DOI:
10.1039/D3DT03509C
(Paper)
Dalton Trans., 2024,
53, 2929-2936
Diphosphene with a phosphineborane tether and its rhodium complex†
Received
21st October 2023
, Accepted 30th November 2023
First published on 11th December 2023
Abstract
A diphosphene ligand possessing a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond and a phosphineborane moiety in its molecule was synthesized. The reaction of the new bidentate diphosphene–phosphineborane ligand with [Rh(cod)2]BF4 provided a cationic diphosphene–rhodium complex with cis-coordination through the η1-PP and η2-BH3 moieties. The complex was applied to the coupling reaction of benzimidazole with cyclohexylallene. The complex also underwent a ligand exchange reaction with N-donor reagents such as N-methylpyrrolidine and N,N,N,N-tetramethylethylenediamine. In particular, the addition reaction of pyridine with the rhodium complex provided an equilibrium mixture of the rhodium complex, pyridine, the diphosphene–phosphineborane ligand, and [Rh(pyridine)2(cod)]BF4.
P bond and a phosphineborane moiety in its molecule was synthesized. The reaction of the new bidentate diphosphene–phosphineborane ligand with [Rh(cod)2]BF4 provided a cationic diphosphene–rhodium complex with cis-coordination through the η1-PP and η2-BH3 moieties. The complex was applied to the coupling reaction of benzimidazole with cyclohexylallene. The complex also underwent a ligand exchange reaction with N-donor reagents such as N-methylpyrrolidine and N,N,N,N-tetramethylethylenediamine. In particular, the addition reaction of pyridine with the rhodium complex provided an equilibrium mixture of the rhodium complex, pyridine, the diphosphene–phosphineborane ligand, and [Rh(pyridine)2(cod)]BF4.
Introduction
Organophosphorus compounds are one of the most common organic ligands for transition metals, yielding molecules with diverse structures in organometallic chemistry.1 A variety of sterically and electronically tuned trivalent phosphorus compounds act as σ-donor ligands through their lone pairs.1c,d On the other hand, low-coordinate phosphorus compounds, such as phosphaalkenes A and phosphinines B, are strong π-acceptors owing to the low-lying π* orbital of the PC bonds (Fig. 1a).2 Some transition metal complexes are utilized in transition-metal-catalyzed organic transformations.1c,2c–f,3,4
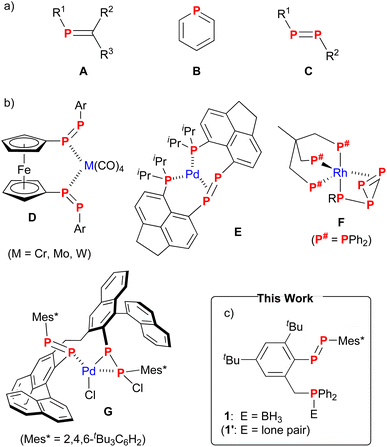 |
| | Fig. 1 (a) Phosphaalkenes A, phosphinines B, and diphosphenes C. (b) Complexes D–G with a multidentate ligand including PP bonds. (c) Diphosphene–phosphineborane ligand 1 (this work). | |
Diphosphenes C with a PP bond in the molecules are also low-coordinate phosphorus compounds that captivated chemists early on.5,6 The LUMO level of the PP π* orbital in diphosphenes is lower than that of the PC π* orbital in phosphaalkenes or phosphinines,7 which should enhance the π-accepting character of the transition metals. We recently demonstrated that a gold complex of diphosphene with a 1,1′-binaphthyl group is an active catalyst for the intramolecular hydroarylation of aryl alkynyl ethers.8 In contrast, monodentate diphosphenes hardly form stable complexes with late transition metals including rhodium(I) and palladium(II), which are known to promote a wide variety of catalytic organic transformations. One solution for generating robust complexes is to increase the coordination ability of diphosphenes by using a multidentate ligand. The combination of a PP bond and other coordinating moieties (e.g., phosphine and amine) holds promise for creating new coordination environments.
Multidentate diphosphene–transition metal complexes are in their infancy compared with their monodentate counterparts that appeared in the 1970s.9,10 Examples of diphosphene complexes with a multidentate ligand include Group 6 carbonyl complexes of bis(diphosphenyl)ferrocene D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and Group 10 complexes with a PP bond and two phosphines12,13 (e.g., E12) (Fig. 1b). Complexes D are synthesized from bis(diphosphene) by a ligand exchange reaction,11 whereas the PP bond in Group 10 complexes is formed by the dimerization of a masked phosphinidene12,13 (phosphanylidene–phosphorane for E12). Rhodium complexes F with a P4R moiety (R = H, alkyl) are obtained by the reaction of a trihydride- (L3RhH3) or an alkyl(ethylene)rhodium (L3RhR(C2H4)) complex with P4.14 The P4R moiety in F acts as a bidentate ligand with an η2-coordinated PP bond in the P3 ring and a phosphido (PR) group. Our recently reported η1-diphosphene/η2-phosphanylphosphido palladium complex G is derived from bis(diphosphene) via chloride migration from palladium to phosphorus.15 To create a diverse array of diphosphene–transition metal complexes with different coordination environments, unsymmetrical bidentate ligands bearing a PP bond are of paramount importance. Such ligands should contribute to expanding the organometallic chemistry of diphosphenes through a direct ligand exchange reaction with a suitable precursor of transition metal complexes. To the best of our knowledge, there are no examples of stable diphosphenes with an additional coordinating moiety, the reason primarily being the inherent synthetic challenges in forging the highly reactive PP bond. This situation sharply contrasts the abundance of multidentate ligands in phosphaalkenes and phosphinines in combination with oxazoline, pyridine, phosphine, etc.16,17 In this regard, we planned to synthesize diphosphene 1′ with a PP bond and a phosphine (Fig. 1c). A phosphinomethyl group was selected as a tether to a benzene ring on the basis of previous reports on bisphosphine ligands with a 2-methylbenzen-diyl (–C6H4CH2–) linker18 and the facile synthesis of bulky aryl groups19 to protect the reactive PP bond. Although we were unable to obtain 1′ despite numerous attempts, we were able to synthesize diphosphene 1 with a phosphineborane moiety. Newly obtained diphosphene–phosphineborane ligand 1 was used in the complexation with rhodium, and the results are described herein.
