
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The nature of metallophilic interactions in closed-shell d8–d8 metal complexes†
Lucas
de Azevedo Santos‡
 a,
Timon
Wagner‡
a,
Klaas
Visscher
a,
Jörn
Nitsch
a,
F. Matthias
Bickelhaupt
*abc and
Célia
Fonseca Guerra
*a
a,
Timon
Wagner‡
a,
Klaas
Visscher
a,
Jörn
Nitsch
a,
F. Matthias
Bickelhaupt
*abc and
Célia
Fonseca Guerra
*a
aDepartment of Chemistry and Pharmaceutical Sciences, AIMMS, Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081 HZ Amsterdam, The Netherlands. E-mail: c.fonsecaguerra@vu.nl; f.m.bickelhaupt@vu.nl; Web: https://www.theochem.nl
bInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
cDepartment of Chemical Sciences, University of Johannesburg, Auckland Park, Johannesburg 2006, South Africa
First published on 18th July 2024
Abstract
We have quantum chemically analyzed the closed-shell d8–d8 metallophilic interaction in dimers of square planar [M(CO)2X2] complexes (M = Ni, Pd, Pt; X = Cl, Br, I) using dispersion-corrected density functional theory at ZORA-BLYP-D3(BJ)/TZ2P level of theory. Our purpose is to reveal the nature of the [X2(CO)2M]⋯[M(CO)2X2] bonding mechanism by analyzing trends upon variations in M and X. Our analyses reveal that the formation of the [M(CO)2X2]2 dimers is favored by an increasingly stabilizing electrostatic interaction when the M increases in size and by more stabilizing dispersion interactions promoted by the larger X. In addition, there is an overlooked covalent component stemming from metal–metal and ligand–ligand donor–acceptor interactions. Thus, at variance with the currently accepted picture, the d8–d8 metallophilicity is attractive, and the formation of [M(CO)2X2]2 dimers is not a purely dispersion-driven phenomenon.
1. Introduction
Metallophilicity is the manifestation of a net attractive intra- or intermolecular interaction between metal centers (M), yielding metal–metal-bonded complexes.1 This metal–metal interaction causes the spontaneous association, i.e., self-assembly, of metal complexes forming aggregates with luminescence properties2 and medicinal applications.3 Earlier work by Hoffman et al. showed that the closed-shell metallophilic interactions between linear d10 metal complexes are covalent due to the donor–acceptor interactions between the filled ndZ2-type HOMO on one M and the (n + 1)s-type LUMO on the other M.4 However, Brands et al. showed that the donor–acceptor interactions cannot overcome the destabilizing steric Pauli repulsion between the two metal centers, and the metallophilic interactions are only attractive due to the additional stabilizing electrostatic interactions.5 In general, the closed-shell d10–d10 metalllophilic interactions have a similar mechanistic picture to other well-known intermolecular interactions, such as hydrogen bonds.6Nonetheless, alternative models have been used to explain metallophilicity. For example, Pyykkö and coworkers concluded that the d10–d10 Au+⋯Au+ attraction in staggered dimers of linear AuPH3Cl complexes is a dispersion-driven phenomenon, whereas the ligand–ligand repulsion hampers the formation of parallel dimers.7 This has been further supported by Chen et al. who found that π–π interactions between face-to-face, eclipsed ligands are repulsive.8 On the other hand, Che et al. attributed the formation of d8–d8 and d10–d10 metal-complexes dimers to stabilizing ligand–ligand dispersion interactions.9
In the present study, we have quantum chemically investigated the bonding mechanism of eclipsed closed-shell d8–d8 metal complexes, using [M(CO)2X2]2 dimers (M = Ni, Pd, Pt; X = Cl, Br, I) as model systems (Scheme 1). As will become clear in the following sections, our bonding analyses reveal that the stability of the [M(CO)2X2]2 dimers stems from significantly attractive electrostatic interactions, on top of dispersion interactions promoted by the ligands. This closed-shell d8–d8 metal–metal interaction is, therefore, similar to that between d10 metal centers, that is, it is also partially covalent in nature and, thus, not a pure dispersion-driven phenomenon.
