
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial carbonyl groups of propylene carbonate facilitate the reversible binding of nitrogen dioxide†
Jessica B.
Clark
 and
Heather C.
Allen
*
and
Heather C.
Allen
*
Department of Chemistry & Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: allen@chemistry.ohio-state.edu
First published on 16th May 2024
Abstract
The interaction of NO2 with organic interfaces is critical in the development of NO2 sensing and trapping technologies, and equally so to the atmospheric processing of marine and continental aerosol. Recent studies point to the importance of surface oxygen groups in these systems, however the role of specific functional groups on the microscopic level has yet to be fully established. In the present study, we aim to provide fundamental information on the interaction and potential binding of NO2 at atmospherically relevant organic interfaces that may also help inform innovation in NO2 sensing and trapping development. We then present an investigation into the structural changes induced by NO2 at the surface of propylene carbonate (PC), an environmentally relevant carbonate ester. Surface-sensitive vibrational spectra of the PC liquid surface are acquired before, during, and after exposure to NO2 using infrared reflection–absorption spectroscopy (IRRAS). Analysis of vibrational changes at the liquid surface reveal that NO2 preferentially interacts with the carbonyl of PC at the interface, forming a distribution of binding symmetries. At low ppm levels, NO2 saturates the PC surface within 10 minutes and the perturbations to the surface are constant over time during the flow of NO2. Upon removal of NO2 flow, and under atmospheric pressures, these interactions are reversible, and the liquid surface structure of PC recovers completely within 30 min.
Introduction
Nitrogen dioxide (NO2) is an anthropogenic atmospheric pollutant that is ubiquitous in urban environments. Due to rapid interconversion with nitrogen monoxide (NO) in the atmosphere, these pollutants are collectively referred to as NOx (NOx = NO + NO2). In the presence of carbon monoxide and/or volatile organic compounds, NOx provides the only known anthropogenic reaction pathway for the production of tropospheric ozone (O3).1,2 Gas-phase reactions of NO2 in the atmosphere have been well studied and are known to have important implications for overall air pollution and climate.1,2 However, consideration of these gas-phase chemical processes alone has led to inaccuracies in atmospheric modeling and predictions such as in the over prediction of HNO3 concentrations.3,4 Such discrepancies have been shown to result, at least in part, from the critical role that NO2 plays in the heterogeneous chemistry occurring at the surface of aerosol particles.4–12Heterogeneous chemistry involving aerosol particles can dramatically impact aerosol properties including their morphology, ability to act as cloud condensation nuclei, and radiation scattering efficiency.2 The complexity of aerosol reactivity and dynamics makes these systems difficult to study experimentally, thus the breadth of these reactions and the mechanisms governing them remain an area of great research interest. In a 2021 study, Abbatt and Liu showed that the formation of sulfate aerosol particles is accelerated under polluted conditions due to the oxidation of SO2 by NO2 at aqueous aerosol interfaces.8 Interfacial reactions of NO2 also provide a pathway for reservoir species of highly-reactive radicals such as OH and Cl to be formed. For example, the reaction of NO2 with chloride salts in marine aerosol and at the air/sea interface forms the Cl reservoir species, NOCl.10,13 Interfacial trapping of NO2 at air/water interfaces has also been identified as an important step in the formation of HONO, a reservoir of OH radical.9
NO2 not only plays a role in the heterogeneous chemistry of aqueous aerosol but also impacts the formation and composition of organic aerosol.14–19 Organic aerosol comprises a significant portion (20–90%) of all submicron aerosol in the troposphere.20–22 This class of aerosol can either be directly emitted into the atmosphere as primary organic aerosol or formed through the oxidation of volatile organic compounds to become secondary organic aerosol (SOA). Recent studies have shown that the formation and composition of SOA can depend strongly on NOx conditions.14–17,23 Shiraiwa and coworkers found that α-pinene and naphthalene SOA generated under low NOx conditions favors the formation of OH radicals and superoxide. The formation of these species was significantly reduced under high NOx conditions where the formation of nitroaromatics and other organic nitrates were favored.15 NOx concentrations can also alter the mechanism of oxidation by which SOA is formed as demonstrated by Lin and coworkers who identified an epoxide intermediate in the formation of isoprene SOA under high NOx conditions.14
Another area that relies on the heterogeneous interfacial chemistry of NO2 is the development of technology to detect and remove NO2 from the atmosphere. Exposure to high levels of NO2 pollution have been linked to many negative health outcomes including lung cancer, heart problems, and asthma,24–27 therefore accurate detection and efficient removal of NO2 from the atmosphere is imperative. Materials including zeolites,28,29 activated carbon,30–33 silica,34 and metal–organic frameworks35 have shown promise in trapping NO2 from exhaust streams. Adsorption of NO2 on each of these substrates depends on both the physical structure and chemical moieties at their interfaces. Activated carbon interfaces provide a low-cost method that repurposes organic waste products like sawdust into NO2 sorbent material.30–32 Carbon surfaces can be functionalized to optimize the surface sites that will promote the greatest NO2 adsorption.32,33 Studies of NO2 adsorption on sawdust activated carbon surfaces linked an increase in NO2 adsorption capacity to an increase in surface oxygen groups.30,31 In the development of high-performance NO2 sensors, interactions of NO2 with surface oxygen groups have resulted in enhancement of sensing properties.36,37 One type of organic field-effect transistor based sensor utilizing poly(methyl methracrylate) and silk fibroin as the dielectric bilayer demonstrated superior sensing abilities compared to sensors with only poly(methyl methracrylate) as the dielectric. This performance enhancement was attributed to strong interactions of NO2 with OH groups on the silk fibroin component of the dielectric.37
From the studies summarized here, it is clear that the presence of oxygen-containing functional groups strongly impacts the heterogeneous chemistry of NO2 at a variety of interfaces. This research aims to provide new fundamental information on functional group-specific interfacial interactions of NO2 in environmentally relevant systems and provide information that has the potential to inform the development of new NO2 detection and removal technologies. For this study, we have selected propylene carbonate (PC) as the representative organic liquid phase. PC is a polar, aprotic carbonate ester that is commonly used as a solvent in electrochemical systems38–40 and in cosmetics.41 The chemical structure of PC is depicted in Fig. 1. Due to its prevalence in commercial products, PC is expected to have a significant environmental fate in the condensed phase and in the atmosphere.41,42 Previous studies have shown that PC is mainly lost in the atmosphere through reaction with radical species such as OH and Cl.41,43 In the condensed phase, PC has been used as a proxy for liquid–liquid phase separated aerosol where it was shown to facilitate significant partitioning of HNO3 into the organic phase.18 In the present work, PC also serves as an atmospheric-proxy compound. The ester functional group of PC is used as a probe to interrogate possible interaction with NO2.
