
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational design of metal hydrides on a defective metal–organic framework HKUST-1 for ethylene dimerization†
Karam
Hashem
 ab,
Ramakrishna
Krishnan
a,
Kuiwei
Yang
a,
Bai Amutha
Anjali
a,
Yugen
Zhang
b and
Jianwen
Jiang
*a
ab,
Ramakrishna
Krishnan
a,
Kuiwei
Yang
a,
Bai Amutha
Anjali
a,
Yugen
Zhang
b and
Jianwen
Jiang
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 117576, Singapore. E-mail: chejj@nus.edu.sg
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pasek Road Jurong Island, 627833, Singapore
First published on 16th January 2024
Abstract
Catalytic ethylene dimerization to 1-butene is a crucial reaction in the chemical industry, as 1-butene is used for the production of most common plastics (e.g., polyethylene). With well-defined tuneable structures and unsaturated active sites, defective metal–organic frameworks have recently emerged as potential catalysts for ethylene dimerization. Herein, we computationally design a series of metal hydrides on defective HKUST-1 namely H-M-DHKUST-1 (M: Co, Ni, Cu, Ru, Rh and Pd), and subsequently assess their catalytic activity for ethylene dimerization by density functional theory calculations. Due to the antiferromagnetic behavior of dimeric metal-based clusters, we comprehensively investigate all possible multiplicity states on H-M-DHKUST-1 and observe multiplicity crossing. The ground-state reaction barriers for four elementary steps (initiation, C–C coupling, β-hydride elimination and 1-butene desorption) are rationalized and C–C coupling is revealed to be the rate-determining step on H-Co-, H-Ni-, H-Ru-, H-Rh- and H-Pd-DHKUST-1. The energy barrier for β-hydride elimination is found to be the lowest on H-Ru- and H-Rh-DHKUST-1, attributed to the weak stability of agostic arrangement; however, the energy barrier for 1-butene desorption is the highest on H-Rh-DHKUST-1. Among the designed H-M-DHKUST-1, Co- and Ni-based ones are predicted to exhibit the best overall catalytic performance. The mechanistic insights from this study may facilitate the development of new MOFs toward efficient ethylene dimerization and other industrially important reactions.
1. Introduction
Light olefins are the primary feedstock of many petrochemical and refinery processes with an annual production of over 160 million tons.1 In particular, ethylene receives the major focus in steam cracking plants and accounts for ∼40% of the global market of light olefins.2 In the last decade, ethylene transformation into more valuable products like linear alpha olefins (LAO) has increased considerably from 7 million metric tons per year (Mtpy) in 2016 to more than 8.5 Mtpy in 2021. Ethylene dimerization into 1-butene is a key process in the production of polyethylene, as 1-butene is used to produce common plastics, e.g., linear low-density polyethylene (LLDPE) and high-density polyethylene (HDPE). Commercially, ethylene dimerization to 1-butene is achieved on homogenous catalysts in the presence of aluminium activators.3–6 These homogeneous Ziegler–Natta catalysts are highly selective, but lack recyclability and require environmentally undesirable solvents and activators. Easily recyclable and separable heterogeneous catalysts have gained prominence in olefin dimerization and oligomerization, particularly for ethylene.7–9 While their development has flourished in recent decades, as listed in Table S1 (ESI†), only a few exhibit competitive reactivity and 1-butene selectivity compared to homogeneous counterparts. For example, alumina and silica-supported metal oxides successfully promote butene production, but often with undesired oligomerization and isomerization, thus hindering selective dimerization to 1-butene; the yield of 1-butene is strictly dependent on the acidity of metal oxides, which causes 1-butene to form isomeric 2-butene.7 As a result, industrial applications of heterogeneous catalysts on such metal oxides have not been realized.Metal–organic frameworks (MOFs) are a class of hybrid nanoporous materials, in which metal clusters are assembled with organic ligands to form extended networks.10 With readily tuneable structures and functionalities, MOFs possess an unprecedentedly large degree of design flexibility from diverse metal and organic building blocks. These salient features have attracted considerable interest in using MOFs for many potential applications. In MOFs, organic linkers or metal nodes can be truncated, thus producing defective MOFs (DMOFs) with multiple pore sizes and/or vacancies. Generally, there are two procedures to synthesize DMOFs: a de novo method by truncating linkers during synthesis and post-synthetic modification of pristine MOFs to eliminate chemisorbed coordination modulators.11 Common DMOFs include HKUST-1,12,13 MOF-5,14 UiO-6615,16 and MIL-101.17–19
With readily accessible open metal sites as well as rich electronic configurations and structural environments, DMOFs have demonstrated high potential for selective ethylene dimerization and oligomerization.20 Despite the inherent instability of HKUST-1,21,22 advances in synthesis technologies persistently facilitate the creation of defective HKUST-1 catalysts.23 Using a mixed ligand approach, Fischer and coworkers introduced defects into Ru-HKUST-1 with one linker (benzene-1,3,5-tricarboxylate) replaced by pyridine-3,5-dicarboxylate, generating coordinatively unsaturated metal sites; then tested the catalytic activity for ethylene dimerization in toluene with the presence of Et2AlCl at 80 °C and 55 bar.24,25 Long-term stability over 24 h was observed, despite a low catalytic activity with a turnover frequency (TOF) of 4.36 h−1; nevertheless, the catalytic activity in the liquid phase was enhanced by increasing the concentration of defective metal sites in Ru-HKUST-1.26 They also synthesized defective monometallic Ru-HKUST-1 (univalent RuII–RuII and multivalent RuII–RuIII), Rh-HKUST-1 and bimetallic Ru/Rh-HKUST-1 by controlling the formation of defects through thermal elimination and/or fragmentation of organic ligands to generate highly accessible unsaturated metal sites of reduced oxidation states.27 Subsequently, Soukri and coworkers created two types of defects in Ru-HKUST-1 and examined their effects on ethylene dimerization and 1-butene selectivity.28 The defective Ru-HKUST-1 was synthesized via incorporating 25% of pyridine-3,5-dicarboxylate ligands in H2 at 150 °C. This led to the formation of highly accessible and robust Ru–H species for ethylene dimerization due to heterolytic H–H bond breaking. After pre-treatment in N2 at 200 °C, ethylene dimerization was tested in H2 at 4.2 MPa and 150 °C. The defective Ru-HKUST-1 showed high selectivity (99%) and stability (>120 h) with TOF > 200 h−1 under optimal conditions. Meanwhile, from density-functional theory (DFT) calculations, they found similar electronic energy profiles for ligand- and thermal-engineered defective Ru-HKUST-1 following the Cossee–Arlman mechanism.29 The rate-determining step (RDS) was predicted to be C–C coupling with energy barriers of 20.9 and 21.4 kcal mol−1 on the ligand- and thermal-engineered defective Ru-HKUST-1, respectively. Furthermore, the activation of β-hydride was found to be barrierless, and the adsorption of ethylene and desorption of 1-butene were predicted with barriers of 6.7 and 26 kcal mol−1.28
While the above handful of studies have demonstrated the catalytic activity of DMOFs for ethylene dimerization, extremely pure 1-butene (99.7%) is required for polyethylene production.30,31 To develop stable, sustainable and economic catalysts with reasonable activity and high 1-butene selectivity, it is indispensable to better understand the mechanism of ethylene dimerization from bottom-up. In this context, computation at the electronic structure level is crucial as it is able to predict catalytic activity and selectivity, and identify preferential reaction pathways and probable products. Such microscopic insights would facilitate designing and tuning high-performance catalysts.
In this study, we computationally design a series of metal hydrides on defective HKUST-1 namely H-M-DHKUST-1 (M: metal atom), then evaluate their catalytic performance for ethylene dimerization through DFT calculations. As shown in Fig. S1 (ESI†), the metals include late transition metals of 3d and 4d, specifically Co, Ni, Cu, Ru, Rh and Pd, because these metals usually exhibit considerable activity for ethylene dimerization via the Cossee–Arlman route.19 Our motivation is to mechanistically understand ethylene dimerization on H-M-DHKUST-1, including the effects of multiplicity, electronic configuration and structural environment. By comparing the performance of different H-M-DHKUST-1, we can identify which metal is the best and possibly design new H-M-DHKUST-1 with low-cost metals for ethylene dimerization and other catalytic reactions.