 | ||
| Scheme 1 The studied [M(CO)2X2]2 dimers. | ||
2. Computational methods
2.1. Computational details
All calculations were carried out using the Amsterdam Modeling Suite (AMS) 2020.101 program.10 All stationary points and energies were obtained using relativistic, dispersion-corrected density functional theory at ZORA-BLYP-D3(BJ)/TZ2P. This approach has been benchmarked in previous work for bond energies of metal–metal complexes using CCSD(T)/CBS with mean absolute error of 0.7 kcal mol−1.5 This approach comprises the BLYP level of the generalized gradient approximation (GGA); exchange functional developed by Becke (B), and the GGA correlation functional developed by Lee, Yang, and Parr (LYP)11 (see Tables S4 and S5 in the ESI,† for the Cartesian coordinates). The DFT-D3(BJ) method developed by Grimme and coworkers,12 which contains the damping function proposed by Becke and Johnson,13 is used to describe non-local dispersion interactions. Scalar relativistic effects are accounted for using the zeroth-order regular approximation (ZORA).14 Molecular orbitals (MO) were expanded in a large, uncontracted set of Slater-type orbitals (STOs) containing diffuse functions: TZ2P.15 The basis set is of triple-ζ quality augmented with polarization functions, i.e., one 3d and one 4f set on C, O, Cl; one 4d and one 4f set on Br; one 5d and one 4f set on I; one 4f set on Ni; one 5p and one 4f set on Pd; one 6p and one 5f set on Pt. All electrons were included in the variational process, i.e., no frozen core approximation was applied. The accuracies of both the fitting scheme and the integration grid (Becke grid) were set to ‘EXCELLENT’. The formation of trans-[M(CO)2X2] monomers and trans-[M(CO)2X2]2 dimers (M = Ni, Pd, Pt; X = Cl, Br, I) is, in most cases, more favorable than the formation of cis-[M(CO)2X2] monomers and cis-[M(CO)2X2]2 dimers (see Table S3 and Fig. S4 for bond energies and geometries, ESI†). The [Ni(CO)2Br2], [Ni(CO)2I2], and [Pd(CO)2I2] monomers, and their respective dimers, are the only exceptions in which the cis forms are more favorable (Table S3, ESI†). Thus, in this study, we always refer to the trans-[M(CO)2X2]2 dimers. We have found here that the global-minimum structures of the [M(CO)2X2]2 dimers are in the staggered conformation by performing a fully relaxed rotation around the M⋯M bond, in which the X–M⋯M–X dihedral angle is varied from 0° to 90° (see Fig. S5 and S6 in the ESI†). All optimized structures were confirmed to be true minima through vibrational analysis (no imaginary frequencies).2.2. Bond analyses
Insight into the bonding mechanism is obtained by analyzing the potential energy curves for the formation of the [M(CO)2X2]2 dimers (M = Ni, Pd, Pt; X = Cl, Br, I). The analyses are done by varying the M⋯M bond distance within the range of 2.5 Å to 4.5 Å, starting from the equilibrium geometry of the complex (rM⋯M,eq) while keeping all other geometrical parameters frozen.The interaction between [X2(CO)2M] and [M(CO)2X2] is analyzed using the activation strain model,16 which is a fragment-based approach to understanding the energy profile associated with a chemical process in terms of the original reactants. Thus, the total bond energy ΔE is decomposed along the M⋯M bond distance rM⋯M (or just at one point along rM⋯M) into the strain energy ΔEstrain(rM⋯M), which is associated with the geometrical deformation of the individual reactants as the process takes place, plus the actual interaction energy ΔEint(rM⋯M) between the deformed reactants [eqn (1)].
| ΔE(rM⋯M) = ΔEstrain(rM⋯M) + ΔEint(rM⋯M) | (1) |
| ΔEint(rM⋯M) = ΔVelstat(rM⋯M) + ΔEPauli(rM⋯M) + ΔEoi(rM⋯M) + ΔEdisp(rM⋯M) | (2) |
The Pauli repulsion energy (ΔEPauli) comprises the destabilizing interactions between the fully occupied orbitals on either fragment and arises from the antisymmetrization of the Hartree wavefunction due to the Pauli principle. The orbital-interaction energy (ΔEoi) accounts for charge transfer, that is, the interaction between occupied orbitals of one fragment with unoccupied orbitals of the other fragment, including the interactions of the highest occupied and lowest unoccupied MOs (HOMO–LUMO), and polarization, that is, empty–occupied orbital mixing on one fragment, due to the presence of another fragment. The dispersion energy ΔEdisp accounts for the dispersion corrections as introduced by Grimme et al.12 To facilitate the analyses, the ASM and EDA were performed using the PyFrag 2019 program.18
3. Results and discussion
3.1. Bond strength and structure
Equilibrium geometries, dimerization energies (ΔE), and the M⋯M bond distances (rM⋯M) of the stable CS symmetrical [M(CO)2X2]2 dimers (M = Ni, Pd, Pt; X = Cl, Br, I) calculated at the ZORA-BLYP-D3(BJ)/TZ2P level of theory are shown in Fig. 1. Upon the formation of the studied dimers, we have identified two main features: (i) the stability of the dimers significantly increases, and the M⋯M bond distance slightly shortens for heavier M; and (ii) the dimers are only slightly stronger but longer for heavier X ligands. For example, along the series from [Ni(CO)2Br2]2 to [Pt(CO)2Br2]2, ΔE becomes more stabilizing from −13.1 kcal mol−1 to −17.6 kcal mol−1 and rM⋯M decreases from 3.269 Å to 3.192 Å (Fig. 1). Along the series from [Pd(CO)2Cl2]2 to [Pd(CO)2I2]2, ΔE varies only from −15.8 kcal mol−1 to −16.6 kcal mol−1 and rM⋯M increases from 3.117 Å to 3.220 Å (Fig. 1). | ||
| Fig. 1 Equilibrium geometries (in Å) and electronic bond energies (in kcal mol−1) of the [M(CO)2X2]2 dimers (M = Ni, Pd, Pt; X = Cl, Br, I). Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
Next, we performed our activation strain analysis16 (ASA) to decompose the bond energies (ΔE) into the strain (ΔEstrain) and interaction (ΔEint) energies (see Computational methods), and the results are shown in Table 1. The trends in the stability of the [M(CO)2X2]2 dimers along M = Ni, Pd, Pt and X = Cl, Br, I are dominated by the stronger ΔEint, whereas the weaker ΔEstrain varies only slightly along the same series. For example, along the series from [Ni(CO)2Br2]2 to [Pt(CO)2Br2]2, ΔEint becomes more stabilizing from −14.0 kcal mol−1 to −18.9 kcal mol−1 and ΔEstrain becomes slightly more destabilizing from 0.9 kcal mol−1 to 1.3 kcal mol−1 (Table 1). Along the series from [Pd(CO)2Cl2]2 to [Pd(CO)2I2]2, ΔEint becomes more stabilizing from −16.6 kcal mol−1 to −17.9 kcal mol−1 and ΔEstrain becomes slightly more destabilizing from 0.8 kcal mol−1 to 1.3 kcal mol−1 (Table 1). Note that the impact on ΔEint and, thus, on the stability of the [M(CO)2X2]2 dimers when varying M is larger than when varying X. As will become clear in the next sections, this is because of the difference in nature between the metal–metal interactions and those involving the ligands. This conclusion emerges from understanding the physical nature behind the observed trends in ΔEint when the metal center M varies along Ni, Pd, and Pt and the ligands X vary along Cl, Br, and I.
