
Reactions of gas-phase uranyl formate/acetate anions: reduction of carboxylate ligands to aldehydes by intra-complex hydride attack†
Amanda R.
Bubas‡
a,
Irena J.
Tatosian§
a,
Anna
Iacovino¶
a,
Theodore A.
Corcovilos
 b and
Michael J.
van Stipdonk
*a
b and
Michael J.
van Stipdonk
*a
aDepartment of Chemistry and Biochemistry, Duquesne University, 600 Forbes Ave, Pittsburgh, PA 15282, USA. E-mail: vanstipdonkm@duq.edu
bDepartment of Physics, Duquesne University, 600 Forbes Ave, Pittsburgh, PA 15282, USA
First published on 10th April 2024
Abstract
In a previous study, electrospray ionization, collision-induced dissociation (CID), and gas-phase ion–molecule reactions were used to create and characterize ions derived from homogeneous precursors composed of a uranyl cation (UVIO22+) coordinated by either formate or acetate ligands [E. Perez, C. Hanley, S. Koehler, J. Pestok, N. Polonsky and M. Van Stipdonk, Gas phase reactions of ions derived from anionic uranyl formate and uranyl acetate complexes, J. Am. Soc. Mass Spectrom., 2016, 27, 1989–1998]. Here, we describe a follow-up study of anionic complexes that contain a mix of formate and acetate ligands, namely [UO2(O2C–CH3)2(O2C–H)]− and [UO2(O2C–CH3)(O2C–H)2]−. Initial CID of either anion causes decarboxylation of a formate ligand to create carboxylate-coordinated U-hydride product ions. Subsequent CID of the hydride species causes elimination of acetaldehyde or formaldehyde, consistent with reactions that include intra-complex hydride attack upon bound acetate or formate ligands, respectively. Density functional theory (DFT) calculations reproduce the experimental observations, including the favored elimination of formaldehyde over acetaldehyde by hydride attack during CID of [UO2(H)(O2C–CH3)(O2C–H)]−. We also discovered that MSn CID of the acetate–formate complexes leads to generation of the oxyl-methide species, [UO2(O)(CH3)]−, which reacts with H2O to generate [UO2(O)(OH)]−. DFT calculations support the observation that formation of [UO2(O)(OH)]− by elimination of CH4 is favored over H2O addition and rearrangement to create [UO2(OH)2(CH3)]−.
Introduction
Electrospray ionization (ESI) provides easy access to a wide range of uranyl (UO22+) complexes for studies of gas-phase structure and reactivity in a species-specific fashion. For example, the use of ESI to transfer the uranium(V) dioxocation, UVO2+, from solution to the gas phase was first reported in 19921 and since then, advances in the fundamental understanding of uranyl coordination chemistry have been made using the ionization method combined with tandem mass spectrometry.2–22 Most importantly, ESI has been used to generate doubly charged UO22+ complexes, such as [UO2(L)n]2+, with L = acetonitrile (acn) or acetone (aco) and n = 4 or 5, for collision-induced dissociation (CID) and ion–molecule reaction experiments.12,13 Use of ESI and tandem mass spectrometry has since been expanded, primarily through the work of Gibson and coworkers, to explore the intrinsic dissociation and ion–molecule reactions of a range of transuranic species.23–32Focusing on U, recent investigations by our laboratory have shown that the MSn capabilities of ion trap instruments can be used to generate novel gas-phase species for studies of intrinsic reactivity.33–44 In one prior study37 the CID pathways of homogeneous anionic uranyl complexes with general formula [UO2(O2C–R)3]−, R = H (formate) or CH3 (acetate) were determined using multiple-stage (MSn) experiments in a linear ion trap (LIT) mass spectrometer. Use of the 2-D ion trap provided access to fragmentation pathways and reactions not observed in earlier studies with 3-D ion traps, which were complicated by a preponderance of product ions obviously generated by interactions with background H2O.
Our previous study demonstrated that anionic complexes containing UO22+ and formate ligands fragmented by loss of CO2, and elimination of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, to create an oxo-hydride product [UO2(O)(H)]−. Analogous complexes that contained acetate ligands instead showed an initial loss of acetyloxyl radical, CH3CO2˙, followed by decarboxylation and elimination of CH4 to generate [UO2(O)]−. The loss of CH4 occurs by an intra-complex proton transfer process that generates UO2+ coordinated by an acetate and acetate enolate ligand. A final CID step caused elimination of ketene (CH2CO) to generate [UO2(O)]−.
O, to create an oxo-hydride product [UO2(O)(H)]−. Analogous complexes that contained acetate ligands instead showed an initial loss of acetyloxyl radical, CH3CO2˙, followed by decarboxylation and elimination of CH4 to generate [UO2(O)]−. The loss of CH4 occurs by an intra-complex proton transfer process that generates UO2+ coordinated by an acetate and acetate enolate ligand. A final CID step caused elimination of ketene (CH2CO) to generate [UO2(O)]−.
Here, we present an expanded study that includes anionic complexes that contain UO22+ coordinated by a mix of acetate and formate ligands. As outlined below, the mixed complexes dissociate to make new species, including several uranyl-hydride ions for which measurements of intrinsic reactivity are relatively rare. Of particular interest was the potential for some species to undergo intra-complex hydride attack steps that convert carboxylate ligands to aldehydes and oxide, which we assume were responsible for the loss of formaldehyde in our earlier investigation of the homogeneous UO2–formate anions. In addition, the outcome of reactions with H2O were determined for a range of dissociation product ions. In this case, our interest was in the tendency for the hydride species to form hydroxides by reaction with H2O and elimination of H2, the hydrolysis of novel UO2–oxo–methide anions with H2O, and the potential for hydrates of UO2–oxo anions to rearrange to generate dihyroxides. To complement the experimental measurements, Kohn–Sham density functional theory (DFT) calculations were used to determine probable reaction pathways and relative energies.
Experimental methods
Sample preparation
Samples of uranyl formate, acetate and d3-acetate were prepared by digesting ca. 2–3 mg of UO3 Strem Chemicals, Newburyport MA with excess of formic or acetic acid individually (purchased from Sigma Aldrich, St. Louis MO) and 400 μL of deionized/distilled H2O contained in a glass scintillation vial. Caution: uranium oxide is radioactive (α- and γ-emitter), and proper shielding, waste disposal and personal protective gear should be used when handling the material.The solutions incubated on a hot plate overnight at 70 °C. 50 μL of the natural or labeled acetate solutions were combined with an equivalent volume of formate solution and diluted to 1 mL total volume using H2O and CH3CH2OH for ESI. CH3CH2OH was used instead of CH3OH for these experiments to avoid any ambiguity when identifying potential molecular O2 adducts to product ions.44–47
Mass spectrometry measurements
All experiments were performed using a ThermoScientific LTQ-XL LIT mass spectrometer (San Jose, CA) modified to allow the mixing of reagents with the helium buffer gas before introduction into the ion trap.43 The uranyl–carboxylate spray solutions were directly infused into the instrument at a flow rate of 5 μl min−1. The LTQ Tune program was used to optimize the atmospheric pressure ionization stack settings (for example, lens voltages, and the offset voltages for transfer quadrupole and octopole) and maximize the transmission of the singly charged ions [UO2(O2C–R)n]−, R = H, CH3 and CD3, to the ion trap mass analyzer. Ultra-high purity He was used as the bath/buffer and collision gas.For CID, precursor ions were isolated using an isolation width of 1.0 to 1.5 mass to charge (m/z) units centered on the 238U isotopic peak. The exact value was adjusted to provide maximum ion intensity while maintaining the isolation of a single isotopic peak. To probe CID behavior in general, the (mass) normalized collision energy (NCE, as defined by ThermoScientific) was set between 5 and 18% (approximately 0.075–0.27 V). The activation Q setting for all experiments was set at 0.30 and a 30 ms activation time was used.