2. Methodology
HKUST-1 consists of a paddle-wheel like secondary building unit (SBU), in which four 1,3,5-benzene tricarboxylate (BTC) linkers are bridged with dimeric Cu atoms (Fig. 1(a)).32 Defective HKUST-1 (DHKUST-1) is created when one BTC linker is missing, as shown in Fig. 1(b) for its cluster model with the cleaved bonds saturated by –CN groups. DHKUST-1 can be experimentally activated by thermal treatment in a H2 atmosphere (Fig. 1(c)) to produce monohydride DHKUST-1 (Fig. 1(d)).11,24,25,28 Furthermore, the Cu atoms in DHKUST-1 can be replaced by different metals.24,25 | ||
| Fig. 1 (a) Crystal structure of HKUST-1. (b) Cluster model of DHKUST-1. (c) Activation of DHKUST-1 to monohydride DHKUST-1 shown in (d). Copper, nitrogen, carbon, oxygen and hydrogen atoms are in brown, blue, grey, red and white, respectively. | ||
We consider six monohydrides H-M-DHKUST-1 with M = Co, Ni, Cu, Ru, Rh and Pd. Simplified as the cluster models, all the H-M-DHKUST-1 were optimized using the M06-L functional, which performs well for transition metals and accounts for medium-range dispersion interactions.33 The polarized split-valence basis set def2-SVP34 was used for p-block atoms (H, C, N and O atoms), and the doubly polarized triple-ζ basis set def2-TZVPP were used for 3d and 4d metals.33,34 The built-in effective core potential in def2-TZVPP was also applied for 4d metals. The nitrogen atoms in –CN were fixed during optimization. Due to the formation of long-range magnetic electron spin ordering with conjugated linkers, MOFs with dimeric transition metals were found to exhibit antiferromagnetic behavior.35–37 Thus, stability calculations were performed to identify the stable electronic configurations of H-M-DHKUST-1. Thereafter, vibrational frequencies were computed at the same level of theory to verify the nature of stationary points (no imaginary frequency for an energy minimum and only one imaginary frequency for a transition state) and provide thermal corrections to thermochemical properties. Each transition state was confirmed to directly connect with the reactant and product through the intrinsic reaction coordinate (IRC) calculations. The reaction pathway of ethylene dimerization based on the Cossee–Arlman route was examined by considering all the reasonable multiplicities to deduce the ground-state intermediates and transition states. To further understand the charges of catalytic sites and charge transfer during the reaction, the Natural Bond Orbital (NBO) charges were evaluated at the same level of theory. The Gibbs energies (G) were estimated at 298.15 K by G = E + Gcorr, where E is the electronic energy and Gcorr is the thermal correction. All the calculations were performed with the Gaussian 16 package.38 The relative Gibbs energy profiles ΔG were constructed by combining all the key intermediates and transition states. Moreover, possible intersystem spin crossing (ISC) was explored from the energy profiles and its effect was quantified.
3. Results and discussion
We first present the stable electronic configurations and NBO charges on six H-M-DHKUST-1 clusters (M: Co, Ni, Cu, Ru, Rh and Pd), as well as the M–H and M–M bond lengths. Then, the relative Gibbs energy profiles for four elementary steps (initiation, C–C coupling, β-hydride elimination and 1-butene desorption) are closely examined and the Gibbs energy barriers are quantified, thus predicting the favourable/unfavourable steps. Furthermore, the catalytic activity of H-M-DHKUST-1 is compared to other MOFs reported in the literature, including MFU-4l(Ni), Ni, Co, Cu and Pd-decorated NU-1000.39–42 Next, the effects of multiplicity on stability and Gibbs energy are discussed for electronic-structural intercorrelations in different intermediates and transition states. Finally, NBO charge distributions and electrostatic correlations are analyzed in order to elucidate the development of atomic charges and their relationships with different metal–substrate interactions.3.1. Stability and NBO charges
As listed in Tables S2–S8 (ESI†), the open-shell electronic configurations are stable on Co, Ni, Ru and Pd based H-M-DHKUST-1 clusters, while the closed-shell configurations are stable on Cu and Rh based clusters. Quintet is the ground-state multiplicity on H-Co-DHKUST-1, triplet on H-Ni-DHKUST-1 and H-Ru-DHKUST-1, open-shell singlet on H-Pd-DHKUST-1, and closed-shell singlet on H-Cu-DHKUST-1 and H-Rh-DHKUST-1, respectively. The electronic energies at different multiplicities are also provided, as well as the energy differences relative to the ground-state multiplicity. Fig. 2 shows the optimized structures with M–H bond lengths on the six H-M-DHKUST-1 clusters. There is a bridged hydride between the dimeric Cu atoms on H-Cu-DHKUST-1, and this hydride is not centered between the two Cu atoms but shifted to one of them giving a partial hetero-valent arrangement of Cu–H–Cu. On the other five H-M-DHKUST-1 clusters, a vertically bonded hydride on one metal is observed and causes apparent hetero-valent configuration of dimeric metals. This information introduces a prior knowledge for the accessibility of hydride for initiating ethylene dimerization. | ||
| Fig. 2 Optimized structures with M–H bond lengths (Å) on H-M-DHKUST-1. Carbon, oxygen and hydrogen atoms are in grey, red and white, respectively. | ||
Table 1 lists the M–H and M–M bond distances on the six H-M-DHKUST-1 clusters. The M–H bond length increases in the order of H-Ni-DHKUST-1 < H-Co-DHKUST-1< H-Pd-DHKUST-1 < H-Rh-DHKUST-1 < H-Cu-DHKUST-1 < H-Ru-DHKUST-1. The longest M–H bond distance is 1.62 Å on H-Ru-DHKUST-1, which implies this cluster is the most readily accessible among the six for the hydride-based reaction. Also listed in Table 1 are the NBO charges on H and M atoms. The NBO charge of the hydride H atom on metal is nearly zero, which was also observed in β-hydride elimination on Au(III) products43 and metal complexes.44,45 This phenomenon is attributed to the high electronegativity of the oxygen atom surrounding the metal; consequently, the charge is redistributed within the cluster. From Bent's rule, increasing the ionic character of the M–O bond would lead to decreasing the d-character of the M–H bond.46 Due to the high electronegativity of oxygen, the M–O bond is expected to attain a high d-character, corresponding to the low d-character of the M–H bond and the small negative charge of the hydride H atom. It is also noticed that the 4d metal attached to hydride carries a smaller NBO charge than the corresponding 3d metal, while the secondary metal atom has a larger NBO charge. Specifically, Rh and Ru have the smallest charges of 0.363e and 0.435e, respectively. The stabilization of hydride on metal and its availability for initiating ethylene dimerization are expected to depend on the type of metal.
