
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the quantum secrets of triel metal triangles: a tale of stability, aromaticity, and relativistic effects†
Sílvia
Escayola
 ab,
Elisa
Jimenez-Izal
bc,
Eduard
Matito
bd,
Jesus M.
Ugalde
bc,
Rafael
Grande-Aztatzi
*be and
Jose M.
Mercero
*bc
ab,
Elisa
Jimenez-Izal
bc,
Eduard
Matito
bd,
Jesus M.
Ugalde
bc,
Rafael
Grande-Aztatzi
*be and
Jose M.
Mercero
*bc
aInstitute of Computational Chemistry and Catalysis and Department of Chemistry, University of Girona, C/M. Aurèlia Capmany, 69, 17003 Girona, Catalonia, Spain
bDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain. E-mail: aztatzi26@gmail.com; jm.mercero@ehu.eus
cKimika Fakultatea, Euskal Herriko Unibertsitatea (UPV/EHU), P.K. 1072, 20080 Donostia, Euskadi, Spain
dIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Euskadi, Spain
eEscuela de Ingeniería y Ciencias, Tecnológico de Monterrey, Av. Eugenio Garza Sada 2501, 64849 Monterrey, Nuevo León, Mexico
First published on 26th March 2024
Abstract
Low lying electronic states of Al3−, Ga3−, In3−, and Tl3− have been characterized using high level multiconfigurational quasi degenerate perturbation theory on the multiconfigurational self-consistent field. Among these species, the singlet  states emerge as the predominant energy minima, displaying remarkable stability. However, within the Tl3− series, our investigation leads to the identification of the high-spin
states emerge as the predominant energy minima, displaying remarkable stability. However, within the Tl3− series, our investigation leads to the identification of the high-spin  , as the most stable spin state, a result corroborated by previous experimental detection via photoelectron spectroscopy. Similarly, we have also identified the singlet state of In3− as the signal detected previously experimentally. By applying Mandado's rules and an array of aromaticity indicators, it is conclusively demonstrated that both the singlet and quintet states exhibit multiple-fold aromaticity, while the triplets exhibit conflicting aromaticity. Furthermore, this investigation highlights the significant impact of relativistic effects, as they enhance the stability of the
, as the most stable spin state, a result corroborated by previous experimental detection via photoelectron spectroscopy. Similarly, we have also identified the singlet state of In3− as the signal detected previously experimentally. By applying Mandado's rules and an array of aromaticity indicators, it is conclusively demonstrated that both the singlet and quintet states exhibit multiple-fold aromaticity, while the triplets exhibit conflicting aromaticity. Furthermore, this investigation highlights the significant impact of relativistic effects, as they enhance the stability of the  state relative to its singlet counterpart. These findings shed new light on the electronic structures and properties of these ions, offering valuable insights into their chemical behavior and potential applications.
state relative to its singlet counterpart. These findings shed new light on the electronic structures and properties of these ions, offering valuable insights into their chemical behavior and potential applications.
1 Introduction
Al3 triangles have widely been studied theoretically and experimentally, in both the ground state and excited states.1–10 However, its anionic form Al3− has been studied to a lesser extent. The first study of Al3− we have found in the literature was conducted by H. Basch.1 He calculated the electronic properties of the anion using MCSCF methods, and a C2v symmetry triangle was reported as the ground state. Later, Taylor et al. and Ganteför et al. studied Al3− using photoelectron spectroscopy.11,12 In 1995, Calaminici et al.2 reported a D3h triangle as the singlet ground state using DFT. In 1998, Baeck and Bartlet3 characterized the low-lying electronic states of Al3− using the CCSD(T), confirming an equilateral triangle as the ground state .
.
A few years later, Kuznetsov and Boldyrev13 extended the all-metal aromaticity concept to the Al3− and Ga3− anions, describing both triangles as π-aromatic systems. Later on, Zhan et al. pointed out that the Al3− triangle was not just π-aromatic,14 and introduced the idea of multiple-fold aromaticity, showing a two-fold aromatic system, π- and σ-aromatic. In 2009, Villalta and Leopold,5 experimentally characterized both the ground and excited states of Al3 and Al3− trimers, confirming the  state as the ground state. These experimental results were rationalized with single-reference DFT and coupled cluster methods by Miller et al.4
state as the ground state. These experimental results were rationalized with single-reference DFT and coupled cluster methods by Miller et al.4
In 2015, we described the lowest-lying electronic states of the Al3− triangle using DFT and multiconfigurational high-level calculations supplemented with quasi-degenerated perturbation theory.15 We showed that not only the ground state, but the triplet and quintet excited states were aromatic. Recently, Al3− aromaticity was analyzed by means of the natural orbital functional (NOF) theory.16
Ga3, In3, and Tl3 trimers have also been previoulsy studied. Pöttgen and coworkers have characterized many crystal structures where Al3,17 Ga3,18–21 and In3,22–24 triangles are present, and there are many other similar structures reported in the literature.25–27 Some molecules where Ga3 triangles are identified have also been synthesized.28–31 Theoretical investigations are also available for Ga3 and In3 in the literature.32–38 Among all these studies, only a few have focused on triangular anions. In 1990, Meier et al.33 studied Ga3− using MRD-CI techniques; few years later, in 1994, Cha et al.39 obtained the photoelectron spectra for Ga3−. In 2002, Kuznetsov and Boldyrev,13 described the aromaticity of the Ga3−. There is a limited number of research works dedicated to the investigation of In3−. Gausa et al. performed photoelectron spectroscopy experiments for Inn− and Tln− clusters.36 The studies regarding the Tl3 are scarce, including those by Gausa et al. and Vijayakumar et al.,40 who used MCSCF methods to characterize Tl3+/− molecules, while Kang41 described the Tl3+ triangle in 1993, and Tsipis et al. discussed NICS evolution in different compounds including the Tl3−.42
The manuscript at hand employees multiconfigurational self-consistent methods to investigate X3− triangles, (where X= Al, Ga, In, and Tl), exploring several properties of the lowest-energy spin states. The study establishes a connection between the experimentally measured photoelectron spectra and the  state of In3− and
state of In3− and  state of Tl3−. Additionally, the aromaticity of the complexes is analyzed using the multicenter index (MCI) and its π-fraction (MCIπ) across different electronic configurations to demonstrate the multiple-fold aromaticity determined by the independent delocalized σ and π bonding systems. The study also employs delocalization indexes43,44 (DI) and adaptive natural density partitioning analysis45 (AdNDP) to obtain an alternative perspective and quantify the aromaticity, and finally, a magnetic criteria is also used, the iso-chemical-shielding surfaces46,47 (ICSS) method.
state of Tl3−. Additionally, the aromaticity of the complexes is analyzed using the multicenter index (MCI) and its π-fraction (MCIπ) across different electronic configurations to demonstrate the multiple-fold aromaticity determined by the independent delocalized σ and π bonding systems. The study also employs delocalization indexes43,44 (DI) and adaptive natural density partitioning analysis45 (AdNDP) to obtain an alternative perspective and quantify the aromaticity, and finally, a magnetic criteria is also used, the iso-chemical-shielding surfaces46,47 (ICSS) method.
