
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT study of the moiré pattern of FeO monolayer on Au(111)†
Eleonora
Ascrizzi
 a,
Jacek
Goniakowski
b,
Jijin
Yang
c,
Stefano
Agnoli
c and
Anna Maria
Ferrari
*a
a,
Jacek
Goniakowski
b,
Jijin
Yang
c,
Stefano
Agnoli
c and
Anna Maria
Ferrari
*a
aDipartimento di Chimica, Università di Torino, via Pietro Giuria 5, I-10125 Turin, Italy. E-mail: anna.ferrari@unito.it
bCNRS, Sorbonne Université, Institut des NanoSciences de Paris, UMR 7588, 4 Place Jussieu, F-75005 Paris, France
cDepartment of Chemical Sciences, University of Padua, via Francesco Marzolo, 1, Padua 35131, Italy
First published on 1st July 2024
Abstract
Metal oxides are a class of material of particular interest for catalytic purposes. Among them the iron oxide as a monolayer supported on gold, FeO/Au, stands out for its capability to promote the CO oxidation and the dissociation of O2 and H2. In this work, we use density functional theory calculations to characterize interfacial properties of this heterostructure. We consider a FeO/Au realistic model system, managing to reproduce the moiré pattern experimentally found. Specific features of the high-symmetry domains of the moiré are identified, providing a robust ground for establishing a structure–activity relationship and guessing how the surface would behave in catalytic conditions. We also describe a strategy to model smaller systems representative of each high-symmetry domains of the moiré, which will be useful in the future to model catalytic reaction mechanisms.
1 Introduction
Metal oxides are a class of materials extensively studied for the crucial role they can play with respect to the transition from fossil to renewable energy sources.1 For example, they can be used as catalysts for the electrochemical or photoelectrochemical water splitting reaction, allowing to obtain hydrogen from sunlight.2,3 In order to be used in a world-wide scale, a catalyst for these reactions should be not too expensive, earth-abundant and have a low environmental impact: these reasons have guided the research interest towards the exploitation of iron oxides, which proved themselves suitable for these tasks in several occasions.4–6In recent years 2D materials have gained attention because of their high surface area and the accessibility of catalytic sites. Furthermore, thin films possess properties that differ from the bulk materials of origin and these could be exploited to obtain increased catalytic activities.7 Among iron oxides materials, FeO as a monolayer supported on Pt stands out to be a suitable catalyst for the CO oxidation.8–10 Given these remarkable results, it would be interesting to see how catalytic properties can be modified by changing the substrate, for example from Pt to Au which is expected to be a less interacting substrate.11
The FeO/Au system has been studied before,11–15 but not to the same extent as the FeO/Pt one10,16–23 and so a complete description of the FeO interaction with Au is still to be reached. It is known that FeO and Au give origin to a moiré pattern characterized by the presence of three high-symmetry domains (fcc, hcp and top), as happens for the similar systems FeO/Pt,24,25 FeO/Pd26 and FeO/Ag,27 with a periodicity of circa 30 Å.13–15 Experimentally, the FeOx/Au system was found able to promote CO oxidation and dissociation of O2 and H2, confirming its suitability for catalytic purposes.11,12 Given the complexity of the FeOx/Au system, only few computational studies have been carried out to rationalize experimental results: Jiang et al.14 and Yu et al.12 performed DFT calculations to shed light on the FeO growth mechanism on Au and on the phase transitions that the system can experience during the interaction with CO, respectively.
Beyond results already reported in the literature, in this paper we aim to acquire precise knowledge of the atomic and electronic structures of the FeO monolayer and of its interaction with the Au substrate. The focus of this work is to identify specific features that are peculiar of each high-symmetry domain: surface electrostatic potential and work function, local film rumpling, interface distance and charge transfer, which may further determine their different behavior towards water and hydrogen molecules, as it is already known in the FeO/Pt system.11,16 In this way, the precise characterization of the three high-symmetry domains of the FeO/Au system provides a robust ground for establishing a structure–activity relationship and guessing how the surface would behave in catalytic conditions. To do so, we simulate an entire FeO/Au moiré superstructure through DFT+U calculations and, because of its challenging size, we construct a set of suitable smaller models representing the three high-symmetry domains of the moiré in a more affordable way. To validate our model we also experimentally acquire STM images of the FeO/Au system. In particular, in Section 2.1 the experimental method is presented, in Section 2.2 the computational settings used in this work are described as well as the structural models considered, in Section 3 the results are presented and discussed. In Section 3.1 experimentally acquired STM images are presented and described. In Section 3.2 the simulated moiré structure is introduced, characterized and compared with experimental data and literature studies. In Section 3.3 the three high-symmetry domains are modelled in more details with three individual small cells.