 | ||
| Fig. 1 Chemical structure of propylene carbonate (PC). | ||
The main goal of this study is to further the understanding of the interactions of NO2 at organic interfaces with oxygen-containing functional groups. To this end, the surface structure of PC, an environmentally relevant liquid, is characterized as a function of time before, during, and after exposure to NO2. Infrared reflection–absorption spectroscopy (IRRAS) enables this characterization by probing vibrational changes that occur as a result of perturbations to the surface. From these studies, we determine the primary site of NO2 interaction with PC and evaluate the recovery of the surface structure in the absence of NO2.
Experimental
Infrared reflection–absorption spectroscopy
IRRAS has been previously employed by this lab to elucidate molecular changes at a variety of interfaces44–50 and is employed here with modifications to allow for studies involving NO2 gas at organic interfaces. Spectra were collected using a PerkinElmer Spectrum 3 FTIR spectrometer equipped with a custom-built mirror array within the sample compartment. In this setup, the unpolarized infrared light from the spectrometer is incident on the first of two gold mirrors (2 in. diameter), which is angled such that the incident light is directed onto the liquid surface at a 48° angle relative to the surface normal. The light that is specularly reflected off the liquid surface is collected with the second gold mirror, which directs the beam into the LN2 cooled HgCdTe (MCT) detector. Surface-sensitive infrared spectra were acquired by calculating reflectance-absorbance (RA) using eqn (1) as follows: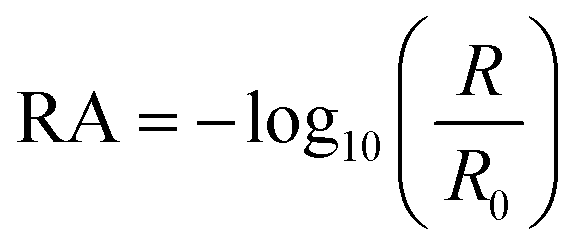 | (1) |
In this equation, R is the reflectance of the sample surface (PC during or after exposure to NO2) and R0 is the reflectance of the reference surface (PC before exposure to NO2). Through this process, the vibrational response of the liquid surface and bulk is effectively subtracted from the vibrational response of the sample after a perturbation to the surface. Both positive and negative peaks are observed in IRRAS spectra due to a convolution of the material's complex index of refraction and the nature of the subtraction of the reference. Generally, positive peaks occur when the reflectivity of the reference is greater than that of the sample, R/R0 < 1. Negative peaks occur when the sample reflectance is greater than that of the reference, thus R/R0 > 1. In the present study, spectra are complicated slightly due to the presence of the infrared-absorbing species NO2 in the gas-phase. This results in vibrational bands of bulk gas-phase NO2 that appear in the IRRAS spectra as positive peaks. IRRAS spectra in this study were taken in single-beam mode with a total 128 accumulations and 4 cm−1 resolution. All experiments were conducted in duplicate.
To study the interaction of NO2 with the surface of PC using IRRAS, a Teflon cell was custom built and used for all studies. The Teflon cell consists of a baseplate that holds the liquid sample and an angled top cover that is fit with optical windows as well as the gas inlet and outlet. The baseplate holds a ∼67 mm diameter glass Petri dish in which the liquid sample is placed. The top cover is constructed such that the two optical windows (37.5 × 4 mm ZnSe, Pike Technologies) sit at a 45° angle. This allows for the spectra to be collected in the desired reflection geometry as well as minimizes reflection losses. The optical windows and the body of the Teflon cell are sealed using perfluoroelastomer (FFKM) o-rings. Gas flows enter and exit through separate Teflon Swagelok connections on the top of the cell. The Teflon cell and the IRRAS assembly are depicted in Fig. 2.
 | ||
| Fig. 2 Schematic of the Teflon cell used for IRRAS experiments of NO2 flowing over the PC surface. Panel A depicts the outside of the Teflon cell with the IR beam from the spectrometer being directed into the cell through the ZnSe windows. In panel B, the inside of the cell is shown and includes the IR beam reflecting off the PC interface within the Petri dish. | ||
Gas flow experiments
Propylene carbonate (≥99.7%, anhydrous) was purchased from Sigma-Aldrich and was used as received. Due to the hygroscopic nature of PC, a single-use syringe was used to transfer all samples from the septum-sealed bottle. As a result, the water content can be reasonably assumed to be consistent with that reported by the manufacturer (≤0.002% by Karl Fischer titration).51 At the beginning of each experiment, 20 mL of PC are placed in the Petri dish within the Teflon cell. The cell is then sealed and purged with 50 mL min−1 N2 (Linde, ultra-high purity 99.999%) and 5 mL min−1 air (Linde, Zero Grade) for at least 18 hours to remove CO2 and limit water adsorbed to the walls of the cell. The flow rates of N2 and air are regulated using mass flow controllers (MKS Instruments, 1479A01511CS1BM), which flow the N2 and air through KOH (Sigma-Aldrich, ACS Reagent Grade) traps to remove excess water before flowing over the PC sample. The sample compartment of the FTIR is also sealed and purged with a separate flow of dry air (Parker Hannifin, 75-62 purge gas generator) to remove atmospheric CO2 and limit water adsorbed to the outside of the ZnSe windows. Following the initial 18 h purge, the N2 flow rate within the cell is increased to 100 mL min−1 and allowed to equilibrate for 30 minutes before initial spectra are acquired. An