| H-M-DHKUST-1 | d M1–H | d M2–H | d M1–M2 | Q H | Q M1 | Q M2 |
|---|---|---|---|---|---|---|
| H-Co-DHKUST-1(q) | 1.49 | 2.905 | 2.45 | 0.001 | 0.537 | 0.985 |
| H-Ni-DHKUST-1(t) | 1.46 | 2.577 | 2.33 | 0.003 | 0.507 | 0.986 |
| H-Cu-DHKUST-1(cs) | 1.59 | 1.692 | 2.41 | −0.053 | 0.786 | 0.800 |
| H-Ru-DHKUST-1(q) | 1.62 | 2.791 | 2.25 | 0.001 | 0.435 | 0.873 |
| H-Rh-DHKUST-1(cs) | 1.55 | 2.910 | 2.40 | 0.047 | 0.363 | 0.808 |
| H-Pd-DHKUST-1(t) | 1.53 | 3.020 | 2.54 | 0.088 | 0.473 | 0.815 |
To predict catalytic properties and design efficient catalysts, it is indispensable to understand the oxidation state and coordination environment of metallic dimer on H-M-DHKUST-1. Tables S9–S14 (ESI†) list the optimized M–M bond distances (Å), NBO charges, and NBO spin density distributions in the ground-state intermediates and transition states. On the bimetallic sites, the metallic interaction can significantly affect multiplicity, which is directly related to the number of unpaired electrons, and this interaction can cause a change in electronic configuration, as reported on perfect HKUST-1 and bimetallic paddle-wheel complexes.47–49 Clusters with open shell configurations such as a quintet on H-Co-DHKUST-1 and H-Ru-DHKUST-1, as well as a triplet on H-Ni-DHKUST-1 and Pd-DHKUST-1, tend to possess an oxidation state of +2 on both metal atoms. The formation of a M–M bond in each metallic dimer consumes one unpaired electron from each metal atom, with two, two, one and one unpaired electrons remaining in each metal atom on H-Co-, H-Ru-, H-Ni- and H-Pd-DHKUST-1 clusters, respectively. Due to the difference in the coordination environment, the electron spin density in the secondary metal atom is found to be larger than in the primary counterpart. Specifically, the electron spin densities are ρCo2 = 2.351, ρRu2 = 2.374, ρNi2 = 1.432 and ρPd2 = 1.166; by contrast ρCo1 = 1.514, ρRu1 = 1.428, ρNi1 = 0.427 and ρPd1 = 0.395. We thus infer there is electron delocalization from the primary to secondary metal atom. However, the closed-shell singlet configuration on H-Cu-DHKUST-1 and H-Rh-DHKUST-1 suggests a completely filled suborbital in each metal atom, where no unpaired electrons exist. It is likely that each metal atom is divalent like Cu2+(d9) and Rh2+(d7), and the two unpaired electrons from each atom interact to form a M–M bond. An alternative is the formation of multivalent metal dimer Cu1+(d10)–Cu23+(d8) and Rh1+(d8)–Rh23+(d6). Overall, the approximate determination of the oxidation state from the NBO electron density distribution provides valuable insight into the electronic structure and coordination environment of a catalyst, which is essential for understanding its catalytic activity.
3.2. Catalytic mechanism and activity
Ethylene dimerization catalysed by defective Ru-HKUST-1 and other similar MOFs was revealed to follow the Cossee–Arlman mechanism.28,50 We also consider this mechanism in the current study on H-M-DHKUST-1. As shown in Fig. 3, there are four elementary steps, namely initiation, C–C coupling, β-hydride elimination and 1-butene desorption. (1) Initiation starts with an ethylene molecule adsorbed on catalyst 1 to form complex 2, which is then hydrogenated to ethyl intermediate 3; (2) upon supplying another ethylene molecule to form intermediate 4, the M-ethyl bond breaks and C–C coupling occurs to generate butyl intermediate 5; (3) after reorientation of butyl intermediate 5 to form 6, β-hydride elimination takes pace to form intermediate 7 with 1-butene adsorbed; (4) the final step is 1-butene desorption and catalyst 1 is recovered. The reaction cycle can be restored with the successive supply of ethylene. | ||
| Fig. 3 Ethylene dimerization to 1-butene via the Cossee–Arlman mechanism with four elementary steps: initiation (including adsorption and hydrogenation), C–C coupling, β-hydride elimination and 1-butene desorption. | ||
To identify the ground-state configurations of intermediates and transition states during ethylene dimerization, we performed deep analysis of the relative Gibbs energies ΔG at different multiplicities on H-M-DHKUST-1 clusters (Tables S15–S20 and Fig. S2–S7, ESI†). The effects of multiplicity on energy will be discussed in detail below (Section 3.3). Table S21 (ESI†) lists the imaginary frequencies of transition states for hydrogenation, C–C coupling and β-hydride elimination. Single imaginary frequency is observed for each transition state, which confirms the nature of each stationary point. Since the stable configurations on H-Cu-DHKUST-1 and H-Rh-DHKUST-1 are closed-shell, stability analysis was further conducted for ethylene dimerization on these two clusters (Table S8, ESI†). Stability calculation of the closed shell singlet on H-Rh-DHKUST-1 reveals that the stable closed-shell configurations are maintained throughout all the intermediates and transition states, while the stable open-shell configurations are observed for species 2, TS2-3, TS4-5, TS6-7 and 7 on H-Cu-DHKUST-1. This finding implies that the electronic structures of intermediates and transition states on the two clusters are substantially different, which would likely cause the two clusters to display different electronic configurations for the reaction. Fig. 4 summarizes the ΔG profiles of ethylene dimerization on the six H-M-DHKUST-1 (M: Co, Ni, Cu, Ru, Rh and Pd). The profiles include four elementary steps for ethylene dimerization: initiation (adsorption and hydrogenation), C–C coupling, β-hydride elimination and 1-butene desorption.
 | ||
| Fig. 4 Relative Gibbs energy ΔG profiles of ethylene dimerization on H-M-DHKUST-1 at 298.15 K. The top illustrates the four elementary steps: initiation (adsorption and hydrogenation), C–C coupling, β-hydride elimination and 1-butene desorption. The Gibbs energy barriers Gb are listed in Table 2. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in ethylene. Moreover, the M–M bonds are shorter on H-Co-DHKUST-1 (2.37 Å), elongated on H-Ni- (2.58 Å), H-Cu- (2.48 Å), H-Ru- (2.32 Å) and H-Rh-DHKUST-1 (2.47 Å), and unchanged on H-Pd-DHKUST-1 (2.56 Å).
C in ethylene. Moreover, the M–M bonds are shorter on H-Co-DHKUST-1 (2.37 Å), elongated on H-Ni- (2.58 Å), H-Cu- (2.48 Å), H-Ru- (2.32 Å) and H-Rh-DHKUST-1 (2.47 Å), and unchanged on H-Pd-DHKUST-1 (2.56 Å).
