
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of poly(N-isopropylacrylamide) cononsolvency in aqueous methanol solutions explored via oxygen K-edge X-ray absorption spectroscopy†
Masanari
Nagasaka
 *ab,
Fumitoshi
Kumaki
c,
Yifeng
Yao
d,
Jun-ichi
Adachi
ce and
Kenji
Mochizuki
d
*ab,
Fumitoshi
Kumaki
c,
Yifeng
Yao
d,
Jun-ichi
Adachi
ce and
Kenji
Mochizuki
d
aInstitute for Molecular Science, Myodaiji, Okazaki 444-8585, Japan. E-mail: nagasaka@ims.ac.jp
bMolecular Science Program, Graduate Institute for Advanced Studies, SOKENDAI, Myodaiji, Okazaki 444-8585, Japan
cPhoton Factory, Institute of Materials Structure Science, High Energy Accelerator Research Organization, 1-1 Oho, Tsukuba, Ibaraki 305-0801, Japan
dDepartment of Chemistry, Zhejiang University, Hangzhou, 310028, P. R. China
eMaterials Structure Science Program, Graduate Institute for Advanced Studies, SOKENDAI, 1-1 Oho, Tsukuba, Ibaraki 305-0801, Japan
First published on 17th April 2024
Abstract
The cononsolvency mechanism of poly(N-isopropylacrylamide) (PNIPAM), dissolving in pure methanol (MeOH) and water (H2O) but being insoluble in MeOH–H2O mixtures, was investigated by O K-edge X-ray absorption spectroscopy (XAS). The cononsolvency emerges from the aggregation of PNIPAM with MeOH clusters, leading to the collapse of the hydrophobic hydration of PNIPAM.
Introduction
PNIPAM is a stimuli-responsive polymer that is sensitive to various chemical environments, such as temperature and pH. This property translates to a low critical solution temperature, where it is dissolvable in solutions at low temperatures, known as a coil state, and is insoluble at high temperatures, known as a globule state.1–3 PNIPAM is dissolved in pure MeOH and H2O at 25 °C but is insoluble in MeOH–H2O mixtures at the same temperature, which is known as cononsolvency.4–6 The coil-to-globule-to-coil transition occurs at the same temperature, where PNIPAM stays in the coil state in pure H2O, then transitions to the globule state by increasing the MeOH molar fraction, and transitions again to the coil state in pure MeOH.7 This behaviour stems from the lower critical solution temperature in aqueous MeOH solution compared to that in pure MeOH and H2O. Cononsolvency is a general phenomenon that emerges not only in polymers, but also in small molecules such as tertiary butyl alcohol in aqueous MeOH solutions.8,9Understanding the mechanism of polymer cononsolvency is paramount for comprehending the phase transition dynamics of biomolecules, including protein folding, DNA packing, and interchain complexation. Therefore, the mechanism of polymer cononsolvency has been investigated in several experimental and theoretical studies.3,10,11 The cononsolvency of PNIPAM in aqueous MeOH solutions is driven by the competition between MeOH and H2O for the hydrogen bond (HB) of PNIPAM; conversely, PNIPAM dissolves in pure MeOH and H2O owing to the cooperative HB of solvent molecules.12 The MeOH–H2O mixed cluster formation drives the cononsolvency of PNIPAM.13,14 The polymer units aggregate owing to several factors, including the preferential binding of MeOH to PNIPAM,15–17 geometric frustration due to the exchange of the molecular interaction of PNIPAM with H2O to that with MeOH,18 an excluded volume effect from the methyl group of MeOH,19 and the disruption of the HB structure of the NH group in PNIPAM with H2O owing to the existence of MeOH.20 The coil-to-globule transition is related to the hydrophobic hydration of the isopropyl group in PNIPAM in the coil state and the collapse of hydration upon the addition of MeOH in the globule state.21,22 However, the cononsolvency mechanism is still unknown because the molecular interactions of PNIPAM with MeOH and H2O have not been fully understood yet.