2 Methods
State-specific multiconfigurational self consistent field (MCSCF)48 complemented by multiconfigurational quasi-degenerate perturbation (MCQDPT)49 methods have been employed to characterize the electronic structure of the different X3− (X= Al, Ga, In and Tl) triangles by using the Karlsruhe Def2-TZVPPD50,51 basis set wich includes pseudopotentials for In52 and Tl.53 Inconsistencies caused by the so-called intruder states, which appear when the perturbation expansion of the reference MCSCF wave function has vanishingly small energy denominators, were remedied by shifting them by 0.02 a.u., as recommended earlier.54 The vibrational frequencies were also calculated at the MCSCF level of theory. The ten valence electrons of the system were included in the active space, which were allowed to distribute themselves in twelve molecular orbitals (MO) (full active space). The selected twelve orbitals are the ones depicted in Fig. 1 and encompass the different-fold aromatic systems as described with elegant simplicity by Dixon et al.14 spin–orbit coupling where estimated using the Pauli–Breit approximation for the coupling Hamiltonian, at the aforementioned level of theory as implemented in the GAMESS US program.55 | ||
| Fig. 1 MCSCF(10,12) natural orbitals (NOs) of the Al3− grouped in the four aromatic independent systems, Ψσ, Ψπ, Ψr and Ψt, σ, π, radial and tangential, respectively. The orbitals for the singlet and the quintet are similar, while, despite of the symmetry change to C2v, the triplet structures triangles maintain a similar form (C2v symmetry equivalent labels are given in parentheses). | ||
The aromaticity of the characterized triangular systems has been assessed by means of different analysis tools, multicenter index (MCI),56 adaptive natural density partitioning (AdNDP)45 and the iso-chemical-shielding surfaces (ICSS) method.46,47 Although these tools are all used to measure the aromaticity in molecules, they differ in their methods. MCI quantifies the degree of magnetic circuit delocalization in a molecule by integrating the magnetic flux density along a closed loop that passes through the aromatic ring, AdNDP uses a natural orbital-based partitioning scheme to analyze electron density by partitioning it into natural orbitals. In conclusion, these methods can be used together to provide a more comprehensive understanding of the electron delocalization in a molecule. MCI and DI have been calculated using the ESI-3D program,44,57 and were computed for all the isomers using their corresponding MCSCF (10,12) wave functions. The atomic overlap matrices, required by ESI-3D to calculate the MCIs, were obtained from the APOST-3D58 software using the topological fuzzy Voronoi (TVFC) cell partition.59–61 To account for the numerical errors in APOST-3D and ESI-3D, we verified that the number of electrons was equal to the sum of all the electron populations of the molecule, the error being always below 0.0016 a.u.
AdNDP and ICSS analysis were carried out at the CAM-B3LYP/Def2-TZVPP level of theory. ICSS method was used via Multiwfn code.62
3 Results
In this manuscript, we will explore the various properties of the lowest-energy spin states, namely, , 3B2 and
, 3B2 and  , of the X3− (where X = Al, Ga, In, and Tl) triangles. The geometries of these structures were optimized at the MCSCF (10,12) level, and their energies were refined at the MCQDPT level of theory. Our results exhibit a substantial multireference character for these structures. The
, of the X3− (where X = Al, Ga, In, and Tl) triangles. The geometries of these structures were optimized at the MCSCF (10,12) level, and their energies were refined at the MCQDPT level of theory. Our results exhibit a substantial multireference character for these structures. The  ground states are mainly dominated by the
ground states are mainly dominated by the  single reference state (with a configuration interaction coefficient (CIC) of around 0.9, see Table S3 in ESI† for the main CICs of the different molecules states), with significant contributions from each of the two configurations featuring degenerate orbitals
single reference state (with a configuration interaction coefficient (CIC) of around 0.9, see Table S3 in ESI† for the main CICs of the different molecules states), with significant contributions from each of the two configurations featuring degenerate orbitals  and
and  , both with identical CIC coefficients (note that the other six electrons are in the Ψσ system, see Fig. 1). The 3B2 states main configuration is the (b1)2(a1)1(b2)1 with a CIC around 0.88. Lastly, the quintets exhibit the smallest multiconfigurational character, with the
, both with identical CIC coefficients (note that the other six electrons are in the Ψσ system, see Fig. 1). The 3B2 states main configuration is the (b1)2(a1)1(b2)1 with a CIC around 0.88. Lastly, the quintets exhibit the smallest multiconfigurational character, with the  configuration being dominant, with a CIC around 0.92.
configuration being dominant, with a CIC around 0.92.
As previously reported in the literature,63,64 the aluminum trimers possess four independent MO sets derived from the s-type and p-type atomic orbitals (AO) (Fig. 1). The Ψσ set is formed by the three s-AOs, the three pz-AOs form the Ψπ, the p orbitals oriented in the radial direction form the Ψr (radial) set, and p-orbitals oriented in the tangential direction the Ψt (tangential) (see Fig. 1). These group-13 X3− triangles possess a valence shell of ten electrons, six of which are located in the Ψσ orbitals, and the remaining four are arranged in four orbitals with similar energies. We observed the same MO sets in all the complexes studied.
