
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpurious proton transfer in hydrogen bonded dimers†
Joanatan
Bautista-Renedo
 *a and
Joel
Ireta
*b
*a and
Joel
Ireta
*b
aDepartamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, Ciudad de México 09340, Mexico. E-mail: joanatan_br@xanum.uam.mx
bDepartamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, Ciudad de México 09340, Mexico. E-mail: iret@xanum.uam.mx
First published on 22nd July 2024
Abstract
In some hydrogen bonded systems, the proton may translocate along the hydrogen bond (hb) upon geometry optimization with electronic structure methods like density functional theory (DFT). Such proton transfer (pt) events, however, may be spurious. In this work, spurious pt events are investigated in a set of hydrogen bonded dimers formed with molecules HXN, where X stands for C, Si, Ge and Sn. It is found that standard approximations to the electronic exchange and correlation (xc) functional either predict spurious pt events or too strong hbs in all the (HXN)2 dimers except the (HCN)2 one. The latter result is revealed by comparing DFT calculations against wave function methods. Such spurious pt events may be avoided by fine-tuning the percentage of exact exchange (ex) in hybrid xc-functionals. It is shown that the minimum amount of ex to avoid a spurious pt event ranged from 8% to 90%, depending on the system, basis set and xc-functional approximation used. However, these fine-tuned xc-functionals inadequately describe the hb in the (HXN)2 dimers. Moreover, it is determined that the spurious pt event originates from a wrong description of the isolated HXN molecules by xc-functionals that do not include ex or a small amount of it. Therefore, it is argued that one can determine if a pt event is spurious by analyzing the geometry and electronic structure of the isolated molecule.
1 Introduction
The hydrogen bond (hb), an interaction between an atom D (donor) and an atom or region A (acceptor) actuated by a H atom covalently bonded to D, influences decisively the properties of matter in which it occurs. The hb is also considered as a preliminary stage of a proton transfer (pt) reaction,1,2 a crucial event involved in many chemical and biological processes.3–6 Therefore, this fundamental interaction has been thoroughly investigated experimentally and theoretically to understand the physicochemical phenomena connected to its origin, and how it eases pt events. The theoretical procedures customarily used to investigate hydrogen bonded systems are electronic structure methods like second order Møller–Plesset perturbation theory (MP2), coupled clusters with singles, doubles and perturbative triple excitations (CCSD(T)),7 and density functional theory (DFT) in its Kohn–Sham formulation.8–10 CCSD(T) is considered the gold standard method for investigating hbs. MP2 is also considered to be a reliable method for studying hbs. However, these methods are computationally very demanding, hence not suitable nowadays, to calculate very large systems (of hundreds or thousands of atoms) or systems modeled using periodic boundary conditions like solids or surfaces. DTF can be used to calculate the systems of such characteristics.11–13 Moreover, depending on the approximation to the exchange and correlation (xc) functional used, DFT may adequately describe hydrogen bonded systems like peptides,14–17 molecular crystals,18–20 or catalytic materials,21–24i.e. complex systems whose physicochemical properties may be connected to hbs and pt events.In the definition of a hb recommended by the international union of pure and applied chemistry,25 the atom D is considered to be more electronegative than the H atom. Nevertheless, it has been shown experimentally and theoretically that hbs can be formed when D is less electronegative than H.26 Investigating HXN dimers (Fig. 1), where X = C, Si, Ge, or Sn, with MP2 and CCSD(T), we have also found evidence for formation of hbs if D is less electronegative than H, as in (HXN)2 where X = Si, Ge or Sn.27 Unexpectedly, as shown in this work, DFT with standard xc-functionals predicts a spontaneous pt event in these dimers upon geometry optimization, except for (HCN)2, the case in which D is more electronegative than H, as in conventional hbs. Spurious pt events upon DFT geometry optimization have also been reported along with the formation of conventional hbs in organic crystals.28 We wonder why standard approximations to the xc-functional fail to describe the hbs in (HXN)2 when D is less electronegative than H and predict spurious pt events, and how to know that a pt event is spurious if CCSD(T) or MP2 calculations are not available.
 | ||
| Fig. 1 Scheme of HXN dimers and their hydrogen bond (hb) interaction. X stands for C, Si, Ge or Sn. | ||
DFT calculations may suffer from large inaccuracies related to the approximation to the xc-functional chosen,29e.g. it is known that xc-functionals that include generalized gradients of the electronic density (GGA) into their formulation, may lead to wrong predictions of pt barriers,30 and spontaneous pt events.28 It has been suggested that such false pt events are connected to the inability of current xc-functionals to correctly predict the linear dependence of the energy with respect to fractional electron numbers,28,29,31,32 which triggers spurious delocalization of electrons or delocalized electrons with too low energies.33 This electron delocalization error can be linked to the underestimation of reaction barriers and excitation energies and overestimations of binding energies in charge transfer complexes,29 which may lead to incorrect identification of protonation sites, especially in acid–base multicomponent crystals.28,34,35 In addition, it has been shown that the electron delocalization error can be reduced incorporating high percentages of exact exchange (ex) in the xc-functionals,28,29,32,33,36,37e.g. about 50% of ex is required for an accurate description of the energetics of charge transfer complexes, and about 40% to avoid spurious pt events in some molecular crystals.
