
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical revelations: the pnictogen effect in linear acetylenes†
Miguel
Gallegos
a,
Vicente
del Amo
 b,
José Manuel
Guevara-Vela
c,
Guillermo
Moreno-Alcántar
d and
Ángel
Martín Pendás
*a
b,
José Manuel
Guevara-Vela
c,
Guillermo
Moreno-Alcántar
d and
Ángel
Martín Pendás
*a
aDepartamento de Química Física y Analítica, Universidad de Oviedo, Oviedo E-33006, Spain. E-mail: ampendas@uniovi.es
bDepartamento de Química Orgánica e Inorgánica, Universidad de Oviedo, Oviedo E-33006, Spain
cDepartamento de Química Física Aplicada, Universidad Autónoma de Madrid, Madrid E-28049, Spain
dDepartment of Chemistry, School of Natural Sciences, Technical University of Munich, Lichtenbergstr. 4, Garching b., München 85748, Germany
First published on 10th February 2024
Abstract
Acetylenes are essential building blocks in modern chemistry due to their remarkable modularity. The introduction of heteroatoms, such as pnictogens (X), is one of the simplest approaches to altering the C≡C bond. However, the chemistry of the resultant dipnictogenoacetylenes (DXAs) is strongly dependent on the nature of X. In this work, rigorous theoretical chemistry tools are employed to shed light on the origin of these differences, providing a detailed evaluation of the impact of X on the geometrical and electronic features of DXAs. Special emphasis is made on the study of the carbene character of the systems through the analysis of the interconversion mechanism between the linear and zigzag isomers. Our results show that second-period atoms behave drastically differently to the remaining X: down the group, a zwitterionic resonance form emerges at the expense of decreasing the carbenoid role, eventually resulting in an electrostatically driven ring closure. Furthermore, our findings pave the way to potentially unveiling novel routes for the promotion of free-radical chemistry.
1. Introduction
Multiple chemical bonds are ubiquitous entities in Chemistry, being present in a wide variety of relevant compounds and materials.1,2 Within these interactions, triple bonds, generally depicted as E≡E, are particularly paradigmatic cases of peculiar chemical moieties, with chemistry and properties that are heavily dependent on the nature of E. This is clearly reflected in the scarcity of easily accessible main-group compounds with E≡E bonds, exhibiting very heterogeneous abundances and properties even across the same group. As an example, whereas carbon based triple bonds (C≡C) are well-known, the first stable molecule bearing a Si≡Si bond was reported for the first time in the early 2000s.3 Besides synthetically challenging, heavy tetrel (i.e. Si, Ge, Sn, and Pb) compounds behave dramatically different than their second-period analogs (e.g. C2H2 and Si2H2 display linear and double bridged geometries in the energy minima, respectively).4 These striking discrepancies have motivated the study of the impact of substituting the element (E) involved in the E≡E scaffolds, with most of the structural differences being attributed to the larger role and flexibility exhibited by their outermost valence orbitals.5 On the other hand, alkynes, being archetypal examples of the simplest of these molecules (with an HE≡EH pattern), are essential building blocks in state-of-the-art synthetic chemistry.6–11 In fact, their remarkable reactivity and modularity make them ideal reagents for the construction of a plethora of intricate and valuable products, including biologically active compounds,12 agrochemicals13 or advance materials,14,15 to name a few. A thoroughly documented review of the pivotal role played by alkynes in modern synthetic strategies is available to the reader in the literature.16,17The chemical reactivity of alkynes is mostly dictated by the high electron density attributed to the C≡C bond which, coupled with its tendency to get delocalized across nearby conjugated systems, allows the formation of new chemical bonds by the interaction, for instance, with Lewis acids, such as metal cations. In this context, alkyne functionalization has gained a lot of attention owing to its ability to alter their chemical properties and reactivity of the latter.17–19 For instance, substitution at the immediately neighboring positions to the C≡C bonds has been widely used as a strategy to enhance and direct such reactivity. Understanding of the electronic factors governing the reactivity of C≡C bonds has recently acquired further relevance owing to the rise of bio-orthogonal chemistry, which required the development of activated substrates capable of operating in the absence of metal catalysts.20,21 Following this trend, the design of activated alkynes has been dominated by two main aspects: (i) the introduction of steric effects over the linear geometry of the alkyne, something which is commonly achieved through the formation of cyclic structures in which the ring strain can disturb the stability of the C≡C interaction and (ii) the electron donating/withdrawing character of the substituents, which polarizes the alkyne electron density. While ring strain has dominated the design of copper-free click chemistry, the use of substituent effects for the activation of C≡C bonds remains relatively unexplored, despite holding great promise.
Indeed, mono-substituted acetylenes, such as aminoalkynes and generally depicted as RR1R2 C–C≡CX (X ≠ H, C), have been recently put into the spotlight owing to their peculiar reactivity and applicability as valuable organic synthons.22–27 Unfortunately, the synthesis of XC≡CX moieties is often challenging, being hampered by particularly pronounced reactivity and promiscuity, making their wide-range application difficult. However, it is known that the nature of the heteroatom determines the stability and properties of these compounds, even when the former belongs to the same group of the periodic table. For instance, obtaining nitrogen-based derivatives of some of these families of compounds is far from trivial, becoming even more challenging in the particular case of disubstituted acetylenes. Actually, the synthesis of the prototypical model of the latter, diaminoalkynes (DAAs) in general and 1,2-diaminoacetylene in particular, is quite challenging, often requiring the introduction of bulky chemical moieties in the skeleton as highlighted in a recently reported and extensive review of the topic.28 Moreover, the resultant DAAs are prone to tautomerism to produce the corresponding ketenimine derivatives, hampering the isolation of the acetylenic isomers of this family of compounds. In contrast, diphosphinoacetylenes (DPAs) are far more common and stable, while being frequently used as valuable ligands in the synthesis of a wide variety of transition-metal compounds.29–31 These, and other, marked differences between DAAs and their heavier analogs, such as DPAs or other dipnictogenoacetylenes (DXAs) in general, are intriguing. Although empirical evidence has sketched some trends in the underlying electronic origin of such divergences, this topic has been very scarcely studied from theoretical perspectives. Additionally, shedding light on to these trends requires the rigor and insights offered by state-of-the-art theoretical chemistry tools, whose large computational cost prevents their extensive applications.
With this motivation in mind, in this work, we use a wide variety of state-of-the-art computational chemistry tools to provide a thorough analysis of the electronic, energetic and geometric features of this peculiar family of compounds. In this way, our study contributes to the elucidation of the effects of introducing heteroatoms on the E≡E scaffold. Special emphasis is made on the impact of changing the nature of the pnictogen on the behavior of the underlying acetylenic structure as well as on the topology of the energy landscape of different DXAs. Furthermore, the tautomerism between two of the most relevant isomers of DXAs is studied in detail by analyzing the ongoing electron rearrangements. Altogether, these results improve the understanding of the periodic properties of pnictogenic-compounds paving the way towards new developments in the rational design of activated alkyne moieties.
The work is organized as follows: first, a brief overview of the theoretical tools employed in this study is presented. Then, the stability of the different isomers of DXAs is analyzed, and the reaction pathways for the interconversion between the linear and zigzag isomers are discussed. Special emphasis is made in this section to the carbene nature of the resultant species, as well as in the Coulombic collapse of the reaction intermediates for heavy pnictogen derivatives. The final section of the manuscript comprises the conclusions that can be drawn from our work.
