
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracking the reaction networks of acetaldehyde oxide and glyoxal oxide Criegee intermediates in the ozone-assisted oxidation reaction of crotonaldehyde†
Alec C.
DeCecco
a,
Alan R.
Conrad
a,
Arden M.
Floyd
a,
Ahren W.
Jasper
 b,
Nils
Hansen
c,
Philippe
Dagaut
d,
Nath-Eddy
Moody
a and
Denisia M.
Popolan-Vaida
*a
b,
Nils
Hansen
c,
Philippe
Dagaut
d,
Nath-Eddy
Moody
a and
Denisia M.
Popolan-Vaida
*a
aDepartment of Chemistry, University of Central Florida, Orlando, FL 32816, USA. E-mail: denisia.popolan-vaida@ucf.edu; Tel: (+1)-407-823-1537
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA
cCombustion Research Facility, Sandia National Laboratories, Livermore, CA 94551, USA
dCentre National de la Recherche Scientifique (CNRS), ICARE, 1C Avenue de la Recherche Scientifique, 45071 Orléans Cedex 2, France
First published on 8th July 2024
Abstract
The reaction of unsaturated compounds with ozone (O3) is recognized to lead to the formation of Criegee intermediates (CIs), which play a key role in controlling the atmospheric budget of hydroxyl radicals and secondary organic aerosols. The reaction network of two CIs with different functionality, i.e. acetaldehyde oxide (CH3CHOO) and glyoxal oxide (CHOCHOO) formed in the ozone-assisted oxidation reaction of crotanaldehyde (CA), is investigated over a temperature range between 390 K and 840 K in an atmospheric pressure jet-stirred reactor (JSR) at a residence time of 1.3 s, stoichiometry of 0.5 with a mixture of 1% crotonaldehyde, 10% O2, at an fixed ozone concentration of 1000 ppm and 89% Ar dilution. Molecular-beam mass spectrometry in conjunction with single photon tunable synchrotron vacuum-ultraviolet (VUV) radiation is used to identify elusive intermediates by means of experimental photoionization energy scans and ab initio threshold energy calculations for isomer identification. Addition of ozone (1000 ppm) is observed to trigger the oxidation of CA already at 390 K, which is below the temperature where the oxidation reaction of CA was observed in the absence of ozone. The observed CA + O3 product, C4H6O4, is found to be linked to a ketohydroperoxide (2-hydroperoxy-3-oxobutanal) resulting from the isomerization of the primary ozonide. Products corresponding to the CIs uni- and bi-molecular reactions were observed and identified. A network of CI reactions is identified in the temperature region below 600 K, characterized by CIs bimolecular reactions with species like aldehydes, i.e., formaldehyde, acetaldehyde, and crotonaldehyde and alkenes, i.e., ethene and propene. The region below 600 K is also characterized by the formation of important amounts of typical low-temperature oxidation products, such as hydrogen peroxide (H2O2), methyl hydroperoxide (CH3OOH), and ethyl hydroperoxide (C2H5OOH). Detection of additional oxygenated species such as alcohols, ketene, and aldehydes are indicative of multiple active oxidation routes. This study provides important information about the initial step involved in the CIs assisted oligomerization reactions in complex reactive environments where CIs with different functionalities are reacting simultaneously. It provides new mechanistic insights into ozone-assisted oxidation reactions of unsaturated aldehydes, which is critical for the development of improved atmospheric and combustion kinetics models.
1. Introduction
The reactions of ozone (O3) with unsaturated organic compounds are known to be one of the main loss processes of these compounds in the atmosphere. These reactions lead to the formation of Criegee intermediates (CIs), an important class of reactive intermediates with strong oxidizing capability, which contribute to the oxidation of atmospheric SOx, NOx, and oxygenated organic compounds, with effects on the atmospheric balance of low-volatility compounds as well as the formation and growth of secondary organic aerosols (SOA).1–18CIs were first postulated by Rudolf Criegee, who uncovered their mechanism of formation in 1949.19,20 The mechanism of ozonolysis reaction involves a three-step sequential process which starts with the 1,3-cycloaddition of O3 to the double bond of unsaturated organic compound leading to the formation of a cyclic trioxolane called the primary ozonide (POZ).21 This step is followed by POZ decomposition into two fragments: a carbonyl compound and a carbonyl oxide with zwitterionic character, the CI. The carbonyl and carbonyl oxide might subsequently recombine in reverse orientation to form a thermodynamically more stable secondary ozonide (SOZ).
The CIs are formed with a wide distribution of excess vibrational energy, and their fate depends on several factors like their size and internal energy, the unimolecular reactions accessible, and availability of co-reactants for bimolecular reactions. The fraction of CIs that are chemically activated and have high internal energy undergo unimolecular reactions, i.e., isomerization or decomposition, leading to the formation of acids and other oxygenated compounds, as well as chain propagation OH radicals. The fraction of CIs that are released with a lower energy content or lose their initial high internal energy through collisions with the bath gas, corresponds to stabilized CIs that have a longer lifetime and may be involved in bimolecular reactions. The yield of chemically activated and stabilized CIs depends on the molecular structure and the carbon number of the parent molecule as well as the bath gas properties and pressure.22–25
Understanding the fate of CIs is essential to determining the impact of the ozonolysis reaction in atmospheric and combustion environments. For instance, in ignition chemistry, the formation of reactive radicals such as OH, which can be obtained via (i) a sequence of reactions, such as O3 → O2 + O and O + RH → OH + R˙ as well as (ii) from CIs unimolecular decomposition, may accelerate ignition while the formation of closed shell species tends to inhibit ignition.26–29 Since ozone has been proposed as a potential additive to reduce emissions and increase the efficiency of internal combustion engines, gaining a fundamental understanding of ozone-assisted combustion, especially in the temperature region where the ozonolysis reactions are the dominant pathways, is of great importance.30 In atmospheric chemistry, CIs play a key role in controlling the atmospheric budget of hydroxyl radical (OH)31–34 and secondary organic aerosols.35 In this context, understanding the mechanism and pathways that dominate the CIs chemistry can answer important questions regarding their impact on atmospheric composition and their role in the formation of SOA.
In this work, the reaction networks of two CIs (CH3CHOO and CHOCHOO) with the same carbon number, but different functionality, formed in the ozone-assisted oxidation reaction of crotonaldehyde are investigated. Crotonaldehyde (CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO) is widely present in the environment. In the troposphere, it originates from both biogenic sources, such as emission from vegetation36,37 and anthropogenic sources, such as emissions from the combustion of biomass and liquid fuels.38–43 The reaction of crotonaldehyde with ozone contributes to the atmospheric budget of hydroxyl radicals through the unimolecular decomposition of CIs and the formation of low-volatility high molecular weight compounds due to bimolecular reactions of CIs with volatile organic compounds, impacting the atmospheric oxidation capacity and local and regional formation of photooxidants. Crotonaldehyde contains two functional groups, i.e., a CC double bond and a CO carbonyl group. Its high reactivity towards ozone is due to the presence of the CC double bond.
CHCHO) is widely present in the environment. In the troposphere, it originates from both biogenic sources, such as emission from vegetation36,37 and anthropogenic sources, such as emissions from the combustion of biomass and liquid fuels.38–43 The reaction of crotonaldehyde with ozone contributes to the atmospheric budget of hydroxyl radicals through the unimolecular decomposition of CIs and the formation of low-volatility high molecular weight compounds due to bimolecular reactions of CIs with volatile organic compounds, impacting the atmospheric oxidation capacity and local and regional formation of photooxidants. Crotonaldehyde contains two functional groups, i.e., a CC double bond and a CO carbonyl group. Its high reactivity towards ozone is due to the presence of the CC double bond.
The reaction of crotonaldehyde with ozone has been investigated both theoretically and experimentally.44–47 The majority of these studies have been focused on determining the rate coefficients and quantifying the substituent effects on the reaction coefficients for different α,β-unsaturated carbonyls such as aldehydes, ketones, esters, and acids. Atkinson et al. used a 175 L Teflon bag to determine the rate constants of the reactions of a series of carbonyls, including crotonaldehyde, with ozone and discussed the substituent effect of –CHO and CH3CO– groups on the rate constants.44
The reaction rate constant for the gas-phase reaction of O3 with unsaturated oxygenates, including crotonaldehyde, has been investigated in an FEP Teflon chamber at ambient temperature and pressure by Grosjean et al.45 The determined rate coefficients were discussed in terms of the reactivity of unsaturated oxygenates toward O3 as a function of the nature, number, and position of the oxygen-containing substituents, i.e., –CHO, –C(O)R, –C(O)OR, and –OC(O)R, as well as the atmospheric implications of such reactions. Sato et al. employed the relative-rate method to measure the rate coefficients for the reactions of O3 with six unsaturated carbonyls, including crotonaldehyde, in the presence of a radical scavenger using a 6 m3 reaction chamber combined with a long-path FTIR system.46 The measured rate coefficients were shown to agree well with those obtained by conventional transition state theory calculations.
Theoretical investigations employing density functional theory have explored the reaction coefficient of the crotonaldehyde ozonolysis reaction and the reaction pathways of the CIs formed in this reaction.47 Energy barriers and reaction energies for CI (CH3CHO and CHOCHOO) reactions with species such as NO, H2O, HCHO and HCOOH were also calculated and compared. Despite previous studies, a detailed description of the mechanism of crotonaldehyde reaction with O3, in particular with respect to the CIs reaction network, is currently lacking. The reaction of crotonaldehyde with O3 is particularly interesting because this reaction leads to the formation of two CIs with the same carbon number, but different functionalities. Fig. 1 shows the initial steps involved in the ozonolysis of crotonaldehyde. The reaction proceeds by cycloaddition of ozone to the CC bond of crotonaldehyde, which results in the formation of a five-membered cyclic POZ. The POZ compound has been reported to be unstable and will rapidly undergo unimolecular reactions.47 The unimolecular decomposition of the POZ proceeds through two reaction pathways. One pathway leads to the formation of acetaldehyde oxide (CH3CHOO, CI-1) and glyoxal (see path (1) in Fig. 1) while the other pathway leads to the formation of glyoxal oxide (CHOCHOO, CI-2) and acetaldehyde (see path (2) in Fig. 1). Alternatively, POZ can undergo unimolecular isomerization and result in the formation of ketohydroperoxide (KHP) products (see path (3) in Fig. 1). Once formed, CIs undergo unimolecular and bimolecular reactions with co-products/co-reactants. Stabilized CI-1 and CI-2 can also recombine in reverse orientation with their carbonyl co-products, i.e., glyoxal and acetaldehyde, respectively, to form a thermodynamically more stable SOZ (see path (4) in Fig. 1).
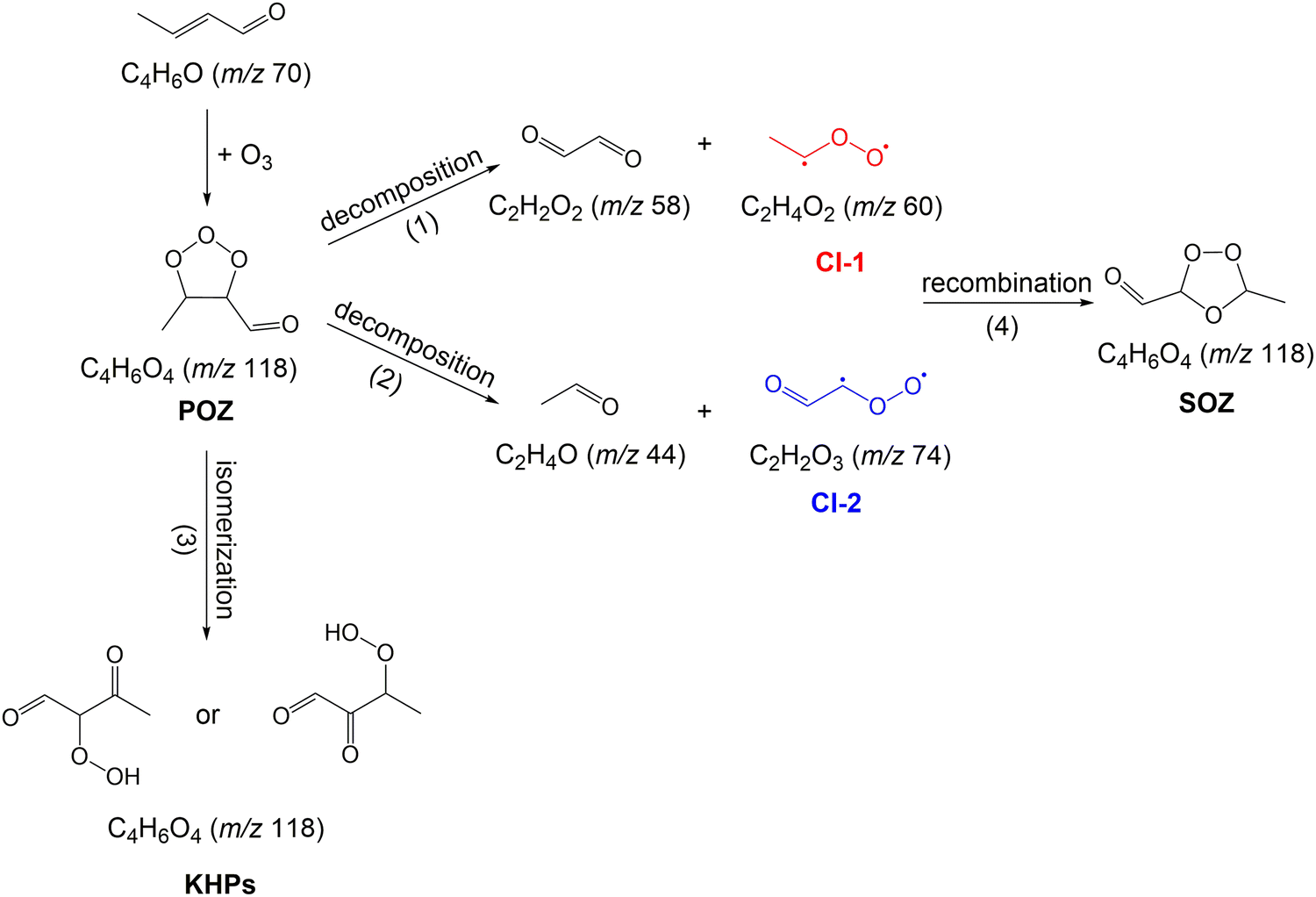 | ||
| Fig. 1 Primary unimolecular processes and bimolecular reactions involved in the ozone-assisted oxidation of crotonaldehyde. The numbers in parentheses correspond to the nominal mass of the respective structure. | ||
CIs have fascinating chemical properties and minimal structural differences can dramatically change their unimolecular and bimolecular reactivity.48–52 For instance, distinct conformational forms of CIs were observed to undergo remarkably different unimolecular decay processes with rates that differ by orders of magnitude. The chemical behavior of the syn- and anti-conformers of acetaldehyde oxide (CH3CHOO), has been reported to be very different. While syn-CH3CHOO follows a fast thermal decomposition pathway that leads to the formation of OH radicals via vinyl hydroperoxide decomposition, anti-CH3CHOO has been reported to rapidly react with atmospheric water vapors and forms a hydroxyalkyl hydroperoxide.53
While many investigations focused on understanding the changes in CIs reactivity induced by structural changes, little is known about the changes induced by different functionalities of CIs with the same carbon numbers. In addition, the role of CIs in the formation of secondary organic aerosols is not completely understood, and only a few studies reported the formation of oligomeric species.48,54,55
The reaction networks of CI-1 and CI-2, which are C2 CIs with different functionalities, are investigated in a jet-stirred reactor equipped with molecular beam sampling capabilities in conjunction with high-resolution mass spectrometry and tunable synchrotron radiation from the Advanced Light Source in Berkeley. While the reaction pathway of CI-1 (CH3CHOO) has been investigated before, including studies in our laboratory,4,5,56–58 and references therein the experimental literature on CHOCHOO reactions is sparse, and mostly based on theoretical predictions.47,59–61 Therefore, our investigations are focused on (i) understanding the reaction network of CHOCHOO, (ii) understanding the reaction network of CH3CHOO when both the primary ozonide (POZ) precursor and the reaction environment are changed with respect with previous studies, and (iii) exploring the CIs induced oligomerization in a system where two CIs with different functionalities are simultaneously reacting.