To probe ion–molecule reactions with (neutral) H2O, specific precursor ions were isolated using widths of between 1 and 2 m/z units and stored in the ion trap for reaction times ranging from 1 ms to 10 s. When examining ion–molecule reactions (IMRs), our intent was to identify the products generated by ion–molecule reactions rather than measure or report rates or rate constants. For both CID and IMR experiments, the mass spectra displayed were created by accumulating and averaging at least 30 isolation, dissociation, and ejection/detection steps.
Density functional theory calculations
While the objective was to generate experimental data regarding the gas-phase reactions of the anionic uranyl–carboxylate complexes and their dissociation products, supporting DFT calculations can provide important insight into reaction mechanisms and relative energetics. Our intent was not to rigorously assess the accuracy of DFT for determining reaction thermochemistry, bond lengths and angles, or bond-dissociation energies, but instead to identify probable reaction pathways “in silico” that reproduce the experimental observations in terms of dissociation or ion–molecule reactions. DFT has been used in several previous studies to probe the properties of gas-phase uranyl species.48–59 For the reactions that were the focus of this study, the structures of the various isomers, reaction intermediates, and transition states, were optimized using the M06-L functional, which was chosen based on the good performance when investigating reaction pathways and energetics for uranyl species in our prior studies.43,45,46 Transition states were identified using the QST2 and QST3 approaches60–63 and were confirmed by means of intrinsic reaction coordinate calculations.To minimize computational cost, initial geometry optimization was performed using the MDF60 pseudopotential and associated basis set on U and the cc-pvtz basis set on all other atoms. Single-point calculations were then performed on the minima and transition state structures (obtained at the M06L/MDF60/cc-pvtz level of theory) using the cc-pvdz-PP64 basis of Peterson (with the MDF60 pseudopotential) on U and aug-cc-pvtz on all other atoms. The Gaussian 16 software package65 was used for all calculations.
It should be noted that spin–orbit corrections were not explicitly included in our calculations. Although spin–orbit effects are not expected to significantly affect the energetics of processes in which there is no change in the formal oxidation state of the heavy metal, substantial changes can occur for reaction energies involving actinide atoms in different oxidation states.66–68 While it is possible that the energies of specific species with U in different oxidation states could be affected by the inclusion of spin–orbit corrections, we feel that this caveat does not influence our interpretation of the DFT investigation of likely reaction mechanisms.
Results and discussion
The ESI mass spectrum generated from a mix of [UO2(O2C–CH3)2] and [UO2(O2C–H)2] in H2O/CH3CH2OH is shown in Fig. S1 of the ESI.† The dominant negative ions created by ESI were [UO2(O2C–H)3]− at m/z 405, [UO2(O2C–H)2(O2C–CH3)]− at m/z 419, [UO2(O2C–H)(O2C–CH3)2]− at m/z 433 and [UO2(O2C–CH3)3]− at m/z 447. The dissociation of [UO2(O2C–H)3]− (m/z 405) and [UO2(O2C–CH3)3]− (m/z 447) were discussed in detail in our earlier report.37 Our focus here was on the dissociation of heterogeneous complexes containing acetate and formate ligands.Tandem MS of [UO2(O2C–H)(O2C–CH3)2]−
The multiple-stage (MSn) CID spectra generated by initially isolating [UO2(O2C–H)(O2C–CH3)2]− at m/z 433 are shown in Fig. 1a–d and 2a and b. Related data generated using analogous species that contained d3-acetate are provided in Fig. S2a–d and S3a and b of the ESI.† A summary of the MSn dissociation pathways for the unlabeled and labeled complex anions are shown in Scheme 1a and b. | ||
| Fig. 1 Product ion spectra derived from MSn CID of [UO2(O2C–H)(O2C–CH3)2]−: (a) CID (MS/MS stage) of [UO2(O2C–H)(O2C–CH3)2]− at m/z 433, (b) CID (MS3 stage) of dissociation product ion at m/z 389, (c) CID (MS4 stage) of dissociation product ion at m/z 345, and (d) CID (MS5 stage) of dissociation product ion at m/z 301. In the spectra, the circles and arrows illustrate the MSn pathway. In each spectrum, the bold peak label indicates the precursor selected for CID while labels in italics represent the products from dissociation or ion–molecule reactions as indicated in the text. | ||
 | ||
| Fig. 2 Product ion spectra derived from MSn CID of [UO2(O2C–H)(O2C–CH3)2]− continued: (a) CID (MS3 stage) of product ion at m/z 387 generated by initial CID of [UO2(O2C–H)(O2C–CH3)2]− at m/z 433, (b) CID (MS4 stage) of dissociation product ion at m/z 345. The bold peak labels indicate the precursor selected for CID while labels in italics represent the products from dissociation or ion–molecule reactions as indicated in the text. | ||
 | ||
| Scheme 1 (a) Summary of the MSn dissociation pathways for (unlabeled) [UO2(O2C–H)(O2C–CH3)2]− and (b) summary of the MSn dissociation pathways for (labeled) [UO2(O2C–H)(O2C–CD3)2]−. | ||
Initial CID (MS/MS stage, Fig. 1a) of [UO2(O2C–H)(O2C–CH3)2]− generated product ions at m/z 389 and 387, corresponding to neutral losses of 44 and 46 mass units (Da) respectively. The elimination of 44 Da to create the ion at m/z 389 is attributed to decarboxylation, which is an effective way to generate organometallic species in the gas phase.69 The loss of 46 Da to create the ion at m/z 387 suggests intra-proton transfer and elimination of neutral formic acid. Because of the low-mass cut-off of the LIT, we could not determine whether [UO2(O2C–H)(O2C–CH3)2]− dissociates to create acetate (m/z 59) or formate (m/z 45) by elimination of neutral [UO2(O2C–H)(O2C–CH3)]− or [UO2(O2C–CH3)]−, respectively.
For CID of the d-labeled version of the anion, [UO2(O2C–H)(O2C–CD3)2]− (m/z 439, Fig. S1a, ESI†) the analogous product ions appear at m/z 395 and 392. Formation of the ion at m/z 395 involves elimination of 44 Da, consistent with decarboxylation, and the shift in product ion mass of 6 units (compared to the un-labeled analogue) indicates the retention of all deuterium atoms in the precursor complex. For the product ion at m/z 392, the shift in mass of 5 Da indicates retention of 5 deuterium atoms, and the shift of neutral loss to 47 Da identifies DO2C–H as the neutral species ejected in the dissociation reaction.
The formation of the product ion at m/z 389 following CID of [UO2(O2C–H)(O2C–CH3)2]− could involve decarboxylation of either a formate or acetate ligand. For the unlabeled complex, decarboxylation of the formate ligand would generate the hydride species [UO2(H)(O2C–CH3)2]−, while loss of CO2 from an acetate ligand would create the organouranyl complex [UO2(O2C–H)(CH3)(O2C–CH3)]−. To determine which dissociation product is generated, the ion at m/z 389 was isolated, without imposed collisional activation, in the ion trap for periods ranging from 1 ms to 1 s for reaction with H2O (ca. 1 × 10−6 torr, Fig. S4a–d of the ESI†). Our hypothesis was that the hydride would react with H2O to create [UO2(OH)(O2C–CH3)2]− at m/z 405 by reaction (1), while the organouranyl species would generate [UO2(OH)(O2C–H)(O2C–CH3)]− at m/z 391 by reaction (2). The product ion spectra in Fig. S4 (ESI†) show that an ion–molecule reaction product at m/z 405 is formed (net addition of 16 u to the precursor at m/z 389), consistent with reaction (1). Subsequent CID of the ion at m/z 405 (Fig. S4c, ESI†) caused the elimination of 60 Da, which is consistent with elimination of acetic acid to create [UO2(O)(O2C–CH3)]− at m/z 345. Overall, the ion–molecule reaction behavior strongly suggests that the dissociation product ion at m/z 389 (Fig. 1a) is [UO2(H)(O2C–CH3)2]− ([UO2(H)(O2C–CD3)2]− for the d-labeled precursor) generated by decarboxylation of a formate ligand.
| [UO2(O2C–R)2]− → [UO2(R)(O2C–CH3)]− + CO2 | (1) |
| [UO2(H)(O2C–CH3)2]− + H2O → [UO2(OH)(O2C–CH3)2]− + H2 | (2) |
| [UO2(CH3)(O2C–H)(O2C–CH3)]− + H2O → [UO2(OH)(O2C–H)(O2C–CH3)]− + CH4 | (3) |
| [UO2(OH)(O2C–CH3)2]− → [UO2(O)(O2C–CH3)]− + CH3COOH | (4) |
CCH2 for the unlabeled ions, OCCD2 for the d-labeled analogues). The elimination of ketene was invoked in our earlier study to explain the MSn fragmentation of [UO2(O2C–CH3)3]−,37 and a similar process has been reported by O’Hair and coworkers for the catalytic dehydration of acetic acid.70 In the present study, elimination of ketene from [UO2(O2C–CH2)(O2C–CH3)]− would create a product ion with composition [UO2(O)(O2C–CH3)]−, and the assignment is consistent with the isotopic labeling experiments (the product ion at m/z 345 shifts to m/z 348 for the d-labeled precursor).