Table 1 presents a summary of the geometrical, energetic, and aromaticity properties obtained for the lowest-lying energy spin-states of the X3− (X = Al, Ga, In and Tl) triangles. The 3A1, 3A2, and 3B1 states were also characterized, but they were found to be higher in energy than the 3B2, and in the interest of brevity, we will focus only on the lowest state, although the details of the 3A1, 3A2, and 3B1 states can be found in the ESI† (see Table S1).
| M | Symm. | ΔE | R 1,21,3 | R 2,3 | MCI | MCIπ | δ(X3−) | δ 1,31,2 | δ 2,3 |
|---|---|---|---|---|---|---|---|---|---|
| Al3− | |||||||||

|
D 3h | 0.0 | 2.558 | 0.48 | 0.23 | 3.11 | 1.07 | ||
| 3B2 | C 2v | 7.36 | 2.543 | 2.762 | 0.35 | 0.22 | 2.95 | 1.00 | 0.92 |

|
D 3h | 12.18 | 2.758 | 0.12 | 0.03 | 2.46 | 0.82 | ||
| Ga3− | |||||||||

|
D 3h | 0.0 | 2.576 | 0.48 | 0.24 | 3.01 | 1.00 | ||
| 3B2 | C 2v | 7.04 | 2.552 | 2.835 | 0.35 | 0.23 | 2.81 | 1.05 | 0.72 |
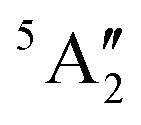
|
D 3h | 11.93 | 2.777 | 0.11 | 0.03 | 2.29 | 0.80 | ||
| In3− | |||||||||

|
D 3h | 0.0 | 2.979 | 0.47 | 0.22 | 2.86 | 0.96 | ||
| 3B2 | C 2v | 2.54 | 2.931 | 3.326 | 0.32 | 0.22 | 2.71 | 1.01 | 0.69 |

|
D 3h | 3.20 | 3.147 | 0.10 | 0.03 | 2.35 | 0.78 | ||
| Tl3− | |||||||||

|
D 3h | 0.0 | 3.129 | 0.46 | 0.22 | 2.68 | 0.89 | ||
| 3B2 | C 2v | −0.71 | 3.049 | 3.441 | 0.29 | 0.22 | 2.56 | 0.97 | 0.63 |

|
D 3h | −2.94 | 3.263 | 0.08 | 0.03 | 2.30 | 0.77 | ||
Upon analyzing the outcomes, it is evident that Al3− and Ga3− not only have a similar size but also exhibit comparable energy differences between different electronic states. Specifically, there is a difference of approximately 7 kcal mol−1 in energy for the 3B2 state and 12 kcal mol−1 for the  state, with respect to the singlets. Conversely, the In3− ion is found to be significantly larger in size, and the energy differences between electronic states are reduced to 2.54 and 3.20 kcal mol−1 for the triplet and quintet states with respect to the singlet. Importantly, for Tl3−, the picture changes, with the quintet being the most stable electronic state followed by the triplet and the singlet, which are around 2.2 and 2.9 kcal mol−1 higher in energy, respectively. Additionally, spin–orbit couplings were measured and found to be small, smaller than 5 cm−1.
state, with respect to the singlets. Conversely, the In3− ion is found to be significantly larger in size, and the energy differences between electronic states are reduced to 2.54 and 3.20 kcal mol−1 for the triplet and quintet states with respect to the singlet. Importantly, for Tl3−, the picture changes, with the quintet being the most stable electronic state followed by the triplet and the singlet, which are around 2.2 and 2.9 kcal mol−1 higher in energy, respectively. Additionally, spin–orbit couplings were measured and found to be small, smaller than 5 cm−1.
Intrigued by the fact that the quintet spin state is the most stable electronic state of Tl3−, vertical detachment energy (VDE) energy calculations were performed to compare them with the photoelectron spectrum reported by Gausa et al.36 Such spectrum exhibited three primary signals, one at 1.5 eV, another at 1.7 eV, and the third around 2.2 eV. We conducted MCQDPT VDE calculations beginning from the 5A1 state, which yielded two degenerated VDEs of 1.30 eV, corresponding to detachment of the e′ electrons, another of 1.66 eV for the detachment of the  electron, and the fourth of 1.77 eV for the
electron, and the fourth of 1.77 eV for the  electron. We also performed partial third-order quasiparticle theory65 (P3) electron propagator calculations (as implemented in Gaussian 1666), which resulted in VDEs of 1.52, 1.76, and 2.12 eV for the quintet, corresponding to e′,
electron. We also performed partial third-order quasiparticle theory65 (P3) electron propagator calculations (as implemented in Gaussian 1666), which resulted in VDEs of 1.52, 1.76, and 2.12 eV for the quintet, corresponding to e′,  , and
, and  electron detachments, respectively. Based on our calculations, we can conclude that the experimentally detected signal corresponds to the quintet electronic state of the Tl3− molecule.
electron detachments, respectively. Based on our calculations, we can conclude that the experimentally detected signal corresponds to the quintet electronic state of the Tl3− molecule.
We also calculated the VDE's for In3− and compared it to the photoelectron spectrum measured by Gausa et al.36 The experimental spectrum exhibited two primary signals, one around 1.65 eV and the other around 1.85 eV. Our MCQDPT calculations resulted in VDEs of 1.70 eV and 1.74 eV, which are consistent with the experimentally measured signals. Additionally, using the Partial Third-order quasiparticle theory65 (P3) electron propagator, we obtained VDEs of 1.69 eV and 1.85 eV, in excellent agreement with the experimental signals. Therefore, it is inferred that the detected signals correspond to the  electronic state of In3−.
electronic state of In3−.
The results for Al3− and Ga3− are consistent with previous theoretical and experimental findings. Villalta et al. experimentally identified a triplet excited state with an obtuse (C2v symmetry) triangle geometry, 3B2 electronic configuration and 9.43 kcal mol−1 higher in energy than the singlet state. Two sides of the triangle have equal lengths, similar to those of the ground state, while the third side is 0.187 Å larger. Our high-level ab initio calculations, as shown in Table 1, are in excellent agreement with their experimental results.5
Note that our calculations indicate highly correlated electronic structures for all the systems considered. However, there is excellent agreement between our results and those obtained from DFT/CCSD(T)4 and CCSD(T)3 calculations, as well as with experimental counterparts.5
It is noteworthy that the quintet state is the most stable electronic state for Tl3−, which is in contrast to the well-studied case of Al3−, where the singlet state is the most stable and exhibits the highest aromaticity when compared with the 3B2 and  states. We have analyzed the aromaticity of Tl3− and compared it with that of the Al3− triangle, to unveil its effect on the stability of the Tl3−.
states. We have analyzed the aromaticity of Tl3− and compared it with that of the Al3− triangle, to unveil its effect on the stability of the Tl3−.
Aromaticity is a concept that lacks a precise definition and does not correspond to any measurable physical property. As a result, it is difficult to identify a specific property that can determine aromaticity.67–69 To address this challenge, numerous indicators have been developed, including the multicenter index (MCI)56 and the Iring70 multicenter index, which are among the most reliable indicators for small systems,71 and are also valid for analyzing metalloaromaticity.72 For triangular systems, it is important to note that the MCI and Iring values are equivalent. In such cases, the MCI and Iring values measure the degree of simultaneous electron sharing between different centers and can be used to estimate aromaticity.
