
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unraveling the electronic structure of LuH, LuN, and LuNH: building blocks of new materials†
Nuno M. S.
Almeida
 ,
Bradley K.
Welch
,
Sasha C.
North
and
Angela K.
Wilson
*
,
Bradley K.
Welch
,
Sasha C.
North
and
Angela K.
Wilson
*
Michigan State University, Department of Chemistry, East Lansing, MI 48864, USA. E-mail: akwilson@msu.edu
First published on 19th March 2024
Abstract
Advances in superconductor technology have been pursued for decades, moving towards room temperature models, such as a postulated nitrogen-doped lutetium hydride network. While experimental observations have been contradictory, insight into the building blocks of potential new superconductor materials can be gained theoretically, unravelling the fascinating electronic structure of these compounds at a molecular level. Here, the fundamental building blocks of lutetium materials (LuH, LuN, and LuNH) have been examined. The structures, spectroscopic constants for the ground and excited states, and the potential energy curves have been obtained for these species using complete active self-consistent field (CASSCF) and multireference configuration interaction with Davidson's correction (MRCI+Q) methods. For LuNH, the energetic properties of its isomers are determined. The bond dissociation energies of the three building blocks are calculated with the state-of-the-art f-block ab initio correlation consistent composite approach (f-ccCA) and the high accuracy extrapolated ab initio thermochemistry (HEAT) scheme. As well, an analysis of different formation pathways of LuNH has been provided.
1 Introduction
From the concept of superconductivity and its first observation in mercury at 4.2 K, scientists have strived to create the first room temperature superconductor.1 The first ambient temperature superconductor would not only revolutionize electronics, but also impact industries, such as quantum computing, and transform the chemical industry. Ambient superconductors will allow for the development of energy efficient computer chips and fusion reactors, and aid in the development of magnets, which can be used in applications such as maglev trains. One very important application would be for particle accelerators, such as the Large Hadron Collider (LHC), which requires 96 tons of liquid helium to cool their current system.2 If this extreme level of cooling was not necessary, the engineering would be simplified.2 An added layer of difficulty in building an ambient superconductor are the required pressures. In the solid state these materials form a lattice that offers no resistance to conducting current. However, large pressures are required to maintain the stability and structure of the material lattices. Under lower pressure conditions (below 200–300 kPa), the lattice can break down, due to the coupling of the electrons and phonons.Early in 2023, a nitrogen-doped lutetium hydride lattice was proposed as an ambient temperature superconductor (294 K and 1–2 Gpa).3 It was reported that the lattice structure is composed of: LuH2±xNy, depicting a dark-blue color, according to the authors consistent with the novel ambient temperature superconductivity. However, this finding did not achieve consensus among the chemistry community, and the publication on the proposed superconducting properties of this structure was retracted in November 2023. Ming et al., synthetized the same compound (LuH2±xNy) utilizing high pressure and high temperature conditions, which was then confirmed by X-ray diffraction.4 The authors found the same color change from dark blue to violet and then pink/red, however superconductivity was not observed above 2 K. Peng et al., have also performed a quantitative temperature-dependent resistance comparison of Lu–N–H and pure lutetium before reaction and concluded that the change is resistance mentioned by Dasenbrock-Gammon et al., is likely caused by a metal-to-poor conductor transition.5 Xingzhou et al. also reported that they could not find evidence of a superconducting transition in all phases from temperatures ranging from 1.8 to 300 K.6 The aforementioned and several other studies have also shown, that the superconductivity properties described by Dasenbrock-Gammon et al.3 cannot be reproduced, hence the original manuscript was retracted.4,5,7–28 However, the electronic properties of the building blocks of these materials have not been explained or understood. As Xingzhou et al. mentioned in their work: “…we identify a notable temperature-induced resistance anomaly of electronic origin in LuH2±xNy, which is most pronounced in the pink phase and may have been erroneously interpreted as a sign of superconducting transition”.6 In addition, the electronic structure of the lattice building blocks has not been detailed properly.
In terms of the building blocks of the nitrogen-doped lutetium hydride lattice, there is little known about it from an electronic structure standpoint. In 1974, Efanttin et al. calculated the electronic spectra of LuH.29 The ground state was established as 11Σ+, the bond length, Re = 1.9111 ± 0.0001 Å, and the ωe of the ground state is 1520 ± 20 cm−1. More recently, gas phase spectroscopic constants have been calculated by Wang et al. by laser ablation with the ωe of the ground state calculated to be 1476 ± 20 cm−1.30
Theoretically, LuH has been studied (mainly for ground state properties), while LuN and LuNH have received less attention. Pantazis et al. calculated the Re of the LuH ground state (11Σ+) using density functional theory (DFT), specifically PBE0 with segmented all-electron relativistically contracted (SARC) basis sets, paired with a Douglas–Kroll–Hess second order Hamiltonian (DKH2) with counterpoise corrections, and rendered the ground state bond length 1.904 Å.31 In addition, the authors estimated the dissociation energy (De) to be 3.12 eV. Later, Jalbout et al. utilized a panoply of DFT functionals, which were then fitted, and predicted the ground state bond length to range between 1.911 to 1.940 Å.32 In this investigation, B3LYP obtained the closest value to experiment, which led to the following spectroscopic constants: 1.911 Å (Re), 1517 cm−1 (ωe), and 20.48 cm−1 (ωeχe). In terms of wavefunction methods, Møller–Plesset second order (MP2) perturbation theory with an all-electron basis set was utilized to investigate ground state properties of LuH resulting in 1.883 Å and 1540 cm−1 for Re and ωe, respectively.33 Dolg and Stoll also calculated spectroscopic constants for the 11Σ+ ground state,34 obtaining 1.95 Å, 1445 cm−1, with CI(SD + Q) and the Wood-Boring pseudopotential for Lu.34 In addition, Cao and Dolg utilized SCF, CCSD, and CCSD(T) paired with natural orbital valence basis sets (small-core pseudopotentials for the lanthanide) to calculate the Re, De and ωe for the 11Σ+ ground state of LuH.35 At the CCSD(T) level, the values were estimated from 1.882–1.914 Å, and the ωe from 1507 and 1577 cm−1 (depending upon the level of core-correlation considered). For De, the values ranged from 3.35 to 3.64 eV.35 Assaf et al. calculated the ground and first excited states of LuH and provided spectroscopic constants for this molecule.36 The authors show the importance of accurately detailing the spin–orbit properties of this system to understand the energetics of the complex.
For LuN and LuNH, computational studies have been done, through mainly to characterize their behavior within a lattice, utilizing DFT, and considering potential solid-state applications. Oualdine et al. investigated the structural, elastic, and electronic properties of LuN using ab initio (solid state) techniques. The authors found that LuN has a semiconductor nature and calculated its band structure and gap.37 Yurdaşan et al. also found LuN to be a promising material for optoelectronic applications.38 More recently, in 2022, Devese et al. probed the defects of LuN1−δ experimentally and computationally.39 The authors calculated the optical bandgap of this material to be 1.7 eV. In addition, that its conductivity can be controlled by nitrogen vacancy doping.
