
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine-tuning of radiative properties by “mild” substituents: searching for a perfectly soft chromophore†
Grażyna
Orzanowska
a,
Claudia
Ryppa
b,
Mathias O.
Senge
 *b and
Jacek
Waluk
*ac
*b and
Jacek
Waluk
*ac
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: jwaluk@ichf.edu.pl
bSchool of Chemistry, Chair of Organic Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, The University of Dublin, 52-160 Pearse Street, Dublin D02R590, Ireland
cFaculty of Mathematics and Science, Cardinal Stefan Wyszyński University, Dewajtis 5, 01-815 Warsaw, Poland
First published on 7th June 2024
Abstract
Controlling spectral properties to achieve desired characteristics is an attractive goal in application-oriented research, e.g., in the design of fluorescence sensors. “Soft” chromophores, molecules with strong spectral responses to internal or external perturbations are good candidates for such studies. In this work, absorption, fluorescence, and magnetic circular dichroism (MCD) spectra were obtained for a series of porphyrins, substituted at the meso-positions with n-hexyl groups. As the number of substituents increases from 1 to 4, significant changes are observed. The intensity of the S0–S1 transition (Qx) in the 0–0 region strongly decreases in mono-substituted porphyrin, but upon additional substitutions it increases to values larger than in the parent, unsubstituted molecule. Such behavior can be explained, using the perimeter model, by changes in the energy splittings between the two highest (HOMO) and two lowest (LUMO) frontier molecular orbitals. Single substitution makes porphyrin a nearly perfect soft chromophore, but upon introduction of a larger number of n-hexyl groups it is transformed into a hard one. DFT simulations incorrectly predict a continuous transition from a soft to hard chromophore, because the calculated ordering of two HOMO orbitals is opposite to that obtained by experiment. On the other hand, for those porphyrins that can be classified as hard chromophores, the calculations nicely reproduce contributions of Franck–Condon and Herzberg–Teller terms to absorption and fluorescence spectra.
Introduction
Rational design of materials considered for applications based on light–matter interactions requires a detailed spectral and photophysical characterization of a potentially useful chromophore. One of the crucial parameters in this respect is the radiative constant of the S0–S1 electronic transition (kr). The value of kr determines the strength of the absorption; it also competes with knr, the sum of the rate constants of processes responsible for nonradiative S1 depopulation. Since the fluorescence quantum yield is proportional to kr, maximizing its value is particularly important for generating efficient fluorophores. This can be achieved, e.g., by substitution that enhances kr. Quite often, however, such modifications involve strongly electron donating or withdrawing moieties and may lead to undesired changes, such as decrease of photostability. Therefore, a chemically “mild” substituent that would affect the radiative properties of a chromophore without changing the other characteristics is an attractive solution.A simple and very useful procedure that allows predicting a spectral response of a cyclic π-electron system to substitution is based on the perimeter model.1 A chromophore of interest is derived from an ideal [n]annulene perimeter by perturbations that may include various effects, e.g., substitution by an electron withdrawing or accepting group. Analytical formulas have been derived that allow predictions of spectral changes caused by a specific perturbation from the knowledge of energy splittings in the two pairs of frontier orbitals: two highest occupied (HOMO) and two lowest unoccupied (LUMO) ones. The magnitude of the splitting can be readily evaluated by inspection of the shapes of the orbitals of the parent perimeter.
The perimeter model points to the class of compounds for which, upon substitution, one can expect large changes in the electronic parameters that characterize electronic absorption, emission, as well as magnetic circular dichroism.1 These compounds belong to the category of the so-called “soft” chromophores, characterized by similar or, at best, equal values of the energy differences between the frontier π orbitals: two highest occupied (ΔHOMO) and two lowest unoccupied ones: (ΔLUMO). For such systems, dramatic effects can be observed, e.g., changes of MCD signs upon single substitution with an “innocuous” methyl group, as reported for methyl-substituted pyrenes.2
Transition from a soft (ΔHOMO ≈ ΔLUMO) to a hard (ΔHOMO ≫ ΔLUMO or ΔHOMO ≪ ΔLUMO) chromophore often results in a large increase of radiative parameters. This has been predicted3 and experimentally demonstrated for porphyrin4–13 and its isomers.14–16 The “hardest” among the isomers is porphycene, whereas parent porphyrin (‘porphine’) is a soft chromophore. The differences in the orbital energy patterns result in the S1(0–0) absorption in porphycene being about 50 times stronger than in porphyrin.17
The frontier orbitals energy pattern in porphyrin does not exactly correspond to an ideal soft chromophore, for which ΔHOMO = ΔLUMO. The present and previous18 DFT calculations, consistent with the observed MCD signs, indicate that HOMO orbitals are split by a small amount, whereas the LUMOs are nearly degenerate, and therefore ΔHOMO > ΔLUMO. One can therefore ask whether, by using appropriate substitution, it is possible to get closer to equal HOMO and LUMO splittings. In this work, we test this possibility by investigating spectroscopic and photophysical properties of the parent porphyrin and five derivatives bearing 1, 2, 3, and 4 n-hexyl groups at the meso-positions (Fig. 1). Alkyl groups act as weak electron donors, which means that they mostly interact with HOMO orbitals, raising their energy. The effect depends on the value of the linear combination of atomic orbitals (LCAO) coefficient at the position of substitution in a particular porphyrin orbital. Two scenarios are possible, depicted in Fig. 2. If the lower occupied frontier orbital (HOMO−1) has a node at the meso-position, the destabilization of the other (HOMO) orbital will lead, upon successive substitutions, to monotonic increase of ΔHOMO, making the chromophore harder and harder. For the opposite orbital ordering the situation is more complicated. The initial substitution may decrease the energy gap, but once the ordering is inverted, ΔHOMO will increase. In this work, we demonstrate for n-hexyl substituted porphyrins, that the latter case occurs. Depending on the number of substituents, the parent porphyrin can be transformed into a nearly perfect soft or a hard chromophore. In contrast, nonradiative properties of the S1 state remain practically unchanged. We also show how the ratio of Franck–Condon and Herzberg–Teller contributions to the probability of weakly allowed S0–S1 and S0–S2 transitions evolves when a chromophore passes from soft to hard regime.
