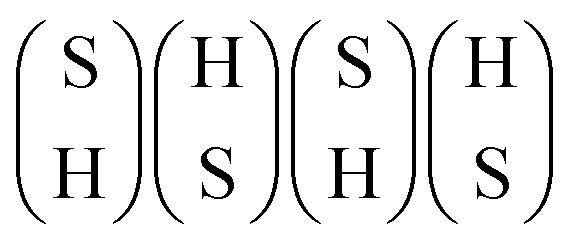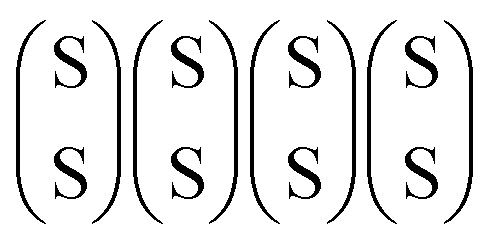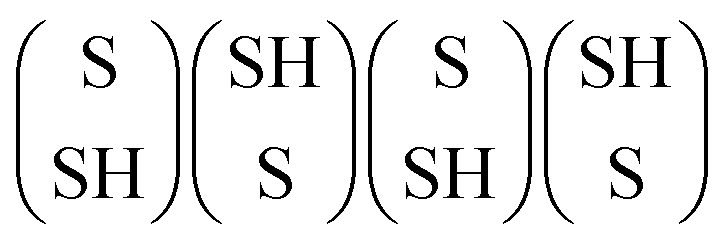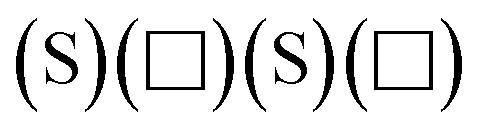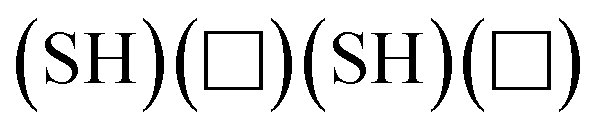Theoretical insight into the rearrangement of sulfur atoms on the Ni- and Cu-doped MoS2 S-edge induced by hydrogen adsorption under HDS reaction conditions†
Received
29th January 2024
, Accepted 25th March 2024
First published on 25th March 2024
Abstract
Density functional theory (DFT) calculations and an atomistic thermodynamic approach were used to study the geometric rearrangement of sulfur atoms on the Ni- and Cu-doped MoS2 S-edge upon hydrogen adsorption. Under HDS conditions, thermodynamically stable hydrogenated structures were identified as SH groups on the undoped S-edge with 100% sulfur coverage, on the Ni-doped S-edge with 50% sulfur coverage and on the Cu-doped S-edge with 25% sulfur coverage. It was found that the rearrangement of the S atoms is essential to reach the most stable state at the edge for the undoped and Ni-doped S-edge. Hydrogen adsorption on the Ni-doped S-edge leads to the greatest amount of S rearrangement (ΔERearrang = 0.93 eV/H2). Our results suggest that under the reaction conditions, the H2 dissociative adsorption process is strongly coupled to the rearrangement of the sulfur atoms. By examining the differential hydrogen adsorption energy on the most stable edge structures, we found a plausible explanation for the trend in the hydrogenation activity of the doped edges. Our results suggest that Ni enhances the hydrogenation activity of the S-edge by decreasing the S–H bond strength, while Cu poisons it by increasing the S–H bond strength.
1. Introduction
Molybdenum disulfide (MoS2) based catalysts are commonly used in oil refineries for hydrotreating, a process that converts crude oil into clean transportation fuels.1,2 Promoting MoS2 with Co or Ni increases the hydrotreating activity by more than one order of magnitude, and the selectivity for the hydrodesulfurization (HDS), hydrodenitrogenation (HDN), and hydrogenation reactions is significantly modified.3–5 The enhanced activity is attributed to the formation of the so-called “Co–Mo–S” phase.6 The Co–Mo–S phase model, now widely accepted and adapted to Ni promotion (Ni–Mo–S phase), proposes that the Co or Ni atoms are located at the edges of the MoS2 slabs.7 In this context, Kibsgaard et al.5 performed a comparative analysis using scanning tunneling microscopy (STM) to study the atomic-scale structure and morphology of MoS2 nanoparticles doped with first-row transition metals (Fe, Co, Ni, and Cu). Their results showed that the addition of all four dopant metals leads to the formation of Co–Mo–S type structures, which take the shape of hexagonally truncated triangular MoS2 nanoparticles. The dopants were found to be preferentially located on the S-edge.
Recent experimental studies have investigated the intrinsic catalytic activity of MoS2 catalysts doped with first-row transition metals in HDS and hydrogenation reactions. These studies have shown that Ni doping enhances the catalytic activity compared to the MoS2 reference, while Cu doping has a negative effect, reducing the catalytic activity.5,8 The active sites on the doped and undoped MoS2 catalysts may involve coordinatively unsaturated sites (CUSs), sulfhydryl (SH) groups, and brim sites. CUSs are vacancies of S2− anions located at the edge or corner of the nanoparticles.9 The SH groups are formed by hydrogen dissociative adsorption on the outermost edge S atoms and behave as Brønsted acid sites.10,11 The brim sites, which can be seen in STM images as a bright brim adjacent to the MoS2 nanoparticle edge, exhibit metallic characteristics because of the presence of one-dimensional electronic edge states.12,13 There is a broad consensus in the literature that the HDS activity of the MoS2-based catalyst proceeds through two parallel pathways: the direct desulfurization (DDS) pathway and the hydrogenation (HYD) pathway.14–17 In the DDS pathway, the sulfur atom is directly removed from the molecule upon hydrogenolysis of the C–S bond, whereas the HYD pathway involves several hydrogenation steps before sulfur extrusion occurs. For sterically hindered alkyl-substituted compounds such as 4,6-dimethyldibenzothiophene (DMDBT), the DDS pathway is often suppressed and the HYD pathway becomes the dominant pathway because it is less sensitive to steric effects. As a result, the HYD pathway becomes increasingly important as crude oils become heavier and contain more refractory compounds.18 Both reaction pathways require the activation of H2. It is proposed that the S–H groups, which are formed by the dissociation of H2 at the edges, drive the hydrogenation steps that precede the sulfur extrusion process.19,20
Determining the state of adsorbed hydrogen on MoS2-based catalysts is fundamental to understand the overall catalytic process of the HDS reaction. Two types of H2 dissociation mechanisms have been proposed: heterolytic dissociation, which produces a molybdenum hydride species and an SH group, and homolytic dissociation, which produces two SH groups. Therefore, several theoretical studies have focused on the activation of H2 by the MoS2 edges.21–26 Some of these studies have reported that H-adsorption induces rearrangement of the edge sulfur atoms. Specifically, the terminal S2 dimers split upon H adsorption, resulting in a significant shift of the S atoms.21,25,26 Tsai et al.27 pointed out that adsorbates often induce significant reorganization of the S atoms at the edge. This generally involves repulsive interactions between neighboring S atoms, as well as the breaking of an S–S bond if the adsorbate is bound to an S2 dimer. Salazar et al.28 combined STM experiments and density functional theory (DFT) calculations to investigate the adsorption of thiophene on S-vacancies at the Mo-edge of MoS2 nanoparticles. The study found that thiophene adsorbs directly on open S-vacancy sites at the corners between the edges. However, adsorption on S-vacancy sites located at the interior of the edge occurs via a concerted displacement of a neighboring S atom. This leads to the simultaneous formation of an S2 dimer and thiophene adsorbed on a double S-vacancy CUS. Their results suggest that these dynamic sulfur rearrangements are generally relevant to HDS reactions of a wider range of compounds. This explains why there is enough space for molecules such as dibenzothiophene to adsorb and undergo DDS on the edges of MoS2. Thus, the adsorption-induced structural response at the MoS2 edges demonstrates the dynamic nature of the catalyst under reaction conditions.
In this work, we investigated the effect of hydrogenation on the energetic stability, structural, and electronic properties of undoped and Ni- and Cu-doped MoS2 S-edges under HDS reaction conditions using DFT calculations and an atomistic thermodynamic approach. We aim to understand how the formation of S–H bonds balances the energy cost associated with the rearrangement of the edge S atoms, and ultimately leads to the overall stabilization of the system. We also investigated the extent to which doping modifies the S–H bond strength, which is a key factor in the hydrogenation functionality of the catalyst.
