
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First-principles studies of enhanced oxygen reduction reactions on graphene- and nitrogen-doped graphene-coated platinum surfaces†
Ho Ngoc
Nam
 *ab,
Quan Manh
Phung
cd,
Pongpol
Choeichom
a,
Yusuke
Yamauchi
bef and
Nagahiro
Saito
*ag
*ab,
Quan Manh
Phung
cd,
Pongpol
Choeichom
a,
Yusuke
Yamauchi
bef and
Nagahiro
Saito
*ag
aInstitute of Materials Innovation, Institutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan. E-mail: honam@sp.material.nagoya-u.ac.jp; hiro@sp.material.nagoya-u.ac.jp
bDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan
cDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan
dInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan
eAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia
fDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul 03722, South Korea
gDepartment of Chemical Systems Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan
First published on 11th March 2024
Abstract
Developing innovative platinum-based electrocatalysts and enhancing their efficiency are crucial for advancing high-performance fuel cell technology. In this study, we employed DFT calculations to provide a theoretical basis for interpreting the impact of graphene coatings on various Pt surfaces on oxygen reduction reaction (ORR) catalytic activity, which are currently applied as protective layers in experiments. We comprehensively assess the geometric and electronic properties of Pt(100), Pt(110), and Pt(111) surfaces in comparison to their graphene-coated counterparts, revealing different adsorption behaviors of O2 across these surfaces. The ORR mechanisms on different Pt surfaces show distinct rate-determining steps, with Pt(111) showing the highest ORR activity, followed by Pt(110) and Pt(100). Graphene coatings play a key role in enhancing charge transfer from the surface, resulting in modifications of O2 adsorption. Despite influencing ORR kinetics, these graphene-coated surfaces demonstrate competitive catalytic activity compared to their bare counterparts. Notably, Pt(111) with a graphene coating exhibits the lowest activation energy among graphene-coated surfaces. Our calculations also suggest that the ORR can occur directly on non-defective Pt@graphene surfaces rather than being restricted to exposed Pt centers due to point defects on graphene. Furthermore, our work highlights the potential of nitrogen doping onto the Pt(111)@C surface to further enhance ORR activity. This finding positions nitrogen-doped Pt@C as a promising electrocatalyst for advancing electrochemical technologies.
1. Introduction
Global warming and the massive decline in fossil fuels in recent years have further proven the importance for humanity to transition towards the use of green, clean, and sustainable energy resources.1 Advanced energy conversion technologies (e.g., solar photovoltaics, thermoelectrics, geothermal power, etc.)2–9 have made a significant contribution to the common effort to produce environmentally sustainable alternative energy sources. Notably, fuel cells have emerged as a promising technology for generating electricity through reactions involving oxygen and hydrogen.10–12 Despite these advancements, a key challenge persists in the development of efficient and durable cathode catalysts for fuel cells. Specifically, the slow kinetics of the oxygen reduction reaction (ORR) occurring in the cathode are a primary bottleneck, resulting in a large proportion of potential loss.13 Therefore, research on high-performance catalysts that can accelerate the ORR is highly required.Despite its high cost and scarcity, platinum (Pt) remains popular as one of the most dynamic ORR electrocatalysts in commercialized proton exchange membrane fuel cells (PEMFCs).13,14 Nevertheless, this type of material has certain drawbacks that limit its performance. According to Sabatier's principle,15 the interaction between the catalyst and the reactant should neither be too weak nor too strong. The pronouncedly strong bond between Pt and oxygen could thereby inhibit the efficient progress of the ORR activity and slow down the overall reaction. For example, it has been reported that alloying transition metals (e.g., Fe, Co, Ni, or Cu) with Pt has the potential not only to reduce material costs but also to help improve the ORR performance of the catalyst.16–18 This might be due to the electronic structure of Pt-based alloys being different from that of pure Pt, resulting in significant changes in how reactants and catalysts bond together and thereby improving ORR activity. Alternatively, materials derived from metal–organic frameworks (MOFs), offering large specific surface areas and different pore sizes,19–21 as well as single-atom catalysts with tunable electronic structures,22–28 are promising for further exploration of ORR electrocatalysis.
Moreover, catalytic durability is the next challenge for PEMFCs. Operating in harsh environments like high temperatures, strong acidic media, or high-potential regions could lead to a significant degradation of Pt catalysts over time.29 Other adverse factors, such as particle coalescence, dissolution, and poisoning, could even make this degradation worse.30,31 As a result, the catalytic activity and overall performance of fuel cells significantly decrease. In previous studies,32,33 MgO and SnO2 used as coatings have shown effectiveness in improving the durability and stability of Pt-based catalysts. Unfortunately, these coatings also hindered ORR activity on the catalyst surface due to the poor conductivity and blocking of the active sites.34 Therefore, enhancing both the catalytic performance and durability of Pt catalysts is crucial.
Graphene is a well-known material for its outstanding electrical conductivity, high surface area, and chemical stability.35 It can form specific solid-state ligand bonds to augment electron transfer with metals.36 Recently, several studies have applied graphene as an effective coating for Pt-based catalysts.37–40 For instance, it was found that Pt-based nanoparticles with graphene encapsulation fabricated via the solution plasma process improved not only the durability but also the efficient ORR activity of the catalysts.34,41 This suggests the great potential of graphene coatings for catalytic applications. Moreover, it was found that nitrogen-doped graphene could be beneficial in enhancing the ORR catalytic performance compared to pure graphene.42 However, the properties of these graphene coatings on Pt surfaces and how they influence the ORR activity of the system have not been clear yet. Previous theoretical studies mainly investigated the ORR mechanism on bare Pt surfaces with various models (e.g., clusters43–45 and slabs46–48) using different exchange–correlation functionals (i.e., PBE,43 PBE-D3,45 SCAN-rVV10,49 PW91,48 B3LYP,50 and PZ51), making comparisons and analysis of the related properties difficult. Furthermore, some studies on the ORR mechanism on graphene-supported Pt nanoparticles52,53 or single Pt atom-supported graphene54,55 have also been done. However, not many studies related to the graphene coating on Pt surfaces have been reported.49 It should be noted that bare Pt surfaces with different energies might have different effects on the geometric and electronic structures of the graphene coatings, leading to significant changes in the ORR activity of catalysts. Therefore, elucidating the role of graphene coatings in the ORR performance is necessary.
In this work, we systematically investigated the mechanism of the oxygen reduction reaction on graphene-coated Pt surfaces. First, a slab model of low-index Pt facets is constructed, and ORR activity on these bare surfaces is investigated. After that, we discuss the changes in the geometric and electronic structure of systems when graphene is coated. The effect of the graphene coating on ORR activity is then revealed via the calculation of charge density and overpotential. Finally, the influence of nitrogen doping on the graphene coating is also discussed, suggesting its potential to reduce the system's overpotential as compared to the pristine graphene layer.
