Kinetics of the reaction of OH with methyl nitrate (223–343 K)†
Received
5th January 2024
, Accepted 1st February 2024
First published on 8th February 2024
Abstract
Rate coefficients (k4) for the reaction of hydroxyl radicals (OH) with methyl nitrate (CH3ONO2) were measured over the temperature range 232–343 K using pulsed laser photolysis to generate OH and pulsed laser-induced fluorescence to detect it in real-time and under pseudo-first-order conditions. In order to optimize the accuracy of the rate coefficients obtained, the concentration of CH3ONO2 (the reactant in excess) was measured on-line by absorption spectroscopy at 213.86 nm for which the absorption cross-section was also measured (σ213.86 = 1.65 ± 0.09 × 10−18 cm2 molecule−1). The temperature-dependent rate coefficient is described by k4(T) = 7.5 × 10−13 exp[(−1034 ± 40)/T] cm3 molecule−1 s−1 with a room temperature rate coefficient of k4(296 ± 2 K) = (2.32 ± 0.12) × 10−14 cm3 molecule−1 s−1 where the uncertainty includes the statistical error of 2σ and an estimation of the potential systematic bias of 5%. This new dataset helps to consolidate the database for this rate coefficient and to reduce uncertainty in the atmospheric lifetime of CH3ONO2. As part of this study, an approximate rate coefficient for the reaction of H-atoms with CH3ONO2 (k9) was also derived at room temperature: k9(298 K) = (1.68 ± 0.45) × 10−13 cm3 molecule−1 s−1.
1 Introduction
Organic nitrates are formed in the atmosphere during the degradation of hydrocarbons in the presence of nitrogen oxides.1 The atmospheric lifetimes of organic nitrates can vary from seconds (e.g. for thermally unstable peroxy-nitrates such as CH3OONO2 at temperatures close to room temperature) to months (e.g. for α-carbonyl peroxy-nitrates such as CH3C(O)OONO2 at temperatures close to 220 K typical for the upper troposphere). In contrast to peroxy-nitrates, alkyl nitrates (such as CH3ONO2) are generally thermally stable at the temperatures of our atmosphere and can be transported over long distances before being oxidized via reaction with OH or photolyzed.1–3 Following OH-induced oxidation of hydrocarbons, alkyl nitrates are formed in a usually minor (termolecular) channel of the reaction of organic peroxy radicals (RO2) with NO:| | | RO2 + NO + M → RONO2 + M | (R1b) |
Alternatively, RONO2 can be formed in the NO3-induced oxidation of unsaturated biogenic hydrocarbons in the presence of O2, which involves electrophilic addition of NO3 to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond followed by further reaction of the nitro-peroxy radicals thus formed.4 The target molecule of this study, CH3ONO2 is formed in the lower atmosphere when CH3O2 radicals (e.g. from methane oxidation) react with NO. The formation of CH3ONO2via this reaction is inefficient, with a yield of <1% at room temperature,5 but with some evidence for an increase in the yield at lower temperatures, commensurate with its formation in a termolecular process:6
C double bond followed by further reaction of the nitro-peroxy radicals thus formed.4 The target molecule of this study, CH3ONO2 is formed in the lower atmosphere when CH3O2 radicals (e.g. from methane oxidation) react with NO. The formation of CH3ONO2via this reaction is inefficient, with a yield of <1% at room temperature,5 but with some evidence for an increase in the yield at lower temperatures, commensurate with its formation in a termolecular process:6
| | | CH3O2 + NO → CH3O + NO2 | (R2a) |
| | | CH3O2 + NO + M → CH3ONO2 + M | (R2b) |
In the lower atmosphere, the fate of CH3O is dominated by reaction with O2 to form HO2 and HCHO. However, in the stratosphere, when O2 concentrations are much lower, some CH3O can react with NO27 and this represents a further source of CH3ONO2:
| | | CH3O + NO2 + M → CH3ONO2 + M | (R3) |
The important chemical loss processes for CH3ONO2 in the atmosphere are photodissociation and reaction with OH, with thermal decomposition and reaction with O3 or NO3 insignificant.1,8 Previous measurements indicate that R4 has a small rate coefficient, of the order of 10−14 cm3 molecule−1 s−1 (see below). The photolysis of CH3ONO2 proceeds via O–N bond fission to form a methoxy radical and NO2, whereas the slow reaction with OH (R4) proceeds via H-abstraction to form an unstable radical that dissociates to formaldehyde and NO2(R5):
| | | OH + CH3ONO2 → CH2ONO2 + H2O | (R4) |
Both HCHO and CH3O react much more rapidly with OH than does CH3ONO2 and secondary chemistry is a potential source of systematic bias in the determination of the rate coefficient (k4) for the title reaction when using absolute rate methods. A further source of systematic bias in an absolute rate coefficient study of this slow reaction (R4) is the presence of reactive impurities in the CH3ONO2 sample. Such problems can be avoided by working at low OH concentrations (and thus low fractional conversion of CH3ONO2 to products) and by carefully purifying the CH3ONO2 sample, thus minimising the levels of reactive impurities (see Section 2).
The rate coefficient for reaction (R4) has been reported by Gaffney et al.,9 Nielsen et al.,10 Talukdar et al.,8 and Shallcross et al.,11 but there are large differences in the rate coefficient at room temperature (a factor of 10) and its temperature dependence which is reported to be both positive and negative (see later). The goal of this study is to generate highly accurate rate coefficients for the title reaction at different (atmospherically relevant) temperatures and to resolve these differences in the literature and thus better constrain the lifetime of CH3ONO2 in the atmosphere.
2 Experimental
Rate coefficients for the reaction of OH with CH3ONO2 were determined using pulsed-laser-photolysis (PLP) pulsed-laser-induced fluorescence (PLIF) under pseudo-first-order conditions ([CH3ONO2] ≫ [OH]) in (mainly) N2 and He bath gases. The concentration of methyl nitrate was measured in situ by optical absorption. The experimental setup has been described in detail elsewhere12 and is only briefly described here.
