Intrinsic conformational preference in the monomeric protein governs amyloid polymorphism†
Received
13th May 2024
, Accepted 15th September 2024
First published on 16th September 2024
Abstract
The inherent stochasticity associated with the hierarchical self-assembly of either native-like or partially-unfolded protein monomers leads to the formation of transient, morphologically-diverse prefibrillar species resulting in structurally-distinct polymorphic protein aggregates. High-resolution structural characterization of mature aggregates has revealed heterogeneous supramolecular packing of protofibrils within amyloid polymorphs. However, little is known about whether initial monomeric protein conformers engender polymorphism at the onset of aggregation. Here, we show that intrinsic conformational preference in aggregation-competent monomeric ovalbumin, an archetypal serpin, dictates fibrillar polymorphism by modulating aggregation pathways. Using fluorescence, FT-IR, and vibrational Raman spectroscopy coupled with dynamic light scattering and electron microscopy, we demonstrate that conformationally-diverse amyloidogenic monomers, formed via an interplay of electrostatic and hydrophobic interactions before the commencement of aggregation, play a crucial role in promoting amyloid polymorphism. Moreover, the monomeric conformational fingerprints, accrued at the onset of aggregation, persist and propagate during the formation of polymorphic amyloids. Our results delineate essential conformational characteristics of the monomeric protein preceding aggregation, which will have broad implications in the mechanistic understanding of amyloid strain diversity observed in disease-related proteins.
1. Introduction
Protein conformational disorders, resulting in the formation of ordered fibrillar amyloids and disordered amorphous aggregates, have been implicated not only in various neurodegenerative diseases but also pose as major bottlenecks in food processing industries as well as in biotechnology-based therapeutic formulations.1–8 Conformational conversion of a monomeric, single polypeptide sequence into polymeric, cross β-sheet-rich amyloid fibrils is mediated via a cascade of protein self-assembly events facilitated by a complex network of non-covalent interactions. Several studies have reported that protein aggregation is a generic phenomenon and every protein can access the ordered, amyloid state irrespective of its native structure and/or the conformation of the aggregation-competent soluble precursors.1,4,6–10 Alterations in solution conditions such as pH, ionic strength, buffer composition, temperature, etc. modulate the fibrillation kinetics resulting in structurally-distinct amyloid polymorphs.11 A recent surge in the elucidation of polymorphic fibrillar architectures at atomic resolution using cryo-electron microscopy,12–15 solid state NMR,16–18 electron paramagnetic resonance,19,20etc. has illuminated that heterogeneity in the numbers of protofibrils coupled with the diversity in their supramolecular packing and varying side chain interactions within the amyloid core leads to amyloid polymorphism comprising fibrils with dissimilar helical pitch and diameter.21 Owing to the inherent complexities and challenges associated with obtaining high-resolution atomic structures, it has been recently demonstrated that hydrogen–deuterium exchange vibrational Raman spectroscopy can illuminate the amyloid structural diversities at the molecular level.22 However, it still remains elusive whether the inherent conformational preference of a protein monomer at a given solution condition, at the onset of aggregation, governs the structural organization within the resulting protofilaments ensuing genesis of the polymorphs. In this work, we have used a model protein, namely ovalbumin, which belongs to the serpin superfamily wherein both α-antitrypsin and neuroserpin have been reported to be associated with Alzheimer-like dementia, hepatocellular carcinoma, and various serpinopathies.23–25 Several reports have demonstrated that ovalbumin forms both amyloid and non-amyloid aggregates with heterogeneous morphologies in vitro depending on the solution conditions.26–28 However, it is not clear yet whether the conformational fingerprint of the monomeric polypeptide chain, attained prior to aggregation as a consequence of solution characteristics, gets recapitulated during the course of aggregation leading to polymorphism. Here, we demonstrate that biophysical tools comprising fluorescence, FT-IR, and vibrational Raman spectroscopy, in conjunction with dynamic light scattering and electron microscopy, allow us to discern conformational characteristics of the monomeric protein and the protein aggregates suggesting that the intrinsic conformational preference of a single-sequence, polypeptide chain governs the altered supramolecular architecture within various polymorphs.
2. Results and discussion
2.1 Bioinformatics analyses of ovalbumin aggregation propensity under different solution conditions
As a prelude, we set out to investigate the aggregation propensity of ovalbumin (Fig. 1a) using well-known bioinformatics predictors, namely AGGRESCAN29 and TANGO.30 Both the analyses (Fig. S1a–c, ESI†) indicated that ovalbumin self-assembly at low pH is facilitated by salt-induced electrostatic screening of positively-charged polypeptides resulting in hydrophobic association between pH-induced molten-globule conformers. Therefore, we conjectured that self-assembly of ovalbumin will be more pronounced at high salt concentrations compared to that at low salt concentrations and hence, a direct correlation between protein aggregation propensity and ionic strength can be drawn experimentally to gain mechanistic insights into the role of electrostatics in governing protein self-assembly.