The loss of 44 Da from [UO2(H)(O2C–CH3)2]− (Fig. 1b) to create the fragment ion at m/z 345 was initially attributed to a second decarboxylation step. However, CID of the analogous ion at m/z 395 derived from the d-labeled complex leads to a product ion at m/z 348, which represents a neutral loss of 47 Da. Therefore, the dissociation step cannot involve decarboxylation. The mass shift to the neutral loss suggests instead that the dissociation reaction involves elimination of acetaldehyde (reaction (4)), which we presume occurs by intra-complex hydride attack. A similar reaction was invoked in our earlier study, in which elimination of OCH2 (30 Da) was observed following CID of [UO2(H)(O2C–H)2]−.
| [UO2(H)(O2C–CH3)2]− → [UO2(O)(O2C–CH3)]− + OC(H)CH3 | (5) |
When using the unlabeled species, subsequent CID (MS3 stage) of the ion at m/z 345 (MS4 stage, Fig. 1c and 2b) generated product ions at m/z 301 and 303. The former is created by elimination of 44 Da, suggesting formation of the oxyl-methide product [UO2(O)(CH3)]− by decarboxylation of the acetate ligand. A neutral loss of 44 Da was also observed following CID of the d-labeled analogue, [UO2(O)(O2C–CD3)]−, at m/z 348 (Fig. 2b), in this case to create a product ion at m/z 304. The shift in mass indicates the retention of 3 D atoms and is consistent with the composition assignment of [UO2(O)(CH3)]− ([UO2(O)(CD3)]− for the labeled precursor). The product ions at m/z 301 (unlabeled precursor) and m/z 304 (d-labeled analogue) were isolated for reaction with H2O (Fig. S5 and S6 of the ESI†). Reaction of the ion at m/z 301 created the product ion at m/z 303, suggesting that the presence of the latter species in the spectrum shown in Fig. 1c is the result of hydrolysis reaction (5) to create [UO2(O)(OH)]−. Isolation of [UO2(O)(CD3)]− for reaction with H2O also leads to formation of the product ion at m/z 303, consistent with a pathway like reaction (5), but with the elimination of CD3H as the neutral species.
| [UO2(O)(CH3)]− + H2O → [UO2(O)(OH)]− + CH4 | (6) |
Subsequent CID of the ions at m/z 301 (MS5 stage, Fig. 1d) caused elimination of 15 Da (CH3) to create the terminal product ion at m/z 286. The same fragment ion was observed following CID of the analogous species derived from the d-labeled precursor (elimination of 18 Da, CD3, Fig. S2b, ESI†). This observation is consistent with a composition assignment of [UO2(O)]− for the terminal product ion generated by MSn CID of the [UO2(O2C–H)(O2C–CH3)2]− precursor ion.
Tandem MS of [UO2(O2C–H)2(O2C–CH3)]−
The multiple-stage (MSn) CID spectra generated by initially isolating [UO2(O2C–H)2(O2C–CH3)]− at m/z 419 are shown in Fig. 3. Related data generated using analogous species that contained d3-acetate are provided in Fig. S7 of the ESI.† A summary of the MSn dissociation pathways for the unlabeled and labeled complex ions are shown in Scheme 2a and b.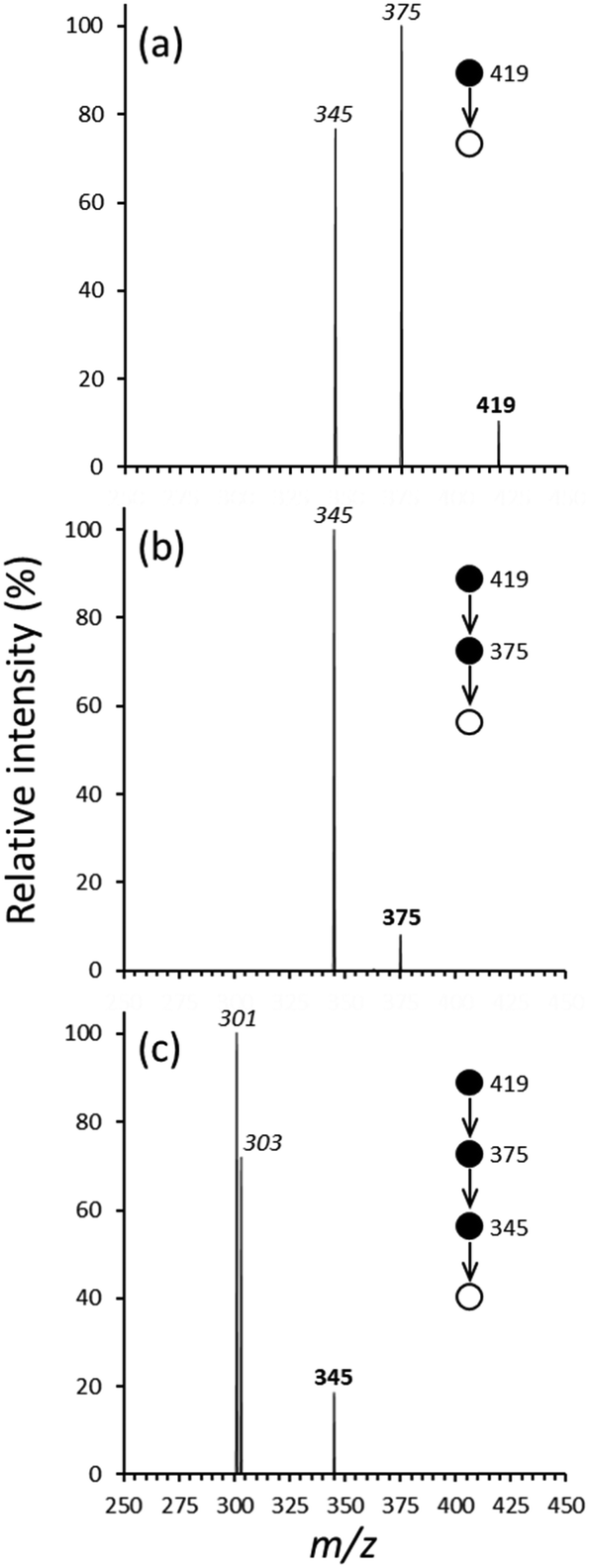 | ||
| Fig. 3 Product ion spectra derived from MSn CID of [UO2(O2C–H)2(O2C–CH3)]−: (a) CID (MS/MS stage) of [UO2(O2C–H)2(O2C–CH3)]− at m/z 419, (b) CID (MS3 stage) of dissociation product ion at m/z 375, (c) CID (MS4 stage) of dissociation product ion at m/z 345. In the spectra, the circles and arrows illustrate the MSn pathway. In each spectrum, the bold peak label indicates the precursor selected for CID while labels in italics represent the products from dissociation or ion–molecule reactions as indicated in the text. | ||
 | ||
| Scheme 2 (a) Summary of the MSn dissociation pathways for (unlabeled) [UO2(O2C–H)2(O2C–CH3)]− and (b) summary of the MSn dissociation pathways for (labeled) [UO2(O2C–H)2(O2C–CD3)]−. | ||
The initial CID (MS/MS stage, Fig. 3a) of [UO2(O2C–H)2(O2C–CH3)]− generated product ions at m/z 375 and 345. Formation of the species at m/z 375 corresponds to elimination of CO2 (44 Da). The species at m/z 345 is consistent with elimination of 44 and 30 Da (74 total). For CID of the d-labeled version of the anion, [UO2(O2C–H)2(O2C–CD3)]− (m/z 422, Fig. S7a, ESI†) the analogous product ions appear at m/z 378 and 348. These product ions are also generated by the elimination of 44 and 74 (net) Da, respectively, indicating that the D-labels on the acetate ligand are retained the product ions.