2. Computational details
STM studies have demonstrated the favorable total substitution of Mo atoms at the S-edge with Fe, Co, Ni, and Cu atoms.5,29 This has also been confirmed by previous DFT studies.8,30 Therefore, our investigation focuses specifically on the S-edge, considering a complete replacement of the outermost Mo atoms with either Ni or Cu atoms. The slab supercell to model the doped S-edge is shown in Fig. 1. This slab exposes the dopant-substituted and fully S-saturated S-edge at the top, and the bare Mo-edge at the bottom before geometric optimization. Periodic boundary conditions were used, with 15 Å of vacuum in the y-direction and 9 Å of vacuum in the z-direction. The cell parameters used were 12.625, 26.845, and 12.068 Å. It is worth noting that the periodicity of the supercell in the x-direction could affect the stable S coverage, the formation of metal–metal dimers, and the S rearrangement. We choose a supercell size of four Mo in the x-direction to make a direct comparison with previous DFT studies on edge doping.8,30,31
 |
| | Fig. 1 Periodic model of the doped MoS2 monolayer exposing the S-edge at the top. The turquoise, yellow, and purple spheres are the molybdenum, sulfur, and dopant atoms, respectively. | |
Previous DFT studies have reported that the effect of transition metal substitution is to modify the binding strength of S at the edge, which is reflected in the S coverage.8,31 A decrease in sulfur coverage is observed as the filling of the 3d band of the dopant metal increases. Several sulfur coverages were investigated, ranging from 12.5% to 100%, corresponding to 1 to 8 sulfur atoms at the edge. The sulfur coverages considered vary for the undoped S-edge from 50% to 100%, for the Ni-doped S-edge from 37.5% to 100%, and the Cu-doped edge from 12.5% to 100%. For the H species adsorbed on the edge, the adsorption of up to four H atoms was considered, corresponding to hydrogen coverages of 25%, 50%, 75%, and 100%. Our focus is only on the fully hydrogenated edges.
Spin-polarized DFT calculations were performed using the Vienna Ab Initio Simulation Package (VASP).32,33 The Perdew–Burke–Ernzerhof exchange–correlation functional with a generalized gradient approximation was used.34 The electron–ion interactions were described employing the projector augmented wave (PAW) method,35 and standard PAW potentials were used for all atoms with the following electron valence distributions: Mo 5s14d5, Ni 4s23d8, Cu 4s13d10, S 3s23p4, and H 1s1. A plane-wave kinetic energy cut-off of 400 eV was employed. To describe the partial occupancies of the electronic states, we used the Gaussian smearing method with a width of 0.05 eV and a convergence criterion of 10−5. Similarly, the conjugate gradient algorithm was applied for ionic relaxation until convergence was reached with a criterion of 10−4 eV. All calculations include dipole corrections due to the nonsymmetric slab supercells.8 A Monkhorst–Pack k-point mesh of 3 × 1 × 1 was used for the slab calculations. All atoms were fully relaxed during the calculations. Bader charge36 and electron density difference (EDD) analyses were performed to evaluate changes in the electronic structure. The EDD was calculated from the following equation:
| | | Δρ = ρ(edge + adsorbate) − ρ(edge) − ρ(adsorbate) | (1) |
where
ρ(edge + adsorbate) is the electron density of the whole system,
ρ(edge) and
ρ(adsorbate) are the electron densities of the edge, and the adsorbate is calculated at the adsorption positions, respectively. Δ
ρ provides information about the electron density distribution upon adsorption, where positive values indicate a density gain and negative values indicate density loss. The VESTA program was used to visualize the electron density difference isosurfaces.
37
The differential hydrogen adsorption energy (ΔEH) for the final adsorbed H was calculated using the following equation:
| |  | (2) |
where
E(edge +
nH) is the total energy for the S-edge structure with
n adsorbed hydrogen atoms,
E(edge + (
n–1)H) is the total energy for (
n–1) adsorbed hydrogen atoms and
E(H
2) is the energy of a gas phase hydrogen molecule.
2.1. Thermodynamic approach
To evaluate the stability of the S-edge at different sulfur coverages, we use the ab initio atomistic thermodynamics formalism proposed by Scheffler and Reuter.38 This formulation assumes that the catalyst surface is in equilibrium with the gas phase and bulk reservoirs within the grand canonical ensemble. The stability of a given MzMoxSy edge composition can be determined by calculating the grand potential Ω(T,p), at temperature T and partial pressure p of the gas phase components. The thermodynamic formula for Ω(T,p) is| |  | (3) |
where  is the DFT total energy of the doped slab, containing x molybdenum atoms, y sulfur atoms, and z dopant atoms (M = Ni or Cu) and
is the DFT total energy of the doped slab, containing x molybdenum atoms, y sulfur atoms, and z dopant atoms (M = Ni or Cu) and  is the DFT total energy of the dopant bulk phase (Ni3S2 and CuS) stable under the reaction conditions.39μs(T,p) is the chemical potential of sulfur, and it is determined by the chemical equilibrium with the gas phase mixture of H2 and H2S, giving
is the DFT total energy of the dopant bulk phase (Ni3S2 and CuS) stable under the reaction conditions.39μs(T,p) is the chemical potential of sulfur, and it is determined by the chemical equilibrium with the gas phase mixture of H2 and H2S, giving| |  | (4) |
ΔEmol is the difference between the DFT total energy of the H2S and H2 molecules  , including zero-point vibrations. The term
, including zero-point vibrations. The term  corresponds to the difference between the thermal contributions to the chemical potential, which is calculated as
corresponds to the difference between the thermal contributions to the chemical potential, which is calculated as  and can be obtained from thermodynamic tables.40 Here, pH2S and pH2 denote the partial pressures of H2S and H2, respectively. The ratio
