
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of energetic positions of electronic states and the exciton dynamics in a π-expanded N-heterotriangulene derivative adsorbed on Au(111)†
Jakob
Steidel
a,
Ina
Michalsky
b,
Mohsen
Ajdari
a,
Milan
Kivala
 b and
Petra
Tegeder
*a
b and
Petra
Tegeder
*a
aRuprecht-Karls-Universität Heidelberg, Physikalisch-Chemisches Institut, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany. E-mail: tegeder@uni-heidelberg.de; Tel: +49 (0) 6221 548475
bRuprecht-Karls-Universität Heidelberg, Organisch-Chemisches Institut, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
First published on 24th May 2024
Abstract
Bridged triarylamines, so-called N-heterotriangulenes (N-HTAs) are promising organic semiconductors for applications in optoelectronic devices. Thereby the electronic structure at organic/metal interfaces and within thin films as well as the electronically excited states dynamics after optical excitation is essential for the performance of organic-molecule-based devices. Here, we investigated the energy level alignment and the excited state dynamics of a N-HTA derivative adsorbed on Au(111) by means of energy- and time-resolved two-photon photoemission spectroscopy. We quantitatively determined the energetic positions of several occupied and unoccupied molecular (transport levels) and excitonic states (optical gap) in detail. A transport gap of 3.20 eV and an optical gap of 2.58 eV is determined, resulting in an exciton binding energy of 0.62 eV. With the first time-resolved investigation on a N-HTA compound we gained insights into the exciton dynamics and resolved processes on the femtosecond to picosecond timescale.
N-Heteropolycyclic aromatic compounds represent a promising molecule class for applications in functional organic materials, since their electronic structure and the resulting individual molecular properties are efficiently tuneable by number and position of nitrogen atoms in the aromatic structural backbone. The isosteric replacement of a C–H unit by N leaves the geometric structure unchanged, while the electronic structure are altered. For instance, they are promising candidates for electron-transporting (n-channel) semiconductors, which are of great interest for organic field effect transistors.1–5 N-heterotriangulenes (N-HTAs), in which the originally propeller-shaped triphenylamine unit is locked into a planar configuration via bridging with appropriate molecular moieties (e.g. carbonyl or dimethylmethylene)6–8 is a class of functional molecules with high potential for optoelectronic materials.9–12 In electron donor/acceptor systems N-HTAs act as the donor compound.13 Recently, we analyzed the electronic and absorption properties of two N-HTA derivatives (N-HTA-550 and N-HTA-557, see Fig. 1) at the interface to Au(111) and within thin molecular films using vibrational and electronic high resolution electron energy loss spectroscopy and quantum chemical calculations. Inter alia, we found that the additional –C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bridge forming the 7-membered ring in N-HTA-557 resulted in a pronounced reduction of the optical gap size by 0.9 eV from 3.4 eV in N-HTA-550 to 2.5 eV in N-HTA-557 due to an increase of the π-conjugated electron system.14 Thus, using structural extensions or substitution patterns opens the opportunity for fine-tuning the electronic properties.7,10,15 Here, we investigated the electronic structure and electronically excited state dynamics of a novel π-expanded N-heterotriangulene derivative (N-HTA-557-P, see Fig. 1), which consists of an electron-deficient pyrazine ring (electron accepting unit)7 adsorbed on Au(111) utilizing two-photon photoemission (2PPE) spectroscopy. 2PPE has been proven to be a powerful tool, which provides the unique opportunity for a quantitative determination of the energetic position of occupied as well as unoccupied molecular electronic states (transport levels) but also excitonic states (optical gaps) and therefore exciton binding energies.16–34 In addition, femtosecond (fs) time-resolved 2PPE allows to gain insights into the electronically excited states dynamics after optical excitation such as exciton formation and decay dynamics.