 | ||
| Fig. 1 Structural formulas of the porphyrins investigated. | ||
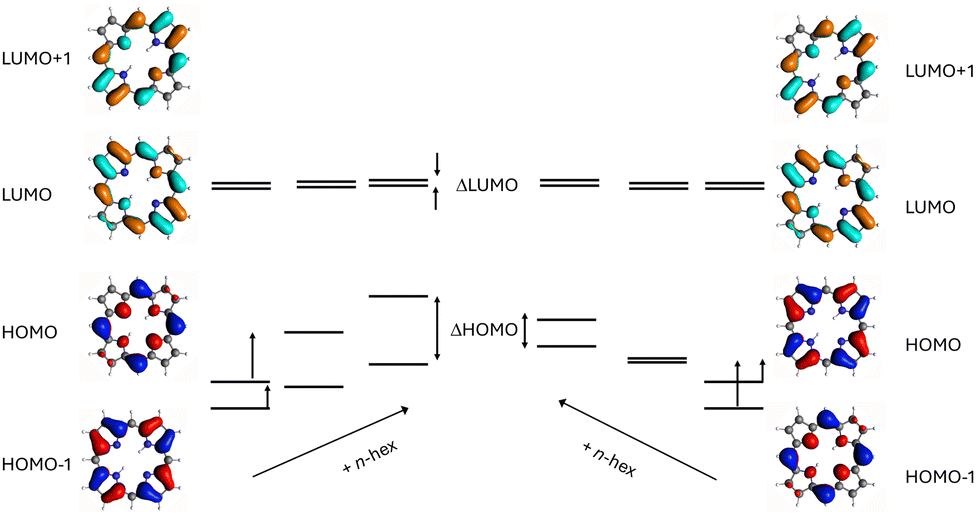 | ||
| Fig. 2 Changes of orbital energies upon substitution with alkyl groups as expected for two possible orderings of HOMO energy levels. | ||
Materials and methods
Syntheses of Pr, Pr-1,19Pr-2a, Pr-2b,20Pr-3,21 and Pr-422 have been reported before.Absorption spectra were measured on Shimadzu UV-3100 and UV-2700 spectrophotometers. Fluorescence spectra were recorded with an Edinburgh FS 090 CDT spectrofluorometer. The same apparatus was used for fluorescence decay measurements, with a 370 nm, 1.5 ns pulse NanoLED (IBH) as the excitation source. FAST software package (Edinburgh Instruments) was used for the determination of decay times. Fluorescence quantum yields were obtained using cresyl violet in ethanol (φfl = 0.55)23 and porphycene in n-hexane (φfl = 0.44)24 as references.
Magnetic circular dichroism (MCD) spectra were initially measured with an OLIS DSM17 spectropolarimeter equipped with a ≈1 T permanent magnet, and then remeasured on a JASCO J-1500 spectropolarimeter, for which the magnetic field of the electromagnet was calibrated using the MCD signal of aqueous CoSO4 solution25 as a standard (nominal field value of 1.5 T, 1.36 T after calibration).
Spectroscopic grade solvents (Merck, UVASOL) have been used. n-Hexane solutions were studied in most cases, except for the parent, unsubstituted porphyrin, for which, due to solubility problems, toluene was used for absorption and MCD studies in the low energy region and for the determination of absorption coefficients.
Quantum-chemical calculations were applied to simulate absorption and MCD spectra. Electronic transition energies were initially calculated using time-dependent density functional theory (TDDFT) with Gaussian 0326 (B3LYP/6-31G(d,p)). Next, the calculations were extended to include MCD, as implemented in the ADF software package.27 BP86-D functional with DZP basis set were used; similar combinations were successful in the analysis of electronic spectra of porphyrinoids.28–30 In all cases, geometry optimization was performed prior to calculating transition energies.