and can be obtained from thermodynamic tables.40 Here, pH2S and pH2 denote the partial pressures of H2S and H2, respectively. The ratio  is used to indicate the relative abundance of H2S and H2 in the gas phase, with larger values indicating sulfur-rich conditions and smaller values indicating strongly reducing conditions where H2 is more abundant than H2S. Under HDS working conditions, the
is used to indicate the relative abundance of H2S and H2 in the gas phase, with larger values indicating sulfur-rich conditions and smaller values indicating strongly reducing conditions where H2 is more abundant than H2S. Under HDS working conditions, the  ratio is typically between 0.01 and 0.05, while the temperature is in the range of 573 K to 700 K. For instance, representative HDS working conditions may include pH2 = 10 bar, pH2S = 0.1 bar, and T = 650 K.25
ratio is typically between 0.01 and 0.05, while the temperature is in the range of 573 K to 700 K. For instance, representative HDS working conditions may include pH2 = 10 bar, pH2S = 0.1 bar, and T = 650 K.25
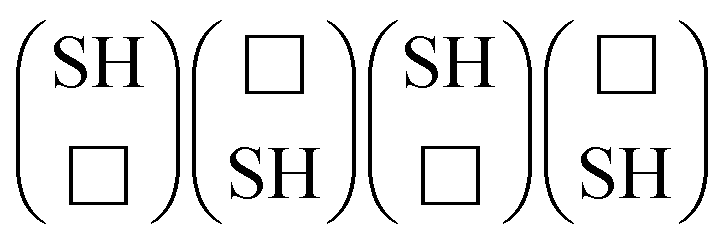
For the hydrogenated edges, the grand potential can be expressed as
| | 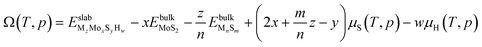 | (5) |
where
| |  | (6) |
3. Results and discussion
3.1. Hydrogen adsorption on an undoped S-edge
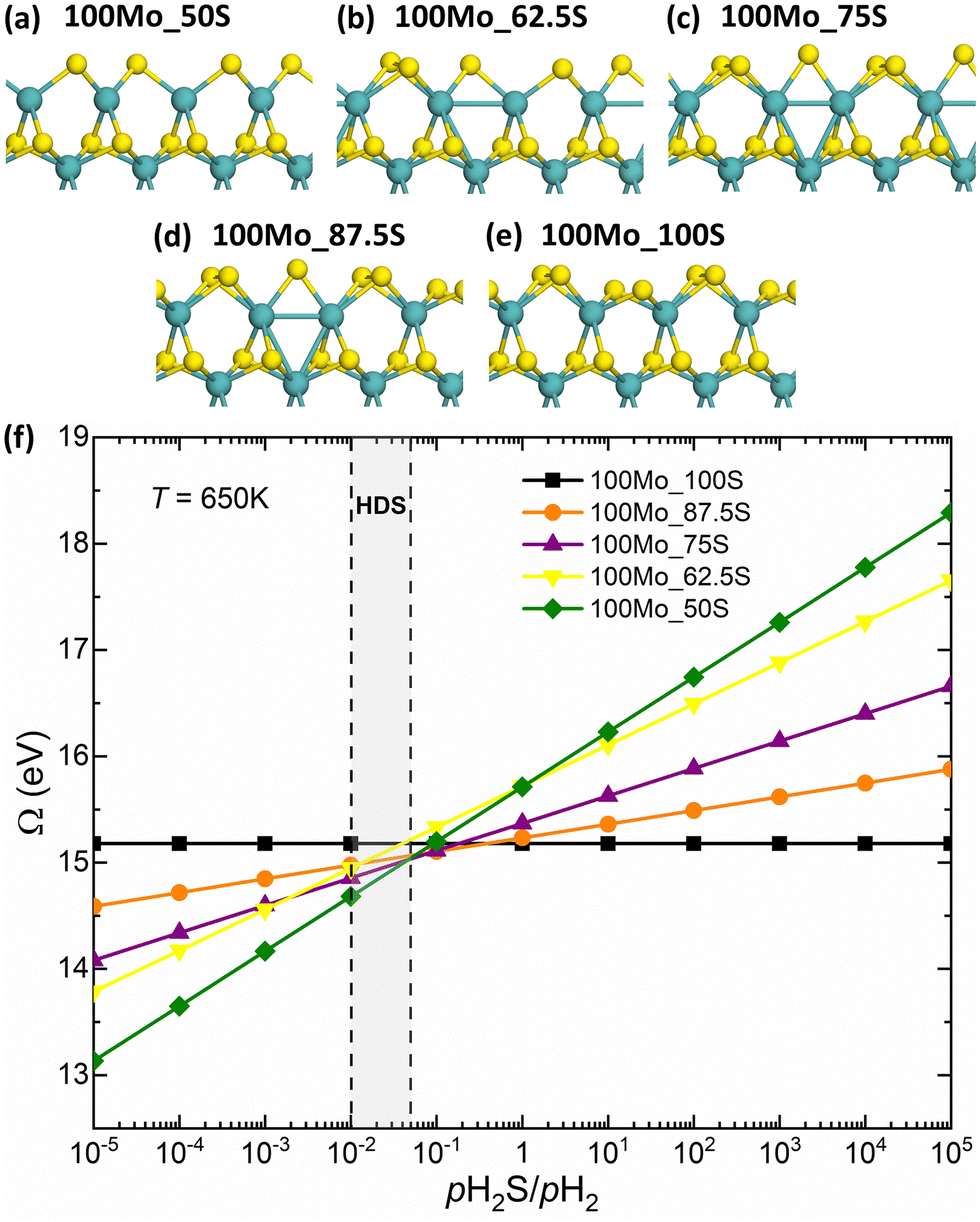
To accurately describe hydrogen adsorption at the edge, the first step is to identify the stable edge configurations under the given reaction conditions. For a given sulfur coverage, several S adsorption configurations are possible. Fig. 2a–e show the most favorable edge configurations of the undoped S-edge for sulfur coverages between 50% and 100%. The edge configurations evaluated for each sulfur coverage are shown in Fig. S1 (ESI†). To evaluate the relative stability of the edge structures, we calculated the grand potential as a function of the pH2S/pH2 partial pressure ratio at 650 K, as shown in Fig. 2f. At high pH2S/pH2 ratios (sulfiding conditions), the most stable sulfur coverage is 100%, with the S atoms forming S2 dimers every two bridging S atoms (100Mo_100S, Fig. 2e). Under typical HDS reaction conditions (T = 650 K, pH2 = 10 bar, pH2S/pH2 = 0.05–0.01), the most stable sulfur coverage is 50%, with the S atoms arranged in a zigzag configuration (100Mo_50S, Fig. 2a), as reported in previous DFT studies.41,42
 |
| | Fig. 2 (a)–(e) Optimized structures of the undoped S-edge for sulfur coverages between 50% and 100%. The turquoise and yellow spheres are the molybdenum and sulfur atoms, respectively. (f) Grand potential as a function of pH2S/pH2 at 650 K. | |
As mentioned above, HDS operating conditions are sulfo-reductive conditions where H2 is more abundant than H2S in the gas phase. Hydrogen can interact with the edges of MoS2 in two ways. First, the reaction of H2 with the edge S atoms can reduce the sulfur coverage by forming H2S. Second, the dissociative adsorption of H2 can lead to the formation of SH or MoH groups. Therefore, we investigated the adsorption of hydrogen on the different structures in Fig. 2. For the undoped S-edge, the fully hydrogenated structures correspond to the dissociative adsorption of two H2 molecules on the edge. The most favorable configurations of the hydrogenated S-edge for sulfur coverages between 50% and 100% are shown in Fig. 3. The edge configurations evaluated for H adsorption at each sulfur coverage are shown in Fig. S2 (ESI†). For the 50% sulfur coverage, the most favorable configuration corresponds to four bridging MoH groups (100Mo_50S_100H, Fig. 3a). This result is in agreement with that reported by Bruix et al.43 for triangular MoS2 nanoparticles exposing S-edges with 50% sulfur coverage. These authors suggested that MoS2-based catalysts featuring edges with undercoordinated Mo atoms can accommodate H atoms in such bridging positions. This behavior is also observed for sulfur coverages between 62.5% and 87.5%. At 62.5% sulfur coverage, the adsorbed H atoms form three bridging MoH groups and one SH group (100Mo_62.5S_100H, Fig. 3b). At 75% sulfur coverage, they form two bridging MoH and two SH groups (100Mo_75S_100H, Fig. 3c). At 87.5% sulfur coverage, one bridging MoH and three SH groups are present (100Mo_87.5S_100H, Fig. 3d). At 100% sulfur coverage, the most favorable configuration corresponds to four alternating SH groups (100Mo_100S_100H, Fig. 3e). It is important to note that all of these edge structures have sulfur atoms arranged in a zigzag configuration without hydrogen adsorption (Fig. S2, ESI†). Thus, these structures can be seen as the result of the adsorption of H or SH groups on the 100Mo_50S structure.
 |
| | Fig. 3 (a)–(e) Optimized structures for different sulfur coverages of the undoped S-edge with 100% of hydrogen coverage. The turquoise, yellow, and white spheres are the molybdenum, sulfur, and hydrogen atoms, respectively. (f) Grand potential as a function of pH2S/pH2 at 650 K and pH2 = 10 bar. | |
Fig. 3f shows the grand potential as a function of the pH2S/pH2 ratio at 650 K for the hydrogenated edge structures. The 100Mo_100S_100H structure shows the highest stability over a wide range of pH2S/pH2 ratios. However, under strong reducing conditions, the 100Mo_50S structure is the most stable. Note that the 100Mo_50S structure is always more stable than the 100Mo_50S_100H structure, indicating that MoH or SH groups would not be present at low sulfur coverages, in agreement with Prodhomme et al.25 Under HDS reaction conditions, the 100Mo_100S_100H structure is the most stable. The formation of the SH groups at 100% sulfur coverage has a strong stabilizing effect on the edge structure. It is worth noting that the hydrogenated structures 100Mo_62.5S_100H, 100Mo_75S_100H, and 100Mo_87.5S_100H are more stable than the 100Mo_50S structure and their stability increases as the sulfur coverage increases. Therefore, the mechanism of H2 dissociation depends on the sulfur coverage of the edge where the dissociation occurs. Homolytic dissociation of H2, leading exclusively to SH groups, is favored only at the fully sulfided S-edge, whereas the heterolytic dissociation of H2, forming MoH and SH groups, is favored at the partially sulfided edges.