C– bridge forming the 7-membered ring in N-HTA-557 resulted in a pronounced reduction of the optical gap size by 0.9 eV from 3.4 eV in N-HTA-550 to 2.5 eV in N-HTA-557 due to an increase of the π-conjugated electron system.14 Thus, using structural extensions or substitution patterns opens the opportunity for fine-tuning the electronic properties.7,10,15 Here, we investigated the electronic structure and electronically excited state dynamics of a novel π-expanded N-heterotriangulene derivative (N-HTA-557-P, see Fig. 1), which consists of an electron-deficient pyrazine ring (electron accepting unit)7 adsorbed on Au(111) utilizing two-photon photoemission (2PPE) spectroscopy. 2PPE has been proven to be a powerful tool, which provides the unique opportunity for a quantitative determination of the energetic position of occupied as well as unoccupied molecular electronic states (transport levels) but also excitonic states (optical gaps) and therefore exciton binding energies.16–34 In addition, femtosecond (fs) time-resolved 2PPE allows to gain insights into the electronically excited states dynamics after optical excitation such as exciton formation and decay dynamics.
 | ||
| Fig. 1 The N-heterotriangulenes N-HTA-550 and N-HTA-557. The π-expanded derivative N-HTA-557-P investigated in the present study. | ||
Here we thoroughly elucidated the electronic structure of N-HTA-557-P/Au(111) in detail, including the determination of transport levels, the optical gap and accordingly the exciton binding energy. In particular for applications of this material in organic field effect transistors or solar cells knowledge about the energetic positions of transport levels (affinity levels and ionization potentials) are extremely important. Moreover, fs-time-resolved 2PPE, allowed to gain insights into the exciton dynamics. Our study represents the first time-resolved investigation on a N-HTA-derived compound, which is of great interest with respect to applications of this donor material in organic solar cells.
To gain insight into the energy level alignment, i.e., the energetic positions of adsorbate-derived occupied as well as unoccupied electronic states we conducted photon-energy-dependent 2PPE measurements.23–26,30–33,35Fig. 2a shows an exemplarily 2PPE spectrum of 5 monolayer (ML) N-HTA-557-P adsorbed on Au(111) recorded with hv = 4.62 eV (for additional 2PPE data see ESI†). Several photoemission features could be detected. On the basis of their photon-energy-dependency they are assigned to occupied and unoccupied molecular orbitals as well as an excitonic state (see Fig. 2b).
 | ||
| Fig. 2 (a) 2PPE spectrum of 5 monolayer (ML) N-HTA-557-P adsorbed on Au(111). The data are fitted with an exponential background and Gaussian-shaped peaks (red curves). The energy axis reveals the final state (EFinal) of photoemitted electrons with respect to the Fermi energy EF (EFinal−EF = Ekin + Φ); thus, the low-energy cutoff corresponds to the work function (Φ) of the adsorbate/substrate system. (b) Photon-energy-dependent peak position extracted to assign peaks observed in the 2PPE spectrum to occupied, unoccupied intermediate or final electronic states. A slope of 1 suggests that a peak originates from an unoccupied intermediate state, a slope of zero from an unoccupied final state (located above the vacuum level), while a slope of 2 is related to peaks originating from occupied states. The slopes from the fits are given next to the respective data. | ||
In Fig. 3, we summarize the level alignments with respect to the vacuum level. Note that the work function of bare Au(111) (ΦAu(111) = 5.5 eV) decreases by 0.7 eV due to the adsorption of 1 ML N-HTA-557-P (see ESI†, Fig. S1 and S2) and stays constant for higher coverages. As discussed before,24,25,30,36,37 depending on the excitation mechanism 2PPE probes transport levels.
