
Shortwave infrared polymethine dyes for bioimaging: ultrafast relaxation dynamics and excited-state decay pathways†
Laura M.
Obloy
 ,
Steffen
Jockusch
and
Alexander N.
Tarnovsky
*
,
Steffen
Jockusch
and
Alexander N.
Tarnovsky
*
Department of Chemistry and the Center for Photochemical Sciences, Bowling Green State University, Bowling Green, OH 43403, USA. E-mail: atarnov@bgsu.edu
First published on 29th May 2024
Abstract
Excited-state relaxation in two prototypical shortwave infrared (SWIR) polymethine dyes developed for bioimaging, heptamethine chromenylium Chrom7 and flavylium Flav7, is studied by means of femtosecond transient absorption with broadband ultraviolet-to-SWIR probing complemented by steady-state and time-resolved fluorescence and phosphorescence measurements. The relaxation processes of the dyes in dichloromethane are resolved with sub-100 fs temporal resolution using SWIR, near-IR, and visible photoexcitation. Different population members of the ground-state inhomogeneous ensemble are found to equilibrate via skeletal deformation changes with time constants of 90 fs and either 230 fs (Chrom7) and 350 fs (Flav7) followed by slower evolution matching the 1-ps timescale of diffusive solvation dynamics. Molecules excited into high-lying singlet electronic states (Sn) by visible excitation repopulate with time constants of 400 fs (Chrom7) and 450 fs (Flav7) the corresponding first excited singlet S1 states, which decay within several hundreds of picoseconds in dichloromethane and chloroform solvents. Vibrational relaxation in S1 for both Chrom7 and Flav7 in dichloromethane occurs with time constants of 350 and 800 fs for excess of vibrational energy of ∼1000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 deposited by near-IR and visible excitation, respectively. Two competing non-radiative processes are present in S1: temperature-independent internal conversion, and thermally-activated twisting about a carbon–carbon bond of the conjugated chain, which is substantial at room temperature but essentially nonreactive, producing traces of isomer product. Intersystem crossing in S1, and thus the triplet quantum yield, is minor. The importance of absorption bands from the excited S1 state in applications requiring high-intensity excitation conditions is discussed.
000 cm−1 deposited by near-IR and visible excitation, respectively. Two competing non-radiative processes are present in S1: temperature-independent internal conversion, and thermally-activated twisting about a carbon–carbon bond of the conjugated chain, which is substantial at room temperature but essentially nonreactive, producing traces of isomer product. Intersystem crossing in S1, and thus the triplet quantum yield, is minor. The importance of absorption bands from the excited S1 state in applications requiring high-intensity excitation conditions is discussed.
Introduction
The development of shortwave infrared (SWIR) emitters for bioimaging applications is of significant interest.1–5 The SWIR portion of the electromagnetic spectrum extends from about 900 to 2500 nm. Compared with in vivo imaging applications in the near-IR range, SWIR imaging presents several substantial advantages6 such as less autofluorescence and scattering produced by the tissue as well as a larger penetration depth into the tissue opening an opportunity for deep tissue imaging. Organic fluorophores exhibit low toxicity and are removed from the body by well-understood mechanisms. Also, they can target specific biomolecules either inherently or by design. These favorable traits make organic fluorophores to be the aspiring and most common imaging agents and fluorescent labels.2,7–10 To improve the efficiency of SWIR fluorophores for bioimaging applications, a comprehensive understanding of the excited-state radiationless relaxation pathways competing with fluorescence in these molecular systems should be gained. This should permit identifying key drawbacks in the fluorophores design and lead to further development of bright and stable organic SWIR emitters with enhanced, tailored photodynamics.Dyes of the polymethine group are one of the most widely used fluorescent agents.7–12 Their ubiquitous use in photographic, laser, and photonic industries was reviewed.9,13–15 In biophysical and biomedical applications, because of photostability as well as intense absorption and fluorescence, the indocarbocyanines, especially Cy3, Cy5, and Cy7 of the Cy series16–22 have become broadly used. They consist of two indolenine heterocyclic end rings connected by 3, 5, or 7 –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) methine units forming the π-conjugated chain. In polymethines, the main absorption band is due to a dipole-allowed ππ* transition between the ground S0 and lowest excited singlet S1 states in the most thermodynamically stable planar trans-isomer (sterically non-hindered dyes23). An extra –CHCH– unit in the conjugation chain produces an approximately 100-nm displacement of the S0–S1 band towards longer wavelengths known as a vinylene shift. Although the absorption of Cy7-like heptamethine dyes reaches the 800-nm range, it does not extend further than ∼850 nm. Further lengthening of the polymethine chain is possible,3,12 but it may enhance the likelihood of thermal instability and photodecomposition, thus hampering the biological imaging applications.24 Extending heterocyclic conjugation, rather than elongating the polymethine chain, is a viable alternative. A recently developed flavylium polymethine dye, Flav7 in Scheme 1,24,25 possesses two dimethyl-flavylium heterocyclic end groups in place of the indolenines in the Cy7-like dyes producing, in comparison, a ∼200-nm shift of absorption and fluorescence spectra into the IR region.
methine units forming the π-conjugated chain. In polymethines, the main absorption band is due to a dipole-allowed ππ* transition between the ground S0 and lowest excited singlet S1 states in the most thermodynamically stable planar trans-isomer (sterically non-hindered dyes23). An extra –CHCH– unit in the conjugation chain produces an approximately 100-nm displacement of the S0–S1 band towards longer wavelengths known as a vinylene shift. Although the absorption of Cy7-like heptamethine dyes reaches the 800-nm range, it does not extend further than ∼850 nm. Further lengthening of the polymethine chain is possible,3,12 but it may enhance the likelihood of thermal instability and photodecomposition, thus hampering the biological imaging applications.24 Extending heterocyclic conjugation, rather than elongating the polymethine chain, is a viable alternative. A recently developed flavylium polymethine dye, Flav7 in Scheme 1,24,25 possesses two dimethyl-flavylium heterocyclic end groups in place of the indolenines in the Cy7-like dyes producing, in comparison, a ∼200-nm shift of absorption and fluorescence spectra into the IR region.
 | ||
| Scheme 1 Structures of the studied dyes (counterion, BF4−). | ||
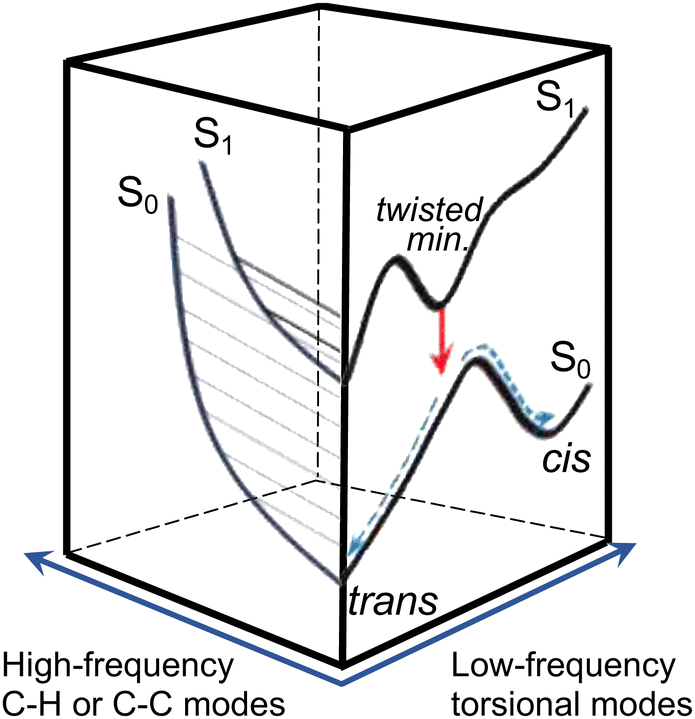
Efficient SWIR fluorescence is critical for bioimaging; however, SWIR fluorophores exhibit low emission quantum yields and it is not clear if other reasons, apart from a small S0–S1 energy gap, affect the S1 deactivation. Flav7 was the brightest organic SWIR emitter at the time of publication24,25 and remains one of the brightest at present. Generally, for polymethines, in agreement with Kasha's rule26 Sn are much less emissive than S1 states, which in the absence of intermolecular relaxation, decay via competing radiative and non-radiative processes of internal conversion (IC), photoisomerization, and intersystem crossing (ISC), Fig. 1a. Photoisomerization, which is temperature and viscosity dependent, is predominant in short-chain and pentamethine cyanines in room-temperature fluid solutions,27–31 but becomes less dominant in heptamethines under the same conditions.32–34 Whereas the geometry restriction imparted at the central region of the heptamethine chain moderately affects the photoisomerization,35–37 conformational restraint imparted along the entire chain is reported to stop the photoisomerization entirely.22 For unrestrained dyes, the Rullière model has been widely invoked in which the S1trans-isomer overcomes the photoisomerization barrier (rate constant, kph) to yield the 90°-twisted structure, which either yields the S0cis-photoisomer or returns to the parent S0trans-form with ϕ and 1 − ϕ branching ratio.27–29 Advanced quantum chemistry computations on streptocyanines38,39 suggest that the initial in-plane symmetric stretching in the Franck–Condon (FC) region on the S1 potential energy surface (PES) is followed by torsional motion dominated by twisting about one of the central carbon–carbon conjugated bonds at the end. This takes the molecule to an extended S1 → S0 conical intersection (CI) seam located close to the 90°-twisted state, but on the trans-side. The recovery of the S0trans-form is thus favored, which is consistent with <10% quantum yields of the photoisomer formation in sterically non-hindered penta- and heptamethines.30–32,40 Because of large kph of these and shorter-chain dyes in fluid solutions, unless ISC is aided by heavy atom substituents, the quantum yield of the formation of the lowest excited triplet T1 state (Φisc) is small. The Φisc values of ∼0.00130,31,33,41–45 are typical; however, 0.0316,17 was recently proposed for Cy3 and Cy5. The twisting pathway leading to the S0trans-form (i.e., unreactive) is thermally-activated and controlled by the photoisomerization barrier and, therefore, should be distinguished from ‘direct’ S1 → S0 IC within the planar trans-form (rate constant, kic). Direct IC is very weakly viscosity and temperature dependent occurring even at low temperatures and/or in highly viscous solutions,29,33,44,46–48 and therefore, does not involve a large structural change. The underlying theory for direct IC is the energy gap law,49 according to which the energy decays intramolecularly from the donor excited electronic state to the high-frequency-mode vibrational states of the acceptor electronic state. When the displacement between the involved PESs is not large, the decay rate constant is predicted to increase exponentially with decreasing energy separation between the electronic states. This behavior was found in several organic systems such as aromatic hydrocarbons,50 linear polyenes, and carotenoids.51–53 However, the exact nature of the accepting modes is still under debate with CC,52,54–57 C–H,57–59 and organic alkenyl C–H22,60 stretching as well as N–H and O–H vibrations61 all having been proposed. Vibrational overlap between the narrowly separated S1 and S0 states becomes substantial and is considered in literature to be the reason for short S1-state lifetimes and low quantum yields of SWIR fluorophores.
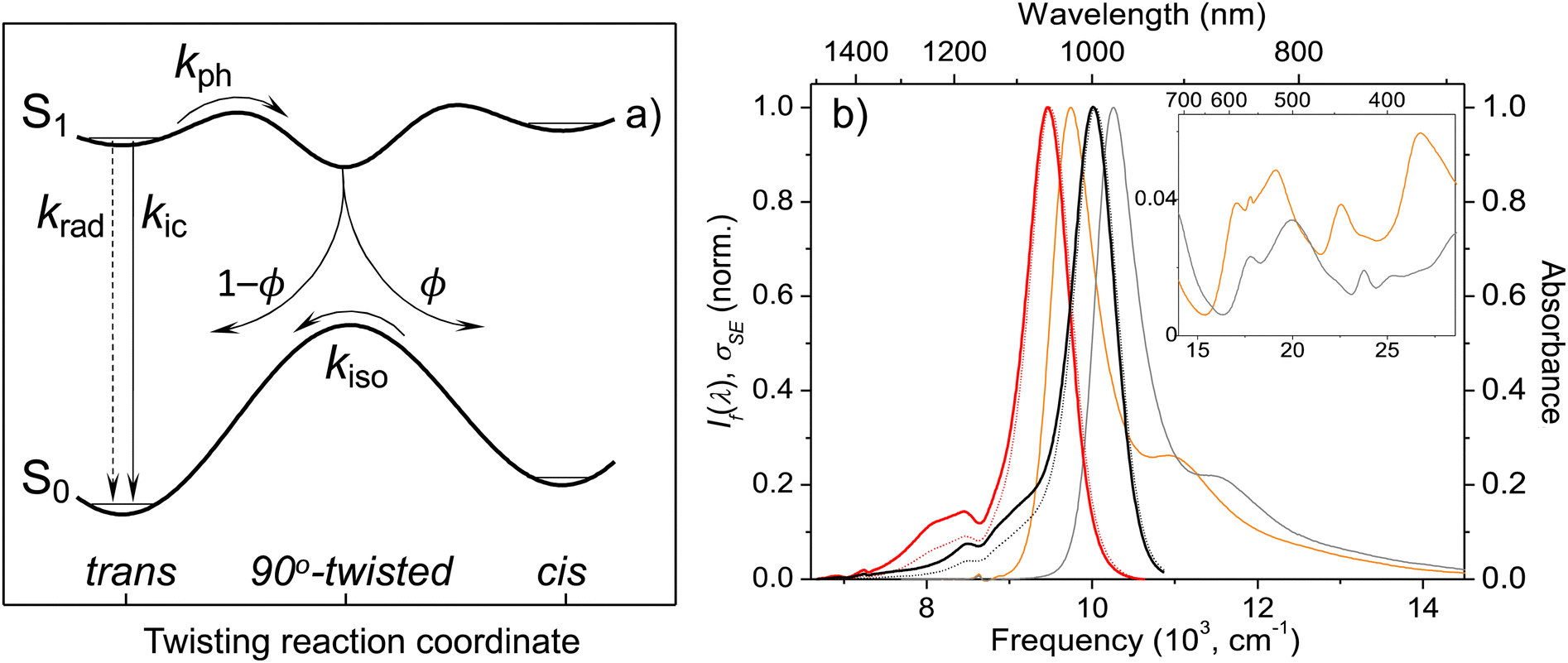 | ||
| Fig. 1 Model potential energy diagram27 used in literature to interpret the photophysics and photochemistry of polymethine dyes, (a) a thermodynamically stable trans-form excited into S1 undergoes radiative decay, direct internal conversion, and thermally activated crossing over the photoisomerization barrier along the twisting reaction coordinate. The corresponding rate constants are krad, kic, and kph. From the 90° twisted S1-state, the molecule branches either into the trans- or cis-S0 forms. The ground-state isomerization rate constant for reformation of the trans-isomer is kiso. Normalized steady-state absorption (orange and grey) and fluorescence25 spectra (If(λ), red and black dotted curves) of Flav7 and Chrom7 in anhydrous CH2Cl2, and the constructed σSE(λ) stimulated emission cross-section spectra (solid red and black curves), (b) the inset shows the short-wavelength absorbance of Flav7 and Chrom7 measured in solutions of larger overall absorbance and appropriately scaled to match the spectra in the main panel. | ||
The main objective of this work is to characterize excited-state relaxation processes following photoexcitation of two SWIR polymethine dyes, Flav7 and closely related Chrom7, where in the latter the phenyl groups are replaced by tert-butyl groups. Experimental work on the excited-state relaxation pathways of SWIR dyes is limited, and further studies on these two systems should lead to a deeper understanding of photophysics and photochemistry of SWIR fluorophores in general. We have used femtosecond transient absorption spectroscopy to capture the evolution of the photoinduced reaction starting from the very initial events. Flav7 and Chrom7 were excited in the (i) low-energy wing of the S0–S1 absorption band, 1042 and 990 nm, respectively, (ii) shoulder of the S0–S1 band, namely 925 nm, and (iii) 500-nm region of the congested S0–Sn absorption bands where the photostability of these dyes was previously evaluated.24,25 In addition, we have performed a series of femtosecond and nanosecond transient absorption as well as steady-state and time-resolved emission measurements at different temperatures to obtain the hitherto unknown information regarding the involvement of photoisomerization in the S1 relaxation and the energetics of the T1 state.
