
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePre-resonance effects in deep UV Raman spectra of normal and deuterated water
B.
Rossi
 a,
M.
Tommasini
b,
P. M.
Ossi
*c and
M.
Paolantoni
d
a,
M.
Tommasini
b,
P. M.
Ossi
*c and
M.
Paolantoni
d
aElettra Sincrotrone Trieste, S.S. 114 km 163.5, Basovizza, 34149 Trieste, Italy
bDipartimento di Chimica, Materiali e Ingegneria Chimica, Politecnico di Milano, Piazza Leonardo da Vinci, 32 – 20133 Milano, Italy
cDipartimento di Chimica, Biologia, Farmacia, Scienze Ambientali, Università degli Studi di Messina, V.le F. Stagno d’Alcontres 131, 98166 Messina, Italy. E-mail: paolo.ossi@polimi.it
dDipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, via Elce di Sotto, 8 - 06123 Perugia (PG), Italy
First published on 22nd July 2024
Abstract
We have investigated the shape of the OH/OD stretching Raman band of water as a function of the excitation wavelength in the deep UV region (200–266 nm). By analyzing the spectral profiles, we highlighted selective pre-resonance effects in the high wavenumber component of the OH/OD stretching band, associated to distorted H-bonded water configurations. A van’t Hoff treatment of the temperature-dependent Raman spectra provides an estimate of the thermal energy associated to the change from ordered (ice-like) to disordered configurations that agrees with values obtained by related methods based on a two-state model of water. These results open the possibility of exploiting the observed pre-resonance deep-UV signal enhancement to investigate H-bonding properties in aqueous media.
Introduction
One of the established models concerning liquid water suggests the existence of two states characterized by different molecular coordination, each with distinct enthalpies.1 This model helps interpret some well-documented water anomalies, such as increased thermal capacity, compressibility, and expansion coefficient with decreasing temperature.2,3 A model based on the coexistence of high-density liquid (HDL) and low-density liquid (LDL) phases and the presence of a liquid–liquid critical point (LLCP) at low temperatures has been recently reviewed.4Raman spectroscopy has a longstanding tradition in investigating the structure of water by observing changes in vibrational behavior in response to various physical and chemical stimuli, such as temperature, pressure, isotopic composition, and added species in solution.5–8 Typically, attention is focused on the OH (OD) stretching band of Raman spectra due to its sensitivity to the above changes. The optical absorption resonances of liquid H2O and D2O are temperature-dependent and lie in the far UV region between 150 and 160 nm.9,10 While the integrated Raman intensity of the OH stretching band is known to undergo a significant enhancement for excitations below 300 nm,11,12 not much attention was paid to the evolution of the band shape. We discussed the effects of the choice of the excitation wavelength on the Raman spectra of water and ice Ih, and snow, at selected temperatures.13 We observed distinct changes in the Raman profile of the OH stretching band moving from visible excitation wavelength (532 nm) to deep UV (200 nm). We interpreted the collected data as signature, at decreasing wavelength of the exciting probe, of the increasing Raman cross-section of water molecules with distorted hydrogen bonds (weakly H-bonded water; high-density liquid, HDL in the literature) relative to that of water molecules with strong hydrogen bonds (strongly H-bonded water; low-density liquid, LDL in the literature).