2. Computational methods and models
Our DFT calculations are performed using the projector-augmented wave56 (PAW) potential as implemented in the Quantum-ESPRESSO package.57 For the PAW potentials, the PSLIBRARY version 1.0.0 was adopted.58 The wave functions and augmentation charge density in terms of a plane-wave basis set were expanded with kinetic energy cut-offs of 50 and 400 Ry, respectively. The spin polarization was taken into account in the calculations. A Gaussian smearing of 0.02 Ry was also added to treat the Fermi surface. For the exchange–correlation, a generalized gradient approximation (GGA) functional in the form of Perdew–Ernzerhof–Burke59 (PBE) functional was employed along with Grimme's van der Waals (vdW) correction60 (D3) to describe the weak dispersion interactions between the adsorbates and the surfaces. It should be noted that the use of different functionals possibly enhances the accuracy of the results but is computationally demanding, which is therefore not considered in this current work. Instead, only the PBE-D3 functional was used throughout the calculations to systematically and comprehensively evaluate how the graphene coating affects ORR activity.To analyze the stability of pristine Pt facets, we estimated their surface formation energy as follows:
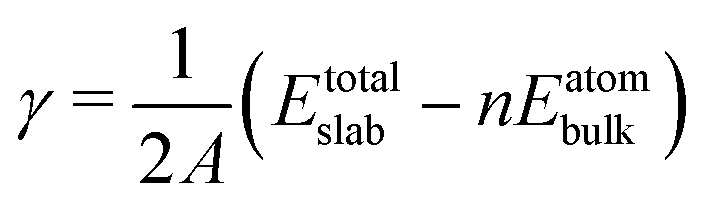 | (1) |
 | (2) |
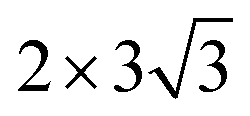 unit cell of graphene; the Pt(110)@C system constructed by interfacing a 2 × 5 unit cell of Pt(110) with a
unit cell of graphene; the Pt(110)@C system constructed by interfacing a 2 × 5 unit cell of Pt(110) with a  unit cell of graphene; and the Pt(111)@C system created by interfacing a 3 × 3 unit cell of Pt(111) with a 3 × 3 unit cell of graphene. The choice of different cell sizes for surfaces is to ensure the lattice mismatch between the Pt surfaces and graphene layers is minimal. These models correspond to a low adsorption coverage of O2. We note that the graphene sheet can have many different orientations on the metal surfaces. In this study, we only adopted orientations that lead to a moderate interaction between graphene and Pt surfaces, which is believed to increase the adsorption energy between graphene and O2.
unit cell of graphene; and the Pt(111)@C system created by interfacing a 3 × 3 unit cell of Pt(111) with a 3 × 3 unit cell of graphene. The choice of different cell sizes for surfaces is to ensure the lattice mismatch between the Pt surfaces and graphene layers is minimal. These models correspond to a low adsorption coverage of O2. We note that the graphene sheet can have many different orientations on the metal surfaces. In this study, we only adopted orientations that lead to a moderate interaction between graphene and Pt surfaces, which is believed to increase the adsorption energy between graphene and O2.
The adsorption energy of an intermediate is defined by:
| Eads = Esystem − (Ecatalyst + Eadsorbate) | (3) |
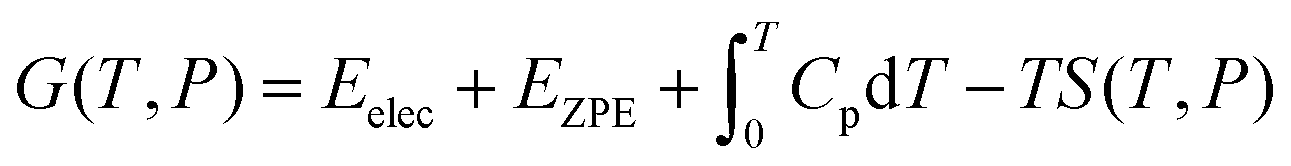 | (4) |
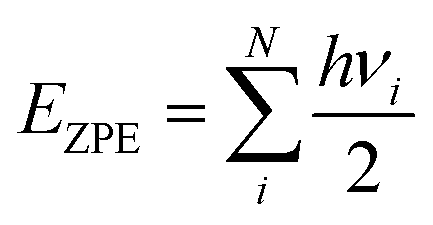 is the zero-point energy with νi being the vibrational frequency of N modes obtained from the finite displacement method in an atomic simulation environment (ASE),63 and Cp is the heat capacity involving translational, rotational, vibrational, electronic parts, and a kB term. It should be noted that for ideal gas cases (e.g., H2 and H2O), translational and rotational degrees of freedom were taken into account. For adsorbates, however, all degrees of freedom of the adsorbate are treated harmonically since they often have no real translational or rotational degrees of freedom. The associative ORR mechanisms are then investigated via the four-electron pathway in an acidic medium (i.e., pH = 0) with intermediates including:64
is the zero-point energy with νi being the vibrational frequency of N modes obtained from the finite displacement method in an atomic simulation environment (ASE),63 and Cp is the heat capacity involving translational, rotational, vibrational, electronic parts, and a kB term. It should be noted that for ideal gas cases (e.g., H2 and H2O), translational and rotational degrees of freedom were taken into account. For adsorbates, however, all degrees of freedom of the adsorbate are treated harmonically since they often have no real translational or rotational degrees of freedom. The associative ORR mechanisms are then investigated via the four-electron pathway in an acidic medium (i.e., pH = 0) with intermediates including:64| * + O2 → *O2 (ΔG0) | (5) |
| *O2 + H+ + e− → *OOH (ΔG1) | (6) |
| *OOH + H+ + e− → *OH + *OH (ΔG2) | (7) |
| *OH + *OH + H+ + e− → *OH + H2O (ΔG3) | (8) |
| *OH + H+ + e− → H2O (ΔG4) | (9) |
| ΔG0 = G*O2 − G* − 2GH2O + 2GH2 − 4.92 | (10) |
 | (11) |
 | (12) |
 | (13) |
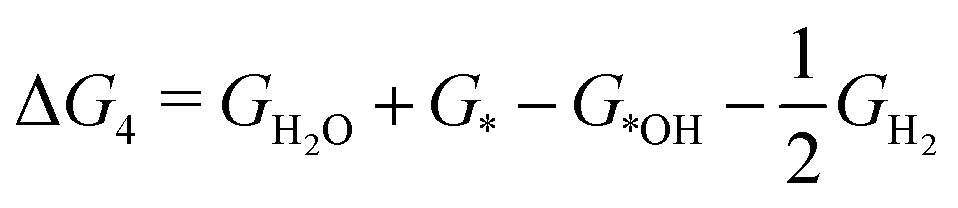 | (14) |
| ηORR = 1.23 − UL | (15) |
| UL = −min(ΔG1, ΔG2, ΔG3, ΔG4) | (16) |
Finally, it is important to note that solvent effects were not considered. However, previous research by Iyemperumal and Deskins67 found that on the Pt(111) surface, solvent effects (especially for small species investigated for the ORR such as O2, O, and OH) are minimal. For example, the solvation energy of O2 on Pt(111) is only 0.01 eV. Additionally, in a recent related work on the ORR performance of a single-layer Pt–graphene hybrid,49 it was found that the use of the implicit solvation model could affect the ORR performance of hybrid Pt–graphene, however, the energy difference is not significant.