Results and discussion
Steady-state absorption and emission: construction of cross-section spectra
The X-ray crystal structure of Chrom7 and JuloFlav5 closely related to Flav7 is all-trans,25 which implies the same conformation in solution based on the literature.23 The UV-vis absorption spectra of Chrom7 and Flav7 in CH2Cl2 exhibit intense bands peaking at 975 and 1027 nm due to the S0–S1 transition, Fig. 1b. The shoulders at shorter wavelengths (∼880 nm in Chrom7 and 915 nm in Flav7) shifted by ∼1230 cm−1 from the S0–S1 band maxima are assigned to the 0-1′ vibronic bands,62 where 0 are 1′ label the optically active vibrational states in S0 and S1, respectively. The frequency at which the approximately mirror-symmetrical fluorescence25 and absorption spectra overlap can serve as an estimate for the S0–S1 energy gap; it is 10140 cm−1 for Chrom7 and 9700 cm−1 for Flav7. The analysis of transient absorption spectra requires the knowledge of S0–S1 absorption (σA) and S1–S0 stimulated emission (σSE) cross-section spectra. The σA spectrum is given as: σA(λ) = ε(λ) × 2303/NA, where NA is the Avogadro number and ε(λ) is the extinction coefficient spectrum. Based on the maximum ε values (Table 1), the maximum σA values are calculated to be 9.57 × 10−16 and 9.22 × 10−16 cm2 for Chrom7 and Flav7, respectively. The σSE(λ) spectra are obtained from the fluorescence spectra when applying necessary scaling corrections for the frequency of the emitted light,63 as detailed in the ESI,† section, and further assuming that the S0–S1 and S1–S0 transition dipole moments are the same.64 The resulting spectra (Fig. 1b) exhibit the σSE(λ) maximum of 1.09 × 10−15 and 1.14 × 10−15 cm2 for Chrom7 and Flav7, respectively.
| Dye | ε (M−1 cm−1) | Φ f (%) | k rad, 108 (s−1) | τ calcf (ps) | τ expf (ps) | |
|---|---|---|---|---|---|---|
| TCSPCb | This work | |||||
| a Ref. 24. b After deconvolution with IRF.25 Determined from S1 ESA decay. Excitation (nm): c 990, d 925, e 1042, f 990, g 1042, all this work. | ||||||
| Chrom7 | 250250 |
1.7a | 1.356 | 125 | 148 | 162c, 158d, 210f |
| 252000a |
||||||
| Flav7 | 241000a |
0.61a | 1.241 | 49 | 68 | 69e, 67d, 77g |
Radiative and total lifetime of the S1 state
The dye S1 lifetime can be calculated using the formula τcalcf = Φf/krad from the known fluorescence quantum yield Φf and the radiative rate constant krad, which in its turn can be calculated using the Strickler–Berg equation:65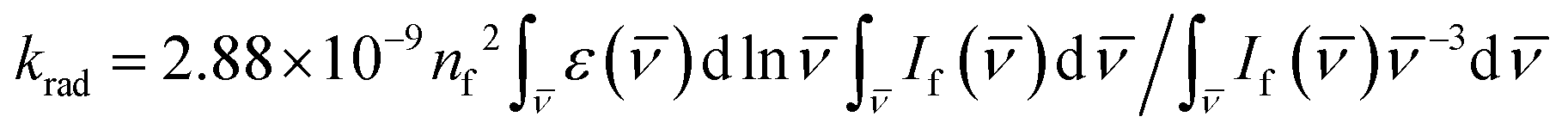 . In the equation, ε(
. In the equation, ε(![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) ) is the S0 → S1 extinction coefficient spectrum, If() is the S1 → S0 fluorescence spectrum, is the frequency (expressed in wavenumber units), and nf is the mean refractive index of the solvent over the fluorescence spectrum. The refractive index of CH2Cl2 in the 1-μm region is about 1.422.66,67 The
) is the S0 → S1 extinction coefficient spectrum, If() is the S1 → S0 fluorescence spectrum, is the frequency (expressed in wavenumber units), and nf is the mean refractive index of the solvent over the fluorescence spectrum. The refractive index of CH2Cl2 in the 1-μm region is about 1.422.66,67 The 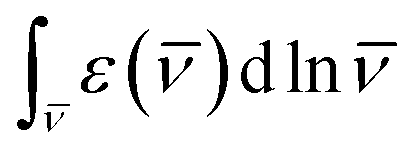 integrals for Flav7 and Chrom7 were evaluated for ≤ 15100 cm−1 to account only for the S0 → S1 transition in question, yielding krad of 1.241 × 108 and 1.356 × 108 s−1, respectively, Table 1.
integrals for Flav7 and Chrom7 were evaluated for ≤ 15100 cm−1 to account only for the S0 → S1 transition in question, yielding krad of 1.241 × 108 and 1.356 × 108 s−1, respectively, Table 1.
Excitation into the low-energy part of the absorption spectrum
Chrom7 was excited at 990 nm and Flav7 at 1042 nm at the low-energy wing of their S0 → S1 0-0′ vibronic transition to minimize contribution from vibrational relaxation to the dynamics. In transient absorption spectra, Fig. 2, the prominent negative ΔA signals are observed at probe wavelengths longer than ∼800-nm. They represent excitation-induced probe gain due to ground-state bleach (GSB) and stimulated emission (SE) caused by the depletion of the S0 population and the population of the S1 state. The positive ΔA signals represent induced absorption (i.e., probe loss) and emerge as a ∼420–780 nm broad band with two maxima, 612 and 657 nm for Chrom7 and 622 and 685 nm for Flav7. They decay concurrently to SE signals and, therefore, are assigned to excited-state absorption (ESA) from S1 to higher Sn states.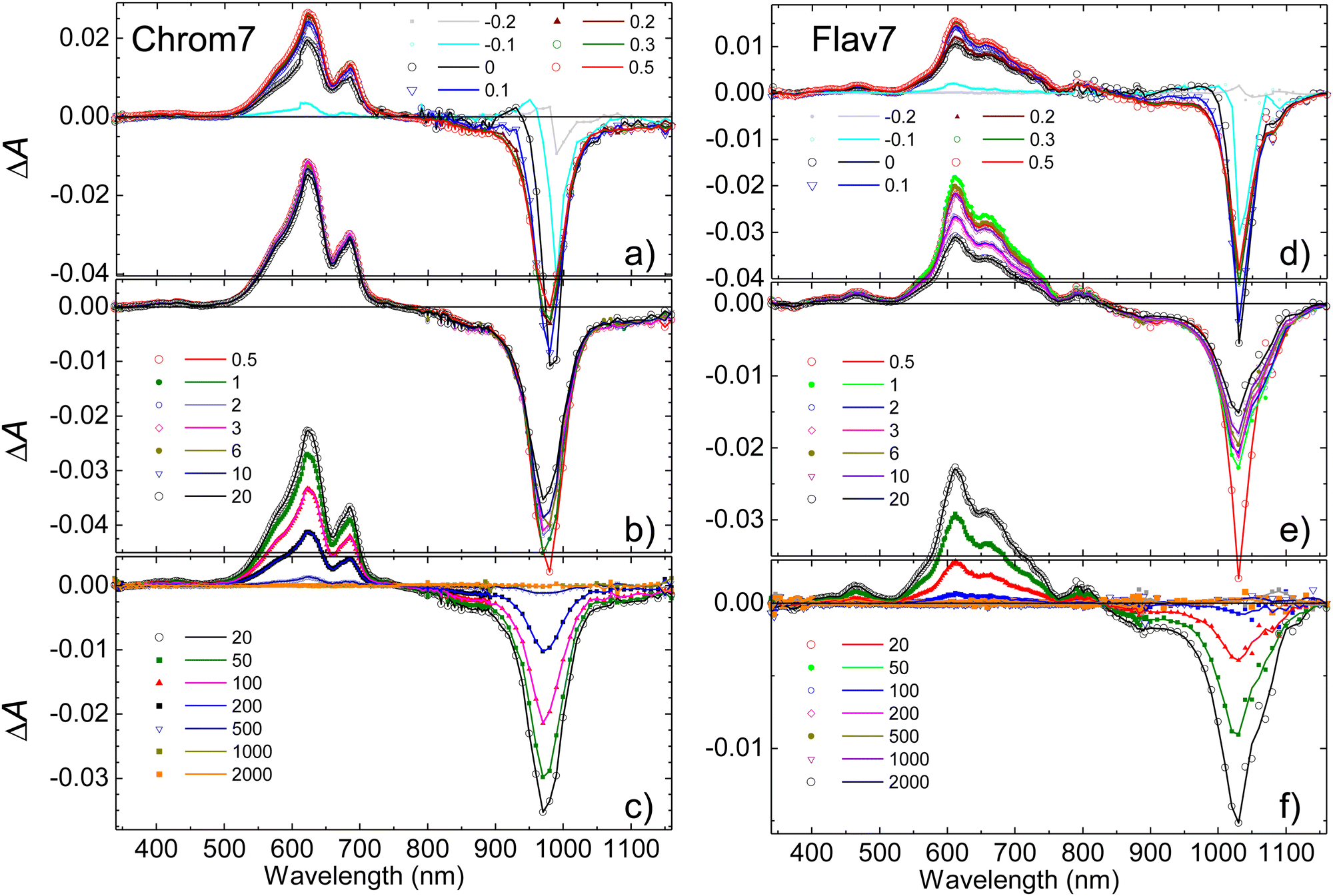 | ||
| Fig. 2 Short- (a) and (d), intermediate- (b) and (e), and long- (c) and (f) time ΔA spectra of 990-nm excited Chrom7 (a)–(c) and 1042-nm excited Flav7 (d)–(f): delay times between excitation and probe pulses expressed in picoseconds are given inside each panel. | ||
The overall photoinduced dynamics can be divided into three different stages. As soon as the 90-fs excitation and probe pulses begin to temporally overlap (delay time, −200 fs), spectrally narrow positive (in the ∼625 nm region) and negative (in the vicinity of the excitation wavelength) ΔA signals are observed for Chrom7 and Flav7, Fig. 3. The initial negative signals broaden and shift to shorter wavelengths as delay times progress to 250 fs. The initial positive signals broaden faster, i.e., within the first 200 fs, while shifting to longer wavelengths. These sharp short-time ΔA signals are characteristic to hole burning dynamics.68–71 From 250 fs to 2 ps, the ΔA spectra undergo only a modest reshaping. Afterwards, the amplitude of ΔA signals decays uniformly and single-exponentially, suggesting that the relaxation is now exclusively determined by the S1 lifetime. The S1 lifetime found for Flav7 is the same as the one reported in time-correlated single photon counting (TCSPC) experiments (68 ps25), whereas the S1 lifetime of 162 ps for Chrom7 is somewhat longer (TCSPC, 148 ps25), Table 1. Confirming the S1 lifetimes using the superior temporal resolution was desirable as it is known that TCSPC may not be accurate enough to measure tens of picoseconds lifetimes of the same temporal duration as the instrument response function (IRF) of the technique.
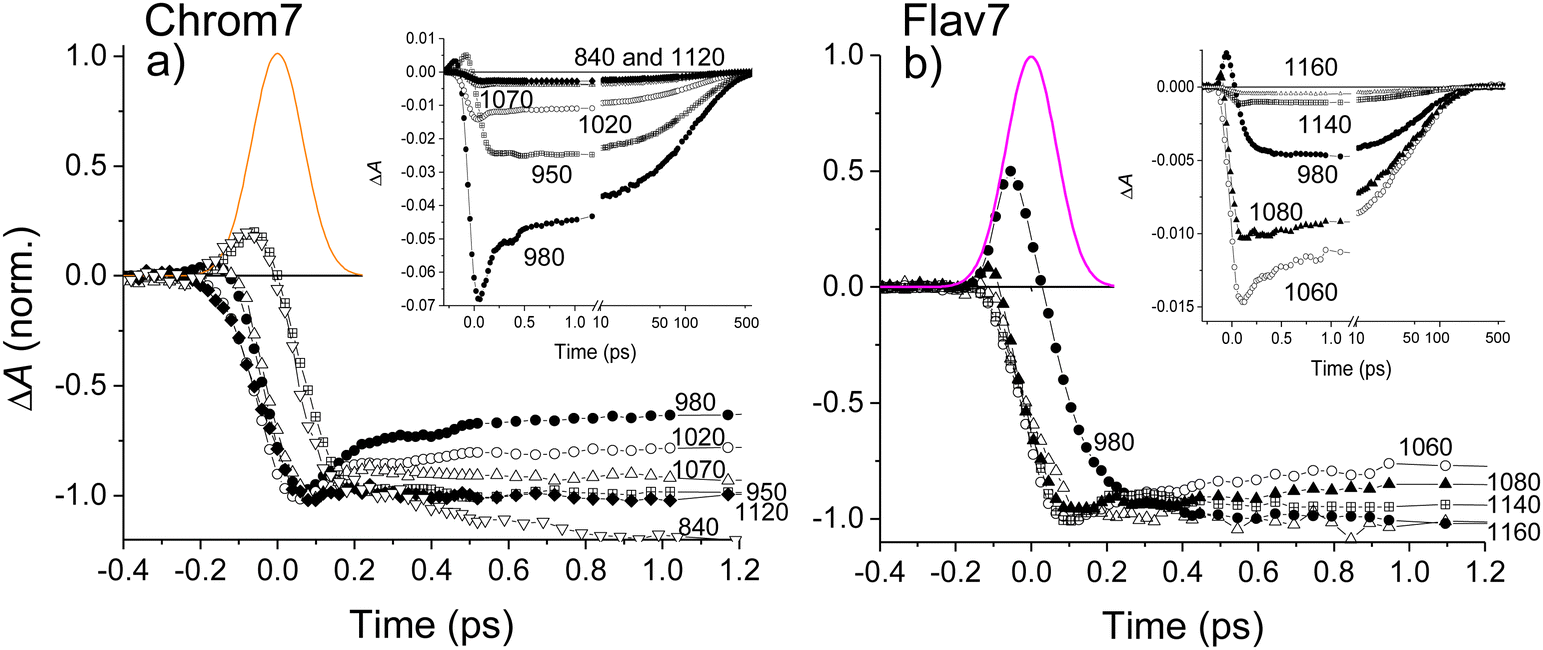 | ||
| Fig. 3 Short-time behavior of ΔA kinetic traces at representative probe wavelengths within S0–S1 absorption and stimulated emission regions for Chrom7 excited at 990 nm, (a) and Flav7 excited at 1042 nm, (b). The kinetic traces are normalized at the maximum short-time negative ΔA signal. Insets: The same ΔA data to 2 ns before normalization. Note the decadic logarithmic time scale after 10 ps. The bell-like curve is the excitation-probe cross-correlation function (CCF, 158 fs fwhm). | ||
When the S1 decay is complete, very weak ΔA signals persisting to the longest investigated 2-ns delay time were observed: induced absorption from 1020 to 1120 nm for Flav7, and induced absorption in the 1020–1070 and 600-nm regions and bleach between 970 and 1020 nm for Chrom7, Fig. S1 (ESI†). Thermal heating and photodegradation can be ruled out as possible reasons for these signals because the excitation energy used was well below 100 nJ pulse−1 and the UV-vis spectra of the solutions measured before and after the experiments showed no changes. Trans–cis photoisomerization or ISC in the trans S1-state yield a long-lived product, the S0cis-isomer or the trans-form in the T1 state, also causing GSB of the parent trans-form. We investigated the transient absorption of Chrom7 solutions saturated with O2 after a nanosecond pulsed 532-nm excitation, Fig. S2 (ESI†). O2 is known to act as a triplet state quencher.72 If T1 → Tn absorption were responsible for the observed transient absorption, the ΔA signal should have decayed within the investigated 4 μs window in the O2-saturated solution compared to the solution in the equilibrium with air at 1 atm (O2 equilibrium concentration in CH2Cl2: 2.2 mmol liter−173). Because this was not observed, Fig. S2 (ESI†), we concluded the long-time ΔA signals in the femtosecond experiments are due to the isomer product. The photoisomer absorption in penta- and heptamethine dyes is typically red-shifted with respect to that of the parent trans-form,30,31,33,74–76 consistent with our assignment. The photoisomer quantum yield of 1.8 × 10−3 and 6.4 × 10−3 for Chrom7 and Flav7, respectively, can be estimated under the assumption that the maximum extinction coefficients for trans and photoisomer forms are similar,30,31,33,74–76 and using the ratio of the 1–2 ns ΔA signals (<0.04 mOD and <0.07 mOD, Fig. S1, ESI†) to the GSB maximum taken to be a half (to exclude the SE contribution) of the maximum negative ΔA signal, −0.042 and −0.023 (Fig. 2) at 300 fs after completion of fast hole-burning dynamics.