2 Experimental and computational method
2.1 Experimental detail
A Au(111) single crystal was firstly cleaned by repeated cycles of Ar+ sputtering (1.5 keV) at room temperature and then annealed at 800 K under ultra-high vacuum conditions for 60 minutes to recover crystallinity. On this surface, a Fe metal film was deposited by physical vapor deposition (PVD), i.e. by evaporating iron atoms with an e-beam evaporator at room temperature. The PVD apparatus consists of a custom modified VG Escalab MK II operating at a base pressure of 10−10 mbar equipped with an e-beam Fe evaporator (EBE-4 Specs GmbH). After the deposition of a submonolayer quantity of iron the sample was annealed at approximately 815 K in an oxygen atmosphere of 5 × 10−7 mbar for 20 minutes resulting in the formation of a FeO wetting layer as confirmed by low energy electron diffraction and X-ray photoemission spectroscopy. The STM data were acquired in constant current mode, with a SPM Aarhus (Specs) instruments at room temperature using a kolibri sensor with chemically etched tungsten tips. The STM topographies were analysed with the WSxM software.282.2 Computational detail and model construction
Calculations for the FeO/Au system were carried out thanks to the Vienna ab initio Simulation package (VASP).29–31 The DFT+U approach was chosen to overcome the intrinsic limitation of the method in describing transition metals centers with localized states. This approach allows to obtain reliable band gap values preventing unphysical overlap between occupied and unoccupied bands and a better agreement between computed redox potentials, energy of oxidation and magnetic moments of Fe cations with respect to experiments.32,33 For the Fe atoms the Dudarev approach34 has been adopted with UFe − JFe = 3 eV. The recommended projector augmented wave (PAW) potentials have been used for all atoms, with a kinetic energy cutoff of 400 eV. Spin–orbit coupling (SOC) is not included in calculations. While SOC has a considerable effect on Au band structure, these modifications around the Fermi energy are negligible and so the reactivity and STM images of the Au layer and the FeO/Au interface (which are the focus of this work) would not change dramatically if SOC was included.35,36 PBEsol has been adopted as functional, given its suitability in describing solid interfaces and its improved capability in computing more accurate lattice constants.37 This is a crucial point as the known overestimation of lattice constants by many other GGA functionals would modify the overall coincidence of FeO on Au and the resulting moiré superstructure. With PBEsol functional 4.08 Å was obtained as bulk gold lattice parameter, as experimentally found.38A layer of FeO(111) is put above three layers of Au(111), with two lower layers of Au kept fixed at bulk positions during the structural optimization procedure. The same procedure has been previously successfully applied for the description of FeO/Au and FeO/Pt moiré superstructures.12,16 Some tests have been performed to assure that three layers of Au are enough to correctly describe the FeO/Au interface, results are collected in Table S1 and Fig. S1 of the ESI.† A vacuum layer of 15 Å has been inserted in between the slabs, with the dipole correction along the non-periodic direction. The resulting structure is shown in Fig. 1 and is characterized by a periodicity of 32 Å, with an angle between FeO and the Au substrate lattices of 9°, in agreement with what was experimentally found.15 The in plane lattice parameter of Au(111) is 2.88 Å, whereas the lattice parameter of the FeO(111) fully relaxed monolayer is 3.22 Å. The lattice mismatch for the FeO monolayer in the FeO/Au moiré model is about 2% along both the a and b directions. Given the large unit cell considered, consisting of 578 atoms, the Brillouin zone has been sampled at the Γ point only. The row-wise antiferromagnetic order has already proven to be the most stable collinear magnetic setting for FeO/Pt21 so we adopted it for FeO/Au. In the moiré superstructure studied in this work an odd number of iron atoms (103) is present and so a magnetic defect, namely a small ferromagnetic zone, can be found in correspondence to the top domain, as already done by Merte et al.16 for FeO/Pt.
 | ||
| Fig. 1 Geometry of the FeO/Au moiré superstructure, with only the first gold layer visible for sake of clarity. Red spheres represent oxygen atoms, yellow spheres gold atoms. Grey and blue spheres represent iron atoms with opposite magnetic moment orientation. Black solid line delimits the cell considered for calculations. The plane highlighted in magenta is the one selected for the plots presented in Fig. 5, 7 and 9. | ||
Bader charges analysis has been performed as implemented in the VASP code39 and STM images have been computed in the framework of the Tersoff–Hamann approximation.40
Three high-symmetry domains can be identified in this moiré superstructure: fcc, hcp and top. In the fcc domain both O and Fe atoms are on top of hollow sites of the Au substrate, in hcp O atoms sit atop of Au atoms of the substrate while Fe atoms are on hollow sites and in the top domain the reverse is true, with Fe atoms atop of Au and O atoms on hollow sites. In order to acquire specific data on the three high-symmetry domains of the moiré and have access to local information, three smaller models have been built, one for each domain. Similar models, consisting of a 2 × 2 cell of FeO(111) on a 2 × 2 cell of Pt(111) have already been used to represent the high-symmetry domains of the FeO/Pt moiré superstructure.10,17 In this work we go beyond this approximation by adapting the in-plane lattice parameter in the three 2 × 2 models, as to reproduce its substantial changes when passing from fcc to hcp and to top domains. More precisely, the mean lattice parameter found for each domain of the moiré, Section 3.2, was then used to build the small models. As in simulation of the full moiré superstructure, also in small models three layers of gold were considered, the lower two fixed. In these models FeO and Au are commensurate to keep the local symmetry: x and y coordinates of Fe atoms are fixed and the substrate is laterally expanded to match the FeO lattice parameter. The optimal Au interplanar spacing has been tuned by relaxing bulk gold for each in-plane lattice parameter considered.