The experimental data in conjunction with ab initio threshold energy calculations facilitated the identification of several reactive intermediates, such as ketohydroperoxide and hydroperoxide species. The results of our studies provide new insights into the ozone-assisted oxidation of crotonaldehyde and inform future developments of more detailed chemical descriptions of the ozone-assisted oxidation of unsaturated aldehydes.
2. Methods
2.1 Experimental method
The experimental setup employed to study the reaction network of CIs formed in ozone-assisted oxidation reaction of crotonaldehyde consists of a jet-stirred reactor (JSR) system, a sampling system, and an analysis system. A detailed description of the experimental setup used in these investigations is given elsewhere.62 Briefly, the gas flow rates are controlled by calibrated MKS mass flow controllers while the liquid crotonaldehyde flow rate is controlled by a syringe pump followed by a vaporization line maintained at 10 K above the boiling point of crotonaldehyde to avoid condensation and mixed with a constant flow of Ar carrier gas. Subsequently, the gasses are injected into the preheating region of the JSR. As the uncertainty in the flow measurements is estimated to be around 0.5% for each mass flow controller, we estimate an uncertainty of about 2% on the residence time. The fuel and oxidizer are kept separate in the concentric quartz tubes of the preheating region prior to mixing at the entrance of the JSR. The heating of the gas flows in the preheating region is achieved by means of an Inconel (Thermocoax) heating resistor and is necessary to prevent temperature gradients inside the reactor. Controlled amounts of O3 are produced by sending a fraction of the oxygen stream into a Corona discharge ozone generator. A 70 cm quartz absorption cell in conjunction with a helium neon UV calibrated lamp and an Ocean Optics spectrometer are used to monitor the concentration of O3 just prior to the preheating zone at a wavelength of 312.57 nm, using the known ozone-adsorption cross section.63The JSR, which is based on the design of Dagaut et al.,64 is a custom-made fused silica sphere with a diameter of 4 cm and a volume of about 33.5 cm3. It has four injectors located in the equatorial plane of the reactor oriented in opposite directions in order to achieve stirring of the reactants in the whole reactor volume. The initial design of Dagaut et al.,64 was slightly modified to accommodate direct sampling into a high-resolution (m/Δm ≈ 4000) reflectron time-of-flight molecular beam mass spectrometer (TOF-MBMS).
The JSR exhaust is coupled to the TOF-MBMS through a quartz cone-like nozzle, with a 40° cone angle and a ∼50 μm orifice diameter at the tip. The temperature inside of the reactor is regulated by a proportional-integral-derivative (PID) controlled tube furnace and monitored by means of an Inconel coated type K thermocouple (Thermocoax) placed in the proximity of the sampling conical nozzle. The reactor temperature is measured with an uncertainty of ±20 K that is in part attributed to the cooling effect of the sampling nozzle. Previous experiments performed to test the reactor temperature homogeneity, by moving the thermocouple along the center line of the reactor, revealed a variation of only ±5 K for dilution levels as low as 80% Ar and residence times up to 4 s, proving the temperature homogeneity of the reactor.
The TOF-MBMS employs vacuum ultraviolet (VUV) photons from the Chemical Dynamics Beamline of the Advanced Light Source65,66 to measure photoionization spectra of the sampled reactant, intermediate, and product species. The synchrotron VUV radiation provides high photon flux (1014 photons per s) and narrow bandwidth energy photons (E/ΔE(fwhm) ≈ 250–400) from 7.4 eV to 30 eV. The narrow bandwidth and the tunability allow for near-threshold ionization, which reduces fragmentation and simplify the interpretation of the mass spectra. In addition, the tunability of the synchrotron radiation enables isomer resolved species identification based on the comparison of the recorded photoionization spectra with the absolute photoionization spectra of individual isomeric species.
The reaction of crotonaldehyde with ozone is investigated over a broad range of temperatures between 390 and 840 K at a constant pressure of 0.92 atm (700 Torr), a reaction time of 1.3 s, and an equivalence ratio of 0.5 with a mixture of 1% crotonaldehyde, 10% O2, and 89% Ar. The ozone concentration was maintained constant at a value of 1000 ppm during all measurements presented in this study. Temperature scans are recorded in 15 to 40 K increments at photon energies of 9.5 eV, 10.5 eV, 11.0 eV, 11.5 eV, and 14.35 eV, in a temperature range from 390 to 840 K while the photoionization efficiency curves in the presence of ozone are recorded at 390 K a temperature at which the majority of hydroperoxide intermediates present the highest signals. For the temperature profile measurements, the ionization energies are carefully selected to capture the behavior of important intermediates and final products as well as to distinguish between the behavior of isomers of the same mass-to-charge ratios. Quantification of the species identified in this work is beyond the scope of this paper, which focuses exclusively on species identification.
2.2 Theoretical method
The photoionization efficiency curves and the ionization energies of many important intermediates formed in the ozone-assisted oxidation reaction of crotonaldehyde are unknown, which hinders their identification. To identify these key intermediates, an automated approach is implemented to systematically explore the conformeric structures of intermediates and to compute up to ∼3Ntorsion locally adiabatic ionization energies62 for the systems of interest, where Ntorsion is the number of rotatable bonds. The cheminformatics tool, Open Babel,67,68 is used to generate initial geometry guesses from the chemical structure. Candidate conformers were then generated by spinning all rotatable bonds by fixed step sizes. A step size of 120° was used for torsions with sp3 hybridization for both central atoms (the majority of cases), and 60° step sizes were used otherwise (e.g., for torsions where one of the rotatable groups is the –CHO group). Each candidate structure was minimized first using Open Babel's automated conformer generating strategy “confab” and then using M06-2X/cc-pVDZ. The structures were checked for duplicates, including mirror images, and higher-level ionization energies were computed for all unique conformers within 3 kcal mol−1 of the lowest-energy conformer at the ∼CCSD(T)/CBS//M06-2X/cc-pVTZ level of theory, where ∼CCSD(T)/CBS indicates that the complete basis set limit correction to CCSD(T)/cc-pVTZ was estimated using MP2 and the cc-pVTZ and cc-pVQZ basis sets, as employed elsewhere.56,62,69 A slightly less computationally demanding approach was used to explore the ring opening products which can have a large number of conformers. For these cases, smaller basis set extrapolations (based on the cc-pVDZ and cc-pVTZ basis sets) were used. The two levels of theory were found to predict adiabatic ionization energies that agreed to be better than 0.05 eV for several test cases. Gaussian 16 was used for the DFT calculations, and Molpro 2022 was used for the CCSD(T) calculations.70–72Often, the locally adiabatic ionization energies are in close agreement with one another and with the global adiabatic ionization energy, as is the case for all the calculated ionization energies reported here. As discussed, and demonstrated in previous publications,73,74 the calculated adiabatic ionization energies are often good predictors of the experimental ionization thresholds (typically within ±0.1 eV) and aid in species identification.
3. Results and discussions
3.1 Ozone-assisted oxidation reaction of crotonaldehyde
The ozone-assisted oxidation reaction of crotonaldehyde is investigated in a temperature range between 390 K and 850 K. The lowest reaction temperature investigated in this study (390 K), is chosen to be slightly above the boiling point of crotonaldehyde (375 K) to maintain crotonaldehyde in the gas phase. The presence of ozone initiates the oxidation of crotonaldehyde already at 390 K. A typical product mass spectrum recorded at 390 K and a photon energy of 11.00 eV, from m/z 20 to 230 is shown in Fig. 2. Groups of peaks containing 2 to 14 heavy atoms (carbon and oxygen atoms) are marked. Note that the maximum number of oxygen atoms is four for all detected products including those with a large number of heavy atoms, i.e. CxHyOz with x + z = 11, 12, and 14. The mass spectrum is characterized by the presence of products indicative of both crotonaldehyde ozonolysis reactions and crotonaldehyde low-temperature oxidation reactions. The mass peaks detected at m/z's that match the chemical composition of the potential CIs reaction products are indicated in the mass spectrum and are color-coded in red or blue corresponding to CI-1 and CI-2 reaction products, respectively. Details about the origin and chemical identity of these products are discussed in Section 3.3. The arrows in the mass spectra indicate the mass peak corresponding to formaldehyde (CH2O, m/z 30.010) and methylhydroperoxide (CH3OOH, m/z 48.021), which are typical low-temperature oxidation products, as well as the mass peak corresponding to crotonaldehyde (CH3CHCHCHO, m/z 70.042).
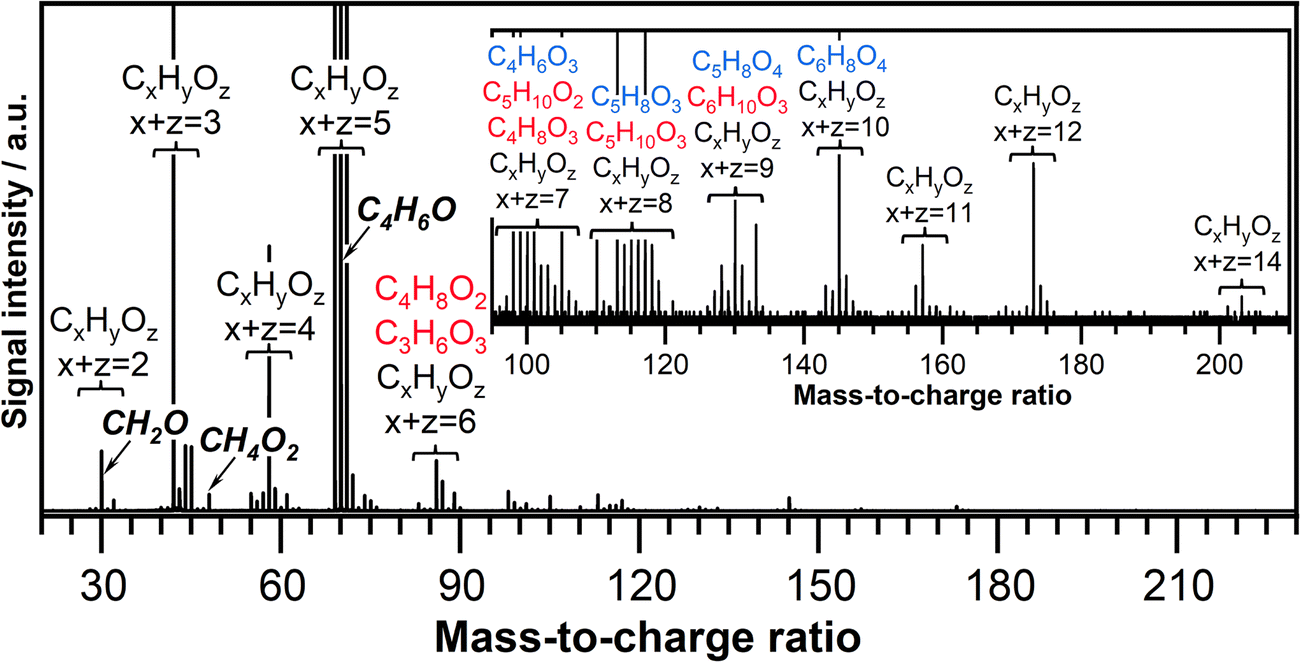 | ||
| Fig. 2 Typical mass spectrum recorded after the ozone-assisted oxidation reaction of crotonaldehyde at a reaction time of 1.3 s, an equivalence ratio of 0.5, a photon energy of 11.0 eV and temperature of 390 K. The inset highlights the 95 amu to 225 amu region of the mass spectrum. The products attributed to the CI-1 reactions are colored in red while the products corresponding to CI-2 reactions are colored in blue. | ||
Fig. 3 shows the temperature profiles of the reactants, i.e., crotonaldehyde and ozone, recorded at a photon energy of 10.50 eV and 14.35 eV, respectively. As mentioned above crotonaldehyde reacts with ozone already at 390 K resulting in an ozonide initiated temperature region (see Fig. 1). Around 500 K a considerable fraction of the crotonaldehyde signal is consumed (see Fig. 3(a)) due to the reaction with atomic oxygen (O) formed because of the O3 thermal decomposition.75,76 Above 500 K, a negative temperature coefficient (NTC) region is observed that corresponds to a decrease in reactivity of crotonaldehyde with increasing temperature. This might be attributed to the fast O atoms recombination resulting in the formation of O2. Previous work by Rousso et al. in which atomic oxygen concentration as a function of temperature was simulated revealed that the concentration of atomic oxygen decreases above 520 K to vanish at around 570 K. The simulated decay in the atomic oxygen concentration correspond to the increase in the crotonaldehyde signal observed under our experimental conditions and support our assumption that the decrease in the overall reactivity of the system is because of the O atoms recombination.75
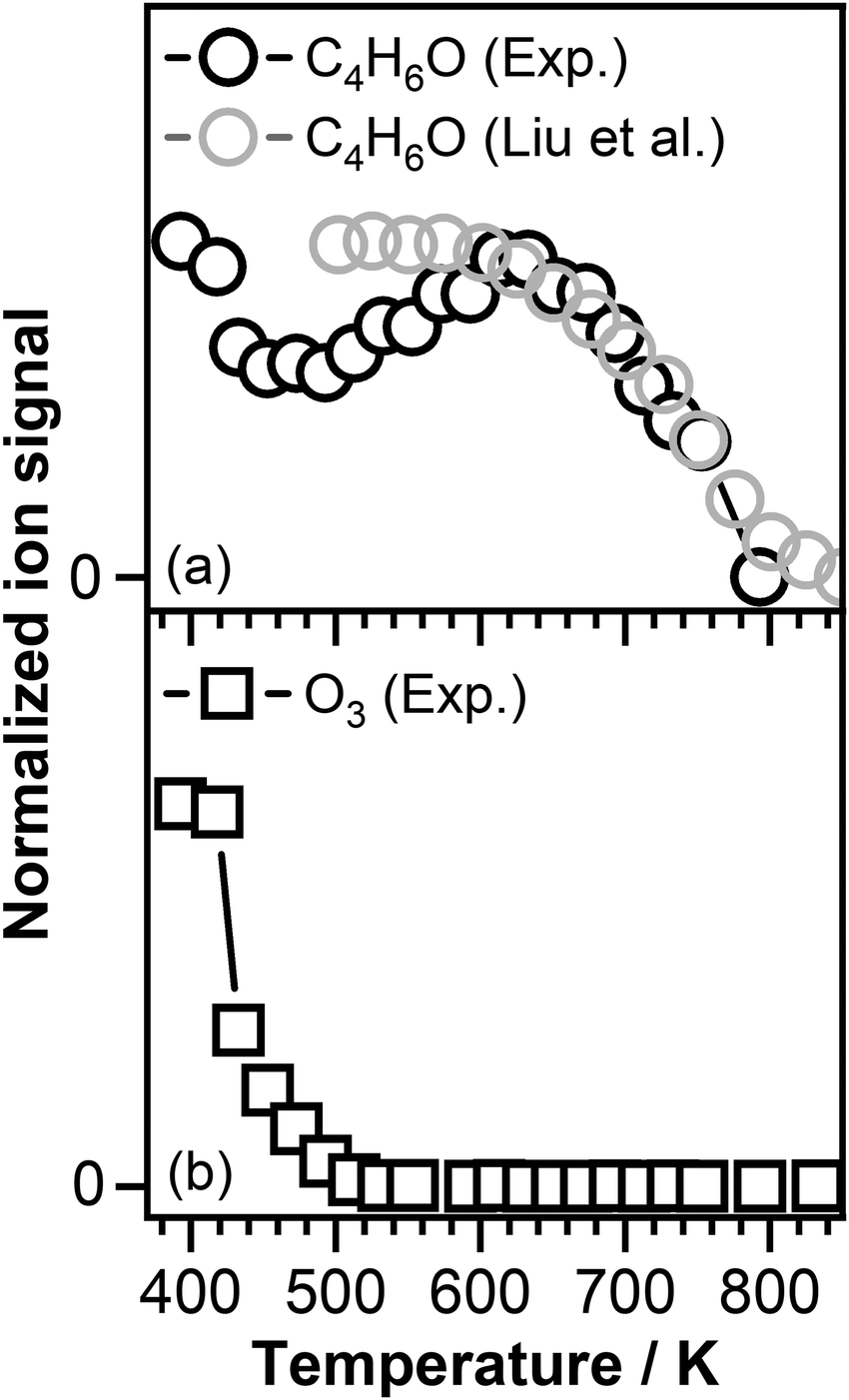 | ||
| Fig. 3 (a) C4H6O signal intensity as a function of temperature recorded in our experiment at a photon energy of 10.50 eV in the presence of ozone and oxygen (black open symbols) compared with the C4H6O signal recorded by Liu et al.77 in the presence of oxygen alone (gray open symbols) and (b) O3 signals intensity as a function of temperature recorded in our experiment at a photon energy of 14.35 eV. | ||
Fig. 3(b) displays the ozone signal intensity as a function of temperature, which indeed confirms the depletion of ozone with increasing temperature and is in agreement with the data reported by Zhao et al. who observed that ozone fully decomposes around 600 K.78 Increasing the temperature above 640 K leads to a dramatic decrease in the intensity of crotonaldehyde signal (see Fig. 3(a)). Recent investigations performed by Liu et al.,77 using a similar experimental arrangement as the one described in this study, revealed that the oxidation of crotonaldehyde in an ozone-free environment occurs at a temperature around 600 K. Therefore, the decrease in the crotonaldehyde signal intensity above 600 K, similar with the one observed by Liu et al. is attributed to the reaction of crotonaldehyde with O2. The gray open symbols in Fig. 3(a) that correspond to the crotonaldehyde temperature profile reported by Liu et al.,77 recorded in the presence of oxygen alone, follows the same temperature dependence as the signal recorded in our experiment above 600 K.