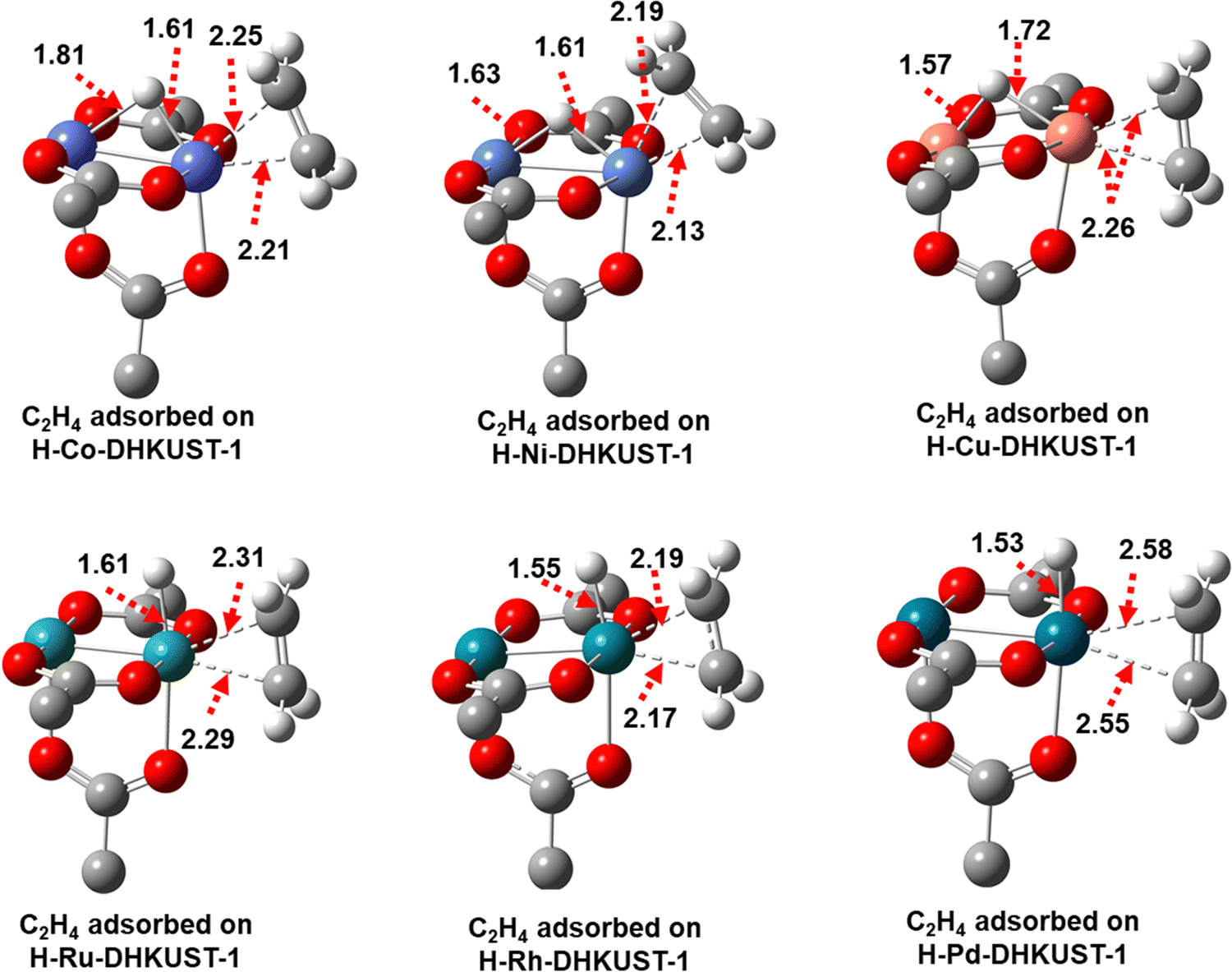 | ||
| Fig. 5 Optimized structures and key bond distances (Å) of intermediate 2 for ethylene adsorption on H-M-DHKUST-1. Carbon, oxygen and hydrogen atoms are in grey, red and white, respectively. | ||
When hydrogenation occurs, the M–H bonds of transitions state TS2-3 are observed in Fig. S8 (ESI†) to become shorter from intermediate 2 on H-Co- (1.48 Å) and H-Ni-DHKUST-1 (1.47 Å), longer on H-Cu-DHKUST-1 (1.83 Å), and remain nearly the same on the other three clusters. Meanwhile, the M–M bonds become longer as listed in Tables S8–S13 (ESI†) on H-Co- (2.46 Å), H-Ru- (2.40 Å) and H-Rh-DHKUST-1 (2.64 Å) from intermediate 2, largely unchanged on H-Cu- (2.50 Å) and H-Pd-DHKUST-1 (2.59 Å), but shorter on H-Ni-DHKUST-1 (2.41 Å). Table 2 lists the Gibbs energy barriers Gb for hydrogenation on the six H-M-DHKUST-1. The Gb are 0.9, 4.7, 4.7, 5.5 and 5.4 kcal mol−1 on Co, Ni, Ru, Rh and Pd-based clusters, respectively, whereas the Gb on H-Cu-DHKUST-1 is significantly higher with a value of 18.4 kcal mol−1. This is consistent with the less accessible hydride on H-Cu-DHKUST-1, in which one of the M–H bond lengths is only 1.57 Å. Overall, for the initiation step including both adsorption and hydrogenation, the largest change in M–M bond length is observed on H-Ru- and H-Rh-DHKUST-1 from 2.25 to 2.40 Å and 2.40 to 2.64 Å, respectively. Thus, H-Ru- and H-Rh-DHKUST-1 possess a greater tolerance than the other four clusters in terms of structural change. On H-Co- and H-Ni-DHKUST-1, the M–M bond lengths are restored to a certain extent due to multiplicity transitions. Specifically, on H-Co-DHKUST-1, the multiplicity is initially quintet intermediate 2 then triplet in TS2-3; the M–M bond length changes from 2.45 to 2.37 then to 2.46 Å. On H-Ni-DHKUST-1, the M–M bond length changes from 2.33 to 2.58 then to 2.41 Å upon multiplicity transition from triplet to open-shell singlet then back to triplet.
| Hydrogenation | C–C coupling | β-H elimination | 1-Butene desorption | Ref. | |
|---|---|---|---|---|---|
| H-Co-DHKUST-1 | 0.9 | 19.3 | 10.4 | 8.4 | This work |
| H-Ni-DHKUST-1 | 4.7 | 22.5 | 9.9 | 3.6 | This work |
| H-Cu-DHKUST-1 | 18.4 | 22.7 | 28.5 | 7.0 | This work |
| H-Ru-DHKUST-1 | 4.7 | 37.3 | 1.6 | 7.7 | This work |
| H-Rh-DHKUST-1 | 5.5 | 39.6 | 0.2 | 17.9 | This work |
| H-Pd-DHKUST-1 | 5.4 | 30.0 | 19.2 | 3.2 | This work |
| MFU-4l(Ni) | 10.7 | 22.0 | 17.3 | −6.1 | 40 and 51 |
| Ni-NU-1000 | 3.7 | 13.7 | 9.6 | 5.7 | 42 |
| Co-NU-1000 | 8.3 | 24.8 | 22.3 | −4.0 | 42 |
| Cu-NU-1000 | 2.3 | 20.9 | 23.8 | −6.0 | 42 |
| Pd-NU-1000 | 3.5 | 12.3 | 1.5 | 16.4 | 42 |
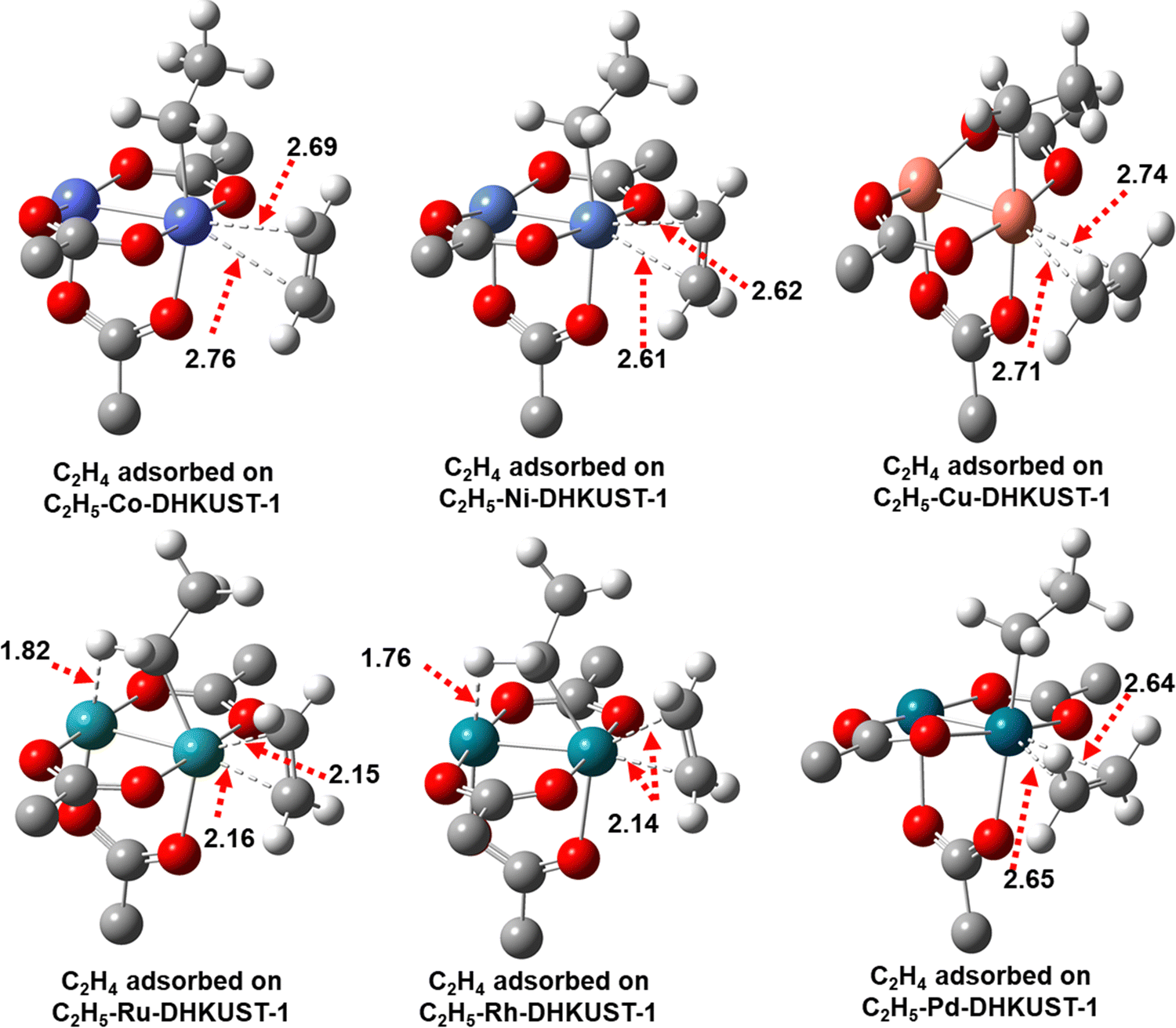 | ||
| Fig. 6 Optimized structures and key bond distances (in Å) of intermediate 4 for the adsorption of second ethylene molecule on ethyl-M-DHKUST-1. Carbon, oxygen and hydrogen atoms are in grey, red and white, respectively. | ||
It is observed in Fig. 6 that the second adsorbed ethylene tends to align horizontally with the metal atoms on H-Cu- and H-Pd-DHKUST-1, and vertically with the metal atoms on the other four clusters. Geometrically, the vertically aligned ethylene is likely to couple with ethyl. In Table 2, the Gibbs energy barriers Gb for C–C coupling are 19.3, 22.5 and 22.7 kcal mol−1 on 3d metal-based H-Co-, H-Ni- and H-Cu-DHKUST-1; on 4d metal-based clusters, the Gb are generally higher. Particularly, Gb on H-Ru- and H-Rh-DHKUST-1 are the highest of 37.3 and 39.6 kcal mol−1 among the six clusters, due to the most stable intermediate 4 on H-Ru- and H-Rh-DHKUST-1. From the optimized structures of transition state TS4-5 in Fig. S9 (ESI†), we observe ethylene reorientation and C–C distance shortening upon C-C coupling. In addition, the M-M bonds are elongated from intermediate 4 to TS4-5, as listed in Tables S8–S13 (ESI†), from 2.44 to 2.61 Å on H-Co-DHKUST-1, 2.36 to 2.47 Å on H-Ni-DHKUST-1, 2.57 to 2.91 Å on H-Cu-DHKUST-1, 2.36 to 2.39 Å on H-Ru-DHKUST-1, 2.42 to 2.71 Å on H-Rh-DHKUST-1, and 2.58 to 2.62 Å on H-Pd-DHKUST-1.
 | ||