11 and Group 10 complexes with a PP bond and two phosphines12,13 (e.g., E12) (Fig. 1b). Complexes D are synthesized from bis(diphosphene) by a ligand exchange reaction,11 whereas the PP bond in Group 10 complexes is formed by the dimerization of a masked phosphinidene12,13 (phosphanylidene–phosphorane for E12). Rhodium complexes F with a P4R moiety (R = H, alkyl) are obtained by the reaction of a trihydride- (L3RhH3) or an alkyl(ethylene)rhodium (L3RhR(C2H4)) complex with P4.14 The P4R moiety in F acts as a bidentate ligand with an η2-coordinated PP bond in the P3 ring and a phosphido (PR) group. Our recently reported η1-diphosphene/η2-phosphanylphosphido palladium complex G is derived from bis(diphosphene) via chloride migration from palladium to phosphorus.15 To create a diverse array of diphosphene–transition metal complexes with different coordination environments, unsymmetrical bidentate ligands bearing a PP bond are of paramount importance. Such ligands should contribute to expanding the organometallic chemistry of diphosphenes through a direct ligand exchange reaction with a suitable precursor of transition metal complexes. To the best of our knowledge, there are no examples of stable diphosphenes with an additional coordinating moiety, the reason primarily being the inherent synthetic challenges in forging the highly reactive PP bond. This situation sharply contrasts the abundance of multidentate ligands in phosphaalkenes and phosphinines in combination with oxazoline, pyridine, phosphine, etc.16,17 In this regard, we planned to synthesize diphosphene 1′ with a PP bond and a phosphine (Fig. 1c). A phosphinomethyl group was selected as a tether to a benzene ring on the basis of previous reports on bisphosphine ligands with a 2-methylbenzen-diyl (–C6H4CH2–) linker18 and the facile synthesis of bulky aryl groups19 to protect the reactive PP bond. Although we were unable to obtain 1′ despite numerous attempts, we were able to synthesize diphosphene 1 with a phosphineborane moiety. Newly obtained diphosphene–phosphineborane ligand 1 was used in the complexation with rhodium, and the results are described herein.
Results and discussion
Synthesis of diphosphene–phosphineborane ligand 1
At the initial stage, we attempted to synthesize dichlorophosphine derivative 2′ from bromo derivative 3′ with a diphenylphosphinomethyl tether (Scheme 1). Compound 3′ was synthesized in two steps from 2-bromo-3,5-di-tert-butyl-1-(hydroxymethyl)benzene (4): the Michaelis–Arbuzov reaction of 4 employing Wang's conditions20 gave phosphine oxide 5 in 84% yield, and then 5 was reduced by triethoxysilane and titanium(IV) tetraisopropoxide to give 3′ in 95% yield (Scheme 1a). When 3′ was reacted with tBuLi in THF at −78 °C and then PCl3 was added, the protonated product 3′-H, in which bromine in 3′ was replaced with hydrogen, was obtained in an almost quantitative yield (data not shown). Changing the reaction conditions such as solvent (Et2O and toluene) and temperature was insufficient to overcome the situation. This result was in sharp contrast to the synthesis of di-tert-butylphenyldichlorophosphines with dialkylamino, aryloxy, and methoxy groups at the methyl tether.19 Müller et al. revealed the molecular structure of (o-lithiobenzyl)dimethylphosphine, in which the phosphine moiety is coordinated to lithium.21 From this, we speculated that related lithium species with the coordination of phosphorus atom would prevent access by phosphorus reagent (PCl3). Next, we turned our attention to borane-protected phosphine derivative 3.22 Compound 3 was obtained in 80% yield by the reaction of 3′ with borane–tetrahydrofuran complex. Alternatively, it was directly obtained by the reaction of 2-bromo-1-bromomethyl-3,5-di-tert-butylbenzene (6) with a borane-protected lithium phosphido, Ph2P(BH3)Li, in 77% yield. After the optimization of the reaction conditions, we isolated dichlorophosphine 2 in 40% yield by the lithium/bromine exchange reaction with tBuLi in Et2O in the presence of 1 equiv. of THF at −105 °C and the addition of PCl3 (Scheme 1b). We noted that a small amount of THF was required for the lithium/bromine exchange reaction. These reaction conditions were applied to the synthesis of 2′ without borane protection from phosphine 3′, but an unidentified mixture was generated without the formation of 2′. Attempts to eliminate borane (BH3) in 2 to give 2′ were also unsuccessful by the reaction with several amines,23 such as N-methylpyrrolidine and 1,4-diazabicyclo[2.2.2]octane (DABCO), or trimethylphosphine in neat or toluene solution, giving a complex mixture. Next, the PP bond was formed by following a previously reported conventional method:5a treatment of 2 with Mes*PHLi to form a P–P single bond and dehydrochlorination with tBu(SiMe3)NLi as a base gave diphosphene–phosphineborane ligand 1 in 30% yield as a yellow solid. This result indicated that phosphineborane remained intact under the reaction conditions. The elimination of borane (BH3) of 1 to obtain diphosphene–phosphine derivative 1′ was unsuccessful again: there was either no reaction or the formation of a complex mixture in the reaction of 1 with N-methylpyrrolidine or DABCO. The 31P NMR signals assignable to the PP bond in 1 were observed at 531.3 and 483.1 ppm (1JPP = 578 Hz), which were comparable to those of unsymmetrically substituted diphosphenes with a Mes* group (e.g., Mes*PPMes:24aδP = 540.4, 467.6, 1JPP = 574 Hz; and Mes*PPBNp:24bδP = 458.7, 528.2, 1JPP = 569 Hz; BNp = 3-methyl-1,1′-binaphthylene-2-yl). The 31P and 11B signals of the phosphineborane moiety at 22.1 and −37.1 ppm, respectively, were slightly shifted from those of 2 (δP = 20.7, δB = −38.2).
