
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theory for nonlinear conductivity switching in semiconducting organic ferroelectrics†
Till
Johann
a,
Weiwei
Xie
 bc,
Sara
Roosta
b,
Marcus
Elstner
bd and
Martijn
Kemerink
*a
bc,
Sara
Roosta
b,
Marcus
Elstner
bd and
Martijn
Kemerink
*a
aInstitute for Molecular Systems Engineering and Advanced Materials, Heidelberg University, Im Neuenheimer Feld 225, 69120 Heidelberg, Germany. E-mail: martijn.kemerink@uni-heidelberg.de
bInstitute of Physical Chemistry (IPC), Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany
cKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), State Key Laboratory of Advanced Chemical Power Sources, College of Chemistry, Nankai University, Tianjin 300071, China
dInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
First published on 25th June 2024
Abstract
In this work, the ferroelectric and semiconducting properties of the organic semiconducting ferroelectric benzotrithiophene tricarboxamide (BTTTA), and especially their nonlinear coupling, are theoretically investigated. BTTTA is an exponent of a small class of semiconducting organic ferroelectrics for which experiments have established a surprising polarization direction dependence of the bulk conductivity at finite fields. First, molecular dynamics (MD) simulations are used to investigate the occurrence and, under the influence of an external electric field, the inversion of the macroscopic electric dipole that forms along the axis of supramolecular columns of BTTTA. The MD results are consistent with the experimentally observed ferroelectric behavior of the material. Building on the MD results, a QM/MM scheme is used to investigate the charge carrier mobility in the quasi-1D BTTTA stacks in the linear and non-linear regimes. Indeed, at finite electric fields, a clear resistance switching effect was observed in the form of a hole mobility that is a factor ∼2 larger for antiparallel orientations of the polarization and field than for a parallel orientation. This phenomenon can be understood as a microscopic ratchet that is based on the non-equilibrium interaction between the (oriented) dipoles and the (direction of the) charge transport.
Introduction
Ferroelectric materials combine a relevance for a range of practical applications with rich, cooperative physics. Although the majority of ferroelectric materials used and investigated to date are of inorganic nature, organic, that is, carbon-based ferroelectrics offer several unique and potentially highly relevant features. In part, these are of pragmatic nature, relating to the abundance and non-toxicity of the constituent elements, the compatibility with large-area, low-cost, measured both in monetary and energetic terms, production techniques and, in the case of polymer or liquid-crystalline materials, a large degree of mechanical flexibility. From a scientific perspective, organic ferroelectrics can be realized on the basis of an astonishingly rich spectrum of principles, ranging from order–disorder-type ferroelectrics based on (partial or full) rotation of dipolar molecules or molecular substituents in a lattice to displacive materials based on dimerization of ionic species, charge transfer or proton tautomerization.1The variety of approaches to realize ferroelectricity in molecular organic materials reflects the freedom of design that is typical for organic chemistry. In the field of organic ferroelectrics, this is beautifully illustrated by the class of discotic liquid crystalline ferroelectrics, of which trialkylbenzene-1,3,5-tricarboxamides (BTA) is the archetypical example. BTA is a C3-symmetric molecule consisting of a benzene core to which three amide groups have been attached to the 1, 3 and 5 positions. Attached to the amide groups are aliphatic tails that make the molecule soluble in organic solvents and, in solid-state thin films, give the material its ferroelectric-friendly liquid-crystalline properties. BTAs are known to self-assemble into one-dimensional columnar structures that are stabilized by threefold H-bonding through the amides and, to a lesser degree, by π-stacking of the conjugated core.2 Although benzene-1,3,5-tricarboxamides were first synthesized in 1915,3 their ferroelectric properties were discovered only in 2010.4 Since then, it was found that the ferroelectric properties of BTAs could be varied in non-trivial ways by varying any of the three ‘zones’ in the molecule. Changing the length or branching of the aliphatic side chains allowed to improve the polarization retention from mere minutes to years.5,6 Changing the dipolar group from a regular (oxygen) amide to a thioamide resulted in a ferrielectric material.7 Finally, exchanging the benzene core for a larger benzotrithiophene unit resulted in the, still C3-symmetric, semiconducting ferroelectric benzotrithiophene tricarboxamide, BTTTA, see Fig. 1. This compound shows, amongst others, a bulk conductivity that, at finite electric fields, depends on the direction of the ferroelectric polarization.8
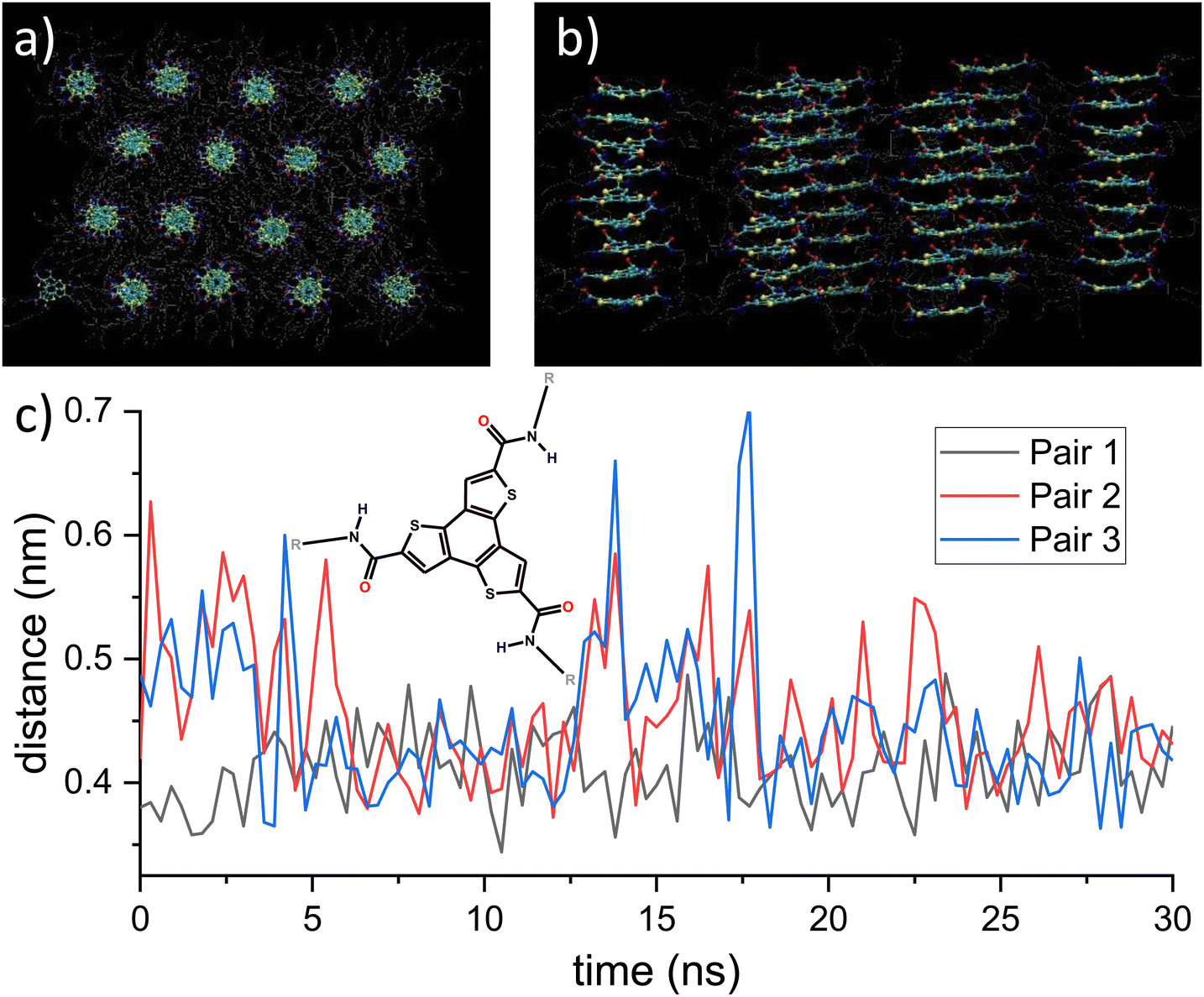 | ||
| Fig. 1 (a) and (b) Top and side view of the BTTTA-C12 lattice structure after NPT equilibration. The BTTTA structure remains in its initial hexagonal structure while distances and dihedral angles reach thermal equilibrium. The structure mostly maintains its H-bonding network. (c) Distances between two sulfur atoms of adjacent molecules. Inset: Structure of BTTTA-C12. The images in (a) and (b) and all of the following molecule renderings were created with VMD.9 | ||
The rectifying and switchable conductivity of BTTTA was previously observed in a few related compounds.10 Although this has so far not been explicitly pursued, this combination of properties is interesting for large area data storage in crossbar arrays, where the binary information can be read-out from the conductivity value, taking away the need to switch (and restore) the ferroelectric polarization state that hampers conventional 2-terminal ferroelectric memories.11 Likewise, said materials may provide a new way forward to actual use of the bulk photovoltaic effect.12,13
Since the direction of current flow and of polarization both lie parallel to the supramolecular column axis, the moving charge carrier sees a periodic but inversion asymmetric energy landscape: when driven away from equilibrium by an external electric field, the system becomes a so-called tilting ratchet that rectifies the external perturbation.14 Apart from the direct electrostatic interaction with the amide dipoles, the lack of inversion symmetry can be expected to manifest itself indirectly through asymmetries in polarizability and structural relaxation, that is, polaronic contributions. In ref. 10 a simple 2-site model was developed in which these effects were lumped together in skewed (reorganization) energy landscape that was used as input in a Marcus-type hopping model. Although this provided some quantification of the intuitive explanation of the phenomenon by a symmetry argument about the non-equivalence of charge carrier motion against or along the polarization direction, a rigorous explanation of the directional conductivity in this type of compounds has so far not been given. Although this is beyond the scope of the present work, having a predictive model would allow to in silico search for materials with a large on/off-ratio for memory applications, prior to actual synthesis.