| M | X | r M⋯M | ΔE | ΔEstrain | ΔEint | ΔVelstat | ΔEPauli | ΔEoi | ΔEdisp |
|---|---|---|---|---|---|---|---|---|---|
| Ni | Cl | 3.199 | −12.6 | 0.6 | −13.2 | −12.3 | 20.3 | −7.6 | −13.6 |
| Br | 3.269 | −13.1 | 0.9 | −14.0 | −14.8 | 21.3 | −8.0 | −15.4 | |
| I | 3.371 | −13.8 | 0.9 | −14.7 | −11.6 | 22.7 | −7.7 | −18.1 | |
| Pd | Cl | 3.117 | −15.8 | 0.8 | −16.6 | −24.1 | 34.8 | −11.4 | −15.8 |
| Br | 3.160 | −15.9 | 1.3 | −17.2 | −23.1 | 35.8 | −12.0 | −17.9 | |
| I | 3.220 | −16.6 | 1.3 | −17.9 | −22.3 | 37.1 | −12.0 | −20.8 | |
| Pt | Cl | 3.157 | −17.1 | 0.9 | −18.0 | −28.0 | 39.5 | −12.9 | −16.6 |
| Br | 3.192 | −17.6 | 1.3 | −18.9 | −27.3 | 41.0 | −13.8 | −18.9 | |
| I | 3.239 | −18.3 | 1.8 | −20.1 | −26.8 | 43.2 | −14.2 | −22.1 | |
3.2. Variation of M
To understand the origin of the increased stabilization of the [M(CO)2X2]2 dimers upon varying M along Ni, Pd, and Pt, we further decomposed the ΔEint into physically meaningful terms, namely the electrostatic interactions (ΔVelstat), steric Pauli repulsion (ΔEPauli), orbital interactions (ΔEoi), and dispersion energy (ΔEdisp) using our energy decomposition analysis17 (EDA; see Computational methods), and the results are shown in Table 1. Our analyses reveal that, for the smaller Ni, we find that ΔEdisp is the largest attractive term in [X2(CO)2M]⋯[M(CO)2X2]. However, as M increases in size, ΔVelstat and ΔEoi become significantly more stabilizing whereas ΔEdisp varies much less, which causes ΔVelstat to dominate and become the most attractive term for Pd and Pt. For example, from [Ni(CO)2Br2]2 to [Pt(CO)2Br2]2, ΔEdisp varies only from −15.4 kcal mol−1 to −18.9 kcal mol−1, whereas ΔVelstat and ΔEoi become significantly more stabilizing from −14.8 kcal mol−1 to −27.3 kcal mol−1 and from −8.0 kcal mol−1 to −13.8 kcal mol−1, respectively (Table 1).For a consistent comparison, we extend our analysis to the entire reaction coordinate, as a function of the M⋯M bond distance (rM⋯M). Since ΔEstrain is small and the ΔE is dominated by ΔEint, the analyses were done while the geometries of the fragments were kept frozen to that of the equilibrium geometries of the [M(CO)2X2]2 dimers. The resulting interaction energy curves ΔEint(rM⋯M) for the representative [Ni(CO)2Br2]2, [Pd(CO)2Br2]2, and [Pt(CO)2Br2]2 series are graphically shown in Fig. 2. Herein, the ΔEint(rM⋯M) curves also become more stabilizing and the energy minimum is shifted towards shorter rM⋯M as M varies along Ni, Pd, and Pt.