 |
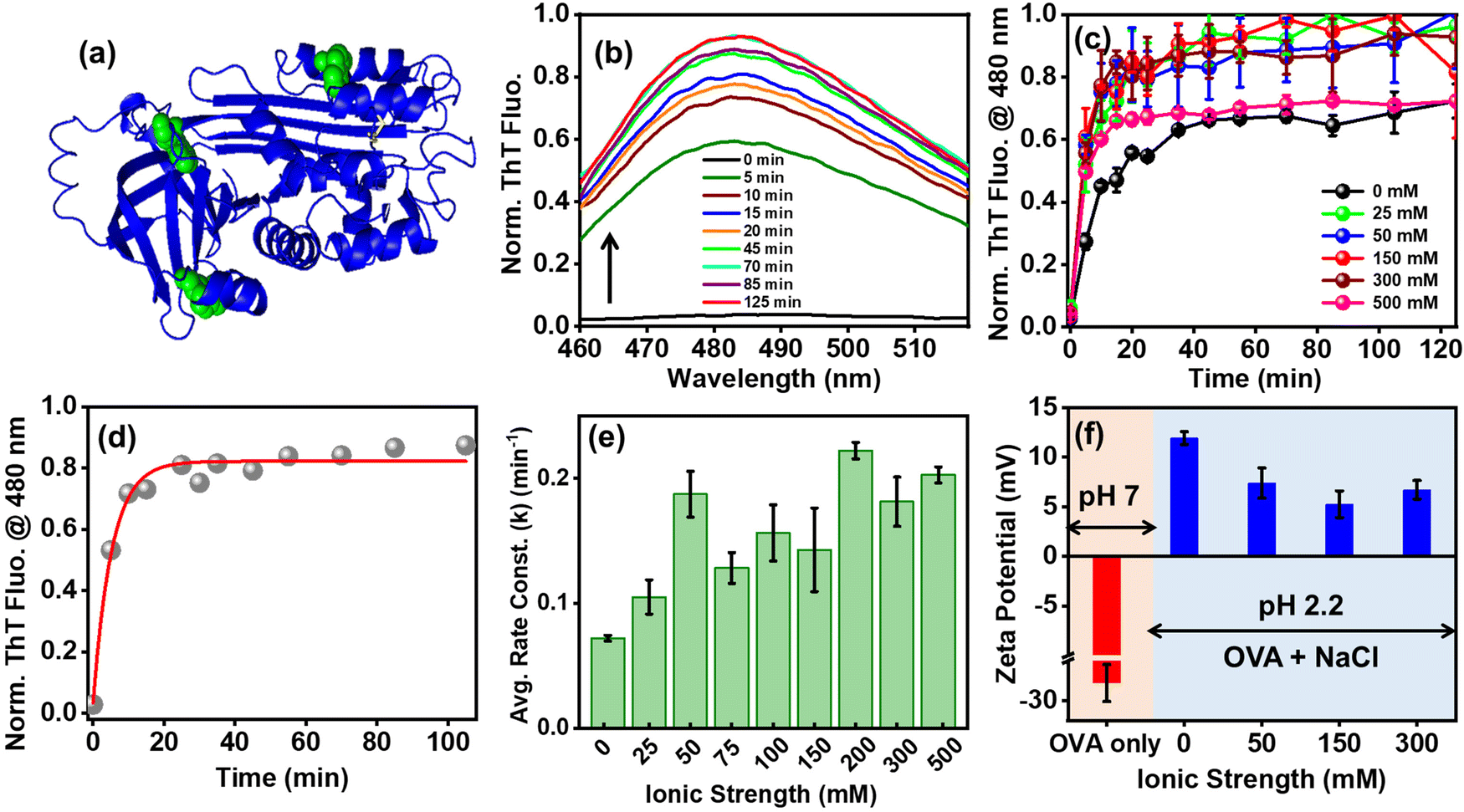
| | Fig. 1 (a) Crystal structure of ovalbumin (PDB ID: 1OVA) generated using PyMOL (Schrodinger Inc., LLC, New York). The protein chain and the tryptophans are shown in blue and green, respectively. (b) Representative time-dependent changes in ThT fluorescence during ovalbumin amyloid aggregation in the presence of NaCl at pH 2.2, 65 °C. The black upward arrow shows an enhancement in ThT emission as a function of time. (c) and (d) Normalized ThT fluorescence kinetics monitored at 480 nm (c) as a function of salt concentration and (d) a representative plot showing fitting of the ThT kinetics data. The gray spheres and the red line denote actual data and mono-exponential fit, respectively. (e) Average rate constants of ovalbumin aggregation as a function of ionic strength. The error bars represent mean ± s.e.m. (n ≥ 6). (f) Zeta (ζ) potential measurements of ovalbumin under native conditions (pH 7) and molten-globule state (pH 2.2) in the absence and in the presence of variable NaCl concentrations at room temperature. The error bars represent mean ± s.e.m. (n ≥ 3). | |
2.2 Kinetics of amyloid assembly as a function of salt concentration
To test our hypothesis, we monitored ovalbumin aggregation kinetics as a function of increasing salt concentration using multiple structural probes. Previous literature reports demonstrated that at low pH (i.e. at pH < pI) and upon heating, ovalbumin spontaneously forms cross β sheet-rich, nanoscopic ordered amyloid aggregates via a nucleation-independent pathway.31,32 To confirm whether variation in the solution ionic strength results in the formation of amyloids, we employed thioflavin-T (ThT), a well-known amyloid marker, which shows an enhancement in its fluorescence intensity at 480 nm.33,34 As expected, we observed a negligible ThT fluorescence at 480 nm at room temperature. Upon incubating the protein at 65 °C, we observed a sharp increase in the ThT fluorescence, devoid of any lag phase, across a range of ionic strengths suggestive of the formation of amyloid aggregates irrespective of the salt concentration (Fig. 1b and c). A closer inspection of the salt-dependent ThT fluorescence intensities obtained during the apparent saturation phase indicated variations that did not show any direct correlation with the increasing ionic strength. Such variations in ThT intensities have been reportedly observed during the course of aggregation of insulin, glucagon, and β2-microglobulin.35 Furthermore, fitting of the ovalbumin amyloid aggregation kinetics, monitored by ThT fluorescence, followed by comparison between the average rate constants revealed modulation in the rates and extent of amyloid aggregation at different salt concentrations (Fig. 1d and e and Table S1, ESI†), which is in contrast to our hypothesis.
The extracted kinetic parameters could be classified into three salt regimes viz. low (0–50 mM), medium (75–150 mM), and high salt (200–500 mM) regions. In the absence of salt, the rate of amyloid formation was the most sluggish (∼70 × 10−3 min−1, Table S1, ESI†) compared to that in the presence of salt. The slow aggregation kinetics in the absence of NaCl is expected and can be ascribed to electrostatic repulsions between the positively-charged polypeptides, which eventually reduces the aggregation propensity. In contrast, the addition of NaCl leads to an increase in the aggregation rates due to electrostatic screening of the charged polypeptides. Therefore, the larger the extent of electrostatic screening due to higher ionic strength, the faster the rate of self-assembly is expected to be. However, our salt concentration-dependent protein aggregation kinetics did not appear to follow this direct monotonic correlation. It should be noted that intermolecular protein interactions in the presence of salt encompass several effects, such as (a) long-range electrostatic interactions due to the net charge of the proteins and surrounding counter-ions, (b) short-range electrostatics whereby charged, well-localized side-chains interact with the bound Na+, and (c) entropic effects accompanied by hydrophobic interactions.36 In the low salt regime, we observed a two-fold increase in the ovalbumin aggregation kinetics until 50 mM of NaCl (from ∼100 × 10−3 min−1 to ∼200 × 10−3 min−1, Fig. 1e and Table S1, ESI†) which dampened in the medium range of ionic strength (∼140 × 10−3 min−1, Fig. 1e and Table S1, ESI†) depicting a significant dip at 75 mM of NaCl followed by a rise again in the high salt regime (∼200 × 10−3 min−1, Fig. 1e and Table S1, ESI†) with a notable enhancement at 200 mM of NaCl. This implied that at low and high salt concentrations, electrostatic screening of the overall positively-charged, molten globule-like polypeptide chains occurs, which governs the ovalbumin amyloid aggregation primarily mediated by hydrophobic interactions. On the other hand, at moderate salt regimes (i.e. at 75–150 mM of NaCl), the electrostatic screening and hydrophobic effect presumably balance each other, which decreases the overall aggregation rate of ovalbumin compared to that observed at low and high salt regimes. This is also in accordance with simple Debye–Hückel screening, which accounts for biomolecular associations at low ionic strength but fails at biologically-relevant salt concentrations wherein the biomolecular charges are effectively electrostatically balanced.37,38 Therefore, Derjaguin–Landau–Verwey–Overbeek (DLVO) theory comes into play, which accounts for both short-range van der Waals attractions coupled with long-range electrostatic repulsions.39–41
2.3 Surface charge screening of protein in the presence of salt
We also performed zeta potential measurements (Fig. 1f) to gain insights into the extent of NaCl-induced electrostatic screening of the polypeptide chains. Our results indicated that the extent of charge screening was similar for both low (50 mM) and high (300 mM) NaCl concentrations, whereas a moderate (150 mM) ionic strength induced a substantial drop in the overall positive charge of ovalbumin. Hence, we proposed that overall charge compensation at moderate NaCl concentrations may counterbalance the hydrophobic effect exhibited by the partially-expanded, molten globule-like conformers of ovalbumin that affects the fibril elongation kinetics and hence, retards the overall aggregation rates, which is also in accordance with our ThT fluorescence data.