2. Methods
The abstract nature of Quantum Chemical Topology (QCT) techniques has slowed down their spread outside the theoretical chemistry realm. However, and unlike the more commonly known orbital-based descriptions,32 QCT provides an extremely rigorous picture of the chemical nature of a system, free of the bias induced by the use of external references. Hence, the current section attempts to provide a grasp of the basic foundations behind the QCT tools employed here, while a more extensive review is available to the reader in the literature.33QCT is a branch of theoretical chemistry which partitions the real space (R3) occupied by a chemical system relying on the topological features of a certain scalar field. In this way, the critical points of a field allow the clear-cut decomposition of R3 in a collection of well-defined domains {Ω} which, by extension, provide a partition of molecular properties into local (e.g. atomic or pairwise) terms. Naturally, different scalar fields can be used resulting in different QCT flavors, as gathered in Fig. 1. Partitioning R3 according to the electron density, ρ(r), yields the so-called Quantum Theory of Atoms in Molecules (QTAIM) developed by R. F. Bader.34 In QTAIM, Ω are attraction basins of ρ(r) corresponding, with the particular exception of non-nuclear attractors, to atoms embedded in a molecule. In this way, atomic and inter-atomic properties readily emerge, for instance, the molecular electron count, N, can be decomposed in localized (atomic) and delocalized (inter-atomic) contributions giving rise to each of the M atomic electron population, NA,
 | (1) |
 | ||
| Fig. 1 (left) Isosurface of the ELF function of the C2H2 molecule, with an isovalue of 0.86 units, showing the spatial distribution of the bonding ELF domains. (right) QTAIM basins of the C2H2 molecule. | ||
For the same reason, the Electron Localization Function (ELF), first introduced by Becke and Edgecombe,35 provides an alternative analysis to QTAIM in terms of electron-pair localization regions. In this way, the ELF is directly linked to the Lewis's valence theory, disguising the space in a collection of core, bonding and lone-pair (valence) basins. Even a simple study of the populations and covariances of the ELF basins has been proven to provide very valuable and easy-to-interpret perspectives on the chemistry of many systems.36 In fact, ELF analyses have gained a lot of popularity outside the QCT field in recent years due to their simplicity and straightforward interpretation.37–39
Finally, the local expectation values of the squared spin operator, 〈SA2〉 = SA(SA + 1), attributed to an atom or chemical fragment can be used to provide a description compatible with the more naïve resonance theory. The local spin concept was introduced by Clark and Davidson in 200140via atomic projection operators as a means to access information about spin localization in singlets (e.g. singlet diradicals), where the spin density vanishes everywhere in space. It later suffered several transformations41 and was also generalized to real space decompositions.42 Recently, some of us have shown43 that the original definition of Clark and Davidson is the one that emerges from the theory of open quantum systems in real space. The local spin can thus be rigorously estimated under a real space perspective from the spinless 2-particle reduced density matrix (ρ2) and the electron density (ρ) as,
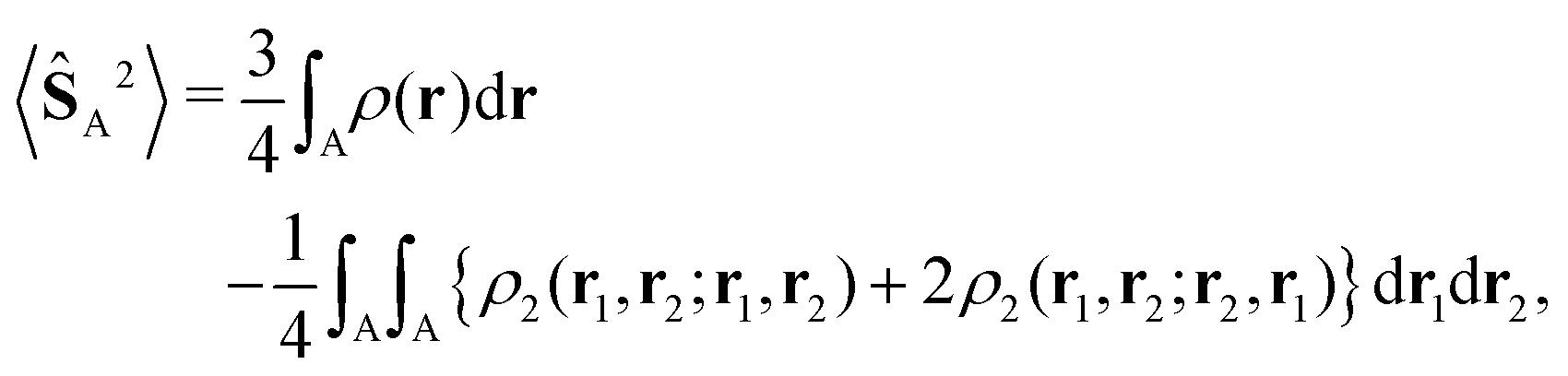 | (2) |
Local spins are valuable descriptors that provide a direct mapping to the explicit electron configurations in molecules. This can be very easily exemplified using the archetypal H2 molecule: as shown in Fig. 2, the overall bonding arises from two ionic (HA− + HB+ and HB− + HA+) and two purely covalent forms (HA + HB). Both ionic forms, comprising either 0 or a couple of paired electrons, have null contributions to the local spin, owing to their singlet spin multiplicity. On the other hand, each atom in the pure covalent distributions behaves as a doublet, leading to  . Altogether, and accounting for equal weights for the ionic and covalent contributions, the expectation value of the local spin of each atom in the simplest mean-field description of H2 becomes
. Altogether, and accounting for equal weights for the ionic and covalent contributions, the expectation value of the local spin of each atom in the simplest mean-field description of H2 becomes  .
.
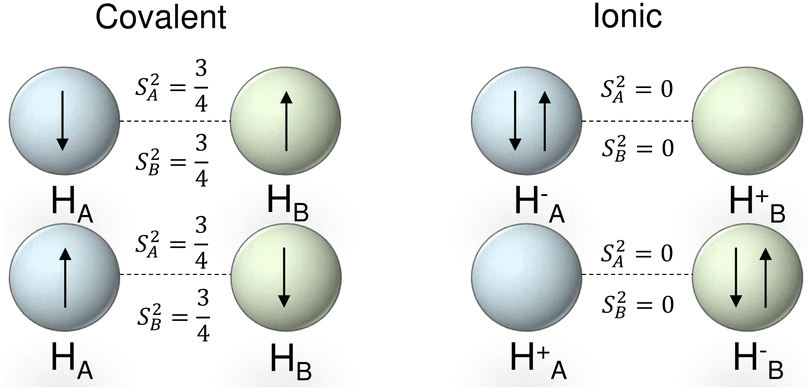 | ||
| Fig. 2 Schematic representation of the covalent and ionic contributions to the bonding of the ground state H2 molecule. S2 shows the expectation value of the local spins of both atoms. | ||
3 Computational details
Geometry optimizations, normal mode analysis, single point calculations and wavefunction generation were achieved using the Gaussian09 quantum chemistry package.44 All calculations, unless otherwise specified, were performed at the DFT level of theory using the well-known B3LYP functional45,46 in combination with the def2-TZVP basis set in the gas phase. Additionally, the GD3 Grimme empirical correction was employed, as implemented in Gaussain09,44 to account for dispersion effects. Such a level of theory was selected on the basis of a small benchmarking study relying on the reproducibility of the energetic and geometrical features of the systems under study (see S1 for more details, ESI†). We also note in passing that the B3LYP functional has been successfully employed in the past to shed light on complex chemical phenomena of a similar nature.47–52 In the particular case of Sb, MWB46 pseudo-potentials53 were employed as included in Gaussian0944 to replicate the effect of the electron cores of the many-electron systems. The relevant structures were characterized as minima or first-order saddle points through the analysis of the eigenvalues of the Hessian Matrix. Gas-phase intrinsic reaction coordinate (IRC) calculations54 were performed as implemented by default in the Gaussian09 code.44 The same level of theory was used throughout. The Local quadratic approximation (LQA)54 was employed to explore extremely flat energy landscapes commonly encountered as one moves down a group.Quantum Theory of Atoms in Molecule (QTAIM) calculations were accomplished using the PROMOLDEN55 and AIMAll56 codes. On the other hand, the computation and analysis of the electron localization function (ELF) was performed using the TopMod package.57 The local spin, 〈SA2〉, was calculated using the in-house made NRDM code. A thorough conformational search was achieved with the CLUSTER program.58 Starting from the energetically minimized linear (acetylene-like) structure of DAA, the conformational space was explored using the default genetic algorithm implemented in the latter package for up to 30 generations. Afterwards, the nature of the resultant structures was evaluated, and those not corresponding to true minima of the Potential Energy Surface (PES) were discarded resulting in five dominant isomers of DAA. Finally, the analogous structures for the remaining DXAs were optimized starting from the previously minimized geometries of DAA.
The energetic and electronic properties are generally reported in kcal mol−1 and electrons, respectively, throughout the upcoming tables and figures of this manuscript. All molecular representations and iso-surfaces were rendered with the Jmol suite.59 For the sake of clarity and simplicity, the symbol X is generally used to denote elements belonging to the pnictogen group (e.g. N, P, and As).