For Pr-2b, two ‘trans’ tautomeric forms are possible; we label them Pr-2b′ and Pr-2b′′. Calculations performed for both tautomers indicate that they are nearly isoenergetic and that their spectral properties should be very similar.
In addition to DFT-based methods, calculations were also done using an extended version of DZDO, an INDO/S-based program, kindly provided by Josef Michl and John Downing.
Results
Absorption spectra
The absorption spectra of all the investigated compounds (Fig. 3) exhibit the pattern typical for porphyrins.31 Four main bands are observed in the low energy region, corresponding to two lowest electronic transitions, L1(Qx) and L2(Qy). For both L1 and L2, the lower energy part corresponds mainly to the 0–0 transition; it is followed by a stronger band, which contains vibronic contributions. Very intense Soret band,32 observed in the blue region contains at least two electronic transitions (B1 and B2). Inspection of Fig. 3 shows that the introduction of consecutive n-hexyl moieties leads to pronounced spectral changes. The Soret band gradually shifts to the red and becomes more intense. The behavior of Q bands is less regular. All four bands gradually shift to lower energies, but the intensity changes for 0–0 transitions are very different from those of the vibronic components. The latter are not much affected by the substitution. On the contrary, intensities of 0–0 transitions exhibit dramatic and non-monotonous changes. Upon single n-hexyl substitution, going from Pr to Pr-1, the 0–0 band practically disappears for Qx and becomes extremely weak for the Qy transition. Adding more substituents, however, results in the gradual increase of the 0–0 band intensity. In