| Fig. 7 Optimized structures and key bond distances (in Å) of intermediate 6 for β-hydride elimination on H-M-DHKUST-1. Carbon, oxygen and hydrogen atoms are in grey, red and white, respectively. | ||
 | ||
| Fig. 8 Optimized structures and key bond distances (in Å) of intermediate 7 on H-M-DHKUST-1. Carbon, oxygen and hydrogen atoms are in grey, red and white, respectively. | ||
By comparing the above four elementary steps (Table 2), we find that the Gb for C–C coupling is the highest on all six clusters, except H-Cu-DHKUST-1 on which the Gb for BHE is the highest. This reveals that BHE is the RDS in ethylene dimerization on H-Cu-DHKUST-1, but C–C coupling is on the other five clusters. Overall, H-Co- and H-Ni-DHKUST-1 are predicted to outperform the other four. It is also instructive to compare the catalytic performance of H-M-DHKUST-1 under this study with other MOF catalysts reported in the literature. As tabulated in Table 2, H-M-DHKUST-1, MFU-4l(Ni), Ni, Co, Cu and Pd-decorated NU-1000 exhibit a similar magnitude of Gb (<10 kcal mol−1) for hydrogenation, however, H-Cu-DHKUST-1 (18.4 kcal mol−1) is an exception to this trend. For C–C coupling, the Gb on H-M-DHKUST-1 are generally higher than MFU-4l(Ni) and metal-decorated NU-1000. In particular, Gb are 37.3, 39.6 and 30.0 kcal mol−1 on the precious metal-based H-Ru-, H-Rh- and H-Pd-DHKUST-1. This reveals that H-M-DHKUST-1 clusters are less active for C–C coupling; comparatively, Ni-NU-1000 and Pd-NU-1000 with square-planar Pd sites are more favoured with Gb of 13.7 and 12.3 kcal mol−1, respectively. For BHE, H-Ru- and H-Rh-DHKUST-1 are predicted to have the lowest Gb of only 1.6 and 0.1 kcal mol−1 respectively, leading to the facile formation of 1-butene. On Pd-NU-1000, Gb of 1.5 kcal mol−1 is also rather low due to agostic arrangement, in contrast to 17.1 kcal mol−1 on the octahedral sites of H-Pd-DHKUST-1 without agostic arrangement.42 However, the desorption of 1-butene on H-Rh-DHKUST-1 and Pd-NU-1000 is impeded by high Gb (17.9 and 1.64 kcal mol−1).
Ni-based catalysts have been extensively examined for ethylene dimerization. The square-planar configuration was revealed to be the stable structural environment on Ni-exchanged MFU-4l and Ni-supported NU-1000.39,51 The Gb on MFU-4l(Ni) were predicted to be 10.7, 22.0 and 17.3 kcal mol−1 for hydrogenation, C–C coupling and BHE,40,51 and lower Gb (3.7, 13.7 and 9.6 kcal mol−1) were estimated on Ni-NU-1000.42 Despite the difference in the electronic environment, H-Ni-DHKUST-1 with an octahedral site possesses similar Gb (4.7, 22.5 and 9.9 kcal mol−1). This could be associated with the difference in the structural environment of the metallic dimer. Comparatively, H-Co-DHKUST-1 displays lower Gb (0.9, 19.3 and 10.4 kcal mol−1) than Co-NU-1000 (8.3, 24.8 and 22.3 kcal mol−1) for hydrogenation, C–C coupling and BHE, suggesting its better catalytic performance and the octahedral site of H-Co-DHKUST-1 is more effective than the trigonal pyramidal site of Co-NU-1000. However, Cu-NU-1000 shows a greater tendency for hydrogenation (2.3 kcal mol−1) and facilitates 1-butene desorption (−6.0 kcal mol−1) compared to H-Cu-DHKUST-1, which requires a higher Gb for the same steps (18.4 and 7.0 kcal mol−1).
It is worthwhile noting that the RDS in a chemical reaction involves more than just the energy barriers associated with elementary steps. In addition to the barriers, other factors also play a crucial role in determining the RDS.52,53 They include the nature of the catalyst, the reactant concentration or pressure, and the specific reaction mechanism. Practically, microkinetic modelling serves as a valuable tool to elucidate intricate reaction mechanisms, optimize reaction conditions, and design catalytic processes. Such modelling offers microscopic insights into various factors influencing reaction rates, unravels complex reaction pathways and enhances our ability to predict and control reaction kinetics, thus facilitating the development of efficient and selective catalysts.54 In our future work, we plan to incorporate microkinetic modelling.