2.1 PLP–PLIF technique
The reaction took place in a cylindrical, jacketed quartz reactor with a volume of approximately 300 cm3. The temperature inside the reactor was controlled by circulating a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ethylene glycol/water mixture (343–251 K) or ethanol (T < 251 K) through an outer-jacket. The gas mixture enters the reactor about 15 cm before the photolysis zone, ensuring thermal equilibrium with the walls of the reactor. The reaction temperature was measured by inserting a recently calibrated K-type thermocouple into the photolysis zone before each experiment at a different temperature. 100 and 1000 torr manometers were used to measure the pressure in the reactor.
:40 ethylene glycol/water mixture (343–251 K) or ethanol (T < 251 K) through an outer-jacket. The gas mixture enters the reactor about 15 cm before the photolysis zone, ensuring thermal equilibrium with the walls of the reactor. The reaction temperature was measured by inserting a recently calibrated K-type thermocouple into the photolysis zone before each experiment at a different temperature. 100 and 1000 torr manometers were used to measure the pressure in the reactor.
OH was generated by excimer laser photolysis (Coherent COMPex 205 F or Pro 201 F, ∼20 ns width) of a precursor gas molecule, either H2O2 (248 nm), HONO (351 nm) or CH3ONO2 itself (248 nm) (see below).
OH radicals were excited at 281.997 nm (A2Σ(ν′ = 1) ← X2Π(ν′′ = 0), Q11 (1) transition) using a Nd-YAG pumped (532 nm, ∼6 ns pulse width) and frequency-doubled dye laser (Quantel Brilliant B, Lambda Physik Scanmate/Scanmate UV with KPD crystal). The resulting fluorescence signal was detected by a photomultiplier tube screened by a long-pass filter (λ > 290 nm). The photolysis and excitation lasers were mainly operated at a repetition rate of 10 Hz using a pulse/delay generator (Stanford Research Systems DG 535), which was also used to control the delay between the photolysis and excitation laser pulses. Prior to the PLP pulse, 20 pulses of the PLIF laser were used to measure a background signal that includes scattered light from the excitation laser and electronic noise. The averaged background signal was subtracted before further processing of the measured data. Each datapoint was averaged between 10 and 40 times, depending on the measurement.
The total flow and pressure in the reactor were chosen such that an average, linear gas-velocity of ≈13–19 cm s−1 was achieved, ensuring that the gas in the photolysis region was replenished between laser pulses. For measurements in helium, the linear velocity was 5 or 6.6 cm s−1 and the laser repetition rate was reduced to 5 Hz, to ensure complete gas exchange.
2.2
In situ absorption spectroscopy: The absorption cross-section of CH3ONO2 at 213.86 nm
As the experiments were performed under pseudo first order conditions, accurate rate coefficients depend on accurate measurement of the concentration of the excess reactant. The concentration of methyl nitrate was therefore determined on-line, downstream of the reactor by optical absorption using a dual-beam absorption cell (path length 34.8 cm) equipped with a low-pressure Zn-lamp and interference filter (214 ± 5 nm) to isolate the 213.86 nm line. The temperature (close to room temperature) and pressure (a few percent lower than in the reactor) in the absorption cell were also measured and used to make corrections for gas-density differences in the reaction and absorption cell. Optical density (OD) due to CH3ONO2 was converted to a concentration using the Beer–Lambert expression (1) and an absorption cross-section that was determined as described below.| | | OD = ln (I0/I) = σ213.86l [CH3ONO2] | (1) |
σ
213.86 is the CH3ONO2 absorption cross-section (cm2 molecule−1), l is the absorption path-length and I0 and I are the transmitted light intensity without and with absorber, respectively and [CH3ONO2] is the concentration of methyl-nitrate (molecule cm−3).
To measure the absorption cross-section, the absorption cell was first filled with N2 to determine I0. Subsequently, static samples of CH3ONO2 in N2 at different pressures were introduced into the cell from a storage bulb and the optical absorption was recorded. Two different storage bulbs (0.482% and 0.728%) made up using samples of CH3ONO2 from two different syntheses (≈ one month apart in time) were used to check for reproducibility. The storage bulbs were prepared using standard manometric methods with 10 and 1000 torr capacitance manometers with quoted accuracies of better than 1‰.
Fig. 1 presents the results from 17 determinations of optical density at room-temperature. The slope of the weighted, linear regression gives an absorption cross-section of σ213.86 = (1.65 ± 0.09) × 10−18 cm2 molecule−1, where the uncertainty is 2σ and includes as assessment of systematic bias (2%) which stems predominantly from uncertainty in the CH3ONO2 mixing ratio and thus [CH3ONO2]. In addition, minor contributions stem from uncertainty in the optical path-length (<1%) and in determination of the optical density (0.05–1.5%). As an interference filter with a finite FWHM was used to isolate the 213.86 nm Zn-line, our cross-section should strictly be regarded as an “effective” cross-section since weaker atomic lines may also be detected. Our cross-section is significantly larger than the value of 3.04 × 10−19 cm2 molecule−1 at 213.99 nm reported by Taylor et al.13 but is similar to a value of 2.16 × 10−18 cm2 molecule−1 at 214.01 nm measured by McMillan et al. and reported by Calvert and Pitts.14 More importantly, our value is in excellent agreement with the more recent determination of Talukdar et al.,15 who obtained σ213.86 = (1.67 ± 0.08) × 10−18 cm2 molecule−1 using a Zn-lamp. We conclude that our cross-section is approriate for converting in situ optical density measurements to accurate concentrations of CH3ONO2.
 |
| | Fig. 1 Optical density (OD) plotted against concentration of methyl nitrate (molecule cm−3) multiplied by path length l (cm). The slope of the corresponding, weighted linear regression yields an effective absorption cross-section of  . The error is 2σ and includes an estimate of systematic uncertainty. . The error is 2σ and includes an estimate of systematic uncertainty. | |
2.3 Determination of impurities in the CH3ONO2 sample
As the reaction between CH3ONO2 and OH is slow, the presence of reactive impurities can bias the rate coefficients obtained to higher values. Likely impurities are CH3OH and HNO3 which are involved in the synthesis of CH3ONO2 as well as NO2 and HONO. Initial measurements revealed the presence of low levels of HNO3 in the CH3ONO2 samples. In order to eliminate this (and other acidic gases) the sample flow was passed through ≈20 cm of PFA tubing containing nylon before entering the reactor. In some experiments (in which methyl nitrate itself was used as OH source) CaCO3 was used to remove HNO3.
