
Excited-state dynamics of 3-hydroxychromone in gas phase†
Li
Zhao
 *a,
Xuehui
Geng
a,
Jiangyue
Wang
a,
Yuxuan
Liu
a,
Wenhui
Yan
a,
Zhijie
Xu
a and
Junsheng
Chen
*b
*a,
Xuehui
Geng
a,
Jiangyue
Wang
a,
Yuxuan
Liu
a,
Wenhui
Yan
a,
Zhijie
Xu
a and
Junsheng
Chen
*b
aCollege of Science, China University of Petroleum (East China) Qingdao 266580, Shandong, China. E-mail: zhaoli282@upc.edu.cn
bNano-Science Center & Department of Chemistry University of Copenhagen Universitetsparken 5, 2100 KøbenhavnØ, Denmark. E-mail: junsheng.chen@chem.ku.dk
First published on 4th July 2024
Abstract
In recent years, 3-hydroxychromone (3-HC) and its derivatives have attracted much interest for their applications as molecular photoswitches and fluorescent probes. A clear understanding of their excited-state dynamics is essential for their applications and further development of new functional 3-HC derivatives. However, the deactivation mechanism of the photoexcited 3-HC family is still puzzling as their spectral properties are sensitive to the surrounding medium and substituents. The excited-state relaxation channels of 3-HC have been a matter of intense debate. In the current work, we thoroughly investigated the excited-state decay process of the 3-HC system in the gas phase using high-level electronic structure calculations and on-the-fly excited-state dynamic simulations intending to provide insight into the intrinsic photochemical properties of the 3-HC system. A new deactivation mechanism is proposed in the gas phase, which is different from that in solvents. The excited-state intramolecular proton transfer (ESIPT) process that occurs in solutions is not preferred in the gas phase due to the existence of a sizable energy barrier (∼0.8 eV), and thus, no dual fluorescence is found. On the contrary, the non-radiative decay process is the dominant decay channel, which is driven by photoisomerization combined with ring-puckering and ring-opening processes. The results coincide with the observations of an experiment performed in a supersonic jet by Itoh (M. Itoh, Pure Appl. Chem., 1993, 65(8), 1629–1634). The current work indicates that the solution environment plays an important role in regulating the excited-state dynamic behaviour of the 3-HC system. This study thus provides theoretical guidance for the rational design and improvement of the photochemical properties of the 3-HC system and paves the way for further investigation into its photochemical properties in complex environments.
Introduction
In recent years, 3-hydroxychromone (3-HC) and its derivatives with dual-band fluorescence have attracted considerable research interest for their use in applications such as molecular photoswitches,1–5 fluorescent molecular probes,6–10 and photochromic materials.11–15 Their dual-band fluorescence originates from their ability to undergo excited-state intramolecular proton transfer (ESIPT) and form intramolecular hydrogen bonds. Upon photoexcitation, the most stable isomer of 3-HC in the ground state (S0), termed cis-enol (Fig. 1), is excited to the first excited state (S1), where it can either relax from the Franck–Condon state to the most stable cis-enol form in S1, or can undergo an ESIPT process to generate a new cis-keto isomer in S1. The cis-enol and cis-keto isomers in S1 can emit fluorescence with well-separated emission bands. After emission, the cis-keto isomer (in S0) subsequently undergoes relaxation and structural reorganization to return to the cis-enol form and complete the photoreaction cycle. This process can be influenced by the solvent polarity16,17 and the type and position of substitutions.13,18 Consequently, the fluorescence peak position and intensity are different under different conditions. A thorough investigation of their excited-state dynamics can provide guidance for further development of 3-HC and its derivatives for advanced applications. | ||
| Fig. 1 The three stable tautomers of 3-HC in S0 and S1 with the general conversion scheme between them after photoexcitation. | ||
A number of experimental and theoretical studies have been undertaken to investigate the photochemical and photophysical properties of 3-HC and its derivatives.11–37 Especially, 3-HC, as the basic unit of its derivatives, is a good model system to study the excited-state dynamics because it can provide elementary information without perturbation from substituents. Clevalier et al.17 found that the photoinduced excited-state dynamics of 3-HC are solvent dependent. In pure water (pH = 7), the dissociation and formation of the short-lived anionic state (deprotonated) is favoured, and this phenomenon has been observed in several 3-HC derivatives.16 However, in organic solvents, the formation of the tautomer excited state (cis-keto and trans-enol) is the main pathway. Nevertheless, the reason for the slow ESIPT process (5.5 ps) in organic solvents remains unclear. Two possible processes have been proposed by Chevalier et al.:17 the intramolecular vibrational relaxation and cis–trans photoisomerization (caused by hydrogen-torsion) processes. Furthermore, to understand them, Perveaux et al.23 carried out a theoretical study to investigate the deactivation process of the 3-HC system. They located a conical intersection within the FC region between the dark S2 (nπ* character) and the bright S1, which opens a channel for the system to re-populate to S2. The new reaction pathway is unreactive with respect to the ESIPT process, and thus a new explanation for the slower ESIPT process has been proposed. Vennapusa et al.30 further investigated the nonadiabatic dynamics of 3-HC using the multiconfiguration time-dependent Hartree (MCTDH) method. A similar deactivation mechanism scheme was proposed. They predicted an average timescale of 84 fs for the fast ESIPT process, and a much slower ESIPT process in S2 with about 19% of the trajectories are remaining in S2 at the end of the simulation (300 fs). Although the above-mentioned theoretical studies were performed in the gas phase, their results support the presence of the ESIPT process, which is observed in the solution phase. However, the experimental results obtained in the gas phase, reported by Itoh,36 concluded that the photoinduced excited-state dynamics of 3-HC are dominated by non-radiative processes instead of involving fluorescent excited-state species generated by the ESIPT process. Hence, the excited-state relaxation pathways of 3-HC in the gas phase remain unclear.
Herein, we employed high-level electronic structure calculations based on the CASPT2//CASSCF method and on-the-fly excited-state dynamic simulations to investigate the intrinsic photoinduced excited-state dynamics of 3-HC in the gas phase. We found that the dark-state S2 has a negligible influence on the deactivation of 3-HC, which can directly transfer to S1 within an ultrafast timescale (∼50 fs), and no “S1 → S2 up-transfer” trajectories were found. The system decays to S0 mainly by a non-radiative process, and two coordinates driven by the cis–trans photoisomerization process and ring-opening motion were found to be the dominant channels in the S1 to S0 relaxation process. The ESIPT process is not preferred due to the presence of a sizable energy barrier (∼0.8 eV), and thus dual fluorescence is not supported. This study provides a detailed understanding of the excited-state dynamics of 3-HC in the gas phase, which can provide theoretical guidance for the further modification of chemical structures to achieve the desired functions.