Herein, the first detailed electronic structure calculations for LuH, LuN, and LuNH (and its isomers) are reported, detailing the ground and excited states of the molecules, along with the different routes of formation of the molecules. Bond dissociation values are also provided with state-of-the-art methodologies: f-block ab initio correlation consistent composite approach (f-ccCA)40 and a type of high accuracy extrapolated ab initio thermochemistry (HEAT).41–44
2 Computational details
Due to the complexity of the electronic structure of lanthanides, multireference wavefunction approaches are typically necessary to accurately describe the fundamental properties of LuH, LuN and LuNH, and also to construct potential energy curves (PECs). MRCI+Q was utilized to describe the ground and several excited states of these molecules. For LuH, the active space considered was: four electrons in ten orbitals. The orbitals included at the atomic level are: five of a1 symmetry (1s (H); 6s, 5dz2, 5dx2−y2, 6pz (Lu)), two of b1 symmetry (5dxz and 6px (Lu)), two of b2 symmetry (5dyz and 6px (Lu)) and one of a2 symmetry (5dxy (Lu)). Due to the smaller active space utilized for this molecule a DKH3 Hamiltonian was employed in MOLPRO 2020.45 Paired with DKH3, a cc-pVQZ-DK346 basis set was used for lutetium, along with aug-cc-pVQZ-DK47 for hydrogen. For LuN, an expansion of the active space to six electrons in fifteen orbitals was necessary: six of a1 symmetry (2pz, 3pz (N); 6s, 5dz2, 5dx2−y2, 6pz (Lu)), four of b1 symmetry (2px, 3px (N); 5dxz and 6px (Lu)), four of b2 symmetry (2py, 3py (N); 5dyz and 6px (Lu)) and one of a2 symmetry (5dxy (Lu)). Since the active space was expanded to fifteen orbitals, it was not possible to employ a fully relativistic Hamiltonian (DKH3), instead, a non-relativistic Hamiltonian was utilized. For Lu, the ECP28MWB segmented pseudopotential was employed at a triple-ζ level.35,48 For nitrogen, aug-cc-pVTZ was utilized.49 Radial distribution functions (RDFs) were calculated for LuH and LuN, showing the importance of the inclusion of 3px,y,z orbitals for the LuN PECs (see Fig. 1(a) and (b)). The 3px,y,z orbitals overlap significantly at the equilibrium region of LuN (and probably LuNH). For both molecules, core correlation was also employed, by including the 4f orbitals (seven extra orbitals). By including these orbitals at the “core” level in the MRCI expansion, single and double excitations are allowed to the active space. Moreover, in previous studies, it was shown that these core orbitals are necessary to reproduce experimental quantities accurately.40,50 | ||
| Fig. 1 (a) Radial distribution functions for LuH, with the bond distance of the molecule plotted along the z-axis, obtained using CCSD(T), the cc-pVTZ-DK3 basis set for Lu, and the cc-pVTZ-DK basis set for H (and using a DKH3 Hamiltonian). (b) Radial distribution functions for LuN, with the bond distance of the molecule plotted along the z-axis, obtained using CCSD(T), the cc-pVTZ-DK3 basis set for Lu, and the aug-cc-pVTZ-DK basis set for N (DKH3 Hamiltonian). | ||
For LuNH PECs, the same orbitals employed for LuN were considered at the CASSCF level, with one more electron originating from the 1s of hydrogen. In addition, the 2pz of nitrogen hybridizes with the 1s of hydrogen, so no additional orbitals were necessary in the active space. At the MRCI level, the three higher orbitals from a1, b1 and b2 symmetries were removed. Having fifteen orbitals and seven electrons in the active space, with additional core orbitals was not feasible at the MRCI level. In addition, at 7.0 Å, (the initial distance the potential energy curves were computed), the three aforementioned orbitals are a hybrid of the 3px,y,z and the 3dz2, 3dxz and 3dyz of nitrogen. However, at equilibrium bond length, the b1 and b2 orbitals lose their 3px,y character and become pure 3dxz and 3dyz respectively. As per LuH and LuN, the 4f orbitals were also included in the core. For the potential energy curves (PECs), the following basis sets were used: ECP28MWB segmented (Lu)35,48 and aug-cc-pVTZ (N,H).49 To optimize, the ground state of LuNH and respective isomers, CCSD(T) with a DKH3 Hamiltonian was utilized, paired with a triple-ζ level basis sets (cc-pVTZ for H and aug-cc-pVTZ for N). Analytical frequencies were calculated, ensuring a minimum in the potential energy surface.49
The spin–orbit corrections for LuH, LuN, and LuNH were calculated with the Breit–Pauli Hamiltonian within the MRCI method. The active space used was the same as previously mentioned for the non-spin orbit calculations, i.e., four electrons in ten orbitals for LuH, six electrons in fifteen orbitals for LuN, and seven electrons in fifteen orbitals for LuNH. Core-correlation was utilized for LuH and LuN, the f-orbitals were included in the “core card”. However, for LuNH the 4f orbitals were not included, due to the added computational cost. For LuH due to the added computational cost, a triple-ζ basis set was utilized for spin–orbit calculations (cc-pVTZ-DK346 (Lu) and aug-cc-pVTZ-DK47 (H), paired with a DKH3 Hamiltonian). For the other species (LuN and LuNH), the same basis set as for the non-spin–orbit calculations was utilized.
For CASSCF and MRCI+Q, equilibrium bond lengths, harmonic, anharmonic and ΔG1/2 were calculated from the PECs of LuH, LuN, and LuNH, and the Numerov/Cooley approach was utilized.51,52 For spin–orbit curves and with fewer sampling energetic points, the Dunham approach was utilized.53
f-ccCA and HEAT were used to obtain thermodynamic quantities for LuH, LuN, and LuNH. For LuN, even though the Hartree–Fock orbitals are not ideal (see Results and Discussion), the higher correlation contributions beyond the CCSD(T) terms for HEAT are able to provide much of the electronic correlation necessary to describe the ground state of this molecule.
HEAT is utilized rather than our own super-ccCA (s-ccCA), as s-ccCA includes a pentuples contribution, which though successful for transition metal species,54 is too expensive computationally for the heavy elements, even at a double-ζ level. HEAT and s-ccCA are similar, though with some differences such as the pentuples contribution (s-ccCA), and the full complete basis set (CBS) extrapolation for the full coupled cluster triples contribution (HEAT).54 Composite schemes such as f-ccCA, s-ccCA, and HEAT mimic the accuracy of computationally more expensive methods, utilizing a series of lower level ab initio calculations, allowing a lower computational cost.40–44,54–58 (An explanation of the composite schemes utilized herein is provided in the ESI†). Due to the large number of valence electrons for these systems for the HEAT protocol, the higher order correlation only consists of CCSDT and Λ-CCSDT(Q). For LuH and LuN, bond dissociation energies and enthalpies of formations were obtained using atomization energies, and standard thermochemical quantities for the atoms. For LuNH, the bond dissociation energies were obtained for three different routes:
| LuNH → LuN + H | (1) |
| LuNH → Lu + NH | (2) |
| LuNH → Lu + N + H | (3) |
The enthalpy of formation was obtained with the atomization scheme (route (3)), as well as from routes (1) and (2) using experimental quantities for Lu, and NH; the computed dissociation energy of LuN was used for route (2).
