
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Collision induced unfolding and molecular dynamics simulations of norovirus capsid dimers reveal strain-specific stability profiles†
Maxim N.
Brodmerkel
 a,
Lars
Thiede
bc,
Emiliano
De Santis
ad,
Charlotte
Uetrecht
bc,
Carl
Caleman
de and
Erik G.
Marklund
*a
a,
Lars
Thiede
bc,
Emiliano
De Santis
ad,
Charlotte
Uetrecht
bc,
Carl
Caleman
de and
Erik G.
Marklund
*a
aDepartment of Chemistry - BMC, Uppsala University, 75123 Uppsala, Sweden. E-mail: erik.marklund@kemi.uu.se
bCSSB Centre for Structural Systems Biology, Deutsches Elektronen-Synchrotron DESY, Leibniz Institute of Virology (LIV), Notkestrasse 85, 22607 Hamburg, Germany
cInstitute of Chemistry and Metabolomics, University of Lübeck, Ratzeburger Allee 160, 23562 Lübeck, Germany
dDepartment of Physics and Astronomy, Uppsala University, 75120 Uppsala, Sweden
eCenter for Free-Electron Laser Science CFEL, Deutsches Elektronen-Synchrotron, DESY, Notkestrasse 85, 22607 Hamburg, Germany
First published on 9th April 2024
Abstract
Collision induced unfolding (CIU) is a method used with ion mobility mass spectrometry to examine protein structures and their stability. Such experiments yield information about higher order protein structures, yet are unable to provide details about the underlying processes. That information can however be provided using molecular dynamics simulations. Here, we investigate the gas-phase unfolding of norovirus capsid dimers from the Norwalk and Kawasaki strains by employing molecular dynamics simulations over a range of temperatures, representing different levels of activation, together with CIU experiments. The dimers have highly similar structures, but their CIU reveals different stability that can be explained by the different dynamics that arises in response to the activation seen in the simulations, including a part of the sequence with previously observed strain-specific dynamics in solution. Our findings show how similar protein variants can be examined using mass spectrometric techniques in conjunction with atomistic molecular dynamics simulations to reveal differences in stability as well as differences in how and where unfolding takes place upon activation.
Introduction
In nature, the majority of proteins exist not as monomeric species but self-assemble into multimeric complexes,1 either as a result of selection pressure associated with protein functions, physical properties such as stability, or as a consequence of non-adaptive evolutionary processes, where interfaces can become entrenched without apparent functional advantages.2 Virus capsids are an example of the former, where rapid self-assembly of monomers results in an encapsulating structure that holds both the genome and essential proteins, and also protects them until their necessary release. This makes virus capsids excellent models for research on protein assembly and stability.3 Noroviruses are the leading cause of viral gastroenteritis,4,5 an acute inflammation of the gastrointestinal tract that can cause severe complications particularly for infants, the elderly or people with compromised immune systems.6 The norovirus capsid has been subject to several studies on the aforementioned assembly and stability,7–10 as they are an essential part of understanding viral mechanisms of infection.11 This is especially true for a foodborne pathogen that has demonstrated extraordinary environmental stability.12,13The viral genome of noroviruses encodes for two structural proteins, the major viral protein 1 (VP1) and minor VP2 capsid protein.12,14,15 The icosahedral capsid, enclosing the single-stranded RNA genome, consists of 90 dimers of VP1, and the exact VP1 sequence is linked to the virus's infection rate.16,17 For that reason, several genogroups and genotypes were categorised based on the VP1 sequence, including the prototypic GI.1 Norwalk strain, and novel emerging strains, such as the GII.17 Kawasaki strain responsible for recent outbreaks in Asia.16,18,19 The general structure of VP1 is subdivided into two domains; the shell (S) domain, facing the inside of the capsid, and the protruding (P) domain, which is outward-facing and further divided into two subdomains, P1 and P2.12,14,15 A comparison of the VP1 dimer of Norwalk and Kawasaki is shown in Fig. 1.
 | ||
| Fig. 1 Structural comparison of the norovirus dimers. The structures of the Norwalk and Kawasaki VP1 dimers look similar at first glance, but have specific differences when the structures are overlaid. The structure of Norwalk was obtained from the protein data bank (PDB) entry 1IHM,20 and Kawasaki model structures were generated using AlphaFold.21 Norwalk consists of 530 amino acid residues per protein chain, whereas Kawasaki comprises a total of 540 residues per chain. The sequence identity between the two VP1 is 46.60% as calculated by using the USCF Chimera tool.22 | ||
Native mass spectrometry (MS), which preserves non-covalent interactions between biomolecules during the experiment, is a powerful method for interrogating protein complexes in general, but virus capsid assembly in particular due to its ability to separate a large range of coexisting proteoforms and assembly intermediates and quantify each one.23 Native MS has been applied to study the stability of norovirus structures and intermediates during its assembly, revealing structural information about the viral capsid and its proteins.7,8 Using ion mobility (IM) spectrometry, where ions interact with an inert buffer gas and are separated as a consequence of their collisions with the gas, proteins with identical or similar mass-to-charge (m/z) ratio, including different conformational states of the same protein, can often be quantified separately, and the different drift times can be converted to collision cross sections (CCSs), which are effective sizes of the ions.24–26 In fact, a CCS depends on the 3D structure of an ion and can therefore be used for modelling purposes or to test structural hypotheses.27–31 By combining these two methods, valuable insights of the intensities for different m/z ratios from MS experiments are complemented with CCSs of the proteins in the gas phase. In collision induced unfolding (CIU), the velocity of the ions are increased prior to the IM by applying an acceleration voltage and making them collide with a gas inside the instrument results in an increased internal energy in the ions, which if high enough, causes the proteins to unfold.32 This is manifested as changes of the CCS at specific voltages, which reveal the transition voltages for the different unfolding intermediates, and provide information about a protein's conformation and stability.33,34 CIU can be used as a fingerprint technique to detect differences between protein variants, but also quantitatively to assess the stabilising effects of different ligands.35,36
Despite their many advantages, and despite potentially high mass- or CCS resolution, CIU and other IM-MS based methods cannot on their own readily give high-resolution information of the spatial structure of proteins, simply because MS data is not closely connected to the exact positions of atoms in a molecule, and a specific CCS is not uniquely given by a single structure. A computational approach to investigate and provide detailed information about the dynamics of gas-phase biomolecules is given by molecular dynamics (MD) simulations. MD allows for the observation of the dynamics of proteins on an atomistic level, thus making it an ideal tool to complement native MS with high-resolution structural information.37 Moreover, MD can be employed to simulate the unfolding of a protein. During a CIU experiment, the unfolding of a protein is triggered by the applied collision energy.33 A common approach to simulate the CIU of a protein using MD is to run sets of simulations over a range of different temperatures, where higher temperatures are used to mimic the activation in the experiments.34,38–40
Here we present CIU experiments complemented with MD simulations using temperature to emulate activation, where VP1 dimer structures of Norwalk and Kawasaki were unfolded in the gas phase in order to elucidate similarities and dissimilarities of the two norovirus strains, and to what extent differences can be probed with CIU experiments. The data was analysed not only to understand at what levels of activation unfolding takes place, but also to see how we can use MD to unravel the structural changes that take place. We find that the dimers of the two strains display different resilience to the activation and also that they differ in how their structures actually break.