2.4 Changes in hydrophobicity during amyloid assembly
Next, we employed a hydrophobic marker, namely 8-anilinonaphthalene-1-sulfonic acid ammonium salt (ANS), to probe the emergence of hydrophobicity during the course of ovalbumin amyloid assembly. Aqueous ANS exhibits a weak fluorescence at ∼505–510 nm but fluoresces strongly with a concomitant blue-shift to ∼475 nm indicating binding of ANS to hydrophobic regions.42 Prior to heating (∼25 °C), we observed ANS emission at ∼480 nm for all the protein samples incubated at varying ionic strength suggestive of ANS being bound to the hydrophobic pockets of ovalbumin (Fig. 2a and Fig. S2a and b, ESI†). Upon raising the temperature to 65 °C for initiating aggregation, we observed a remarkable enhancement in the ANS fluorescence intensity within 10 minutes of heating, which remained almost constant even upon reaching an apparent saturation (Fig. 2b and Fig. S2c and d, ESI†). A careful look at the ANS fluorescence kinetics as a function of NaCl concentration (Fig. S3a and Table S1, ESI†) revealed that the rate of increase in ANS emission was slower in the absence of NaCl compared to that observed in the presence of salt (Fig. 2b inset). Furthermore, a comparison between the average rate constants extracted at different NaCl concentrations depicted that the rate of emergence of hydrophobicity remained almost similar across several NaCl concentrations, albeit significantly slower (ranging between ∼13 to ∼40-fold) compared to that observed at 150 and 300 mM (Fig. 2c and d and Table S1, ESI†). Such large discrepancy could be attributed to the predominance of entropic effects, i.e. hydrophobic interactions operative during ovalbumin aggregation in the presence of 150 and 300 mM of NaCl. Moreover, a comparison between the salt-mediated ovalbumin aggregation kinetics monitored by ThT and ANS (Table S1, ESI†) suggested that the temporal evolution of cross β-sheet-rich fibrils was overall slower compared to the hydrophobicity-mediated polypeptide association kinetics (Fig. 1e and 2c, d) that were extremely prominent in the presence of 150 and 300 mM of salt. Therefore, we proposed that in addition to conformational conversion-mediated amyloid fibrillation of ovalbumin, genesis of hydrophobically-collapsed, unstructured oligomeric species with little or no β-sheet content may concurrently drive ovalbumin aggregation at 150 and 300 mM of salt.
 |
| | Fig. 2 (a) Representative time-dependent changes in ANS fluorescence emission during ovalbumin amyloid aggregation in the presence of NaCl at pH 2.2, 65 °C. The black upward arrow shows an enhancement in ANS emission intensity as a function of time. (b) A representative normalized ANS fluorescence kinetics, monitored at 475 nm, whereby the gray spheres and the red line denote actual data and mono-exponential fit, respectively. The inset shows the initial sharp rise in ANS fluorescence upon initiating ovalbumin aggregation in the absence of salt and in the presence of varying ionic strength. (c) and (d) Average rate constants, extracted from ANS fluorescence kinetics, during ovalbumin aggregation as a function of ionic strength and (d) its magnified depiction at a few selected salt concentrations. The error bars represent mean ± s.e.m. (n ≥ 6). | |
2.5 Changes in the protein hydrodynamic sizes during amyloid aggregation
Next, we directed our efforts to estimate the overall sizes of ovalbumin monomer, plausible oligomers, and aggregates as a function of ionic strength using the dynamic light scattering (DLS) technique. At room temperature, we observed that in the absence of NaCl, the hydrodynamic diameter of ovalbumin is ∼5 nm, thus corroborating earlier reports.43 Upon addition of variable concentrations of NaCl into ovalbumin solution at room temperature (∼25 °C), the hydrodynamic diameter of the protein remained almost constant irrespective of the solution ionic strength until 300 mM (Fig. 3a and Fig. S3b, ESI†). However, the width of the hydrodynamic diameter profile broadened slightly with an increase in NaCl concentration denoting heterogeneity in the protein sample plausibly due to the presence of a small proportion of soluble protein oligomers. Moreover, at 500 mM NaCl, we observed a substantial population of large-sized intermediates (hydrodynamic diameter ∼60–100 nm) formed at room temperature (Fig. 3a and Fig. S3b, ESI†). Upon heating at 65 °C and in the absence of salt, we observed a gradual transition of ovalbumin monomers into oligomers (especially dimers) with size ranging between ∼6–9 nm. Thereafter, the hydrodynamic size of the oligomers attained a plateau within our experimental timescale (Fig. 3b). In the presence of 50 mM NaCl, as the temperature was increased, we observed the formation of dimers (∼10 nm) within 10 minutes of incubation that formed large aggregates (∼18–20 nm) upon reaching the saturation phase (Fig. 3c). These results are in accordance with a previous literature report.31 At medium ionic strength, i.e. at 150 mM NaCl, we observed the formation of large oligomers (∼45 nm) along with larger aggregates (>500 nm) within 10 minutes of heating. Thereafter, we observed an increase in the formation of even larger, polydisperse aggregates (≥1 μm) at the expense of the large oligomers as aggregation progressed (Fig. S3c, ESI†). At high salt concentration, i.e. at 300 mM NaCl, we observed a rapid formation of heterogeneous, large-sized aggregates (≥1 μm) within 10 minutes of heating that exhibited a time-dependent increment in size (Fig. 3d). Taken together, our DLS data indicated that the overall size of the aggregates increases with an increase in NaCl concentration although both of our ThT and ANS fluorescence data indicate that the aggregation rates are faster at low and high ionic strength compared to that observed in the medium salt regime. Additionally, based on the DLS peak profiles, the ovalbumin aggregates at 50 mM NaCl appeared to exhibit lower polydispersity implying lesser heterogeneity within the aggregates compared to that observed under medium and high salt regimes. These observations further led us to probe into the morphological characteristics of the nanoscopic, ordered amyloid aggregates.