Here, we use a combination of molecular dynamics (MD) and density functional theory (DFT) to rigorously address the problem of the nonlinear charge transport in the BTTTA system. Specifically, the MD simulations are used to investigate the supramolecular organization of BTTTA, including the polarization, as a function of temperature and external field, reproducing experimentally found behavior. The rectifying, polarization-dependent conductivity was investigated using a quantum mechanics/molecular mechanics (QM/MM) embedding scheme, i.e., only a part of the crystal is treated using quantum mechanics, the remainder of the system is treated classically. This scheme provides the first theoretical confirmation that indeed the ferroelectric polarization couples to the charge carrier mobility. In agreement with experiments, the nonlinear mobility for an antiparallel (parallel) relative orientation of polarization and field gives rise to an enhanced (reduced) charge carrier mobility.
Models and computational details
Molecular dynamics
The study of the ferroelectric properties of the BTTTA and BTA systems will be based on molecular dynamics (MD) simulations, which have previously provided a good agreement with experimental observations for this class of materials.6,15,16 In a second step, the MD model is combined with a non-adiabatic quantum mechanical propagation scheme to investigate the charge transfer properties of BTTTA.BTA and BTTTA monomer structures were created with the Avogadro software.17 The Gromacs software was used for MD simulations.18 Topologies were created with the OPLS forcefield, which is known for its superior performance regarding H-bonds.19 BTTTA partial charges yield a dipole moment of 3.47D per amide group, which is close to 4 D obtained from DFT calculations.8 Three-dimensional monoclinic periodic boundary conditions were applied with the non-perpendicular angle at 60°, which causes a hexagonal structure, as can be seen in Fig. 1a. The particle mesh Ewald method is used to treat long-range electrostatics. After initial energy minimization, the equilibration and production MD simulations were performed at 300 K and 1 bar using the Parrinello–Rahman barostat together with the Nosé–Hoover temperature coupling with a coupling constant of 0.2 ps. Intracolumnar and intercolumnar distances were reached through the NPT equilibration after the initial configuration was created according to X-ray parameters.8 10 ns of NPT was used as equilibration and subsequent simulation of 20 ns to obtain averages of quantities of interest, unless longer times were needed, as noted.
The externally applied electric field was modeled with the built-in Gromacs method, which employs an additional electrostatic energy term in the force field, proportional to the partial charge on each atom. The BTTTA liquid crystal system consisted of 4-by-4 columns with 9 molecules per column for the ferroelectric switching simulations and 27 molecules per column for the CT simulations. Furthermore, simulations with a single BTTTA-C0 column in vacuum were performed to calculate switching energy barriers.
Charge transfer
To study the resistance switching effect, we used a multiscale methodology for charge transfer that we developed in recent years for the simulation of rapid electron and exciton transfer processes across various domains, including electron transfer in DNA photolyase,20–22 and electron and exciton transfer in organic materials.23–26 For details of the methodology please see ref. 25 and 26 Specifically, charge transport was treated by modeling the dynamics of a single excess charge carrier in a mixed quantum classical simulation,24,25,27,28 where the wavefunction of the excess charge carrier is propagated explicitly and coupled nonadiabatically to the classical, molecular environment using a non-adiabatic molecular dynamics scheme based on Tully's fewest switches algorithm (FSSH) with corrections for decoherence and trivial crossings.25 This non-adiabatic molecular dynamics (NAMD) simulation employs the common QM/MM separation scheme, where the full molecular system is divided into a quantum mechanical and a classical zone. This way, most of the system dynamics are modeled with the MD force field, and only the excess charge carrier has to be described quantum mechanically. The quantum part is computed using a fragmentation scheme, which makes it computationally very efficient. The site energies of the molecular fragments and the coupling between those are computed using the density-functional-tight-binding (DFTB) approximation of DFT.29–33 This method expresses the DFT Hamiltonian as a Taylor expansion of the density around a reference density, which leads to several levels of DFTB, depending on the order.Previous works have applied these methods to non-polar, crystalline materials such anthracene, but also to liquid crystals like hexabenzocoronene (HBC).23 BTTTA is quite different from those materials since it possesses polar amide groups that create H-bonds to connect adjacent molecules to form the supramolecular columns, in contrast to HBC. In the latter, the columns are held together solely by π–π interactions. Validation steps that were taken to assure the accuracy of the QM/MM approach are discussed in the ESI,† Section S4.
Since the charge transfer is one dimensional along the columns, the QM zone is taken as one of the columns, given by 25 molecules, where the molecules at the edge of the box boundary are not included. The aliphatic tails are also put into the MM zone since they do not participate in the frontier molecular orbitals (MOs). Charge transport is described by the dynamics of a single positive excess charge carrier. For this hole, only the highest lying MOs are included in the Hilbert space, the remaining electronic degrees of freedom of the system are electronically inert and modeled with MD. Often, only the HOMO orbital per fragment is used, but in the case of BTTTA, DFTB calculations have revealed that the HOMO and HOMO-1 frontier orbitals are energetically degenerate, which necessitated the inclusion of both in the Hilbert space, with the help of a state-tracking algorithm.