3. Results and discussion
3.1. Geometric and electronic structures of bare Pt and Pt@C surfaces
First, we examine the structures and the corresponding surface energies of bare Pt facets, as illustrated in Fig. 1. Notably, Pt(111) emerges as the most stable facet, having a surface energy of 0.120 eV Å−2 owing to its larger planar density. In contrast, Pt(110) and Pt(100) exhibit significantly higher surface energies (0.174 and 0.162 eV Å−2, respectively), reflecting their lower planar density and higher number of dangling bonds. The less stable Pt(110) and Pt(100) surfaces are thereby ready to form additional bonds with adsorbates to stabilize their structures. As a result, the geometry of graphene coatings on Pt(100) and Pt(110) could be different from that on Pt(111). Because of the interaction of Pt(100) and Pt(110) surfaces with the graphene top layer [see Fig. 2(a and b)], their bonds exhibit considerable strength with an average distance of approximately 2.1–2.2 Å, indicative of some covalent character as shown by the electron localization function calculations (Fig. S1, ESI†). Remarkably, the graphene layer undergoes distortion from its planarity. This reduces the stress due to lattice misfit between graphene and the metal surfaces. The maximum buckling in the carbon layer reaches around 0.33 Å on Pt(100) and Pt(110), while the C–C bond distance ranges from 1.467 to 1.570 Å. In contrast, Pt(111) exhibits higher surface stability, resulting in the physisorption of graphene [Fig. 2(c)]. The Pt(111)–graphene distance is 2.7 Å, which is in fair agreement with the distance estimated in previous work by Wang et al.68 (i.e., 3.3 Å). The small mismatch between the Pt(111) unit cell and graphene (1.08%) minimizes the distortion, allowing graphene to maintain planarity, albeit with an increased C–C bond distance from 1.424 Å (in isolated graphene) to 1.556 Å. In fact, the adsorption of graphene on Pt(111), in several Moiré patterns (this work),
(this work),  , and
, and  , has been experimentally shown to be very weak.69,70
, has been experimentally shown to be very weak.69,70
 | ||
| Fig. 1 The calculated surface energy corresponds to three different low-index Pt facets, including Pt(100), Pt(110), and Pt(111). | ||
 | ||
| Fig. 2 The side and top views of graphene-coated platinum models used for calculations include (a) Pt(100)@C, (b) Pt(110)@C, and (c) Pt(111)@C. The purple and grey balls represent C and Pt atoms, respectively. The distortion of graphene surfaces due to Pt–C covalent bonds induces two different sp2 and sp3-like hybridizations of carbon. | ||
Moreover, we also look at the electronic structure of these surfaces. The density of states (DOS) along with the partial density of states (pDOS) is depicted in Fig. 3. According to the d-band center theory proposed by Nørskov et al.,61,62,71 the adsorption energy of the adsorbate is related to the relative energy between the d-states and the Fermi level. Simply put, the higher the d-states, the more empty the anti-bonding states, and the stronger the adsorption. The calculated energy of the d-band center (εd) stands at −2.637, −2.570, and −2.729 eV for Pt(100), Pt(110), and Pt(111), respectively. The estimated εd of Pt(111) closely aligns with values previously estimated by Hammer et al.72 The highest d-band center of Pt(110) indicates that this surface should have the strongest adsorption, whereas the adsorption ability of Pt(111) should be the weakest (see Table 1 for the case of O2). This tendency matches well the trend of the surface energy above, i.e., the least stable surface has the highest adsorption energy.
 | ||
| Fig. 3 Total DOSs and partial DOSs of (a) bare Pt and (b) Pt@C surfaces. The Fermi level (εF, black dashed line) is found at 0 eV. Positions of the C p-band center (εp, orange vertical line), Pt p-band center (εp, green vertical line), and Pt d-band center (εd, blue vertical line) are marked for comparison. | ||
| Surface | E ads (eV) | d X–O (Å) [X = Pt, C] | d O–O (Å) | ΔQ |
|---|---|---|---|---|
| a Ref. 51. b Ref. 49. c Ref. 74. d Ref. 75. | ||||
| Pt(100)@C | −1.02 | 1.476 | 1.486 | −0.95 |
| Pt(110)@C | −0.59 | 1.480 | 1.490 | −0.91 |
| Pt(111)@C | −0.32 | 1.477 | 1.489 | −0.97 |
| Pt(100) | −1.22 | 1.983 | 1.370 | −0.49 |
| Pt(110) | −1.52 | 1.968 | 1.368 | −0.53 |
| Pt(111) | −0.70 (−0.72,a −0.84,b −1.11b) | 2.036 | 1.358 | −0.48 |
| Graphene | −0.12 (−0.13,c −0.15d) | 3.380 | 1.230 | −0.09 |
As graphene is coated on Pt surfaces, the d-band center slightly shifts to values of −2.712, −2.675, and −2.781 eV for Pt(100)@C, Pt(110)@C, and Pt(111)@C, respectively [see Fig. 3(b)]. This indicates that, if O2 could penetrate the graphene layer to interact with the Pt surface (see ref. 73), the anticipated interaction would be weaker than that observed on the pristine Pt surface. However, since adsorbates directly interact with the graphene surface in Pt@C, the specific location of d-band centers is less relevant. Indeed, the adsorption of O2 on Pt(100)@C is the strongest (Table 1), whereas the d-band center of Pt in Pt(100)@C is not the highest. Similarly, the p-band centers of Pt in Pt@C (−1.234, −0.987, and −1.270 eV for Pt(100)@C, Pt(110)@C, and Pt(111)@C, respectively) also do not correlate well with the adsorption energy tendency. We instead look at the carbon p-band center as an alternative descriptor for the adsorption on non-metallic surfaces. The calculated p-band center of C is at −3.030, −3.138, and −2.954 eV for Pt(100)@C, Pt(110)@C, and Pt(111)@C, respectively. It is then assumed that Pt(110)@C has weaker adsorption than the other surfaces. However, the results of adsorption energy (see Table 1) invalidate this assumption, e.g., the Pt(110)@C surface adsorbs stronger than Pt(111)@C. This observation can be easily understood as the adsorption of O2 not only depends on the interaction between O2 and graphene but also on the interaction between the (mobile) graphene layer and the top layer of Pt. Indeed, upon O2 adsorption, the distance between graphene and the Pt surfaces significantly changes (vide infra). Therefore, solely looking at the p-band center will not give the correct trend of O2 adsorption on these graphene-coated surfaces. Furthermore, due to the buckling of graphene layers on Pt(110) and Pt(100), the electronic structure of graphene is expected to change, in which the hybridization of some carbon atoms deviates from a perfect sp2 towards sp3. Such a change in hybridization has been linked to the ability of graphene to bind to O2. The change of hybridization is also associated with the formation of additional Pt–C bonds (Fig. S1, ESI†). We therefore referred to these carbons as “sp3-like”, as their character should lie between sp2 and sp3.
To conclude, our data clearly show that, whereas the d-band center of Pt is a good indicator of the adsorption strength on the bare Pt surfaces, neither it nor the p-band center of graphene can be used to explain the adsorption trend of O2 on the Pt@C surfaces.