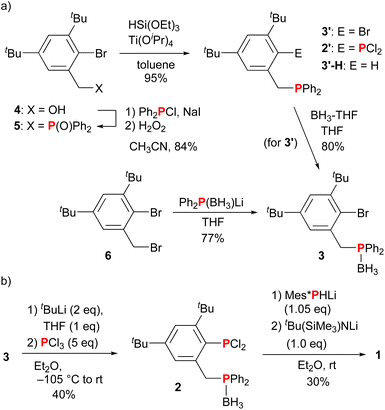 |
| | Scheme 1 (a) Synthesis of phosphine 3′ and phosphineborane 3. (b) Synthesis of diphosphene–phosphineborane ligand 1. | |
X-ray crystallographic analysis of 1 (Fig. 2a) showed that the P2 atom of the PP bond and the P3 atom of the phosphinomethyl group are in a trans relationship with respect to central benzene R. The BH3 group and the tert-butyl group at the para position of benzene R are oriented in the same direction. The P–P bond length (2.0406(8) Å) and the P–P–C bond angles (98.73(5)° and 100.39(6)°) are comparable to those of Mes*-substituted diphosphenes (Table S2†). The P–B bond length (1.924(2) Å) is also comparable to the (PhMe2C)Ph2P–BH3 bond length (1.935(2) Å).25 The IR spectrum of 1 exhibited moderately strong absorptions at 2400 and 2338 cm−1 owing to B–H stretching vibrations (Fig. S25†). The UV–vis spectrum measured in CH2Cl2 solution showed absorptions at 464 nm (ε, 400) and 338 nm (4190) as a shoulder (Fig. 3a), which were assigned to the n+–π* and π–π* transitions of the PP bond, respectively.5a,26
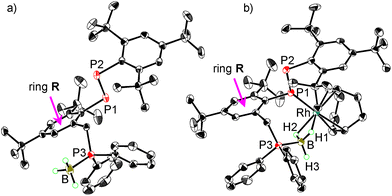 |
| | Fig. 2 Molecular structures of (a) diphosphene–phosphineborane ligand 1 and (b) diphosphene–rhodium complex 7. Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms except BH3, anion BF4, and solvated dichloroethanes in 7 are omitted for clarity. | |
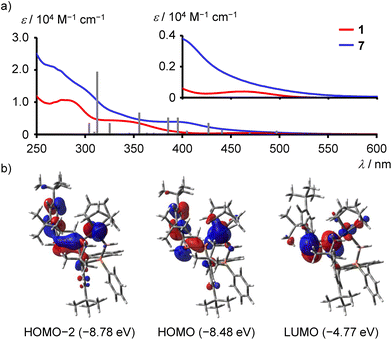 |
| | Fig. 3 (a) UV–vis absorption spectra of 1 (red) and 7 (blue) in CH2Cl2 solution. Gray bars represent calculated oscillator strengths of 7 longer than 300 nm. (b) Selected molecular orbitals of 7 (Isovalue = 0.03). | |
Complexation of 1 with rhodium
Diphosphene–phosphineborane ligand 1 was directly used for complexation because rhodium complexes with a bidentate phosphine–phosphineborane ligand are known (Scheme 2).27 Treatment of 1 with 1 equiv. of [Rh(cod)2]BF4 in CH2Cl2 afforded diphosphene–rhodium complex 7 in 80% yield as a red-orange solid (Scheme 2a). This is the first example of a diphosphene–rhodium complex directly synthesized from a stable diphosphene. In contrast, treatment of 1 with 1 equiv. of [Ir(cod)2]BF4 in CD2Cl2 resulted in a complex mixture as judged from the 1H and 31P NMR spectra of the crude mixture. The 31P NMR signals of 7 were observed at 439.8, 348.7, and 4.6 ppm. The two former chemical shifts and their coupling constant (1JPP = 544 Hz) supported the η1-coordination of the PP bond to a rhodium atom.5a,8 In addition, the signal at 348.7 ppm indicated the additional coupling to rhodium (1JPRh = 149 Hz). This value was larger than that of the η2-PP bond of complex F (1JPRh = ca. 20 Hz)14a owing to the coordination of the lone pair of phosphorus atom with high s contribution, and was comparable to those of phosphine–rhodium complexes 8a (145 Hz)27a and 9b (140 Hz).27b The 31P signal of phosphineborane in 7 (4.6 ppm) was shifted upfield relative to that of 1 (22.1 ppm), whereas the 11B chemical shift (−36.6 ppm) was comparable to that of 1 (−34.8 ppm). The coordination behavior of the BH3 moiety was similar to that of 8a and 9b.27 The BH3 signal in the 1H NMR spectrum was observed at −0.60 ppm as a broad doublet (J = 105 Hz) with the integral of three protons (Fig. S36†). This result indicated that the BH3 group is fluxional in solution via a P–B bond rotation at room temperature. When a CD2Cl2 solution of 7 was cooled to −70 °C, the signal broadened, indicating coalescence. The peak observed at −0.60 ppm shifted to −1.47 ppm at −80 °C and then split into two independent signals −1.12 and −1.91 ppm upon further cooling to −90 °C. The two protons coordinated to rhodium at low temperatures under the non-equivalent environment. The third proton of BH3, which was observed around 2.4 ppm, could not be detected owing to the overlap to the CH2 signal of cod. The IR spectrum of 1 exhibited weak absorptions at 2432, 2075, and 2008 cm−1 (Fig. S25†). The former frequency was due to the remaining B–H bond, whereas the latter two frequencies were due to the Rh–H bond in the solid state. The Rh−H vibration of HRh(CO)2(PPh3)2 was reported as 2038 cm−1.28 Theoretical calculations of the cation moiety of 7 at M06/SDD (Rh) and 6-31G(d) (other atoms) level of theory supported the B–H stretching (2449 cm−1), the Rh–H antisymmetric stretching (2233 cm−1), and the Rh–H symmetric stretching (2166 cm−1) vibrations, where the values were scaled by 0.9585.29 Although these calculated values deviated somewhat from the experimental ones, other vibrations were not estimated in the range of 2900 to 2000 cm−1.