3.3. Effects of multiplicity
As pointed out above, it is important to investigate the effects of multiplicity on catalytic properties. Tables S14–S19 and Fig. S2–S7 (ESI†) compare the Gibbs energies at different multiplicities on H-M-DHKUST-1 clusters. Due to the binding and coordination of reacting species with catalysts, intersystem crossing (ISC) may occur and two energy surfaces at different multiplicities compete for the ground state.51,55–58 It is observed that the ground-state multiplicity on H-M-DHKUST-1 shows a decreasing trend upon moving through the same row of 3d transition metals. On H-Co-DHKUST-1, quintet and triplet are the dominant ground-state multiplicities in most intermediate and transition states, except for septet in intermediate 5 (Fig. S2, ESI†). Triplet-singlet crossing is seen on H-Ni-DHKUST-1 (Fig. S3, ESI†), while singlet (open and closed shells) is the only ground-state multiplicity on H-Cu-DHKUST-1 (Fig. S4, ESI†). A similar trend exists on the 4d transition metal H-M-DHKUST-1. On H-Ru-DHKUST-1, there is an ISC between quintet and triplet, though the quintet is more prevalent (Fig. S5, ESI†). On H-Rh-DHKUST-1, closed-shell singlet and triplet states compete with each other (Fig. S6, ESI†). However, triplet is the common ground-state multiplicity on H-Pd-DHKUST-1 with an open-shell singlet existing only in intermediate 6 (Fig. S7, ESI†).Ethylene dimerization is remarkably associated with the interplay between the electronic configuration and structural environment of a catalyst. A singlet-to-triplet ISC was reported on MFU-4l(Ni) from square-planar to tetrahedral for ethylene dimerization and oligomerization.51 Similarly, the ISC between triplet and singlet is also observed on H-Ni-DHKUST-1. As illustrated in Fig. S3 (ESI†), triplet persists in most of the intermediates and transition states, but it switches to open-shell singlet in intermediates 2 and 7 upon the adsorption of ethylene and 1-butene, respectively. Compared to triplet, singlet exhibits lower Gibbs energy (0.9 kcal mol−1 in intermediate 2 and 1.7 kcal mol−1 in intermediate 7). The reason is that H-Ni-DHKUST-1 with an octahedral site requires a 7-fold-like arrangement upon coordinating with ethylene molecule and 1-butene (Fig. 5 and 8), thus adopting singlet as the ground state with bridged-like hydride. This configuration allows easy hydrogenation (with low Gb of 4.7 kcal mol−1), as well as facile 1-butene desorption (with low Gb of 3.6 kcal mol−1). In this context, it is helpful to understand the significance of ISC during ethylene adsorption and 1-butene desorption to practically tailor the catalytic performance toward ethylene dimerization.51,59
The electronic-structural intercorrelations at different multiplicities are also observed on other H-M-DHKUST-1 except H-Cu-DHKUST-1. For instance, the agostic arrangement of β-hydride intermediate 6 on H-Co-DHKUST-1 demonstrates intercorrelation between the electronic state and structural environment. As shown in Fig. 9, the triplet, quintet and septet structures sustain the agostic arrangement of trigonal pyramidal assembly, and M1–M2 and M1–βH bonds are observed to be elongated with increasing multiplicity. Among the three structures, the septet (Fig. 9(c)) is found to be the ground state (−35.8 kcal mol−1), while triplet (Fig. 9(a)) and quintet (Fig. 9(b)) have higher relative Gibbs energy with −29.4 kcal mol−1 and −32.7 kcal mol−1, respectively. Thus, the septet essentially represents intermediate 6 on H-Co-DHKUST-1 and the Gb for BHE is thus 10.4 kcal mol−1. As discussed above, the butyl intermediate of non-agostic arrangement is alternatively stabilized on singlet and triplet structures on H-Cu- and H-Pd-DHKUST-1 (Fig. S11 and S12, ESI†), whereas the agostic assembly of β-hydride intermediate is not converged. Additionally, there is quintet-to-triplet crossing on H-Ru-DHKUST-1 (Fig. S2 and S5, ESI†), while triplet-to-singlet crossing is seen on H-Rh- and H-Pd-DHKUST-1 (Fig. S6 and S7, ESI†).
 | ||
| Fig. 9 Intermediate 6 on H-Co-DHKUST-1 with different multiplicities (a) triplet, (b) quintet and (c) septet. The septet displays the lowest Gibbs energy with an agostic arrangement, while the triplet and quintet exhibit an agostic arrangement with 6.4 and 3.1 kcal mol−1 higher Gibbs energy, respectively. | ||
Fig. 10 further highlights the role of multiplicity during BHE on H-Rh-DHKUST-1. The closed-shell singlet state is found to allow for pentacyclic stabilization of intermediate 6 and transition state TS6-7 in a synergistic manner of both metal atoms. Comparatively, the triplet adopts an agostic arrangement but is 2.4 and 3.0 kcal mol−1 lower in Gibbs energy (Fig. S6, ESI†). With this small energy difference, BHE could be sidestepped by other reaction pathways unless the secondary metal atom is fully saturated.28 This observation underpins the importance of complete saturation of the secondary metal atom in dimeric metal catalysts for ethylene dimerization. Overall, ISC is revealed to be crucial to determine the reaction pathway. Nevertheless, the significance between difference multiplicities is not highly appreciable among analogous geometries, because the energy difference is usually only within a few kilocalories. As shown on H-Pd-HKUST-1, the energy profiles of singlet and triplet states are nearly identical (Fig. S7, ESI†).
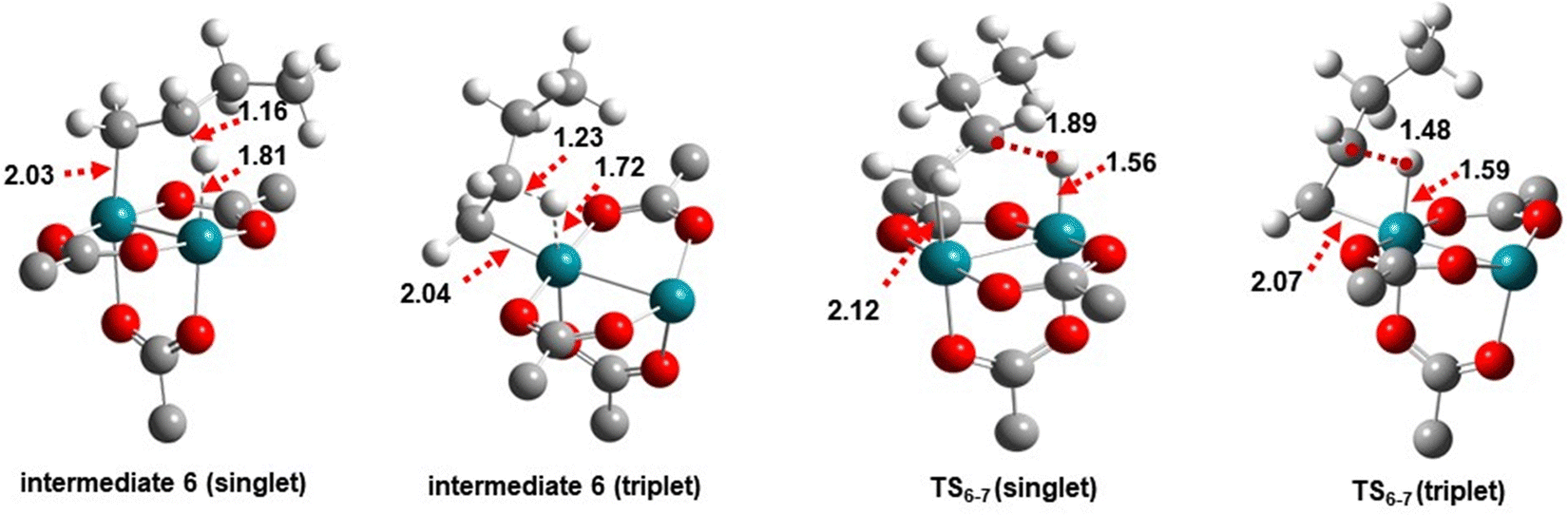 | ||
| Fig. 10 Intermediate 6 and transition state TS6-7 on H-Rh-DHKUST-1 with closed-shell singlet and triplet states. The closed-shell singlet displays a pentacyclic arrangement, while the triplet exhibits an agostic arrangement but is 2.4 and 3.0 kcal mol−1 lower in Gibbs energy, respectively. | ||