3 Results and discussions
3.1 STM measurement
A typical STM topography is reported in Fig. 2, which shows the Au(111) substrate fully covered by a monolayer FeO film. On a large scale, it is possible to distinguish a hexagonal modulation of the film corrugation (≈50 pm) with a pitch of about 3.1 nm, i.e. the so called moiré superstructure, whereas on a smaller scale also the surface unit cell of the FeO(111) structure becomes apparent, consisting of a hexagonal pattern with a spacing of 3.0 Å. As thoroughly reported in the previous literature,11–14 the long range modulation of the STM contrast of the moiré superstructure is strongly dependent on the tip conditions (i.e. shape and possible presence of adsorbates) and tunneling parameters (i.e. bias and current). | ||
| Fig. 2 STM image at low (a) and high (b) magnification of one monolayer of FeO deposited on the Au(111) surface. The inset in (b) shows a further zoom in on the FeO moiré superstructure. Tunneling parameters: (a) (−1.4 V, 1.5 nA) (b) (−0.9 V, 1 nA). The scale bar is reported on the bottom left corner of the images. | ||
As a matter of fact, Fig. 3 shows that when the tunneling bias is switched from 0.6 V (empty states, bottom part of the image) to −0.6 V (filled states), a clear change of the topographic contrast takes place, and the moiré, which before was visible as an array of bright dots, is seen as dark round regions separated by brighter edges.
 | ||
| Fig. 3 STM image of the FeO/Au(111) surface: during the acquisition at the point indicated by the white dashed line the tunneling bias was switched from +0.6 V (bottom part) to −0.6 V (upper part). Tunneling current 1 nA, scale bar on the bottom left of the image. | ||
3.2 The moire model
The moiré superstructure of FeO/Au depicted in Fig. 1 has been characterised by means of local Fe–Fe distance l (which gives an estimation of the local lattice parameter), film rumpling ΔzFe–O = zO − zFe, interface distance ΔzFeO–Au = zFe + zO)/2 −zAu, Fe–O distance dFe–O, atomic charges q and Fe magnetic moment μ. The results collected in Table 1 correspond to averages over the central atom of each high-symmetry domain and its six closest Fe, O and Au neighbors (see Fig. 4).| Domain | l [Å] | ΔzFe–O [Å] | ΔzFeO–Au [Å] | d Fe–O [Å] | q Fe [e] | q O [e] | μ Fe [μB] |
|---|---|---|---|---|---|---|---|
| fcc | 3.06 | 0.63 | 2.52 | 1.89 | 1.32 | −1.04 | ±3.73 |
| hcp | 3.21 | 0.40 | 2.69 | 1.90 | 1.29 | −1.12 | ±3.69 |
| top | 3.23 | 0.39 | 2.85 | 1.90 | 1.31 | −1.13 | ±3.73 |
 | ||
| Fig. 4 The three high-symmetry domains that can be found in the FeO/Au moiré: (a) fcc, (b) hcp, (c) top, with only the first gold layer visible for sake of clarity. Red spheres represent oxygen atoms, yellow spheres gold atoms. Grey and blue spheres represent iron atoms with opposite magnetic moment orientation. The magenta circles indicate the central region of each domain and identify the atoms whose properties are collected in Table 1. | ||
As expected for metal oxide monolayers deposited on a transition metal substrate, the FeO film on Au is characterized by a significant rumpling with oxygen atoms relaxing outwards.41 However, the three domains are quite different from each other as regards the lattice parameter, rumpling and interface distance, while the Fe–O distance, atomic charges and magnetic moments remain almost the same. In particular, the fcc domain stands out to be the one with the smallest l, the highest ΔzFe–O and the lowest ΔzFeO–Au. In contrast, the top one is the one with the largest l, the lowest ΔzFe–O and the largest ΔzFeO–Au. Finally, the hcp domain displays intermediate properties. These results show that changes of film rumpling accommodate the differences of its lattice parameter, such that Fe–O bond lengths are the same in the three domains. Regarding this aspect of the local film structure, the fcc domain clearly stands out (the smallest l, the largest ΔzFe–O), while the hcp and top ones appear quite similar to each other, the only parameter discriminating between them is the interface distance ΔzFeO–Au, see Table 1.
Regarding electronic characteristics, the difference electron density map presented in Fig. 5 highlights electron redistributions that take place upon formation of the FeO/Au interface from unsupported FeO film and the bare Au substrate. The film-substrate interaction alters mainly the Fe dz2 and O pz orbitals, but modifications appear to be different in the three high-symmetry domains. The Fe dz2 orbitals are systematically depopulated, but the effect is by far the most pronounced in the fcc domain. The modification of O pz orbitals is relatively weak in the top and hcp domains, while in the fcc one they become visibly populated. More importantly, Fig. 5 reveals a substantial charge accumulation at the FeO/Au interface, represented by red zones between the oxide film and the metal substrate. It corresponds to an electron transfer from the FeO film into the Au substrate which, based on Au atomic charges, amounts in average to −0.21 e/FeO. Also in this case, this electron transfer is much more pronounced in the fcc domain, where it also associated with the most visible modifications of charge density at Au atoms in the surface plane of gold.