Calculation details
All CASSCF and CASPT2 calculations were performed using the OpenMolcas suite.38 All DFT and TDDFT calculations were executed in the Gaussian 09 program package.39 The geometry optimization of S0 and the two lowest excited state (S1, S2) minima and the minimum energy conical intersections (MECIs) were performed using the complete active space self-consistent field (CASSCF) method. The higher excited states were not included given that they have little influence on the decay process (lying > 1.0 eV above S2, as shown in Table S1, ESI†). Initially, the active space was chosen as sixteen electrons distributed in thirty orbitals, including seven doubly occupied π orbitals, one doubly occupied n orbital and five unoccupied π* orbitals. By eliminating the orbitals with occupations close to 2.00 or 0.00, the active space was then reduced to 12 electrons distributed in 10 orbitals. More information about the included orbitals is provided in Fig. S1 (ESI†). The reaction pathways were constructed using the linearly interpolated internal coordinate (LIIC) method between the Franck–Condon point (FC) and MECIs. Considering that the CASSCF method is unable to provide accurate energy information due to the lack of dynamic electron correlation, all energies (including the vertical excitation energies of the critical points and potential energy profiles) were calculated again using the complete active space perturbation theory to second order (CASPT2) method. An imaginary shift of 0.2 a.u. was employed to avoid intruder-state issues.40 The 6-31G* basis set was employed throughout this work. All CASSCF and CASPT2 calculations were performed using a three-state-average with equal weights for each state. We tested different basis sets, including cc-pVTZ, cc-pVDZ, 6-31G** and 6-31G, and different active spaces, such as (16,13), (10,8), (8,8) and (6,6), to confirm the reliability of the method employed in the static electronic structure calculations. This helps to find an appropriate calculation level for the following dynamics simulations. In addition, the DFT//B3LYP//6-31G** and TDDFT//B3LYP//6-31G** methods were also employed to optimize the stable structures and calculate the vertical excitation energies, respectively.The photoinduced deactivation dynamics of 3-HC were followed by performing the on-the-fly surface hopping dynamics simulation procedure NAIMD-DICP, which has been successfully employed to study the deactivation process of many different molecular systems.41–46 The nuclear trajectory propagation was integrated with the velocity Verlet algorithm.47 The transition probability between different electronic states was estimated based on the Zhu–Nakamura theory,48–51 which was compared with a random number generated between 0 and 1 to determine whether to hop or not. The velocity adjustment was immediately executed to maintain the energy conservation after a success hop. Considering that S2 is nearly degenerate with S1, we initiated 50 dynamics simulation trajectories from S2. The maximum simulation time was about 1 ps with a step of 0.5 fs, which decreased to 0.1 fs when approaching the conical intersection region. The decreased step size caused the total simulation time of the trajectories, which experienced non-adiabatic transitions, to be less than 1 ps. The adiabatic energies, gradients and non-adiabatic couplings were computed using the SA3-CASSCF(8,8)//6-31G* method, which has been confirmed to be suitable for dynamics simulations by balancing the accuracy and computational cost (Table S3, ESI†). Furthermore, 35 additional trajectories starting from S1 were simulated to avoid any potential bias or influence from the initial state on the dynamics simulation results. The S0 → S1 and S1 → S2 processes were allowed in both dynamics simulations (S1 or S2 as the initial state). The initial conditions were generated at 0 K based on the harmonic Wigner distribution,52,53 with the frequencies and normal modes in the ground state calculated using SA3-CASSCF(12,10)//6-31G* method. The reliability of the CASSCF method for dynamic simulations has also been confirmed. As shown in Fig. S4 (ESI†), the relative energy information for the various critical points obtained by the CASSCF method agreed with that obtained by the CASPT2 method. Additionally, the topological structures of the potential energy curves constructed between the FC point and the conical intersection points obtained by the CASSCF method (Fig. S12, ESI†) agreed with those obtained by the CASPT2 method (Fig. 2b). Moreover, to compare the effect of the calculation methods on the dynamic results, we further ran five additional trajectories using the Newton-X package54 combined with TDDFT calculations. The time step and the maximum simulation time were set to 0.5 fs and 1 ps, respectively.
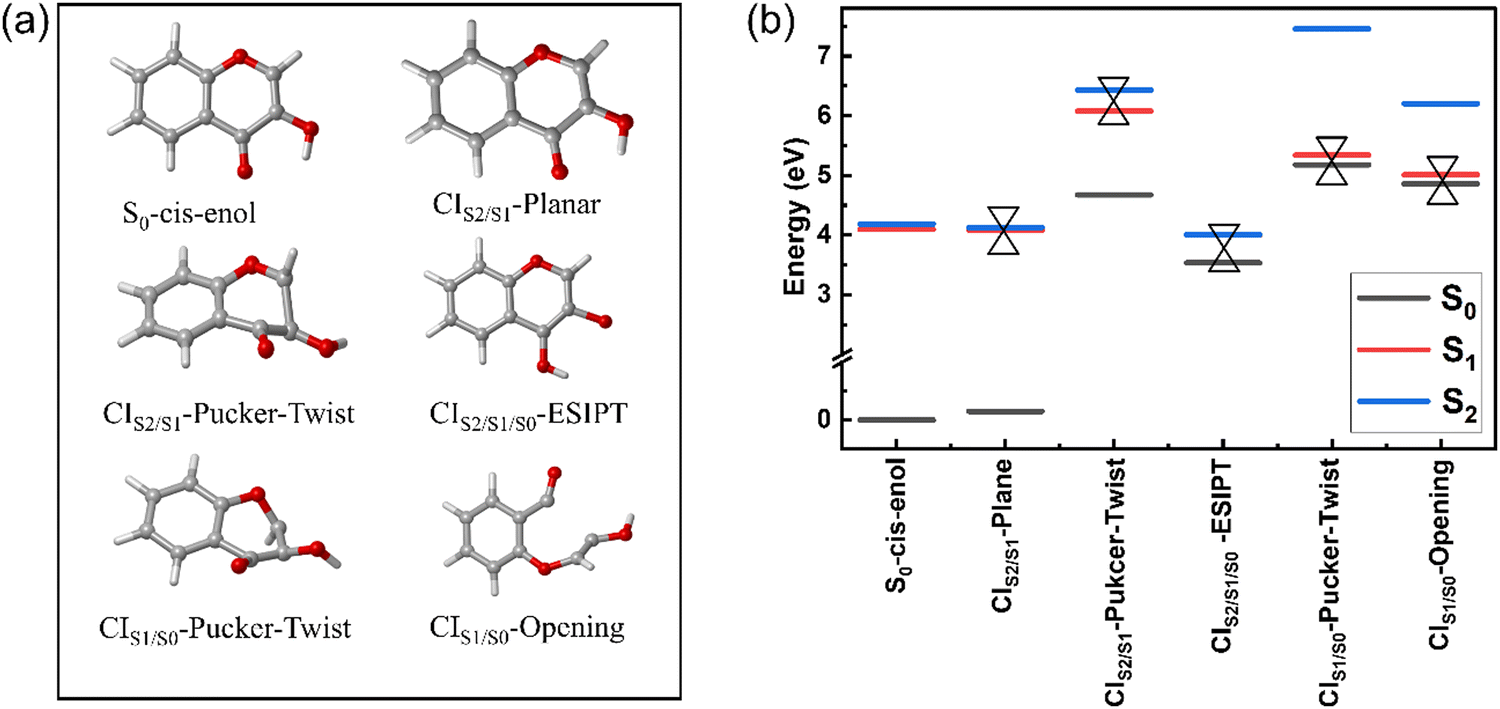 | ||
| Fig. 2 (a) S0-cis-enol and five MECIs between two adjacent electronic states optimized with the SA3-CASSCF(12,10)//6-31G* method and (b) energies of the MECIs relative to the ground state energy of S0-cis-enol. | ||
Results and discussion
Stationary points and energies