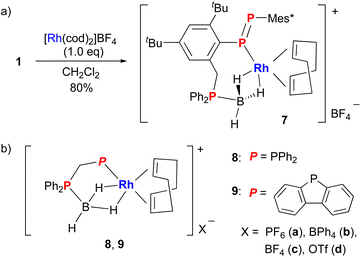 |
| | Scheme 2 (a) Synthesis of diphosphene–rhodium complex 7. (b) Rhodium complexes 8 and 9 with a phosphine–phosphineborane ligand. | |
Recrystallization of 7 from a 1,2-dichloroethane/diisopropyl ether solution at −25 °C gave single crystals suitable for X-ray crystallographic analysis (Fig. 2b). Hydrogen atoms on BH3 were directly located in the difference Fourier map and refined isotropically. In the same manner as 1, the P2 atom of the PP bond and the P3 atom of the diphenylphosphinomethyl group are in a trans relationship with respect to central benzene R. One molecule each of diphosphene–phosphineborane ligand 1 and cod coordinates to rhodium, and 1 achieves cis-coordination through a phosphorus atom (P1) of diphosphene in an η1 fashion and the BH3 moiety in an η2 fashion. The two B–H–Rh three-center two-electron bonds in 7 are similar to those in 8c27a and 9c.27b The P1–Rh–B angle (92.97(9)°) in 7 is larger than that in 8c (87.26(8)°) or 9c (87.35(4)°) owing to the different carbon linkers (three carbon atoms in 7versus one carbon atom in 8 and 9). The PP bond length of 7 (2.0413(13) Å) is comparable to the bond length of free ligand 1 (2.0406(8) Å), retaining the PP double-bond character, and is slightly longer than those of η1-diphosphene Group 11 complexes (1.975(5)–2.0269(16) Å).8,30 The Rh–P1 bond length of 7 (2.2694(8) Å) lies between that of 8c (2.2743(7) Å) and that of 9c (2.2483(4) Å) (Table 1). To evaluate the effect of the nature of the coordinated phosphorus atom (PPAr vs. PPh2) and the length of the linker (methylene vs. 2-methylbenzen-diyl), theoretical calculations were carried out at M06/SDD (Rh) and 6-31G(d) (other atoms) level of theory for the cation moieties of complexes 7 and 8c together with 7′ with hydrogen atoms instead of five tert-butyl groups in 7; 10 with the PPPh group and a methylene linker; and 11 with the PPh2 group and a 2-methylbenzen-diyl linker (Table 1). The Rh–P1 bonds in the diphosphene complexes with a PPPh moiety (2.294 Å in 7′ and 2.310 Å in 10) are shorter than those in the phosphine complexes (2.390 Å in 11 and 2.324 Å in 8c) regardless of the linker. Changing the methylene linker into a 2-methylbenzen-diyl linker shortened the Rh–P1 bonds in the diphosphene complexes (2.294 Å in 7′vs. 2.310 Å in 10), whereas the opposite trend was observed in the phosphine complexes (2.390 Å in 11vs. 2.324 Å in 8c). Although the reason for the difference is unclear at present, the rhodium complex with a 2-methylbenzen-diyl linker may be strongly affected by the steric effect of the substituents on the phosphorus atom. In fact, phosphine complex 11 with a 2-methylbenzen-diyl linker has the longest Rh–P1 bond (2.390 Å) that markedly differed from the others. The Wiberg bond index of the Rh–P1 bond ranges from 0.501 to 0.558, indicating comparable Rh–P bond strengths between the diphosphenes and the phosphines. On the other hand, the different bonding character of the P1 atom in the diphosphenes (two coordinate) and the phosphines (three coordinate) affected the Rh–P bond properties. NBO analysis showed the localized lone-pair character of P1 (1.60e, sp0.34) in 7 and the Rh–P1 bond character (1.95e, sd7.33 for Rh and sp2.04 for P) in 8c.
Table 1 Selected structural parameters of rhodium complexes with a bidentate ligand including a phosphineborane moiety
The photophysical properties of 7 were evaluated from the UV–vis spectrum measured in CH2Cl2 solution. The UV–vis spectrum showed featureless and broad band tailing to 570 nm with a shoulder at 398 nm (Fig. 3a). TD–DFT calculations of 7 at TD-M06/SDD (Rh) and 6-311+G(2d,p) (other atoms) level of theory showed 10 transitions larger than 350 nm with a small oscillator strength (f = 0.0033–0.0649, Table S8†) including the HOMO–LUMO transition at 497.5 nm (f = 0.0084). The HOMO is the lone-pair n+ orbital of the PP bond and the d orbital of rhodium, whereas the LUMO is the dominant contribution of the π* orbital of the PP bond (Fig. 3b). The character of the low-lying LUMO of the PP bond was maintained even in 7. The π orbital of the PP bond was found in HOMO−2 together with the contribution of the d orbital of rhodium and the π orbital of the benzene ring of the Mes* group.