 | ||
| Fig. 3 Energy level diagram of N-HTA-557-P adsorbed on Au(111) for N-HTA-557-P coverages above 2 ML with respect to the vacuum level. The blue levels are the ionization potentials and the red ones are the electron affinities, which are in the molecular orbital picture represented by the energies of the HOMO, LUMO, UMOs (unoccupied molecular orbital) or OMO (occupied molecular orbital), respectively. The dashed lines indicate vibronic transition. EF is the Au(111) Fermi level. | ||
Unoccupied single-electron states can be populated via an electron transfer from the metal to the N-HTA-557-P molecule creating a negative ion resonance and occupied single-electron states are ionized, creating a positive ion resonance. Thus, we obtain the electron affinities (EA) and the ionization potentials (IP) of the investigated molecule. In addition, in 2PPE also intramolecular electronic excitation is possible, which corresponds to exciton generation, i.e. formation of an electron–hole pair, in which the molecule remains overall neutral. The minimal energy required for this process is the optical gap (Eopt), which corresponds to the excitation energy of the lowest excited singlet state. Note, that the two excitation mechanisms in 2PPE can be distinguished via coverage-dependent measurements, since 2PPE is surface sensitive and probes only the topmost layers. Thus at higher coverages a contribution in the 2PPE spectrum of unoccupied electronic states populated via electron-transfer from the metallic substrate is not observed and accordingly optical gaps can be determined. Eopt is smaller than the difference between the lowest IP and the highest EA. The so-called transport gap is given as Etransp = IP−EA. The difference between the transport gap and the optical gap corresponds to the exciton binding energy Etransp−Eopt = EB.19,38
For the N-HTA-557-P we obtain a transport gap of 3.20 eV (IP = 6.37 eV, EA = 3.17 eV), and an optical gap of 2.58 eV, the excitation energy of the lowest excited singlet state. Hence, the exciton binding energy (EB) of this lowest excitonic state amounts to 620 meV. For comparison, N-HTA-557-P in solution (CH2Cl2) possesses an optical gap of 2.61 eV (see ESI,† Fig. S3, ESI†). The similar value indicates that the adsorbed molecules (5 ML) are decoupled from the metallic substrate and weak adsorbate-adsorbate (lateral) interactions. In addition, we identified two further unoccupied molecular states (unoccupied molecular orbital: UMO), an intermediate (at 1.17 eV) and a final state (0.11 eV above Evac) as well as an occupied molecular state (occupied molecular orbital: OMO) at 8.74 eV. All other 2PPE features are assigned to vibronic contributions related to the LUMO, and UMO2 as well as to the HOMO. They exhibit an equidistance energy different of around 200 meV, which can be related to an excitation of ν(C–C) and δ(C–H) vibrational modes of the N-HTA-557 moiety of N-HTA-557-P located at 1636 cm−1 (see Fig. S4 and Table S1, ESI†). Vibronic features in 2PPE data have recently been also proposed for another N-heterocyclic compound adsorbed on Au(111) supported by scanning tunneling spectroscopy results.33 For lower N-HTA-557-P coverages, i.e., around 1 ML conributions from the Au(111) surface, the d-bands, the shifted surface state and the first image potential state are visible in the 2PPE data (see ESI†, Fig. S1). The differences in the energetic positions of the unoccupied and occupied electronic molecular states as a function of coverage are rather small and lie within the error bars. This indicates relatively weak adsorbate/substrate interactions. In addition, no dispersing molecular states at the metal/organic interface have been observed in our angle-resolved 2PPE measurements, which for instance have been identified for other N-heteropolycycles adsorbed on Au(111).32,33 Thus we exclude hybridization between molecular states with metal bands. The most striking difference between the 2PPE data of the monolayer and higher coverages (≥2 ML) is the increased amount of vibronic contributions due to decoupling effects from the metal substrate.
