
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature dissociation of CO2 molecules on vicinal Cu surfaces†
Takanori
Koitaya
 *a,
Yuichiro
Shiozawa
b,
Yuki
Yoshikura
b,
Kozo
Mukai
b,
Shinya
Yoshimoto
b and
Jun
Yoshinobu
*b
*a,
Yuichiro
Shiozawa
b,
Yuki
Yoshikura
b,
Kozo
Mukai
b,
Shinya
Yoshimoto
b and
Jun
Yoshinobu
*b
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-Ku, Kyoto 606-8502, Japan. E-mail: koitaya.takanori.5j@kyoto-u.ac.jp
bThe Institute for Solid State Physics, The University of Tokyo, 5-1-5, Kashiwanoha, Kashiwa, Chiba 277-8581, Japan. E-mail: junyoshi@issp.u-tokyo.ac.jp
First published on 6th March 2024
Abstract
The reaction of carbon dioxide on the vicinal Cu surfaces at low temperatures was investigated by infrared reflection absorption spectroscopy, scanning tunneling microscopy, X-ray photoelectron spectroscopy, and quadrupole mass spectrometry. Dissociation of CO2 molecules into CO on the Cu(997) and Cu(977) surfaces was observed at temperatures between 80 K and 90 K, whereas it did not occur on Cu(111) under a similar condition. CO and physisorbed CO2 were the main adsorbates during the reaction. In contrast, the amount of atomic oxygen on the surface was small. The dissociation of CO2 was promoted by the small amount of oxygen produced by the CO2 dissociation on the Cu surfaces. This leads to the induction period in the CO2 reaction; the initial reaction rate on the clean Cu surfaces was low, and the coadsorbed oxygen enhanced the dissociation reactivity of CO2. Mass analysis of desorption species during the reaction revealed that the observed CO formation on the vicinal Cu surface is mainly caused by an oxygen-exchange reaction with residual CO in an ultra-high vacuum chamber.
Introduction
Catalytic transformation of carbon dioxide is a challenging topic in C1 chemistry.1,2 The interaction of CO2 with the catalytic surfaces is essential for the activation of CO2 molecules due to its chemical inertness. Thus, the adsorption and reaction of CO2 on metal and metal-oxide surfaces have been intensively studied to reveal the nature of the interaction between adsorbed CO2 and solid surfaces.3–6CO2 adsorption on the metallic copper surfaces is an important topic for methanol synthesis from CO2 and H2 on Cu/ZnO catalysts.7 The flat single-crystal Cu surfaces are less reactive for the CO2 reaction. Under an ultrahigh-vacuum (UHV) condition, only physisorbed molecules are observed at temperatures below 80 K.3,8–10 In contrast to the flat surfaces, higher reactivity of vicinal or polycrystalline Cu surfaces for CO2 has been reported experimentally. The chemisorbed (bent) CO2 molecules were observed by XPS on the polycrystalline Cu,11 and dissociation into CO was detected by temperature-programmed desorption (TPD) on Cu(332)12 and Cu(311)13 surfaces at temperatures between 95 and 200 K. However, there is a report that adsorbed CO2 molecules on the Cu(211) surface do not react and completely desorb after heating the sample to 120 K.14 The van der Waals density functional (vdW DF) calculations have shown that the activation energy for CO2 dissociation on vicinal Cu surfaces is larger than 0.67 eV.15 This theoretical result would suggest that the dissociation hardly occurs on the clean Cu surfaces at low temperatures, especially below 100 K. To untangle the controversial results on the CO2 reactivity on vicinal Cu surfaces, further investigations are required to reveal whether CO2 molecules can dissociate on the Cu surfaces and which factor is important to the CO2 reaction on the low-temperature Cu surfaces. In this study, we investigated the reaction of CO2 on the Cu(111), Cu(997), and Cu(977) surfaces at low temperatures using infrared reflection absorption spectroscopy (IRAS), X-ray photoelectron spectroscopy (XPS), in situ quadrupole mass spectroscopy (QMS), and scanning tunneling microscopy (STM).
The CO2 dissociation into CO on the vicinal Cu surfaces at 83 K was detected by IRAS and STM. XPS measurements of the reaction show characteristic uptake curves of the CO produced on the surface; the dissociation rate is increased as a function of elapsed time until reaching near-saturation coverage of CO. The XPS and QMS experiments indicate that the CO2 dissociation observed in this study leads to an oxygen-exchange reaction between CO2 and CO.
Experimental
All experiments were performed under UHV conditions. The Cu(111), Cu(997), and Cu(977) samples (10 mm diameter disk) were purchased from the surface preparation laboratory. Cu(997) consists of nine-atom wide (111) terraces separated by monoatomic (111) steps and thus can be labelled as 9(111) × (111) using a microfacet notation. On the other hand, the Cu(977) surface is referred to as 8(111) × (100). The terrace widths of Cu(997) and Cu(977) are 18.7 Å and 17.0 Å, respectively. Before each experiment, these Cu surfaces were cleaned by several cycles of Ne+ ion sputtering and annealing in the 673–700 K range. The temperature of the sample was measured using a K-type thermocouple attached to the side of the Cu crystal by a Ta foil. The samples were cooled by liquid nitrogen.IRAS measurements were performed in a UHV chamber with a base pressure of 1.3 × 10−8 Pa. IRAS spectra were measured using an FT-IR spectrometer (Bruker IFS66v/S) with a mercury–cadmium–telluride (MCT) detector. All the spectra were obtained with 4 cm−1 resolution. To investigate the thermal reaction of inert CO2 on the Cu surfaces, it is essential to pay careful attention to the possibility of experimental artifacts.6 A high-purity isotopic 13CO2 gas (>99.999% purity) was used for IRAS experiments to differentiate reaction products from the background adsorption of residual 12CO in the UHV chamber. During a series of IR measurements, all the filaments, including a hot-cathode vacuum gauge in the chamber, were turned off to exclude the possibilities of the CO2 reaction at a hot filament and electron-induced reactions on the Cu surface. In the IRAS experiments, CO2 gas was introduced into the UHV chamber through a pulse gas dosing system. The gas was first filled in a gas line at a constant pressure of 133 Pa. The gas line is connected to the UHV chamber via a pulse valve (Parker Hannifin Pulse Valves Series 99). CO2 molecules were dosed onto the sample by opening the pulse valve for a certain period (5 ms). The exposure of gaseous molecules was controlled by the number of pulses. The exposure per one pulse shot has been estimated to be 0.065 L from an uptake curve of cyclohexane on Rh(111) [1 L = 1 × 10−6 Torr s = 1.33 × 10−4 Pa s].16
The synchrotron-radiation XPS (SR-XPS) measurements were performed using a UHV chamber (base pressure = 1 × 10−8 Pa) at a soft X-ray undulator beamline (BL-13B) of Photon Factory in Tsukuba, Japan. All XPS spectra in this paper were obtained at a sample temperature of 85 K using a hemispherical electron analyzer (SPECS, Phoibos 100) at a normal emission angle with a photon energy of 630 eV. The total instrumental energy resolution was 0.2 eV. The CO2 and CO coverages in the SR-XPS experiments were estimated from C 1s area intensity, which is calibrated by the intensity of a (1.4 × 1.4) CO superstructure (θCO = 0.52 ML; ML = molecules per one surface Cu atom) on Cu(997).17
The laboratory XPS measurements with an Al Kα X-ray source (hν = 1486.6 eV) were performed in a UHV chamber equipped with a QMS (Balzers, Prisma QME200) and a hemispherical electron analyzer (Scienta Omicron, R3000). The QMS measurements of desorbed species during the surface reaction were also conducted using this chamber. The isotope molecules, C18O2 (18O purity: 94 atom%), were used in the QMS experiments to detect oxygen-containing reaction products from the CO2 dissociation.