3.2. O2 adsorption on Pt and Pt@C surfaces
In Table 1, we present the adsorption energy of O2 on both bare graphene, Pt surfaces, and graphene-coated Pt surfaces, along with the corresponding bond distances between O, Pt, and C atoms. The stable adsorption configurations are visually illustrated in Fig. 4.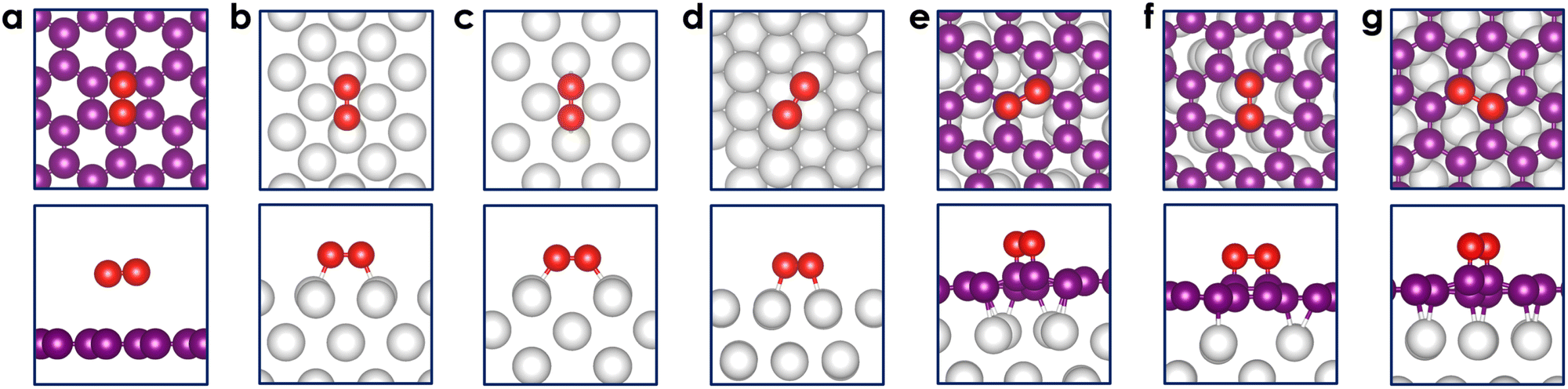 | ||
| Fig. 4 Stable configurations of O2 adsorption on (a) graphene, (b) Pt(100), (c) Pt(110), (d) Pt(111), (e) Pt(100)@C, (f) Pt(110)@C, and (g) Pt(111)@C surfaces from top and side views. The red, purple, and grey balls represent O, C, and Pt atoms, respectively. | ||
On graphene, the adsorption is primarily physisorption, characterized by a modest adsorption energy of only −0.12 eV and a relatively long distance between O2 and graphene (∼3.4 Å). Our calculated adsorption energy is in excellent agreement with a recent experimental value of −0.15 eV76 as well as a diffusion Monte Carlo result (−0.14 eV).77 The O2 molecule is found to be parallel to the surface and maintains its triplet spin state. The O–O equilibrium bond distance is 1.23 Å, being close to the bond length of 1.21 Å in its isolated state. The resulting charge transfer between O2 and graphene remains minimal, amounting to less than 0.1 electrons [see also Fig. 5(c)]. It should be noted that while the O2 singlet can be chemisorbed on graphene (with a C–O bond length of ∼1.5 Å), it is highly unstable. The adsorption energy of a singlet-O2 on graphene was estimated to be highly positive (∼1.8 eV).75
 | ||
| Fig. 5 (a) Interaction between C(sp3)–O(sp3), (b) Pt(dz2)–O(sp3) and Pt(dxz)–O(sp3). (c) Charge transfers between O2 and three surfaces: graphene, Pt(111), and Pt(111)@C. Color code: green: gaining electron region, blue: losing electron region (isosurface value of 0.002 e bohr−3). The red, purple, and grey balls represent O, C, and Pt atoms, respectively. | ||
On the bare Pt surfaces, O2 is strongly chemisorbed, with an adsorption energy of −0.7 eV to −1.5 eV. In all cases, O2 is parallel to the surfaces and is characterized as a closed-shell singlet species. As previously discussed, the Pt(111) surface, being the most stable among the three bare surfaces, displays the weakest adsorption. The calculated value of −0.7 eV is in good agreement with previously reported values ranging from −0.72 eV to −1.11 eV.49,51 Notably, O2 is found to bridge two adjacent Pt atoms, also referred to as the top–bridge–top (t–b–t) site.78 At this site, the distance between O2 and the surface measures approximately ∼2.0 Å, with the O–O bond distance elongated to ∼1.36 Å. A charge transfer from Pt(111) to the π* orbital of O2 is estimated at 0.5 electrons [see Fig. 5(b)]. This observation clearly indicates that the character of the adsorbed O2 molecule is somewhere between that of an isolated O2 and a superoxo O2− species.
In comparison to the Pt(111) surface, the adsorption behavior of O2 on Pt(110) and Pt(100) is quite similar. The distance between O2 and the surfaces is slightly shorter, ranging from 1.97 to 1.98 Å, and the O–O bond exhibits a slight weakening with a bond distance of 1.37 Å. Remarkably, the binding of O2 on Pt(110) is significantly stronger, manifesting as an adsorption energy of −1.5 eV. On this surface, O2 could be adsorbed along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] or [001] direction, referred to as top–short-bridge–top (t–sb–t) or top–long-bridge–top (t–lb–t) configurations, respectively. The latter configuration is found to be more stable than the former by 0.2 eV. It is important to note that a clean Pt(110) surface may undergo reconstruction, forming a missing-row type structure.79–81 Such structural changes facilitate dissociative chemisorption of O2,82 though this aspect is not considered in this work. Also, a recent Raman spectroscopy experiment showed that the oxygen reduction reaction (ORR) on the three Pt facets under acidic conditions most likely proceeds through molecular oxygen instead of atomic oxygen.83 On the unreconstructed Pt(100) surface, O2 adsorbs at the bridge site (t–b–t configuration) with an adsorption energy of −1.22 eV, closely matching the results obtained in the work of Duan and Wang48 employing the PW91 functional (−1.10 eV).
0] or [001] direction, referred to as top–short-bridge–top (t–sb–t) or top–long-bridge–top (t–lb–t) configurations, respectively. The latter configuration is found to be more stable than the former by 0.2 eV. It is important to note that a clean Pt(110) surface may undergo reconstruction, forming a missing-row type structure.79–81 Such structural changes facilitate dissociative chemisorption of O2,82 though this aspect is not considered in this work. Also, a recent Raman spectroscopy experiment showed that the oxygen reduction reaction (ORR) on the three Pt facets under acidic conditions most likely proceeds through molecular oxygen instead of atomic oxygen.83 On the unreconstructed Pt(100) surface, O2 adsorbs at the bridge site (t–b–t configuration) with an adsorption energy of −1.22 eV, closely matching the results obtained in the work of Duan and Wang48 employing the PW91 functional (−1.10 eV).
The adsorption of O2 on graphene-coated surfaces is also summarized in Table 1 and Fig. 4(e–g). The important finding is that, in contrast to the bare graphene surface, O2 chemisorbs on graphene-coated Pt surfaces with adsorption energies ranging from −0.3 eV to −1.0 eV. These negative values indicate the potential of Pt@C as a promising candidate for the ORR. Various configurations of O2 on the three Pt@C were investigated, revealing a preference for the parallel orientation to the graphene layer in a top–top (t–t) configuration. The distance between carbon and oxygen is ∼1.48 Å, while the O–O bond length increases to around 1.49 Å. The C–O bond distance closely mirrors the values typically found in epoxides, whereas the O–O bond length resembles that of peroxide. As compared to the bare Pt surfaces, the strength of the O–O bond on Pt@C is clearly weaker. This difference can be attributed to the enhanced directional nature of the C(sp3)–O(sp3) bond, as illustrated in Fig. 5(a). On the other hand, the Pt–O bond manifests as either Pt(dz2)–O(sp3) (displaying less directionality) or Pt(dxz)–O(sp3) (exhibiting more directionality).