3 Results and discussion
3.1 Lutetium hydride (LuH)
The lutetium hydride curves are depicted in Fig. 2. The first two dissociation channels are considered for this molecule: Lu (2D) + H (2S), which render the following states:| 1,3[Σ+, Π, Δ] |
| 1,3[Σ+, Π] |
 | ||
| Fig. 2 LuH MRCI + Q potential energy curves (kcal mol−1) as a function of the Lu–H distance. | ||
Attractive states of singlet and triplet multiplicity are depicted. The bond length (Re), harmonic (ωe), anharmonic (ωeχe), ΔG1/2, and excitation energies (Te) are shown in Table 1. The CI vectors coinciding with the investigated states are displayed in Table S1 (ESI†). The ground state of LuH is a 1Σ+. At equilibrium bond length, the valence 5d1 electron from lutetium migrates to the 1s of hydrogen, filling the shell (1s2). MRCI + Q and CCSD(T) are both in agreement with predictions of the bond length of the complex to be 1.920 Å. In addition, the calculated ωe, from the PECs is 1503.0 cm−1, comparing favorably to our coupled-cluster predictions, 1487.1 cm−1 (obtained with analytical gradients). In addition, both our equilibrium bond length and harmonic frequencies compare well to experimental values of Efanttin et al. and Wang et al.29,30 Our ωe obtained with CCSD(T), with a DKH3 Hamiltonian and triple-ζ basis set is the closest value to experiment (1487.1 vs. 1476 ± 20 cm−1). However, the DFT bond lengths shown in Table 1 (calculated by Pantazis et al. and Jalbout et al. with a panoply of functionals), range from 1.904 to 1.940 Å, demonstrating the sensitivity of the bond length predictions with respect to functional choice, which can dramatically impact the bond length of lanthanide hydrides.31,32 The MP2 bond length is also quite far from experiment (1.883 vs. 1.9111 Å).33 Coupled-cluster predictions show sensitivity to core correlation, when considering different active and inactive orbitals within the core (below valence) set of orbitals. In addition, counterpoise corrections were also considered for these calculations.35 One of the main conclusions of Cao et al., based on the range of coupled-cluster predictions focused on the fact that the 4f orbitals are crucial to obtain correct results. The bond lengths obtained from Cao and Dolg fall between 1.882 to 1.914 Å.34 Finally, the ic-MRCI prediction from Assaf et al. slightly underestimate the bond length of the ground state, though is on par with experiment.36
| States | Methods | R e (Å) | ω e (cm−1) | ω e χ e (cm−1) | ΔG1/2 (cm−1) | T e (cm−1) |
|---|---|---|---|---|---|---|
| a Values calculated in this study. CCSD(T) with a DKH3 Hamiltonian. cc-pVTZ-DK3 for Lu and cc-pVTZ-DK for H. b The data from prior studies reports the exact number of digits as provided in the original sources. | ||||||
| X1Σ+ | CASSCF | 1.982 | 1404.8 | 17.6 | 1370.5 | 0 |
| MRCI+Q | 1.920 | 1503.0 | 18.3 | 1467.9 | 0 | |
| CCSD(T)a | 1.920 | 1487.1 | — | — | 0 | |
PBE031![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
1.904 | — | — | — | 0 | |
| DFT functionals32 | 1.911–1.940 | 1511.8–1557.6 | 14.0–20.48 | — | 0 | |
| MP233 | 1.883 | 1540 | — | — | 0 | |
| CCSD(T)35 | 1.882–1.914 | 1507–1577 | — | — | 0 | |
| CI(SD+Q)34 | 1.95 | 1445 | — | — | 0 | |
| ic-MRCI36 | 1.906 | 1539.24 | 18.69 | — | 0 | |
| Exp.30 | — | 1476 ± 20 | — | — | 0 | |
| Exp.29 | 1.9111 | 1520 ± 20 | 22 ± 1 | — | 0 | |
| 13Δ | CASSCF | 2.035 | 1313.1 | 25.8 | 1256.4 | 14426.5 |
| MRCI+Q | 1.966 | 1421.7 | 22.2 | 1375.0 | 12738.3 | |
| ic-MRCI36 | 1.962 | 1436.42 | 21.71 | — | 12653 | |
| 13Π | CASSCF | 1.990 | 1386.5 | 23.7 | 1336.5 | 12763.0 |
| MRCI+Q | 1.949 | 1448.4 | 22.8 | 1402.0 | 12963.1 | |
| ic-MRCI36 | 1.938 | 1469.7 | 22.18 | — | 12828 | |
| 13Σ+ | CASSCF | 2.013 | 1336.5 | 24.6 | 1283.3 | 14435.0 |
| MRCI + Q | 1.949 | 1426.6 | 22.5 | 1380.3 | 14418.3 | |
| ic-MRCI36 | 1.945 | 1453.25 | 30.55 | — | 14116 | |
| 11Δ | CASSCF | 2.056 | 1303.2 | 36.0 | 1220.7 | 17369.0 |
| MRCI + Q | 1.994 | 1389.3 | 28.8 | 1327.6 | 16528.1 | |
| ic-MRCI36 | 1.987 | 1401.07 | 27.60 | — | 16335 | |
| 11Π | CASSCF | 2.037 | 1271.9 | 57.5 | 1132.4 | 18954.1 |
| MRCI+Q | 1.952 | 1396.0 | 32.9 | 1323.0 | 16808.8 | |
| ic-MRCI36 | 1.942 | 1400.68 | 31.23 | — | 16843 | |
| 23Π | CASSCF | 1.988 | 1442.0 | 68.7 | 1262.8 | 19743.9 |
| MRCI+Q | 1.889 | 1566.5 | 32.8 | 1492.7 | 18688.0 | |
| ic-MRCI36 | 1.879 | 1575.38 | 25.42 | — | 18514 | |
| 21Σ+ | CASSCF | 2.056 | 1299.5 | 36.4 | 1218.5 | 17381.1 |
| MRCI+Q | 1.941 | 1420.1 | 32.7 | 1346.1 | 19309.1 | |
| ic-MRCI36 | 1.928 | 1427.95 | 30.17 | — | 19364 | |
| 21Π | CASSCF | 2.012 | 1479.5 | — | — | 23138.9 |
| MRCI+Q | 1.941 | 1494.4 | 46.8 | 1384.6 | 23786.7 | |
| ic-MRCI36 | 1.916 | 1488.29 | 35.34 | 23620 | ||
The first excited state of the complex is a 3Δ, followed closely by a 3Π, and these states lie 12738.3 and 12963.1 cm−1, respectively, above the ground state according to MRCI+Q. The 3Δ has two electrons coupled in the 1s of hydrogen and two unpaired electrons on the 6s and 5d of lutetium. However, the 3Π state has one electron on the 6px or 6py of lutetium, while two electrons are also coupled in the hydrogen's 1s orbital. The first 3Σ+ has one electron on the 6s and one on 6pz of lutetium. The first singlet excited state only appears at 16528.1 cm−1 according to MRCI+Q, and it is a 1Δ. The 1Δ, is an open shell singlet, i.e., it has one electron in the 6s of lutetium of alpha spin and a beta spin electron on the 5d orbital. The next state, 23Π is ∼2100 cm−1 above 11Δ and has its bond length shortened by ∼0.1 Å (1.889 Å according to MRCI+Q). This is the first triplet state that places one electron in the 5dxz or 5dyx of lutetium. The last two bonding states are the 21Σ+ and 21Π. The 21Σ+ is at 19309.1 cm−1 and is a multireference state, i.e., placing one alpha electron in the 6pz, a beta electron on the 6s and vice-versa. The last plotted state (21Π) is also quite multireference, alternating between alpha and beta electrons on the 6px, 6py, and 6s orbitals of lutetium.
The ground and the first excited states were considered for spin–orbit contributions for LuH and are displayed in Fig. 3. The following 2S+1Λ states were selected: X1Σ+ → X1Σ+0+, 13Π → 3Π0+,3Π0−,3Π1,3Π2, 13Δ → 3Δ1,3Δ2,3Δ3, 13Σ+ → 3Σ+0−,3Σ+1, and 11Δ → 1Δ2. The spin orbit constants and composition of states are shown in Table S2 (ESI†).
 | ||
| Fig. 3 LuH MRCI + Q spin–orbit potential energy curves (kcal mol−1) as a function of the Lu–H distance. | ||
The ground state is well separated from the first excited state, 13Π0−. When corrected at a spin–orbit level, the 13Δ is not the first excited state anymore. In fact, the 13Π0− and 13Δ are separated by 700 cm−1. Furthermore, the three components of the 13Δ state are separated by 2138.8 cm−1, which shows the impact of spin–orbit for these species. Also, by comparing the composition of the 13Δ1 and 13Π1, it is noted that these states are severely mixed. The Ω = 2 states are also mixed between 13Δ2 and 13Π2 but not to the same extent as Ω = 1 states. The first component of the 13Π is at 12133.2 cm−1 and the four components of this state are also separated by 2370.1 cm−1. The 13Σ+1 and 13Σ+0− are at 15248.8 and 15401.1 cm−1 respectively. The last considered states in Fig. 3 is the 1Δ2, which lies at 16944.2 cm−1. Even though the 1Δ2 is well separated from the rest it is still mixed with the 13Δ and 13Π.