Investigation of catalytic properties of diphosphene–rhodium complex 7
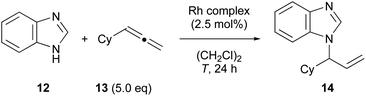
We investigated the catalytic properties of newly obtained diphosphene–rhodium complex 7. To our regret, complex 7 did not act as an active catalyst in the hydroboration of vinylarene with catecholborane27c or the intramolecular hydroamination of 5-(N-benzylamino)pentene.31 Nevertheless, after investigating several reactions, we found that complex 7 promoted the coupling reaction of benzimidazole (12) with cyclohexylallene (13) although the yield was low (Table 2):32 mixing 12 and 5 equiv. of 13 in the presence of 2.5 mol% of complex 7 in 1,2-dichloroethane (DCE) at 80 °C for 24 h afforded coupling product 14 in 35% yield (entry 1). The yield was decreased to 17% and 0% when the temperature was lowered to 70 °C and 60 °C, respectively (entries 2 and 3). No product 14 was obtained when [Rh(cod)2]BF4 was used instead of 7 (entry 4). We confirmed that a new phosphorus species was generated by the reaction of 7 with 12 (7.5 equiv.) in DCE as judged from the 31P NMR spectrum measured at room temperature. On the other hand, complex 7 was stable at room temperature in the presence of 13 (10 equiv.) but was converted into a new species at 80 °C. None of these new products could be characterized. Because the ligand exchange reaction with the diphosphene ligand in 7 proceeded smoothly (vide infra), other phosphorus species generated from diphosphene–phosphineborane ligand 1 under the reaction conditions were likely the true active ligand.
Table 2 Coupling reaction of benzimidazole (12) with cyclohexylallene (13)
|
|
| Entry |
Rh complex |
T (°C) |
Yielda (%) |
|
Isolated yield.
|
| 1 |
7
|
80 |
35 |
| 2 |
7
|
70 |
17 |
| 3 |
7
|
60 |
0 |
| 4 |
[Rh(cod)2]BF4 |
80 |
0 |
Reaction of 7 with N-donor reagents
The fact that diphosphene–rhodium complex 7 easily reacts with benzimidazole (12) implies that other nitrogen-containing compounds should also be active reagents for 7. If the phosphineborane moiety in 7 is converted into a phosphine, an electronically different diphosphene–rhodium complex with more robust coordination could be obtained. Thus, we investigated the deboranation reaction of 7. Treating N-methylpyrrolidine (20 equiv.) with a CD2Cl2 solution of 7 in an NMR tube and allowing the reaction mixture to stand for 4 h gave diphosphene–phosphineborane ligand 1 in a quantitative yield (Scheme 3a, Fig. S37†). The quantitative dissociation of 1 was also observed in the reaction with triethylamine (20 equiv.). The reaction took a longer time than that with N-methylpyrrolidine (approximately 2 days, Fig. S38†). In both reactions, the generated rhodium species could not be sufficiently characterized because rhodium complexes bearing these tertiary amines and cod were not reported.33 Although the reaction mechanism is unclear at present, the ligand exchange reaction between 1 and amines should be promoted by the high electron-accepting character of diphosphene because the LUMO is the dominant contribution of the π* orbital of the PP bond (Fig. 3b). To characterize the rhodium species after the ligand exchange reaction, N,N,N,N-tetramethylethylenediamine (tmeda) was used. The reaction of 7 with tmeda (20 equiv.) in CD2Cl2 resulted in the formation of a 1:1 mixture of diphosphene–phosphineborane ligand 1 and [Rh(tmeda)(cod)]BF4 (Scheme 3b, Fig. S39†) as judged from the 1H NMR spectrum.34 The result clearly showed that the dissociation of diphosphene–phosphineborane ligand 1 was faster than that of cod. The facile dissociation of 1 with tertiary amines prompted us to evaluate the coordination ability of diphosphene–phosphineborane ligand 1 toward rhodium. As a result, we found that the reaction of diphosphene–rhodium complex 7 with pyridine (py) gave an equilibrium mixture of 7, pyridine, diphosphene–phosphineborane ligand 1, and [Rh(py)2(cod)]BF4 (15; Scheme 3c, Fig. 4a).34 Starting diphosphene–rhodium complex 7 was recovered after removal of unreacted pyridine under reduced pressure from a mixture of 7 and pyridine (20 equiv.) in CD2Cl2 solution, and its 1H NMR spectrum was measured in CD2Cl2 (Fig. 4b). Treatment of 1 with 15 also afforded a mixture of 1, 15, 7, and pyridine (Fig. 4c). The equilibrium constant (Keq = ([1]·[15])/([7]·[py]2) was estimated to be 1.0(2) mol−1 L−1 by pyridine titration of a CD2Cl2 solution of 7 (Table S1†). Thus, the coordination ability of 1 is comparable to that of pyridine even though 1 exhibits bidentate coordination. The weak coordination ability of the PP bond (instability of the obtained rhodium complex) impedes the formation of a stable rhodium complex with a monodentate diphosphene ligand by the ligand exchange reaction. Meanwhile, no ligand exchange occurred in the reaction with acetonitrile (Scheme 3d, Fig. S41†).
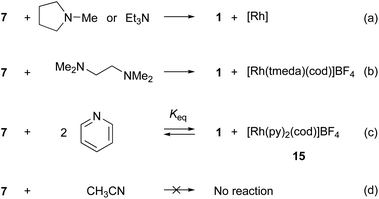 |
| | Scheme 3 Reactions of diphosphene–rhodium complex 7 with N-donor reagents. (tmeda = N,N,N,N-tetramethylethylenediamine, cod = 1,5-cyclooctadiene, py = pyridine); Keq = ([1]·[15])/([7]·[py]2). | |
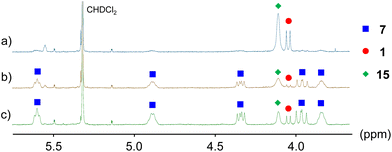 |
| | Fig. 4
1H NMR spectra measured in CD2Cl2 (a) after the reaction of 7 with pyridine (20 equiv.); (b) after removal of unreacted pyridine from the reaction mixture; and (c) after the treatment of 1 with [Rh(py)2(cod)]BF4 (15, 1 equiv.). | |
Conclusions
Diphosphene 1 with a phosphineborane tether was synthesized without affecting the phosphineborane to form a PP bond. Diphosphene–rhodium complex 7 was isolated with the aid of the phosphineborane moiety. X-ray crystallographic analysis and NMR spectroscopy of 7 revealed a bidentate cis-coordination through the PP bond in an η1 fashion and the BH3 moiety in an η2 fashion. Complex 7 catalyzed the coupling reaction of benzimidazole with cyclohexylallene although the reactive PP bond was likely converted into another phosphorus species under the reaction conditions. Theoretical calculations showed the dominant contribution of the π* orbital of the PP bond even in complex 7, and its high electron-accepting character should promote facile ligand exchange reaction with N-donor reagents despite the bidentate coordination of 1. In particular, the reaction of diphosphene–rhodium complex 7 with pyridine gave an equilibrium mixture of 7, pyridine, diphosphene–phosphineborane ligand 1, and [Rh(py)2(cod)]BF4 (15) owing to the weak coordination ability of 1 toward rhodium. Although further improvements in the fascinating catalytic activity of the diphosphene–transition metal complexes are required, our fundamental studies on bidentate diphosphene–rhodium complex 7 are expected to contribute to catalytic chemistry in the near future. We are actively investigating the coordination behavior toward other transition metals and developing other types of multidentate ligands with the PP bond for use in catalytic organic transformations.