In this study, we investigated the molecular interactions of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in PNIPAM with MeOH and H2O from the energy shifts of the CO π* peaks in the O K-edge XAS of PNIPAM along with molecular dynamics (MD) simulations and inner-shell calculations. XAS is advantageous for the element-selective structural analysis of light elements (C, N, O, etc.), as shown by its applicability to liquids and solutions.23 However, XAS measurements in the transmission mode are difficult because soft X-rays are strongly absorbed by air and liquids. Recently, we performed operando XAS measurements of the chemical processes in solution by developing a transmission-type liquid cell, including a precise thickness control technique.24,25 The CO π* peak (532 eV) in PNIPAM can be observed via O K-edge XAS by separation of the contribution from solvents MeOH and H2O, whose first peaks are around 535 eV.26,27
O group in PNIPAM with MeOH and H2O from the energy shifts of the CO π* peaks in the O K-edge XAS of PNIPAM along with molecular dynamics (MD) simulations and inner-shell calculations. XAS is advantageous for the element-selective structural analysis of light elements (C, N, O, etc.), as shown by its applicability to liquids and solutions.23 However, XAS measurements in the transmission mode are difficult because soft X-rays are strongly absorbed by air and liquids. Recently, we performed operando XAS measurements of the chemical processes in solution by developing a transmission-type liquid cell, including a precise thickness control technique.24,25 The CO π* peak (532 eV) in PNIPAM can be observed via O K-edge XAS by separation of the contribution from solvents MeOH and H2O, whose first peaks are around 535 eV.26,27
Results and discussion
Fig. 1 shows the O K-edge XAS spectra of PNIPAM in aqueous MeOH solutions (MeOH)x(H2O)1−x with different molar fractions at 25 °C. The XAS experiments were performed using a transmission-type liquid cell24,25 at the soft X-ray beamline BL-7A of the Photon Factory, Institute of Materials Structure Science, High Energy Accelerator Research Organization (KEK-PF).28 The CO π* peaks of PNIPAM were observed at approximately 532 eV, and the strong absorbances of solvents MeOH and H2O were observed at the higher energy side. Note that soft X-rays above 535 eV cannot transmit the liquid layers because the thick liquid layers were prepared for the increase of the absorbance of the CO π* peaks of PNIPAM. PNIPAM with 50 mg mL−1 was dissolved in solutions considering the detection limit of CO π* peaks in solvents MeOH and H2O (Section S1, ESI†). PNIPAM is insoluble at 0.4 > x > 0.1 (Section S2, ESI†). The CO π* peaks were not observed at x = 0.4 and x = 0.2 because the concentrations of PNIPAM are below the detection limit due to the formation of PNIPAM aggregates, whose details are discussed in Section S1 of the ESI.†