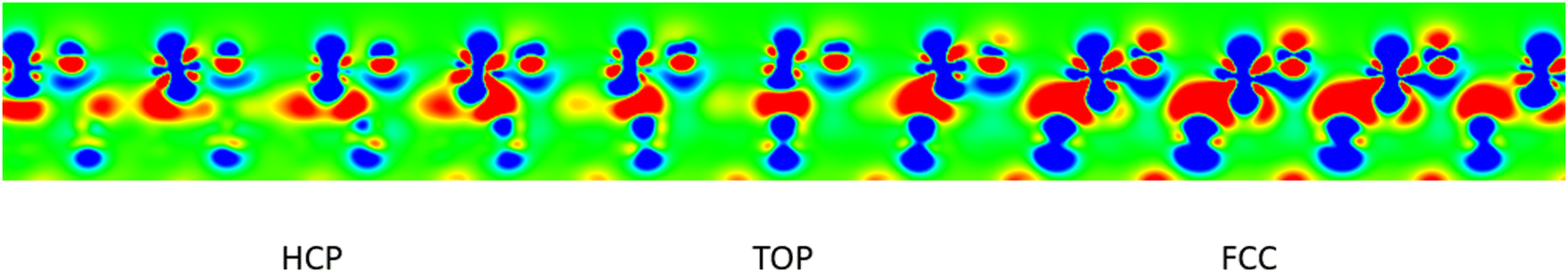 | ||
| Fig. 5 Difference electron density map between the FeO/Au moiré, the FeO slab and the Au slab, for a plane passing through Fe and O atoms of the three high-symmetry domains. Regions of electron excess and deficiency are depicted in red and blue, respectively. Only the FeO film and the first layer of the Au substrate are shown in this Figure for sake of clarity. | ||
These results show that in the fcc domain, the FeO film interacts most strongly with the Au substrate, whereas in top and hcp ones the interaction is remarkably weaker. The observation is additionally supported by the behaviour of interface distances in the three domains, Table 1. These differences are expected to impact the local film stability, interface charge transfer and work function in the three domains, but it is not possible to quantify these effects from the full moiré superstructure. This topic will be discussed in Section 3.3, in which smaller models representing the three high-symmetry domains are described.
The results presented in Table 1 can be compared with those obtained for similar FeO/Pt and FeO/Pd systems. Some similarities can be observed, as fcc is recognized to be the most stable domain and the one interacting the most strongly with the substrate, while top is the one that interacts to a lesser extent, also in FeO/Pt and FeO/Pd.19–21,23,26 In particular, as in our study, also in FeO/Pt and FeO/Pd the local lattice parameter l increases from fcc to hcp to top. Nonetheless, l values in FeO/Au are larger than those reported for FeO/Pt and FeO/Pd (e.g. fcc/hcp/top computed l is 3.00/3.09/3.18 Å for FeO/Pd,26 3.01/3.08/3.15 Å for FeO/Pt,20,23 while in this work it is 3.06/3.21/3.23 for FeO/Au). This difference can be assigned to the larger lattice parameter of Au (4.08 Å) compared to Pt and Pd (3.92 Å and 3.89 Å respectively), influencing the whole modulation across the moiré. However, the trend of increasing l from fcc to hcp to top is the same in all the considered systems.
Regarding film rumpling, its relative strength is also the same in the three considered systems, with the fcc domain systematically exposing the largest ΔzFe–O, followed by the hcp and the top ones.20,21,23,26 In terms of their values, the ΔzFe–O is systematically smaller in FeO/Au, (computed ΔzFe–O in fcc/hcp/top domains is 0.74/0.64/0.47 Å for FeO/Pd,26 0.77/0.71/0.58 Å for FeO/Pt20 while in this work is 0.63/0.40/0.39 Å for FeO/Au). This is in part due to a smaller mismatch between FeO and Au lattices but, since film rumpling is also induced by an electrostatic interaction of Fe and O ions with the negatively charged substrate,41 the smaller ΔzFe–O values may also reflect a weaker oxide–metal interaction, accompanied by a smaller interface charge transfer. This observation is supported by the corresponding values of interface distances (fcc/hcp/top computed interface distance is 2.59/2.59/2.70 Å for FeO/Pd,26 2.61/2.62/2.77 Å for FeO/Pt,23 while in this work it is 2.52/2.69/2.85 Å for FeO/Au), which are systematically much longer in the present FeO/Au system. The fcc domain, in which ΔzFeO–Au is visibly shorter than the corresponding ΔzFeO–Pt and ΔzFeO–Pd, constitutes an interesting exception. It points again to the particularity of the fcc domain in FeO/Au that, not only structurally stands strongly out with respect to the hcp and top ones, but also does not follow the trends found for the FeO on the Pt and Pd substrates.
We have simulated STM images of the FeO/Au moiré system and results are shown in Fig. 6 and 7. The images in Fig. 6 have been computed at different positive and negative values of bias and taken at heights between 2.5 and 3.5 Å above the FeO layer. The line profiles of the partial charge density (bias +0.3 V) presented in Fig. 7 are plotted for two different density values, in a plane passing through Fe and O atoms in the three high-symmetry domains of the moiré.