Upon binding with O2, two carbon atoms change their hybridization to sp3, leading to a further distortion of the graphene layer and inducing additional sp3 carbons. The C–O covalent bonds not only alter the structure of the graphene layer but also influence the interaction between the graphene layer and Pt. For instance, while the interaction between Pt(111) and graphene is diffusive, the introduction of O2 adsorption is responsible for the formation of more covalent bonds between C-sp3 and Pt. In terms of the electronic structure, the adsorbed O2 molecule can be characterized as a superoxide O2− species, as we found that 0.9–1.0 electrons were transferred from the surface to O2 [Fig. 5(c)]. More interestingly, the charge transfer process involves not only the graphene layer but also the first and second layers of the Pt surface.
3.3. ORR mechanism on Pt and Pt@C surfaces
The associative ORR mechanisms on six surfaces including Pt(100), Pt(110), Pt(111), and their corresponding graphene-coated counterparts, Pt(100)@C, Pt(110)@C, Pt(111)@C are summarized in Fig. 6. Following the well-established computational protocol for the ORR, we only focus on four intermediates along the reaction: *O2, *O–*OH, *OH–*OH, and *OH. Specifically, *O2 is the starting active species of the ORR on all surfaces. We noted that OOH+, as discussed in a recent work of Gueskine et al.,84 is not a viable species in aqueous acidic media. They calculated that the proton exchange reaction O2 + H3O+ → OOH+ + H2O is endothermic by 2.65 eV. Therefore, OOH+ should not be the starting active species, as its concentration is negligible. They also pointed out that numerous researchers have fallen into this trap by picking up OOH+ as the starting active species of ORR simulations. | ||
| Fig. 6 (a) Free energy diagram and structures of ORR intermediates on Pt(100)@C (green steps line), Pt(110)@C (blue steps line), and Pt(111)@C (orange steps line) at URHE = 0 V, where URHE is the potential of the electrode relative to the RHE. The red, purple, grey, and white balls represent O, C, Pt, and H atoms. Free energy of the corresponding bare Pt surfaces (grey steps line) is inserted for comparison. (b) Free energy profile of the ORR on the graphene-coated surfaces at URHE = 0 V (in black), URHE = 1.23 V (in red), and limiting potential UL (in blue). | ||
Although OOH+ is not the starting active species, the formation of *OOH on the surface is still plausible. This intermediate could be directly formed from the first hydrogenation of *O2. Thus, we also calculated the *OOH species on all surfaces (see Fig. S2, ESI†). On all surfaces, the first hydrogenation of *O2 can yield either *OOH or *O–*OH. Interestingly, *O–*OH is systematically more stable than *OOH by ∼2.0 eV on Pt(100) and Pt(100)@C; ∼1.0 eV on Pt(110) and Pt(110)@C; and ∼1.4 eV on Pt(111) and Pt(111)@C. On the Pt(111) surface, a similar value of 1.51 eV was also found by Duan and Wang.85 On a carbon alloy catalyst, Chai et al.86 also reported the instability of *OOH. Given the significant stability of *O–*OH, it is likely that once *OOH is formed, it is rapidly decomposed into *O–*OH. Indeed, on the Pt(111) surface, the activation energy for the *OOH dissociation reaction is negligible (0.05 eV).85
Let us first discuss the reactivity of the three bare Pt surfaces. Here, we focus only on the associative mechanism involving the *O–*OH intermediate. The mechanism with the *OOH species is discussed in the ESI.† In general, all surfaces possess similar ORR mechanisms, summarized as O2 + 4(H+ + e−) → *O2 + 4(H+ + e−) → *O + *OH + 3(H+ + e−) → *OH + *OH + 2(H+ + e−) → H2O + *OH + (H+ + e−) → 2H2O. Each surface exhibits different gaps between the reaction intermediates, resulting in different rate-determining steps (RDSs). At the electrode potential URHE (versus the reversible hydrogen electrode (RHE)) of zero, the free energy profiles are all downhill along the reaction pathway, i.e., all reduction steps are exergonic. For example, on Pt(111), the reduction free energies are −1.74, −0.71, −0.92, and −1.04 eV (Fig. S2, ESI†). The gaps between the intermediates can then be adjusted by changing the working potential. Specifically, the Gibbs free energy of the intermediates shifts by an amount of −neURHE, where n represents the number of electrons. On Pt(111), at the limiting potential UL = 0.71 V, the reduction free energies change to −1.03, 0, −0.21, and −0.33 eV (Fig. S3, ESI†). Beyond this electrode potential, one of the reduction steps (the second reduction step *O–*OH + (H+ + e−) → *OH–*OH) becomes endergonic. When the electrode potential URHE is increased to 1.23 V, this reaction step has a reaction free energy of 0.52 eV, which corresponds to the RDS. This result is in line with the study of Duan and Wang,48 in which the authors found the same RDS with an activation energy of 0.79 eV.
Hereafter, we will focus our discussion on the ORR at an electrode potential URHE of 1.23 V. For the Pt(110) surface, the RDS is the last reduction (*OH hydrogenation). The result agrees with the calculations reported by Dong et al.,83 where they estimated an activation free energy of 1.18 eV. In our study, we estimated a slightly lower activation free energy of 0.96 eV. For the Pt(100) surface, the RDS is related to the third reduction step (*OH–*OH + (H+ + e−) → *OH + H2O), which has an activation free energy of 0.98 eV. The overpotential can then be calculated as the activation free energy of the RDS divided by e or the difference between 1.23 V and the limiting potential (see eqn (15)). Therefore, the overpotential for the ORR on Pt(100), Pt(110), and Pt(111) surfaces is estimated to be 0.98, 0.96, and 0.52 V, respectively. This suggests a decreasing ORR activity trend for Pt surfaces in the order of (111) > (110) > (100). Notably, this result is in good agreement with both prior experimental findings87 and recent results reported by Dong et al.83 utilizing Raman spectroscopy. In the same work, the authors also performed a similar computational study. According to their presented data, the RDS on all surfaces is interpreted as the last hydrogenation step: *OH + (H+ + e−) → * + H2O. The different RDSs between our and their data are unclear to us but probably come from the fact that (i) a significantly smaller model was employed in their models, therefore accounting for higher adsorption coverage, and (ii) different configurations were used. For instance, the higher adsorption coverage leads to unfavored chemisorption, e.g., the adsorption free energy on Pt(100) is positive.
The ORR mechanisms on Pt(100)@C, Pt(110)@C, and Pt(111)@C surfaces are depicted in Fig. 6. Similar to the Pt bare surfaces, the associated mechanism involves the same intermediates. To the best of our knowledge, only one theoretical study has examined the ORR mechanism of a graphene-coated Pt surface.49 In their work, Choi et al.49 found stability in a hybrid system of graphene and a monolayer of the Pt(100)-like surface, which is active for the ORR. Their identified RDS was the third hydrogenation step (*OH–*OH + (H+ + e−) → *OH + H2O). As we employed a Pt(100) surface with several layers, we found the RDS to be the last hydrogenation, namely *OH + (H+ + e−) → H2O. The activation free energy for this step is estimated to be 1.03 eV, which is slightly higher than the activation free energy for the RDS on bare Pt(100) (i.e., 0.98 eV). This result suggests that Pt(100)@C should exhibit ORR reactivity comparable to Pt(100). Our results also nicely corroborate the experimental observations of Abdelhafiz et al.88 regarding the ORR activity of thin graphene–Pt hybrid catalysts.