3.2 Lutetium nitride (LuN)
The lutetium nitride molecule PECs are depicted in Fig. 4. From an electronic standpoint, this molecule is a lot more challenging to analyze when compared to LuH. The lowest dissociation channels of LuN are: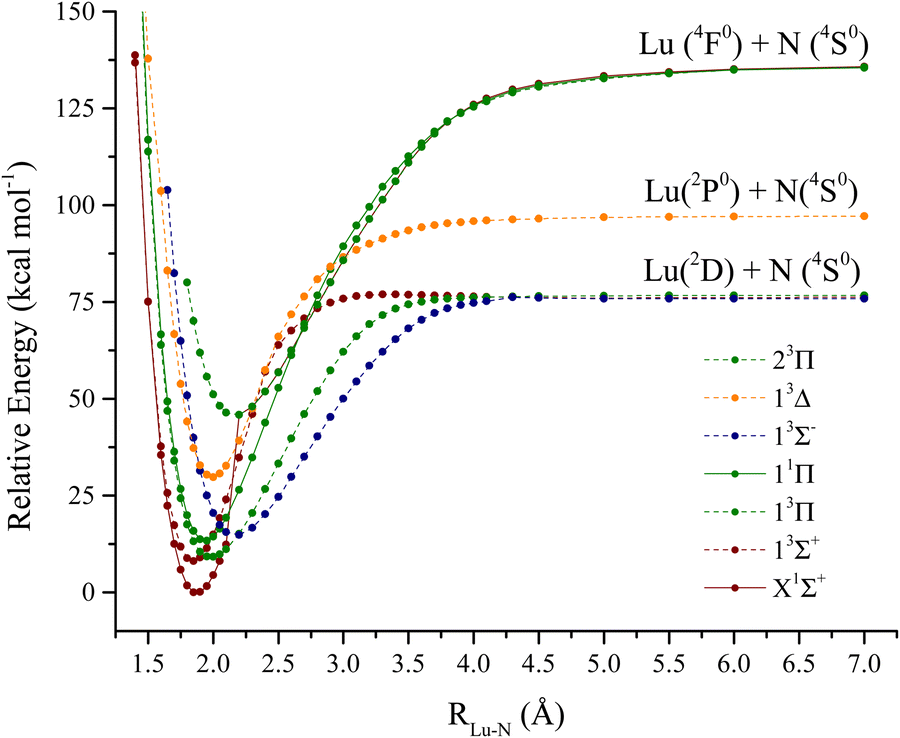 | ||
| Fig. 4 LuN MRCI + Q potential energy curves (kcal mol−1) as a function of Lu–N distance. | ||
Lu (2D) + N(4S0):
| 3,5[Σ−, Π, Δ] |
Lu (2P0) + N(4S0):
| 3,5[Σ−, Π] |
And
Lu (4F0)+ N(4S0):
| 1,3,5,7[Σ−, Π, Δ, Φ] |
The spectroscopic constants calculated for LuN are shown in Table 2 and the calculated CI vectors are provided in Table S3 (ESI†). Even though, from the considered dissociation channels there are not any Σ+ states, when considering the lowest three dissociation channels of LuN (by Wigner–Witmer rules), the ground state is a 1Σ+! There is crossing between 2.4–2.5 Å, from a higher dissociation channel (possibly the ionic channel), the 1Δ state switches to the 1Σ+ ground state. This state is formed by nitrogen abstracting three electrons from lutetium, the ground state is Lu3+N3−, rather than Lu2+N2−. The latter is the electronic structure of the aforementioned 1Δ state. As shown by the CI vectors in Table S3 (ESI†), the ground state is composed of three different components (Lu3+ N3−); Hartree–Fock can only describe one, which in this case is not one of the main CI vectors predicted by CAS/MRCI. In Fig. 5, the Hartree–Fock valence orbitals (except for the 2s of nitrogen) responsible for the bonding are shown. In addition, the LUMO orbital was also included for comparison. Furthermore, a comparison with the natural orbitals obtained with CASSCF is provided. At the Hartree–Fock level, the ground state determinant corresponds to filled 2px,y, of nitrogen (HOMO−1 and HOMO−2), while the HOMO orbital is the distorted 6s of lutetium. This configuration has a very low probability (CI ∼ 0.11) when compared to the natural orbital occupations. Furthermore, an orbital rotation of the 2pz orbital with the 6s was performed; however, the energy of the X1Σ+ state became a lot higher. When CASCSF is utilized (and furthermore MRCI), there are three determinants responsible for the ground state. These consist of: six electrons in the 2px,y,z orbitals from nitrogen, and two combinations of alpha and beta electrons on the 2pz of nitrogen and 6s of lutetium. The difference between CCSD(T) and CASSCF/MRCI+Q bond lengths arises from a multireference ground state (1Σ+), along with incorrect orbitals, which cannot be described correctly by single reference methods. Despite the discrepant equilibrium bond lengths, the predicted frequencies by CASSCF, MRCI + Q and CCSD(T) are close in energy.
| States | Methods | R e (Å) | ω e (cm−1) | ω e χ e (cm−1) | ΔG1/2 (cm−1) | T e (cm−1) |
|---|---|---|---|---|---|---|
| X1Σ+ | CASSCF | 1.908 | 773.5 | 6.6 | 765.6 | 0 |
| MRCI+Q | 1.872 | 799.9 | 6.8 | 777.9 | 0 | |
| CCSD(T) | 2.010 | 757.2 | — | — | 0 | |
| 13Σ+ | CASSCF | 1.874 | 803.3 | 6.7 | 782.8 | 2898.4 |
| MRCI+Q | 1.844 | 812.6 | 4.1 | 801.0 | 2865.2 | |
| CCSD(T) | 1.840 | 802.6 | — | — | 3452.5 | |
| 13Π | CASSCF | 2.093 | 526.1 | 2.5 | 522.1 | 685.1 |
| MRCI+Q | 1.978 | 562.7 | 1.5 | 556.9 | 3255.5 | |
| 11Π | CASSCF | 1.968 | 666.3 | 3.0 | 658.4 | 4476.2 |
| MRCI+Q | 1.937 | 726.3 | 4.7 | 722.3 | 4716.4 | |
| 13Σ− | CASSCF | 2.219 | 475.0 | 1.7 | 468.6 | 1111.5 |
| MRCI+Q | 2.175 | 517.4 | 2.8 | 512.1 | 5199.7 | |
| 13Δ | CASSCF | 2.134 | 339.1 | 6.2 | 415.1 | 17556.5 |
| MRCI+Q | 1.993 | 743.6 | 4.6 | 733.2 | 10460.5 | |
| 23Π | CASSCF | 1.977 | 737.0 | 3.5 | 724.1 | 8076.8 |
| MRCI+Q | 2.167 | 533.0 | 2.6 | 528.2 | 16025.7 |
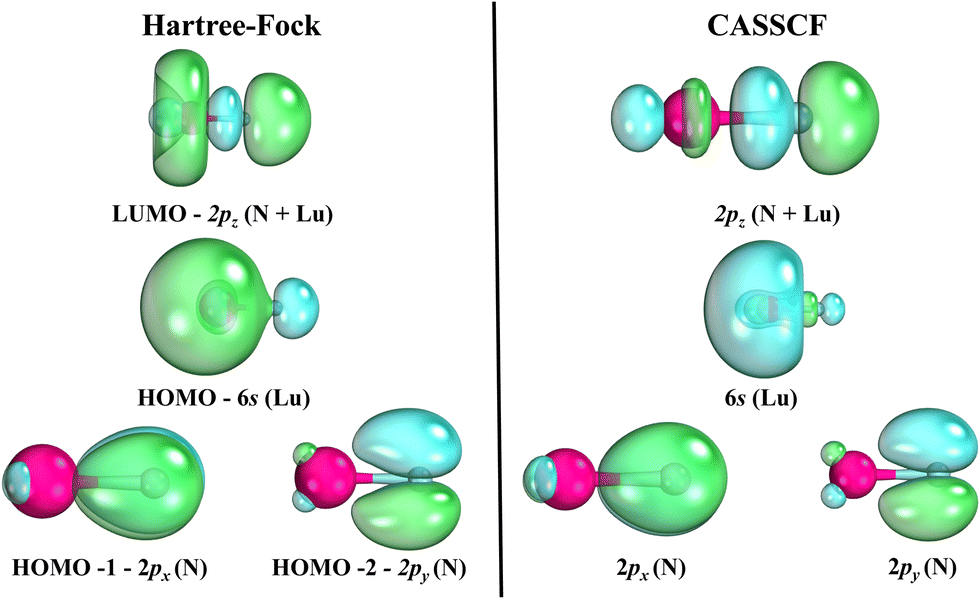 | ||
| Fig. 5 Hartree–Fock orbitals versus selected CASSCF orbitals. For purposes of visualization, the Hartree–Fock orbitals were obtained with cc-pVDZ-DK3 (Lu) and aug-cc-pVDZ (N) at the equilibrium bond length for this method (2.010 Å). The CASSCF orbitals were collected from the PECs at 1.85 Å. | ||
Regarding the excited states of LuN, the lowest six states were described with CASSCF/MRCI+Q and the 13Σ+ state was also described with CCSD(T). For LuN and in contrast to LuH, the excited states are not accessed from the internuclear region of the ground state, i.e. not the Franck–Condon region, which pertains to vertical transitions from the initial state (for example the ground state). In addition, there are also multiple crossings, which will be explained further. The first excited state is a 3Σ+, also a product of a higher dissociation channel, which occurs between 2.5 Å and 2.4 Å. This state is constructed by placing an unpaired electron on the nitrogen 2pz orbital and lutetium 6s, which leads to a bond that is Lu2+N2− in nature. The bond length of the 13Σ+ is of the same magnitude of the X1Σ+, 1.844 Å according to MRCI+Q. For CCSD(T), the predicted bond length is 1.840 Å, which compares quite well to MRCI+Q. The main component of this state corresponds to a CI vector of 0.89, obtained from the CI vectors, making it largely single reference in nature as compared to the ground state, so it can also be described by single reference methods. The next excited state, 3Π is separated only by ∼300 cm−1 from 13Σ+ according to MRCI + Q, however at the CASSCF level, the 3Π is the first excited state. Due to having unpaired electrons in the nitrogen's 2px or 2py orbitals, the 3Π bond length is elongated to 1.978 Å. The next state in Table 2 is 11Π, which is an “open shell” singlet. The 11Π is the singlet version of the first 3Π, however, this state has alpha or beta electrons in the 6s and 2px or 2py of lutetium and nitrogen, respectively. The bond length of the 11Π is also elongated when compared to the ground state to 1.978 Å, at the MRCI + Q level. The first 3Σ− state is only separated by 400 cm−1 above the 11Π. This state is the first one to have two unpaired electrons in nitrogen's p orbitals. This triplet is formed by placing one unpaired electron in each of the 2px and 2py orbitals. This state is quite single reference; its main CI component is 0.94. However, due to these two unpaired electrons in nitrogen orbitals, there is less orbital overlap, which elongates the Lu–N bond to 2.175 Å according to MRCI+Q. The last two states in Table 2 are triplets, the first of Δ symmetry and the 23Π. CASSCF predicts the 23Π to be lower in energy than the 13Δ, however, MRCI + Q has the order reversed. The 23Π has two main components in which two electrons are placed in the 2pz (nitrogen), two in the 6s of lutetium and one alpha and one beta electrons in the 2px and 2py orbitals of nitrogen. In addition, the 23Π is also one of the longest states investigated for LuN, 2.167 Å at MRCI + Q level. In contrast, the 13Δ six different CI contributions to the state, make this state extremely multireference in nature.