Pr-4, this effect is so strong that the intensity of the S1(0–0) band becomes higher than that of the vibronic component. Similarly, the two bands contributing to Qy become comparable in intensity: the intensity ratio (measured at the band maximum) is 0.79, compared to 0.18 in unsubstituted Pr and 0.12 in Pr-1.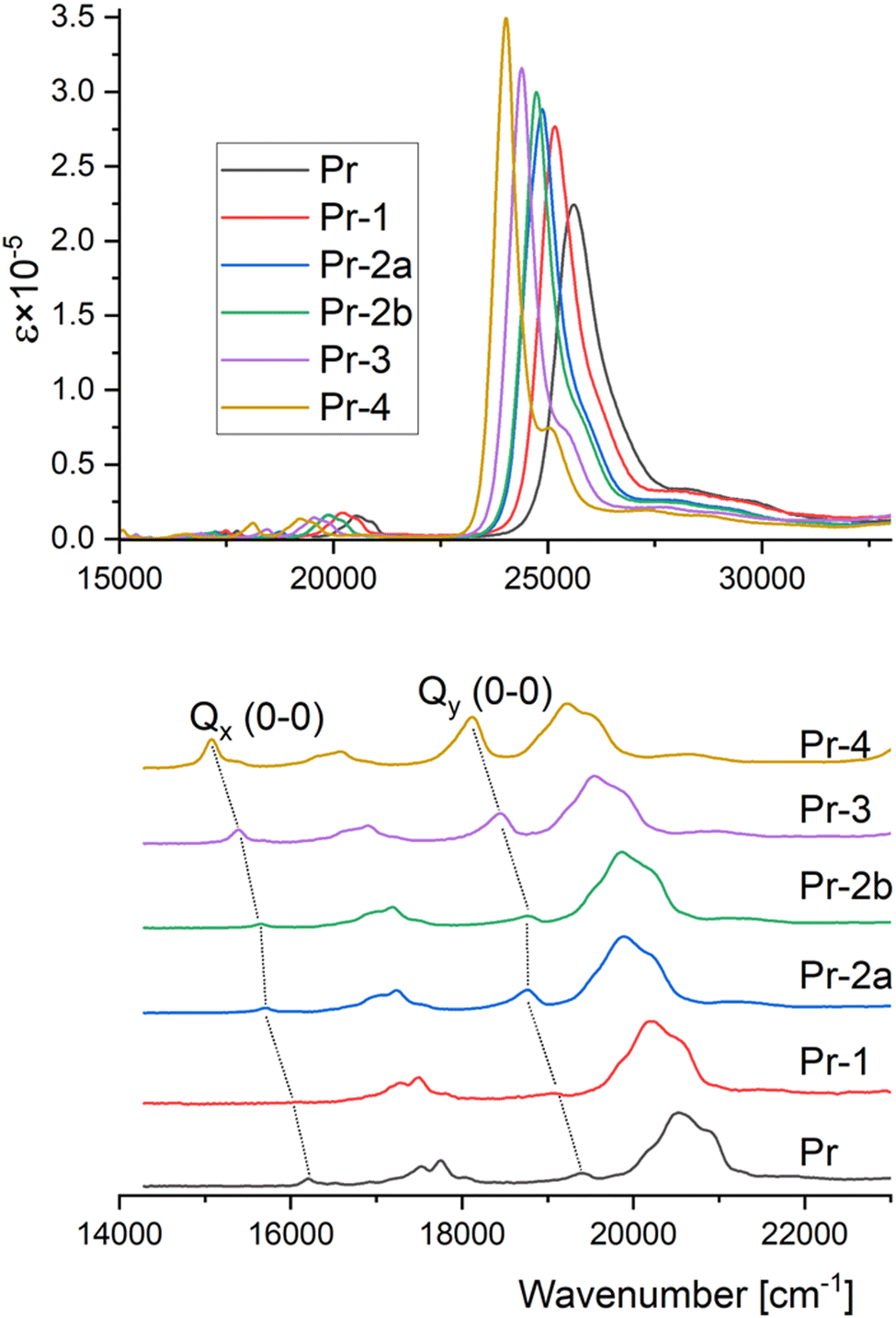 | ||
| Fig. 3 Comparison of absorption spectra of the porphyrins studied (n-hexane solutions at 293 K, toluene for Pr). | ||
Fluorescence spectra
Comparison of the fluorescence spectra (Fig. 4) reveals trends similar to those observed in absorption. The relative intensity of the 0–0 band strongly varies across the series: as in absorption, it is the highest in Pr-4 and lowest in Pr-1. The spectra of the two doubly substituted porphyrins are very similar, which suggests additive, relative position-independent contributions from the substituents. Analogous behavior is observed in absorption, as revealed by comparison of the spectra of Pr-2a and Pr-2b.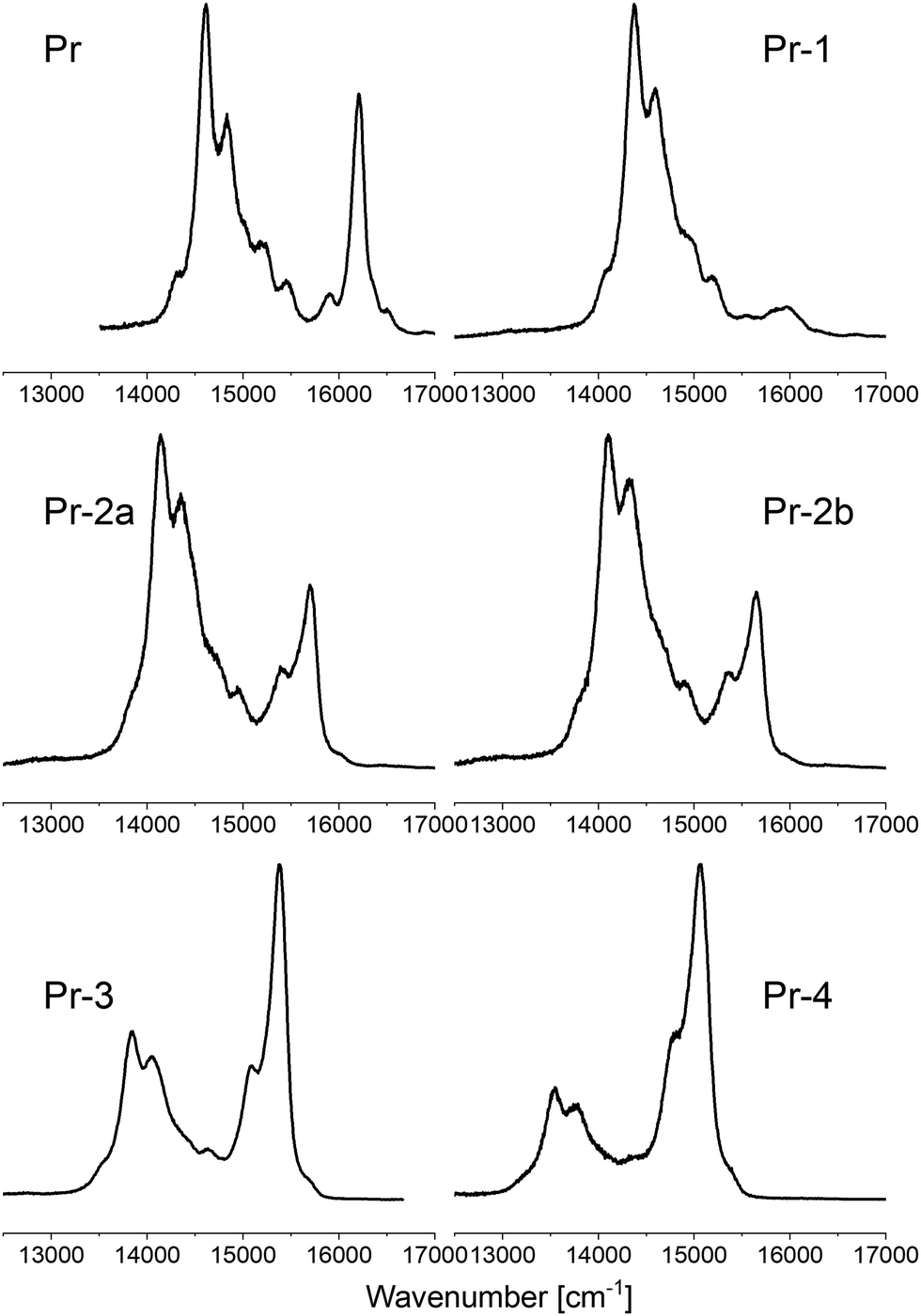 | ||
| Fig. 4 Comparison of fluorescence spectra for Pr and hexyl-substituted porphyrins (n-hexane solutions at 293 K). | ||
The fluorescence quantum yield (Table 1) varies along the series. The lowest emission intensity is observed for Pr-1, whereas Pr-4 shows the highest values. Fluorescence quantum yields of Pr-2a, Pr-2b, and Pr are practically the same. Comparison of the emission spectral profiles shows that the monotonic increase in the quantum yield upon passing from Pr-1 to Pr-4 is due to the growing intensity of the 0–0 region, whereas the vibronic part remains practically unchanged. Such behavior reflects the trend observed in absorption.