As can be seen in Fig. 3, the presence of ozone triggers the oxidation of crotonaldehyde at much lower temperatures then O2 alone. According to the oxidation scheme displayed in Fig. 1, the initial step in the crotonaldehyde ozonolysis reaction involves the formation of a primary ozonide (POZ, 5-methyl-1,2,3-trioxolane-4-carbaldehyde, m/z 118.026) by O3 cycloaddition across the olefinic CC bond of crotonaldehyde. Once formed, the POZ can either undergo unimolecular isomerization leading to the formation of ketohydroperoxides (2-hydroperoxy-3-oxobutanal or 3-hydroperoxy-2-oxobutanal) or undergo unimolecular decomposition. The unimolecular decomposition of the POZ can proceed through two different channels, producing two pairs of aldehyde – CI compounds, i.e., glyoxal + CH3CHOO and acetaldehyde + CHOCHOO. Subsequently, the aldehyde compounds and the CIs might recombine to form a secondary ozonide, (SOZ, 5-methyl-1,2,4-trioxolane-3-carbaldehyde, m/z 118.026). Consequently, the small signal observed at m/z 118.026 in the product mass spectrum could be attributed to the POZ, KHP, or SOZ. Fig. 4 displays the photoionization efficiency (PIE) curve of the signal recorded at m/z 118.026 (C4H6O4) at a reaction temperature of T = 390 K. As can be seen in Fig. 4, the ionization threshold of the signal recorded at m/z 118.026 appears to be around 9.50 ± 0.05 eV.
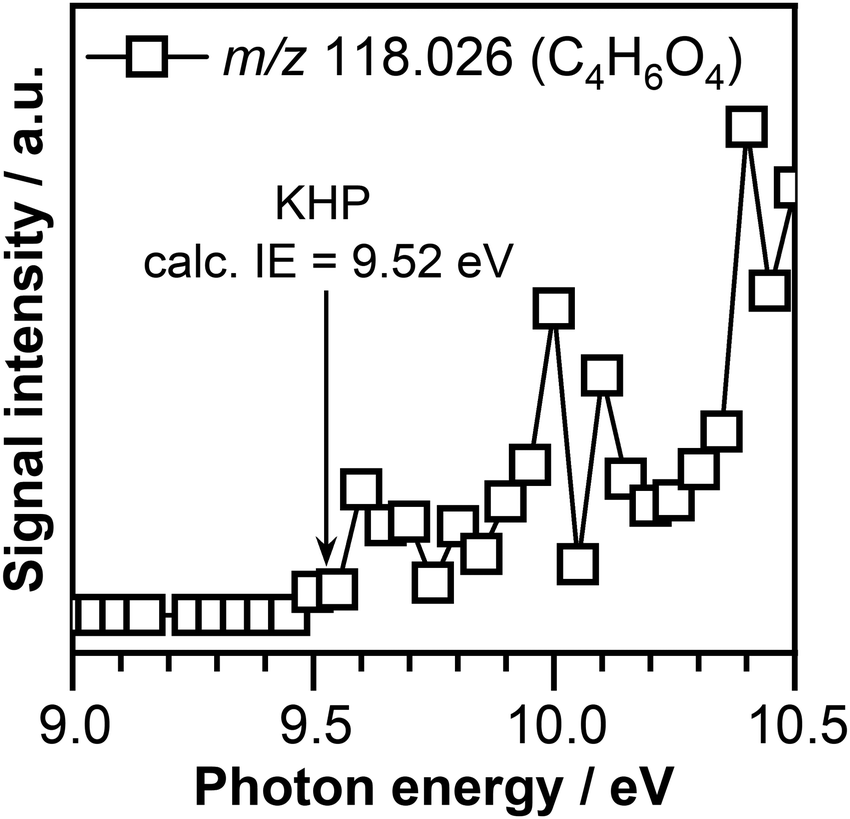 | ||
| Fig. 4 Photoionization efficiency curve of m/z 118.026 (C4H6O4) recorded at a temperature of 390 K. The calculated adiabatic ionization energy of 2-hydroperoxy-3-oxobutanal KHP is indicated by the arrow. | ||
The adiabatic ionization energies of the POZ and SOZ calculated in this work at the ∼CCSD(T)/CBS//M06-2X/cc-pVTZ level of theory are 9.58 eV and 9.64 eV, respectively. While the calculated adiabatic energy of SOZ is slightly above the experimentally observed ionization threshold, the adiabatic ionization energy of the POZ matches within the experimental errors the observed ionization threshold. However, based on our experimental conditions and previous observations, POZ is not expected to be stable and therefore is not expected to contribute to the observed signal.69,79
Interestingly, the calculated adiabatic ionization energy of the 2-hydroperoxy-3-oxobutanal KHP of 9.52 eV is in good agreement with the experimentally observed ionization threshold. The calculated adiabatic ionization energy of 3-hydroperoxy-2-oxobutanal KHP of 9.33 eV is far below the observed ionization threshold of the C4H6O4 signal. Therefore, part of the signal recorded at m/z 118.026 (C4H6O4) is attributed to the presence of the 2-hydroperoxy-3-oxobutanal KHP formed as a result of unimolecular isomerization of the POZ. This observation is consistent with the detection of the KHP isomer of POZ in similar ozonolysis experiments of trans-2-butene56 and ethylene.69 While POZ is not expected to be stable under our experimental conditions, we cannot exclude the contribution of SOZ to the signal recorded at m/z 118.026.
3.2 Unimolecular reactions of Criegee intermediates
As mentioned above, the unimolecular decomposition of the POZ (m/z 118.027) leads to the formation of two different carbonyl oxides. One POZ decomposition channel is responsible for the formation of CH3CHOO (CI-1, acetaldehyde oxide, m/z 60.021) and CHOCHO (glyoxal, m/z 58.005), while the other decomposition channel is responsible for the formation of CHOCHOO (CI-2, glyoxal oxide, m/z 74.004) and CH3CHO (acetaldehyde, m/z 44.026) compounds. CH3CHOO and CHOCHOO are recognized to be the smallest CIs that have two conformers (anti- and syn-CI).The mass spectra recorded at reaction temperatures below 550 K display mass peaks at m/z 44.026 (C2H4O), 58.005 (C2H2O2), and 60.021 (C2H4O2), but not at m/z 74.004 (C2H2O3). To determine the chemical identity of these mass peaks, the PIE curves of these signals are recorded at a temperature of 390 K and analyzed. The signals recorded at m/z 44.026 and m/z 58.005 are attributed to acetaldehyde and glyoxal, respectively based on the recorded PIE curves that match the PIEs reported in the literature for these species (see Fig. S1(a) and (b) in the ESI†).
The ionization threshold of the signal recorded at m/z 60.021 is observed at a photon energy of 9.95 ± 0.05 eV (see the PIE in Fig. S1(c) in the ESI†) followed by a sharper rise near 10.70 ± 0.05 eV. The detection of CI-1 or its vinylhydroperoxide isomer is excluded as the onset of the PIE curve of the C2H4O2 signal is above the ionization energies reported in the literature for these species.80–82 Based on the observed ionization threshold, which is in agreement with the ionization threshold of 9.98 eV calculated for glycolaldehyde,69 part of the signal recorded at m/z 60.021 is attributed to the formation of glycolaldehyde, a species formed as a result of CI-1 isomerization via a hydroperoxide channel. The isomerization of CH3CHOO to glycolaldehyde has been observed by our group in previous experiments investigating the reaction network of CH3CHOO formed in the ozone-assisted oxidation reaction of trans-2-butene.56 The signal increase above 10.70 ± 0.05 eV observed in Fig. S1(c) (ESI†) correlates with the presence of acetic acid (IE = 10.65 eV83). While no evidence for 1,2-dioxetane was found (IE = 9.35 eV62), we cannot exclude the presence of small amounts of methyl formate (IE = 10.83 eV84).
Based on the adiabatic ionization energy calculated in this work, CI-2 (CHOCHOO) is expected to ionize around 10.28 eV. However, as mentioned above, within our detection limit no signal is identified at m/z 74.004 over the range of temperatures, equivalence ratios, and reaction times, investigated in this study. The lack of signal at m/z 74.004 corresponding to CHOCHOO, suggest rapid reactions, which might lead to CI-2 depletion due to the low activation energy for reaction in the timescale of our experiment. Calculations performed by Zhong et al. predict that CI-2 can undergo unimolecular isomerization leading to the formation of a hydroperoxide that subsequently decomposes leading to OH formation. The isomerization-decomposition pathway leading to OH formation was found to be energetically more favorable than isomerization pathway expected to produce dioxirane.85
Temperature profiles measured at 10.50 eV and 11.00 eV, displayed in Fig. 5(a), confirm the contribution of several isomers to the signal recorded at C2H4O2. For instance, the temperature profile measured at 10.50 eV has a different shape than the temperature profile measured at 11.00 eV and presents a signal only in the temperature region below 600 K that correlates with the temperature region dominated by the ozone-assisted reactions. In contrast, the signal recorded at 11.00 eV is shifted to higher temperature with respect to the signal recorded at 10.50 eV and after a decay around 600 K is rising with increasing temperature, indicative of a product(s) formed as a result of low-temperature oxidation of crotonaldehyde.
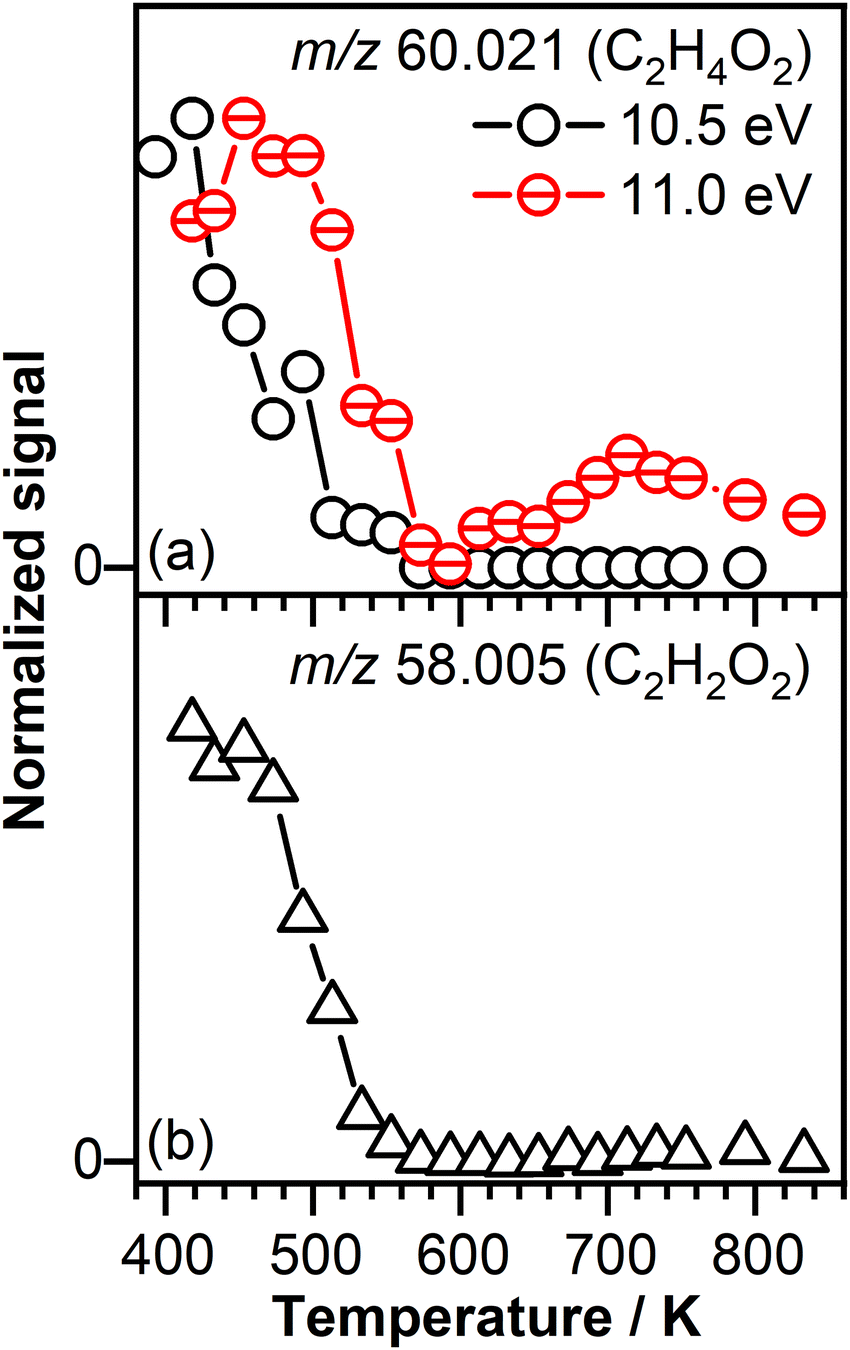 | ||
| Fig. 5 (a) C2H4O2 signal intensity represented as a function of temperature recorded at a photon energy of 10.50 eV and 11.00 eV, respectively. (b) C2H2O2 signal intensity represented as a function of temperature recorded at a photon energy of 11.00 eV. | ||
Fig. 5(b) shows the temperature profile of the glyoxal product, expected to originate from the unimolecular decomposition of the POZ through the CI-1 reaction channel. The lack of signal in the temperature range in which ozone thermally decomposes confirms that glyoxal is an ozonolysis product, potentially originating from the POZ unimolecular decomposition.