initial spectrum of PC with 100 mL min−1 N2 and 5 mL min−1 air flow is taken at the start of each trial and used as the reference (R0) to calculate RA for all subsequent spectra taken in the trial. Following this initial spectrum, spectra are taken every 10 min for at least 30 min while still flowing 100 mL min−1 N2 and 5 mL min−1 air. These spectra are used as controls for the PC surface before exposure to NO2. After the initial spectra are acquired, NO2 (Linde, 51 ppm in N2) is set to a flow rate of 40 mL min−1 using a third mass flow controller (MKS Instruments, 1479A01511CS1BM). The NO2 flows from the mass-flow controller through a nylon trap to remove HNO3 that forms from water contamination in the cylinder and/or adsorbed to the walls of the delivery line. The NO2 flow then joins the N2 and air flows before entering the Teflon cell and flowing over the PC surface. The concentration of NO2 that is delivered to the liquid surface is determined to be 5.1 ppm. Details of the NO2 concentration calculation can be found in the ESI† along with the FTIR spectrum of gas-phase NO2 (Fig. S1, ESI†). Spectra are acquired every 10 min after turning on the NO2 for a total elapsed time of 60 min at which point the NO2 flow is turned off while leaving the N2 and air flows unchanged. Spectra are then taken 5 minutes after the NO2 flow is turned off and then in 10 min intervals for a total of 30 minutes as the NO2 is purged from the gas cell. This set of spectra is referred to as “purge spectra” as they demonstrate changes occurring to the PC surface as the NO2 gas is purged from the system with N2 and air. A schematic of the gas flow system used in these experiments is depicted in Fig. 3. | ||
| Fig. 3 Schematic of gas lines used to purify and deliver gases to Teflon cell for spectroscopic measurements. | ||
Results and discussion
The interaction of NO2 with organic interfaces is known to play a critical role in the heterogeneous chemistry of the atmosphere as well as in the development of cutting-edge NO2 sensing and trapping technologies. The specific role that surface functional groups play in these processes remains elusive. Here, we present an investigation into the interaction of NO2 at the interface of the organic liquid propylene carbonate (PC), a species commonly used as representative atmospheric organic.18,41,43 Surface-sensitive IRRAS spectra were taken of the PC surface in three phases; before, during, and after exposure to NO2. All spectra were pre-processed by applying a Savitzky–Golay filter and fitting the spectra to a common baseline (see ESI† for details of the spectral pre-processing). In the initial phase, 100 mL min−1 N2 and 5 mL min−1 air were flowed over the PC surface and time-resolved spectra were acquired for up to 60 min before NO2 was introduced. The first spectrum acquired in this initial set of spectra was used as the reference (R0) to calculate RA for all subsequent spectra. The IRRAS spectrum of PC after 30 min elapsed in the initial phase is plotted as spectrum A in Fig. 4. No notable peaks are present in this spectrum, indicating that the flow of N2 and air does not change the vibrational modes of PC at the surface (see Fig. S2 in the ESI† for a plot of all initial spectra).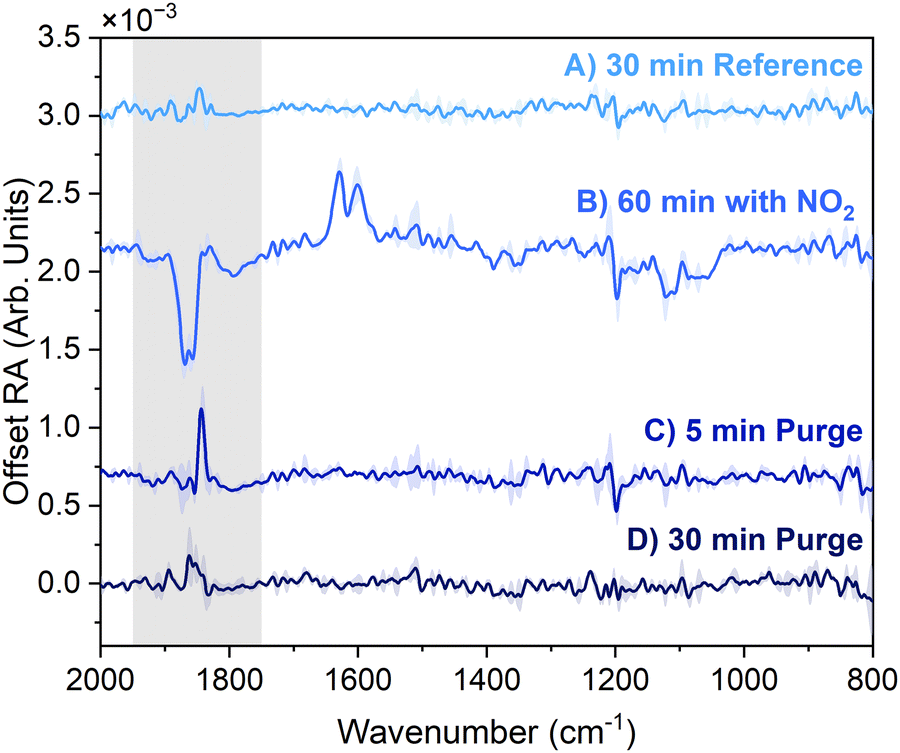 | ||
| Fig. 4 IRRAS spectra of the surface of PC before, during, and after exposure to 5.1 ppm NO2. (A) 30 min reference (B) 60 min with NO2 (C) 5 min of purging NO2 with N2 and air. (D) 30 min of purging NO2 with N2 and air. Spectra plotted are an average of two trials and the standard deviation is plotted as shading. Spectra are offset for clarity and the carbonyl region (1950–1750 cm−1) is highlighted with grey shading. | ||
In the second phase of the experiment, 40 mL min−1 of 50 ppm NO2 was allowed to mix with the initial flow to deliver 5.1 ppm NO2 to the PC surface. The spectrum of the PC surface with NO2 flowing for 60 min is plotted as spectrum B in Fig. 4. Comparison of spectrum A and spectrum B demonstrates that the flow of NO2 over the PC surface causes significant changes to the interfacial vibrational modes of PC. The vibrational mode of PC that is most strongly affected by the presence of NO2 is the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretch, as observed in the appearance of the intense negative band at ∼1860 cm−1 in spectrum B. Other peaks of PC are also affected, but to a lesser extent. These peaks occur in the low frequency region of the spectrum (<1500 cm−1) and are assigned to combinations of vibrations within the ring structure of PC, including O–C, C–H, and C–C modes.38,52 Assignment of the low frequency peaks observed in the IRRAS spectrum during NO2 flow can be found in the ESI† (see Fig. S3 and Table S1). The small changes observed in the low frequency modes are expected to be a result of the strong correlation of the modes within the ring structure. The asymmetric stretching band of gas-phase NO2 is observed in spectrum B as P and R rotational branches centered at 1600 and 1628 cm−1, respectively.53
O) stretch, as observed in the appearance of the intense negative band at ∼1860 cm−1 in spectrum B. Other peaks of PC are also affected, but to a lesser extent. These peaks occur in the low frequency region of the spectrum (<1500 cm−1) and are assigned to combinations of vibrations within the ring structure of PC, including O–C, C–H, and C–C modes.38,52 Assignment of the low frequency peaks observed in the IRRAS spectrum during NO2 flow can be found in the ESI† (see Fig. S3 and Table S1). The small changes observed in the low frequency modes are expected to be a result of the strong correlation of the modes within the ring structure. The asymmetric stretching band of gas-phase NO2 is observed in spectrum B as P and R rotational branches centered at 1600 and 1628 cm−1, respectively.53