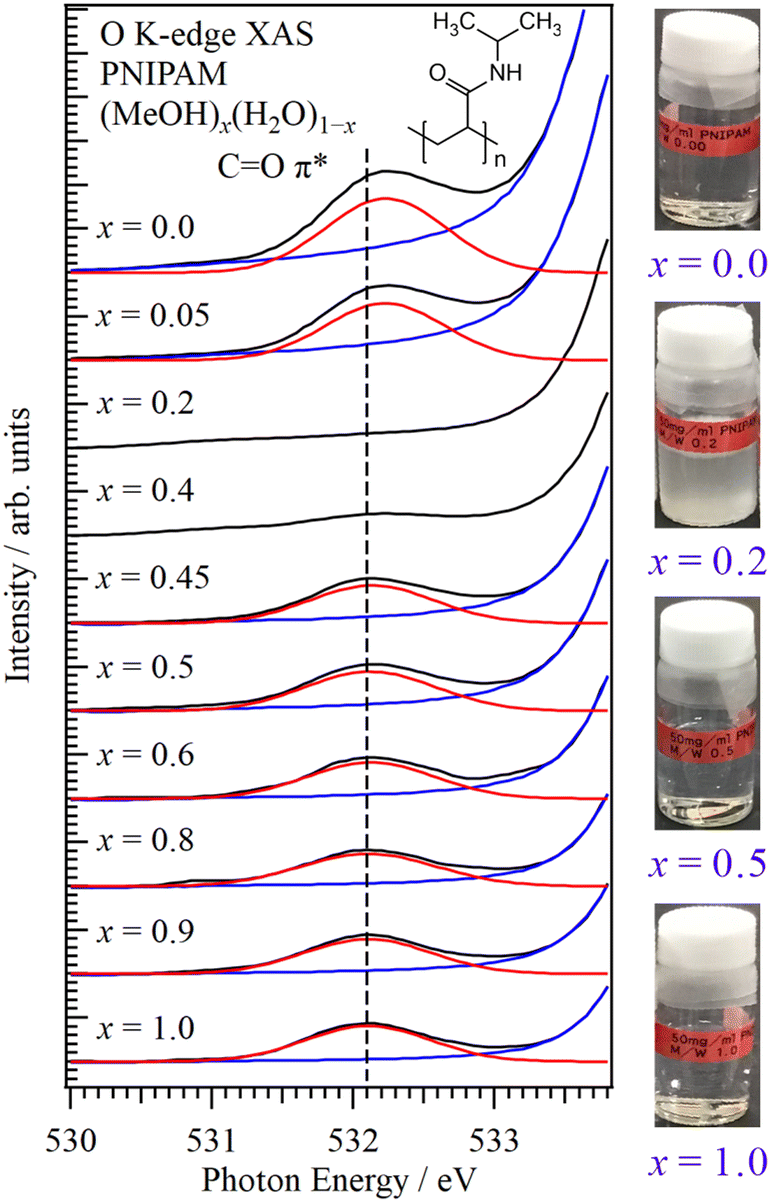 | ||
| Fig. 1 O K-edge XAS spectra of PNIPAM in aqueous MeOH solutions at different molar fractions. The energetic position of the CO π* peak at x = 1.0 is indicated by the dashed line. The photos of PNIPAM in aqueous MeOH solutions are shown in the inset. | ||
Fig. 2 shows the energy shift of the CO π* peak in PNIPAM as a function of the MeOH molar fraction. The energetic positions of the CO π* peaks were obtained by the fitting procedures shown in Fig. 1. We confirmed that the spectral fluctuations did not affect the energetic positions of the CO π* peaks. The CO π* peaks were not observed at 0.4 > x > 0.1, denoting the cononsolvency region. The errors of the energetic positions were within 17 meV by the calibrations of the photon energies. By increasing the H2O molar fraction in the MeOH-rich region (x > 0.4), the CO π* peaks exhibited higher energy shifts compared to that of pure MeOH (x = 1.0) as a slope of 51 meV/(1 − x). The highest energy shift corresponding to the CO π* peak at x = 0.45 was 27 meV. In contrast, in the H2O-rich region (0.1 > x), notably higher energy shifts were recorded for the CO π* peaks; specifically, the energy shift of the CO π* peak in pure H2O (x = 0.0) was 127 meV. Although PNIPAM exhibits identical dissolution behaviours in MeOH and H2O at the macroscopic scale, the molecular interactions of PNIPAM with MeOH and H2O significantly differ at the microscopic scale, explaining the cononsolvency.
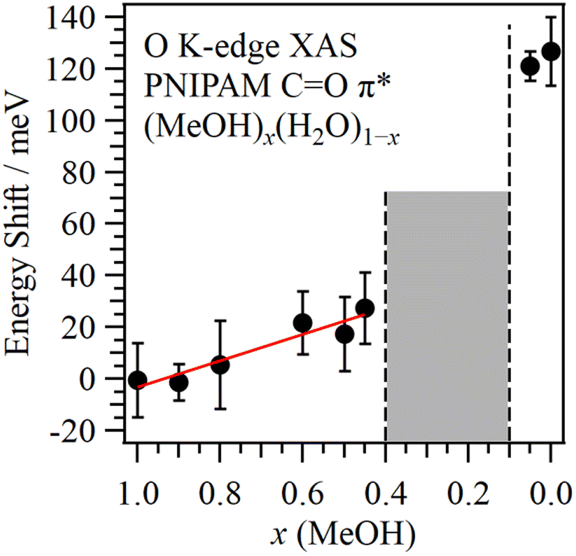 | ||
| Fig. 2 Energy shift of the CO π* peak in PNIPAM as a function of MeOH molar fraction from pure MeOH (x = 1.0). The middle concentration region denotes cononsolvency. | ||
To reveal the origin of the energy shift of the CO π* peak in PNIPAM, the structure of a single atactic 40-mer PNIPAM chain dissolved in 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 solvent molecules of MeOH and H2O was investigated by MD simulations using the GROMACS 2021.3 package.29 The radius of gyration (Rg) was determined for different molar fractions of aqueous MeOH solutions to investigate the coil and globule states. The Rg values at x = 1.0 (pure MeOH), x = 0.2, and x = 0.0 (pure H2O) were calculated by averaging all the trajectories (Section S3, ESI†). Note that x = 0.2 was selected because it is near the middle region of cononsolvency (0.4 > x > 0.1). Table 1 lists the Rg values at different molar fractions. At x = 1.0, Rg = 1.99, indicating that the PNIPAM chain spread in pure MeOH. Because Rg = 1.78 at x = 0.0, the polymer units of PNIPAM were inferred to remain close to each other in pure H2O. The smallest Rg value (= 1.71) was measured at x = 0.2, indicating that PNIPAM exhibited a globule state.