Upon hydrogen adsorption, the S2 dimers split, resulting in a shift of the S atoms. Therefore, we calculated the dissociative adsorption energy of H2 and the energy cost associated with the rearrangement of S atoms for the 100Mo_100S_100H structure. The S atoms rearrangement energy (ΔERearrag) was calculated as the energy difference between the frozen edge in the geometry after hydrogen adsorption and the edge in its relaxed geometry before hydrogen adsorption. The dissociative adsorption energy of two H2 molecules is exothermic by −2.27 eV (−1.14 eV/H2) and the corresponding S rearrangement energy is 1.34 eV (0.67 eV/H2) (Table 1). Thus, hydrogen adsorption involves a dynamic movement of the edge atoms in the process, which can be attributed to the large bending flexibility of the metal-sulfur bonds at the edges.21
Table 1 The dissociative adsorption energy of H2 and the corresponding S rearrangement energy for the most stable edge structures under reaction conditions
| Edge configuration |
ΔEads (H2) (eV/H2) |
ΔERearrag (eV/H2) |
| 100Mo_100S_100H |
−1.14 |
0.67 |
| 100Ni_50S_100H |
−0.78 |
0.93 |
| 100Cu_25S_50H |
−1.84 |
0.18 |
The difference in the stability of the hydrogen species adsorbed at the edge may lie in the electronic properties, specifically in the reducibility of the Mo atoms involved. The stabilization of hydrogen as a proton species is formally an oxidation process and requires the reduction of Mo atoms, whereas the stabilization of hydrogen as a hydride species is a reduction process and involves the oxidation of Mo atoms.44Table 2 shows the Bader charges (q) of the edge atoms for the non-hydrogenated and hydrogenated structures with 50% and 100% sulfur coverage. The various adsorbed species at the edge with their respective formal charges are H−, SH−, S2−, and S22−. When H adsorbs to form SH groups in the 100Mo_50S structure, the excess electrons partially reduce the neighboring Mo atoms by about 0.17 e. Conversely, H adsorption as a hydride oxidizes the Mo atoms by about 0.12 e. Each H atom carries a negative charge of −0.31 e. For the S-dimer 100Mo_100S structure, the charge on the S atoms is found to be more negative as S2− anions (−0.57 e) than as S22− dimers (−0.26 e), consistent with the fact that the S atoms in the dimers are low-valence sulfur species, S−.45 Thus, by comparing the S-dimer and the S-bridge 100Mo_100S structures, it is observed that the replacement of the S22− dimers by two S2− anions has an oxidizing effect on the neighboring Mo atoms (each Mo atom loses about 0.08 e). For the 100Mo_100S_100H structure, it was found that the charge on the Mo atoms is almost the same as in the 100Mo_100S structure containing S22− dimers (+1.25 e). This may be because the charge on the S atoms of the S22− dimers is the same as that of the SH− species (−0.26 e). Also, the charge of the S2− anions is almost the same in both structures (−0.56 e). Therefore, the formation of SH groups in the S-dimer 100Mo_100S structure does not involve the reduction of Mo atoms. However, if the S-bridge 100Mo_100S structure is taken as a reference for the non-hydrogenated structure, the reduction of the Mo atoms is observed upon the formation of SH groups (each Mo atom gains 0.07 e). This process is also accompanied by the oxidation of the S atoms belonging to the SH groups. Recently, Khare et al.44 pointed out that a higher electron density on Mo atoms should favor hydride formation, while a lower electron density on Mo atoms should favor sulfhydryl (SH) formation. This could explain the remarkable stability of the SH group formation on the 100Mo_100S structure compared to the 100Mo_50S structure. On the S-edge, Mo atoms with lower coordination tend to localize a higher electron density. Therefore, the formation of SH groups on the 100Mo_50S structure would reduce the Mo atoms to a less favorable oxidation state. However, the adsorption of H as hydride species on the 100Mo_50S structure is also less stable.
Table 2 Bader charges (q) for the non-hydrogenated and hydrogenated structures with 50% and 100% sulfur coverage for the undoped S-edge
| Edge configuration |
q
Mo (e)
|
q
S (e)
|
q
S(S2) (e)
|
q
S(SH) (e)
|
q
H (e)
|
q
H(SH) (e)
|
|
q
Mo charge of the edge Mo atoms.
q
S charge of the single S atoms.
q
S(S2) charge of the S atom in the dimer.
q
S(SH) charge of the S atom in the SH group.
q
H charge of the single H atoms.
q
H(SH) charge of the H atom in the SH group.
|
100Mo_50S  |
+1.15 |
−0.68 |
— |
— |
— |
— |
100Mo_50S_100H (S–H)  |
+0.98 |
— |
— |
−0.47 |
— |
+0.02 |
100Mo_50S_100H (Mo–H)  |
+1.27 |
−0.59 |
— |
— |
−0.31 |
— |
100Mo_100S (S-dimer)  |
+1.24 |
−0.57 |
−0.26 |
— |
— |
— |
100Mo_100S (S-bridge)  |
+1.32 |
−0.48 |
— |
— |
— |
— |
100Mo_100S_100H  |
+1.25 |
−0.56 |
— |
−0.29 |
— |
+0.03 |
As mentioned above, the hydrogenated structures 100Mo_50S_100H and 100Mo_100S_100H can be seen as a result of the adsorption of H or SH groups, respectively, onto the 100Mo_50S structure. To explain their different stability, it is necessary to understand the nature of the bonding between the H− and SH− species and the 100MoS_50S edge structure. We calculated the electron density difference to illustrate the redistribution of electron density due to the interaction between these species and the edge. In the EDD plots in Fig. 4, the blue regions represent electron density accumulation and the red regions represent electron density depletion. As shown in Fig. 4(a) and (b), the electron density accumulates between Mo–SH bonds but depletes between Mo–H bonds, suggesting a stronger interaction between SH− species and Mo atoms. This may explain why stabilizing hydrogen as sulfhydryl groups at the S-edge is more thermodynamically favorable than stabilizing as hydrides.
 |
| | Fig. 4 Top view of the EDD plots for the (a) 100Mo_50S_100H and (b) 100Mo_100S_100H structures. The blue and red regions represent the electron density gain and loss, respectively. The EDD was plotted using an isodensity level of 0.0035 e borh−3. | |
3.2. Hydrogen adsorption on Ni-doped S-edge
To begin with the study of hydrogen adsorption on the Ni-doped S-edge, we first evaluated different configurations for S adsorption for sulfur coverages between 37.5% and 100%. Fig. 5a–f show the most favorable edge configurations. The edge configurations evaluated for each sulfur coverage are shown in Fig. S3 (ESI†). For sulfur coverages between 37.5% and 75%, Ni atoms tend to form Ni–Ni dimers as a result of the adsorption of S2 dimers between them. This local edge reconstruction causes the Ni atoms to form a local structure with the S atoms that closely resembles a square planar environment, as previously reported in DFT studies.8,30 Therefore, at low sulfur coverages, the S2 dimers are located between two Ni–Ni dimers, while at high sulfur coverages, the S2 dimers are located on top of the Ni atoms. This keeps the coordination number of the Ni atoms close to 4. Fig. 5g shows the grand potential as a function of the pH2S/pH2 ratio at 650 K for the Ni-doped edge structures. It can be seen that under strong sulfiding conditions, the most stable sulfur coverage is 100% (100Ni_100S, Fig. 5f), while under strong reducing conditions the most stable sulfur coverage is 37.5% (100Ni_37.5S, Fig. 5a). Over a wide range of pH2S/pH2 ratios, including HDS reaction conditions, the most stable sulfur coverage is 50% (100Ni_50S, Fig. 5b).
 |
| | Fig. 5 (a)–(f) Optimized structures of the Ni-doped S-edge for sulfur coverages between 37.5% and 100%. The turquoise, blue, and yellow spheres are the molybdenum, nickel, and sulfur atoms, respectively. (g) Grand potential as a function of pH2S/pH2 for the Ni-doped S-edge at 650 K. | |
Experimental studies have shown that SH groups are the stable form of activated H on Ni-doped MoS2.46,47 To evaluate the formation of SH groups on the Ni-doped S-edge, we calculated the adsorption of hydrogen on the edge structure with 50% sulfur coverage, which is reported in Fig. 5 as the most stable under HDS reaction conditions. We also included the edge structure with 37.5% sulfur coverage to account for the formation of a sulfur vacancy at the edge. Fig. 6a–d show the two configurations evaluated for H adsorption for sulfur coverages of 37.5% and 50%. For instance, in the bridge-labeled configurations, the S atoms are adsorbed in a bridge position, while in the dimer-labeled configurations, the sulfur atoms are adsorbed as S2 dimers between Ni–Ni dimers. Fig. 6d shows the grand potential as a function of the pH2S/pH2 ratio at 650K for these hydrogenated edge structures. It can be seen that the 100Ni_50S_100H_bridge structure is the most stable in almost the entire range of pH2S/pH2 ratios studied. It should be noted that the 100Ni_50S_100H_dimer structure is less stable than the 100Ni_50S structure, indicating that stable SH groups are not present in this configuration under reaction conditions. Therefore, the 100Ni_50S structure undergoes significant S rearrangement upon hydrogen adsorption. We found that the dissociative adsorption energy of two H2 molecules to obtain the 100Ni_50S_100H_bridge structure is exothermic to −1.55 eV (−0.78 eV/H2), and the energy cost associated with the S rearrangement is 1.85 eV (0.93 eV/H2) (Table 1). Furthermore, by removing an S atom from the edge, the most stable hydrogenated structure is 100Ni_37.5S_75H_bridge. Note that this structure is more stable than the 100Ni_50S structure under reducing conditions, including HDS conditions, but its stability decreases when moving to sulfiding conditions. However, in the absence of hydrogen, the S-bridge configurations are less stable than the S-dimer configurations (Fig. S3, ESI†). Therefore, the formation of SH groups at the Ni-doped edge has a strong stabilizing effect despite the significant rearrangement of the S atoms. With this result, we could speculate that the dissociative adsorption of H2 and the displacement of S atoms may occur with a concerted mechanism. However, a detailed kinetic analysis of such a concerted displacement is beyond the scope of the present work.