Ultrafast transient absorption experiments were also performed for both dyes in CHCl3 to compare and determine the S1 lifetimes. The fits of the ΔA kinetic traces acquired within the region of ESA from the S1 state, Fig. S3 (ESI†), yielded the lifetime of 210 ± 3 ps for Chrom7 and 77 ± 2 ps for Flav7 (22 °C, Table 1).
Ultrafast transient absorption: 925-nm excitation into the vibronic shoulder
In response to the abrupt solute charge redistribution caused by excitation, various re-equilibration processes occur on ultrafast timescales to a varying degree such as solvation dynamics and solute vibrational relaxation. Rearrangement of the nearby solvent to accommodate the new solute charge distribution may initially occur as fast as 20–60 fs depending on solvent, and typically lasts about a few picoseconds. Vibrational energy redistribution in the polyatomic solutes may be as fast as 30–100 fs71,77–79 to several hundreds of femtoseconds, and is followed by picosecond energy transfer to the surrounding solvent.80–82 To gauge the contribution of these processes into the initial dynamics, we utilized 925-nm excitation into the 0-1′ vibronic shoulder and probed the S1 ESA region, Fig. 4. This excitation produces the excess vibrational energy of 700 cm−1 for Chrom7 and 1200 cm−1 for Flav7 in their S1 states with respect to the red-wing excitation. The ΔA spectra of both dyes excited at 925-nm initially (−200 fs) exhibit sharp ESA features, which broaden on their red side as the excitation and probe pulses better temporally overlap. At 100 fs, the ESA spectra already look like those in Fig. 3, displaying the 612-nm (Chrom7) and 622 nm (Flav7) bands, but the weaker bands at 657 nm (Chrom7) and 685 nm (Flav7) fully develop at ∼1–2 ps via red-side broadening and blue-side narrowing, Fig. S4 (ESI†). Afterwards, the ΔA spectra remained unaltered in shape and decayed single-exponentially with the S1 lifetimes, Table 1.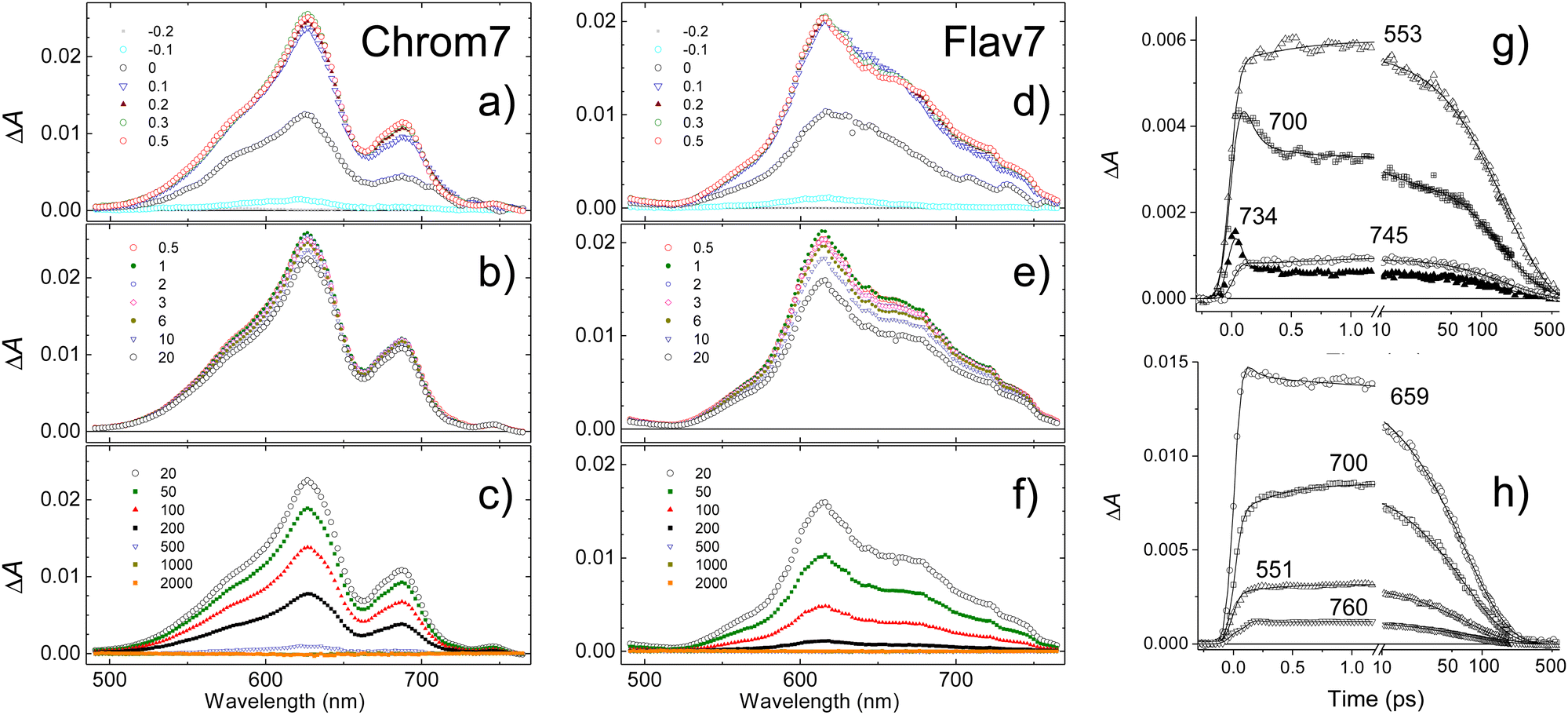 | ||
| Fig. 4 Transient absorption spectra of Chrom7 (a)–(c) and Flav7 (d)–(f) in CH2Cl2 at short (a) and (d), intermediate (b) and (e), and long (c) and (f) times following 90-fs 925-nm excitation. Delay times between the excitation and probe pulses are given in the legends. The representative ΔA kinetic traces at several probe wavelengths and the fits with CCF deconvolution are shown for Chrom7 in (g) and Flav7 in (h). Note the decadic logarithmic time scale after 10 ps. | ||
Hole burning
The spectrally sharp features in short-time ΔA spectra (Fig. 2) are due to hole burning and hole replica. If the transition energies of solute molecules are inhomogeneously distributed, only a fraction of these molecules whose transition energies are equal to the excitation photon energy, will be photoexcited.83 As a result, a population hole is burned in the thermally-equilibrated ground-state solute–solvent configurational distribution and can be seen as the series of narrow vibronic lines missing from the initial solute absorption or, specifically in transient absorption experiments, as the series of spectrally narrow GSB signals. The population excited into the FC state initially represents the replica of the ground-state hole: a narrow ensemble localized on the multidimensional solute–solvent PES of the excited state. The hole replica is seen through spectrally narrow negative (SE) and positive (ESA) ΔA signals. Thus, the short- and long-wavelength sides of the initial, negative sharp feature at 990 nm in Chrom7 and 1042 nm in Flav7 (Fig. 2) are due to the S0 absorption hole and the S1 stimulated emission hole replica. These ΔA features rapidly broaden on the high-energy side from 990 to 980 nm in Chrom7 and 1023 to 1015 nm in Flav7 between −100 and 100 fs. Analogous dynamics was observed previously in several dyes.84,85 When excitation is at the low-energy side of absorption spectrum,84–86 as the re-equilibration begins on the S0 solute–solvent configurational potential the GSB develops on the high-energy side of the initial hole. At the same time, the hole replica is positioned in the minimum of the solute–solvent configurational potential and, consequently, hole replica SE does not show time-dependent shift, Fig. 5a. Hole replica ESA is also stationary. The shift of the positive ΔA maxima from 621 to 623 nm for Chrom7 and 608 to 612 nm for Flav7 between −100 and 100 fs is due to the developing bleach of the S0–Sn absorption, which contributes more at the high-energy side of the ESA band (Fig. 1b and 2). Whereas initially fast, the S0 hole broadens to ∼2 ps.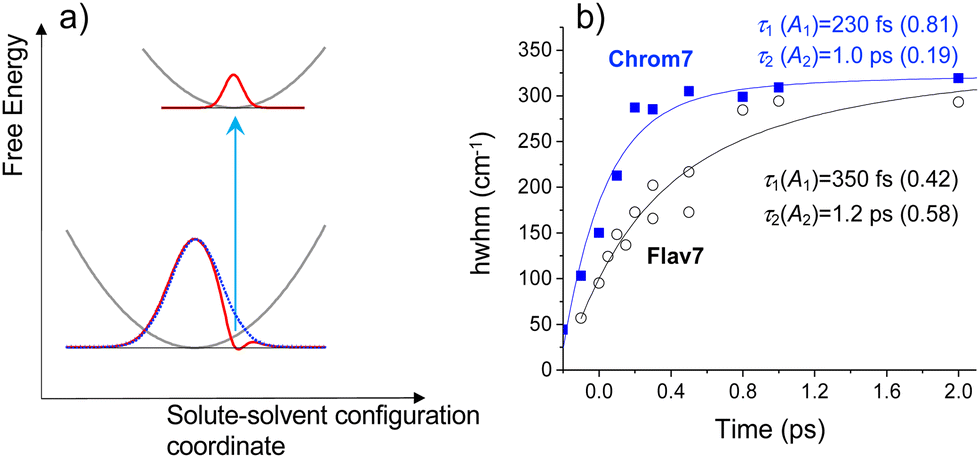 | ||
| Fig. 5 (a) Schematic diagram for the hole relaxation when excitation is into the low-energy wing of the linear absorption spectrum and excites only a narrow range of resonant sites.84–86 The distribution of excited-state population may only broaden without shift, but the ground-state population bleaches to fill the hole. In (b), for Chrom7 (excitation, 990 nm) and Flav7 (excitation, 1042 nm), the time evolution of the hwhm bandwidth evaluated at the short-wavelength side of the main negative ΔA band is fitted to a two-exponential A1exp(−t/τ1) + A2exp(−t/τ2) function, with the fit curves and best-fit parameters shown. | ||
The hole dynamics can be quantitative characterized by analyzing the time-dependent GSB hole bandwidth (specifically, the half-width at half-maximum, hwhm).87,88 Assuming that the hole replica is stationary, the hwhm evaluated at the short-wavelength side of the 990- and 1042-nm ΔA features yields the time evolution of the ground-state hole. The bandwidth broadens with time for both Flav7 and Chrom7 from about 50 to 290 cm−1, which behavior can be described by a double exponential with time constants of τ1 = 230 fs and τ2 = 1.0 ps for Chrom7 and τ1 = 350 fs and τ2 = 1.2 ps for Flav7, Fig. 5. These time constants are also found in ΔA kinetic traces within the S0–S1 absorption, Fig. 3 and Fig. S5–S8 (ESI†), but not outside this region, Fig. S7–S9 (ESI†). At wavelengths nearby the excitation wavelength (e.g., 980 nm, Chrom7, and 1060 nm, Flav7) these time constants describe the hole refilling (decay), whereas at those away (e.g., 840 nm, Chrom7, and 980 nm, Flav7) they describe the hole broadening (rise), Fig. 3. Within the outermost blue wing of the S0–S1 absorption spectrum, a faster initial hole dynamics is observed (−100 and 0 fs ΔA spectra, Fig. S10, ESI†), for which the fits yield a time constant of τ1 = 90 ± 30 fs for both Flav7 and Chrom7, Table S1 (ESI†). In an inhomogeneous ensemble, different members may have different lifetimes; probing across the blue wing interrogates the different, evidently steeper, portion of the S0 solute–solvent configurational potential, Fig. 5a. In the SE range, the double exponential decay is practically absent, e.g., at 1070 and 1120 nm for Chrom7 and 1140 and 1160 nm for Flav7 (Fig. 3), consistent with the notion that the hole replica is positioned at the minimum of the excited-state solute–solvent configurational potential. The lack of red shift of the SE maximum is evidence that solvent stabilization of the excited S1-solute is minor in these dyes.
Following 925-nm excitation, the high-energy-side ESA hwhm for Flav7 and Chrom7 evolves with τ1 ∼120 fs, which is the same as the 90 ± 30 fs time constant within the accuracy, Table S1 (ESI†). This similarity may be explained by the fact that in cyanine dyes the S0 surfaces are typically much steeper than the S1 surfaces,38 and therefore, although the S1 hole replica is not stationary following 0-1′ excitation, the hwhm dynamics is predominantly determined by the gradient of the S0 solute–solvent configurational potential. The second component of the hole dynamics following 925-nm excitation remains about 1 ps for both dyes (Fig. S11–S16, ESI†).
Let us begin by interpreting τ2 = 1 ps, which is independent of the dye nature and excitation wavelengths, and therefore, can be assigned to nonspecific solvent motion. This component matches the slow, 1.02 ps79 time constant in the spectral response function for CH2Cl2 obtained with a coumarin solute and assigned to diffusive solvation dynamics. Regarding τ1, there is no match of either 230 or 350 fs to the fast 144 fs79 time constant due to inertial solvent dynamics in the same spectral response function. The 230 or 350 fs time constants are also significantly shorter than the average solvation time of CH2Cl2, 0.56 ps.79 There is also no agreement with time constants of 0.57 and 2.21 ps due to solvent reorientation reported for neat CH2Cl2 in ultrafast Raman-induced Kerr effect studies.89 Therefore, the initial hole dynamics cannot be considered to be due to inertial solvation dynamics alone and also is not reducible to the rearrangement of the CH2Cl2 molecules through their rotational motion. Instead, one can propose the S0 site distribution with low-frequency (95–145 cm−1) skeletal deformations, such as torsional and bending vibrational modes known90 to be optically active in polymethines. The 230- and 350-fs relaxation times are firmly within vibrational periods of these modes. An inhomogeneous ensemble in which different members exhibit a small, varying degree of deviation from the planarity has often been invoked to explain the absorption spectrum broadening in cyanine dyes.23,91,92 Also, the link between torsional motion and hole burning dynamics was reported in barrierless excited-state relaxation of 1,1′-diethyl-2,2′-cyanine.69,93 Skeletal deformations with somewhat higher vibrational frequencies of about 350 cm−1 may be responsible for the 90-fs hole dynamics.
To determine vibrational relaxation rate constants for Chrom7 and Flav7 in the S1 state, the ESA ΔA kinetic traces were fitted to a sum of multiexponential decay functions while including the known hole relaxation time constants. For 0-1′ 925-nm excitation, this procedure yielded ∼350-fs time constants for both dyes, Fig. S15 and S16 (ESI†). Spectral narrowing/broadening is a well-known indicator of vibrational energy flow,80–82 and therefore, we also examined the time evolution of the ESA fwhm. For Flav7, a similar 323-fs time constant was found, Fig. S11 (ESI†). We note that vibrational relaxation manifests itself as the growth of the weaker peak relative to the major one in the ESA spectra (cf. the 300- and 500-fs ΔA spectra, Fig. S17c and f, ESI†). A similar spectral change occurs during the fast hole dynamics, Fig. S10 (ESI†), which suggest that the involved low-frequency skeletal deformations also participate in the vibrational relaxation process. Vibrational relaxation was not observed after 990- and 1042-nm excitation.