No changes in the fluorescence lifetime (τ) could be observed. All the measured values are in the range of 7.8–8.1 ns and are considered to be the same within experimental error. Since the fluorescence quantum yields are rather low, this observation indicates that the rates of nonradiative S1 depopulation (Table S2, ESI†), which govern the decay time, do not change with substitution. In contrast, the emission quantum yields scale with kr, since φfl = kr × τ.
MCD spectra
All the investigated porphyrins exhibit the same pattern of MCD signs: −, +, −, + for the L1, L2, B1, and B2 transitions (Fig. 5). According to the perimeter model, such a sequence corresponds to ΔHOMO > ΔLUMO. The signals in the low energy region are much weaker than for the Soret bands. Most interesting is the intensity ratio of the 0–0 transitions in Qx and Qy to the corresponding vibronic bands. A similar pattern as in absorption and fluorescence spectra is observed. The S1(0–0) transition is extremely weak in Pr-1, but introduction of consecutive n-hexyl moieties leads to its large increase with respect to the vibronic counterpart, so that it becomes dominant in Pr-3 and Pr-4. A similar behavior is observed for S2. It is noteworthy that the S1(0–0) transition in Pr-1 is unique with respect to its positive MCD sign, since all other compounds display negative MCD signals for this transition. In contrast, Pr-1 is no exception regarding the vibronic features of S1, revealing the same negative sign as all other porphyrins investigated. | ||
| Fig. 5 Comparison of MCD spectra for the porphyrins studied (n-hexane solutions at 293 K, except for Pr, for which absorption and MCD in the low energy range were measured in toluene, and for the S1(0–0) transition in Pr-1 measured in dichloromethane). | ||
Calculations
It has been established that, in order to properly describe the weak S0–S1 (Qx) transition in free base porphyrin, two contributions have to be taken into account: Franck–Condon (FC) and Herzberg–Teller (HT). The former is responsible for the 0–0 region, whereas the HT terms are dominant in the vibronic features that form the second band, stronger in both absorption and emission. The Qx band served as the case study in theoretical works that included both Herzberg–Teller and Duschinsky effects.33,34 The spectra could be reproduced very well by calculations.Our present results clearly show that the perturbation of the porphyrin macrocycle by the meso-substitution strongly affects the FC intensity, whereas the HT contributions remain approximately constant. This is observed in the three types of spectra that we recorded: absorption (Fig. 3), fluorescence (Fig. 4), and MCD (Fig. 5). All show (for both Qx and Qy bands) significant changes in the intensity ratio of the 0–0 band with respect to the vibronic features. The 0–0 region is very sensitive to substitution, whereas the intensity of the vibronic bands remains approximately constant. Interestingly, the changes are not monotonic: the first substitution leads to a decrease of FC vs. HT contributions, but upon introducing two or more n-hexyl groups, the intensity in the 0–0 region steadily grows. In the perimeter model, this implies that the ΔHOMO − ΔLUMO difference decreases upon passing from Pr to Pr-1, and then continuously increases. Such behavior is compatible with the orbital energy ordering shown in the right part of Fig. 2. However, both B3LYP and BP86-D DFT functionals used in this work predict the other sequence. Therefore, they are unable to reproduce the decrease of 0–0 band intensity in Pr-1 with respect to Pr. Instead, they predict continuous growth concomitant with the increasing number of substituents (Fig. S2–S7, ESI†). On the other hand, lower intensity in Pr-1 than in Pr is correctly predicted by INDO/S (Fig. S2 and S3, ESI†). This is because of the calculated energy ordering of the frontier occupied orbitals, opposite to that predicted by DFT, which agrees with the experimental findings. In turn, the INDO/S method fails to reproduce the intensity increase starting from double substitution: the calculated S1 ← S0 oscillator strengths start growing in Pr-3, but even for Pr-4 they are lower that the value computed for Pr (Fig. S4–S7, ESI†).