The DFTB treatment starts with the reference density ρ0 of neutral atoms as an approximation for the actual density of the neutral molecular system. This might seem like an improper approximation since the amide groups of BTTTA are polar - the polarity is essential for the resistance switching effect. However, this is unproblematic because the interaction between the partial charges of the amides with the excess carrier are included in the QM/MM interaction term, and because we rewrite the DFT energy of the QM zone in terms of the forcefield energy. Second order DFTB represents the change of the Hamiltonian that the hole evolves with, due to the change in partial charges because of the presence of the hole. Because the BTTTA molecule is relatively large, these changes are rather small. This means that the second order contributions are negligible compared to the first order terms. We will hence use the DFTB1 scheme for our simulations. The closed boundary conditions of the QM zone enable to easily implement an external field into the QM propagator by simply shifting the diagonals of the DFTB Hamiltonian according to the field strength E as ΔHnm = ndmmE with n and dmm the molecule number and the intermolecular distance along the column. This model then yields the time evolution of the hole wavefunction ψ(t).
For zero external fields, the charge flow will enter a diffusive regime, and the carrier mobility μ can be extracted via the Einstein formula23
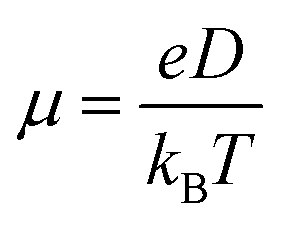 | (1) |
 | (2) |
The mean-square-displacement (MSD) 〈Δx2〉 is linear in time in the diffusive regime, and can be calculated as
| 〈Δx2〉 = ∑(xA(t) − x0)2PA(t) | (3) |
Since the resistance switching effect is a nonlinear phenomenon that occurs only at strong enough electric fields, we cannot use the MSD to extract the mobility in that regime. Instead, for finite external fields E, the mobility can be extracted from the drift motion of the center of mass position 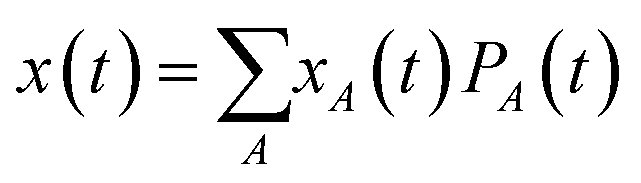 of the charge carrier:24
of the charge carrier:24
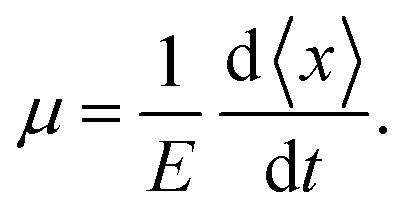 | (4) |
Experimentally, the resistance switching effect is observed at around 20 V μm−1. Although sufficient to induce nonlinearities in the conductivity, such fields do not significantly perturb the dihedral angles in the MD simulation, vide infra, so we can set the MD electric force field to zero during the charge transfer simulations.
We calculated the reorganization energy λ of the BTTTA molecule in the NAMD simulation by forcing the hole to stay on one molecule and subtract the resulting equilibrated ionization energy from the neutral one. This yields a value of λDFTB ≈ 0.12 eV, see ESI† Fig. S11. However, a benchmark test with Gaussian revealed that DFTB underestimates λ. In a computational study, it was found that the ωB97XD functional produces the most accurate values for the reorganization energy, especially for large polar molecules with a high wavefunction delocalization on the molecular core.34 Using this functional and the 6-31G* basis set, the reorganization energy was calculated with the four point method. The calculation resulted in a value of λ = 0.4 eV, which necessitates the use of an implicit relaxation method. In this method, the quantum force from the wavefunction on the atoms of the charge carrying molecule is ignored, so that no intramolecular relaxation occurs. Instead, this effect is treated by adding a shift on the diagonals of the Hamiltonian, given by the product between the charge and the reorganization energy.26 This allows us to use the more reliable value of λ from the Gaussian DFT calculation. Note that there is still a force from the charge carrier on the MM component, namely the electrostatic force due to changed partial charges. Moreover, the velocity adjustment along the nonadiabtic vector after surface hopping is replaced with a Bolzmann correction for hopping probability. This treatment is more efficient, as it avoids the time-consuming computation of multi-dimensional nonadiabatic coupling vectors (see ref. 26 for more details).
Results and discussion
Equilibration
The initial static structure is equilibrated in two steps. First, we perform an energy minimization via steepest descent until the largest force in the system is below the tolerance range of 10 kJ nm−1 mol−1. This procedure leads to the formation of the typical triple helix H-bonding network, see Fig. S1 in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 Second, upon subjecting the BTTTA system to the NPT ensemble, the density quickly increases, then fluctuates around an equilibrium value of around 1000 kg m−3, see Fig. S1 (ESI†). The equilibrated box size is 210 nm3, which together with the remnant dipole moment of 960 D yields a polarization of 15.3 mC m−2. This value is similar to but somewhat smaller than the experimental value of 25 mC m−2. The latter is what one would expect from the lattice geometry, with an out-of-plane dihedral angle of 40° and a total dipole moment of 12D per molecule.8 Since the calculated geometry and dihedral angle are very close to the experimental ones, and the calculated dipole moment per molecule is 10.4D, one would expect a calculated polarization of 25 × 10.4/12 = 21.7 mC m−2 in the crystal. The reason for this discrepancy lies in a screening of charges due to the molecular environment in the simulation. This deviation should not change the qualitative outcomes of the further simulations though. The thermally equilibrated structure mostly maintained its hydrogen-bonded network, and the columnar morphology remained intact as can be seen in Fig. 1. The dihedral distribution takes a Gaussian shape, centered at 40° with some broadening due to thermal fluctuations, see Fig. S1(b) in the ESI.† However, even in thermal equilibrium, there can be significant structural dynamics due to temporary breaking of H-bonds, see Fig. 1(c). If the corresponding amide groups form a hydrogen bond, the distance fluctuates only weakly around its equilibrium value. However, individual H-bonds can be broken for a few ns, see e.g. the trace labeled pair 3 at ∼14 and ∼17.5 ns, before they self-heal into the bonded configuration. This behavior is related to the self-assembly properties of hydrogen bonded supramolecular polymers like BTTTA and BTA, for which this has also been observed in MD simulations.35 ESI† Section S1 provides results of several tests we ran to assure robustness of the results with respect to simulation details.
