by CEA and CNES. MP and JZ acknowledge support from the Ministry of Education, Youth and Sports of the Czech Republic (grant no. LTC20062). This article is based upon work from COST Action CA18212 – Molecular Dynamics in the GAS phase (MD-GAS), supported by COST (European Cooperation in Science and Technology).Notes and references
- N. Marcelino, J. Cernicharo, M. Agúndez, E. Roueff, M. Gerin, J. Martín-Pitado, R. Mauersberger and C. Thum, Astrophys. J., 2007, 665, L127 CrossRef CAS.
- M. Agúndez, J. Cernicharo and M. Guélin, Astron. Astrophys., 2015, 577, L5 CrossRef PubMed.
- S. Manigand, A. Coutens, J.-C. Loison, V. Wakelam, H. Calcutt, H. S. P. Müller, J. K. Jørgensen, V. Taquet, S. F. Wampfler, T. L. Bourke, B. M. Kulterer, E. F. van Dishoeck, M. N. Drozdovskaya and N. F. W. Ligterink, Astron. Astrophys., 2021, 645, A53 CrossRef CAS.
- C. Cabezas, M. Agúndez, Y.-T. Liu, Y. Endo and J. Cernicharo, Mon. Not. R. Astron. Soc., 2023, 524, 491–497 CrossRef CAS.
- C. Nixon, D. Jennings, B. Bézard, S. Vinatier, N. Teanby, K. Sung, T. Ansty, P. Irwin, N. Gorius and V. Cottini, Astrophys. J. Lett., 2013, 776, L14 CrossRef.
- C. Li, X. Zhang, P. Gao and Y. Yung, Astrophys. J. Lett., 2015, 803, L19 CrossRef.
- N. A. Lombardo, C. A. Nixon, T. K. Greathouse, B. Bézard, A. Jolly, S. Vinatier, N. A. Teanby, M. J. Richter, P. J. Irwin and A. Coustenis, Astrophys. J. Lett., 2019, 881, L33 CrossRef CAS.
- E. Herbst, E. Roueff and D. Talbi, Mol. Phys., 2010, 108, 2171–2177 CrossRef CAS.
- Z. Lin, D. Talbi, E. Roueff, E. Herbst, N. Wehres, C. A. Cole, Z. Yang, T. P. Snow and V. M. Bierbaum, Astrophys. J., 2013, 765, 80 CrossRef.
- J. M. C. Rawling, D. A. Williams, S. Viti and C. Cecchi-Pestellini, Mon. Not. R. Astron. Soc.: Lett., 2013, 436, L59–L63 CrossRef.
- K. M. Hickson, V. Wakelam and J.-C. Loison, Mol. Astrophys., 2016, 3–4, 1–9 Search PubMed.
- M. J. McEwan, G. B. Scott and V. G. Anicich, Int. J. Mass Spectrom. Ion Processes, 1998, 172, 209–219 CrossRef CAS.
- J.-C. Loison and A. Bergeat, Phys. Chem. Chem. Phys., 2009, 11, 655–664 RSC.
- P. D. Kelly, C. C. Bright, S. J. Blanksby, G. da Silva and A. J. Trevitt, J. Phys. Chem. A, 2019, 123, 8881–8892 CrossRef CAS PubMed.
- V. Richardson, C. Alcaraz, W. D. Geppert, M. Polášek, C. Romanzin, D. Sundelin, R. Thissen, P. Tosi, J. Žabka and D. Ascenzi, Chem. Phys. Lett., 2021, 775, 138611 CrossRef CAS.
- V. Richardson, D. Ascenzi, D. Sundelin, C. Alcaraz, C. Romanzin, R. Thissen, J.-C. Guillemin, M. Polášek, P. Tosi, J. Žabka and W. D. Geppert, Front. Astron. Space Sci., 2021, 8, 158 Search PubMed.
- D. Sundelin, D. Ascenzi, V. Richardson, C. Alcaraz, M. Polášek, C. Romanzin, R. Thissen, P. Tosi, J. Žabka and W. D. Geppert, Chem. Phys. Lett., 2021, 777, 138677 CrossRef CAS.
- V. Vuitton, R. Yelle, S. Klippenstein, S. Hörst and P. Lavvas, Icarus, 2019, 324, 120–197 CrossRef CAS.
- P. D. Godfrey, R. D. Brown, B. J. Robinson and M. W. Sinclair, Astrophys. J. Lett., 1973, 13, 119 CAS.
- J. E. Dickens, W. M. Irvine, C. H. DeVries and M. Ohishi, Astrophys. J., 1997, 479, 307–312 CrossRef CAS PubMed.
- B. E. Turner, R. Terzieva and E. Herbst, Astrophys. J., 1999, 518, 699 CrossRef CAS.
- H. Bock and R. Dammel, Chem. Ber., 1987, 120, 1961–1970 CrossRef CAS.
- C. Alcaraz, C. Nicolas, R. Thissen, J. Žabka and O. Dutuit, J. Phys. Chem. A, 2004, 108, 9998–10009 CrossRef CAS.