000 solvent molecules of MeOH and H2O was investigated by MD simulations using the GROMACS 2021.3 package.29 The radius of gyration (Rg) was determined for different molar fractions of aqueous MeOH solutions to investigate the coil and globule states. The Rg values at x = 1.0 (pure MeOH), x = 0.2, and x = 0.0 (pure H2O) were calculated by averaging all the trajectories (Section S3, ESI†). Note that x = 0.2 was selected because it is near the middle region of cononsolvency (0.4 > x > 0.1). Table 1 lists the Rg values at different molar fractions. At x = 1.0, Rg = 1.99, indicating that the PNIPAM chain spread in pure MeOH. Because Rg = 1.78 at x = 0.0, the polymer units of PNIPAM were inferred to remain close to each other in pure H2O. The smallest Rg value (= 1.71) was measured at x = 0.2, indicating that PNIPAM exhibited a globule state.
O group of PNIPAM from MD simulations
| R g/nm | O w | O m | |
|---|---|---|---|
| x = 0.0 | 1.78 ± 0.19 | 2.10 ± 0.03 | — |
| x = 0.2 | 1.71 ± 0.17 | 1.65 ± 0.01 | 0.34 ± 0.01 |
| x = 1.0 | 1.99 ± 0.16 | — | 1.35 ± 0.01 |
The coordination numbers of the HB structures of MeOH and H2O to the CO group of PNIPAM were obtained by radial distribution function (RDF) analysis (Section S4, ESI†). As shown in Table 1, the coordination number of MeOH (Om) to PNIPAM was 1.35 in pure MeOH (x = 1.0), indicating that one or two MeOH molecules were connected to the CO group in PNIPAM. The two H2O molecules formed an HB structure with PNIPAM in pure H2O (x = 0.0) because the coordination number of H2O (Ow) to PNIPAM was 2.10. Because Ow = 1.65 and Om = 0.34 at x = 0.2, the H2O molecules could form HB structures with PNIPAM more easily compared to MeOH molecules. Approximately two solvent molecules were connected to the CO group in PNIPAM at x = 0.2, owing to a total coordination number of 1.99. Upon increasing the molar fraction of H2O from pure MeOH, the HB structures with MeOH are substituted to those with H2O. Specifically, the HB structures originally formed between PNIPAM and one or two MeOH molecules were replaced by interactions involving two H2O molecules.
Fig. 3 shows the O K-edge inner-shell spectra of the HB structures of PNIPAM and N-isopropylacrylamide (NIPAM), which is the polymer unit of PNIPAM, using the program package GSCF3.30,31 The oxygen atom in the central polymer unit was considered only in the inner-shell calculations of PNIPAM. The structures of NIPAM and 5-mer PNIPAM chain were optimized including the solvent effect of MeOH using the polarizable