Methods
Protein mass spectrometry analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 hours at 4 °C to get concentrated noroVLPs. Next, the concentrated noroVLPs were purified using CsCl equilibrium gradient ultracentrifugation at 35000 rpm (SW56 rotor, Beckman) for 18 hours at 4 °C. Finally, noroVLPs were pelleted for 2 hours at 40000 rpm (TLA55 rotor, Beckman) at 4 °C and dissolved in PBS buffer at pH 7.4.
000 MWCO, Sartorius, Göttingen, Germany). These buffer conditions were chosen to dissociate dimers reliably from the intact capsids. Calibrants for CCS calculations from traveling wave ion mobility mass spectrometry (TWIMS) were alcohol dehydrogenase (ADH) from yeast (Sigma Life Sciences), pyruvate kinase (PK) from rabbit muscle (Sigma Life Sciences) concanavalin A (Conc A) from jack bean (Sigma Life Sciences) and bovine serum albumin (BSA, Carl Roth). These were exchanged using Micro Bio-spin 6 Columns (6000 MWCO; Bio-Rad) at 1000 × g and 4 °C into 100 mM AmAc pH 7.4. Measurements were performed at approximately 14–26 μM VP1 and 20 μM calibrants. Native MS and travelling wave IMS (TWIMS) measurements were performed on a G1 Synapt HDMS (Waters, Manchester, UK and MS Vision, Almere, the Netherlands). Capillaries were made by pulling borosilicate glass tubes (inner diameter 0.68 mm, outer diameter 1.2 mm with filament, World Precision Instruments, Sarasota, FL, USA) using a two-step program in a micropipette puller (Sutter Instruments, Novato, CA, USA) with a squared box filament (2.5 × 2.5 mm). Subsequently, capillaries were gold-coated using a sputter coater (Safematic, Zizers, Switzerland, process pressure 5 × 10−2 mbar, process current 30.0 mA, coating time 100 s, 3 runs to vacuum limit 3 × 10−2 mbar argon).
000 rpm for 2 hours at 4 °C to get concentrated noroVLPs. Next, the concentrated noroVLPs were purified using CsCl equilibrium gradient ultracentrifugation at 35000 rpm (SW56 rotor, Beckman) for 18 hours at 4 °C. Finally, noroVLPs were pelleted for 2 hours at 40000 rpm (TLA55 rotor, Beckman) at 4 °C and dissolved in PBS buffer at pH 7.4.
000 MWCO, Sartorius, Göttingen, Germany). These buffer conditions were chosen to dissociate dimers reliably from the intact capsids. Calibrants for CCS calculations from traveling wave ion mobility mass spectrometry (TWIMS) were alcohol dehydrogenase (ADH) from yeast (Sigma Life Sciences), pyruvate kinase (PK) from rabbit muscle (Sigma Life Sciences) concanavalin A (Conc A) from jack bean (Sigma Life Sciences) and bovine serum albumin (BSA, Carl Roth). These were exchanged using Micro Bio-spin 6 Columns (6000 MWCO; Bio-Rad) at 1000 × g and 4 °C into 100 mM AmAc pH 7.4. Measurements were performed at approximately 14–26 μM VP1 and 20 μM calibrants. Native MS and travelling wave IMS (TWIMS) measurements were performed on a G1 Synapt HDMS (Waters, Manchester, UK and MS Vision, Almere, the Netherlands). Capillaries were made by pulling borosilicate glass tubes (inner diameter 0.68 mm, outer diameter 1.2 mm with filament, World Precision Instruments, Sarasota, FL, USA) using a two-step program in a micropipette puller (Sutter Instruments, Novato, CA, USA) with a squared box filament (2.5 × 2.5 mm). Subsequently, capillaries were gold-coated using a sputter coater (Safematic, Zizers, Switzerland, process pressure 5 × 10−2 mbar, process current 30.0 mA, coating time 100 s, 3 runs to vacuum limit 3 × 10−2 mbar argon).
Molecular dynamics simulations
The structures extracted from the solution simulations were not placed under PBC, as to mimic perfect vacuum conditions, and the steepest descent algorithm was used to minimise the energy of the system in vacuo. In order to emulate activation and potential unfolding of the protein structures, we simulated each of the seven replicas at multiple temperatures: {300, 400, …, 900} K. The specific temperatures were adjusted with the Berendsen thermostat over a short 10 ps MD simulation with a time step of 0.5 fs and a coupling constant of τ equal to 0.1 ps, where all bonds were constrained with the LINCS algorithm.60 Eventually, 30 ns of production simulations with a 2 fs time step were started to capture the dynamics of the virus dimers at the specified temperatures.
Results and discussion
Experimental unfolding patterns
The unfolding processes of both VP1 dimers were characterised using TWIMS. While the full capsid and the P dimer have both been subjected to ion mobility,8,64 this is the first time the VP1 homodimer has been in the focus. The unfolding pattern of VP1 of GI. 1 Norwalk and GII. 17 Kawasaki can be seen in the CIU fingerprints (Fig. 2). | ||
| Fig. 2 Ion mobility of Norwalk and Kawasaki VP1 dimers. A CIU fingerprints of 22+ charge states of GI.1 Norwalk and GII.17 Kawasaki were observed from 5 to 100 V. See Fig. S3 (ESI†) for other charge states. Norwalk and Kawasaki display distinct unfolding patterns, where Norwalk shows multiple conformational changes, suggesting higher susceptibility to collisional unfolding. Dashed lines indicate CIU50 for conformational transitions as identified with CIUSuite 2. B Representative mass spectra of both dimers with selected charge states annotated. CIU fingerprints and CIU50 for these charge states can be found in Fig. S2 and Table S1 (ESI†). | ||
Here, both dimers start with similar CCSs slightly above 6000 Å2. Both show an equal behaviour up until 40–60 V collision energy. Here, the CCS of Norwalk increases abruptly, suggesting a conformational transition. Using the CIUSuite 2 software43 to analyse the mobility data opened the possibility to calculate the CIU50 (Table S1, ESI†). This is done by assigning features to the data sets, representing distinct conformations (Fig. S1, ESI†) and subsequently determining the collisional voltage where 50 percent of transition to a larger conformation has occurred (Fig. S2, ESI†).43 For Norwalk the software calculates the first CIU50 at 51 V, which is in agreement with the CIU fingerprint (Fig. 2). The additional CIU50 for Norwalk are 71 V and 90 V. In comparison, Kawasaki has a transition around 70 V, which shows fine structure resulting in two transitions in the program. Norwalk going through multiple conformational changes, compared to a single one for Kawasaki, suggests a higher susceptibility of Norwalk to collisional unfolding and consequently less stability.