To compare the aromaticity of different rings, MCI value needs to be normalized to the number of atoms (n), MCI1/n, as MCI is known to be dependent on ring size.73,74 The normalized MCI1/3 value for benzene is 0.65 e, (obtained using CAM-B3LYP/6-311G(d,p)75). For Al3−, the normalized MCI value is larger, 0.85 e, while the inclusion of electron correlation reduces the MCI1/3 value to 0.79. Therefore, it is clear that these triangles are aromatic. As we will be working with molecules of the same size, we will focus on the MCI indices, see Table 1. When analyzing them, we observe large values for both the singlet and triplets, whereas the MCI values of the quintets are comparatively lower but not negligible, i.e., MCI1/3 are 0.49 and 0.43 for Al3− and Tl3− respectively.
To better understand the origin of aromaticity, we looked at the π and σ-aromaticities separately, (see MCIπ in Table 1, and note that MCIσT = MCI − MCIπ). While the origin of the π-aromaticity is directly linked with the Ψπ MOs set, the σT-aromaticity (σ total aromaticity) can arise from the other three different sets, Ψσ, Ψr, and Ψt, shown in Fig. 1. We found that in the  ground states, MCIσT was slightly larger than MCIπ. In the 3B2 states, an electron was excited from the radial
ground states, MCIσT was slightly larger than MCIπ. In the 3B2 states, an electron was excited from the radial  NO, to the tangential b2, resulting in a reduction of MCIσT, with values ranging from 0.07 e for Tl3− to 0.13 e for Al3−. In the quintets, another electron is excited from the π
NO, to the tangential b2, resulting in a reduction of MCIσT, with values ranging from 0.07 e for Tl3− to 0.13 e for Al3−. In the quintets, another electron is excited from the π  NO to the second degenerate tangential NO, leaving the
NO to the second degenerate tangential NO, leaving the  ,
,  and both e′ MOs singly occupied. As a result, both MCIπ and MCIσT were reduced (see Table 1).
and both e′ MOs singly occupied. As a result, both MCIπ and MCIσT were reduced (see Table 1).
Together with the MCI, DI can help us construct a vision of the electronic structure of the system. In Table 1, the total DI δ(X3−) are shown. These numbers give us an estimation of the number of electron pairs delocalized in the system. According to these indexes, the delocalization is significant, very large in the singlets, and smaller for the triplets and quintets. The δ(X3−) indexes for the quintets are noteworthy, ranging from 2.46 for Al3− to 2.30 for the Tl3−. For the D3h symmetry states, the properties of the three bonds are equal, so are the atom-pair DI, however for the C2v symmetry triplets, we have two long and one short bonds with different atom-pair DI as it can be seen in Table 1.
In order to gain a deeper understanding of the origin of the DI, we have separated the contribution of each molecular orbital sets (described in Fig. 1) to the atom-pair DI. The resulting figures are collected in Table 2 (note that for the sake of brevity the two extreme cases are shown, Ga3− and In3− data is summarized in the ESI,† see Table S2). An examination of the  DIs, describe that 1.5 electron-pairs (0.48 × 3) are delocalized in the Ψσ set, alongside two delocalized electron pairs one in the π- and the second in the r-set. The main difference for the Tl3− is the reduction of the delocalization of the Ψσ set. In the context of triplets, the delocalization of the Ψσ and Ψπ sets is very similar to that of the singlet states, while the delocalization of the radial set reduces to 0.24 electron-pairs with a simultaneous increase in the delocalization of the Ψt-set. This behavior is what we could expect since an electron from the Ψr-set is excited to one of the Ψt sets. Finally, in the
DIs, describe that 1.5 electron-pairs (0.48 × 3) are delocalized in the Ψσ set, alongside two delocalized electron pairs one in the π- and the second in the r-set. The main difference for the Tl3− is the reduction of the delocalization of the Ψσ set. In the context of triplets, the delocalization of the Ψσ and Ψπ sets is very similar to that of the singlet states, while the delocalization of the radial set reduces to 0.24 electron-pairs with a simultaneous increase in the delocalization of the Ψt-set. This behavior is what we could expect since an electron from the Ψr-set is excited to one of the Ψt sets. Finally, in the  states, Ψπ-delocalization is also reduced and Ψt DI increases again as a consequence of the excitation of one of the π-electrons to the second Ψt tangential set.
states, Ψπ-delocalization is also reduced and Ψt DI increases again as a consequence of the excitation of one of the π-electrons to the second Ψt tangential set.
| Al3− | Tl3− | |||||
|---|---|---|---|---|---|---|
| MO sets |
|
3B2 |
|
|
3B2 |
|
| Ψ σ | 0.48 | 0.52/0.46 | 0.40 | 0.21 | 0.27/0.15 | 0.20 |
| Ψ π | 0.33 | 0.29/0.38 | 0.16 | 0.32 | 0.32/0.29 | 0.17 |
| Ψ r | 0.30 | 0.08/0.08 | 0.06 | 0.30 | 0.10/0.11 | 0.10 |
| Ψ t | −0.02 | 0.10/0.03 | 0.20 | 0.02 | 0.17/0.03 | 0.24 |




The adaptive natural density partitioning analysis (AdNDP) method45 has also been applied to our molecules. AdNDP localizes the valence electrons in 1-center 2-electron bonds (1c–2e), 2-center 2-electron bonds (2c–2e) and n-center 2-electron bonds (nc–2e). For the singlet states, AdNDP produces three possible bonding pictures, as shown in Fig. 2. The first one consists of three lone pairs (occupation numbers, ON = 1.78 |e|), and two 3c–2e bonds (ON = 1.99 |e|) of σ and π nature. An analogous picture is obtained instead by localizing the electronic density into 2c–2e bonds (ON = 1.99) along with the same two 3c–2e bonds (ON = 1.99 |e|). Lastly, the electronic density can be fully delocalized into 3c–2e bonds (ON = 1.99 |e|) (see Fig. 2C). These five 3c–2e AdNDP bonds and the  , e′, e′ (of the Ψσ set), the
, e′, e′ (of the Ψσ set), the  (of the Ψπ set) and
(of the Ψπ set) and  (of the Ψr set) natural orbitals shown in Fig. 1 are similar in shape. The latter description is in agreement with the high delocalization found in these molecules. The co-existence of the two alternative localization schemes (A and B in Fig. 2) and the fully delocalized scheme (C in Fig. 2) suggests that there is a resonance between these solutions, reflecting the delocalized nature of the electronic density in these molecules, a fact directly related to the aromaticity. For the triplet states, we can identify the 3c–2e bonds related with the Ψσ and Ψπ sets, and the 3c–1e bonds related to Ψr and Ψt sets, while for the quintets, the Ψσ are related with the 3c–2e bonds and the four 3c–1e bonds are related with the π-, r-sets, and the last two with the tangential sets respectively (see Fig. 2).