The three stable tautomers of 3-HC are in S0, which are termed trans-enol, cis-enol and cis-keto, as shown in Fig. 1. All the optimized tautomers in S0 are planar with the absolute dihedral angle values of O3C8C9H5 and C7C9O2H6 equal to 180° and 0°, respectively. Our calculations match well with previous theoretical work as they share similar values for most of the related geometrical parameters (Table 1). The most stable tautomer in S0 is the cis-enol tautomer with an intramolecular hydrogen bond O1H6 (2.199 Å) between the hydroxyl and carbonyl group. Upon photoexcitation, the hydrogen bond is strengthened with a decrease in the bond length to 2.147 Å. The strengthening of the hydrogen bond has been reported based on DFT and TDDFT calculations.8,11,20,23,32 The cis-keto tautomer (S0) is generated by the ESIPT process of the cis-enol form, with the breaking of the O2H6 bond and formation of the O1H6 bond, which is located 0.64 eV above the S0-cis-enol tautomer.| Geo. | C9O2 | O2H6 | O1H6 | C7O1 | C7C9O2H6 | O3C8C9H5 | S0 → S1 | S0 → S2 |
|---|---|---|---|---|---|---|---|---|
| 20TDDFT//DFT//B3LYP//TZVP; 23TDDFT//DFT//PBE0//cc-PVDZ; 32TDDFT//DFT//B3LYP//6-31G*a DFT//B3LYP//6-31G**.b TDDFT//B3LYP//6-31G**; and f means the transition oscillator strength. | ||||||||
| S0-cis-enol | 1.349 | 0.952 | 2.199 | 1.235 | 0.0 | 180.0 | 4.09 | 4.18 |
| S0-cis-enola | 1.351 | 0.983 | 2.044 | 1.238 | 0.0 | 180.0 | 4.14 (f = 0.08)b | 4.20 (f = 0.00)b |
| Exp. 3.96;17 3.81;19 3.7736 | ||||||||
| S0-cis-enol20 | 1.351 | 0.976 | 2.097 | 1.232 | 0.0 | 180.0 | — | — |
| S0-cis-enol23 | 1.341 | 0.982 | 2.000 | 1.233 | 0.0 | 180.0 | 4.11 | 4.23 |
| S0-cis-enol32 | 1.351 | 0.983 | 2.043 | 1.238 | 0.0 | 180.0 | 4.02 | 4.12 |
| S1-cis-enol | 1.344 | 0.953 | 2.147 | 1.248 | 0.0 | 180.0 | 3.78 (0.12) b | — |
| S1-cis-enol20 | 1.319 | 1.002 | 1.871 | 1.248 | 0.0 | 180.0 | — | — |
| S1-cis-enol23 | 1.309 | 1.026 | 1.695 | 1.251 | 0.0 | 180.0 | — | — |
| S1-cis-enol32 | 1.368 | 0.969 | 2.370 | 1.334 | 19.8 | 180.0 | — | — |
| S0-cis-keto | 1.212 | 2.153 | 0.957 | 1.323 | 0.0 | 180.0 | 2.98 | 3.77 |
| S0-cis-keto20 | 1.265 | 1.812 | 1.008 | 1.312 | 0.0 | 180.0 | — | — |
| S1-cis-keto | 1.384 | 2.349 | 0.949 | 1.347 | 0.0 | 180.0 | 1.63 | — |
| S1-cis-keto20 | 1.335 | 2.143 | 0.975 | 1.341 | 0.0 | 180.0 | — | — |
| S1-cis-keto32 | 1.344 | 2.083 | 0.982 | 1.342 | 0.0 | 180.0 | — | — |
| S0-trans-enol | 1.350 | 0.947 | 3.655 | 1.209 | 180.0 | 180.0 | 3.98 | 4.27 |
| S0-trans-enol20 | 1.359 | 0.965 | 3.684 | 1.220 | 180.0 | 180.0 | — | — |
| S1-trans-enol | 1.356 | 0.947 | 3.666 | 1.359 | 180.0 | 180.0 | 2.73 | — |
| S1-trans-enol20 | 1.370 | 0.964 | 3.649 | 1.313 | 180.0 | 180.0 | — | — |
| S1-trans-enol23 | 1.356 | 0.965 | 3.655 | 1.312 | 180.0 | 180.0 | — | — |
For the minimum in S1, compared to that in S0, the main changes in the cis-enol tautomer are the elongation of the C7O1 bond by 0.01 Å and shortening of the O1H6 bond by about 0.02 Å. The fluorescence energy is 3.78 eV. Consequently, the cis-keto form becomes the most stable tautomer, which is termed as S1-cis-keto. The most obvious change in S1-cis-keto compared with S0-cis-keto is the bond length of the C9O2 carbonyl group, which is extended from 1.212 Å (in S0) to 1.384 Å (in S1). The geometrical changing trend agrees with previous reports.20,32 The trans-enol is generated from the twisting motion of the C9O2 bond in cis-enol. The intramolecular hydrogen bond breaks as the O1H6 bond length increases to 3.655 Å. After photoexcitation, the C7O1 bond length increases by about 0.15 Å, which is longer than that in previous reports.20,32
The calculated vertical excitation energies from S0 to S1 and S2 are listed in Table 1 and displayed in Fig. 1. These results are highly consistent with those obtained with the CASPT2//SA3-CASSCF(16,13)//6-31G**, CASPT2//SA3-CASSCF(12,10)//cc-pVDZ, and CASPT2//SA3-CASSCF(12,10)//cc-pVTZ methods, as shown in Table S2 (ESI†). The most relevant molecular orbitals are provided in Fig. S2 (ESI†). The S1 state is a “bright” state with ππ* character, with the transition oscillator strength of 0.08 calculated using the TDDFT method. The vertical excitation energy of S0-cis-enol to S1 is 4.09 eV, which agrees well with the value of 4.11 eV obtained using the TDDFT method23,32 and experimental values of 3.77–3.96 eV.17,19,36 The S2 state of S0-cis-enol is “dark” with nπ* character and the transition oscillator strength of 0.00. The vertical excitation energy to S2 is 4.18 eV, which is consistent with the reported value of 4.23 eV.23 At the FC point, the S1 and S2 states are degenerate with an energy gap of less than 0.1 eV.32 This degeneracy (∼0.3 eV) is present in the results calculated using the CASSCF method (Table S2, ESI†). Thus, the good agreement between our calculation results and previous theoretical and experimental values indicates the reliability of the employed calculation methods and models. The vertical excitation energy of S0-cis-keto from S0 to S1 and S2 is 2.98 eV and 3.77 eV, respectively. For the S0-trans-enol tautomer, the excitation energy to the S1 and S2 states is 3.98 and 4.27 eV, respectively.