The difficulty in predicting the sign of ΔHOMO − ΔLUMO in a soft chromophore has been discussed before.35 In line with the present results, inversion of MCD signs observed for several substituted zinc porphyrins was correctly predicted by INDO/S, but not DFT calculations. The problem is caused by similar HOMO−1 and HOMO energies (difference of about 0.1 eV). Fortunately, the splitting of LUMO and LUMO+1 in all presently studied porphyrins is even lower (0.02–0.04 eV), so that all DFT calculations yield ΔHOMO > ΔLUMO, the ordering that agrees with the experimentally observed pattern of MCD signs for Q and Soret transitions. The only exception is the positive MCD observed for an extremely weak 0–0 band of Pr-1 (Fig. 5), a molecule which is the closest to an ideal soft chromophore.
The experimentally observed transition energy shifts to the red with the increasing number of substituents are well predicted by both DFT models (Fig. 6). The general trend in absorption intensity changes is also well reproduced, except for Pr-1, as discussed above.
 | ||
| Fig. 6 Calculated FC (top, red) and FCHT (bottom, black) spectra, normalized to the maximum, convoluted with a Gaussian of 135 cm−1 width (FWHM). Right, absorption, left, fluorescence. | ||
Simulations of absorption and fluorescence spectra were performed using the algorithm proposed in the case study of porphyrin, now implemented in Gaussian.33 This allowed a comparison of FC and HT contributions along the series (Fig. 6). The calculations predict a monotonic increase of FC intensity in the 0–0 region with respect to the vibronic bands that mainly consist of HT induced contributions. Except for Pr-1, the agreement with experiment is good (Fig. S8–S13, ESI†), especially for Pr-3 and Pr-4, molecules which exhibit the largest ΔHOMO − ΔLUMO values and are therefore the hardest chromophores along the series.
We have also simulated the MCD spectra, using DFT (BP86-D functional) and INDO/S (Fig. S2–S7, ESI†). Similar patterns as in the absorption calculations were obtained. The sign sequence was computed correctly by both models (except from INDO/S calculations for L1 and L2 in Pr-3 and Pr-4), but the intensity changes were not. DFT predicted continuous MCD increase of Qx and Qy MCD intensities along the series, whereas INDO/S correctly reproduced the decrease upon passing from Pr to Pr-1, but the experimentally observed increase for the growing number of n-hexyl moieties was not predicted. However, INDO/S fared much better in the Soret region, for which DFT calculations predicted a number of transitions located lower than the Soret bands; weak in absorption, but closely spaced. This proximity of states led to several calculated strong MCD signals with intensity similar to that of the Soret transitions. Such pattern is not observed experimentally (Fig. 5). In contrast, in agreement with the experiment, INDO/S calculations correctly predicted that the MCD is dominated by two strong signals from the Soret bands.
Discussion and conclusions
The experimental results, obtained by using three different techniques, reveal a characteristic pattern of spectroscopic evolution with the increasing number of alkyl substituents. The observed changes in absorption, fluorescence, and MCD spectra can be readily interpreted based on the simple, but powerful perimeter model. Unsubstituted porphyrin, a soft chromophore, comes very close to be an ideal case of ΔHOMO = ΔLUMO upon a single substitution with an alkyl residue. Adding more substituents leads to ΔHOMO > ΔLUMO with a continuous increase of the difference in orbital energy splittings. In other words, depending on the number of substituents, a soft chromophore can be made either softer or harder. One should note that these changes influence not only the transition intensities, but the color of absorbed or emitted light, because of different responses of FC and HT to substitution. Thus, fluorescence becomes more light red in Pr-3 and Pr-4 due to the increasing FC contribution in the 0–0 region.The computational results demonstrate that a proper calculation of orbital energy ordering remains a challenge when the orbitals lie close to each other. As shown for unsubstituted Pr, different methods can predict different orderings. Thus, it would be instructive to check various DFT functionals in this regard.
Approaching an ideal soft chromophore via electron-donating substitution has an alternative. If an electron withdrawing group is used, it will mostly interact with one of the LUMO orbitals, increasing the ΔLUMO gap. In a sense, the situation is then simpler than that involving HOMO orbitals. Since the two LUMOs are nearly degenerate, a perturbation will increase the splitting for each of both possible orbital energy orderings.
One can think of diverse applications for a perfectly soft chromophore. It should be extremely sensitive to small environmental effects, thus being a good candidate for an optical sensor. Usually, such devices are based on signal intensity changes. We note that, because of different responses of FC and HT contributions to external perturbations, working in ratiometric mode should be possible. In addition, not only the intensity, but also the spectral profile and, in the case of MCD, the sign of the signal can be changed. Therefore, (possibly very small) changes in the vicinity of the chromophore could be probed by different spectral techniques, increasing the measurement accuracy and sensitivity of such a device.
An ideal soft chromophore could also be very helpful in theoretical studies of vibronic effects in electronic transitions. An example of a problem that calls for deeper analysis is provided by the MCD spectrum of Pr-1, which reveals, for the same S1 ← S0 electronic transition, opposite signs in the 0–0 and vibronic regions.