3.3 Bimolecular reactions of the stabilized CIs
Criegee intermediates are very reactive species and are known to participate in different types of bimolecular reactions like cycloaddition to unsaturated or carbonyl compounds, insertion into O–H bonds, radical recombination, i.e., attack of a radical on the C atom or terminal O atom of the carbonyl oxide group, and combination with other singlet zwitterions/biradicals (dimer formation).5 Given the complex reactive environment within the JSR, CI-1 (CH3CHOO) and CI-2 (CHOCHOO) are expected to react with species like water, aldehydes (formaldehyde, acetaldehyde, and propionaldehyde), alcohols (methanol, ethanol, and propanol), ketones (acetone), carboxylic acids (formic acid and acetic acid), alkenes (ethene and propene), and hydroperoxides (hydrogen peroxide, methyl hydroperoxide, and ethyl hydroperoxide), which are formed as co-products in the ozone-assisted oxidation reaction of crotonaldehyde. In addition, CI-1 and CI-2 might also react with ozone. Table 1 summarizes the CI reactions targeted in this study and the ionization energies of the corresponding products either calculated in this work or adopted from the literature. The specific isomers that are not detected under our experimental conditions are italicized.| CH3CHOO/CHOCHOO reactions | |||||
|---|---|---|---|---|---|
| m/z | Composition | Formula | IE (eV) | Reaction | |
| Aldehydes | 90.032 | C3H6O3 | cyc-CH(CH3)OOCH2O– | 9.41* | CH3CHOO + H2CO |
| HOCH2OC(CH3)O |
10.20* | ||||
| HOCH(CH3)OCHO | 10.46* | ||||
| 104.010 | C3H4O4 | cyc-CH(CHO)OOCH 2 O– | 9.69 | CHOCHOO + H2CO |
|
| 104.047 | C4H8O3 | cyc-CH(CH 3 )OOCH(CH 3 )O– | 9.23* | CH3CHOO + CH3HCO |
|
| 118.027 | C4H6O4 | cyc-CH(CHO)OOCH(CH 3 )O– | 9.64 | CHOCHOO + CH3HCO |
|
| 118.063 | C5H10O3 | cyc-CH(CH 3 )OOCH(CH 2 CH 3 )O– | 9.24 | CH3CHOO + CH3CH2HCO |
|
| CH3C(O)OCH(OH)CH2CH3 |
9.83 | ||||
| CH3CH(OH)OC(O)CH2CH3 |
9.81 | ||||
| 132.042 | C5H8O4 | cyc-CH(CHO)OOCH(CH 2 CH 3 )O– | 9.53 | CHOCHOO + CH3CH2HCO |
|
| 130.062 | C6H10O3 | Add to CO: cyc-CH(C3H5)OOCH(CH3)O– |
9.63 | CH3CHOO + CH3CHCHCHO |
|
| Add to double bond: cyc-CH(CH3)OOCH(CH3)CH(HCO)– | 8.96 | ||||
| Add to double bond: cyc-CH(CH3)CH(CH3)OOCH(HCO)– | 9.10 | ||||
| Ring opening product: CHOCH(OH)CH(CH3)C(O)CH3 |
9.33 | ||||
| 144.042 | C6H8O4 | Add to CO: cyc-CH(C3H5)OOCH(HCO)O– |
9.90 (9.35) | CHOCHOO + CH3CHCHCHO |
|
| Add to double bond: cyc-CH(HCO)OOCH(CH3)CH(HCO)– | 9.23 | ||||
| Add to double bond: cyc-CH(CH3)CH(HCO)OOCH(HCO)– | 9.49 | ||||
| Alkenes | 88.052 | C4H8O2 | cyc-CH(CH 3 )OOCH 2 CH 2 – | 8.92* | CH3CHOO + H2CCH2 |
CH
2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) CHCH(OOH)CH
3 CHCH(OOH)CH
3
|
9.36* | ||||
| CH3CHCHCH2OOH |
9.41* | ||||
| 102.031 | C4H6O3 | cyc-CH(CHO)OOCH2CH2– | 9.35 | CHOCHOO + H2CCH2 |
|
| CH2CHCH(OOH)CHO |
9.34 | ||||
|
CHOCHCHCH
2
(OOH)
|
9.65 (9.45) | ||||
| 102.068 | C5H10O2 | cyc-CH 2 CH(CH 3 )OOCH(CH 3 )– | 8.76 | CH3CHOO + H2CCH2CH3 |
|
| cyc-CH(CH 3 )CH 2 OOCH(CH 3 )– | 8.88 | ||||
|
CH
3
CH(OOH)CHCHCH
3
|
9.16 (9.28) | ||||
|
CH
3
CHCH–CH(OOH)CH3 |
9.17 | ||||
| CH 3 CH(OOH)C(CH 2 )CH 3 | 9.30 | ||||
|
CH
3
CHC(CH
2
OOH)CH
3
|
8.85 | ||||
| 116.047 | C5H8O3 | cyc-CH 2 CH(CH 3 )OOCH(HCO)– | 9.15 | CHOCHOO + H2CCH2CH3 |
|
| cyc-CH2(CH3)CH2OOCH(HCO)– | 9.31 | ||||
| Ketones | 118.063 | C5H10O3 | cyc-CH 2 (CH 3 )OOC(CH 3 ) 2 O– | 8.99 | CH3CHOO + H3CCOCH3 |
| 132.042 | C5H8O4 | cyc-CH 2 (CHO)OOC(CH 3 ) 2 O– | 9.35 | CHOCHOO + H3CCOCH3 | |
| Acids | 101.995 | C3H2O4 | OCHCH(OOH)OCHO(CH 3 ) | 9.90 | CHOCHOO + HCOOH |
| 120.042 | C4H8O4 |
CH
3
CH(OOH)OC(O)
|
9.13 | CH3CHOO + H3CCOOH | |
| Alcohols | 106.062 | C4H10O3 | CH 3 CH(OOH)OCH 2 CH 3 | 8.91 | CH3CHOO + CH3CH2OH |
| 134.058 | C5H10O4 | OCHCH(OOH)OCH 2 CH 2 CH 3 | 9.45 (9.27) | CHOCHOO + CH3CH2CH2OH | |
| H2O | 78.031 | C2H6O3 | CH 3 CH(OH)OOH | 9.55* | CH3CHOO + H2O |
| O3 | 121.985 | C2H2O6 | CH 3 CH(OH)OOH | 10.42 | CHOCHOO + O3 |
Within the detection limit of our experiment (about 1 ppm), no reaction products corresponding to the reactions of CI-1 and CI-2 with water, ozone, alcohols, carboxylic acids, or hydroperoxides are observed in the range of experimental conditions investigated in this work. In particular, the lack of signals at m/z 78.031 (C2H6O3) and 92.011 (C2H4O4) corresponding to CI-1 + water and CI-2 + water products, respectively, is surprising as CIs are known to react with water molecules.56,86–89 The reaction of CI-1 with water has been investigated both experimentally and theoretically, including studies performed by our group, and found to lead to the formation of hydroxyethyl hydroperoxide (CH3CH(OH)OOH, m/z 78.031).56,89–92
The lack of signal at m/z 78.031 corresponding to hydroxyethyl hydroperoxide in this experiment could be due to its thermal decomposition at 390 K, which is the lowest reaction temperature measured in this work and is required to keep the crotonaldehyde reactant in the gas phase. This is consistent with the lack of hydroxyethyl hydroperoxide signal at temperatures above 350 K in the ozone-assisted oxidation reaction of trans-2-butene. Theoretical calculations performed by Wang et al. predict that fragments like acetaldehyde and hydrogen peroxide are produced as a result of hydroxyethyl hydroperoxide unimolecular decomposition.47 However, other unimolecular or bimolecular pathways might be responsible for hydroxyethyl hydroperoxide's low concentration.
Thermal decomposition might also explain the lack of signal at m/z 92.011 (C2H4O4) corresponding to the product formed as a result of CI-2 reaction with water. Wang et al. predicted that the unimolecular decomposition of 1-hydroperoxyprop-2-en-1-ol (CHO(OH)CHOOH) formed after reaction of CI-2 with water lead to the formation of glyoxal and hydrogen peroxide,47 both observed and identified in our product mass spectra. The detection of glyoxal in the product mass spectra cannot be solely attributed to the unimolecular decomposition of 1-hydroperoxyprop-2-en-1-ol as other reaction channels can lead to its formation.
O) bond of an aldehyde or ketone is recognized as a key step in the Criegee mechanism of ozonolysis.11,21 When CIs react with unsaturated aldehydes the reaction can proceed by CIs cycloaddition across either the olefinic CC bond or carbonyl –C(O) bond of the unsaturated aldehyde.93 Apart from the parent molecule, i.e., crotonaldehyde, three additional aldehyde compounds, i.e., formaldehyde, acetaldehyde, and propionaldehyde, are detected as co-products in the ozone-assisted oxidation of crotonaldehyde. In addition, one ketone, i.e., acetone is detected and identified as a co-product.
Reaction of CIs with formaldehyde. Previous studies performed in our laboratory using the same experimental arrangement and slightly different experimental conditions revealed that CI-1 formed in the ozone-assisted oxidation of trans-2-butene reacts with formaldehyde and leads to the formation of a secondary ozonide, cyc-CH(CH3)OOCH2O–, as shown in (R1).56
| CH3CHOO + HCHO ⇄ cyc-CH(CH3)OOCH2O– | (R1) |
Therefore, it is expected that the product mass spectra recorded in the temperature region below 600 K will contain a mass peak at m/z 90.031 (C3H6O3). Indeed, the product mass spectra recorded in this experiment display a peak at m/z 90.031. The corresponding PIE curve of m/z 90.031 (Fig. 6(a)) is in agreement with the formation of a cyc-CH(CH3)OOCH2O– compound as the ionization threshold of the C3H6O3 signal of 9.45 ± 0.05 eV agrees well with the theoretically calculated ionization energy of 9.41 eV.56 The breakpoints observed at 10.20 ± 0.05 eV and 10.50 ± 0.05 eV in the PIE curve of the signal measured in this work, similar to those observed in the trans-2-butene experiment, indicate that part of the signal recorded at m/z 90.031 might be attributed to the contribution of the secondary ozonide ring opening products, i.e., HOCH2OC(CH3)O and HOCH(CH3)OCHO. These assignments are supported by calculated ionization energies shown in Fig. 6(a). Furthermore, the PIE curve measured in this experiment at m/z 90.031 resembles the characteristics of the PIE curve measured in the trans-2-butene experiment. In Fig. 6(a), for comparison, the signal recorded at m/z 90.031 in this work (open squares) is represented together with the signal recorded for the same mass-to-charge ration in the trans-2-butene experiment (red curve).
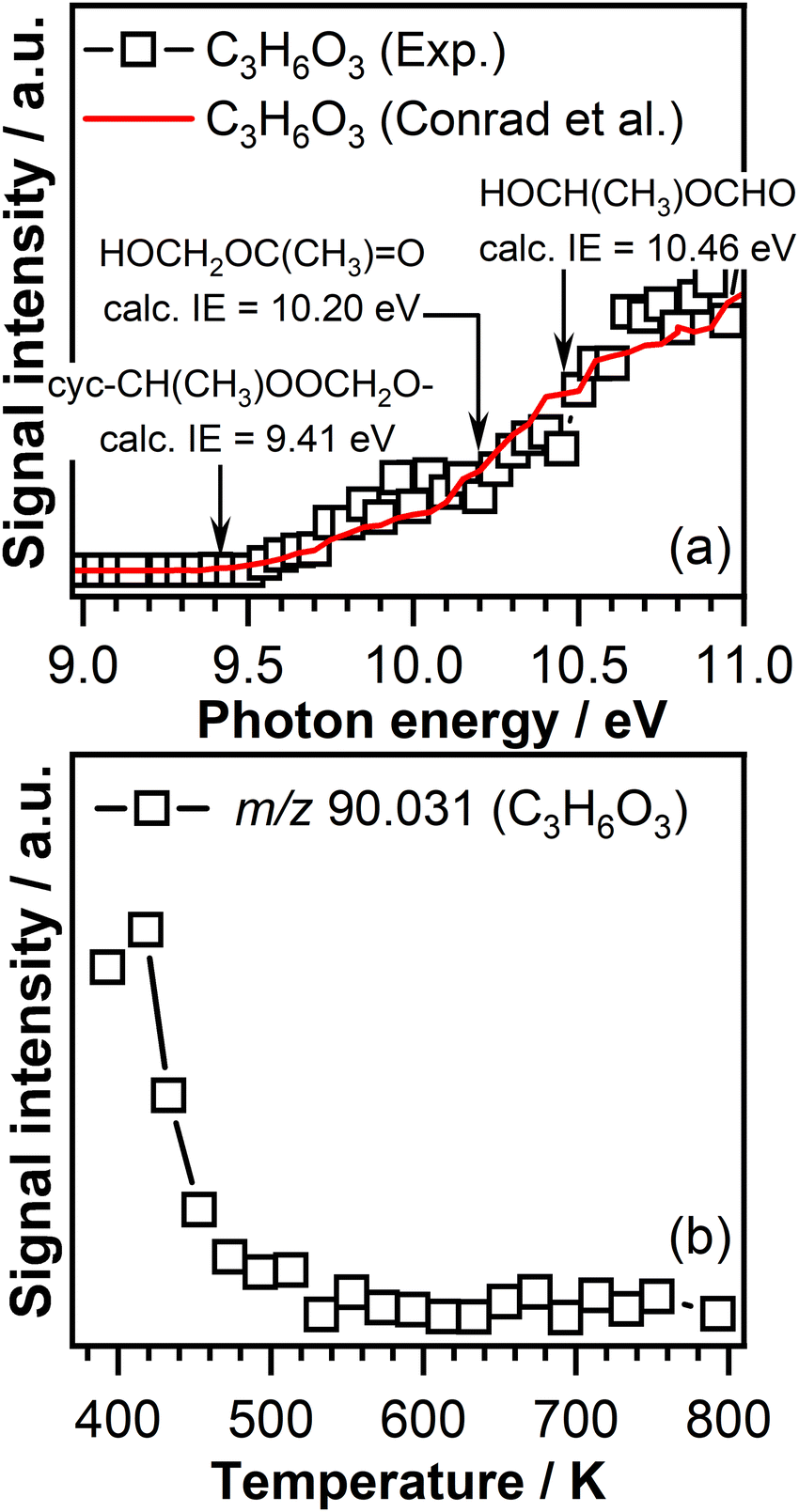 | ||
| Fig. 6 (a) Photoionization efficiency curve of m/z 90.031 (C3H6O3) signal measured at a temperature of 390 K (open symbol) and compared with spectra from Conrad et al. (red curve). (b) C3H6O3 signal intensity represented as a function of temperature recorded at a photon energy of 10.50 eV. | ||
Fig. 6(b) shows the C3H6O3 signal intensity as a function of temperature recorded at a photon energy of 10.50 eV. A signal is observed only in the temperature range below 600 K. The lack of C3H6O3 signal in the temperature region above 600 K where ozone thermally decomposes confirms that the formation of this product is due to the ozone-initiated reactions.