The STM experiments of the Cu(997) surface were conducted in another UHV chamber with a low-energy electron diffraction (LEED) apparatus, QMS, and STM (Createc). The density of kink sites of the clean Cu(997) surface was estimated to be less than 0.5% from the STM images of the bare Cu(997) surface. CO2 was dosed on the Cu(997) surface at 80 K and 92 K, followed by post-reaction STM experiments at sample temperatures between 77 K and 80 K. The STM images were obtained using an electrochemically polished tungsten tip.
Results and discussion
Fig. 1(a) shows a series of IRAS spectra on Cu(997) at 83 K as a function of 13CO2 exposure. At exposure below 7 L, two peaks were observed at 2050 and 2067 cm−1, assigned to 13CO stretching vibrations on the atop site. These peaks were observed at higher wavenumbers than those on the flat Cu(111) surfaces (2031 cm−1).18 The STM experiments shown later (Fig. 2) indicate that the CO molecules produced at low coverage preferentially adsorb on the kink sites, followed by adsorption at the step sites. Therefore, the peak at 2067 cm−1, which is more intense at 2 L, is assigned to the 13CO at the kink, and the peak at 2050 cm−1 can be attributed to the isolated molecules at step sites. At an exposure of 20 L, a new peak of 13CO appeared at 2046 cm−1, and this peak was shifted to a lower wavenumber with increasing exposure (2038 cm−1 at 46 L). Judging from the STM images (Fig. 2), the coverage of 13CO was still low at this exposure, and thus, we attributed this peak to step 13CO species at higher coverage. At the exposure of 46 L, the antisymmetric stretching mode of physisorbed 13CO2 was observed at 2290 cm−1. The asymmetric Fano-like line shape indicates the coupling between the molecular vibrational motion and electronic states of the Cu substrate.17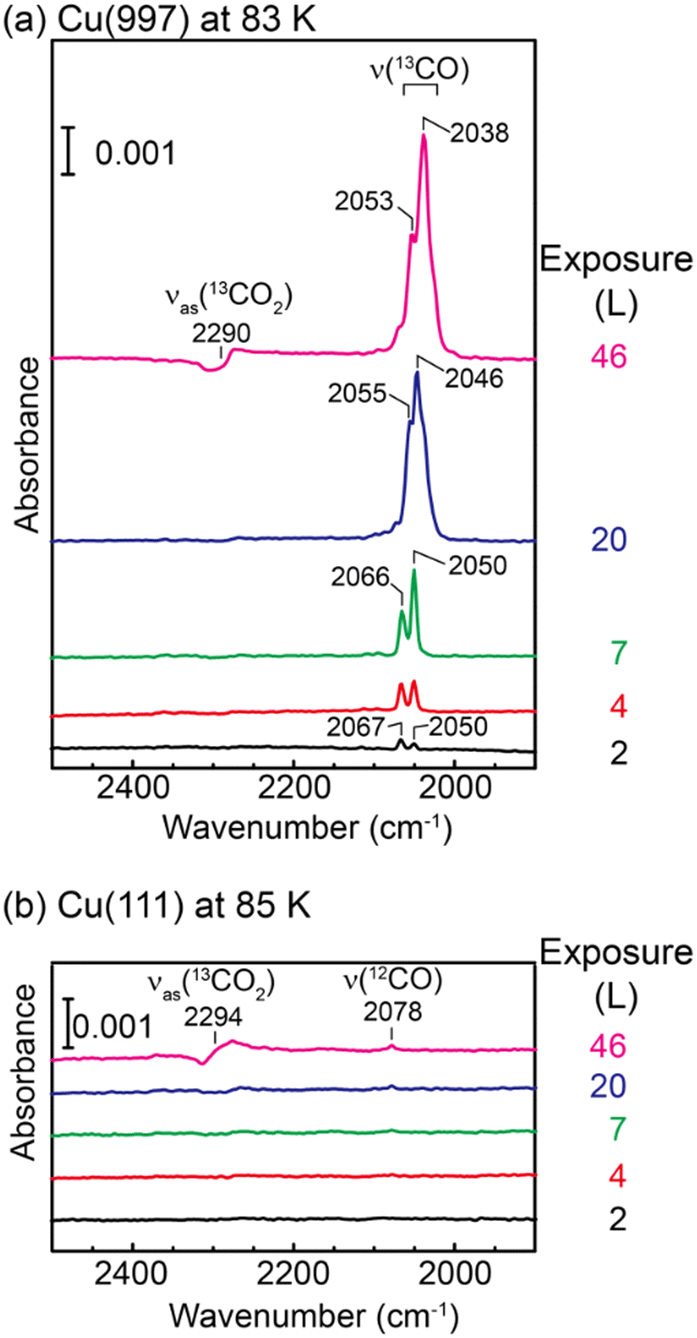 | ||
| Fig. 1 A series of IRAS spectra of (a) Cu(997) at 83 K and (b) Cu(111) at 85 K as a function of 13CO2 exposure. | ||
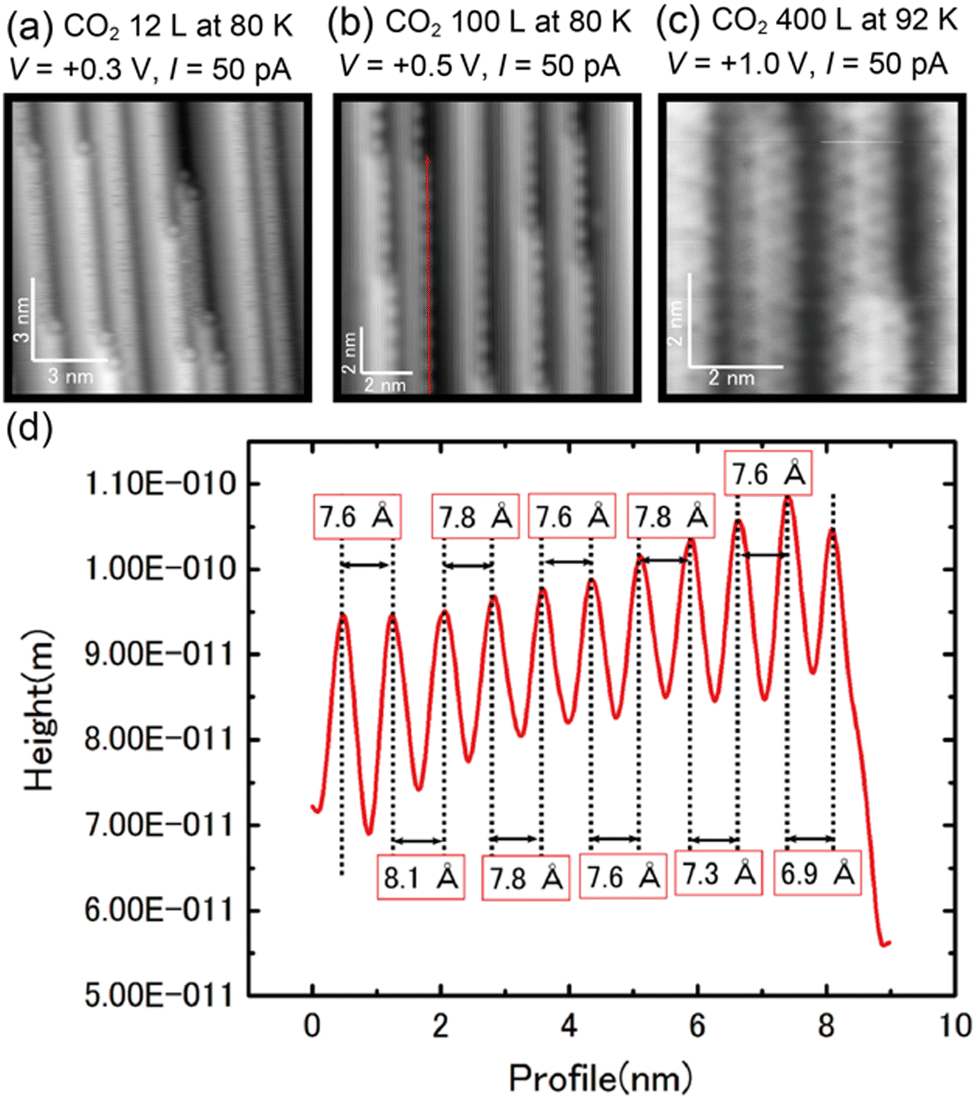 | ||
| Fig. 2 (a)–(c) STM images of the Cu(997) surface as a function of CO2 exposure. (d) Line profile along the red arrow in (b). | ||
In contrast, IRAS spectra of Cu(111) at 85 K (Fig. 1(b)) show no vibrational mode of adsorbed 13CO. The main peak at 46 L is assigned to the Fano-like 13CO2 antisymmetric stretching mode, and a small peak at 2078 cm−1 is assigned to 12CO.19 The observation of the 12CO molecules is due to the adsorption of background residual 12CO gas in the UHV chamber.
The IRAS spectra shown in Fig. 1 are clear evidence of dissociation of CO2 on the vicinal Cu(997) surface at 83 K. Using isotope 13CO2 molecules, the possibility of the adsorption of residual 12CO molecules in the UHV chamber is ruled out. In addition, the control experiments on the Cu(111) surface exclude the possibility of experimental artifacts such as the adsorption of 13CO impurity in the 13CO2 gas and electron-stimulated dissociation of CO2, which was suggested by XPS of CO2 on Cu surfaces.14,20 Therefore, CO2 molecules are dissociated on the Cu(997) surface at 83 K. The low-temperature dissociation indicates a low activation energy for this reaction.