In this work, the spurious pt events observed in the above mentioned HXN dimers are thoroughly investigated through DFT geometry optimizations. In order to do this, different approximations to the xc-functional are tested. It is shown that standard approximations to the xc-functional (GGAs, meta-GGAs, which include a dependence of the Laplacian of the density and/or the kinetic energy density, hybrid GGAs, with and without range separation and hybrid meta-GGAs) predict spurious pt events in these dimers. The tested functionals are the Perdew, Burke and Ernzerhof (PBE) GGA,38 the strongly-constrained and appropriately normed (SCAN) meta-GGA39–41 and its Laplacian-dependent deorbitalized variant (SCAN-L),42 the Minnessota M06-L meta-GGA,43 the PBE0 global hybrid functional, which have 25% of ex, and two versions of the Heyd–Scuseria–Ernzerhof range-separated hybrid functionals (HSE03 and HSE06),44–48 which have 25% of ex at short range, the LC-PBE range-separated hybrid functional that includes 100% of ex at long range and the Minnessota M06-2X hybrid meta-GGA which includes 54% of ex.49 Next, we gradually increase the ex contribution in PBE0 and the HSE family to find the amount of ex needed to avoid the pt in the investigated dimers. It is found that this amount is system and basis set dependent, however, beyond 44% of ex the pt is no longer observed in any of the investigated dimers. Additionally, it is shown that the maximum amount of xc needed to avoid pt increases as the short range in the HSE functional is reduced. Furthermore, we find that the amount of ex needed to avoid pt does not guarantee an adequate description of hb properties like their strength (Ehb), i.e. the dimer association energy, the X–H vibrational frequencies (νX–H) or the X⋯H hb distance (dhb). Lastly, we show that by analysing the local softness (s(r)), an intrinsic reactivity index that measures the capacity of the system to receive or donate electrons at a given molecular site,50,51 and the magnitude of X–H bond distance (dX–H), one can determine whether a spurious pt event is going to happen, even without performing a DFT calculation of the hydrogen bonded dimer, i.e. solely analyzing the monomer electronic structure and geometry. Furthermore, it is argued that these results give us insight into how the formation of a hb eases a proton transfer process, and about the ability of xc-functionals to describe non-conventional hbs.
2 Computational details
HXN monomers and dimers at angles of interaction of 180° are fully optimized using DFT with Grimme's-D3 dispersion correction52 and the next set of xc-functionals: PBE, PBE0, LC-PBE, HSE03, HSE06, M06-L and M06-2X, while for SCAN and SCAN-L the Grimme's-D2 dispersion correction53 is used as no D3 parameters for these functionals are implemented in the code used for the calculations. It is well known that Minnesota functionals are grid-sensitive, and for this reason, an extra-fine integration grid was used as well as for all the tested functionals.54 This extra-fine grid assures a convergence in energy of ∼10−8 a.u.55 We also calculate (HXN)2, and its monomers, using MP2 with a frozen core and resolution of the identity approximations (RI-MP2) for comparison purposes. In all calculations the triple-zeta basis set of Ahlrichs with fuzzy functions def2-TZVPD,56,57 the Dunning aug-cc-PVYZ basis sets with Y = D, T, and Q and the aug-cc-PVYZ-PP basis set for Sn are used.58–60 Hereinafter, the latter basis set is solely referred to as aug-cc-PVYZ. The counterpoise method for correcting the basis set superposition error (BSSE) is used for calculating Ehb. Conformations with a distance A⋯H shorter than X⋯H upon geometry optimization are considered to result from a pt event. All electronic structure calculations and vibrational frequencies are carried out with the NWChem 6.8 code,55 while the Multiwfn v3.8 program61 is used for the analysis of properties related to the electron density. For geometry optimization calculations, it is considered that the structure has reached the minimum of the potential energy surface (PES) if the maximum and root mean square gradients are equal to or smaller than 1.5 × 10−5 and 1.0 × 10−5 in atomic units, respectively. Moreover, for the optimized structures obtained utilizing the def2-TZVPD basis set, it is confirmed that structures are at the minimum of PES corroborating the absence of imaginary frequencies.It is worth mentioning that the implementation of HSE03 in the NWChem code, here denoted as HSE03NW, is slightly different from the original HSE03. The separation between long and short range in the HSE functionals is controlled by the w parameter, whose values are w = 0.11 bohr−1 in HSE06, and w = 0.15 bohr−1 in HSE03. However, in NWChem w = 0.33 bohr−1 is used in HSE03NW. As the larger the w value the smaller the short range, which is the section in which the ex is mixed with the GGA exchange, HSE03NW can be considered of ultra short range. We have tested both versions of the HSE03 functional. To determine the amount of ex to avoid pt we modify systematically the amount of ex in PBE0, HSE06, HSE03 and HSE03NW. To avoid confusions, the functionals in which we modify the percentage of ex are renamed m-PBE0, m-HSE03, m-HSE03NW and m-HSE06.
To determine whether a pt could happen we calculate the quantity s(r) (eqn (1)) for the HXN isolated monomers. Under finite differences and the rigid electronic band approach s(r) can be estimated according to:51,62,63
 | (1) |
Although s(r) is usually applied for elucidating the reactivity of organic molecules, proteins and surfaces,51,64–67 it has also being correlated with the ability of a chemical group to form hbs,23 and here we show that it can help to elucidate whether a pt could occur upon geometry optimization, without carrying the geometry optimization.