Comparing the optical gaps sizes of the N-HTAs (see Fig. 1), N-HTA-550 (3.4 eV), N-NTA-557 (2.5 eV), and N-HTA-557-P (2.58 eV), in N-NTA-557 the additional –CC– bridge forming the 7-membered ring lead to a reduction of the optical gap by 0.9 eV compared to N-HTA-550 due to the extension of the π-conjugated electron system.14 In N-HTA-557-P the 7-membered ring is formed via a –C–C– single bond (N-HTA 557-SB), which then is π-expanded with a pyrazine-containing unit (P). Quantum chemical calculations have been shown that the N-HTA 557-SB possesses a similar size of the optical gap as the one for N-HTA-550.14 Thus, the introduction of the pyrazine-containing moiety in N-HTA 557-SB forming N-HTA-557-P results again in a pronounced reduction of the optical gap.
We used femtosecond time-resolved 2PPE to study how the electronic coupling between the N-HTA-557-P molecules and the metal substrate influences the dynamics of optically excited states such as excitons in N-HTA-557-P. For low coverages (1–2 ML) none of the electronically excited states, e.g. the LUMO or excitonic states, exhibit a detectable lifetime, thus τ < 10 fs. The reason for these ultrashort lifetimes is a strong electron coupling enabling an efficient electron transfer from the molecule back to the metal. Such short lifetime in the order of a few femtoseconds have been found also for other adsorbates on metal surfaces.35,39–41 In contrast, for higher coverages longer lifetimes are usually observed.
This is the case for our findings as can be seen Fig. 4a for 15 ML N-HTA-557-P on Au(111) in a two-dimensional false colored plot representation taken at a pump photon energy of hν1 = 3.08 eV and a probe photon energy hν2 = 3.92 eV. Such representations visualize the normalized correlated dichromatic 2PPE signal at a given final state energy (EFinal) with respect to EF as a function of pump–probe delay. Positive pump–probe delays imply that the pump pulse hν1 reaches the sample before the probe pulse hν2, and vice versa for negative delays. A long-lived contribution can clearly be seen in the energy region around 4.9 eV, thus the energy at which we identified the first excitonic state. To receive the lifetimes of the involved state a cross-correlation (XC) curve as shown in Fig. 4b has been employed. To fit the XC curve, we used a sech2-function representing the laser pulse duration convoluted with a response function of the intermediate state. A superposition of two exponential decays with different time constants, τ1 and τ2, describes the time-resolved photoemission data well (see solid line in Fig. 4b). The time constants are τ1 = 312 ± 18 fs and τ2 = 3.22 ± 0.15 ps at a coverage of 15 ML. Coverage-dependent measurements (see Fig. 4c) clearly demonstrate that the lifetimes increase with rising coverage. Coverage-dependent lifetimes can be explained by the availability of two relaxation channels.24–26,40,42 An intrinsic channel due to the decay in the bulk material and a distance-dependent (external) channel resulting from a quenching process by the metallic substrate, i.e., transfer of the electrons to the metal. Note that, in literature no time-dependent measurements on the N-HTAs excited states dynamics are available. However, by comparing with our previous time-resolved 2PPE results obtained from other electron donating materials24,25 we consider the following processes: Excitation with a photon energy of 3.08 eV leads to the creation of hot (Eopt = 2.58 eV), delocalized excitons. These excitons relax and localize on an ultrafast time-scale (τ1 = 312 ± 18 fs). A very fast relaxation of hot excitons has also been found in other organic semiconductors.43–46 The longer lived component (τ2 = 3.22 ± 0.15 ps) may be related to the decay of the exciton to the ground state. However, it could also be connected to the lifetime of polarons as well as electrons bound at defect sites. Note, that no energetic stabilization is observed, which would be expected for polaron formation or population of defect sites.