 | ||
| Fig. 6 STM images for the FeO/Au moiré superstructure computed in this work at different biases. Symbols mark the three high-symmetry domains: □ fcc, Δ hcp, and ○ top. Computed images are taken at 2.5 Å above the FeO layer. | ||
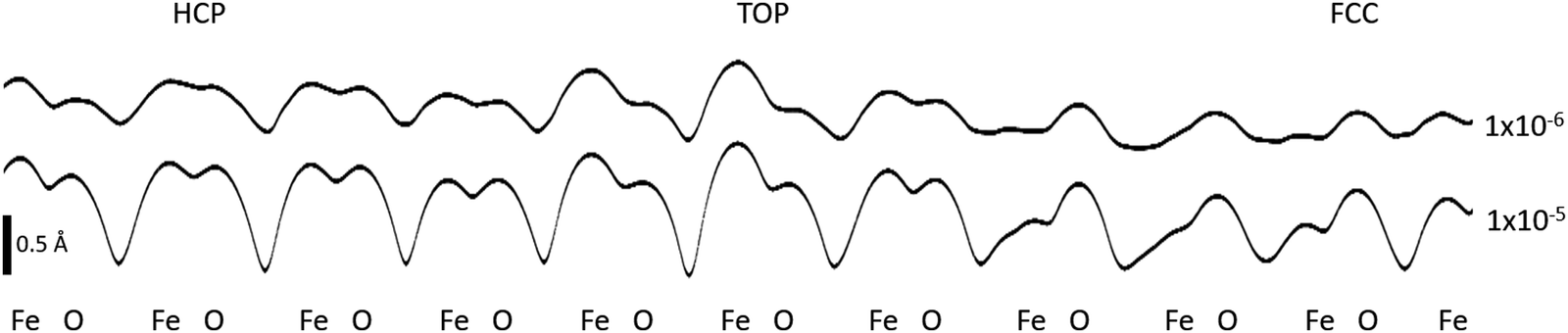 | ||
| Fig. 7 Line profiles extracted form the moiré STM image at +0.3 V, showing the variation across the three high-symmetry domains of the moiré. Isolines are taken at 10−5 and 10−6 e/Å3. | ||
Our calculations account fairly well for the overall contrast modulation observed in Fig. 3 and in other experimental studies,11,14,15 with fcc being the darkest region, top the brightest and hcp the one with an intermediate contrast, regardless the value of bias, Fig. 6. Also the line profiles, Fig. 7, show that the average apparent height is the smallest in the fcc domain and the largest in the top one, roughly following the mean geometric heights of surface ions in the three domains (the same relative height is found in the FeO/Pt moiré superstructure20). Our computed STM images reproduce in a excellent way the experimental STM image reported in Fig. 3 and appear to be sensitive to the applied bias, without unfortunately reproducing the inversion of contrast passing from negative to positive biases. However in the work from Yang et al. the top region is found to be the brightest at both negative and positive biases (see Fig. 2 of ref. 15) as in our simulated images.
On the computational side, the accuracy of the theoretical method is limited by the Tersoff–Hamann approximation we are working with.40 The STM images are obtained by sampling the electron density of the FeO/Au system only, whereas the experimentally measured contrast is driven by the chemical interaction between surface atoms and the specific tip state,20 which is completely neglected in this computational framework. This dependency of the contrast from the tip leads to a great variability also in experimentally acquired STM images, emphasizing the complexity and sensitivity of the technique. Specific tip states can be considered in first principle calculations in the framework of the Bardeen's method42 or Chen's derivative method,43 explicitly modelling the atoms of the tip and overcoming the limitation of the Tersoff–Hamann approximation. These approaches are however more suited for the study of a localised point of interest rather than for obtaining the STM image of a whole surface as we desire for the FeO/Au moiré superstructure.44,45 Considering the large dimension of our cell and the diverse morphology it presents along the lattice parameter, the STM simulation would require a complex modelling of the substrate/tip interaction, which which would be difficult to construct and would exceed our computational power.
Interestingly, the origin of the calculated atomically-resolved contrast is different in the three domains as depicted in Fig. 7. In the fcc one, surface oxygen atoms produce a much more pronounced contrast than the Fe atoms, which is consistent with the large film rumpling and their relative geometric height in this domain. In contrast, in the top domain, the STM contrast is mainly due to surface Fe ions. In this region the geometric effect is less strong (film rumpling is much smaller), such that the electronic effect, associated to a large density of unoccupied Fe states (see Fig. 8), contributes also to the STM contrast. In the hcp domain, the Fe and O atoms display a similar apparent height deriving from the partial charge density: both of them should be visible in STM images. No assignment of the atomically-resolved contrast to particular surface ions was proposed in other experimental studies.11,14,15
 | ||
| Fig. 8 PDOS (projected density of states) of the central atoms at the three symmetry regions: blue line for fcc, green line for top and yellow line for hcp. Solid lines represent Fe PDOS, dashed lines O PDOS. Alpha and beta states have been summed in this plot. The Fermi energy (Ef) is marked with a red line. | ||
Comparing with the similar and more studied FeO/Pt system, considering both experimental and theoretical studies, we can see that the modulation across the moiré is essentially the same.10,16,19,20,23 On FeO/Pt an unambiguous determination of the three different domains of the moiré was achieved by Merte et al., who have inserted defects in the different domains in order to identify them in STM images.19,20 They found that in their and in most studies, the top domain is the brightest at positive and slightly negative biases, while it becomes dark at negative bias values.20 It is more difficult to discriminate in between the hcp and fcc domains: fcc has the lowest work function and so it is often recognized to be the darkest region, but it is not easy to differentiate its contrast from the one of hcp, especially at low bias values.19,20 Furthermore, FeO/Pt STM images were recognized to be “mixed”, meaning that atomic contrast in STM images corresponds to either surface O or Fe atoms, depending on the high-symmetry domain. In particular protrusions due to Fe atoms were found in the top domain, to O atoms in the fcc domain, while in the hcp one a nearly equal tip height was found over surface O and Fe atoms.20
All these characteristics are consistent with our present results on FeO/Au, thus validating our approach. The main difference is that the calculated STM contrast for FeO/Au is the same for every bias value. Aiming not only to rationalize the experimental STM images, but also to enquire on the origin of the contrast modulation, the electrostatic potential above the FeO/Au surface has been computed, and it is shown in a plane passing through the three high-symmetry domains of the moiré in Fig. 9.