Ultrafast transient absorption. 500-nm Sn excitation
To provide an estimate for the lifetimes of highly-excited Sn electronic states, excitation of Chrom7 and Flav7 was carried out at 500 nm. The S1 ESA ΔA signals rise with a significant delay within the first picosecond after excitation, Fig. 6 and Fig. S18, S19 (ESI†). The 300-fs ΔA spectra are broadened, Fig. S20 (ESI†). The population in the S1 state becomes relaxed and thermalized at 6 ps because afterwards the ΔA spectral shape remains unaltered with time and similar to that after S1 excitation. The amplitude of ΔA signals decays to the noise level with the S1 lifetime, Table 1.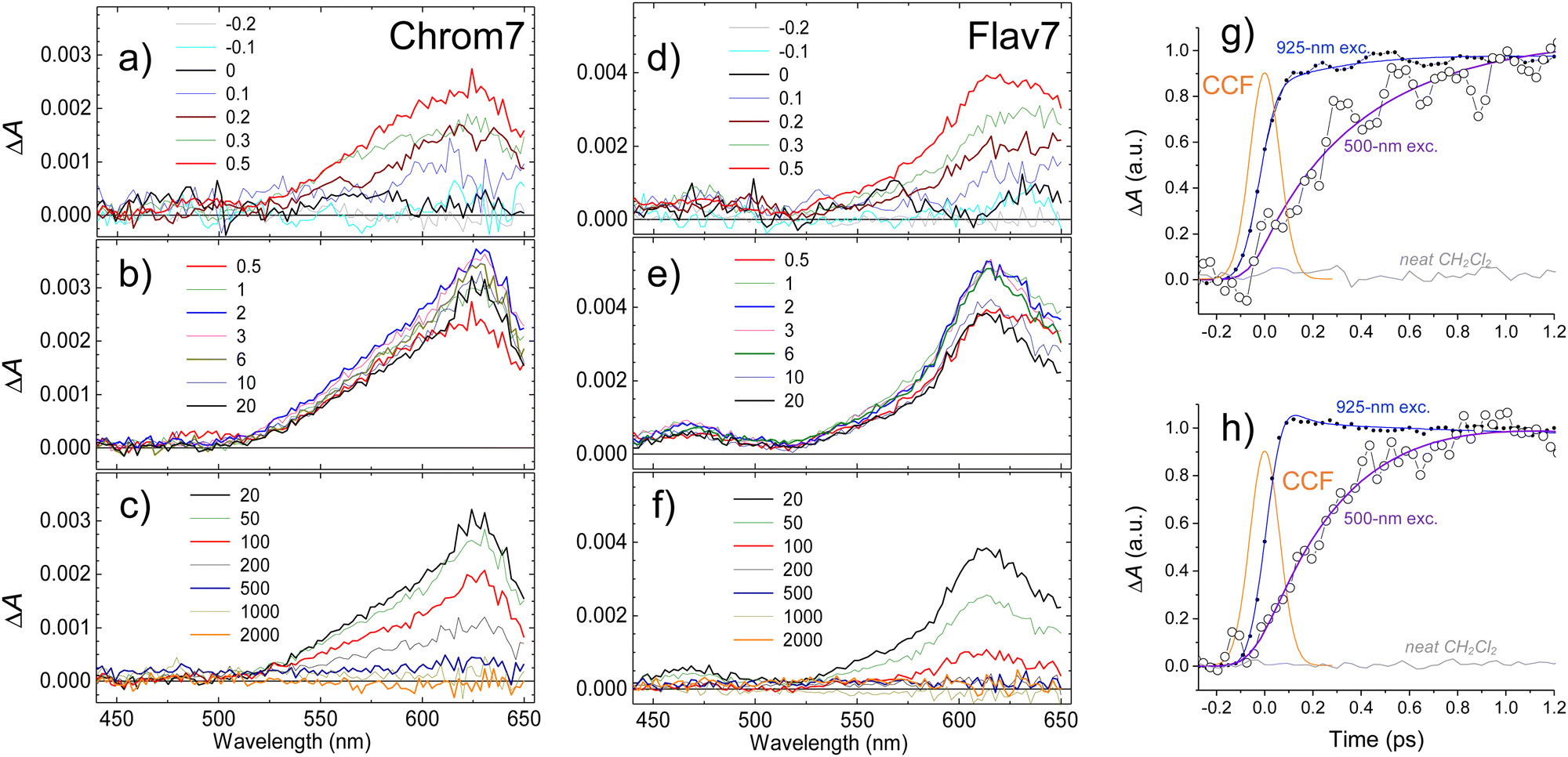 | ||
| Fig. 6 Transient absorption spectra of Chrom7 (a)–(c) and Flav7 (d)–(f) in CH2Cl2 at short (a) and (d), intermediate (b) and (e), and long (c) and (f) times following 500-nm excitation. Delay times between the excitation and probe pulses are given in the legends. Representative ΔA kinetics traces (symbols) at the probe wavelength of 575 nm for Chrom7 in (g) and 645 nm for Flav7 in (h) are compared with the data (symbols) measured following 925-nm excitation within the same ESA range: 553 nm in (g), and 659 nm in (h). The neat CH2Cl2 yields a negligibly small ΔA signal (as shown) under the same excitation conditions. The CCF between 500-nm excitation and probe pulses is Gaussian-shaped with a 145-fs fwhm, (g) and (h). | ||
The entire ΔA kinetic traces can be described by a triexponential decay model using deconvolution with the CCF. In addition to the S1 lifetime, two other time constants describe the initial ΔA signal buildup and the spectral narrowing, Fig. S21 (ESI†). The buildup is interpreted as the S1 population rise following IC from the Sn state (time constant, 0.40 ± 0.05 ps for Chrom7 and 0.45 ± 0.05 ps for Flav7). Sub-picosecond Sn lifetimes were reported for other polymethines.94 The narrowing (time constant, ∼0.8 ps, both dyes) is due vibrational relaxation in the S1 well bottom. Following Sn → S1 IC, ∼10000 cm−1 excess vibrational energy is deposited in the S1 state for Chrom7 and Flav7, which is ∼10 times more than produced by 925-nm excitation. Vibrational relaxation times are expected to be faster in higher vibrational states as reported for the indotricarbocyanine HITCI.95,96 This expectation is transferrable provided that with an increase of vibrational energy the microscopic picture of energy transfer remains the same: e.g., initially from solute's higher- to lower-frequency modes and then to solvent low-frequency modes, and other mechanisms, e.g., from solute's high-frequency modes directly into high-frequency modes of the solvent, do not intervene.97 The opposite trend in the vibrational relaxation times we observed (0.8 vs. 0.35 ps for 500- and 925-nm excitation, respectively) is because they characterize the arrival of hot population into the S1 well bottom and embody the transit times through all intermediate vibrational states involved.
Vibrational coherences
Following S0 → S1 excitation of Chrom7 and Flav7, the oscillatory ΔA signals occur predominantly in the S1 ESA region with the decoherence time constants of about 500 fs, Table S2 (ESI†). Following subtraction of exponential decays determined from the ΔA fits, the oscillatory signals can be isolated and further analyzed by the fast-Fourier transform (FFT). The FFT analysis revealed several oscillation frequencies of 52, 139, and 278 cm−1 for Chrom7 (excited at 990 nm) and 208 and 121 cm−1 for Flav7 (excited at 1042 nm), Fig. S22a, b and S23a, b (ESI†). Upon 925-nm excitation of Chrom7, 104 and 208 cm−1 FFT bands, in addition to the one at 52 cm−1, were detected (Fig. S23g and h, ESI†). For Flav7, a 225 cm−1 FFT band (Fig. S22g and h, 925-nm excitation, ESI†) is thought to be the same as the 208 cm−1 band observed following 1042-nm excitation based on the accuracy of data analysis.The coherent motion observed most likely takes place in the S1 and not S0 state because the absorption from S1 predominates throughout the ESA probing region. Ultrafast pulse excitation prepares the molecule in the FC electronic state in a superposition of optically active vibrational levels called a wavepacket. The vibrational wavepacket oscillates between the inner and outer turning points of the excited-state potential. As a result, the oscillations occur with a π-shift in the red and blue wings of the S1 ESA band, as observed, Fig. S24 (ESI†). Excitation at the low-energy side of the absorption spectrum is known to suppress vibrational coherence in the S1 state,98,99 which agrees with the oscillations for Chrom7 and Flav7 in this case being less pronounced than for 0-1′ 925-nm excitation. Upon 500-nm Sn excitation of Chrom7, the sub-1 ps ΔA signal rise is superimposed with 139-cm−1 modulations, Fig. S25 (ESI†). Product vibrational coherence may result from crossing of PESs itself100 or be transferred from the initial state following its preparation by a short excitation pulse. In polymethine dyes, vibrational coherence in S1 following IC from Sn was observed for the first-time, to our knowledge, in indolenine HDITC following the deposition into S1 chemically significant ∼1.5 eV of energy,101 similar to the amount for Chrom7. No coherence was observed after 500-nm excitation of Flav7.
Vibrational wavepackets were ubiquitously observed in polymethine dyes following ultrafast pulse excitation.93,98,99,101–109 Whereas in many instances the coherence observed was merely due to ‘spectator’ optically-active vibrational levels, for Cy5 the 273 cm−1 vibrational mode connecting the planar FC and photoisomerization transition state structures was claimed to be involved.105 Several studies demonstrated that vibrational coherence can control the photoisomerization quantum yield,110–112 which was interpreted as altering the excited-state relaxation path towards the CI seam separating the parent and the photoisomer forms.90 In 1,1′-diethyl-2,2′-cyanine, vibrational coherences in Raman-active 200 cm−1 butterfly-like symmetric stretching as well as 130 and 300 cm−1 bending modes play a role.111,112 The prominent 52, 139, 104, 208, and 278 cm−1 vibrational modes observed in our work are in the same frequency range as the modes previously proposed to be involved in the photoisomerization pathway.
Lowest-energy triplet states
We performed phosphorescence measurements of Chrom7 and Flav7 in a frozen solvent matrix at 77 K following 532-nm 7-ns pulsed excitation to locate the energy position of lowest-energy triplet T1 states. No phosphorescence was observed in ethanol glass. A heavy-atom containing matrix (CD3I) was then used to increase the ISC and radiative phosphorescence rates. Also, deuterated solvents may reduce possible non-radiative deactivation of low-energy excited states via coupling to C–H vibrational modes of the surrounding medium and are often preferred for near-IR luminescence measurements. The excitation of Chrom7 in a CD3I matrix at 77 K resulted in weak phosphorescence with a maximum at 1305 nm, Fig. 7a. The ∼1260-nm shoulder is assigned to 1O2 emission,72 whereas the feature at 1110 nm is due to contributions from scattered fundamental (1064 nm) laser light and CD3I matrix emission, Fig. S26 (ESI†). The phosphorescence intensity decayed monoexponentially with a lifetime of 12 μs, Fig. 7b. Nanosecond pulsed excitation of Flav7 in a CD3I matrix (77 K) either at 532 nm or 1064 nm did not result in any detectable phosphorescence in the 1000–1400 nm range. A possible reason is that Flav7 phosphorescence occurs at wavelengths longer than the 1400 nm PMT detection limit. This implies a lower T1 state in Flav7 than in Chrom7, which is consistent with the lower S1 state in Flav7 than in Chrom7. The absence of phosphorescence from Flav7 might also have been caused by poor triplet state population and/or a very low phosphorescence radiative rate constant. The first cause is unlikely; provided that about a factor of 2.5 difference in the S1 lifetimes between Chrom7 and Flav7 transfers into the S1 → T1 ISC quantum yields, the T1 population in Flav7 would still be measurable. The second explanation is also unlikely because of the similar radiative rate constants in these two dyes (Table 1), which implies a similar electronic nature of the low-energy excited states.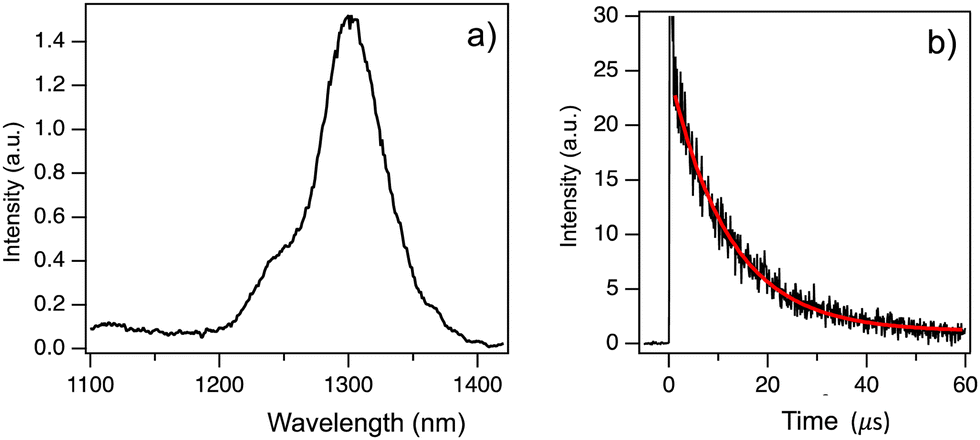 | ||
| Fig. 7 Phosphorescence spectrum for Chrom7 in a CD3I matrix at 77 K, (a) The phosphorescence was recorded in the 4–19 μs time interval after pulsed laser excitation (532 nm, 7-ns pulse width). The phosphorescence decay (black) was measured at a 1300-nm wavelength and can be fitted to a single exponential decay (red) with a 12 μs lifetime, (b). | ||
The energy of the T1 state may in principle be determined from the position of the highest-energy phosphorescence vibronic band.56 If, however, the phosphorescence spectrum does not show sufficiently resolved vibronic structure, the ‘onset’ of the short-wavelength portion of the phosphorescence spectrum may be used to estimate the upper limit of the T1 state energy.56 Neither of these two approaches work for Chrom7 because of the lack of vibronic structure and the presence of O2 emission. Instead, a 0.9 level at which the 0–0′ vibronic bands of absorption and fluorescence spectra cross (S0–S1 energy gap), Fig. 1, was adopted for the 1305-nm phosphorescence band, which yields the intersection at 1290 nm and thus the T1 energy at 7750 cm−1 above S0. Based on the 1400-nm detection limit, the Flav7 T1 state is placed at 7140 cm−1 or lower.
Temperature dependence of the S1 state lifetime
The fluorescence quantum yields, Φf, and the S1-state lifetime, τf for Chrom7 and Flav7 rise upon lowering the temperature before reaching a plateau, Fig. S27–S29 (ESI†), and Fig. 8. The total non-radiative rate constant in the S1 state, knr, is given by:27,33,46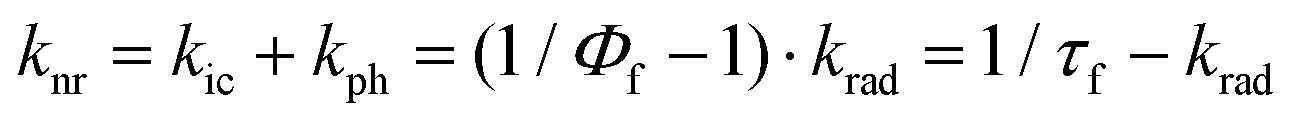 . Here, krad and kic are the practically temperature independent radiative and direct IC rate constants, and kph is the rate constant for photoisomerization barrier crossing, which is temperature and solvent viscosity dependent according to the following equation: kph = Dς−aexp(−E0/RT). Here, R is the universal gas constant, T is the absolute temperature, E0 is the intrinsic barrier height, ς is the solvent viscosity, and D and a are the parameters, where a (0 ≤ a ≤ 1) depends on the size and shape of the intrinsic barrier,113–115 reaching the low limit for larger barriers.116,117 The approximation used in many studies48,116–121 is that in a narrow temperature range, ς = ς0exp(Eς/RT),122 where ς0 is the constant, and Eς is the solvent viscosity activation energy. As a result, kph obeys the Arrhenius equation, kph = Aexp(−Ea/RT), with the preexponential factor A and the activation energy Ea given by Ea = E0 + aEς. When plotted against T−1, knr should exhibit the slope defined by Ea before reaching a plateau defined by kic at low T. The knr values determined from the measured Φf and τf are fitted to a sum of kph and kic, Fig. 8. At 22 °C, the fits for Chrom7 yield kic = 4.03 × 109 s−1, which is larger than kph = 5.72 × 108 s−1 by a factor of 7, but for Flav7 the difference is not dramatic as kic = 8.89 × 109 s−1 is larger than kph = 3.88 × 109 s−1 only by a factor of 2.3. The fits yield reasonable A = 6.97 × 1012 and 5.29 × 1012 s−1 for Chrom7 and Flav7, respectively, as well as Ea = 5.20 ± 0.75 and 4.26 ± 0.74 kcal mol−1, which are much larger that Eς of CHCl3. The latter is estimated to be 1.78 kcal mol−1 based on viscosities123 reported between −25 and 50 °C, which is about the temperature range investigated in this work.