The formation of the product ion at m/z 375 following CID of [UO2(O2C–H)2(O2C–CH3)]− could also involve decarboxylation of either a formate or acetate ligand, as considered above for the dissociation of [UO2(O2C–H)(O2C–CH3)2]−. Ion–molecule reactions (Fig. S8a–d of the ESI†) were again used to determine whether formation of the product ion at m/z 375 involves decarboxylation of a formate or acetate ligand. The reaction of the ion at m/z 375 with H2O resulted in the generation of an ion–molecule reaction product at m/z 391 (net addition of 16 Da), consistent with the hydrolysis of a hydride species by reaction (6).
| [UO2(H)(O2C–H)(O2C–CH3)]− + H2O → [UO2(OH)(O2C–H)(O2C–CH3)]− + H2 | (7) |
Subsequent CID of the ion–molecule reaction product at m/z 391 caused the elimination of 46 Da, consistent with elimination of formic acid to create [UO2(O)(O2C–CH3)]− at m/z 345. A minor product ion at m/z 347 corresponds to elimination of 44 Da (CO2), which we assume generates [UO2(O)(H)(O2C–H)]−. These observations identify the dissociation product ion at m/z 375 (Fig. 3a) as [UO2(H)(O2C–H)(O2C–CH3)]− ([UO2(H)(O2C–H)(O2C–CD3)]− for the d-labeled precursor).
When using the unlabeled species, subsequent CID (MS3 stage) of the ion at m/z 375 (Fig. 3b) leads to a product ion at m/z 345 (loss of 30 Da). The elimination of 30 Da is attributed to loss of formaldehyde to generate [UO2(O)(O2C–CH3)]−. As for the MSn dissociation of [UO2(O2C–H)(O2C–CH3)2]− as described above, CID of the ion at m/z 345 caused decarboxylation to create [UO2(O)(CH3)]− at m/z 301, which undergoes hydrolysis to generate [UO2(O)(OH)]− at m/z 303. As described above, subsequent CID of the ion at m/z 301 (MS5 stage, Fig. 1d) caused elimination of 15 Da (CH3) to create the terminal product ion at m/z 286.
Computational investigation of intra-complex hydride attack
Of particular interest in this study was the apparent elimination of formaldehyde and/or acetaldehyde during CID of the anionic formate/acetate complexes. We initiated our computational investigation of the presumed hydride attack mechanism with the dissociation of [UO2(H)(O2C–H)2]−, which was investigated experimentally in our previous study. Relevant minima and transition state structures for the dissociation of [UO2(H)(O2C–H)2]− are provided in Fig. S9 of the ESI.† The zero-point corrected electronic energies and free energies for the respective species are provided in Table S1 of the ESI.† A reaction energy diagram for the CID of [UO2(H)(O2C–H)2]− is shown in Fig. 4. The global minimum identified for [UO2(H)(O2C–H)2]− (structure I) features two bidentate formate ligands, and a pseudo-pentagonal bipyramidal coordination geometry. Intra-complex hydride attack requires conversion to an intermediate structure, II, which includes one monodentate formate ligand. This type of isomerization has been reported for other metal-carboxylate species71,72 and a prior infrared multiple-photon dissociation spectroscopy study demonstrated variable denticity in anionic uranyl–carboxylate species.73 From II, formation of an ion–molecule complex between [UO2(O)(O2C–H)]− and formaldehyde (structure III) can occur through transition state structure TS II→III. Elimination of formaldehyde from III creates [UO2(O)(O2C–H)]− (structure IV). The hydride attack pathway lies significantly lower in energy than the dissociation reaction in which formate would be generated by elimination of (neutral) [UO2(H)(O2C–H)]. | ||
| Fig. 4 A reaction energy diagram for the CID of [UO2(H)(O2C–H)2]−. | ||
The product ion spectrum generated by CID of the analogous acetate complex [UO2(H)(O2C–CH3)2]− (m/z 389) and the presumed elimination of acetaldehyde to create [UO2(O)(O2C–CH3)]− at m/z 345, was shown in an earlier section (Fig. 2a and Scheme 1). The relevant minima and transition state structures for the dissociation of [UO2(H)(O2C–CH3)2]− are provided in Fig. S10 of the ESI.† The zero-point corrected electronic energies and free energies for the respective species are provided in Table S2 of the ESI.† As indicated in the reaction energy diagram for [UO2(H)(O2C–CH3)2]− (Fig. S10 of the ESI†), the intra-complex hydride attack follows a pathway like the one identified and described above for the CID of [UO2(H)(O2C–H)2]−, and ultimately generates [UO2(O)(O2C–CH3)]− (m/z 345, structure IV-ac in Fig. S9, ESI†). Here too, the hydride attack pathway is lower in energy than the decomposition to produce acetate anion and (neutral) [UO2(H)(O2C–CH3)]−.
As discussed in an earlier section, the dissociation of [UO2(H)(O2C–H)(O2C–CH3)]− (m/z 375, initially created by CID of the [UO2(O2C–H)2)(O2C–CH3)]− precursor at m/z 419, generated a product ion at m/z 345. The neutral loss in this process corresponds to 30 Da and is consistent with elimination of formaldehyde, and the single dissociation product suggests that elimination of formaldehyde is energetically favored over acetaldehyde in the mixed carboxylate/hydride species.
Relevant minima and transition state structures for the dissociation of [UO2(H)(O2C–H)(O2C–CH3)]− are provided in Fig. S12 of the ESI.† The zero-point corrected electronic energies and free energies for the respective species are provided in Table S3 of the ESI.† A reaction energy diagram for the CID of [UO2(H)(O2C–H)(O2C–CH3)]− is shown in Fig. 5.
 | ||
| Fig. 5 A reaction energy diagram for the CID of [UO2(H)(O2C–H)(O2C–CH3)]−. | ||
The global minimum identified for [UO2(H)(O2C–H)(O2C–CH3)]− (structure V, Fig. 5) again features bidentate coordination by the carboxylate ligands. Hydride attack upon the acetate ligand requires isomerization of V to structure VI, which includes a monodentate acetate ligand. Creation of the ion–molecule complex between [UO2(O)(O2C–H)]− and acetaldehyde (structure VII) is achieved through transition state TS VI→VII. Elimination of neutral formaldehyde creates [UO2(O)(O2C–H)]− (structure VIII).
Hydride attack upon the formate ligand of [UO2(H)(O2C–H)(O2C–CH3)]− instead requires isomerization of V to structure IX. Creation of the ion–molecule complex between [UO2(O)(O2C–CH3)]− and formaldehyde (structure X) is achieved through transition state TS IX→X. Elimination of neutral formaldehyde creates [UO2(O)(O2C–CH3)]− (structure XI). While the post-attack complex VII, and formation of the terminal product VIII, is predicted to be lower in energy than the analogous structures X and XI on the formaldehyde loss pathway, the energy of TS IX→X is lower than the energy of TS VI→VII, consistent with the experimental observation that elimination of formaldehyde is favored over acetaldehyde following CID of [UO2(H)(O2C–H)(O2C–CH3)]−. We note again that both hydride attack pathways lie well below the energy required to generate either formate or acetate anion.