The experimental data here indicate strain-specific stability differences, which has been shown in previous works for the full capsids of the respective noroVLP. Kawasaki was observed to be more resistant to harsh environmental conditions than Norwalk.7,12 This may in part explain some differences between the unfolding patterns in the IM experiments, as harsh buffer conditions were used to disassemble the capsids. Interestingly, this also agrees with a recent study on the mechanical properties of noroVLP. Atomic force microscopy unveiled differences in the elasticity of the capsids, also demonstrating Kawasaki to be more stable than Norwalk.10 A caveat here is of course the difference from gas phase, where we test unfolding, and in-solution testing. In the gas phase, hydrophobic contacts are weakened and hydrophilic ones are strengthened. Nevertheless, our observation of strain-specific dimer stability differences is consistent with the notion that the stability of the full capsid originates in the stability of VP1, which is the protomer of the norovirus virion and the dominating protein in the natural capsid.
Theoretical CCS can be computed directly from MD trajectories, allowing for a connection between the high level of structural detail in MD and IM-MS experiments. To this end, we calculated the time evolution of the CCS for each temperature, averaged over the replicas, both for Norwalk and Kawasaki (Fig. 3). Temperatures up to 600 K did not result in any large increase of the CCS. In fact, initially, the CCSs decreased, most likely related to the structures being exposed to vacuum, resulting in side chains collapsing onto the protein surface.67 We have seen a similar trend in our recent study and in published literature, linked to vacuum-compaction being the reason for an initial decrease of the CCS.55,65,68,69 After 30 ns of MD simulation, both Norwalk and Kawasaki structures from 300 to 500 K arrive at similar CCSs (Fig. 3). These values are somewhat lower than what we see in the CIU experiments at low activation voltages (Fig. 2). The discrepancy might be explained by the tendency of the projection approximation, which IMPACT relies on, to slightly underestimate CCSs.70 We therefore assume that the structures are indeed largely intact in the CIU up to the first unfolding step, apart from minor changes mostly on the protein surface arising from the adaptation to a solvent-free environment.65,69
 | ||
| Fig. 3 Time-evolution of the CCS. The top panels shows the CCSs over time, averaged over replicas, for the Norwalk and Kawasaki at different temperatures, and the bottom panels show the CCSs averaged for each time slot and temperature together with the respective standard error of the mean. The individual CCS values shown in the bottom panels are shown in Table S2 (ESI†). Up to 600 K, the dimers resist unfolding. At 700 K Kawasaki retains a low CCS, but for Norwalk the CCS indicates unfolding. At temperatures above 700 K both protein variants unfold, but Norwalk to a higher degree than Kawasaki. Interestingly, Kawasaki indicates an initial drop of CCS, even at 900 K, whereas Norwalk shows an immediate increase of CCS at 900 K. | ||
At 600 K Kawasaki is still at a CCS similar to those at lower temperatures, whereas the CCS is notably higher and increasing for Norwalk at that temperature, indicating differences in stability for the two dimers. This becomes even more evident when comparing the trends for Norwalk and Kawasaki at 700 K. Whilst we can observe an elevated and increasing CCS-trend for Kawasaki, Norwalk displays a more pronounced CCS increase from the start and over the 30 ns of simulation. At 800 and 900 K, both dimers appear to unfold, again with Norwalk reaching higher CCSs than Kawasaki. The strain-specificity shown for the experimental CCS values described above are further in agreement with the MD simulations. The experimental CCS values show a similar pattern to the time-evolution of theoretical CCS calculations (Fig. 3), especially for 700 K and Norwalk.
For Norwalk and Kawasaki at temperatures up to 500 K, initially larger CCS values around 5200 Å2 are seen for time slot 1, decreasing over time for the remaining time slots and matching those published for MS experiments of the Norwalk capsid proteins8 and our experimental data (Fig. 2). At 700 K, Kawasaki demonstrates a slight CCS increase of approximately 1%, whereas the CCS of Norwalk increases from time slot 1 to 6 by roughly 10% at the same temperature. At temperatures 800 and 900 K, both Norwalk and Kawasaki seem to undergo unfolding based on the rapid increase of the average CCS over the time slots. Interestingly, Norwalk increases by approximately 17% from time slot 1 to 6, both at 800 and 900 K, with 900 K data showing overall larger CCS values as during 800 K simulations. The Kawasaki dimer displays a 17% increase of CCS at 800 K as well, but from a lower starting value in time slot 1, and by approximately 23% when comparing the CCS values at 900 K for time slot 1 to time slot 6.
Overall, splitting the MD simulation data into time slots, and comparing the averaged CCS values for those time slots between each other could funnel the information of the underlying dynamics in a more comprehensive way. Moreover, this allows us to view the structures in the time slots as series of states the protein structures pass through as they unfold, with the potential to reveal an unfolding mechanism to compare to experiments.