. Here, krad and kic are the practically temperature independent radiative and direct IC rate constants, and kph is the rate constant for photoisomerization barrier crossing, which is temperature and solvent viscosity dependent according to the following equation: kph = Dς−aexp(−E0/RT). Here, R is the universal gas constant, T is the absolute temperature, E0 is the intrinsic barrier height, ς is the solvent viscosity, and D and a are the parameters, where a (0 ≤ a ≤ 1) depends on the size and shape of the intrinsic barrier,113–115 reaching the low limit for larger barriers.116,117 The approximation used in many studies48,116–121 is that in a narrow temperature range, ς = ς0exp(Eς/RT),122 where ς0 is the constant, and Eς is the solvent viscosity activation energy. As a result, kph obeys the Arrhenius equation, kph = Aexp(−Ea/RT), with the preexponential factor A and the activation energy Ea given by Ea = E0 + aEς. When plotted against T−1, knr should exhibit the slope defined by Ea before reaching a plateau defined by kic at low T. The knr values determined from the measured Φf and τf are fitted to a sum of kph and kic, Fig. 8. At 22 °C, the fits for Chrom7 yield kic = 4.03 × 109 s−1, which is larger than kph = 5.72 × 108 s−1 by a factor of 7, but for Flav7 the difference is not dramatic as kic = 8.89 × 109 s−1 is larger than kph = 3.88 × 109 s−1 only by a factor of 2.3. The fits yield reasonable A = 6.97 × 1012 and 5.29 × 1012 s−1 for Chrom7 and Flav7, respectively, as well as Ea = 5.20 ± 0.75 and 4.26 ± 0.74 kcal mol−1, which are much larger that Eς of CHCl3. The latter is estimated to be 1.78 kcal mol−1 based on viscosities123 reported between −25 and 50 °C, which is about the temperature range investigated in this work.
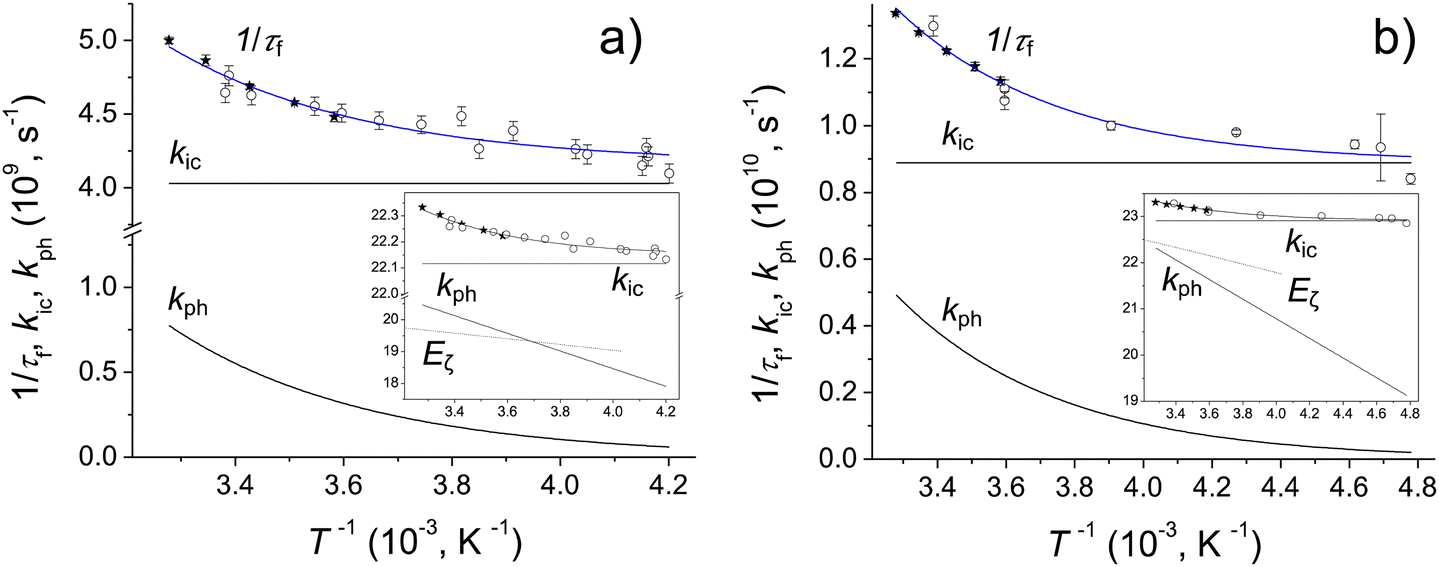 | ||
| Fig. 8 Chrom7, (a) and Flav7, (b) in CHCl3: influence of temperature on the inverse S1-state lifetime, where τf values were measured by femtosecond transient absorption (open circles) and calculated from the fluorescence quantum yields (solid circles) assuming that krad is the same as in CH2Cl2, Table 1, and the best fits of the 1/τf data using the Arrhenius kph and temperature independent kic and krad rate constants. The insets show the rate constants and CHCl3 viscosity, labelled Eς (for comparison, the ς values were scaled by a constant factor), plotted on a natural logarithmic scale vs. 103T−1. | ||
Fig. 9 summarizes the S1 radiationless relaxation mechanism we propose for the SWIR polymethine dyes studied in this work, which involves competing energy gap and isomerization decay pathways. Several conclusions can be made. First, the application of energy gap theories for unconstrained SWIR polymethine dyes, which does not account for kph while silently assuming that the nonradiative decay is entirely due to direct IC,20,124 may be in error. Compared to the fit124 of knr alone to the energy gap model, plotting knr–kph produces a better fit. Second, the difference in room-temperature lifetimes for Chrom7 and Flav7 upon going from CH2Cl2 to CHCl3 (Table 1) cannot be attributed to the gap law alone because the solvatochromic shifts involved are small (∼3 nm). Shorter S1 lifetimes in CH2Cl2 are consistent with the presence of the photoisomerization pathway less hindered by a less viscous solvent (0.42 and 0.56 cP for CH2Cl2 and CHCl3, respectively, 20 °C). Third, it is reasonable to assume that the solvent contribution to Ea is similar for Flav7 and Chrom7 because the measured activation energies are not much different.114–117 This suggests that the intrinsic photoisomerization barrier is somewhat larger in Chrom7 than Flav7. This is consistent with Chrom7 having a bulkier 2-alkyl group as compared to a phenyl group in Flav7. A slowing down of the S1 photoisomerization with increasing bulkiness of the twisting fragment was previously observed.125,126
 | ||
| Fig. 9 The radiationless relaxation mechanism of the SWIR polymethine dyes studied in this work with the competing pathways of direct IC, which takes place within the planar S1trans-form and obeys the energy gap law, and photoisomerization, which is thermally activated. The photoisomerization motion predominantly samples the trans-side of the twisted S1 minimum and is essentially nonreactive. | ||
Finally, one can compare kph, kic, and Ea values found in this work with those reported in the S1 state for heptamethine cyanine dyes. For HITCI (in DMSO unless indicated otherwise), direct IC is slow, kic = 2.8 × 108 s−1,33 as expected because its absorption maximum is blue shifted by ∼260 nm compared to Flav7 and Chrom7. The photoisomerization activation energies, 5.3 (HITCI33), 5.0 (DTTCI34), and 6.0 (IR-14034) kcal mol−1 as well 3.5 (DTTCI34), 4.5 (IR-14034), and 5.74 (DOTCI48) kcal mol−1 in C2H5OH are comparable with the Ea values in Flav7 and Chrom7. Similar activation energies were measured for photoisomerization of dicarbocyanine dyes.48,76 Photoisomerization is the major S1 relaxation pathway in heptamethine cyanine dyes, e.g., the kphτf quantum yield is ∼0.7 in HITCI (kph = 3.3 × 108 s−133). Photoisomer quantum yields are about 0.08 in penta- and heptamethine cyanines,30–32,40 which based on kphτf in these dyes yields a branching ratio ϕ of ∼0.1, Fig. 1a. The values of kph in Chrom7 and Flav7 are significant (5.72 × 108 s−1 and 3.88 × 109 s−1), so that kphτf becomes 0.12 and 0.3, that is still significant, and which for the photoisomer quantum yields estimated, 1.8 × 10−3 and 6.4 × 10−3, suggests the branching ratio of 0.014 and 0.02 in Chrom7 and Flav7, respectively, Table S3 (ESI†). This is 10 times smaller compared with penta- and heptamethine dyes. Quantum chemical computations of polymethine dyes suggest the presence of the S1/S0 CI seam on the trans-side in the vicinity of the S1-state twisted minimum.38,39 One then can propose that Chrom7 and Flav7, after overcoming the photoisomerization barrier, reach the seam region where local topology even more strongly favors the S0-trans nonreactive channel over S0-cis product formation.
Highly excited states and implications for bioimaging
After SE becomes spectrally separated from the absorption and unaffected over time in the femto-to-picosecond range, the σSE(λ) spectrum can be constructed from the steady-state fluorescence spectrum,127,128Fig. 1. When vibrational relaxation in S0 is faster compared with the S1 lifetime and in the absence of an appreciable photochemical change, which are valid assumptions based on the data, the ΔA(λ, t) spectrum is: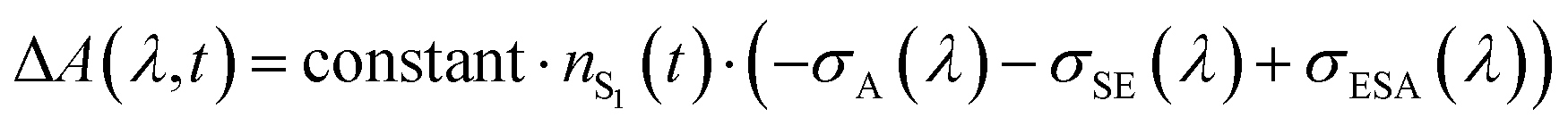 , where nS, (t) is the S1 population at a delay time t. The measured ΔA(λ,t) spectrum can be scaled to a sum of σA(λ) and σSE(λ) with the minimal uncertainty using the wavelength range where the spectrum and the sum closely match each other (implying that σESA(λ) ∼ 0 in this range) and change rapidly; the excess of the scaled ΔA(λ, t) over σA(λ) + σSE(λ) outside the matching range yields the unknown σESA(λ) spectrum. The said wavelength range is present between 875–970 nm for Chrom7 and 960–1020 nm for Flav7. Consequently, the σESA(λ) spectra were determined, Fig. 10, with the accuracy of the σESA(λ) values estimated to be better than 50%.
, where nS, (t) is the S1 population at a delay time t. The measured ΔA(λ,t) spectrum can be scaled to a sum of σA(λ) and σSE(λ) with the minimal uncertainty using the wavelength range where the spectrum and the sum closely match each other (implying that σESA(λ) ∼ 0 in this range) and change rapidly; the excess of the scaled ΔA(λ, t) over σA(λ) + σSE(λ) outside the matching range yields the unknown σESA(λ) spectrum. The said wavelength range is present between 875–970 nm for Chrom7 and 960–1020 nm for Flav7. Consequently, the σESA(λ) spectra were determined, Fig. 10, with the accuracy of the σESA(λ) values estimated to be better than 50%.
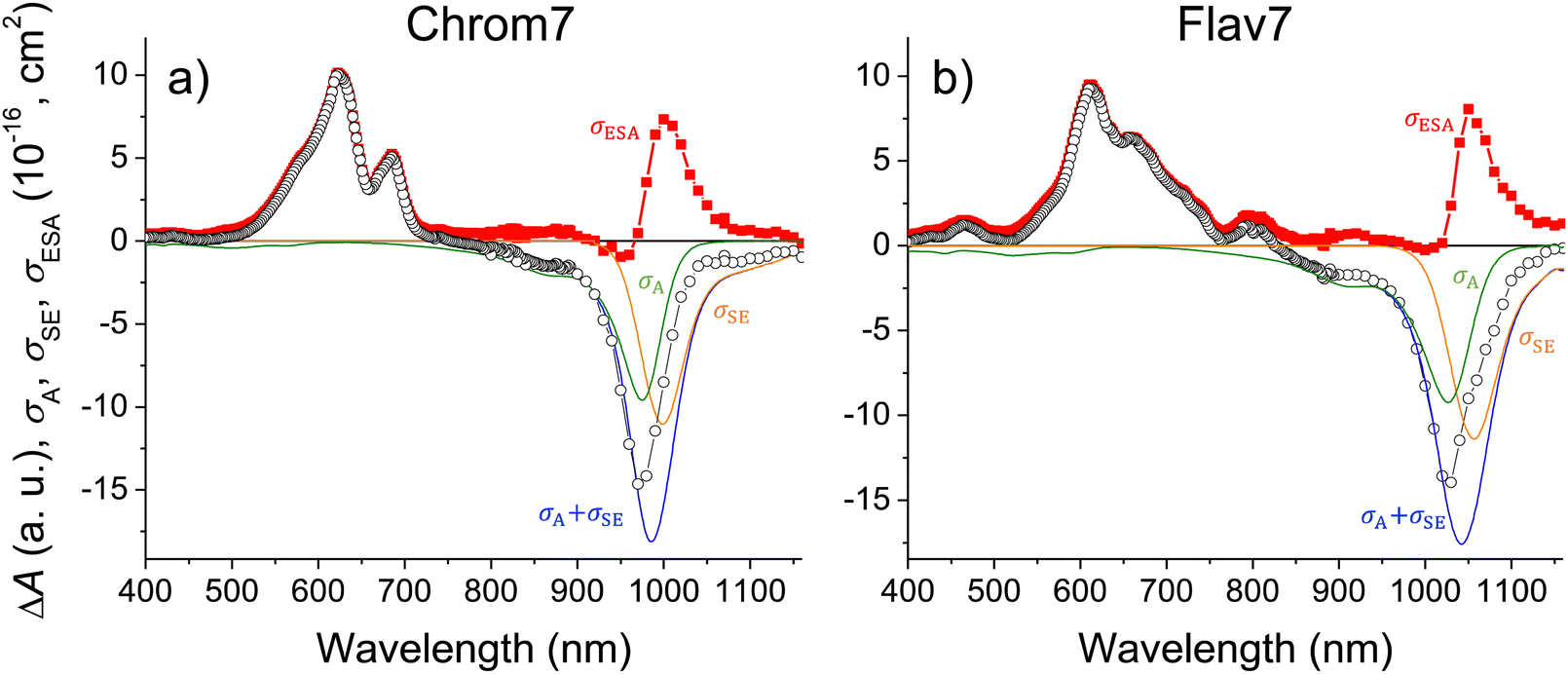 | ||
| Fig. 10 Chrom7 excited at 990 nm, (a) and Flav7 excited at 1042 nm, (b) for which the 50-ps ΔA spectrum from Fig. 2 is scaled (open symbols) to the σA(λ) + σSE(λ) sum (with the individual σA(λ) and σSE(λ) contributions shown); their difference (solid symbols) is equal to the σESA(λ) spectrum of ESA from S1. The σESA(λ) spectral shape is found to be constant from ∼100 ps to delay times as short as 500 fs, Fig. S30 (ESI†). | ||
Both Chrom7 and Flav7 exhibit an intense ESA band in the SWIR range (1000 and 1050 nm, respectively), which is somewhat red-shifted with respect to the S0–S1 absorption maximum, as well as weak ESA in the near-IR region, Fig. 10. We attribute these two ESA features to the terminal S2 and S3 states, although we cannot exclude the possibility of singlet excited states lying even lower. Blue-shifted, intense ESA (Fig. 10) resembles that in other symmetric long-chain cyanines129 and assigned to the terminal S4,5 states (dual peaks at 683 and 622 nm for Chrom7, and 657 and 612 nm for Flav7). Incidentally, the peak separation gap of 1435 and 1120 cm−1 is not overwhelmingly larger than the ∼1230 cm−1 frequency of the optically active vibrational mode of the S0–S1 transition, Fig. 1. The transition energies of ESA from the equilibrated S1 state, when they are adjusted for the S0–S1 energy gap (10140 and 9700 cm−1 for Chrom7 and Flav7) are expected to match the S0–Sn band positions. We found the terminal S2 and S4,5 states to be close (within 120–240 cm−1 for S2 and 300–800 cm−1 for S4,5) to the visible band maxima in the linear absorption spectrum, Fig. S31 (ESI†). We note that the ESA bands are much narrower than the diffuse S0–Sn absorption bands. Similar effects were observed for ESA of coumarin C153130 and HITCI,131 and may be attributed to either fewer vibrational modes being optically active in S1 compared to S0130 and/or the presence of several closely-lying excited states where intense absorption from S1 is only allowed to occur into one of them.132 The energy diagrams for Chrom7 and Flav7 are summarized in Fig. 11.
