
Why does the orientation of azulene affect the two-photon activity of a porphyrinoid–azulene system?†
Swati Singh
Rajput
 a,
Nikita
Raghuvanshi
bc,
Tejendra
Banana
a,
Pooja
Yadav
a and
Md. Mehboob
Alam
*a
a,
Nikita
Raghuvanshi
bc,
Tejendra
Banana
a,
Pooja
Yadav
a and
Md. Mehboob
Alam
*a
aDepartment of Chemistry, Indian Institute of Technology Bhilai, Durg, Chhattisgarh-491001, India. E-mail: mehboob@iitbhilai.ac.in
bCentre for Basic Sciences, Pt. Ravishankar Shukla University, Raipur, Chhattisgarh, India
cSchool of Studies in Chemistry, Pt. Ravishankar Shukla University, Raipur, India
First published on 3rd May 2024
Abstract
Attaching a dipolar molecule in a symmetric system induces a major change in the electronic structure, which may be reflected as the enhancement of the optical and charge-transfer properties of the combined system as compared to the pristine ones. Furthermore, the orientation of the dipolar molecule may also affect the said properties. This idea is explored in this work by taking porphyrinoid molecules as the pristine systems. We attached azulene, a dipolar molecule, at various positions of five porphyrinoid cores and studied the effect on charge-transfer and one- and two-photon absorption properties using the state-of-the-art RICC2 method. The attachment of azulene produces two major effects – firstly it introduces asymmetry in the system and, secondly, being dipolar, it makes the resultant molecule dipolar/quadrupolar. Porphyrin, N-confused porphyrin, sub-porphyrin, sapphyrin, and hexaphyrin are used as core porphyrinoid systems. The change in charge-transfer has been studied using the orbital analysis and charge-transfer distance parameter for the first five singlet states of the systems. The effect of orientation of azulene on the said properties is also explored. The insights gained from our observations are explored further at the dipole and transition dipole moment levels using a three-state model.
1 Introduction
There has been a great deal of research done on macrocyclic complexes in the field of bioinorganic chemistry.1–4 Porphyrin systems are some of the most extensively studied macrocyclic systems.5–9 They are 26 π-electron conjugated heterocyclic aromatic systems with applications in photodynamic therapy,10–13 fluorescence imaging,14–16 catalysis,17–19 anti-inflammatory therapy,20,21etc. In recent years, synthetic analogues of porphyrins, i.e. “contracted” and “expanded” porphyrins, have received considerable attention from researchers.22–24 Contracted porphyrins are formed by elimination of a meso-methine carbon from the pyrollic unit and the expanded porphyrins are formed by addition of a meso-methine carbon or an extra pyrrole ring forming a macrocyclic system with minimum 17 atoms. Sapphyrin aka pentaphyrin-(1.1.1.1.0) is the first reported expanded porphyrin that was experimentally synthesised by Woodward et al. in 1966.25 Expanded porphyrins because of having a higher number of rings as compared to porphyrin possess longer π-conjugation, resulting in bathochromic shift in their absorption spectra.26Nonlinear optical (NLO) activity of expanded porphyrins has been widely studied by different research groups.27–39 Recently, in 2021, Desmedt et al. theoretically studied the effect of meso substitution on the first hyperpolarizability of hexaphyrin derivatives.32 They reported that meso substitution introduced strain in hexaphyrin derivatives leading to enhanced NLO response. In 2005, Rath et al. reported enhanced third-order NLO properties of core-modified aromatic hexaphyrin 26π-electron systems.33 In the same year, Ahn et al.34 experimentally studied the relation between aromaticity/antiaromaticity and two-photon (TP) activity of two hexaphyrin derivatives, i.e., [26]- and [28]-hexaphyrin(1.1.1.1.1.1)s. They reported that the aromatic system has a larger TP activity than the antiaromatic one. Later, in 2008, Yoon et al. reported a relationship between the TP activity and aromaticity index of four pentapyrrolic expanded porphyrins.35 Apart from hexaphyrins, other expanded porphyrins were also explored for NLO properties. For example, in 2014 Sharma et al. experimentally studied the TP activity of naphtho-bipyrrole-derivatives of sapphyrin and reported that the corresponding perchlorate salt shows higher TP absorption.36 Like expanded porphyrins, the NLO properties of contracted porphyrins such as subporphyrins have also been studied.37–39 Tsurumaki et al. in 2008 reported the large TP activity of hexa substituted sub-porphyrins.38 In the same year Inokuma et al. reported a large enhancement in the TP activity of meso-substituted subporphyrins upon increasing the arm-length.39