 | ||
| Fig. 2 Interaction energies (in kcal mol−1) as a function of the M⋯M distance (in Å) starting from the equilibrium geometries of the representative [M(CO)2Br2]2 dimers (M = Ni, Pd, Pt), while the geometry of the monomers is kept frozen. Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
Next, we analyze the EDA terms as a function of rM⋯M and the resulting diagrams for the representative [Ni(CO)2Br2]2, [Pd(CO)2Br2]2, and [Pt(CO)2Br2]2 series are graphically shown in Fig. 3. Our findings show that the increased stability of the [M(CO)2Br2]2 dimers as M varies along Ni, Pd, Pt is due to a greater electrostatic attraction between the monomers, as the electrostatic interaction curves ΔVelstat(rM⋯M) become significantly more stabilizing along the same series (Fig. 3). This is because the [X2(CO)2M]⋯[M(CO)2X2] electrostatic attraction is largely affected by the charge distributions around the metal centers within the monomers. In essence, there is a more effective electron–nuclei overlap as the electron density (ρ) of [M(CO)2Br2]2 becomes more diffuse, and the nucleus of the metal center increases in size (i.e., has more protons) along the same series. For example, at the same point of the reaction coordinate, e.g., rM⋯M = 3.5 Å, the negatively charged ρ of one fragment is more diffuse around the metal center and extends further towards the nucleus of the metal center of the other fragment, which becomes more positively charged as M varies along Ni, Pd, and Pt (see Fig. 4). As the two monomers approach each other, this attractive electron–nuclei overlap more quickly increases for heavier M and, consequently, the slope of the descending ΔVelstat(rM⋯M) curves increases along Ni, Pd, and Pt, shifting the equilibrium geometries of the [M(CO)2Br2]2 dimers to a shorter rM⋯M as M varies along the same series.
 | ||
| Fig. 3 Energy decomposition analysis (in kcal mol−1) as a function of the M⋯M distance (in Å) for the representative [M(CO)2Br2]2 dimers (M = Ni, Pd, Pt), while the geometry of the monomers is kept frozen. Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
 | ||
| Fig. 4 Attractive overlap between the negatively charged electron densities and the positively charged nuclei (density contours from 0.0001 to 0.5000 a.u.) for the representative [M(CO)2Br2]2 dimers (M = Ni, Pd, Pt) at a consistent M⋯M bond distance (rM⋯M = 3.5 Å). Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
Together with ΔVelstat(rM⋯M), the orbital interactions ΔEoi(rM⋯M) and the dispersion energy curves ΔEdisp(rM⋯M) follow the same trend as ΔEint(rM⋯M) and become more stabilizing along M = Ni, Pd, Pt (Fig. 3). This effect is more pronounced in ΔEoi(rM⋯M) which arises, in part, from the donation of charge from the metal dZ2-type HOMO of one M(CO)2X2 monomer into the metal s-type LUMO of the other monomer and will be explained in detail later (see Scheme 2). The ΔEoi(rM⋯M) curves are weakly attractive at longer rM⋯M and, as soon as the dZ2-HOMO and s-LUMO start to overlap at shorter rM⋯M, they become significantly stabilizing (Fig. 3). The ΔEdisp(rM⋯M) curves, on the other hand, are relatively strong already at longer rM⋯M, but neither become much more stabilizing at shorter rM⋯M nor significantly vary along M = Ni, Pd, Pt. In other words, the ΔEdisp term significantly contributes to the stability of the [M(CO)2X2]2 dimers but is almost insensitive to variations of the metal centers.
 | ||
| Scheme 2 Schematic molecular orbital diagram of the metal–metal donor–acceptor interactions in [M(CO)2X2]2 dimers. The DFT orbitals are shown in Fig. S3 of the ESI.† | ||
3.3. Variation of X
To understand why the ΔEint, and thus the [M(CO)2X2]2 dimers, becomes slightly more stable upon varying X along Cl, Br, and I, we first analyze the EDA terms at the equilibrium geometries. Contrary to the variation along M = Ni, Pd, Pt, our analyses reveal that ΔEdisp becomes more stabilizing, whereas ΔVelstat and ΔEoi do not significantly change when X varies along Cl, Br, and I. For example, from [Pd(CO)2Cl2]2 to [Pd(CO)2I2]2, ΔEdisp becomes more stabilizing from −15.8 kcal mol−1 to −20.8 kcal mol−1, whereas ΔVelstat and ΔEoi vary from −24.1 kcal mol−1 to −22.3 kcal mol−1 and −11.4 kcal mol−1 to −12.0 kcal mol−1, respectively (Table 1).Next, we analyze the ΔEint and the EDA terms as a function of rM⋯M, and the resulting diagrams for the representative [Pd(CO)2Cl2]2, [Pd(CO)2Br2]2, and [Pd(CO)2I2]2 series are graphically shown in Fig. 5 and 6. The ΔEint(rM⋯M) curves also become more stabilizing, and the energy minimum is shifted towards longer rM⋯M as X varies along Cl, Br, and I. Our analyses show that the increased stability of the [Pd(CO)2X2]2 dimers as X varies along Cl, Br, and I is due to a greater attractive dispersion interaction promoted by the ligands as they increase in size along the same series. Note that the ΔEdisp(rM⋯M) curves become significantly more stabilizing along [Pd(CO)2Cl2]2, [Pd(CO)2Br2]2, and [Pd(CO)2I2]2 (Fig. 6). Nevertheless, the equilibrium rM⋯M slightly expands because the Pauli repulsion curves ΔEPauli(rM⋯M) are steeper than the ΔEdisp(rM⋯M) curves and push the equilibrium rM⋯M to longer values as X varies along Cl, Br, and I (Fig. 6).