15 Second, upon subjecting the BTTTA system to the NPT ensemble, the density quickly increases, then fluctuates around an equilibrium value of around 1000 kg m−3, see Fig. S1 (ESI†). The equilibrated box size is 210 nm3, which together with the remnant dipole moment of 960 D yields a polarization of 15.3 mC m−2. This value is similar to but somewhat smaller than the experimental value of 25 mC m−2. The latter is what one would expect from the lattice geometry, with an out-of-plane dihedral angle of 40° and a total dipole moment of 12D per molecule.8 Since the calculated geometry and dihedral angle are very close to the experimental ones, and the calculated dipole moment per molecule is 10.4D, one would expect a calculated polarization of 25 × 10.4/12 = 21.7 mC m−2 in the crystal. The reason for this discrepancy lies in a screening of charges due to the molecular environment in the simulation. This deviation should not change the qualitative outcomes of the further simulations though. The thermally equilibrated structure mostly maintained its hydrogen-bonded network, and the columnar morphology remained intact as can be seen in Fig. 1. The dihedral distribution takes a Gaussian shape, centered at 40° with some broadening due to thermal fluctuations, see Fig. S1(b) in the ESI.† However, even in thermal equilibrium, there can be significant structural dynamics due to temporary breaking of H-bonds, see Fig. 1(c). If the corresponding amide groups form a hydrogen bond, the distance fluctuates only weakly around its equilibrium value. However, individual H-bonds can be broken for a few ns, see e.g. the trace labeled pair 3 at ∼14 and ∼17.5 ns, before they self-heal into the bonded configuration. This behavior is related to the self-assembly properties of hydrogen bonded supramolecular polymers like BTTTA and BTA, for which this has also been observed in MD simulations.35 ESI† Section S1 provides results of several tests we ran to assure robustness of the results with respect to simulation details.
Ferroelectric switching
Prior to performing switching simulations of BTTTA, we performed benchmark simulations on BTA, for which MD data are available.15 In short, our simulations reproduce those by Cornelissen et al., including the finding that the dominant polarization reversal mode is the so-called z-flip, a rotation of the dihedral angle by 80°, from 40° to −40°, and not the full-flip, which would be a 180° rotation from 40° to −140°.15 Further details can be found in the ESI† Section S2.As for BTA, equilibrated structures are used as the starting point for the switching simulations of BTTTA in finite electric fields. The crystal box was put into the ground state via a steepest-descent energy minimization, which caused all columns to take a 0:3 state with negative polarization. Then, this structure was equilibrated in an NPT ensemble for 10 ns, with the polarization remaining at the large negative value. The negatively polarized system was then propagated a further 100 ns in NPT, with a positive external electric field. In contrast to experimental observations, relatively strong fields ∼0.4 V nm−1 have, on this time scale, no influence on the dihedral distribution. The reason for the stronger stability in the MD simulation lies in the switching mechanism that is mostly intrinsic in simulations while in experiments, it is typically extrinsic due to the presence of electrodes, impurities or structural defects, which act as nucleation sources that reduce the flipping activation energy.15 If the field strength is increased, switching is observed, as discussed in the following.
Fig. 2a shows representative transients of the dipole moment of individual columns during switching at intermediate fields. Typically, a column remains in the 0:3 state for a while, until suddenly most amides switch into the 3:0 configuration: in general, the strong intra-columnar interactions cause the amide groups to switch collectively. If the field is very strong (2 V nm−1, not shown), all columns switch immediately. As the field gets weaker, the typical time until the switch happens grows, and some columns stay in the 0:3 configuration for the full duration of the simulation. For intermediate and small fields, a metastable state is often produced as the result of switching a part of a full column, which is visible as a plateau at intermediate polarization value. As not all amides have flipped, the H-bonding network is incomplete until, after several ns, the molecules twist and the dihedrals rearrange to produce a proper network again.
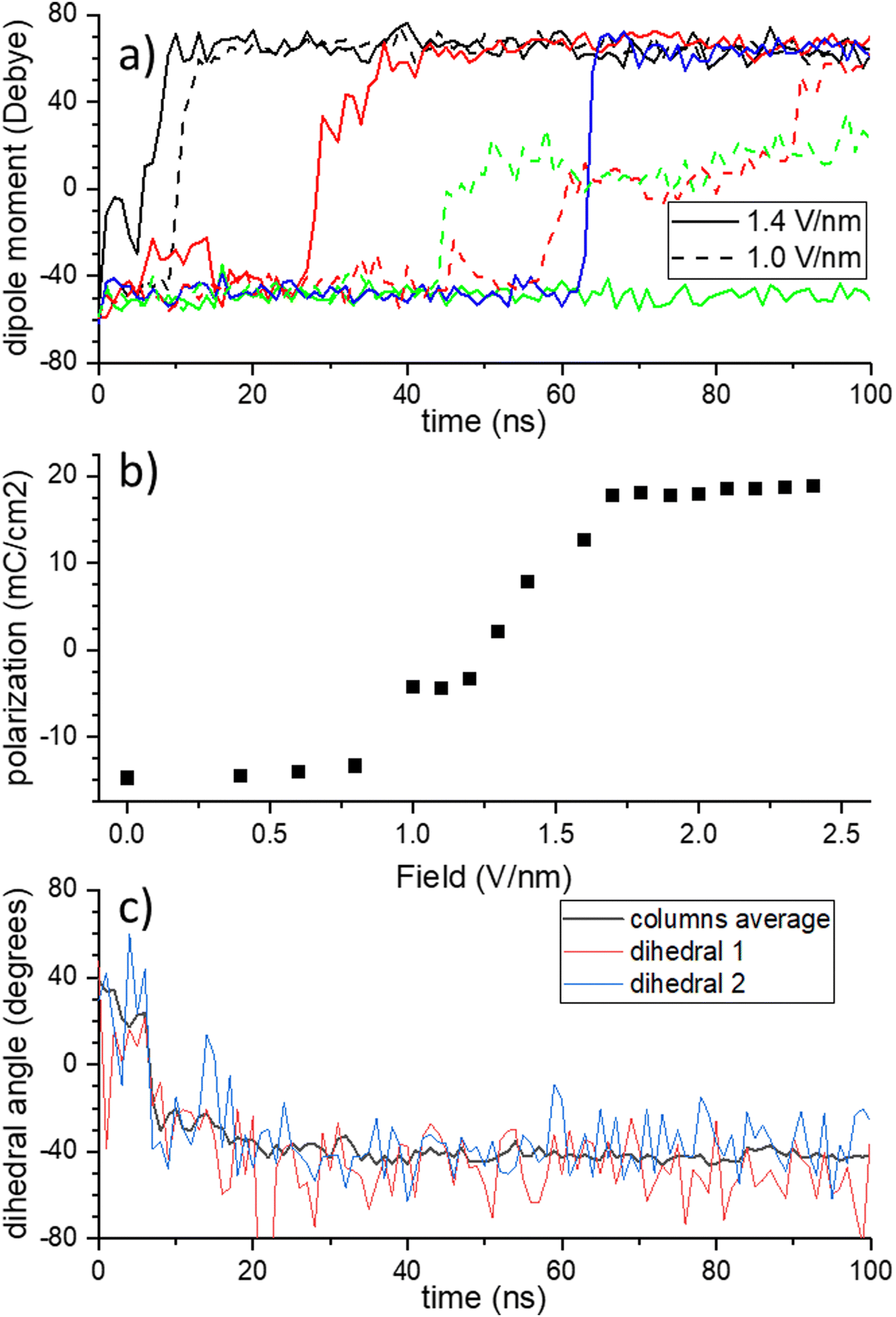 | ||
| Fig. 2 Ferroelectric switching in BTTTA. (a) Dipole moment of individual columns. Full lines: E = 1.4 V nm−1, dashed lines: E = 1 V nm−1. (b) Full dipole moment of the simulation box after 100 ns NPT with constant field as function of the field. (c) Dynamics of individual and average dihedral angles. | ||
Summing the columnar dipole moments over the full simulation box, a single side of the characteristic ferroelectric hysteresis curve is obtained, see Fig. 2b, with a coercive field Ec ≈ 1.2 V nm−1. Note that the slight difference in polarization at zero field, i.e. the remnant polarization, and the saturation polarization at large fields is due to the linear contribution of the electric field, which pushes the dihedrals further than they would be in the ground state configuration. To check for consistency of our simulations with generally observed experimental trends for this class of disordered liquid crystalline ferroelectrics, we compared the frequency- and temperature dependence of the coercive field to the thermally activated - nucleation limited switching (TANLS) model in ESI† Section S3.36 Indeed, the simulations show decent agreement between the analytical fits and the theoretical data. The same holds for the field dependence of the switching time τ, that is the timescale that it takes for the whole system to switch its polarization P(t) from negative to positive values and that is usually described by the semiempirical Merz-law τ ∝ exp(E0/E) with a characteristic field.36 From the fit to the TANLS model, the energy barrier for polarization reversal can be estimated, yielding a value of wTANLSb = 0.29 eV nm−3.