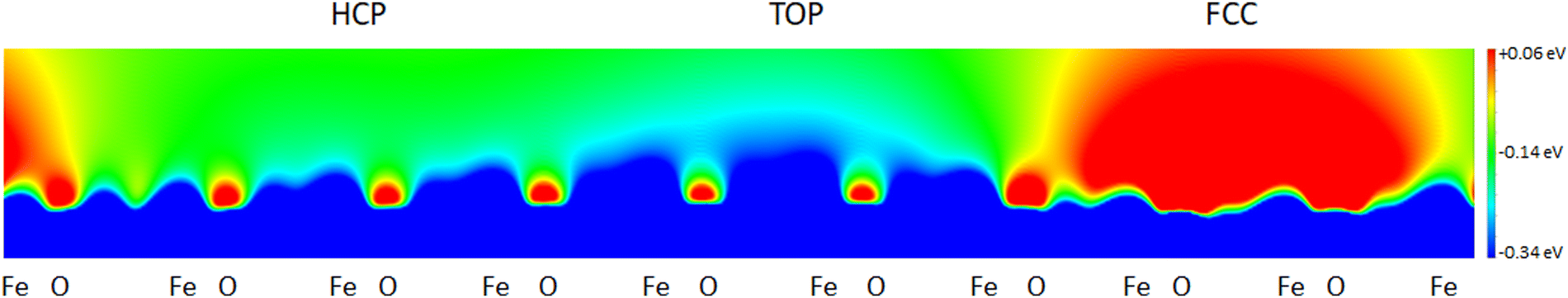 | ||
| Fig. 9 Electrostatic potential of the FeO/Au surface plotted on a plane passing through Fe and O atoms in the three high-symmetry regions of the moiré. | ||
The fcc domain is characterized by a large zone of surface electrostatic potential that is strongly repulsive for electrons (red), while the top one produces an overall attractive electrostatic effect (blue). This makes electron tunneling more difficult in the fcc domain (where the local work function is expected to be the largest) than in the top one (where it is expected to be the smallest). Since a larger work function generally results in a lower tunneling current,46 the modulation of the surface electrostatic potential could be held responsible for the overall modulation of the STM contrast, and the dark (bright) appearance of the fcc (top) domains in Fig. 6.
3.3 2 × 2 models
Computational results for the full moiré superstructure do not give access some of the local properties such as the work function Φ, the interface charge transfer CT and the adhesion energy Ead between the FeO film and the Au substrate. All of these features are important not only to further interpret the STM contrast but also for a first assessment of reactivity differences in the three high-symmetry domains. Small 2 × 2 models of the fcc, hcp and top domains have been built, with in-plane lattice parameters l estimated at the three high-symmetry domains of the moiré superstructure (Table 1). The structural and electronic characteristics are reported in Table 2.| Domain | l [Å] | ΔzFe–O [Å] | ΔzFeO–Au [Å] | d Fe–O [Å] | q Fe [e] | q O [e] | μ Fe [μB] | E ad [eV] | CT | ΔΦ [eV] |
|---|---|---|---|---|---|---|---|---|---|---|
| fcc | 3.06 | 0.68 | 2.48 | 1.87 | +1.31 | −1.03 | ±3.71 | −0.66 | −0.28 | +0.31 (+5.44) |
| hcp | 3.21 | 0.44 | 2.53 | 1.90 | +1.24 | −1.11 | ±3.65 | −0.56 | −0.13 | −0.35 (+5.24) |
| top | 3.23 | 0.32 | 2.80 | 1.89 | +1.27 | −1.14 | ±3.66 | −0.53 | −0.12 | −0.79 (+5.16) |
Even at this level of approximation the resemblance with data in Table 1 is remarkable, since not only all the trends are correctly reproduced, but also the computed values of ΔzFe–O, ΔzFeO–Au, dFeO, q and μ are quite similar to what was found in the moiré superstructure, assuring that these small models can indeed convincingly represent the three domains. As described in Table 1 for the full moiré superstructure, also in these smaller models the two principal features that discriminate the three domains are ΔzFe–O and ΔzFeO–Au, while dFe–O, q and μ are the same in all the high-symmetry domains. The additional features presented in Table 2 are Ead, CT and Φ. CT has a negative sign as expected for metal oxide monolayers deposited on a transition metal support.41 Because of high electronegativity of the Pt substrate, electrons move from cations of the oxide film to the substrate and the overall structural response to this interfacial charge transfer is the rumpling of FeO, with oxygens relaxing outwards. As a consequence, the strengths of film rumpling and interface charge transfer are correlated, with their maximal values found in the fcc domain. The most negative Ead found in the fcc domain confirms that there the film-substrate interaction is the strongest. This fact is additionally corroborated by the the smallest value of ΔzFeO–Au and the shortest l. In contrast, Ead is the least negative in the top domain. This confirms the weak interaction with the substrate, consistent with the smallest CT and the largest ΔzFeO–Au. Finally, the value ΔΦ reported in Table 2 represents the work function difference between the FeO/Au system and the bare Au(111) surface, presented in brackets in Table 2. Again, the fcc domain stands out, being the only one with a positive ΔΦ value and a Φ considerably larger than that in the hcp and top domains, suggesting that the dipole moment associated to the film rumpling determines the modulation of the local work function. We notice that the domains with the largest and the smallest work function (fcc and top, respectively), correspond well to those with the most repulsive and attractive surface electrostatic potential in Fig. 9, respectively.