The reaction of CI-2 with formaldehyde is also expected to lead to the formation of a secondary ozonide, cyc-CH(CHO)OOCH2O–:
| CHOCHOO + HCHO ⇄ cyc-CH(CHO)OOCH2O– | (R2) |
The adiabatic ionization energy calculated in this work for the cyc-CH(CHO)OOCH2O– product is 9.69 eV. A very small signal at mass m/z 104.010 has been detected in the product mass spectra. However, the measured signal is close to the detection limit of our experiment and the measured PIE is not conclusive enough to unambiguously assign the signal recorded at m/z 104.010 to a product formed due to the reaction of CI-2 with formaldehyde. The small signal recorded at m/z 104.010 might be due to the unimolecular decomposition of cyc-CH(CHO)OOCH2O–. Calculations performed by Wang et al.47 found that products like glyoxal and formic acid are expected after the reaction of CI-2 with formaldehyde and cyc-CH(CHO)OOCH2O– unimolecular decomposition. Both species are detected and identified under our experimental conditions, however the presence of these species in the product mass spectra cannot be solely attributed to unimolecular decomposition cyc-CH(CHO)OOCH2O– as other reaction channels can lead to their formation.
Reaction of CIs with acetaldehyde. The CI-1 and CI-2 are also expected to react with acetaldehyde, and lead to the formation of cyclic adducts, SOZs, according to the following:
| CH3CHOO + CH3CHO ⇄ cyc-CH(CH3)OOCH(CH3)O– | (R3) |
| CHOCHOO + CH3CHO ⇄ cyc-CH(CHO)OOCH(CH3)O– | (R4) |
The product mass spectra recorded at temperatures below 600 K display signals at m/z 104.047 (C4H8O3) and m/z 118.026 (C4H6O4) that might correspond to the mass of potential products formed by the CI-1 + acetaldehyde and CI-2 + acetaldehyde reactions, respectively. Fig. 7 displays the PIE curve of m/z 104.047 (C4H8O3) recorded at a reaction temperature of 390 K. The temperature profile of the C4H8O3 product measured at 10.50 eV (shown in Fig. S2(a) in the ESI†) displays a signal only in the region where ozone is present, while the intensity of the signal decreases with increasing temperature to vanish around 550 K. This behavior is indicative of a product that is formed due to ozone reactions. Previous experiments performed in our laboratory to understand the reaction network of CH3CHOO under slightly different experimental conditions did not find any evidence for the formation of SOZs.56
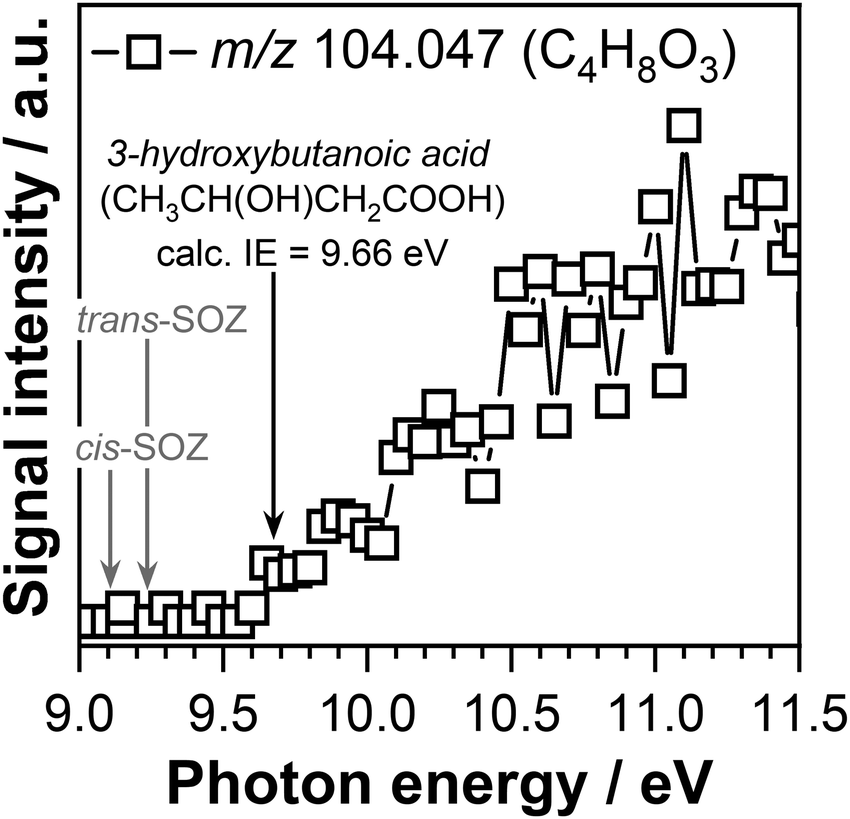 | ||
| Fig. 7 Photoionization efficiency curve of m/z 104.047 (C4H8O3) signal measured at a temperature of 390 K. The adiabatic ionization energy of 3-hydroxybutanoic acid calculated in this work is indicated with a black arrow. Gray arrows indicate the calculated adiabatic ionization energies of the cis-SOZ and trans-SOZ adopted from ref. 56. | ||
The calculated adiabatic ionization energy of trans-SOZ and cis-SOZ of 9.23 eV and 9.10 eV reported in our previous studies do not to match the ionization threshold of 9.60 ± 0.05 eV observed in this work for the C4H8O3 (m/z 104.047) signal. However, the observed experimental ionization threshold matches the adiabatic ionization energy of 9.66 eV calculated in this work for 3-hydroxybutanoic acid. Interesting, the temperature profile of the m/z 104.047 mass peak recorded at 10.50 eV presents a signal only in the temperature region below 600 K, the temperature range where ozone is present. This observation indicates that 3-hydroxybutanoic acid is formed through a reaction pathway that involves OH and O3 reaction, but not CI-1 (according to the Fig. S3 in the ESI†).
The recombination reaction of CI-2 and acetaldehyde, which are the products of POZ unimolecular decomposition, is expected to lead to the formation of the initial SOZ (C4H6O4, m/z 118.026). The origin of the signal observed in our experiment at m/z 118.026 is discussed in Section 3.1.
Reaction of CIs with propionaldehyde and acetone. The product mass spectrum recorded at temperatures below 600 K also displays mass peaks at m/z 118.063 (C5H10O3) and 132.042 (C5H8O4) that might be formed as a result of the reaction of another aldehyde co-product, i.e. propionaldehyde with CI-1 and CI-2, respectively. However, these mass peaks can also be attributed to the reaction of CIs with acetone, as propionaldehyde (CH3CH2CHO) and acetone (CH3COCH3) are structural isomers, and both have been detected and identified as co-products under our experimental conditions. Consequently, the signals observed at m/z 118.063 (C5H10O3) and 132.042 (C5H8O4) might be due to the reaction of CI-1 and CI-2 with propionaldehyde and/or acetone, respectively. The reactions of both propionaldehyde and acetone with CIs are expected to proceed by 1,3-cycloaddition of CIs across the carbonyl –C(
O) double bound to form cyclic ozonides intermediates as follow:| CH3CHOO + CH3CH2CHO ⇄ cyc-CH(CH3)OOCH(CH2CH3)O– | (R5) |
| CH3CHOO + CH3COCH3 ⇄ cyc-CH(CH3)OOC(CH3)2O– | (R6) |
| CHOCHOO + CH3CH2CHO ⇄ cyc-CH(CHO)OOCH(CH2CH3)O– | (R7) |
| CHOCHOO + CH3COCH3 ⇄ cyc-CH(CHO)OOC(CH3)2O– | (R8) |
CIs are known to preferentially react with aldehydes over ketones. To our knowledge neither the reaction of CI-1 or CI-2 with propionaldehyde or acetone have been investigated. Experimental and theoretical investigations have been performed to understand the reaction behavior of the smallest CI (CH2OO) with acetone and aldehydes like acetaldehyde, formaldehyde, and propionaldehyde. The SOZs stabilization and the mechanism of these reactions was found to depend on the reaction temperature and pressure.11,94,95
The PIE curves of the C5H10O3 (m/z 118.063) and C5H8O4 (m/z 132.042) signals are shown in Fig. 8. The ionization threshold of the C5H10O3 signal displayed in Fig. 8(a) is observed around 9.70 ± 0.05 eV. Indicated by a gray arrow in Fig. 8(a) is the adiabatic ionization energy of the cyc-CH(CH3)OOC(CH3)2O– ozonide, formed in CI-1 + acetone reaction, calculated in this work. The calculated adiabatic ionization energy of 8.99 eV is far below the measured ionization threshold, indicating that this compound does not contribute to the observed signal. The calculated adiabatic ionization energy of the cyc-CH(CH3)OOCH(CH2CH3)O– ozonide, formed as a result of CI-1 + propionaldehyde reaction, has a value of 9.24 eV, which is close to the measured ionization threshold, but still below the measured ionization threshold of C5H10O3 signal. Previous investigations in which the reaction of CIs with aldehydes was investigated revealed that the cyclic ozonide can undergo ring opening and lead to the formation of linear compounds.54,56 The adiabatic ionization energy of the CH3C(O)OCH(OH)CH2CH3 and CH3CH(OH)OC(O)CH2CH3 ring opening adducts calculated in this work are 9.83 eV and 9.81 eV, respectively.
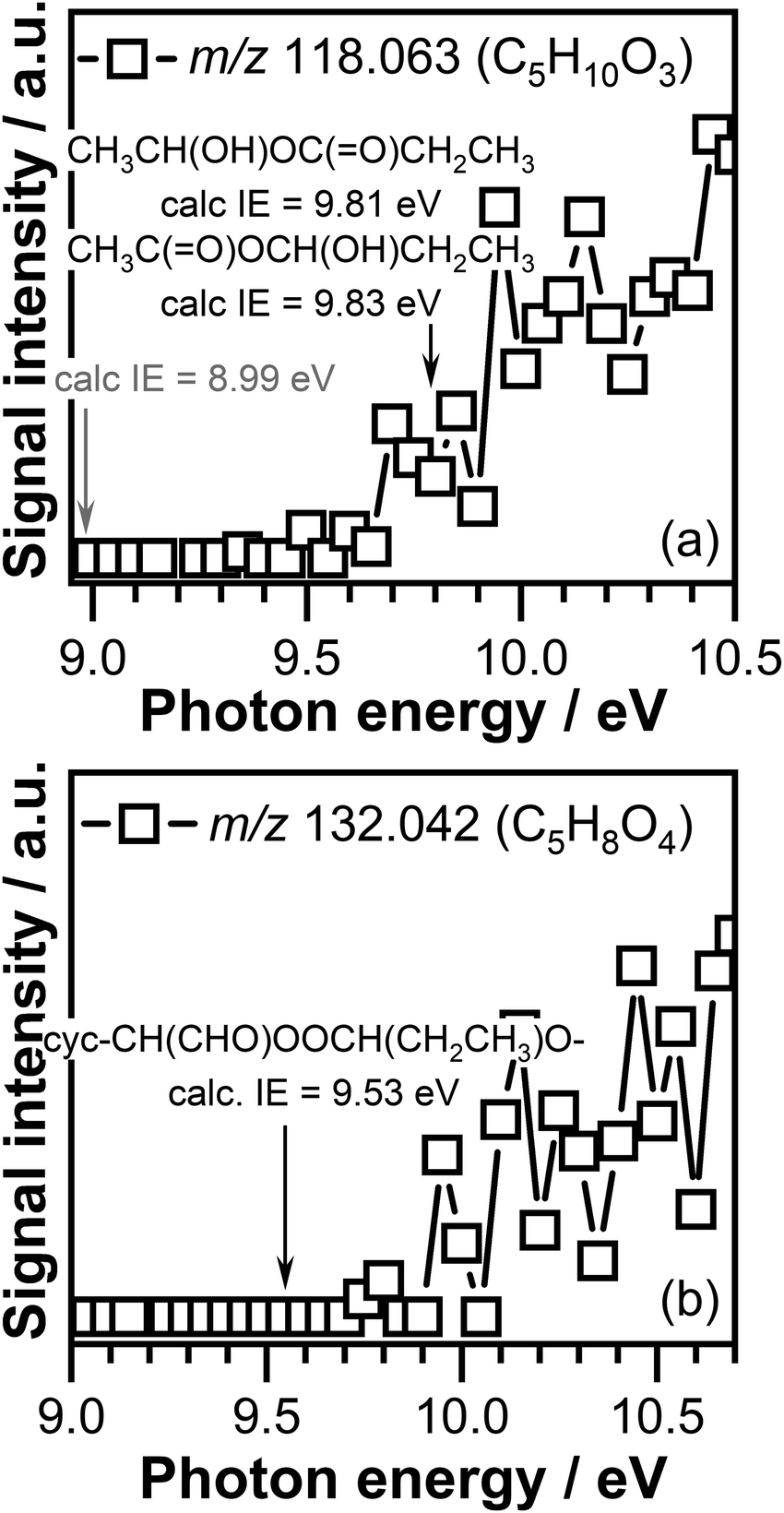 | ||
| Fig. 8 Photoionization efficiency curve of the (a) C5H10O3 (m/z 118.063) signal and (b) C5H8O4 (m/z 132.042) signal measured at a temperature of 390 K. The adiabatic ionization energy of the potential CIs reaction products calculated in this work are indicated with an arrow. The gray arrow indicates the adiabatic ionization energy of the cyc-CH(CH3)OOC(CH3)2O– ozonide of the CI-1 + acetone reaction. | ||
Within experimental error the measured ionization threshold of the C5H10O3 (m/z 118.063) signal matches the adiabatic ionization energy values calculated for the ring opening products. As the calculated values are very similar, we are unable to distinguish between the contribution of these isomers to the observed C5H10O3 (m/z 118.063) signal. Consequently, the observed C5H10O3 signal can be attributed to either the CH3C(O)OCH(OH)CH2CH3 compound or the CH3CH(OH)OC(O)CH2CH3 compound, or both.
The ionization threshold measured for the C5H8O4 (m/z 132.042) signal, was observed around 9.75 ± 0.05 eV (see Fig. 8(b)). This value is much higher than the calculated adiabatic energy of the cyclic ozonide cyc-CH(CHO)OOC(CH3)2O– of the CI-2 + acetone product of 9.35 eV, indicating that this compound does not contribute to the signal observed at m/z 132.042 (C5H8O4). The calculated ionization energy value of 9.53 eV of the cyclic ozonide cyc-CH(CHO)OOCH(CH2CH3)O– product, formed in the CI-2 + propionaldehyde reaction is about 0.2 eV lower than the measured ionization threshold of C5H8O4 signal, which is greater than expected uncertainties of our experimental and theoretical procedures. Experimental photoionization thresholds are typically less sharp for larger species than for small species, considering that large species can exist in multiple low-lying conformers with weak Franck–Condon overlaps. Combined with a low ionization cross-section and signal close to the detection threshold, characteristic to reactive intermediates, that might lead to a detectable signal at energies slightly above the ionization threshold. Therefore, the C5H8O4 (m/z 132.042) signal might be attributed in part to the cyc-CH(CHO)OOCH(CH2CH3)O– cyclic ozonide. Calculations performed in this work also revealed that the ring opening products of this cyc-CH(CHO)OOCH(CH2CH3)O– cyclic ozonide, i.e., CHOC(O)OCH(OH)CH2CH3 and CHOCH(OH)OC(O)CH2CH3 are unstable and will decompose upon ionization via CO2 or HCO loss.