Following each new synthesis of CH3ONO2, levels of HNO3, HONO, CH3OH and NO2 were determined using a combination of IR and UV/Vis absorption spectroscopy. An FTIR (Bruker Vector 22) spectrometer coupled to an absorption cell of 45 cm pathlength16 and equipped with an external MCT detector was used to determine impurity levels of HNO3 and CH3OH in a flowing mixture of CH3ONO2 in N2, as used in the kinetic experiments. Generally, 32 spectra at a resolution of 1 cm−1 (acquisition time ≈ 1 min) were co-added to measure absorption in the 700–3750 cm−1 range. HONO and NO2 impurities were investigated using multi-pass (l = 880 cm) UV/Vis absorption spectroscopy (346–518 nm) using a H4-lamp light source coupled to a CCD detector (Andor DU420A-OE) and a 0.5 m monochromator (BM50) with 300 lines mm−1 grating. The spectral resolution (FWHM of a Hg line from a low-pressure discharge lamp) was 0.5 nm.
The results of the impurity determinations (none were detected and upper limits were established) are summarized in Table 1, which shows that the rate coefficients obtained were not significantly influenced by impurities in the CH3ONO2 sample.
Table 1 Impurities and their contribution to the OH decay coefficient
| Impurity |
Fractiona |
k
impurity
|
Contribution to k′ (%) |
|
Relative concentration of impurity (in %) of the CH3ONO2 concentration.
Rate coefficient (cm3 molecule−1 s−1) at room temperature.
Calculated for a pressure of 135 torr.
|
| HNO3 |
<0.01 |
1.5 × 10−13 |
<0.06 |
| CH3OH |
<0.01 |
9.0 × 10−13 |
<0.38 |
| HONO |
<0.03 |
6.0 × 10−12 |
<6.80 |
| NO2 |
<0.03 |
4.2 × 10−12c |
<4.78 |
2.4 OH sources
During the course of this study, three different sources of OH were tested. The photolysis of HONO at 351 nm (R6) and the photolysis of H2O2 at 248 nm (R7) both generate OH in the ground vibrational state.17 The photolysis of CH3ONO2 at 248 nm is an indirect source of OH15via formation of H atoms (R8b) and (R9).| | | HONO + hν (351 nm) → OH + NO | (R6) |
| | | H2O2 + hν (248 nm) → 2OH | (R7) |
| | | CH3ONO2 + hν (248 nm) → CH3O + NO2 | (R8a) |
| | | CH3ONO2 + hν (248 nm) → H + CH2ONO2 | (R8b) |
| | | H + CH3ONO2 → OH + CH3ONO | (R9) |
In Section 3.2 and 3.3, we describe how the OH-decays obtained using the 248 nm photolysis laser (i.e., (R7) or (R8b) and (R9)) are strongly influenced by secondary reactions involving CH3O and NO2 resulting from the photolysis of CH3ONO2. Only data obtained using the photolysis of HONO at 351 nm (a wavelength at which CH3ONO2 no longer absorbs significantly) were used to derive the rate coefficient for the title reaction.
2.5 Chemicals
The synthesis of CH3ONO2, the nitration of methanol with HNO3, was carried out according to Black and Babers18 but with a modification of the purification step. ∼62 mL of Nitric acid (HNO3; Sigma-Aldrich, ≥65%) was pre-cooled (ice bath) in a 250 ml Erlenmeyer flask. ∼31 mL of Methanol (CH3OH; Sigma-Aldrich, ≥99.9%) was pre-cooled in a 100 mL Erlenmeyer flask. ∼62 mL of sulphuric acid (Roth, 98%) was added to the nitric acid. The remaining sulphuric acid was added to the methanol using a dropping funnel with vigorous stirring, whereby a temperature of 14 °C was not exceeded. The CH3OH/H2SO4 mixture was then added. The temperature of the mixture increased to about 45 °C and was subsequently kept between 32 °C and 35 °C for about 15 minutes. The mixture was then rapidly cooled in an ice-water bath for 5 minutes and then transferred to a separating funnel. The aqueous phase and part of the organic phase were discarded.
The unwashed methyl nitrate was transferred to a bubbler and N2 was bubbled through the liquid for about 15 min to remove any dissolved methanol. The liquid was then vacuum-distilled through a drying tube with CaCl2 (Roth, ≥94%, dehydrated) before the transmitted gas was trapped in a cold-finger at liquid nitrogen temperature. The methyl nitrate was then subjected to repeated freeze–pump–thaw cycles at −196 °C. Nitrogen (N2, Air-Liquide, 99.999%) and helium (He, Westfalen, 99.999%) were used as buffer gases in the experiments. HONO was prepared in situ by adding 5–10 drops of 0.1 M NaNO2 solution (Roth, ≥98%) into a 20 wt% H2SO4 solution (Roth, 98%). Hydrogen peroxide (H2O2; AppliChem, 35%) was vacuum distilled to >90% purity.