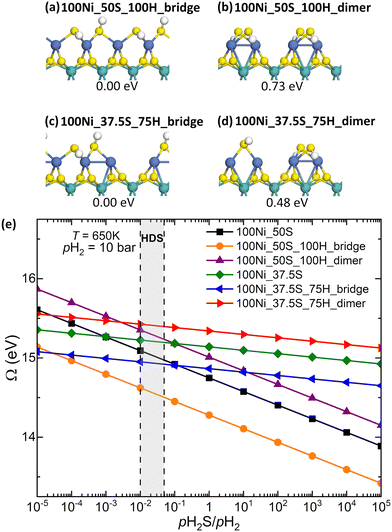 |
| | Fig. 6 Optimized structures for different configurations of the 100Ni_50S_100H structure, (a) 100Ni_50S_100H_bridge and (b) 100Ni_50S_100H_dimer, and the 100Ni_37.5S_75H structure, (c) 100Ni_37.5S_75H_bridge and (d) 100Ni_37.5S_75H_dimer. For a given sulfur coverage, the configurations of hydrogen adsorption are shown in order of decreasing stability. The turquoise, blue, yellow, and white spheres are the molybdenum, nickel, sulfur, and hydrogen atoms, respectively. € Grand potential as a function of pH2S/pH2 at 650 K and pH2 = 10 bar. | |
To gain insight into the underlying reason for the difference in stability between the 100Ni_50S_100H_dimer and 100Ni_50S_100H_bridge structures, Bader charge and electron density difference analyses were performed. Table 3 presents the Bader charges (q) of the edge atoms for the non-hydrogenated and hydrogenated structures of the dimer and bridge configurations with 50% sulfur coverage. When comparing the 100Ni_50S_dimer and 100Ni_50S_bridge structures, the oxidation of the Ni atoms is observed by replacing the S22− dimers with two S22− anions (each Ni atom loses about 0.07 e). This is also reflected in the calculated charges around the different sulfur species; the charge of the S2− anions (−0.31 e) is more negative than around the S22− dimers (−0.18 e). The formation of the SH groups has a weak oxidizing effect on the Ni atoms compared to the 100Ni_50S_dimer structure. For the 100Ni_50S_100H_dimer structure, the Ni atoms are oxidized by about 0.03 e upon hydrogen adsorption. In the 100Ni_50S_100H_bridge structure, each Ni atom loses about 0.04 e. However, the reduction of Ni atoms during the formation of SH groups is observed if the 100Ni_50S_bridge structure is taken as a reference for the non-hydrogenated structure. As in the undoped edge, this process also involves the oxidation of the S atoms belonging to the SH groups. Fig. 7a and b show the EDD plots for the 100Ni_50S_100H_bridge and 100Ni_50S_100H_dimer structures, respectively, using the densities of the SH groups as adsorbate references. In the 100Ni_50S_100H_bridge structure, there is a significant accumulation of electron density between the Ni–S bond, suggesting a strong interaction between the SH− species and the Ni atoms.48 In the 100Ni_50S_100H_dimer structure, a depletion region is observed close to the Ni atoms, which is not present in the 100Ni_50S_100H_bridge structure. Therefore, a large electron density redistribution occurs upon the rearrangement of S atoms. The movement of the S atoms during H adsorption may be driven by the electron density redistribution to reduce the repulsive interactions between the SH groups.
Table 3 Bader charges (q) of the non-hydrogenated and hydrogenated structures with 50% sulfur coverage for the Ni-doped S-edge structures
| Edge configuration |
q
Ni (e)
|
q
S (e)
|
q
S(S2) (e)
|
q
S(SH) (e)
|
q
H(SH) (e)
|
100Ni_50S_dimer  |
+0.43 |
— |
−0.18 |
— |
— |
100Ni_50S_bridge  |
+0.50 |
−0.31 |
— |
— |
— |
100Ni_50S_100H_dimer  |
+0.46 |
— |
— |
−0.23 |
+0.03 |
100Ni_50S_100H_bridge  |
+0.47 |
— |
— |
−0.27 |
+0.02 |
 |
| | Fig. 7 Top view of the EDD plots for the (a) 100Ni_50S_100H_bridge and (b) 100Ni_50S_100H_dimer structures. The blue and red regions represent the electron density gain and loss, respectively. The EDD was plotted using an isodensity level of 0.003 e borh−3. | |
3.3. Hydrogen adsorption on Cu-doped S-edge
Fig. 8a–h show the most favorable configurations for the Cu-doped S-edge with sulfur coverages between 12.5% and 100%. The edge configurations evaluated for each sulfur coverage are shown in Fig. S4 (ESI†). At low sulfur coverages (12.5% and 37.5%), Cu atoms tend to form Cu–Cu dimers with only one S atom adsorbed between them. For sulfur coverages between 37.5% and 75%, the edge structures contain S2 dimers. Each S atom of the dimer is bonded to a single Cu atom. This keeps the coordination number of Cu atoms close to 3, and the local structure that Cu atoms form with S atoms resembles a trigonal planar environment. At high sulfur coverages (87.5% and 100%), the edge structures are more complex. Here, each S atom of the dimer is bonded to an S atom from the edge. Fig. 8i shows the grand potential as a function of the pH2S/pH2 ratio at 650 K for the Cu-doped edge structures. It can be seen that under strong sulfiding conditions, the most stable sulfur coverage is 50% (100Cu_50S, Fig. 8d). On the other hand, over a wide range of pH2S/pH2 ratios, including reducing, HDS, and moderate sulfiding conditions, the most stable sulfur coverage is 25% (100Cu_25S, Fig. 8b). This result is consistent with the result reported by Tsai et al.31 They found that group 11 metal dopants tend to have the lowest sulfur coverage, group 8 to 10 metal dopants have higher sulfur coverage, and group 5 to 7 metal dopants have the highest sulfur coverage.
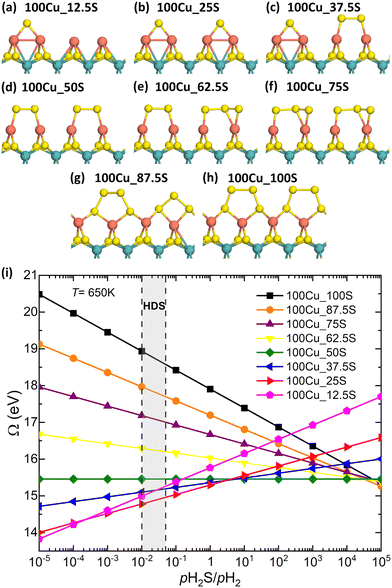 |
| | Fig. 8 (a)–(h) Optimized structures of the Cu-doped S-edge for sulfur coverages between 12.5% and 100%. The turquoise, orange, and yellow spheres are the molybdenum, copper, and sulfur atoms, respectively. (i) Grand potential as a function of pH2S/pH2 for the Cu-doped S-edge at 650 K. | |
To study the Cu-doped hydrogenated edge, we calculated the hydrogen adsorption on the edge structure with 25% sulfur coverage, which is shown in Fig. 8 as the most stable under HDS reaction conditions. We also included the edge structure with 12.5% sulfur coverage to account for the formation of a sulfur vacancy at the edge. For the Cu-doped S-edge, the fully hydrogenated edge results from the dissociative adsorption of one H2 molecule at the edge. Fig. 9a and b show the side views for the optimized structures for H adsorption for sulfur coverages of 12.5% and 25%. Fig. 9c shows the grand potential as a function of the pH2S/pH2 partial pressure ratio at 650 K. It can be seen that the 100Cu_25S_50H structure is the most stable over the whole range of pH2S/pH2 ratios studied. On the other hand, the 100Cu_12.5S_25H structure is more stable than the 100Cu_25S structure over a wide range of pH2S/pH2 ratios, including reducing, HDS, and moderate sulfiding conditions. Again, we found that the formation of SH groups has a strong stabilizing effect on the edge structure. We also found that the dissociative adsorption energy of an H2 molecule is exothermic to −1.84 eV, and the S rearrangement energy is 0.18 eV (Table 1). Therefore, when H is adsorbed, the rearrangement of the S atoms is negligible. Table 4 shows the Bader charges (q) of the edge atoms for the non-hydrogenated and hydrogenated structures with 25% sulfur coverage. In the 100Cu_25S_50H structure, the formation of the SH groups has a weak reducing effect on the Cu atoms compared to the 100Cu_25S structure (each Cu atom gains 0.04 e). The oxidation of S atoms belonging to SH groups is also observed.