4. Results and discussion
4.1. Relative stability of DXA isomers
Let us start by exploring how the relative stability of different DXA isomers evolves with the nature of the heteroatom. As detailed in Computational details, a conformational search was performed starting from the plain acetylene structure of DAA and yielding a total of five main isomers, as shown in Fig. 3. We note in passing that two main groups can be found within the latter: structures having an X–X connectivity along with these isomers exhibiting explicit C–C bonding, shown in blue and red, respectively, in Fig. 3 being the latter the only ones formally related to the common acetylenic skeleton studied in this work.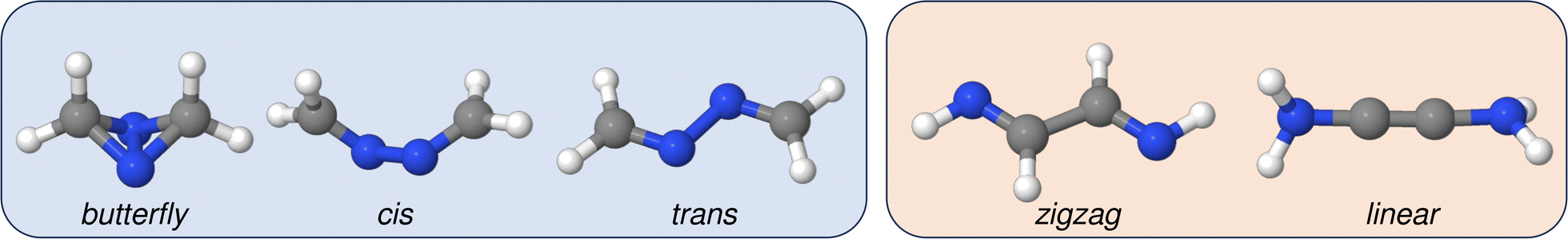 | ||
| Fig. 3 Butterfly, cis, trans, zigzag and linear isomers of DAA. Representations rendered with Jmol.59 The species corresponding to X–X and C–C connectivities are highlighted in blue and red, respectively. | ||
The relative stability of these five DXA isomers, gathered in Fig. 4, shows quite interesting trends. First of all, it is worth noting that the acetylene-like linear scaffold does not represent the most stable species in any of the here explored DXAs. As for DAA, the cis, trans and linear DAA isomers display similar stabilities, whereas it is the zigzag structure, bearing two imine moieties, the most stable out of all the studied ones. On the other hand, the bridge-like butterfly geometry is notably less stable than any of the other species, being +30 kcal mol−1 above the linear DAA, taken as a reference. Substituting the N atom for a heavier pnictogen dramatically distorts the topology of the energy landscape, resulting in diametrically opposed relative stabilities. In fact, the remaining DXAs show very uniform trends, with a structural stability dictated by the sequence butterfly > trans > cis ≈ zigzag > linear. We note in passing that similar trends in the relative stability of the DXAs isomers have been found at different levels of theory (see S1, ESI†), further validating the reliability of the here reported results.
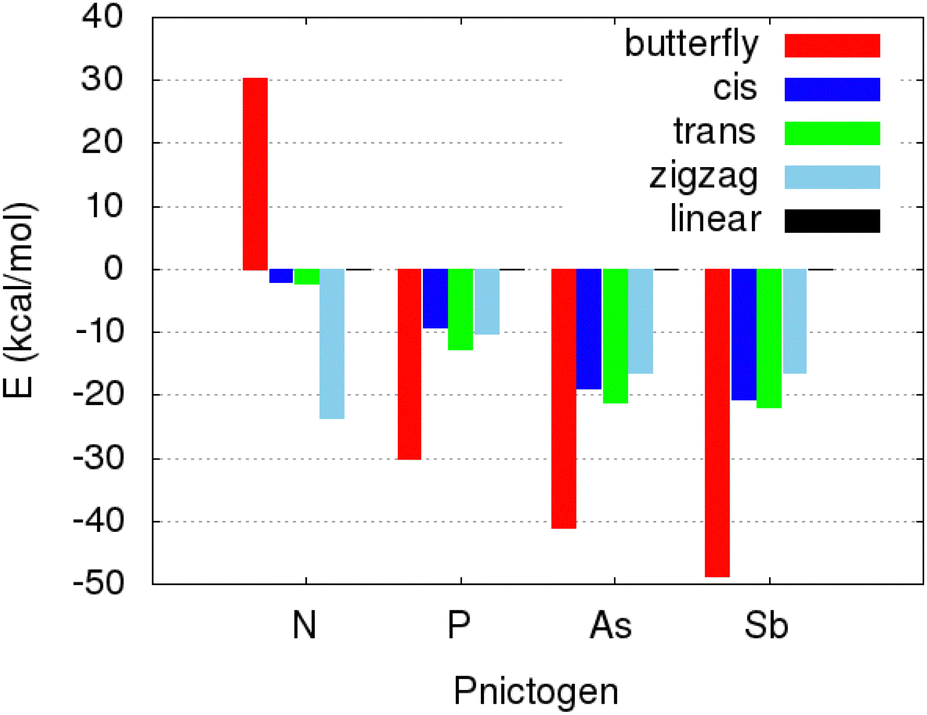 | ||
| Fig. 4 Relative stability, in terms of the electronic energy, of the here considered isomers of DXAs for different pnictogens (X). All the energies are reported, in kcal mol−1, relative to the linear (acetylenic) structure. | ||
These large discrepancies found when going beyond the second period are in well agreement with previously reported trends in the tetrel family, where heavy analogs tend to form more exotic structures than those exhibited by lighter elements.4,60–66 Such an abrupt change in the geometrical features when going beyond the second period is often explained attending to the increasing contribution of high angular momentum flexible orbitals to the underlying chemical bonds.
At this point, it is worth mentioning that the reactivity of DAA and related compounds is commonly understood accounting for the interplay between the more common acetylenic structure and a hidden dicarbenoid/diiminium scaffold, as represented by the resonance forms shown in Fig. 5. Interestingly, this hypothetical intermediate structure is also structurally related to the zig-zag conformer found in the C–C connected isomers.
 | ||
| Fig. 5 Acetylenic (left) and dicarbenoid (right) resonance forms of DAA. | ||
In fact, such a dicarbene character has been reported for other alkyne derivatives,67–69 being highly enhanced by the inclusion of heteroatoms in the α or β positions or when the C≡C interaction is embedded in a non-linear chemical environment.68 In such a way, distortion of the linear alkyne geometry should promote the reactivity at the C atoms, leading to highly active species which can be trapped upon interaction with electron deficient compounds such as Lewis acids. Based on our findings, and in agreement with these trends, the presence of highly localized and directional lone electron pairs, such as those found in N, would ease the distortion of the linear geometries. Such deformation, would in turn localize some electron density to the C atom, increasing the reactivity of the latter and explaining the low stability and promiscuity of DAA, specially when devoid from additional substituents. Altogether, these findings, consistent with the unusually large stability of the zigzag DAA isomer, point out that the contribution of the dicarbenoid form to the whole resonance hybrid of DXAs should decrease when moving down the group.
4.2 Mechanistic study of the linear and zigzag tautomerism
From the previous section, it is evident that the aforementioned dicarbenoid character could be playing an active role in the stabilization of the zigzag isomer and, more importantly, its tautomerism with the linear (acetylenic) analog. Trying to further gain insights on to the origin of the marked differences between N and the heavier pnictogens, we have decided to explore the zigzag/linear tautomerism mechanism between these two structures, paying special attention to the role played by the potential carbenoid character of DXAs. Given that all the pnictogens from the third period or above are expected to exhibit a similar behavior, DAA and DPA will be used as prototypical models, whereas the results for the remaining DXAs are explicitly gathered in the ESI.†Fig. 6 and 7 comprise the stationary states (reactants, transition states, intermediates and products) involved in the DAA and DPA tautomerism reactions whereas Fig. 8 shows the reaction energy profiles for both chemical transformations. In both cases, the tautomerism mechanism seems to proceed by means of a step-wise intra-molecular proton (H+)/hydride (H−) transfer reaction between the XH2 (X = N, P) moieties and the neighboring sp C atom.