The surface adsorbates by the CO2 exposure were further investigated by STM. Fig. 2(a) shows an STM image of Cu(997) after 12 L CO2 exposure at 80 K. Several protrusions were observed at kink sites after the CO2 exposure. Based on the IRAS results in Fig. 1(a), the protrusions are assigned to CO adsorbed at the kink sites. Note that the percentage of kink sites was estimated to be less than 0.5% on the Cu(997) clean surface. The CO molecules preferentially adsorbed at kink sites on the Cu(997) surface. In addition, line-noise-like features were often observed at step sites, which were not observed on the clean Cu(997) and the terrace sites on the CO2-exposed Cu(997). Thus, this is probably due to the diffusion of CO molecules along the step. At higher exposure (100 L), the ordered protrusions are observed along the steps (Fig. 2(b)). The line profile along the step (red arrow in Fig. 2(b)) is shown in Fig. 2(d). The average distance between neighboring protrusions was estimated to be 7.7 Å, which is three times longer than the nearest-neighbor distance between surface Cu atoms of the Cu(997) surface (2.56 Å). The obtained images after CO2 exposure are similar to the STM images of the Cu(997) surface after CO exposure (see the ESI,† Fig. S1). This also supports the assignment that the observed protrusions are attributed to the CO molecules. The observed peak wavenumbers in IRAS indicate that CO molecules are adsorbed on the atop site. Therefore, the step CO molecules at the atop sites form a one-dimensional (1 × 3) superstructure. Further exposure of CO2 (400 L) leads to the observation of depressions on the terrace sites. The difference in the images at lower exposures and higher exposure is due to the difference in the state of the tip apex. According to the previous STM study on CO adsorbed on Cu(111), a CO molecule is imaged as a protrusion (depression) with a CO-adsorbed tip (a bare metal tip), respectively.21 Therefore, the produced CO molecules could also occupy the terrace sites. The present STM result indicates that CO, which is produced at active sites, can diffuse to the terrace and does not suppress further the dissociation reaction of CO2. Significant surface reconstruction was not observed after the CO2 reaction. However, fuzzy protrusions at kink sites were often observed after the CO2 exposure (see the ESI,† Fig. S2). This structure could be attributed to oxygen adsorption at the Cu kink sites, where the atomic oxygen was produced by the CO2 dissociation.