3 Results and discussion
In all experiments, the methyl nitrate concentration was 5–6 orders of magnitude larger than that of OH (typically 1 × 1010 molecule cm−3) and pseudo-first order conditions prevailed. The decay of OH can therefore be described by| | | [OH]t = [OH]0exp(−k′t) | (2) |
where [OH]t is the OH concentration at time t after the photolysis laser pulse, [OH]0 is the initial OH concentration and k′ is the first-order decay coefficient. The latter is defined as:where k4 is the bimolecular rate coefficient (cm3 molecule−1 s−1) of the reaction of methyl nitrate with OH and kd (s−1) accounts for diffusion of OH from the detection zone and its reaction with H2O2 or HONO.
3.1 Generation of OH from HONO photolysis at 351 nm
Fig. 2 shows an example of the OH fluorescence signal plotted against time after the excimer laser pulse. The typical HONO concentration in this experiment is approx. 1.5 × 1013 molecule cm−3, which results in an initial OH concentration of ∼1 × 1010 molecule cm−3. As expected (eqn (2)), the decay of OH is exponential (over 2 orders of magnitude). Decay coefficients (k′, s−1), obtained by least-squares-fitting of eqn (2) to the OH-decays, are plotted against the respective concentration of CH3ONO2 in Fig. 3. A linear dependence of k’ on the CH3ONO2 concentration (eqn (3)) was observed under all experimental conditions, enabling derivation of k4(T). Variability in the y-axis intercept of ∼200–250 s−1 reflects slight changes in the HONO concentration between experiments at different temperatures. Prior experiments indicate that the contribution of diffusion to k′ is circa 50–100 s−1, depending mainly on the pressure (or bath gas) and the flow rate. As the rate coefficient for the reaction between OH and HONO is 6.0 × 10−12 cm3 molecule−1 s−1 (at room temperature), the approximate HONO concentration can be calculated to be < 4 × 1013 molecule cm−3. This is consistent with the concentration obtained using the weak absorption (OD < 1 × 10−3) observed at 213.86 nm (σ = 1.8 × 10−18 cm−3 molecule5) when adding HONO in the absence of CH3ONO2.
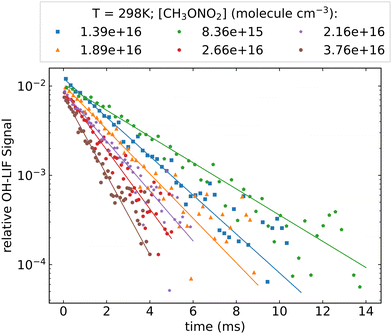 |
| | Fig. 2 OH-LIF signal (datapoints) with mono-exponential fits (solid lines) plotted against time after the photolysis laser pulse. Data were obtained using HONO as OH source at room temperature, the pressure was 134.0 torr (N2 bath-gas). | |
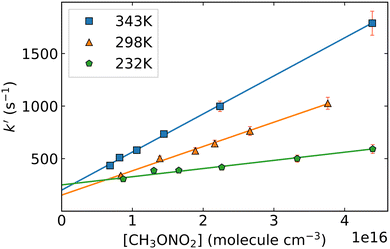 |
| | Fig. 3 First-order OH decay-coefficient (k′) plotted against the corresponding methyl nitrate concentration [CH3ONO2] (data obtained using HONO as the OH precursor). The slope of the linear regression (orange: k(298 K) = (2.31 ± 0.15) × 10−14 cm3 molecule−1 s−1; green: k(232 K) = (7.78 ± 0.92) × 10−15 cm3 molecule−1 s−1; k(343 K) = (3.62 ± 0.22) × 10−14 cm3 molecule−1 s−1) is the rate coefficient k4 under these experimental conditions. Error bars represent 2σ statistical uncertainty. | |
Within experimental uncertainty, variation of the pressure or bath-gas (N2 or He) (for a given temperature) had no effect on the rate coefficient, which is thus independent of pressure or collision partner within the range of pressures (69–400 torr N2, 100 torr He) investigated in this study. Variation of the excimer-laser fluence over a factor 30 also had no significant influence (±3%) on the OH decay coefficient, showing that reactions of OH with primary and secondary products of the title reaction do not contribute significantly to its loss. A complete list of the values of k4 obtained under various experimental conditions is given in Table 2 and plotted in Arrhenius form in Fig. 4. The regression through the data points is described by k4(T) = 7.5 × 10−13 exp[(−1034 ± 40)/T]. The average, room-temperature (296 ± 2) K rate coefficient is 2.32 × 10−14 cm3 molecule−1 s−1. The total uncertainty associated with the rate coefficients obtained (and derived from the Arrhenius expression), is dominated by uncertainty in the concentration of CH3ONO2 and is estimated to be ∼ 5% (see ESI,† Section S4).