Computational investigation of reaction of [UO2(O)(CH3)]− with H2O
Our last objective was to investigate the hydrolysis of [UO2(O)(CH3)]− to create [UO2(O)(OH)]− as shown in Fig. 1c and Fig. S5 (ESI†). Of interest here was the potential competition between hydrolysis reaction (5), and reaction (7), which involves formation of a hydrate, followed by isomerization to create a dihydroxide.| [UO2(O)(CH3)]− + H2O → [UO2(OH)2(CH3)]− | (8) |
Relevant minima and transition state structures for the reactions of [UO2(O)(CH3)]− (Fig. 6, structure XII) with H2O are provided in Fig. S13 of the ESI.† The zero-point corrected electronic energies and free energies for the respective species are provided in Table S4 of the ESI.† A reaction energy diagram for hydrolysis of, or addition of H2O to, [UO2(O)(CH3)]− by reactions (5) or (6), respectively, is shown in Fig. 6.
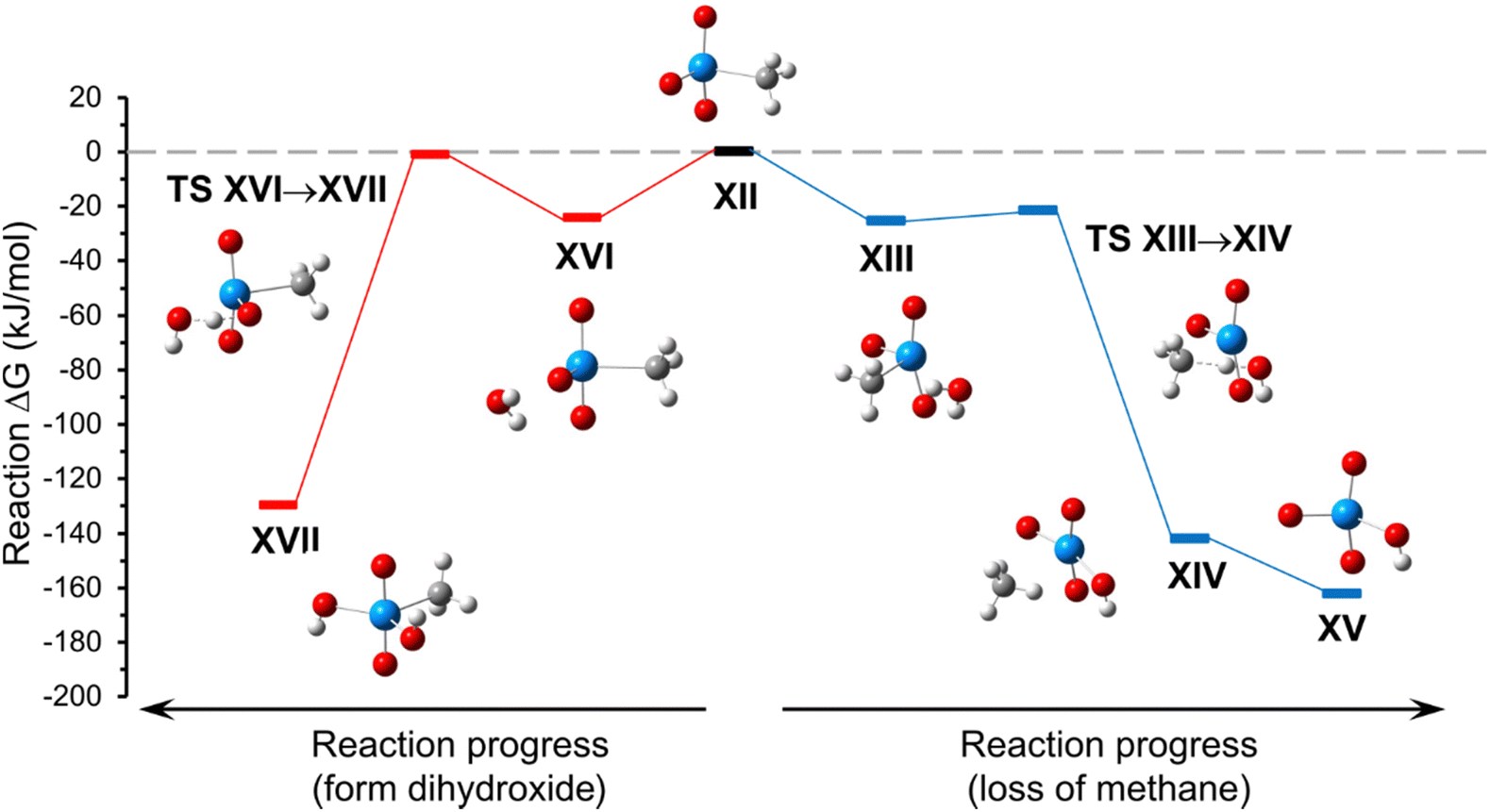 | ||
| Fig. 6 A reaction energy diagram for hydrolysis of, or addition of H2O to, [UO2(O)(CH3)]− by reactions (5) or (6), respectively, described in the text. | ||
As shown in Fig. 6, hydrolysis by reaction (5), and H2O addition and isomerization by reaction (6) proceed through H2O adduct structures that differ in the relative position of orientation of the H2O ligand. For the hydrolysis pathway (reaction (5)), the adduct structure XIII the H2O and CH3 ligands occupy adjacent equatorial coordination sites. In this case, proton transfer to generate the ion–molecule complex between [UO2(O)(OH)]− and CH4 (structure XIV) occurs through transition state structure TS XIII→XIV, and loss of CH4 is computed to proceed spontaneously to generate [UO2(O)(OH)]− (structure XV). For the H2O addition pathway with isomerization (reaction (6)), the adduct structure XVI includes the oxo and H2O ligands that occupy adjacent equatorial coordination sites. Proton transfer to generate the terminal dihydroxide product [UO2(OH)2(CH3)]− (structure XVII) occurs through transition state structure TS XVI→XVII.
Overall, the computed energies suggest that formation of either terminal product (XIV and XVII) should be energetically favorable. However, the energy of TS XVI→XVII is nearly at the energy of reactants (XII and H2O), and significantly higher than the proton transfer step of hydrolysis reaction (5) (TS XIII→XIV), which suggests that reaction (6) is kinetically less favored. The DFT calculations therefore support the experimental observation that the hydrolysis of [UO2(O)(CH3)]− to create [UO2(O)(OH)]− is spontaneous, indicating that the process is significantly favored over adduct formation with subsequent isomerization to make [UO2(CH3)(OH)2]−.
Conclusions
To summarize, we have conducted a follow-up study of the MSn CID of [UO2(O2C–CH3)2(O2C–H)]− and [UO2(O2C–CH3)(O2C–H)2]−, and the reactions of product ions with H2O. Reaction pathways were determined with the aid of deuterium labeling. Regardless of the precursor ion examined, initial CID causes decarboxylation of a formate ligand to create UO2-hydride with carboxylate ligands. Using d-labeled acetate, we determined that subsequent CID of the hydride complexes causes elimination of acetaldehyde or formaldehyde, depending on the precursor complex. The elimination of aldehyde as neutral species is consistent with reactions that include intra-complex hydride attack upon bound acetate or formate ligands. Density functional theory calculations reproduce the experimental observations, including favored elimination of formaldehyde over acetaldehyde by hydride attack during CID of [UO2(H)(O2C–CH3)(O2C–H)]−. To the best of our knowledge, this represents the first experimental and computational investigation of the intrinsic reactivity of this type of uranyl-hydride species. The results presented here are interesting given the desire to develop strategies for the selective conversion of carboxylic acids to aldehydes.74–76We also discovered that MSn CID of the acetate–formate complexes leads to generation of the oxyl-methide species, [UO2(O)(CH3)]−. The formation of the methide was confirmed using isotopically labeled acetate. Reaction of [UO2(O)(CH3)]− with H2O generates [UO2(O)(OH)]−. DFT calculations confirm that formation of [UO2(O)(OH)]− by elimination of CH4 would be spontaneous and favored over H2O addition and rearrangement to create the dihydroxide species [UO2(OH)2(CH3)]−.