Quantitative analysis of the CO2 dissociation was carried out by SR-XPS. Fig. 3(a) and (b) show a series of XPS C 1s and O 1s spectra of Cu(997) as a function of elapsed time measured under a constant CO2 pressure (PCO2 = 1.8 × 10−6 Pa). During the measurements, the sample temperature was maintained at 85 K. A peak at 284.3 eV in the C 1s spectra is attributed to a small amount of atomic carbon contamination (0.02 ML) of the Cu sample. Under the constant CO2 pressure, a peak at 291.4 eV appeared at the exposure time of 60 s, which is assigned to physisorbed CO2. This peak gradually increased in intensity with the increase in reaction time. The peak of physisorbed CO2 disappeared when the chamber was evacuated, indicating that CO2 molecules are in an adsorption–desorption equilibrium at 85 K. At t > 2000 s, new peaks appeared at 286.1 eV with broader peaks at higher binding energies. These peaks are attributed to the main peak and the shake-up satellites of adsorbed CO on Cu.22 In fact, the positions of the main and satellite peaks are the same as the reference spectrum of CO on Cu(997) at a saturation coverage (0.52 ML). The observation of CO is consistent with the IRAS and STM measurements. The O 1s spectra in Fig. 3(b) also show the adsorption of CO2 and the formation of CO. In addition, a peak at 530.8 eV was observed as time elapsed, indicating that there is an oxygen-containing coadsorbed species other than CO2 and CO. As discussed later, this peak can be attributed to atomic oxygen coadsorbed with CO and CO2. It should be noted that the peak intensity of atomic oxygen was significantly smaller than that of the CO peaks; coverages of atomic oxygen estimated from O 1s spectra in Fig. 3(b) were less than 0.03 ML. This result indicates that oxygen atoms do not remain on the surface after the CO2 dissociation or migrate into subsurface/bulk sites. Similar experiments were conducted at higher pressure (5.3 × 10−6 Pa). A series of SR-XPS spectra is shown in Fig. 3(c) and (d). The surface reaction process was the same as that at lower pressure; first, the physisorption of CO2 occurred, followed by a gradual increase of surface CO coverage.
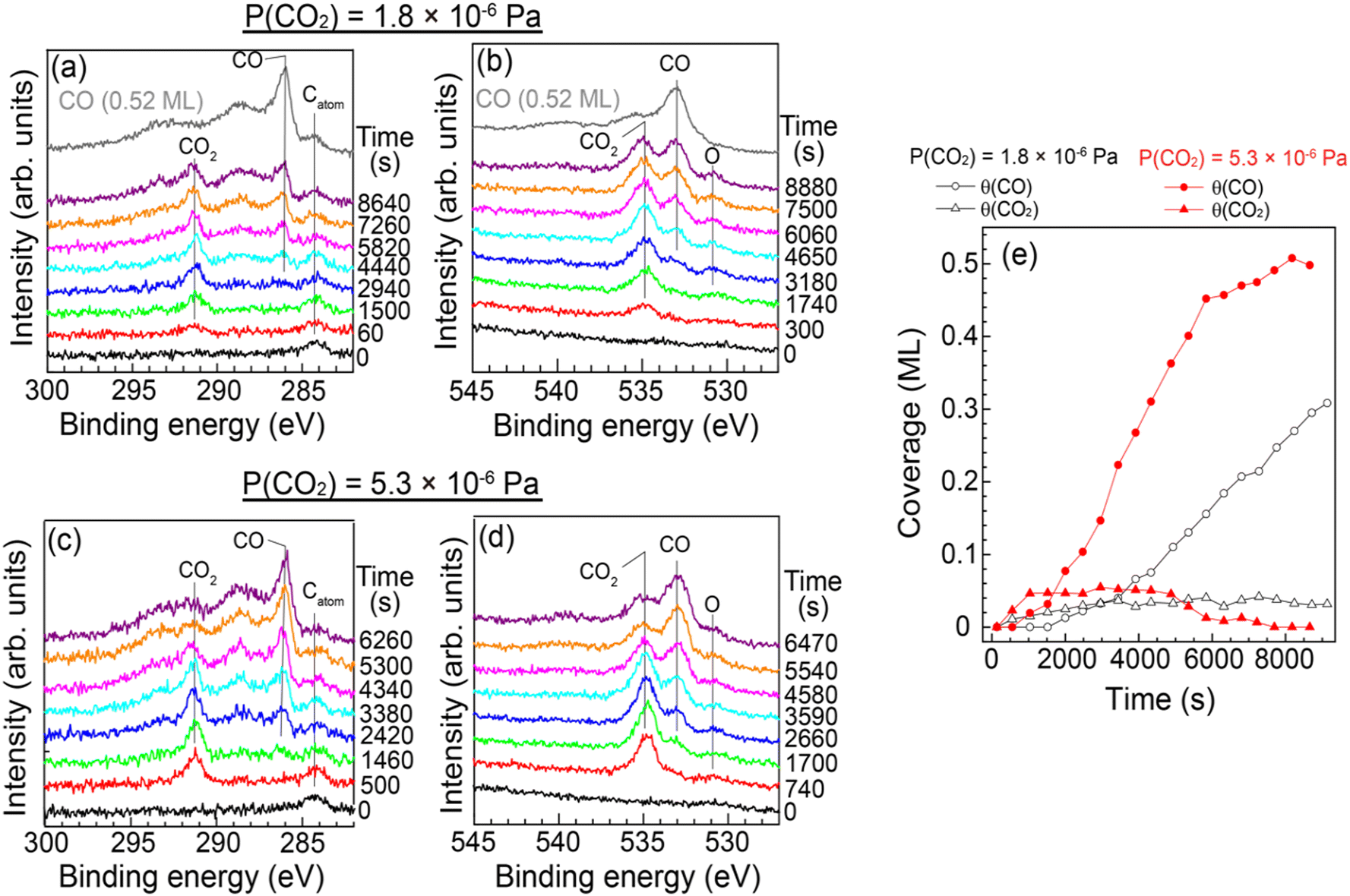 | ||
| Fig. 3 (a) C 1s and (b) O 1s SR-XPS spectra on Cu(997) at 85 K as a function of CO2 exposure time together with the reference spectra of adsorbed CO on Cu(997) at saturation coverage (topmost). During the measurement, CO2 pressure was maintained at 1.8 × 10−6 Pa. (c) C 1s and (d) O 1s SR-XPS spectra on Cu(997) at 85 K at a CO2 pressure of 5.3 × 10−6 Pa. (e) Coverages of adsorbed CO (circles) and CO2 (triangles) estimated from C 1s area intensity at 1.8 × 10−6 Pa (open symbols) and 5.3 × 10−6 Pa (filled symbols). | ||
The CO and CO2 coverages during the dissociation reaction were estimated from C 1s area intensities (Fig. 3(e)). The CO2 coverage was gradually increased from 0.015 ML at 60 s to 0.04 ML at t > 3000 s at PCO2 = 1.8 × 10−6 Pa. As mentioned above, CO2 is in the adsorption–desorption equilibrium at 85 K. Thus, the increase of the CO2 coverage indicates the formation of additional adsorption sites with slightly higher adsorption energy for CO2 than the clean Cu(997) surface. On the other hand, the adsorbed CO molecules appeared at t ∼ 2000 s, and the CO coverage increased linearly at t > 4000 s. The observed result indicates an induction period before acceleration to the CO2 dissociation. At higher CO2 pressure (5.3 × 10−6 Pa), the equilibrium CO2 coverage was slightly increased (0.05 ML) compared with the lower pressure. The CO formation is faster than that at the lower CO2 pressure, and the CO coverage increased to 0.5 ML at t ∼ 8000 s. The saturation coverage of CO on Cu(997) is 0.52 ML, indicating that the CO2 dissociation is not suppressed by coadsorbed CO. This may be due to the rapid diffusion of CO on the Cu surface as suggested by the STM experiments; the formed CO at an active site easily diffused to other adsorption sites even at ∼85 K.