Finally, we would like to underscore the validity of simple models to understand phenomena which cannot be correctly treated by models that formally are considered more developed. In the present work, using only DFT for spectral predictions leads to inconsistencies between experiment and theory. On the other hand, the perimeter model not only explains the observed spectral changes well, but also gives an understanding of why DFT calculations fail in certain cases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Polish National Science Center grant no. 2019/35/B/ST4/00297, by a grant from the PL-Grid infrastructure, by the Interdisciplinary Centre for Mathematical and Computational Modelling University of Warsaw (ICM UW) under computational allocation no. G96-1824, and by a grant from Science Foundation Ireland (SFI award 21/FFP-A/9469, PORPHYSHAPE).Notes and references
- J. Michl, Magnetic Circular-Dichroism of Aromatic-Molecules, Tetrahedron, 1984, 40, 3845–3934 CrossRef CAS.
- J. Waluk and J. Michl, Determination of Positional Isomers of Methylpyrenes and Other Polycyclic Hydrocarbons by Magnetic Circular Dichroism, Anal. Chem., 1981, 53, 236–239 CrossRef CAS.
- J. Waluk and J. Michl, The perimeter model and magnetic circular dichroism of porphyrin analogues, J. Org. Chem., 1991, 56, 2729–2735 CrossRef CAS.
- J. D. Keegan, A. M. Stolzenberg, Y. C. Lu, R. E. Linder, G. Barth, A. Moscowitz, E. Bunnenberg and C. Djerassi, Magnetic circular dichroism studies. 60. Substituent-induced sign variation in the magnetic circular dichroism spectra of reduced porphyrins. 1. Spectra and band assignments, J. Am. Chem. Soc., 1982, 104, 4305–4317 CrossRef CAS.
- J. D. Keegan, A. M. Stolzenberg, Y. C. Lu, R. E. Linder, G. Barth, A. Moscowitz, E. Bunnenberg and C. Djerassi, Magnetic circular dichroism studies. 61. Substituent-induced sign variation in the magnetic circular dichroism spectra of reduced porphyrins. 2. Perturbed molecular orbital analysis, J. Am. Chem. Soc., 1982, 104, 4317–4329 CrossRef CAS.
- C. Djerassi, Y. Lu, A. Waleh, A. Y. L. Shu, R. A. Goldbeck, L. A. Kehres, C. W. Crandell, A. G. H. Wee, A. Knierzinger, R. Gaeteholmes, G. H. Loew, P. S. Clezy and E. Bunnenberg, Magnetic Circular-Dichroism Series. 65. Sign Variation in the Magnetic Circular-Dichroism Spectra of Free-Base Porphyrins Having a Single Pi-Acceptor Pyrrole Ring Substituent – Structure Implications, J. Am. Chem. Soc., 1984, 106, 4241–4258 CrossRef.
- R. A. Goldbeck, B. R. Tolf, E. Bunnenberg and C. Djerassi, Substituent conformational effects in the magnetic circular dichroism and absorption spectra of free-base carbonyl porphyrins, J. Am. Chem. Soc., 1987, 109, 28–32 CrossRef CAS.
- A. Gorski, M. Kijak, E. Zenkevich, V. Knyukshto, A. Starukhin, A. Semeikin, T. Lyubimova, T. Roliński and J. Waluk, Magnetic Circular Dichroism of meso-Phenyl-Substituted Pd-Octaethylporphyrins, J. Phys. Chem. A, 2020, 124, 8144–8158 CrossRef CAS PubMed.
- A. Ceulemans, W. Oldenhof, C. Görller-Walrand and L. G. Vanquickenborne, Gouterman's “four-orbital” model and the MCD spectra of high-symmetry metalloporphyrins, J. Am. Chem. Soc., 1986, 108, 1155–1163 CrossRef CAS.
- R. A. Goldbeck, Sign variation in the magnetic circular dichroism spectra of π-substituted porphyrins, Acc. Chem. Res., 1988, 21, 95–101 CrossRef CAS.
- N. Kobayashi and K. Nakai, Applications of magnetic circular dichroism spectroscopy to porphyrins and phthalocyanines, Chem. Commun., 2007, 4077–4092 RSC.
- J. Mack, M. J. Stillman and N. Kobayashi, Application of MCD spectroscopy to porphyrinoids, Coord. Chem. Rev., 2007, 251, 429–453 CrossRef CAS.
- H. M. Rhoda, J. Akhigbe, J. Ogikubo, J. R. Sabin, C. J. Ziegler, C. Brückner and V. N. Nemykin, Magnetic Circular Dichroism Spectroscopy of meso-Tetraphenylporphyrin-Derived Hydroporphyrins and Pyrrole-Modified Porphyrins, J. Phys. Chem. A, 2016, 120, 5805–5815 CrossRef CAS PubMed.
- J. Waluk, M. Müller, P. Swiderek, M. Köcher, E. Vogel, G. Hohlneicher and J. Michl, Electronic states of porphycenes, J. Am. Chem. Soc., 1991, 113, 5511–5527 CrossRef CAS.