(of the Ψr set) natural orbitals shown in Fig. 1 are similar in shape. The latter description is in agreement with the high delocalization found in these molecules. The co-existence of the two alternative localization schemes (A and B in Fig. 2) and the fully delocalized scheme (C in Fig. 2) suggests that there is a resonance between these solutions, reflecting the delocalized nature of the electronic density in these molecules, a fact directly related to the aromaticity. For the triplet states, we can identify the 3c–2e bonds related with the Ψσ and Ψπ sets, and the 3c–1e bonds related to Ψr and Ψt sets, while for the quintets, the Ψσ are related with the 3c–2e bonds and the four 3c–1e bonds are related with the π-, r-sets, and the last two with the tangential sets respectively (see Fig. 2).
 | ||
Fig. 2 AdNDP analysis of Al3− , 3B2 and , 3B2 and  states. states. | ||
Finally, the iso-chemical-shielding surfaces (ICSS) method46,47 was also used to quantify the direction and scale of the anisotropy effect (see Fig. 3). This approach can characterize aromaticity and antiaromaticity. The shielding lobes (represented in blue) surrounded by donuts like deshielding loops (represented in green) in the planar plane, describe aromatic systems, while the opposite represents an antiaromatic system. Thus, according to the ICSS, the singlet and quintet states exhibit an aromatic character, while the triplet is antiaromatic.
 | ||
| Fig. 3 ICSS plots for Al3− singlet, triplet, and quintet respectively. Shielding surface is shown in blue and de-shielding surfaces in green. Isovalues for the isosurfaces are shown in parentheses. The same ICSS description is found for the rest of Ga3−, In3−, and Tl3− triangles. | ||
4 Discussion
After conducting a thorough analysis of the aromaticity of these structures, we have been able to rationalize it using Mandado's rules76 who established general rules for predicting the aromaticity based on the separate α/β electron counting. Systems with (2n + 1) α- (or β-) electrons are expected to be aromatic, whereas those containing 2n α- (or β-) electrons are antiaromatic (n = 0, 1, 2).In Table 3 we have summarized the outcomes after applying Mandado's rules for α and β electrons on each of the four independent MOs sets (see Fig. 1). Starting with the singlet, we have 3 α and β electrons in the Ψσ set, which follows Mandado's rule (2 × 1 + 1) either for an α- and β-aromatic system. The σ-aromaticity of this MO set is corroborated by the MCI, DI, and AdNDP analyses shown above. The Ψπ set is doubly occupied, thus also meets Mandado's rule (2 × 0 + 1) for α and β electrons, being this set the one that provides π-aromaticity. Similarly, Ψr provides additional σ-aromaticity in agreement with MCI, DI, and AdNDP results. Therefore the singlets present two σ- and a π-aromatic systems, overall being an aromatic molecule which is in agreement with ICSS description (see Fig. 3).
| Multiplicity | Ψ σ | Ψ π | Ψ r | Ψ t | Summary | |
|---|---|---|---|---|---|---|

|
(α or β) | 3 (σA) | 1 (πA) | 1 (σA) | 0 | 2 × σA, πA |
| 3B2 | α | 3 (σA) | 1 (πA) | 1 (σA) | 1 (σ Baird AA) | 2 × σA, πA, σ Baird AA |
| β | 3 (σA) | 1 (πA) | 0 | 0 | σA, πA | |

|
α | 3 (σA) | 1 (πA) | 1 (σA) | 2 (σ Baird A) | 2 × σA, σ Baird A, πA |
| β | 3 (σA) | 0 | 0 | 0 | σA | |
Moving to the 3B2 states, the description of the Ψσ and Ψπ molecular sets is identical to that of the singlet described above. However the picture changes for Ψr, since now it is singly occupied with an α electron, so is the Ψt. We can attribute α-aromaticity to the Ψr, (2 × 0 + 1), but we should be careful when analyzing Ψt. While conducting an analytical assessment, it is plausible to inadvertently apply the (2 × 0 + 1) rule and consider Ψt aromatic, however, is crucial to note that within the tangential set, due to the odd (D3h) symmetry MOs are ordered as two degenerate orbitals caped by a non-degenerate one,77 (see Fig. 4). One of these degenerate MOs contains a single electron, being the other degenerate tangential orbital empty. Therefore, a combination of Mandado's and Baird's rules is warranted, resulting in the (2n) rule for aromatic compounds and the (2n + 1) rule for antiaromatic compounds (observe the MO orbital arrangement for odd symmetry molecules shown in Fig. 4). According to Mandado's rule extension, the Ψt contribution is α-antiaromatic (2 × 0 + 1). Accordingly, the triplet presents what is known as conflicting aromaticity,78 where Ψσ and Ψr present σ-aromatic contribution, the Ψπ aromatic contributions while the Ψt contribution is antiaromatic. The latter contribution could explain the deshielding cones of the 3B2 after applying a magnetic field (see Fig. 3). Atom-pair DIs have different values when comparing the two symmetric bonds with the third bond, in the case of Al3−, the δi,j corresponding to the long bond is slightly smaller than the values of the shorter bonds (1.00 vs. 0.92 a.u.), and this difference increases as increases the size of the atoms with values of 0.97 and 0.63 a.u. for Tl3−. This DI partial alternation pattern can be associated with antiaromaticity.79 AdNDP also agrees with the antiaromaticity description for this state. Analyzing the AdNDP objects (or bonds), we can associate one of 3c–1e objects with the radial aromaticity, which meets Mandado's aromaticity rule, and a single 3c–1e object associated to the tangential aromaticity describing an antiaromatic system for this state.
 | ||
| Fig. 4 Baird aromaticity representation for tangential MOs set. On the left-hand side, the arrangement corresponding to a D3h odd symmetry system is shown, and the arrangement corresponding to even symmetry (D4h) is on the right. Notice that Bairds classical aromaticity rule can be straightforwardly applied to even symmetry systems (4n) while due to the MOs rearrangement in odd symmetry systems, the Baird rule will turn into the (2n) rule. | ||
Finally, focusing on the quintets, we have the same description for the Ψσ as in the cases above, providing σ-aromaticity character to the molecule. The rest of the electrons are α electrons distributed as follows: one in Ψπ and Ψr sets, which according to Mandado's rules, provide π-(the former) and σ-(the later) aromaticity to the system, and the last two, are in the Ψt set, one electron in each of the degenerate orbitals, which agrees with Baird's and Mandado's rules combination (2 × 1) for aromaticity. This description is in agreement with MCI (note that, though, MCI values are much smaller than those of the singlet and triplet). DI also agrees with this description, giving reasonable values for the π-, radial and tangential sets, but smaller than the singlet and triplet DIs. Finally, AdNDP reports four objects formed by one electron and three centers, where two of these centers are associated with the tangential set, thus are α-Baird aromatic, and the other two sets are describing α π- and α σ-aromaticity.
