
Computational insights into the binding modes, keto–enol tautomerization and stereo-electronically controlled decarboxylation of oxaloacetate in the active site of macrophomate synthase†
Xinyi
Li
a,
Fa-Guang
Zhang
 b,
Jun-An
Ma
*b and
Yongjun
Liu
*a
b,
Jun-An
Ma
*b and
Yongjun
Liu
*a
aSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, Shandong 250100, China. E-mail: yongjunliu_1@sdu.edu.cn
bDepartment of Chemistry, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Frontiers Science Center for Synthetic Biology (Ministry of Education), Tianjin University, Tianjin 300072, China. E-mail: majun_an68@tju.edu.cn
First published on 2nd April 2024
Abstract
Oxaloacetic acid (OAA) is a β-ketocarboxylic acid, which plays an important role as an intermediate in some metabolic pathways, including the tricarboxylic acid cycle, gluconeogenesis and fatty acid biosynthesis. Animal studies have indicated that supplementing oxaloacetic acid shows an increase of lifespan and other substantial health benefits including mitochondrial DNA protection, and protection of retinal, neural and pancreatic tissues. Most of the chemical transformations of OAA in the metabolic pathways have been extensively studied; however, the understanding of decarboxylation of OAA at the atomic level is relatively lacking. Here, we carried out MD simulations and combined quantum mechanical/molecular mechanical (QM/MM) calculations as an example to systematically elucidate the binding modes, keto–enol tautomerization and decarboxylation of OAA in the active site of macrophomate synthase (MPS), which is a Mg(II)-dependent bifunctional enzyme that catalyzes both the decarboxylation of OAA and [4+2] cycloaddition of 2-pyrone with the decarboxylated intermediate of OAA (pyruvate enolate). On the basis of our calculations, it was found that the Mg2+-coordinated oxaloacetate may exist in enol forms and keto forms. The four keto forms can be transformed into each other by simply rotating the C2–C3 single bond, nevertheless, the keto–enol tautomerization strictly requires the assistance of pocket water molecules. In addition, the decarboxylation is stereo-electronically controlled, i.e., it is the relative orientation of the terminal carboxyl anion that determines the rate of decarboxylation. As such, the chemistry of oxaloacetate in the active site of MPS is complex. On one hand, the most stable binding mode (K-I) may undergo enol–keto tautomerization to isomerize to the enol form, which may further react with the second substrate; on the other hand, K-I may isomerize to another binding mode K-II to proceed decarboxylation to generate pyruvate enolate and CO2. Starting from K-I, the enol–keto tautomerization corresponds to a barrier of 16.2 kcal mol−1, whereas the decarboxylation is associated with an overall barrier of 19.7 kcal mol−1. These findings may provide useful information for understanding the chemistry of OAA and the catalysis of related enzymes, and they are basically in agreement with the available experimental kinetic data.
1. Introduction
Oxaloacetate (OAA) plays a central role as a metabolic intermediate, which is mainly involved in gluconeogenesis, citric acid cycle, amino acid synthesis and fatty acid synthesis.1 OAA exists in every cell in the body, and has important biochemical and physiological effects on living organisms. For example, OAA is required for the first step of gluconeogenesis; it takes part in the aerobic decomposition of sugar in the citric acid cycle, where it condenses with acetyl-CoA to form citrate and CoA with the catalysis of citrate synthase; it is a potent inhibitor of mitochondrial complex II and plays an important role in central metabolism; and as a key regulator of mitochondrial metabolism, it inhibits the succinate dehydrogenase.2,3 Recently, OAA was also found to have a wide range of pharmacological effects that may be used in the treatment of many diseases.4OAA is both an α-keto acid and a β-keto acid, which can undergo a series of chemical conversions in the body with the catalysis of many enzymes. In the citric acid cycle, OAA can sequentially convert to citrate, isocitrate, α-ketoglutarate, succinyl-CoA, succinate, fumarate and malate.3 In the urea cycle and amino acid metabolism, aspartate is transaminated to form oxaloacetate with the catalysis of aspartate transaminase (AST). In gluconeogenesis, the pyruvate carboxylase and phosphoenolpyruvate carboxykinase catalyze the decarboxylation of OAA.5 Of the variety of OAA-involving reactions, decarboxylation has always been the issue of concerns,6 because it is one of the most common processes in nature, and all of the carbon dioxide evolved in fermentation and respiration is generated by the decarboxylation of organic acids. Besides, decarboxylation is also a key step for the biosynthesis of terpenoids, steroids, and neurotransmitter amino compounds. Numerous decarboxylase enzymes serve as key components of aerobic and anaerobic carbohydrate metabolism and amino acid conversion. According to the IUPAC classification, there are at least 90 different classes of decarboxylases.7 The enzyme-catalyzed decarboxylation usually requires either an organic cofactor such as biotin, flavin, NAD, pyridoxal 5′-phosphate and thiamin pyrophosphate, or an inorganic cofactor such as an iron or zinc complex.8,9 Only a few decarboxylase enzymes do not use any cofactors.10,11 These decarboxylases employ different mechanisms to perform the decarboxylation of a variety of substrates, and the mechanisms of metal-dependent decarboxylases have drawn much attention. Since the decarboxylation reaction requires the cleavage of the C–C single bond, the stabilization of the negative charge in the carbanion upon the elimination of carbon dioxide is always necessary. In the active sites of the decarboxylases with binding organic or metal cofactors, the negative charge can be neutralized by these cofactors, and in the active sites of cofactor-free decarboxylases, the nearby residues play a similar role by providing a proton or forming a Schiff base.
Previous studies have revealed that amines and divalent metal ions can facilitate the decarboxylation of OAA to generate pyruvate and carbon dioxide, while decarboxylases could significantly increase the reaction rate.7,12,13 Oxaloacetate decarboxylases have been shown to require divalent metal ions such as Mn2+ or Mg2+ as the cofactors, in which the enzyme-bound metal iron chelates to the α-carboxyl and keto-carbonyl of the oxaloacetate. The metal ions are also expected to stabilize the transition state with carbanionic character.14
The enzymes reported to have activity of decarboxylation of oxaloacetate are malic enzyme,15 pyruvate kinase16 and oxaloacetate decarboxylase.17 Malic enzyme (ME) catalyzes both the decarboxylation of malate to generate pyruvate and CO2 and the reverse carboxylation reaction that introduces CO2 as the carboxyl group to pyruvate. Pyruvate kinase catalyzes the metal ion-dependent decarboxylation of oxaloacetate in the same site as the phosphor transfer reaction in the enzyme. Oxaloacetate decarboxylase (OXAD) catalyzes the irreversible decarboxylation of oxaloacetate to generate pyruvate and CO2. It is worth mentioning that oxaloacetate is not the in vivo natural substrate of malic enzyme and pyruvate kinase.
Macrophomate synthase (MPS) is an enzyme that catalyzes a complex multi-step reaction of α-pyrones with oxaloacetate, involving two C–C bond forming steps and two decarboxylations to afford benzoic acid derivatives (Scheme 1a).18,19 The two C–C bond formation steps were firstly speculated to be a Diels–Alder reaction, and it was later confirmed to follow a stepwise Michael–Aldol mechanism.20 So far, there is still no definitive evidence to give a consistent conclusion. Another focal point of the debate is whether the decarboxylation of OAA is prior to the C–C bond formation. It is worth noting that Jorgensn et al.20 have studied the MPS-catalyzed [4+2]-cycloaddition of OAA with 2-pyrone by performing QM/MM calculations, however, they simply employed the pyruvate enolate as the dienophile. That is to say, it was based on the premise that the oxaloacetate directly undergoes the decarboxylation without considering the reaction sequence of decarboxylation vs. cycloaddition. Actually, the experimental examination has suggested that a portion of oxaloacetate is present in its enol form (ca. 6%) in the active site of MPS,21 which implies the possibility of oxaloacetate enolate directly participating in the cycloaddition with 2-pyrone. Indeed, previous experimental studies indicated that the change of the substrate from oxaloacetate to pyruvate significantly slows down the reaction rate (kcat for oxaloacetate = 0.46 s−1vs. kcat for pyruvate = 0.027 s−1).19,22 In addition, the decarboxylation of oxaloacetate to pyruvate was previously observed in the absence of 2-pyrone, but in the presence of 2-pyrone, there was no clear evidence to confirm the formation of pyruvate by in situ NMR detection.21 These results strongly imply that pyruvate is probably not a real intermediate of the MPS-catalyzed [4+2] cycloaddition. Our recent studies23–25 in a series of decarboxylative Michael or Mannich or Aldol addition reactions of β-keto acids demonstrated that the C–C bond formation step always takes place before the decarboxylation process. More importantly, Huang's group recently identified that the direct Mannich-type reaction of pyrrolidine-β-keto acid without decarboxylation serves as an important step in the cocaine biosynthetic pathway.26,27 Therefore, deciphering the decarboxylation of OAA is the first step in understanding the MPS-catalyzed reactions. More detailed and in-depth mechanistic explorations of the reaction sequence of decarboxylation of OAA vs. [4+2]-cycloaddition are still required. Unlike metal-dependent malic enzyme and oxaloacetate decarboxylase, MPS has high specificity for Mg(II).22 In addition to catalyzing [4+2]-cycloaddition of OAA and 2-pyrone, MPS has also been proven to function simply as a decarboxylase in the absence of the second substrate 2-pyrone.28,29 Therefore, MPS is another enzyme with the activity of catalyzing the decarboxylation of OAA. The Ose group has reported the crystal structure of MPS in complex with pyruvate and Mg2+ at 1.7 Å resolution (Scheme 1b),30 enabling us to theoretically investigate the enzyme-catalyzed decarboxylation of OAA. According to previous studies,31 the decarboxylation of β-keto acid is stereo-electronically controlled by the dihedral angle between the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the cleaved C–C single bond, i.e., if the breaking C–C single bond is roughly orthogonal to the plane of carbonyl CO, the decarboxylation would correspond to a continuous overlap of the π-orbital of carbonyl CO with the breaking C–C σ-bond via hyperconjugation and resonance (Scheme 1c), lowering the barrier of decarboxylation. In contrast, the parallel conformation of the CO bond with the C–C single bond is unfavorable for decarboxylation. In the active site of MPS, the terminal carboxylate of the Mg2+-coordinated OAA may have different binding conformations, in particular, it may form hydrogen bonds with the nearby residues and solvent water. These factors may all contribute to the complexity of the decarboxylation reaction.
O bond and the cleaved C–C single bond, i.e., if the breaking C–C single bond is roughly orthogonal to the plane of carbonyl CO, the decarboxylation would correspond to a continuous overlap of the π-orbital of carbonyl CO with the breaking C–C σ-bond via hyperconjugation and resonance (Scheme 1c), lowering the barrier of decarboxylation. In contrast, the parallel conformation of the CO bond with the C–C single bond is unfavorable for decarboxylation. In the active site of MPS, the terminal carboxylate of the Mg2+-coordinated OAA may have different binding conformations, in particular, it may form hydrogen bonds with the nearby residues and solvent water. These factors may all contribute to the complexity of the decarboxylation reaction.