Table 2 Experimental conditions and rate coefficients (k4) for the reaction OH + CH3ONO2
|
T (K) |
p (torr) |
k
4 (10−14 cm3 molecule−1 s−1) |
Δk4a (10−14 cm3 molecule−1 s−1) |
Bath gas |
| Notes: All rate coefficients were obtained using the 351 nm photolysis of HONO as OH-source. Uncertainty is 2σ, statistical only. |
| 223.3 |
101.2 |
0.84 |
0.07 |
He |
| 231.7 |
134.6 |
0.78 |
0.09 |
N2 |
| 239.6 |
134.3 |
0.87 |
0.09 |
N2 |
| 247.3 |
135.7 |
1.05 |
0.10 |
N2 |
| 251.5 |
134.8 |
1.28 |
0.05 |
N2 |
| 259.7 |
127.0 |
1.42 |
0.07 |
N2 |
| 266.0 |
136.3 |
1.49 |
0.10 |
N2 |
| 268.7 |
135.2 |
1.56 |
0.10 |
N2 |
| 276.2 |
134.8 |
1.69 |
0.09 |
N2 |
| 282.2 |
135.1 |
1.86 |
0.12 |
N2 |
| 289.0 |
135.1 |
2.07 |
0.15 |
N2 |
| 294.2 |
101.6 |
2.28 |
0.11 |
He |
| 294.5 |
135.5 |
2.21 |
0.20 |
N2 |
| 295.2 |
98.8 |
2.14 |
0.13 |
N2 |
| 295.8 |
133.5 |
2.39 |
0.08 |
N2 |
| 296.2 |
99.1 |
2.41 |
0.15 |
N2 |
| 296.7 |
201.0 |
2.48 |
0.15 |
N2 |
| 297.5 |
135.1 |
2.34 |
0.17 |
N2 |
| 297.7 |
400.4 |
2.12 |
0.30 |
N2 |
| 298.0 |
134.0 |
2.31 |
0.15 |
N2 |
| 298.2 |
68.5 |
2.23 |
0.18 |
N2 |
| 317.8 |
135.2 |
2.78 |
0.19 |
N2 |
| 342.7 |
135.4 |
3.62 |
0.22 |
N2 |
 |
| | Fig. 4 Arrhenius plot of k4versus inverse temperature. In blue: k4 from generation of OH from HONO photolysis at 351 nm. In red, the exponential fit to the data with 2σ confidence limit. In green: k4 from generation of OH from CH3ONO2 photolysis at 248 nm. In purple: k4 from generation of OH from CH3ONO2 photolysis at 248 nm considering only data at t > 1.7 ms. | |
3.2 Generation of OH from CH3ONO2 or H2O2 photolysis at 248 nm (298 K)
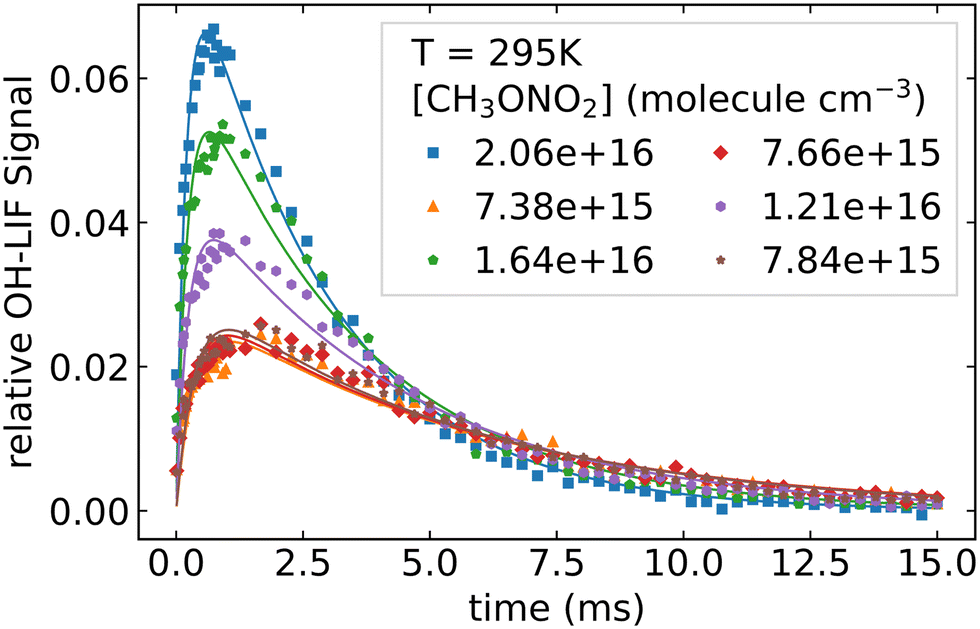
As described above, the photolysis of methyl nitrate at 248 nm indirectly produces OH. Based on the CH3ONO2 concentration, the laser fluence and the photolysis quantum yields at 248 nm,15 between 8 × 108 and 2 × 109 molecule cm−3 of OH are generated via R8 and R9 when [CH3ONO2] is varied from 7.4 × 1015 to 2.1 × 1016 molecule cm−3. At the same time 1.4 × 1011 to 3.9 × 1011 molecule cm−3 of NO2 and CH3O are formed.
Fig. 5 displays OH time profiles from the photolysis of six different concentrations of CH3ONO2. The OH decays are bi-exponential, with well separated production and loss terms. The rapid production of OH (via R9) is followed by a slower decay as OH is lost (mainly) via R4. The solid lines are fits to the data according to:
| |  | (4) |
where

is the pseudo-first order term dominated by H-atom loss
via its reactions with CH
3ONO
2(R9). In principle, values of
k4 and
k9 can be extracted from the data by plotting

and
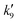 versus
versus the concentration of CH
3ONO
2 as displayed in
Fig. 6. From the regression slopes we derive
k4(298 K) = (1.37 ± 0.19) × 10
−14 and
k9(298 K) = (1.68 ± 0.45) × 10
−13 cm
3 molecule
−1 s
−1. The
y-axis intercept of 75 ± 22 s
−1 for the plot of
 versus
versus [CH
3ONO
2] is consistent with diffusive losses of OH. The much larger intercept for the plot of
 versus
versus [CH
3ONO
2] stems from H-atom diffusion and possibly reaction of H atoms with O
2 present as impurity.