 | ||
| Fig. 6 Stationary points involved in the DAA tautomerism reaction. | ||
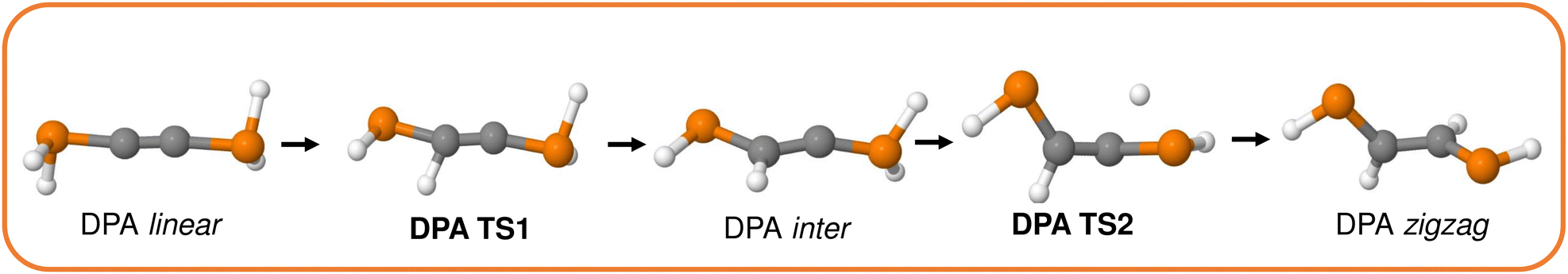 | ||
| Fig. 7 Stationary points involved in the DPA tautomerism reaction. | ||
 | ||
| Fig. 8 Reaction profile for the DXA tautomerism, all values are reported in terms of the Gibbs free energy at 298.15 K in kcal mol−1. The energies are computed relative to that of the isolated references. | ||
A brief inspection of the geometries of the stationary points involved reveals some notorious structural differences, something which becomes specially pronounced in the actual reaction intermediate. More specifically, the DAA intermediate displays a remarkably bent structure around the spectator C atom, characteristic of a sp2 hybridization. This feature, which is also observed to a lower extent in DPA, points out to the formation of a carbene-like structure, in agreement with chemical intuition and previously reported results. Additionally, the activated complexes involved in the first step of the DPA tautomerism strongly resemble the corresponding product, something which is also found for the remaining heavy DXAs (see S2.1, ESI†). Unlike in the case of DAA, this observation highlights the very late nature of the transition states of the former. Actually, these findings are in perfect agreement with the Hammond postulate, as reflected in the energetics of the first H transfer of the reaction energy profile, comprised in Fig. 8 and S2.1 (ESI†). Further insights into the nature of the underlying reaction mechanisms can be obtained from the analysis of the QTAIM electronic metrics throughout the reaction, as gathered in Fig. 10 and S2.2 (ESI†). The nomenclature used throughout the latter, with A and B labels being used to refer to the H, C and X atoms involved in the first and second H transfers, respectively, is shown in Fig. 9.
 | ||
| Fig. 9 Nomenclature used to refer to the C, H and X atoms directly involved in the first (A) and second (B) H transfers under consideration, shown in red and green, respectively. The schematics for DAA (left), DPA (center) and DAsA (right) are collected. | ||
 | ||
| Fig. 10 Evolution of the atomic charges, Q, (top), localization index, LI, (middle) and delocalization index, DI, (bottom) for the main atoms and bonds participating in the first H transfer involved in the DAA (left) and DPA (right) tautomerism reactions. All values are reported in electrons. For Q and LI, the values are reported relative to the reactant complex. The A and B labels are used to indicate those C, H and X atoms directly involved in the first and second H transfers, respectively, as shown in Fig. 9. The black vertical line shows the transition state region. | ||
In the case of DAA, the CA–CB bond experiences a very rapid weakening, as evidenced by the decrease in the electron delocalization, measured by DI (C–C), from 2.4 to just 1.2 electrons in a single step. Such an abrupt drop in the electron population shared between both C atoms would correspond to the annihilation of two standard covalent bonds, going thus from a triple to almost a single covalent interaction. This process is accompanied by a noticeable strengthening of the C–N bonds, something which is particularly pronounced for the atomic pair directly involved in the H transfer (NA–CA) showing DI values of up to 1.7 electrons characteristic of an N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The latter effect is further evidenced by a moderate decrease in the local electron density around the CA atom, resulting in a noticeable increase in its atomic charge of about 0.30 electrons. Such an electronic impoverishment seems to arise almost solely from a drop in its localized electron count, as shown in green in the middle left panel of Fig. 10. In contrast, the neighboring CB atom (not directly involved in the first H transfer) experiences a very large electron localization of almost 0.40 electrons, as highlighted by the monotonously increasing trend shown by the LI (CB), in blue. The latter finding arises as a straightforward manifestation of the presence of a strong electron-rich carbenoid character in the product of the first H transfer of DAA, compatible with the formation of a carbene at the CB atom of DAA inter. Furthermore, the first H transfer in DAA seems to proceed in an almost perfectly concerted way, with the synchronous NA–HA and CA–HA bond cleavage and formation, respectively. In fact, this is clearly reflected in the evolution of the electron delocalization, comprised in the left panel of Fig. 12, which shows that, in the TS, the HA atom exhibits two half-formed covalent bonds with ≈0.5 electrons each.
C double bond. The latter effect is further evidenced by a moderate decrease in the local electron density around the CA atom, resulting in a noticeable increase in its atomic charge of about 0.30 electrons. Such an electronic impoverishment seems to arise almost solely from a drop in its localized electron count, as shown in green in the middle left panel of Fig. 10. In contrast, the neighboring CB atom (not directly involved in the first H transfer) experiences a very large electron localization of almost 0.40 electrons, as highlighted by the monotonously increasing trend shown by the LI (CB), in blue. The latter finding arises as a straightforward manifestation of the presence of a strong electron-rich carbenoid character in the product of the first H transfer of DAA, compatible with the formation of a carbene at the CB atom of DAA inter. Furthermore, the first H transfer in DAA seems to proceed in an almost perfectly concerted way, with the synchronous NA–HA and CA–HA bond cleavage and formation, respectively. In fact, this is clearly reflected in the evolution of the electron delocalization, comprised in the left panel of Fig. 12, which shows that, in the TS, the HA atom exhibits two half-formed covalent bonds with ≈0.5 electrons each.
On the other hand, DPA seems to undergo a different electron redistribution throughout the tautomerism: the CA–CB delocalization drops only to about 1.5 electrons, corresponding to an intermediate bond order between a C–C and a CC scenario. At the same time, besides the previously mentioned electron localization experienced by the spectator CB atom, the localized electron count of the main PA atom gets notably enhanced at the expense of decreasing that of its neighboring analog, PB (shown in black and red, respectively). Moreover, such a drift in the electron localization promotes a net transfer of electron density between both P atoms of about −0.8 and +0.4 electrons, evidencing an increase in the Zwitterionic nature of the resultant intermediate. It is worth noticing that a similar electronic redistribution is found for DAsA and DSbA, as reflected in Fig. S14–S16 (ESI†). Additionally, the drop in the CA–CB delocalized electron population accompanying the first H transfer becomes less pronounced throughout the P > As > Sb series, with limiting values of 1.50, 1.75 and 2.25 electrons for the reaction intermediates. Moreover, and unlike in the case of DAA, the very late nature of the first transition state in DPA results in almost fully depleted and formed PA–HA and CA–HA bonds, respectively. These trends are further reinforced by the DI metrics, comprised in the right panel of Fig. 12, which reveal that the activated complex exhibits fully formed CA–HA contacts, as evidenced by the almost 0.8 electrons shared between both atoms at this state of the reaction.