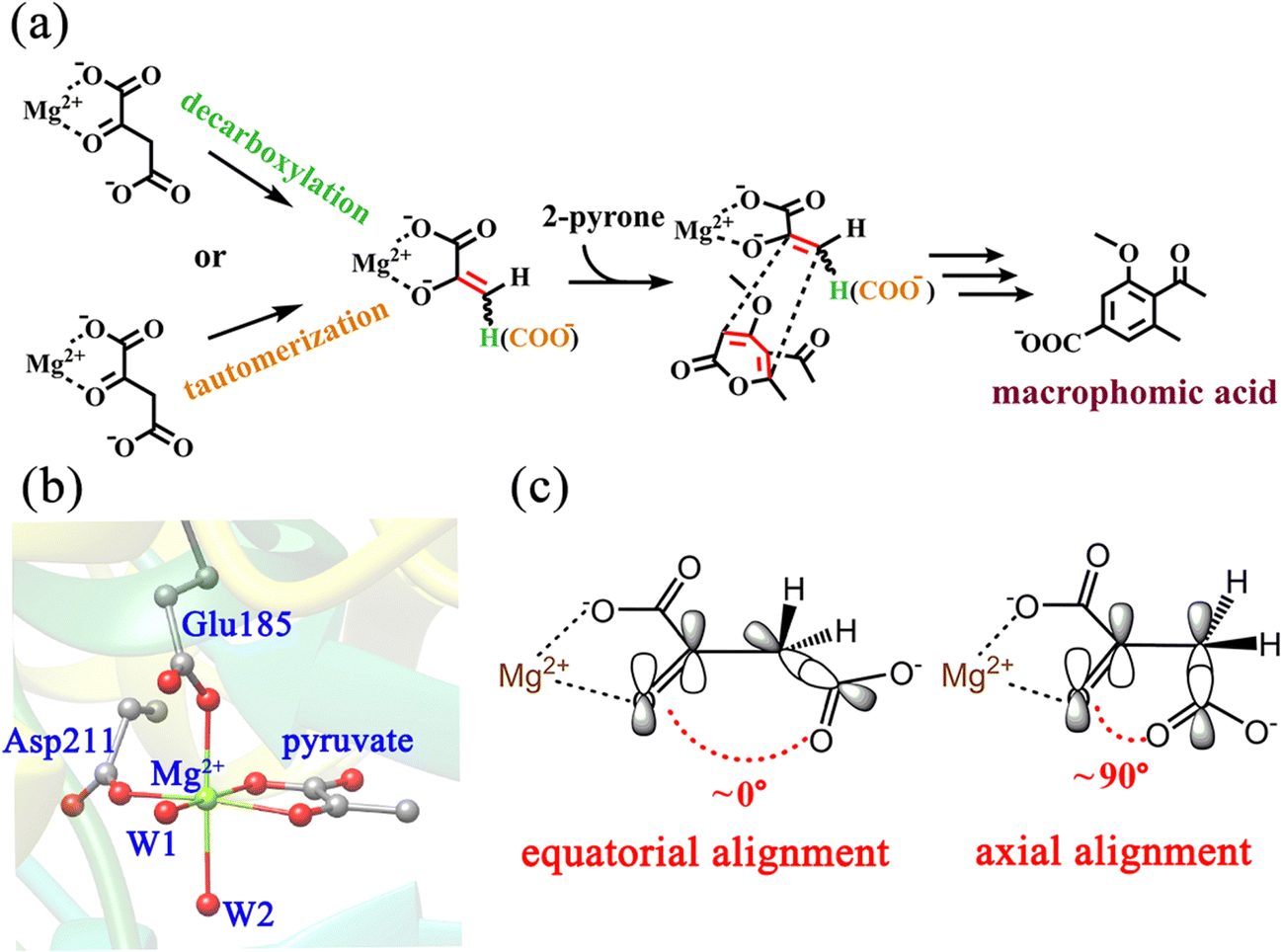 | ||
| Scheme 1 (a) Decarboxylation and tautomerization of OAA, and subsequent cycloaddition catalyzed by MPS in the biosynthesis of macrophomic acid; (b) active site of the crystal structure of MPS in complex with pyruvate (PDB code: 1IZC);30 and (c) orbital alignments during the decarboxylation of OAA with the axial and equatorial terminal carboxylate. | ||
In general, very little is known about the MPS-catalyzed decarboxylation of OAA at the atomic level, and some key issues are still poorly understood, for example, the binding modes of the substrate, the details of keto–enol tautomerization and decarboxylation. To address these issues, we have performed MD simulations and a series of combined quantum mechanics/molecular mechanics (QM/MM) calculations. Our calculations revealed that the energy barrier of the decarboxylation step is closely related to the conformation of the terminal carboxylate in OAA. Among the four binding modes of OAA, only one conformation is favorable for decarboxylation. Furthermore, the investigation on the [4+2]-cycloaddition reaction catalyzed by MPS is going on in our group.
2. Methods
2.1. Setup of the system and MD simulations
On the basis of the crystal structure of MPS (PDB code: 1IZC),30 the computational model was built. Fig. S1a (ESI†) shows the whole hexameric crystal structure of MPS, which contains six chains. Since the active site is located at the interface of a three-fold-related protomer, we selected one chain (Chain A) from one protomer and parts of the neighboring chain (Chain B) in our model. To investigate the decarboxylation step, the Mg2+-bound pyruvate in the crystal was manually modified to oxaloacetate.According to the pKa values calculated by the PROPKA program32 and visual inspection using VMD software,33 the protonation states of all titratable residues were determined. All aspartic acid and glutamic acid residues were present in the deprotonated states. Histidines 12, 48, 73, 86, 91 and 177 were protonated at the δ position, while histidines 48, 82, 90, 105, 125, 203, 229 and 285 were protonated at the ε position. Besides, all lysine and arginine residues were treated as protonated. The missing hydrogen atoms were added using the HBUILD module in the CHARMM package.34 The whole system was solvated by a water sphere with a radius of 40 Å, and was neutralized by thirteen randomly added sodium ions. Subsequently, the solvated system underwent a series of energy minimizations for equilibration. For the following MD simulations, the relaxed system was first heated from 0 to 300 K for 200 ps with a time step of 1 fs, and then was subjected to a 35-ns molecular dynamics (MD) simulation with stochastic boundary conditions using the CHARMM22/CMAP all-atom force field35 performed using the CHARMM package. The RMSD for the backbone atoms of the protein in MD simulation is displayed in Fig. S2 (ESI†), which shows that the system has basically reached equilibrium after 10 ns.
2.2. QM/MM methodology
A representative snapshot of the equilibrated system at 20 ns in MD simulations was taken as the initial structure of subsequent QM/MM calculations. To prove its representativeness, four other snapshots from the MD trajectory at 15, 25, and 30 ns were also optimized, which show minor displacements for their backbones and active sites (Fig. S3, ESI†). All QM/MM calculations were performed using the Chemshell program,36 which combines the Turbomole program37 for the QM region calculations and the DL_POLY program38 for the MM region calculations. By the analysis of the crystal structure,30 the pyruvate is hydrogen-bonded to the main chain of Gly210 and the side chain of Arg101, and the side chains of the second shell residues His125 and Ser148 form hydrogen bonds with the carboxyl oxygen of Asp211 and the Mg-coordinated water molecule (W2), respectively. Besides, the hydrophobic residue Phe149 is located near the first coordination layer, which may affect the binding of the substrates. The above-mentioned residues, the first coordination layer (Mg2+, Asp211, Glu185, and two water molecules) and the substrate oxaloacetate were assigned to the QM region for exploring the decarboxylation step (Fig. S4, ESI†), while the rest of the system was assigned to the MM region. It should be mentioned that the QM regions in the QM/MM calculations were adjusted as needed, which are shown in the ESI† (Fig. S4). The electronic embedding scheme was implemented to describe the polarizing effect of the MM point charges of the force field on the QM region.39 The boundary of QM and MM parts was treated by the hydrogen link atoms with the charge shift model.40 The region within 16 Å of Mg2+ was designated as the active region, which was fully relaxed in QM/MM calculations. Considering that the rest of the active region may have a minor influence on the reaction, they were kept frozen during the QM/MM calculations.In our work, geometry optimizations were carried out with the hybrid delocalized internal coordinate (HDLC) optimizer,41 and the minima search was performed using the quasi-Newton limited memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm.42,43 To determine the transition state, an assumed structure was first derived from the highest point of the scanned potential energy surface, and it was further optimized using the partitioned rational function optimization (P-RFO) algorithm.44 The unrestricted hybrid UB3LYP density functional with the 6-31G(d,p) basis set (referred as B1) was used to describe the QM region atoms,45 whereas the MM region was described by the CHARMM22/CMAP force field.35 The single-point energy calculations were carried out using the larger basis set 6-311++G(d,p) (referred to as B2).45 In addition, the frontier orbital diagrams were made using Multiwfn46 combined with VMD software.33