The spin–orbit curves of LuN are displayed in Fig. 6. Due to the complexity of these curves, and because the excitations are not vertical, only the ground and first three excited states are calculated. The spin–orbit spectroscopic constants and composition of the four considered states are shown in Table S4 (ESI†). The 2S+1Λ states split into the Ω-states as follows: X1Σ+ → X1Σ+0+, 13Σ+ → 13Σ+0−,13Σ+1, 13Π → 3Π0+,3Π0−,3Π1,3Π2, and 11Π → 11Π1. Even though the curves were not extended beyond 2.05 Å due to the multiple crossings, the four components of the 23Π were also included in the calculation to have a more complete mixture of states and compositions. In addition, the 23Π state was not tabulated, but its data was added to the state composition row in Table S4 (ESI†).
 | ||
| Fig. 6 LuN MRCI + Q spin–orbit potential energy curves (kcal mol−1) as a function of Lu–N distance. | ||
Contrary to LuH, the mixing of the Ω-states of LuN at the equilibrium bond length is much less extensive. However, there are multiple avoided crossings for this complex. The 13Σ+1/13Π1 and 13Σ+0−/13Π0− avoid each other as shown in Fig. 6. Even though the ground state is separated by 3215.3 cm−1 from the first excited state (13Σ+0−), the rest of the considered states are quite close in energy. The 13Π components are separated by 734 cm−1. It is interesting to note that the bond lengths of the different components also range from 2.002 Å (13Π1) to 1.942 Å (13Π0−). Without spin–orbit estimations, it is not possible to accurately describe these states. Unfortunately, after 2.1/2.2 Å there are multiple crossings that do not allow for further extension of the PECs.
3.3 Lutetium imidogen (LuNH)
Combining the properties of LuH and LuN allows an understanding of the bonding properties of LuNH. To form this triatomic, all the possible isomers were considered. Herein, the following questions are raised: does lutetium bind to nitrogen and hydrogen, or just nitrogen? Which spin is the lowest, and how are the bonds of the triatomic formed? Is the lowest geometry linear or bent?The electronic landscape of the bond formation of LuNH is shown in Fig. 7. The spectroscopic constants of the ground and first two excited states are shown in Table 3 and CI vectors in Table S5 (ESI†). The states considered for this complex at the CASSCF level comprise all of the excitations until the 6px,y,z of lutetium of singlet and triplet spin is reached, which include 32 states. The lowest geometry consists of nitrogen as a central atom bonded to hydrogen and lutetium and it is perfectly linear (180° between Lu–N–H). The PECs were created with MRCI+Q for the ground and first excited state, and by fixing the N–H bond, utilizing the CCSD(T)/TZ with a DKH3 Hamiltonian bond length. The N–H bond length is 1.036 Å. The Lu–N distance was then stretched to seven angstroms, and the energy profile was analyzed. For this triatomic, as for LuN, there is a crossing from a higher ionic channel. However, for LuNH it occurs at ∼3 Å, switching the 2Δ state to a 2Σ+. The ground state has an unpaired electron on the 6s of lutetium, due to the movement of one of the lutetium valence electrons to the hydrogen, filling its 1s orbital. The bond distance of the first excited state is elongated to ∼2.1 Å and it is formed by moving one unpaired electron to the 2px or 2py of nitrogen rather than to the 6s of lutetium.
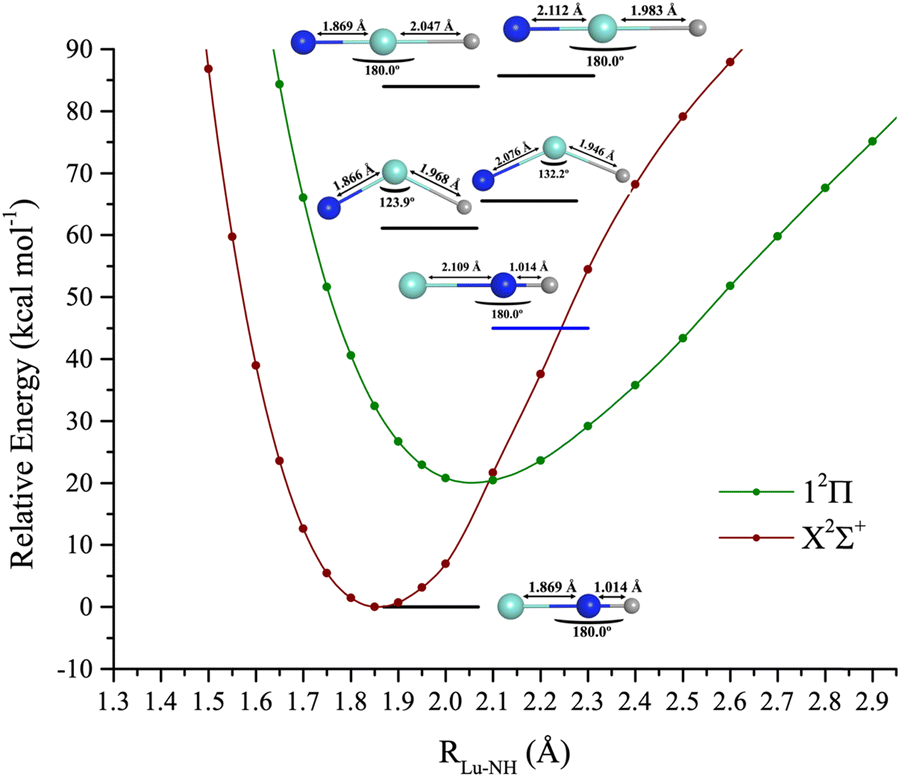 | ||
| Fig. 7 Lu–NH MRCI + Q potential energy curves (kcal mol−1) as a function of the Lu–N distance. The blue horizontal bar corresponds to a CASSCF relative energy of the ground CASSCF state 2Σ+. The black horizontal bars correspond to CCSD(T)/DKH3 relative energies to the ground CCSD(T)/DKH3 state 2Σ+. | ||
| States | Methods | R e (Å) | ω e (cm−1) | ω e χ e (cm−1) | ΔG1/2 (cm−1) | T e (cm−1) |
|---|---|---|---|---|---|---|
| a Lu–NH symmetric stretch frequency. b Lu–N–H bending frequency. c N–H asymmetric stretch frequency. | ||||||
| X2Σ+ | CASSCF | 1.882 | 775.3a | −17.8 | 821.0 | 0 |
| MRCI+Q | 1.858 | 809.8a | −5.7 | 835.3 | 0 | |
| CCSD(T)* | 1.869 | 552.8b | — | — | — | |
| 560.1b | — | — | — | |||
| 816.1a | — | — | — | |||
| 3569.4c | — | — | — | |||
| 12Π | CASSCF | 2.108 | 651.0 | 9.6 | 650.2 | 1240.1 |
| MRCI+Q | 2.056 | 603.1 | 2.1 | 598.3 | 7021.6 | |
| 14Σ+ | CASSCF | 2.109 | 631.6 | 9.3 | 634.3 | 15801.2 |
Both structures are quite single reference as shown by the CI vector in Table S5 (ESI†). The first quartet (14Σ+) was not possible to determine at the MRCI level due to the time required to solve the different MRCI states, however, it was still estimated at the CASSCF level, represented by a blue horizontal bar in Fig. 7. The 14Σ+ lies at ∼45 kcal mol−1 and is the second excited state, after the 12Π. The 14Σ+ has two main components, the first by having unpaired electrons on the 2px (N), 6px and 6s of (Lu), and second on the 2py (N), 6py and 6s of (Lu). The Lu–N bond length is elongated to 2.109 Å.