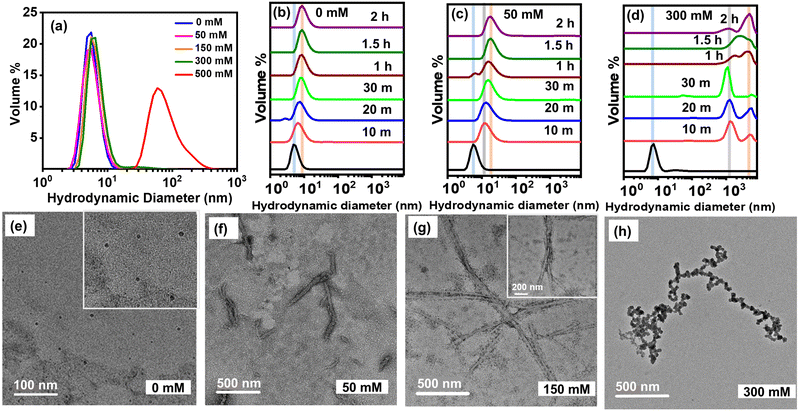 |
| | Fig. 3 (a) Hydrodynamic diameter of ovalbumin in the absence and in the presence of variable concentrations of NaCl at room temperature prior to triggering aggregation. (b)–(d) Time-dependent changes in hydrodynamic diameter of ovalbumin oligomers and aggregates, formed at pH 2.2, 65 °C, in the absence (0 mM) and in the presence of NaCl (50 and 300 mM). (e)–(h) Transmission electron microscopy (TEM) images of ovalbumin polymorphic aggregates formed after 2 hours of incubation at pH 2.2, 65 °C (e) in the absence of NaCl (0 mM), the inset shows a magnified view of the spherical oligomers, and (f)–(h) in the presence of variable ionic strength. The inset in (g) shows long intertwined amyloid fibrils formed in the presence of 150 mM of NaCl. | |
2.6 Nanoscale morphological characterization of aggregates
Next, we characterized the morphologies of ovalbumin amyloid aggregates (Fig. 3e–h and Fig. S3d–f, ESI†) using transmission electron microscopy (TEM). At 0 mM NaCl, we observed the presence of spherical, morphologically-unstructured oligomers (Fig. 3e and inset) that we refer to as type A polymorphs in this study, whereas at 50 mM NaCl, we observed short, fibrillar aggregates (denoted as type B polymorphs) (Fig. 3f and Fig. S3d, ESI†). Upon increasing the NaCl concentration to 150 mM, we observed long, branched as well as intertwined fibrils whose surfaces appeared to be sparsely adorned with spherical, unstructured aggregates (type C polymorph) (Fig. 3g and Fig. S3e, ESI†). Interestingly at 300 mM NaCl, the aggregates appeared to be tubular; however, a closer inspection suggested the predominance of amorphous aggregates, which seemingly appended onto the fibrillar species giving a beads-on-a-string appearance (type D polymorph) (Fig. 3h and Fig. S3f, ESI†). The formation of amorphous aggregates at higher ionic strength has been suggested during the amyloid aggregation of insulin, β2-microglobulin, etc.44 All of our aforementioned results indicate that while higher ionic strength of the solution facilitates ovalbumin self-assembly, microscopic images reveal the presence of amorphous aggregates in addition to fibrillar species. This may be attributed to two plausible reasons (a) higher ionic strength stabilizes the low pH-induced, partially-expanded monomeric state to a greater extent due to enhanced electrostatic screening, which augments both intra- and intermolecular interactions and/or (b) higher salt concentrations probably promote the formation of amorphous aggregates of ovalbumin, rich in β-content, concurrently with ordered, amyloid fibrils as observed for SH3 and β2-microglobulin aggregation at low pH.45,46
2.7 Hydrolysis of protein during aggregation
It has been proposed that heating at low pH may lead to hydrolysis of the polypeptide chains into shorter peptidyl fragments and the subsequent fibrillar morphologies are influenced by the hydrolysis kinetics that may induce fragmentation of fibrils.26 However, the mechanism of protein hydrolysis remains unknown. We analyzed our aggregates (pH 2.2, 65 °C, in the absence and presence of NaCl) on SDS-PAGE in a time-dependent manner and the results indeed depicted hydrolysis of ovalbumin during aggregation. However, we did not observe any hydrolysis of the native protein while performing the aggregation assay at pH 7, 65 °C, in the absence and presence of NaCl (Fig. S4a, ESI†). Additionally, we investigated the amyloid aggregation tendency of native protein at pH 7, 65 °C in the presence of salt using ThT fluorescence for 2 hours. The changes in ThT emission were measurable but insignificant compared to that observed for low pH-induced aggregates indicating low or negligible aggregation propensity of ovalbumin at pH 7 (Fig. S4b, ESI†). Such findings precluded us from conducting any further experiments at pH 7 with other structural probes used in this study. Therefore, in order to further discern the heterogeneity within the secondary structural contents across these diverse polymorphs and to validate whether there is a direct correlation between the extent of secondary structural elements and the observed polymorphism, we took advantage of vibrational spectroscopic techniques that are extremely sensitive towards the molecular architecture.32,47–49
2.8 Structural insights into protein aggregation using Raman spectroscopy
We next directed our efforts towards delineating conformational heterogeneity within these distinct amyloid polymorphs using vibrational Raman spectroscopy that provides a wealth of molecular information on the polypeptide backbone as well as on a few key residues comprising aromatic side-chains such as tryptophan, tyrosine, etc.32,50Fig. 4a represents the characteristic conformational signature of the amyloid polymorphs formed in the absence and in the presence of variable NaCl concentrations. We monitored the changes in the polypeptide backbone markers viz. amide I and amide III as well as alterations in the band positions and intensities of tyrosines and tryptophans.