When comparing these results with a previous literature study on small models for the three high symmetry domains of FeO/Pt21 we can see that also in this case the trends for l, ΔzFe–O, ΔzFeO–Au, Ead and Φ are the same for the three domains. The one thing that stands out the most when comparing the two works, is that in FeO/Pt the hcp domain has characteristics (such as ΔzFe–O and the Φ) which are closer to fcc, while in FeO/Au the hcp domain seems to be more similar to top. Again, as observed for the moiré superstructure, in FeO/Au ΔzFe–O for hcp and top is lower with respect to the value computed for FeO/Pt (i.e. computed ΔzFe–O for FeO/Pt smaller models fcc/hcp/top is 0.68/0.65/0.51 Å,21 while in this work the computed ΔzFe–O for FeO/Au smaller models is 0.68/0.44/0.32 Å). This results also in a lower ΔΦ in FeO/Au compared to FeO/Pt (i.e. computed ΔΦ for FeO/Pt smaller models fcc/hcp/top is +0.49/+0.13/−0.60 eV21 while in this work the computed ΔΦ for FeO/Au smaller models is +0.31/−0.35/−0.79 eV). This behaviour may derive from Au larger parameter with respect to Pt, but also by a lower charge transfer from FeO to Au. This can be true especially at the top and hcp domains, in which a lower charge transfer may be the cause of the lower rumpling and ΔΦ found out when comparing FeO/Au to FeO/Pt.
From data collected in Table 2, we can conclude that the fcc domain has the strongest interaction with the substrate and this determines ΔzFeO–Au to be the shortest and the CT towards gold to be largest. The ΔzFe–O here is the largest because it is a structural response to the interfacial CT.41 As seen before, the opposite can be said for top, while hcp has intermediate characteristics. These features are different in the three high symmetry domains and are the cause of different Φ and different contrast in STM.
4 Conclusions
In this work we characterized the FeO/Au system, comparing our results to the more studied FeO/Pt and FeO/Pd systems to validate our method. We studied the moiré superstructure as a whole and three smaller models of the three high-symmetry domains in order to acquire specific information. We reproduced experimental STM images, rationalized the origin of their contrast and how it changes with respect to the bias and the tunneling distance. Different specific features identified in the three high-symmetry domains, such as surface electrostatic potential and work function, local film rumpling, interface distance and charge transfer, may suggest different adsorption and reactivity properties across the FeO/Au moiré, as happens for FeO/Pt.11,16In general FeO/Au results to be similar to FeO/Pt and FeO/Pd. Nonetheless, some differences as the lower rumpling, the work function and their correlation with the surface electrostatic potential can be important in determining the surface reactivity towards water and the possible exploitation of this system as a catalyst for the electrochemical water splitting reaction. Further studies are needed to have a better understanding of the reactivity towards water, both from an experimental and computational point of view.
In this work we have proposed a strategy to build smaller models for the three high-symmetry domains, keeping the local characteristics that can be found in the entire structure. This approach could be useful in the future to allow the simulation of different adsorption mechanisms that would be prohibitive considering a system of the size of the entire moiré.
Author contributions
Conceptualization, A. M. Ferrari, S. Agnoli. Data curation, E. Ascrizzi, J. Yang. Formal analysis, E. Ascrizzi, J. Goniakowski, A. M. Ferrari. Funding acquisition, A. M. Ferrari, S. Agnoli. Investigation, E. Ascrizzi. Methodology, E. Ascrizzi, J. Goniakowski, A. M. Ferrari. Project administration, A. M. Ferrari, S. Agnoli. Resources, A. M. Ferrari, J. Goniakowski. Software, E. Ascrizzi, J. Goniakowski. Supervision, A. M. Ferrari, J. Goniakowski, S. Agnoli. Validation, A. M. Ferrari, J. Goniakowski, S. Agnoli. Visualization, E. Ascrizzi, J. Yang, J. Goniakowski. Writing – original draft, E. Ascrizzi. Writing – review & editing, A. M. Ferrari, J. Goniakowski, S. Agnoli.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, by the Italian Ministry of University and Research (MUR), funded by the European Union - NextGenerationEU- Project Title “Microscopic approach to Understand Synergies in Electrocatalysis” - Project No. 2022E5L4Y2 - CUP: C53D23003780006. We acknowledge support from Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001). J. Yang ackowledges the China scholarship council for the financial support No. 202006880005. The access to the ARCHER2 UK National Supercomputing Service (https://www.archer2.ac.uk) is also gratefully acknowledged.References
- I. Concina, Z. H. Ibupoto and A. Vomiero, Adv. Energy Mater., 2017, 7, 1700706 CrossRef.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- T. Jafari, E. Moharreri, A. S. Amin, R. Miao, W. Song and S. L. Suib, Molecules, 2016, 21, 900 CrossRef PubMed.
- J.-W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu, A. Javey, J. Guo and D. Wang, Nat. Commun., 2015, 6, 1–5 Search PubMed.
- J. S. Kang, Y. Noh, J. Kim, H. Choi, T. H. Jeon, D. Ahn, J.-Y. Kim, S.-H. Yu, H. Park, J.-H. Yum, W. Choi, D. C. Dunand, H. Choe and Y.-E. Sung, Angew. Chem., Int. Ed., 2017, 56, 6583–6588 CrossRef CAS PubMed.
- B. H. R. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- C. J. Heard, J. Čejka, M. Opanasenko, P. Nachtigall, G. Centi and S. Perathoner, Adv. Mater., 2019, 31, 1801712 CrossRef PubMed.
- Q. Fu, W.-X. Li, Y. Yao, H. Liu, H.-Y. Su, D. Ma, X.-K. Gu, L. Chen, Z. Wang, H. Zhang, B. Wang and X. Bao, Science, 2010, 328, 1141–1144 CrossRef CAS PubMed.