The isomers of the ground state structure were also investigated. For this step, all of the isomers were optimized at CCSD(T), utilizing a DKH3 Hamiltonian, with a triple-ζ basis set. These include: (a) the linear structure, with lutetium as the central atom, S = 1/2, 3/2, and, (b) the bent structure (Cs) also S = 1/2, 3/2 (see Table S6, ESI† for geometry details). There is an interesting trend among the isomers. For the doublets (S = 1/2), the Lu–N is ∼0.3 Å shorter than for the quartet (S = 3/2) counterpart. The state above 14Σ+ is the first state with Cs symmetry (12A′), at 61.1 kcal mol−1 or 21373.5 cm−1. The Cs quartet is 1542.4 cm−1 above the doublet and the Lu–N bond length is elongated by ∼0.2 Å and the Lu–H shortened by 0.22 Å. For the linear structures, here treated as a C2v point group, the doublet and quartet structures are only separated by 600 cm−1. However, the Lu–N bond is still quite elongated by ∼0.3 Å. The N–H bond is again shortened by ∼0.06 Å. The 2S+1Λ spin–orbit states of the X2Σ+ → X2Σ+1/2 and 12Π → 12Π3/2,12Π1/2 were also considered. These two states are displayed in Fig. S1 (ESI†) and their spectroscopic constants, and composition of states are shown in Table S7 (ESI†). The ground state is X2Σ+1/2, followed by excited states at 12Π3/2 and 12Π1/2. The 12Π3/2 and 12Π1/2 are separated by 417.9 cm−1.
3.5 Bond dissociation energies (BDEs) of LuH, LuN and LuNH
Until now, the different states and bond patterns of LuH, LuN, and LuNH have been elucidated. However, it is also important to calculate bond dissociation energies. The dissociation energies of LuH and LuN, along with the different energetics routes for breaking the LuNH bonds with f-ccCA and HEAT are shown in Table 4. The breakdown of the energetics arising from each of the different terms of f-ccCA and HEAT are shown in Tables S8 and S9 (ESI†). For LuH, f-ccCA and HEAT energies are similar, with a difference of only 1.29 kcal mol−1. However, for LuN, the difference in dissociation energy is much larger when comparing HEAT and f-ccCA, at 5.45 kcal mol−1. From Tables S8 and S9 (ESI†), the two main differences from f-ccCA and HEAT arise from the core–valence term and the contribution from the quadruple excitations in HEAT, calculated at the λ-CCSD(T)/double-ζ level. The core–valence correlation for HEAT is extrapolated to the CBS utilizing energies determined at the triple- and -quadruple-ζ basis set level, while for f-ccCA, the core–valence correlation is solely obtained at a double-ζ level. Another contributing factor for the energetic difference is attributed to the full triples contribution at the CBS (from triple- and quadruple-ζ basis sets) limit, with −10.03 kcal mol−1 for HEAT, while f-ccCA does not include full triple corrections, only perturbative triples. Furthermore, the quadruple excitation contribution is quite large at 3.38 kcal mol−1. Even though the LuN ground state is a 11Σ+, it is by no means a single reference state, which requires additional contributions from higher excitations to accurately predict its thermodynamic properties. As the pentuple contributions are quite computationally demanding, as noted earlier, the contribution from the pentuples (i.e., as in s-ccCA) was not calculated, but its value may still be significant, particularly considering the size of the quadruple contribution. For the NH fragment, f-ccCA and HEAT render similar D0 values, with a difference between f-ccCA and HEAT energies of 1.03 kcal mol−1.| f-ccCA D0 | HEAT D0 | |
|---|---|---|
| LuH | 72.96 | 71.67 |
| LuN | 83.95 | 78.50 |
| NH | 77.50 | 78.47 |
| LuNH routes | ||
| (1) LuNH → LuN + H | 106.96 | 111.05 |
| (2) Lu + NH | 112.86 | 111.76 |
| (3) LuNH → Lu + N + H | 190.36 | 190.23 |
| Dissociation energy via route 1 + LuN D0 | 196.81 | 189.55 |
For LuNH, three dissociation pathways were probed (routes (1)–(3)). The first route entailed breaking the N–H bond from LuN (LuN + H). The second route was to break the NH fragment from lutetium (Lu + NH) and the third route was a total atomization scheme, i.e., Lu + N + H. For the first route, f-ccCA and HEAT predictions do not agree energetically, the difference between methods is 4.09 kcal mol−1. It is interesting to note that the quadruples contribution for the λ-CCSDT(Q) from HEAT is −2.91 kcal mol−1, to break the LuN–H bond. However, the core–valence correlation calculated from HEAT is only 0.01 kcal mol−1versus 1.36 kcal mol−1 for f-ccCA, which accounts for a part of the overall energetic differences predicted by the methods. The full triples correction at the triple-ζ level renders 8.74 kcal mol−1. This value is the primary difference between the f-ccCA and HEAT energies. The isolated fragment requires 77.50 and 78.47 kcal mol−1 for f-ccCA and HEAT, respectively, to break the N–H bond. However, when complexed with lutetium, this bond becomes much stronger, i.e. breaking the N–H bond within the complex requires 35.36 and 32.58 kcal mol−1, as predicted by f-ccCA and HEAT respectively.
For route (2), breaking the NH fragment from lutetium (Lu + NH), HEAT predicts the D0 to be 111.76 kcal mol−1, while f-ccCA predicts 112.86 kcal mol−1. When comparing the energy contributions in Tables S8 and S9 (ESI†), the core valence energetic contributions are quite different. For f-ccCA, the contribution is positive, 1.36 kcal mol−1, but for HEAT, it is negative, −0.21 kcal mol−1. For this route, the quadruples contribution is quite small (0.41 kcal mol−1) compared to the contributions for LuN or LuN + H. The full triples correction is much smaller (−0.68 kcal mol−1) than for route (1). For both composite methods, it is easier to break the LuN–H bond, than the Lu–NH bond. However, for f-ccCA, the difference between routes is 6.1 kcal mol−1, while for HEAT, the difference is only 0.71 kcal mol−1. The higher order coupled–cluster correlations (full triples and higher) play a crucial role in obtaining full energetic contributions, for highly multireference ground states such as LuN.
For route (3), for the total atomization approach, both HEAT and f-ccCA result in a very similar D0 (∼190 kcal mol−1). However, if the dissociation energy is calculated via route (1), i.e., summing the atomization of LuN to the value obtained through route (1) (D0 LuNH → LuN + H + D0 LuN), f-ccCA predicts the D0 to be 196.81 kcal mol−1, while HEAT results in an energy of 189.55 kcal mol−1, which is near to the total atomization energy predicted via route (3).
The total atomization energies calculated in route (3) are the common ground when comparing the different dissociation energies.
4 Conclusions
In this project, for the first time (to our knowledge), the elucidation of the electronic structure of multiple building blocks of a lutetium nitride doped network have been described. The PECs of LuH, LuN, and LuNH are described in detail including an accounting for spin–orbit effects. The LuH molecule ground state is a well-separated ground (X1Σ+) and it is formed by pairing two electrons on the 1s orbital of hydrogen and 6s of lutetium. For lutetium hydride, after constructing the Breit–Pauli Hamiltonian, severe mixing of the Ω = 1 states of the 13Π, 13Δ, 13Σ+ was observed, which consequently shifted the first excited state of this complex; now a 13Π0− followed by 13Δ1. In fact, the three components of the 13Δ state are separated by 2138.8 cm−1 at a spin–orbit level.Lutetium nitride is a more complex molecule from the electronic structure standpoint. The ground state is constructed by three different components, which makes single reference methodologies breakdown. The ground state of LuN is also a 11Σ+, which originates from a crossing with a potential ionic channel. The excited states of this molecule are not in the Frank–Condon region, however, herein are still characterized by MRCI + Q, and at spin–orbit level. For LuN, there is a lot less mixing when comparing the composition tables of this molecule versus LuH, and there are multiple crossings.
For LuNH, the ground and two first excited states were investigated. The ground state is a 12Σ+, which also comes from a higher dissociation channel. It is observed that the linear molecule, with the nitrogen as the center atom is the most stable structure, followed by the bent (N–Lu–H) and linear (N–Lu–H).
An in-depth analysis of the different thermochemical routes for bond breaking of the following complexes: LuH, LuN and LuNH bonds was performed with f-ccCA and HEAT schemes. Both f-ccCA and HEAT energetics agree for LuH and NH, but are ∼5.3 kcal mol−1 different for LuN. The HEAT composite scheme considers quadruple excitation contributions through λ-CCSDT(Q), in this case at a double-ζ level that show the importance of these higher-level contributions. In addition, the full triples contribution is quite important, especially for LuN. The HEAT and f-ccCA composite schemes show that LuNH → LuN + H bond requires the least energy to break (route (1)). However, the full triple and λ-CCSDT(Q) energetic contributions are crucial for accurate energetics profiles when breaking the LuN bond. In addition, both methods agree in the energy predictions using the total atomization scheme LuNH → Lu + N + H at 190.36 and 190.23 kcal mol−1 for f-ccCA and HEAT, respectively.