The extent of perturbation to the carbonyl stretching mode when the surface of PC is exposed to NO2 points to a selective interaction of NO2 with this moiety. We have considered the possibility of this change resulting from a permanent reaction as well as from favorable non-covalent interactions between the dipoles of NO2 and PC. In an investigation of oleic acid monolayers exposed to 1000 ppm NO2, there was no evidence of bond cleavage or addition across the double bond. The major effect of NO2 on the monolayers was found to be an isomerization of the double bond.54 It is therefore unlikely for NO2 to cause a significant reaction with PC in the present study considering PC has similar functional groups and was exposed to a much lower NO2 concentration. Additionally, NO2 has been shown to have slight hydrophobic character at aqueous interfaces and tends to orient with the oxygen atoms pointed towards the air and the nitrogen toward the surface.9,55 Considering this orientation, we are confident that the spectral changes observed here are a result of non-covalent interactions, namely van der Waals and dipole–dipole interactions, between NO2 and the carbonyl of PC.
To determine if this perturbation to the carbonyl persists in the absence of NO2 flow, purge spectra were acquired. After flowing NO2 over the PC surface for 60 min, the flow was turned off and it was purged from the sample cell with the initial flow rates of N2 and air. The spectra of the PC surface after 5 and 30 min of purging are presented in Fig. 4 as spectrum C and D, respectively. After 30 min of purging, the surface of PC completely returns to its structure before exposure to NO2, providing evidence that the perturbations to the PC surface are reversible and the surface is not permanently altered. We then assert that the carbonyl of PC facilitates the reversible binding of NO2 at the interface.
We also observe that the spectral changes induced by NO2 at the PC surface are established within the first 5 min of exposure and are constant with time. Time-resolved spectra of the PC surface during the 60 min NO2 exposure are presented in Fig. 5a. The carbonyl band is present at the earliest time point (5 min) and does not change appreciably, in intensity or peak shape, for the duration of the exposure. The NO2 band intensity reaches an equilibrium after 10 minutes. Combined, these observations indicate that NO2 saturates the PC surface within 10 min and the surface structure remains constant as NO2 continues to flow. No peaks were observed that increased in intensity over time, providing additional evidence against the formation and subsequent build-up of a reaction product over time.
 | ||
| Fig. 5 Time-resolved IRRAS spectra of the PC surface during flow of 5.1 ppm NO2 (panel A). Comparison of IRRAS spectrum of PC surface exposed to NO2 for 60 min and the bulk ATR-FTIR spectrum of pure PC (panel B). All spectra are an average of two trials and the standard deviation is plotted as shading. ATR spectrum is scaled by 10−3 in order to plot on same scale as IRRAS spectrum. Dashed vertical reference lines are included every 200 cm−1 in panel B to aid in the comparison. | ||
To further elucidate the surface structure of PC caused by NO2 exposure, IRRAS spectra of PC at 60 min NO2 flow are compared to bulk ATR-FTIR spectra of PC in Fig. 5b (see ESI† for details of the ATR-FTIR experiment). In the low frequency “fingerprint” region (<1500 cm−1), the IRRAS and ATR peaks are well overlapped, supporting our previous assignment. The largest discrepancy between the bulk and interfacial spectra is in the carbonyl stretching band. In the ATR, which serves as the representative bulk PC spectrum, the carbonyl band is centered at 1780 cm−1. Assignment of this band is confirmed through comparison with previous reports of FTIR spectra of bulk PC.38,52,56 In the interfacial IRRAS spectra, this peak is significantly blue-shifted and occurs at ∼1860 cm−1. This shift is due to several factors including convolution of the real and imaginary components of the complex index of refraction. This phenomenon occurs in reflection spectra when a material has a very strong absorption band, which causes a change in the intensity of the light as well as a change in the reflectivity of the interface. The results of this phenomena are differences in peak position and intensity for the strongly affected bands in the reflection spectra (such as the carbonyl) compared to linear bulk methods.57 Binding interactions at the air/water interface have yielded similar spectral shifts due to perturbation of the solvation environment of interfacial carbonyl modes.48 For example, binding of arginine to the headgroup of 1,2-dipalmitoyl-sn-glycero-3-phosphatidic acid films induces dehydration of the carbonyl groups leading to a blue-shift of the carbonyl mode in the IRRAS spectra.48 It is therefore highly plausible that the blue-shift observed here is at least partially due to the disruption and subsequent rearrangement of the PC liquid structure induced by association of NO2 with the carbonyl.