Very recently Yang et al.40 observed that introducing azulene in a pristine porphyrin system leads to redistribution of charges, thereby drastically changing the second order NLO response. This is a very simple strategy to affect the charge-distribution and hence the NLO properties of porphyrins. A similar strategy is employed in this work to enhance the NLO properties of expanded-, contracted-, and isomeric-porphyrin systems. We have considered two expanded-, one contracted- and one isomeric-porphyrin systems and theoretically studied the effect on their charge-transfer, one-photon, and two-photon activities when azulene is attached at various positions.
2 Computational details
In this work, five different expanded, contracted, and isomeric porphyrin derivatives, namely hexaphyrin (Hx), sapphyrin (S), sub-porphyrin (SP), N-confused porphyrin (NCP), and porphyrin (Pr), are considered, to which azulene is attached at different positions and orientations. The ground state geometry of all the pristine porphyrins and porphyrinoid–azulene systems are optimized in the vacuum phase at the B3LYP41–43/6-311+G(d,p)44 level of theory using the Gaussian 16 program.45 The optimized geometries are further used for vibrational frequency calculations at the same level of theory. No imaginary frequency is found for any of them confirming their validity as the minimum energy geometry on their respective potential energy surfaces. One- and two-photon absorption46–49 (OPA and TPA) cross-sections for the first five singlet states are computed at the RICC2/cc-pVDZ50,51 level of theory as implemented in the Turbomole 7.352,53 package. Furthermore, to study the charge-transfer character of the electronic excitations in the said systems, natural transition orbitals (NTOs)54–56 are generated at the RICC2/cc-pVDZ level of theory using the Turbomole 7.3 package, and the density-difference plot and charge-transfer distance parameter are computed at the MN1557,58/6-311+G(d,p) level of theory as implemented in the Gaussian 16 program package. Our choice of MN15 exchange–correlation functional is based on a recent work conducted by Grabarz et al.,59 where they studied various density functional approximations for the prediction of excited-state properties of 85 fluorescent dyes. They found that the results predicted using the MN15 functional for both the ground and the excited states are comparable to those obtained using the CC2 model. This reflects that this functional is the best for handling both the single- and multi-reference systems even if they have non-covalent interactions. Additionally, it is well-known that a long-range exchange (as present in the MN15 functional) is required for a correct description of the charge-transfer process.60 To pinpoint the factor(s) responsible for the variation in TP activity61–63 among the considered systems a three-state model (3SM) within the generalized few-state models (GFSM) is employed. The parameters such as excitation energy, ground to excited state transition dipole moments, excited-excited state transition moments, and dipole moments of different states are calculated at the RICC2/cc-pVDZ level of theory using the Turbomole7.3 program package.523 Results and discussion
3.1 Structure and electronic properties
We start our discussion by describing important geometrical characteristics of porphyrinoid–azulene systems considered for this study. The five pristine porphyrin (PP) systems, namely sub-porphyrin (SP), porphyrin (Pr), N-confused porphyrin (NCP), sapphyrin (S) and hexaphyrin (Hx), are considered. These are altered by attaching azulene at various positions, as shown in Fig. 1.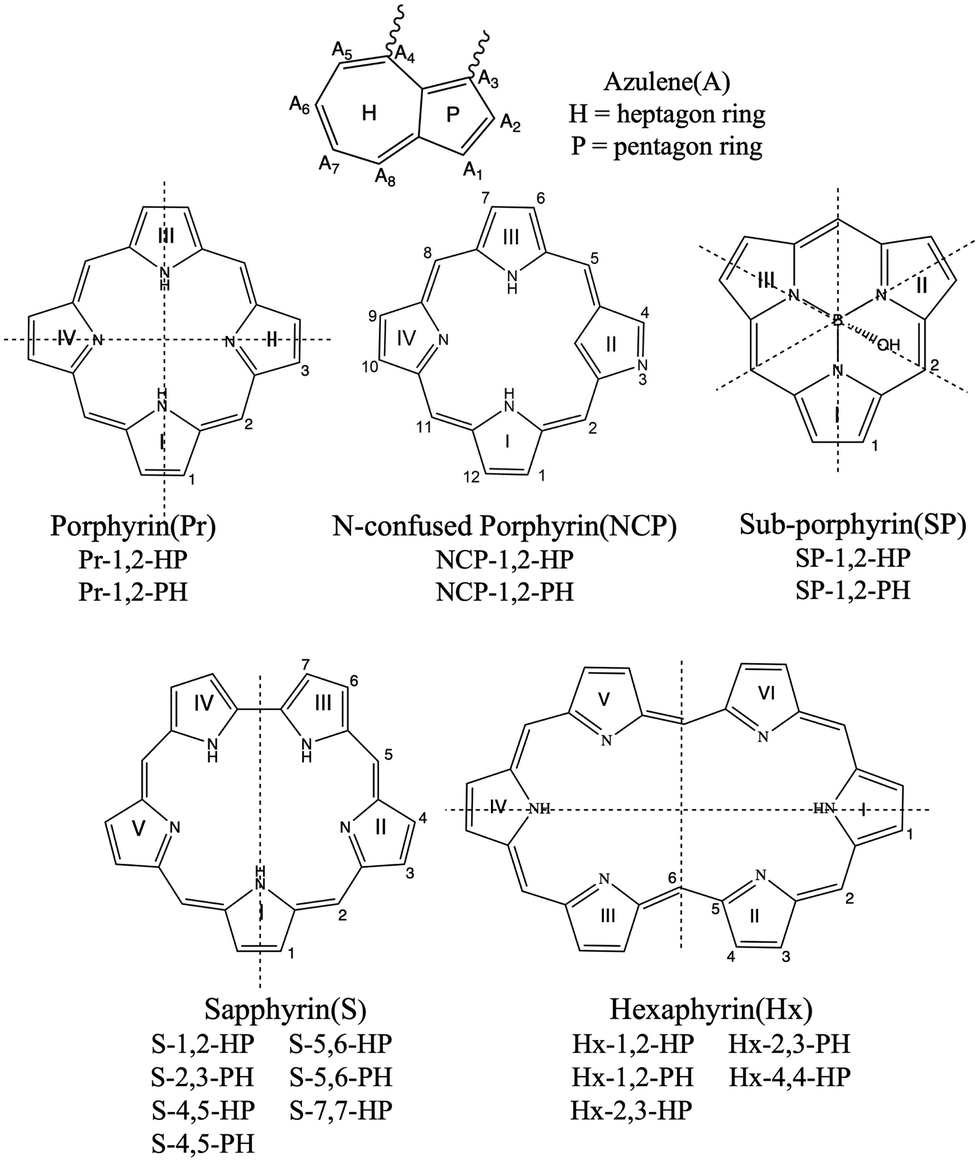 | ||