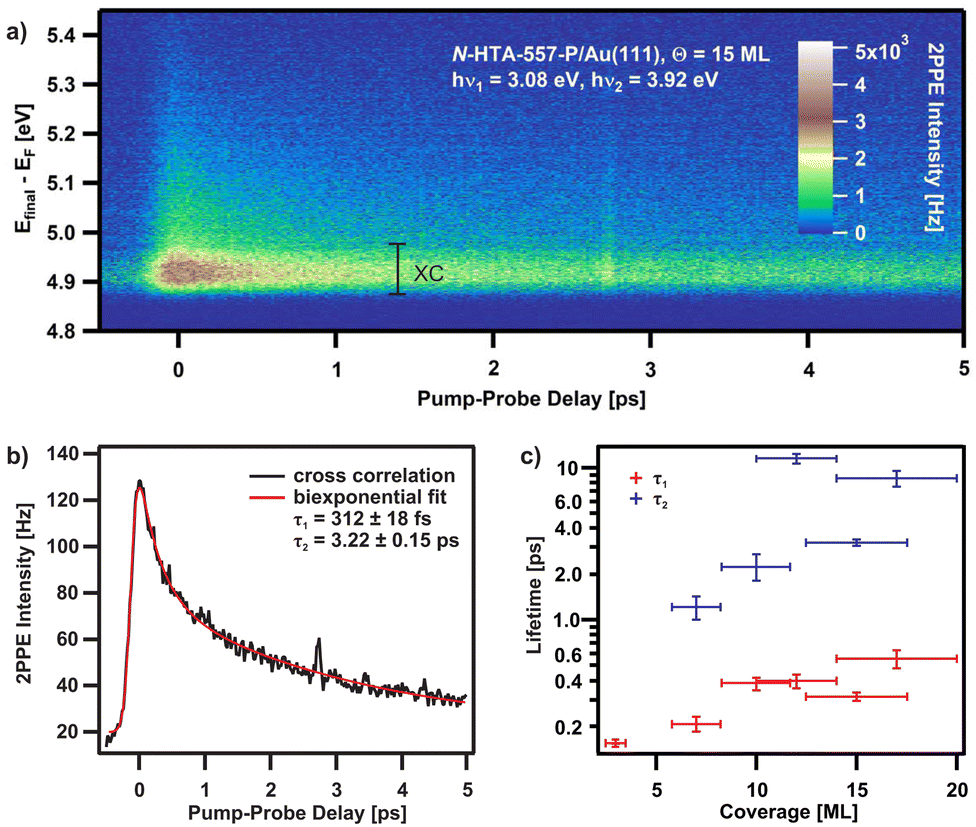 | ||
| Fig. 4 (a) 2D-plot of the 2PPE intensity as a function of final-state energy and the pump–probe delay for 15 ML N-HTA-557-P on Au(111). (b) The cross-correlation (XC) curves fitted by a sech2 function convoluted by a double exponential decay (XC energy range in a). (c) Coverage dependent lifetimes. | ||
In summary, we employed energy- and time-resolved two-photon photoemission spectroscopy to quantitatively determine the energetic positions of unoccupied and occupied electronic states as well as an excitonic state and to resolve the exciton dynamics in a novel π-expanded N-heterotriangulene derivative adsorbed on Au(111). A transport gap of 3.20 eV and an optical gap of 2.58 eV have been identified, resulting in an exciton binding energy of 0.62 eV. Hot and delocalized excitons have found to localize on a femtosecond timescale followed by an exciton decay within picoseconds. The quantitative determination of the energetic positions of molecular electronic levels, the achieved detailed understanding of the electronic structure and insights into the exciton dynamics is an essential prerequisite for the design of new functional organic molecules for (opto) electronic devices.
Author contributions
J. S. performed the 2PPE measurements and analysed the results. I. M. synthesized the H-NTA derivative. M. A. conducted the IR measurements and analysed the results. P. T. wrote the manuscript. M. K. supervised I. M. and P. T. supervised J. S. as well as M. A. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the German Research Foundation (DFG) through the collaborative research center CRC 1249 ′′N-Heteropolycycles as Functional Materials′′ (Project Number 281029004-SFB 1249, projects A05 and B06) is gratefully acknowledged.References
- H. Klauk, U. Zschieschang, J. Pflaum and M. Halik, Nature, 2007, 445, 745–748 CrossRef CAS PubMed.