Expanding on this investigation, we explore a range of temperatures (283–348 K) and wavelengths (200 nm–514 nm) and compare the behavior of D2O to that of H2O at selected temperatures (ca. 300 K) as a function of the excitation wavelength. The gathered data confirm and reinforce our earlier findings:13 the contribution to the OH stretching band from the population of water molecules with weak H-bonds is selectively enhanced when water is probed with deep UV excitation. Despite this selective enhancement, a two-state approximation applied to the current data yields an enthalpy change that does not depend on the excitation wavelength. Values of 1.4 ± 0.1 kcal mol−1 and 1.6 ± 0.1 kcal mol−1 were obtained for H2O and D2O, respectively, associated with the thermal energy needed to go from ordered (ice-like) to disordered configurations, in agreement with previous estimates.14,15
Experimental
The Raman spectra of liquid water were collected using double-distilled and deionised Milli-Q water and deuterium oxide 99.9 atom% D (Merck) placed into suitable suprasil quarz cuvettes. The ice Ih samples have been obtained by spontaneous freezing of Milli-Q water in the cuvette at −20 °C.Multi-wavelength UV Resonance Raman (UVRR) measurements were carried at the BL10.2-IUVS beamline of Elettra Sincrotrone Trieste (Italy).16 The excitation wavelengths at 200, 226, 240 and 250 nm were provided by the synchrotron source by setting the gap aperture of the undulator device and monochromatizing the incoming synchrotron radiation through a Czerny–Turner monochromator (750 nm focal length Acton SP2750, Princeton Instruments, Acton, MA, USA) equipped with holographic gratings with 1800 and 3600 grooves per mm. Two passively Q-switched laser systems emitting at wavelengths of 213 nm and 266 nm were used as sources for collecting UVRR spectra with these excitation energies. The Raman signal of the samples was collected in back-scattered geometry, analysed via a single-pass Czerny–Turner spectrometer (Trivista 557, Princeton Instruments, 750 mm of focal length) equipped with a holographic grating at 1800 g mm−1 and detected using an UV-enhanced CCD camera. The resolution, depending on the excitation wavelength, was set at 2.1, 1.9, 1.7, 1.5, 1.4 and 1.2 cm−1/pixel at 200, 213, 226, 240, 250 and 266 nm, respectively. The calibration of the spectrometer was performed using cyclohexane (spectroscopic grade, Sigma Aldrich). Except for the excitation wavelength at 200 nm, the UV Raman spectra were recorded both in vertical–vertical (VV) and horizontal–vertical (HV) polarization.
A sample holder equipped with a thermal bath coupled to a resistive heating system was used to control the temperature of the samples during the collection of the Raman spectra measurements of liquid water (temperature stability of ±0.1 K). The UVRR spectra of ice Ih were recorded at a fixed temperature between −4 and −6 °C to preserve the sample from any microstructural change. A constant flux of gaseous nitrogen was kept on the sample cells during the measurements of ice to avoid the freezing of atmospheric water vapor on the cuvette window.
Raman spectra with excitation in the visible (VIS) range were collected in the 2800–3800 cm−1 range (spectral resolution of 2 cm−1) at different temperatures (10–75 °C) using the apparatus described elsewhere.17 In particular, the 514.5 nm line of an argon ion laser (Coherent model Innova 90) was employed as a light source (typical power ∼500 mW). The spectra were collected using an ISA Jobin-Yvon model U1000 double monochromator (1 m focal length holographic gratings) and a photon counting detection device. This involved a thermoelectrically cooled photomultiplier (Hamamatsu model 943XX), computer-controlled by the ISA Jobin-Yvon SpectraMax package. The spectra were collected in 90 degree scattering geometry with vertical–vertical (VV) and horizontal–vertical (HV) polarization configurations, using standard polarization optics (Melles-Griot). Liquid CCl4 was used to calibrate the spectrometer. The temperature of the sample was controlled by a Haake F6 ultrathermostat (precision of 0.1 °C).
The shape of the Raman spectra collected with excitation in the VIS and UV range was corrected to account for the ν4 dependence of the scattering efficiency. The first moment (M1) of the Raman spectra reported in Fig. 1 was computed according to 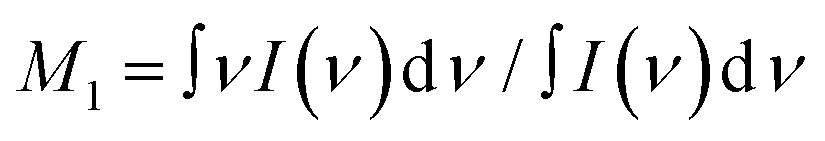 .