Author contributions
ARB: investigation, methodology and validation writing – review and editing. IJT: investigation and methodology. AI: investigation and methodology. TAC: conceptualization, methodology, writing – review and editing. MJV: conceptualization, methodology, writing – original draft, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. V. S and T. A. C. acknowledge support for this work from the School of Science and Engineering (SOSE) at Duquesne University. Laboratory space renovation was made possible through support by the National Science Foundation through grant CHE-0963450. A. R. B and A. I. acknowledge SOSE, Duquesne University and the National Science Foundation (CHE-1659823 and CHE-1950585) for support of their summer undergraduate research. Quantum mechanical calculations were performed using the resources of the Duquesne University Center for Computational Studies, and the National Science Foundation (CHE-1726824). This work was also supported in part by the Robert Dean Loughney Faculty Development Endowment of Duquesne University.Notes and references
- G. R. Agnes and G. Horlick, Electrospray mass spectrometry as a technique for elemental analysis: preliminary results, Appl. Spectrosc., 1992, 46, 401–406 CrossRef CAS.
- C. Moulin, N. Charron, G. Plancque and H. Virelizier, Speciation of Uranium by Electrospray Ionization Mass Spectrometry: Comparison with Time-Resolved Laser-Induced Fluorescence, Appl. Spectrosc., 2000, 54, 843–848 CrossRef CAS.
- N. G. Tsierkezos, J. Roithova, D. Schroder, M. Oncak and P. Slavicek, Can electrospray mass spectrometry quantitatively probe speciation? Hydrolysis of uranyl nitrate studied by gas-phase methods, Inorg. Chem., 2009, 48, 6287–6296 CrossRef CAS PubMed.
- G. S. Groenewold, A. K. Gianotto, K. C. Cossel, M. J. Van Stipdonk, D. T. Moore, N. Polfer, J. Oomens, W. A. de Jong and L. Visscher, Vibrational spectroscopy of mass-selected [UO2(ligand)n]2+ complexes in the gas phase: comparison with theory, J. Am. Chem. Soc., 2006, 128, 4802–4813 CrossRef CAS PubMed.
- G. S. Groenewold, J. Oomens, W. A. de Jong, G. L. Gresham, M. E. McIlwain and M. J. Van Stipdonk, Vibrational spectroscopy of anionic nitrate complexes of UO22+ and Eu3+ isolated in the gas phase, Phys. Chem. Chem. Phys., 2008, 10, 1192–1202 RSC.
- G. S. Groenewold, M. J. Van Stipdonk, W. A. de Jong, J. Oomens, G. L. Gresham, M. E. McIlwain, D. Gao, B. Siboulet, L. Visscher, M. Kullman and N. Polfer, Infrared spectroscopy of dioxouranium(V) complexes with solvent molecules: effect of reduction, ChemPhysChem., 2008, 9, 1278–1285 CrossRef CAS PubMed.
- G. S. Groenewold, M. J. van Stipdonk, J. Oomens, W. A. de Jong and M. E. McIlwain, The gas-phase bis-uranyl nitrate complex [(UO2)2(NO3)5]−: Infrared spectrum and structure, Int. J. Mass Spectrom., 2011, 308, 175–180 CrossRef CAS.
- S. Pasilis, Á. Somogyi, K. Herrmann and J. E. Pemberton, Ions generated from uranyl nitrate solutions by electrospray ionization (ESI) and detected with fourier transform ion-cyclotron resonance (FT-ICR) mass spectrometry, J. Am. Soc. Mass Spectrom., 2006, 17, 230–240 CrossRef CAS PubMed.
- S. P. Pasilis and J. E. Pemberton, Speciation and coordination chemistry of uranyl(VI)-citrate complexes in aqueous solution, Inorg. Chem., 2003, 42, 6793–6800 CrossRef CAS PubMed.
- D. Rios, G. Schoendorff, M. J. Van Stipdonk, M. S. Gordon, T. L. Windus, J. K. Gibson and W. A. de Jong, Roles of acetone and diacetone alcohol in coordination and dissociation reactions of uranyl complexes, Inorg. Chem., 2012, 51, 12768–12775 CrossRef CAS PubMed.
- G. Schoendorff, W. A. de Jong, M. J. Van Stipdonk, J. K. Gibson, D. Rios, M. S. Gordon and T. L. Windus, On the formation of “hypercoordinated” uranyl complexes, Inorg. Chem., 2011, 50, 8490–8493 CrossRef CAS PubMed.
- M. J. Van Stipdonk, W. Chien, V. Anbalagan, K. Bulleigh, D. Hanna and G. S. Groenewold, Gas-phase complexes containing the uranyl ion and acetone, J. Phys. Chem. A, 2004, 108, 10448–10457 CrossRef CAS.
- M. J. Van Stipdonk, W. Chien, K. Bulleigh, Q. Wu and G. S. Groenewold, Gas-phase uranyl-nitrile complex ions, J. Phys. Chem. A, 2006, 110, 959–970 CrossRef CAS PubMed.
- D. Das, S. Kannan, D. K. Maity and M. G. B. Drew, Steric effects on uranyl complexation: synthetic, structural, and theoretical studies of carbamoyl pyrazole compounds of the Uranyl(VI), Ion. Inorg. Chem., 2012, 51, 4869–4876 CrossRef CAS PubMed.
- P. D. Dau, J. Su, H.-T. Liu, D.-L. Huang, J. Li and L.-S. Wang, Photoelectron spectroscopy and the electronic structure of the uranyl tetrachloride dianion: UO2Cl42−, J. Chem. Phys., 2012, 137, 064315 CrossRef PubMed.
- P. D. Dau, J. Su, H.-T. Liu, J.-B. Liu, D.-L. Huang, J. Li and L.-S. Wang, Observation and investigation of the uranyl tetrafluoride dianion (UO2F42−) and its solvation complexes with water and acetonitrile, Chem. Sci., 2012, 3, 1137–1146 RSC.
- W.-L. Li, J. Su, T. Jian, G. V. Lopez, H.-S. Hu, G.-J. Cao, J. Li and L.-S. Wang, Strong electron correlation in UO2−: A photoelectron spectroscopy and relativistic quantum chemistry study, J. Chem. Phys., 2014, 140, 094306 CrossRef PubMed.
- J. Su, P. D. Dau, Y.-H. Qiu, H.-T. Liu, C.-F. Xu, D.-L. Huang, L.-S. Wang and J. Li, Probing the Electronic Structure and Chemical Bonding in Tricoordinate Uranyl Complexes UO2X3− (X = F, Cl, Br, I): Competition between Coulomb Repulsion and U–X Bonding, Inorg. Chem., 2013, 52, 6617–6626 CrossRef CAS PubMed.
- B. T. McGrail, G. E. Sigmon, L. J. Jouffret, C. R. Andrews and P. C. Burns, Raman Spectroscopic and ESI-MS Characterization of Uranyl Peroxide Cage Clusters, Inorg. Chem., 2014, 53, 1562–1569 CrossRef CAS PubMed.
- C.-L. Xiao, C.-Z. Wang, L. Mei, X.-R. Zhang, N. Wall, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, Europium, uranyl, and thorium-phenanthroline amide complexes in acetonitrile solution: an ESI-MS and DFT combined investigation, Dalton Trans., 2015, 44, 14376–14387 RSC.
- X.-H. Kong, Q.-Y. Wu, X.-R. Zhang, C. Wang, K.-Q. Hu, Z.-F. Chai, C.-M. Ni and W.-Q. Shi, Coordination behavior of uranyl with PDAM derivatives in solution: Combined study with ESI-MS and DFT, J. Mol. Liq., 2020, 300, 112287 CrossRef CAS.
- Z. Qin, S. Shi, C. Yang, J. Wen, J. Jia, X. Zhang, H. Yu and X. Wang, The coordination of amidoxime ligands with uranyl in the gas phase: a mass spectrometry and DFT study, Dalton Trans., 2016, 45, 16413–16421 RSC.
- D. Rios, M. C. Michelini, A. F. Lucena, J. Marçalo and J. K. Gibson, On the origins of faster oxo exchange for uranyl(V) versus plutonyl(V), J. Am. Chem. Soc., 2012, 134, 15488–15496 CrossRef CAS PubMed.