 |
| | Fig. 9 Optimized structures for the (a) 100Cu_25S_50H and (b) 100Cu_12.5S_25H edge structures. The turquoise, orange, yellow, and white spheres are the molybdenum, copper, sulfur, and hydrogen atoms, respectively. (c) Grand potential as a function of pH2S/pH2 at 650 K and pH2 = 10 bar. | |
Table 4 Bader charges (q) of the non-hydrogenated and hydrogenated structures with 25% sulfur coverage for the Cu-doped S-edge
| Edge configuration |
q
Cu (e)
|
q
S (e)
|
q
S(SH) (e)
|
q
H(SH) (e)
|
100Cu_25S  |
+0.48 |
−0.51 |
— |
— |
100Cu_25S_50H  |
+0.44 |
— |
−0.36 |
+0.01 |
3.4. Insight into the hydrogenation properties of the doped S-edges
An important factor to examine is the adsorption strength of hydrogen on the doped edges. Kibsgaard et al.5 pointed out that the variations in the binding of H could be an overriding factor determining both the DDS and HYD pathways, since a too weak/strong binding of H would significantly impede the key reaction steps involving the transfer of H from the edges. On the other hand, SH groups are known to exhibit Brønsted acidic character.49,50 The strength of Brønsted acidity is related to the strength of H adsorption. Therefore, a weaker S–H bond results in a hydrogen atom that is more acidic and more easily deprotonated.51–54Table 5 shows the calculated differential hydrogen adsorption energy (ΔEH), for the final adsorbed H on the 100Ni_50S_100H_bridge, 100Mo_100S_100H and 100Cu_25S_50H structures. It was found that the hydrogen adsorption strength at the S-edge shows the following trend: Cu-doped > undoped > Ni-doped. This trend is opposite to that observed for hydrogenation catalytic activity,5,8 suggesting that lower activity may be associated with stronger H bonding. The hydrogen atom on the 100Ni_50S_100H_bridge structure has the lowest ΔEH. This would be the most Brønsted acidic hydrogen and the most likely to be removed from the edge. In contrast, the hydrogen atom on the 100Cu_25S_50H structure is the most strongly bound to the edge and therefore should be the least acidic and the most difficult to remove. We also calculated the Bader charge of the S atoms belonging to the SH group. The negative charge on the S atom decreases in the following order: Cu-doped > undoped > Ni-doped. Within these results, we speculate that the Ni doping could decrease the basicity of the S atoms compared to the undoped S-edge, leading to an increase in the Brønsted acidity of the S–H groups. Consequently, this would enhance the proton transfer ability in the SH groups, which is a key for an active hydrogenation catalyst. In contrast, the Cu doping could lead to a poisoning effect by increasing the basicity of the S atoms. This results in strongly adsorbed H, which cannot act as a reactive H species. It is important to note that the Brønsted acid nature of the SH groups should be confirmed by the adsorption of basic molecules such as ammonia, pyridine, and lutidine.51,54–57 However, this is outside the scope of the present study.
Table 5 The differential hydrogen adsorption energy (ΔEH) for the final adsorbed hydrogen and the Bader charge on the S atoms of the SH group for the undoped and doped S-edge
| Edge configuration |
ΔEH (eV) |
q
S(SH) (e)
|
| 100Ni_50S_100H_bridge |
−0.48 |
−0.27 |
| 100Mo_100S_100H |
−0.60 |
−0.29 |
| 100Cu_25S_50H |
−0.90 |
−0.36 |
4. Conclusions
In this work, we have carried out a comprehensive theoretical analysis of S atom rearrangement on the Ni and Cu-doped MoS2 S-edge upon hydrogen adsorption under HDS reaction conditions. Our results show that SH groups are the stable form of hydrogen adsorption and have a strong stabilization effect on the edges. For the undoped S-edge, the most stable structure corresponds to 100% coverage of sulfur and hydrogen. This structure shows a shift of the S atoms caused by the splitting of the S2 dimers. For the Ni-doped S-edge, the most stable structure is one with 50% sulfur and 100% hydrogen coverage. This particular structure results from a significant rearrangement of the edge S atoms upon hydrogen adsorption. For the Cu-doped S-edge, the most stable structure is achieved with 25% sulfur and 50% hydrogen coverage. This structure shows minimal rearrangement of the S atoms. Therefore, the dynamic nature of the S atoms is demonstrated by the structural response of the edges to H adsorption. This suggests that the edge equilibrium structure can be significantly perturbed by the reaction conditions.
Using hydrogen adsorption strength as a simple probe of hydrogenation activity, our results suggest that the promoting effect of Ni may be associated with decreased S–H bond strength, whereas the poisoning effect of Cu may be associated with increased S–H bond strength. For the HDS reaction, it is essential that the hydrogen adsorption is exothermic to ensure that reactive hydrogen species are available at the edge. However, the hydrogen adsorption should not be too exothermic as this would reduce the availability of these hydrogen species for the hydrogenation steps.
Conflicts of interest
There are no conflicts to declare.
References
-
H. Topsøe, B. S. Clausen and F. E. Massoth, Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996, p. 459 Search PubMed.
-
Catalysis by Transition Metal Sulphides: From Molecular Theory to Industrial Application, ed. H. Toulhoat and P. Raybaud, Editions Technip, Paris, 2013 Search PubMed.
- X. Li, A. Wang, M. Egorova and R. Prins, Kinetics of the HDS of 4,6-dimethyldibenzothiophene and its hydrogenated intermediates over sulfided Mo and NiMo on γ-Al2O3, J. Catal., 2007, 250, 283–293 CrossRef CAS.
- E. Krebs, B. Silvi, A. Daudin and P. Raybaud, A DFT study of the origin of the HDS/HydO selectivity on Co(Ni)MoS active phases, J. Catal., 2008, 260, 276–287 CrossRef CAS.
- J. Kibsgaard, A. Tuxen, K. G. Knudsen, M. Brorson, H. Topsøe, E. Lægsgaard, J. V. Lauritsen and F. Besenbacher, Comparative atomic-scale analysis of promotional effects by late 3d-transition metals in MoS2 hydrotreating catalysts, J. Catal., 2010, 272, 195–203 CrossRef CAS.
- H. Topsøe, B. S. Clausen, R. Candia, C. Wivel and S. Mørup, In situ Mössbauer emission spectroscopy studies of unsupported and supported sulfided CoMo hydrodesulfurization catalysts: Evidence for and nature of a CoMoS phase, J. Catal., 1981, 68, 433–452 CrossRef.
- N. Y. Topsøe and H. Topsøe, Characterization of the structures and active sites in sulfided CoMo/Al2O3 and NiMo/Al2O3 catalysts by NO chemisorption, J. Catal., 1983, 84, 386–401 CrossRef.
- R. Arancon, M. Saab, A. Morvan, A. Bonduelle-Skrzypczak, A.-L. Taleb, A.-S. Gay, C. Legens, O. Ersen, K. Searles, V. Mougel, A. Fedorov, C. Copéret and P. Raybaud, Combined Experimental and Theoretical Molecular Approach of the Catalytically Active Hydrotreating MoS2 Phases Promoted by 3d Transition Metals, J. Phys. Chem. C, 2019, 123, 24659–24669 CrossRef CAS.
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoat, Ab Initio Study of the H2–H2S/MoS2 Gas–Solid Interface: The Nature of the Catalytically Active Sites, J. Catal., 2000, 189, 129–146 CrossRef CAS.