Comparing the aromaticity of the different compounds, we can say that it reduces when increasing the size of the atom, but still, we can state that the  molecule exhibits some degree of aromaticity, albeit significantly lower compared to the
molecule exhibits some degree of aromaticity, albeit significantly lower compared to the  . However, the high-spin
. However, the high-spin  state is the lowest in energy for Tl3−, and its photo-electron spectra matches the experimental spectra available for Tl3−. Surprisingly, the stability of this state contradicts the conventional belief that higher aromaticity leads to increased stability, as the singlet state is considerably more aromatic than the quintet. Despite different indicators also suggest that the quintet state is aromatic, its aromaticity remains inferior to that of the singlet state. The stability of the quintet could be attributed to the relativistic effects present in the Tl3− molecule, which equalize the
state is the lowest in energy for Tl3−, and its photo-electron spectra matches the experimental spectra available for Tl3−. Surprisingly, the stability of this state contradicts the conventional belief that higher aromaticity leads to increased stability, as the singlet state is considerably more aromatic than the quintet. Despite different indicators also suggest that the quintet state is aromatic, its aromaticity remains inferior to that of the singlet state. The stability of the quintet could be attributed to the relativistic effects present in the Tl3− molecule, which equalize the  ,
,  , and both degenerate e′ orbitals in energies. As a consequence of this stabilization, are then filled up according to Hund's rule, with one electron each, being the relativistic effects more important than the stability provided by the aromaticity in the singlet state. Previous studies by Pino-Rios et al.80 have also reported a loss of aromaticity due to relativistic effects, although they attributed it to spin–orbit coupling. However, the spin–orbit coupling for the Tl3− triangles under investigation was found to be relatively small (less than 5 cm−1).
, and both degenerate e′ orbitals in energies. As a consequence of this stabilization, are then filled up according to Hund's rule, with one electron each, being the relativistic effects more important than the stability provided by the aromaticity in the singlet state. Previous studies by Pino-Rios et al.80 have also reported a loss of aromaticity due to relativistic effects, although they attributed it to spin–orbit coupling. However, the spin–orbit coupling for the Tl3− triangles under investigation was found to be relatively small (less than 5 cm−1).
To account for the relativistic effects, the Tl3− states where recalculated at the MCSCF (10,12) level of theory incorporating dynamic correlation through CASPT2, and employing the zeroth-order regular approximated (ZORA)81 method with the SARC-ZORA-TZVPP82 basis set as implemented in ORCA.83 The geometries obtained with ZORA closely resemble the previous ones, with bond lengths of 3.147 and 3.282 Å for the singlet and the quintet respectively, the singlet being 2.91 kcal mol−1 higher in energy.
states where recalculated at the MCSCF (10,12) level of theory incorporating dynamic correlation through CASPT2, and employing the zeroth-order regular approximated (ZORA)81 method with the SARC-ZORA-TZVPP82 basis set as implemented in ORCA.83 The geometries obtained with ZORA closely resemble the previous ones, with bond lengths of 3.147 and 3.282 Å for the singlet and the quintet respectively, the singlet being 2.91 kcal mol−1 higher in energy.
We conducted additional calculations by modulating the relativistic effects, altering the value of the speed of the light used in the ZORA Hamiltonian (note that increasing the speed of the light value in the Hamiltonian reduces the relativistic effects). These effects are clearly depicted in Fig. 5. As the speed of light is increased to larger values, the energy difference between the two states diminishes. When the speed of light is increased sufficiently, to the point where relativistic effects are neglected, both states become nearly energetically degenerated, indicating that relativistic effects favor the quintet with respect to the singlet.
 | ||
Fig. 5 Relative Energy change of Tl3− with respect to logarithm of the speed of the light (in a.u.) used in the relativistic calculations. Note that the point corresponding to the real speed of the light (log10(137.0359895)) is indicated with a c label. with respect to logarithm of the speed of the light (in a.u.) used in the relativistic calculations. Note that the point corresponding to the real speed of the light (log10(137.0359895)) is indicated with a c label. | ||
5 Conclusions
High-level ab-initio quantum mechanics calculations have been performed to characterize the lowest-lying states of the X3− triangles, X = Al, Ga, In, and Tl.We have assign to  and to
and to  the experimental signals detected by Gausa et al. back in 1990, and we attribute the stability of the
the experimental signals detected by Gausa et al. back in 1990, and we attribute the stability of the  to relativistic effects.
to relativistic effects.
Analyzing the electronic structures of these triangular structures, and applying Mandado's aromaticity/antiaromaticity rules, we have been able to rationalize the aromaticity of these molecules, which are in agreement with the different electron density methods employed (MCI, DI, and AdNDP) and the ICSS, which represents the response of the molecule to a external magnetic field of the molecule. The separate analysis of the α and β-electrons made possible to explain the antiaromatic character (the deshielding cone in the ICSS) of the 3B2 as a consequence of the single α electron present in the Ψt orbital set.