Fig. 4 shows the formation rates of CO estimated from the first derivative of the CO coverage change as a function of the reaction time displayed in Fig. 3(e). It clearly shows that the reaction rate increased gradually at low CO coverage. The result indicates that the clean Cu(997) is less reactive for CO2 dissociation compared with Cu(997) with the adsorbates, and that the low reactivity of the clean surface leads to the induction period. Thus, CO2 dissociation is facilitated by coadsorbed species. This may be consistent with previous theoretical calculations, which reveal that activation energy for the CO2 dissociation on the clean Cu stepped surfaces is higher than 0.67 eV, indicating that the dissociation rarely occurs at 85 K.15 The higher CO2 pressure (5.3 × 10−6 Pa) leads to a faster increase in the reaction rate, followed by a decrease at t > 3500 s. The gradual reduction of the reaction rate is probably due to the decrease of the active sites or hindrance of diffusion of the reaction product by adsorbed CO.
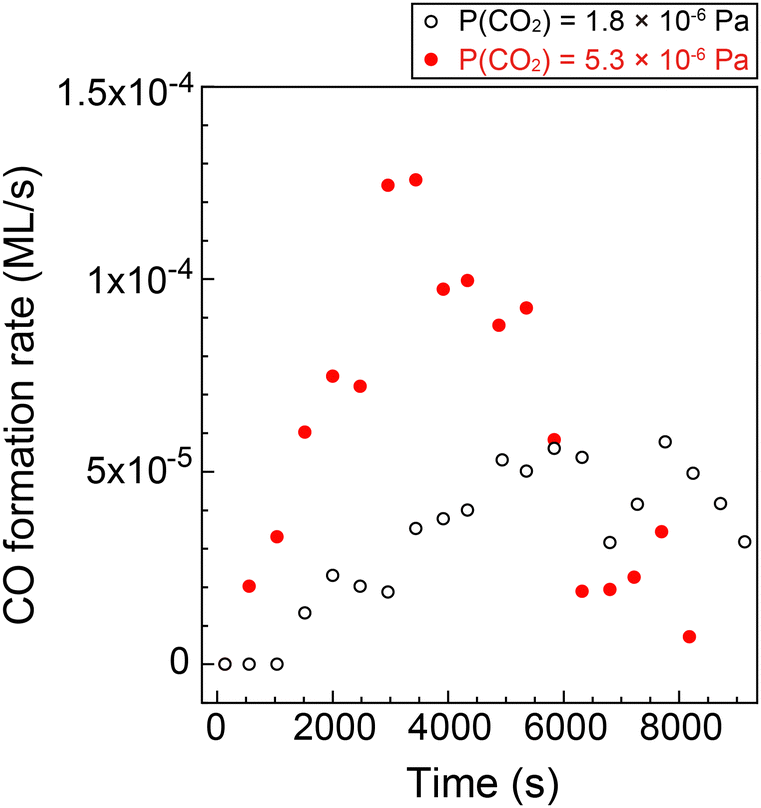 | ||
| Fig. 4 CO formation rate on Cu(997) at 85 K as a function of the reaction time at CO2 pressures of 1.8 × 10−6 Pa (open black circles) and 5.3 × 10−6 Pa (filled red circles). The rates were estimated from CO coverage shown in Fig. 4(e). | ||
The small amount of oxygen atoms on the surface (<0.03 ML) after the CO2 dissociation suggests that the atomic oxygen produced was desorbed from the surface, probably via reaction with other molecules or atoms. Thus, we investigated the reaction products desorbed from the Cu(997) surface by QMS to reveal the details of the CO formation reaction from CO2. The experimental setup is shown in Fig. 5(a). The Cu(997) surface at 82 K first faced the opposite direction to QMS. The isotopic C18O2 gas was introduced into the UHV chamber at a constant pressure (1 × 10−5 Pa). At 100 seconds after starting CO2 flow into the chamber, the sample faced the QMS to detect reaction products desorbed from the surface. The QMS signals of selected masses are shown in the ESI† (Fig. S3). Note that reaction products were scarce compared with the feed gases, and it is difficult to distinguish the QMS signal of the product from those of background gases. Thus, the relative intensity of the QMS signals, i.e., the intensity ratio of the QMS signal to the main m/z = 48 (C18O2) signal, is also shown in Fig. S3 (ESI†). The relative intensity of m/z = 46 (C16O18O) was increased when the sample faced the QMS (t = 100 s).
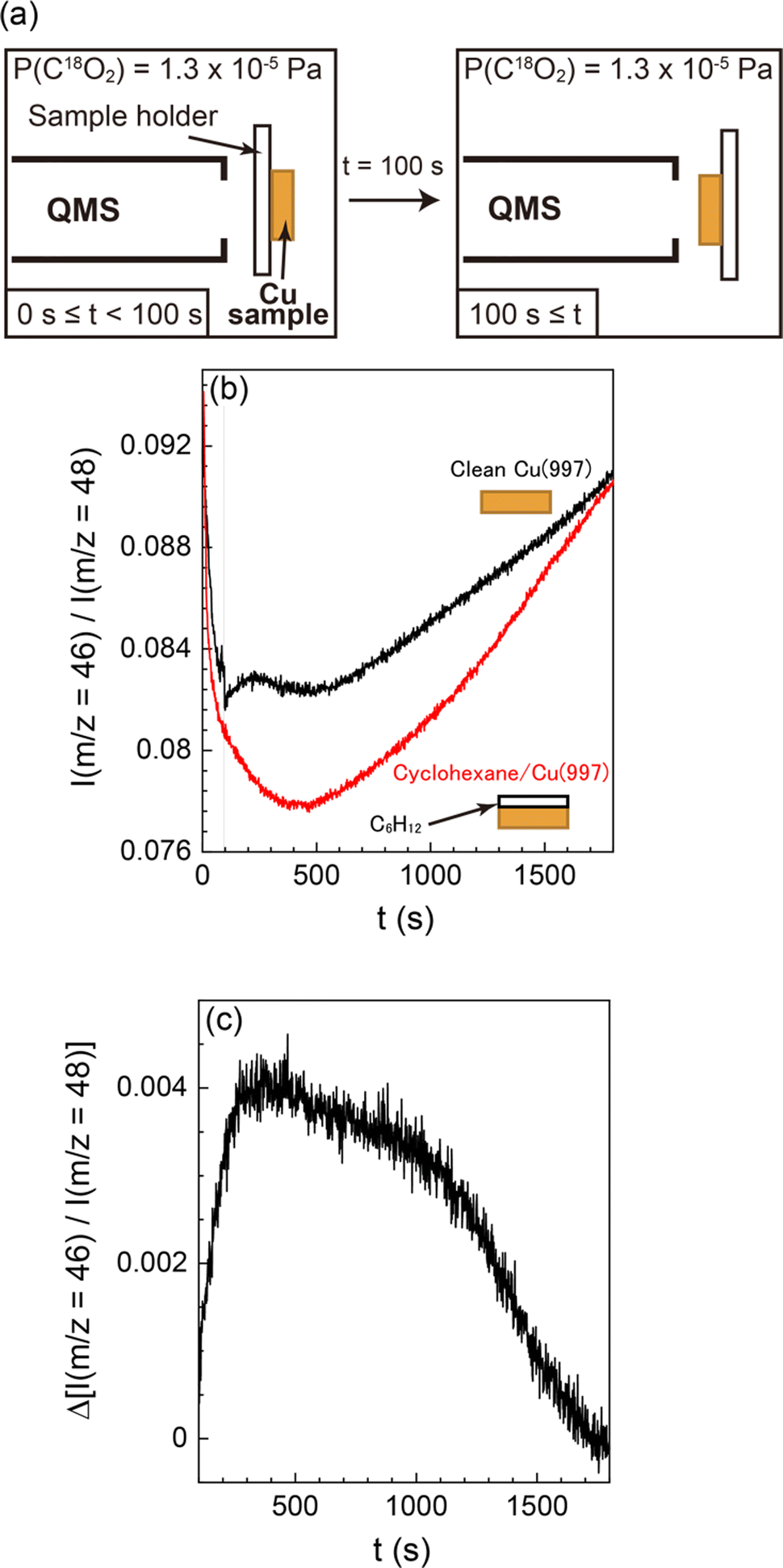 | ||
| Fig. 5 (a) Experimental setup of QMS measurements of desorbed reaction products. First, the Cu(997) surface faced the opposite direction from QMS. At t = 0 s, C18O2 gas (1 × 10−5 Pa) was introduced in the UHV chamber, then facing the Cu(997) surface in the QMS direction at t = 100 s. (b) QMS signal ratio between m/z = 46 and 48 as a function of CO2 exposure time using the bare Cu(997) (black) and the deactivated cyclohexane-covered Cu(997) (red) surfaces. (c) A differential of the QMS signals shown in (b). The surface temperature was maintained at 82 K. | ||