- N. Y. Topsøe, H. Topsøe and F. E. Massoth, Evidence of Brønsted acidity on sulfided promoted and unpromoted Mo/Al2O3 catalysts, J. Catal., 1989, 119, 252–255 CrossRef.
- N. Salazar, S. B. Schmidt and J. V. Lauritsen, Adsorption of nitrogenous inhibitor molecules on MoS2 and CoMoS hydrodesulfurization catalysts particles investigated by scanning tunneling microscopy, J. Catal., 2019, 370, 232–240 CrossRef CAS.
- M. V. Bollinger, K. W. Jacobsen and J. K. Nørskov, Atomic and electronic structure of MoS2 nanoparticles, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 085410 CrossRef.
- J. V. Lauritsen, M. V. Bollinger, E. Lægsgaard, K. W. Jacobsen, J. K. Nørskov, B. S. Clausen, H. Topsøe and F. Besenbacher, Atomic-scale insight into structure and morphology changes of MoS2 nanoclusters in hydrotreating catalysts, J. Catal., 2004, 221, 510–522 CrossRef CAS.
- F. Bataille, J.-L. Lemberton, P. Michaud, G. Pérot, M. Vrinat, M. Lemaire, E. Schulz, M. Breysse and S. Kasztelan, Alkyldibenzothiophenes Hydrodesulfurization-Promoter Effect, Reactivity, and Reaction Mechanism, J. Catal., 2000, 191, 409–422 CrossRef CAS.
- M. Egorova and R. Prins, Hydrodesulfurization of dibenzothiophene and 4,6-dimethyldibenzothiophene over sulfided NiMo/γ-Al2O3, CoMo/γ-Al2O3, and Mo/γ-Al2O3 catalysts, J. Catal., 2004, 225, 417–427 CrossRef CAS.
- J. V. Lauritsen, M. Nyberg, J. K. Nørskov, B. S. Clausen, H. Topsøe, E. Lægsgaard and F. Besenbacher, Hydrodesulfurization reaction
pathways on MoS2 nanoclusters revealed by scanning tunneling microscopy, J. Catal., 2004, 224, 94–106 CrossRef CAS.
- E. M. Morales-Valencia, C. O. Castillo-Araiza, S. A. Giraldo and V. G. Baldovino-Medrano, Kinetic Assessment of the Simultaneous Hydrodesulfurization of Dibenzothiophene and the Hydrogenation of Diverse Polyaromatic Structures, ACS Catal., 2018, 8, 3926–3942 CrossRef CAS.
- A. S. Walton, J. V. Lauritsen, H. Topsøe and F. Besenbacher, MoS2 nanoparticle morphologies in hydrodesulfurization catalysis studied by scanning tunneling microscopy, J. Catal., 2013, 308, 306–318 CrossRef CAS.
- N. Y. Topsoe and H. Topsoe, FTIR Studies of Mo/Al2O2-Based Catalysts, J. Catal., 1993, 139, 641–651 CrossRef CAS.
- S. S. Grønborg, N. Salazar, A. Bruix, J. Rodríguez-Fernández, S. D. Thomsen, B. Hammer and J. V. Lauritsen, Visualizing hydrogen-induced reshaping and edge activation in MoS2 and Co-promoted MoS2 catalyst clusters, Nat. Commun., 2018, 9, 2211 CrossRef PubMed.
- L. S. Byskov, M. Bollinger, J. K. Nørskov, B. S. Clausen and H. Topsøe, Molecular aspects of the H2 activation on MoS2 based catalysts—the role of dynamic surface arrangements, J. Mol. Catal. A: Chem., 2000, 163, 117–122 CrossRef CAS.
- S. Cristol, J. F. Paul, E. Payen, D. Bougeard, S. Clémendot and F. Hutschka, Theoretical Study of the MoS2 (100) Surface: A Chemical Potential Analysis of Sulfur and Hydrogen Coverage. 2. Effect of the Total Pressure on Surface Stability, J. Phys. Chem. B, 2002, 106, 5659–5667 CrossRef CAS.
- A. Travert, H. Nakamura, R. A. van Santen, S. Cristol, J.-F. Paul and E. Payen, Hydrogen Activation on Mo-Based Sulfide Catalysts, a Periodic DFT Study, J. Am. Chem. Soc., 2002, 124, 7084–7095 CrossRef CAS PubMed.
- M. Sun, A. Nelson and J. Adjaye,
Ab initio DFT study of hydrogen dissociation on MoS2, NiMoS, and CoMoS: mechanism, kinetics, and vibrational frequencies, J. Catal., 2005, 233, 411–421 CrossRef CAS.
- P.-Y. Prodhomme, P. Raybaud and H. Toulhoat, Free-energy profiles along reduction pathways of MoS2 M-edge and S-edge by dihydrogen: A first-principles study, J. Catal., 2011, 280, 178–195 CrossRef CAS.
- R. Kronberg, M. Hakala, N. Holmberg and K. Laasonen, Hydrogen adsorption on MoS2-surfaces: a DFT study on preferential sites and the effect of sulfur and hydrogen coverage, Phys. Chem. Chem. Phys., 2017, 19, 16231–16241 RSC.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Understanding the Reactivity of Layered Transition-Metal Sulfides: A Single Electronic Descriptor for Structure and Adsorption, J. Phys. Chem. Lett., 2014, 5, 3884–3889 CrossRef CAS PubMed.
- N. Salazar, S. Rangarajan, J. Rodríguez-Fernández, M. Mavrikakis and J. V. Lauritsen, Site-dependent reactivity of MoS2 nanoparticles in hydrodesulfurization of thiophene, Nat. Commun., 2020, 11, 4369 CrossRef PubMed.
- J. Lauritsen, J. Kibsgaard, G. Olesen, P. Moses, B. Hinnemann, S. Helveg, J. Nørskov, B. Clausen, H. Topsoe and E. Lagsgaard, Location and coordination of promoter atoms in Co- and Ni-promoted MoS2-based hydrotreating catalysts, J. Catal., 2007, 249, 220–233 CrossRef CAS.
- E. Krebs, B. Silvi and P. Raybaud, Mixed sites and promoter segregation: A DFT study of the manifestation of Le Chatelier's principle for the Co(Ni)MoS active phase in reaction conditions, Catal. Today, 2008, 130, 160–169 CrossRef CAS.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Rational design of MoS2 catalysts: tuning the structure and activity via transition metal doping, Catal. Sci. Technol., 2015, 5, 246–253 RSC.
- G. Kresse and J. Hafner,
Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- K. Momma and F. Izumi,
VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- K. Reuter and M. Scheffler, Composition, structure, and stability of RuO2 (110) as a function of oxygen pressure, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406 CrossRef.
- R. R. Chianelli, T. A. Pecoraro, T. R. Halbert, W. H. Pan and E. I. Stiefel, Transition metal sulfide catalysis: Relation of the synergic systems to the periodic trends in hydrodesulfurization, J. Catal., 1984, 86, 226–230 CrossRef CAS.
- M. W. Chase Jr., NIST-JANAF Thermochemical Tables, J. Phys. Chem. Ref. Data, Monogr., 1998, 9, 1–1951 Search PubMed.
- H. Schweiger, P. Raybaud, G. Kresse and H. Toulhoat, Shape and Edge Sites Modifications of MoS2 Catalytic Nanoparticles Induced by Working Conditions: A Theoretical Study, J. Catal., 2002, 207, 76–87 CrossRef CAS.
- A. Nelson, M. Sun and A. Junaid, On the structure and composition of the phosphosulfide overlayer on Ni2P at hydrotreating conditions, J. Catal., 2006, 241, 180–188 CrossRef CAS.
- A. Bruix, J. V. Lauritsen and B. Hammer, Effects of particle size and edge structure on the electronic structure, spectroscopic features, and chemical properties of Au(111)-supported MoS2 nanoparticles, Faraday Discuss., 2016, 188, 323–343 RSC.
- R. Khare, R. Weindl, A. Jentys, K. Reuter, H. Shi and J. A. Lercher, Di- and Tetrameric Molybdenum Sulfide Clusters Activate and Stabilize Dihydrogen as Hydrides, JACS Au, 2022, 2, 613–622 CrossRef CAS PubMed.
- J. Duchet, E. van Oers, V. de Beer and R. Prins, Carbon-supported sulfide catalysts, J. Catal., 1983, 80, 386–402 CrossRef CAS.