3.4. NBO charge distributions and electrostatic correlations
To gain in-depth insights into the development of atomic charges and their relationships with different metal-substrate interactions, as shown in Fig. 11, we analyse NBO charge distributions at six atoms (M1, M2, H, Cα, Cβ and Cv in Fig. S1, ESI†) during dimerization. The charge variations of the metallic dimer (M1 and M2) and hydride (H) were scrutinized upon interacting with carbon atoms at three positions (Cα, Cβ and Cv). The secondary metal M2 is found to exhibit a consistent cationic nature on all the H-M-DHKUST-1 clusters. On Ru- (Fig. 11(d)) and Rh-clusters (Fig. 11(e)), a decrease in charge for M2 is apparent in intermediate 4 due to interaction with ethyl, as discussed above (Fig. 6). The hydride (H) is observed to depart from a neutral-like state upon hydrogenation, but restores it after eliminated from Cβ atom during BHE. Concurrently, ethylene hydrogenation and 1-butene formation are associated with negligibly weak cationic M1 in transition states TS2-3 and TS6-7 on all the H-M-DHKUST-1 clusters, but reaching minimum charges on H-Ru- and H-Rh-DHKUST-1 (0.030e and 0.053e). In general, it is demonstrated that the primary metal M1 interacting with ethylene is less cationic on H-Ru- (Fig. 11(d)) and H-Rh-DHKUST-1 (Fig. 11(e)). However, Co (Fig. 11(a)) and Ni (Fig. 11(b)) atoms possess larger NBO charges. Apparently, the higher cationic nature of M1 in ethyl intermediate 3 is compensated by a decrease in Cα and Cβ charges. This is also shown by butyl intermediates 5 and 6 at high multiplicity (septet) on H-Co-DHKUST-1 (Fig. 11(a)). The double bond is revealed to develop with the decreasing anionic nature of Cβ until reaching a free 1-butene molecule. | ||
| Fig. 11 NBO charges of M1, M2, H, Cα, Cβ and Cv atoms during dimerization on (a) H-Co-, (b) H-Ni-, (c) H-Cu, (d) H-Ru-, (e) H-Rh-, and (f) H-Pd-DHKUST-1. | ||
Finally, to elucidate the impact of electrostatic interaction on the activity of H-M-DHKUST-1 clusters, we examine how the electronegativity of the primary metal is correlated with the Gibbs energy barrier of the RDS (i.e., C–C coupling). As shown in Fig. 12, the Gibbs energy barrier is observed to vary linearly with electronegativity. This is consistent with an earlier discussion, i.e., the cluster with 4d metal exhibits a higher energy barrier than the counterpart with 3d metal. This insight is useful toward the design of new catalysts for efficient C–C coupling in ethylene dimerization and other reactions.
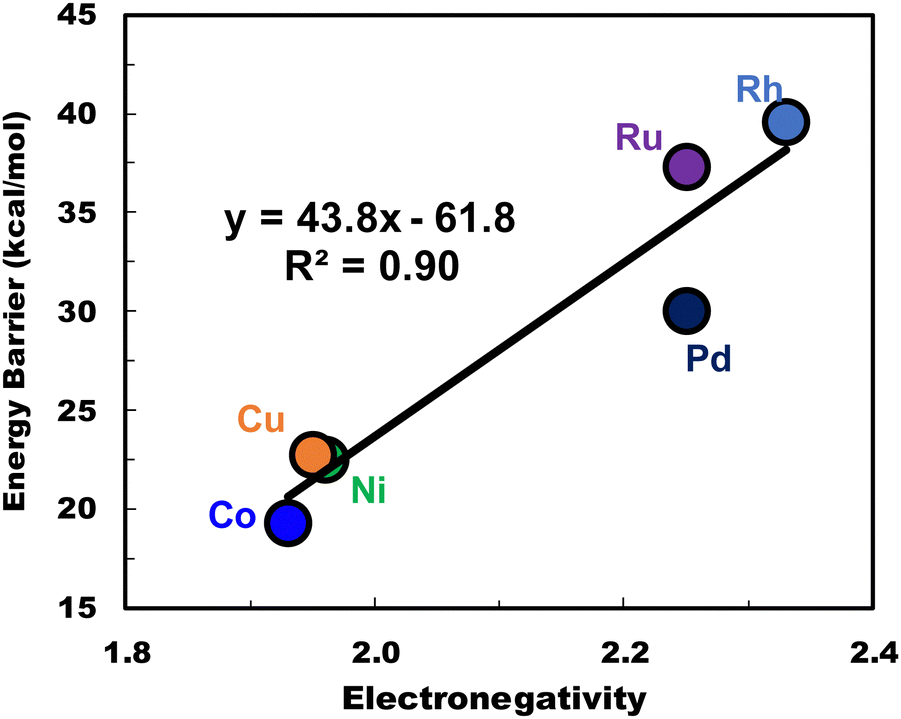 | ||
| Fig. 12 Correlation between the electronegativity of primary metal and the Gibbs energy barrier of C–C coupling on H-M-DHKUST-1 clusters. | ||
4. Conclusions
We have computationally designed metal hydrides on defective HKUST-1 (H-M-DHKUST-1) (M: Co, Ni, Cu, Ru, Rh and Pd) and investigate their catalytic performance for ethylene dimerization. Four elementary steps in the Cossse-Arlman route are considered including initiation, C–C coupling, β-hydride elimination and 1-butene desorption. For each step, the Gibbs energy profiles are comprehensively calculated at all the possible multiplicities. Interestingly, intersystem spin crossing is observed on the six H-M-DHKUST-1 clusters, revealing the importance of electronic-structural intercorrelations in ethylene dimerization. Compared to 3d metal (Co, Ni and Cu) based H-M-DHKUST-1, 4d metal (Ru, Rh and Pd) based ones are predicted to be less favorable in C–C coupling but more active in BHE. In particular, the Gibbs energy barriers are only 1.6 and 0.2 kcal mol−1 for BHE on H-Ru- and H-Rh-DHKUST-1, respectively. However, the energy barrier for 1-butene desorption is high at 17.9 kcal mol−1 on H-Rh-DHKUST-1 due to the strong stability of adsorbed 1-butene. The rate determining step is determined to be C–C coupling on H-Co-, H-Ni-, H-Ru-, H-Rh- and H-Pd-DHKUST-1 with Gibbs energy barriers of 19.3, 22.5, 37.3, 39.6 and 30.0 kcal mol−1, respectively. The barriers on H-Ru- and H-Pd-DHKUST-1 are the highest due to the interference of the secondary metal atom. Nevertheless, BHE is the rate determining step on H-Cu-DHKUST-1 with a barrier of 28.5 kcal mol−1, as a result of stabilization of non-agostic arrangement. Overall, H-Co- and H-Ni-DHKUST-1 are predicted to outperform the other four counterparts. This computational study provides not only microscopic insights into the catalytic activity and the performance of bimetallic H-M-DHKUST-1, but also quantitative understanding of the effects of the electronic configuration and structural environment of H-M-DHKUST-1 on ethylene dimerization. These bottom-up insights are useful to facilitate the rational design of defective MOF catalysts for important catalytic reactions.Data availability
Data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the A*STAR LCER-FI project (LCERFI01-0033 U2102d2006), the Ministry of Education of Singapore and the National University of Singapore (C-261-000-207-532/C-261-000-777-532 and R-279-000-574-114) for financial support.References
- U. N. E. Programme, Global Chemicals Outlook II: From Legacies to Innovative Solutions: Implementing the 2030 Agenda for Sustainable Development, 2019.
- M. Bender, ChemBioEng Rev., 2014, 1, 136–147 CrossRef CAS.
- J. Skupinska, Chem. Rev., 2002, 91, 613–648 CrossRef.
- K. I. Dement’ev, A. D. Sagaradze, P. S. Kuznetsov, T. A. Palankoev and A. L. Maximov, Ind. Eng. Chem. Res., 2020, 59, 15875–15883 CrossRef.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- T. Agapie, Coord. Chem. Rev., 2011, 255, 861–880 CrossRef CAS.