2.3. Ligand Gaussian accelerated MD (LiGaMD) simulations
LiGaMD47,48 implemented in Amber2249 was performed to investigate different low-energy binding conformations of oxaloacetate and their interconversion. A harmonic boost potential was applied to both the total potential energy of the ligand and remaining potential energy of the entire system to enhance the sampling of the substrate without predefined reaction coordinates. The LiGaMD simulations proceeded with 6.0 ns classical MD simulations to collect the potential statistics, 94.0 ns GaMD equilibration after adding the boost potential, and then three independent 600 ns production runs. LiGaMD production simulation frames were saved every 1 ps for analysis. The two-dimensional (2D) potential of the mean force (PMF) profile was drawn with reweighting using the cumulant expansion to the second order.50 The bin size was set to 5.0° for the dihedrals.3. Results and discussion
3.1. Binding modes of oxaloacetate
To better understand the catalysis of MPS, the binding modes of OAA in the activate site were first investigated. The oxaloacetate is bidentate coordinated with Mg2+ using its C1-carboxylate and C2-carbonyl. According to the crystal and our analysis, the Mg2+-coordinated OAA may exist in its enol form or keto form. In the keto form, the terminal carboxylate may rotate along the C2–C3 bond, which may lead to different binding conformations. By scanning the dihedral φC1–C2–C3–C4 of OAA, four local minima were optimized, as shown in Fig. 1. These four keto structures are mainly distinguished by the relative orientation of C4-carboxylate. Among them, the binding mode of K-I corresponds to the lowest relative energy, which means that oxaloacetate should mainly exist in this form. To check whether K-I, K-II, K-III and K-IV are dynamically stable and whether they can easily transform into each other, we performed a 600-ns LiGaMD simulation using K-I as the starting structure. Fig. 2 presents the 2D potential of the mean force (PMF) profile. According to the dihedrals of φC2–C3–C4–O1 and φC1–C2–C3–C4, the four keto forms can be located on the PMF profile. Moreover, we can also see that these keto forms can transform into each other by simply rotating the dihedrals of φC1–C2–C3–C4 and φC2–C3–C4–O1. According to the PMF profile, the conversion from K-I to K-II should correspond to the lowest energy barrier. To obtain more accurate energy barriers, we also performed QM/MM calculations on the four keto structures (Fig. S5, ESI†). The energy profiles are shown in Fig. 3. In general, the results of QM/MM calculations are basically consistent with those of LiGaMD simulations.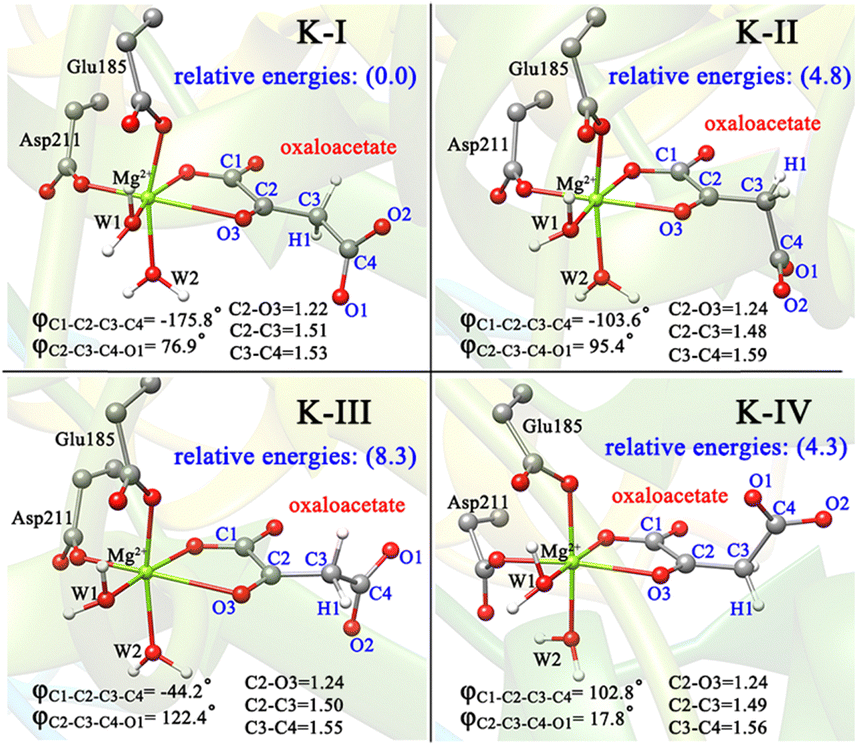 | ||
| Fig. 1 QM/MM-optimized structures of four binding modes of OAA in keto forms, named K-I, K-II, K-III and K-IV. The data in brackets are the relative energies in kcal mol−1. All distances are given in angstrom. | ||
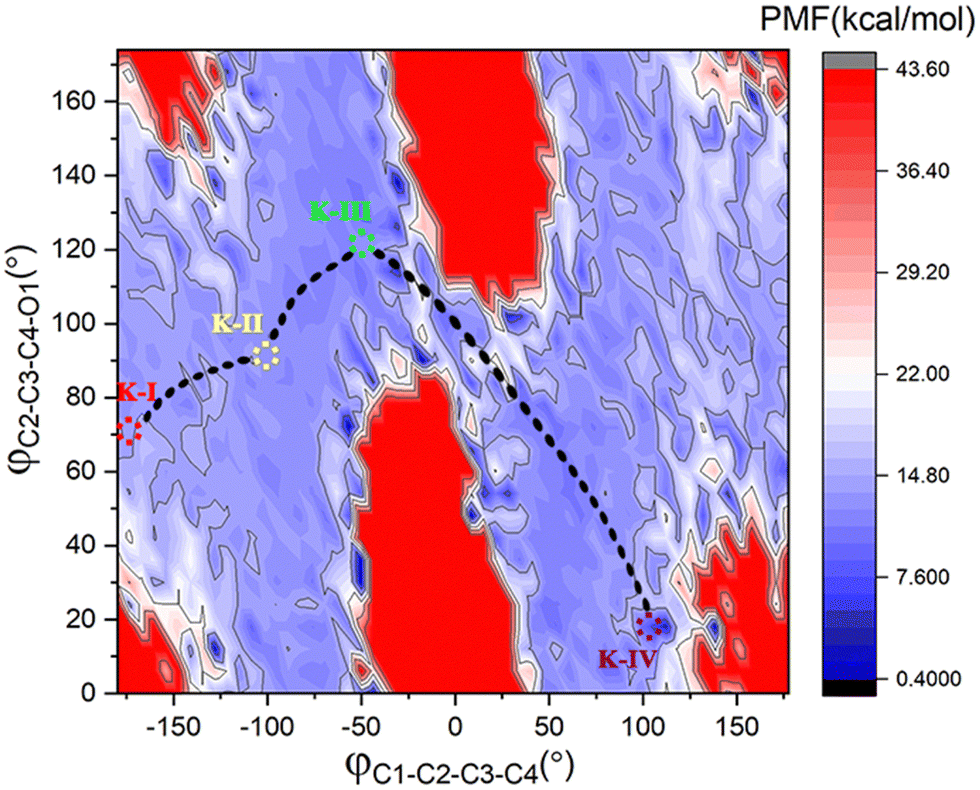 | ||
| Fig. 2 2D PMF profile with the dihedrals of φC2–C3–C4–O1 and φC1–C2–C3–C4 as reaction coordinates from 600-ns LiGaMD simulations of K-I at 300 K and 1.0 atm. | ||
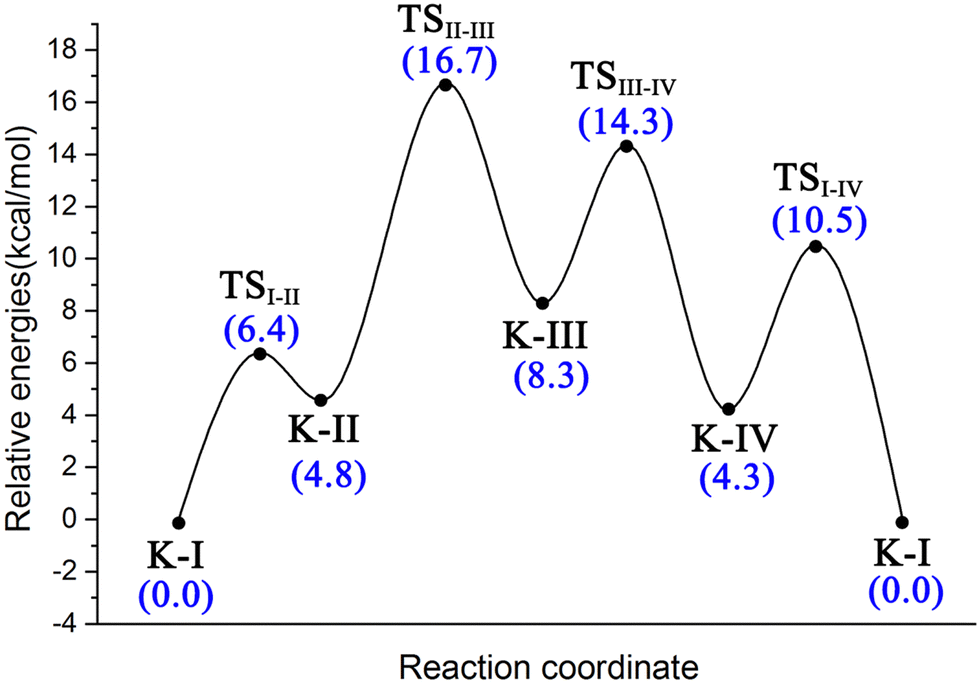 | ||
| Fig. 3 Energy profile for the isomerization of four binding conformations of oxaloacetate from K-I to K-II, K-III and K-IV calculated by using QM/MM calculations in conjunction with scanning φC1–C2–C3–C4. | ||
OAA may also coordinate with Mg2+ in the enol form, thus, both the Z- and E-isomers of oxaloacetate were considered, and four representative conformations are constructed, as shown in Fig. 4a. As displayed in (Z)-E-A and (E)-E-B′, the terminal carboxylate groups are almost parallel to the plane of the CC double bond, whereas in the (Z)-E-A′ and (E)-E-B, the carboxylate groups are almost perpendicular to the plane of the CC double bond. According to our QM/MM optimizations, the (E)-E-B′ was not recognized as a minimum, because it always converged into the more stable (E)-E-B. It is understandable because the carboxyl group of oxaloacetate forms hydrogen bonding interaction with the surrounding water molecules, making (E)-E-B′ an unstable conformation. As such, (Z)-E-A, (Z)-E-A′ and (E)-E-B are three enolate structures that could be recognized by QM/MM optimizations. Their relative energies and key parameters are displayed in Fig. 4b and Fig. S6 (ESI†). One can see that (Z)-E-A is 3.5 kcal mol−1 higher than the most stable binding mode of K-I, while (Z)-E-A′ and (E)-E-B are respectively 2.6 and 16.8 kcal mol−1 higher than (Z)-E-A. The structural parameters in Fig. 1 and 4b show that the four keto forms correspond to a shorter C2–O3 bond (1.22–1.24 Å, CO) and a longer C2–C3 bond (1.48–1.51 Å, C–C bond), and two enol forms correspond to a longer C2–O3 bond (1.34–1.40 Å, C–O bond) and a shorter C2–C3 bond (1.34–1.36 Å, CC bond).
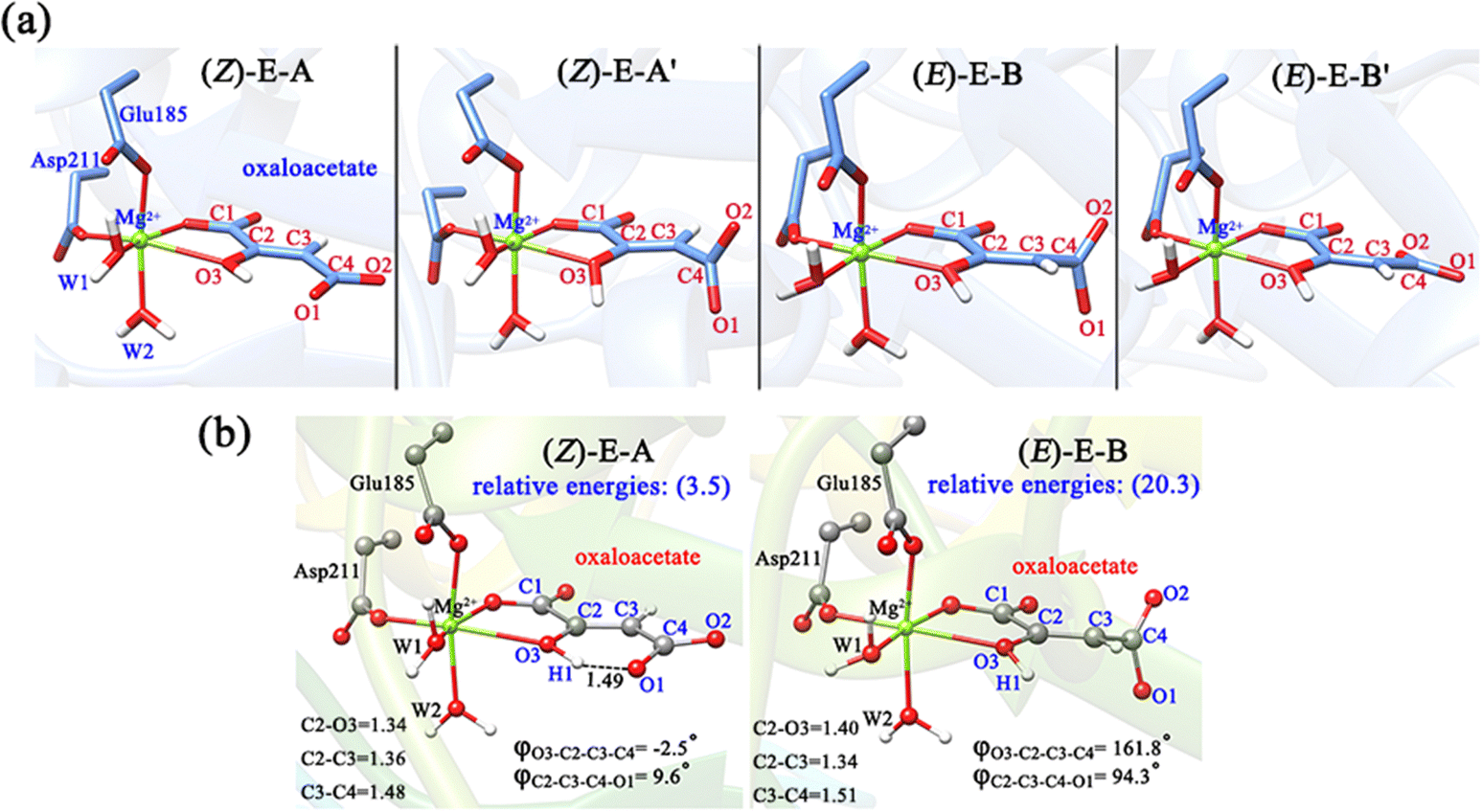 | ||
| Fig. 4 (a) Primarily constructed four binding modes of OAA in enol forms, including two (Z)- and two (E)-conformations; (b) two stable conformations confirmed by MD simulations, which were further optimized by QM/MM, named (Z)-E-A and (E)-E-B. The data in brackets are the relative energies (kcal mol−1) to K-I in Fig. 1. All distances are given in angstrom. | ||
Furthermore, we also conducted 120-ns classical MD simulations to test their stabilities (Fig. S7, ESI†). Our MD simulations show that (Z)-E-A is stable, and the dihedral of φC2–C3–C4–O1 always stabilizes at 10°. However, (Z)-E-A′ is quite unstable, and the dihedral of φC2–C3–C4–O1 rapidly changes from ∼90° to 10°; therefore, (Z)-E-A′ is a dynamically unstable structure, which is consistent with the higher relative energy of (Z)-E-A′ compared with (Z)-E-A in the QM/MM optimization. In general, the K-I form was calculated to be the dominant binding mode, which should account for the largest proportion of OAA in the active site of MPS.