Representative structures and comparison of unfolding time slots
With the simulation data split into six time slots, we clustered all structures that would fall within such a time slot in order to obtain a single representative structure for each slot, which we use to track and visualise the unfolding process. The structures for each temperature are presented in Fig. S4–S17 (ESI†). The RMSDs for the representative structures were calculated (Table 1) using the structure for time slot 1 as reference, which allows for an estimation of the deviation between the structures over the course of the simulations.| Time slots | RMSD per time slot for Norwalk relative to time slot 1 [Å] | ||||||
|---|---|---|---|---|---|---|---|
| 300 K | 400 K | 500 K | 600 K | 700 K | 800 K | 900 K | |
| Time slot 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Time slot 2 | 1.24 | 3.14 | 4.43 | 6.09 | 9.56 | 28.64 | 43.39 |
| Time slot 3 | 2.60 | 2.32 | 4.58 | 6.30 | 13.60 | 32.40 | 51.09 |
| Time slot 4 | 1.47 | 3.17 | 4.83 | 6.55 | 14.30 | 37.84 | 51.39 |
| Time slot 5 | 2.54 | 3.19 | 4.74 | 6.74 | 16.65 | 41.54 | 55.76 |
| Time slot 6 | 2.60 | 3.23 | 4.75 | 7.09 | 22.97 | 44.96 | 64.66 |
| Time slot 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Time slot 2 | 2.05 | 3.00 | 2.21 | 4.47 | 4.29 | 10.10 | 20.21 |
| Time slot 3 | 1.26 | 2.94 | 2.28 | 4.71 | 5.84 | 14.91 | 31.90 |
| Time slot 4 | 2.10 | 3.02 | 4.14 | 4.87 | 7.38 | 16.99 | 33.57 |
| Time slot 5 | 2.10 | 3.05 | 4.05 | 4.79 | 7.53 | 20.47 | 37.56 |
| Time slot 6 | 2.06 | 3.11 | 4.68 | 4.89 | 7.73 | 20.96 | 59.29 |
From the CCS calculations for 300 to 500 K, Norwalk dimers are seemingly stable and do not change their CCS much. This is confirmed by looking at the representative structures for these temperatures, shown in Fig. 4 and Fig. S4–S6 (ESI†). The RMSD calculations further support these observations, where the deviations increase with higher temperatures (Table 1). Especially the RMSD values for 500 K show that the structures corresponding to time slot 2 to 6 are deviating by at least 4 Å from the representative structure for time slot 1, indicating that more and more differences between these structures exist. Whilst the conformation of the Norwalk structure for time slot 1 at 600 K (Fig. S7, ESI†) resembles those at lower temperatures, a clear difference can be seen compared to time slot 2, where the structure of chain A is seemingly different as that of chain B. With an RMSD of 6.09 Å between these time slots, the deviation is most likely associated with parts of chain A starting to protrude from the rest of the structure. However, unfolding seems to not take place here on these time scales, as the deviations of the remaining time slots do not reflect large parts of the structure to change, which can also be seen looking at the respective structures in Fig. S7 (ESI†), and supported by the calculated CCSs. Yet again, 700 K seems to mark a limit for when the Norwalk dimer succumbs to the unfolding forces. Inspecting the structures in Fig. S8 (ESI†), already time slot 1 shows signs of unfolding, restricted to the same extruded area of chain A seen at 600 K (Fig. S7, ESI†). Starting from the termini, the structures show a gradual unfolding of the Norwalk dimer, explaining the steep increase of the RMSD from time slot 1, where after 10 ns of simulation, chain B began to unfold as well. Overall, the unfolding of the dimer is shown here to occur evenly over the simulation time. At 800 K (Fig. S9, ESI†), the Norwalk dimers start with an overall shape that resembles for the most part that of the native dimer, but from time slot 2 and onwards the structures are unfolded with no similarity of the original Norwalk dimer. With the RMSD ranging up to above 40 Å, the unfolding process seems to progress rapidly, which explains the observed CCS trends. At 900 K, unfolding occurs from the beginning of the simulations, where the structures are seemingly being ‘pulled’ apart as time progresses (Fig. S10, ESI†). From the large deviations presented in Table 1, together with the CCS trends at 900 K, the unfolding of Norwalk is shown to be happening promptly and chaotically, with the only clearly discernible mechanism is an unraveling starting at the termini.
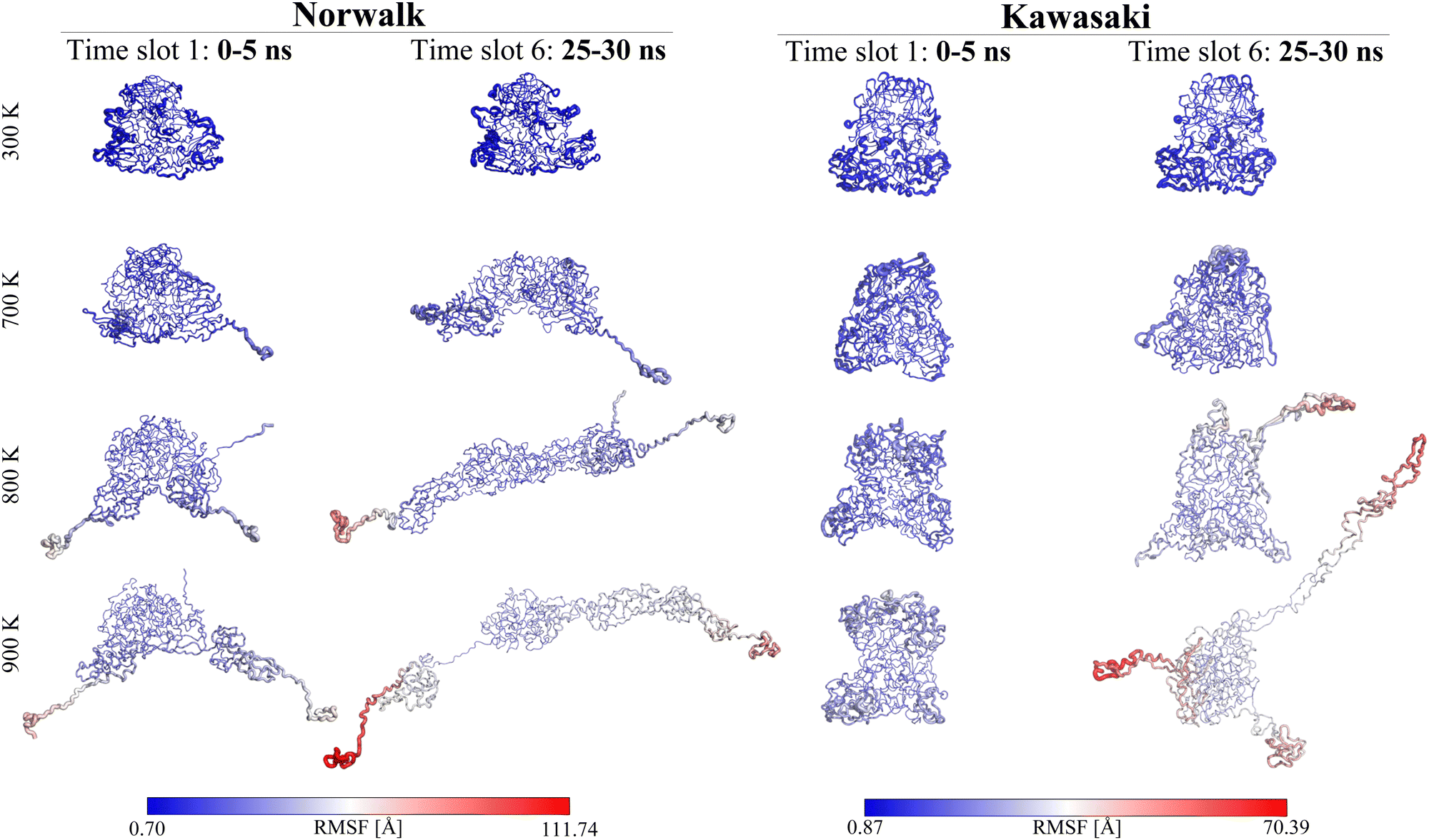 | ||
| Fig. 4 RMSF per residue projected on selected structures. The RMSF calculations were projected onto the selected representative structures, using the ribbon thickness and colour to show RMSF values. At 300 K, Kawasaki shows almost identical representative structures at 300 K for the first and last 5 ns of MD simulations, whereas Norwalk is showing slight distortions for time slot 6. At temperatures 700+ K, unfolding can be observed for Norwalk, whilst Kawasaki still shows a globular-like shape. Moreover, the representative structures suggest different regions within the proteins the unfolding to occur from. Norwalk displays unfolding to be most likely originating from the termini, with the N-terminus starting to unfold at 700 K. For the other dimer, Kawasaki, 800 K is required to induce unfolding, starting from residues within the P2-domain of the protein. For more details on all representative structures, please see Fig. S4–S17 (ESI†). | ||