 |
| | Fig. 4 (a) Raman spectra of ovalbumin aggregates formed after 2 hours of incubation at pH 2.2, 65 °C in the absence (0 mM) and in the presence of variable ionic strength (50, 150, and 300 mM). The black short-dotted line refers to the peak at ∼1003 cm−1 due to phenylalanine that is taken as an internal standard. (b) A magnified view of the amide I and amide III regions of ovalbumin polymorphs, formed in the absence and in the presence of varying NaCl concentrations, are shown here for better clarity to depict the peak shift and peak narrowing. All of the Raman spectra are intensity-normalized at the respective peak maximum. (c) and (d) Representative Gaussian deconvolutions of the amide I (1640–1700 cm−1) and amide III (1200–1280 cm−1) regions of aggregates to analyze the percentage of secondary structural elements in ovalbumin polymorphs. The black lines in both the spectra represent the actual data, whereas the wine and the purple lines represent the cumulative fits of the amide I and III, respectively. The other colored and shaded Gaussian peaks represent various secondary structural elements. | |
2.8.1 Insights into alterations in the protein amide backbone.
A careful scrutiny of the amide I band (Fig. 4a and b) followed by deconvolution (Fig. 4c and Fig. S5, ESI†) and percentage analysis (Table S2a, ESI†) of secondary structural content of all the aggregates revealed the following: (a) all of the polymorphs showed an increase in β sheet content at the expense of α helices, (b) it became sharper progressively with an increase in ionic strength; however, aggregates formed at 150 mM depicted the lowest full width at half-maximum (FWHM) suggesting that the heterogeneity within the amyloids was the least amongst the ones formed at all other salt concentrations. A similar scrutiny of the amide III band followed by deconvolution and percentage analysis (Fig. 4d, Fig. S5, and Table S2b, respectively, ESI†) depicted an increase in the β sheet content and disordered coils at the expense of α helices, similar to that of the amide I band. A detailed comparison between the secondary elements of the polymorphs (Table S2b, ESI†) suggested that aggregates formed at 50 and 150 mM NaCl (polymorphs B and C, respectively) are substantially richer in β sheets with a prominent peak at ∼1240 cm−1 indicating conformational rearrangements into antiparallel β sheets as reported earlier for ovalbumin and insulin filaments.32,51,52 On the contrary, aggregates formed at 0 and 300 mM NaCl (polymorphs A and D, respectively) are richer in disordered structures, thus corroborating the predominant presence of amorphous aggregates. These observations further emphasize that ionic strength-induced modulation in aggregation kinetics plays an important role in structural distributions within the polymorphs.
2.8.2 Insights into residue-specific changes during self-assembly.
Following the conformational analysis of the polymorphs’ backbone, we next analyzed the band intensities of the tyrosine Fermi doublet and its ratio (I852/I827), which indicate the strength of hydrogen bonding between the phenolic hydroxyl and the surrounding water molecules.50,53 We observed a decrease in the ratio for aggregates as a function of NaCl concentration (Table S2c, ESI†) denoting that tyrosines get sequestered into aggregates in the presence of NaCl and thus, the hydrogen bonding weakens. Similar ratiometric analyses for the tryptophan Fermi doublet (I1360/I1340), which suggests strength of hydrophobic interactions between the indole side chain and the neighboring aliphatic groups of other residues, depicted a slight increase in the ratio at 150 mM NaCl (Table S2c, ESI†) indicating burial of tryptophans into aggregates, which is facilitated in the presence of NaCl compared to that formed in the absence of salt. Overall, ratiometric analyses of both tyrosines and tryptophans indicated sequestration of both the residues within the aggregates in the presence of salt. However, we would like to note that the inferences drawn here are an average estimate because ovalbumin contains three tryptophans and ten tyrosines.32 Taken together, our Raman spectroscopic analyses of all the aggregate polymorphs indicated sequestration of tyrosines and tryptophans into the aggregates’ core comprising β sheets and disordered structures that vary depending on the NaCl concentration.
2.9 Secondary structural insights into polymorphic aggregates using ATR-FTIR spectroscopy
Next, we asked whether the formation of well-organized, polymorphic amyloid fibrils and amorphous aggregates at various NaCl concentrations is predetermined at the onset of aggregation. More specifically, whether the pre-heated ovalbumin solutions, incubated at variable ionic strength, facilitate the accumulation of aggregation-competent species at room temperature prior to aggregation, which render the aggregation pathways predisposed either towards amyloid- or amorphous protein self-assembly. To address this question, we took advantage of attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy, which is a sensitive spectroscopic technique that provides secondary structural information on the amide backbone viz. amide I (1600–1700 cm−1) and amide II (1500–1580 cm−1) at a molecular level and can distinguish between inter- and intramolecular hydrogen bonding responsible for varied patterns of β-sheet conformations.47,48 In particular, the amide I band (arising due to carbonyl ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) is the most intense band and is sensitive to the protein backbone geometry, dipole–dipole interactions, and hydrogen bonding patterns.47,54 Additionally, ATR-FTIR provides a unique opportunity to gather useful spectral information from insoluble aggregates at a very low concentration.47 All of the protein samples were incubated at pH 2.2 in the absence and in the presence of varying amounts of salt at ∼25 °C (see Experimental section). Prior to collecting the FT-IR spectra, we utilized the DLS technique to check whether the protein remains monomeric even at higher concentrations (≥1 mM). A hydrodynamic diameter of ∼5 nm suggested that ovalbumin remains predominantly monomeric (Fig. S6a, ESI†). However, we cannot rule out the possibility of trace amounts of oligomers present in the samples that remained undetectable by DLS. The IR spectra of all the samples (solvent subtracted and baseline corrected) are depicted in Fig. 5a and the amide I and amide II spectral features are shown separately for better clarity (Fig. 5b and c). At the outset, we characterized the native ovalbumin at pH 7 in the absence of salt, which showed amide I absorption bands (second derivative spectrum) at 1625 and 1652 cm−1 that are characteristic of the native β-sheet and α-helix, respectively (Fig. 5d and Table S3, ESI†). Upon incubation of ovalbumin at pH 2.2 in the absence of NaCl at room temperature, we observed two prominent absorption bands at 1620 cm−1 and 1648 cm−1 suggesting the presence of intermolecular β-sheet-rich and structurally-disordered species, respectively. Additionally, a minor absorption band at 1659 cm−1 was observed, which is characteristic of α-helix. Upon incubating ovalbumin solution with 50 mM NaCl at pH 2.2 and at room temperature, we observed a significant reduction in the amide I band intensity at 1648 cm−1 and 1659 cm−1 with a concomitant rise in intensity at 1621 cm−1 indicating conformational rearrangements within the ovalbumin molecules leading to an accumulation of intermolecularly hydrogen bonded, antiparallel β-sheet-rich intermediates/protofibrils at the expense of disordered coils and α-helices, respectively (Fig. 5d). The emergence of antiparallel β-sheet-rich pre-fibrillar species was further established by the appearance of an absorption band at 1693 cm−1, similar to that observed for lysozyme amyloids formed at low pH.55 As the salt concentration was increased, we observed a similar amide I band profile for ovalbumin solution incubated with 150 mM NaCl (Fig. 5d) suggestive of the formation of a conformationally-similar, aggregation-competent species at 25 °C as that observed at 50 mM NaCl. Upon raising the ionic strength further to 300 mM NaCl, we observed