 |
| | Fig. 5 OH formation and decay when different concentrations of CH3ONO2 were photolyzed at 248 nm. | |
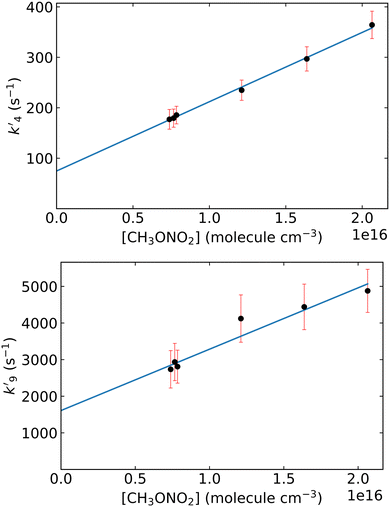 |
| | Fig. 6 First-order rate coefficients k′ for OH generation and loss derived from the bi-exponential fits (eqn (4)) to OH profiles obtained by photolysis of CH3ONO2 at 248 nm and plotted against the corresponding methyl nitrate concentration [CH3ONO2] in molecule cm−3. Error bars represent 2σ statistical uncertainty. The solid lines are linear regressions where the slopes correspond to the rate coefficients k4 (upper panel) and k9 (lower panel). | |
The value of k4 thus obtained is a factor ≈ 1.6 lower than the average room-temperature value (2.32 × 10−14 cm3 molecule−1 s−1) obtained in the experiments in which OH was generated by the 351 nm photolysis of HONO. A different data treatment, in which only the latter part of the bi-exponential profile (t > 1700 μs) was analysed (i.e. after all H atoms have been converted to OH via reaction with CH3ONO2) results in a rate coefficient of (1.65 ± 0.10) × 10−14 cm3 molecule−1 s−1, which is also lower (factor 1.4) than that obtained using HONO photolysis. As the photolysis of CH3ONO2 at 248 nm results mainly in the formation of CH3O and NO2 and OH reacts rapidly with both (k(OH + NO2) = 3.6 × 10−12 cm3 molecule−1 s−15 and k(OH + CH3O) = 3 × 10−11 cm3 molecule−1 s−119 at 298 K and 100 torr), the unavoidable presence of CH3O and NO2 would be expected to result in an overestimation of the rate coefficient via this method. In the ESI† we use numerical simulations to examine the impact of secondary reactions and the effect of simultaneous detection of both OH and CH3O (resulting in “OH” profiles that are not perfectly bi-exponential). Clearly, the photolysis of CH3ONO2 with complex secondary chemistry does not lend itself to accurate measurement of the OH-rate coefficient. As shown in Fig. 6, the linear relationship between k’9 and [CH3ONO2] enables us to derive an approximate rate coefficient (k9) for the OH-forming reaction of H + CH3ONO2. Our result is k9(298 K) = (1.68 ± 0.45) × 10−13 cm3 molecule−1 s−1, where the uncertainty is 2σ, statistical only. This result is in good agreement with that reported by Talukdar et al.15 using an almost identical method and similar analysis.
The photolysis of H2O2 is commonly used as a source of prompt OH radicals in PLP–PLIF experiments. The generation of OH via 248 nm photolysis of H2O2 in the presence of CH3ONO2 does however not result in exponential OH decays. The more complex behaviour compared to that observed when using HONO at 351 nm as OH precursor is related to the photolysis of CH3ONO2 at 248 nm (see above). Values of k′ obtained using this method were strongly dependent on the 248 nm pulse energy and no linear relationship with the concentration of methyl nitrate was found. Clearly, as described above for experiments in the absence of H2O2, the photolysis of methyl nitrate at 248 nm precludes a straightforward analysis of the OH-profiles thus obtained.
3.3 Comparison of k4 with literature values and the CH3ONO2 atmospheric lifetime
The low rate coefficient for the reaction of OH with CH3ONO2 is consistent with the reaction proceeding via H-atom abstraction from the methyl group, with each C–H bond contributing about 1 × 10−14 cm3 molecule−1 s−1 to the rate coefficient at room temperature. Kinetic isotope effects8 confirm that H-atom abstraction is the only significant pathway for the title reaction in the temperature range representative of our atmosphere.
In order to compare our rate coefficients and their temperature dependence with literature values, we consider only our data measured using the photolysis of HONO at 351 nm, which are not biased by complex secondary processes involving photo-fragments of CH3ONO2. Table 3 and Fig. 7 compare the room temperature rate coefficients and Arrhenius expression obtained in this work with those reported in the literature.
Table 3 Comparison with previous determinations of k4
| Source |
k
4 (cm3 molcule−1 s−1) |
T (K) |
Method |
| PLP–PLIF = Pulsed laser photolysis/pulsed laser induced fluorescence; DF-RF = discharge flow/resonance fluorescence; PR-RA = pulse radiolysis/resonant absorption; RR = relative rate. |
| This work |
(2.32 ± 0.04) × 10−14 |
296 ± 2 |
PLP–PLIF |
| 7.5 × 10−13 × exp[(−1034 ± 40)/T] |
223–343 |
|
| Gaffney et al.9 |
(3.4 ± 0.4) × 10−14 |
298 |
DF-RF |
| Nielsen et al.22 |
(3.2 ± 0.5) × 10−13 |
298 ± 2 |
PR-RA |
| 8.8 × 10−15 × exp[(1050 ± 180)/T] |
298–393 |
|
| (3.1 ± 0.7) × 10−13 |
298 ± 2 |
RR |
| Talukdar et al.8 |
(2.36 ± 0.16) × 10−14 |
298 |
PLP–PLIF |
| 8.2 × 10−13 × exp[(−1020 ± 60)/T] |
221–414 |
|
| Shallcross et al.23 |
(4.7 ± 1.0) × 10−14 |
298 |
DF-RF |
| 4.1 × 10−13 × exp[(−604 ± 121)/T] |
298–423 |
|
| Kerr and Stocker20 |
(3.8 ± 1.0) × 10−13 |
303 ± 2 |
RR |
| Kakesu et al.3 |
(3.0 ± 0.7) × 10−14 |
307 ± 3 |
RR |
 |
| | Fig. 7 Arrhenius plot of k4versus inverse temperature. The rate coefficients and experimental conditions of the present work can be found in Table 3. | |
Absolute rate coefficient measurements.
When considering only those studies that used absolute methods, our room-temperature rate coefficient is essentially identical to that of Talukdar et al.,8 but factors of 13, 1.4 and 2 lower than those reported by Nielsen et al.,10 Gaffney et al.9 and Shallcross et al.,11 respectively. For slow reactions such as OH + CH3ONO2, reactive impurities and secondary reactions of OH (e.g. with products or photo-fragments) can bias the rate coefficients derived from absolute methods to higher values. In the study of Nielsen et al. (the only one to report a negative dependence of k4 on temperature), the pulse-radiolysis technique generated not only very high concentrations of OH (∼1013 molecule cm−3) but also would have caused substantial fragmentation of CH3ONO2 (e.g. to CH3O and NO2). This probably explains the very large rate coefficient and anomalous temperature dependence observed.