- F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC.
- U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef CAS PubMed.
- Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed.
- U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- N. Hammer, T. A. Schaub, U. Meinhardt and M. Kivala, Chem. Rec., 2015, 15, 1119–1131 CrossRef CAS PubMed.
- I. Michalsky, V. Gensch, C. Walla, M. Hoffmann, F. Rominger, T. Oeser, P. Tegeder, A. Dreuw and M. Kivala, Chem. – Eur. J., 2022, 28, e202200326 CrossRef CAS PubMed.
- J. Wagner, P. Z. Crocomo, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Angew. Chem., Int. Ed., 2022, 61, e202202232 CrossRef CAS PubMed.
- U. Meinhardt, F. Lodermeyer, T. A. Schaub, A. Kunzmann, P. O. Dral, A. C. Sale, F. Hampel, D. M. Guldi, R. D. Costa and M. Kivala, RSC Adv., 2016, 6, 67372–67377 RSC.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- T. A. Schaub, K. Padberg and M. Kivala, J. Phys. Org. Chem., 2020, 33, e4022 CrossRef CAS.
- M. Krug, M. Wagner, T. A. Schaub, W. Zhang, C. M. Schüßlbauer, J. D. R. Ascherl, P. W. Münich, R. R. Schröder, F. Gröhn and P. O. Dral, et al. , Angew. Chem., Int. Ed., 2020, 59, 16233–16240 CrossRef CAS PubMed.
- T. Kader, B. Stöger, J. Fröhlich and P. Kautny, Chem. – Eur. J., 2019, 25, 4412–4425 CrossRef CAS PubMed.
- M. Ajdari, R. Pappenberger, C. Walla, M. Hoffmann, I. Michalsky, M. Kivala, A. Dreuw and P. Tegeder, J. Phys. Chem. C, 2023, 127, 542–549 CrossRef CAS.
- N. Deng and G. Zhang, Org. Lett., 2019, 21, 5248–5251 CrossRef CAS PubMed.
- X.-Y. Zhu, Surf. Sci. Rep., 2004, 56, 1–83 CrossRef CAS.
- X.-Y. Zhu, J. Phys. Chem. B, 2004, 108, 8778–8793 CrossRef CAS.
- C. Lindstrom and X.-Y. Zhu, Chem. Rev., 2006, 106, 4281–4300 CrossRef CAS PubMed.
- M. Muntwiler and X.-Y. Zhu, Dynamics at Solid State Surfaces and Interfaces, Wiley-VCH, 2010 Search PubMed.
- C. Bronner, M. Schulze, S. Hagen and P. Tegeder, New J. Phys., 2012, 14, 043023 CrossRef.
- C. Bronner, G. Schulze, K. J. Franke, J. I. Pascual and P. Tegeder, J. Phys.: Condens. Matter, 2011, 23, 484005 CrossRef PubMed.
- N. Armbrust, J. Güdde, P. Jakob and U. Höfer, Phys. Rev. Lett., 2012, 108, 056801 CrossRef CAS PubMed.
- C. Bronner, S. Stremlau, M. Gille, F. Brauße, A. Haase, S. Hecht and P. Tegeder, Angew. Chem., Int. Ed., 2013, 52, 4422–4425 CrossRef CAS PubMed.
- L. Bogner, Z. Yang, S. Baum, R. F. M. Corso, P. Bäuerle, K. J. Franke, J. I. Pascual and P. Tegeder, J. Phys. Chem. C, 2016, 120, 27268–27275 CrossRef CAS.
- L. Bogner, Z. Yang, M. Corso, R. Fitzner, P. Bäuerle, K. J. Franke, J. I. Pascual and P. Tegeder, Phys. Chem. Chem. Phys., 2015, 17, 27118–27126 RSC.