| Fig. 1 All pristine porphyrin–azulene systems considered for this study. | ||
Azulene is a 10-π-electron aromatic system consisting of a heptagon (H) and a pentagon (P) ring. It is attached to each PP-unit through the curly bonds shown in the top panel of Fig. 1. Based on the symmetry of PP units all possibilities of attaching azulene are considered for this study. We named the molecules PP-m,n-HP and PP-m,n-PH, where PP represents the above-mentioned acronym for the pristine porphyrin unit and ‘m’ and ‘n’ represent the position in the PP unit (shown in Fig. 1) at which heptagon (or pentagon) and pentagon (or heptagon) rings are attached respectively. For example, S-4,5-PH represents a sapphyrin system where the pentagon ring of azulene is attached at position 4 and the heptagon ring is attached at position 5 of the sapphyrin core. Note that some structures are not possible due to the overlap of one of the H-atoms in azulene with that in the PP unit. Therefore, such structures are not considered. NCP being an asymmetric system provides several possibilities for attaching azulene. In order to keep the data analysis simple, we considered only one pair of NCP–azulene systems i.e., NCP-1,2-HP and NCP-1,2-PH.
3.2 One-photon absorption (OPA)
The OPA64–66 profiles of the first five excited singlet states of azulene, five PP systems, and the 18 porphyrinoid–azulene systems calculated at the RICC2/cc-pVDZ level of theory are presented in Fig. 2–7. We observed that OPA spectra of azulene have a single peak at 245 nm resulting from S0 → S4 transitions. This peak is dominated by HOMO−1 → LUMO and HOMO → LUMO+1 orbital transitions. These orbitals are distributed over the entire ring structure of azulene. For both Pr and NCP, three peaks appear due to S0 → Sn (n = 3, 4, and 5) transitions at 352, 343, and 295 nm (in Pr) and at 378, 356, and 339 nm (in NCP). | ||
| Fig. 2 OPA profile of azulene, calculated at the RICC2/cc-pVDZ level of theory. | ||
 | ||
| Fig. 3 Top panel presents the one-photon absorption spectra of Pr, Pr-1,2-HP, and Pr-1,2-PH systems calculated at the RICC2/cc-pVDZ level of theory. A Gaussian envelope of FWHM = 10 nm is utilized for plotting the graphs. The dominant orbitals involved in the transitions are given in the inset. Bottom panel presents NTOs of Pr-1,2-HP and Pr-1,2-PH. | ||
 | ||
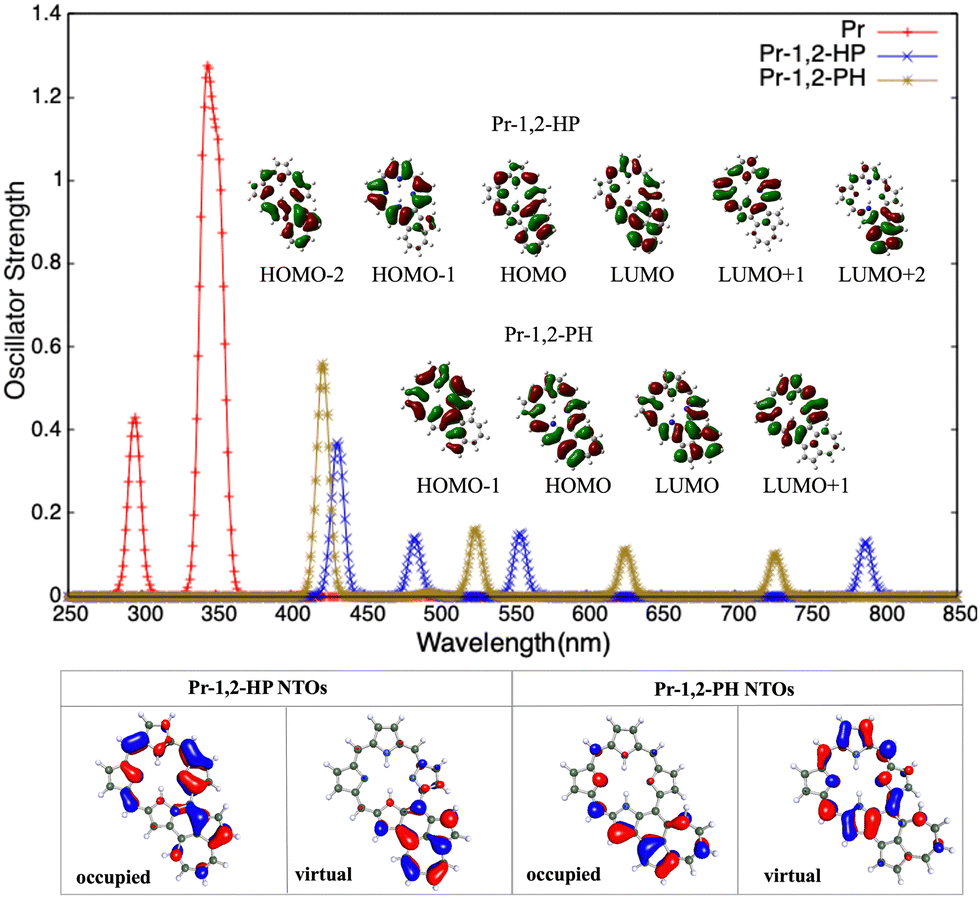
| Fig. 4 Top panel presents the one-photon absorption spectra of NCP, NCP-1,2-HP, and NCP-1,2-PH systems calculated at the RICC2/cc-pVDZ level of theory. A Gaussian envelope of FWHM = 10 nm is utilized for plotting the graphs. The dominant orbitals involved in the transitions are given in the inset. Bottom panel presents NTOs of NCP-1,2-HP and NCP-1,2-PH. | ||
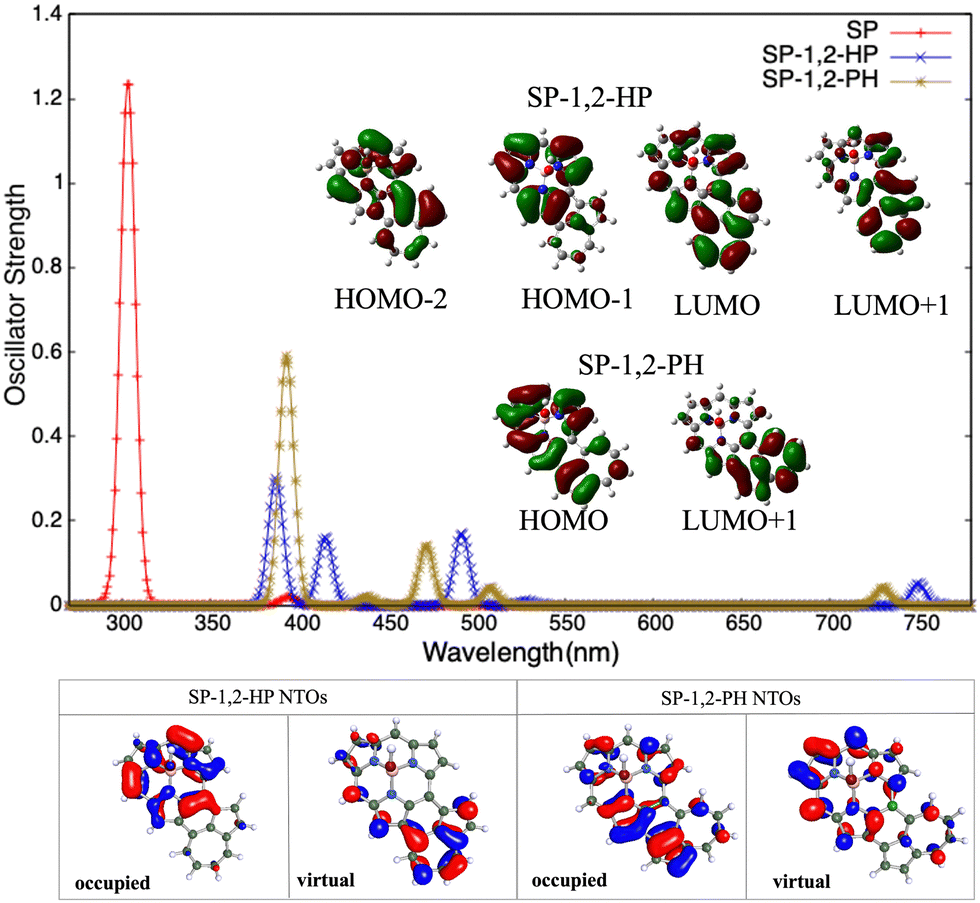 | ||
| Fig. 5 Top panel presents the one-photon absorption spectra of SP, SP-1,2-HP, and SP-1,2-PH systems calculated at the RICC2/cc-pVDZ level of theory. A Gaussian envelope of FWHM = 10 nm is utilized for plotting the graphs. The dominant orbitals involved in the transitions are given in the inset. Bottom panel presents NTOs of SP-1,2-HP and SP-1,2-PH. | ||
 | ||
| Fig. 6 One-photon absorption spectra of S–azulene systems calculated at the RICC2/cc-pVDZ level of theory. A Gaussian envelope of FWHM = 10 nm is utilized for plotting the graphs. | ||
 | ||
| Fig. 7 One-photon absorption spectra of Hx–azulene systems calculated at the RICC2/cc-pVDZ level of theory. A Gaussian envelope of FWHM = 10 nm is utilized for plotting the graphs. | ||
On the other hand, for S, SP, and Hx, only two closely-spaced peaks appear corresponding to S0 → Sn (n = 3, 4) transitions. These peaks appear at 386 and 380 nm (for S), at 304 and 303 nm (for SP), and at 463 and 447 nm (for Hx). Evidently, addition of the azulene ring to PP units leads to major changes in the OPA profiles, which are further affected by the orientation of the azulene group (i.e., HP or PH connectivity). We observed that the PP–azulene systems always have red shifted OPA spectra as compared to those of the respective pristine PP systems. For example, for Pr-1,2-HP and Pr-1,2-PH the peaks are well separated and appear at (432, 484, 555, and 788 nm) and (422, 525, 626, and 727 nm) respectively.