The CCSs for the Kawasaki dimer structures are similar between 300 and 600 K (Fig. 3 and Table S2, ESI†). A similar observation can be made by looking at the RMSDs per time slot (Table 1). We can rationalise this by comparing the representative structures at those temperatures (Fig. S11 and S14, ESI†). Here, whilst a small RMSD increase is seen from 300 to 500 K, the relatively small values most likely indicate minor structural deviations. Whilst the structures for 600 K demonstrate similar shapes and sizes, the RMSD values show deviations from the time slot 1 structure of 4.47 Å or more, indicating increasingly larger differences than for the lower temperatures. Larger structural differences are eventually seen for the representative structures at 700 K (Fig. S15, ESI†), which is further reflected by the RMSD increasing to 7.73 Å. As such, 700 K appears to be the threshold for unfolding in our simulations. Looking in more detail at the structures at 800 K (Fig. S16, ESI†), where time slot 1 displays a structure that resembles the native dimer structure the most, unfolding of the Kawasaki dimer happens from time slot 2 and onwards, seemingly starting from chain A in time slot 2, and for chain B in later time slots. The RMSD increases throughout the time slots at 800 K, up to approximately 21 Å. Overall, the structures for 800 K suggest unfolding over 30 ns of MD simulation, and moreover, that the unfolding is seemingly starting from specific areas of the protein, whilst the core of the protein retains a more globular and compact, yet disorganised structure. At the highest simulated temperature, 900 K, the representative structures and the RMSDs between them suggest severe structural rearrangements (Fig. S17, ESI†). Whilst initially the dimers still seem to have a globular-like structure, for time slot 2 to 5, not only is the unfolding visualised, but one can observe large perturbations of the structure stepping from one time slot to the next. With the calculated deviations between the representative structures starting from 20.21 Å between time slot 1 and 2, and ranging up to almost 60 Å at the end of the MD simulations, showing drastic unfolding of the dimer at 900 K.
Interrogating the unfolding pattern tracking residue dynamics and distances
Assigning a representative structure per time slot for each temperature data set, together with the initial CCS calculation, allowed us to present the dynamics of the dimers at the specified temperatures as a series of potential unfolding states. Here, whilst the dimers are shown to be rather stable at temperatures ranging from 300 to 600 K, the higher temperatures, especially 800 and 900 K, are seemingly inducing enough energy to cause the structural unfolding of VP1. The propagation of the overall dimer shape could be inferred from looking at the representative structures across the time slots and temperatures, but the dynamics at a finer level is harder to discern. To further pinpoint such details, the RMSF was calculated for each protein chain (Fig. S18–S31, ESI†), and compared together with distance and contact maps, all calculated as averages for each time slot (Fig. S32–S45, ESI†).Looking at the RMSFs for Norwalk at 300 to 500 K (Fig. S18–S20, ESI†), one can observe similarities between the time slots. Whilst the RMSFs seem to increase with higher temperature, the values likely reflect an overall increase in mobility, especially around the termini, rather than unfolding. This is further supported by the almost identical patterns of the distance and contact maps for all time slots (Fig. S32–S34, ESI†). At 600 K, the majority of the protein is still displaying a similar RMSF trend over all time slots, both for chain A and B (Fig. S21, ESI†). Noteworthy are the high RMSF values for the N-termini of both chains, whereas the C-terminus of chain A is seemingly more mobile at 600 K than for chain B, displayed as well in the distance maps (Fig. S35, ESI†). The representative structure for Norwalk time slot 2 at 600 K (Fig. S7, ESI†) shows an extruded part of the protein chain, which is given by the N-terminus, and therefore could indicate that area to be a potential starting point of the unfolding process. As the N-terminus of chain B shows basically an identical RMSF over all time slots, we could conclude that chain B, whilst being highly mobile, is still stable enough and does not start to unfold at 600 K. At 700 K, the RMSF trends for the time slots are starting to differentiate from each other (Fig. S22, ESI†), furthermore confirming, as suggested by the CCS and RMSD values at that temperature, that 700 K is the threshold temperature to induce unfolding for Norwalk. In detail, whilst both termini of chain A are still displaying large RMSF values, we can see other parts of the protein becoming more mobile as well. A prominent peak is given for residues between approximately Ala120 and Ile139, comprising a loop between β-strandE and α-helix3 within the S-domain of the protein chain.20 This area is increased in mobility the most over time, as compared to the rest of the protein (excluding termini), which is further shown in the distance and contact maps (Fig. S36, ESI†). Towards 800 and 900 K, disturbances in the Norwalk dimers become more evident. The RMSFs in Fig. S23 (ESI†) exhibit, next to the prominent peaks as observed already at 700 K, further differences from one time slot to the next. High RMSF values are seen throughout each chain, where additional peaks, for example shown around residue Asn300 within the P2-domain, are becoming more prominent as the simulation progresses. The distance maps in Fig. S37 (ESI†) provide further information of the unfolding, where the distances increase whilst the contact patterns become progressively more diffuse. Moreover, differences in the unfolding dynamics of the chains can be seen, where for example the distances between residues of chain B to each other are not increasing as much as for chain A, potentially indicating a more compact conformation of chain B at 800 K than chain A, whilst residues within chain A are increasingly departing from each other. At 900 K, the RMSFs suggest large distortions and mobility throughout the protein chains (Fig. S24, ESI†), with chain A and B progressively being more “chaotic” and dissimilar. High fluctuations are recorded for several areas of the protein chains, where interestingly the N-terminus of chain A actually shows smaller values than for 800 K, whilst the same residues in chain B are reaching RMSF values above 70 Å. Moreover, as shown in Fig. S38 (ESI†), where unfolding is already present for time slot 1, the distances and contacts for time slot 3 to 6 are very similar, which could indicate that 900 K is inducing unfolding too rapidly and highly incoherently for it to provide useful information to track the unfolding process with any confidence.