Reaction of CIs with crotonaldehyde. The product mass spectra recorded at temperatures below 600 K show mass peaks at m/z 130.062 (C6H10O3) and 144.042 (C6H8O4) that might correspond to products formed in the reactions of CI-1 and CI-2 with crotonaldehyde, respectively. The temperature profile of the C6H10O3 and C6H8O4 compounds (shown in Fig. S2(b) and (c), ESI†) display signals already at 390 K that monotonically decrease with increasing temperature to vanish around 550 K, which corresponds to the temperature to which ozone almost fully decomposes. This indicates that the C6H10O3 and C6H8O4 products are formed by ozone-assisted oxidation reactions.
As mentioned above, both the olefinic CC and carbonyl –C(O) bonds of crotonaldehyde are susceptible to CIs attack via cycloaddition to form five-membered ring adducts. CIs may also insert their terminal oxygen atom or insert themself into the C–H bond of crotonaldehyde. No information is available about the reactions of CI-1 or CI-2 with crotonaldehyde. The reaction of the smallest Criegee intermediate, CH2OO, with the smallest unsaturated aldehyde, i.e., acrolein has been investigated by Sun et al.93 This study revealed that the CIs addition reactions are energetically more favorable than the insertion reactions, with calculations indicating that the most competitive reaction channel is the 1,3-cycloaddition of CH2OO across the CO double bond to form a cyclic ozonide. In addition, the branching ratio of collisional stabilized SOZ was found to depend on the reaction pressure and was observed to increase with increasing pressure.
The addition of CI-1 and CI-2 to the CC or –C(O) bonds of crotonaldehyde are expected to lead to the formation of the following adducts:
| CH3CHOO + CH3CHCHCHO ⇄ cyc-CH(C3H5)OOCH(CH3)O– | (R9) |
| CH3CHOO + CH3CHCHCHO ⇄ cyc-CH(CH3)OOCH(CH3)CH(HCO)– | (R10) |
| CH3CHOO + CH3CHCHCHO ⇄ cyc-CH(CH3)CH(CH3)OOCH(HCO)– | (R11) |
| CHOCHOO + CH3CHCHCHO ⇄ cyc-CH(C3H5)OOCH(HCO)O– | (R12) |
| CHOCHOO + CH3CHCHCHO ⇄ cyc-CH(HCO)OOCH(CH3)CH(HCO)– | (R13) |
| CHOCHOO + CH3CHCHCHO ⇄ cyc-CH(CH3)CH(HCO)OOCH(HCO)– | (R14) |
Fig. 9 displays the PIE curves of the C6H10O3 and C6H8O4 compounds recorded at 390 K. The ionization threshold of the C6H10O3 signal, a potential product of CI-1 reaction with crotonaldehyde, is observed at 9.30 ± 0.05 eV (see Fig. 9(a)). The adiabatic ionization energies calculated in this work for the cyc-CH(C3H5)OOCH(CH3)O–, cyc-CH(CH3)OOCH(CH3)CH(HCO)–, and cyc-CH(CH3)CH(CH3)OOCH(HCO)– cyclic adducts (see (R9)–(R14) reactions) are 9.63 eV, 8.96 eV, and 9.10 eV, respectively. As can be seen in Fig. 9(a), no signal corresponding to the cyc-CH(CH3)OOCH(CH3)CH(HCO)– adduct, that can be formed by CI-1 addition to the olefinic CC bond of crotonaldehyde, is detected under our experimental conditions as no signal is present in the energy range (8.96 eV) where this compound is expected to be ionized.
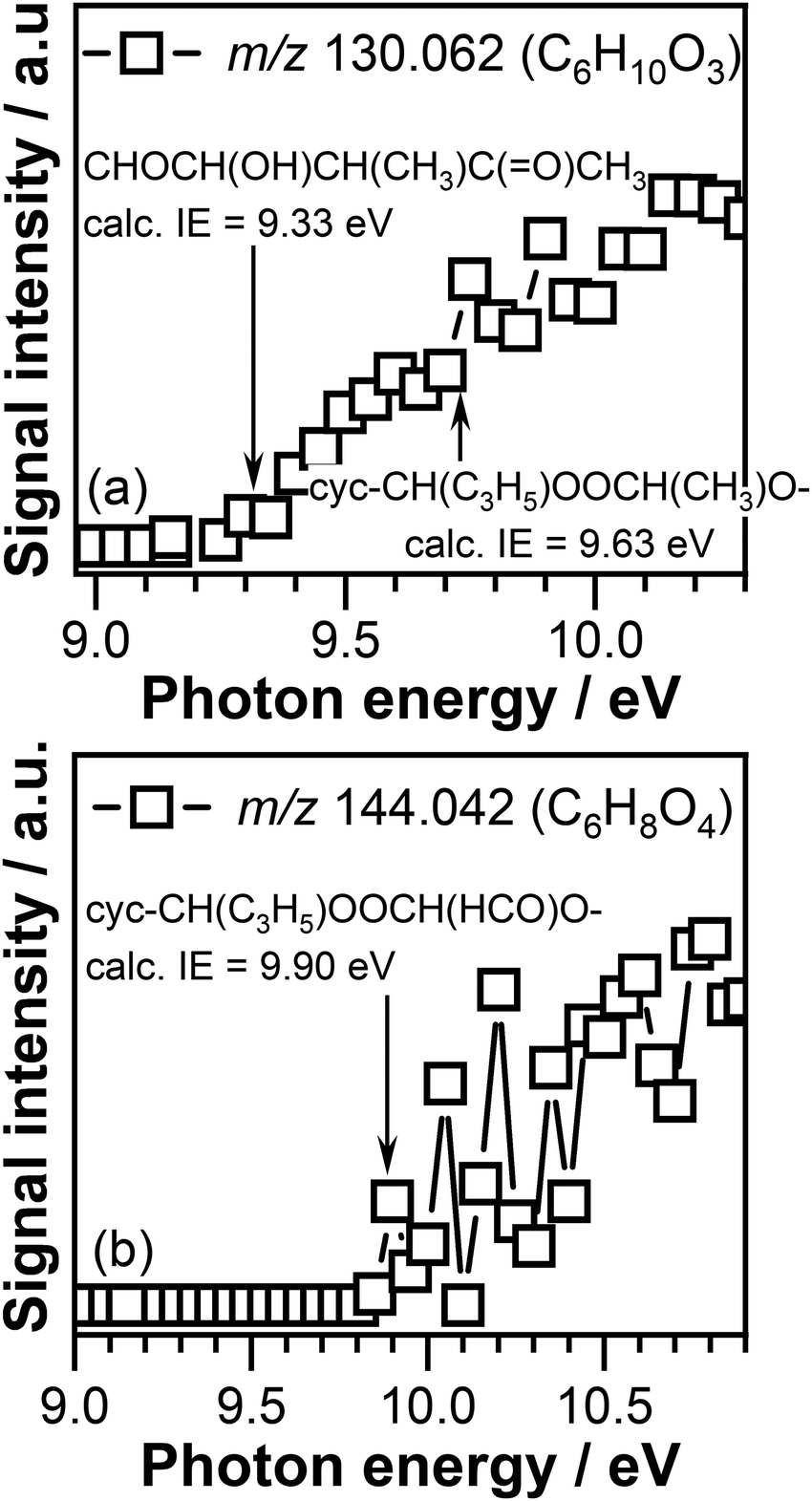 | ||
| Fig. 9 Photoionization efficiency curve of the signal recorded at (a) m/z 130.062 and (b) m/z 144.042 at a temperature of 390 K. The arrows indicate the adiabatic energy for the CHOCH(OH)CH(CH3)C(O)CH3, cyc-CH(C3H5)OOCH(HCO)O–, and cyc-CH(C3H5)OOCH(CHO)O– compounds, calculated in this work. | ||
The adiabatic ionization energy of the cyc-CH(CH3)CH(CH3)OOCH(HCO)– adduct of 9.10 eV, another product that can potentially form as a result of CI-1 cycloaddition across the CC olefinic bond of crotonaldehyde, is about 0.2 eV below the experimentally observed ionization threshold of 9.30 ± 0.05 eV. Due to the large size of this molecule that might result in a photoionization threshold that is typically less sharp than for small species, as explained above, we cannot exclude that part of the observed signal to be due to the presence of cyc-CH(CH3)CH(CH3)OOCH(HCO)– adduct. This observation is consistent with the results of Sun et al. who reported the preferential addition of the CH2OO Criegee intermediate to acrolein olefinic CC bound.93 In addition, the adiabatic ionization energy of one of the cyc-CH(CH3)CH(CH3)OOCH(HCO)– ring opening products, i.e., CHOCH(OH)CH(CH3)C(O)CH3, of 9.33 eV calculated in this work, perfectly matches the measured ionization threshold of the C6H10O3 signal, clearly indicating that this product likely contributes to the observed signal.
In addition, the calculated adiabatic ionization energy of cyc-CH(C3H5)OOCH(CH3)O– of 9.63 eV appears to match the breakpoint observed in the measured signal at 9.70 ± 0.05 eV. This indicates that part of the observed signal might correspond to the adduct formed as a result of CI-1 addition to the –C(O) carbonyl bond of crotonaldehyde. However, without a complete PIE of this product no definitive conclusions can be drawn about the contribution of the cyc-CH(C3H5)OOCH(CH3)O– compound.
The ionization threshold of the C6H8O4 signal that corresponds to the mass of potential CI-2 + crotonaldehyde reaction products, displayed in Fig. 9(b), is observed at 9.85 ± 0.05 eV. This value is above the adiabatic ionization energy of the cyc-CH(HCO)OOCH(CH3)CH(HCO)– and cyc-CH(CH3)CH(HCO)OOCH(HCO)– products of 9.23 eV and 9.49 eV, respectively, calculated in this work for the products formed as a result of CI-2 cycloaddition to the CC bond of crotonaldehyde. Interesting enough, the calculated adiabatic energy of the cyc-CH(C3H5)OOCH(HCO)O– adduct of 9.90 eV (a low but not the lowest energy conformer of cyc-CH(C3H5)OOCH(HCO)O–) formed by the CI-2 addition to the –C(O) bonds of crotonaldehyde matches the experimentally measured ionization threshold. The preference of the glyoxal oxide Criegee intermediate for the –C(O) bond of crotonaldehyde over the CC bond is interesting, and further investigations are required to understand the details of this reaction.
C bond to form a 1,2-dioxolane. The cyclic peroxide formed in CI + alkene reactions are known to be significantly more stable than the cyclotrioxolane (the “primary ozonide”, POZ) formed in the ozonolysis of alkenes. Stationary point calculations performed by Vereecken et al. at the M06-2X/aVTZ and CCSD(T)/aVTZ//M06-2X levels, for several reactions of Criegee intermediates with alkenes, revealed that these reactions exhibit weakly bound pre-reactive complexes and barriers to cycloaddition that are lower in energy than the corresponding barriers for addition of ozone to alkenes, and in particular cases lower than the energy of the reactants.96
The reaction of CIs with alkene was first observed and reported by Story and Burgess, who investigated the ozonolysis reaction with tetramethylethylene.97 Since then, products corresponding to the reaction of CIs with alkenes have been observed and reported by various laboratories including our own.54,56,98
The mass spectra recorded at temperatures below 600 K display peaks at m/z 88.052 (C4H8O2) and 102.032 (C4H6O3), corresponding to the mass of the potential products of the reaction of ethene (C2H4) with CI-1 and CI-2, respectively. Both products display a signal in the temperature region where ozone is present (see Fig. S2(d) and (e), ESI†), indicative of products that are formed as a result of ozone driven reactions. The C4H8O2 (m/z 88.052) product indicative of CI-1 + ethene reactions have been reported in previous studies in which the ozone-assisted oxidation reaction of methylcrotonate has been investigated.99 However, the exact chemical identity of the C4H8O2 signal has not been confirmed in this study, and the assignment of the peak was exclusively based on the expected reaction of CIs with alkenes and the presence of a signal exclusively in a temperature region where ozone is present, indicative of ozone initiated oxidation pathways. In addition, the C4H8O2 product was observed by our group in the ozonolysis of trans-2-butene.56 The observed C4H8O2 signal has been attributed in part to but-1-enyl-3-hydroperoxide based on the experimentally observed ionization threshold.
The ionization threshold of the C4H8O2 signal measured in this experiment at a reaction temperature of 390 K is observed around 9.50 ± 0.05 eV (see Fig. 10(a)). However, the signal detected at this energy is close to detection limit of our experiment. Calculations performed in this work reveals that the adiabatic ionization energy of cyc-CH2CH2CH3CHOO– adduct formed in the reaction of CI-1 with ethene has an ionization energy of 8.92 eV, which is far below the measured ionization energy of the C4H8O2 signal, indicating that cyc-CH2CH2CH3CHOO– is not stable under our experimental conditions. Once formed cyc-CH2CH2CH3CHOO– can undergo ring opening to form hydroxide adducts, i.e. 3-hydroperoxybut-1-ene (CH2CHCH(OOH)CH3) or but-2-enyl-1-hydroperoxide (CH3CHCHCH2OOH). The adiabatic ionization energy calculated in this work for 3-hydroperoxybut-1-ene of 9.36 eV is slightly below the measured ionization threshold of the C4H8O2 signal. Within experimental uncertainty, the calculated value of 9.41 eV of the 2-enyl-1-hydroperoxide adduct matches the experimentally observed ionization threshold of the C4H8O2 signal. The breakpoint observed in the PIE curve of the C4H8O2 signal (see Fig. 10(a)) around 10.20 ± 0.05 eV matches the ionization energy of 10.17 eV reported in the literature for butanoic acid.100 Interesting enough, the PIE curve measured in our experiment resembles the characteristics of the PIE curve reported by Zhai et al.101 for the C4H8O2 signal, which was attributed to the contribution of but-1-enyl-3-hydroperoxide, but-2-enyl-1-hydroperoxide, and butanoic acid. Similar to our experiment a clear onset in the PIE signal of the C4H8O2 signal was observed only at an ionization energy of 9.50 ± 0.05 eV, with a very small signal in the energy region where hydroperoxide products are expected to be ionized. Based on these observations we attributed the C4H8O2 signal to the presence of but-2-enyl-1-hydroperoxide, and butanoic acid.