- A. Gorski, E. Vogel, J. L. Sessler and J. Waluk, Magnetic circular dichroism of octaethylcorrphycene and its doubly protonated and deprotonated forms, J. Phys. Chem. A, 2002, 106, 8139–8145 CrossRef CAS.
- A. Gorski, E. Vogel, J. L. Sessler and J. Waluk, Magnetic circular dichroism of neutral and ionic forms of octaethylhemiporphycene, Chem. Phys., 2002, 282, 37–49 CrossRef CAS.
- J. Waluk, Spectroscopy and tautomerization studies of porphycenes, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- H. Cortina, M. L. Senent and Y. G. Smeyers, Ab initio comparative study of the structure and properties of H2-porphin and H2-phthalocyanine. The electronic absorption spectra, J. Phys. Chem. A, 2003, 107, 8968–8974 CrossRef CAS.
- A. Wiehe, C. Ryppa and M. O. Senge, A Practical Synthesis of Meso-monosubstituted, β-Unsubstituted Porphyrins, Org. Lett., 2002, 4, 3807–3809 CrossRef CAS PubMed.
- S. Hatscher and M. O. Senge, Synthetic access to 5,10-disubstituted porphyrins, Tetrahedron Lett., 2003, 44, 157–160 CrossRef CAS.
- S. Horn and M. O. Senge, The Intermolecular Pauson–Khand Reaction of meso-Substituted Porphyrins, Eur. J. Org. Chem., 2008, 4881–4890 CrossRef CAS.
- A. Wiehe, Y. M. Shaker, J. C. Brandt, S. Mebs and M. O. Senge, Lead structures for applications in photodynamic therapy. Part 1: Synthesis and variation of m-THPC (Temoporfin) related amphiphilic A2BC-type Porphyrins, Tetrahedron, 2005, 61, 5535–5564 CrossRef CAS.
- J. Olmsted III, Calorimetric Determinations of Absolute Fluorescence Quantum Yields, J. Phys. Chem., 1979, 83, 2581–2584 CrossRef.
- M. Kijak, K. Nawara, A. Listkowski, N. Masiera, J. Buczyńska, N. Urbańska, G. Orzanowska, M. Pietraszkiewicz and J. Waluk, 2+2 Can Make Nearly a Thousand! Comparison of Di- and Tetra-Meso-Alkyl-Substituted Porphycenes, J. Phys. Chem. A, 2020, 124, 4594–4604 CrossRef CAS PubMed.
- B. Holmquist, Magnetic circular dichroism, Methods Enzymol., 1986, 130, 270–291 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- S. Sripothongnak, C. J. Ziegler, M. R. Dahlby and V. N. Nemykin, Controllable and Reversible Inversion of the Electronic Structure in Nickel N-Confused Porphyrin: A Case When MCD Matters, Inorg. Chem., 2011, 50, 6902–6909 CrossRef CAS PubMed.
- C. J. Ziegler, N. R. Erickson, M. R. Dahlby and V. N. Nemykin, Magnetic Circular Dichroism Spectroscopy of N-Confused Porphyrin and Its Ionized Forms, J. Phys. Chem. A, 2013, 117, 11499–11508 CrossRef CAS PubMed.
- G. A. Peralta, M. Seth and T. Ziegler, Magnetic circular dichroism of porphyrins containing M = Ca, Ni, and Zn. A Computational study based on time-dependent density functional theory, Inorg. Chem., 2007, 46, 9111–9125 CrossRef CAS PubMed.
- M. Gouterman, Spectra of porphyrins, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- J.-L. Soret, Analyse spectrale: Sur le spectre d’absorption du sang dans la partie violette et ultra-violette, C. R. Acad. Sci., 1883, 97, 1269–1270 Search PubMed.
- F. Santoro, A. Lami, R. R. Improta, J. Bloino and V. Barone, Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg–Teller effect: The Qx band of porphyrin as a case study, J. Chem. Phys., 2008, 128, 224311 CrossRef PubMed.
- B. Minaev, Y. H. Wang, C. K. Wang, Y. Luo and H. Ågren, Density functional theory study of vibronic structure of the first absorption Q band in free-base porphin, Spectrochim. Acta, Part A, 2006, 65, 308–323 CrossRef PubMed.
- P. Kowalska, M. D. Peeks, T. Roliński, H. L. Anderson and J. Waluk, Detection of a weak ring current in a nonaromatic porphyrin nanoring using magnetic circular dichroism, Phys. Chem. Chem. Phys., 2017, 19, 32556–32565 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Description of computational procedures, atomic coordinates obtained for optimized geometries, calculated orbital energy splittings, orbital parentage of the lowest energy singlet state, rates of nonradiative and nonradiative depopulation of S1; comparison of experimental and calculated energies for Q and Soret transition, comparison of absorption and MCD spectra simulated using BP86-D and INDO/S methods, comparison of experimentally measured absorption with the simulated ones that take into account both FC and HT contributions. See DOI: https://doi.org/10.1039/d4cp01502a |
| This journal is © the Owner Societies 2024 |