3.2. Keto–enol tautomerization
As previously reported,51 the enol and keto forms of oxaloacetate may be interconverted via keto–enol tautomerization, which has received considerable interest for its biochemical importance. Nevertheless, most of the past research52–54 focused on exploring the acid-catalyzed keto–enol tautomerization of OAA (non-enzymatic reaction) by using the simple models in the gas phase and solution, which cannot well simulate the chemistry of oxaloacetate in the active site of enzymes. Herein, we further explored the keto–enol tautomerization of OAA by performing QM/MM calculations. For comparison, the direct proton/hydrogen transfer from C3-methylene to C2-carbonyl was investigated, in which all the four keto forms were used as the reactants. Our results showed that the direct keto–enol tautomerizations always correspond to very high energy barriers (>53.5 kcal mol−1, Fig. S8, ESI†). This result is consistent with the previous theoretical study on keto–enol tautomerization.51Since previous studies have suggested that the solvent water can significantly promote the keto–enol tautomerization of β-keto acids,51,55 two computational models that contain one and two water molecules were further constructed, as shown in Fig. 5. Our calculations showed that the water molecules can greatly reduce the energy barriers of the keto–enol tautomerization. Fig. 5a and b show the keto–enol tautomerization of two keto forms (K-IV and K-I) mediated by one and two water molecules, respectively, which correspond to the relatively lower barriers. With the assistance of one water molecule, the energy barrier of tautomerization can be reduced to 36.6 kcal mol−1, as displayed in the tautomerization from K-IV1w to E-A1w (Fig. 5a), which is the lowest energy barrier for the one water molecule-mediated tautomerization. The other two models, K-I1w and K-III1w correspond to the barriers of above 45 kcal mol−1 (Fig. S9, ESI†). In addition, the keto–enol tautomerization mediated by two water molecules was also investigated. Our results show that the energy barrier can be reduced to 16.2 kcal mol−1 (from K-I2w to E-A2w, Fig. 5b). Note that the other two models correspond to the energy barriers of 32.8 and 24.4 kcal mol−1 (Fig. S10, ESI†). Therefore, it can be concluded that water molecules can not only stabilize the transition states and intermediates by forming the hydrogen bonding network with the polar functional groups of oxaloacetate, but also serve as a necessary mediator to facilitate the keto–enol tautomerization. We also considered the model that contains three water molecules in the keto–enol tautomerization (Fig. S11, ESI†). However, in the optimized structure, the third water molecule (W5) is not in the proton/hydrogen transfer chain, and it would provide little assistance for keto–enol tautomerization.
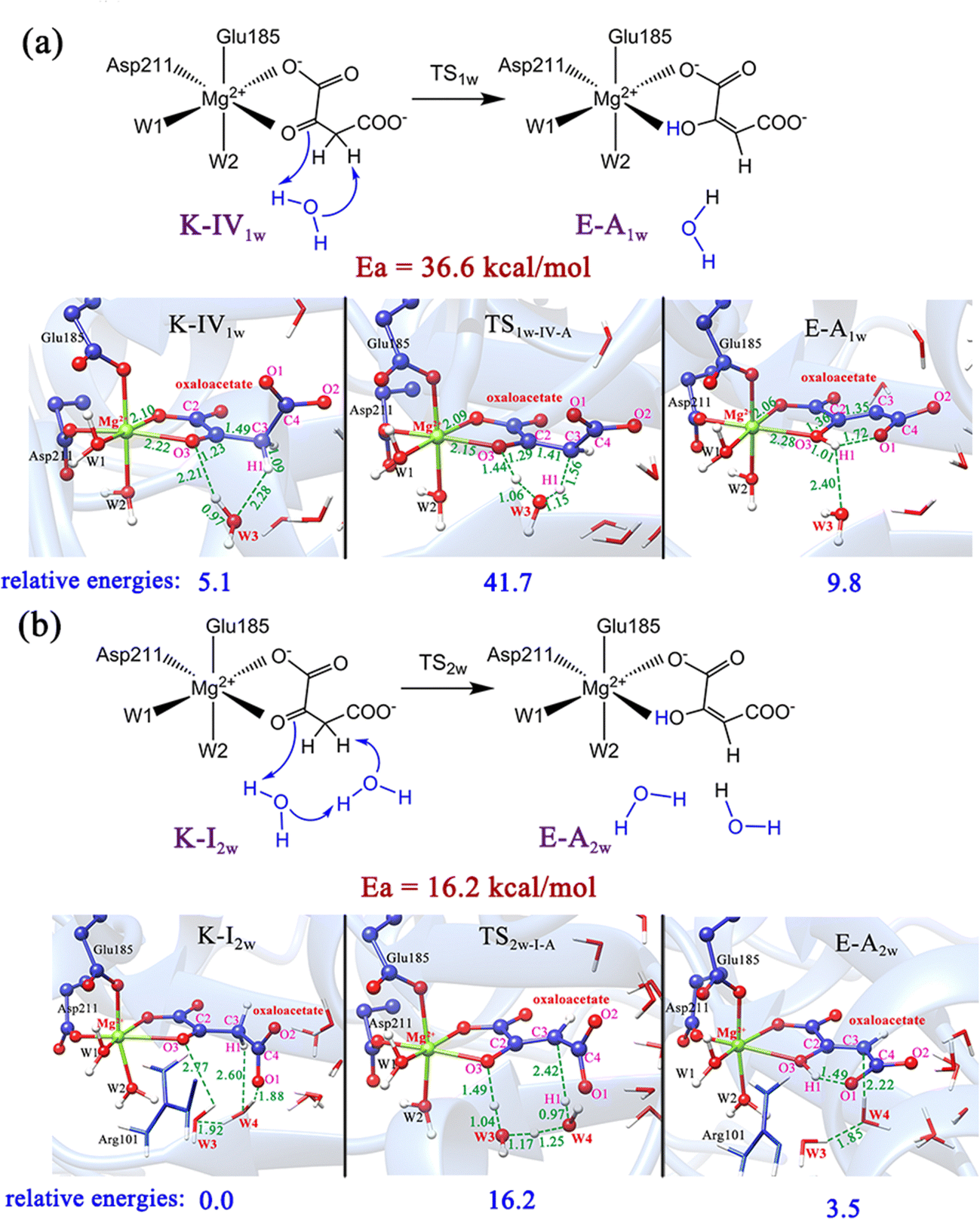 | ||
| Fig. 5 Schematic illustration and optimized structures of keto–enol tautomerization from K-IV to E-A with the assistance of one water molecule (a), and from K-I to E-A with the assistance of two water molecules (b). All distances are given in angstrom and relative energies are in kcal mol−1. | ||
3.3. Decarboxylation of OAA
O bond, the two atomic p orbitals in the π-orbital of carbonyl and the soon forming p orbital by breaking the C–C σ-bond have a continuous orbital overlap, which significantly stabilizes the pyruvate carbanion and facilitates decarboxylation. The same result was confirmed by our calculations. For the binding modes of K-I and K-III, featured by the equatorial carboxylate relative to carbonyl CO, the direct decarboxylation was calculated to be difficult (Fig. 6a, b and Fig. S12a, ESI†). While in the case of the axially positioned carboxyl in K-IV (Fig. S12b, ESI†), the decarboxylation also does not seem to be so facile. It is understandable, because the residue Gln183 causes the steric hindrance, hindering the cleavage of the C–C bond and the departure of CO2.
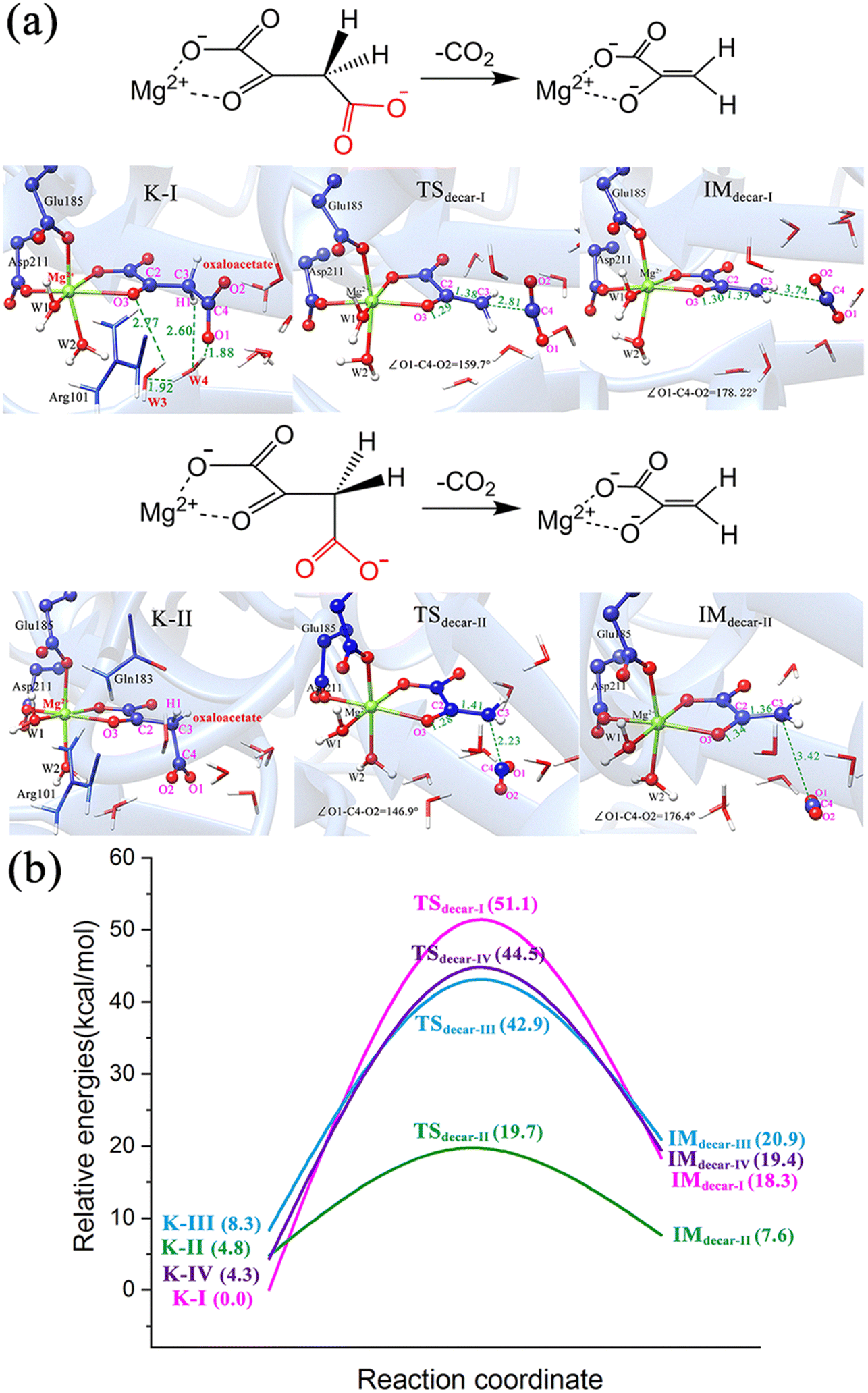 | ||
| Fig. 6 (a) Schematic illustration and optimized structures of direct decarboxylation of K-I and K-II. All distances are given in angstrom. (b) Potential energy profiles for the direct decarboxylation of four keto forms of oxaloacetate. | ||
To get deeper insights into the direct decarboxylation of two keto modes of oxaloacetate (K-I and K-II), the scanned energy profiles of decarboxylation using distances of C3⋯C4 as reaction coordinates are displayed in Fig. 7. It can be seen that the transition state of decarboxylation of K-II is reached much earlier than that of K-I, and the total energies of K-II are always lower than those of K-I. This larger energy difference of the system can be mainly attributed to the different orbital overlaps during the elongation of the C3–C4 bond. Therefore, K-I should be first isomerized into K-II to facilitate the decarboxylation. Fig. 7c shows the changes of bond orders of C2–C3 and C3–C4 during decarboxylation. During the elongation of the C3–C4 bond, the bond orders of C3–C4 in K-I are always larger than those of K-II, whereas the bond orders of C2–C3 in K-I are always lower than those of K-II, which means that during the decarboxylation, the C2–C3 bond is stronger than that in K-I with the same reaction coordinate. These results are consistent with the more orbital overlap of carbonyl CO with C2 and C3 atoms in the decarboxylation of K-II.