The Kawasaki data shows similar RMSFs from 300 K to 500 K (Fig. S25–S27, ESI†), as was observed for Norwalk as well. Together with the distance and contact maps (Fig. S39–S41, ESI†), this suggests similar underlying dynamics across these temperatures. Whilst a slight increase in RMSF can be seen in at 600 K, the distances indicate no obvious differences of the dynamics between the six time slots, and as such complement what was seen for the average CCS and RMSD at 600 K. Moreover, both chains are still exhibiting similar dynamics throughout, even at 600 K. At 700 K, we can see that the dynamics of the residues are increasing over the MD simulations (Fig. S29, ESI†). Especially at later simulation times, for time slot 5 to 6, peaks throughout the protein chains reach up to 20 Å. Interestingly, the termini show relatively low RMSF values, a very different observation as compared to Norwalk, where the termini, especially the N-termini, are displaying high mobility. Turning to the contact maps for Kawasaki at 700 K in (Fig. S43, ESI†), we can further see that the structures, whilst being highly mobile, are seemingly able to withstand unfolding, as no obvious increase in distance maps can be seen. Moreover, the contact patterns for the time slots at 700 K are displaying similar patterns, indicating the absence of induced unfolding. At 800 K however, the CCS of Kawasaki increases, accompanied by RMSF increases for that temperature (Fig. S30, ESI†). The termini show high RMSFs that increase over time, with new parts of the protein becoming progressively more dynamic. Especially parts of the P2-domain, around residue Ala350, present a prominent, increasing peak over time. That area is seen to unfold in the representative Kawasaki structures for time slot 2 and above at 800 K, which is reflected by the increasing distances involving the corresponding residues (Fig. S44, ESI†). We described a similar observation in a recent study, where we experimentally investigated different P dimers with hydrogen–deuterium exchange together with MD simulations.71 Amongst the investigated proteins was the P dimer of the Kawasaki strain, and the results suggested increased dynamics both during the experiments as well as during the simulations for the same area we observe the unfolding to start from in this investigation. Interestingly, Kawasaki is seemingly unfolding from a completely different part of the protein than Norwalk, which appears to start unfolding from the N-termini already at 700 K. The Kawasaki contact patterns fade for higher temperatures, suggesting gradual unfolding, with structural differences within or between replicas, and a more subtle trend than Norwalk. Eventually, at the highest temperature of 900 K, two areas of the structure have large RMSFs (Fig. S31, ESI†). They comprise largely residues of the S- and P2-domains, spanning residues from approximately Ala100 to Cys200, as well as Trp300 till Phe400, which were mobile also for Norwalk. Interestingly, the latter part includes a short loop for which strain-specific dynamics has been observed, and where glycan binding is known to take place as part of the infection process.71 The RMSF increases over time, per time slot, and reaches values above 40 Å, which is evidently linked to large distances being observed in the corresponding contact maps as a consequence of the unfolding (Fig. S45, ESI†). However, the distance maps of Kawasaki appear to suggest a more gradual unfolding process, as the distances increase steadily, yet for the same specific parts of the protein, rather than all over the structure.
Conclusions
In this study we investigated how we can use CIU and MD to find and explain differences in dynamics and stability between different variants of the same protein. Specifically, we interrogated the VP1 dimers from two norovirus strains with minor structural differences, but with decidedly distinct physical properties on the capsid level. The CIU experiments revealed differences in the unfolding dynamics, with the Kawasaki variant not only requiring more activation to start unfolding, but also going through fewer conformational transitions overall. MD simulations of the same proteins, where elevated temperatures mimicked the activation in the experiments, gave insights into the conformational changes the protein underwent during the unfolding. We employed a decomposition of our MD simulations into time slots, which we think can be developed further, to gain useful information for and from CIU experiments, utilising the mutually complementary nature of MD and IM-MS. First, we note that a high temperature is necessary to induce unfolding on relative short timescales of 30 ns. Moreover, the temperature thresholds at which the first instances of unfolding were observed were different for the two dimers, consistent with the different profiles obtained from CIU. For example, for Norwalk, we have seen that 700 K is enough to induce unfolding, and higher temperatures make it progressively more chaotic, which makes it difficult to pinpoint and describe the full pathway. Kawasaki requires 800 K and above to undergo unfolding, as is suggested by the representative structures and RMSF calculation (Fig. 4), whereas at 700 K, Kawasaki is stable enough to withstand unfolding. In addition to the caveats outlined earlier, it is important to stress that classical force fields might not represent high (or low) temperatures accurately, which entails that the exact dependence on temperature can have poor correspondence to actual temperatures in an experiment. Compounding this, we use very high temperatures to compensate for the short time scales compared to the millisecond-timescales in IM-MS, which risk affecting the apparent stability of unfolding intermediates, and the unfolding pathways thus risk becoming artificially “smooth”. We also remind ourselves that as proteins unfold in experiment there is the possibility of scrambling of protons across the structure, which is not accommodated in our simulations, which also limits how confidently we can draw conclusions about the most unfolded structures. This is particularly true for multimeric proteins such as our dimers, where charges are known to partition asymmetrically when the proteins dissociate,72 typically leading to an asymmetric dissociation where one highly charged subunit unfolds completely, leaving the rest of the complex more intact and at a lower m/z ratio. This complicates the correspondence between simulations and experiments further. It is challenging to incorporate such dynamics in MD. Important advances have been made recently,73,74 but it remains a difficult endeavour and to our knowledge there is to date no established means for accomplishing it that is thermodynamically let alone kinetically accurate. Notwithstanding such obstacles, the simulations should provide a qualitatively correct view of the unfolding, at least for moderately unfolded states, especially with respect to the weakest points of the proteins and the relative stability between the variants.With the two dimers having similar sizes and shapes, we see that their structural responses to the activation required to induce unfolding are different. The starting point of the unfolding process, for example, differs between Norwalk and Kawasaki; Norwalk is starting to unfold starting from the N-termini, whereas Kawasaki is unfolding from parts of the P2-domain. In a recent study, we have shown that residues in that part of Kawasaki, around residue Ala350, are displaying large dynamics,71 a similar observation we make during the unfolding simulated here. It is interesting to see how features of those in vitro dynamics carry over to the gas phase. The importance of the N-termini for capsid stability has been previously investigated.7,9 A higher instability of the critical N-terminus may also influence the overall decreased stability of the Norwalk capsid compared to Kawasaki as shown in other studies.7,10,12 In light of the new data presented here together with earlier results, the VP1 dimer is becoming the likely origin of the strain-dependent distinction in physical properties of the fully assembled capsids. To our knowledge, we present here the first study of combining both experimental and theoretical research on the unfolding of norovirus dimers, providing valuable insights into their stability and details about their behaviour and thus adding to the compounding evidence on previously identified structural linchpins. And as such we also demonstrate how CIU and MD can be combined to shed more light on protein systems than either technique in isolation. We firmly believe that studies of this kind will gradually advance our understanding of how proteins respond to the experimental conditions and how they can be experimentally interrogated, and that future advances in computational techniques and algorithms as well as in experimental methods and instrumentation will accelerate that process.