Minimum energy conical intersections (MECIs)
Fig. 2 displays the five MECIs located between two adjacent electronic states and their energies relative to the energy of S0-cis-enol, which are in line with those calculated using the CASPT2//CASSCF(16,13)//6-31G**, CASPT2//SA3-CASSCF(12,10)//cc-pVDZ and CASPT2//SA3-CASSCF(12,10)//cc-pVTZ methods (Fig. S3, ESI†). The key geometrical parameters and specific energy information are summarized in Table 2. All MECIs were confirmed by the CASPT2 method with an energy gap less than 0.4 eV. Herein, we employed CIm/n-X to describe their features, where m and n indicate the two adjacent electronic states and X describes the structural feature (e.g. planar, pucker, ESIPT and twist). CIS2/S1-planar is located at 4.08 eV above the ground state minimum. The structure of CIS2/S1-planar is similar to that of S0-cis-enol, except that the bond lengths of C9O2 and C7O1 are shorter in CIS2/S1-planar. Perveaux et al.23 and Vennapusa et al.32 identified a similar structure in their TDDFT study, which was proposed as the channel for the re-population to S2. CIS2/S1-pucker-twist is located at 6.07 eV above S0-cis-enol, featuring a strongly twisted C9O2 bond and puckering motion of the ring. The related dihedral angle of C7C9O2H6 changes to −174.2° and the C8H5 bond is almost perpendicular to the heterocyclic ring. However, its high energy makes it difficult to reach. CIS2/S1/S0-ESIPT is a three-state CI, characterized by an intramolecular proton transfer process from O2 to O1. Its energy gap for S1/S0 and S2/S0 is 0.4 eV and 0.55 eV, respectively. In terms of geometry, the C9O2 and C7O1 bonds show obvious elongation, changing from a double bond character to a single bond character, which is similar to the keto-type MECI.32 CIS1/S0-pucker-twist was found at 5.33 eV above S0-cis-enol. Similar to the case of CIS2/S1-pucker-twist, it is characterized with strong out-of-plane deformation of the C8H5 bond and rotation around the C9O2 bond. The related dihedral angle of O3C8C9H5 and C7C9O2H6 is 133.3° and 130.9°, respectively. CIS1/S0-opening is 5.01 eV above S0-cis-enol. It is mainly characterized by bond breakage between the C7 and C9 atoms, with an increase in their distance to 2.625 Å. Furthermore, it contains a pronounced twisting motion around the C9O2 bond, with the dihedral angle C7C9O2H6 of −69°. For the first time, we located the last four MECIs here, which may play important roles in the deactivation of 3-HC in the gas phase.| Geo. | C9O2 | O2H6 | O1H6 | C7C9 | C7O1 | C7C9O2H6 | O3C8C9H5 | REa | ΔS1−S0 | ΔS2−S1 |
|---|---|---|---|---|---|---|---|---|---|---|
| a The relative energy (RE) between listed geometries and the energy of S0-cis-enol. | ||||||||||
| CIS2/S1-Plane | 1.333 | 0.956 | 2.101 | 1.495 | 1.220 | 0.0 | 180.0 | 4.08 | 3.94 | 0.05 |
| CoIn23 | 1.349 | 0.970 | 2.077 | 1.448 | 1.228 | 0.0 | 180.0 | — | — | — |
| Enol-MECI32 | 1.347 | 0.982 | 2.088 | 1.466 | 1.287 | 0.0 | 180.0 | 3.90 | — | — |
| CIS2/S1/S0-ESIPT | 1.554 | 2.638 | 0.943 | 1.361 | 1.381 | 0.0 | 180.0 | 3.99 | 0.40 | 0.15 |
| Keto-MECI32 | 1.352 | 2.274 | 0.968 | 1.408 | 1.362 | 0.0 | 180.0 | 3.26 | — | — |
| CIS2/S1-Pucker-Twist | 1.352 | 0.948 | 3.800 | 1.371 | 1.502 | −174.2 | −145.7 | 6.07 | 1.40 | 0.35 |
| CIS1/S0-Pucker-Twist | 1.393 | 0.946 | 3.676 | 1.310 | 1.378 | 133.3 | 130.9 | 5.33 | 0.16 | 2.11 |
| CIS1/S0-Opening | 1.331 | 0.948 | 3.834 | 2.625 | 1.183 | −69.0 | −177.2 | 5.01 | 0.15 | 1.19 |
Excited-state nonadiabatic dynamics
To gain insight about the specific roles of the MECIs in the deactivation process of photoexcited 3-HC, we carried out the excited-state dynamic simulations starting from S2. Fig. 3(a) shows the time-dependent fractional occupations of S0, S1 and S2, where 49 out of 50 trajectories were used for the following data analysis. We ruled out one trajectory with the non-conservation of total energy. Within the first 100 fs, more than 60% of the trajectories decayed to S1. All trajectories depopulated completely from S2 after 600 fs. We did not observe any “S1 → S2 up transfer” process during the whole simulation. Moreover, the S1 → S2 up transfer process was not present in the dynamic simulations starting from S1 (see sections below). At 150 fs, some trajectories approached the conical intersection region and the S1 → S0 decay process started. At the end of the simulation, the population of S0 reached 40% and about 60% of the trajectories remained in S1. This implies that there are additional decay channels with longer lifetimes and 800 fs is not enough for simulating these channels. | ||
| Fig. 3 (a) Time-dependent occupation of S2, S1 and S0 of the 3-HC system; (b) key bond lengths of the S2 → S1 hopping points; and (c) key bond lengths of the S1 → S0 hopping points. The green, orange and purple stars represent the C9O2, O1H6 and C7O1 bond lengths at the FC point, respectively. | ||
The excited-state lifetime of S1 and S2 can be estimated by fitting their time-dependent population with exponential decay functions. Based on the above-mentioned discussion, S1 and S2 are two degenerate states. The decay channel of S2 most likely is via fast the S2 → S1 internal conversion process. Hence, we used a monoexponential decay function to fit the time-dependent population of S2 (fitted up to 800 fs). We determined the lifetime of S2 to be 118 fs, which is within the typical timescale of the internal conversion process.8,55,56 We then applied bootstrapping to extract 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sets of data, each containing 1701 values. Subsequently, we fitted each set and obtained a 95% confidence interval of [118.90685135, 119.00291629], which is consistent with our fitting results. A plateau is present in the S2 population (Fig. 3a), which is attributed to the molecular structure undergoing a slight torsion in the C9O2 bond at the beginning of the simulation. Subsequently, this torsion returned to a planar configuration, which is a process that takes some time. The population of S1 builds up in the first ∼200 fs due to the S2 → S1 relaxation. The build-up process should have the same time constant as the lifetime of S2. There are several channels that can contribute to the decay process of S1 including isomerization, ESIPT and radiative processes. These processes are different in terms of energy and molecular configuration. Most likely, they will have different decay time constants, which causes S1 to show multiple lifetimes. Thus, to obtain this information quantitatively, we fitted the time-dependent population of S1 (P1(t)) with the sum of an exponential growth function (build-up process of S1) and a bi-exponential decay function.