3 Results
In Tables 1 and 2 are listed the dhb and Ehb values obtained with the tested xc-functionals and the aug-cc-PVQZ and def2-TZVPD basis sets. The results obtained with aug-cc-PVDZ and aug-cc-PVTZ are listed in Tables S1 and S2 of the ESI.† For comparison purposes, values obtained with MP2 and RI-MP2 are also listed along with those reported in ref. 27 calculated with MP2 and CCSD(T) at the complete basis set extrapolation (CBS). First, we notice that all xc-functionals together with the dispersion correction and any of the basis sets used predict no pt events in (HCN)2, the dimer in which D is more electronegative than H. This result is in accordance with the MP2 and RI-MP2 results (Table 1 and Table S1 in the ESI†). For the other dimers (Table 1 and Tables S1 and S2 in the ESI†) where D is less electronegative than H, pt events may be obtained upon geometry optimization depending on the functional and/or basis set used. For example, for (HSiN)2 PBE-D3 and SCAN-L-D2 predict a pt event disregarding the basis set utilized, however SCAN-D2 predicts a pt event only if the aug-cc-PVDZ basis set is used. Similarly, for (HGeN)2, PBE-D3 and SCAN-L-D2 predict pt events independently of the basis set used, as well as SCAN-D2 with all the basis sets but the aug-cc-PVTZ. For (HSnN)2 all the combinations of xc-functional/basis-set predict pt events, except if the M06-2X-D3 xc-functional is used with any of the basis sets or LC-PBE-D3 with aug-cc-PVQZ or def2-TZVPD. Nevertheless, MP2 and RI-MP2 predict no pt events for the investigated dimers independently of the basis set used.| Methoda | aug-cc-PVQZ | def2-TZVPD | ||
|---|---|---|---|---|
| d hb | E hb | d hb | E hb | |
| a Energies are basis set superposition error corrected. b Complete basis set extrapolation (CBS) of MP2 single point calculations on MP2/aug-cc-pVDZ optimized geometries. Results from ref. 27. c MP2/aug-cc-pVDZ optimized geometries. d CBS of CCSD(T) single point calculations on MP2/aug-cc-pVDZ optimized geometries. Results from ref. 27 | ||||
| PBE-D3 | 2.18 | −4.83 | 2.21 | −4.83 |
| SCAN-D2 | 2.19 | −5.52 | 2.20 | −4.07 |
| SCAN-L-D2 | 2.13 | −5.70 | 2.13 | −4.74 |
| M06-L-D3 | 2.17 | −4.01 | 2.16 | −4.23 |
| PBE0-D3 | 2.18 | −4.98 | 2.19 | −4.98 |
| PBE0 | 2.19 | −4.59 | 2.23 | −4.60 |
| HSE03-D3 | 2.22 | −4.69 | 2.23 | −4.69 |
| HSE03NW-D3 | 2.20 | −4.81 | 2.23 | −4.81 |
| HSE06-D3 | 2.22 | −4.68 | 2.23 | −4.68 |
| LC-PBE-D3 | 2.06 | −6.08 | 2.07 | −6.16 |
| M06-2X-D3 | 2.21 | −4.52 | 2.24 | −4.45 |
| MP2 | 2.19 | −4.80 | ||
| RI-MP2 | 2.22 | −4.59 | ||
| MP2/CBSb | 2.15c | −4.88 | ||
| CCSD(T)/CBSd | −4.68 | |||
| System/methoda | aug-cc-PVQZ | def2-TZVPD | ||
|---|---|---|---|---|
| d hb | E hb | d hb | E hb | |
| a Basis set superposition error corrected values obtained using the def2-TZVPD basis set. b pt stands for proton transfer. c Complete basis set extrapolation (CBS) of MP2 single point calculations on MP2/aug-cc-pVDZ optimized geometries. Results from ref. 27. d MP2/aug-cc-pVDZ optimized geometries. Results from ref. 27. e CBS of CCSD(T) single point calculations on MP2/aug-cc-pVDZ optimized geometries. Results from ref. 27. | ||||
| (HSiN)2 | ||||
| PBE-D3 | –pt– | –pt– | ||
| SCAN-D2 | 1.85 | −7.90 | 1.91 | −5.09 |
| SCAN-L-D2 | –pt– | –pt– | ||
| M06-L-D3 | 2.07 | −5.06 | 2.10 | −5.18 |
| PBE0-D3 | 1.95 | −7.00 | 1.96 | −7.11 |
| PBE0 | 1.96 | −6.41 | 1.98 | −6.41 |
| HSE03-D3 | 2.01 | −6.36 | 2.02 | −6.31 |
| HSE03NW-D3 | 1.92 | −6.44 | 1.94 | −6.56 |
| HSE06-D3 | 2.01 | −6.37 | 2.03 | −6.48 |
| LC-PBE-D3 | 1.82 | −9.59 | 1.84 | −9.72 |
| M06-2X-D3 | 2.08 | −6.79 | 2.12 | −7.10 |
| MP2 | 2.22 | −3.75 | ||
| RI-MP2 | 2.29 | −3.40 | ||
| MP2/CBSc | 2.21d | −3.68 | ||
| CCSD(T)/CBSe | −4.97 | |||
| (HGeN)2 | ||||
| PBE-D3 | –pt– | –pt– | ||
| SCAN-D2 | –pt– | –pt– | ||
| SCAN-L-D2 | –pt– | –pt– | ||
| M06-L-D3 | 2.02 | −4.75 | 2.03 | −5.46 |
| PBE0-D3 | 1.91 | −6.64 | 1.93 | −6.87 |
| PBE0 | 1.93 | −5.94 | 1.95 | −5.99 |
| HSE03-D3 | 1.97 | −5.99 | 1.99 | −5.92 |
| HSE03NW-D3 | 1.85 | −6.13 | 1.88 | −6.35 |
| HSE06-D3 | 1.98 | −6.00 | 2.00 | −6.25 |
| LC-PBE-D3 | 1.82 | −9.21 | 1.85 | −9.51 |
| M06-2X-D3 | 2.10 | −6.32 | 2.13 | −6.75 |
| MP2 | 2.01 | −3.18 | ||
| RI-MP2 | 2.21 | −3.03 | ||
| MP2/CBSc | 2.09d | −3.58 | ||
| CCSD(T)/CBSe | −4.91 | |||
| (HSnN)2 | ||||
| PBE-D3 | –pt– | –pt– | ||
| SCAN-D2 | –pt– | –pt– | ||
| SCAN-L-D2 | –pt– | –pt– | ||
| M06-L-D3 | –pt– | –pt– | ||
| PBE0-D3 | –pt– | –pt– | ||
| PBE0 | –pt– | –pt– | ||
| HSE03-D3 | –pt– | –pt– | ||
| HSE03NW-D3 | –pt– | –pt– | ||
| HSE06-D3 | –pt– | –pt– | ||
| LC-PBE-D3 | 1.65 | −9.17 | 1.71 | −9.04 |
| M06-2X-D3 | 2.00 | −5.72 | 2.11 | −5.46 |
| MP2 | 1.70 | −2.11 | ||
| RI-MP2 | 2.10 | −3.04 | ||
| MP2/CBSc | 1.91d | −3.49 | ||
| CCSD(T)/CBSe | −2.67 | |||
The dhb and Ehb values obtained using either def2-TZVPD or aug-cc-PVQZ basis sets and any of the tested functionals differ little between them, except if these basis sets are utilized together with the SCAN-D3 or SCAN-L-D2 xc-functionals. The latter combinations lead to differences between 1 and 3 kcal mol−1 for Ehb. Although differences in dhb are small, 0.06 Å at most (Tables 1 and 2). Yet, the results obtained with these two basis sets are consistent regarding the prediction of pt events. The other basis sets, aug-cc-PVDZ and aug-cc-PVTZ, give somewhat erratic results concerning pt events, particularly if these are used together with the SCAN-D2 or the LC-PBE-D3 xc-functionals. Hereinafter we use primarily the def2-TZVPD basis sets and solely key results are corroborated repeating the calculations using the aug-cc-PVQZ basis set.