These observations evidence that, in agreement with our initial hypothesis, DAA and DPA (along with the remaining DXAs) follow different mechanistic pathways for the tautomerism process, as proposed in Fig. 11 and 15. We note in passing that the intricacies of the here presented transformations cannot be adequately represented by classical electron push–pull diagrams and so the here proposed mechanisms are just an educated approximation. For DAA, the electron density attributed to the original C≡C bond would shift to produce an imine group with the simultaneous intra-molecular proton-like H transfer to the sp2 CB atom. The latter rearrangement is likely to be assisted by the highly contributing ionic forms of the reactant. Simultaneously, this process triggers an electron flow chain reaction, depicted in Fig. 11, which ultimately results in the inevitable localization of electron density around the spectator CB atom, affording, thus, a carbene-like intermediate, in accordance with previously reported studies.67–69 Such a carbene character is further supported by the ELF's topology of the DAA intermediate, shown in the left panel in Fig. 13, which reveals the presence of a very well-defined (highly localized and particularly prominent) valence basin on the CB atom, V(CB), with an estimated population of ≈2.1 electrons (S2.3, ESI†). Actually, the topology of the latter is identical to that shown by an amino-stabilized singlet (S = 0) carbene, see Tables S45 and S46 in S5.1 (ESI†), with an estimated population of 2.2 electrons for V(CB).
 | ||
| Fig. 11 Mechanistic proposal for the DAA tautomerism. Dashed arrows are used to indicate the electron rearrangement involved in the interconversion between different resonance structures. The terms Neutral and Ionic are used to refer to different resonance structures of the DAA linear isomer. | ||
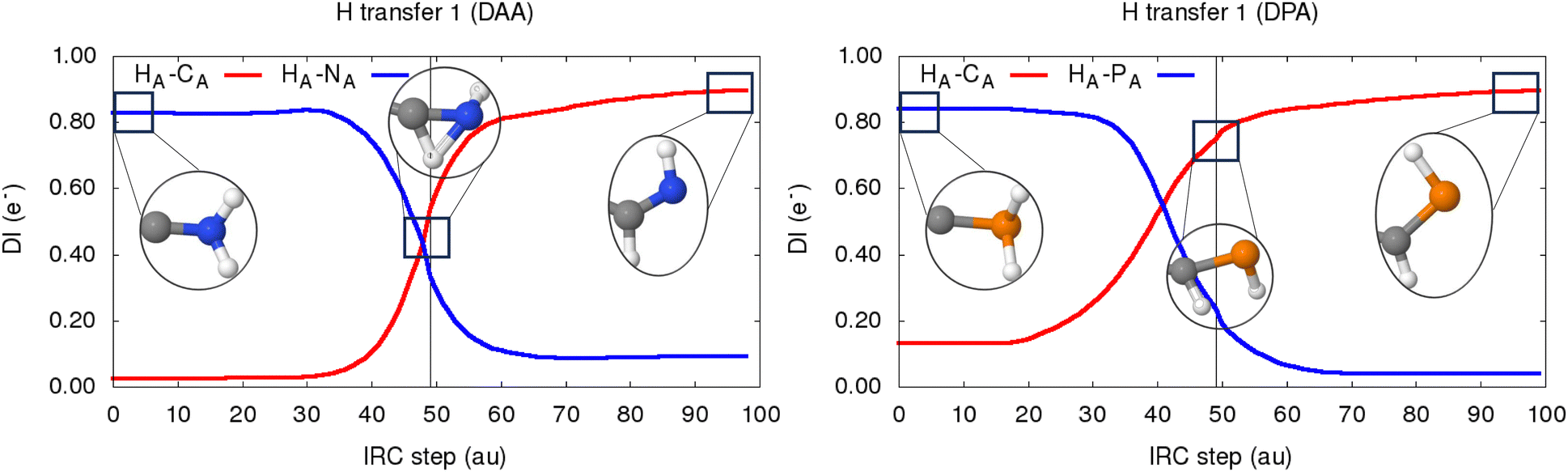 | ||
| Fig. 12 Evolution of the number of delocalized electrons, as measured by the Delocalization Index (DI), between the XA–HA and CA–HA atomic pairs throughout the first H transfer of the DAA (left) and DPA (right) tautomerisms. | ||
 | ||
| Fig. 13 ELF = 0.86 isosurfaces of the DAA (left) and DPA (right) intermediates formed upon the first H transfer reaction. | ||
Trying to shed light on to the true nature of such reaction intermediate, the evolution of the local spin of the CB atom, 〈SCB2〉, was monitored, as shown in Fig. 14. At this point, it should be mentioned that even a naive classical picture allows us to get a grasp of the expectation values of the corresponding local spins. As such, standard homopolar covalent bonds contribute with  to the local spin of an atom. Whereas a radical (↑ or ↓), singlet carbene (↑↓) and triplet carbene (↑↑ or ↓↓) would account for
to the local spin of an atom. Whereas a radical (↑ or ↓), singlet carbene (↑↓) and triplet carbene (↑↑ or ↓↓) would account for 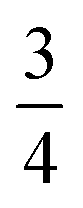 , 0 and 2, respectively. It is worth noting that in real systems the spin statistics will be biased by different phenomena, such as the shift of the ionic contributions to the bonding arising from the electronegativity differences between the bonded atoms, which will perturb the aforementioned 〈SA2〉 values. Starting from an almost pure covalent bonding
, 0 and 2, respectively. It is worth noting that in real systems the spin statistics will be biased by different phenomena, such as the shift of the ionic contributions to the bonding arising from the electronegativity differences between the bonded atoms, which will perturb the aforementioned 〈SA2〉 values. Starting from an almost pure covalent bonding  , as shown in the left panel of Fig. 14, the CB atom of DAA undergoes a very notorious decrease in its local spin. Indeed, the latter drops down to about 〈S2〉 ≈ 1.10 in the DAA intermediate. The partial depletion of some of the original covalent bonds involving the CB atom, coupled with the enhancement of these resonance forms with a dominant electron pairing, would explain such a notorious impoverishment in the local spin values. As such, these findings, being in perfect agreement with the expected values for an amino-stabilized carbene (〈SCB2〉 ≈ 1.0, see Table S50, ESI†), evidence the formation of a singlet (S = 0) carbene character at the CB throughout the first H transfer of the DAA tautomerism. For the same reason, the HA atom being transferred in the processes undergoes a subtle increase in its local spin, going from a polar chemical interaction to the ideal covalent bond attributed to the C–H pair,
, as shown in the left panel of Fig. 14, the CB atom of DAA undergoes a very notorious decrease in its local spin. Indeed, the latter drops down to about 〈S2〉 ≈ 1.10 in the DAA intermediate. The partial depletion of some of the original covalent bonds involving the CB atom, coupled with the enhancement of these resonance forms with a dominant electron pairing, would explain such a notorious impoverishment in the local spin values. As such, these findings, being in perfect agreement with the expected values for an amino-stabilized carbene (〈SCB2〉 ≈ 1.0, see Table S50, ESI†), evidence the formation of a singlet (S = 0) carbene character at the CB throughout the first H transfer of the DAA tautomerism. For the same reason, the HA atom being transferred in the processes undergoes a subtle increase in its local spin, going from a polar chemical interaction to the ideal covalent bond attributed to the C–H pair,  , as shown in orange in the right panel of Fig. 14.
, as shown in orange in the right panel of Fig. 14.
 | ||
| Fig. 14 (left) Evolution of the local spin, 〈SA2〉, of the CB throughout the first (H 1) and second (H 2) H transfers involved in the DAA and DPA tautomerism reactions. (right) Evolution of the local spin of the HA and HB atoms throughout the first (1) and second (2) H transfers involved in the DAA and DPA tautomerism reactions. For the H atom, a representation of the ELF = 0.9 isosurface of the local maxima and minima in the evolution of 〈SA2〉, marked with black squares, is also shown. | ||
On the other hand, the more accessible lone electron pair of the PB atom of the spectator PH2 moiety would assist the H transfer in DPA. In this scenario, using a simple electron push–pull diagram allows us to quickly obtain a mechanistic proposal yielding a keteniminium intermediate (DPA inter Ionic B in Fig. 15) with a prominent charge separation which would be in equilibrium with its carbenoid analog, as shown in Fig. 15.