000 sets of data, each containing 1701 values. Subsequently, we fitted each set and obtained a 95% confidence interval of [118.90685135, 119.00291629], which is consistent with our fitting results. A plateau is present in the S2 population (Fig. 3a), which is attributed to the molecular structure undergoing a slight torsion in the C9O2 bond at the beginning of the simulation. Subsequently, this torsion returned to a planar configuration, which is a process that takes some time. The population of S1 builds up in the first ∼200 fs due to the S2 → S1 relaxation. The build-up process should have the same time constant as the lifetime of S2. There are several channels that can contribute to the decay process of S1 including isomerization, ESIPT and radiative processes. These processes are different in terms of energy and molecular configuration. Most likely, they will have different decay time constants, which causes S1 to show multiple lifetimes. Thus, to obtain this information quantitatively, we fitted the time-dependent population of S1 (P1(t)) with the sum of an exponential growth function (build-up process of S1) and a bi-exponential decay function.
P 1(t) = −A1 exp(t − t0/τ1) + A2 exp(t − t0/τ2)+ A3 exp(t − t0/τ3), where A1, A2 and A3 represent the amplitude for different processes, t0 is time zero, and τ1, τ2, and τ3 are the fitted lifetimes. Here, we fixed the growth lifetime of S1 to be 118 fs, which is the same as the decay lifetime of S2. We obtained τ2 = 110 fs, τ3 ≫ 800 fs, which indicates that there are at least two different decay processes for S1. As illustrated in Fig. 1 and the calculated multiple MECIs (Fig. 2), this is straightforward for the presence of multiple decay channels for S1. However, it is rather unusual for the lifetimes of the different decay channels to show such a big difference.
Thus, to understand the different decay channels, initially we analyzed the structural change for the fast decay processes, i.e., the hopping events of S2 → S1 and S1 → S0, as shown in Fig. 3(b) and (c), respectively. Here, we focus on the key bond lengths, where in the case of both S2 → S1 and S1 → S0, the C7O1 and C9O2 bond lengths show a clear elongation (from 1.204 Å and 1.347 Å at FC to over 1.400 Å at the hoping events, respectively). The most obvious change was the elongation of the hydrogen bond between the H6 and O1 atoms (from 2.199 Å at FC to over 2.200 Å at the hoping events). These geometrical changes indicate that a twisting motion of the C9O2 bond is present in the hopping events.
The time-dependent key geometrical parameters (Fig. 4) provide information about the structural changes during the hopping events. They offer the possibility to examine whether the ESIPT process occurs during the simulation, given that the ESPIT process is characterized by the breaking of the O2H6 bond and the formation of the O1H6 bond. However, we did not observe this change, as shown in Fig. 4(a); instead, the O2H6 bond remained constant at around 1 Å and the O1H6 distance increased due to the twisting motion of the C9O2 bond. Hence, we can conclude that the ESIPT process was absent within the simulation time range. To provide a clear picture of the roles played by the twisting motion in the deactivation processes, we plotted the dihedral angles of C7C9O2H6 and O3C8C9H5 from all 49 trajectories (Fig. 4(b) and (c), respectively) with the S2 → S1 and S1 → S0 hopping events. All the S2 → S1 hopping events occur around the dihedral angles of C7C9O2H6 of 0° and O3C8C9H5 of ±180°, indicating that the twisting motion of the C9O2 bond does not play an important role in the relaxation process of S2. Alternatively, the S1 → S0 hopping events occur around the dihedral angles of C7C9O2H6 of ±100° and O3C8C9H5 of ±140°, suggesting that the S1 → S0 decay process is characterized by a twisting motion of the C9O2 bond and clear deformation of the heterocyclic ring around the C8 atom.
 | ||
| Fig. 4 Variation in (a) O2H6 and O1H6 bond lengths and (b) dihedral angle of C7C9O2H6 and (c) O3C8C9H5 as a function of the simulation time. The blue and rosy balls represent the S2 → S1 and S1 → S0 hopping events, respectively. | ||
Moreover, we summarized the changes in the bond lengths of C7C9 and C7O1 during the simulation time in Fig. S5 (ESI†). The C7O1 bond length fluctuated around 1.3 Å throughout the simulation period, while the C7C9 bond length showed a larger range of variation, mostly above 1.45 Å. Moreover, two trajectories of C7C9 bond cleavage were observed. These hopping events are directly linked with or went through the relevant MECIs. To visualize this, we summarized the key parameters of the S2 → S1 and S1 → S0 hopping events together with the five MECIs in Fig. 5. Almost all the S2 → S1 hopping points are located around CIS2/S1-plane, but far away from CIS2/S1-pucker-twist. This suggests that only CIS2/S1-plane will be easily accessible during the S2 → S1 relaxation dynamics, which is in good agreement with the fact that the two states are degenerate and the fast internal conversion process. The S1 → S0 hopping points are scattered around the two S1/S0 MECI points related with the pucker-twisting and ring-opening motion, which accounts for the ultrafast decay process of the S1 population. These hopping events are far away from CIS2/S1/S0-ESIPT, indicating that the bond twisting and ring-opening motion rather than the ESIPT process play important roles in the deactivation process of S1. Our results from the simulations using the Newton-X package combined with TDDFT methods indicate that the 3-HC molecule tends to undergo deactivation through C9O2 bond torsion, as shown in Fig. S6 (ESI†).