- D. Rios, P. X. Rutkowski, D. K. Shuh, T. H. Bray, J. K. Gibson and M. J. Van Stipdonk, Electron transfer dissociation of dipositive uranyl and plutonyl coordination complexes, J. Mass Spectrom., 2011, 46, 1247–1254 CrossRef CAS PubMed.
- D. Rios, P. X. Rutkowski, M. J. Van Stipdonk and J. K. Gibson, Gas-phase coordination complexes of dipositive plutonyl, PuO22+: chemical diversity across the actinyl series, Inorg. Chem., 2011, 50, 4781–4790 CrossRef CAS PubMed.
- P. X. Rutkowski, D. Rios, J. K. Gibson and M. J. Van Stipdonk, Gas-phase coordination complexes of UVIO22+, NpVIO22+, and PuVIO22+ with dimethylformamide, J. Am. Soc. Mass Spectrom., 2011, 22, 2042–2048 CrossRef CAS PubMed.
- P. D. Dau, D. Rios, Y. Gong, M. C. Michelini, J. Marçalo, D. K. Shuh, M. Mogamman, M. J. Van Stipdonk, T. A. Corcovilos, J. K. Martens, J. Oomens, B. Redlich and J. K. Gibson, Synthesis and hydrolysis of uranyl, neptunyl and plutonyl gas-phase complexes exhibiting discrete actinide-carbon bonds, Organometallics, 2016, 35, 1228–1240 CrossRef CAS.
- V. Vallet, Y. Gong, M. Saab, F. Réal and J. K. Gibson, Carbon-sulfur bond strength in methanesulfinate and benzenesulfinate ligands directs decomposition of Np(v) and Pu(v) coordination complexes, Dalton Trans., 2020, 49, 3293–3303 RSC.
- D. Rios, M. C. Michelini, A. F. Lucena, J. Marçalo, T. H. Bray and J. K. Gibson, Gas-Phase Uranyl, Neptunyl, and Plutonyl: Hydration and Oxidation Studied by Experiment and Theory, Inorg. Chem., 2012, 51, 6603–6614 CrossRef CAS PubMed.
- P. D. Dau, M. Vasiliu, R. E. Wilson, D. A. Dixon and J. K. Gibson, Hydrolysis of Metal Dioxides Differentiates d-block from f-block Elements: Pa(V) as a 6d Transition Metal; Pr(V) as a 4f “Lanthanyl”, J. Phys. Chem. A, 2020, 124, 9272–9287 CrossRef CAS PubMed.
- T. Jian, X. Yu, D. Dan, T. E. Albrecht-Schmitt, J. Autschbach and J. K. Gibson, Gas-Phase Complexes of Americium and Lanthanides with a Bis-triazinyl Pyridine: Reactivity and Bonding of Archetypes for F-Element Separations, J. Phys. Chem. A, 2020, 124, 2982–2990 CrossRef CAS PubMed.
- J. Jian, S.-X. Hu, W.-L. Li, M. J. van Stipdonk, J. Martens, G. Berden, J. Oomens, J. Li and J. K. Gibson, Uranyl/12-crown-4 Ether Complexes and Derivatives: Structural Characterization and Isomeric Differentiation, Inorg. Chem., 2018, 57, 4125–4134 CrossRef CAS PubMed.
- M. J. Van Stipdonk, M. del Carmen Michelini, A. Plaviak, D. Martin and J. K. Gibson, Formation of bare UO22+ and NUO+ by fragmentation of gas-phase uranyl-acetonitrile complexes, J. Phys. Chem. A, 2014, 118, 7838–7846 CrossRef CAS PubMed.
- M. J. Van Stipdonk, C. O’Malley, A. Plaviak, D. Martin, J. Pestok, P. A. Mihm, C. G. Hanley, T. A. Corcovilos, J. K. Gibson and B. J. Bythell, Dissociation of gas-phase, doubly-charged uranyl-acetone complexes by collisional activation and infrared photodissociation, Int. J. Mass Spectrom., 2016, 396, 22–34 CrossRef CAS.
- M. J. Van Stipdonk, M. del Carmen Michelini, A. Plaviak, D. Martin and J. K. Gibson, Formation of bare UO22+ and NUO+ by fragmentation of gas-phase uranyl-acetonitrile complexes, J. Phys. Chem. A, 2014, 118, 7838–7846 CrossRef CAS PubMed.
- M. J. Van Stipdonk, C. O’Malley, A. Plaviak, D. Martin, J. Pestok, P. A. Mihm, C. G. Hanley, T. A. Corcovilos, J. K. Gibson and B. J. Bythell, Dissociation of gas-phase, doubly-charged uranyl-acetone complexes by collisional activation and infrared photodissociation, Int. J. Mass Spectrom., 2016, 396, 22–34 CrossRef CAS.
- E. Perez, C. Hanley, S. Koehler, J. Pestok, N. Polonsky and M. Van Stipdonk, Gas phase reactions of ions derived from anionic uranyl formate and uranyl acetate complexes, J. Am. Soc. Mass Spectrom., 2016, 27, 1989–1998 CrossRef CAS PubMed.
- M. J. Van Stipdonk, C. Hanley, E. Perez, J. Pestok, P. Mihm and T. A. Corcovilos, Collision-Induced Dissociation of uranyl-methoxide and uranyl-ethoxide cations: formation of UO2H+ and uranyl-alkyl product ions, Rapid Commun. Mass Spectrom., 2016, 30, 1879–1890 CrossRef CAS PubMed.
- M. Van Stipdonk, A. Bubas, I. Tatosian, E. Perez, N. Polonsky, L. Metzler and A. Somogyi, Formation of [UVOF4]− by collision-induced dissociation of a [UVIO2(O2)(O2C-CF3)2]− precursor, Int. J. Mass Spectrom., 2018, 424, 58–64 CrossRef CAS.
- M. J. Van Stipdonk, A. Iacovino and I. Tatosian, Influence of background H2O on the collision-induced dissociation products generated from [UO2NO3]+, J. Am. Soc. Mass Spectrom., 2018, 29, 1416–1424 CrossRef CAS PubMed.
- I. J. Tatosian, A. C. Iacovino and M. J. Van Stipdonk, CID of [UVIO2(ClO4)]+ revisited: production of [UVIO2(Cl)]+ and subsequent hydrolysis to create [UVIO2(OH)]+, Rapid Commun. Mass Spectrom., 2018, 32, 1085–1091 CrossRef CAS PubMed.
- M. J. Van Stipdonk, I. J. Tatosian, A. C. Iacovino, A. R. Bubas, L. Metzler, M. C. Sherman and A. Somogyi, Gas-phase deconstruction of UO22+: Mass spectrometry evidence for generation of [OUVICH]+ by collision-induced dissociation of [UVIO2(C ≡ CH)]+, J. Am. Soc. Mass Spectrom., 2019, 30, 796–805 CrossRef CAS PubMed.
- M. Van Stipdonk, E. Perez, C. Hanley, I. Tatosian, A. Bubas and S. Kline, Formation and Hydrolysis of Gas-phase [UVIO2(R)]+ [R = CH3, CH2CH3, CH = CH2 and C6H5], J. Mass Spectrom., 2019, 54, 780–789 CrossRef PubMed.
- L. J. Metzler, C. T. Farmen, T. A. Corcovilos and M. J. Van Stipdonk, Intrinsic Chemistry of [OUCH]+: Reactions with H2O, CH3C ≡ N and O2, Phys. Chem. Chem. Phys., 2021, 23, 4475–4479 RSC.
- G. S. Groenewold, K. C. Cossel, G. L. Gresham, A. K. Gianotto, A. D. Appelhans, J. E. Olson, M. J. Van Stipdonk and W. Chien, Binding of Molecular O2 to Di- and Tri-Ligated [UO2]+, J. Am. Chem. Soc., 2006, 128, 3075–3084 CrossRef CAS PubMed.