- B. Cunha de Miranda, C. Romanzin, S. Chefdeville, V. Vuitton, J. Žabka, M. Polášek and C. Alcaraz, J. Phys. Chem. A, 2015, 119, 6082–6098 CrossRef CAS PubMed.
- L. Nahon, N. de Oliveira, G. Garcia, J.-F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, J. Synchrotron Radiat., 2012, 19, 508–520 CrossRef CAS PubMed.
- P. C. Burgers, J. L. Holmes and J. K. Terlouw, J. Am. Chem. Soc., 1984, 106, 2762–2764 CrossRef CAS.
- B. Mercier, M. Compin, C. Prevost, G. Bellec, R. Thissen, O. Dutuit and L. Nahon, J. Vac. Sci. Technol., 2000, 18, 2533–2541 CrossRef CAS.
- L. Minnhagen, J. Opt. Soc. Am., 1973, 63, 1185–1198 CrossRef CAS.
- A. Kramida, Y. Ralchenko, J. Reader and N. I. S. T. A. S. D. Team, NIST Atomic Spectra Database (version 5.8), 2023 Search PubMed.
- E. Teloy and D. Gerlich, Chem. Phys., 1974, 4, 417–427 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian∼16 Revision D.01, Gaussian Inc., Wallingford CT, 2019 Search PubMed.
- B. Ruscic and D. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.122p of the Thermochemical Network, 2020 Search PubMed.
- I. Corral, O. Mó and M. Yánez, Int. J. Quantum Chem., 2003, 91, 438–445 CrossRef CAS.
- G. Bouchoux, F. Penaud-Burruyer and M. T. Nguyen, J. Am. Chem. Soc., 1993, 115, 9728–9733 CrossRef CAS.
- G.-X. Liu, Y.-H. Ding, Z.-S. Li, X.-R. Huang and C.-C. Sun, J. Mol. Struct., 2001, 548, 191–218 CrossRef CAS.
- J. Holmes, C. Aubry and P. Mayer, Assigning Structures to Ions in Mass Spectrometry, CRC Press, Taylor and Francis Group, Boca Raton, FL (USA), 2006 Search PubMed.
- G. Bouchoux and M. Eckert-Maksic, Mass Spectrom. Rev., 2018, 37, 139–170 CrossRef CAS PubMed.
- J. Chamot-Rooke, P. Mourgues, G. van der Rest and H. E. Audier, Int. J. Mass Spectrom., 2003, 226, 249–269 CrossRef CAS.
- K. M. Ervin and P. Armentrout, J. Chem. Phys., 1985, 83, 166–189 CrossRef CAS.
- C. Nicolas, C. Alcaraz, R. Thissen, J. Žabka and O. Dutuit, Planet. Space Sci., 2002, 50, 877–887 CrossRef CAS.
- G. Gioumousis and D. Stevenson, J. Chem. Phys., 1958, 29, 294–299 CrossRef CAS.
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183–5185 CrossRef CAS.
- V. Wakelam, I. Smith, E. Herbst, J. Troe, W. Geppert, H. Linnartz, K. Öberg, E. Roueff, M. Agündez, P. Pernot, H. Cuppen, J.-C. Loison and D. Talbi, Space Sci. Rev., 2010, 156, 13–72 CrossRef CAS.
- A. Tsikritea, J. A. Diprose, T. P. Softley and B. R. Heazlewood, J. Chem. Phys., 2022, 157, 060901 CrossRef CAS PubMed.
- NIST Computational Chemistry Comparison and Benchmark Database, 2020, https://cccbdb.nist.gov/.
- M. Nguyen, J. Rademakers and J. Martin, Chem. Phys. Lett., 1994, 221, 149–155 CrossRef CAS.
- C. H. Yuen, M. A. Ayouz, N. Balucani, C. Ceccarelli, I. F. Schneider and V. Kokoouline, Mon. Not. R. Astron. Soc., 2019, 484, 659–664 CrossRef CAS.
- C. R. Markus, S. Thorwirth, O. Asvany and S. Schlemmer, Phys. Chem. Chem. Phys., 2019, 21, 26406–26412 RSC.
- H. Luthra, V. Wakelam, M. K. Sharma and S. Chandra, Mon. Not. R. Astron. Soc., 2023, 521, 2181–2186 CrossRef CAS.
- Z. Yang, I. A. Medvedkov, S. J. Goettl and R. I. Kaiser, J. Phys. Chem. Lett., 2023, 14, 8500–8506 CrossRef CAS PubMed.
- D. Alberton, L. Bizzocchi, N. Jiang, M. Melosso, V. M. Rivilla, A. P. Charmet, B. M. Giuliano, P. Caselli, C. Puzzarini, S. Alessandrini, L. Dore, I. Jimenez-Serra and J. Martin-Pintado, Astron. Astrophys., 2023, 669, A93 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook – Standard Reference Database n. 69, 2023, https://webbook.nist.gov Search PubMed.
- V. Taquet, E. Wirström and S. Charnley, Astrophys. J., 2016, 821, 46 CrossRef.
- M. Jin and R. T. Garrod, Astrophys. J., Suppl. Ser., 2020, 249, 26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02002b |
| This journal is © the Owner Societies 2024 |