The def2-TZVPD basis set together with the tested xc-functionals describe adequately the hb in (HCN)2, within an error bar of 0.6 kcal mol−1 with respect to CCSD(T)/CBS for the hb strength, and of 0.1 Å with respect to MP2/aug-cc-pVDZ results for dhb, except for LC-PBE-D3 that predicts a too strong hb, with Ehb ∼ 1.5 kcal mol−1 larger than the CCSD(T)/CBS result. For the other dimers ((HXN)2 where X = Si, Ge or Sn), however, the GGA PBE-D3 predicts pt events. The meta-GGAs-D3 either predict Ehb within an error bar of 0.6 kcal mol−1 and dhb within an error bar of 0.3 Å, or pt events; i.e. SCAN-L-D3 predicts pt events in the three dimers, SCAN-D3 in two ((HGeN)2 and (HSnN)2), and M06-L-D3 in one ((HSnN)2). Hybrid xc-functionals together with the D3 dispersion correction predict either too strong hbs, with error bars larger than 1.3 kcal mol−1, as in (HSiN)2 and (HGeN)2, or pt events, as in (HSnN)2, except for LC-PBE-D3 that does not predict pt events in any case. Yet, the largest deviations from the CCSD(T)/CBS Ehb values are obtained with LC-PBE-D3. These deviations lead to errors in the order of the expected values. The hybrid meta-GGA M06-2X together with the dispersion correction D3 also predicts no pt events, however, it greatly overestimates the hb strength with errors that can even be of the same magnitude as the Ehb value obtained with CCSD(T)/CBS. For MP2 the description of these hbs in which D is less electronegative than H is also challenging. The MP2/aug-cc-PVQZ error can be as large as 1.7 kcal mol−1, with respect to CCSD(T), in the calculations of Ehb. This error may increase in some dimers if RI-MP2/def2-TZVPD is used, e.g. for (HGeN)2 it amounts to 1.88 kcal mol−1. Still, neither MP2 nor RI-MP2 predict pt events.
According to the previous results only hybrid functionals with ex larger than 50% (LC-PBE and M06-2X) do not predict any spurious pt events in the investigated dimers. However, these functionals greatly overestimate the Ehb. To determine the minimum amount of ex that is required to avoid spurious pt events, we have systematically modified the percentage of ex in PBE0, HSE03, HSE03NW and HSE06 (see Fig. S1 in the ESI†). It is found that the minimum amount of ex to avoid pt is system dependent, e.g. for (HSiN)2 and (HGeN)2 less than 10% of ex in m-HSE06, m-PBE0 and m-HSE03, and between 13% and 15% in m-HSE03NW is required (see Table 3). However, that amount increases significantly for (HSnN)2, between 40% and 44% in m-HSE06, m-PBE0 and m-HSE03, and up to 90% in m-HSE03NW. In all these cases, m-HSE03NW requires the largest amount of ex to avoid the pt event. This result suggest that for range separated xc-functionals the smaller the short range the larger the ex needed to avoid the pt. Yet, Ehb is overestimated using the xc-functionals modified with the minimum amount of ex listed in Table 3, and dhb is underestimated. Increasing the amount of ex can improve the prediction of Ehb solely with the m-HSE03 functional, and with the other functionals the overestimation is increased. Nevertheless, the description of dhb will be improved with any of the xc-functionals tested if ex is increased (see Fig. S1 in the ESI†), except in (HSnN)2 calculated with m-HSE03 or m-HSE03NW. For that system, the dhb values obtained with the minimum amount of ex required to avoid pt, is already quite close to the one obtained with MP2. The amount of ex to avoid spurious pt events is also basis set dependent, although only slightly, e.g. the amount of ex increases between 1% and 4% if the calculations are performed with aug-cc-PVQZ instead of def2-TZVPD (Table 3).