- V. S. Bryantsev, K. C. Cossel, M. S. Diallo, W. A. Goddard, III, W. A. de Jong, G. S. Groenewold, W. Chien and M. J. Van Stipdonk, 2-Electron 3-Atom Bond in Side-on (η2) Superoxo Complexes: U(IV) and U(V) Dioxo Monocations, J. Phys. Chem. A, 2008, 112, 5777–5780 CrossRef CAS PubMed.
- C. M. Leavitt, V. S. Bryantsev, W. A. de Jong, M. S. Diallo, W. A. Goddard III, G. S. Groenewold and M. J. Van Stipdonk, Addition of H2O and O2 to Acetone and Dimethylsulfoxide Ligated Uranyl(V) Dioxocations, J. Phys. Chem. A, 2009, 113, 2350–2358 CrossRef CAS PubMed.
- A. F. Lucena, J. M. Carretas, J. Marçalo, M. C. Michelini, Y. Gong and J. K. Gibson, Gas-phase reactions of molecular oxygen with uranyl(V) anionic complexes – synthesis and characterization of new superoxides of uranyl(VI), J. Phys. Chem. A, 2015, 119, 3628–3635 CrossRef CAS PubMed.
- S.-X. Hu, J. Jian, J. Li and J. K. Gibson, Destruction of the uranyl moiety in a U(V) “cation-cation” interaction, Inorg. Chem., 2019, 58, 10148–10159 CrossRef CAS PubMed.
- V. Vallet, U. Wahlgen and I. Grenthe, Probing the nature of chemical bonding in uranyl(VI) complexes with quantum chemical methods, J. Phys. Chem. A, 2012, 116, 12373–12380 CrossRef CAS PubMed.
- W. A. De Jong, E. Apra, T. L. Windus, J. A. Nichols, R. J. Harrison, K. E. Gutowski and D. A. Dixon, Complexation of the Carbonate, Nitrate, and Acetate Anions with the Uranyl Dication: Density Functional Studies with Relativistic Effective Core Potentials, J. Phys. Chem. A, 2005, 109, 11568–11577 CrossRef CAS PubMed.
- M. García-Hernández, C. Willnauer, S. Krüger, L. V. Moskaleva and N. Rösch, Systematic DFT Study of Gas Phase and Solvated Uranyl and Neptunyl Complexes [AnO2X4]n (An = U, Np; X = F, Cl, OH, n = −2; X = H2O, n = +2), Inorg. Chem., 2006, 45, 1356–1366 CrossRef PubMed.
- D. Rios, M. C. Michelini, A. F. Lucena, J. Marçalo, T. H. Bray and J. K. Gibson, Gas-Phase Uranyl, Neptunyl, and Plutonyl: Hydration and Oxidation Studied by Experiment and Theory, Inorg. Chem., 2012, 51, 6603–6614 CrossRef CAS PubMed.
- P. D. Dau, P. B. Armentrout, M. C. Michelini and J. K. Gibson, Activation of carbon dioxide by a terminal uranium–nitrogen bond in the gas-phase: a demonstration of the principle of microscopic reversibility, Phys. Chem. Chem. Phys., 2016, 18, 7334–7340 RSC.
- D. S. Emanuela, M. Maria del Carmen and R. Nino, Methane C–H Bond Activation by Gas-Phase Th+ and U+: Reaction Mechanisms and Bonding Analysis, Organometallics, 2009, 28, 3716–3726 CrossRef.
- M. Maria del Carmen, R. Nino and S. Emilia, How can uranium ions (U+, U2+) activate the O-H bond of water in the gas phase?, Angew. Chem., Int. Ed., 2006, 45, 1095–1099 CrossRef PubMed.
- E. Di Santo, M. Santos, M. C. Michelini, J. Marcalo, N. Russo and J. K. Gibson, Gas-Phase Reactions of the Bare Th2+ and U2+ Ions with Small Alkanes, CH4, C2H6, and C3H8: Experimental and Theoretical Study of Elementary Organoactinide Chemistry, J. Am. Chem. Soc., 2011, 133, 1955–1970 CrossRef CAS PubMed.
- L. Shi, P. Li, M.-G. Guo and T. Gao, Reaction mechanisms and topological analyses for the C–H activation of ethylene by uranium atom using density functional theory, Comput. Theor. Chem., 2020, 1190, 113022 CrossRef CAS.
- M. C. Michelini, J. Marçalo, N. Russo and J. K. Gibson, Gas-phase reactions of uranate ions, UO2−, UO3−, UO4− and UO4H−, with methanol: a convergence of experiment and theory, Inorg. Chem., 2010, 49, 3836–3850 CrossRef CAS PubMed.
- C. Peng and H. B. Schlegel, Combining synchronous transit and quasi-Newton methods for finding transition states, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- K. Fukui, The Path of Chemical-Reactions – The IRC Approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, An Improved Algorithm for Reaction-Path Following, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, Reaction-Path Following in Mass-Weighted Internal coordinates, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- K. A. Peterson, Correlation consistent basis sets for actinides. I. The Th and U atoms, J. Chem. Phys., 2015, 142, 074105 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- N. Kaltsoyannis, Transuranic Computational Chemistry, Chem. – Eur. J., 2018, 24, 2815–2825 CrossRef CAS PubMed.
- J. T. Pegg, A. E. Shields, M. T. Storr, D. O. Scanlon and N. H. de Leeuw, Noncollinear Relativistic DFT+U Calculations of Actinide Dioxide Surfaces, J. Phys. Chem. C, 2019, 123, 356–366 CrossRef CAS.
- U. D. Wdowik, P. Piekarz, D. Legut and G. Jagło, Effect of Spin–Orbit and on-Site Coulomb Interactions on the Electronic Structure and Lattice Dynamics of Uranium Monocarbide, Phys. Rev. B, 2016, 94, 054303 CrossRef.
- R. A. J. O’Hair and N. J. Rijs, Gas Phase Studies of the Pesci Decarboxylation Reaction: Synthesis, Structure, and Unimolecular and Bimolecular Reactivity of Organometallic Ions, Acc. Chem. Res., 2015, 48, 329–340 CrossRef PubMed.
- T. Waters, R. A. J. O’Hair and A. G. Wedd, Catalytic gas phase dehydration of acetic acid to ketene, Int. J. Mass Spectrom., 2003, 228, 599–611 CrossRef CAS.
- E. L. Piacentino, K. Parker, T. M. Gilbert, R. A. J. O’Hair and V. Ryzhov, Chem. – Eur. J., 2019, 25, 9959–9966 CrossRef CAS PubMed.
- K. L. Vikse, G. N. Khairallah and R. A. J. O’Hair, Organometallics, 2012, 31, 7467–7475 CrossRef CAS.
- G. S. Groenewold, W. A. de Jong, J. Oomens and M. van Stipdonk, Variable denticity in carboxylate binding to the uranyl dication, J. Am. Soc. Mass Spectrom., 2010, 21, 719–727 CrossRef CAS PubMed.
- B. Han, C. Ren, M. Jian and L. Wu, Titanium-Catalyzed Exhuastive Reduction of Oxo-Chemical, Angew. Chem., Int. Ed., 2022, 61, e202209232 CrossRef CAS PubMed.
- A. W. Iosub, S. Moravčik, C.-J. Wallentin and J. Bergman, Nickel-Catalyzed Selective Reduction of Carboxylic Acids to Aldehydes, Org. Lett., 2019, 21, 7804–7808 CrossRef CAS PubMed.
- R. A. W. Johnstone, in Comprehensive Organic Synthesis: Reduction of Carboxylic Acids to Aldehydes by Metal Hydrides, ed. B. M. Trost and I. Fleming, Pergamaon, Oxford, 1991, vol 8, pp. 259–281 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00823e |
| ‡ Present address: Department of Chemistry, University of Utah, Salt Lake City, UT, 84112 USA. |
| § Present address: American Preparatory Academy, Draper, UT, 84020 USA. |
| ¶ Present address: PPG Industries, Allison Park PA, 15101 USA. |
| This journal is © the Owner Societies 2024 |