To the best of our knowledge this is the first ab initio study of the different fragments of the nitrogen-doped lutetium hydride network. Herein, the foundations of bond formation of the different molecules that compose the LuNHx lattice, along with bond lengths, harmonic and anharmonic frequencies, and accurate thermodynamic dissociation energies of LuH, LuN and LuNH are provided. This investigation will provide experimentalists with detailed information of the building blocks of a nitrogen-doped lutetium hydride network, enabling a full understanding of its electronic structure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation [grant no CHE-2154526]. This work utilized computational facilities at the Institute for Cyber-Enabled Research (ICER) at Michigan State University as well as via ACCESS (Computational Support via CHE-230100) and the computational resources of the Pittsburgh Supercomputing Center.References
- H. K. Omes, Further experiments with liquid helium. C. On the change of electric resistance of ure metals at very low temperatures etc. IV. The Resistance of Pure Mercury at Helium Temperatures, Commun. Phys. Lab. Univ. Leiden, 1911, 120b Search PubMed.
- D. Castelvecchi, How Would Room-Temperature Superconductors Change science?, Nature, 2023, 621, 18–19 CrossRef CAS PubMed.
- N. Dasenbrock-Gammon, E. Snider, R. McBride, H. Pasan, D. Durkee, N. Khalvashi-Sutter, S. Munasinghe, S. E. Dissanayake, K. V. Lawler, A. Salamat and R. P. Dias, RETRACTED ARTICLE: Evidence of Near-Ambient Superconductivity in a N-Doped Lutetium Hydride, Nature, 2023, 615, 244–250 CrossRef CAS PubMed.
- X. Ming, Y.-J. Zhang, X. Zhu, Q. Li, C. He, Y. Liu, T. Huang, G. Liu, B. Zheng, H. Yang, J. Sun, X. Xi and H.-H. Wen, Absence of Near-Ambient Superconductivity in LuH2 ± xNy, Nature, 2023, 620, 72–77 CrossRef CAS PubMed.
- D. Peng, Q. Zeng, F. Lan, Z. Xing, Y. Ding and H. Mao, The Near-Room-Temperature Upsurge of Electrical Resistivity in Lu–H–N is not Superconductivity, but a Metal-To-Poor-Conductor Transition, Matter Radiat. Extremes, 2023, 8, 58401 CrossRef CAS.
- X. Xing, C. Wang, L. Yu, J. Xu, C. Zhang, M. Zhang, S. Huang, X. Zhang, Y. Liu, B. Yang, X. Chen, Y. Zhang, J. Guo, Z. Shi, Y. Ma, C. Chen and X. Liu, Observation of Non-Superconducting Phase Changes in Nitrogen Doped Lutetium Hydrides, Nat. Commun., 2023, 14, 5991 CrossRef CAS PubMed.
- P. P. Ferreira, L. J. Conway, A. Cucciari, S. Di Cataldo, F. Giannessi, E. Kogler, L. T. F. Eleno, C. J. Pickard, C. Heil and L. Boeri, Search for Ambient Superconductivity in the Lu-N-H System, Nat. Commun., 2023, 14, 5367 CrossRef CAS PubMed.
- C. H. Roman Lucrezi, P. P. Ferreira, M. Aichhorn and C. Heil, Temperature and Quantum Anharmonic Lattice Effects in Lutetium Trihydride: Stability and Superconductivity, Nat. Commum., 2024, 15, 1712 CrossRef PubMed.
- O. Moulding, S. Gallego-Parra, Y. Gao, P. Toulemonde, G. Garbarino, P. De Rango, S. Pairis, P. Giroux and M.-A. Méasson, Pressure Induced Formation of Cubic Lutetium Hydrides Derived from Trigonal LuH3, Phys. Rev. B., 2023, 108, 214505 CrossRef CAS.
- X. Zhao, P. Shan, N. Wang, Y. Li, Y. Xu and J. Cheng, Pressure Tuning of Optical Reflectivity in LuH2, Sci. Bull., 2023, 68, 883–886 CrossRef CAS PubMed.
- Y.-J. Zhang, X. Ming, Q. Li, X. Zhu, B. Zheng, Y. Liu, C. He, H. Yang and H.-H. Wen, Pressure Induced Color Change and Evolution of Metallic Behavior in Nitrogen-Doped Lutetium Hydride, Sci. China: Phys., Mech. Astron., 2023, 66, 287411 CAS.
- S. Cai, J. Guo, H. Shu, L. Yang, P. Wang, Y. Zhou, J. Zhao, J. Han, Q. Wu, W. Yang, T. Xiang, H. Mao and L. Sun, No Evidence of Superconductivity in a Compressed Sample Prepared from Lutetium Foil and H2/N2 Gas Mixture, Matter Radiat. Extremes, 2023, 8, 48001 CrossRef CAS.
- S.-W. Kim, L. J. Conway, C. J. Pickard, G. L. Pascut and B. Monserrat, Microscopic Theory of Colour in Lutetium Hydride, Nat. Commun., 2023, 14, 7360 CrossRef CAS PubMed.
- X. Tao, A. Yang, S. Yang, Y. Quan and P. Zhang, Leading Components and Pressure-Induced Color Changes in N-Doped Lutetium Hydride, Sci. Bull., 2023, 68, 1372–1378 CrossRef CAS PubMed.
- R. Lv, W. Tu, D. Shao, Y. Sun and W. Lu, Physical Origin of Color Changes in Lutetium Hydride Under Pressure, Chin. Phys. Lett., 2023, 40, 117401 CrossRef.
- P. Li, J. Bi, S. Zhang, R. Cai, G. Su, F. Qi, R. Zhang, Z. Wei and Y. Cao, Transformation of Hexagonal Lu to Cubic LuH 2 + x Single-Crystalline Films, Chin. Phys. Lett., 2023, 40, 087401 CrossRef.
- P. Shan, N. Wang, X. Zheng, Q. Qiu, Y. Peng and J. Cheng, Pressure-Induced Color Change in the Lutetium Dihydride LuH2, Chin. Phys. Lett., 2023, 40, 046101 CrossRef.
- Z. Liu, Y. Zhang, S. Huang, X. Ming, Q. Li, C. Pan, Y. Dai, X. Zhou, X. Zhu, H. Yan and H.-H. Wen, Pressure-Induced Color Change Arising from Transformation Between Intra- and Inter-Band Transitions in LuH2 ± xNy, Sci. China: Phys., Mech. Astron., 2024, 67, 227411 CAS.
- J. Guo, S. Cai, D. Wang, H. Shu, L. Yang, P. Wang, W. Wang, H. Tian, H. Yang, Y. Zhou, J. Zhao, J. Han, J. L. Q. Wu, Y. Ding, W. Yang, T. Xiang, H. Mao and L. Sun, Robust Magnetism Against Pressure in Non-Superconducting Samples Prepared from Lutetium Foil and H2/N2 Gas Mixture, Chin. Phys. Lett., 2023, 40, 097401 CrossRef.
- Đ. Dangić, P. Garcia-Goiricelaya, Y.-W. Fang, J. Ibañez-Azpiroz and I. Errea, Ab Initio Study of the Structural, Vibrational, and Optical Properties of Potential Parent Structures of Nitrogen-Doped Lutetium Hydride, Phys. Rev. B, 2023, 108, 064517 CrossRef.
- N. Wang, J. Hou, Z. Liu, P. Shan, C. Chai, S. Jin, X. Wang, Y. Long, Y. Liu, H. Zhang, X. Dong and J. Cheng, Percolation-Induced Resistivity Drop in Cold-Pressed LuH2, Sci. China: Phys., Mech. Astron., 2024, 66, 297412 Search PubMed.
- Z. Li, X. He, C. Zhang, K. Lu, B. Min, J. Zhang, S. Zhang, J. Zhao, L. Shi, Y. Peng, S. Feng, Z. Deng, J. Song, Q. Liu, X. Wang, R. Yu, L. Wang, Y. Li, J. D. Bass, V. Prakapenka, S. Chariton, H. Liu and C. Jin, Superconductivity Above 70 K Observed in Lutetium Polyhydrides, Sci. China: Phys., Mech. Astron., 2023, 66, 267411 CAS.
- M. Liu, X. Liu, J. Li, J. Liu, Y. Sun, X.-Q. Chen and P. Liu, Parent Structures of Near-Ambient Nitrogen-Doped Lutetium Hydride Superconductor, Phys. Rev. B, 2023, 108, L020102 CrossRef CAS.
- S. Zhang, J. Bi, R. Zhang, P. Li, F. Qi, Z. Wei and Y. Cao, Electronic and Magnetic Properties of Lu and LuH2, AIP Adv., 2023, 13, 65117 CrossRef CAS.