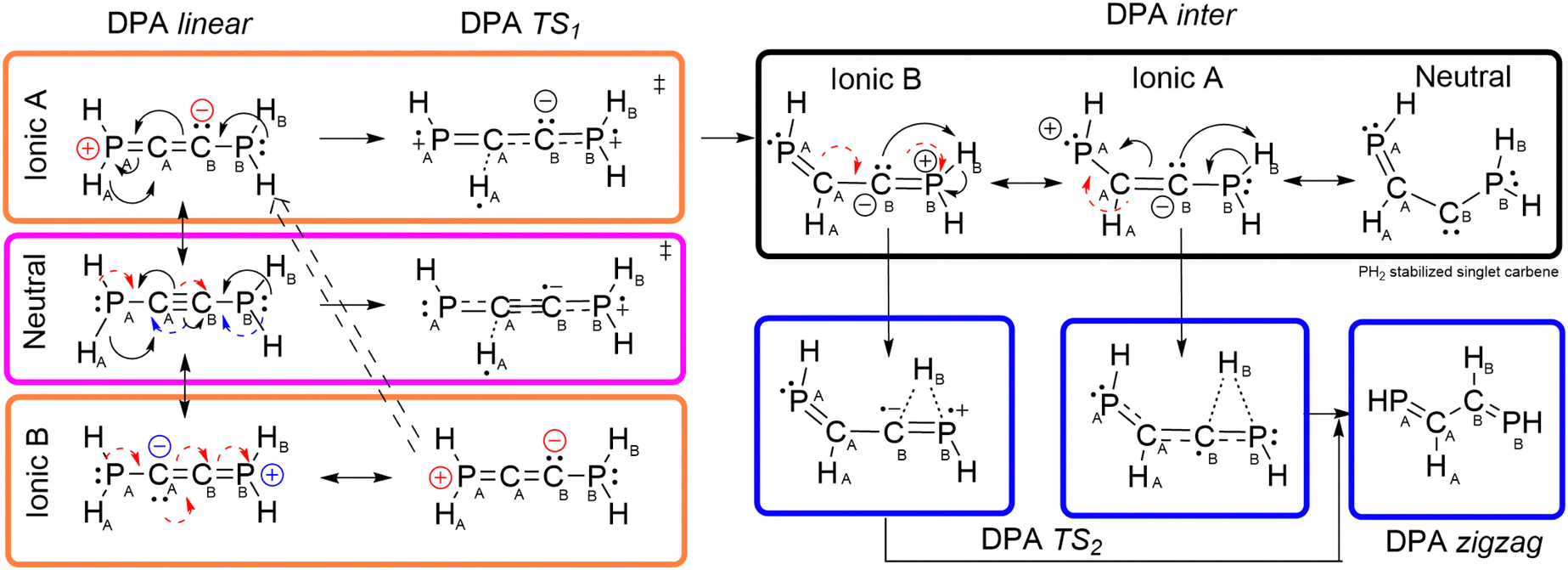 | ||
| Fig. 15 Mechanistic proposal for the DPA tautomerism. Dashed arrows are used to indicate the electron rearrangement involved in the interconversion between different resonance structures. The terms Neutral and Ionic are used to refer to different resonance structures of the DPA linear isomer. | ||
In this regard, the much smaller and distorted V(CB) ELF basin, shown in the right panel of Fig. 13 and integrating to about 1.8 electrons (S2.3, ESI†), coupled to the considerably less bent structure of the C–C–PH2 scaffolds, suggests a lower dominance of the carbene structure for the stability of the DPA intermediate. Similar findings are observed for the DAsA and DSbA analogs, showing distorted and underpopulated basins for the CB atom in the reaction intermediates (see S2.3, ESI†). Actually, these trends are also in agreement with the evolution of the local spins in DPA, shown in magenta in Fig. 14. Interestingly, the CB carbon experiences only a slight drop in its local spin, being very far from the expected values for a singlet phosphino carbene (see Table S50, ESI†). The latter seems to be quenched by the very polar C–P bonds, assisting the dominant zwitterionic form of the DXA species.
A significant observation emerges from the local spin behavior of the migrating H atoms during the involved H transfers. In contrast to DAA, where 〈SH2〉 exhibits a steady increase, this quantity shows a notable spike near the DPA transition states. This spike, with limiting values of approximately 0.48 a.u., is associated with the formation of a covalent CA–HA bonding from a hydride-like H atom. In this scenario, the ionic contribution of the proton-like H+ bonding increases at the expense of the originally dominant hydride-like form (H−). Despite both forms having null contributions to local spin (〈SH+2〉 = 〈SH−2〉 = 0), their interchange promotes electron delocalization, decreasing fully localized electron counts. This interplay results in the formation of a fully delocalized H radical with  , causing the observed spike in 〈SH2〉. This reasoning aligns with trends in the ELF field shown in Fig. 14, leading to unbound V(HA) basins with an estimated population of approximately 1.6 electrons (see Table S9, ESI†). The shift in the reaction mechanism is further emphasized by the distinct character of the H atoms in DAA and DPA, behaving essentially as H+ and H−, respectively, as evidenced by their local charges of +0.38 and −0.50 electrons, respectively, in the linear isomers. This hydride character extends to other pnictogens, with atomic charges of −0.28 and −0.32 electrons for H atoms in the linear isomers of DAsA and DSbA, respectively.
, causing the observed spike in 〈SH2〉. This reasoning aligns with trends in the ELF field shown in Fig. 14, leading to unbound V(HA) basins with an estimated population of approximately 1.6 electrons (see Table S9, ESI†). The shift in the reaction mechanism is further emphasized by the distinct character of the H atoms in DAA and DPA, behaving essentially as H+ and H−, respectively, as evidenced by their local charges of +0.38 and −0.50 electrons, respectively, in the linear isomers. This hydride character extends to other pnictogens, with atomic charges of −0.28 and −0.32 electrons for H atoms in the linear isomers of DAsA and DSbA, respectively.
Besides supporting our mechanistic proposals, the latter findings pave the way towards unveiling novel chemical routes for the obtention of radical species. To the best of our knowledge, this radical character in DXAs has not been experimentally reported yet, however, we expect carbanion-like scaffolds to undergo similar spikes in their spin when transitioning from a highly polar bond to a more covalent-rich interaction. The emerging radical character that accompanies this transformation could give rise to highly reactive pseudo-intermediaries, opening the door to previously unknown patterns of reactivity.
Accounting for the very different nature of DAA and the remaining DXA intermediates, the second H transfer to yield the final zigzag structure would follow drastically different mechanistic proposals: in DAA, the lone electron pair of the carbene traps HB of the neighboring NH2 moiety to yield a second imine scaffold and leaving behind a plain CA–CB single bond. This is in consonance with the evolution of the QTAIM metrics, S2.2 (ESI†), showing a remarkable boost in the DI (CB–NB) values at the expense of decreasing the electron population of the carbenoid CB and the C–C interaction. Instead, the lone electron pair of the PA atom in DPA induces an electron redistribution cascade to strengthen the PB–CB and PA–CA bonds in the final product while weakening the C–C interaction, see S2.2 (ESI†). Such a shift results in a prominent increase in the localized electron count on the PB atom and, with it, a local accumulation of electron density on this (previously cation-like) P atom, in agreement with the previously proposed mechanistic paths, shown in Fig. 15.
4.3. Electrostatically driven ring closure
The large charge separation attributed to the increase of the Zwitterionic character of the previously shown reaction intermediate when moving down the group opens the way towards alternative tautomerism reactions. Indeed, in the particular case of the DAsA and DSbA reactions, the product of the first H transfer depicted in Fig. 8 is not a true minimum of the PES. Instead, the latter structure tends to collapse in an electrostatically driven process to yield a cyclic isomer. In this section, we will study this additional ring closure phenomenon and its impact on the net linear to zigzag isomerization, using the DAsA as a prototypical model.For such a purpose, extended IRC calculations were performed starting from the TS structures of the first H transfer involved in the DAsA tautomerism, as comprised in Fig. S7 (S2.1, ESI†). The evolution of the geometrical features throughout the latter reveals that, beyond the plateau of the plain reaction intermediates, the system undergoes a very pronounced decrease in the electronic energy as represented in Fig. 16. Such an extra stabilization, accounting for ≈35 kcal mol−1 for both DAsA and DSbA, corresponds to a barrierless transformation to yield a stable three-membered ring, resembling the more common aziridines, as gathered in the embedded panel of Fig. 16.