- E. Schachtl, J. S. Yoo, O. Y. Gutiérrez, F. Studt and J. A. Lercher, Impact of Ni promotion on the hydrogenation pathways of phenanthrene on MoS2/γ-Al2O3, J. Catal., 2017, 352, 171–181 CrossRef CAS.
- W. Luo, H. Shi, E. Schachtl, O. Y. Gutiérrez and J. A. Lercher, Active Sites on Nickel-Promoted Transition-Metal Sulfides That Catalyze Hydrogenation of Aromatic Compounds, Angew. Chem., Int. Ed., 2018, 57, 14555–14559 CrossRef CAS PubMed.
- X. Hong, K. Chan, C. Tsai and J. K. Nørskov, How Doped MoS2 Breaks Transition-Metal Scaling Relations for CO2 Electrochemical Reduction, ACS Catal., 2016, 6, 4428–4437 CrossRef CAS.
- N. Y. Topsoe and H. Topsoe, FTIR Studies of Mo/Al2O3-Based Catalysts, J. Catal., 1993, 139, 641–651 CrossRef CAS.
- G. Berhault, M. Lacroix, M. Breysse, F. Maugé, J.-C. Lavalley, H. Nie and L. Qu, Characterization of Acidic Sites of Silica-Supported Transition Metal Sulfides by Pyridine and 2,6 Dimethylpyridine Adsorption: Relation to Activity in CH3SH Condensation, J. Catal., 1998, 178, 555–565 CrossRef CAS.
- Á. Logadóttir, P. G. Moses, B. Hinnemann, N.-Y. Topsøe, K. G. Knudsen, H. Topsøe and J. K. Nørskov, A density functional study of inhibition of the HDS hydrogenation pathway by pyridine, benzene, and H2S on MoS2-based catalysts, Catal. Today, 2006, 111, 44–51 CrossRef.
- S. Rangarajan and M. Mavrikakis, DFT Insights into the Competitive Adsorption of Sulfur- and Nitrogen-Containing Compounds and Hydrocarbons on Co-Promoted Molybdenum Sulfide Catalysts, ACS Catal., 2016, 6, 2904–2917 CrossRef CAS.
- H. Cai, R. Schimmenti, H. Nie, M. Mavrikakis and Y.-H. C. Chin, Mechanistic Role of the Proton–Hydride Pair in Heteroarene Catalytic Hydrogenation, ACS Catal., 2019, 9, 9418–9437 CrossRef CAS.
- D. Lin and S. Rangarajan, A DFT study of site-dependent energetics of hexagonal MoS2 nanoparticles under varying reaction conditions, Surf. Sci., 2023, 729, 122231 CrossRef CAS.
- B. Temel, A. K. Tuxen, J. Kibsgaard, N.-Y. Topsøe, B. Hinnemann, K. G. Knudsen, H. Topsøe, J. V. Lauritsen and F. Besenbacher, Atomic-scale insight into the origin of pyridine inhibition of MoS2-based hydrotreating catalysts, J. Catal., 2010, 271, 280–289 CrossRef CAS.
- S. S. Grønborg, N. Salazar, A. Bruix, J. Rodríguez-Fernández, S. D. Thomsen, B. Hammer and J. V. Lauritsen, Visualizing hydrogen-induced reshaping and edge activation in MoS2 and Co-promoted MoS2 catalyst clusters, Nat. Commun., 2018, 9, 2211 CrossRef PubMed.
- N. Salazar, S. B. Schmidt and J. V. Lauritsen, Adsorption of nitrogenous inhibitor molecules on MoS2 and CoMoS hydrodesulfurization catalysts particles investigated by scanning tunneling microscopy, J. Catal., 2019, 370, 232–240 CrossRef CAS.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Oscar
Hurtado-Aular
b,
José Luis
Peña-Mena
a,
Rafael
Añez
a and
Aníbal
Sierraalta
*a,
Oscar
Hurtado-Aular
b,
José Luis
Peña-Mena
a,
Rafael
Añez
a and
Aníbal
Sierraalta

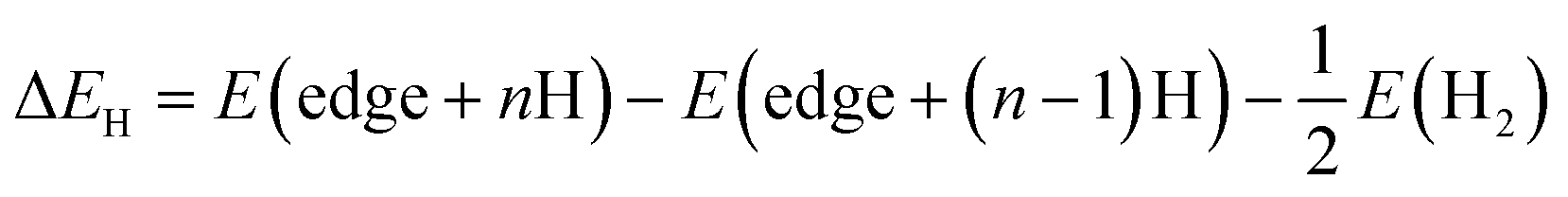
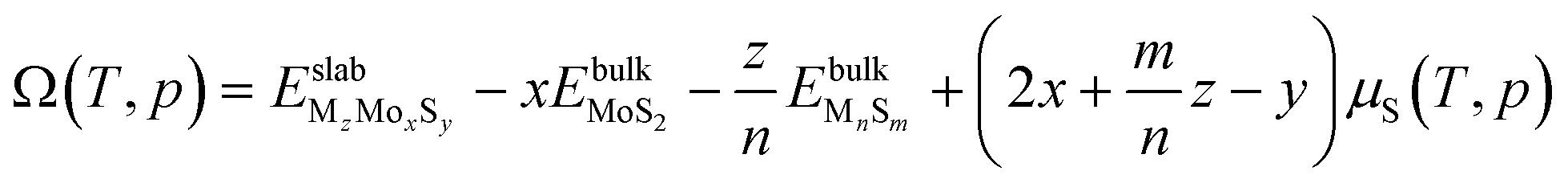
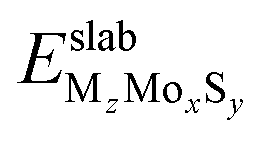 is the DFT total energy of the doped slab, containing x molybdenum atoms, y sulfur atoms, and z dopant atoms (M = Ni or Cu) and
is the DFT total energy of the doped slab, containing x molybdenum atoms, y sulfur atoms, and z dopant atoms (M = Ni or Cu) and 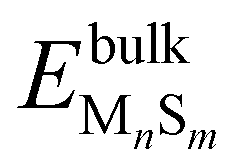 is the DFT total energy of the dopant bulk phase (Ni3S2 and CuS) stable under the reaction conditions.39μs(T,p) is the chemical potential of sulfur, and it is determined by the chemical equilibrium with the gas phase mixture of H2 and H2S, giving
is the DFT total energy of the dopant bulk phase (Ni3S2 and CuS) stable under the reaction conditions.39μs(T,p) is the chemical potential of sulfur, and it is determined by the chemical equilibrium with the gas phase mixture of H2 and H2S, giving
 , including zero-point vibrations. The term
, including zero-point vibrations. The term  corresponds to the difference between the thermal contributions to the chemical potential, which is calculated as
corresponds to the difference between the thermal contributions to the chemical potential, which is calculated as  and can be obtained from thermodynamic tables.40 Here, pH2S and pH2 denote the partial pressures of H2S and H2, respectively. The ratio
and can be obtained from thermodynamic tables.40 Here, pH2S and pH2 denote the partial pressures of H2S and H2, respectively. The ratio  is used to indicate the relative abundance of H2S and H2 in the gas phase, with larger values indicating sulfur-rich conditions and smaller values indicating strongly reducing conditions where H2 is more abundant than H2S. Under HDS working conditions, the
is used to indicate the relative abundance of H2S and H2 in the gas phase, with larger values indicating sulfur-rich conditions and smaller values indicating strongly reducing conditions where H2 is more abundant than H2S. Under HDS working conditions, the  ratio is typically between 0.01 and 0.05, while the temperature is in the range of 573 K to 700 K. For instance, representative HDS working conditions may include pH2 = 10 bar, pH2S = 0.1 bar, and T = 650 K.25
ratio is typically between 0.01 and 0.05, while the temperature is in the range of 573 K to 700 K. For instance, representative HDS working conditions may include pH2 = 10 bar, pH2S = 0.1 bar, and T = 650 K.25