Author contributions
A. T.: Conceptualization, data curation, formal analysis, funding acquisition, investigation, project administration, validation, visualization, writing – original draft, writing – review and editing. S. T.: Data curation, formal analysis, investigation, visualization. K. K.: Funding acquisition, supervision, writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B: #22H02081 and #22K19035 to K. K.; and C: #21K05071 to A. T.) from the JSPS (Japan). The authors thank Professor Ikuko Miyahara (Osaka Metropolitan University) for assistance in X-ray crystallographic analysis; Professor Hajime Kameo (Osaka Metropolitan University) for assistance in the measurement of IR spectra; and Professor Hideki Fujiwara and Professor Daisuke Sakamaki (Osaka Metropolitan University) for allowing access to the UV-vis spectrophotometer. All theoretical calculations were carried out at the Research Centre (21-IMS-C029) for Computational Science (Japan).
References
-
(a)
Organophosphorus Chemistry: From Molecules to Applications, ed. V. Iaroshenko, Wiley-VCH, 2019 Search PubMed
 ;
(b)
Phosphorus: Chemistry, Biochemistry and Technology 6th, ed. D. E. C. Corbridge, CRC Press Taylor & Francis Group, 2013 Search PubMed ;
(c)
Phosphorus(III) Ligands in Homogenous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley, 2012 Search PubMed ;
(d)
Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, 2008 Search PubMed .
;
(b)
Phosphorus: Chemistry, Biochemistry and Technology 6th, ed. D. E. C. Corbridge, CRC Press Taylor & Francis Group, 2013 Search PubMed ;
(c)
Phosphorus(III) Ligands in Homogenous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley, 2012 Search PubMed ;
(d)
Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, 2008 Search PubMed .
-
(a) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604 CrossRef CAS PubMed ;
(b) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef CAS ;
(c) C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187–202 CrossRef ;
(d) F. Ozawa and Y. Nakajima, Chem. Rec., 2016, 16, 2314–2323 CrossRef CAS PubMed ;
(e) N. T. Coles, A. S. Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729 CrossRef CAS ;
(f) A. Ziółkowska, J. Doroszuk and Ł. Ponikiewski, Organometallics, 2023, 42, 505–537 CrossRef .
- Selected examples of phosphaalkene complexes:
(a) W. Keim, R. Appel, S. Gruppe and F. Knoch, Angew. Chem., Int. Ed. Engl., 1987, 26, 1012–1013 CrossRef ;
(b) T. Minami, H. Okamoto, S. Ikeda, R. Tanaka, F. Ozawa and M. Yoshifuji, Angew. Chem., Int. Ed., 2001, 40, 4501–4503 CrossRef CAS ;
(c) F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968–10969 CrossRef CAS PubMed ;
(d) A. Ionkin and W. Marshall, Chem. Commun., 2003, 710–711 RSC ;
(e) M. Freytag, S. Ito and M. Yoshifuji, Chem. – Asian J., 2006, 1, 693–700 CrossRef CAS PubMed ;
(f) J. Dugal-Tessier, G. R. Dake and D. P. Gates, Org. Lett., 2010, 12, 4667–4669 CrossRef CAS PubMed ;
(g) Y.-H. Chang, Y. Nakajima and F. Ozawa, Organometallics, 2013, 32, 2210–2215 CrossRef CAS ;
(h) K. Takeuchi, Y. Tanaka, I. Tanigawa, F. Ozawa and J.-C. Choi, Dalton Trans., 2020, 49, 3630–3637 RSC .
- Selected examples of phosphinine complexes:
(a) B. Breit, Chem. Commun., 1996, 2071–2072 RSC ;
(b) B. Breit, R. Winde, T. Mackewitz, R. Paciello and K. Harms, Chem. – Eur. J., 2001, 7, 3106–3121 CrossRef CAS PubMed ;
(c) M. T. Reetz and X. Li, Angew. Chem., Int. Ed., 2005, 44, 2962–2964 CrossRef CAS PubMed ;
(d) J. J. M. Weemers, W. N. P. van der Graaff, E. A. Pidko, M. Lutz and C. Müller, Chem. – Eur. J., 2013, 19, 8991–9004 CrossRef CAS PubMed ;
(e) R. J. Newland, M. F. Wyatt, R. L. Wingad and S. M. Mansell, Dalton Trans., 2017, 46, 6172–6176 RSC ;
(f) R. J. Newland, J. M. Lynam and S. M. Mansell, Chem. Commun., 2018, 54, 5482–5485 RSC ;
(g) M. Rigo, E. R. M. Habraken, K. Bhattacharyya, M. Weber, A. W. Ehlers, N. Mézailles, J. C. Slootweg and C. Müller, Chem. – Eur. J., 2019, 25, 8769–8779 CrossRef CAS PubMed ;
(h) E. C. Trodden, M. P. Delve, C. Luz, R. J. Newland, J. M. Andresen and S. M. Mansell, Dalton Trans., 2021, 50, 13407–13411 RSC .