 | ||
| Fig. 5 Interaction energies (in kcal mol−1) as a function of the M⋯M distance (in Å) starting from the equilibrium geometries of the representative [Pd(CO)2X2]2 dimers (X = Cl, Br, I), while the geometry of the monomers is kept frozen. Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
 | ||
| Fig. 6 Energy decomposition analysis (in kcal mol−1) as a function of the M⋯M distance (in Å) for the representative [Pd(CO)2X2]2 dimers (X = Cl, Br, I), while the geometry of the monomers is kept frozen. Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
The ΔVelstat(rM⋯M) curves are almost insensitive to variations of X in the [Pd(CO)2X2]2 dimers and become only slightly more stabilizing along X = Cl, Br, I (Fig. 6). This is because, as aforementioned, the attractive electron–nuclei overlap occurs mainly at the M⋯M bond region and does not significantly change as the X ligands increase in size along X = Cl, Br, I. Instead, the increased size of X causes the ρ of the monomers to extend further along the CO⋯X region, where there is not a pronounced electron–nuclei overlap (see Fig. S1, ESI†). Consequently, the electrostatic attraction between the monomers in the [M(CO)2X2]2 only marginally increases when X varies along Cl, Br, and I.
Our analyses along variations of M and X have revealed two important interactions for the bonding mechanism and, thus, stability of the [M(CO)2X2]2 dimers. On the one hand, there are electrostatic interactions that increase in relevance as the metal becomes bigger. On the other hand, there are attractive dispersion interactions stemming from the ligands. Despite this difference in nature between the metal–metal and ligand–ligand interactions, they also share some similarities. Similar to the trend upon varying M along Ni, Pd, and Pt, the ΔEoi(rM⋯M) curves also become more stabilizing when X varies along Cl, Br, and I in the [Pd(CO)2X2]2 dimers (Fig. 6). This is due to the stabilizing ligand–ligand donor–acceptor interaction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O π*-type (π*CO) LUMO of one monomer with the X lone-pair-type (LPX) HOMO of the other monomer (see Scheme 3). In the following section, we explain why the ΔEoi(rM⋯M) curves become more stabilizing along M = Ni, Pd, Pt and X = Cl, Br, I in the [M(CO)2X2]2 dimers.
O π*-type (π*CO) LUMO of one monomer with the X lone-pair-type (LPX) HOMO of the other monomer (see Scheme 3). In the following section, we explain why the ΔEoi(rM⋯M) curves become more stabilizing along M = Ni, Pd, Pt and X = Cl, Br, I in the [M(CO)2X2]2 dimers.
 | ||
| Scheme 3 Schematic molecular orbital diagram of the ligand–ligand donor–acceptor interactions in [M(CO)2X2]2 dimers. The DFT orbitals are shown in Fig. S3 of the ESI.† | ||
3.4. Metal–metal versus ligand–ligand donor–acceptor interactions
In previous sections, we showed that the stability of the [M(CO)2X2]2 dimers is mainly governed by the electrostatic attraction, which becomes more important for larger M and dispersive interactions promoted by the ligands. However, both metal–metal and ligand–ligand interactions have a covalent component stemming from donor–acceptor interactions that has been hitherto overlooked. Note that the ΔEoi between the monomers in [M(CO)2X2]2 dimers, which accounts for donor–acceptor interactions, becomes more stabilizing along M = Ni, Pd, Pt and X = Cl, Br, I (vide supra). Next, we explain the physical nature behind these trends.The donor–acceptor interactions in the [M(CO)2X2]2 dimers comprise of two main components, namely, the metal–metal dZ2-HOMO–s-LUMO and the ligand–ligand π*CO-LUMO–LPX-HOMO interactions (see Schemes 2 and 3). The relevance of a donor–acceptor interaction can be estimated by the magnitude of its orbital stabilization, which is proportional to its HOMO–LUMO overlap squared (S2) divided by its respective orbital energy gap (Δε) [see eqn (3)]. For a consistent comparison, we report these values for the metal–metal and ligand–ligand donor–acceptor interactions at rM⋯M = 3.5 for all [M(CO)2X2]2 dimers in Table 2.