a few prominent bands at 1625, 1636, and 1649 cm−1 at room temperature (Fig. 5d). While the peaks at 1625 and 1649 cm−1 could be ascribed to the presence of a significant population of native β-sheet and disordered structures within the pre-fibrillar species, the band at 1636 cm−1 remains unassigned. As we raised the temperature to initiate aggregation, we monitored time-dependent changes in the amide I bands at various salt concentrations (Fig. 5e–g and Fig. S6b–e, ESI†). A critical look at the temporal variations in the amide I regions indeed revealed that the vibrational signatures at 50 and 150 mM NaCl, obtained at room temperature, were mostly retained during aggregation at higher temperature (65 °C) albeit with differences in the intensities. For instance, the peak at 1620 cm−1 (intermolecular β-sheet-rich species) at 150 mM NaCl remained prominent and almost constant as aggregation progressed but that at 50 mM NaCl showed a significant increment as a function of time (Fig. 5e and f and Fig. S6b–e, ESI†). Similarly, the peak at 1649 cm−1 (structurally-disordered species) observed at both 0 and 300 mM of NaCl at room temperature persisted during the course of aggregation (Fig. 5g and Fig. S6e, ESI†) although a small, broad peak at 1620 cm−1 was observed intermittently at 300 mM NaCl indicating that a smaller population of intermolecularly hydrogen-bonded β sheet-rich species were also present in addition to primarily disordered aggregates. All of these observations indicate that the kinetic partitioning between amorphous and ordered, fibrillar aggregates governs the conformational heterogeneity within the morphologically-diverse aggregates. Overall, the FTIR data of the pre-heated ovalbumin samples reveal that at both 0 and 300 mM, a predominance of disordered and native-like protein structure at room temperature is observed that eventually leads to the formation of primarily amorphous, disordered aggregates, namely polymorphs A and D, respectively (Fig. 3e and h), thus corroborating our aforementioned Raman spectroscopic analyses. On the other hand, samples incubated at 50 and 150 mM NaCl form antiparallel, β-sheet-rich prefibrillar conformers at room temperature that result in the formation of ordered amyloid fibrils upon raising the temperature (polymorphs B and C Fig. 3f and g), corroborating the Raman spectral data. Taken together, a direct correlation between the appearance of specific bands in the amide I region at the onset of aggregation and in various aggregate polymorphs during the course of aggregation can be drawn which suggests that the intrinsic conformational preference of a polypeptide chain at a given solution condition perpetuates, which governs polymorphism during self-assembly processes.
O stretch) is the most intense band and is sensitive to the protein backbone geometry, dipole–dipole interactions, and hydrogen bonding patterns.47,54 Additionally, ATR-FTIR provides a unique opportunity to gather useful spectral information from insoluble aggregates at a very low concentration.47 All of the protein samples were incubated at pH 2.2 in the absence and in the presence of varying amounts of salt at ∼25 °C (see Experimental section). Prior to collecting the FT-IR spectra, we utilized the DLS technique to check whether the protein remains monomeric even at higher concentrations (≥1 mM). A hydrodynamic diameter of ∼5 nm suggested that ovalbumin remains predominantly monomeric (Fig. S6a, ESI†). However, we cannot rule out the possibility of trace amounts of oligomers present in the samples that remained undetectable by DLS. The IR spectra of all the samples (solvent subtracted and baseline corrected) are depicted in Fig. 5a and the amide I and amide II spectral features are shown separately for better clarity (Fig. 5b and c). At the outset, we characterized the native ovalbumin at pH 7 in the absence of salt, which showed amide I absorption bands (second derivative spectrum) at 1625 and 1652 cm−1 that are characteristic of the native β-sheet and α-helix, respectively (Fig. 5d and Table S3, ESI†). Upon incubation of ovalbumin at pH 2.2 in the absence of NaCl at room temperature, we observed two prominent absorption bands at 1620 cm−1 and 1648 cm−1 suggesting the presence of intermolecular β-sheet-rich and structurally-disordered species, respectively. Additionally, a minor absorption band at 1659 cm−1 was observed, which is characteristic of α-helix. Upon incubating ovalbumin solution with 50 mM NaCl at pH 2.2 and at room temperature, we observed a significant reduction in the amide I band intensity at 1648 cm−1 and 1659 cm−1 with a concomitant rise in intensity at 1621 cm−1 indicating conformational rearrangements within the ovalbumin molecules leading to an accumulation of intermolecularly hydrogen bonded, antiparallel β-sheet-rich intermediates/protofibrils at the expense of disordered coils and α-helices, respectively (Fig. 5d). The emergence of antiparallel β-sheet-rich pre-fibrillar species was further established by the appearance of an absorption band at 1693 cm−1, similar to that observed for lysozyme amyloids formed at low pH.55 As the salt concentration was increased, we observed a similar amide I band profile for ovalbumin solution incubated with 150 mM NaCl (Fig. 5d) suggestive of the formation of a conformationally-similar, aggregation-competent species at 25 °C as that observed at 50 mM NaCl. Upon raising the ionic strength further to 300 mM NaCl, we observed a few prominent bands at 1625, 1636, and 1649 cm−1 at room temperature (Fig. 5d). While the peaks at 1625 and 1649 cm−1 could be ascribed to the presence of a significant population of native β-sheet and disordered structures within the pre-fibrillar species, the band at 1636 cm−1 remains unassigned. As we raised the temperature to initiate aggregation, we monitored time-dependent changes in the amide I bands at various salt concentrations (Fig. 5e–g and Fig. S6b–e, ESI†). A critical look at the temporal variations in the amide I regions indeed revealed that the vibrational signatures at 50 and 150 mM NaCl, obtained at room temperature, were mostly retained during aggregation at higher temperature (65 °C) albeit with differences in the intensities. For instance, the peak at 1620 cm−1 (intermolecular β-sheet-rich species) at 150 mM NaCl remained prominent and almost constant as aggregation progressed but that at 50 mM NaCl showed a significant increment as a function of time (Fig. 5e and f and Fig. S6b–e, ESI†). Similarly, the peak at 1649 cm−1 (structurally-disordered species) observed at both 0 and 300 mM of NaCl at room temperature persisted during the course of aggregation (Fig. 5g and Fig. S6e, ESI†) although a small, broad peak at 1620 cm−1 was observed intermittently at 300 mM NaCl indicating that a smaller population of intermolecularly hydrogen-bonded β sheet-rich species were also present in addition to primarily disordered aggregates. All of these observations indicate that the kinetic partitioning between amorphous and ordered, fibrillar aggregates governs the conformational heterogeneity within the morphologically-diverse aggregates. Overall, the FTIR data of the pre-heated ovalbumin samples reveal that at both 0 and 300 mM, a predominance of disordered and native-like protein structure at room temperature is observed that eventually leads to the formation of primarily amorphous, disordered aggregates, namely polymorphs A and D, respectively (Fig. 3e and h), thus corroborating our aforementioned Raman spectroscopic analyses. On the other hand, samples incubated at 50 and 150 mM NaCl form antiparallel, β-sheet-rich prefibrillar conformers at room temperature that result in the formation of ordered amyloid fibrils upon raising the temperature (polymorphs B and C Fig. 3f and g), corroborating the Raman spectral data. Taken together, a direct correlation between the appearance of specific bands in the amide I region at the onset of aggregation and in various aggregate polymorphs during the course of aggregation can be drawn which suggests that the intrinsic conformational preference of a polypeptide chain at a given solution condition perpetuates, which governs polymorphism during self-assembly processes.