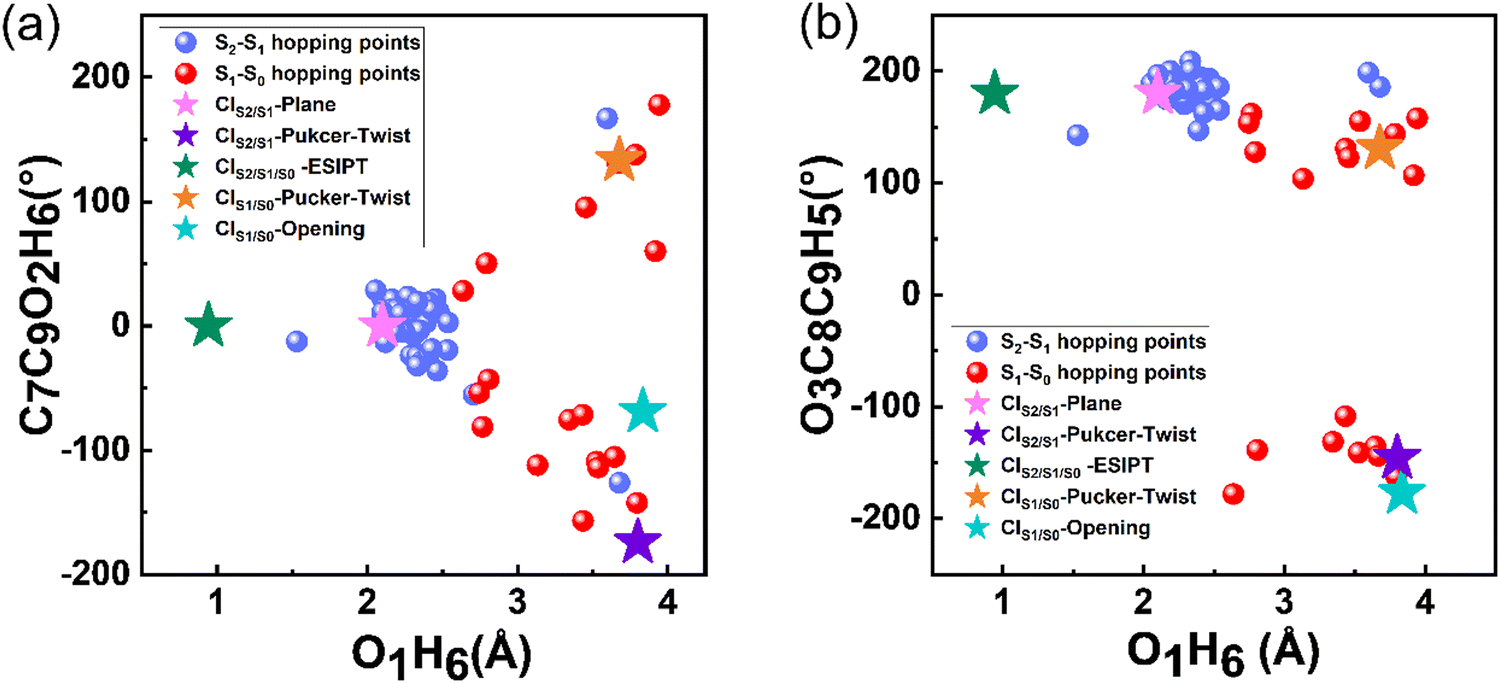 | ||
| Fig. 5 Diagram of the dihedral angles of C7C9O2H6 (ordinate) and O1H6 (abscissa) (a) and C3C8O9H5 (ordinate) and O1H6 (abscissa) (b) of the S2 → S1 and S1 → S0 hopping events. The stars with different colours represent the five MECIs. | ||
To avoid introducing any bias by using S2 as the starting point for the simulation, we performed dynamics simulation using S1 as the starting point. Specifically, 35 trajectories were employed for the simulation, among which one was discarded due to a violation of energy conservation. Note that 20 trajectories completed deactivation back to S0, and the remaining 14 trajectories were still in S1 at the end of the simulation (Fig. S7, ESI†). The results of the dynamics simulations initiated from S1 (Fig. S8, ESI†) are similar to the findings from the simulations initiated from S2, except that the S2 → S1 internal conversion process was absent. Neither S1 → S2 up-transfer nor ESIPT was observed during the dynamic simulations. Hence, the dynamics of S1 is independent of the initial state.
Deactivation mechanism and comparison with experiments
Based on the excited-state dynamic simulations, we found that the ESIPT process did not show a direct contribution to the S1 → S0 relaxation of 3-HC within the simulation time. Thus, to understand the reason for this, we constructed the energy profiles with the LIIC method connecting the FC point and three S1/S0 MECIs (Fig. 6). These energy profiles were then confirmed using the CASPT2//SA3-CASSCF(16,13)//6-31G**, CASPT2//SA3-CASSCF(12,10)//cc-pVDZ and CASPT2//SA3-CASSCF(12,10)//cc-pVTZ methods, as shown in Fig. S9–S11 (ESI†). A large energy barrier (∼1.63 eV) is present along the ESIPT pathway, which prevented the ESIPT process from occurring in our simulation timescale. However, the energy barrier may be overestimated with the LIIC method, and thus we re-optimized the excited-state transition state by picking the highest energy point on the LIIC path, as displayed by the red star in Fig. 6(a). The energy barrier decreased to 0.8 eV, which is still too high to overcome. Thus, to validate the high energy barrier for ESIPT, we employed a scan of the bond length of O1H6 to obtain the potential energy curve for S1, as shown in Fig. S13(a) (ESI†). This approach corroborated our previous findings, with the energy barrier for ESIPT estimated to be approximately 0.7 eV, which aligns closely with the revised barrier obtained from the excited-state transition-state re-optimization. Different from CIS2/S1/S0-ESIPT, there is a potential well along the other two paths (CIS1/S0-pucker-twist and CIS1/S0-opening, Fig. 6(b) and (c), respectively). The existence of the potential well can cause the system to be easily trapped in it during the relaxation process in S1, and the photoisomerization pathway from the cis-enol to trans-enol form is about 0.08 eV, which was obtained by scanning the C7C9O2H6 dihedral angle change from 0° to 180°, as shown in Fig. S13(b) (ESI†). At the end of our simulation, about 57% of all the trajectories remained in S1. This means the lifetime of S1 can be much longer than our simulation time (800 fs), as indicated by the fitted second lifetime of ≫ 800 fs. These trajectories can be categorised into two branches based on their geometrical information (Fig. S14, ESI†). One part remains a semi-planar configuration with the O1H6 bond length shorter than 2.75 Å and the dihedral angle of C7C9O2H6 smaller than 30°. The other part displays a torsion of the C9O2 bond, which is trapped in the potential well along the twisting motion-driven deactivation channel, as shown in Fig. 6(b). Apart from the two branches showing difference in geometry, all structures of the remaining trajectories have lower energy than the FC point in S1 and MECIs between S1 and S0 (Fig. S15, ESI† and Fig. 2(b)), which means that these trajectories can be activated via thermal energy and decay back to S0 (e.g. via ESIPT process) on a longer timescale. Furthermore, the energy barrier presented at the ESIPT pathway may be lowered by the surrounding solvent molecules, which makes the ESIPT process possible in the solution phase, as observed experimentally.16,17 | ||
| Fig. 6 Energy profiles constructed using the LIIC method connecting the Franck–Condon geometry and (a) CIS2/S1/S0-ESIPT, black star: S1 energy of the FC point, red star: S1 energy of the transition state between cis-enol and CIS2/S1/S0-ESIPT, blue star: CIS2/S1/S0-ESIPT; (b) CIS1/S0-pucker-twist; and (c) CIS1/S0-opening computed at the CASPT2//SA3-CASSCF(12,10)//6-31G* level. | ||