.
 | ||
| Fig. 1 Raman and UVRR spectra of H2O (a) and D2O (b) recorded at room temperature using different excitation wavelengths (spectra have been vertically shifted for clarity). The first moment (M1) of the OH-stretching band (c) and OD-stretching band (d) as a function of the excitation wavelength. | ||
Results and discussion
Fig. 1(a and b) displays the spectra of liquid water recorded at room temperature in the OH stretching region at various excitation wavelengths.The OH stretching band is broad and structured, reflecting the spread of intermolecular interactions within liquid water. Typically, the OH groups involved in stronger H-bonds vibrate at lower frequencies. More specifically, the spectra display three main components at ca. 3200 cm−1 (i), 3400 cm−1 (ii), and 3650 cm−1 (iii) that can be respectively assigned to (i) the stretching of OH groups embedded in more ordered (ice-like) configurations, (ii) the stretching of OH groups present in distorted H-bonds, (iii) the stretching of free OH bonds.18–20 The attribution of the (i) 3200 cm−1 component remains controversial. It was often linked to a collective intermolecular mode in which OH oscillators embedded in tetrahedral configurations with linear H-bonds effectively couple17 and references therein. It has also been assigned to the Fermi resonance (FR) between the stretching fundamental and the bending overtone.5,21,22–25 Even though theoretical studies suggest that the contribution of the FR is essential to reproduce experimental Raman features, it appears that the intermolecular coupling alone is largely responsible for the increase of the intensity in the low-frequency portion (<3300 cm−1) of the Raman spectrum and for its bimodal character.24,25 Additionally, as already remarked,24 the bending overtone in the liquid occurs at ca. 3250 cm−1 and overlaps with the red-shifted vibration of the OH groups that are more strongly H-bonded. Thus, the FR would affect more specifically the more ordered (ice-like) configurations characterized by linear H-bonds. The increased coupling between the stretching and bending modes for the water molecules involved in stronger H-bonds has been emphasized recently.26 Overall, it seems reasonable to attribute the 3250 cm−1 component to vibration modes of more ordered water structures whose Raman activity depends on both intermolecular coupling and FR effect.
The comparison of Fig. 1(a) evidences an overall intensity redistribution towards higher wavenumbers with decreasing the excitation wavelength, as previously reported.13 Since the band is structured and asymmetric, its dependence on the excitation wavelength can be suitably monitored by calculating the band's first moment (M1)27 , as reported in Fig. 1(c). M1 changes significantly with excitation wavelength: it strongly increases at shorter wavelengths in the deep UV region in connection with the occurrence of pre-resonance effects. Specifically, OH groups vibrating at higher frequencies (associated with disordered molecular configurations and weaker H-bonds) experience a larger pre-resonance enhancement than those vibrating at lower frequencies (associated with more ordered structures and stronger H-bonds).
, as reported in Fig. 1(c). M1 changes significantly with excitation wavelength: it strongly increases at shorter wavelengths in the deep UV region in connection with the occurrence of pre-resonance effects. Specifically, OH groups vibrating at higher frequencies (associated with disordered molecular configurations and weaker H-bonds) experience a larger pre-resonance enhancement than those vibrating at lower frequencies (associated with more ordered structures and stronger H-bonds).
To gain more insights into this phenomenon, an analogous comparison performed for OD stretching of deuterated water is reported in Fig. 1b. Similarly to the case of H2O, also the spectrum of D2O at 514 nm shows three main components. The low-wavenumber component (ca. 2400 cm−1) can be associated to ordered (ice-like) configurations, which experience stronger H-bonds and more effective vibrational coupling.21,22 The intermediate component (ca. 2500 cm−1) can be associated with more distorted configurations, in which H-bonds partially weaken and the vibrational coupling reduces. The high-wavenumber shoulder (ca. 2650 cm−1) can be associated to almost free OD groups (free ODs). For H2O and D2O the intensity distribution in the VIS Raman spectrum is significantly different. This observation can be mainly ascribed to variations of both intra- and inter-molecular vibrational coupling upon H/D substitution.28 Nevertheless, as observed for H2O, the decrease of the excitation wavelength causes a noticeable redistribution of the band towards higher wavenumber (Fig. 1b and d), due to the relative enhancement of the high-frequency vibrations assigned to broken and distorted H-bonds.