To validate that the increase of m/z = 46 relative intensity results from the surface reaction, a similar experiment was performed using a deactivated Cu(997) surface fully covered by multilayer cyclohexane (Fig. S4, ESI†). The relative intensities of m/z = 46 on both surfaces are shown in Fig. 5(b). The normalized m/z = 46 signal was steeply decreased at t < 400 s. This is due to the gradual increase of the m/z = 48 signal during this period (Fig. S4a, ESI†). The normalized signal was then increased at t > 500 s, possibly due to the reaction of C18O2 at the QMS ionization chamber or the ionization pressure gauge. Such behaviour was also observed in other possible reaction products (m/z = 20 (H218O), 28 (C16O), and 44 (C16O2) in Fig. S4b, ESI†). Thus, C18O2 molecules could be reacted with residual gases in the UHV chamber, such as H2 and CO, probably at the hot filaments. However, the effect of such undesired reactions occurring at sites other than the Cu surface can be excluded by conducting the control experiment using the deactivated (cyclohexane-covered) surface under the same reaction condition.
There is a clear difference between the two surfaces; the increase in the relative intensity was observed on the “bare” Cu(997) surface, whereas there was no response in the signal when the cyclohexane-covered Cu(977) surface faced the QMS. This indicates that the increase in the relative intensity is due to the surface reaction on Cu(997). The signal difference of the two surfaces is shown in Fig. 5(c). The signal difference is monotonically increased up to t ∼ 400 s, followed by a gradual decrease to zero at t ∼ 1700 s. The observed time-dependent QMS signal of the desorbed C16O18O shows a similar trend with the increase of the CO coverage by CO2 dissociation in the SR-XPS experiments (Fig. 3). This suggests that the formation of C16O18O is concomitant with the CO formation on the surface. The C16O18O molecules are likely to be produced via oxidation of background C16O molecules in the UHV chamber. Therefore, a possible reaction mechanism of CO2 in this experiment is an oxygen-exchange reaction with CO molecules on Cu(997): C18O2 (dosed gas) + C16O (background gas) → C18O (ad) + C16O18O (gas). The oxygen-exchange reaction suggested by the QMS results is consistent with the SR-XPS results; only a small amount of atomic oxygen was detected on the Cu(997) surface during the reaction.
The CO2 dissociation was also observed on the Cu(977) surface. The reactivity is similar to that of Cu(997), and the induction period was also observed on Cu(977) (see the ESI,† Fig. S5). Therefore, the CO2 dissociation reaction is not sensitive to the structure of the Cu step facets. Fig. 6(a) shows O 1s XPS spectra of Cu(977) measured during CO2 dissociation at 3 × 10−6 Pa. The physisorbed CO2 was first observed, followed by the growth of CO peaks at ∼533 eV, which is similar to the Cu(997) surface. In addition, the effect of coadsorbed oxygen in the CO2 dissociation was investigated. Prior to the CO2 reaction, oxygen was dosed on the Cu(977) surface at 88 K. The O 1s spectrum of the oxygen-preadsorbed Cu(977) shows broad peaks between 528 eV and 532 eV. The oxygen coverage was estimated to be 0.06 ML from the peak intensity of the O 1s spectrum. After dosing CO2 gas, the physisorbed CO2 peak was first observed, and its intensity is slightly larger than those on the bare Cu(977) surface. Thus, preadsorbed oxygen creates more stable adsorption sites for CO2. The gradual increase of CO2 coverage in the SR-XPS experiment (Fig. 3) is probably due to the adsorption of a small amount of oxygen during the reaction. More importantly, the CO peak is clearly observed at t = 1445 s on the oxygen-preadsorbed Cu(977) surface, which is much faster that than on the clean Cu(977). This indicates that the coadsorbed oxygen promotes CO2 dissociation. After the formation of CO, an O 1s peak was observed at ∼531 eV, which can be assigned to atomic oxygen. The position of this peak is similar to that observed in the SR-XPS experiments (Fig. 3(b) and (d)). The chemical state of oxygen is changed from the initial O/Cu(977) surface by coadsorption with CO2 or CO.