| System | Methoda | % ex | d hb | E hb |
|---|---|---|---|---|
| a Data in parenthesis are obtained with the aug-cc-PVQZ basis set. The Ehb values are basis set superposition error corrected. | ||||
| (HSiN)2 | m-HSE06-D3 | 7(7) | 1.79(1.76) | −6.43(−6.49) |
| m-HSE03-D3 | 8 | 1.81 | −6.42 | |
| m-HSE03NW-D3 | 13 | 1.75 | −6.47 | |
| m-PBE0 | 8 | 1.80 | −6.06 | |
| (HGeN)2 | m-HSE06-D3 | 7(9) | 1.69(1.75) | −6.13(−6.17) |
| m-HSE03-D3 | 8 | 1.72 | −6.12 | |
| m-HSE03NW-D3 | 15 | 1.74 | −6.14 | |
| m-PBE0 | 8 | 1.75 | −5.66 | |
| (HSnN)2 | m-HSE06-D3 | 40(44) | 1.90(1.88) | −5.38(−5.51) |
| m-HSE03-D3 | 44 | 1.91 | −5.36 | |
| m-HSE03NW -D3 | 90 | 2.01 | −5.49 | |
| m-PBE0 | 43 | 1.81 | −5.70 | |
Analyzing the description of the optimized HXN isolated-molecules obtained with different amounts of ex, we find that the dX–H (Table 4 and Table S3 in the ESI†) and the νX–H (Table S4 in ESI†) values deviate significantly from the corresponding MP2 values if no ex is included, as when using PBE. For example, PBE predicts dX–H to be between 0.7% and 2.9% longer than the MP2 values, and between 2.8% and 12.4% smaller νX–H frequencies than the MP2 ones. Increasing ex, the overestimation of dX–H and the underestimation of νX–H by DFT may even turn into underestimation and overestimation, respectively, as when ex is increased beyond 40%. The dX–H and νX–H values are somewhat indicative of how strong is the proton bonded to the X atom. Thus, the latter results suggest that the proton in the HXN molecules becomes more labile as the amount of ex included in the xc-functional is reduced, hence more prone to be transferred upon hydrogen bonding. Therefore, it seems that the spurious pt event in (HXN)2 predicted by DFT is partly connected to an inadequate description of the X–H bond in the isolated HXN molecules when no ex, or a small amount of it, is included in the xc-functional. That is, to the prediction by DFT of longer X–H covalent bonds and smaller νX–H frequencies than the expected ones, particularly for the monomers in which X is less electronegative than H.
| System | % exa | s i(r) | d X–H | L |
|---|---|---|---|---|
| a 0% and 100% of ex correspond to calculations with PBE and MP2, respectively. Otherwise, m-HSE06-D3 is used. The def2-TZVPD basis set is used in these calculations, except for the data in parenthesis, which are obtained with the aug-cc-PVQZ basis set. | ||||
| HCN | 0 | 0.047(0.035) | 1.076(1.075) | 5.1(3.8) |
| 10 | 0.038 | 1.073 | 4.1 | |
| 20 | 0.031 | 1.070 | 3.3 | |
| 25 | (0.02) | (1.068) | (2.1) | |
| 40 | 0.022 | 1.065 | 2.3 | |
| 50 | (0.012) | (1.061) | (1.3) | |
| 100 | 0.005 | 1.068 | 0.0 | |
| HSiN | 0 | 0.063(0.065) | 1.504(1.502) | 9.5(9.8) |
| 10 | 0.058 | 1.497 | 8.7 | |
| 20 | 0.053 | 1.491 | 7.9 | |
| 25 | (0.052) | (1.486) | (7.7) | |
| 40 | 0.045 | 1.479 | 6.7 | |
| 50 | (0.043) | (1.473) | (6.3) | |
| 100 | 0.008 | 1.488 | 0.0 | |
| HGeN | 0 | 0.064(0.064) | 1.538 (1.535) | 9.8(9.8) |
| 10 | 0.060 | 1.530 | 9.2 | |
| 20 | 0.055 | 1.525 | 8.4 | |
| 25 | (0.055) | (1.519) | (8.4) | |
| 40 | 0.048 | 1.513 | 7.3 | |
| 50 | (0.045) | (1.506) | (6.8) | |
| 100 | 0.011 | 1.519 | 0.0 | |
| HSnN | 0 | 0.068(0.062) | 1.703(1.703) | 11.6 (10.6) |
| 10 | 0.062 | 1.695 | 10.5 | |
| 20 | 0.057 | 1.688 | 9.6 | |
| 25 | (0.056) | (1.686) | (9.4) | |
| 40 | 0.050 | 1.676 | 8.4 | |
| 50 | (0.044) | (1.672) | (7.4) | |
| 100 | 0.016 | 1.655 | 0.0 | |
It may be then possible to determine if a pt event is spurious by analyzing the isolate molecule, i.e., before hydrogen bonding. For example, if the deviation of dX–H with respect to the MP2 result is larger than 1.3% a pt event will happen upon hydrogen bonding, and if such deviation is smaller than 0.7% it will not (see Table S3 in the ESI†). However, if the deviation is between 1.3% and 0.7%, the outcome cannot be predicted. A similar conclusion can be reached analyzing the deviations of νX–H from the MP2 values; i.e., if the deviation ≤−6% a pt event will happen, and if the deviation ≥−2.5% it will not. Still, for some deviation intervals one cannot predict if the pt event will happen or not.