Fig. 2 shows a direct comparison between the OH stretching profiles of H2O and D2O at two excitation wavelengths, after rescaling the wavenumber axis by the ratio of the gas-phase antisymmetric stretching of the two species (0.742).28 Looking at the VIS Raman spectra, it can be observed that the low wavenumber component, mainly ascribed to more ordered tetrahedral structures, is relatively more intense in D2O than H2O. Differences in vibrational features between the two species were interpreted considering that the OH stretching modes are more anharmonic and delocalized in H2O than in D2O.28 As a result, vibrations in D2O are more localized, resembling to a greater extent those of the gas phase molecules. Noticeably, due to the larger relative increase of the component related to distorted configurations, the profiles of D2O and H2O become more similar to each other with the use of UV excitation (Fig. 2).
 | ||
| Fig. 2 Comparison between the UVRR spectra of H2O and D2O collected using 213 and 514 nm as excitation wavelengths. For a better comparison, the wavenumber-axis for D2O was rescaled by the ratio of gas-phase antisymmetric stretching bands (0.742). | ||
To better visualize excitation-induced band shape variations, we attempted an internal normalization procedure. The VIS Raman spectrum (514 nm excitation line) was taken as a reference and we subtracted it from the corresponding UV Raman spectra, after rescaling the spectra on the low-wavenumber tail. The rescaling factor was set to obtain the minimum area non-negative difference spectra (DS), as exemplified in Fig. 3. The method is formally analogous to that used to extract the so-called solute-correlated (SC) spectra in aqueous solutions.29,30 The method here proposed is based on the idea that with respect to the VIS Raman spectrum, the overall intensity increases when the excitation wavelength is lowered. Additionally, it considers that the enhancement is relatively greater for the weakly H-bonded components. Thus, the DS highlight the spectral distribution that is more affected by the pre-resonance and can be used for comparative purposes.
 | ||
| Fig. 3 Low-frequency normalized Raman spectra collected using 213, 266 and 514 nm as excitation wavelengths for H2O (a) and D2O (b). Representative difference spectra (DS) calculated subtracting the VIS Raman spectrum (514 nm excitation line), taken as a reference, from the UV Raman spectra, at two different excitation wavelengths (213 and 266 nm) for H2O (c) and D2O (d) (see text for details). The blue coloured area in panels (a) and (b) represents the DS spectrum calculated for 213 nm-excited spectra. The DS have been normalized (norm-DS) by dividing for the total area of the corresponding reference spectrum. Inset: Excitation wavelength dependence of the area of the norm-DS for H2O and D2O. | ||
The resulting DS (Fig. 3) are peaked at around 3500 cm−1 and 2500 cm−1 for H2O and D2O, respectively, indicating that the Raman cross-section of the OH/OD groups involved in distorted and broken H-bonds is preferentially enhanced. For both samples the relative enhancement (inset of Fig. 3) increases by lowering the excitation wavelength. We remark that, based on the proposed analysis, the relative changes are always larger for D2O. Since the lowest energy UV absorption band of water blue-shifts moving from H2O to D2O,9,10 the greater effect observed for D2O might be related to changes in vibrational features induced by the isotopic substitution.
Taken together, the data on H2O and D2O support the idea that the occurrence of a specific pre-resonance effect, where weakly H-bonded OHs experience a larger intensity increase, is a general phenomenon.