- F. Xie, T. Lu, Z. Yu, Y. Wang, Z. Wang, S. Meng and M. Liu, Lu–H–N Phase Diagram from First-Principles Calculations, Chin. Phys. Lett., 2023, 40, 057401 CrossRef.
- Y. Sun, F. Zhang, S. Wu, V. Antropov and K.-M. Ho, Effect of Nitrogen Doping and Pressure on the Stability of LuH3, Phys. Rev. B, 2023, 108, L020101 CrossRef CAS.
- K. P. Hilleke, X. Wang, D. Luo, N. Geng, B. Wang, F. Belli and E. Zurek, Structure, Stability, and Superconductivity of N-Doped Lutetium Hydrides at kbar Pressures, Phys. Rev. B, 2023, 108, 014511 CrossRef CAS.
- Z. Huo, D. Duan, T. Ma, Z. Zhang, Q. Jiang, D. An, H. Song, F. Tian and T. Cui, First-Principles Study on the Conventional Superconductivity of N-Doped fcc -LuH3, Matter Radiat. Extremes, 2023, 8, 38402 CrossRef CAS.
- C. Effantin and J. D’Incan, Spectre Électronique de la Molécule LuH, Can. J. Phys., 1973, 51, 1394–1402 CrossRef CAS.
- X. Wang, L. Andrews, I. Infante and L. Gagliardi, Matrix Infrared Spectroscopic and Computational Investigation of Late Lanthanide Metal Hydride Species MHx (H2)y (M = Tb–Lu, x = 1–4, y = 0–3), J. Phys. Chem. A, 2009, 113, 12566–12572 CrossRef CAS PubMed.
- D. A. Pantazis and F. Neese, All-Electron Scalar Relativistic Basis Sets for the Lanthanides, J. Chem. Theory Comput., 2009, 5, 2229–2238 CrossRef CAS PubMed.
- A. F. Jalbout, X.-H. Li and H. Abou-Rachid, Analytical Potential Energy Functions and Theoretical Spectroscopic Constants for MX/MX – (M = Ge, Sn, Pb; X = O, S, Se, Te, Po) and LuA (A = H, F) Systems: Density Functional Theory Calculations, Int. J. Quantum Chem., 2007, 107, 522–539 CrossRef CAS.
- J. K. Laerdahl, K. Fægri, L. Visscher and T. Saue, A Fully Relativistic Dirac–Hartree–Fock and Second-Order Moller–Plesset Study of the Lanthanide and Actinide Contraction, J. Chem. Phys., 1998, 109, 10806–10817 CrossRef CAS.
- M. Dolg and H. Stoll, Pseudopotential Study of the Rare Earth Monohydrides, Monoxides and Monofluorides, Theor. Chim. Acta, 1989, 75, 369–387 CrossRef CAS.
- X. Cao and M. Dolg, Valence Basis Sets for Relativistic Energy-Consistent Small-Core Lanthanide Pseudopotentials, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS.
- J. Assaf, F. El Haj Hassan, É. C. M. Nascimento and A. Haydar, Theoretical Study of LuH Molecule: Potential Energy Curves, Spectroscopic Constants and Spin-orbit Couplings, Comput. Theor. Chem., 2018, 1128, 31–41 CrossRef CAS.
- A. Oualdine, A. Bentouaf, A. Chebli, B. Nouamane, A. Z. Bouyakoub and B. Aïssa, Structural, Elastic, and Electronic Properties of CeN and LuN Using: Ab Initio Study, J. Supercond. Novel Magn., 2018, 31, 3323–3330 CrossRef CAS.
- N. B. Yurdaşan, S. E. Gülebağlan, A. Y. Tunali and G. B. Akyüz, DFT Investigations on Structural, Electronic and Vibrational Properties of LuN Under High Pressure, Yuzuncu Yil Univ. J. Inst. Nat. Appl. Sci., 2021, 26, 45–52 Search PubMed.
- S. Devese, K. Van Koughnet, R. G. Buckley, F. Natali, P. P. Murmu, E.-M. Anton, B. J. Ruck and W. F. Holmes-Hewett, Probing the Defect States of LuN1−δ: An Experimental and Computational Study, AIP Adv., 2022, 12, 35108 CrossRef.
- N. M. S. Almeida, T. R. L. Melin, S. C. North, B. K. Welch and A. K. Wilson, Ab Initio Composite Strategies and Multireference Approaches for Lanthanide Sulfides and Selenides, J. Chem. Phys., 2022, 157, 24105 CrossRef PubMed.
- A. Tajti, P. G. Szalay, A. G. Császár, M. Kállay, J. Gauss, E. F. Valeev, B. A. Flowers, J. Vázquez and J. F. Stanton, HEAT: High Accuracy Extrapolated Ab Initio Thermochemistry, J. Chem. Phys., 2004, 121, 11599–11613 CrossRef CAS PubMed.
- Y. J. Bomble, J. Vázquez, M. Kállay, C. Michauk, P. G. Szalay, A. G. Császár, J. Gauss and J. F. Stanton, High-Accuracy Extrapolated Ab Initio Thermochemistry. II. Minor Improvements to the Protocol and a Vital Simplification, J. Chem. Phys., 2006, 125, 064108 CrossRef PubMed.
- M. E. Harding, J. Vázquez, B. Ruscic, A. K. Wilson, J. Gauss and J. F. Stanton, High-Accuracy Extrapolated Ab Initio Thermochemistry. III. Additional Improvements and Overview, J. Chem. Phys., 2008, 128, 114111 CrossRef PubMed.
- J. H. Thorpe, C. A. Lopez, T. L. Nguyen, J. H. Baraban, D. H. Bross, B. Ruscic and J. F. Stanton, High-Accuracy Extrapolated Ab Initio Thermochemistry. IV. A Modified Recipe for Computational Efficiency, J. Chem. Phys., 2019, 150, 224102 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro Quantum Chemistry Package, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- Q. Lu and K. A. Peterson, Correlation Consistent Basis Sets for Lanthanides: The Atoms La–Lu, J. Chem. Phys., 2016, 145, 054111 CrossRef PubMed.
- N. B. Balabanov and K. A. Peterson, Systematically Convergent Basis Sets for Transition Metals. I. All-electron Correlation Consistent Basis Sets for the 3d Elements Sc–Zn, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed.
- X. Cao and M. Dolg, Segmented Contraction Scheme for Small-Core Lanthanide Pseudopotential Basis Sets, J. Mol. Struct., 2002, 581, 139–147 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- N. M. S. Almeida, T. R. L. Melin and A. K. Wilson, Multireference Calculations on the Ground and Lowest Excited States and Dissociation Energy of LuF, J. Chem. Phys., 2021, 154, 244304 CrossRef CAS PubMed.
- J. W. Cooley, An Improved Eigenvalue Corrector Formula for Solving the Schrödinger Equation for Central Fields, Math. Comput., 1961, 15, 363 Search PubMed.
- B. Numerov, Note on the Numerical Integration of d2x/dt2 = f(x,t), Astron. Nachr., 1927, 230, 359 CrossRef.
- J. L. Dunham, The Energy Levels of a Rotating Vibrator, Phys. Rev., 1932, 41, 721–731 CrossRef CAS.
- B. K. Welch, N. M. S. Almeida and A. K. Wilson, Super ccCA (s-ccCA): An Approach for Accurate Transition Metal Thermochemistry, Mol. Phys., 2021, 119, e1963001 CrossRef.
- P. Patel, T. R. L. Melin, S. C. North and A. K. Wilson, in Ab initio composite methodologies: Their significance for the chemistry community, ed. C. C. Dixon, Elsevier, 2021, vol. 17, pp. 113–161 Search PubMed.
- C. Peterson, D. A. Penchoff and A. K. Wilson, Ab Initio Approaches for the Determination of Heavy Element Energetics: Ionization Energies of Trivalent Lanthanides (Ln = La–Eu), J. Chem. Phys., 2015, 143, 194109 CrossRef PubMed.
- C. Peterson, D. A. Penchoff and A. K. Wilson, Prediction of Thermochemical Properties Across the Periodic Table: A Review of the correlation consistent Composite Approach (ccCA) Strategies and Applications, Annu. Rep. Comput. Chem., 2016, 12, 3–45 Search PubMed.
- M. L. Laury, N. J. DeYonker, W. Jiang and A. K. Wilson, A Pseudopotential-Based Composite Method: The Relativistic Pseudopotential Correlation Consistent Composite Approach for Molecules Containing 4d Transition Metals (Y-Cd), J. Chem. Phys., 2011, 135, 214103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00533c |
| This journal is © the Owner Societies 2024 |