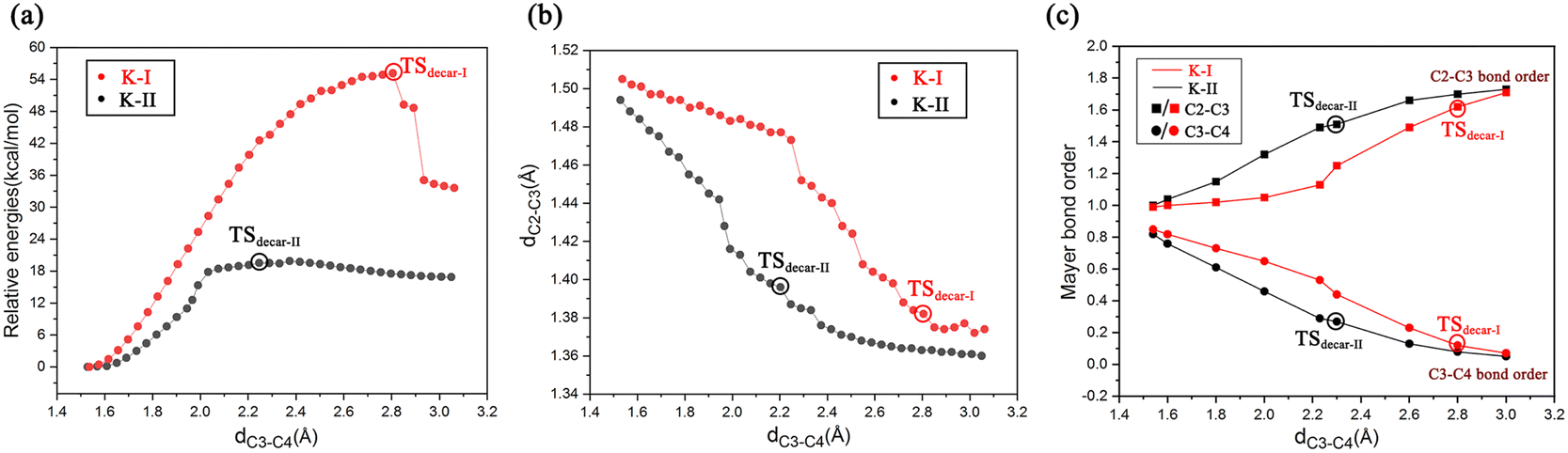 | ||
| Fig. 7 (a) Scanned energy profiles for the decarboxylation of binding modes of K-I and K-II using the distances of C3⋯C4 as reaction coordinates; (b) changes in the distance of C2⋯C3 during the elongation of C3⋯C4 (decarboxylation); and (c) changes of bond orders of C2–C3 and C3–C4 during decarboxylation using the distance of C3⋯C4 as reaction coordinate. | ||
In order to intuitively observe the orbital overlap of the carbonyl CO with C2 and C3 atoms, the frontier orbitals of OAA during the elongation of the C3–C4 bond were calculated, as shown in Fig. 8. One can see that, during the decarboxylation with the same C3–C4 bond distances in K-II and K-I, there is more orbital overlap of two p orbitals of carbonyl CO with the C3 atomic orbital in K-II. However, in K-I, at the early stage of carboxylation, the orbital overlap of the two p orbitals of carbonyl CO with the C3 atomic orbital is quite small, which leads to always smaller bond order of C2–C3 than in K-II. In addition, the smaller C3–C4 bond order should correspond to the larger C2–C3 bond order. Thus, during the decarboxylation, the system energy of K-II can be significantly lowered by the orbital overlap of carbonyl CO and C3/C4 atoms.
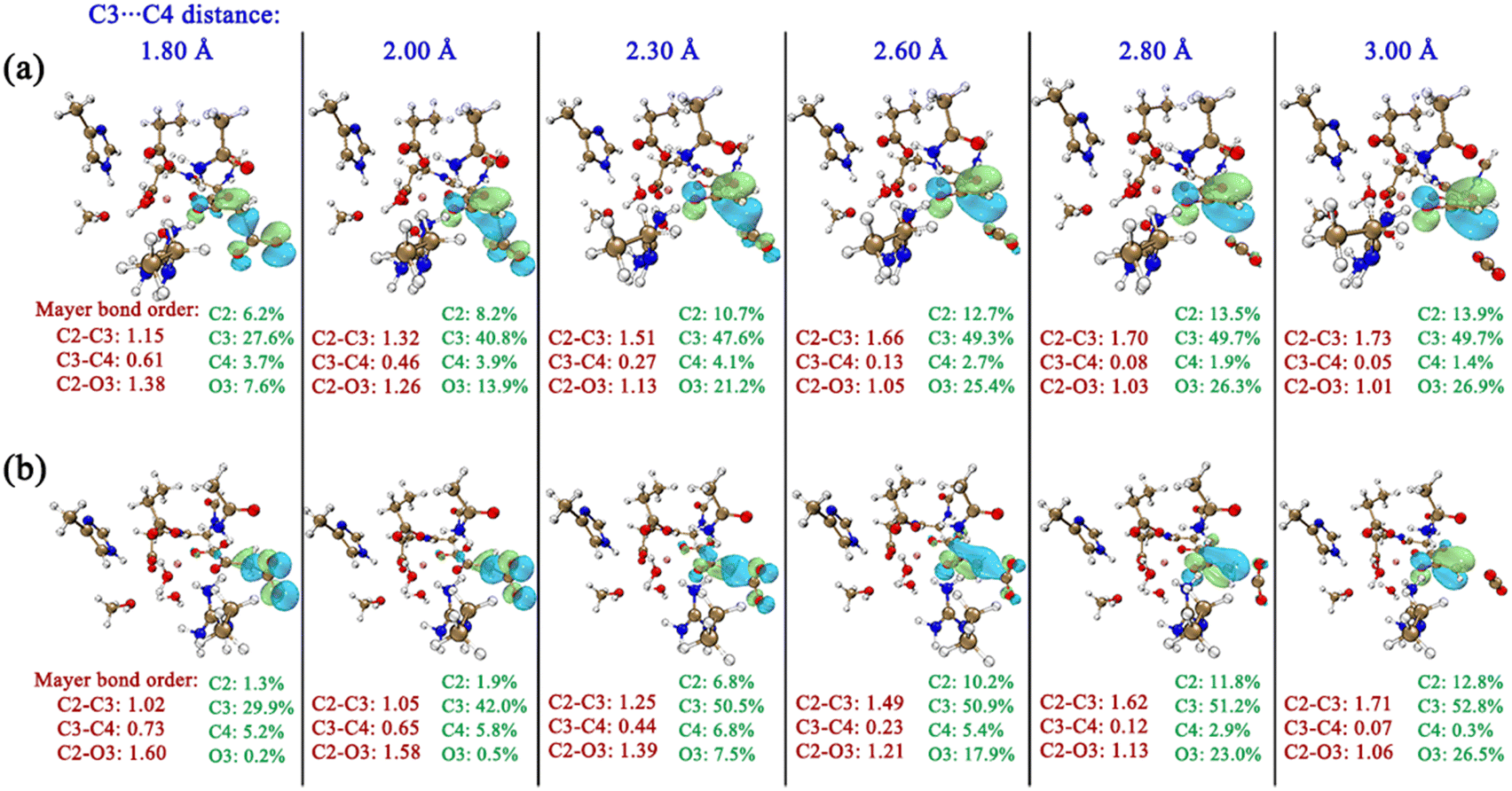 | ||
| Fig. 8 Changes of bond orders of C2–C3, C3–C4 and C2–O3 and evolutions of frontier orbitals of OAA during decarboxylation using the distances of C3–C4 as reaction coordinates for K-II (a) and K-I (b). Only the contributions of related key atoms (C2, C3, C4 and O3) to the orbitals are shown. | ||
In general, for enzymatic decarboxylation, many factors may influence the overall reaction barriers. Since the decarboxylation of β-keto acid is stereo-electronically controlled, we should pay more attention to the binding conformations of the β-keto acids in the active sites of enzymes. Note that for the non-enzymatic reactions, the substrate is relatively free, and the decarboxylation of β-keto acids should not be stereo-electronically controlled.