Fig. 4 compares VV and HV spectra of H2O collected with 213 and 250 nm excitation wavelengths. Here, DS were calculated by taking the spectra obtained with 250 nm excitation as a reference. The analysis indicates that the pre-resonance only marginally affects the shape of the HV (anisotropic) profile when compared to the VV one, leading to a relatively weak normalized DS (norm-DS). The low-frequency ice-like component is strongly depressed in the HV profile, and the band mainly accounts for OHs present in distorted and broken H-bond configurations. As such, we expect the pre-resonance enhancement to be rather constant across the HV profile, as observed.
 | ||
| Fig. 4 VV (a) and HV (b) spectra of H2O collected with 213 and 250 nm as excitation wavelengths. In panels (c) and (d) we report the corresponding spectral differences DS calculated subtracting the 250 nm-excited Raman spectrum, taken as a reference, from the 213 nm-excited Raman spectra. The DS spectra have been rescaled by dividing for the total area of the corresponding reference spectrum (norm-DS). | ||
Also the Raman spectra of ice Ih do not show any appreciable change in their band shapes as a function of the excitation wavelength (Fig. 5). This is expected based on the dominance of OHs forming strong H-bonds in tetrahedral configurations, which are not selectively enhanced by the above pre-resonance effects.
 | ||
| Fig. 5 Raman spectra of ice Ih collected using different excitation wavelengths. The profiles have been normalized to the maximum intensity and vertically shifted for a better comparison. | ||
To further investigate the effect induced by UV excitation on the OH stretching band, we extended the above-proposed analysis to the spectra of H2O recorded at different temperatures (Fig. 6). With the increase in temperature, the components related to distorted and broken H-bonds gain intensity at the expense of the low-wavenumber component (3250 cm−1), related to tetrahedral configurations forming unstrained H-bonds. Raman spectra recorded at different excitation wavelengths and temperatures are displayed in Fig. 6a and b, after rescaling on the low-frequency tail. Moving from 283 to 348 K, the resulting difference spectrum (Fig. 6c) shifts from ca. 3400 cm−1 to 3500 cm−1. This is consistent with the idea that the average H-bonding energy of the enhanced OH sub-population further reduces at higher temperatures. Notice that the relative enhancement (Fig. 5d) is temperature-independent, suggesting that the Raman cross-section at a given wavelength does not depend strongly on the redistribution occurring within distorted and broken H-bonds.
 | ||
| Fig. 6 (a) and (b) Low-frequency normalized Raman spectra collected with excitation wavelength at 213 and 514 nm for H2O at 283 K (a) and 348 K (b), respectively; the blue-coloured area represents the DS spectra. (c) Representative difference spectra (DS), calculated by subtracting the VIS Raman spectrum (taken as a reference) from the UV Raman spectrum (see text for details). Each DS has been normalized (norm-DS) by dividing it by the total area of the corresponding reference spectrum. (d) Temperature dependence of the area of norm-DS calculated for 213 nm-excited spectra of H2O. | ||
The temperature dependence of the OH profile can be interpreted by considering distinct components (multi-component models) or a continuous distribution of H-bonds (continuous models). Even within the framework of a continuous model, a van’t Hoff analysis based on a pseudo two-state approximation has proven suitable to estimate the enthalpy variation (ΔH) when moving from ordered (ORD) to disordered (DIS) configurations. ORD refers to OHs in tetrahedral (ice-like) structures, whereas DIS involves OHs forming distorted and broken H-bonds.14,15,17 A reference frequency (νREF) of about 3350 cm−1, placed between the two main spectral components of the H2O VIS Raman spectra, was considered to define the two sub-ensembles, using a simple integration method to quantify the amount of ORD and DIS species.14,17 Considering various Raman profiles (i.e., VV, VV + HV, isotropic) and different reference frequencies (νREF = 3350 ± 50 cm−1), ΔH values of 1.3–1.7 kcal mol−1 were derived.14,17 The resulting ΔH is only a fraction of the H-bonding energy of water (5.7 kcal mol−1), evaluated from its sublimation enthalpy,5,31 and can be considered an estimate of the energy cost to modify the local structure of liquid water, from tetrahedral to more distorted or broken configurations.14,17
Here, to verify the possibility of using UV Raman data to estimate the H-bond reorganization enthalpy, we applied the same integration method to T-dependent spectra collected for H2O and D2O at different excitation wavelengths, as reported in Fig. 7.