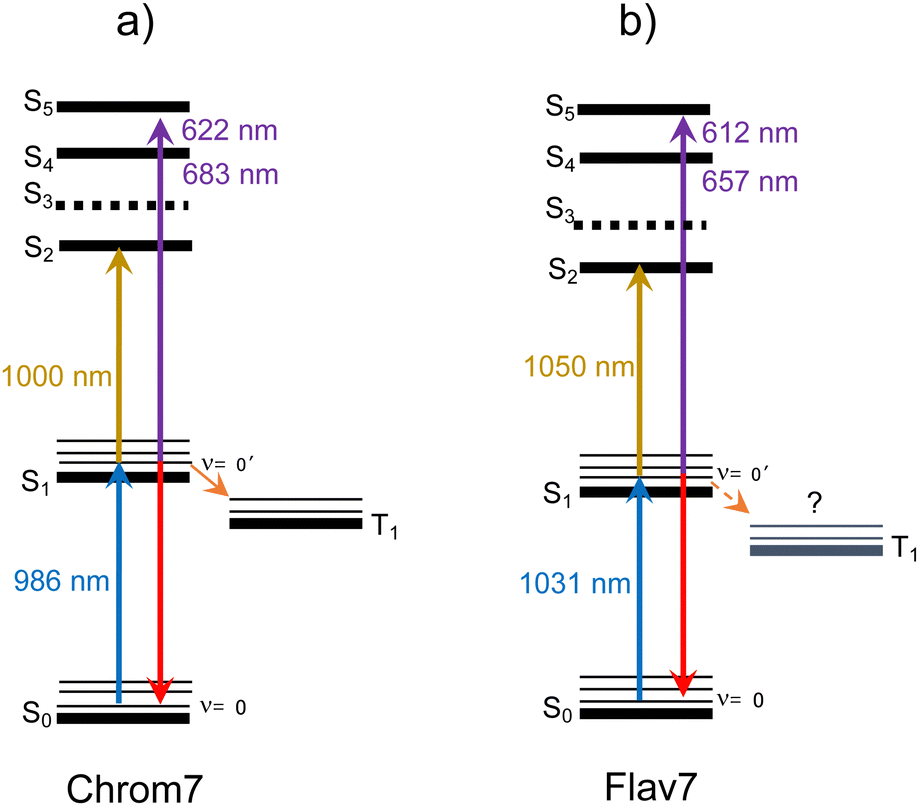 | ||
| Fig. 11 Energy diagram for Chrom7, (a) and Flav7, (b) based on this work. The S0–S1 energy gap is 10140 and 9700 cm−1 based on the overlap of the 0–0′ absorption and fluorescence bands at 986 and 1031 nm. The S1 → S2 and S0 → S1 transitions occur at nearly the same energy, and the S5 states are found to be within 100 cm−1 of each other at ∼26150 cm−1. The S3 position is only tentatively shown because near-IR ESA attributed to the S1 → S3 transition is weak and diffuse. The T1 state is at 7750 cm−1 above S0 for Chrom7 and is assumed, see the text, at 7140 cm−1 or lower for Flav7. | ||
The σESA(λ) values in two major bands are 8.1 × 10−16 cm2 at 1050 nm and 9.2 × 10−16 cm2 at 612 nm for Flav7 and 7.3 × 10−16 cm2 at 1000 nm and σESA = 9.8 × 10−16 cm2 at 622 nm for Chrom7. Consideration of the ESA spectrum is important for Flav7 and Chrom7, as well as generally for molecular probes in high-intensity photonic applications and measurements, e.g., fluorescence correlation spectroscopy18,133,134 where the fluorophore S0 → S1 excitation rate constant (intensity times cross-section) may become comparable with the inverse S1 lifetime. The S1 population may then be promoted by secondary excitation into one of the higher electronically excited states, which usually is much more prone to molecular decomposition compared to S1. Also, Sn may exhibit a very different photoreactivity. Facile photoisomerization135,136 and ISC137 in Sn states of polymethine dyes were reported. High-intensity irradiation, such as 25 kW peak power focused into a micrometer spot typical for two-photon microscopy,138 of Flav7 and Chrom7 in the 1050- and 1000-nm region should cause, at least, the S0 → S1 → S2 excitation based on comparable intensity times cross-section products of the S0 → S1 and S1 → S2 steps. In the case of pulsed visible irradiation utilized in confocal microscopy, biphotonic excitation may also be expected if the used irradiation wavelength overlaps the visible ESA. On the other hand, irradiation of Flav7 between 820 and 1020 nm and Chrom7 between 730 and 910 nm limits the population promotion further than the S1 state because ESA in these regions is weak.
Conclusions
We have characterized the ultrafast dynamics as well as photophysical and photochemical properties of a chromenylium and a flavylium polymethine dye the latter of which is one of the brightest organic SWIR emitters reported to date. By using different excitation wavelengths, the separation of dynamic inhomogeneous broadening of vibronic spectra and intramolecular vibrational relaxation was made possible, as well as the characterization of ultrafast deactivation of highly excited electronic states. The complexity of deactivation of the first excited singlet state (S1) is revealed by excited-state lifetime and time-resolved phosphorescence measurements at different temperatures. Intersystem crossing is found not to impact room-temperature excited-state relaxation. The results highlight the presence of two competing radiationless relaxation mechanisms in S1. One with the minimal temperature sensitivity is internal conversion via energy gap law. Another is twisting about one of the carbon–carbon conjugated bonds, which has an excited-state Arrhenius activation energy comparable to that of photoisomerization in symmetric penta- and heptamethine cyanines and permits traces of isomer products. Direct internal conversion predominates thermally-activated twisting in the studied SWIR dyes at room temperature, but the latter pathway is substantial, constituting ∼15–40% of the total relaxation rate constant, suggesting that the SWIR fluorophore brightness can be further improved by conformational constraints. The knowledge of excited-state absorption is important in the design of fluorophores for biological and medical applications because it may reduce their brightness thereby limiting their potential in imaging application. The understanding of photoproperties is overall crucial in the design of organic fluorophores.Experimental
Materials
Anhydrous dichloromethane (EMD Millipore, ACS grade) was used as a solvent, and in a few cases, chloroform was used. Chrom7 and Flav7 were synthetized by and received from Dr E. Sletten (UCLA).24,25 UV-vis absorption spectra of the solutions were measured using a Varian Cary 50 Bio spectrophotometer.Photoluminescence experiments
Temperature dependent fluorescence spectra following 900-nm irradiation were recorded on a FLS1000 spectrometer (Edinburgh Analytical Instruments) in conjunction with a H10330B-45 near-IR sensitive PMT (Hamamatsu). The sample solutions in 1 × 1 cm2 quartz cells were deoxygenated before measurements. CHCl3 (Acros, spectroscopic grade) was used as a solvent instead of CH2Cl2 because of a higher boiling point of the former. In conjunction with these experiments, steady-state absorption spectra were recorded on an Agilent 8453 spectrometer equipped with a Peltier temperature-controlled sample holder (PTP-1, PerkinElmer) in 1-cm pathlength quartz cells.Phosphorescence measurements were performed on a modified Fluorolog-3 spectrometer (HORIBA Jobin Yvon) in conjunction with a near-IR sensitive photomultiplier (H10330A-45, Hamamatsu). A Spectra-Physics GCR-150-30 Nd:YAG laser (532 nm, 7-ns pulse width or 1064 nm, 10-ns pulse width) was used for excitation (<2 mJ pulse−1). For recording time-resolved phosphorescence spectra, the signal from the near-IR sensitive PMT was time gated using a SR250 Boxcar Integrator (Stanford Research Systems) and digitized in the Fluorolog-3 SPECTRACQ unit through a DM303M Voltage Input Module (HORIBA Jobin Yvon). Phosphorescence decay was measured with the near-IR sensitive PMT and stored using a digital oscilloscope (TDS 360, Tektronics). The sample solutions were placed in 4-mm (outer diameter) quartz tubes, deoxygenated by purging with N2 gas and frozen in an optically-transparent quartz liquid N2 Dewar (77 K) located inside the sample chamber of the spectrometer.
Nanosecond transient absorption
The previously described set-up139 used 532-nm 7-ns excitation pulses produced by the GCR-150-30 Nd:YAG laser and an Xe-arc lamp as a probing source. Solutions of Chrom7 were prepared in 1 × 1 cm2 quartz cells at concentrations such that the absorbance was ∼0.3 at the excitation wavelength.Ultrafast transient absorption
The details of the set-up and experimental procedures can be found in ESI,† section. Briefly, the set-up used140,141 is based on a regeneratively amplified Ti:sapphire laser system producing a 1 kHz train of 90 fs (fwhm) pulses with 0.9 mJ of energy centered at 800 nm. The 50% portion of the amplified output is sent to an optical parametric amplifier (OPA) to produce 1042-, 990-, and 925-nm and 500-nm pulses used for sample excitation. The remaining half was used either to produce a broadband white-light continuum (wlc) probe light in the 340–935 nm range or to pump another OPA to generate a single-wavelength probe light tunable from 800 to 1160 nm. The polarization plane of the excitation light was set to the magic angle (54.7°) with respect to that of the probe light to eliminate rotational reorientation effects from transient absorption signals. The probe and excitation beams overlapped at a 6° angle at the sample position, being 60 and 200 μm in diameter. The excitation energy was attenuated to around 45–60 nJ pulse−1 to ensure that single-photon excitation is responsible for the measured ΔA data, Fig. S32 (ESI†). The experiments were performed at 22 °C unless stated otherwise and utilized a 2-mm pathlength rotating cell. The absorption spectra of Flav7 and Chrom7 samples obeyed the Lambert–Beer law and showed no change after laser experiments. The ΔA data were corrected for group-velocity dispersion in the wlc probe light with a 20-fs accuracy using electronic instantaneous response of the CH2Cl2 solvent142,143 present within the initial ∼170 fs after excitation, Fig. S33a–c (ESI†). The CCF between the excitation pulse and spectral components of the wlc pulse (Gaussian-shaped with fwhm of 125 fs for 990- and 1042-nm excitation, and 145 fs for 925-nm excitation) was determined using the same solvent response as described in the literature.142 The CCF between 500-nm and wlc probe pulses (Gaussian, 145-fs fwhm, Fig. S33d, ESI†) was determined using the solvent anti-Stokes ΔA signal at 433 nm at time zero144 due to the Raman-active C–H stretching mode of CH2Cl2 (3045 cm−1145). When probing in the near-IR using the OPA, the time zero and CCF (fwhm, 155 fs) were determined via sum-frequency mixing of the excitation and probe light in a 0.1-mm thick, type-I BBO crystal, Fig. S34 (ESI†). The representative ΔA kinetic traces were fitted with CCF deconvolution to a sum of up to six exponential decay functions, 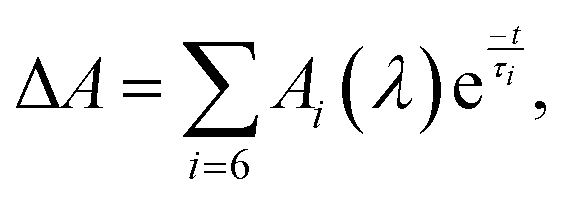 where Ai(λ) is the amplitude and τi is the time constant of the ith component. From multiexponential fits with CCF deconvolution the time resolution of the experiments reported herein is 40 fs. FFT analysis was performed on the residuals of multiexponential fits.
where Ai(λ) is the amplitude and τi is the time constant of the ith component. From multiexponential fits with CCF deconvolution the time resolution of the experiments reported herein is 40 fs. FFT analysis was performed on the residuals of multiexponential fits.
The same set-up with wlc probing was used for low-temperature S1-state lifetime measurements. Excitation was at 1042 nm (Flav7) and 990 nm (Chrom7). CHCl3 was used as a solvent (ACS-grade anhydrous, Fischer Scientific). The samples were kept in a 1 × 1 × 4 cm3 quartz cell immersed into a Dewar condenser filled with acetone cooled by liquid N2, where the temperature was monitored with a thermocouple.
Author contributions
L. M. Obloy: femtosecond pump–probe experiments, data analysis, and manuscript writing; S. Jockusch: temperature-dependent absorption and emission experiments and data analysis; A. N. Tarnovsky: work proposal and organization, femtosecond pump–probe experiments, data analysis, and manuscript writing. All the authors participated in the discussions and contributed writing and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
In memory of T. K. Razumova (1932–2023), whose work advanced the understanding of photoprocesses in polymethine dyes and who was the graduate advisor of ANT. The authors acknowledge Emily D. Cosco and Ellen M. Sletten for providing Flav7 and Chrom7 and critical reading of the manuscript. The authors are also thankful to George T. Lawton for his help with the 925-nm transient absorption experiments. This work is supported by the National Science Foundation (CHE-2102619, CHE-0923360, and CHE-1626420).Notes and references
- E. Thimsen, B. Sadtler and M. Y. Berezin, Shortwave-infrared (SWIR) Emitters for Biological Imaging: a Review of Challenges and Opportunities, Nanophotonics, 2017, 6, 1043–1054 CrossRef CAS.
- S. M. Usama and K. Burgess, Hows and Whys of Tumor-Seeking Dyes, Acc. Chem. Res., 2021, 54, 2121–2131 CrossRef CAS.
- V. G. Bandi, M. P. Luciano, M. Saccomano, N. L. Patel, T. S. Bischof, J. G. P. Lingg, P. T. Tsrunchev, M. N. Nix, B. Ruehle, C. Sanders, L. Riffle, C. M. Robinson, S. Difilippantonio, J. D. Kalen, U. Resch-Genger, J. Ivanic, O. T. Bruns and M. J. Schnermann, Targeted Multicolor In Vivo Imaging over 1000 nm Enabled by Nonamethine Cyanines, Nat. Methods, 2022, 19, 353–358 CrossRef CAS.
- M. P. Coogan and V. Fernandes-Moreira, Progress with, and Prospects for, Metal Complexes in Cell Imaging, Chem. Commun., 2014, 50, 384–399 RSC.
- S. Li, J. Wei, Q. Yao, X. Song, J. Xie and H. Yang, Emerging Ultrasmall Luminescent Nanoprobes for In Vivo Bioimaging, Chem. Soc. Rev., 2023, 52, 1672–1696 RSC.
- M. J. Schnermann, Organic Dyes for Deep Bioimaging, Nature, 2017, 551, 176–177 CrossRef CAS.
- A. Waggoner, Fluorescent labels for proteomics and genomics, Curr. Opin. Chem. Biol., 2006, 10, 62–66 CrossRef CAS.
- B. A. Armitage, Cyanine Dye-DNA Interactions: Intercalation, Groove Binding, and Aggregation, Top. Curr. Chem., 2005, 253, 55–76 CrossRef CAS.
- M. Levitus and S. Ranjit, Cyanine Dyes in Biophysical Research: The Photophysics of Polymethine Fluorescent Dyes in Biomolecular Environments, Q. Rev. Biophys., 2011, 44, 123–151 CrossRef CAS.
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Cyanine Polyene Reactivity: Scope and Biomedical Applications, Org. Biomol. Chem., 2015, 13, 7584–7598 RSC.
- A. S. Tatikolov, Polymethine Dyes as Spectral-Fluorescent Probes for Biomacromolecules, J. Photochem. Photobiol., C, 2012, 13, 55–90 CrossRef CAS.
- J. L. Bricks, A. D. Kachkovskii, Y. L. Slominskii, A. O. Gerasov and S. V. Popov, Molecular Design of Near Infrared Polymethine Dyes: A Review, Dyes Pigm., 2015, 121, 238–255 CrossRef CAS.
- J. Fabian, H. Nakazumi and M. Matsuoka, Near-Infrared Absorbing Dyes, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. P. Behera, Cyanines during the 1990's: A Review, Chem. Rev., 2000, 100, 1973–2011 CrossRef CAS.
- N. Tyutyulkov, J. Fabian, A. Melhorn, F. Dietz and A. Tadjer, Polymethine Dyes: Structure and Properties, St. Kliment Ohridski University Press, Bulgaria, 1991, pp. 1–250 Search PubMed.
- K. Jia, Y. Wan, A. Xia, S. Li., F. Gong and G. Yang, Characterization of Photoinduced Isomerization and Intersystem Crossing of the Cyanine Dye Cy3, J. Phys. Chem. A, 2007, 111, 1593–1597 CrossRef CAS.
- Z. Huang, D. Ji, S. Wang, A. Xia, F. Koberling, M. Patting and R. Erdmann, Spectral Identification of Specific Photophysics of Cy5 by Means of Ensemble and Single Molecule Measurements, J. Phys. Chem. A, 2006, 110, 45–50 CrossRef CAS.