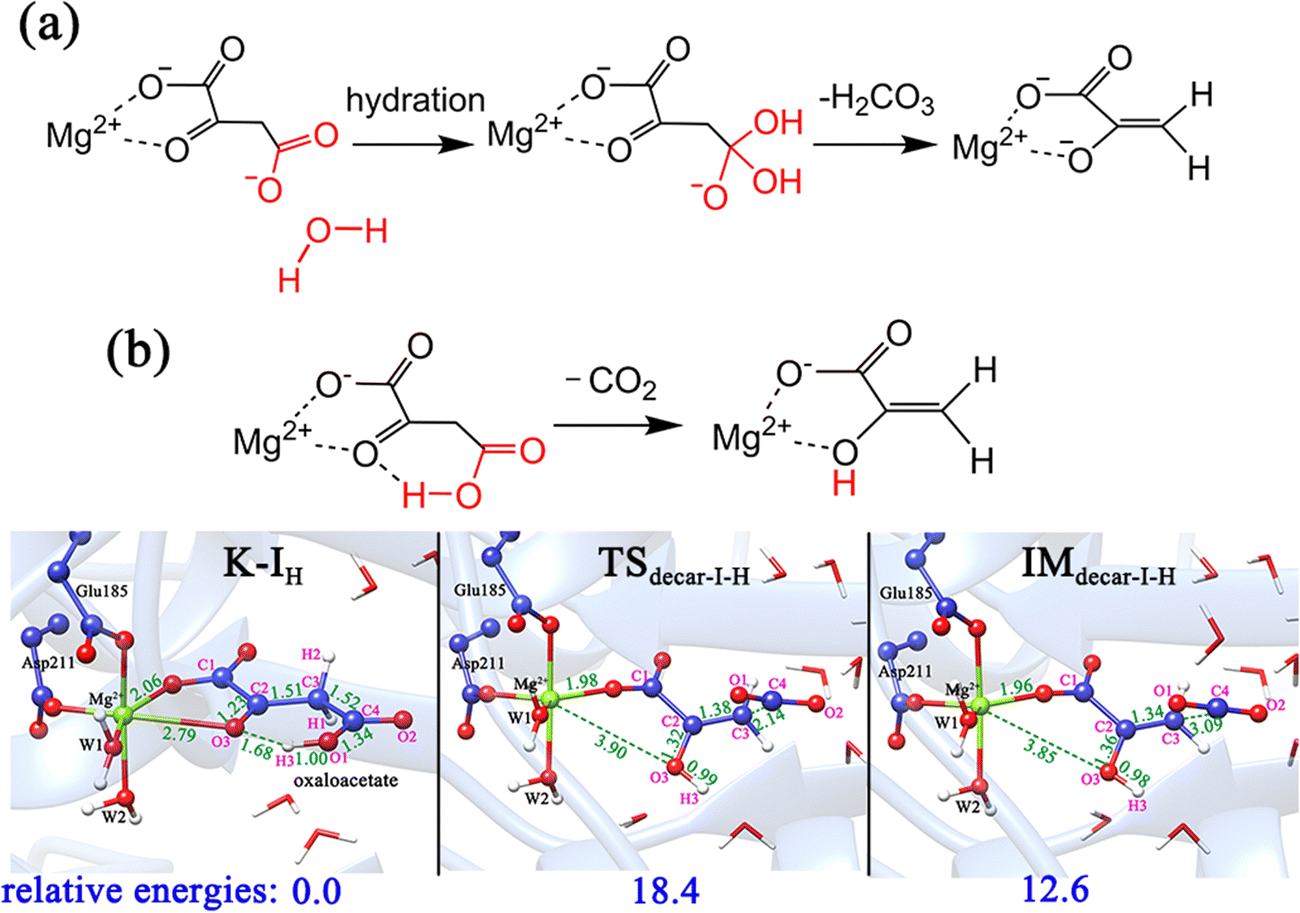 | ||
| Fig. 9 (a) Hydration-assisted decarboxylation of OAA; (b) proton migration-assisted decarboxylation of OAA. All distances are given in angstrom and relative energies are in kcal mol−1. | ||
In addition, according to the previous studies10,59 regarding the proton-migration decarboxylation mechanism of β-keto acid, we also constructed a model by introducing a proton to the terminal carboxylate (Fig. 9b) to test the possibility. This decarboxylation mechanism was calculated to correspond to a relatively low barrier of 18.4 kcal mol−1. It is reasonable, because the rate of decarboxylation is affected by the stabilizing effect of the residual species, and during the proton migration decarboxylation, the developed negative charge on the oxygen anion of the formed pyruvate enolate will be neutralized by the coming proton. However, the protonation state of oxaloacetate is mainly dependent on the environmental pH value. According to the calculated pKa value (3.7) of the C4-carboxyl group,56 the doubly deprotonated oxaloacetate may predominate in the optimal physiological pH 7.0 of the MPS enzyme. Thus, in an acidic environment, oxaloacetate is more likely to proceed decarboxylation via the proton migration-assisted decarboxylation mechanism. These results suggest that many factors, including the binding mode of oxaloacetate, the orientation of the terminal carboxylate group and the protonation state, all have important influence on the decarboxylation process. At the physiological pH 7.0, the oxaloacetate should firstly transform from the most stable binding mode of K-I to K-II, and then undergo decarboxylation.
The formed pyruvate enolate tends to undergo protonation to yield pyruvate, as in many cases studied in the past.60,61 Previous kinetic analysis of Zn(II)- or Mg(II)-bound oxaloacetate complexes in solution showed that the protonation rate of pyruvate enolate is quite rapid compared to the rate of decarboxylation.62 We have investigated this process in the enzyme. Our calculations reveal that if a hydronium ion presents near the enolpyruvate, the protonation process is so facile that it is difficult to recognize the transition state structure, as illustrated in Fig. S14 (ESI†), that is, when the second substrate pyrone has not yet entered into the active site, a fast protonation of enolpyruvate formed by decarboxylation would take place.
Therefore, there may exist a complicated catalysis of MPS in the active site before 2-pyrone enters. The binding mode of K-I with the lowest relative energy may account for the majority. On one hand, K-I would go through the keto–enol tautomerization with two water molecules to generate enolate via a barrier of 16.2 kcal mol−1, which is reactive for the cycloaddition. On the other hand, the K-I could also undergo decarboxylation, but it should first isomerize to K-II. And a total energy barrier of 19.7 kcal mol−1 is required for decarboxylation from the most stable K-I binding mode, which was calculated to be higher than that of keto–enol tautomerization (19.7 vs. 16.2 kcal mol−1). Thus, before the entrance of the second substrate 2-pyrone, different Mg-bound oxaloacetate complexes may simultaneously exist, and they can be interconverted into each other.
3.4. Influence of the second substrate on keto–enol tautomerization and decarboxylation of OAA
The formation of the final product macrophomic acid requires the participation of the second substrate 2-pyrone to perform the [4+2]-cycloaddition. However, it is unknown whether the decarboxylation and keto–enol tautomerization of OAA are affected by the entrance of 2-pyrone. Therefore, we further constructed two models (Fig. 10 and 11) to check whether the presence of the second substrate affects the keto–enol tautomerization and decarboxylation. In the first model, 2-pyrone has entered into the active site, but the keto–enol tautomerization and decarboxylation of oxaloacetate have not yet been completed. Our calculations showed that when 2-pyrone enters, the solvent water molecules originally locating at this position have easily diffused into the outer layer and form hydrogen bond networks (Fig. 10). Our above calculation results have confirmed that the keto–enol tautomerization strictly requires the mediation of water molecules. However, in Fig. 10, there is no water molecule to assist the keto–enol tautomerization, thus, the Mg2+-oxaloacetate can not be converted to the enol form. Besides, the decarboxylation process was also discussed here. As mentioned earlier, only the binding configuration of K-II is conducive to the decarboxylation, and the following study is also based on it. Fig. 11 shows that, since the C3–C4 bond is basically perpendicular to the carbonyl CO plane, the cleavage of the C3–C4 bond would be hindered due to the presence of 2-pyrone, as indicated by the much higher barrier of decarboxylation. From our calculations, it can be concluded that both keto–enol tautomerization and decarboxylation of oxaloacetate would be hindered by the entrance of the second substrate. Therefore, the keto–enol tautomerization and decarboxylation should be completed before the entrance of 2-pyrone.
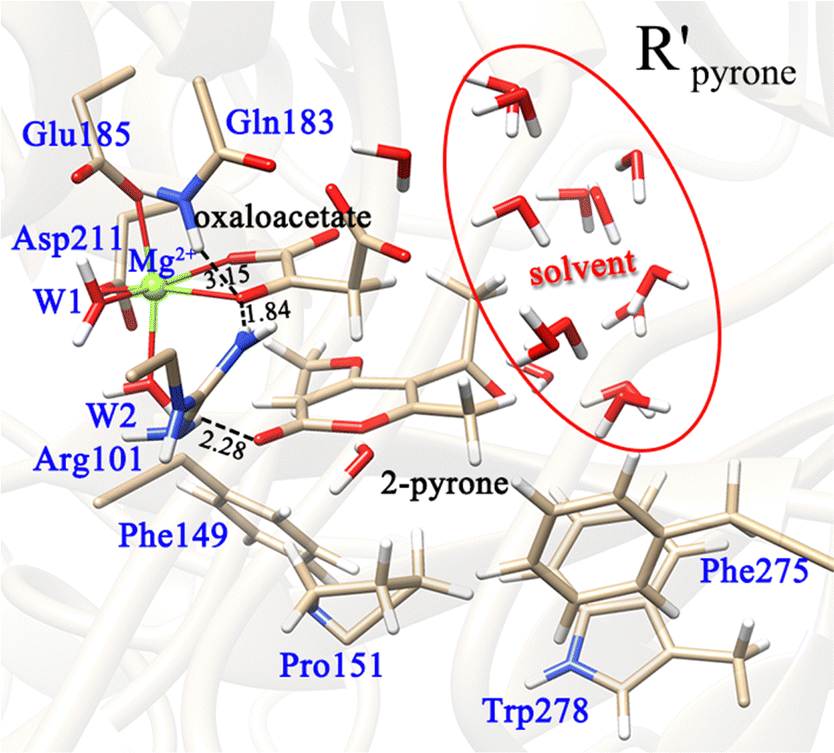 | ||
| Fig. 10 Optimized reactant complex, in which both OAA and 2-pyrone bind in the active site. Note that the entrance of 2-pyrone is prior to the keto–enol tautomerization or decarboxylation of OAA. All distances are given in angstrom. | ||
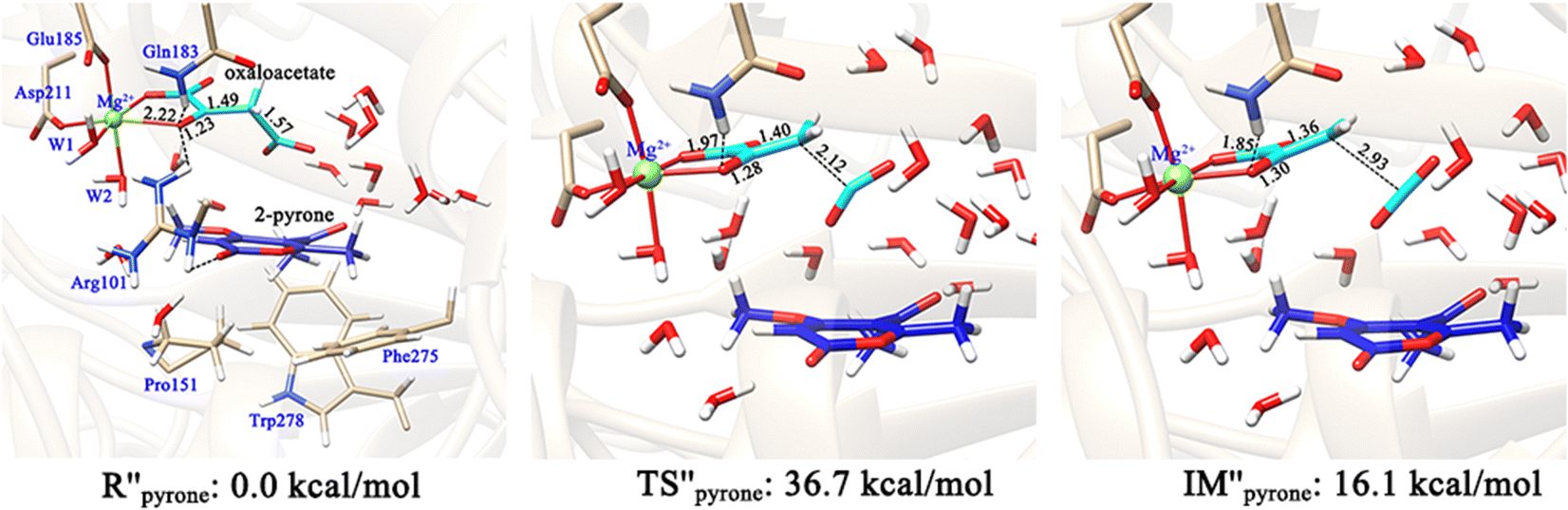 | ||
| Fig. 11 Optimized structures of involved species in the decarboxylation of OAA in the keto form. In this reactant model, both the 2-pyrone and OAA bind in the active site. All distances are given in angstrom and all energies are given in kcal mol−1. | ||
4. Conclusions
In this work, the binding modes, keto–enol tautomerization, and decarboxylation of OAA have been explored by MD simulations and QM/MM calculations. On the basis of our calculations, a relatively complete picture regarding the chemistry of OAA with the catalysis of MPS has been given, as shown in Fig. 12.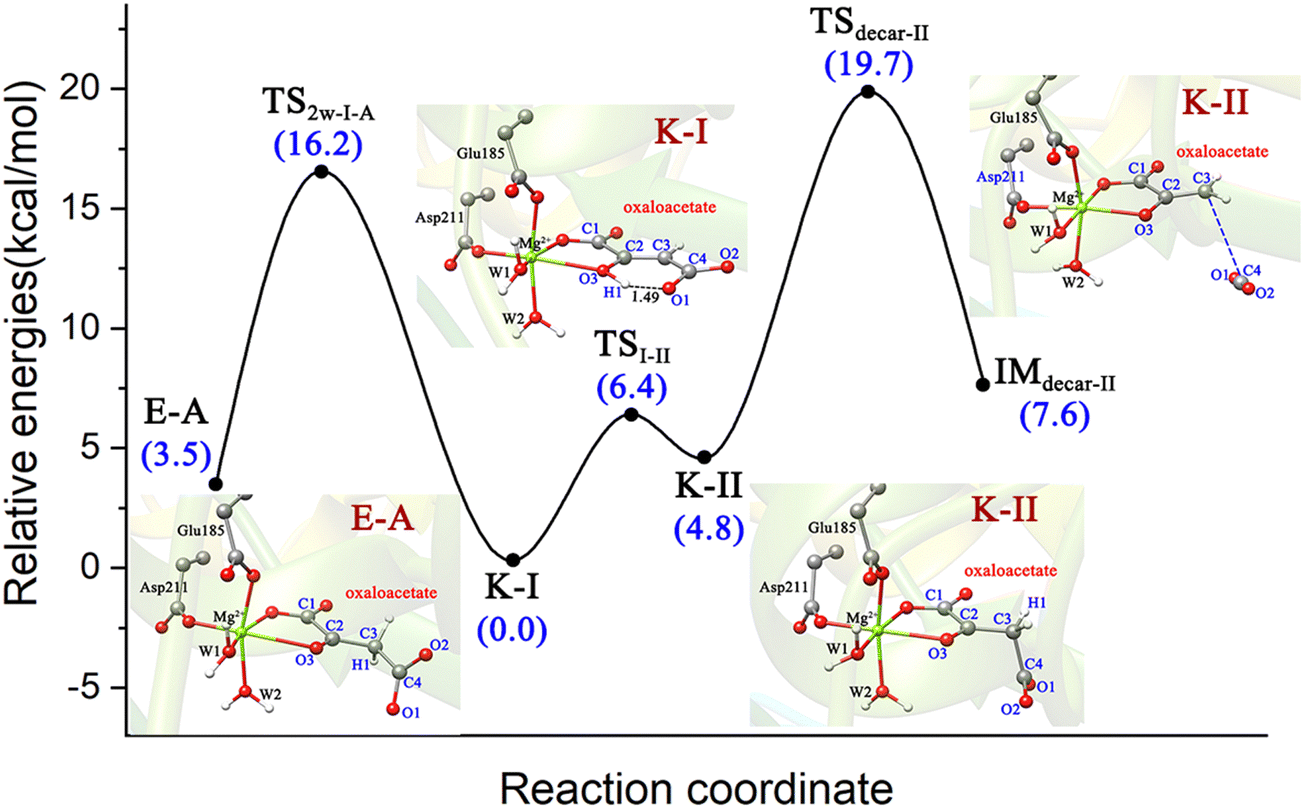 | ||
| Fig. 12 The keto–enol tautomerization and decarboxylation of OAA in the active site of MPS. | ||