continuum model (Section S5, ESI†). The HB model structures of NIPAM and PNIPAM were obtained from the distances and angles of the HB structures determined by two-dimensional RDF analysis (Section S6, ESI†). Note that the inner-shell calculations of the HB model structures are qualitatively effective for discussing the relations of the energy shifts of the CO π* peaks with the changes of the HB structures because the energy shifts reflect the intermolecular interactions.25
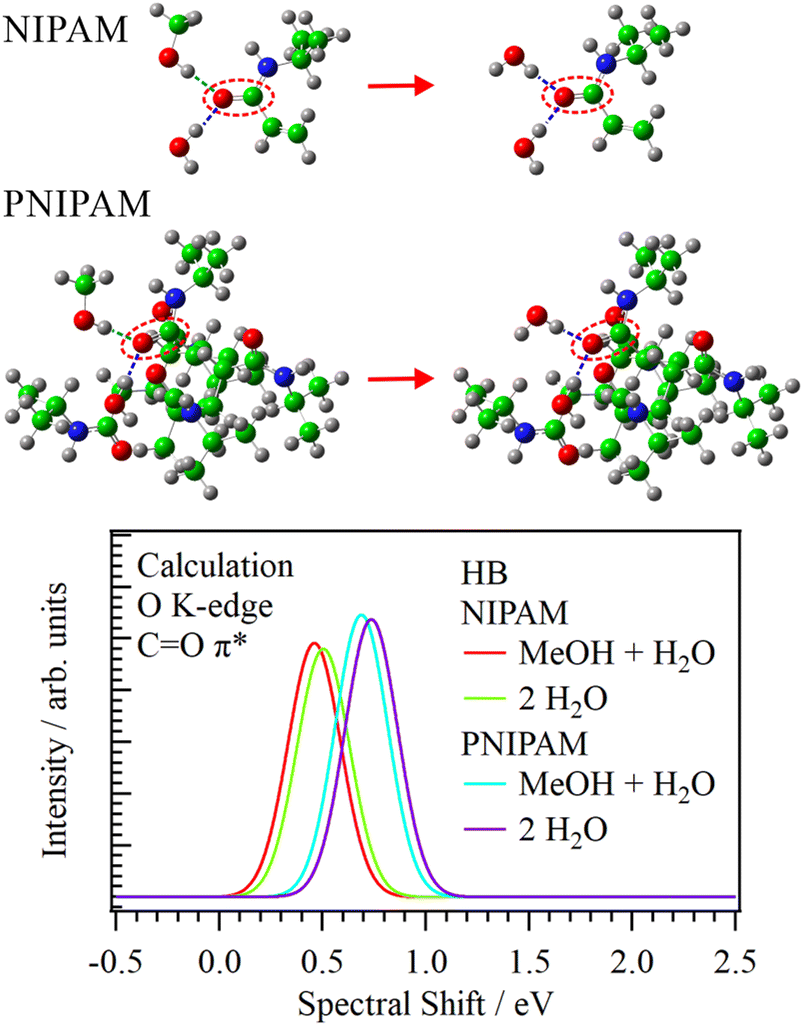 | ||
| Fig. 3 Calculated O K-edge inner-shell spectra of several HB structures of the CO group in NIPAM or PNIPAM with H2O and MeOH molecules. The inset shows the HB structures of NIPAM or PNIPAM with H2O and MeOH molecules. | ||
The CO π* peaks in both NIPAM and PNIPAM showed higher energy shifts when the coordination numbers of the HB structures were increased and the HB structures involving MeOH were substituted with H2O (Section S7, ESI†). In the inner-shell spectra of NIPAM, the CO π* peak in the HB structure of NIPAM with two H2O molecules showed a higher energy shift (43 meV) compared to that of MeOH + H2O. The energy shift of the CO π* peak in the HB structures of PNIPAM from MeOH + H2O to two H2O was 48 meV, approximating that of NIPAM. In contrast, in the same HB structures (MeOH + H2O), the CO π* peak in PNIPAM showed a tremendously higher energy shift (228 meV) than that in NIPAM. Note that the CO π* peak in PNIPAM includes the interaction between the polymer units of PNIPAM owing to structural optimization, and that in NIPAM has no interaction of the polymer units, which resembles the chain structures of PNIPAM in liquid MeOH.
In the MeOH-rich region (x > 0.4), the CO π* peak in the O K-edge XAS spectra showed a higher energy shift upon increasing the H2O molar fraction as a slope of 51 meV/(1 − x). In the inner-shell calculations, the energy shift of the CO π* peak in the HB structures of PNIPAM from MeOH + H2O to two H2O was 48 meV. Thus, the higher energy shift of the CO π* peak in the MeOH-rich region indicated that the HB structure of the CO group in PNIPAM with MeOH was substituted with its H2O equivalent by increasing the H2O molar fraction. The increase of the coordination number of MeOH and H2O also affected the higher energy shift of the CO π* peak. These results were consistent with the MD simulations, in which the HB structures of PNIPAM with one or two MeOH molecules shifted to those with two H2O molecules by increasing the H2O molar fraction.