As compared to the extensively studied drosophila system of supramolecular ferroelectrics, BTA, BTTTA has a larger core and, hence, the amide groups are further away from the center. This causes a smaller rotation angle of adjacent molecules with respect to each other. While neighboring molecules are rotated by 60° in BTA, in BTTTA the value is 40°. Therefore, an amide group in BTTTA has a different distance to the two closest amide groups in each of its neighboring molecules in the column. That is, in BTA, each amide has 4 nearest neighbor amide groups (2 above, 2 below), to 2 of which it forms a H-bond; in BTTTA, it has only 2 nearest neighbors, namely those to which it is hydrogen-bound. One might therefore expect that the z-flip does not exist in BTTTA. The alternative switching mechanics in BTTTA would hence be a full flip, which for BTA was found to have a higher energy barrier than the z-flip. The authors of ref. 8 assume this is the explanation for the larger coercive field that was measured in BTTTA versus BTA. Indeed, our simulations confirm this higher coercive field. Surprisingly, Fig. 2c shows that the dihedral angles flip from an initial 40° to −40°, thus performing a z-flip. As explained above, this behavior is not possible if the molecular cores stay static during the flip and indeed it is accompanied by a quick change of molecular geometry of individual molecules, see Fig. S7 (ESI†), followed by a process of rearrangements of disc twist angles (and hence sulfur-sulfur distances) of the whole column over a period of tens of ns for a 9-molecule column, see Fig. S8 (ESI†). The BTTTA cores twist during the switching. They rotate in such a way that the amide bonding pairs that would be far away after the z-flip get close to each other and enable a new formation of a H-bonding network in this way.
This process is captured in the schemes in Fig. 3. The three pictures show a column before (oxygens point up), during and several ns after (oxygens point down) the switch. In the intermediate state, some amide groups have already turned and form a sub-network. In the final structure, the z-component of the dipole moments is inverted. Because the flipping mode is a z-flip instead of a full flip, the helicity of the system is inverted. The final structure has a defect above the second molecule from the bottom. This defect is a result of the twisting dynamics of the molecular cores during the switch and consists of a H-bond between two”wrong” amides. Not all three amide pairs are bound but only one pair, which results in a translational shift between the molecules. This defect can disappear on its own after a few ns, as discussed in Fig. 1c.
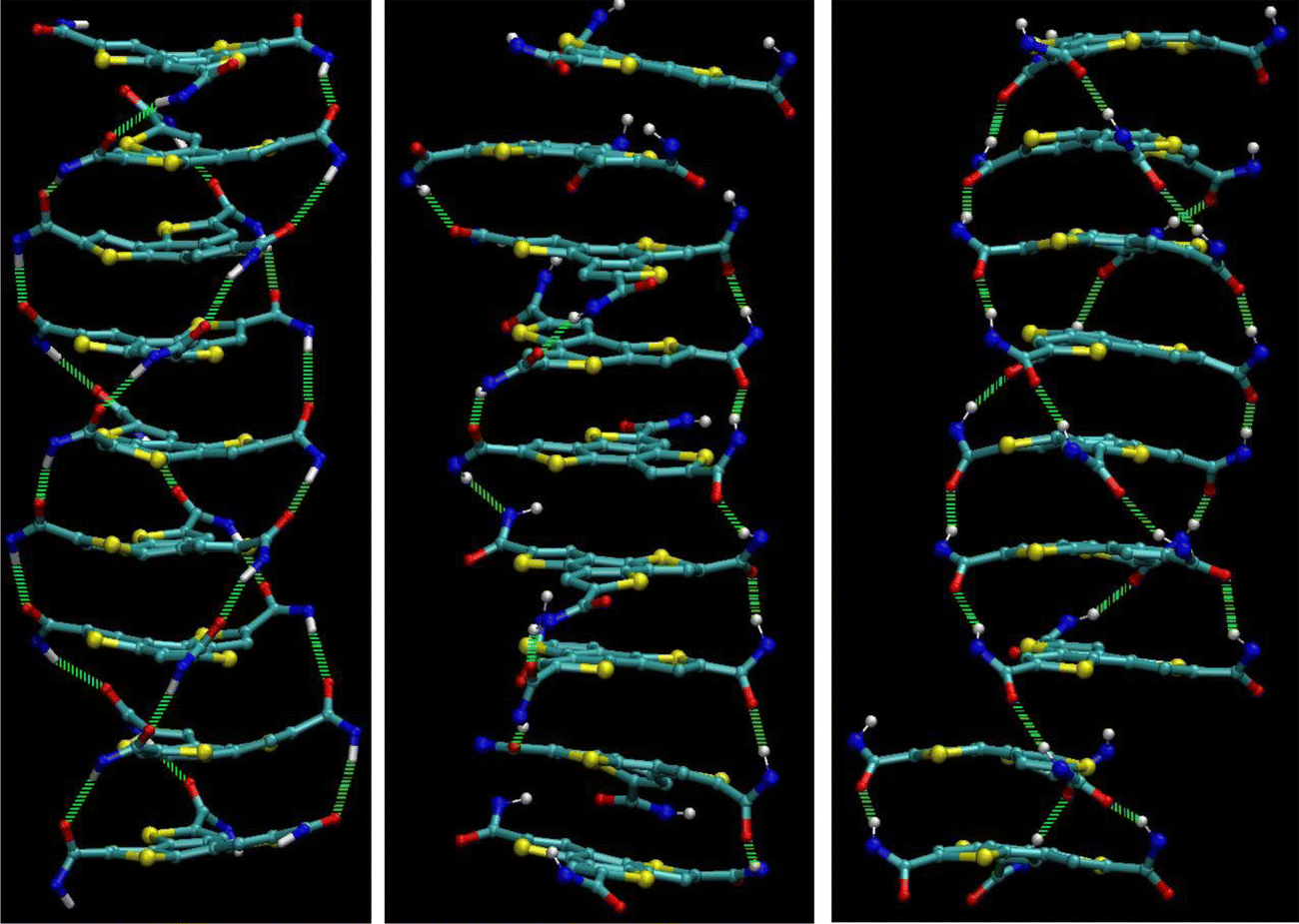 | ||
| Fig. 3 Ferroelectric switching in BTTTA-C12. Left, middle and right panels are prior, during and after polarization reversal from down to up, respectively. White: hydrogen, red: oxygen, blue: nitrogen, yellow: sulfur, light green: H-bonds. For clarity, the aliphatic chains are not shown. | ||
Using position restraints of Gromacs, we are able to observe the behavior of BTTTA in a hypothetical scenario of fixed molecular cores. We can use this feature to test the switching if the disc twist is not allowed, by restraining the molecular cores while allowing the amide groups to rotate. In this case, switching indeed required higher fields than before. At E = 2.3 V nm−1, many dihedrals are still in their initial configuration. However, importantly, some of the columns perform a full flip, from 40° to −140°, forming a new H-bonding network with the same helicity as in the initial state. This adds to our results, as it shows that the z-flip with disc twist requires a lower activation energy than a full-flip.