 |
| | Fig. 5 Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra (normalized) of the native (pH 7) and molten-globule states of ovalbumin in the absence (0 mM) and in the presence of variable NaCl concentrations (50, 150, and 300 mM) prior to aggregation at room temperature depicting (a) amide I and amide II, (b) amide I (1600–1700 cm−1) only, and (c) amide II (1500–1580 cm−1) regions. Changes in the second-derivative spectra of the amide I region of (d) native (pH 7) and molten-globule (pH 2.2) ovalbumin in the absence of salt (0 mM) and as a function of salt concentration at room temperature, and (e)–(g) ovalbumin aggregates formed at pH 2.2, 65 °C as a function of salt concentration at different time intervals. All of the experiments were repeated at least three times. | |
3. Conclusions
In summary (Fig. 6), we have demonstrated that by varying the physico-chemical properties of a solution such as pH and ionic strength, polymorphic amyloid aggregates are formed that can be distinguished based on their varying ThT-binding affinities, hydrophobicity, and aggregation kinetics accompanied by morphological diversity. Elucidation of structural details of these polymorphs using vibrational Raman spectroscopy revealed progressive sequestration of tyrosines and tryptophans into β sheet-rich aggregates, albeit discernible variations within the secondary structures as a function of salt concentration were observed which show good agreement with the polymorphism. FTIR spectroscopy provided key molecular insights into the ionic strength-dependent conformational attributes of the polypeptide chain accumulated prior to aggregation, which eventually predominate, perpetuate, and dictate alterations in the supramolecular architecture ensuing polymorphs. To the best of our knowledge, this is the first experimental demonstration of the monomeric conformational fingerprints persisting and propagating during amyloid formation. We believe that such molecular insights will aid in the conformational control of the amyloidogenic proteins and design of suitable anti-amyloid therapeutics to address amyloid strain diversity.
 |
| | Fig. 6 Schematic representation of aggregate polymorphism depicting variations in the secondary structure of the amyloidogenic monomeric precursor and the ensuing respective aggregates. | |
Author contributions
M. B. conceived and designed the research, A. G. performed research, A. G. and M. B. analyzed data, and A. G. and M. B. wrote the article.
Data availability
Experimental details including materials and methods, additional figures depicting bioinformatics analyses, fluorescence, Raman, and FT-IR spectral data, SDS-PAGE results, fitting of the kinetic data as well as tables comprising secondary structural content and recovered kinetic parameters, and additional TEM images are available in the ESI.†
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
M. B. thanks the Science and Engineering Research Board (SERB) for the POWER Research Grant (SPG/2021/001574) and A. G. thanks TIET Patiala for the teaching associateship. We are also grateful to TIET Patiala for the infrastructural facilities and for ATR-FTIR facility at the CEEMS (TIET Patiala), Prof. Bonamali Pal (TIET Patiala) for allowing us to use the DLS instrument, and the TEM facility at IISER Mohali. We also thank Dr Anamika Avni (Prof. Samrat Mukhopadhyay's lab, IISER Mohali) for helping us with the Raman microscopy experiments and sample preparations for TEM. We gratefully acknowledge the members of the Bhattacharya lab (TIET Patiala) for reading the manuscript critically.
References
- M. Wilkinson, Y. Xu, D. Thacker, A. I. P. Taylor, D. G. Fisher, R. U. Gallardo, S. E. Radford and N. A. Ranson, Cell, 2023, 186, 5798–5811 CrossRef CAS PubMed.
- M. M. Rahman, R. S. Pires, A. Herneke, V. Gowda, M. Langton, H. Biverstal and C. Lendel, Sci. Rep., 2023, 13, 985 CrossRef CAS PubMed.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek R, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- N. Louros, J. Schymkowitz and F. Rousseau, Nat. Rev. Mol. Cell Biol., 2023, 24, 912–933 CrossRef CAS PubMed.
- K. A. Murray, C. J. Hu, S. L. Griner, H. Pana, J. T. Bowler, R. Abskharon, G. M. Rosenberga, X. Cheng, M. Seidler and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206240119 CrossRef CAS PubMed.
- G. Meisl, X. Yang, C. M. Dobson, S. Linse and T. P. J. Knowles, Chem. Sci., 2017, 8, 4352–4362 RSC.
- D. Willbold, B. Strodel, G. F. Schröder, W. Hoyer and H. Heise, Chem. Rev., 2021, 121, 8285–8307 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- M. Calamai, F. Chiti and C. M. Dobson, Biophys. J., 2005, 89, 4201–4210 CrossRef CAS.
- N. Rezaei-Ghaleh, M. Zweckstetter, D. Morshedi, A. Ebrahim-Habibi and M. Nemat-Gorgani, Biopolymers, 2009, 91, 28–36 CrossRef CAS PubMed.
- A. Farzadfard, A. Kunka, T. O. Mason, J. A. Larsen, R. K. Norrild, E. T. Dominguez, S. Ray and A. K. Buell, Chem. Sci., 2024, 15, 2528–2544 RSC.
- T. Heerde, D. Schütz, Y. J. Lin, J. Münch, M. Schmidt and M. Fändrich, Nat. Commun., 2023, 14, 4293 CrossRef CAS PubMed.
- Q. Li, W. M. Babinchak and W. K. Surewicz, Nat. Commun., 2021, 12, 1620 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Nat. Commun., 2019, 10, 4760 CrossRef PubMed.