These are largely red shifted with respect to porphyrin. Upon changing the orientation of azulene from HP to PH we observed a decrease in the oscillator strength of the most intense peak. This can be correlated with the change in charge-transfer due to the change in interaction of H/P rings of azulene with pyrrole ring(s) of Pr. In Pr-1,2-PH, the electron-rich P-ring of azulene is in the vicinity of the electron-rich pyrrole ring (ring I) of Pr. This results in an electrostatic repulsion between the two causing charge-separation. On the other hand, in Pr-1,2-HP the same interaction is attractive in nature, resulting in less charge-separation. This is supported by the respective transition moment values (Table S2 in ESI†) in the two cases. An analysis of the orbitals involved in these transitions reflects that excitations have a charge-transfer character, involving charge transfer between the tetrapyrrole and azulene moieties. For example, in Pr-1,2-HP the most OPA active peak (432 nm) has contributions from HOMO−2 → LUMO (31%), HOMO−1 → LUMO (14%), HOMO−1 → LUMO+2 (12%), and HOMO → LUMO+1 (12%). Among these orbital transitions, the last three transitions clearly show a charge-transfer between porphyrin and azulene cores. Similarly, in Pr-1,2-PH the highest peak (422 nm) is dominated by HOMO−1 → LUMO (33%), and HOMO → LUMO+1 (26%) transitions. These orbital transitions involve charge-transfer from porphyrin to azulene core as well as from the azulene to porphyrin core. This is further visible in the involved NTOs. It is worth mentioning that the orbital involved in the Pr unit is similar to those in chlorophyll resulting from the common tetrapyrrole structure, and this can be validated using Gouterman's four-orbital model.67,68 Similarly, for NCP-1,2-HP and NCP-1,2-PH, the peaks appear at (447, 495, 572, and 807 nm) and (441, 545, 676, and 754 nm) respectively. These peaks are also largely red-shifted with respect to the OPA peaks in NCP. However, upon switching from HP to PH orientation we observed that unlike the case of Pr–azulene here NCP-1,2-HP has a larger oscillator strength than NCP-1,2-PH. This can be explained in the same way as we did for Pr–azulene systems. However, now the confused N-atom plays a significant role. In NCP-1,2-HP the N-atom of ring II is close to the electron-rich P ring of azulene causing repulsion and hence enhanced charge-separation, which is reversed in NCP-1,2-PH. This is further supported by larger values of respective transition moments in NCP-1,2-HP than in NCP-1,2-PH (Table S2 in ESI†). The orbitals involved in these two systems show that the most active OPA peak in NCP-1,2-HP (447 nm) is dominated by HOMO−2 → LUMO (25%) and HOMO−1 → LUMO (24%) orbital transitions, whereas in NCP-1,2-PH the same (441 nm) is dominated by HOMO−2 → LUMO+1 (32%), HOMO−1 → LUMO+1 (19%), and HOMO → LUMO+1 (15%) transitions. In addition to the participation of azulene in charge-transfer, the effect of its orientation is also evident in this case. For example, for NCP-1,2-HP the orbital pictures show that there is a charge-transfer from the NCP core to the azulene core and the same in NCP-1,2-PH is from the azulene core to the NCP core. This is further supported by the NTOs involved in these two cases.
For SP-1,2-HP and SP-1,2-PH, the peaks appear at (387, 415, 492, and 750 nm) and (393, 472, 508, and 730 nm) respectively. They are red shifted with respect to the most intense OPA peak in pristine SP. SP–azulene systems are different from Pr and NCP in the sense that now the B-atom is directly connected to three N-atoms of pyrrole rings, making it non-planar. The presence of the B-atom with a vacant p-orbital decreases the electron density on the said rings significantly and hence changes the above-mentioned electrostatic interactions. Most likely, the presence of the B-atom increases the charge-separation in SP–azulene systems, which is reflected in their higher transition moment values as compared to those of Pr– and NCP–azulene systems. The highest peak of SP-1,2-HP (387 nm) is due to HOMO−2 → LUMO (35%) and HOMO−1 → LUMO+1 (26%) transitions, whereas the same for SP-1,2-PH (393 m) is due to HOMO → LUMO+1 (47%) transition only. The orbitals involved in these transitions reveal that the red shift for both SP-1,2-HP and SP-1,2-PH is a result of charge-transfer from the SP core to the azulene core as further supported by the NTOs. Similar variations are observed in S–azulene and Hx–azulene systems too. For brevity, the images of the orbitals involved and NTO plots for these systems are provided in the ESI.† The OPA profiles of pristine S, pristine Hx, S–azulene, and Hx–azulene systems are shown in Fig. 6 and 7 respectively.
In order to study the effect of the orientation of azulene in PP–azulene systems on the charge-transfer character of the electronic excitations, we plotted the difference-density plot69–71 obtained by subtracting the electron density in ground and the respective excited states having the largest oscillator strength.
This gives a clear idea of variation in charge-transfer character of the electronic excitations among different molecules. The density-difference plots and dCT72,73 values of some representative systems are given in Fig. 8. The variation in the electron density difference plot upon changing the orientation of azulene is visible in all the PP–azulene systems. For example, in Pr–azulene systems, when the orientation is changed from HP to PH, there is a clear shift of difference-density towards the electron-rich P ring of azulene. Similarly, in NCP–azulene systems the density difference is more on the pyrrole ring with the confused N-atom in NCP and the electron-rich P ring in azulene. The said effect is quantitatively analysed by calculating the barycenter of positive and negative electron densities and the corresponding charge-transfer distance (dCT = R+ − R−) in these systems. The location of the center of positive (R+) and negative (R−) electron densities and distance are also shown in Fig. 8. We observed that in Pr-1,2-HP the dCT vector is along one of the C–C bonds in the Pr unit, whereas the same in Pr-1,2-PH makes an acute angle about the same C–C bond. The value of dCT is also increased more than two-times between the said pair of molecules. This variation is explicable by the change in the direction of dipole moment and interactions between the pyrrole ring of Pr and H/P rings of azulene as mentioned earlier. In NCP–azulene systems too, there is a large increase in the magnitude of dCT when orientation of azulene is changed from HP to PH. The change is opposite to what is observed in Pr–azulene systems and is also consistent with the variation of transition dipole moment involved in the most intense OPA peak of these systems supporting our explanation based on interaction between different rings we presented earlier. In SP–, S–, and Hx–azulene pairs, a reverse trend is observed i.e., the value of dCT is decreased when the orientation of azulene is changed from HP to PH.