In the O K-edge XAS spectra, the CO π* peak of PNIPAM in pure H2O (x = 0.0) showed a higher energy shift of 127 meV from that of pure MeOH (x = 1.0), which could not be explained by the simple substitution of the HB structures from MeOH to H2O. In the inner-shell calculation, the energy shift of the CO π* peak from NIPAM to PNIPAM was 228 meV in the same HB structures, indicating that the higher energy shift was induced by the increase of the interaction between polymer units. Because PNIPAM is dissolved in pure MeOH and H2O, it was considered that the PNIPAM chains expanded in both MeOH and H2O solvents; however, this study proposes that PNIPAM forms rounded structures in pure H2O, whereas it forms chain structures in pure MeOH. These results were consistent with the MD simulations, in which the Rg value in pure H2O was inferior to that in pure MeOH.
Liquid MeOH forms one-dimensional chain structures.32 The CO group in PNIPAM formed HB structures with MeOH, and the isopropyl group in PNIPAM exhibited hydrophobic interactions with the methyl group in MeOH. Because liquid MeOH has a chain structure, the PNIPAM chains spread through the hydrophobic interactions of the MeOH chains. In contrast, H2O molecules formed a three-dimensional HB network and exhibited hydrophobic hydration of the isopropyl group in PNIPAM. PNIPAM shows rounded structures with the hydrophobic hydration of PNIPAM. Because the interaction between polymer units in pure H2O is larger than that in pure MeOH, the CO π* peak at pure H2O shows a higher energy shift (127 meV) compared to that in pure MeOH.
The structures of aqueous MeOH solutions were extensively investigated by several experimental and theoretical studies, which are summarized in Section S8 of the ESI.† We have also revealed from the C K-edge XAS of aqueous MeOH solutions that the hydrophobic clusters of MeOH are formed in the middle concentration region (0.7 > x > 0.3).27 Because MeOH shifts from chain structures to cluster ones by increasing the H2O molar fraction from pure MeOH, the polymer units of PNIPAM tend to gather by the hydrophobic interaction with MeOH clusters, leading to the deposition of PNIPAM at x = 0.4. This finding also supports that PNIPAM aggregates include 80% solvent MeOH and H2O.7 This was consistent with the smallest Rg value (x = 0.2) observed in the MD simulation. PNIPAM maintains a coil state in pure H2O (x = 0.0) owing to the hydrophobic hydration of PNIPAM. By increasing the molar fraction of MeOH from pure H2O, the hydrophobic hydration of PNIPAM was disrupted and the hydrophobic interaction of PNIPAM with MeOH increased,21,22 inducing cononsolvency at 0.4 > x > 0.1.
Conclusions
The mechanism of PNIPAM cononsolvency in aqueous MeOH solutions was investigated from the molecular interactions of the CO group in PNIPAM with MeOH and H2O using O K-edge XAS. The CO π* peak in pure H2O shows an extremely higher energy shift from pure MeOH, which cannot be explained by the simple substitution of the HB structure from MeOH to H2O. The inner-shell calculation proposed that it is due to the increase of the interaction between polymer units, indicating PNIPAM in pure H2O is the rounded structure with the hydrophobic hydration of the isopropyl group in PNIPAM. Although PNIPAM is dissolved in pure MeOH and H2O, this study proposes that PNIPAM forms rounded structures in pure H2O, whereas it forms chain structures in pure MeOH. At 0.4 > x > 0.1, the hydrophobic hydration of PNIPAM is interrupted by the addition of MeOH, whereas the hydrophobic interaction of PNIPAM with MeOH is enhanced. The polymer units of PNIPAM aggregated via the MeOH hydrophobic clusters, inducing cononsolvency. The concentration change of PNIPAM might lead to the different mechanism of cononsolvency, which would be the subject of future study.
The phase transition dynamics of polymers, such as cononsolvency and low critical solution temperature,1–3 can be investigated using XAS from the molecular interactions of different groups in the polymers with solvent molecules. Thus, the element-selective analysis using XAS can be applied to the phase transition dynamics of biomolecules, such as protein folding, DNA packing, and interchain complexation.
Author contributions
Masanari Nagasaka: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing – original draft, writing – review & editing; Fumitoshi Kumaki: investigation, data curation, formal analysis, methodology; Yifeng Yao: investigation, data curation, formal analysis, methodology; Jun-ichi Adachi: investigation, methodology, validation; Kenji Mochizuki: investigation, data curation, formal analysis, funding acquisition, resources, validation, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. JP19H02680) and the National Natural Science Foundation of China (Grant No. 22273083 and 22250610195). The experiments were performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2021G047). The inner-shell calculations were performed using the facilities of the Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C187). We would like to thank Editage (https://www.editage.jp) for English language editing.References
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- T. Amiya, Y. Hirokawa, Y. Hirose, Y. Li and T. Tanaka, J. Chem. Phys., 1987, 86, 2375–2379 CrossRef CAS.