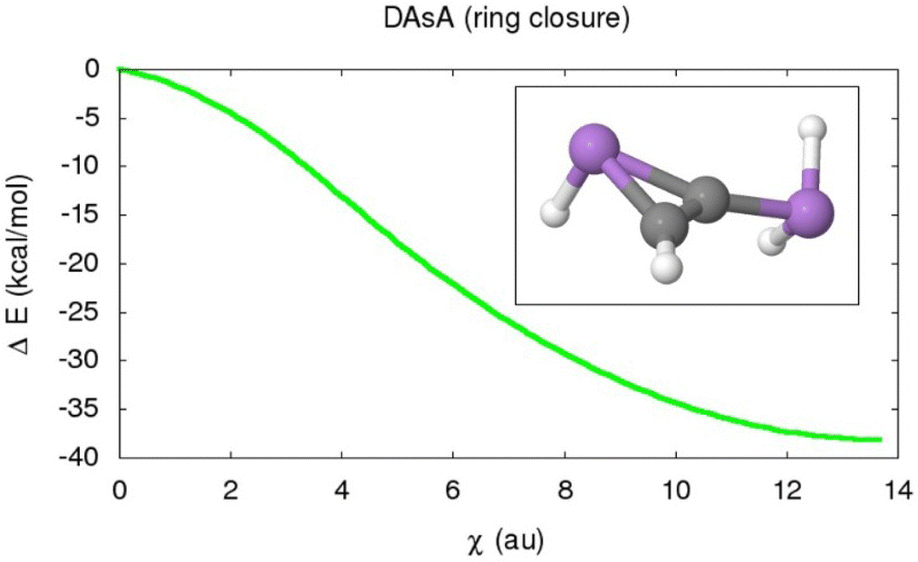 | ||
| Fig. 16 Evolution of the electronic energy of the DAsA reaction intermediate throughout the ring-closure phenomenon to form a stable cyclic structure. The x-axis is reported in terms of the evolution of the reaction coordinate attributed to the ring closure reaction (χ), reported in atomic units. | ||
Additionally, the resultant mono-bridged ring resembles previously reported stable isomers of heavy tetrel derivatives.63 On the other hand, the lack of a saddle point connecting the open and closed DAsA intermediate points out that their interconversion may be driven by the mere Coulombic attraction arising from the prominent buildup of partial charges.
The evolution of the total and localized electron counts, shown in Fig. 17, reveals that the ring closure is assisted by the fuzzy lone electron pair of the AsA atom. Actually, the latter undergoes a quite remarkable decrease in its local electron density, as reflected by the increase in the partial charge of about 0.17 electrons. This finding is accompanied by a noticeable interplay of the localized electron counts of both As atoms: LI (AsB) is enhanced at the expense of decreasing the LI (AsA) and LI (CB) values.
 | ||
| Fig. 17 Evolution of the change in atomic charges (left) and localization index (right) for the main atoms and bonds involved in the DAsA ring closure phenomenon. All values are reported, in electrons, relative to the starting non-cyclic intermediate. The nomenclature used to refer to the atoms is shown in Fig. 9. | ||
Furthermore, the cyclization phenomenon reinforces the C–C interaction while weakening the CB–AsB and CA–AsA bonds, as pointed out by the large fluctuations found in the electron delocalization, comprised in Table 1. Altogether, these results, also observed in the case of DSbA (see S2.2, ESI†), allow us to depict a reasonable mechanism for this transformation, as represented in Fig. 18. The original Zwitterionic (open) intermediate would be in equilibrium with a 1,3-dipole resulting in a prominent charge separation between the AsA and CB fragments. In such a resonance form, the negative formal charge at the former will get partially delocalized to form the emerging AsA–CB bond. This process, which will eventually result in the ring closure, promotes the accumulation of localized electron density at the AsB atom at the expense of weakening the original CB–AsB and CA–AsA interactions which become single C–X bonds, with DIs slightly below 1.0 electrons (see S2.2, ESI†).
| Atom | ΔQ | ΔLI | Bond | ΔDI |
|---|---|---|---|---|
| AsA | 0.17 | −0.21 | CA–AsA | −0.30 |
| AsB | −0.06 | 0.16 | CA–CB | 0.25 |
| CA | 0.02 | 0.00 | CB–AsB | −0.19 |
| CB | −0.09 | −0.16 |
 | ||
| Fig. 18 Mechanistic proposal for the electrostatically driven ring closure of the DAsA reaction intermediate. The term Ionic is used to refer to a particular resonance structure of DAsA. | ||
The latter mechanistic proposal, consistent with the QTAIM metrics, proves that, as expected, this ring closure tautomerism is electrostatically driven, explaining the lack of an actual reaction barrier. Furthermore, the cyclization of the intermediate reinforces the C–C interaction by about 0.25 electrons leading to an almost pure CC bond (DIs of 2.00 electrons, see Fig. S22 and S23, ESI†). This observation would annihilate the previously observed carbene character, in agreement with the aforementioned drop in the electron localization at the C atom. Indeed, this is further evidenced by the ELF topology of the cyclic intermediate, right panel of Fig. 19, showing how V(C), characteristic of the carbene, disappears upon cyclization.
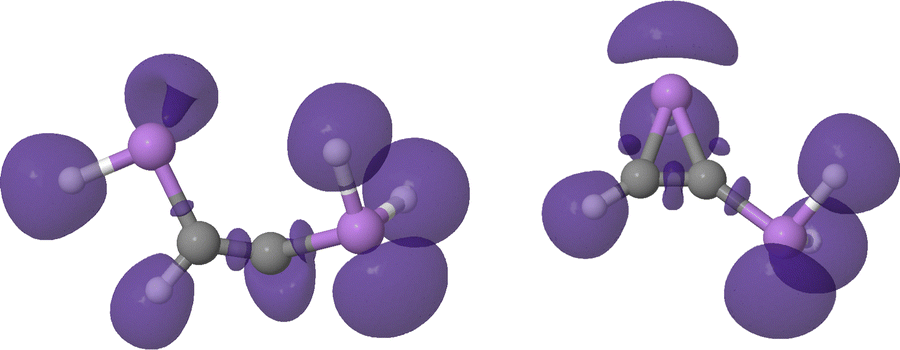 | ||
| Fig. 19 ELF = 0.86 isosurfaces of the open (left) and cyclic (right) intermediate structure of the DAsA tautomerism reaction. | ||
5. Conclusions
Acetylenes are fundamental organic synthons in state-of-the-art chemistry, being crucial building blocks for the synthesis of a wide range of valuable compounds. Decorating their skeleton with different functional groups, such as heteroatom-containing scaffolds, can dramatically tune their reactivity and stability, something which becomes particularly handy for the obtention of tailor-made molecules. However, the features and properties of these systems vary strongly when moving down a group, with the second-period elements behaving in a diametrically different way when compared to their heavier analogs. In this work, several high-level theoretical chemistry tools are used to shed light on to the origin of these differences, using dipnictogenoacetylenes (DXAs) as prototypical models.As for the potential energy landscape, the structural isomers of DAA show completely different relative stabilities when compared to the remaining DXAs. Whereas the doubly bridged butterfly isomer is the most stable conformation for the latter, the global energy minimum for DAA was found to be the unusual zigzag structure, resembling the dicarbenoid/diiminium scaffold often claimed to be responsible for the peculiar reactivity of DAAs. It should be noted that the linear isomer, corresponding to the common acetylenic scaffold, did not represent the most stable species for any of the DXAs.
Additional insights into the origin of these marked differences were obtained through the computation of the interconversion pathways between the linear and zigzag conformers. The study of the tautomerism reactions revealed that DAA and other DXAs proceed by means of step-wise proton-like (H+) and hydride-like (H−) reaction mechanisms, respectively. The intra-molecular H migrations are assisted by the original C≡C electron density which promotes a notorious accumulation of localized electron count at the β carbon (not directly involved in the first H migration). Such an electron enrichment results in the formation of a singlet carbene in the reaction intermediate, clearly evidenced by the ELF and local spin calculations. Moving down the group enhances the zwitterionic character of the DXAs at the expense of reducing their carbenoid nature. Actually, these trends result in a significant build up of local charges in the product of the first H transfer, increasing across the N > P > As > Sb series. For As and above, the charge separation becomes large enough to yield an electrostatically driven ring closure, resulting in the formation of highly stabilized three-membered heavy aziridine-like derivatives.
Finally, the transition from a weakly bounded hydride (H−) in the X–H bonding of heavy DXAs to the vanilla C–H covalent bonding in the zigzag isomers inevitably results in the spurious formation of a short-lived delocalized H˙ radical. Hence, not only do our results shed light on the pnictogen effects on simple acetylene derivatives, but they also pave the way towards new routes for the obtention of tailor-made radical species with potential synthetic applicability.
Author contributions
All authors contributed equally to the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish MICIU, grants PID2021-122763NB-I00 and PID2020-113473GB-I00, for financial support. M. G. specially thanks the Spanish MICIU for the predoctoral FPU grant, FPU19/02903.References
- C. Weetman, Chem. – Eur. J., 2020, 27, 1941–1954 CrossRef PubMed.
- J. Klekota and F. P. Roth, Bioinformatics, 2008, 24, 2518–2525 CrossRef CAS PubMed.
- A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed.
- M. Bogey, H. Bolvin, C. Demuynck and J. Destombes, Phys. Rev. Lett., 1991, 66, 413–416 CrossRef CAS PubMed.