| ΔEoi ∝ S2/Δε | (3) |
| M | X | ΔEoi | Metal–metal | Ligand–ligand | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | Δεa | 103×S2/Δε | %b | S | Δεa | 103×S2/Δε | %b | |||
| a See HOMO and LUMO energies in Tables S1 and S2 (ESI). b Contribution of the associated orbital stabilization to the total metal–metal + ligand–ligand orbital stabilization. | ||||||||||
| Ni | Cl | −3.8 | 0.11 | 8.2 | 1.5 | 53 | 0.07 | 3.8 | 1.3 | 47 |
| Br | −4.9 | 0.09 | 7.8 | 1.0 | 36 | 0.08 | 3.5 | 1.8 | 64 | |
| I | −5.8 | 0.08 | 7.3 | 0.9 | 25 | 0.09 | 3.1 | 2.6 | 75 | |
| Pd | Cl | −4.7 | 0.18 | 8.4 | 3.9 | 67 | 0.08 | 3.4 | 1.9 | 33 |
| Br | −5.6 | 0.17 | 8.1 | 3.6 | 58 | 0.09 | 3.1 | 2.6 | 42 | |
| I | −6.5 | 0.16 | 7.7 | 3.3 | 47 | 0.10 | 2.7 | 3.7 | 53 | |
| Pt | Cl | −5.8 | 0.20 | 8.2 | 4.9 | 67 | 0.09 | 3.3 | 2.5 | 33 |
| Br | −6.8 | 0.18 | 8.1 | 4.0 | 55 | 0.10 | 3.0 | 3.3 | 45 | |
| I | −8.0 | 0.17 | 7.8 | 3.7 | 44 | 0.11 | 2.6 | 4.7 | 56 | |
The ΔEoi(rM⋯M) curves for the [M(CO)2X2]2 dimers become more stabilizing along M = Ni, Pd, Pt because the metal–metal donor–acceptor interactions between the dZ2-type HOMO of one monomer and the s-type LUMO of the other monomer are strengthened along the same series. When the metal center increases in size along Ni, Pd, and Pt, the bond overlap S between the dZ2-HOMO and the s-LUMO increases as both orbitals become more diffuse, resulting in larger orbital stabilization and, thus, stronger donor–acceptor interactions along the same series. For example, in the series of [Ni(CO)2Br2]2, [Pd(CO)2Br2]2, and [Pt(CO)2Br2]2 at rM⋯M = 3.5, S increases from 0.09 to 0.17 to 0.18, as both the dZ2-HOMO and the s-LUMO further extend towards the other metal center (see Fig. 7a, but also the overlap densities in Fig. S2a, ESI†). Consequently, S2/Δε times 103 increases from 1.0 to 3.6 to 4.0 along the same series (Table 2).
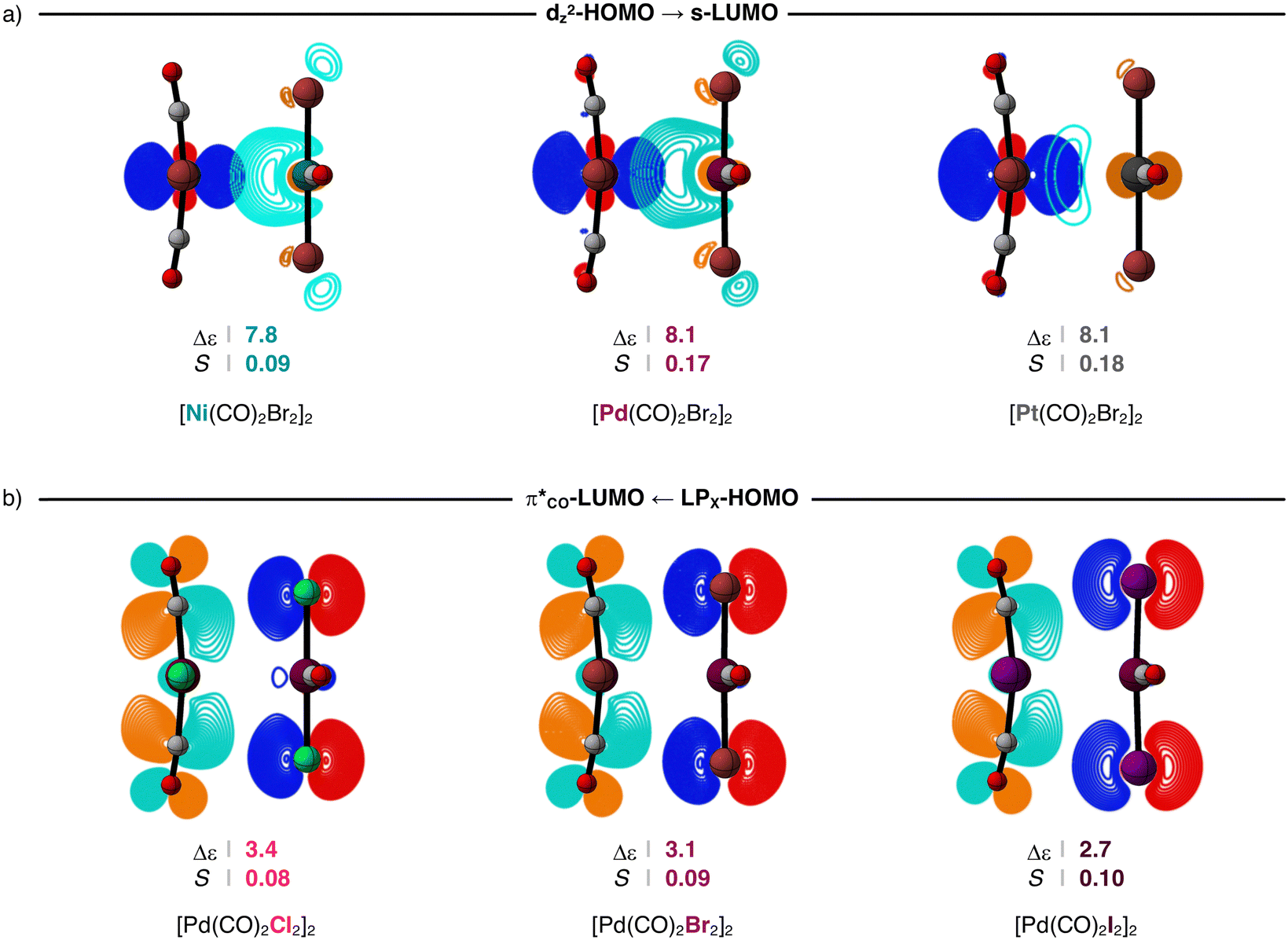 | ||
| Fig. 7 Stabilizing HOMO–LUMO energy gap (in eV) and orbital overlap for (a) the metal–metal and (b) ligand–ligand donor–acceptor interactions for the representative [M(CO)2Br2]2 (M = Ni, Pd, Pt) and [Pd(CO)2X2]2 (X = Cl, Br, I) dimers at a consistent M⋯M bond distance (rM⋯M = 3.5 Å; contour plots from 0.9 to 0.04 a.u.). Computed at ZORA-BLYP-D3(BJ)/TZ2P. | ||
The ΔEoi term in the [M(CO)2X2]2 dimers is not fully dominated by the metal–metal donor–acceptor interactions and is, in part, made by ligand–ligand donor–acceptor interactions. This is confirmed by the trends in the ΔEoi(rM⋯M) curves that also become more stabilizing along X = Cl, Br, I (Fig. 6). The bond overlap S for the π*CO-LUMO–LPX-HOMO interaction is, in general, significantly smaller than that of the metal–metal donor–acceptor interactions (Table 2). However, as the X ligands become larger and less electronegative along Cl, Br, and I, the LPX-HOMO extends further towards the π*CO-LUMO and becomes higher in energy (see Fig. S2b and Table S2 for the orbital energies, ESI†). This results in a larger S and a smaller Δε between the π*CO-LUMO and the LPX-HOMO. For example, in the series of [Pd(CO)2Cl2]2, [Pd(CO)2Br2]2, and [Pd(CO)2I2]2 at rM⋯M = 3.5, S increases from 0.08 to 0.09 to 0.10 and Δε decreases from 3.4 eV to 3.1 eV to 2.7 eV. Consequently, S2/Δε times 103 increases from 1.9 to 2.6 to 3.7 along the same series (Table 2).