The other temperature-dependent studies report a positive dependence on temperature, with values of E/R (in K) varying from −1054 (this work) to −1020 (Talukdar et al.8) and −604 (Shallcross et al.11). While the two studies using the “wall-free” method of PLP–PLIF agree extremely well, the flow tube study of Shallcross et al. has a substantially weaker dependence on temperature. Close examination of their data set (5 data points all at T > 298 K) reveals considerable scatter in k4, which is also seen in their plots of k′ versus [CH3ONO2]. We speculate that the difference in slope reflects changes in the wall loss rates of OH, which were determined at room temperature only. As mentioned above, absolute rate studies of slow reactions require that very pure samples are used. In this study and that of Talukdar et al.,8 spectroscopic checks were made to exclude the presence of specific reactive impurities. In the studies of Gaffney et al.9 and Shallcross et al.,11 the sample purity was determined to be > 99%, but no attempt was made to detect impurities such as HNO3, NO2 or CH3OH. Perhaps more pertinent however, is the fact that the two studies that utilized in situ (optical absorption) measurement of CH3ONO2 are in excellent agreement. In situ measurement of the concentration of the excess reactant removes uncertainty associated with calculating [CH3ONO2] from partial flows, pressures and variations in the mixing-ratios in storage bulbs. The fact that both this work and Talukdar et al.8 measured absorption cross-sections for CH3ONO2 at 213.86 nm that are in almost perfect agreement strongly suggests that the excellent agreement in the values of k4 reported by these two groups is not fortuitous, but a result of multi-diagnostics and careful execution of the experiments, leading to derivation of the true rate coefficient.
Relative rate coefficient measurements.
In principle, the relative rate measurements (all close to room temperature) should not be biased by reactive impurities in the CH3ONO2 sample, yet the results of the three relative-rate studies are highly divergent. The very large rate coefficients measured by Nielsen et al.10 and Kerr and Stocker20 can be traced back to an inappropriate choice of reference reactants, which ideally should react with rate coefficients that are similar to the target molecule. The use of 2-methylpropane (kOH+2-methylpropane = 2.1 × 10−12 cm3 molecule−1 s−15) and ethene (kOH+ethene = 7.8 × 10−12 cm3 molecule−1 s−1 at 1 bar5) by Nielsen et al.10 and Kerr and Stocker,20 respectively, would have made relative depletion rates of the reference and target molecule difficult to measure and also prone to other loss processes of CH3ONO2. In the relative rate study of Kakesu et al.,3 the more slowly reacting reference trace-gases, methane (kOH+methane = 6.4 × 10−15 cm3 molecule−1 s−15) and ethane (kOH+ethane = 2.4 × 10−13 cm3 molecule−1 s−15) were used. Those authors derived a rate coefficient at 307 K of (3.0 ± 0.7) × 10−14 cm3 molecule−1 s−1. Within combined uncertainty, this value agrees with the absolute rate coefficient derived in the present study for the same temperature: of k4(307 K) = (2.7 ± 0.4) × 10−14 cm3 molecule−1 s−1.
Atmospheric lifetime of CH3ONO2 with respect to reaction with OH.
As the rate coefficient for the title reaction is independent of pressure under all conditions of the Earth's lower atmosphere (i.e. the troposphere and lower stratosphere) we use the Arrhenius expression derived from our experiments using HONO as OH source: k4(T) = 7.5 × 10−13 exp(−1034/T) to calculate the lifetime of CH3ONO2 with respect to OH loss. For this we assume an altitude independent, global mean OH concentration of 1 × 106 molecule cm−3.21 The lifetime of CH3ONO2 w.r.t. OH reaction at different altitudes is plotted in Fig. S5 (ESI†) and varies from circa 500 days at ground level to > 1000 days at 10 km. Photolysis rate coefficients for CH3ONO2 vary from ∼3 × 10−7 s−1 to 5 × 10−7 s−1 in the boundary layer and lower stratosphere,15 respectively, resulting in lifetimes with respect to photodissociation of close to 1 month. Our data thus confirms the conclusion of Talukdar et al.15 showing that reaction with OH is a minor sink of CH3ONO2 in the atmosphere. This is in contrast to the conclusions of Nielsen et al.10 and Kerr and Stocker20 who (based on too-high rate coefficients) suggested that reaction with OH is important.
4 Conclusions
Motivated by greatly divergent reports of the rate coefficient for the reaction of OH with CH3ONO2, we have determined the rate coefficient using pulsed laser photolysis-laser induced fluorescence. High-quality kinetic data were obtained when measures were taken to avoid the photolysis of CH3ONO2. Our result, a positive dependence of the rate coefficient on temperature described by k4(T) = 7.5 × 10−13 exp[(−1034 ± 40)/T], is in close agreement with that reported by Talukdar et al..8 Both studies used in situ measurement of [CH3ONO2] using independently determined absorption cross-sections that agreed with 1.2%. The divergent results obtained by other groups are rationalised in terms of choice of OH source or reference reactant and data corrections.
Author contributions
This work was carried out as part-fulfilment of CF's master's degree in chemistry at the Julius-Maximilians-University, Würzburg, Germany. CF and FB conducted the experiments, CF evaluated the data and prepared the manuscript. JC conceptualized the experiment, JC and JL contributed to writing the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Open Access funding provided by the Max Planck Society.
Notes and references
- J. M. Roberts, Atmos. Environ., Part A, 1990, 24, 243 CrossRef.
- E. Atlas, Nature, 1988, 331, 426 CrossRef CAS.
- M. Kakesu, H. Bandow, N. Takenaka, Y. Maeda and N. Washida, Int. J. Chem. Kinet., 1997, 29, 933 CrossRef CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Atmos. Chem. Phys., 2017, 17, 2103 CrossRef CAS PubMed.