Author contributions
Maxim N. Brodmerkel: conceptualization, methodology, formal analysis, investigation, validation, writing – original draft, visualization. Lars Thiede: methodology, formal analysis, investigation, writing – editing. Emiliano De Santis: methodology, writing – review & editing. Charlotte Uetrecht: writing – review & editing, supervision, funding acquisition. Carl Caleman: conceptualization, writing – review & editing, supervision, funding acquisition. Erik G. Marklund: conceptualization, resources, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX), the National Academic Infrastructure for Supercomputing in Sweden (NAISS), former Swedish National Infrastructure for Computing (SNIC), and the provided computational resources under projects SNIC 2022-22-854, 2022-22-925, 2022-22-947, 2022-5-415 and 2022-23-57. EGM and CC are supported by Project grants from the Swedish Research Council (2020-04825 and 2018-00740). EDS, CU, CC and EGM acknowledge support from a Röntgen Ångström Cluster grant provided by the Swedish Research Council and the Bundesministerium für Bildung und Forschung (2021-05988, 05K22PSA). MNB, EDS, CU, CC and EGM acknowledge support from the MS SPIDOC consortium, funded by the European Horizon 2020 research and innovation programme under grant agreement 801406. CU and LT acknowledge funding by EIC Pathfinder Open Ariadne-Vibe (964553). CC further acknowledges the Helmholtz Association through the Center of Free-electron Laser Science at DESY. Lastly, we thank Kristina Lorenzen and the XBI BioLab for instrument access.Notes and references
- D. S. Goodsell and A. J. Olson, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 105–153 CrossRef CAS PubMed.
- G. K. A. Hochberg, Y. Liu, E. G. Marklund, B. P. H. Metzger, A. Laganowsky and J. W. Thornton, Nature, 2020, 588, 503–508 CrossRef CAS PubMed.
- N. F. Steinmetz, T. Lin, G. Lomonossoff and J. Johnson, Structure-based engineering of an icosahedral virus for nanomedicine and nanotechnology, Springer, 2009 Search PubMed.
- E. J. Elliott, BMJ, 2007, 334, 35–40 CrossRef PubMed.
- R. I. Glass, U. D. Parashar and M. K. Estes, N. Engl. J. Med., 2009, 361, 1776–1785 CrossRef CAS PubMed.
- N. Zachos, in Viral Gastroenteritis, ed. L. Svensson, U. Desselberger, H. B. Greenberg and M. K. Estes, Academic Press, Boston, 2016, 1.1, pp. 1–21.
- G. K. Shoemaker, E. van Duijn, S. E. Crawford, C. Uetrecht, M. Baclayon, W. H. Roos, G. J. Wuite, M. K. Estes, B. V. Prasad and A. J. Heck, Mol. Cell. Proteomics, 2010, 9, 1742–1751 CrossRef CAS PubMed.
- C. Uetrecht, I. M. Barbu, G. K. Shoemaker, E. Van Duijn and A. J. Heck, Nat. Chem., 2011, 3, 126–132 CrossRef CAS PubMed.
- R. Pogan, V. U. Weiss, K. Bond, J. Dülfer, C. Krisp, N. Lyktey, J. Müller-Guhl, S. Zoratto, G. Allmaier and M. F. Jarrold, et al. , Vaccines, 2020, 9, 8 CrossRef PubMed.
- Y. Feng, R. Pogan, L. Thiede, J. Müller-Guhl, C. Uetrecht and W. H. Roos, Viruses, 2023, 15, 1482 CrossRef CAS PubMed.
- C. Scheller, F. Krebs, R. Minkner, I. Astner, M. Gil-Moles and H. Wätzig, Electrophoresis, 2020, 41, 1137–1151 CrossRef CAS PubMed.
- R. Pogan, J. Dülfer and C. Uetrecht, Curr. Opin. Virol., 2018, 31, 59–65 CrossRef CAS PubMed.
- L. Barclay, G. Park, E. Vega, A. Hall, U. Parashar, J. Vinjé and B. Lopman, Clin. Microbiol. Infect., 2014, 20, 731–740 CrossRef CAS PubMed.
- M. E. Hardy, FEMS Microbiol. Lett., 2005, 253, 1–8 CrossRef CAS PubMed.
- G. I. Parra, Virus Evol., 2019, 5, vez048 CrossRef PubMed.
- E. Robilotti, S. Deresinski and B. A. Pinsky, Clin. Microbiol. Rev., 2015, 28, 134–164 CrossRef CAS PubMed.
- C. Preeti, D. G. Miranda, I. P. Gabriel, C.-W. C. Martin, G. Kim, M. Vito, W. Qiuhong, A. W. Peter, K. Kazuhiko and V. Harry, et al. , J. Gen. Virol., 2019, 100, 1393–1406 CrossRef PubMed.
- K. Bányai, M. K. Estes, V. Martella and U. D. Parashar, The Lancet, 2018, 392, 175–186 CrossRef PubMed.
- Y. Liao, L. Xue, J. Gao, Y. Zuo, Y. Liang, Y. Jiang, W. Cai, J. Yang, J. Zhang and Y. Ding, et al. , Gut Pathog., 2022, 14, 31 CrossRef CAS PubMed.
- B. V. Prasad, M. E. Hardy, T. Dokland, J. Bella, M. G. Rossmann and M. K. Estes, Science, 1999, 286, 287–290 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Ždek and A. Potapenko, et al. , Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- C. Uetrecht and A. J. Heck, Angew. Chem., Int. Ed., 2011, 50, 8248–8262 CrossRef CAS PubMed.
- F. Lanucara, S. W. Holman, C. J. Gray and C. E. Eyers, Nat. Chem., 2014, 6, 281–294 CrossRef CAS PubMed.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. Benesch, Structure, 2015, 23, 791–799 CrossRef CAS PubMed.
- V. Gabélica and E. Marklund, Curr. Opin. Chem. Biol., 2018, 42, 51–59 CrossRef PubMed.
- E. Mack, J. Am. Chem. Soc., 1925, 47, 2468–2482 CrossRef CAS.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS PubMed.
- M. Kaldmäe, C. Sahin, M. Saluri, E. G. Marklund and M. Landreh, Prot. Sci., 2019, 28, 1024–1030 CrossRef PubMed.
- M. Landreh, C. Sahin, J. Gault, S. Sadeghi, C. L. Drum, P. Uzdavinys, D. Drew, T. M. Allison, M. T. Degiacomi and E. G. Marklund, Anal. Chem., 2020, 92, 12297–12303 CrossRef CAS PubMed.
- S. B. A. Turzo, J. T. Seffernick, A. D. Rolland, M. T. Donor, S. Heinze, J. S. Prell, V. H. Wysocki and S. Lindert, Nat. Commun., 2022, 13, 4377 CrossRef CAS PubMed.
- K. B. Shelimov, D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1997, 119, 2240–2248 CrossRef CAS.
- S. M. Dixit, D. A. Polasky and B. T. Ruotolo, Curr. Opin. Chem. Biol., 2018, 42, 93–100 CrossRef CAS PubMed.