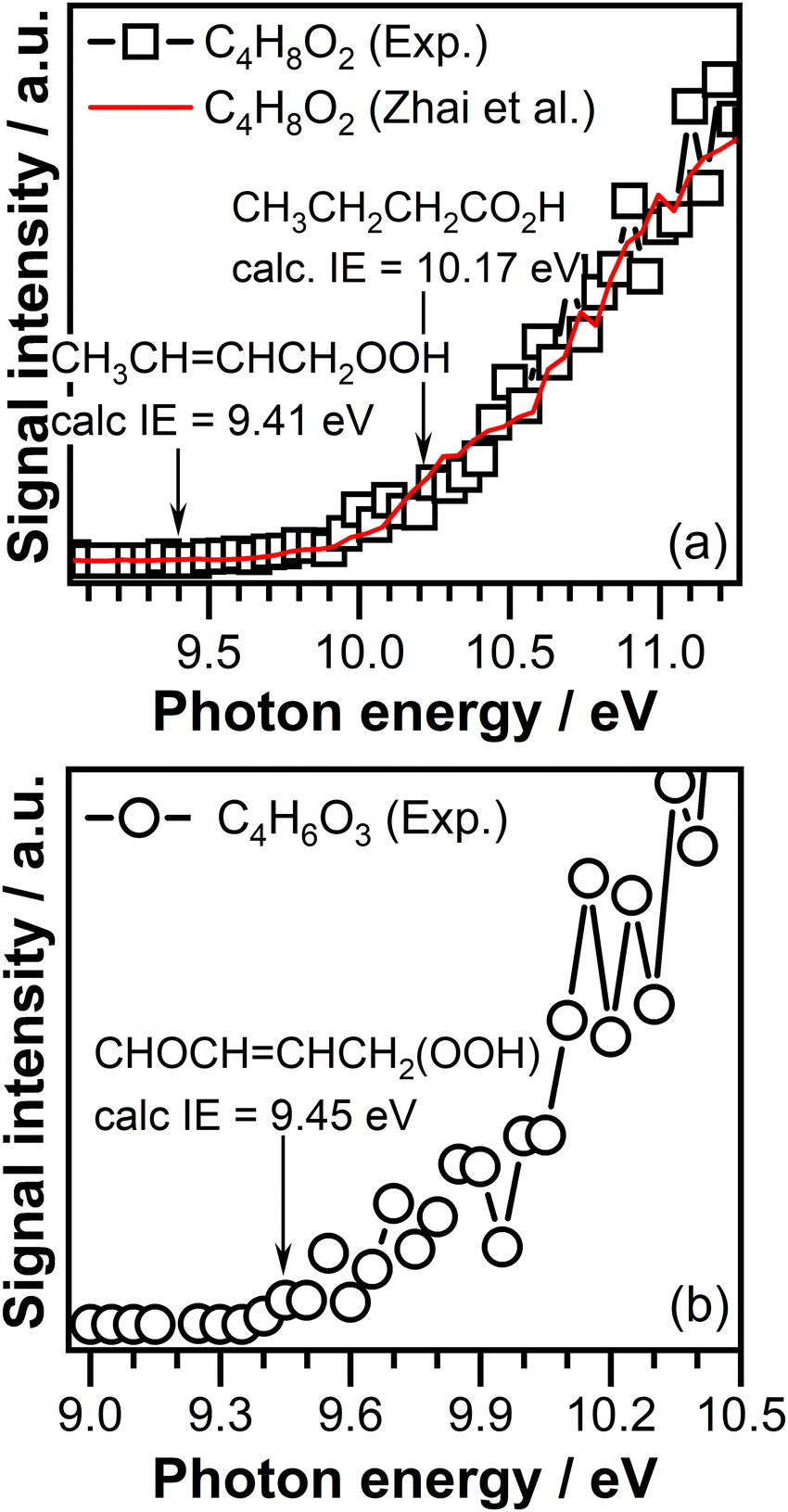 | ||
| Fig. 10 Photoionization efficiency curve of the (a) m/z 88.052 (C4H8O2) signal compared with the photoionization ionization curve of the C4H8O2 signal reported by Zhai et al.101 and (b) m/z 102.032 (C4H6O3) signal recorded at a reaction temperature of 390 K. The arrows in panel (a) indicate the ionization energy of but-2-enyl-1-hydroperoxide calculated in this work and the ionization energy of butanoic acid.100 The arrows in panel (b) indicate the ionization energy of cyc-CH2CH2CHOCHOO– cyclic compound and its ring opening adduct CH2CHCH(OOH)CHO, calculated in this work. | ||
Fig. 10(b) shows the PIE curve of the C4H6O3 (m/z 102.032) signal. The ionization threshold of this signal is 9.40 ± 0.05 eV. Calculations performed in this work revelated that the adiabatic ionization energy of the cyc-CH2CH2CHOCHOO– adduct that can be formed as a result of CI-2 addition across the double bond of ethene has a value of 9.35 eV, which is consistent with the observed ionization threshold. However, calculations reveal that the adiabatic ionization energy of the CH2CHCH(OOH)CHO and CHOCHCHCH2(OOH) hydroperoxides that might be formed due to the cyc-CH2CH2CHOCHOO– ring opening are 9.34 eV and 9.65 eV, respectively. Conformers with ionization energies down to 9.45 eV have been found for the CHOCHCHCH2(OOH) hydroperoxide. As the calculated adiabatic energies of the potential products are very similar and consistent with the observed ionization threshold of the C4H6O3 signal, the observed signal can be attributed to any of these products. Additional investigations are required to understand the origin of the C4H6O3 signal.
The product mass spectra recorded at temperatures below 600 K is also characterized by the presence of signals at m/z 102.068 (C5H10O2) and m/z 116.047 (C5H8O3) that might correspond to the products formed due to the reaction of CI-1 and CI-2 with propene (CH3CHCH2), respectively. However, the ionization threshold of the C5H10O2 (m/z 102.068) of 10.05 ± 0.05 eV is much higher than the calculated adiabatic energy of the any potential products, i.e., cyc-CH2CH(CH3)OOCH(CH3)– (8.76 eV), cyc-CH(CH3)CH2OOCH(CH3)– (8.88 eV), CH3CH(OOH)CHCHCH3 (9.16 eV, with conformers at 9.28 eV), CH3CHCH–CH(OOH)CH3 (9.17 eV), CH3CH(OOH)C(CH2)CH3 (9.30 eV), and CH3CHC(CH2OOH)CH3 (8.85 eV). Therefore, the formation of significant amounts of products as a result of the CI-1 + propene reaction is excluded under our experimental conditions.
The PIE curve of the C5H8O3 (m/z 116.047) signal is shown in Fig. 11. The ionization threshold of this signal is observed around 9.30 ± 0.05 eV. This value matches the calculated adiabatic ionization energy of 9.31 eV of the cyc-CH2(CH3)CHOOCH(HCO)– cyclic compound formed by CI-2 addition across the CC double bound of propene. The PIE of the C5H8O3 signal shown in Fig. 11 has a breakpoint around 9.75 ± 0.05 eV indicating that more than one compound contributes to the observed signal.
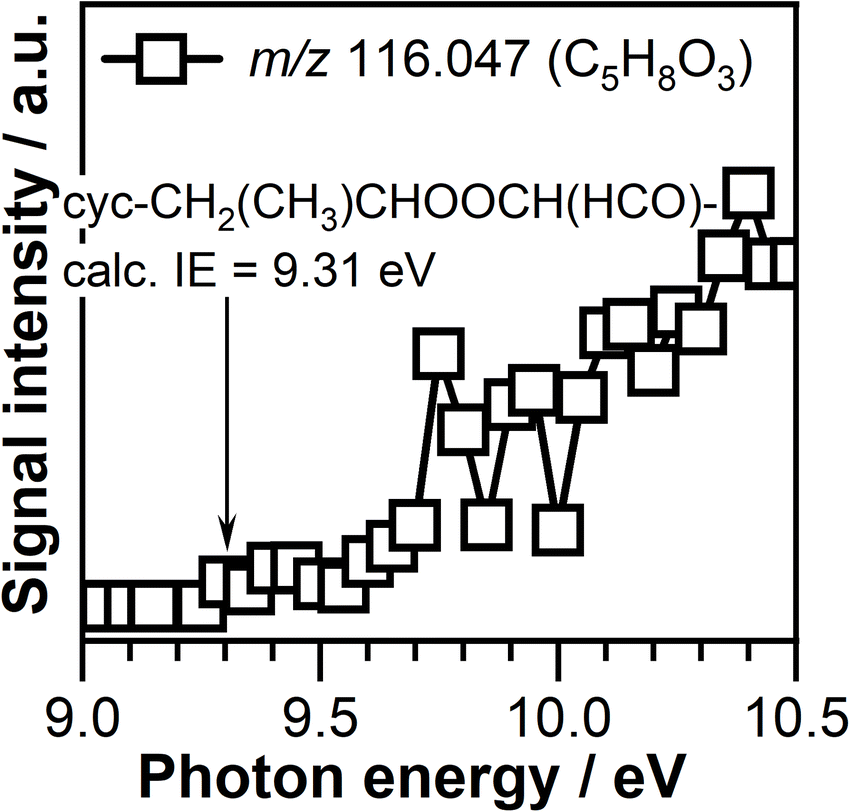 | ||
| Fig. 11 Photoionization efficiency curve of m/z 116.047 (C5H8O3) signal measured at a temperature of 390 K. The arrow indicates the adiabatic ionization energy of the cyc-CH2(CH3)CHOOCH(HCO) compound. | ||
As mentioned above, the product mass spectra recorded at temperatures below 600 K are characterized by clusters of peaks of up to 14 heavy atoms. In addition, the product mass spectra are abundant in high molecular weight peaks at odd masses. This suggests that high molecular weight products, i.e., oligomers might be formed but are not stable under our experimental conditions. In addition, dissociative photoionization might be also responsible for the fragmentation of high molecular weight products.
4. Conclusions
The reaction network of two Criegee intermediates with different functionalities, i.e., aldehyde oxide and glyoxal oxide, formed in the ozone-assisted oxidation reaction of crotonaldehyde is investigated in a jet-stirred reactor in conjunction with photoionization molecular beam mass spectrometry in a range of temperatures between 390 K and 840 K. Products corresponding to both CIs unimolecular and bimolecular reaction channels are detected and identified based on experimental ionization efficiency curve and ab initio threshold energy calculations. The presence of ozone is observed to trigger the oxidation of crotonaldehyde already at 390 K, the lowest temperature required to keep crotonaldehyde in the gaseous phase. This temperature is about 200 K below the temperature where crotonaldehyde was observed to be oxidized in the presence of oxygen alone.77 The temperature region below 600 K is characterized by the presence of peaks characteristics to ozonolysis and OH-driven low-temperature oxidation reactions. The low-temperature oxidation channel is responsible for the formation of important amounts of hydroperoxides like H2O2, CH3OOH, and CH3CH2OOH, as well as alcohols, ketones, carboxylic acids, and aldehydes.The crotonaldehyde + O3 adduct is identified as a ketohydroperoxide (2-hydroperoxy-3-oxobutanal), while the observation of glycolaldehyde is attributed to the unimolecular isomerization of acetaldehyde oxide CIs. The lack of signal indicative of Criegee intermediates suggests that these compounds either undergo fast reactions or the stabilization reactions of the activated complex are less favorable in the temperature range investigated in this study.
Despite a reactive environment rich in water, hydroperoxides, aldehydes, alcohols, ketones, carboxylic acids, and alkenes co-products only reaction products corresponding to the reaction of CIs with aldehydes (formaldehyde, acetaldehyde, and crotonaldehyde) and alkenes (ethene and propene) are observed under our experimental conditions. The detection of reaction products with up to 14 heavy atoms indicates that CIs assisted oligomerization might be occurring under our experimental conditions. The lack of clear peaks at masses corresponding to the CIs subsequent addition might be in part attributed to the fact that some of these products are not stable under our experimental conditions or undergo dissociative photoionization. That is confirmed by the presence of molecular peaks at high masses with an unpaired number of electrons. These findings are of particular interest as the CIs induced oligomerization reactions are considered to contribute to the SOA atmospheric budget. Additional investigations are required to fully understand the kinetic behavior of the C2 Criegee intermediates with different functionalities.
In addition, this study provides insights into a less characterized low temperature combustion enhancement pathway facilitated by the ozone. This is important as ozone is one of the major products in non-equilibrium plasma assisted combustion for fuel lean homogeneous charge compression ignition enhancement.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Early Career Research program under Award Number DE-SC0023004. A. C. D. acknowledges partial support from UCF Office of Research through the Seed Funding Program. A. M. F. and N.-E. M. acknowledge partial support from UCF Office of Undergraduate Research through the UCF Student Research Grant and the UCF Summer Undergraduate Research Fellowship. N. H. and A. W. J. are supported by the Gas-Phase Chemical Physics program of the Chemical Science, Geosciences, and Biosciences Division of the US Department of Energy Basic Energy Sciences. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC (NTESS), a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy's National Nuclear Security Administration (DOE/NNSA) under contract DE-NA0003525. This written work is authored by an employee of NTESS. The employee, not NTESS, owns the right, title and interest in and to the written work and is responsible for its contents. Any subjective views or opinions that might be expressed in the written work do not necessarily represent the views of the U.S. Government. The publisher acknowledges that the U.S. Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this written work or allow others to do so, for U.S. Government purposes. The DOE will provide public access to results of federally sponsored research in accordance with the DOE Public Access Plan. P. D. acknowledges support from the CAPRYSSES project (ANR-11-LABX-006-01) funded by ANR through the PIA (Programme d’Investissement d’Avenir). We gratefully acknowledge computing resources provided by the Laboratory Computing Resource Center at Argonne National Laboratory. The Advanced Light Source is a U.S. Department of Energy (DOE) Scientific User Facility supported by the Director, Office of Science, Basic Energy Sciences Program under Contract Number DE-AC02-05CH11231.References
- R. A. Cox and S. A. Penkett, Nature, 1971, 230, 321–322 CrossRef CAS PubMed.
- R. A. Cox and S. A. Penkett, J. Chem. Soc., Faraday Trans. 1, 1972, 68, 1735–1753 RSC.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2014, 16, 1704–1718 RSC.
- D. L. Osborn and C. A. Taatjes, Int. Rev. Phys. Chem., 2015, 34, 309–360 Search PubMed.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- R. L. Caravan, M. F. Vansco and M. I. Lester, Commun. Chem., 2021, 4, 44 CrossRef CAS PubMed.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. Anwar, H. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., Int. Ed., 2014, 53, 4547–4550 CrossRef CAS PubMed.
- R. Chhantyal-Pun, M. R. McGillen, J. M. Beames, M. A. H. Khan, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Angew. Chem., Int. Ed., 2017, 56, 9044–9047 CrossRef CAS PubMed.
- W.-m Wei, X. Yang, R.-h Zheng, Y.-d Qin, Y.-k Wu and F. Yang, Comput. Theor. Chem., 2015, 1074, 142–149 CrossRef CAS.
- A. Jalan, J. W. Allen and W. H. Green, Phys. Chem. Chem. Phys., 2013, 15, 16841–16852 RSC.
- R. M. I. Elsamra, A. Jalan, Z. J. Buras, J. E. Middaugh and W. H. Green, Int. J. Chem. Kinet., 2016, 48, 474–488 CrossRef CAS.
- M. R. McGillen, B. F. E. Curchod, R. Chhantyal-Pun, J. M. Beames, N. Watson, M. A. H. Khan, L. McMahon, D. E. Shallcross and A. J. Orr-Ewing, ACS Earth Space Chem., 2017, 1, 664–672 CrossRef CAS.
- S. V. Tadayon, E. S. Foreman and C. Murray, J. Phys. Chem. A, 2018, 122, 258–268 CrossRef CAS PubMed.
- Y.-H. Lin, C. Yin, W.-H. Lin, Y.-L. Li, K. Takahashi and J. J.-M. Lin, J. Phys. Chem. Lett., 2018, 9, 7040–7044 CrossRef CAS PubMed.
- W. Chao, Y.-H. Lin, C. Yin, W.-H. Lin, K. Takahashi and J. J.-M. Lin, Phys. Chem. Chem. Phys., 2019, 21, 13633–13640 RSC.
- C. Cabezas and Y. Endo, Phys. Chem. Chem. Phys., 2020, 22, 13756–13763 RSC.
- S. Enami and A. J. Colussi, J. Phys. Chem. A, 2017, 121, 5175–5182 CrossRef CAS PubMed.