Using the Gromacs energy functionality, the energy barrier for polarization reversal (in the unconstrained system) can be explicitly calculated as the height of the peak in the total system potential energy during the flip, see Fig. S9 in the ESI.† The value wb = 0.15 eV nm−3 is in reasonable agreement with the value estimated above from the fit to the TANLS model of wTANLSb = 0.29 eV nm−3.
Summarizing the MD simulations of BTA and especially BTTTA, we note that these are in good qualitative and semi-quantitative agreement with the available experimental data and previous simulations. For BTTTA, an unexpected polarization reversal mechanism, in which the dihedral angle of the amide groups flip from an initial 40° to −40°, is predicted.
Charge transfer and resistance switching
For the QM/MM charge transport simulations, 300 different initial conditions were created in NPT MD simulations using a stack of 27 molecules by taking snapshots every 3.33 ps, for 1 ns simulation in total. All subsequent simulations use a timestep of 0.1 fs for the QM propagation. All charge transport simulations were performed for full polarization pointing in the negative z-direction. Further details including couplings and energies of the frontier orbitals are shown in the ESI† Section S4, Fig. S10–S12.25Zero field diffusive motion
To set a baseline for the subsequent simulations of drift motion and to connect to previous work, first zero field simulations were performed. In absence of a field, the charge motion should be polarization direction independent, and no directionality was observed. The QM zone was taken as a single column and the initial state of the excess charge carrier given by the HOMO orbital of the molecule at the edge. Quantities of interest were averaged over 300 trajectories. The results, see Fig. 4, show an initial thermalization process which takes around 300 fs, followed by a diffusive motion as indicated by a linear increase in MSD, as defined by eqn (3), versus time. For the shown data, the slope of the MSD corresponds to a zero field mobility μ0 = 1.4 ± 0.1 cm2 Vs−1, a value that is close to that of hexabenzocoronene.23 Simulations were repeated using different columns as the QM zone, yielding a mean zero-field mobility of μ0 = 0.5 ± 0.1 cm2 Vs−1, see ESI† Fig. S13. The MSD slope and the thermalization time were found to significantly differ for different columns. The reason are structural imperfections of the columns, which hider charge transport and can be different for different columns. As discussed above, such geometrical imperfections can last for several ns, so they can pose a problem for all of the 300 initial conditions in a given column. This is also the reason why the MSD is not perfectly linear. Morphological defects trap the charge carrier at a certain site for an extended period of time, which hinders the diffusive motion. Similar trapping behavior also explains the discrepancy between the experimental (μ0 ∼ 10−4 cm2 Vs−1) and simulated mobilities. In the experiment, due to the inherently imperfect long-range order of the material, there will be even more pronounced structural defects in the column morphology, acting as traps for the charge carrier. Since the charge transport is quasi one dimensional, such trapping will have a strong, detrimental influence on the macroscopic mobility.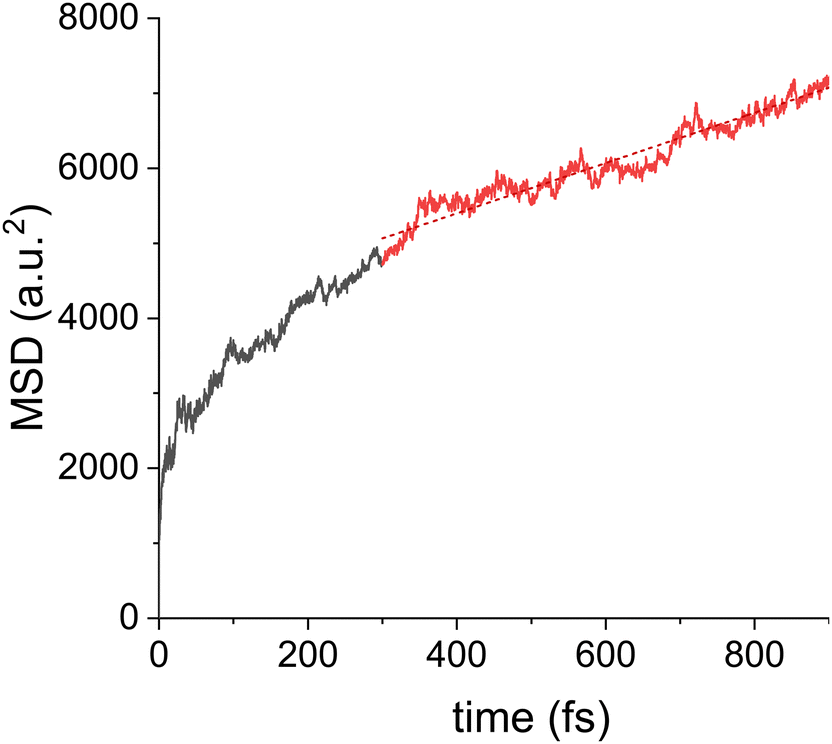 | ||
| Fig. 4 Mean-square-displacement (MSD) of the hole wavefunction during the NAMD simulation without external field, averaged over 300 trajectories in a single column. After an initial thermalization process (black line), the motion enters the diffusive regime where the MSD grows approximately linearly (solid red line), as indicated by the (dashed red) trend line. | ||
Finite field drift motion
For transport at finite fields, we use the same simulation setup as in the zero field case but include the shift ΔHnn = ndmmE of the Hamiltonian diagonals. To simulate the ON/OFF-state case, the charge is initially put at the bottom/top (in z-direction) of the (downward polarized) column, and the onsite energies decrease/increase in z-direction. 300 trajectories were simulated for each direction, which was repeated for six different columns in the middle of the crystal box. Further details of these simulations are discussed in ESI† Section S4.Fig. 5 shows representative dynamics of the center of charge of the excess carrier. Initially, the evolution of the wavefunction is dominated by a thermalization process, which results from the initial condition of being located on a single site, cf.Fig. 4, and can vary from column to column, see ESI† Fig. S15 and S16. After several hundred fs, the charge enters the drift regime, and the mobility can be obtained from a linear fit. In the ON state (blue line in Fig. 5), in which the current and polarization directions are antiparallel, higher mobilities could be observed, in agreement with experiment.8 We obtain a value of μon/μoff = 1.7 + 0.4 and a mean mobility of 0.98 cm2 Vs−1, averaged over 6 ON and 6 OFF simulations. As for the zero-field simulations, significant deviations between columns are to be expected since the morphological state of the column has a strong influence on the carrier dynamics, and since the timescale of the morphological changes is much larger than the practically accessible timescale of the charge transfer simulations. Nevertheless, the mean calculated mobility at finite field (∼1 cm2 Vs−1) exceeds the zero-field value (∼0.5 cm2 Vs−1) by about a factor 2, indicating a finite field activation. Such behavior is anticipated as the polarization-dependent conductivity can only occur in the nonlinear regime where the field is no longer small and, in the case of dynamically and statically disordered materials as BTTTA, contributes to the charge carrier mobility.