 | ||
| Fig. 7 Temperature-evolution of Raman spectra collected with excitation wavelength at 200, 266, and 514 nm for H2O (a)–(c) and D2O (d)–(f). The profiles have been normalized to the total intensity of the OH/OD band. | ||
Table 1 reports the resulting ΔH values, obtained by the van’t Hoff treatment of the ORD/DIS ratio, evaluated considering a νREF of 3350 cm−1 and 2440 cm−1 for H2O and D2O, respectively. Similar ΔH values were derived for the two species regardless of the excitation wavelengths. Specifically, values of 1.3–1.6 kcal mol−1 were obtained for H2O (average ΔH = 1.4 ± 0.1 kcal mol−1), while values of 1.4–1.8 kcal mol−1 were obtained for D2O (average ΔH = 1.6 ± 0.1 kcal mol−1).
| Excitation wavelength (nm) | ΔH (kcal mol−1) H2O | ΔH (kcal mol−1) D2O |
|---|---|---|
| 200 (VV + HV profile) | 1.3 ± 0.1 | 1.4 ± 0.1 |
| 213 | 1.5 ± 0.1 | — |
| 240 | 1.4 ± 0.1 | 1.6 ± 0.1 |
| 266 | 1.6 ± 0.1 | 1.7 ± 0.1 |
| 514 | 1.4 ± 0.1 | 1.8 ± 0.1 |
| Average | 1.4 ± 0.1 | 1.6 ± 0.1 |
Hence, although the overall shape of the spectrum changes with the excitation wavelength, the same thermal-induced reorganization process can be probed. This is consistent with the previous suggestion that the Raman cross-section at a given wavelength is weakly temperature-dependent. Additionally, the increase of ΔH going from H2O to D2O agrees with a previous estimate and with the observation that the H-bonding energy increases upon H/D isotopic substitution.5,15 Notably, the latent heat of melting is 1.4 kcal mol−1 for H2O ice and 1.5 kcal mol−1 for D2O ice.5,14
Conclusions
In conclusion, we analyzed the changes in the band shape of the OH/OD stretching Raman band of liquid water as a function of deep UV excitation wavelength and temperature. We evidenced a selective pre-resonance enhancement for the water molecules forming weak/distorted H-bonds relative to those forming strong H-bonds within ordered (ice-like) configurations. Despite excitation-induced spectral modifications, a van’t Hoff treatment of Raman data leads to H-bond enthalpy changes (ΔH) for both H2O and D2O that do not depend on the excitation wavelength. The resulting ΔH values, 1.4 ± 0.1 kcal mol−1 (H2O) and 1.6 ± 0.1 kcal mol−1 (D2O), relate the thermal energy associated to the reorganization from ordered (ice-like) to disordered configurations and support previous estimates.14,15Overall, these findings suggest the possibility of exploiting the signal enhancement that occurs in the deep UV region to investigate H-bonding properties in aqueous media. Since the enhancement is related to the H-bonding state of water molecules, in principle, the Raman signal arising from water molecules specifically affected by the solute could be selectively enhanced by using deep UV excitation.
Data availability
Data for this article, including Raman spectra in digital format, are available at Zenodo at https://doi.org/10.5281/zenodo.12701034.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron radiation facilities and for financial support under the SUI internal project. We thank Alessandro Gessini and Francesco D’Amico for assistance in using beamline IUVS. M. P. thanks the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 - VITALITY - CUP J97G22000170005.References
- P. H. Poole, F. Sciortino, U. Essmann and H. E. Stanley, Phase behaviour of metastable water, Nature, 1992, 360(6402), 324–328, DOI:10.1038/360324a0.
- J. R. Errington and P. G. Debenedetti, Relationship between structural order and the anomalies of liquid water, Nature, 2001, 409(6818), 318–321, DOI:10.1038/35053024.