- R. Criegee and G. Wenner, Liebigs Ann. Chem., 1949, 564, 9–15 CrossRef CAS.
- R. Criegee, Justus Liebigs Ann. Chem., 1953, 583, 1–36 CrossRef CAS.
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Phys. Chem. Chem. Phys., 2011, 13, 10848–10857 RSC.
- G. T. Drozd and N. M. Donahue, J. Phys. Chem. A, 2011, 115, 4381–4387 CrossRef CAS PubMed.
- M. J. Newland, B. S. Nelson, A. Muñoz, M. Ródenas, T. Vera, J. Tárrega and A. R. Rickard, Phys. Chem. Chem. Phys., 2020, 22, 13698–13706 RSC.
- M. J. Newland, C. Mouchel-Vallon, R. Valorso, B. Aumont, L. Vereecken, M. E. Jenkin and A. R. Rickard, Atmos. Chem. Phys., 2022, 22, 6167–6195 CrossRef CAS.
- F. Foucher, P. Higelin, C. Mounaïm-Rousselle and P. Dagaut, Proc. Combust. Inst., 2013, 34, 3005–3012 CrossRef CAS.
- J.-B. Masurier, F. Foucher, G. Dayma and P. Dagaut, Proc. Combust. Inst., 2015, 35, 3125–3132 CrossRef CAS.
- J. B. Masurier, F. Foucher, G. Dayma and P. Dagaut, Energy Fuel, 2013, 27, 5495–5505 CrossRef CAS.
- J. B. Masurier, F. Foucher, G. Dayma and P. Dagaut, Appl. Energy, 2015, 160, 566–580 CrossRef CAS.
- W. Sun, X. Gao, B. Wu and T. Ombrello, Prog. Energy Combust. Sci., 2019, 73, 1–25 CrossRef.
- J. H. Kroll, J. S. Clarke, N. M. Donahue and J. G. Anderson, J. Phys. Chem. A, 2001, 105, 1554–1560 CrossRef CAS PubMed.
- J. H. Kroll, S. R. Sahay, J. G. Anderson, K. L. Demerjian and N. M. Donahue, J. Phys. Chem. A, 2001, 105, 4446–4457 CrossRef CAS.
- F. Liu, J. M. Beames, A. S. Petit, A. B. McCoy and M. I. Lester, Science, 2014, 345, 1596–1598 CrossRef CAS PubMed.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, J. Chem. Phys., 2016, 144, 061102 CrossRef PubMed.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- V. A. Isidorov, I. G. Zenkevich and B. V. Ioffe, Atmos. Environ. (1967), 1985, 19, 1–8 CrossRef CAS.
- P. Ciccioli, E. Brancaleoni, A. Cecinato, R. Sparapani and M. Frattoni, J. Chromatogr. A, 1993, 643, 55–69 CrossRef CAS.
- G. Creech, R. T. Johnson and J. O. Stoffer, J. Chromatogr. Sci., 1982, 20, 67–72 CAS.
- H. Nishikawa, T. Hayakawa and T. Sakai, Analyst, 1987, 112, 859–862 RSC.
- M. Gambino, R. Cericola, P. Corbo and S. Iannaccone, J. Eng. Gas Turbines Power, 1993, 115, 747–749 CrossRef CAS.
- F. Lipari, J. M. Dasch and W. F. Scruggs, Environ. Sci. Technol., 1984, 18, 326–330 CrossRef CAS PubMed.
- P. Sinharoy, S. L. McAllister, M. Vasu and E. R. Gross, Environmental Aldehyde Sources and the Health Implications of Exposure, Springer, Singapore, 2019 Search PubMed.
- C. Fan, C. Song, G. Lv, G. Wang, H. Zhou and X. Jing, Appl. Therm. Eng., 2018, 129, 1382–1391 CrossRef CAS.
- R. Atkinson, S. M. Aschmann, A. M. Winer and J. N. Pitts Jr, Int. J. Chem. Kinet., 1981, 13, 1133–1142 CrossRef CAS.
- E. Grosjean and D. Grosjean, Int. J. Chem. Kinet., 1998, 30, 21–29 CrossRef CAS.
- K. Sato, B. Klotz, T. Taketsugu and T. Takayanagi, Phys. Chem. Chem. Phys., 2004, 6, 3969–3976 RSC.
- N. Wang, F. Wei, J. Sun, B. Wei, Q. Mei, Z. An, M. Li, Z. Qiu, X. Bo, J. Xie, J. Zhan and M. He, Chemosphere, 2021, 281, 130996 CrossRef CAS PubMed.
- Y. Sakamoto, R. Yajima, S. Inomata and J. Hirokawa, Phys. Chem. Chem. Phys., 2017, 19, 3165–3175 RSC.
- L. Vereecken, A. Novelli and D. Taraborrelli, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- V. P. Barber, S. Pandit, A. M. Green, N. Trongsiriwat, P. J. Walsh, S. J. Klippenstein and M. I. Lester, J. Am. Chem. Soc., 2018, 140, 10866–10880 CrossRef CAS PubMed.
- M. F. Vansco, B. Marchetti, N. Trongsiriwat, T. Bhagde, G. Wang, P. J. Walsh, S. J. Klippenstein and M. I. Lester, J. Am. Chem. Soc., 2019, 141, 15058–15069 CrossRef CAS PubMed.
- R. L. Caravan, M. F. Vansco, K. Au, M. A. H. Khan, Y.-L. Li, F. A. F. Winiberg, K. Zuraski, Y.-H. Lin, W. Chao and N. Trongsiriwat, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9733–9740 CrossRef CAS PubMed.
- J. Jr-Min Lin and W. Chao, Chem. Soc. Rev., 2017, 46, 7483–7497 RSC.
- A. C. Rousso, N. Hansen, A. W. Jasper and Y. Ju, Phys. Chem. Chem. Phys., 2019, 21, 7341–7357 RSC.
- R. L. Caravan, T. J. Bannan, F. A. F. Winiberg, M. A. H. Khan, A. C. Rousso, A. W. Jasper, S. D. Worrall, A. Bacak, P. Artaxo, J. Brito, M. Priestley, J. D. Allan, H. Coe, Y. Ju, D. L. Osborn, N. Hansen, S. J. Klippenstein, D. E. Shallcross, C. A. Taatjes and C. J. Percival, Nat. Geosci., 2024, 17, 219–226 CrossRef CAS.
- A. R. Conrad, N. Hansen, A. W. Jasper, N. K. Thomason, L. Hidaldo-Rodrigues, S. P. Treshock and D. M. Popolan-Vaida, Phys. Chem. Chem. Phys., 2021, 23, 23554–23566 RSC.
- R. Chhantyal-Pun, M. A. H. Khan, C. A. Taatjes, C. J. Percival, A. J. Orr-Ewing and D. E. Shallcross, Int. Rev. Phys. Chem., 2020, 39, 385–424 Search PubMed.
- C. A. Taatjes, Annu. Rev. Phys. Chem., 2017, 68, 183–207 CrossRef CAS PubMed.
- D. Grosjean, J. Air Waste Manage. Assoc., 1990, 40, 1664–1669 CrossRef CAS.
- N. Wang, J. Sun, B. Wei, Q. Mei, Z. An, F. Wei, M. Li, Z. Qiu, X. Bo, J. Xie, J. Zhan and M. He, Atmos. Environ., 2021, 254, 118392 CrossRef CAS.
- W. Xiao, S. Sun, S. Yan, W. Wu and J. Sun, Chemosphere, 2022, 303, 135142 CrossRef CAS PubMed.
- K. Moshammer, A. W. Jasper, D. M. Popolan-Vaida, A. Lucassen, P. Diévart, H. Selim, A. J. Eskola, C. A. Taatjes, S. R. Leone, S. M. Sarathy, Y. Ju, P. Dagaut, K. Kohse-Höinghaus and N. Hansen, J. Phys. Chem. A, 2015, 119, 7361–7374 CrossRef CAS PubMed.
- J. Orphal, J. Staehelin, J. Tamminen, G. Braathen, M.-R. De Backer, A. Bais, D. Balis, A. Barbe, P. K. Bhartia, M. Birk, J. B. Burkholder, K. Chance, T. von Clarmann, A. Cox, D. Degenstein, R. Evans, J.-M. Flaud, D. Flittner, S. Godin-Beekmann, V. Gorshelev, A. Gratien, E. Hare, C. Janssen, E. Kyrölä, T. McElroy, R. McPeters, M. Pastel, M. Petersen, I. Petropavlovskikh, B. Picquet-Varrault, M. Pitts, G. Labow, M. Rotger-Languereau, T. Leblanc, C. Lerot, X. Liu, P. Moussay, A. Redondas, M. Van Roozendael, S. P. Sander, M. Schneider, A. Serdyuchenko, P. Veefkind, J. Viallon, C. Viatte, G. Wagner, M. Weber, R. I. Wielgosz and C. Zehner, J. Mol. Spectrosc., 2016, 327, 105–121 CrossRef CAS.
- P. Dagaut, M. Cathonnet, J. P. Rouan, R. Foulatier, A. Quilgars, J. C. Boettner, F. Gaillard and H. James, J. Phys. E: Sci. Instrum., 1986, 19, 207–209 CrossRef CAS.
- P. A. Heimann, M. Koike, C. W. Hsu, D. Blank, X. M. Yang, A. G. Suits, Y. T. Lee, M. Evans, C. Y. Ng, C. Flaim and H. A. Padmore, Rev. Sci. Instrum., 1997, 68, 1945–1951 CrossRef CAS.
- S. R. Leone, M. Ahmed and K. R. Wilson, Phys. Chem. Chem. Phys., 2010, 12, 6564–6578 RSC.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- W. Sun and Y. Ju, J. Plasma Fusion Res., 2013, 89, 208–219 CAS.
- A. C. Rousso, N. Hansen, A. W. Jasper and Y. Ju, J. Phys. Chem. A, 2018, 122, 8674–8685 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- H.-J. Werner, P. J. Knowles, P. Celani, W. Györffy, A. Hesselmann, D. Kats, G. Knizia, A. Köhn, T. Korona, D. Kreplin, R. Lindh, Q. Ma, F. R. Manby, A. Mitrushenkov, G. Rauhut, M. Schütz, K. R. Shamasundar, T. B. Adler, R. D. Amos, J. Baker, S. J. Bennie, A. Bernhardsson, A. Berning, J. A. Black, P. J. Bygrave, R. Cimiraglia, D. L. Cooper, D. Coughtrie, M. J. O. Deegan, A. J. Dobbyn, K. Doll, M. Dornbach, F. Eckert, S. Erfort, E. Goll, C. Hampel, G. Hetzer, J. G. Hill, M. Hodges, T. Hrenar, G. Jansen, C. Köppl, C. Kollmar, S. J. R. Lee, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, B. Mussard, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. A. Peterson, K. Pflüger, R. Pitzer, I. Polyak, P. Pulay, M. Reiher, J. O. Richardson, J. B. Robinson, B. Schröder, M. Schwilk, T. Shiozaki, M. Sibaev, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, J. Toulouse, M. Wang, M. Welborn and B. Ziegler, MOLPRO, version, a package of ab initio programs, https://www.molpro.net Search PubMed.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, III, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- F. N. Egolfopoulos, N. Hansen, Y. Ju, K. Kohse-Höinghaus, C. K. Law and F. Qi, Prog. Energy Combust. Sci., 2014, 43, 36–67 CrossRef.
- N. Hansen, T. A. Cool, P. R. Westmoreland and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2009, 35, 168–191 CrossRef CAS.
- A. C. Rousso, A. W. Jasper, Y. Ju and N. Hansen, J. Phys. Chem. A, 2020, 124, 9897–9914 CrossRef CAS PubMed.
- H. Liao, S. Kang, N. Hansen, F. Zhang and B. Yang, Combust. Flame, 2020, 214, 277–286 CrossRef CAS.
- Z. Liu, X. Fan, H. Chen, Q. Hou, H. Liao, J. Yang, L. Zhao, F. Zhang and B. Yang, Proc. Combust. Inst., 2023, 39, 435–444 CrossRef.
- H. Zhao, X. Yang and Y. Ju, Combust. Flame, 2016, 173, 187–194 CrossRef CAS.
- M. Pfeifle, Y.-T. Ma, A. W. Jasper, L. B. Harding, W. L. Hase and S. J. Klippenstein, J. Chem. Phys., 2018, 148, 174306 CrossRef PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177–180 CrossRef CAS PubMed.
- F. Liu, Y. Fang, M. Kumar, W. H. Thompson and M. I. Lester, Phys. Chem. Chem. Phys., 2015, 17, 20490–20494 RSC.
- C. Huang, B. Yang and F. Zhang, J. Chem. Phys., 2019, 150, 164305 CrossRef PubMed.
- S. Leach, M. Schwell, H.-W. Jochims and H. Baumgärtel, Chem. Phys., 2006, 321, 171–182 CrossRef CAS.
- J. Wang, B. Yang, T. A. Cool and N. Hansen, Int. J. Mass Spectrom., 2010, 292, 14–22 CrossRef CAS.
- L. Zhong, Y. Gao, X. Chen, W. Yao and S. Li, Struct. Chem., 2014, 25, 1405–1414 CrossRef CAS.
- A. B. Ryzhkov and P. A. Ariya, Phys. Chem. Chem. Phys., 2004, 6, 5042–5050 RSC.
- J. M. Anglada, J. González and M. Torrent-Sucarrat, Phys. Chem. Chem. Phys., 2011, 13, 13034–13045 RSC.
- K. E. Leather, M. R. McGillen, M. C. Cooke, S. R. Utembe, A. T. Archibald, M. E. Jenkin, R. G. Derwent, D. E. Shallcross and C. J. Percival, Atmos. Chem. Phys., 2012, 12, 469–479 CrossRef CAS.
- W. Chao, J.-T. Hsieh, C.-H. Chang and J. J.-M. Lin, Science, 2015, 347, 751 CrossRef CAS PubMed.
- C. Cabezas and Y. Endo, J. Chem. Phys., 2018, 148, 014308 CrossRef PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- L. Vereecken, A. Novelli, A. Kiendler-Scharr and A. Wahner, Phys. Chem. Chem. Phys., 2022, 24, 6428–6443 RSC.
- C. Sun, S. Zhang, J. Yue and S. Zhang, J. Phys. Chem. A, 2018, 122, 8729–8737 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, E. P. F. Lee, J. M. Dyke, D. W. K. Mok, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2012, 14, 10391–10400 RSC.
- R. Kaipara and B. Rajakumar, J. Phys. Chem. A, 2018, 122, 8433–8445 CrossRef CAS PubMed.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2014, 16, 4039–4049 RSC.
- P. R. Story and J. R. Burgess, J. Am. Chem. Soc., 1967, 89, 5726–5727 CrossRef CAS.
- Z. J. Buras, R. M. I. Elsamra, A. Jalan, J. E. Middaugh and W. H. Green, J. Phys. Chem. A, 2014, 118, 1997–2006 CrossRef CAS PubMed.
- X. He, N. Hansen and K. Moshammer, J. Phys. Chem. A, 2020, 124, 7881–7892 CrossRef CAS PubMed.
- J. J. Butler, M. L. Fraser-Monteiro, L. Fraser-Monteiro, T. Baer and J. R. Hass, J. Phys. Chem., 1982, 86, 747–752 CrossRef CAS.
- Y. Zhai, Q. Xu, S. Ruan, L. Zhang, C. Xie, Z. Wang and S. M. Sarathy, Combust. Flame, 2023, 249, 112601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01942c |
| This journal is © the Owner Societies 2024 |