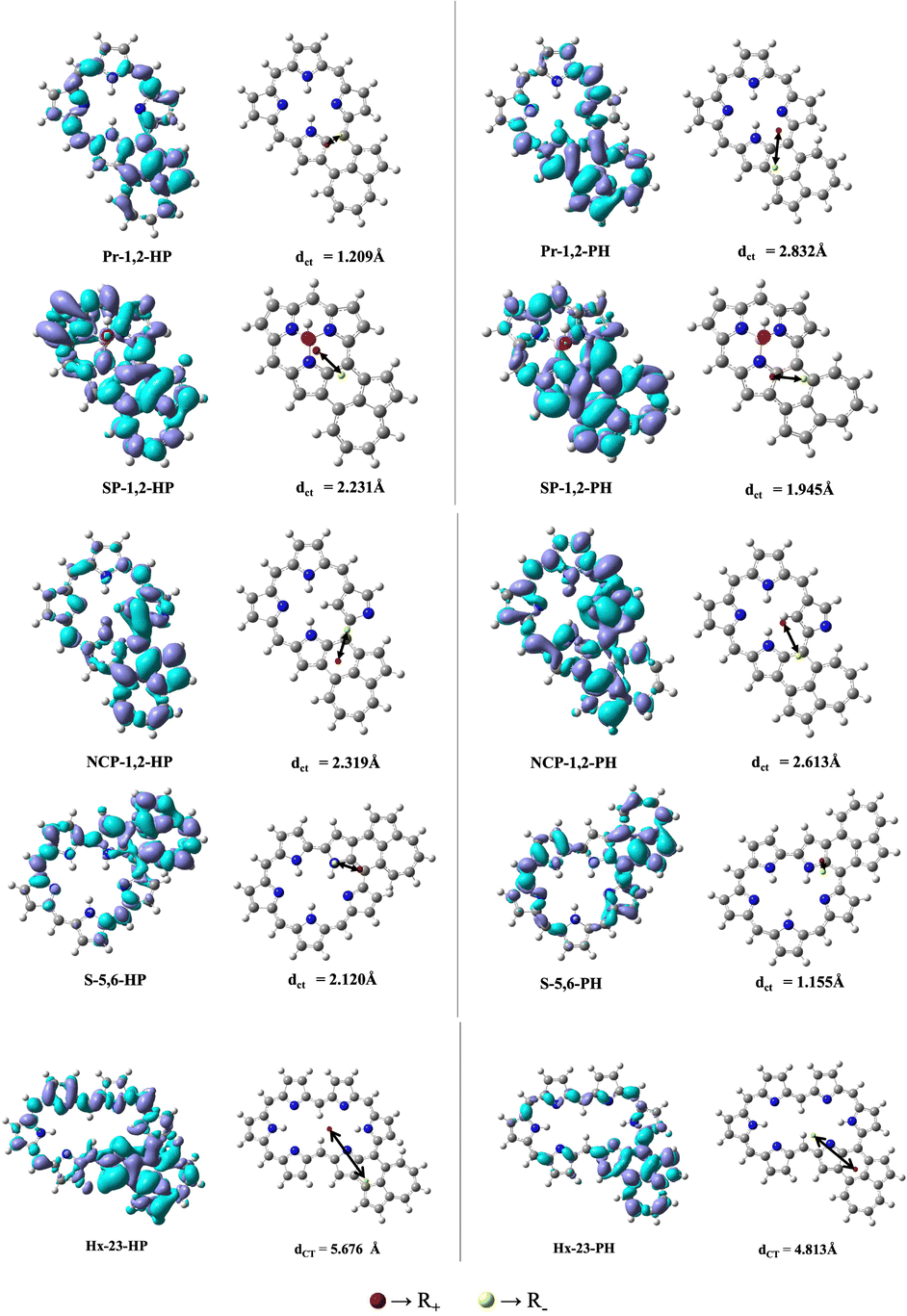 | ||
| Fig. 8 Plot of the difference in total density Δρ(r) and the distance between the barycenters dCT (Å) calculated at the MN15/6-311+G(d,p) level. For the Δρ(r) plot an isocontour value of 0.0004 a.u. is taken. | ||
3.3 Effect of orientation of azulene on two-photon activity
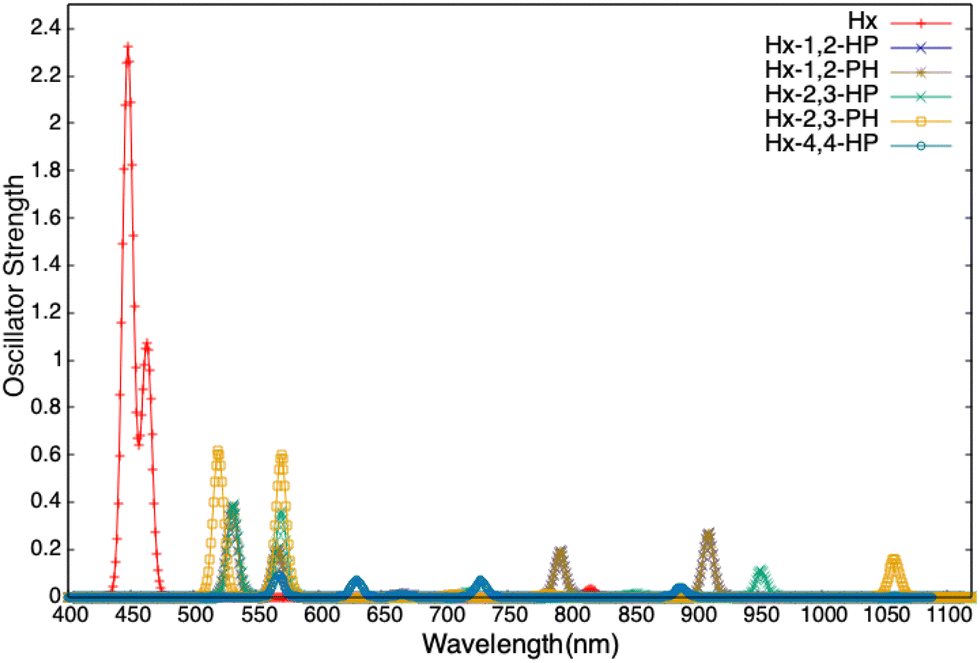
Attaching an azulene to porphyrinoid systems introduces asymmetry and changes the directions of various transition dipole moments. This is reflected in the TPA activity of the PP–azulene systems. For example, due to centrosymmetric structure both pristine porphyrin and hexaphyrin systems possess zero two-photon transition strength (δRespTPA), but when azulene is attached to them the resulting systems become polar as well as TPA active. Similarly, the TPAs of other systems are affected by azulene attachment. The relevant data are provided in the ESI.† In this section, we rather focus on another interesting aspect viz. variation of δRespTPA with the orientation of the azulene ring in PP–azulene systems. The values of δRespTPA for the first five singlet excited states of all 18 systems calculated at the RI-CC2/cc-pVDZ level of theory are presented in Fig. 9. It is evident from the plots that the orientation of azulene affects the TPA of these systems significantly.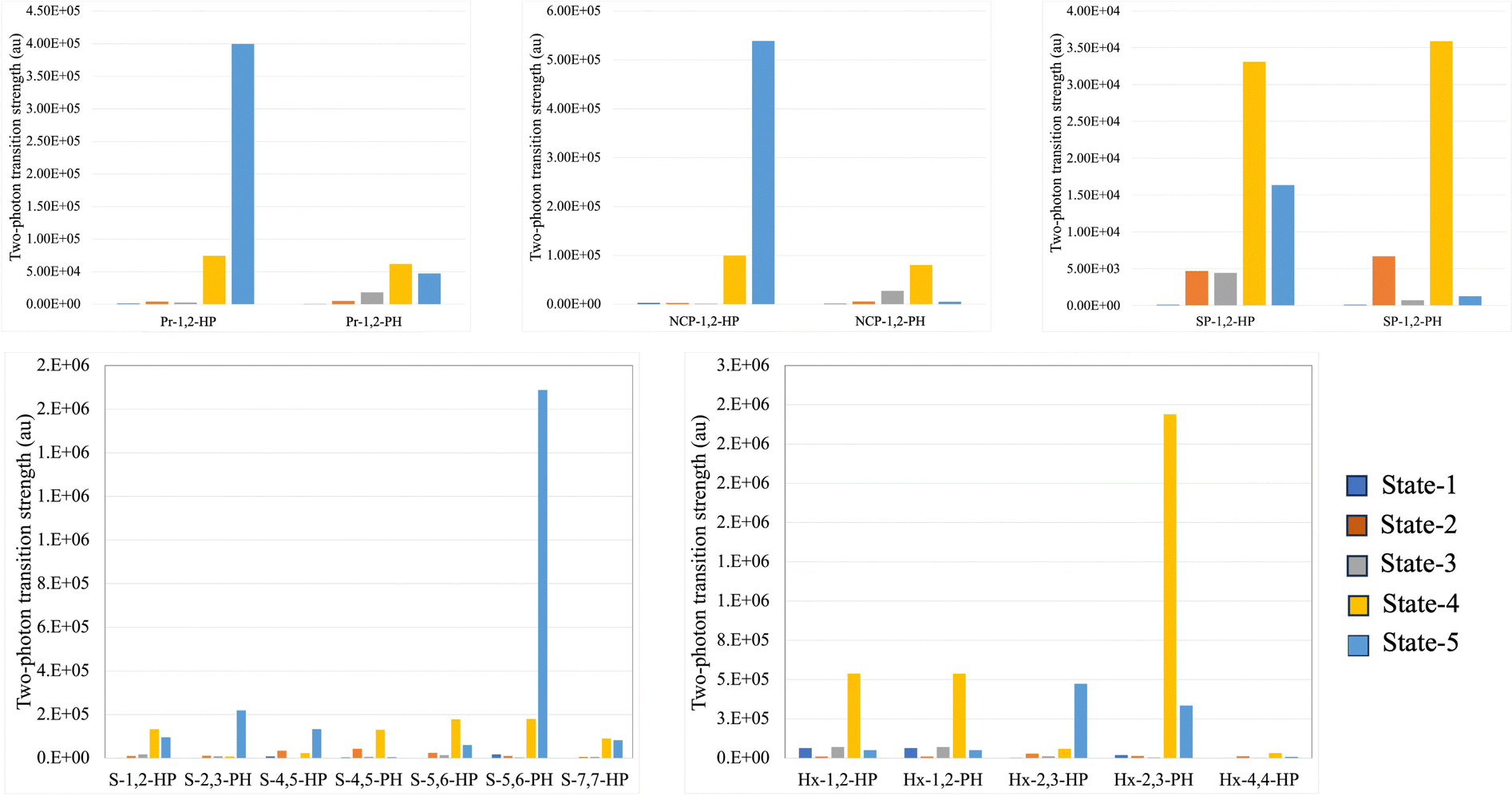 | ||
| Fig. 9 TPA transition strength of the first five singlet excited states of all the systems computed at the RICC2/cc-pVDZ level of theory. | ||
In both Pr–azulene and NCP–azulene systems, the molecules with HP orientation of azulene have a larger δRespTPA (for the most TP active state) than the ones with PH orientation of azulene. However, in other systems it is the other way round. For example, in Pr-1,2-HP, δRespTPA for S0 → S5 transition has a value of 4.0 × 105 a.u., whereas the same in Pr-1,2-PH is reduced to 5.0 × 104 a.u. In NCP-1,2-HP, δRespTPA is 5.5 × 105 a.u. for the S0 → S5 state and the same in NCP-1,2-PH is negligibly small. Similarly, in S–azulene and Hx–azulene systems, large variations in δRespTPA are observed in the following pairs (S-5,6-HP, S-5,6-PH) and (Hx-2,3-HP, Hx-2,3-PH). The said effect is less pronounced in other studied systems. Note that in SP–azulene, the S5 state did not converge and hence we could manage to get the values of δRespTPA for up to the S4 state. The effect of azulene in these systems is very small. In SP-1,2-HP the S0 → S4 state has the largest δRespTPA value of 3.3 × 104 a.u. and the same in SP-1,2-PH is 3.6 × 104 a.u.