Summarizing, the  electronic states present multiple-fold aromaticity, specifically 3-fold aromaticity, being the origin the Ψσ and Ψr sets, providing σ-aromaticity, and the Ψπ which provides π-aromaticity to all the studied singlet states. The triplets are π-aromatic but they do also present certain σ-aromaticity, originated from the inner Ψσ and the α electron present in the Ψr. However, the antiaromatic character of the α electron in the Ψt causes a magnetic shielding/deshielding response associated with an antiaromatic system, as reported by ICSS. The quintets also present some aromatic character, which despite being smaller than that of the singlet and triplet, it is not negligible. According to Mandado's rules, the Ψσ set is α/β σ-aromatic, the Ψπ set α π-aromatic, and the Ψt set α σ-Baird aromatic, in agreement with the diamagnetic response of the quintet.
electronic states present multiple-fold aromaticity, specifically 3-fold aromaticity, being the origin the Ψσ and Ψr sets, providing σ-aromaticity, and the Ψπ which provides π-aromaticity to all the studied singlet states. The triplets are π-aromatic but they do also present certain σ-aromaticity, originated from the inner Ψσ and the α electron present in the Ψr. However, the antiaromatic character of the α electron in the Ψt causes a magnetic shielding/deshielding response associated with an antiaromatic system, as reported by ICSS. The quintets also present some aromatic character, which despite being smaller than that of the singlet and triplet, it is not negligible. According to Mandado's rules, the Ψσ set is α/β σ-aromatic, the Ψπ set α π-aromatic, and the Ψt set α σ-Baird aromatic, in agreement with the diamagnetic response of the quintet.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank for technical and human support provided by IZO-SGI (SGIker) of UPV/EHU and European funding (ERDF and ESF) and the DIPC. Financial support comes from Eusko Jaurlaritza (Basque Government) through the project IT588-22 and from Grant No. PID2020-114754GAI00 provided by MCIN/AEI/10.13039/501100011033. The authors thankfully acknowledge also the computer resources at MareNostrum and the technical support provided by the Barcelona Supercomputing Center (Grant No. QHS-2022- 2-002 and QHS-2022-3-0015). E. M. acknowledges funding from Agencia Española de Investigación, “FEDER Una manera de hacer Europa” (PID2022-140666NB-C21), the Donostia International Physics Center (DIPC-INV-003132) and Eusko Jaurlaritza (PIBA_2023_1_0055). Open Access funding provided by University of Basque Country (UPV/EHU).References
- H. Basch, Chem. Phys. Lett., 1987, 136, 289–293 CrossRef CAS.
- P. Calaminici, N. Russo and M. Toscano, Z. Phys. D – Atoms Mol. Clust., 1995, 33, 281–288 CrossRef CAS.
- K. K. Baeck and R. J. Bartlett, J. Chem. Phys., 1998, 109, 1334–1342 CrossRef CAS.
- S. R. Miller, N. E. Schultz, D. G. Truhlar and D. G. Leopold, J. Chem. Phys., 2009, 130, 024304 CrossRef PubMed.
- P. W. Villalta and D. G. Leopold, J. Chem. Phys., 2009, 130, 024303 CrossRef PubMed.
- T. H. Upton, Phys. Rev. Lett., 1986, 56, 2168–2171 CrossRef CAS PubMed.
- M. F. Jarrold, J. E. Bower and J. S. Kraus, J. Chem. Phys., 1987, 86, 3876–3885 CrossRef CAS.
- S. Li, R. J. Van Zee and W. Weltner, Chem. Phys. Lett., 1996, 262, 298–302 CrossRef CAS.
- G. Pacchioni, P. Fantucci and J. Koutecký, Chem. Phys. Lett., 1987, 142, 85–91 CrossRef CAS.
- J. Tse, J. Mol. Struct. THEOCHEM, 1988, 165, 21–35 CrossRef.
- K. J. Taylor, C. L. Pettiette, M. J. Craycraft, O. Chesnovsky and R. E. Smalley, Chem. Phys. Lett., 1988, 152, 347–352 CrossRef CAS.
- G. Ganteför, M. Gausa, K. H. Meiwes-Broer and H. O. Lutz, Z. Phys. D – Atoms Mol. Clust., 1988, 9, 253–261 CrossRef.
- A. E. Kuznetsov and A. I. Boldyrev, Struct. Chem., 2002, 13, 141–148 CrossRef CAS.
- C. G. Zhan, F. Zheng and D. A. Dixon, J. Am. Chem. Soc., 2002, 124, 14795 CrossRef CAS PubMed.
- J. M. Mercero, E. Matito, F. Ruipérez, I. Infante, X. Lopez and J. M. Ugalde, Chem. – Eur. J., 2015, 21, 9610–9614 CrossRef CAS PubMed.
- J. M. Mercero, R. Grande-Aztatzi, J. M. Ugalde and M. Piris, Advances in Quantum Chemistry, Elsevier, 2023, p. S0065327623000217 Search PubMed.
- B. Gerke and R. Pöttgen, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 121–124 CrossRef CAS.
- S. Seidel, R. D. Hoffmann and R. Pöttgen, Monatsh. Chem., 2014, 145, 1043–1049 CrossRef CAS.
- B. Gerke, R. D. Hoffmann and R. Pöttgen, Z. Anorg. Allg. Chem., 2013, 639, 2444–2449 CrossRef CAS.
- B. Gerke, A. Korthaus, O. Niehaus, F. Haarmann and R. Pöttgen, Z. Naturforsch., B: J. Chem. Sci., 2016, 71, 567–577 CrossRef CAS.
- S. Seidel and R. Pöttgen, Z. Kristallogr. – Cryst. Mater., 2020, 235, 53–57 CrossRef CAS.
- R. Pöttgen, J. Alloys Compd., 1995, 226, 59–64 CrossRef.
- R. Pöttgen, R.-D. Hoffmann and G. Kotzyba, Z. Anorg. Allg. Chem., 1998, 624, 244–250 CrossRef.
- M. F. Zumdick, G. A. Landrum, R. Dronskowski, R. D. Hoffmann and R. Pöttgen, J. Solid State Chem., 2000, 150, 19–30 CrossRef CAS.
- O. Sichevych, Y. Prots, W. Schnelle, M. Schmidt and Y. Grin, Z. Kristallogr. – New Cryst. Struct., 2006, 221, 263–264 CAS.
- F. Haarmann, Y. Prots, S. Göbel and H. G. von Schnering, Z. Kristallogr. – New Cryst. Struct., 2006, 221, 257–258 CAS.
- M. Wagner, R. Cardoso-Gil, N. Oeschler, H. Rosner and Y. Grin, J. Mater. Res., 2011, 26, 1886–1893 CrossRef CAS.
- X.-W. Li, W. T. Pennington and G. H. Robinson, J. Am. Chem. Soc., 1995, 117, 7578–7579 CrossRef CAS.
- Y. Xie, P. R. Schreiner, H. F. Schaefer, X. W. Li and G. H. Robinson, J. Am. Chem. Soc., 1996, 118, 10635–10639 CrossRef CAS.
- G. H. Robinson, Acc. Chem. Res., 1999, 32, 773–782 CrossRef CAS.