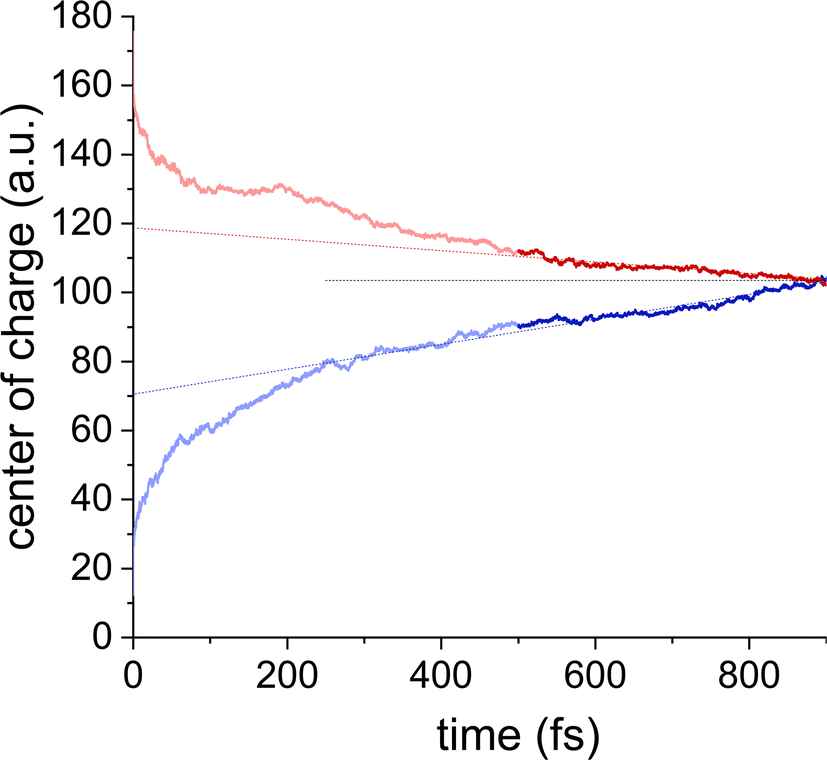 | ||
| Fig. 5 Resistance switching in BTTTA. After an initial thermalization process up to ∼300 fs, the motion enters the drift regime. The mobility obtained from the slope of the center of charge of the hole wavefunction is larger in the ON (blue, lower curve) than the OFF direction (red, upper curve) by a factor ∼2, as can be seen by the linear (dotted) trend lines. The data shown are for a single, representative, simulation. | ||
Conclusion and outlook
We have theoretically investigated the nonlinear charge transport in an organic semiconducting ferroelectric, BTTTA. In agreement with experiments, a hysteretic macroscopic polarization was found in MD simulations, the field- and temperature-dependencies of which were found to follow (semi-empirical) trends that are typically observed experimentally for this material class. Surprisingly, polarization inversion was found to proceed through an inversion of only the z-component of the amide dipole moment, as opposed to the geometrically expected full inversion of the amide. As a consequence, the helical chirality of the supramolecular column inverts upon switching.The nonlinear charge transport in the BTTTA system was investigated using a QM/MM scheme. The main result of this work is that the (direction of) ferroelectric polarization is found to couple to the charge carrier mobility once the externally applied electric field is no longer small. In agreement with experiments, the nonlinear hole mobility for an antiparallel (parallel) relative orientation of polarization and field gives rise to an enhanced (reduced) mobility. The calculated on/off ratio is about a factor 2. As anticipated for charge transport in a disordered organic semiconductor, the mean mobility in the nonlinear regime exceeds that in the linear regime; in the present simulations also by a factor ∼2.
The simulations presented herein provide the first fundamental description of the phenomenology described experimentally in ref. 8 and 10 As such, it is also the first description of its kind of a ‘supramolecular tilting ratchet’,14 formed by the charge carrier being forced to move, by electrostatic ‘tilting’, in the periodic but inversion-asymmetric energy landscape that arises from its coupling to the polarized supramolecular stack. Although the calculations performed here focus on a specific material, no material-specific approximations or assumptions were made and the results, therefore, reflect a fundamental symmetry argument and should be generic for a wide class of materials and be extendable to other driving forces. Specifically, we anticipate a similar rectification of motion to arise when charges are introduced in a non-equilibrium state of the system through photoexcitation, giving rise to a bulk photovoltaic effect in an organic material.37
Data availability
The data supporting this article have been included as part of the manuscript and its ESI.†Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
The authors are grateful to “Deutsche Forschungsgemeinschaft” supporting this project (SFB1249). The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through Grant No. INST 40/575-1 FUGG (JUSTUS 2 cluster). W. X. thanks the National Natural Science Foundation of China (No. 22203047). M.K. thanks the Carl Zeiss Foundation for financial support.References
- K. Asadi, Organic Ferroelectric Materials and Applications, Woodhead Publishing Series in Electronic and Optical Materials, Elsevier, 1st edn, 2021 Search PubMed.
- C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Cooperativity Scale: A Structure–Mechanism Correlation in the Self-Assembly of Benzene-1,3,5-Tricarboxamides, Acc. Chem. Res., 2017, 50(8), 1928–1936, DOI:10.1021/acs.accounts.7b00176.
- S. Cantekin, T. F. A. Greef and A. R. A. de; Palmans, Benzene-1,3,5-Tricarboxamide: A Versatile Ordering Moiety for Supramolecular Chemistry, Chem. Soc. Rev., 2012, 41(18), 6125–6137, 10.1039/C2CS35156K.
- C. F. C. Fitié, W. S. C. Roelofs, M. Kemerink and R. P. Sijbesma, Remnant Polarization in Thin Films from a Columnar Liquid Crystal, J. Am. Chem. Soc., 2010, 132(20), 6892–6893, DOI:10.1021/ja101734g.
- I. Urbanaviciute, X. Meng, T. D. Cornelissen, A. V. Gorbunov, S. Bhattacharjee, R. P. Sijbesma and M. Kemerink, Tuning the Ferroelectric Properties of Trialkylbenzene-1,3,5-Tricarboxamide (BTA), Adv. Electron. Mater., 2017, 3(7), 1600530, DOI:10.1002/aelm.201600530.
- I. Urbanaviciute, S. Bhattacharjee, M. Biler, J. A. Lugger, T. Cornelissen, P. Norman, M. Linares, R. Sijbesma and M. Kemerink, Suppressing Depolarization by Tail Substitution in an Organic Supramolecular Ferroelectric, Phys. Chem. Chem. Phys., 2019, 21(4), 2069–2079, 10.1039/C8CP06315J.
- I. Urbanaviciute, M. Garcia-Iglesias, A. Gorbunov, E. W. Meijer and M. Kemerink, Ferro- and Ferrielectricity and Negative Piezoelectricity in Thioamide-Based Supramolecular Organic Discotics, Phys. Chem. Chem. Phys., 2023, 25(25), 16930–16937, 10.1039/D3CP00982C.
- N. M. Casellas, I. Urbanaviciute, T. D. Cornelissen, J. Augusto Berrocal, T. Torres, M. Kemerink and M. García-Iglesias, Resistive Switching in an Organic Supramolecular Semiconducting Ferroelectric, Chem. Commun., 2019, 55, 8828–8831, 10.1039/C9CC02466B.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14(1), 33–38, DOI:10.1016/0263-7855(96)00018-5.