- F. M. Winnik, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415–2416 CrossRef CAS.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948–952 CrossRef CAS.
- G. Zhang and C. Wu, Phys. Rev. Lett., 2001, 86, 822–825 CrossRef CAS PubMed.
- K. Mochizuki and K. Koga, Phys. Chem. Chem. Phys., 2016, 18, 16188–16195 RSC.
- K. Mochizuki, S. R. Pattenaude and D. Ben-Amotz, J. Am. Chem. Soc., 2016, 138, 9045–9048 CrossRef CAS PubMed.
- C. Scherzinger, A. Schwarz, A. Bardow, K. Leonhard and W. Richtering, Curr. Opin. Colloid Interface Sci., 2014, 19, 84–94 CrossRef CAS.
- S. Bharadwaj, B.-J. Niebuur, K. Nothdurft, W. Richtering, N. F. A. van der Vegt and C. M. Papadakis, Soft Matter, 2022, 18, 2884–2909 RSC.
- F. Tanaka, T. Koga and F. M. Winnik, Phys. Rev. Lett., 2008, 101, 028302 CrossRef PubMed.
- G. Zhang and C. Wu, J. Am. Chem. Soc., 2001, 123, 1376–1380 CrossRef CAS.
- J. Pang, H. Yang, J. Ma and R. Cheng, J. Phys. Chem. B, 2010, 114, 8652–8658 CrossRef CAS PubMed.
- J. Walter, J. Sehrt, J. Vrabec and H. Hasse, J. Phys. Chem. B, 2012, 116, 5251–5259 CrossRef CAS PubMed.
- D. Mukherji, C. M. Marques and K. Kremer, Nat. Commun., 2014, 5, 4882 CrossRef PubMed.
- F. Rodríguez-Ropero, T. Hajari and N. F. A. van der Vegt, J. Phys. Chem. B, 2015, 119, 15780–15788 CrossRef PubMed.
- A. Pica and G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 25601–25608 RSC.
- K. Mochizuki, J. Phys. Chem. B, 2020, 124, 9951–9957 CrossRef CAS PubMed.
- C. Dalgicdir, F. Rodríguez-Ropero and N. F. A. van der Vegt, J. Phys. Chem. B, 2017, 121, 7741–7748 CrossRef CAS PubMed.
- I. Bischofberger, D. C. E. Calzolari, P. De Los Rios, I. Jelezarov and V. Trappe, Sci. Rep., 2014, 4, 4377 CrossRef CAS PubMed.
- K. Kyriakos, M. Philipp, C.-H. Lin, M. Dyakonova, N. Vishnevetskaya, I. Grillo, A. Zaccone, A. Miasnikova, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Macromol. Rapid Commun., 2016, 37, 420–425 CrossRef CAS PubMed.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- M. Nagasaka, H. Yuzawa and N. Kosugi, Anal. Sci., 2020, 36, 95–105 CrossRef CAS PubMed.
- M. Nagasaka and N. Kosugi, Chem. Lett., 2021, 50, 956–964 CrossRef CAS.
- T. Tokushima, Y. Horikawa, Y. Harada, O. Takahashi, A. Hiraya and S. Shin, Phys. Chem. Chem. Phys., 2009, 11, 1679–1682 RSC.
- M. Nagasaka, K. Mochizuki, V. Leloup and N. Kosugi, J. Phys. Chem. B, 2014, 118, 4388–4396 CrossRef CAS PubMed.
- K. Amemiya, H. Kondoh, T. Yokoyama and T. Ohta, J. Electron Spectrosc. Relat. Phenom., 2002, 124, 151–164 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- N. Kosugi and H. Kuroda, Chem. Phys. Lett., 1980, 74, 490–493 CrossRef CAS.
- N. Kosugi, Theor. Chim. Acta, 1987, 72, 149–173 CrossRef CAS.
- T. Yamaguchi, K. Hidaka and A. K. Soper, Mol. Phys., 1999, 96, 1159–1168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00676c |
| This journal is © the Owner Societies 2024 |