As the formation of a hb will tend to affect the strength of the X–H bond in the HXN molecules, likely due to a redistribution of the charge density upon hydrogen bonding, one may wonder if there is a correlation between the capacity of the N atom and that of the X–H bond, in the isolated molecule, to donate/accept charge, respectively, and an eventual pt event. The latter because the acceptor atom's capacity to donate charge has been correlated with its ability to form a hydrogen bond.23 Thus, we have estimated the capacity of the isolated HXN (monomer) to donate/accept electronic charge by means of s(r). The donating capacity, (s(r)−), is calculated considering the highest-energy occupied-orbitals, at the N atom, pointing along the direction of the hb formation. For calculating the accepting capacity, (s(r)+) are considered the lowest-energy empty-orbitals, at the X–H bond, pointing along the direction of the hb formation (Fig. 2). For comparison purposes Δμ > 0 is considered in the calculation of both s(r)− and s(r)+. We estimate how much electronic charge could transfer in the process of the formation of the hb overlapping s(r)− with s(r)+. For that, we plot together s(r)− and s(r)+ at HXN (upper part of Fig. 2). Next, two replicas are made and located one to the left and one to the right as depicted in the lower part of Fig. 2. The origin of the combined plot is at the N atom of the molecule located to the left. The molecule to the right is located in a position such that its H atom is at the distance dN⋯H = 0.68(rN + rH) from the origin of the combined plots rN and rH stand for the van der Waals radius of N and H, respectively. These van der Waals radius are determined integrating the electronic density obtained from a DFT calculation with the corresponding percentage of ex. For each case, the van der Waals radius is considered to be the radius of the sphere in which the electronic density integrates to the 99.9% of the value it should integrate in an infinite sphere. In such arrangement, the tails of s(r)− and s(r)+ overlap (see Fig. S2 in ESI†). The values of s(r) at the intersecting point (si(r), Fig. 2) are listed in Table 4. This value reduces as the percentage of ex is increased. We find that if si(r) is larger than 0.060 (in units of Å−3 eV−1) a spurious pt event will happen upon hydrogen bonding, and if it is lower or equal to 0.05 it will not. However, if si(r) is between 0.06 and 0.05 it cannot be predicted whether a spurious pt event will happen or not. We note that combining si(r) and dX–H we can identify all the spurious pt events; i.e., we find that if the quantity L = si(r)dX–H × 100, here called the lability index, is larger than 9.2 (in units of Å−2 eV−1) a spurious pt event will be observed, at least in the systems investigated here (see Table 4). It is worth mentioning that L calculated with MP2 is ∼0.0 for all the systems, which suggests that when fully considering the ex the possibility of a pt event upon hydrogen bonding practically vanishes, in the systems investigated here. Moreover if the lability index is calculated with the aug-cc-PVQZ basis sets (see Table 4) the same conclusion is reached.
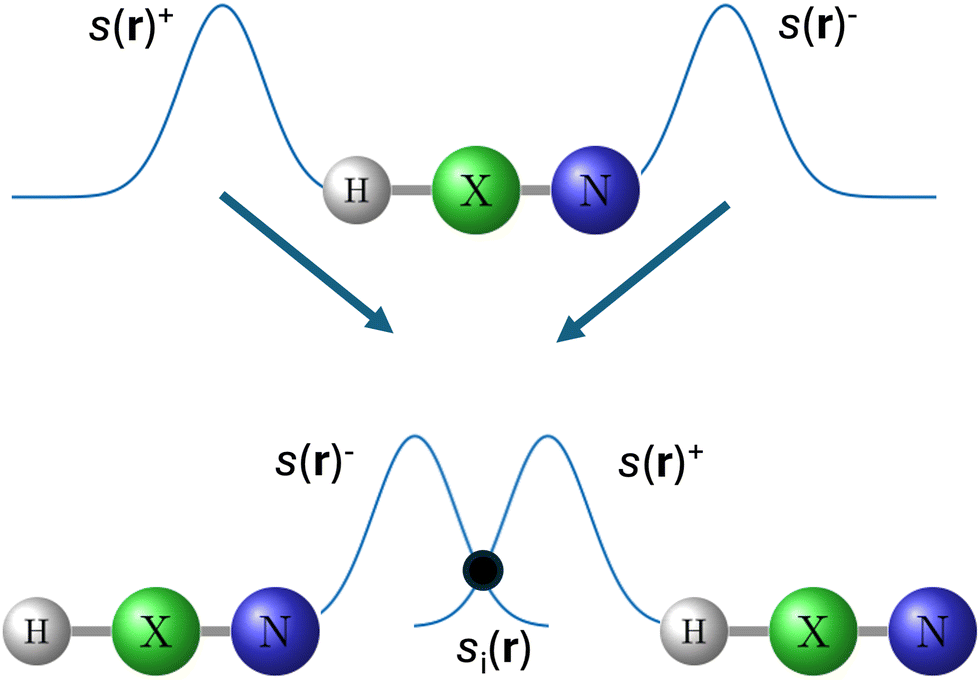 | ||
| Fig. 2 Upper scheme, local softness s(r)− (donating capacity) and s(r)+ (accepting capacity) at the HXN isolated molecule. Lower scheme, replicas of the HXN isolated molecule located at the distance dN⋯H in which s(r)− and s(r)+ overlap. Considering the origin at the N atom to the left, dN⋯H is calculated as dN⋯H = 0.68(rN + rH), where rN and rH stand for the van der Waals radius of N and H, respectively. si(r) stands for the value in which s(r)− = s(r)+. X stands for C, Si, Ge or Sn. | ||
4 Discussion
In DFT calculations, one may usually associate a spurious pt event in hydrogen bonded systems with a bad description of the hb by the xc-functional. This bad description may be due to the charge delocalization error common to many of the current xc-functionals. The results presented above, however, reveal that spurious pt events may actually be connected to a bad description of the X–H bond in the non-hydrogen bonded monomer by the xc-functional, particularly when X is more electronegative than H, and not to the nature of the hb formed; i.e., conventional or not conventional. The latter is corroborated calculating L, that depends only on quantities obtained from the isolated, non-hydrogen bonded, molecules. This empirical index combines a parameter connected to the strength of the X–H bond (dX–H), and an estimation of how much charge could be transferred upon hydrogen bonding (si(r)), a quantity that depends on the charge donor/acceptor capabilities of the isolated molecule. Thus, it seems that for no ex or a small amount of it considered in the xc-functional, the X–H may be predicted to be too elongated (i.e. weakened) and with an enhanced capability for accepting charge, that together with a great ability for donating charge of the N atom predicted with a given xc-functional, combine to ease the pt event upon hydrogen bonding. All these characteristics diminish as the percentage ex included in the xc-functional is increased, up to a point in which the pt event is no longer feasible. Nevertheless, preventing the pt event by increasing the percentage of ex tends to deteriorate the description of the hb.5 Conclusions
Studying the proton translocation along the hb through geometry optimization with DFT, we find that the amount of ex required to avoid a spurious pt event in the (HXN)2 dimers is system, basis set and xc-functional approximation dependent. Also, it is demonstrated that the spurious pt event is connected to an inadequate description of the isolated HXN molecule by a given xc-functional, not to the type of the hb that is expected to be formed. Once the description of the isolated molecule is improved by increasing the amount of ex included in the xc-functional, the spurious pt event is avoided. Still, even though the pt transfer is avoided, the hb may not be well described by the xc-functional. Our results may help to elucidate whether a pt event is spurious or not in DFT studies of more complex systems.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to acknowledge funding from CONAHCYT, projects A1-S-42775 and 1561802. J. B.-R. acknowledges CONAHCYT for the fellowship “estancias postdoctorales por México 2022(3).” Authors thank LANCAD and CONAHCYT for the computer time granted in the supercomputer Yoltla at the LSVP at UAM-Iztapalapa.Notes and references
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- S. J. Grabowski, Molecules, 2020, 25, 4668 CrossRef CAS PubMed.