- P. G. Debenedetti, F. Sciortino and G. H. Zerze, Second critical point in two realistic models of water, Science, 2020, 369(6501), 289–292, DOI:10.1126/science.abb9796.
- P. Gallo, K. Amann-Winkel, C. A. Angell, M. A. Anisimov, F. Caupin and C. Chakravarty, et al., Water: A Tale of Two Liquids, Chem. Rev., 2016, 116(13), 7463–7500, DOI:10.1021/acs.chemrev.5b00750.
- D. Eisenberg and W. Kauzmann, The Structure and Properties of Water, Oxford University Press, London, England, 1969. Available from: https://academic.oup.com/book/34782 Search PubMed.
- G. E. Walrafen, M. R. Fisher, M. S. Hokmabadi and W. H. Yang, Temperature dependence of the low- and high-frequency Raman scattering from liquid water, J. Chem. Phys., 1986, 85(12), 6970–6982 CrossRef CAS.
- M. E. Gallina, P. Sassi, M. Paolantoni, A. Morresi and R. S. Cataliotti, Vibrational analysis of molecular interactions in aqueous glucose solutions. Temperature and concentration effects, J. Phys. Chem. B, 2006, 110(17), 8856–8864 CrossRef CAS PubMed.
- Q. Sun, Raman spectroscopic study of the effects of dissolved NaCl on water structure, Vib. Spectrosc., 2012, 62, 110–114 CrossRef CAS.
- Y. Ozaki, Y. Morisawa and I. Tanabe, ATR-far-ultraviolet spectroscopy: a challenge to new σ chemistry, Chem. Soc. Rev., 2024, 53, 1730–1768 RSC.
- Y. Ozaki, Y. Morisawa, A. Ikehata and N. Higashi, Far-Ultraviolet Spectroscopy in the Solid and Liquid States: A Review, Appl. Spectrosc., 2012, 66(1), 1–25, DOI:10.1366/11-06496.
- S. A. Asher and J. L. Murtaugh, UV Raman Excitation Profiles of Imidazole, Imidazolium, and Water, Appl. Spectrosc., 1988, 42(1), 83–90, DOI:10.1366/0003702884428653.
- S. R. Ahmad and A. Iles, Pre-resonance Raman excitation profile of the 3400 cm-1 mode of liquid water, J. Raman Spectrosc., 2001, 32(8), 649–655 CrossRef CAS.
- E. Maggiore, M. Tortora, B. Rossi, M. Tommasini and P. M. Ossi, UV resonance Raman spectroscopy of weakly hydrogen-bonded water in the liquid phase and on ice and snow surfaces, Phys. Chem. Chem. Phys., 2022, 24(17), 10499–10505 RSC.
- J. D. Smith, C. D. Cappa, K. R. Wilson, B. M. Messer, R. C. Cohen and R. J. Saykally, Energetics of Hydrogen Bond Network Rearrangements in Liquid Water, Science, 2004, 306(5697), 851–853, DOI:10.1126/science.1102560.
- J. D. Smith, C. D. Cappa, K. R. Wilson, R. C. Cohen, P. L. Geissler and R. J. Saykally, Unified description of temperature-dependent hydrogen-bond rearrangements in liquid water., Proc. Natl. Acad. Sci. U. S. A., 2005, 102(40), 14171–14174 CrossRef CAS PubMed.
- B. Rossi, C. Bottari, S. Catalini, F. D’Amico, A. Gessini and C. Masciovecchio, In Synchrotron-based ultraviolet resonance Raman scattering for material science. ed V. P. Gupta, Y. Ozaki, Molecular and Laser Spectroscopy, Elsevier, 2020, Ch. 13, pp. 447–482. Available from: https://www.sciencedirect.com/science/article/pii/B9780128188705000137 Search PubMed.