Since the ΔEoi term is not made of one single component, our analyses show that there is an interplay between the metal–metal and ligand–ligand donor–acceptor interactions in the [M(CO)2X2]2 dimers along M = Ni, Pd, Pt and along X = Cl, Br, I. Therefore, to understand the relative importance of the metal–metal and ligand–ligand interactions, we computed the magnitude of their orbital stabilization according to eqn (3) and estimated their relative contribution to the total orbital stabilization (metal–metal + ligand–ligand). The results are collected in Table 2.
When the metal centers are large and the ligands are small and more electronegative, the donor–acceptor metal–metal interactions are strengthened and dominate over the weakened ligand–ligand interactions. For example, for [Pt(CO)2Cl2]2 at rM⋯M = 3.5 Å, the metal–metal interactions are up to 67% of the total orbital stabilization. On the other hand, the ligand–ligand donor–acceptor interactions only dominate when the metal center is small and the ligands are large and less electronegative, like in the [Ni(CO)2I2]2 dimer, in which the ligand–ligand donor–acceptor interactions are up to 75% of the total orbital stabilization at rM⋯M = 3.5 Å (Table 2).
4. Conclusions
The stability of square planar [X2(CO)2M]⋯[M(CO)2X2] dimers increases as the metal centers M vary along Ni, Pd, and Pt, and the X ligands vary along Cl, Br, and I. Our quantum chemical bonding analyses show that the dimers are formed due to stabilizing electrostatic interactions on top of stabilizing dispersion interactions promoted by the ligands. Both interactions become more stabilizing as M and X increase in size. In addition, our analyses revealed an overlooked covalent component stemming from metal–metal and ligand–ligand donor–acceptor interactions. These findings emerge from our quantitative Kohn–Sham molecular orbital analyses using dispersion-corrected relativistic density functional theory.The stability of the studied [M(CO)2X2]2 dimers increases as M varies along Ni, Pd, and Pt as the electron density of one monomer becomes more diffuse around M and more effectively interpenetrates towards the nucleus of M on the other monomer, resulting in a stronger electron–nuclei electrostatic attraction. Larger X ligands further increase the stability of [M(CO)2X2]2 due to a greater dispersion interaction as X increases in size along Cl, Br, and I. The dispersion component in [X2(CO)2M]⋯[M(CO)2X2], which is almost insensitive to variations in the metal center, only dominates when M is small, and X is large.
The overlooked covalent component in [X2(CO)2M]⋯[M(CO)2X2] comprises the metal–metal donor–acceptor interaction of the ndz2-type HOMOs of one monomer with (n + 1)s-type LUMOs of the other monomer as well as the ligand–ligand donor–acceptor interaction of the CO π*-type LUMO on one monomer with the X lone-pair-type HOMO on the other monomer. The metal–metal donor–acceptor interaction dominates when M is large, resulting in a larger stabilizing ndz2-HOMO–(n + 1)s-LUMO overlap. On the other hand, the ligand–ligand donor–acceptor interactions dominate when M is small and X becomes less electronegative. In this situation, the metal–metal donor–acceptor interaction is weakened, and the ligand–ligand donor–acceptor interaction is favored due to a smaller π*CO-LUMO–LPX-HOMO energy gap.