(1) The Mg2+-coordinated OAA may exist in its enol form or keto form, and one keto form (K-I) accounts for the largest proportion, which corresponds to the lowest binding energy. As for the enol forms, only one Z-isomer and one E-isomer were confirmed to be dynamically stable and local minima by MD simulations and QM/MM optimizations. For the keto binding modes, four keto forms can be transformed into each other by rotating the C2–C3 single bond. However, the keto–enol tautomerization strictly requires the assistance of water molecules.
(2) The energy barriers of decarboxylation are closely related to the binding conformation of OAA. For the four keto forms, only one keto form (K-II) corresponds to a lower energy barrier (14.9 kcal mol−1), whereas the K-I, which is the most stable binding mode, corresponds to a barrier of 51.1 kcal mol−1 for decarboxylation. This huge energy barrier difference of decarboxylation is mainly caused by the stereo-electronic effect of the terminal carboxylate. Only when OAA exists in its optimal conformation, in which the breaking C3–C4 σ-bond and π-orbital of C2-carbonyl keep a continuous orbital overlap, can the decarboxylation occur easily. Specifically, in the binding mode of K-II, the breaking C3–C4 bond is approximately orthogonal to the plane of the carbonyl C2O bond, which is favorable for decarboxylation.
(3) The chemistry of OAA in the active site of MPS is complex, which would depend on the specific reaction conditions and whether the second substrate (2-pyrone) enters the reaction center. On one hand, the most stable binding mode of K-I can undergo enol–keto tautomerization to isomerize to the enol form (Z)-E-A, which further reacts with the second substrate; on the other hand, K-I can isomerize to K-II to proceed decarboxylation to generate the pyruvate enolate and CO2. Starting from K-I, the enol–keto tautomerization corresponds to a barrier of 16.2 kcal mol−1, whereas the decarboxylation is associated with an overall barrier of 19.7 kcal mol−1. Thus, in the absence of second substrate 2-pyrone, MPS is a typical decarboxylase.
(4) Both the enol–keto tautomerization and decarboxylation would be affected by the entrance of 2-pyrone. If the entrance of the second substrate 2-pyrone is prior to the enol–keto tautomerization, the enol–keto tautomerization will be blocked by the entrance of 2-pyrone. And the decarboxylation of OAA is also hindered due to the steric hindrance during the removal of carbon dioxide.
In general, our calculations reveal that β-keto acids may adopt different binding conformations with the metal ion in the active site of enzymes, and the chemistry of β-keto acids is complex. As the dominant reaction, the decarboxylation is stereo-electronically controlled, thus, we should pay more attention to the binding conformation of these β-keto acids. This work not only provides theoretical guidance for the catalysis of MPS as a decarboxylase, it is also a reference for the decarboxylation and keto–enol tautomerization of similar systems of α- and β-keto acids.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2019YFA0905100), the National Natural Science Foundation of China (22073056) and the Key-Area Research and Development Program of Guangdong Province (2020B0303070002).References
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, 4th edn, W. H. Freeman & Company, New York, 2005 Search PubMed.
- B. D. Fink, F. Bai, L. Yu, R. D. Sheldon, A. Sharma, E. B. Taylor and W. I. Sivitz, Oxaloacetic acid mediates ADP-dependent inhibition of mitochondrial complex II-driven respiration, J. Biol. Chem., 2018, 293, 19932–19941 CrossRef CAS PubMed.
- A. Cash, Oxaloacetic acid supplementation as a mimic of calorie restriction, Open Longevity Sci., 2009, 3, 22–27 CrossRef CAS.
- H. A. Yamamoto and P. V. Mohanan, Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice, Toxicol. Lett., 2003, 143, 115–122 CrossRef CAS PubMed.
- F. Camposx, T. Sobrino, P. Ramos-Cabrer and J. Castillo, Oxalacetate: a novel neuroprotective for acute ischemic stroke, Int. J. Biochem. Cell Biol., 2012, 44, 262–265 CrossRef PubMed.
- X. Cheng, Coupling and Decarboxylation Mechanism of Oxaloacetic Acid and Ethylenediamine: A Theoretical Investigation, J. Phys. Org. Chem., 2019, 32, e3955 CrossRef.
- T. Li, L. Huo, C. Pulley and A. Liu, Decarboxylation Mechanisms in Biological System, Bioorg. Chem., 2012, 43, 2–14 CrossRef CAS PubMed.
- X. Sheng and F. Himo, Mechanisms of metal-dependent non-redox decarboxylases from quantum chemical calculations, Comput. Struct. Biotechnol. J., 2021, 19, 3176–3186 CrossRef CAS PubMed.
- X. Sheng, K. Plasch, S. E. Payer, C. Ertl, G. Hofer, W. Keller, S. Braeuer, W. Goessler, S. M. Glueck, F. Himo and K. Faber, Reaction Mechanism and Substrate Specificity of Iso-orotate Decarboxylase: A Combined Theoretical and Experimental Study, Front. Chem., 2018, 6, 608 CrossRef CAS PubMed.
- X. Sheng and F. Himo, Mechanism of 3-methylglutaconyl CoA decarboxylase AibA/AibB: Pericyclic reaction versus direct decarboxylation, Angew. Chem., 2020, 132, 23173–23177 CrossRef.
- X. Sheng, M. E. S. Lind and F. Himo, Theoretical study of the reaction mechanism of phenolic acid decarboxylase, FEBS J., 2015, 282, 4703–4713 CrossRef CAS PubMed.
- M. Birus and D. L. Leussing, Comparison of an Enzymatic Reaction and Its Model: Metal Ion Promoted Decarboxylation of Oxalacetate, Inorg. Chem., 1982, 21, 374–380 CrossRef CAS.
- N. K. Thalji, W. E. Crowe and G. L. Waldrop, Kinetic Mechanism and Structural Requirements of the Amine-Catalyzed Decarboxylation of Oxaloacetic Acid, J. Org. Chem., 2009, 74, 144–152 CrossRef CAS PubMed.
- G. G. Chang and L. Tong, Structure and function of malic enzymes, a new class of oxidative decarboxylases, Biochemistry, 2003, 42, 12721–12733 CrossRef CAS PubMed.
- P. D. Sender, M. G. Martín, S. Peirú and C. Magni, Characterization of an oxaloacetate decarboxylase that belongs to the malic enzyme family, FEBS Lett., 2004, 570, 217–222 CrossRef CAS PubMed.
- D. M. Kiick and W. W. Cleland, Steady-State Kinetic Studies of the Metal Ion-Dependent Decarboxylation of Oxalacetate Catalyzed by Pyruvate Kinase, Arch. Biochem. Biophys., 1989, 270, 647–654 CrossRef CAS PubMed.
- P. Dahinden, Y. Auchli, T. Granjon, M. Taralczak, M. Wild and P. Dimroth, Oxaloacetate decarboxylase of Vibrio cholerae: purification, characterization, and expression of the genes in Escherichia coli, Arch. Microbiol., 2005, 183, 121–129 CrossRef CAS PubMed.