Although having a smaller affinity towards O2 compared to the Pt(100)@C surface, the ORR mechanism on the Pt(110)@C surface does not deviate much from Pt(100)@C. Most importantly, the Pt(110)@C surface also identifies the last hydrogenation reaction as the RDS, with an activation free energy of approximately 0.82 eV. Therefore, we predict that the ORR reactivity of Pt(110)@C should be slightly better than that of Pt(100)@C. In contrast, the ORR mechanism on Pt(111)@C turns out to deviate the most from that on Pt(111) as well as the other graphene-coated surfaces. First, the adsorption free energy is significantly smaller. Second, we found two competing RDSs, the second and last hydrogenation steps, with pretty similar activation energies, 0.78 eV and 0.71 eV. Based on the activation free energy of the RDS, we predict the overpotential values of Pt(100)@C, Pt(110)@C, and Pt(111)@C to be 1.03, 0.82, and 0.78 V. Thus, the ORR reactivity of graphene-coated surfaces can be ordered as Pt(111)@C > Pt(110)@C > Pt(100)@C. This order parallels the order found on the bare surfaces.
Among the six surfaces studied, the bare Pt(111) surface turns out to have the smallest overpotential (i.e., 0.53 V), closely followed by Pt(111)@C (i.e., 0.78 V) and Pt(110)@C (i.e., 0.82 V). In contrast, the Pt(100)@C surface displays significantly larger overpotential values (approximately 1.0 V). Although the results suggest that Pt(111)@C possesses slightly lower catalytic reactivity compared to the pristine Pt(111) surface, our findings highlight the dual role of graphene. On the one hand, graphene enhances the durability and stability of the catalyst by serving as a physical barrier, preventing Pt aggregation.88–90 On the other hand, graphene-coated Pt surfaces can still catalyze the ORR with a competing catalytic reactivity compared to the pristine Pt surface. Notably, introducing defects in graphene, leading to Pt exposure to O2, can further enhance the activity of Pt@C.90
3.4. ORR mechanism on the N-doped graphene-coated Pt surface
Considering the higher reactivity observed in the Pt(111)@C surface, coupled with the established effectiveness of nitrogen-doped carbon materials as metal-free catalysts for the oxygen reduction reaction (ORR),91 we briefly discuss the ORR mechanism on a nitrogen-doped Pt(111)@C surface (also referred to as graphitic-N defects),92 denoted as Pt(111)@C/N. Accordingly, the doping of nitrogen on the graphene coating is anticipated to exhibit better ORR activity due to (i) a higher concentration of protons, which can facilitate four-electron reduction, and (ii) enhanced O2 adsorption capabilities.93The calculated ORR mechanism on Pt(111)@C/N, in comparison with the Pt(111)@C surface, is illustrated in Fig. 7. First, we investigated different locations of nitrogen on graphene. We found that nitrogen is preferably located on the hollow site of the top Pt layer, thus preserving its sp2 character. Consequently, the three carbon atoms binding with this nitrogen (denoted as CN) reside on the top sites of the Pt layer. Upon bonding with Pt, the hybridization of these carbons changes to sp3. Here, we only explored the adsorption of O2 in the vicinity of the nitrogen and CN atoms, aiming at probing the impact of nitrogen. Most importantly, we found that O2 neither adsorbs on nitrogen nor these three CN atoms due to a lack of dangling bonds. This behavior is different from the adsorption of O2 on nitrogen-doped carbon materials, in which the active sites are CN.94 On this Pt(111)@C/N surface, the most favorable adsorption configuration is observed when O2 adopts an end-on (or Pauling) configuration on the carbon atoms bonding to CN. Similar to the behavior observed for O2 on Pt(111)@C, O2 on Pt(111)@C/N can be characterized as a superoxide O2−, as approximately 0.8 electrons are transferred from the surface to O2 [see Fig. 8(a)]. The slightly reduced charge transfer on Pt(111)@C/N compared to Pt(111)@C (i.e., 0.98 electrons) probably comes from the end-on adsorption configuration. The adsorption energy on Pt(111)@C/N is around −0.3 eV, which is approximate to the adsorption energy on Pt(111)@C.
 | ||
| Fig. 7 Comparison of the ORR mechanism on (a) Pt(111)@C and (b) Pt(111)@C/N surfaces at five intermediates (ii–vi). The green, red, purple, grey, and white balls represent N, O, C, Pt, and H atoms, respectively. | ||
 | ||
| Fig. 8 (a) Charge transfers between O2 and the Pt(111)@C/N surface. Color code: green: gaining electron region, blue: losing electron region (isosurface value of 0.002 e bohr−3). (b) Free energy profile of the ORR on the Pt(111), Pt(111)@C, and Pt(111)@C/N surfaces at URHE = 0 V. (c) Free energy profile of the ORR on the Pt(111)@C/N surface at URHE = 0 V (in black), URHE = 1.23 V (in red), and limiting potential UL (in blue). | ||
The major distinction between the ORR on Pt(111)@C/N and Pt(111)@C lies in the hydrogenation product of *O2. As mentioned above, the ORR mechanism on Pt(111)@C is expected to proceed via the *O–*OH intermediate, as it is significantly more stable than the *OOH intermediate. On Pt(111)@C/N, only *OOH is formed, a feature attributed to the inability of N and CN to provide dangling bonds to oxygen. As a result, the ORR mechanism on Pt(111)@C/N takes a distinct pathway involving *OOH, *O–H2O, and *OH–H2O intermediates [Fig. 7(b)]. The second hydrogenation from *OOH to *O–H2O is similar to findings observed in an iron-embedded N-doped graphene catalyst.95 The third hydrogenation *O–H2O + (H+ + e−) → *OH–H2O turns out to be the RDS, with an activation free energy of 0.73 eV. Finally, the remaining *OH species undergoes hydrogenation, leading to the release of water molecules from the surface. The activation free energy of 0.73 eV is slightly smaller than that of the Pt(111)@C surface (i.e., 0.78 eV), suggesting a small enhancement in ORR catalytic reactivity on the N-doped surface [Fig. 8(b and c)]. These results affirm the potential of the as prepared N-doped graphene-encapsulated PtFe alloy core–shell catalyst in our lab previously.34,41
4. Conclusions
In summary, we performed first-principles calculations to gain insight into the geometric and electronic properties of bare Pt surfaces, graphene- and N-doped graphene-coated Pt surfaces, as well as the mechanism of oxygen reduction reactions on these surfaces. We show that O2 is strongly adsorbed on the bare Pt surfaces, whereas the adsorption on graphene-coated surfaces is slightly weaker. We then found that different bare Pt surfaces behaved differently in ORR catalytic activities, with the most efficient facets ordered as Pt(111) > Pt(110) > Pt(100). On the other hand, due to having a lower affinity towards O2, the graphene-coated surfaces display different rate-determining steps. By considering only the activation energies of the rate-determining steps, the catalytic activity follows the order of Pt(111)@C > Pt(110)@C > Pt(100)@C. Estimated overpotentials indicated comparable ORR catalytic activity between the graphene-coated surfaces and bare Pt surfaces. Our calculations also highlight the dual role of the graphene coating. On the one hand, it improves the stability of the catalyst, for instance, by inhibiting the aggregation of Pt nanoparticles. On the other hand, it can actively participate in the reaction mechanism. Finally, we also pointed out that the introduction of nitrogen on the Pt(111)@C surface could potentially enhance the ORR activity by reducing the overpotential. The activity of Pt(111)@C and Pt(111)@C/N, together with the fact that they possess good stability owing to the graphene layer, have demonstrated the potential of graphene-encapsulated Pt-based nanoparticle catalysts for the ORR.34,41Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the project JPNP20003, commissioned by the New Energy and Industrial Technology Development Organization (NEDO), Japan.Notes and references
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 CrossRef.