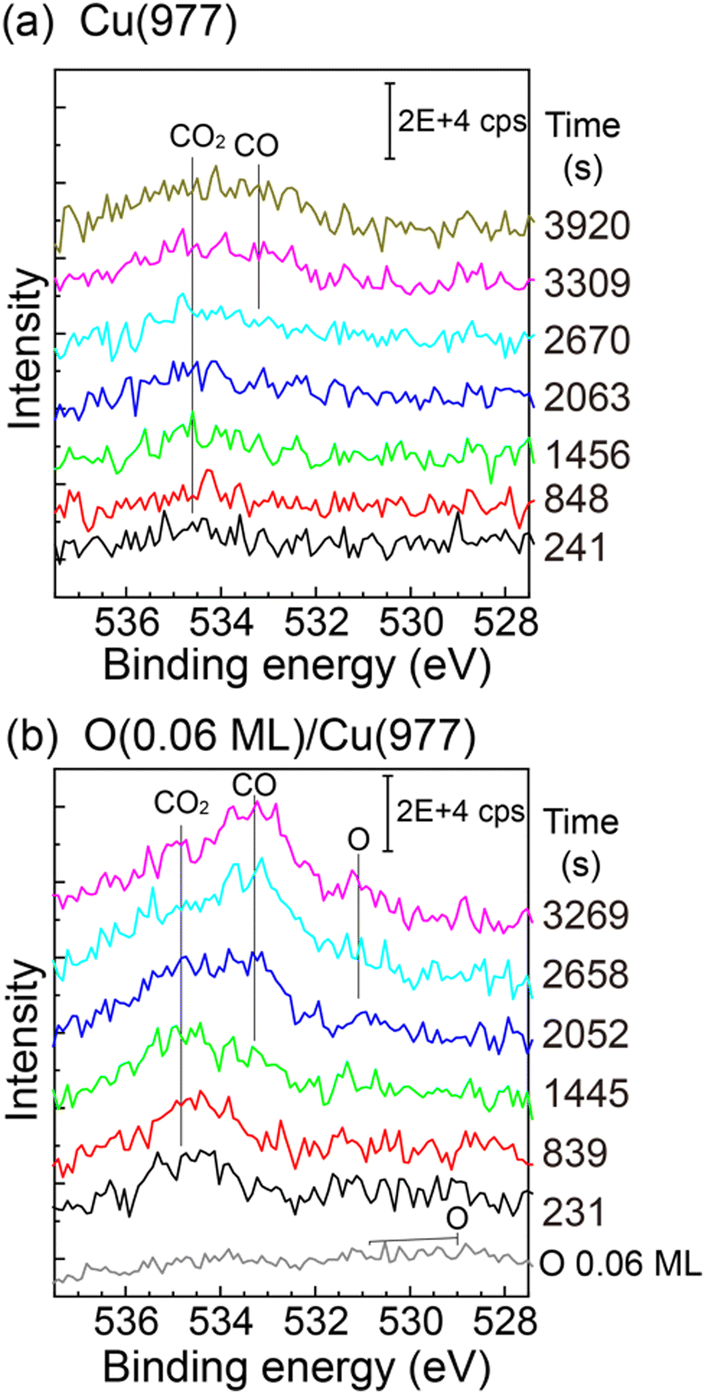 | ||
| Fig. 6 O 1s XPS spectra on (a) Cu(977) and (b) oxygen-preadsorbed Cu(977) at 88 K as a function of CO2 exposure. CO2 pressure was maintained at 3 × 10−6 Pa during the measurements. The oxygen-preadsorbed Cu(977) was prepared before the CO2 exposure by dissociative adsorption of O2 at 88 K. The spectra were subtracted by the O 1s spectrum of clean Cu(977) to remove background Cu auger signals. | ||
The observed promotional effect depends significantly on the coverage of preadsorbed oxygen. Fig. 7 shows the CO coverages as a function of the reaction time (88 K, PCO2 = 3 × 10−6 Pa) at different preadsorbed-oxygen coverages. The results on the bare Cu(977) and O(0.06 ML)/Cu(977) surfaces show the significant shortening of the induction period by the existence of a small amount of oxygen. Preadsorption of further oxygen (θO = 0.11 and 0.14 ML) leads to a decrease in the reaction rates compared with that at θO = 0.06 ML. A small amount of oxygen required to promote the CO2 dissociation suggests that the possible reaction site is an oxygen-adsorbed step or kink site, and that oxygen-adsorbed terrace sites suppress the dissociation reaction or diffusion of CO produced at the active sites to inactive vacant sites.
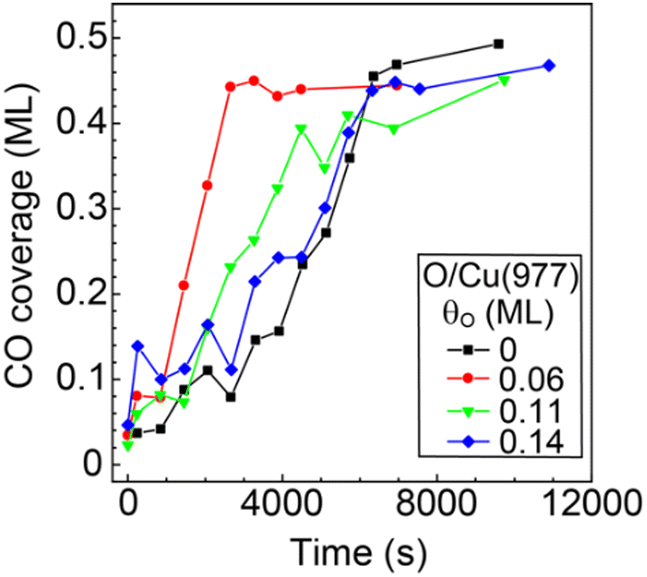 | ||
| Fig. 7 CO coverage produced from CO2 dissociation on bare Cu(977) and oxygen-preadsorbed Cu(977) as a function of the reaction time. The sample temperature and CO2 pressure were maintained at 88 K and 3 × 10−6 Pa during the reaction. | ||
By a series of present experiments, the dissociation of the CO2 molecules through the oxygen-exchange reaction with CO on the Cu(997) and Cu(977) surfaces was clearly observed. In addition, atomic oxygen adsorbed on the stepped Cu surface promoted the CO2 dissociation at low temperatures (80–92 K). Recently, the activation of CO2 molecules has also been studied under near-ambient pressure (NAP) conditions above room temperature. On the Cu(997) surface between 299 K and 340 K, the CO2 dissociation and formation of CO, O, and CO3 are observed at a CO2 pressure of 80 Pa.23,24 Kim et al. have revealed that the CO2 dissociation into CO and O is also observed on Cu(997) at 300 K in the presence of 100 Pa CO2.25 In addition, high-pressure (HP) STM shows surface roughening and formation of partially oxidized Cu nanoclusters on the Cu(997) surface during the reaction.25 Under a NAP condition, CO2 is also dissociated into CO and O at step sites of the Cu(100) surface at 370 K in 40 Pa CO2 gas.26 An important point is that oxygen remains on Cu surfaces as atomic oxygen or CO3 under the NAP conditions, whereas oxygen coverage on the Cu(997) surface was very low (less than 0.03 ML) under the UHV conditions investigated in this study.