- H. Oikawa, K. Watanabe, K. Yagi, S. Ohashi, T. Mie, A. Ichihara and M. Honma, Macrophomate synthase: unusual enzyme catalyzing multiple reactions from pyrones to benzoates, Tetrahedron Lett., 1999, 40, 6983–6986 CrossRef CAS.
- K. Watanabe, H. Oikawa, K. Yagi, S. Ohashi, T. Mie, A. Ichihara and M. Hornma, Macrophomate Synthase: Characterization, Sequence, and Expression in Escherichia coli of the Novel Enzyme Catalyzing Unusual Multistep Transformation of 2-Pyrones to Benzoates, J. Biochem., 2000, 127, 467–473 CrossRef CAS PubMed.
- C. R. W. Guimarães, M. Udier-Blagović and W. L. Jorgensen, Macrophomate synthase: QM/MM simulations address the Diels− Alder versus Michael-Aldol reaction mechanism, J. Am. Chem. Soc., 2005, 127, 3577–3588 CrossRef PubMed.
- J. M. Serafimov, T. Westfeld, B. H. Meier and D. Hilvert, Trapping and structural elucidation of an intermediate in the macrophomate synthase reaction pathway, J. Am. Chem. Soc., 2007, 129, 9580–9581 CrossRef CAS PubMed.
- K. Watanabe, T. Mie, A. Ichihara, H. Oikawa and M. Honma, Detailed reaction mechanism of macrophomate synthase: extraordinary enzyme catalyzing five-step transformation from 2-pyrones to benzoates, J. Biol. Chem., 2000, 275, 38393–38401 CrossRef CAS PubMed.
- S. Li and J.-A. Ma, Core-structure-inspired asymmetric addition reactions: enantioselective synthesis of dihydrobenzoxazinone- and dihydroquinazolinone-based anti-HIV agents, Chem. Soc. Rev., 2015, 44, 7439–7448 RSC.
- H.-N. Yuan, S. Wang, J. Nie, W. Meng, Q. Yao and J.-A. Ma, Hydrogen-Bond-Directed Enantioselective Decarboxylative Mannich Reaction of β-Ketoacids with Ketimines: Application to the Synthesis of Anti-HIV Drug DPC 083, Angew. Chem., Int. Ed., 2013, 52, 3869–3873 CrossRef CAS PubMed.
- J.-P. Niu, J. Nie, S. Li and J.-A. Ma, Organocatalytic asymmetric synthesis of β,β-diaryl ketones via one-pot tandem dehydration/1,6-addition/decarboxylation transformation of β-keto acids and 4-hydroxybenzyl alcohols, Chem. Commun., 2020, 56, 8687–8690 RSC.
- Y.-J. Wang, J.-P. Huang, T. Tian, Y. Yan, Y. Chen, J. Yang, J. Chen, Y.-C. Gu and S.-X. Huang, Discovery and Engineering of the Cocaine Biosynthetic Pathway, J. Am. Chem. Soc., 2022, 144, 22000–22007 CrossRef CAS PubMed.
- Y.-J. Wang, T. Tain, J.-Y. Yu, J. Li, B. Xu, J. Chen, J. C. D’Auriad, J.-P. Huang and S.-X. Huang, Genomic and structural basis for evolution of tropane alkaloid biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2302448120 CrossRef CAS PubMed.
- H. Oikawa, Involvement of the Diels–Alderases in the Biosynthesis of Natural Products, Bull. Chem. Soc. Jpn., 2005, 78, 537–554 CrossRef CAS.
- J. M. Serafimov, Mechanistic investigations of macrophomate synthase, Doctoral Dissertation, ETH Zurich, 2007.
- T. Ose, K. Watanabe, T. Mie, M. Honma, H. Watanabe, M. Yao, H. Oikawa and I. Tanaka, Insight into a natural Diels-Alder reaction from the structure of macrophomate synthase, Nature, 2003, 422, 185–189 CrossRef CAS PubMed.
- A. J. Borg, K. Beerens, M. Pfeiffer, T. Desmet and B. Nidetzky, Stereo-electronic control of reaction selectivity in short-chain dehydrogenases: Decarboxylation, epimerization, and dehydration, Curr. Opin. Chem. Biol., 2021, 61, 43–52 CrossRef CAS PubMed.
- M. H. Olsson, C. R. Søndergard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. A. Swaminathan and M. Karplus, CHARMM: A program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjovoll, A. Fahmi, A. Schafer and C. Lennartz, QUASI: A General Purpose Implementation of the QM/MM Approach and its Application to Problems in Catalysis, THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Electronic Structure Calculations on Workstation Computers: The Program System Turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- W. Smith and T. R. Forester, DL_POLY_2.0: A General-purpose Parallel Molecular Dynamics Simulation Package, J. Mol. Graph., 1996, 14, 136–141 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, Hybrid Models for Combined Quantum Mechanical and Molecular Mechanical Approaches, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- A. H. de Vries, P. Sherwood, S. J. Collins, A. M. Rigby, M. Rigutto and G. J. Kramer, Zeolite Structure and Reactivity by Combined Quantum-Chemical-Classical Calculations, J. Phys. Chem. B, 1999, 103, 6133–6141 CrossRef CAS.
- S. R. Billeter, A. J. Turner and W. Thiel, Linear Scaling Geometry Optimisation and Transition State Search in Hybrid Delocalised Internal Coordinates, Phys. Chem. Chem. Phys., 2000, 2, 2177–2186 RSC.
- J. Nocedal, Updating Quasi-Newton Matrices with Limited Storage, Math. Comput., 1980, 35, 773–782 CrossRef.
- D. C. Liu and J. Nocedal, On the Limited Memory BFGS Method for Large Scale Optimization, Math. Prog., 1989, 45, 503–528 CrossRef.
- A. Banerjee, N. Adams, J. Simons and R. Shepard, Search for Stationary Points on Surfaces, J. Phys. Chem., 1985, 89, 52–57 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. Lu and F. W. J. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Y. L. Miao, A. Bhattarai and J. A. Wang, Ligand Gaussian Accelerated Molecular Dynamics (LiGaMD) for Characterization of Ligand Binding Thermodynamics and Kinetics, Biophys. J., 2021, 120, 97a CrossRef.
- Y. L. Miao, A. Bhattarai and J. N. Wang, Ligand Gaussian Accelerated Molecular Dynamics (LiGaMD): Characterization of Ligand Binding Thermodynamics and Kinetics, J. Chem. Theory Comput., 2020, 16, 5526–5547 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Y. Ben-Shalom, J. T. Berryman, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, N. Forouzesh, G. Giambasu, T. Giese, M. K. Gilson, H. Gohlke, A. W. Götz, J. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O’Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, J. Wang, H. Wei, X. Wu, Y. Wu, Y. Xiong, Y. Xue, D. M. York, S. Zhao, Q. Zhu and P. A. Kollman, Amber 2023, University of California, San Francisco, 2023 Search PubMed.
- Y. L. Miao, W. Sinko, L. Pierce, D. Bucher, R. C. Walker and J. A. McCammon, Improved Reweighting of Accelerated Molecular Dynamics Simulations for Free Energy Calculation, J. Chem. Theory Comput., 2014, 10, 2677–2689 CrossRef CAS PubMed.
- T. Kaweetirawatt, T. Yamaguchi, T. Higashiyama, M. Sumimoto and K. Hori, Theoretical study of keto–enol tautomerism by quantum mechanical calculations (the QM/MC/FEP method), J. Phys. Org. Chem., 2012, 25, 1097–1104 CrossRef CAS.
- C. D’Cunha, A. N. Morozov and D. C. Chatfield, Theoretical study of HOCl-catalyzed keto-enol tautomerization of β-cyclopentanedione in an explicit water environment, J. Phys. Chem. A, 2013, 117, 8437–8448 CrossRef PubMed.
- C. S. Cucinotta, A. Ruini, A. Catellani and A. Stirling, Ab initio molecular dynamics study of the keto-enol tautomerism of acetone in solution, ChemPhysChem, 2006, 7, 1229–1234 CrossRef CAS PubMed.
- S. F. Tayyari, F. Naghavi, S. Pojhan, R. W. McClurg and R. E. Sammelson, Conformational analysis, tautomerization, IR, Raman, and NMR studies of benzyl acetoacetate, J. Mol. Struct., 2011, 987, 241–254 CrossRef CAS.
- G. Alagona, C. Ghio and P. I. Nagy, The catalytic effect of water on the keto-enol tautomerism. Pyruvate and acetylacetone: a computational challenge, Phys. Chem. Chem. Phys., 2010, 12, 10173–10188 RSC.
- A. K. Weiss, R. Wurzer, P. Klapec, M. P. Eder, J. R. Loeffler, S. von Grafenstein, S. Monteleone, K. R. Liedl, P. Jansen-Durr and H. Gstach, Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1), Molecules, 2021, 26, 5009 CrossRef CAS PubMed.
- X. Sheng, W. Zhu, J. Huddleston, D. F. Xiang, F. M. Raushel, N. G. Richards and F. Himo, A combined experimental-theoretical study of the LigW-catalyzed decarboxylation of 5-carboxyvanillate in the metabolic pathway for lignin degradation, ACS Catal., 2017, 7, 4968–4974 CrossRef CAS.
- S. O. Mundle, G. Lacrampe-Couloume, B. S. Lollar and R. Kluger, Hydrolytic decarboxylation of carboxylic acids and the formation of protonated carbonic acid, J. Am. Chem. Soc., 2010, 132, 2430–2436 CrossRef CAS PubMed.
- M. Staikova, M. Oh and D. J. Donaldson, Overtone-induced decarboxylation: A potential sink for atmospheric diacids, J. Phys. Chem. A, 2005, 109, 597–602 CrossRef CAS PubMed.
- P. Baker, J. Carere and S. Y. Seah, Probing the molecular basis of substrate specificity, stereospecificity, and catalysis in the class II pyruvate aldolase, BphI, Biochemistry, 2011, 50, 3559–3569 CrossRef CAS PubMed.
- D. Rea, R. Hovington, J. F. Rakus, J. A. Gerlt, V. Fülüp, T. D. Bugg and D. I. Roper, Crystal Structure and Functional Assignment of YfaU, a Metal Ion Dependent Class II Aldolase from Escherichia coli K12, Biochemistry, 2008, 47, 9955–9965 CrossRef CAS PubMed.
- T. M. Dougherty and W. W. Cleland, pH studies on the chemical mechanism of rabbit muscle pyruvate kinase. 1. Alternate substrates oxalacetate, glycolate, hydroxylamine, and fluoride, Biochemistry, 1985, 24, 5870–5875 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Hexameric crystal structure and active site of MPS; solvated model of pretreatment; RMSD for the backbone atoms in 35-ns MD simulation; overlay of four QM/MM-optimized structures derived from snapshots of MD trajectory; selected QM regions; optimized transition states involved in isomerization from K-I to K-II, K-III and K-IV; a QM/MM-optimized and dynamically unstable enolate structure; time evolution of the dihedral of C2–C3–C4–O1 during 120-ns MD simulations of (Z)-E-A and (Z)-E-A′; optimized structures of keto–enol tautomerization with the assistance of zero, one and two water molecules; optimized structure of K-IV including three intermediate water molecules; optimized structures for decarboxylation of K-III and K-IV and hydrolytic decarboxylation of K-I and K-II; protonation process of the enolpyruvate intermediate; and Cartesian coordinates of QM regions of key transition states and intermediates. See DOI: https://doi.org/10.1039/d4cp00716f |
| This journal is © the Owner Societies 2024 |