-
(a) L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS ;
(b) T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395–1408 RSC ;
(c) M. C. Simpson and J. D. Protasiewicz, Pure Appl. Chem., 2013, 85, 801–815 CrossRef CAS ;
(d) M. Yoshifuji, Eur. J. Inorg. Chem., 2016, 607–615 CrossRef CAS ;
(e) L. Weber, F. Ebeler and R. S. Ghadwal, Coord. Chem. Rev., 2022, 461, 214499 CrossRef CAS .
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS .
-
(a) A. H. Cowley, A. Decken, N. C. Norman, C. Krüger, F. Lutz, H. Jacobsen and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 3389–3390 CrossRef CAS ;
(b) C. Dutan, S. Shah, R. C. Smith, S. Choua, T. Berclaz, M. Geoffroy and J. D. Protasiewicz, Inorg. Chem., 2003, 42, 6241–6251 CrossRef CAS PubMed ;
(c) T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582–12588 CrossRef CAS PubMed ;
(d) B. Lu, L. Wang, X. Jiang, G. Rauhut and X. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202217353 CrossRef CAS PubMed .
- A. Tsurusaki, R. Ura and K. Kamikawa, Organometallics, 2020, 39, 87–92 CrossRef CAS .
-
(a) J. C. Green, M. L. H. Green and G. E. Morris, J. Chem. Soc., Chem. Commun., 1974, 212–213 RSC ;
(b) P. S. Elmes, M. L. Scudder and B. O. West, J. Organomet. Chem., 1976, 122, 281–288 CrossRef CAS .
- For recent investigations on diphosphene complexes:
(a) C. Taube, J. Fidelius, K. Schwedtmann, C. Ziegler, F. Kreuter, L. Loots, L. J. Barbour, R. Tonner-Zech, R. Wolf and J. J. Weigand, Angew. Chem., Int. Ed., 2023, 62, e202306706 CrossRef CAS PubMed ;
(b) A. Schmer, D. Welideniya, T. Terschüren, G. Schnakenburg, J. Daniels, A. Bauza, A. Frontera and R. Streubel, Dalton Trans., 2021, 50, 17892–17896 RSC ;
(c) A. Schmer, A. Bauza, G. Schnakenburg, A. Frontera and R. Streubel, Dalton Trans., 2021, 50, 2131–2137 RSC ;
(d) D. Dhara, D. Scheschkewitz, V. Chandrasekhar, C. B. Yildiz and A. Jana, Chem. Commun., 2021, 57, 809–812 RSC .
- N. Nagahora, T. Sasamori, Y. Watanabe, Y. Furukawa and N. Tokitoh, Bull. Chem. Soc. Jpn., 2007, 80, 1884–1900 CrossRef CAS .
-
(a) B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Angew. Chem., Int. Ed., 2012, 51, 10150–10153 CrossRef CAS PubMed ;
(b) B. A. Surgenor, B. A. Chalmers, K. S. A. Arachchige, A. M. Z. Slawin, J. D. Woollins, M. Bühl and P. Kilian, Inorg. Chem., 2014, 53, 6856–6866 CrossRef CAS PubMed .
- S. C. Kosnik, J. F. Binder, M. C. Nascimento, A. Swidan and C. L. B. Macdonald, Chem. – Eur. J., 2019, 25, 1208–1211 CrossRef CAS PubMed .
-
(a) P. Barbaro, A. Ienco, C. Mealli, M. Peruzzini, O. J. Scherer, G. Schmitt, F. Vizza and G. Wolmershäuser, Chem. – Eur. J., 2003, 9, 5195–5210 CrossRef CAS PubMed ;
(b) P. Barbaro, M. Caporali, A. Ienco, C. Mealli, M. Peruzzini and F. Vizza, Eur. J. Inorg. Chem., 2008, 1392–1399 CrossRef CAS .
- R. Ura, A. Tsurusaki and K. Kamikawa, Dalton Trans., 2022, 51, 2943–2952 RSC .
- Examples of rhodium complexes of phosphaalkenes:
(a) R. S. Jensen, K. Umeda, M. Okazaki, F. Ozawa and M. Yoshifuji, J. Organomet. Chem., 2007, 692, 286–294 CrossRef CAS ;
(b) A. Hayashi, M. Okazaki and F. Ozawa, Organometallics, 2007, 26, 5246–5249 CrossRef CAS ;
(c) T. Matsumoto, T. Sasamori, H. Miyake and N. Tokitoh, Organometallics, 2014, 33, 1341–1344 CrossRef CAS ;
(d) P. M. Miura-Akagi, M. L. Nakashige, C. K. Maile, S. M. Oshiro, J. R. Gurr, W. Y. Yoshida, A. T. Royappa, C. E. Krause, A. L. Rheingold, R. P. Hughes and M. F. Cain, Organometallics, 2016, 35, 2224–2231 CrossRef CAS ;
(e) P. Gupta, T. Taeufer, J.-E. Siewert, F. Reiß, H.-J. Drexler, J. Pospech, T. Beweries and C. Hering-Junghans, Inorg. Chem., 2022, 61, 11639–11650 CrossRef CAS PubMed .
- Examples of rhodium complexes of phosphinines:
(a) B. Schmid, L. M. Venanzi, T. Gerfin, V. Gramlich and F. Mathey, Inorg. Chem., 1992, 31, 5117–5122 CrossRef CAS ;
(b) X. Sava, N. Mézailles, N. Maigrot, F. Nief, L. Ricard, F. Mathey and P. Le Floch, Organometallics, 1999, 18, 4205–4215 CrossRef CAS ;
(c) A. C. Carrasco, E. A. Pidko, A. M. Masdeu-Bultó, M. Lutz, A. L. Spek, D. Vogt and C. Müller, New J. Chem., 2010, 34, 1547–1550 RSC ;
(d) L. E. E. Broeckx, M. Lutz, D. Vogt and C. Müller, Chem. Commun., 2011, 47, 2003–2005 RSC ;
(e) X. Chen, Z. Li and H. Grützmacher, Chem. – Eur. J., 2018, 24, 8432–8437 CrossRef CAS PubMed ;
(f) R. J. Newland, J. M. Lynam and S. M. Mansell, Chem. Commun., 2018, 54, 5482–5485 RSC ;
(g) K. Masada, S. Kusumoto and K. Nozaki, Angew. Chem., Int. Ed., 2022, 61, e202117096 CrossRef CAS PubMed .