- S. Hammes-Schiffer, J. Phys. Chem. B, 2021, 125, 3725–3726 CrossRef CAS PubMed.
- I. Belevich, M. Verkhovsky and M. Wikstrom, Nature, 2006, 440, 829–832 CrossRef CAS PubMed.
- S. Adam and A.-N. Bondar, PLoS One, 2018, 13, 1–28 Search PubMed.
- H. Ishikita and K. Saito, J. R. Soc., Interface, 2014, 11, 20130518 CrossRef PubMed.
- R. Sedlak, T. Janowski, M. Pitonak, J. Rezac, P. Pulay and P. Hobza, J. Chem. Theory Comput., 2013, 9, 3364–3374 CrossRef CAS PubMed.
- C. Tuma, A. Boese and N. Handy, Phys. Chem. Chem. Phys., 1999, 1, 3939–3947 RSC.
- J. Ireta, J. Neugebauer and M. Scheffler, J. Phys. Chem. A, 2004, 108, 5692–5698 CrossRef CAS.
- Y. Zhao and D. Truhlar, J. Chem. Theory Comput., 2005, 1, 415–432 CrossRef CAS PubMed.
- J. C. A. Prentice, J. Aarons, J. C. Womack, A. E. A. Allen, L. Andrinopoulos, L. Anton, R. A. Bell, A. Bhandari, G. A. Bramley, R. J. Charlton, R. J. Clements, D. J. Cole, G. Constantinescu, F. Corsetti, S. M.-M. Dubois, K. K. B. Duff, J. M. Escartn, A. Greco, Q. Hill, L. P. Lee, E. Linscott, D. D. O'Regan, M. J. S. Phipps, L. E. Ratcliff, A. R. Serrano, E. W. Tait, G. Teobaldi, V. Vitale, N. Yeung, T. J. Zuehlsdorff, J. Dziedzic, P. D. Haynes, N. D. M. Hine, A. A. Mostofi, M. C. Payne and C.-K. Skylaris, J. Chem. Phys., 2020, 152, 174111 CrossRef CAS PubMed.
- W. Dawson, A. Degomme, M. Stella, T. Nakajima, L. E. Ratcliff and L. Genovese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1574 Search PubMed.
- A. Nakata, J. S. Baker, S. Y. Mujahed, J. T. L. Poulton, S. Arapan, J. Lin, Z. Raza, S. Yadav, L. Truflandier, T. Miyazaki and D. R. Bowler, J. Chem. Phys., 2020, 152, 164112 CrossRef CAS PubMed.
- L. Ismer, J. Ireta, S. Boeck and J. Neugebauer, Phys. Rev. E, 2005, 71, 031911 CrossRef PubMed.
- L. Ismer, J. Ireta and J. Neugebauer, J. Phys. Chem. B, 2008, 112, 4109–4112 CrossRef CAS PubMed.
- J. Nochebuena, A. Ramirez and J. Ireta, Int. J. Quantum Chem., 2015, 115, 1613–1620 CrossRef CAS.
- J. Ireta, Int. J. Quantum Chem., 2012, 112, 3612–3617 CrossRef CAS.
- M. Neumann and M. Perrin, J. Phys. Chem. B, 2005, 109, 15531–15541 CrossRef CAS PubMed.
- J. M. del Campo and J. Ireta, Phys. Chem. Chem. Phys., 2021, 23, 11931–11936 RSC.
- N. E. Gonzalez-Diaz, R. Lopez-Rendon and J. Ireta, J. Phys. Chem. C, 2019, 123, 2526–2532 CrossRef CAS.
- J. K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef CAS PubMed.
- C. Cuautli, J. S. Valente, J. C. Conesa, M. V. Ganduglia-Pirovano and J. Ireta, J. Phys. Chem. C, 2019, 123, 8777–8784 CrossRef CAS.
- G. Castro, J. S. Valente, M. Galvan and J. Ireta, Phys. Chem. Chem. Phys., 2022, 24, 23507–23516 RSC.
- J. Ireta, F. Aparicio, M. Viniegra and M. Galvan, J. Phys. Chem. B, 2003, 107, 811–818 CrossRef CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- S. Civis, M. Lamanec, V. Spirko, J. Kubista, M. Spet'ko and P. Hobza, J. Am. Chem. Soc., 2023, 145, 8550–8559 CrossRef CAS PubMed.
- J. M. Bautista-Renedo, H. Reyes-Pérez, E. Cuevas-Yañez, C. Barrera-Díaz, N. González-Rivas and J. Ireta, RSC Adv., 2019, 9, 5937–5941 RSC.
- L. M. LeBlanc, S. G. Dale, C. R. Taylor, A. D. Becke, G. M. Day and E. R. Johnson, Angew. Chem., Int. Ed., 2018, 57, 14906–14910 CrossRef CAS PubMed.