Exposure of the PC surface to NO2 also causes the carbonyl band in the IRRAS spectra to take on a different peak shape in comparison to the bulk solution spectrum. In the ATR spectra that reports on the solution phase, the observed carbonyl band consists of a single, sharp peak centered at 1780 cm−1. A weaker Fermi resonance band causes a broadening of this peak on the high frequency side.52,56,58 In contrast, the carbonyl band observed in the IRRAS spectra during NO2 flow has a doublet peak structure, with peaks of nearly equal intensity centered at 1869 and 1857 cm−1. Similar splitting of carbonyl bands in FTIR spectra has been observed for systems in which the carbonyl experiences a distribution of interactions (e.g. hydrogen bonding and ion solvation) causing changes to the vibrational environment.59–61 Propylene carbonate exhibits a 15–40 cm−1 splitting of the carbonyl band as a result of solvating various ions.56,61 Brooksby and Fawcett show that the carbonyl band of pure PC is a single peak at 1795 cm−1 in the ATR-FTIR spectrum. Association of Li+ ions with the carbonyl in solution results in the splitting of the band into two peaks centered at 1770 and 1792 cm−1.56 We expect that the carbonyl doublet peak structure observed in these experiments is due to NO2 binding to the carbonyl in different symmetries. It is also possible that this splitting is due to two populations of carbonyl groups: one that is directly associated with NO2 and another that is not but has a disrupted solvation structure due to the presence of NO2. This doublet peak structure of the carbonyl band was also observed intermittently during control studies with diethyl sebacate (DES) as the liquid phase. In half of the studies conducted, DES exhibited a perturbation to only the carbonyl band, providing support to the conclusion that NO2 interacts in a carbonyl specific manner with ester functional groups. The results of the DES control studies can be found in the ESI† (see Fig. S4 and S5).
The flow of NO2 has been shown in previous works to repel water from the surface of 1,4-dioxane/water solutions due to the mildly hydrophobic nature of the gas.55 Here, we have taken careful measures to limit water contamination in the pure PC solutions and confirmed using Raman spectra that the OH stretch was below the limit of detection (see ESI† including Fig. S6). However, PC has also been shown to facilitate microscopic aggregation of water within the solvent,62,63 therefore there is a slight possibility of very small amounts of water contamination. We then consider the possibility that the spectral changes observed when the PC surface is exposed to NO2 are a result of dehydration. To test this possibility, the spectral changes resulting from flowing 100 mL min−1 N2 and 5 mL min−1 air over a 0.091 mole fraction (χwater) water in PC solution were investigated. Spectra were acquired in 10 min increments for 120 min. RA was calculated for every time point using the initial spectrum as R0 and the result is plotted in Fig. 6a. These spectra demonstrate changes to PC vibrational modes as water is removed from the surface due to the N2 and air flow. All PC modes undergo significant changes that increase in intensity over time. Comparison of the changes in the carbonyl band caused by exposure to NO2 and separately by dehydration (Fig. 6b), shows that NO2 causes a significantly different peak shape that is blue-shifted from the peak caused by dehydration. Thus, it is clear the changes to the PC interfacial structure when NO2 is present are not simply caused by dehydration. Exposure of the PC/water solution interface to NO2 was also studied and any changes that may have been caused by NO2 were obscured by the large changes caused by dehydration (see Fig. S7 in the ESI†). The results support our conclusions that the spectral changes induced by the presence of NO2 over the PC surface are a result of NO2 forming strong interactions with the carbonyl of PC.
 | ||
| Fig. 6 Time-resolved IRRAS spectra of a 0.091 mole fraction water/PC solution as N2 and air flows over the surface (panel A). Comparison of the PC spectral changes resulting from 90 min of dehydration of the water/PC solution interface (panel B, red) with those caused by 60 min NO2 flow at the pure PC surface (panel B, blue). All spectra are an average of two trials and the standard deviation is plotted as shading. The 60 min NO2 flow spectrum (panel B, blue) is scaled by 10 for comparison. The carbonyl region (1950–1750 cm−1) is highlighted with grey shading. | ||
Conclusions
In this work, we characterize the structural changes that occur as 5.1 ppm NO2 is flowed over the surface of propylene carbonate using surface-sensitive infrared reflection–absorption spectroscopy. IRRAS spectra reveal large changes to the interfacial vibrational modes of PC upon introduction of NO2. The carbonyl stretching mode of PC is most affected by this perturbation, resulting in an intense negative band with a doublet peak structure. Interfacial changes induced by PC were shown to be constant over time and NO2 was shown to saturate the PC surface within 10 min. The surface of PC recovers fully within 30 min of stopping the NO2 flow. These observations lead to the conclusion that the carbonyl of PC facilitates reversible non-covalent interactions with NO2 at the gas/liquid interface. Binding of NO2 at the surface induces a disruption of the PC liquid structure at the interface and establishes a distribution of binding symmetries.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge support from the National Science Foundation through grant No. CHE-2102313. The authors would like to thank Dr David Limmer for insightful discussions about the interpretation of experimental results through the theoretical lens. The authors would also like to thank Dr Narendra Adhikari for helpful advice in experimental troubleshooting.References
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, CA, 2000 Search PubMed.
- G. Ritchie, Atmospheric Chemistry: From The Surface To The Stratosphere, World Scientific Publishing, New Jersey, 2016 Search PubMed.
- R. B. Chatfield, Anomalous HNO3/NOx Ratio of Remote Tropospheric Air: Conversion of Nitric Acid to Formic Acid and NOx?, Geophys. Res. Lett., 1994, 21(24), 2705–2708, DOI:10.1029/94GL02659.
- G. M. Underwood, T. M. Miller and V. H. Grassian, Transmission FT-IR and Knudsen Cell Study of the Heterogeneous Reactivity of Gaseous Nitrogen Dioxide on Mineral Oxide Particles, J. Phys. Chem. A, 1999, 103(31), 6184–6190, DOI:10.1021/jp991586i.
- K. Stemmler, M. Ndour, Y. Elshorbany, J. Kleffmann, B. D’Anna, C. George, B. Bohn and M. Ammann, Light Induced Conversion of Nitrogen Dioxide into Nitrous Acid on Submicron Humic Acid Aerosol, Atmos. Chem. Phys., 2007, 7(16), 4237–4248, DOI:10.5194/acp-7-4237-2007.