- T. Fanjul, G. Eastham, J. Floure, S. J. K. Forrest, M. F. Haddow, A. Hamilton, P. G. Pringle, A. G. Orpen and M. Waugh, Dalton Trans., 2013, 42, 100–115 RSC .
-
(a) K. Kamijo, A. Otoguro, K. Toyota and M. Yoshifuji, Bull. Chem. Soc. Jpn., 1999, 72, 1335–1342 CrossRef CAS ;
(b) S. Kawasaki, T. Fujita, K. Toyota and M. Yoshifuji, Bull. Chem. Soc. Jpn., 2005, 78, 1082–1090 CrossRef CAS .
- Y. Ma, F. Chen, J. Bao, H. Wei, M. Shi and F. Wang, Tetrahedron Lett., 2016, 57, 2465–2467 CrossRef CAS .
- G. Müller, H.-P. Abicht, M. Waldkircher, J. Lachmann, M. Lutz and M. Winkler, J. Organomet. Chem., 2001, 622, 121–134 CrossRef .
- For an example of the introduction of a phosphine moiety after the halogen/lithium exchange from a compound with a borane-protected phosphinomethyl group: Q. Shen, T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 6586–6596 CrossRef CAS PubMed .
-
(a) G. C. Lloyd-Jones and N. P. Taylor, Chem. – Eur. J., 2015, 21, 5423–5428 CrossRef CAS PubMed ;
(b) A. Zhdanko, B. A. van der Worp and S. Lemaire, Eur. J. Org. Chem., 2022, e202200130 CrossRef CAS .
-
(a) M. Yoshifuji, K. Shibayama, N. Inamoto, T. Matsushita and K. Nishimoto, J. Am. Chem. Soc., 1983, 105, 2495–2497 CrossRef CAS ;
(b) A. Tsurusaki, R. Ura and K. Kamikawa, Dalton Trans., 2018, 47, 4437–4441 RSC .
- L. Routaboul, F. Toulgoat, J. Gatignol, J.-F. Lohier, B. Norah, O. Delacroix, C. Alayrac, M. Taillefer and A.-C. Gaumont, Chem. – Eur. J., 2013, 19, 8760–8764 CrossRef CAS PubMed .
-
(a) K. Miqueu, J.-M. Sotiropoulos, G. Pfister-Guillouzo, H. Ranaivonjatovo and J. Escudié, J. Mol. Struct., 2001, 545, 139–146 CrossRef ;
(b) T. Sasamori, N. Takeda and N. Tokitoh, J. Phys. Org. Chem., 2003, 16, 450–462 CrossRef CAS ;
(c) Y. Amatatsu, J. Phys. Chem. A, 2008, 112, 8824–8828 CrossRef CAS PubMed ;
(d) H.-L. Peng, J. L. Payton, J. D. Protasiewicz and M. C. Simpson, J. Phys. Chem. A, 2009, 113, 7054–7063 CrossRef CAS PubMed See also references cited therein.
-
(a) M. Ingleson, N. J. Patmore, G. D. Ruggiero, C. G. Frost, M. F. Mahon, M. C. Willis and A. S. Weller, Organometallics, 2001, 20, 4434–4436 CrossRef CAS ;
(b) D. H. Nguyen, H. Lauréano, S. Jugé, P. Kalck, J.-C. Daran, Y. Coppel, M. Urrutigoity and M. Gouygou, Organometallics, 2009, 28, 6288–6292 CrossRef CAS ;
(c) D. H. Nguyen, J. Bayardon, C. Salomon-Bertrand, S. Jugé, P. Kalck, J.-C. Daran, M. Urrutigoity and M. Gouygou, Organometallics, 2012, 31, 857–869 CrossRef CAS ;
(d) C. Salomon-Bertrand, J. Bayardon, H. Lauréano, S. Jugé, J.-C. Daran and M. Gouygou, J. Organomet. Chem., 2021, 938, 121753 CrossRef CAS .
- A. M. Trzeciak and J. J. Ziółkowski, J. Organomet. Chem., 1992, 429, 239–244 CrossRef CAS .
- M. K. Kesharwani, B. Brauer and J. M. L. Martin, J. Phys. Chem. A, 2015, 119, 1701–1714 CrossRef CAS PubMed .
-
(a) D. V. Partyka, M. P. Washington, T. G. Gray, J. B. Updegraff III, J. F. Turner II and J. D. Protasiewicz, J. Am. Chem. Soc., 2009, 131, 10041–10048 CrossRef CAS PubMed ;
(b) X. Pan, L. Zhang, Y. Zhao, G. Tan, H. Ruan and X. Wang, Chin. J. Chem., 2020, 38, 351–355 CrossRef CAS ;
(c) S. Lauk, M. Zimmer, B. Morgenstern, V. Huch, C. Müller, H. Sitzmann and A. Schäfer, Organometallics, 2021, 40, 618–626 CrossRef CAS .
- Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570–1571 CrossRef CAS PubMed .
- K. Xu, N. Thieme and B. Breit, Angew. Chem., Int. Ed., 2014, 53, 2162–2165 CrossRef CAS PubMed .
- The reaction of [Rh(cod)2]BF4 with N-methylpyrrolidine or triethylamine gave a complex mixture.
- [Rh(tmeda)(cod)]BF4 and [Rh(py)2(cod)]BF4 (15) were synthesized by the reaction of [Rh(cod)2]BF4 with tmeda and pyridine, respectively (see ESI).†.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Shingo
Takechi
and
Ken
Kamikawa
*,
Shingo
Takechi
and
Ken
Kamikawa