To explain these observations, we employed a three-state model (3SM) within the framework of the generalized few-state model (GFSM)74 developed for the RI-CC2 method. Within the three-state model, one intermediate state (i) is involved in addition to the ground state (0) and the final two-photon active state (f). In this model, the expression for two-photon transition strength is given as:75,76
 | (1) |
 , E0p is the excitation energy for S0 → Sp transition, and θrspq represents the angle between transition dipole moment vectors μpq and μrs.
, E0p is the excitation energy for S0 → Sp transition, and θrspq represents the angle between transition dipole moment vectors μpq and μrs.
The results are presented in Fig. 10. It is evident that in all cases, 3SM with S1 as the intermediate state correctly reproduces the variation of δTPA among the pair of molecules considered. However, in case of SP-1,2-HP and SP-1,2-PH the agreement is only qualitative. The component-wise analysis of 3SM results reflects that in all the cases δ0fii is the most contributing term except for Hx-2,3-HP, where δ0fff is the most contributing term.
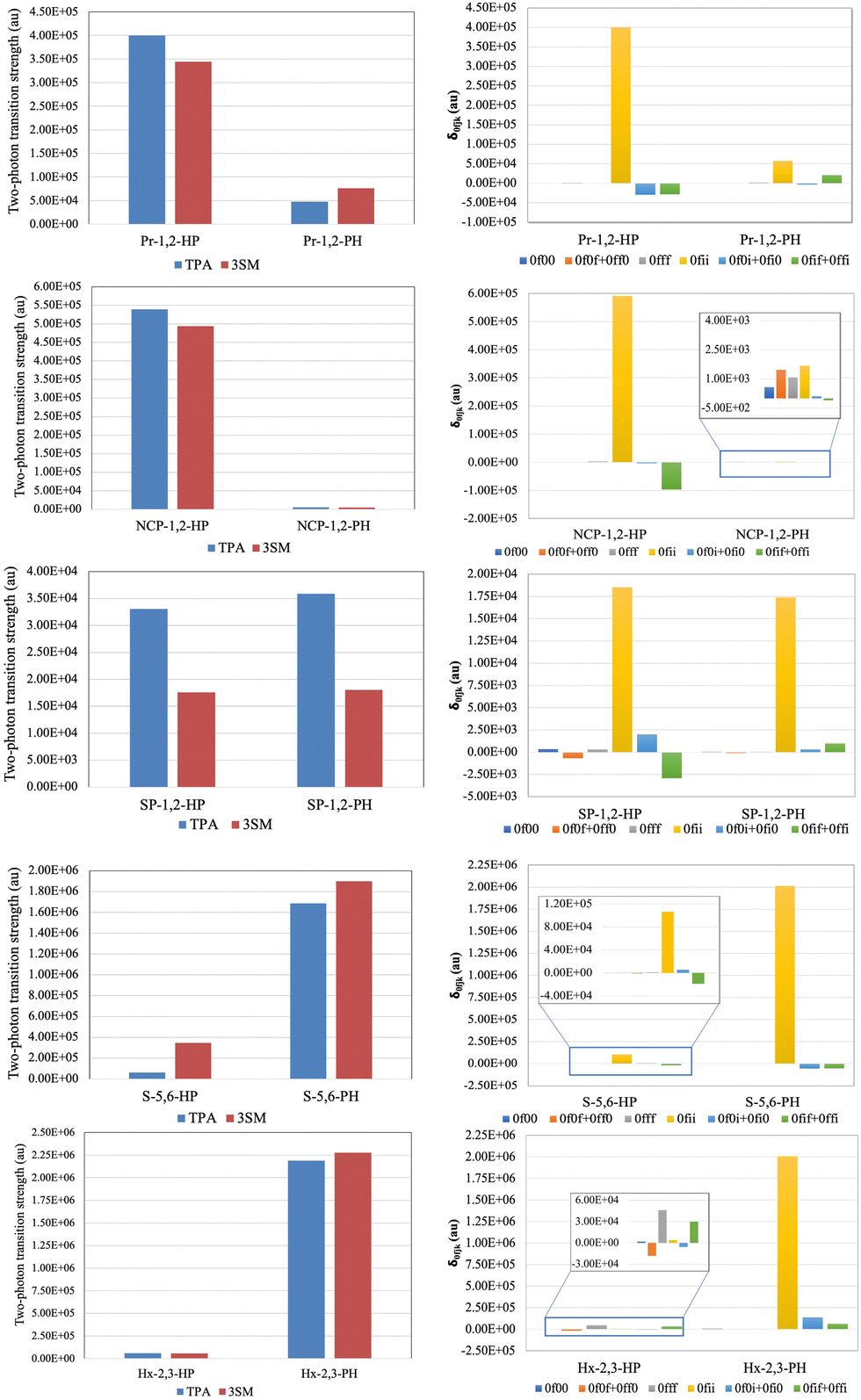 | ||
| Fig. 10 Two-photon transition strength calculated using response theory and 3SM, and the contribution of different δ0fjk involved in 3SM. | ||
In each pair of molecules, the one having larger δTPA also has a larger value of δ0fii, except in the SP-azulene pair. In SP-1,2-HP δ0fii is larger than that in SP-1,2-PH. However, because of the negative contributions from δ0fif and δ0ffi the value of δ3SMTPA is smaller in SP-1,2-HP than that in SP-1,2-PH. An analysis of involved transition dipole moment vectors in δ0fii reveals that the effect of orientation of azulene on different transition dipole moments varies from system to system. For example, in Pr–azulene systems when the orientation of azulene is changed from HP to PH, the dipole moment of the excited state (μff) increases, whereas the same in NCP–azulene systems is reversed. However, in both these systems, the dipole moments of ground and intermediate states as well as the values of transition moments μif, μ0i, and μi0 decrease significantly. Owing to this variation both Pr-1,2-PH and NCP-1,2-PH become less TP active than Pr-1,2-HP and NCP-1,2-HP respectively.