- M. Victoria, N. Haegel, I. M. Peters, R. Sinton, A. Jäger-Waldau, C. del Canizo, C. Breyer, M. Stocks, A. Blakers, I. Kaizuka, K. Komoto and A. Smets, Joule, 2021, 5, 1041–1056 CrossRef CAS.
- H. N. Nam, K. Suzuki, A. Masago, T. Q. Nguyen, H. Shinya, T. Fukushima and K. Sato, Appl. Phys. Lett., 2022, 120, 143903 CrossRef CAS.
- H. N. Nam, Q. M. Phung, K. Suzuki, H. Shinya, A. Masago, T. Fukushima and K. Sato, J. Mater. Chem. A, 2024, 12, 451–459 RSC.
- H. N. Nam, Q. M. Phung, K. Suzuki, A. Masago, H. Shinya, T. Fukushima and K. Sato, ACS Appl. Mater. Interfaces, 2023, 15, 43871–43879 CrossRef CAS PubMed.
- H. N. Nam, K. Suzuki, A. Masago, H. Shinya, T. Fukushima and K. Sato, Jpn. J. Appl. Phys., 2023, 62, 020904 CrossRef.
- H. N. Nam, K. Suzuki, T. Q. Nguyen, A. Masago, H. Shinya, T. Fukushima and K. Sato, Phys. Rev. B, 2022, 105, 075205 CrossRef CAS.
- H. N. Nam, R. Yamada, H. Okumura, T. Q. Nguyen, K. Suzuki, H. Shinya, A. Masago, T. Fukushima and K. Sato, Phys. Chem. Chem. Phys., 2021, 23, 9773–9784 RSC.
- M. T. Islam, M. Nabi, M. Arefin, K. Mostakim, F. Rashid, N. Hassan, S. Rahman, S. McIntosh, B. Mullins and S. Muyeen, Heliyon, 2022, 8, e11836 CrossRef PubMed.
- H. Yuan, Y. Hou, I. M. Abu-Reesh, J. Chen and Z. He, Mater. Horiz., 2016, 3, 382–401 RSC.
- J. Li, W. Xia, Y. Guo, R. Qi, X. Xu, D. Jiang, T. Wang, Y. Sugahara, J. He and Y. Yamauchi, Chem. Eng. J., 2023, 477, 146841 CrossRef CAS.
- Y. Zhao, H. Nara, D. Jiang, T. Asahi, S. M. Osman, J. Kim, J. Tang and Y. Yamauchi, Small, 2023, 19, 2304450 CrossRef CAS PubMed.
- M. Liu, X. Xiao, Q. Li, L. Luo, M. Ding, B. Zhang, Y. Li, J. Zou and B. Jiang, J. Colloid Interface Sci., 2022, 607, 791–815 CrossRef CAS PubMed.
- S. G. Ji, H. C. Kwon, T.-H. Kim, U. Sim and C. H. Choi, ACS Catal., 2022, 12, 7317–7325 CrossRef CAS.
- M. Che, Catal. Today, 2013, 218, 162–171 CrossRef.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- W. Chen, J. Huang, J. Wei, D. Zhou, J. Cai, Z.-D. He and Y.-X. Chen, Electrochem. Commun., 2018, 96, 71–76 CrossRef CAS.
- W.-J. Lee, S. Bera, H.-J. Woo, W. Hong, J.-Y. Park, S.-J. Oh and S.-H. Kwon, Chem. Eng. J., 2022, 442, 136123 CrossRef CAS.
- X. Xiao, L. Zou, H. Pang and Q. Xu, Chem. Soc. Rev., 2020, 49, 301–331 RSC.
- S. Wu, X. Qu, J. Zhu, X. Liu, H. Mao, K. Wang, G. Zhou, J. Chi and L. Wang, J. Alloys Compd., 2023, 172518 Search PubMed.
- X. Chen, Y. Li and X. Zhao, Surf. Interfaces, 2023, 38, 102821 CrossRef CAS.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang and D. Su, et al. , J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 2075 CrossRef CAS PubMed.
- N. Wang, S. Gan, Y. Mao, J. Xiao, C. Xu and T. Zhou, Phys. Chem. Chem. Phys., 2024, 26, 2449–2456 RSC.
- W. Zhang, J.-K. Pan, Y.-F. Yu, X.-J. Zhang, J.-H. Wang, W.-X. Chen and G.-L. Zhuang, Phys. Chem. Chem. Phys., 2023, 25, 11673–11683 RSC.
- Y. Li, Y. Feng, D. Zheng, X. Zhao, Y. Zhou, X. Fu and X. Chen, J. Chem. Eng., 2023, 476, 146753 CrossRef CAS.
- Z. Xue, X. Zhang, J. Qin and R. Liu, J. Energy Chem., 2021, 55, 437–443 CrossRef CAS.
- X. Lv, W. Wei, H. Wang, B. Huang and Y. Dai, Appl. Catal., B, 2020, 264, 118521 CrossRef CAS.
- R. K. Ahluwalia, D. D. Papadias, N. N. Kariuki, J.-K. Peng, X. Wang, Y. Tsai, D. G. Graczyk and D. J. Myers, J. Electrochem. Soc., 2018, 165, F3024–F3035 CrossRef CAS.
- M. F. Labata, G. Li, J. Ocon and P.-Y. A. Chuang, J. Power Sources, 2021, 487, 229356 CrossRef CAS.
- A. G. Manjón, M. Vega-Paredes, V. Berova, T. Gänsler, T. Schwarz, N. A. R. Rivas, K. Hengge, T. Jurzinsky and C. Scheu, Nanoscale, 2022, 14, 18060–18069 RSC.
- Q. Li, L. Wu, G. Wu, D. Su, H. Lv, S. Zhang, W. Zhu, A. Casimir, H. Zhu and A. Mendoza-Garcia, et al. , Nano Lett., 2015, 15, 2468–2473 CrossRef CAS PubMed.
- N. Aoki, H. Inoue, H. Kawasaki, H. Daimon, T. Doi and M. Inaba, J. Electrochem. Soc., 2018, 165, F737 CrossRef CAS.
- J.-H. Park, K. Kim, X. Wang, M. Huda, Y. Sawada, Y. Matsuo, N. Saito and M. Kawasumi, J. Power Sources, 2023, 580, 233419 CrossRef CAS.
- H.-J. Choi, S.-M. Jung, J.-M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS.
- M. B. Burkholder, F. B. A. Rahman, E. H. Chandler Jr, J. Regalbuto, B. Gupton and J. M. M. Tengco, Carbon Trends, 2022, 100196 CrossRef CAS.
- H. Yamada, H. Kato and K. Kodama, J. Electrochem. Soc., 2020, 167, 084508 CrossRef CAS.
- S. He, Y. Liu, H. Zhan and L. Guan, ACS Catal., 2021, 11, 9355–9365 CrossRef CAS.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J.-H. Jang, Y.-H. Cho, T. Lim, Y.-E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- H. Kim, A. W. Robertson, S. O. Kim, J. M. Kim and J. H. Warner, ACS Nano, 2015, 9, 5947–5957 CrossRef CAS PubMed.
- P. Q. Phan, R. Naraprawatphong, P. Pornaroontham, J. Park, C. Chokradjaroen and N. Saito, Mater. Adv., 2021, 2, 322–335 RSC.