Data availability
The data that support the findings of this study are available in the ESI,† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Netherlands Organization for Scientific Research (NWO) for its support. This work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.References
- (a) H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746 CrossRef PubMed; (b) H. Schmidbaur and A. Schier, Organometallics, 2015, 34, 2048 CrossRef CAS; (c) N. Zhou, C. Zou, S. Suo, Y. Liu, J. Lin, X. Zhang, M. Shi, X. Chang and W. Lu, Dalton Trans., 2023, 52, 550 Search PubMed; (d) E. Priola, G. Mahmoudi, J. Andreo and A. Frontera, Chem. Commun., 2021, 57, 7268 RSC; (e) J. Lin, M. Xie, X. Zhang, Q. Gao, X. Chang, C. Zou and W. Lu, Chem. Commun., 2021, 57, 1627 RSC.
- (a) J. Nitsch, F. Lacemon, A. Lorbach, A. Eichhorn, F. Cisnetti and A. Steffen, Chem. Commun., 2016, 52, 2932 RSC; (b) M. El Sayed Moussa, S. Evariste, H. L. Wong, L. Le Bras, C. Roiland, L. Le Polles, B. Le Guennic, K. Costuas, V. W.-W. Yam and C. Lescop, Chem. Commun., 2016, 52, 11370 RSC; (c) Y. Zhang, J. Miao, J. Xiong, K. Li and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202113718 CrossRef CAS PubMed; (d) Q. Hu, C. Zhang, X. Wu, G. Liang, L. Wang, X. Niu, Z. Wang, W.-D. Si, Y. Han, R. Huang, J. Xiao and D. Sun, Angew. Chem., Int. Ed., 2023, 62, e202217784 CrossRef CAS PubMed; (e) R. Kobayashi, T. Yumura, H. Imoto and K. Naka, Chem. Commun., 2021, 57, 5382 RSC.
- (a) E. Barreiro, J. S. Casas, M. D. Couce, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, A. Sánchez, J. Sordo, J. M. Varela and E. M. V. A. López, Eur. J. Inorg. Chem., 2011, 1322 CrossRef CAS; (b) E. Barreiro, J. S. Casas, M. D. Couce, A. Sánchez, A. Sánchez-González, J. Sordo, J. M. Varela and E. M. Vázquez López, J. Inorg. Biochem., 2010, 104, 551 CrossRef CAS PubMed.
- (a) P. K. Mehrotra and R. Hoffmann, Inorg. Chem., 1978, 17, 2187 CrossRef CAS; (b) A. Dedieu and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 2074 CrossRef CAS; (c) Y. Jiang, S. Alvarez and R. Hoffmann, Inorg. Chem., 1985, 24, 749 CrossRef CAS.
- M. B. Brands, J. Nitsch and C. Fonseca Guerra, Inorg. Chem., 2018, 57, 2603 CrossRef CAS PubMed.
- L. de Azevedo Santos, T. C. Ramalho, T. A. Hamlin and F. M. Bickelhaupt, Chem. – Eur. J., 2023, 29, e202203791 CrossRef CAS PubMed.
- (a) P. Pyykkö and Y. F. Zhao, Angew. Chem., Int. Ed. Engl., 1991, 30, 604 CrossRef; (b) J. Li and P. Pyykkö, Chem. Phys. Lett., 1992, 197, 586 CrossRef CAS; (c) P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133 CrossRef; (d) P. Pyykkö, N. Runeberg and F. Mendizabal, Chem. – Eur. J., 1997, 3, 1451 CrossRef; (e) P. Pyykkö, Chem. Rev., 1997, 97, 597 CrossRef PubMed.
- J.-P. Zhang, Y.-B. Wang, X.-C. Huang, Y.-Y. Lin and X.-M. Chen, Chem. – Eur. J., 2005, 11, 552 CrossRef CAS PubMed.
- Q. Wan, J. Yang, W.-P. To and C.-M. Che, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2019265118 CrossRef CAS PubMed.
- (a) AMS2020.101, SCM Theoretical Chemistry, Vrije Universiteit Amsterdam, The Netherlands, https://www.scm.com Search PubMed; (b) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142 CrossRef CAS PubMed.
- (a) P. Vermeeren, T. A. Hamlin and F. M. Bickelhaupt, Chem. Commun., 2021, 57, 5880 RSC; (b) P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649 CrossRef CAS PubMed.
- (a) F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 1–86 Search PubMed; (b) T. A. Hamlin, P. Vermeeren, C. Fonseca Guerra and F. M. Bickelhaupt, in Complementary Bonding Analysis, ed. S. Grabowski, De Gruyter, Berlin, 2021, pp. 199–212 Search PubMed.
- (a) X. Sun, T. M. Soini, L. P. Wolters, W.-J. van Zeist, C. Fonseca Guerra, T. A. Hamlin and F. M. Bickelhaupt, PyFrag, Vrije Universiteit Amsterdam, The Netherlands, 2019 Search PubMed; (b) X. Sun, T. M. Soini, J. Poater, T. A. Hamlin and F. M. Bickelhaupt, J. Comput. Chem., 2019, 40, 2227 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00250d |
| ‡ L. A. S and T. W. contributed equally to this work. |
| This journal is © the Owner Societies 2024 |