This variation may be correlated with the interactions mentioned in earlier discussion. However, one should be careful in using such interaction based discussion when it comes to TPA or other non-linear optical processes. In such processes, several transition dipole moments including those between two different excited states are involved. These transition dipole moments may not be directly linked with the physical interactions between different parts of the molecule anticipated from the ground state geometry. The variation of different transition dipole moments in all the pairs considered is shown in Fig. 11. Thus, we see that the addition of azulene in porphyrinoid systems affects the charge-transfer process significantly through the change in the values of various transition dipole moments, which is different in contracted- and expanded-porphyrinoid systems.
 | ||
| Fig. 11 Variation of different transition dipole moments with orientation of the azulene group. | ||
4 Conclusions
The effect of orientation of azulene in PP–azulene systems (where PP refers to porphyrinoid systems such as porphyrin and expanded- and contracted-porphyrins) on their charge-transfer and optical properties is explored computationally in this work. For this purpose, porphyrin (Pr), N-confused porphyrin (NCP), sub-porphyrin (SP), sapphyrin (S), and hexaphyrin (Hx) systems are considered and one azulene unit is attached to them at various positions. The addition of azulene to these PP systems leads to a two-fold change. Firstly, it breaks the symmetry of the systems (except N-confused porphyrin, which is itself asymmetric) and secondly, due to its dipolar nature azulene adds polarity to the non-polar systems by altering their electronic configuration, resulting in an increased charge-transfer. Owing to these two effects a significant change in the electronic structure and charge-transfer and optical properties of the PP–azulene systems is observed. Using the state-of-the-art RICC2 method in conjunction with the cc-pVDZ basis set, we studied the UV-vis absorption, two-photon absorption, and charge-transfer in the PP–azulene systems. We observed that irrespective of the core porphyrinoid system, addition of azulene leads to a large red-shift in the OPA spectra, thereby shifting the same to the near infrared region. Furthermore, the orientation of azulene causes a drastic change in the direction of charge-transfer, which is visible through the orbital pictures involved in a particular transition. In one orientation the charge-transfer is from the azulene to porphyrinoid unit and in the other orientation it is reversed. We further observed that this effect is different in contracted- and expanded porphyrine systems, which is quantitatively analysed using the charge-transfer distance parameter (dCT). In Pr– and NCP–azulene systems, the change in orientation of azulene from HP to PH increases the value of dCT, whereas the same in SP–, S– and Hx–azulene systems decreases it. This effect is also visible in their density-difference plots.In both contracted and expanded PP–azulene systems a change in orientation of the azulene unit may lead to a massive change in their TPA activity. In Pr– and NCP–azulene systems the change in orientation from HP to PH causes an order of magnitude or more decrease in δTPA, whereas the same remains unaffected in SP–azulene systems. Contrastingly, in S– and Hx–azulene systems a reverse trend is observed. An analysis involving a three-state model (with S1 as the intermediate state) reveals that this is because of the change in the values of dipole moments of TP active excited states and those of transition dipole moments involving the S1 state. At the end, we conclude that our strategy of attaching azulene could be used to make a PP–azulene system highly TP active in the near IR region, which could be useful for various NLO applications, including in photodynamic therapy (PDT). The usefulness of these PP–azulene systems as photosensitizers in TP-PDT is under investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Wroclaw Center for Networking and Supercomputing for granting computational resources.References
- R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS.
- R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288–10317 CrossRef CAS PubMed.
- D. K. Deda, B. A. Iglesias, E. Alves, K. Araki and C. R. S. Garcia, Molecules, 2020, 25, 2080 CrossRef CAS PubMed.
- N. Tsolekile, S. Nelana and O. S. Oluwafemi, Molecules, 2019, 24, 2669 CrossRef CAS PubMed.
- X. Xue, A. Lindstrom and Y. Li, Bioconjugate Chem., 2019, 30, 1585–1603 CrossRef CAS PubMed.
- M. Imran, M. Ramzan, A. K. Qureshi, M. A. Khan and M. Tariq, Biosensors, 2018, 8, 95 CrossRef CAS PubMed.
- C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- B. M. Amos-Tautua, S. P. Songca and O. S. Oluwafemi, Molecules, 2019, 24, 2456 CrossRef CAS PubMed.
- J. Tian, B. Huang, M. H. Nawaz and W. Zhang, Coord. Chem. Rev., 2020, 420, 213410 CrossRef CAS.
- D. Xu, Q. Duan, H. Yu and W. Dong, J. Mater. Chem. B, 2023, 11, 5976–5989 RSC.
- B. Babu, J. Mack and T. Nyokong, Dalton Trans., 2020, 49, 15180–15183 RSC.
- D. Pan, P. Liang, X. Zhong, D. Wang, H. Cao, W. Wang, W. He, Z. Yang and X. Dong, ACS Appl. Bio Mater., 2019, 2, 999–1005 CrossRef CAS PubMed.
- Y. Cao, D. Wei, L. Yang, Z. Luo, P. Yu, H. Li, C. Zhang, X. Liu, F. Wu, M. Wu and Y. Zeng, Adv. Healthcare Mater., 2022, 11, 2102526 CrossRef CAS PubMed.
- W. Chen, J. Zhao, M. Hou, M. Yang and C. Yi, Nanoscale, 2021, 13, 16197–16206 RSC.
- L. Feng, K.-Y. Wang, E. Joseph and H.-C. Zhou, Trends Chem., 2020, 2, 555–568 CrossRef CAS.
- N. Zion, J. C. Douglin, D. A. Cullen, P. Zelenay, D. R. Dekel and L. Elbaz, Adv. Funct. Mater., 2021, 31, 2100963 CrossRef CAS.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS PubMed.
- D. Jelić, I. Tatić, M. Trzun, B. Hrvačić, K. Brajša, D. Verbanac, M. Tomašković, O. Čulić, R. Antolović, I. Glojnarić and I. Weygand-Đurašević, Eur. J. Pharmacol., 2012, 691, 251–260 CrossRef PubMed.
- D. Sengupta, S. Das, D. Sharma, S. Chattopadhyaya, A. Mukherjee, Z. H. Mazumdar, B. Das, S. Basu and M. Sengupta, ChemMedChem, 2022, 17, e202100550 CrossRef CAS PubMed.