- J. Bai, Q. Zhu, Z. Lv, H. Dong, J. Yu and L. Dong, Int. J. Hydrogen Energy, 2013, 38, 1413–1418 CrossRef CAS.
- P. Rodrguez-Kessler and A. Rodrguez-Domnguez, J. Chem. Phys., 2015, 143, 184312 CrossRef PubMed.
- L. Sementa, O. Andreussi, W. A. Goddard III and A. Fortunelli, Catal. Sci. Technol., 2016, 6, 6901–6909 RSC.
- P. L. Rodrguez-Kessler, A. Muñoz-Castro, A. R. Rodrguez-Domnguez and J. L. Cabellos, Phys. Chem. Chem. Phys., 2023, 25, 4764–4772 RSC.
- A. Panchenko, M. Koper, T. Shubina, S. Mitchell and E. Roduner, J. Electrochem. Soc., 2004, 151, A2016 CrossRef CAS.
- B. Han, V. Viswanathan and H. Pitsch, J. Phys. Chem. C, 2012, 116, 6174–6183 CrossRef CAS.
- Z. Duan and G. Wang, J. Phys. Chem. C, 2013, 117, 6284–6292 CrossRef CAS.
- J. I. Choi, H. S. Kim, Y.-J. Sohn, S.-D. Yim, F. M. Alamgir and S. S. Jang, ACS Appl. Nano Mater., 2021, 4, 1067–1075 CrossRef CAS.
- J. A. Keith, G. Jerkiewicz and T. Jacob, ChemPhysChem, 2010, 11, 2779–2794 CrossRef CAS PubMed.
- A. Eichler and J. Hafner, Phys. Rev. Lett., 1997, 79, 4481 CrossRef CAS.
- D.-H. Lim and J. Wilcox, J. Phys. Chem. C, 2012, 116, 3653–3660 CrossRef CAS.
- N. Jin, J. Han, H. Wang, X. Zhu and Q. Ge, Int. J. Hydrogen Energy, 2015, 40, 5126–5134 CrossRef CAS.
- R. Zhang, N. Shehzad, L. Zhang, A. Ali, B. Amin and I. Shahid, Chem. Phys., 2023, 570, 111888 CrossRef CAS.
- J.-K. Sun, Y.-W. Pan, M.-Q. Xu, L. Sun, S. Zhang, W.-Q. Deng and D. Zhai, Nano Res., 2023, 1–8 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- A. Dal Corso, Comput. Mater. Sci., 2014, 95, 337–350 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Advances in catalysis, Elsevier, 2000, vol. 45, pp. 71–129 Search PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer and C. Hargus, et al. , J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- C. Fu, C. Liu, T. Li, X. Zhang, F. Wang, J. Yang, Y. Jiang, P. Cui and H. Li, Comput. Mater. Sci., 2019, 170, 109202 CrossRef CAS.
- J.-W. Chen, Z. Zhang, H.-M. Yan, G.-J. Xia, H. Cao and Y.-G. Wang, Nat. Commun., 2022, 13, 1734 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. K. Iyemperumal and N. A. Deskins, ChemPhysChem, 2017, 18, 2171–2190 CrossRef CAS PubMed.
- Q. Wang, R. Pang and X. Shi, J. Phys. Chem. C, 2015, 119, 22534–22541 CrossRef CAS.
- A. J. Martnez-Galera, N. Nicoara, J. I. Martnez, Y. J. Dappe, J. Ortega and J. M. Gómez-Rodrguez, J. Phys. Chem. C, 2014, 118, 12782–12788 CrossRef.
- M. Cattelan, G. Peng, E. Cavaliere, L. Artiglia, A. Barinov, L. T. Roling, M. Favaro, I. Pš, S. Nappini and E. Magnano, et al. , Nanoscale, 2015, 7, 2450–2460 RSC.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Phys. Rev. Lett., 1996, 76, 2141 CrossRef CAS PubMed.
- Y. Yao, Q. Fu, Y. Zhang, X. Weng, H. Li, M. Chen, L. Jin, A. Dong, R. Mu and P. Jiang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17023–17028 CrossRef CAS PubMed.
- H. Yan, B. Xu, S. Shi and C. Ouyang, J. Appl. Phys., 2012, 112, 104316 CrossRef.
- H. Guang, M. Aoki, S. Tanaka and M. Kohyama, Trans. Mater. Res. Soc. Jpn., 2013, 38, 477–480 CrossRef CAS.
- F. R. Bagsican, A. Winchester, S. Ghosh, X. Zhang, L. Ma, M. Wang, H. Murakami, S. Talapatra, R. Vajtai and P. M. Ajayan, et al. , Sci. Rep., 2017, 7, 1774 CrossRef PubMed.
- H. Shin, Y. Luo, A. Benali and Y. Kwon, Phys. Rev. B, 2019, 100, 075430 CrossRef CAS.
- Z. Shi, J. Zhang, Z.-S. Liu, H. Wang and D. P. Wilkinson, Electrochim. Acta, 2006, 51, 1905–1916 CrossRef CAS.
- A. Lahee, W. Allison, R. Willis and K. Rieder, Surf. Sci., 1983, 126, 654–660 CrossRef CAS.
- G. Kellogg, Phys. Rev. Lett., 1985, 55, 2168 CrossRef CAS PubMed.
- E. C. Sowa, M. Van Hove and D. Adams, Surf. Sci., 1988, 199, 174–182 CrossRef CAS.
- A. Walker, B. Klötzer and D. King, J. Chem. Phys., 1998, 109, 6879–6888 CrossRef CAS.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu and C. T. Williams, et al. , Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- V. Gueskine, A. Singh, M. Vagin, X. Crispin and I. Zozoulenko, J. Phys. Chem. C, 2020, 124, 13263–13272 CrossRef CAS.
- Z. Duan and G. Wang, Phys. Chem. Chem. Phys., 2011, 13, 20178–20187 RSC.
- G.-L. Chai, Z. Hou, D.-J. Shu, T. Ikeda and K. Terakura, J. Am. Chem. Soc., 2014, 136, 13629–13640 CrossRef CAS PubMed.
- A. M. Gómez-Marn, R. Rizo and J. M. Feliu, Catal. Sci. Technol., 2014, 4, 1685–1698 RSC.
- A. Abdelhafiz, A. Vitale, P. Buntin, B. deGlee, C. Joiner, A. Robertson, E. M. Vogel, J. Warner and F. M. Alamgir, Energy Environ. Sci., 2018, 11, 1610–1616 RSC.
- C. D. Jaimes-Paez, E. Morallón and D. Cazorla-Amorós, Energy, 2023, 278, 127888 CrossRef CAS.
- D. Liu, J. Zhang, D. Liu, T. Li, Y. Yan, X. Wei, Y. Yang, S. Yan and Z. Zou, J. Phys. Chem. Lett., 2022, 13, 2019–2026 CrossRef CAS PubMed.
- Q. Lv, W. Si, J. He, L. Sun, C. Zhang, N. Wang, Z. Yang, X. Li, X. Wang and W. Deng, et al. , Nat. Commun., 2018, 9, 3376 CrossRef PubMed.
- A. Martn-Recio, C. Romero-Muñiz, P. Pou, R. Pérez and J. M. Gómez-Rodrguez, Nanoscale, 2016, 8, 17686–17693 RSC.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- A. F. Z. Abidin and I. Hamada, Surf. Sci., 2022, 724, 122144 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00269e |
| This journal is © the Owner Societies 2024 |