- Y.-N. Sun, Z.-H. Qin, M. Lewandowski, E. Carrasco, M. Sterrer, S. Shaikhutdinov and H.-J. Freund, J. Catal., 2009, 266, 359–368 CrossRef CAS.
- L. Giordano, M. Lewandowski, I. M. N. Groot, Y.-N. Sun, J. Goniakowski, C. Noguera, S. Shaikhutdinov, G. Pacchioni and H.-J. Freund, J. Phys. Chem. C, 2010, 114, 21504–21509 CrossRef CAS.
- Y. Li, K. C. Adamsen, L. Lammich, J. V. Lauritsen and S. Wendt, ACS Nano, 2019, 13, 11632–11641 CrossRef CAS PubMed.
- L. Yu, Y. Liu, F. Yang, J. Evans, J. A. Rodriguez and P. Liu, J. Phys. Chem. C, 2015, 119, 16614–16622 CrossRef CAS.
- N. A. Khan and C. Matranga, Surf. Sci., 2008, 602, 932–942 CrossRef CAS.
- Y. Jiang, S. Bu, D. Zhou, X. Shi, F. Pan, Q. Ji and T. Niu, J. Phys. Chem. C, 2021, 125, 24755–24763 CrossRef CAS.
- S. Yang, J. Gou, P. Cheng, L. Chen and K. Wu, Phys. Rev. Mater., 2020, 4, 074004 CrossRef CAS.
- L. R. Merte, R. Bechstein, G. Peng, F. Rieboldt, C. A. Farberow, H. Zeuthen, J. Knudsen, E. Lægsgaard, S. Wendt, M. Mavrikakis and F. Besenbacher, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- F. Ringleb, Y. Fujimori, H.-F. Wang, H. Ariga, E. Carrasco, M. Sterrer, H.-J. Freund, L. Giordano, G. Pacchioni and J. Goniakowski, J. Phys. Chem. C, 2011, 115, 19328–19335 CrossRef CAS.
- M. Ritter, W. Ranke and W. Weiss, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 7240–7251 CrossRef CAS.
- L. R. Merte, J. Knudsen, L. C. Grabow, R. T. Vang, E. Lægsgaard, M. Mavrikakis and F. Besenbacher, Surf. Sci., 2009, 603, L15–L18 CrossRef CAS.
- L. R. Merte, L. C. Grabow, G. Peng, J. Knudsen, H. Zeuthen, W. Kudernatsch, S. Porsgaard, E. Lægsgaard, M. Mavrikakis and F. Besenbacher, J. Phys. Chem. C, 2011, 115, 2089–2099 CrossRef CAS.
- L. Giordano, G. Pacchioni, J. Goniakowski, N. Nilius, E. D. L. Rienks and H.-J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 075416 CrossRef.
- L. Giordano, G. Pacchioni, J. Goniakowski, N. Nilius, E. D. L. Rienks and H.-J. Freund, Phys. Rev. Lett., 2008, 101, 026102 CrossRef PubMed.
- W. Zhang, Z. Li, Y. Luo and J. Yang, J. Phys. Chem. C, 2009, 113, 8302–8305 CrossRef CAS.
- W. Weiss and W. Ranke, Prog. Surf. Sci., 2002, 70, 1–151 CrossRef CAS.
- Y. J. Kim, C. Westphal, R. X. Ynzunza, H. C. Galloway, M. Salmeron, M. A. Van Hove and C. S. Fadley, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, R13448 CrossRef CAS.
- H. Zeuthen, W. Kudernatsch, G. Peng, L. R. Merte, L. K. Ono, L. Lammich, Y. Bai, L. C. Grabow, M. Mavrikakis, S. Wendt and F. Besenbacher, J. Phys. Chem. C, 2013, 117, 15155–15163 CrossRef CAS.
- M. Lewandowski, T. Pabisiak, N. Michalak, Z. Miłosz, V. Babačič, Y. Wang, M. Hermanowicz, K. Palotaás, S. Jurga and A. Kiejna, Nanomaterials, 2018, 8, 828 CrossRef PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78(1), 013705 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- M. Shishkin and H. Sato, J. Chem. Phys., 2019, 151(2), 024102 CrossRef CAS PubMed.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- P. Romaniello and P. L. de Boeij, J. Chem. Phys., 2005, 122(16), 164303 CrossRef CAS PubMed.
- H. A. Alluhaybi, S. K. Ghoshal, B. O. Alsobhi and W. N. W. Shamsuri, Comput. Condens. Matter, 2019, 18, e00360 CrossRef.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- R. W. G. Wyckoff and R. W. Wyckoff, Crystal structures, Interscience publishers, New York, 1963, vol. 1 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805–813 CrossRef CAS PubMed.
- J. Goniakowski and C. Noguera, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155433 CrossRef.
- J. Bardeen, Phys. Rev. Lett., 1961, 6, 57–59 CrossRef CAS.
- C. J. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 8841–8857 CrossRef PubMed.
- S. Duan, G. Tian and X. Xu, JACS Au, 2023, 3, 86–92 CrossRef CAS PubMed.
- A. Gustafsson and M. Paulsson, Phys. Rev. B, 2016, 93, 115434 CrossRef.
- G. Binnig, H. Rohrer, Ch Gerber and E. Weibel, Phys. Rev. Lett., 1982, 49, 57–61 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Tests to demonstrate how calculation are affected by the number of Au layers are reported. See DOI: https://doi.org/10.1039/d4cp01546k |
| This journal is © the Owner Societies 2024 |