- M. Paolantoni, N. Faginas Lago, M. Alberti and A. Laganà, Tetrahedral ordering in water: Raman profiles and their temperature dependence, J. Phys. Chem. A, 2009, 113(52), 15100–15105 CrossRef CAS PubMed.
- A. Di Michele, M. Freda, G. Onori, M. Paolantoni, A. Santucci and P. Sassi, Modulation of hydrophobie effect by cosolutes, J. Phys. Chem. B, 2006, 110(42), 21077–21085 CrossRef PubMed.
- B. Rossi, C. Bottari, L. Comez, S. Corezzi, M. Paolantoni and A. Gessini, et al., Structural and molecular response in cyclodextrin-based pH-sensitive hydrogels by the joint use of Brillouin, UV Raman and Small Angle Neutron Scattering techniques, J. Mol. Liq., 2018, 271, 738–746 CrossRef CAS.
- C. Bottari, L. Comez, M. Paolantoni, S. Corezzi, F. D’Amico and A. Gessini, et al., Hydration properties and water structure in aqueous solutions of native and modified cyclodextrins by UV Raman and Brillouin scattering, J. Raman Spectrosc., 2018, 49(6), 1076–1085 CrossRef CAS.
- L. De Marco, K. Ramasesha and A. Tokmakoff, Experimental Evidence of Fermi Resonances in Isotopically Dilute Water from Ultrafast Broadband IR Spectroscopy, J. Phys. Chem. B, 2013, 117(49), 15319–15327, DOI:10.1021/jp4034613.
- K. Ramasesha, L. De Marco, A. Mandal and A. Tokmakoff, Water vibrations have strongly mixed intra- and intermolecular character, Nat. Chem., 2013, 5(11), 935–940, DOI:10.1038/nchem.1757.
- B. M. Auer and J. L. Skinner, IR and Raman spectra of liquid water: Theory and interpretation, J. Chem. Phys., 2008, 128(22), 224511, DOI:10.1063/1.2925258.
- A. A. Kananenka and J. L. Skinner, Fermi resonance in OH-stretch vibrational spectroscopy of liquid water and the water hexamer, J. Chem. Phys., 2018, 148(24), 244107, DOI:10.1063/1.5037113.
- T. Takayama, T. Otosu and S. Yamaguchi, Theoretical and experimental OD-stretch vibrational spectroscopy of heavy water, J. Chem. Phys., 2024, 160(10), 104504, DOI:10.1063/5.0200623.
- H. Shen, X. Shen and Z. Wu, Simulating the isotropic Raman spectra of O–H stretching mode in liquid H2O based on a machine learning potential: the influence of vibrational couplings, Phys. Chem. Chem. Phys., 2023, 25(41), 28180–28188, 10.1039/D3CP03035K.
- M. Paolantoni, P. Sassi, A. Morresi and R. S. Cataliotti, Raman noncoincidence effect on OH stretching profiles in liquid alcohols, J. Raman Spectrosc., 2006, 37(4), 528–537, DOI:10.1002/jrs.1427.
- L. De Marco, W. Carpenter, H. Liu, R. Biswas, J. M. Bowman and A. Tokmakoff, Differences in the Vibrational Dynamics of H2O and D2O: Observation of Symmetric and Antisymmetric Stretching Vibrations in Heavy Water, J. Phys. Chem. Lett., 2016, 7(10), 1769–1774 CrossRef CAS PubMed.
- F. Matroodi, C. Bottari, B. Rossi, A. Mannu, M. Paolantoni and A. Mele, Hydration water and ionic aggregation in aqueous solutions of imidazolium-based protic ionic liquids, J. Mol. Liq., 2024, 395, 123881 CrossRef CAS.
- D. Ben-Amotz, Hydration-Shell Vibrational Spectroscopy, J. Am. Chem. Soc., 2019, 141(27), 10569–10580 CrossRef CAS PubMed.
- F. H. Stillinger, Water Revisited, Science, 1980, 209(4455), 451–457, DOI:10.1126/science.209.4455.451.
| This journal is © the Owner Societies 2024 |