- J. Widengren and P. Schwille, Characterization of Photoinduced Isomerization and Back-Isomerization of the Cyanine Dye Cy5 by Fluorescence Correlation Spectroscopy, J. Phys. Chem. A, 2000, 104, 6416–6428 CrossRef CAS.
- J. A. Carr, D. Franke, J. R. Caram, C. F. Perkinson, M. Saif, V. Askoxylakis, M. Datta, D. Fukumura, M. G. Jain, M. G. Bawendi and O. T. Bruns, Shortwave Infrared Fluorescence Imaging with the Clinically Approved Near-Infrared Dye Indocyanine Green, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4465–4470 CrossRef CAS.
- L. Štacková, E. Muchová, M. Russo, P. Slavíček, P. Štacko and P. Klán, Deciphering the Structure-Property Relations in Substituted Heptamethine Cyanines, J. Org. Chem., 2020, 85, 9776–9790 CrossRef.
- Q. Zheng, S. Jockusch, Z. Zhou, R. B. Altman, J. D. Warren, N. J. Turro and S. C. Blanchard, On the Mechanisms of Cyanine Fluorophore Photostablization, J. Phys. Chem. Lett., 2012, 3, 2200–2203 CrossRef CAS.
- S. S. Matikonda, G. Hammersley, N. Kumari, L. Grabenhorst, V. Glembockyte, P. Tinnefeld, J. Ivanic, M. Levitus and M. J. Schnermann, Impact of Cyanine Conformational Restraint in the Near-Infrared Range, J. Org. Chem., 2020, 85, 5907–5915 CrossRef CAS.
- A. M. Kolesnikov and F. A. Mikhailenko, The Conformations of Polymethine Dyes, Russ. Chem. Rev., 1987, 56, 275–287 CrossRef.
- E. D. Cosco, J. R. Caram, O. T. Bruns, D. Franke, R. A. Day, E. P. Farr, M. G. Bawendi and E. M. Sletten, Flavylium Polymethine Fluorophores for Near- and Shortwave Infrared Imaging, Angew. Chem., Int. Ed., 2017, 56, 13126–13129 CrossRef CAS.
- E. D. Cosco, B. A. Arús, A. L. Spearman, T. L. Atallah, I. Lim, O. S. Leland, J. R. Caram, T. S. Bischof, O. T. Bruns and E. M. Sletten, Bright Chromenylium Polymethine Dyes Enable Fast, Four-Color In Vivo Imaging with Shortwave Infrared Detection, J. Am. Chem. Soc., 2021, 143, 6836–6846 CrossRef CAS.
- M. Kasha, Characterization of Electronic Transitions in Complex Molecules, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- C. Rullière, Laser Action and Photoisomerisation of 3,3′-Diethyl Oxadicarbocyanine iodide (DODCI): Influence of Temperature and Concentration, Chem. Phys. Lett., 1976, 43, 303–308 CrossRef.
- F. Momicchioli, I. Baraldi and G. Berthier, Theoretical Study of Trans-Cis Photoisomerism in Polymethine Cyanines, Chem. Phys., 1988, 123, 103–122 CrossRef CAS.
- G. Ponterini and F. Momicchioli, Trans–Cis Photoisomerization Mechanisms of Carbocyanines: Experimental Check of Theoretical Models, Chem. Phys., 1991, 151, 111–126 CrossRef CAS.
- D. N. Dempster, T. Morrow, R. Rankin and G. F. Thompson, Photochemical Characteristics of Cyanine Dyes. Part 1. – 3,3′-Diethyloxadicarbocyanine Iodide and 3,3′-Diethylthiadicarbocyanine Iodide, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 1479–1496 RSC.
- V. A. Kuzmin and A. P. Darmanyan, Study of Sterically Hindered Short-Lived Isomers of Polymethine Dyes by Laser Photolysis, Chem. Phys. Lett., 1978, 54, 159–163 CrossRef CAS.
- J.-P. Fouassier, D.-J. Lougnot and J. Faure, Transient Absorption in a Polymethine Laser Dye, Chem. Phys. Lett., 1975, 35, 189–194 CrossRef CAS.
- J.-P. Fouassier, D.-J. Lougnot and J. Faure, Étude Photophysique D’une Série de Cyanines. I. – Processus D’évolution des États Excites, J. Chem. Phys., 1977, 74, 23–31 CAS.
- J.-P. Fouassier, D.-J. Lougnot and J. Faure, Photoisomerization Processes in the IR-140 Laser Dye, Opt. Commun., 1977, 23, 393–397 CrossRef CAS.
- D. J. Lougnot, P. Brunero, J.-P. Fouasier and J. Faure, Etude Physicochimique d’Une Serie de Cyanines. Partie IV: Rigidification, Photoisomerization et Effet Laser, J. Chem. Phys., 1982, 79, 343–349 CAS.
- B. Kopainsky, P. Qiu and W. Kaiser, Lifetime, Photostability, and Chemical Structure of IR Heptamethine Cyanine Dyes Absorbing Beyond 1 μm, Appl. Phys. B, 1982, 29, 15–18 CrossRef.
- W. Kranitzky, B. Kopainsky and W. Kaiser, A New Infrared Laser Dye of Superior Photostability Tunable to 1.24 μm with Picosecond Excitation, Opt. Commun., 1980, 36, 149–152 CrossRef.
- A. Sanchez-Galvez, P. Hunt, M. A. Robb, M. Olivucci, T. Vreven and H. B. Schlegel, Ultrafast Radiationless Deactivation of Organic Dyes: Evidence for a Two-State Two-Mode Pathway in Polymethine Cyanines, J. Am. Chem. Soc., 2000, 122, 2911–2924 CrossRef CAS.
- P. A. Hunt and M. A. Robb, Systematic Control of Photochemistry: The Dynamics of Photoisomerization of a Model Cyanine Dye, J. Am. Chem. Soc., 2005, 127, 5720–5726 CrossRef CAS.
- X. R. Zhu and J. M. Harris, Studies of Excited-State Absorption and Photoisomerization of Cyanine Dyes by Using Laser-Induced Anharmonic Thermal Gratings, Chem. Phys., 1990, 142, 301–309 CrossRef CAS.
- J. C. Mialocq, P. Goujon and M. Arvis, Etude par Spectroscopy Picoseconde et par Photolyse Eclair Conventionnelle de Polymethine-Cyanines, J. Chem. Phys., 1979, 76, 1067–1075 CAS.
- C. Carre, C. Reichardt and D. J. Lougnot, Étude Physicochimique d’Une Série de Cyanines: – Partie VII: Rendement en état Triplet et Photosensibilisation de l’oxygène Singulet, J. Chem. Phys., 1987, 84, 575–585 Search PubMed.
- F. Köhn, J. Hofkens, R. Gronheid, M. Van der Auweraer and F. C. De Schryver, Parameters Influencing the On- and Off-Times in the Fluorescence Intensity Traces of Single Cyanine Dye Molecules, J. Phys. Chem. A, 2002, 106, 4808–4814 CrossRef.
- A. K. Chibisov, G. V. Zakharova and H. Görner, Effects of Substituents in the Polymethine Chain on the Photoprocesses in Indodicarbocyanine Dyes, J. Chem. Soc., Faraday Trans., 1996, 92, 4917–4925 RSC.
- H. Gratz, A. Penzkofer, C. Abels, R.-M. Szeimies, M. Landthaler and W. Bäumler, Photo-Isomerisation, Triplet Formation, and Photo-Degradation Dynamics of Indocyanine Green Solutions, J. Photochem. Photobiol., A, 1999, 128, 101–109 CrossRef CAS.
- A. T. Eske and K. R. Naqvi, Viscosity Dependence of the Fluorescence Lifetimes of Cryptocyanine, Pinacyanol and DDI, Chem. Phys. Lett., 1979, 63, 128–132 CrossRef CAS.
- A. K. Chibisov, G. V. Zakharova, H. Görner, Y. A. Sogulyaev, I. L. Mushkalo and A. I. Tolmachev, Photorelaxation Processes in Covalently Linked Indocarbocyanine and Thiacarbocyanine Dyes, J. Phys. Chem., 1995, 99, 886–893 CrossRef CAS.
- P. F. Aramendía, R. M. Negri and E. S. Román, Temperature Dependence of Fluoresence and Photoisomerization in Symmetric Carbocyanines. Influence of Medium Viscosity and Molecular Structure, J. Phys. Chem., 1994, 98, 3165–3173 CrossRef.
- R. Engleman and J. Jortner, The Energy Gap Law for Radiationless Transitions in Large Molecules, Mol. Phys., 1980, 18, 145–164 CrossRef.
- W. Siebrand and D. F. Williams, Radiationless Transitions in Polyatomic Molecules. III. Anharmonicity, Isotope Effects, and Singlet-to-Ground-State Transitions in Aromatic Hydrocarbons, J. Chem. Phys., 1968, 49, 1860–1871 CrossRef CAS.
- U. Dinur and B. Scharf, Radiationless Transitions in Linear Polyenes, J. Chem. Phys., 1983, 79, 2600–2608 CrossRef CAS.
- V. Chynwat and H. A. Frank, The Application of the Energy Gap Law to the S1 Energies and Dynamics of Carotenoids, Chem. Phys., 1995, 194, 237–244 CrossRef CAS.
- M. Mimuro, S. Seiji Akimoto, S. Shinichi Takaichi and I. Yamazaki, Effect of Molecular Structures and Solvents on the Excited State Dynamics of the S2 State of Carotenoids Analyzed by the Femtosecond Up-Conversion Method, J. Am. Chem. Soc., 1997, 119, 1452–1453 CrossRef CAS.
- H. A. Frank, V. Chynwat, R. Z. B. Desamero, R. Farhoosh, J. Erickson and J. Bautista, On the Photophysics and Photochemical Properties of Carotenoids and their Role as Light-Harvesting Pigments in Photosynthesis, Pure Appl. Chem., 1997, 69, 2117–2124 CrossRef CAS.
- G. Orlandi, F. Zerbetto and M. Z. Zgierski, Theoretical Analysis of Spectra of Short Polyenes, Chem. Rev., 1991, 91, 867–891 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry. An Introduction, University Science Books, 2009, pp. 1–1084 Search PubMed.
- Y.-C. Wei, K.-H. Kuo, Y. Chi and P.-T. Chou, Efficient Near-Infrared Luminescence of Self-Assembled Platinum(II) Complexes: From Fundamentals to Applications, Acc. Chem. Res., 2023, 56, 689–699 CrossRef CAS.
- W. Siebrand, Radiationless Transitions in Polyatomic Molecules. I. Calculation of Franck–Condon Factors, J. Chem. Phys., 1967, 46, 440–447 CrossRef CAS.
- W. Siebrand, Radiationless Transitions in Polyatomic Molecules. II. Triplet-Ground-State Transitions in Aromatic Hydrocarbons, J. Chem. Phys., 1967, 47, 2411–2422 CrossRef CAS.
- H. C. Friedman, E. D. Cosco, T. L. Atallah, S. Jia, E. M. Sletten and J. R. Caram, Establishing Design Principles for Emissive Organic SWIR Chromophores from Energy Gap Laws, Chem, 2021, 7, 3359–3376 CAS.
- E. S. Medvedev and V. I. Osherov, Radiationless Transitions in Plyatomic Molecules, Springer-Verlag, Berlin Heidelberg, 1995, p. 88 Search PubMed.
- H. Mustroph and A. Towns, Fine Structure in Electronic Spectra of Cyanine Dyes: Are Sub-Bands Largely Determined by a Dominant Vibration or a Collection of Singly Excited Vibrations?, ChemPhysChem, 2018, 19, 1016–1023 CrossRef CAS.
- G. Angulo, G. Grampp and A. Rosspeintner, Recalling the Appropriate Representation of Electronic Spectra, Spectrochim. Acta, Part A, 2006, 65, 727–731 CrossRef.
- V. A. Gilbert and J. Baggott, Essentials of Molecular Photochemistry, Blackwell Scientific, Oxford, 1991, pp. 1–538 Search PubMed.
- S. J. Strickler and R. A. Berg, Relationship between Absorption Intensity and Fluorescence Lifetime of Molecules, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
- J. E. Saunders, C. Sanders, H. Chen and H.-P. Loock, Refractive Indices of Common Solvents and Solutions at 1550 nm, Appl. Opt., 2016, 55, 947–953 CrossRef CAS.
- X. Li, C. Wang, L. Ma and L. Liu, Ellipsometry-Transmission Measurement of the Complex Refractive Indices for a Series of Organic Solvents in the 200–1700 nm Spectral Range, Infrared Phys. Technol., 2022, 125, 104313 CrossRef CAS.
- C. H. Brito Cruz, J. P. Gordon, P. C. Becker, R. L. Fork and C. V. Shank, Dynamics of Spectral Hole Burning, IEEE J. Quantum. Electron., 1988, 24, 261–265 Search PubMed.
- B. Dietzek, A. Yartsev and A. N. Tarnovsky, Watching Ultrafast Barrierless Excited-State Isomerization of Pseudocyanine in Real Time, J. Phys. Chem. B, 2007, 111, 4520–4526 CrossRef CAS.
- S. A. Kovalenko, N. P. Ernsting and J. Ruthmann, Femtosecond Hole-Burning Spectroscopy of the Dye DCM in Solution: the Transition from the Locally Excited to a Charge-Transfer State, Chem. Phys. Lett., 1996, 258, 445–454 CrossRef CAS.
- S. A. Kovalenko, J. Ruthmann and N. P. Ernsting, Femtosecond Hole-Burning Spectroscopy with Stimulated Emission Pumping and Supercontinuum Probing, J. Chem. Phys., 1998, 109, 1894–1900 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Physical Mechanisms of Generation and Deactivation of Singlet Oxygen, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel Dekker, 1993, p. 290 Search PubMed.
- L. A. Shvedova, A. S. Tatikolov, S. M. Makin, N. N. Romanov and V. A. Kuzmin, Flash Photolysis of Tricarbocyanine Dyes, Russ. Chem. Bull., 1979, 28, 696–701 CrossRef.
- A. N. Tarnovsky, T. K. Razumova, E. P. Shchelkina and T. V. Veselova, Photophysical, Photochemical, and Lasing Characteristics of Symmetric and Asymmetric Di- and Tricarbocyanine Dyes, Opt. Spectrosc., 1993, 74, 65–78 Search PubMed [Opt. Spektrosk. (USSR), 1993, 74, 107–129].
- T. K. Razumova and A. N. Tarnovsky, Isomerization Processes in the Ground and First Excited States of 1-1’-Diethyl-2,2’-Dicarbocyanine Iodide (DDI), Opt. Spectrosc., 1995, 78, 56–63 Search PubMed [Opt. Spektrosk. (USSR), 1995, 78, 65–72].
- J. S. Baskin, H.-Z. Yu and A. H. Zewail, Ultrafast Dynamics of Porphyrins in the Condensed Phase: I. Free Base Tetraphenylporphyrin, J. Phys. Chem. A, 2002, 42, 9837–9844 CrossRef.
- S. A. Kovalenko, R. Schanz, H. Hennig and N. P. Ernsting, Cooling Dynamics of an Optically Excited Molecular Probe in Solution from Femtosecond Broadband Transient Absorption Spectroscopy, J. Chem. Phys., 2001, 115, 3256–3273 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- T. Elsaesser and W. Kaiser, Vibrational and Vibronic Relaxation of Large Polyatomic Molecules in Liquids, Annu. Rev. Phys. Chem., 1991, 42, 83–107 CrossRef CAS.
- E. L. Sibert III, S. G. Ramesh and T. S. Gulmen, Vibrational Relaxation of OH and CH Fundamentals of Polar and Nonpolar Molecules in the Condensed Phase, J. Phys. Chem. A, 2008, 112, 11291–11305 CrossRef.
- D. J. D. Miller, Vibrational Energy Relaxation and Structural Dynamics of Heme Proteins, Annu. Rev. Phys. Chem., 1991, 42, 581–614 CrossRef.
- R. I. Personov, Site Selection Spectroscopy of Complex Molecules in Solutions and its Applications, in Spectroscopy and Excitation Dynamics of Condensed Molecular Systems, ed. V. M. Agranovich and R. M. Hochstrasser, 1983, ch. 10, pp. 555–619 Search PubMed.