- C. E. Kolb, R. A. Cox, J. P. D. Abbatt, M. Ammann, E. J. Davis, D. J. Donaldson, B. C. Garrett, C. George, P. T. Griffiths, D. R. Hanson, M. Kulmala, G. McFiggans, U. Pöschl, I. Riipinen, M. J. Rossi, Y. Rudich, P. E. Wagner, P. M. Winkler, D. R. Worsnop and C. D. O’Dowd, An Overview of Current Issues in the Uptake of Atmospheric Trace Gases by Aerosols and Clouds, Atmos. Chem. Phys., 2010, 10(21), 10561–10605, DOI:10.5194/acp-10-10561-2010.
- L. Li, M. R. Hoffmann and A. J. Colussi, Role of Nitrogen Dioxide in the Production of Sulfate during Chinese Haze-Aerosol Episodes, Environ. Sci. Technol., 2018, 52(5), 2686–2693, DOI:10.1021/acs.est.7b05222.
- T. Liu and J. P. D. Abbatt, Oxidation of Sulfur Dioxide by Nitrogen Dioxide Accelerated at the Interface of Deliquesced Aerosol Particles, Nat. Chem., 2021, 13(12), 1173–1177, DOI:10.1038/s41557-021-00777-0.
- G. Murdachaew, M. E. Varner, L. F. Phillips, B. J. Finlayson-Pitts and R. B. Gerber, Nitrogen Dioxide at the Air–Water Interface: Trapping, Absorption, and Solvation in the Bulk and at the Surface, Phys. Chem. Chem. Phys., 2012, 15(1), 204–212, 10.1039/C2CP42810E.
- D. D. Weis and G. E. Ewing, The Reaction of Nitrogen Dioxide with Sea Salt Aerosol, J. Phys. Chem. A, 1999, 103(25), 4865–4873, DOI:10.1021/jp984488q.
- L. F. Phillips, Atmospheric Reactions on Electrically Charged Surfaces, Phys. Chem. Chem. Phys., 2013, 15(26), 10749–10752, 10.1039/C3CP51171E.
- G. Rubasinghege and V. H. Grassian, Role(s) of Adsorbed Water in the Surface Chemistry of Environmental Interfaces, Chem. Commun., 2013, 49(30), 3071–3094, 10.1039/C3CC38872G.
- B. J. Finlayson-Pitts, Reaction of NO2 with NaCl and Atmospheric Implications of NOCl Formation, Nature, 1983, 306(5944), 676–677, DOI:10.1038/306676a0.
- Y.-H. Lin, H. Zhang, H. O. T. Pye, Z. Zhang, W. J. Marth, S. Park, M. Arashiro, T. Cui, S. H. Budisulistiorini, K. G. Sexton, W. Vizuete, Y. Xie, D. J. Luecken, I. R. Piletic, E. O. Edney, L. J. Bartolotti, A. Gold and J. D. Surratt, Epoxide as a Precursor to Secondary Organic Aerosol Formation from Isoprene Photooxidation in the Presence of Nitrogen Oxides, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(17), 6718–6723, DOI:10.1073/pnas.1221150110.
- K. C. Edwards, A. L. Klodt, T. Galeazzo, M. Schervish, J. Wei, T. Fang, N. M. Donahue, B. Aumont, S. A. Nizkorodov and M. Shiraiwa, Effects of Nitrogen Oxides on the Production of Reactive Oxygen Species and Environmentally Persistent Free Radicals from α-Pinene and Naphthalene Secondary Organic Aerosols, J. Phys. Chem. A, 2022, 126(40), 7361–7372, DOI:10.1021/acs.jpca.2c05532.
- X. Zang, Z. Zhang, Y. Zhao, G. Li, H. Xie, W. Zhang, G. Wu, X. Yang and L. Jiang, Effects of NO2 and SO2 on the Secondary Organic Aerosol Formation from β-Pinene Photooxidation, J. Environ. Sci., 2024, 136, 151–160, DOI:10.1016/j.jes.2022.10.040.
- P. J. Ziemann and R. Atkinson, Kinetics, Products, and Mechanisms of Secondary Organic Aerosol Formation, Chem. Soc. Rev., 2012, 41(19), 6582–6605, 10.1039/C2CS35122F.
- B. L. Deming and P. J. Ziemann, Measurements of the Partitioning of Nitric Acid and Sulfuric Acid in Aqueous/Organic Phase-Separated Systems, Environ. Sci.: Atmos., 2021, 1(2), 93–103, 10.1039/D0EA00003E.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Recent Advances in Understanding Secondary Organic Aerosol: Implications for Global Climate Forcing, Rev. Geophys., 2017, 55(2), 509–559, DOI:10.1002/2016RG000540.
- D. M. Murphy, D. J. Cziczo, K. D. Froyd, P. K. Hudson, B. M. Matthew, A. M. Middlebrook, R. E. Peltier, A. Sullivan, D. S. Thomson and R. J. Weber, Single-Particle Mass Spectrometry of Tropospheric Aerosol Particles, J. Geophys. Res.: Atmos., 2006, 111(D23), D23S32/1–D23S32/15, DOI:10.1029/2006JD007340.
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes, Geophys. Res. Lett., 2007, 34(13), L13801/1–L13801/6, DOI:10.1029/2007GL029979.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Organic Aerosol and Global Climate Modelling: A Review, Atmos. Chem. Phys., 2005, 5(4), 1053–1123, DOI:10.5194/acp-5-1053-2005.
- D. Zhao, S. H. Schmitt, M. Wang, I.-H. Acir, R. Tillmann, Z. Tan, A. Novelli, H. Fuchs, I. Pullinen, R. Wegener, F. Rohrer, J. Wildt, A. Kiendler-Scharr, A. Wahner and T. F. Mentel, Effects of NOx and SO2 on the Secondary Organic Aerosol Formation from Photooxidation of α-Pinene and Limonene, Atmos. Chem. Phys., 2018, 18(3), 1611–1628, DOI:10.5194/acp-18-1611-2018.
- G. B. Hamra, F. Laden, A. J. Cohen, O. Raaschou-Nielsen, M. Brauer and D. Loomis, Lung Cancer and Exposure to Nitrogen Dioxide and Traffic: A Systematic Review and Meta-Analysis, Environ. Health Perspect., 2015, 123(11), 1107–1112, DOI:10.1289/ehp.1408882.