- B. Li, P. Ge, K. A. Murray, P. Sheth, M. Zhang, G. Nair, M. R. Sawaya, W. S. Shin, D. R. Boyer, S. Ye, D. S. Eisenberg, Z. H. Zhou and L. Jiang, Nat. Commun., 2018, 9, 3609 CrossRef.
- T. Theint, P. S. Nadaud, D. Aucoin, J. J. Helmus, S. P. Pondaven, K. Surewicz, W. K. Surewicz and C. P. Jaroniec, Nat. Commun., 2017, 8, 753 CrossRef PubMed.
- M. R. Elkins, T. Wang, M. Nick, H. Jo, T. Lemmin, S. B. Prusiner, H. F. DeGrado, J. Stöhr and M. Hong, J. Am. Chem. Soc., 2016, 138, 9840–9852 CrossRef CAS PubMed.
- R. Tycko and R. B. Wickner, Acc. Chem. Res., 2013, 46, 1487–1496 CrossRef CAS PubMed.
- C. Jang, D. P. Barron, L. Duo, C. Ma, H. Seabaugh and Z. Guo, ACS Chem. Neurosci., 2024, 15, 86–97 CrossRef CAS.
- M. A. Walti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Böckmann, P. Guntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef CAS PubMed.
- R. Chakarborty and K. Chattopadhyay, ACS Chem. Neurosci., 2019, 10, 1135–1136 CrossRef.
- A. Avni, A. Joshi and S. Mukhopadhyay, J. Phys. Chem. Lett., 2023, 14, 5592–5601 CrossRef CAS PubMed.
- B. Gooptu and D. A. Lomas, Annu. Rev. Biochem., 2009, 78, 147–176 CrossRef CAS PubMed.
- A. S. Knaupp and S. P. Bottomley, IUBMB Life, 2009, 61, 1–5 CrossRef CAS.
- U. I. Ekeowa, J. Freeke, E. Miranda, B. Gooptu, M. F. Bush, J. Pérez, J. Teckman, C. V. Robinson and D. A. Lomas, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17146–17151 CrossRef CAS PubMed.
- C. Lara, S. Gourdin-Bertin, J. Adamcik, S. Bolisetty and R. Mezzenga, Biomacromolecules, 2012, 13, 4213–4221 CrossRef CAS PubMed.
- K. J. A. Jansens, K. Brijis, J. Stetefeld, J. A. Delcour and M. G. Scanlon, ACS Omega, 2017, 2, 4612–4620 CrossRef CAS PubMed.
- S. Tufail, M. A. Sherwani, S. Shoaib, S. Azmi, M. Owais and N. Islam, J. Biol. Chem., 2018, 293, 11310–11324 CrossRef CAS.
- O. Conchillo-Solé, N. S. D. Groot, X. F. Avilés, J. Vendrell, X. Daura and S. Ventura, BMC Bioinf., 2007, 8, 65 CrossRef.
- A. M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS PubMed.
- M. Bhattacharya and P. Dogra, Langmuir, 2015, 31, 8911–8922 CrossRef CAS PubMed.
- M. Bhattacharya, N. Jain, P. Dogra, S. Samai and S. Mukhopadhyay, J. Phys. Chem. Lett., 2013, 4, 480–485 CrossRef CAS.
- M. Groenning, J. Chem. Biol., 2010, 3, 1–18 CrossRef PubMed.
- A. I. Sulatskaya, A. V. Lavysh, A. A. Maskevich, I. M. Kuznetsova and K. K. Turoverov, Sci. Rep., 2017, 7, 2146 CrossRef PubMed.
- A. K. Buell, P. Hung, X. Salvatella, M. E. Welland, C. M. Dobson and T. P. J. Knowles, Biophys. J., 2013, 104, 1116–1126 CrossRef CAS PubMed.
- F. Zhang, M. W. A. Skoda, R. M. J. Jacobs, S. Zorn, R. A. Martin, C. M. Martin, G. F. Clark, S. Weggler, A. Hildebrandt, O. Kohlbacher and F. Schreiber, Phys. Rev. Lett., 2008, 101, 148101 CrossRef CAS.
- P. Debye and E. Hückel, Phys. Z., 1923, 9, 185–206 Search PubMed.
- M. Boström, D. R. Williams and B. W. Ninham, Phys. Rev. Lett., 2001, 87, 168103 CrossRef.
- B. Derjaguin and L. Landau, Acta Physicochim. URSS, 1941, 14, 633–662 Search PubMed.
-
E. J. W. Verwey and J. Th. G. Overbeek, Theory of the stability of lyophobic colloids, Elsevier, Amsterdam, The Netherlands, 1948 Search PubMed.
- S. Zhang, D. A. Genov, Y. Wang, M. Liu and X. Zhang, Phys. Rev. Lett., 2008, 101, 1481011 Search PubMed.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- K. Huang, N. C. Maiti, N. B. Phillips, P. R. Carey and M. A. Weiss, Biochemistry, 2006, 45, 10278–10293 CrossRef CAS PubMed.
- J. Zurdo, J. I. Guijarro, J. L. Jiménez, H. R. Saibil and C. M. Dobson, J. Mol. Biol., 2001, 311, 325–340 CrossRef CAS.
- T. Ikenoue, Y. H. Lee, J. Kardos, H. Yagi, T. Ikegami, H. Naiki and Y. Goto, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6654–6659 CrossRef CAS PubMed.
- J. M. Ruysschaert and V. Raussens, Methods Mol. Biol., 2018, 1777, 69–81 CrossRef CAS.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyka and M. Baranskaa, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- S. D. Moran and M. T. Zanni, J. Phys. Chem. Lett., 2014, 5, 1984–1993 CrossRef CAS PubMed.
- R. Tuma, J. Raman Spectrosc., 2005, 36, 307–319 CrossRef CAS.
- P. C. Painter and J. L. Koenig, Biopolymers, 1976, 15, 2155–2166 CrossRef CAS PubMed.
- T. Zako, M. Sakono, N. Hashimoto, M. Ihara and M. Maeda, Biophys. J., 2009, 96, 3331–3340 CrossRef PubMed.
-
D. Němeček and G. J. Thomas Jr., in Handbook of Molecular Biophysics, ed. H. G. Bohr, Wiley-VCH, Weinhem, Germany, 2009 Search PubMed.
- G. Zandomeneghi, M. R. H. Krebs, M. G. Mccammon and M. Fandrich, Protein Sci., 2004, 13, 3314–3321 CrossRef CAS.
- R. Sarroukh, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biochim. Biophys. Acta, 2013, 1828, 2328–2338 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*