- K. M. Schueller, H. F. Mull, J. T. Turney and H. F. Schaefer, Isr. J. Chem., 2023, 63, e202300033 CrossRef CAS.
- K. Gilmore and I. V. Alabugin, Chem. Rev., 2011, 111, 6513–6556 CrossRef CAS PubMed.
- U. Wille, Chem. Rev., 2012, 113, 813–853 CrossRef PubMed.
- Y. Yamamoto, Chem. Soc. Rev., 2014, 43, 1575–1600 RSC.
- R. Chinchilla and C. Nájera, Chem. Rev., 2013, 114, 1783–1826 CrossRef PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed.
- G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- T. T. Talele, J. Med. Chem., 2020, 63, 5625–5663 CrossRef CAS PubMed.
- C. Lamberth, Bioorg. Med. Chem., 2009, 17, 4047–4063 CrossRef CAS PubMed.
- Y. Gao, C. Feng, T. Seo, K. Kubota and H. Ito, Chem. Sci., 2022, 13, 430–438 RSC.
- Y. Liu, J. W. Y. Lam and B. Z. Tang, Natl. Sci. Rev., 2015, 2, 493–509 CrossRef CAS.
- M. M. Heravi, M. Dehghani, V. Zadsirjan and M. Ghanbarian, Curr. Org. Synth., 2019, 16, 205–243 CrossRef CAS PubMed.
- A. Ramani, B. Desai, M. Patel and T. Naveen, Asian J. Org. Chem., 2022, 11, e202200047 CrossRef CAS.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS PubMed.
- A. Ramani, B. Desai, B. Z. Dholakiya and T. Naveen, Chem. Commun., 2022, 58, 7850–7873 RSC.
- Q. Wang, S. Chittaboina and H. Barnhill, Lett. Org. Chem., 2005, 2, 293–301 CrossRef CAS.
- X. Yang, L. P. Beroske, J. Kemmink, D. T. Rijkers and R. M. Liskamp, Tetrahedron Lett., 2017, 58, 4542–4546 CrossRef CAS.
- A. Mukherjee, R. B. Dateer, R. Chaudhuri, S. Bhunia, S. N. Karad and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 15372–15375 CrossRef CAS PubMed.
- K. Mishiro, T. Kimura, T. Furuyama and M. Kunishima, Org. Lett., 2019, 21, 4101–4105 CrossRef CAS PubMed.
- M. Z. C. Hatit, C. P. Seath, A. J. B. Watson and G. A. Burley, J. Org. Chem., 2017, 82, 5461–5468 CrossRef CAS PubMed.
- M. Z. C. Hatit, J. C. Sadler, L. A. McLean, B. C. Whitehurst, C. P. Seath, L. D. Humphreys, R. J. Young, A. J. B. Watson and G. A. Burley, Org. Lett., 2016, 18, 1694–1697 CrossRef CAS PubMed.
- J. Kim and S. S. Stahl, J. Org. Chem., 2015, 80, 2448–2454 CrossRef CAS PubMed.
- S. Conejero, J. Díez, M. P. Gamasa, J. Gimeno and S. García-Granda, Angew. Chem., Int. Ed., 2002, 41, 3439–3442 CrossRef CAS.
- L. Hackl, L. Körner and M. Tamm, Chem. Lett., 2021, 50, 1428–1440 CrossRef CAS.
- A. J. Carty, N. K. Hota, T. W. Ng, H. A. Patel and T. J. O. Connor, Can. J. Chem., 1971, 49, 2706–2711 CrossRef CAS.
- N. Terashima, Y. Sakata, T. Meguro, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 14003–14006 RSC.
- T. Görlich, D. S. Frost, N. Boback, N. T. Coles, B. Dittrich, P. Müller, W. D. Jones and C. Müller, J. Am. Chem. Soc., 2021, 143, 19365–19373 CrossRef PubMed.
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1345 Search PubMed.
- P. L. A. Popelier, Challenges and Advances in Computational Chemistry and Physics, Springer International Publishing, 2016, pp. 23–52 Search PubMed.
- R. Bader and R. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1990 Search PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and R. J. Gillespie, The ELF Topological Analysis Contribution to Conceptual Chemistry and Phenomenological Models, 2007 Search PubMed.
- J. Contreras-García, Á. Martín Pendás, B. Silvi and J. M. Recio, J. Phys. Chem. Solids, 2008, 69, 2204–2207 CrossRef.
- J. Melin and P. Fuentealba, Int. J. Quantum Chem., 2003, 92, 381–390 CrossRef CAS.
- J. Andrés, V. S. Safont, M. Oliva, K. L. Caster and F. Goulay, Int. J. Quantum Chem., 2022, 122, e26892 CrossRef.
- A. E. Clark and E. R. Davidson, J. Chem. Phys., 2001, 115, 7382–7392 CrossRef CAS.
- I. Mayer, Chem. Phys. Lett., 2007, 440, 357–359 CrossRef CAS.
- E. Ramos-Cordoba, E. Matito, I. Mayer and P. Salvador, J. Chem. Theory Comput., 2012, 8, 1270–1279 CrossRef CAS PubMed.
- Á. Martín Pendás and E. Francisco, Phys. Chem. Chem. Phys., 2021, 23, 8375–8392 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. H. Hertwig and W. Koch, Chem. Phys. Lett., 1997, 268, 345–351 CrossRef CAS.
- D. Jiang, M. Fu, Y. Zhang, Q. Li, K. Guo, Y. Yang and L. Zhao, J. Comput. Chem., 2019, 41, 279–289 CrossRef PubMed.
- S. Chen, L. Liu, X. Gao, Y. Hua, L. Peng, Y. Zhang, L. Yang, Y. Tan, F. He and H. Xia, Nat. Commun., 2020, 11, 4651 CrossRef CAS PubMed.
- B. Lin, H. Liu, I. Karki, E. C. Vik, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem., Int. Ed., 2023, 62, e202304960 CrossRef CAS PubMed.
- C. Trujillo, G. Sánchez-Sanz, I. Alkorta and J. Elguero, Struct. Chem., 2016, 28, 345–355 CrossRef.
- J. Brzeski, Polyhedron, 2022, 227, e116145 CrossRef.
- T. Tandarić and R. Vianello, J. Phys. Chem. A, 2018, 122, 1464–1471 CrossRef PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- S. Maeda, Y. Harabuchi, Y. Ono, T. Taketsugu and K. Morokuma, Int. J. Quantum Chem., 2014, 115, 258–269 CrossRef.
- A. Martín Pendás and E. Francisco, Promolden. A QTAIM/IQA code (Avaliable from the authors upon request) Search PubMed.
- T. A. Keith, AIMALL, TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- R. P. F. Kanters and K. J. Donald, J. Chem. Theory Comput., 2014, 10, 5729–5737 CrossRef CAS PubMed.
- Jmol: an open-source Java viewer for chemical structures in 3D. https://www.jmol.org/.
- D. E. Goldberg, D. H. Harris, M. F. Lappert and K. M. Thomas, J. Chem. Soc., Chem. Commun., 1976, 261–262 RSC.
- D. E. Goldberg, P. B. Hitchcock, M. F. Lappert, K. M. Thomas, A. J. Thorne, T. Fjeldberg, A. Haaland and B. E. R. Schilling, J. Chem. Soc., Dalton Trans., 1986, 2387–2394 RSC.
- K. Raghavachari, J. Chem. Phys., 1988, 88, 1688–1702 CrossRef CAS.
- M. Lein, A. Krapp and G. Frenking, J. Am. Chem. Soc., 2005, 127, 6290–6299 CrossRef CAS PubMed.
- H. F. Mull, P. R. Franke, C. Sargent, G. E. Douberly, J. M. Turney and H. F. S. III, Mol. Phys., 2021, 119, 21–22 CrossRef.
- G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC.
- G. N. Srinivas and M. Schwartz, Inorg. Chim. Acta, 2009, 362, 2172–2176 CrossRef CAS.
- G. Haberhauer and R. Gleiter, J. Am. Chem. Soc., 2013, 135, 8022–8030 CrossRef CAS PubMed.
- P. V. Bijina and C. H. Suresh, ChemPhysChem, 2018, 19, 3266–3272 CrossRef CAS PubMed.
- D. C. Georgiou, L. Zhao, D. J. D. Wilson, G. Frenking and J. L. Dutton, Chem. – Eur. J., 2017, 23, 2926–2934 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06324k |
| This journal is © the Owner Societies 2024 |