- G. Favarato, H. R. Anderson, R. Atkinson, G. Fuller, I. Mills and H. Walton, Traffic-Related Pollution and Asthma Prevalence in Children. Quantification of Associations with Nitrogen Dioxide, Air Qual., Atmos. Health, 2014, 7(4), 459–466, DOI:10.1007/s11869-014-0265-8.
- A. Larkin, J. A. Geddes, R. V. Martin, Q. Xiao, Y. Liu, J. D. Marshall, M. Brauer and P. Hystad, Global Land Use Regression Model for Nitrogen Dioxide Air Pollution, Environ. Sci. Technol., 2017, 51(12), 6957–6964, DOI:10.1021/acs.est.7b01148.
- R. D. Brook, S. Rajagopalan, C. A. Pope, J. R. Brook, A. Bhatnagar, A. V. Diez-Roux, F. Holguin, Y. Hong, R. V. Luepker, M. A. Mittleman, A. Peters, D. Siscovick, S. C. Smith, L. Whitsel and J. D. Kaufman, Particulate Matter Air Pollution and Cardiovascular Disease, Circulation, 2010, 121(21), 2331–2378, DOI:10.1161/CIR.0b013e3181dbece1.
- J.-M. Goupil, J.-F. Hemidy and D. Cornet, Adsorption of NO2 on Modified Y Zeolites, Zeolites, 1982, 2(1), 47–50, DOI:10.1016/S0144-2449(82)80040-7.
- R. W. Triebe and F. H. Tezel, Adsorption of Nitrogen, Carbon Monoxide, Carbon Dioxide and Nitric Oxide on Molecular Sieves, Gas Sep. Purif., 1995, 9(4), 223–230, DOI:10.1016/0950-4214(95)00017-6.
- R. Pietrzak, Sawdust Pellets from Coniferous Species as Adsorbents for NO2 Removal, Bioresour. Technol., 2010, 101(3), 907–913, DOI:10.1016/j.biortech.2009.09.017.
- P. Nowicki and R. Pietrzak, Carbonaceous Adsorbents Prepared by Physical Activation of Pine Sawdust and Their Application for Removal of NO2 in Dry and Wet Conditions, Bioresour. Technol., 2010, 101(15), 5802–5807, DOI:10.1016/j.biortech.2010.02.098.
- M. Jeguirim, M. Belhachemi, L. Limousy and S. Bennici, Adsorption/Reduction of Nitrogen Dioxide on Activated Carbons: Textural Properties versus Surface Chemistry – A Review, Chem. Eng. J., 2018, 347, 493–504, DOI:10.1016/j.cej.2018.04.063.
- M. Belhachemi, M. Jeguirim, L. Limousy and F. Addoun, Comparison of NO2 Removal Using Date Pits Activated Carbon and Modified Commercialized Activated Carbon via Different Preparation Methods: Effect of Porosity and Surface Chemistry, Chem. Eng. J., 2014, 253, 121–129, DOI:10.1016/j.cej.2014.05.004.
- B. Levasseur, A. M. Ebrahim and T. J. Bandosz, Interactions of NO2 with Amine-Functionalized SBA-15: Effects of Synthesis Route, Langmuir, 2012, 28(13), 5703–5714, DOI:10.1021/la300371m.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Reversible Adsorption of Nitrogen Dioxide within a Robust Porous Metal–Organic Framework, Nat. Mater., 2018, 17(8), 691–696, DOI:10.1038/s41563-018-0104-7.
- Q. Ding, Z. Zhou, H. Wang, Z. Wu, K. Tao, B.-R. Yang, X. Xie, J. Fu and J. Wu, Self-Healable, Recyclable, Ultrastretchable, and High-Performance NO2 Sensors Based on an Organohydrogel for Room and Sub-Zero Temperature and Wireless Operation, SmartMat, 2023, 4(1), e1141, DOI:10.1002/smm2.1141.
- X. Li, W. Shi, X. Yu and J. Yu, Performance Improvement of Organic Field-Effect Transistor Based Nitrogen Dioxide Gas Sensor Using Biocompatible PMMA/Silk Fibroin Bilayer Dielectric, J. Mater. Sci.: Mater. Electron., 2015, 26(10), 7948–7954, DOI:10.1007/s10854-015-3448-7.
- D. Battisti, G. A. Nazri, B. Klassen and R. Aroca, Vibrational Studies of Lithium Perchlorate in Propylene Carbonate Solutions, J. Phys. Chem., 1993, 97(22), 5826–5830, DOI:10.1021/j100124a007.
- D. Aurbach and A. Zaban, Impedance Spectroscopy of Lithium Electrodes: Part 1. General Behavior in Propylene Carbonate Solutions and the Correlation to Surface Chemistry and Cycling Efficiency, J. Electroanal. Chem., 1993, 348(1), 155–179, DOI:10.1016/0022-0728(93)80129-6.
- K. Kondo, M. Sano, A. Hiwara, T. Omi, M. Fujita, A. Kuwae, M. Iida, K. Mogi and H. Yokoyama, Conductivity and Solvation of Li+ Ions of LiPF6 in Propylene Carbonate Solutions, J. Phys. Chem. B, 2000, 104(20), 5040–5044, DOI:10.1021/jp000142f.
- I. Barnes, P. Wiesen and M. Gallus, Kinetics and Mechanism of the OH-Radical and Cl-Atom Oxidation of Propylene Carbonate, RSC Adv., 2016, 6(100), 98234–98242, 10.1039/C6RA21952G.
- PubChem. Propylene Carbonate. https://pubchem.ncbi.nlm.nih.gov/compound/7924.
- T. V.-T. Mai, H. T. Nguyen and L. K. Huynh, Atmospheric Chemistry of the Reaction between Propylene Carbonate and OH Radical: An Ab Initio RRKM-Based Master Equation Study, Chem. Phys. Lett., 2020, 739, 137020, DOI:10.1016/j.cplett.2019.137020.
- M. G. Vazquez de Vasquez, K. A. Carter-Fenk, L. M. McCaslin, E. E. Beasley, J. B. Clark and H. C. Allen, Hydration and Hydrogen Bond Order of Octadecanoic Acid and Octadecanol Films on Water at 21 and 1 °C, J. Phys. Chem. A, 2021, 125(46), 10065–10078, DOI:10.1021/acs.jpca.1c06101.