- J. Mack, Chem. Rev., 2017, 117, 3444–3478 CrossRef CAS PubMed.
- T. Sarma and P. K. Panda, Chem. Rev., 2017, 117, 2785–2838 CrossRef CAS PubMed.
- J. L. Sessler, S. J. Weghorn and S. J. Weghorn, Chem. Rev., 2017, 117, 2201–2202 CrossRef CAS PubMed.
- R. Woodward, Special Publication, 1966.
- S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 CrossRef CAS PubMed.
- S. Mori and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8030–8031 CrossRef CAS PubMed.
- D. Koszelewski, A. Nowak-Król, M. Drobizhev, C. J. Wilson, J. E. Haley, T. M. Cooper, J. Romiszewski, E. Górecka, H. L. Anderson, A. Rebane and D. T. Gryko, J. Mater. Chem. C, 2013, 1, 2044–2053 RSC.
- J. M. Lim, Z. S. Yoon, J.-Y. Shin, K. S. Kim, M.-C. Yoon and D. Kim, Chem. Commun., 2008, 261–273 RSC.
- B. Li, P. Sathishkumar, F. L. Gu and C. Zhu, J. Phys. Chem. A, 2020, 124, 955–965 CrossRef CAS PubMed.
- K. Garg, R. Shanmugam and P. C. Ramamurthy, Carbon, 2017, 122, 307–318 CrossRef CAS.
- E. Desmedt, T. Woller, J. L. Teunissen, F. De Vleeschouwer and M. Alonso, Front. Chem., 2021, 9, 786036 CrossRef CAS PubMed.
- H. Rath, J. Sankar, V. PrabhuRaja, T. K. Chandrashekar, A. Nag and D. Goswami, J. Am. Chem. Soc., 2005, 127, 11608–11609 CrossRef CAS PubMed.
- T. K. Ahn, J. H. Kwon, D. Y. Kim, D. W. Cho, D. H. Jeong, S. K. Kim, M. Suzuki, S. Shimizu, A. Osuka and D. Kim, J. Am. Chem. Soc., 2005, 127, 12856–12861 CrossRef CAS PubMed.
- Z. S. Yoon, D.-G. Cho, K. S. Kim, J. L. Sessler and D. Kim, J. Am. Chem. Soc., 2008, 130, 6930–6931 CrossRef CAS PubMed.
- T. Sarma, P. T. Anusha, A. Pabbathi, S. Venugopal Rao and P. K. Panda, Chem. – Eur. J., 2014, 20, 15561–15570 CrossRef CAS PubMed.
- S. Shimizu, Chem. Rev., 2017, 117, 2730–2784 CrossRef CAS PubMed.
- E. Tsurumaki, Y. Inokuma, S. Easwaramoorthi, J. Lim, D. Kim and A. Osuka, Chem. – Eur. J., 2009, 15, 237–247 CrossRef CAS PubMed.
- Y. Inokuma, S. Easwaramoorthi, S. Jang, K. Kim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 4840–4843 CrossRef CAS PubMed.
- C.-C. Yang, L. Li, W. Q. Tian, W.-Q. Li and L. Yang, Phys. Chem. Chem. Phys., 2022, 24, 13275–13285 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- S. P. T. Matsuda, W. K. Wilson and Q. Xiong, Org. Biomol. Chem., 2006, 4, 530–543 RSC.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- M. M. Alam, M. Chattopadhyaya, S. Chakrabarti and K. Ruud, Acc. Chem. Res., 2014, 47, 1604–1612 CrossRef CAS PubMed.
- M. M. Alam and K. Ruud, Mol. Phys., 2020, 118, e1777335 CrossRef.
- S. Pascal, S. David, C. Andraud and O. Maury, Chem. Soc. Rev., 2021, 50, 6613–6658 RSC.
- S. S. Rajput, R. Zaleśny and M. M. Alam, J. Phys. Chem. A, 2023, 127, 7928–7936 CrossRef CAS PubMed.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- R. Zaleśny, M. M. Alam, P. N. Day, K. A. Nguyen, R. Pachter, C. K. Lim, P. N. Prasad and H. Ågren, J. Mater. Chem. C, 2020, 8, 9867–9873 RSC.
- TURBOMOLE V7.3 2018, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.com.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- A. I. Krylov, J. Chem. Phys., 2020, 153, 080901 CrossRef CAS PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- H. S. Yu, X. He and D. G. Truhlar, J. Chem. Theory Comput., 2016, 12, 1280–1293 CrossRef CAS PubMed.
- A. M. Grabarz and B. Ośmiałowski, Molecules, 2021, 26, 7434 CrossRef CAS PubMed.
- A. Migliore, J. Chem. Theory Comput., 2019, 15, 4915–4923 CrossRef CAS PubMed.
- E. F. Petrusevich, B. Ośmiałowski, R. Zaleśny and M. M. Alam, J. Phys. Chem. A, 2021, 125, 2581–2587 CrossRef CAS PubMed.
- M. Chattopadhyaya, M. M. Alam and S. Chakrabarti, J. Phys. Chem. A, 2011, 115, 2607–2614 CrossRef CAS PubMed.
- B. Tejendra, S. S. Rajput and M. M. Alam, Chem. Phys. Chem., 2023, e202300710 Search PubMed.
- M. Guinra, N. Djiedeu, D. Bongué and C. Assongo Kenfack, Comput. Theor. Chem., 2022, 1217, 113910 CrossRef CAS.
- M. Uudsemaa, A. Trummal, S. de Reguardati, P. R. Callis and A. Rebane, Phys. Chem. Chem. Phys., 2017, 19, 28824–28833 RSC.
- S. S. Rajput and M. Alam, Chem. Phys. Chem., 2022, 23, e202200529 CrossRef CAS PubMed.
- M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, 1978, pp. 1–165 Search PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- A. Perfetto, F. Maschietto and I. Ciofini, J. Photochem. Photobiol. A: Chem, 2019, 383, 111978 CrossRef CAS.
- F. A. Korsaye, A. de la Lande and I. Ciofini, J. Comput. Chem., 2022, 43, 1464–1473 CrossRef CAS PubMed.
- L. Huet, A. Perfetto, F. Muniz-Miranda, M. Campetella, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2020, 16, 4543–4553 CrossRef CAS PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- C. Adamo, T. Le Bahers, M. Savarese, L. Wilbraham, G. García, R. Fukuda, M. Ehara, N. Rega and I. Ciofini, Coord. Chem. Rev., 2015, 304–305, 166–178 CrossRef CAS.
- P. Cronstrand, Y. Luo and H. Ågren, Chem. Phys. Lett., 2002, 352, 262–269 CrossRef CAS.
- M. T. Beerepoot, M. M. Alam, J. Bednarska, W. Bartkowiak, K. Ruud and R. Zaleśny, J. Chem. Theory Comput., 2018, 14, 3677–3685 CrossRef CAS PubMed.
- M. Chołuj, R. Behera, E. F. Petrusevich, W. Bartkowiak, M. M. Alam and R. Zaleśny, J. Phys. Chem. A, 2022, 126, 752–759 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Orbital diagrams, (2) absorption wavelength and orbital contribution, (3) natural transition orbital plots, (4) Cartesian coordinates of ground state optimized geometry of all the systems, (5) magnitudes of different transition dipole moment vectors, and (6) two-photon absorption cross-section. See DOI: https://doi.org/10.1039/d4cp00438h |
| This journal is © the Owner Societies 2024 |