- A. V. Gorbunov, M. G. Iglesias, J. Guilleme, T. D. Cornelissen, W. S. C. Roelofs, T. Torres, D. González-Rodríguez, E. W. Meijer and M. Kemerink, Ferroelectric Self-Assembled Molecular Materials Showing Both Rectifying and Switchable Conductivity, Sci. Adv., 2017, 3(9), e1701017, DOI:10.1126/sciadv.1701017.
- K. Asadi, M. Li, N. Stingelin, P. W. M. Blom and D. M. de Leeuw, Crossbar Memory Array of Organic Bistable Rectifying Diodes for Nonvolatile Data Storage, Appl. Phys. Lett., 2010, 97(19), 193308, DOI:10.1063/1.3508948.
- S. Y. Yang, J. Seidel, S. J. Byrnes, P. Shafer, C.-H. Yang, M. D. Rossell, P. Yu, Y.-H. Chu, J. F. Scott, J. W. Ager, L. W. Martin and R. Ramesh, Above-Bandgap Voltages from Ferroelectric Photovoltaic Devices, Nat. Nanotechnol., 2010, 5(2), 143–147, DOI:10.1038/nnano.2009.451.
- C. Zhang, K. Nakano, M. Nakamura, F. Araoka, K. Tajima and D. Miyajima, Noncentrosymmetric Columnar Liquid Crystals with the Bulk Photovoltaic Effect for Organic Photodetectors, J. Am. Chem. Soc., 2020, 142(7), 3326–3330, DOI:10.1021/jacs.9b12710.
- P. Reimann, Brownian Motors: Noisy Transport Far from Equilibrium, Phys. Rep., 2002, 361(2), 57–265 CrossRef CAS.
- T. D. Cornelissen, M. Biler, I. Urbanaviciute, P. Norman, M. Linares and M. Kemerink, Kinetic Monte Carlo Simulations of Organic Ferroelectrics, Phys. Chem. Chem. Phys., 2019, 21(3), 1375–1383, 10.1039/C8CP06716C.
- I. Urbanaviciute, X. Meng, M. Biler, Y. Wei, T. Cornelissen, S. Bhattacharjee, M. Linares and M. Kemerink, Negative Piezoelectric Effect in an Organic Supramolecular Ferroelectric, Mater. Horiz., 2019, 6(8), 1688–1698, 10.1039/C9MH00094A.
- T. A. Team Avogadro - Free cross-platform molecular editor. Avogadro. https://avogadro.cc/(accessed 2024-04-02).
- P. Bauer, B. Hess and E. Lindahl, GROMACS 2022.2 Manual, 2022 DOI:10.5281/zenodo.6637572.
- R. S. Paton and J. M. Goodman, Hydrogen Bonding and π-Stacking: How Reliable Are Force Fields? A Critical Evaluation of Force Field Descriptions of Nonbonded Interactions, J. Chem. Inf. Model., 2009, 49(4), 944–955, DOI:10.1021/ci900009f.
- G. Lüdemann, I. A. Solov’yov, T. Kubař and M. Elstner, Solvent Driving Force Ensures Fast Formation of a Persistent and Well-Separated Radical Pair in Plant Cryptochrome, J. Am. Chem. Soc., 2015, 137(3), 1147–1156, DOI:10.1021/ja510550g.
- T. Kubař and M. Elstner, Coarse-Grained Time-Dependent Density Functional Simulation of Charge Transfer in Complex Systems: Application to Hole Transfer in DNA, J. Phys. Chem. B, 2010, 114(34), 11221–11240, DOI:10.1021/jp102814p.
- G. Lüdemann, P. B. Woiczikowski, T. Kubař, M. Elstner and T. B. Steinbrecher, Charge Transfer in E. Coli DNA Photolyase: Understanding Polarization and Stabilization Effects via QM/MM Simulations, J. Phys. Chem. B, 2013, 117(37), 10769–10778, DOI:10.1021/jp406319b.
- A. Heck, J. J. Kranz, T. Kubař and M. Elstner, Multi-Scale Approach to Non-Adiabatic Charge Transport in High-Mobility Organic Semiconductors, J. Chem. Theory Comput., 2015, 11(11), 5068–5082, DOI:10.1021/acs.jctc.5b00719.
- A. Heck, J. J. Kranz and M. Elstner, Simulation of Temperature-Dependent Charge Transport in Organic Semiconductors with Various Degrees of Disorder, J. Chem. Theory Comput., 2016, 12(7), 3087–3096, DOI:10.1021/acs.jctc.6b00215.
- W. Xie, D. Holub, T. Kubař and M. Elstner, Performance of Mixed Quantum-Classical Approaches on Modeling the Crossover from Hopping to Bandlike Charge Transport in Organic Semiconductors, J. Chem. Theory Comput., 2020, 16(4), 2071–2084, DOI:10.1021/acs.jctc.9b01271.
- S. Roosta, F. Ghalami, M. Elstner and W. Xie, Efficient Surface Hopping Approach for Modeling Charge Transport in Organic Semiconductors, J. Chem. Theory Comput., 2022, 18(3), 1264–1274, DOI:10.1021/acs.jctc.1c00944.
- J. C. Tully, Molecular Dynamics with Electronic Transitions, J. Chem. Phys., 1990, 93(2), 1061–1071, DOI:10.1063/1.459170.
- T. Kubař and M. Elstner, What Governs the Charge Transfer in DNA? The Role of DNA Conformation and Environment, J. Phys. Chem. B, 2008, 112(29), 8788–8798, DOI:10.1021/jp803661f.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Self-Consistent-Charge Density-Functional Tight-Binding Method for Simulations of Complex Materials Properties, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(11), 7260–7268, DOI:10.1103/PhysRevB.58.7260.
- T. A. Niehaus, Approximate Time-Dependent Density Functional Theory, J. Mol. Struct. THEOCHEM, 2009, 914(1), 38–49, DOI:10.1016/j.theochem.2009.04.034.
- M. Elstner and G. Seifert, Density Functional Tight Binding, Philos. Trans. R. Soc., A, 2014, 372(2011), 20120483, DOI:10.1098/rsta.2012.0483.
- M. Gaus, Q. Cui and M. Elstner, Density Functional Tight Binding: Application to Organic and Biological Molecules, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4(1), 49–61, DOI:10.1002/wcms.1156.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrică, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W.-z Yu and T. Frauenheim, DFTB +, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations, J. Chem. Phys., 2020, 152(12), 124101, DOI:10.1063/1.5143190.
- C. Brückner and B. Engels, A Theoretical Description of Charge Reorganization Energies in Molecular Organic P-Type Semiconductors, J. Comput. Chem., 2016, 37(15), 1335–1344, DOI:10.1002/jcc.24325.
- K. K. Bejagam, G. Fiorin, M. L. Klein and S. Balasubramanian, Supramolecular Polymerization of Benzene-1,3,5-Tricarboxamide: A Molecular Dynamics Simulation Study, J. Phys. Chem. B, 2014, 118(19), 5218–5228, DOI:10.1021/jp502779z.
- T. D. Cornelissen, I. Urbanaviciute and M. Kemerink, Microscopic Model for Switching Kinetics in Organic Ferroelectrics Following the Merz Law, Phys. Rev. B, 2020, 101(21), 214301, DOI:10.1103/PhysRevB.101.214301.
- K. T. Butler, J. M. Frost and A. Walsh, Ferroelectric Materials for Solar Energy Conversion: Photoferroics Revisited, Energy Environ. Sci., 2015, 8, 838–848, 10.1039/C4EE03523B.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01632g |
| This journal is © the Owner Societies 2024 |