- A. A. Enders, J. B. Clark, S. M. Elliott and H. C. Allen, New Insights into Cation- and Temperature-Driven Protein Adsorption to the Air–Water Interface through Infrared Reflection Studies of Bovine Serum Albumin, Langmuir, 2023, 39(15), 5505–5513, DOI:10.1021/acs.langmuir.3c00249.
- L. F. Voss, M. F. Bazerbashi, C. P. Beekman, C. M. Hadad and H. C. Allen, Oxidation of Oleic Acid at Air/Liquid Interfaces, J. Geophys. Res.: Atmos., 2007, 112(D6), D06209/1–D06209/9, DOI:10.1029/2006JD007677.
- N. C. Auvil, M. G. Vazquez de Vasquez and H. C. Allen, Zinc–Carboxylate Binding in Mixed Octadecanoic Acid and Octadecanol Monolayers on Proxy Seawater Solution Surfaces, ACS Earth Space Chem., 2021, 5(10), 2947–2956, DOI:10.1021/acsearthspacechem.1c00272.
- J. F. Neal, W. Zhao, A. J. Grooms, A. H. Flood and H. C. Allen, Arginine–Phosphate Recognition Enhanced in Phospholipid Monolayers at Aqueous Interfaces, J. Phys. Chem. C, 2018, 122(46), 26362–26371, DOI:10.1021/acs.jpcc.8b03531.
- M. Xu, R. Spinney and H. C. Allen, Water Structure at the Air−Aqueous Interface of Divalent Cation and Nitrate Solutions, J. Phys. Chem. B, 2009, 113(13), 4102–4110, DOI:10.1021/jp806565a.
- M. R. Sierra-Hernández and H. C. Allen, Incorporation and Exclusion of Long Chain Alkyl Halides in Fatty Acid Monolayers at the Air−Water Interface, Langmuir, 2010, 26(24), 18806–18816, DOI:10.1021/la1032674.
- Propylene carbonate anhydrous, 99.7 108-32-7. https://www.sigmaaldrich.com/US/en/product/sial/310328?gclid=Cj0KCQiAyracBhDoARIsACGFcS7weCHAT5_OlIWtLy6JvNcz06-j4DeGkq5UR4t4DxbnhJnzKzSJ-YMaAmauEALw_wcB&gclsrc=aw.ds (accessed 2022-12-05).
- G. J. Janz, J. Ambrose, J. W. Coutts and J. R. Downey, Raman Spectrum of Propylene Carbonate, Spectrochim. Acta, Part A, 1979, 35(2), 175–179, DOI:10.1016/0584-8539(79)80181-6.
- I. Wängberg, T. Etzkorn, I. Barnes, U. Platt and K. H. Becker, Absolute Determination of the Temperature Behavior of the NO2 + NO3 +(M) ↔ N2O5 + (M) Equilibrium, J. Phys. Chem. A, 1997, 101(50), 9694–9698, DOI:10.1021/jp972203o.
- M. D. King, A. R. Rennie, C. Pfrang, A. V. Hughes and K. C. Thompson, Interaction of Nitrogen Dioxide (NO2) with a Monolayer of Oleic Acid at the Air–Water Interface – A Simple Proxy for Atmospheric Aerosol, Atmos. Environ., 2010, 44(14), 1822–1825, DOI:10.1016/j.atmosenv.2010.01.031.
- G. Murdachaew, M. E. Varner, W. E. van der Veer, R. B. Gerber and L. F. Phillips, Raman Spectroscopy of Solutions and Interfaces Containing Nitrogen Dioxide, Water, and 1,4 Dioxane: Evidence for Repulsion of Surface Water by NO2 Gas, J. Chem. Phys., 2014, 140(18), 184702, DOI:10.1063/1.4874640.
- P. A. Brooksby and W. R. Fawcett, Infrared (Attenuated Total Reflection) Study of Propylene Carbonate Solutions Containing Lithium and Sodium Perchlorate, Spectrochim. Acta, Part A, 2006, 64(2), 372–382, DOI:10.1016/j.saa.2005.07.033.
- F. Mirabella, Modern Techniques in Applied Molecular Spectroscopy; Techniques in Analytical Chemistry, Wiley, New York, NY, 1998 Search PubMed.
- M. G. Giorgini, K. Futamatagawa, H. Torii, M. Musso and S. Cerini, Solvation Structure around the Li+ Ion in Mixed Cyclic/Linear Carbonate Solutions Unveiled by the Raman Noncoincidence Effect, J. Phys. Chem. Lett., 2015, 6(16), 3296–3302, DOI:10.1021/acs.jpclett.5b01524.
- R. N. Lewis, R. N. McElhaney, W. Pohle and H. H. Mantsch, Components of the Carbonyl Stretching Band in the Infrared Spectra of Hydrated 1,2-Diacylglycerolipid Bilayers: A Reevaluation, Biophys. J., 1994, 67(6), 2367–2375, DOI:10.1016/S0006-3495(94)80723-4.
- A. Blume, W. Huebner and G. Messner, Fourier Transform Infrared Spectroscopy of 13C:O Labeled Phospholipids Hydrogen Bonding to Carbonyl Groups, Biochemistry, 1988, 27(21), 8239–8249, DOI:10.1021/bi00421a038.
- H. L. Yeager, J. D. Fedyk and R. J. Parker, Spectroscopic Studies of Ionic Solvation in Propylene Carbonate, J. Phys. Chem., 1973, 77(20), 2407–2410, DOI:10.1021/j100639a008.
- J. B. Clark, T. Bowling-Charles, S. Jabeen Proma, B. Biswas, D. T. Limmer and H. C. Allen, Structural Evolution of Water-in-Propylene Carbonate Mixtures Revealed by Polarized Raman Spectroscopy and Molecular Dynamics, Phys. Chem. Chem. Phys., 2023, 25(35), 23963–23976, 10.1039/D3CP02181E.
- L. Dei and S. Grassi, Peculiar Properties of Water as Solute, J. Phys. Chem. B, 2006, 110(24), 12191–12197, DOI:10.1021/jp060633l.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01382d |
| This journal is © the Owner Societies 2024 |