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- V. Hulea, J. Catal., 2004, 225, 213–222 CrossRef CAS.
- M. Lallemand, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2006, 301, 196–201 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- N. Nijem, K. Fürsich, H. Bluhm, S. R. Leone and M. K. Gilles, J. Phys. Chem. C, 2015, 119, 24781–24788 CrossRef CAS.
- W. Zhang, M. Kauer, P. Guo, S. Kunze, S. Cwik, M. Muhler, Y. Wang, K. Epp, G. Kieslich and R. A. Fischer, Eur. J. Inorg. Chem., 2017, 925–931 CrossRef CAS.
- J. Ren, D. E. C. Rogers, T. Segakweng, H. W. Langmi, B. C. North, M. Mathe and D. Bessarabov, Int. J. Mater. Res., 2014, 105, 89–93 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier and V. Van Speybroeck, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- D. D. Borges, S. Devautour-Vinot, H. Jobic, J. Ollivier, F. Nouar, R. Semino, T. Devic, C. Serre, F. Paesani and G. Maurin, Angew. Chem., Int. Ed., 2016, 55, 3919–3924 CrossRef CAS PubMed.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- C. L. Zhang, T. Zhou, Y. Q. Li, X. Lu, Y. B. Guan, Y. C. Cao and G. P. Cao, Small, 2023, 19, e2205898 CrossRef PubMed.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Chem. Commun., 2020, 56, 10409–10418 RSC.
- S. R. Marri, N. Chauhan, R. K. Tiwari, J. Kumar and J. N. Behera, Inorg. Chim. Acta, 2018, 478, 8–14 CrossRef CAS.
- P. Maniam and N. Stock, Inorg. Chem., 2011, 50, 5085–5097 CrossRef CAS PubMed.
- Y. C. Tan and H. C. Zeng, Adv. Funct. Mater., 2017, 27, 1703765 CrossRef.
- W. R. Heinz, T. Kratky, M. Drees, A. Wimmer, O. Tomanec, S. Gunther, M. Schuster and R. A. Fischer, Dalton Trans., 2019, 48, 12031–12039 RSC.
- W. R. Heinz, I. Agirrezabal-Telleria, R. Junk, J. Berger, J. Wang, D. I. Sharapa, M. Gil-Calvo, I. Luz, M. Soukri, F. Studt, Y. Wang, C. Woll, H. Bunzen, M. Drees and R. A. Fischer, ACS Appl. Mater. Interfaces, 2020, 12, 40635–40647 CrossRef CAS PubMed.
- O. Kozachuk, I. Luz, F. X. Llabres i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 Search PubMed.
- W. Zhang, M. Kauer, O. Halbherr, K. Epp, P. Guo, M. I. Gonzalez, D. J. Xiao, C. Wiktor, L. I. I. X. Fx, C. Woll, Y. Wang, M. Muhler and R. A. Fischer, Chem. – Eur. J., 2016, 22, 14297–14307 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria, I. Luz, M. A. Ortuno, M. Oregui-Bengoechea, I. Gandarias, N. Lopez, M. A. Lail and M. Soukri, Nat. Commun., 2019, 10, 2076 CrossRef PubMed.
- E. Arlman and P. Cossee, J. Catal., 1964, 3, 99–104 CrossRef CAS.
- L. Lobbert, S. Chheda, J. Zheng, N. Khetrapal, J. Schmid, R. Zhao, C. A. Gaggioli, D. M. Camaioni, R. Bermejo-Deval, O. Y. Gutierrez, Y. Liu, J. I. Siepmann, M. Neurock, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2023, 145, 1407–1422 CrossRef PubMed.
- B. Yeh, S. Chheda, S. D. Prinslow, A. S. Hoffman, J. Hong, J. E. Perez-Aguilar, S. R. Bare, C. C. Lu, L. Gagliardi and A. Bhan, J. Am. Chem. Soc., 2023, 145, 3408–3418 CrossRef CAS PubMed.
- S. S. Chui, S. M. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- L. Shen, S. W. Yang, S. Xiang, T. Liu, B. Zhao, M. F. Ng, J. Goettlicher, J. Yi, S. Li, L. Wang, J. Ding, B. Chen, S. H. Wei and Y. P. Feng, J. Am. Chem. Soc., 2012, 134, 17286–17290 CrossRef CAS PubMed.
- C. H. Hendon and A. Walsh, Chem. Sci., 2015, 6, 3674–3683 RSC.
- M. A. S. Francisco, F. Fantuzzi, T. M. Cardozo, P. M. Esteves, B. Engels and R. R. Oliveira, Chem. – Eur. J., 2021, 27, 12126–12136 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- V. Bernales, A. B. League, Z. Li, N. M. Schweitzer, A. W. Peters, R. K. Carlson, J. T. Hupp, C. J. Cramer, O. K. Farha and L. Gagliardi, J. Phys. Chem. C, 2016, 120, 23576–23583 CrossRef CAS.
- D. E. Ortega and R. A. Matute, Catal. Sci. Technol., 2021, 11, 2422–2432 RSC.
- E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dinca, ACS. Cent. Sci., 2016, 2, 148–153 CrossRef CAS PubMed.
- J. Ye, L. Gagliardi, C. J. Cramer and D. G. Truhlar, J. Catal., 2018, 360, 160–167 CrossRef CAS.
- M. Castineira Reis, C. S. Lopez, E. Kraka, D. Cremer and O. N. Faza, Inorg. Chem., 2016, 55, 8636–8645 CrossRef CAS PubMed.
- T. Debnath, T. Ash, T. Banu and A. K. Das, Theor. Chem. Acc., 2016, 135, 175 Search PubMed.
- P. L. Theofanis and W. A. Goddard, Organometallics, 2011, 30, 4941–4948 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, 2005 Search PubMed.
- J. Pirillo and Y. Hijikata, Inorg. Chem., 2020, 59, 1193–1203 CrossRef CAS PubMed.
- R. Hrdina, Eur. J. Inorg. Chem., 2021, 501–528 CrossRef CAS.
- A. Raghavan and T. Ren, Organometallics, 2019, 38, 3888–3896 CrossRef CAS.
- C. Wang, G. Li and H. Guo, Catalysts, 2023, 13, 640 CrossRef CAS.
- J. L. Mancuso, C. A. Gaggioli, L. Gagliardi and C. H. Hendon, J. Phys. Chem. C, 2021, 125, 22036–22043 CrossRef CAS.
- C. Stegelmann, A. Andreasen and C. T. Campbell, J. Am. Chem. Soc., 2009, 131, 8077–8082 CrossRef CAS PubMed.
- B. Peters, Reaction Rate Theory and Rare Events, Elsevier, 2017 Search PubMed.
- A. H. Motagamwala and J. A. Dumesic, Chem. Rev., 2021, 121, 1049–1076 CrossRef CAS PubMed.
- Y. Homma and T. Ishida, Chem. Mater., 2018, 30, 1835–1838 CrossRef CAS.
- M. Dommaschk, C. Schutt, S. Venkataramani, U. Jana, C. Nather, F. D. Sonnichsen and R. Herges, Dalton Trans., 2014, 43, 17395–17405 RSC.
- H. Brandenburg, J. Krahmer, K. Fischer, B. Schwager, B. Flöser, C. Näther and F. Tuczek, Eur. J. Inorg. Chem., 2018, 576–585 CrossRef CAS.
- A. A. Farcas and A. Bende, Molecules, 2019, 24, 4249 CrossRef PubMed.
- R. J. Comito, E. D. Metzger, Z. Wu, G. Zhang, C. H. Hendon, J. T. Miller and M. Dincă, Organometallics, 2017, 36, 1681–1683 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Heterogeneous catalysts for ethylene dimerization; transition metals; stability and multiplicity tests; optimized M–M bond distances and NBO analysis; Gibbs energies at different multiplicities; frequencies and structures of transition states; coordinates. See DOI: https://doi.org/10.1039/d3cp06257k |
| This journal is © the Owner Societies 2024 |