- N. Wiberg, T. Blank, K. Amelunxen, H. Nöth, J. Knizek, T. Habereder, W. Kaim and M. Wanner, Eur. J. Inorg. Chem., 2001, 1719–1727 CrossRef CAS.
- K. Balasubramanian and P. Feng, Chem. Phys. Lett., 1988, 146, 155–161 CrossRef CAS.
- U. Meier, S. D. Peyerimhoff and F. Grein, Z. Phys. D – Atoms Mol. Clust., 1990, 17, 209–224 CrossRef CAS.
- G. Li, L. Meng, H. Zhang, X. Li and Y. Zeng, Phys. Chem. Chem. Phys., 2020, 22, 18071–18077 RSC.
- P. Y. Feng and K. Balasubramanian, Chem. Phys., 1989, 138, 89–98 CrossRef CAS.
- M. Gausa, G. Ganteför, H. O. Lutz and K. H. Meiwes-Broer, Int. J. Mass Spectrom. Ion Processes, 1990, 102, 227–237 CrossRef CAS.
- W.-Q. Zhang, G.-F. Zhao, J.-M. Sun, L.-L. Zhi and Y.-Z. Gu, Chem. Phys., 2009, 361, 44–48 CrossRef CAS.
- S. Shi, Y. Liu, Y. Li, B. Deng, C. Zhang and G. Jiang, Comput. Theor. Chem., 2016, 1079, 47–56 CrossRef CAS.
- C. Y. Cha, G. Ganteför and W. Eberhardt, J. Chem. Phys., 1994, 100, 995–1010 CrossRef CAS.
- M. Vijayakumar and K. Balasubramanian, J. Chem. Phys., 1992, 97, 7474–7488 CrossRef CAS.
- S. Kang, Bull. Korean Chem. Soc., 1993, 14, 191–195 CAS.
- A. C. Tsipis, I. G. Depastas and C. A. Tsipis, Symmetry, 2010, 2, 284–319 CrossRef CAS.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304–314 CrossRef CAS.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2006, 135, 325–345 RSC.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 468–475 CrossRef CAS.
- A. C. Wahl and G. Das, in Methods of Electronic Structure Theory, ed. H. F. Shaefer, III, Plenum Press, New York, 1977 Search PubMed.
- H. Nakano, J. Chem. Phys., 1993, 99, 7983–7992 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef CAS.
- B. Metz, M. Schweizer, H. Stoll, M. Dolg and W. Liu, Theor. Chim. Acta, 2000, 104, 22–28 CrossRef CAS.
- H. A. Witek, Y. Choe, J. P. Finley and K. Hirao, J. Comput. Chem., 2002, 23, 957–966 CrossRef CAS PubMed.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville, S. Irle, J. Ivanic, K. Kowalski, S. S. Leang, H. Li, W. Li, J. J. Lutz, I. Magoulas, J. Mato, V. Mironov, H. Nakata, B. Q. Pham, P. Piecuch, D. Poole, S. R. Pruitt, A. P. Rendell, L. B. Roskop, K. Ruedenberg, T. Sattasathuchana, M. W. Schmidt, J. Shen, L. Slipchenko, M. Sosonkina, V. Sundriyal, A. Tiwari, J. L. Galvez Vallejo, B. Westheimer, M. Wloch, P. Xu, F. Zahariev and M. S. Gordon, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS.
- E. Matito, ESI-3D: Electron Sharing Indices Program for 3D Molecular Space Partitioning, 2006, https://ematito.dipc.org/programs.html.
- P. Salvador and E. Ramos-Cordoba, APOST-3D Program, 2014, Institut de Química Computacional i Catàlisi. Universitat de Girona (Spain) Search PubMed.
- I. Mayer and P. Salvador, Chem. Phys. Lett., 2004, 383, 368–375 CrossRef CAS.
- P. Salvador and E. Ramos-Cordoba, J. Chem. Phys., 2013, 139, 071103 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. P. Sergeeva, B. B. Averkiev and A. I. Boldirev, in Metal–Metal Bonding, ed. G. Parkin, Springer-Verlag, Berlin, Germany, 2010, vol. 136 of Structure and Bonding, pp. 275–306 Search PubMed.
- J. M. Mercero, I. Infante and J. M. Ugalde, in Aromaticity and Metal Clusters, ed. P. K. Chattaraj, CRC Press, Boca Raton, FL, USA, 2011, ch. 16, pp. 323–337 Search PubMed.
- J. V. Ortiz, J. Chem. Phys., 1996, 104, 7599–7605 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision B.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- M. Solà, Front. Chem., 2017, 5, 5–8 Search PubMed.
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023, 14, 5569–5576 RSC.
- M. Solà, I. Fernández and G. Merino, Chem. Sci., 2023, 14, 9628–9629 RSC.
- M. Giambiagi, M. S. De Giambiagi, C. D. Dos Santos Silva and A. P. De Figueiredo, Phys. Chem. Chem. Phys., 2000, 2, 3381–3392 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, J. Comput. Chem., 2008, 29, 1543–1554 CrossRef CAS PubMed.
- F. Feixas, E. Matito, J. Poater and M. Solà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 105–122 CAS.
- J. Cioslowski, E. Matito and M. Solà, J. Phys. Chem. A, 2007, 111, 6521–6525 CrossRef CAS PubMed.
- E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, in Aromaticity, ed. I. Fernandez, Elsevier, 2021, pp. 235–259 Search PubMed.
- M. Mandado, A. M. Graña and I. Pérez-Juste, J. Chem. Phys., 2008, 129, 164114 CrossRef PubMed.
- J. M. Mercero, I. Infante and J. M. Ugalde, in Aromaticity and Metal Clusters, ed. P. K. Chattaraj, CRC Press, Florida, 2011, ch. 16, pp. 323–338 Search PubMed.
- D. Chen, D. W. Szczepanik, J. Zhu and M. Solà, Chem. Commun., 2020, 56, 12522–12525 RSC.
- F. Feixas, E. Matito, M. Solà and J. Poater, Phys. Chem. Chem. Phys., 2010, 12, 7126–7137 RSC.
- R. Pino-Rios, A. Vásquez-Espinal, L. Alvarez-Thon and W. Tiznado, Phys. Chem. Chem. Phys., 2020, 22, 22973–22978 RSC.
- C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- D. A. Pantazis and F. Neese, Theor. Chem. Acc., 2012, 131, 1292 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00484a |
| This journal is © the Owner Societies 2024 |