The reaction mechanism of CO2 on Cu surfaces observed under NAP conditions differed from those observed in this study under the UHV conditions. The observed difference between the UHV and the NAP experiments can be attributed to the difference in the activation energies of reactions. The previous theoretical study has shown that the activation energy for the CO2 dissociation into CO and O on vicinal Cu surfaces is larger than 0.67 eV,15 indicating that this reaction hardly occurs on clean Cu vicinal surfaces at low temperatures. Thus, the simple CO2 dissociation reaction can occur only on the Cu surfaces above room temperature. However, based on the present experimental results, once such a rare dissociation event occurs at low temperature, atomic oxygen produced on the Cu surfaces can promote CO2 dissociation. Note that the oxygen-exchange reaction could take place under both UHV and NAP conditions. Another possible reason for the different reaction pathways under NAP and UHV conditions might be the difference in the surface structures during the reactions; the HP-STM study shows the formation of Cu nanoclusters on the surface under the NAP conditions,25 whereas the structural change of the Cu(997) was not observed at 80–92 K under the UHV conditions (Fig. 2). Therefore, the nature of active sites for the CO2 dissociation can depend on the sample temperature that affects diffusivity of atoms and molecules and stability of reaction products on the Cu surfaces. Further theoretical studies on CO2 dissociation at kink sites on Cu and the pre-adsorbed oxygen effect would be helpful in understanding the experimental results.
Conclusions
The dissociation of CO2 on the vicinal Cu surfaces at low temperatures was investigated using several experimental techniques. Dissociation of CO2 molecules into CO on the Cu(997) and Cu(977) surfaces was observed at temperatures between 80 K and 90 K. In contrast, the reaction of CO2 did not occur on the flat Cu(111), indicating that the defect sites, such as steps and kinks, play a key role in CO2 dissociation. The XPS experiments show that CO and physisorbed CO2 are the main adsorbates during the reaction. On the other hand, the amount of atomic oxygen on the surface was significantly smaller than that of CO. Analysis of desorbed reaction products during the reaction of isotopically labeled CO2 revealed that the observed CO formation on the vicinal Cu surface is mainly due to the oxygen-exchange reaction with residual CO in a UHV chamber. The reaction of CO2 on the vicinal Cu surface has an induction period; the initial reaction rate on the clean Cu surfaces was low, and the reactivity was gradually increased during the reaction. Once such a rare CO2 dissociation event occurs at low temperature, atomic oxygen produced on the Cu surfaces may promote CO2 dissociation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Advanced-Catalytic-Transformation program for Carbon utilization (ACT-C) of the Japan Science and Technology Agency (JST) (Grant No. JPMJCR12YU), JST CREST (JPMJCR20R4) and the Grants-in-Aid for Scientific Research (No. 22K05030) from the Japanese Society for the Promotion of Sciences (JSPS). The synchrotron-radiation XPS experiments were conducted under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2012-S2-006). We are also grateful to the staff members of the Photon Factory for their technical support.References
- M. Aresta, Carbon Dioxide as Chemical Feedstock, WILEY-VCH Verlag GmbH and Co. KGaA, Weinheim, 2010 Search PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- F. Solymosi, J. Mol. Catal., 1991, 65, 337–358 CrossRef CAS.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- U. Burghaus, Catal. Today, 2009, 148, 212–220 CrossRef CAS.
- U. Burghaus, Prog. Surf. Sci., 2014, 89, 161–217 CrossRef CAS.
- J. Nakamura, Y. Choi and T. Fujitani, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- P. B. Rasmussen, P. A. Taylor and I. Chorkendorff, Surf. Sci., 1992, 269, 352–359 CrossRef.
- J. A. Rodriguez, W. D. Clendening and C. T. Campbell, J. Phys. Chem., 1989, 93, 5238–5248 CrossRef CAS.
- P. A. Taylor, P. B. Rasmussen and I. Chorkendorff, J. Vac. Sci. Technol., A, 1992, 10, 2570–2575 CrossRef CAS.
- K. H. Ernst, D. Schlatterbeck and K. Christmann, Phys. Chem. Chem. Phys., 1999, 1, 4105–4112 RSC.
- I. A. Bönicke, W. Kirstein and F. Thieme, Surf. Sci., 1994, 307, 177–181 CrossRef.
- S. S. Fu and G. A. Somorjai, Surf. Sci., 1992, 262, 68–76 CrossRef CAS.
- P. R. Davies and J. M. Keel, Surf. Sci., 2000, 469, 204–213 CrossRef CAS.
- F. Muttaqien, Y. Hamamoto, K. Inagaki and Y. Morikawa, J. Chem. Phys., 2014, 141, 034702 CrossRef PubMed.
- T. Koitaya, S. Shimizu, K. Mukai, S. Yoshimoto and J. Yoshinobu, J. Chem. Phys., 2012, 136, 214705 CrossRef PubMed.
- T. Koitaya, Y. Shiozawa, K. Mukai, S. Yoshimoto and J. Yoshinobu, J. Chem. Phys., 2016, 144, 054703 CrossRef PubMed.
- P. Hollins and J. Pritchard, Surf. Sci., 1979, 89, 486–495 CrossRef CAS.
- B. E. Hayden, K. Kretzschmar and A. M. Bradshaw, Surf. Sci., 1985, 155, 553–566 CrossRef CAS.
- V. M. Browne, A. F. Carley, R. G. Copperthwaite, P. R. Davies, E. M. Moser and M. W. Roberts, Appl. Surf. Sci., 1991, 47, 375–379 CrossRef CAS.
- L. Bartels, G. Meyer and K. H. Rieder, Appl. Phys. Lett., 1997, 71, 213–215 CrossRef CAS.
- H. Tillborg, A. Nilsson and N. Martensson, J. Electron Spectrosc. Relat. Phenom., 1993, 62, 73–93 CrossRef CAS.
- T. Koitaya, S. Yamamoto, Y. Shiozawa, K. Takeuchi, R. Y. Liu, K. Mukai, S. Yoshimoto, K. Akikubo, I. Matsuda and J. Yoshinobu, Top. Catal., 2016, 59, 526–531 CrossRef CAS.
- T. Koitaya, S. Yamamoto, Y. Shiozawa, Y. Yoshikura, M. Hasegawa, J. Y. Tang, K. Takeuchi, K. Mukai, S. Yoshimoto, I. Matsuda and J. Yoshinobu, ACS Catal., 2019, 9, 4539–4550 CrossRef CAS.
- J. Kim, Y. Yu, T. W. Go, J. J. Gallet, F. Bournel, B. S. Mun and J. Y. Park, Nat. Commun., 2023, 14, 3273 CrossRef CAS PubMed.
- B. Hagman, A. Posada-Borbon, A. Schaefer, M. Shipilin, C. Zhang, L. R. Merte, A. Hellman, E. Lundgren, H. Gronbeck and J. Gustafson, J. Am. Chem. Soc., 2018, 140, 12974–12979 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06336d |
| This journal is © the Owner Societies 2024 |