A schematic overview of the excited-state dynamics of the 3-HC system is presented in Fig. 7. Upon photoexcitation to S2, the system relaxes to S1via a barrierless bond alternation process. Within our simulation time, 39% and 4% of the total simulated trajectories from S1 decay to the ground state via CIS1/S0-pucker-twist, and CIS1/S0-opening, respectively, and about 26% of these trajectories finally generate the trans-enol product in S0. The presence of a large energy barrier hinders the ESIPT process. Hence, the nonradiative deactivation of the photoexcited 3-HC system in the gas phase is mainly induced by bond twisting motion and ring-opening motion. However, these motions may be restricted in solvents due to the steric hindrance effect, forcing the system to relax via the ESIPT process. As discussed above, a large energy barrier is present in the ESIPT pathway, and thus thermal activation can enable this process to occur at a slow speed, e.g., on the picosecond timescale, which has been observed experimentally in the solution phase. However, the destruction of intramolecular hydrogen bonding due to torsional motion may slowdown the ESIPT process. Thus, the two ESIPT time constants observed in solution can be explained. Previous experiments mainly focused on studying the influence of protonic and polarity on the behavior of the excited-state dynamics of the 3-HC system. Alternatively, we suggest that future experimental studies pay more attention to the effect of solution viscosity on the deactivation mechanism of the 3-HC system.
 | ||
| Fig. 7 Schematic overview of the excited-state dynamics of the 3-HC system following excitation to the S2 state. | ||
Conclusion
In the current work, we investigated the excited-state deactivation process of the 3-HC system in the gas phase using high-level electronic structure calculations and on-the-fly excited-state dynamic simulations. Based on the calculations, we found that the 3-HC system prefers decaying to S0 by a nonradiative process rather than fluorescence. The results coincide with the experimental observations. The non-radiative process is mainly driven by the twisting of the C9O2 bond and the deformation of the heterocyclic ring. We did not observe the ESIPT process in our dynamic simulations due to the presence of a large energy barrier in this pathway. Alternatively, the ESIPT process of 3-HC has been observed in solvent. This difference is partly because the twisting motion and the ring deformation would be restricted due to the steric-hindrance effect from solvent molecules. In addition, the energy barrier for ESIPT may decrease and the 3-HC system would gain enough thermal energy under the experimental condition (room temperature) to overcome the energy barrier. This ESIPT process would happen at a slow speed, while the destruction of intramolecular hydrogen bonding due to torsional motion may further slowdown the ESIPT process. This could explain the fact that two ESIPT reaction constants were observed in the solution phase. The present work proposes a comprehensive description of the intrinsic photophysical and photochemical properties of 3-HC, which not only provides a foundation for the further investigation of the effect of environment, but also theoretical guidance for the rational design of molecular photoswitches. Furthermore, given that the photoinduced dynamic behaviour of 3-HC and its derivatives shows strong dependence on the environment, it is worthwhile to further explore their solute–solvent hydrogen-bonding interactions in different solvents.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21803077), the Fundamental Research Funds for the Central Universities (No. 24CX03009A, and 23CX03016A) and Novo Nordisk Foundation (NNF22OC0073582).References
- A. D. Dunkelberger, R. D. Kieda, J. Y. Shin, R. Rossi Paccani, S. Fusi, M. Olivucci and F. F. Crim, J. Phys. Chem. A, 2012, 116, 3527 CrossRef CAS PubMed.
- M. Andresen, M. C. Wahl, A. C. Stiel, F. Grater, L. V. Schafer, S. Trowitzsch, G. Weber, C. Eggeling, H. Grubmuller, S. W. Hell and S. Jakobs, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13070 CrossRef CAS PubMed.
- R. Mitric, U. Werner and V. Bonacic-Koutecky, J. Chem. Phys., 2008, 129, 164118 CrossRef PubMed.
- C. J. Xia, D. S. Liu and H. C. Liu, Optik, 2012, 123, 1307 CrossRef CAS.
- T. Wendler, C. Schutt, C. Nather and R. Herges, J. Org. Chem., 2012, 77, 3284 CrossRef CAS PubMed.
- J. S. Chen, M. H. Yuan, J. P. Wang, Y. Yang and T. S. Chu, J. Phys. Chem. A, 2014, 118, 8986 CrossRef CAS PubMed.
- J. S. Chen, P. W. Zhou, S. Q. Yang, A. P. Fu and T. S. Chu, Phys. Chem. Chem. Phys., 2013, 15, 16183 RSC.
- W. Zhang, L. Zhao, B. W. Laursen and J. Chen, Phys. Chem. Chem. Phys., 2022, 24, 26731 RSC.
- J. S. Chen, T. S. Chu, R. Z. Liu and Y. Yang, Theor. Chem. Acc., 2014, 133, 1411 Search PubMed.
- L. Zhao, S. Jiang, Y. He, L. Wu, T. D. James and J. Chen, Phys. Chem. Chem. Phys., 2024, 26, 13506 RSC.
- S. Basu, S. Mondal and D. Mandal, J. Chem. Phys., 2010, 132, 034701 CrossRef PubMed.
- S. Chaudhuri, K. Basu, B. Sengupta, A. Banerjee and P. K. Sengupta, Luminescence, 2008, 23, 397 CrossRef CAS PubMed.
- A. P. Demchenko, K. C. Tang and P. T. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC.