- A. Stein, F. Maass and P. Tegeder, J. Phys. Chem. C, 2017, 121, 18075–18083 CrossRef CAS.
- D. Gerbert, F. Maass and P. Tegeder, J. Phys. Chem. C, 2017, 121, 15696–15701 CrossRef CAS.
- D. Gerbert and P. Tegeder, J. Phys. Chem. Lett., 2017, 8, 4685–4690 CrossRef CAS PubMed.
- W. Bronsch, T. Wagner, S. Baum, M. Wansleben, K. Zielke, E. Ghanbari, M. Györök, A. Navarro-Quezada, P. Zeppenfeld and M. Weinelt, et al. , J. Phys. Chem. C, 2019, 123, 7931–7939 CrossRef CAS.
- A. Stein, D. Rolf, C. Lotze, C. Czekelius, K. J. Franke and P. Tegeder, J. Phys.: Condens. Matter, 2019, 31, 044002 CrossRef PubMed.
- M. Ajdari, A. Stein, M. Hoffmann, M. Müller, U. H. F. Bunz, A. Dreuw and P. Tegeder, J. Phys. Chem. C, 2020, 124, 7196–7204 CrossRef CAS.
- A. Stein, D. Rolf, C. Lotze, B. Günther, L. H. Gade, K. J. Franke and P. Tegeder, J. Phys. Chem. Lett., 2021, 12, 947–951 CrossRef CAS PubMed.
- A. Stein, D. Rolf, C. Lotze, S. Feldmann, D. Gerbert, B. Günther, A. Jeindl, J. J. Cartus, O. T. Hofmann and L. H. Gade, et al. , J. Phys. Chem. C, 2021, 125, 19969–19979 CrossRef CAS PubMed.
- K. Stallberg, A. Namgalies, S. Chatterjee and U. Höfer, J. Phys. Chem. C, 2022, 126, 12728–12734 CrossRef CAS.
- C. Bronner, M. Schulze, S. Hagen and P. Tegeder, New J. Phys., 2012, 14, 043032 CrossRef.
- E. Varene, I. Martin and P. Tegeder, J. Phys. Chem. Lett., 2011, 2, 252–256 CrossRef CAS.
- C. Bronner, D. Gerbert, A. Broska and P. Tegeder, J. Phys. Chem. C, 2016, 120, 26168–26172 CrossRef CAS.
- M. Knupfer, Appl. Phys. A: Mater. Sci. Process., 2003, 77, 623–626 CrossRef CAS.
- A. Yang, S. T. Shipman, S. Garrett-Roe, J. Johns, M. Strader, P. Szymanski, E. Muller and C. B. Harris, J. Phys. Chem. C, 2008, 112, 2506 CrossRef CAS.
- G. Dutton, D. P. Quinn, C. D. Lindstrom and X.-Y. Zhu, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045441 CrossRef.
- S. Hagen, Y. Luo, R. Haag, M. Wolf and P. Tegeder, New J. Phys., 2010, 12, 125022 CrossRef.
- E. Varene, L. Bogner, C. Bronner and P. Tegeder, Phys. Rev. Lett., 2012, 109, 7601 CrossRef PubMed.
- M. Marks, S. Sachs, C. Schwalb, A. Schöll and U. Höfer, J. Chem. Phys., 2013, 139, 124701 CrossRef CAS PubMed.
- E. Engel, M. Koschorreck, K. Leo and M. Hoffmann, Phys. Rev. Lett., 2005, 95, 157403 CrossRef CAS PubMed.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2009, 131, 16869–16880 CrossRef CAS PubMed.
- K. Chen, A. Barker, M. Reish, K. Gordon and J. M. Hodgkiss, J. Am. Chem. Soc., 2013, 135, 18502–18512 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, two-photon photoemission data, UV/vis absorption and emission data as well as infrared vibrational data. See DOI: https://doi.org/10.1039/d4cp01713g |
| This journal is © the Owner Societies 2024 |