- C. Eldrid, T. Cragnolini, A. Ben-Younis, J. Zou, D. P. Raleigh and K. Thalassinos, Anal. Chem., 2022, 94, 16113–16121 CrossRef CAS PubMed.
- A. Laganowsky, E. Reading, T. M. Allison, M. B. Ulmschneider, M. T. Degiacomi, A. J. Baldwin and C. V. Robinson, Nature, 2014, 510, 172–175 CrossRef CAS PubMed.
- D. D. Vallejo, C. K. Jeon, K. F. Parson, H. R. Herderschee, J. D. Eschweiler, D. I. Filoti and B. T. Ruotolo, Anal. Chem., 2022, 94, 6745–6753 CrossRef CAS PubMed.
- E. G. Marklund and J. L. Benesch, Curr. Opin. Struct. Biol., 2019, 54, 50–58 CrossRef CAS PubMed.
- Z. Hall, A. Politis, M. F. Bush, L. J. Smith and C. V. Robinson, J. Am. Chem. Soc., 2012, 134, 3429–3438 CrossRef CAS PubMed.
- S.-H. Chen and D. H. Russell, J. Am. Soc. Mass Spectrom., 2015, 26, 1433–1443 CrossRef CAS PubMed.
- C. E. Bartman, H. Metwally and L. Konermann, Anal. Chem., 2016, 88, 6905–6913 CrossRef CAS PubMed.
- G. Hansman, K. Natori, T. Oka, S. Ogawa, K. Tanaka, N. Nagata, H. Ushijima, N. Takeda and K. Katayama, Arch. Virol., 2005, 150, 21–36 CrossRef CAS PubMed.
- G. S. Hansman, H. Saito, C. Shibata, S. Ishizuka, M. Oseto, T. Oka and N. Takeda, J. Clin. Microbiol., 2007, 45, 1347–1349 CrossRef CAS PubMed.
- D. A. Polasky, S. M. Dixit, S. M. Fantin and B. T. Ruotolo, Anal. Chem., 2019, 91, 3147–3155 CrossRef CAS PubMed.
- M. T. Marty, A. J. Baldwin, E. G. Marklund, G. K. A. Hochberg, J. L. P. Benesch and C. V. Robinson, Anal. Chem., 2015, 87, 4370–4376 CrossRef CAS PubMed.
- K. Thalassinos, M. Grabenauer, S. E. Slade, G. R. Hilton, M. T. Bowers and J. H. Scrivens, Anal. Chem., 2009, 81, 248–254 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- V. Gabelica, A. A. Shvartsburg, C. Afonso, P. Barran, J. L. Benesch, C. Bleiholder, M. T. Bowers, A. Bilbao, M. F. Bush and J. L. Campbell, et al. , Mass Spectrom. Rev., 2019, 38, 291–320 CrossRef CAS PubMed.
- Y. Matsushima, M. Ishikawa, T. Shimizu, A. Komane, S. Kasuo, M. Shinohara, K. Nagasawa, H. Kimura, A. Ryo and N. Okabe, et al. , Eurosurveillance, 2015, 20, 1–12 CrossRef PubMed.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- E. G. Marklund, D. S. Larsson, D. van der Spoel, A. Patriksson and C. Caleman, Phys. Chem. Chem. Phys., 2009, 11, 8069–8078 RSC.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- K. A. Feenstra, B. Hess and H. J. Berendsen, J. Comput. Chem., 1999, 20, 786–798 CrossRef CAS PubMed.
- A. Patriksson, E. Marklund and D. van der Spoel, Biochemistry, 2007, 46, 933–945 CrossRef CAS PubMed.
- T. Mandl, C. Östlin, I. E. Dawod, M. N. Brodmerkel, E. G. Marklund, A. V. Martin, N. Timneanu and C. Caleman, J. Phys. Chem. Lett., 2020, 11, 6077–6083 CrossRef CAS PubMed.
- M. N. Brodmerkel, E. De Santis, C. Uetrecht, C. Caleman and E. G. Marklund, Curr. Res. Struct. Biol., 2022, 4, 338–348 CrossRef CAS PubMed.
- M. N. Brodmerkel, E. De Santis, C. Caleman and E. G. Marklund, Protein J., 2023, 1–14 Search PubMed.
- A. Sinelnikova, T. Mandl, H. Agelii, O. GrÅnäs, E. G. Marklund, C. Caleman and E. De Santis, Biophys. J., 2021, 120, 3709–3717 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. J. Berendsen, J. v Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Domanski, D. L. Dotson, S. Buchoux, I. M. Kenney and O. Beckstein, Proceedings of the 15th Python in Science Conference, 2016, pp. 98–105 Search PubMed.
- J. Z. Bereszczak, I. M. Barbu, M. Tan, M. Xia, X. Jiang, E. van Duijn and A. J. Heck, J. Struct. Biol., 2012, 177, 273–282 CrossRef CAS PubMed.
- D. van der Spoel, E. G. Marklund, D. S. D. Larsson and C. Caleman, Macromol. Biosci., 2011, 11, 50–59 CrossRef CAS PubMed.
- T. M. Allison, P. Barran, S. Cianférani, M. T. Degiacomi, V. Gabelica, R. Grandori, E. G. Marklund, T. Menneteau, L. G. Migas, A. Politis, M. Sharon, F. Sobott, K. Thalassinos and J. L. P. Benesch, Anal. Chem., 2020, 92, 10872–10880 CrossRef CAS PubMed.
- K. Breuker and F. W. McLafferty, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18145–18152 CrossRef CAS PubMed.
- M. Landreh, E. G. Marklund, P. Uzdavinys, M. T. Degiacomi, M. Coincon, J. Gault, K. Gupta, I. Liko, J. L. P. Benesch, D. Drew and C. V. Robinson, Nat. Commun., 2017, 8, 13993 CrossRef CAS PubMed.
- A. D. Rolland and J. S. Prell, Trends Anal. Chem., 2019, 116, 282–291 CrossRef CAS PubMed.
- J. L. P. Benesch and B. T. Ruotolo, Curr. Opin. Struct. Biol., 2011, 21, 641–649 CrossRef CAS PubMed.
- J. Dülfer, H. Yan, M. N. Brodmerkel, R. Creutznacher, A. Mallagaray, T. Peters, C. Caleman, E. G. Marklund and C. Uetrecht, Molecules, 2021, 26, 2125 CrossRef PubMed.
- J. C. Jurchen and E. R. Williams, J. Am. Chem. Soc., 2003, 125, 2817–2826 CrossRef CAS PubMed.
- L. Konermann, J. Phys. Chem. B, 2017, 121, 8102–8112 CrossRef CAS PubMed.
- R. Marchese, R. Grandori, P. Carloni and S. Raugei, J. Am. Soc. Mass Spectrom., 2012, 23, 1903–1910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06344e |
| This journal is © the Owner Societies 2024 |