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Science, 2008, 321, 792–794 CrossRef CAS PubMed.
- G. F. Mangiatordi, E. Bremond and C. Adamo, J. Chem. Theory Comput., 2012, 8, 3082–3088 CrossRef CAS PubMed.
- D. Flait and M. Head-Gordon, J. Phys. Chem. Lett., 2018, 9, 6280–6288 CrossRef PubMed.
- K. R. Bryenton, A. A. Adeleke, S. G. Dale and E. R. Johnson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 13, e1631 Search PubMed.
- X. Zheng, M. Liu, E. R. Johnson, J. Contreras-García and W. Yang, J. Chem. Phys., 2012, 137, 214106 CrossRef PubMed.
- J. Blahut, J. R. Stocek, M. Sala and M. Dracinsky, J. Magn. Reson., 2022, 345, 107334 CrossRef CAS PubMed.
- M. Husak, S. Sajbanova, J. Klimes and A. Jegorovc, Acta Crystallogr., B: Struct. Sci., Cryst. Eng. Mater., 2022, 78, 781–788 CrossRef CAS.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2014, 16, 1150–1160 RSC.
- B. G. Janesko, Chem. Soc. Rev., 2021, 50, 8470–8495 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- J. Cirera and E. Ruiz, J. Phys. Chem., 2020, 124, 5053–5058 CrossRef CAS PubMed.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- D. Mejia-Rodriguez and S. B. Trickey, Phys. Rev. A, 2017, 96, 052512 CrossRef.
- Y. Wang, X. Jin, H. S. Yu, D. G. Truhlar and X. He, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8487–8492 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 21996 CrossRef.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 120, 7274–7280 CrossRef CAS PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. Yang and R. G. Parr, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 6723–6726 CrossRef CAS PubMed.
- K. D. Brommer, M. Galvan, A. Dal Pino Jr and J. Joannopoulos, Surf. Sci., 1994, 314, 57–70 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Dasgupta and J. M. Herbert, J. Comput. Chem., 2017, 38, 869–882 CrossRef CAS PubMed.
- E. Aprà, E. J. Bylaska, W. A. de Jong, N. Govind, K. Kowalski, T. P. Straatsma, M. Valiev, H. J. J. van Dam, Y. Alexeev, J. Anchell, V. Anisimov, F. W. Aquino, R. Atta-Fynn, J. Autschbach, N. P. Bauman, J. C. Becca, D. E. Bernholdt, K. Bhaskaran-Nair, S. Bogatko, P. Borowski, J. Boschen, J. Brabec, A. Brunauer, E. Cauët, Y. Chen, G. N. Chuev, C. J. Cramer, J. Daily, M. J. O. Deegan, T. H. Dunning Jr., M. Dupuis, K. G. Dyall, G. I. Fann, S. A. Fischer, A. Fonari, H. Früchtl, L. Gagliardi, J. Garza, N. Gawande, S. Ghosh, K. Glaesemann, A. W. Götz, J. Hammond, V. Helms, E. D. Hermes, K. Hirao, S. Hirata, M. Jacquelin, L. Jensen, B. G. Johnson, H. Jónsson, R. A. Kendall, M. Klemm, R. Kobayashi, V. Konkov, S. Krishnamoorthy, M. Krishnan, Z. Lin, R. D. Lins, R. J. Littlefield, A. J. Logsdail, K. Lopata, W. Ma, A. V. Marenich, J. Martin del Campo, D. Mejia-Rodriguez, J. E. Moore, J. M. Mullin, T. Nakajima, D. R. Nascimento, J. A. Nichols, P. J. Nichols, J. Nieplocha, A. Otero-de-la Roza, B. Palmer, A. Panyala, T. Pirojsirikul, B. Peng, R. Peverati, J. Pittner, L. Pollack, R. M. Richard, P. Sadayappan, G. C. Schatz, W. A. Shelton, D. W. Silverstein, D. M. A. Smith, T. A. Soares, D. Song, M. Swart, H. L. Taylor, G. S. Thomas, V. Tipparaju, D. G. Truhlar, K. Tsemekhman, T. Van Voorhis, A. Vázquez-Mayagoitia, P. Verma, O. Villa, A. Vishnu, K. D. Vogiatzis, D. Wang, J. H. Weare, M. J. Williamson, T. L. Windus, K. Wolinski, A. T. Wong, Q. Wu, C. Yang, Q. Yu, M. Zacharias, Z. Zhang, Y. Zhao and R. J. Harrison, J. Chem. Phys., 2020, 152, 184102 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- J. Dunning and H. Thom, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. A. Kendall, J. Dunning, H. Thom and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. I. Perea-Ramírez, A. Guevara-García and M. Galván, J. Mol. Model., 2018, 24, 1–8 CrossRef PubMed.
- M. Cohen, M. Gandugliapirovano and J. Kudrnovsky, Phys. Rev. Lett., 1994, 72, 3222–3225 CrossRef CAS PubMed.
- J. Ireta, M. Galvan, K. Cho and J. Joannopoulos, J. Am. Chem. Soc., 1998, 120, 9771–9778 CrossRef CAS.
- F. Aparicio, J. Ireta, A. Rojo, L. Escobar, A. Cedillo and M. Galvan, J. Phys. Chem. B, 2003, 107, 1692–1697 CrossRef CAS.
- F. Aparicio, N. Gonzalez-Rivas, J. Ireta, A. Rojo, L. I. Escobar, A. Cedillo and M. Galvan, Int. J. Quantum Chem., 2012, 112, 3618–3623 CrossRef CAS.
- C. Muñoz Gutierrez, F. Adasme-Carreño, J. Alzate-Morales and J. Ireta, Phys. Chem. Chem. Phys., 2023, 25, 23885–23893 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00907j |
| This journal is © the Owner Societies 2024 |