- IUPAC, Task Group on Atmospheric Chemical Kinetic Data Evaluation, (Ammann, M., Cox, R.A., Crowley, J.N., Herrmann, H., Jenkin, M.E., McNeill, V.F., Mellouki, A., Rossi, M. J., Troe, J. and Wallington, T. J.). Last access Sept. 2023, (https://iupac.aeris-data.fr/).
- N. Butkovskaya, A. Kukui and G. Le Bras, J. Phys. Chem. A, 2012, 116, 5972 CrossRef CAS PubMed.
- F. Flocke, E. Atlas, S. Madronich, S. M. Schauffler, K. Aikin, J. J. Margitan and T. P. Bui, Geophys. Res. Lett., 1998, 25, 1891 CrossRef CAS.
- R. K. Talukdar, S. C. Herndon, J. B. Burkholder, J. M. Roberts and A. R. Ravishankara, J. Chem. Soc., Faraday Trans., 1997, 93, 2787 RSC.
- J. S. Gaffney, R. Fajer, G. I. Senum and J. H. Lee, Int. J. Chem. Kinet., 1986, 18, 399 CrossRef CAS.
- O. J. Nielsen, H. W. Sidebottom, M. Donlon and J. Treacy, Chem. Phys. Lett., 1991, 178, 163 CrossRef CAS.
- D. E. Shallcross, P. Biggs, C. E. Canosa-Mas, K. C. Clemitshaw, M. G. Harrison, M. Reyes López Alañón, J. A. Pyle, A. Vipond and R. P. Wayne, J. Chem. Soc., Faraday Trans., 1997, 93, 2807 RSC.
- M. Wollenhaupt, S. A. Carl, A. Horowitz and J. N. Crowley, J. Phys. Chem., 2000, 104, 2695 CrossRef CAS.
- W. D. Taylor, T. D. Allston, M. J. Moscato, G. B. Fazekas, R. Kozlowski and G. A. Takacs, Int. J. Chem. Kinet., 1980, 12, 231 CrossRef CAS.
-
J. G. Calvert and J. N. J. Pitts, Photochemistry, J. Wiley, New York, 1966 Search PubMed.
- K. R. Talukdar, J. B. Burkholder, M. Hunter, M. K. Gilles, J. M. Roberts and A. R. Ravishankara, J. Chem. Soc., Faraday Trans., 1997, 93, 2797 RSC.
- C. B. M. Groß, T. J. Dillon, G. Schuster, J. Lelieveld and J. N. Crowley, J. Phys. Chem. A, 2014, 118, 974 CrossRef PubMed.
- G. L. Vaghjiani and A. R. Ravishankara, J. Chem. Phys., 1990, 92, 996 CrossRef CAS.
- A. P. Black and F. H. Babers, Org. Synth., 1939, 19, 64 CrossRef CAS.
- W. Tsang and R. F. Hampson, J. Phys. Chem. Ref. Data, 1986, 15, 1087 CrossRef CAS.
- J. A. Kerr and D. W. Stocker, J. Atmos. Chem., 1986, 4, 253 CrossRef CAS.
- J. Lelieveld, S. Gromov, A. Pozzer and D. Taraborrelli, Atmos. Chem. Phys., 2016, 16, 12477 CrossRef CAS.
- O. J. Nielsen, H. W. Sidebottom, M. Donlon and J. Treacy, Chem. Phys. Lett., 1991, 178, 163 CrossRef CAS.
- D. Shallcross, C. Canosa-Mas, K. Clemitshaw, M. Harrison, J. Pyle and R. Wayne, J. Chem. Soc., Faraday Trans., 1997, 93, 2807 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp00054d |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Fabienne
Baumann
,
Fabienne
Baumann

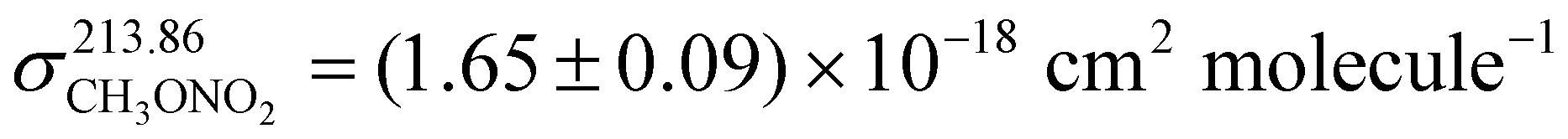 . The error is 2σ and includes an estimate of systematic uncertainty.
. The error is 2σ and includes an estimate of systematic uncertainty.



 is the pseudo-first order term dominated by H-atom loss via its reactions with CH3ONO2(R9). In principle, values of k4 and k9 can be extracted from the data by plotting
is the pseudo-first order term dominated by H-atom loss via its reactions with CH3ONO2(R9). In principle, values of k4 and k9 can be extracted from the data by plotting  and
and 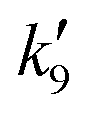 versus the concentration of CH3ONO2 as displayed in Fig. 6. From the regression slopes we derive k4(298 K) = (1.37 ± 0.19) × 10−14 and k9(298 K) = (1.68 ± 0.45) × 10−13 cm3 molecule−1 s−1. The y-axis intercept of 75 ± 22 s−1 for the plot of
versus the concentration of CH3ONO2 as displayed in Fig. 6. From the regression slopes we derive k4(298 K) = (1.37 ± 0.19) × 10−14 and k9(298 K) = (1.68 ± 0.45) × 10−13 cm3 molecule−1 s−1. The y-axis intercept of 75 ± 22 s−1 for the plot of  versus [CH3ONO2] is consistent with diffusive losses of OH. The much larger intercept for the plot of
versus [CH3ONO2] is consistent with diffusive losses of OH. The much larger intercept for the plot of  versus [CH3ONO2] stems from H-atom diffusion and possibly reaction of H atoms with O2 present as impurity.
versus [CH3ONO2] stems from H-atom diffusion and possibly reaction of H atoms with O2 present as impurity.