- C. Dyrager, A. Friberg, K. Dahlén, M. Fridén-Saxin, K. Börjesson, L. M. Wilhelmsson, M. Smedh, M. Grøtli and K. Luthman, Chem. – Eur. J., 2009, 15, 9417 CrossRef CAS PubMed.
- A. S. Klymchenko, C. Kenfack, G. Duportail and Y. Mely, J. Chem. Sci., 2007, 119, 83 CrossRef CAS.
- R. Das, A. S. Klymchenko, G. Duportail and Y. Mely, Photochem. Photobiol. Sci., 2009, 8, 1583 CrossRef CAS PubMed.
- K. Chevalier, A. Grun, A. Stamm, Y. Schmitt, M. Gerhards and R. Diller, J. Phys. Chem. A, 2013, 117, 11233 CrossRef CAS PubMed.
- C. Sun, X. Su, Q. Zhou and Y. Shi, Org. Chem. Front., 2019, 6, 3093 RSC.
- G. A. Brucker and D. F. Kelley, J. Phys. Chem., 1987, 91, 2862 CrossRef CAS.
- A. Stamm, M. Weiler, A. Bracher, K. Schwing and M. Gerhards, Phys. Chem. Chem. Phys., 2014, 16, 21795 RSC.
- J. D. Huang, J. Zhang, D. Chen and H. Ma, Org. Chem. Front., 2017, 4, 1812 RSC.
- K. R. Nandipati, Z. Lan, H. Singh and S. Mahapatra, J. Chem. Phys., 2017, 146, 214304 CrossRef CAS PubMed.
- A. Perveaux, M. Lorphelin, B. Lasorne and D. Lauvergnat, Phys. Chem. Chem. Phys., 2017, 19, 6579 RSC.
- Y. Yang, Z. Tang, P. Zhou, Y. Qi, Y. Wang and H. Wang, J. Mol. Liq., 2018, 260, 447 CrossRef CAS.
- J. Liu, X. Chen, Y. Zhang, G. Gao, Z. Xueyan, S. Hou and Y. Hou, New J. Chem., 2018, 42, 12918 RSC.
- G. A. Ndongo, M. O. Boyomo and P. A. Owono, J. Mol. Model., 2018, 24, 92 CrossRef PubMed.
- K. R. Nandipati, A. K. Kanakati, H. Singhb and S. Mahapatra, Phys. Chem. Chem. Phys., 2019, 21, 20018 RSC.
- K. Kaur, J. Tomar and M. Bansal, Can. J. Chem., 2019, 97, 584 CrossRef CAS.
- J. S. Lim, H. S. You, S.-Y. Kim, J. Kim, Y. C. Park and S. K. Kim, J. Chem. Phys., 2019, 151, 244305 CrossRef PubMed.
- N. Anand, S. V. K. Isukapalli and S. R. Vennapusa, J. Comput. Chem., 2020, 41, 1068 CrossRef CAS PubMed.
- P. Nag and S. R. Vennapusa, J. Chem. Phys., 2020, 153, 084306 CrossRef CAS PubMed.
- P. Nag, N. Anand and S. R. Vennapusa, J. Chem. Phys., 2021, 155, 094301 CrossRef CAS PubMed.
- P. Nag and S. R. Vennapusa, J. Photochem. Photobiol., A, 2022, 431, 114024 CrossRef CAS.
- P. Nag and S. R. Vennapusa, J. Photochem. Photobiol., A, 2022, 427, 113767 CrossRef CAS.
- P. K. Sengupta and M. Kasha, Chem. Phys. Lett., 1979, 68, 382 CrossRef CAS.
- M. Itoh, Pure Appl. Chem., 1993, 65, 1629 CrossRef CAS.
- S. Ash, S. P. De, H. Beg and A. Misra, Mol. Simul., 2011, 37, 914 CrossRef CAS.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. D. Vico, I. F. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. L. Manni, H. Lischka, D. X. Ma, P. Å. Malmqvist, T. Muller, A. Nenov, M. Olivucci, T. B. Pedersen, D. L. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. S. Segarra-Martí, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Angeli, J. Comput. Chem., 2009, 30, 1319 CrossRef CAS PubMed.
- L. Zhao, G. Xu, Y. Guo, H. Zheng, P. Dong and J. Chen, J. Photochem. Photobiol., A, 2023, 434, 114255 CrossRef CAS.
- L. Zhao, P. Zhou, X. Liu, H. Zheng, K. Zhan, J. Luo and B. Liu, Spectrochim. Acta, Part A, 2021, 244, 118884 CrossRef CAS PubMed.
- L. Zhao, H. Zheng, K. Zhan, Y. Guo, B. Liu and G. Xu, J. Phys. Chem. A, 2021, 125, 165–174 CrossRef CAS PubMed.
- L. Zhao, J. Liu and P. Zhou, J. Chem. Phys., 2018, 149, 034309 CrossRef PubMed.
- L. Zhao, P. W. Zhou, B. Li, A. H. Gao and K. L. Han, J. Chem. Phys., 2014, 141, 235101 CrossRef PubMed.
- A. H. Gao, B. Li, P. Y. Zhang and K. L. Han, J. Chem. Phys., 2012, 137, 204305 CrossRef PubMed.
- W. C. Swope, H. C. Andersen, P. H. Berens and K. R. Wilson, J. Chem. Phys., 1982, 76, 637 CrossRef CAS.
- C. Zhu and H. Nakamura, J. Chem. Phys., 1992, 97, 8497 CrossRef CAS.
- C. Zhu and H. Nakamura, J. Chem. Phys., 1993, 98, 6208 CrossRef CAS.
- C. Zhu and H. Nakamura, J. Chem. Phys., 1994, 101, 10630 CrossRef CAS.
- C. Zhu and H. Nakamura, J. Chem. Phys., 1995, 102, 7448 CrossRef CAS.
- L. Sun and W. L. Hase, J. Chem. Phys., 2010, 133, 044313 CrossRef PubMed.
- S. Kube, C. Lasser and M. Weber, J. Comput. Phys., 2009, 228, 1947 CrossRef CAS.
- M. Barbatti, M. Ruckenbauer and F. Plasser, et al., Newton-X: a surface-hopping; program for nonadiabatic molecular dynamics, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CAS.
- M. Bixon and J. Jortner, J. Chem. Phys., 1968, 48, 715 CrossRef CAS.
- S. Jiang, Y. He, J. H. Brandt, L. Zhao and J. Chen, J. Phys. Chem. Lett., 2023, 14, 10482 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further information regarding the orbitals included in the active spaces, the orbitals involved in forming the first three excited singlet states, the comparison of key geometrical parameters among different calculation methods, and the Cartesian coordinates about the stable structures and MECIs are provided. See DOI: https://doi.org/10.1039/d4cp01190b |
| This journal is © the Owner Societies 2024 |