- H. Murakami, S. Kinoshita, Y. Hirata, T. Okada and N. Mataga, Transient Hole-Burning and Time-Resolved Fluorescence Spectra of Dye Molecules in Solution: Evidence for Ground-state Relaxation and Hole-filling Effect, J. Chem. Phys., 1992, 97, 7881–7888 CrossRef CAS.
- S. Kinoshita, H. Itoh, H. Murakami, H. Miyasaka, T. Okada and N. Mataga, Solvent Relaxation Effect on Transient Hole-Burning Spectra of Organic Dyes, Chem. Phys. Lett., 1990, 166, 123–127 CrossRef CAS.
- S. Kinoshita, Theory of Transient Hole-burning Spectrum of Molecules in Solution, J. Chem. Phys., 1989, 91, 5175–5183 CrossRef CAS.
- K. Nishiyama and T. Okada, Relaxation of Inhomogeneous Spectral Band Width of Dye Molecules in Polar Solvents Studied by Time-Resolved Hole and Fluorescence Spectroscopy, J. Phys. Chem. A, 1997, 101, 5729–5735 CrossRef CAS.
- K. Nishiyama and T. Okada, Relaxation Dynamics of Inhomogeneous Spectral Width in Binary Solvents Studied by Transient Hole-Burning Spectroscopy, J. Phys. Chem. A, 1998, 102, 9729–9733 CrossRef CAS.
- H. Shirota and E. W. Castner Jr., Molecular Dynamics and Interactions of Aqueous and Dichloromethane Solutions of Polyvinylpyrrolidone, J. Chem. Phys., 2006, 125, 034904 CrossRef.
- B. Dietzek, B. Brüggemann, P. Persson and A. Yartsev, On the Excited-state Multi-Dimensionality in Cyanines, Chem. Phys. Lett., 2008, 455, 13–19 CrossRef CAS.
- I. Renge, Mechanisms of Solvent Shifts, Pressure Shifts, and Inhomogeneous Broadening of the Optical Spectra of Dyes in Liquids and Low-Temperature Glasses, J. Phys. Chem. A, 2000, 104, 7452–7463 CrossRef CAS.
- I. Renge and U. P. Wild, Solvent, Temperature, and Excitonic Effects in the Optical Spectra of Pseudoisocyanine Monomer and J-Aggregates, J. Phys. Chem. A, 1997, 101, 7977–7988 CrossRef CAS.
- B. Dietzek, A. N. Tarnovsky and A. Yartsev, Visualizing Overdamped Wavepacket Motion: Excited-state Isomerization of Pseudocyanine in Viscous Solvents, Chem. Phys., 2009, 357, 54–62 CrossRef CAS.
- C. A. Guarin, J. P. Villabona-Monsalve, R. López-Arteaga and J. Peon, Dynamics of the Higher Lying Excited States of Cyanine Dyes. An Ultrafast Fluorescence Study, J. Phys. Chem. B, 2013, 117, 7352–7362 CrossRef CAS.
- I. Martini and G. V. Hartland, Ultrafast Investigation of Vibrational Relaxation in the S1 Electronic State of HITC, J. Phys. Chem., 1996, 100, 19764–19770 CrossRef CAS.
- I. Martini and G. V. Hartland, Relaxation Dynamics in the First Excited Singlet State of a Cyanine Dye: HITC, Chem. Phys. Lett., 1996, 258, 180–186 CrossRef CAS.
- C. T. Middleton, B. Cohen and B. Kohler, Solvent and Solvent Isotope Effects on the Vibrational Cooling Dynamics of a DNA Base Derivative, J. Phys. Chem. A, 2007, 111, 10460–10467 CrossRef CAS.
- T.-S. Yang, M.-S. Chang, R. Chang, M. Hayashi, S. H. Lin, P. Vöhringer, W. Dietz and N. F. Scherer, Femtosecond Pump-probe Study of Molecular Vibronic Structures and Dynamics of a Cyanine Dye in Solution, J. Chem. Phys., 1999, 110, 12070–12081 CrossRef CAS.
- D. K. Das, K. Makhal and D. Goswami, Observing Ground State Vibrational Coherence and Excited State Relaxation Dynamics of a Cyanine Dye in Pure Solvents, Phys. Chem. Chem. Phys., 2018, 20, 13400–13411 RSC.
- J. M. Jean and G. R. Fleming, Competition between Energy and Phase Relaxation in Electronic Curve Crossing Processes, J. Chem. Phys., 1995, 103, 2092–2101 CrossRef CAS.
- T. Fuji, H. J. Ong and T. Kobayashi, Real-time Observation of Vibrational Coherence Persisting after Internal Conversion and Vibrational Relaxation in Cyanine Dye Molecules, Chem. Phys. Lett., 2003, 380, 135–140 CrossRef CAS.
- F. W. Wise, M. J. Rosker and C. L. Tang, Oscillatory Femtosecond Relaxation of Photoexcited Organic Molecules, J. Chem. Phys., 1987, 86, 2827–2832 CrossRef CAS.
- A. Yu, C. A. Tolbert, D. A. Farrow and D. M. Jonas, Solvatochromism and Solvation Dynamics of Structurally Related Cyanine Dyes, J. Phys. Chem. A, 2002, 106, 9407–9419 CrossRef.
- J.-C. Gumy, O. Nicolet and E. Vauthey, Investigation of the Solvation Dynamics of an Organic Dye in Polar Solvents using the Femtosecond Transient Grating Technique, J. Phys. Chem. A, 1999, 103, 10737–10743 CrossRef CAS.
- M. M. Bishop, J. D. Roscioli, S. Ghosh, J. J. Mueller, N. C. Shepherd and W. F. Beck, Vibrationally Coherent Preparation of the Transition State for Photoisomerization of the Cyanine Dye Cy5 in Water, J. Phys. Chem. B, 2015, 119, 6905–6915 CrossRef CAS.
- S. M. Hart, J. L. Banal, M. Bathe and G. S. Schlau-Cohen, Identification of Nonradiative Decay Pathways in Cy3, J. Phys. Chem. Lett., 2020, 11, 5000–5007 CrossRef CAS.
- Z. Wei, T. Nakamura, S. Takeuchi and T. Tahara, Tracking of the Nuclear Wavepacket Motion in Cyanine Photoisomerization by Ultrafast Pump-Dump-Probe Spectroscopy, J. Am. Chem. Soc., 2011, 133, 8205–8210 CrossRef CAS.
- C. J. Bardeen, Q. Wang and C. V. Shank, Femtosecond Chirped Pulse Excitation of Vibrational Wave Packets in LD690 and Bacteriorhodopsin, J. Phys. Chem. A, 1998, 102, 2759–2766 CrossRef CAS.
- T. Teramoto and T. Kobayashi, Multiple Mode Coupling in Cy3 Molecules by Impulsive Coherent Vibrational Spectroscopy Using a Few-Cycle Laser Pulse, Phys. Chem. Chem. Phys., 2010, 12, 13515–13518 RSC.
- B. Dietzek, T. Pascher and A. Yartsev, Tracking Ultrafast Excited-State Bond-Twisting Motion in Solution Close to the Franck-Condon Point, J. Phys. Chem. B, 2007, 111, 6034–6041 CrossRef.
- B. Dietzek, B. Brüggemann, T. Pascher and A. Yartsev, Pump-Shaped Dump Optimal Control Reveals the Nuclear Reaction Pathway of Isomerization of a Photoexcited Cyanine Dye, J. Am. Chem. Soc., 2007, 129, 13014–13021 CrossRef CAS.
- B. Dietzek, B. Brüggemann, T. Pascher and A. Yartsev, Mechanisms of Molecular Response in the Optimal Control of Photoisomerization, Phys. Rev. Lett., 2006, 97, 258301(4) CrossRef.
- D. H. Waldeck, Photoisomerization Dynamics of Stilbenes, Chem. Rev., 1991, 91, 415–436 CrossRef CAS.
- G. R. Fleming, Chemical Applications of Ultrafast Spectroscopy, Oxford University Press, 1986, p. 186 Search PubMed.
- P. Hänggi, P. Talkner and M. Borkovec, Reaction- Rate Theory: Fifty Years after Kramers, Rev. Mod. Phys., 1990, 62, 251–341 CrossRef.
- S. P. Velsko, D. H. Waldeck and G. R. Fleming, Breakdown of Kramers Theory Description of Photochemical Isomerization and the Possible Involvement of Frequency Dependent Friction, J. Chem. Phys., 1983, 78, 249–258 CrossRef CAS.
- K. Hara and S. Akimoto, High-Pressure Study of the Viscosity Effect on the Ground-State Isomerization of 3,3′-Diethyloxadicarbocyanine Iodide (DODCI), J. Phys. Chem., 1991, 95, 5811–5814 CrossRef CAS.
- V. Sundström and T. Gillbro, Effects of Solvent on TMP Photophysics. Transition from no Barrier to Barrier Case, Induced by Solvent Properties, J. Chem. Phys., 1984, 81, 3463–3474 CrossRef.
- E. Åkesson, V. Sundström and T. Gillbro, Solvent-Dependent Barrier Heights of Excited-State Photoisomerization Reactions, Chem. Phys. Lett., 1985, 121, 513–522 CrossRef.
- G. Ponterini and M. Caselli, Photoisomerization Dynamics of 3,3′-Diethyl Oxacarbocyanine. Intramolecular and Solvent Viscosity Effects, Ber. Bunsenges. Physik. Chem., 1992, 96, 564–573 CrossRef CAS.
- J. Korppi-Tommola, A. Hakkarainen, T. Hukka and J. Subbi, An Isomerization Reaction of a Cyanine Dye in n-Alcohols: Microscopic Friction and an Excited-State Barrier Crossing, J. Phys. Chem. A, 1991, 95, 8482–8491 CrossRef CAS.
- N. Hirai and H. Henry Eyring, Bulk Viscosities of Liquids, J. Appl. Phys., 1958, 29, 810–816 CrossRef CAS.
- D. R. Lide, RC Handbook of Chemistry and Physics, CRC Press LLC, Boca Raton, 90th edn, 1990, pp. 6–209 Search PubMed.
- H. C. Friedman, E. D. Cosco, T. L. Atallah, S. Jia, E. M. Sletten and J. R. Caram, Establishing Design Principles for Emissive Organic SWIR Chromophores from Energy Gap Laws, Cell, 2021, 7, 3359–3376 CAS.
- E. Åkesson, A. Hakkarainen, E. Laitinen, V. Helenius, T. Gillbro, J. Korppi-Tommola and V. Sundström, Analysis of Microviscosity and Reaction Coordinate Concepts in Isomerization Dynamics Described by Kramers’ Theory, J. Chem. Phys., 1991, 95, 6508–6523 CrossRef.
- S. Murphy and G. B. Schuster, Electronic Relaxation in a Series of Cyanine Dyes: Evidence for Electronic and Steric Control of the Rotational Rate, J. Phys. Chem., 1995, 99, 8516–8518 CrossRef CAS.
- D. Bingemann and N. P. Ernsting, Femtosecond Solvation Dynamics Determining the Band Shape of Stimulated Emission from a Polar Styryl Dye, J. Chem. Phys., 1995, 102, 2691–2700 CrossRef CAS.
- S. H. Ashworth, T. Hasche, M. Woerner, E. Riedle and T. Elsaesser, Vibronic Excitations of Large Molecules in Solution Studied by Two-Color Pump–Probe Experiments on the 20 fs Time Scale, J. Chem. Phys., 1996, 104, 5761–5769 CrossRef CAS.
- Y. H. Meyer, M. Pittman and P. Plaza, Transient Absorption of Symmetrical Carbocyanines, J. Photochem. Photobiol., A, 1998, 114, 1–21 CrossRef CAS.
- S. A. Kovalenko, J. Ruthmann and N. P. Ernsting, Ultrafast Stokes Shift and Excited-State Transient Absorption of Coumarin 153 in Solution, Chem. Phys. Lett., 1997, 271, 40–50 CrossRef CAS.
- A. Müller, J. Schulz-Hennig and H. Tashiro, Excited State Absorption of 1,3,3,1′,3′,3′-Hexamethylindotricarbocyanine Iodide: A Quantitative Study by Ultrafast Absorption Spectroscopy, Appl. Phys., 1977, 12, 333–339 Search PubMed.
- R. S. Lepkowicz, O. V. Przhonska, J. M. Hales, D. J. Hagan, E. W. Van Stryland, M. V. Bondar, Y. L. Slominsky and A. D. Kachkovski, Excited-State Absorption Dynamics in Polymethine Dyes Detected by Polarization-Resolved Pump-Probe Measurements, Chem. Phys., 2003, 286, 277–291 CrossRef CAS.
- E. Sandberg, J. Piguet, U. Kostiv, G. Baryshnikov, H. Liu and J. Widengren, Photoisomerization of Heptamethine Cyanine Dyes Results in Red-Emissive Species: Implications for Near-IR, Single-Molecule, and Super-Resolution Fluorescence Spectroscopy and Imaging, J. Phys. Chem. B, 2023, 127, 3208–3222 CrossRef CAS.
- E. Sandberg, J. Piguet, H. Liu and J. Widengren, Combined Fluorescence Fluctuation and Spectrofluorometric Measurements Reveal a Red-Shifted, Near-IR Emissive Photo-Isomerized Form of Cyanine 5, Int. J. Mol. Sci., 2023, 24, 1990 CrossRef CAS.
- K. Furuta, M. Fuyuki and A. Wada, Multiphoton Reaction of DTTCI Observed by Femtosecond Pump-probe and Two-pulse Correlation Measurements, Chem. Phys., 2013, 418, 42–46 CrossRef CAS.
- M. Fuyuki, K. Furuta and A. Wada, New Photo-Isomerization Path of Indocyanine Green in Condensed Phase Investigated by Two-Pump Excitation, Chem. Phys. Lett., 2010, 499, 121–125 CrossRef CAS.
- R. W. Redmond, I. E. Kochevar, M. Kreig, G. Smith and W. G. J. McGimpsey, Excited State Relaxation in Cyanine Dyes: A Remarkably Efficient Reverse Intersystem Crossing from Upper Triplet Levels, J. Phys. Chem. A, 1997, 101, 2773–2777 CrossRef CAS.
- E. M. C. Hillman, Optical Brain Imaging In Vivo: Techniques and Applications from Animal to Man, J. Biomed. Opt., 2007, 12, 1–28 CrossRef.
- Y. Yagci, S. Jockusch and N. J. Turro, Mechanism of Photoinduced Step Polymerization of Thiophene by Onium Salts: Reactions of Phenyliodinium and Diphenylsulfinium Radical Cations with Thiophene, Macromolecules, 2007, 40, 4481–4485 CrossRef CAS.
- S. K. Pal, A. S. Mereshchenko, E. V. Butaeva, P. Z. El-Khoury and A. N. Tarnovsky, Global Sampling of the Photochemical Reaction paths of Bromoform by Ultrafast Deep-UV through Near-IR Transient Absorption and ab initio Multiconfigurational Calculations, J. Chem. Phys., 2013, 138, 124501 CrossRef CAS.
- F. T. Gemeda, V. Vorobyev and A. N. Tarnovsky, Ultrafast Solution-Phase Photophysical and Photochemical Dynamics of Hexaiodobismuthate(III), the Heart of Bismuth Halide Perovskite Solar Cells, J. Phys. Chem. B, 2022, 126, 1254–1267 CrossRef CAS.
- S. A. Kovalenko, A. L. Dobryakov, J. Ruthmann and N. P. Ernsting, Femtosecond Spectroscopy of Condensed Phases with Chirped Supercontinuum Probing, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 2369–2384 CrossRef CAS.
- K. Ekvall, P. van der Meulen, C. Dhollande, L.-E. Berg, S. Pommeret, R. Naskreski and J.-C. Mialocq, Cross Phase Modulation Artifact in Liquid Phase Transient Absorption Spectroscopy, J. Appl. Phys., 2000, 87, 2340–2352 CrossRef CAS.
- S. A. Kovalenko, N. P. Ernsting and J. Ruthmann, Femtosecond Hole-Burning Spectroscopy of the Dye DCM in Solution: the Transition from the Locally Excited to a Charge-Transfer State, Chem. Phys. Lett., 1996, 258, 445–454 CrossRef CAS.
- H. J. Marrinan and N. Sheppard, Relative Intensities of the Raman Lines of Carbon Tetrachloride, Chloroform, and Methylene Chloride, J. Opt. Soc. Am., 1954, 44, 815–819 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01411a |
| This journal is © the Owner Societies 2024 |